User login
Alpha-1 antitrypsin deficiency is a common but underrecognized genetic condition that increases the risk of chronic obstructive pulmonary disease (COPD) and liver disease. Primary care providers can play a critical role in detecting it and managing patients who have it.
RECOGNIZED CASES ARE THE TIP OF THE ICEBERG
First described in 1963,1 alpha-1 antitrypsin deficiency is estimated to affect 100,000 Americans, fewer than 15,000 of whom have received a clinical diagnosis. As further evidence of its underrecognition,2–7 many patients experience long delays between their first symptoms and the diagnosis. Early studies indicated that the average diagnostic delay was 7.2 years,4 and the latest studies, as recent as 2013, indicate a similar diagnostic delay.7
Furthermore, many patients see multiple healthcare providers before receiving the correct diagnosis. A 1994 survey by this author4 found that 43.7% of patients who had severe deficiency of alpha-1 antitrypsin saw at least three physicians before the correct diagnosis was made.
Why is the disease underrecognized?
Several reasons may account for underrecognition of this disease. Many clinicians—including, unfortunately, many pulmonologists—do not know much about it,7,8 do not adhere to clinical guidelines,9,10 or harbor the misperception that there is no therapy available and, therefore, no compelling reason to make a diagnosis.7
Regarding inadequate knowledge, in a study by Taliercio, Chatburn, and this author,8 internal medicine residents scored only 63% correct on a 10-question quiz on diagnostic features of alpha-1 antitrypsin deficiency. There was no evidence of a training effect—senior residents scored no higher than interns.
Similarly, when Greulich et al7 surveyed German and Italian internists, general practitioners, and pulmonologists, one-fourth to one-half of them (depending on specialty and country) stated that they knew either very little or nothing at all about alpha-1 antitrypsin deficiency. In addition, 7% to 8% agreed with the statement, “There is no treatment available for this disease.”7
Nonadoption of clinical guidelines has been widely recognized in medicine and is evident in the failure to implement various recommended practices,9,10 such as low-stretch ventilation for acute respiratory distress syndrome and prophylaxis against deep vein thrombosis.
Finding the rest of the iceberg
Efforts to enhance compliance with guidelines on testing for alpha-1 antitrypsin deficiency have included using the electronic medical record to prompt physicians to test appropriate candidates.11–13
Jain et al13 examined the effect of installing such a prompting system to remind physicians to test for alpha-1 antitrypsin deficiency in patients with airflow obstruction that does not reverse with a bronchodilator—a recognized indication for testing for this disease according to standards endorsed by the American Thoracic Society and European Respiratory Society.14 At baseline, only 4.7% of appropriate candidates were being tested; after a prompt was installed in the electronic medical record, the rate rose to 15.1%, still a minority of candidates.
Another strategy is to empower respiratory therapists who perform pulmonary function tests to invite patients to be tested if their pulmonary function tests show postbronchodilator airflow obstruction. Rahaghi et al15 showed that using this strategy, 20 (0.63%) of 3,152 patients who were found to have fixed airflow obstruction when they underwent pulmonary function testing were newly diagnosed with severe deficiency of alpha-1 antitrypsin. Other targeted detection studies in patients with COPD estimated the prevalence of alpha-1 antitrypsin deficiency at up to 12%.3
PHYSIOLOGY AND PATHOPHYSIOLOGY OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Alpha-1 antitrypsin is a single-chain, 394-amino acid glycoprotein with three carbohydrate side chains found at asparagine residues along the primary structure.16
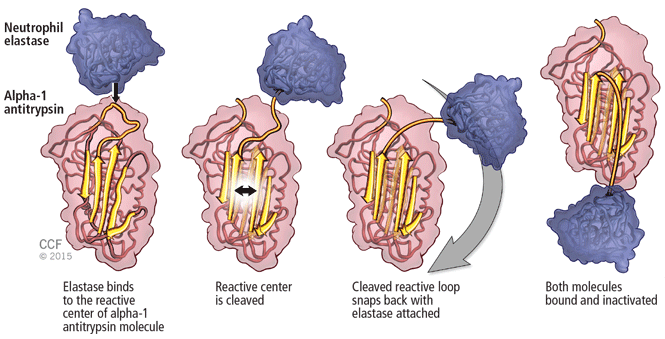
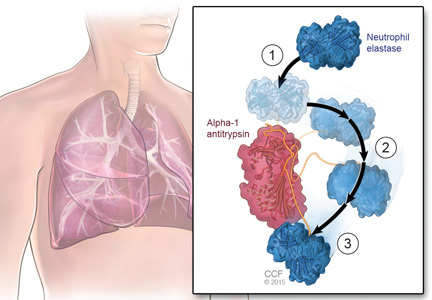
A major physiologic function of this molecule is to bind neutrophil elastase, which it does avidly. In a “mousetrap-like” mechanism,16 an active site on the alpha-1 antitrypsin molecule captures the neutrophil elastase and is cleaved, releasing steric energy in the molecule, catapulting the neutrophil elastase to the opposite side of the alpha-1 antitrypsin molecule, and inactivating it (Figure 1).
MM is normal, ZZ is not
Alpha-1 antitrypsin deficiency is inherited as an autosomal-codominant condition.17
The SERPINA1 gene, which codes for alpha-1 antitrypsin, is located on the long arm of the 14th chromosome, and more than 150 alleles of this gene have been identified to date. The normal allele is denoted M, and the allele most commonly associated with severe deficiency is denoted Z. People who are homozygous for the M allele (ie, normal) are called PI*MM (PI stands for “protease inhibitor”), and those who are homozygous for the Z allele are PI*ZZ. More than 90% of patients with severe alpha-1 antitrypsin deficiency are PI*ZZ.18
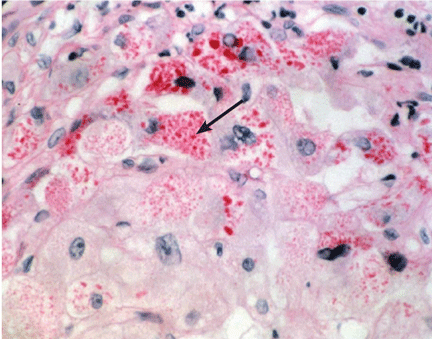
The Z allele has a single amino acid substitution (glutamic acid-to-lysine at position 342), which results in abnormal folding and formation of polymers of the Z molecule within hepatocytes.19,20 These polymers are recognized on liver biopsy as periodic acid-Schiff diastase-resistant eosinophilic inclusion bodies on histologic staining (Figure 2).
With alpha-1 antitrypsin trapped as Z-molecule polymers in the liver, the amount in the bloodstream falls, and there is a consequent decrease in the amount available in the lung to oppose the proteolytic burden of neutrophil elastase, especially in people who smoke or work in dusty environments.21
Tan et al22 have shown that some of the polymerized Z protein can escape the liver and circulate in the blood and that alveolar macrophages may also produce Z polymers. These Z polymers are chemotactic for neutrophils,23 so that their presence in the lung fuels the inflammatory cascade by recruiting more neutrophils to the lung, thereby increasing the proteolytic burden to the lung and increasing the risk of emphysema. Z monomers that do circulate can bind neutrophil elastase, but their binding avidity to neutrophil elastase is substantially lower than that of M-type alpha-1 antitrypsin.
CLINICAL MANIFESTATIONS
Alpha-1 antitrypsin deficiency of the PI*ZZ type is associated with two major clinical manifestations:
- Emphysema, resulting from the loss of proteolytic protection of the lung by alpha-1 antitrypsin (a toxic loss of function), and
- Liver diseases such as cirrhosis and chronic hepatitis, which result from abnormal accumulation of alpha-1 antitrypsin within hepatocytes (a toxic gain of function), and hepatoma.17
Other clinical manifestations of PI*ZZ alpha-1 antitrypsin deficiency include panniculitis and an association with cytoplasmic antineutrophil cytoplasmic antibody-positive vasculitis.17
Some uncertainty exists regarding the risk associated with the PI*MZ heterozygous state because there has been no systematic longitudinal study of people with this genotype. However, the weight of available experience suggests that PI*MZ individuals who have never smoked are not at increased risk of developing emphysema.24
Findings from a national registry: PI*ZZ COPD resembles ‘usual’ COPD
Distinguishing patients with alpha-1 antitrypsin deficiency from those with “usual” COPD (ie, without alpha-1 antitrypsin deficiency) can be difficult, as shown in data from the National Heart, Lung, and Blood Institute’s Alpha-1 Antitrypsin Deficiency Registry study.18 This multicenter, longitudinal, observational study contains the largest well-characterized cohort with severe deficiency of alpha-1 antitrypsin (PI*ZZ, PI*ZNull, etc), with 1,129 patients.
Pulmonary function test results were consistent with emphysema in most of the patients in the registry. Mean postbronchodilator pulmonary function values (± standard error of the mean) were:
- Forced expiratory volume in 1 second (FEV1) 46.7% of predicted (± 30%)
- Ratio of FEV1 to forced vital capacity 42.9% (± 20.4% )
- Mean diffusing capacity for carbon monoxide 50.3% of predicted (± 22.5%).
Like many patients with usual COPD, 60% of the registry patients demonstrated a component of airway reactivity, with significant reversal of airflow obstruction over three spirometries after receiving a dose of an inhaled bronchodilator (characterized by a 12% and 200-mL postbronchodilator rise in FEV1). Moreover, 78 patients had normal lung function.
Symptoms also resembled those in patients with usual emphysema, chronic bronchitis, or both. On enrollment in the registry, 83.9% of the patients had shortness of breath on exertion, 75.5% had wheezing with upper respiratory infections, 65.3% had wheezing without upper respiratory infection, 67.6% had recent debilitating chest illness, 42.4% had “usual” cough, and 49.6% had annual cough and phlegm episodes.
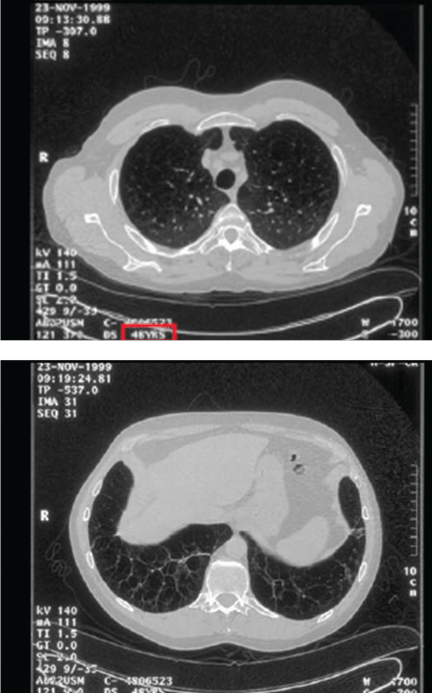
Imaging findings. Although the classic teaching is that emphysema due to alpha-1 antitrypsin deficiency produces lower-lobe hyperlucency on plain films, relying on this sign would lead to underrecognition, as 36% of PI*ZZ patients have apical-predominant emphysema on chest computed tomography,24 which resembles the usual centriacinar emphysema pattern. Figure 3 shows axial computed tomographic scans through the apices and the bases of the lungs of a patient with alpha-1 antitrypsin deficiency.

In view of these difficulties, guidelines from the American Thoracic Society and European Respiratory Society14 endorse testing for alpha-1 antitrypsin deficiency in all adults who have symptoms and fixed airflow obstruction (Table 1).
CONSEQUENCES OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Two large screening studies2,3,25,26 followed people who were identified at birth as having alpha-1 antitrypsin deficiency to examine the natural course of the disease.
The larger of the two studies27 tested 200,000 Swedish newborns. Follow-up of this cohort to age 35 indicated that 35-year-old never-smoking PI*ZZ individuals have normal lung function and no excess emphysema on computed tomography compared with normal peers matched for age and sex.27 In contrast, the few PI*ZZ ever-smokers demonstrated a lower level of transfer factor and significantly more emphysema on computed tomography than normal (PI*MM) never-smokers.
Faster decline in lung function
Data from the National Heart, Lung, and Blood Institute registry indicate that, on average, people with severe alpha-1 antitrypsin deficiency lose lung function faster than people without the disease.28 Specifically, in never-smokers in the registry, the average rate of FEV1 decline was 67 mL/year, and among ex-smokers, it was 54 mL/year. Both of these values exceed the general age-related rate of FEV1 decline of approximately 20 to 25 mL/year in never-smoking, normal adults. Among current smokers in the registry with severe alpha-1 antitrypsin deficiency, the rate of FEV1 decline was 109 mL/year.
Rates of FEV1 decline over time vary among groups with differing degrees of airflow obstruction. For example, PI*ZZ patients with moderate COPD (stage II of the four-stage Global Initiative for Chronic Obstructive Lung Disease classification system) lose lung function faster than patients with either milder or more severe degrees of airflow obstruction.29
As with COPD in general, exacerbations of COPD in people with severe deficiency of alpha-1 antitrypsin are associated with worsened clinical status. In one series,30 54% of 265 PI*ZZ patients experienced an exacerbation in the first year of follow-up, and 18% experienced at least three. Such exacerbations occurred in December and January in 32% of these individuals, likely due to a viral precipitant.
Increased mortality
Severe deficiency of alpha-1 antitrypsin is associated not only with severe morbidity but also death. In the national registry, the overall rate of death was 18.6% at 5 years of follow-up, or approximately 3% per year.28
A low FEV1 at entry was a bad sign. Patients entering the registry with FEV1 values below 15% of predicted had a 36% mortality rate at 3 years, compared with 2.6% in those whose baseline FEV1 exceeded 50% of predicted.
Underlying causes of death in registry participants included emphysema (accounting for 72% of deaths) and cirrhosis (10%),31 which were the only causes of death more frequent than in age- and sex-matched controls. In a series of never-smokers who had PI*ZZ alpha-1 antitrypsin deficiency,32 death was less frequently attributed to emphysema than in the national registry (46%) and more often attributed to cirrhosis (28%), indicating that never-smokers may more frequently escape the ravages of emphysema but experience a higher rate of developing cirrhosis later in life.33
DIAGNOSING ALPHA-1 ANTITRYPSIN DEFICIENCY
Available blood tests for alpha-1 antitrypsin deficiency include:
The serum alpha-1 antitrypsin level, most often done by nephelometry. Normal serum levels generally range from 100 to 220 mg/dL.
Phenotyping, usually performed by isoelectric focusing, which can identify different band patterns associated with different alleles.
Genotyping involves determining which alpha-1 antitrypsin alleles are present, most often using polymerase chain reaction testing targeting the S and Z alleles and occasionally set up to detect less common alleles such as F and I.17
Gene sequencing is occasionally necessary to achieve an accurate, definitive diagnosis.
Free, confidential testing is available
Clinical testing most often involves checking both a serum level and a phenotype or genotype. Such tests are often available in hospital laboratories and commercial laboratories, with testing also facilitated by the availability of free testing kits from several manufacturers of drugs for alpha-1 antitrypsin deficiency.
The Alpha-1 Foundation (www.alpha1.org)34 also offers a free, home-based confidential testing kit through a research protocol at the Medical University of South Carolina (alphaone@musc.edu) called the Alpha-1 Coded Testing (ACT) study. Patients can receive a kit and lancet at home, submit the dried blood-spot specimen, and receive in the mail a confidential serum level and genotype.
The availability of such home-based confidential testing allows patients to seek testing without a physician’s order and makes it easier for facilitated allied health providers, such as respiratory therapists, to recommend testing in appropriate clinical circumstances.15
TREATMENT OF ALPHA-1 ANTITRYPSIN DEFICIENCY
The treatment of patients with severe deficiency of alpha-1 antitrypsin and emphysema generally resembles that of patients with usual COPD. Specifically, smoking cessation, bronchodilators, occasionally inhaled steroids, supplemental oxygen, preventive vaccinations, and pulmonary rehabilitation are indicated as per usual clinical assessment.
Lung volume reduction surgery, which is beneficial in appropriate subsets of COPD patients, is generally less effective in those with severe alpha-1 antitrypsin deficiency,35 specifically because the magnitude of FEV1 increase and the duration of such a rise are lower than in usual COPD patients.
Augmentation therapy
Specific therapy for alpha-1 antitrypsin deficiency currently involves weekly intravenous infusions of purified, pooled human-plasma-derived alpha-1 antitrypsin, so-called augmentation therapy. Four drugs have been approved for use in the United States:
- Prolastin-C (Grifols, Barcelona, Spain)
- Aralast NP (Baxalta, Bonneckborn, IL)
- Zemaira (CSL Behring, King of Prussia, PA)
- Glassia (Baxalta, Bonneckborn, IL, and Kamada, Ness Ziona, Israel).
All of these were approved for use in the United States on the basis of biochemical efficacy. Specifically, infusion of these drugs has been shown to raise serum levels above a protective threshold value (generally considered 57 mg/dL, the value below which the risk of developing emphysema increases beyond normal).
Randomized controlled trials36,37 have addressed the efficacy of intravenous augmentation therapy, and although no single trial has been definitive, the weight of evidence shows that augmentation therapy can slow the progression of emphysema. For example, in a study by Dirksen et al,37 augmentation therapy was associated with a slower progression of emphysema as assessed by the rate of loss of lung density on computed tomography.
On the basis of the available evidence, the American Thoracic Society and European Respiratory Society14 have recommended augmentation therapy in individuals with “established airflow obstruction from alpha-1 antitrypsin deficiency.”14 Their guidelines go on to say that the evidence that augmentation therapy is beneficial “is stronger for individuals with moderate airflow obstruction (eg, FEV1 35%–60% of predicted) than for those with severe airflow obstruction. Augmentation therapy is not currently recommended for individuals without emphysema.”
The guidelines recognize that although augmentation therapy does not satisfy the usual criteria for cost-effectiveness (< $50,000 per quality-adjusted life year) due to its high cost (approximately $100,000 per year if paid for out of pocket),38 it is recommended for appropriate candidates because it is the only available specific therapy for severe deficiency of alpha-1 antitrypsin.
Novel therapies
In addition to current treatment approaches of augmentation therapy, a number of novel treatment strategies are being investigated, several of which hold much promise.
Gene therapy, using adeno-associated virus to transfect the normal human gene into individuals with severe deficiency of alpha-1 antitrypsin, has been undertaken and is currently under study. In addition, a variety of approaches to interdict production of abnormal Z protein from the liver are being examined, as well as inhaled hyaluronic acid to protect the lung.
- Laurell C, Eriksson A. The electrophoretic alpha-1 globulin pattern of serum in alpha-1 antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15:132–140.
- Aboussouan LS, Stoller JK. Detection of alpha-1 antitrypsin deficiency: a review. Respir Med 2009; 103:335–341.
- Stoller JK, Brantly M. The challenge of detecting alpha-1 antitrypsin deficiency. COPD 2013; 10(suppl 1):26–34.
- Stoller JK, Smith P, Yang P, Spray J. Physical and social impact of alpha 1-antitrypsin deficiency: results of a survey. Cleve Clin J Med 1994; 61:461–467.
- Stoller JK, Sandhaus RA, Turino G, Dickson R, Rodgers K, Strange C. Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest 2005; 128:1989–1994.
- Campos MA, Wanner A, Zhang G, Sandhaus RA. Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest 2005; 128:1179–1186.
- Greulich T, Ottaviani S, Bals R, et al. Alpha1-antitrypsin deficiency—diagnostic testing and disease awareness in Germany and Italy. Respir Med 2013; 107:1400–1408.
- Taliercio RM, Chatburn RL, Stoller JK. Knowledge of alpha-1 antitrypsin deficiency among internal medicine house officers and respiratory therapists: results of a survey. Respir Care 2010; 55:322–327.
- Rubenfeld GD, Cooper C, Carter G, Thompson BT, Hudson LD. Barriers to providing lung-protective ventilation to patients with acute lung injury. Crit Care Med 2004; 32:1289–1293.
- Cabana MD, Rand CS, Powe NR, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA 1999; 282:1458–1465.
- Rahaghi F, Ortega I, Rahaghi N, et al. Physician alert suggesting alpha-1 antitrypsin deficiency testing in pulmonary function test (PFT) results. COPD 2009; 6:26–30.
- Campos M, Hagenlocker B, Martinez N, et al. Impact of an electronic medical record clinical reminder to improve detection of COPD and alpha-1 antitrypsin deficiency in the Veterans Administration (VA) system (abstract). Am J Respir Crit Care Med 2011;183:A5356. www.atsjournals.org/doi/pdf/10.1164/ajrccm-conference.2011.183.1_MeetingAbstracts.A5356. Accessed May 24, 2016.
- Jain A, McCarthy K, Xu M, Stoller JK. Impact of a clinical decision support system in an electronic health record to enhance detection of alpha(1)-antitrypsin deficiency. Chest 2011;140:198–204.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Rahaghi FF, Sandhaus RA, Brantly ML, et al. The prevalence of alpha-1 antitrypsin deficiency among patients found to have airflow obstruction. COPD 2012; 9:352–358.
- Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency—a model for conformational diseases. N Engl J Med 2002; 346:45–53.
- Stoller JK, Aboussouan LS. A review of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2012; 185:246–259.
- McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of Alpha 1-Antitrypsin Deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Chest 1997; 111:394–403.
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992; 357:605–607.
- Lomas DA, Finch JT, Seyama K, Nukiwa T, Carrell RW. Alpha 1-antitrypsin Siiyama (Ser53-->Phe). Further evidence for intracellular loop-sheet polymerization. J Biol Chem 1993; 268:15333–15335.
- Mayer AS, Stoller JK, Bucher Bartelson B, James Ruttenber A, Sandhaus RA, Newman LS. Occupational exposure risks in individuals with PI*Z alpha(1)-antitrypsin deficiency. Am J Respir Crit Care Med 2000; 162:553–558.
- Tan L, Dickens JA, Demeo DL, et al. Circulating polymers in alpha1-antitrypsin deficiency. Eur Respir J 2014; 43:1501–1504.
- Parmar JS, Mahadeva R, Reed BJ, et al. Polymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysema. Am J Respir Cell Mol Biol 2002; 26:723–730.
- Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in alpha1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med 2014; 189:419–427.
- Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med 2004; 170:1172–1178.
- Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976; 294:1316–1321.
- O’Brien ML, Buist NR, Murphey WH. Neonatal screening for alpha1-antitrypsin deficiency. J Pediatr 1978; 92:1006–1010.
- Piitulainen E, Montero LC, Nystedt-Duzakin M, et al. Lung function and CT densitometry in subjects with alpha-1-antitrypsin deficiency and healthy controls at 35 years of age. COPD 2015; 12:162–167.
- The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Respir Crit Care Med 1998; 158:49–59.
- Dawkins PA, Dawkins CL, Wood AM, Nightingale PG, Stockley JA, Stockley RA. Rate of progression of lung function impairment in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1338–1344.
- Needham M, Stockley RA. Alpha 1-antitrypsin deficiency. 3: clinical manifestations and natural history. Thorax 2004; 59:441–445.
- Tomashefski JF Jr, Crystal RG, Wiedemann HP, Mascha E, Stoller JK. The bronchopulmonary pathology of alpha-1 antitrypsin (AAT) deficiency: findings of the Death Review Committee of the National Registry for Individuals with Severe Deficiency of Alpha-1 Antitrypsin. Hum Pathol 2004; 35:1452–1461.
- Tanash HA, Nilsson PM, Nilsson JA, Piitulainen E. Clinical course and prognosis of never-smokers with severe alpha-1-antitrypsin deficiency (PiZZ). Thorax 2008; 63:1091–1095.
- Walsh JW, Snider GL, Stoller JK. A review of the Alpha-1 Foundation: its formation, impact, and critical success factors. Respir Care 2006; 51:526–531.
- Rokadia HK, Stoller JK. Surgical and bronchoscopic lung volume reduction treatment for a-1 antitrypsin deficiency. Clin Pulm Med 2015; 22:279–285.
- Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1345–1353.
- Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160:1468–1472.
- Gildea TR, Shermock KM, Singer ME, Stoller JK. Cost-effectiveness analysis of augmentation therapy for severe alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2003; 167:1387–1392.
Alpha-1 antitrypsin deficiency is a common but underrecognized genetic condition that increases the risk of chronic obstructive pulmonary disease (COPD) and liver disease. Primary care providers can play a critical role in detecting it and managing patients who have it.
RECOGNIZED CASES ARE THE TIP OF THE ICEBERG
First described in 1963,1 alpha-1 antitrypsin deficiency is estimated to affect 100,000 Americans, fewer than 15,000 of whom have received a clinical diagnosis. As further evidence of its underrecognition,2–7 many patients experience long delays between their first symptoms and the diagnosis. Early studies indicated that the average diagnostic delay was 7.2 years,4 and the latest studies, as recent as 2013, indicate a similar diagnostic delay.7
Furthermore, many patients see multiple healthcare providers before receiving the correct diagnosis. A 1994 survey by this author4 found that 43.7% of patients who had severe deficiency of alpha-1 antitrypsin saw at least three physicians before the correct diagnosis was made.
Why is the disease underrecognized?
Several reasons may account for underrecognition of this disease. Many clinicians—including, unfortunately, many pulmonologists—do not know much about it,7,8 do not adhere to clinical guidelines,9,10 or harbor the misperception that there is no therapy available and, therefore, no compelling reason to make a diagnosis.7
Regarding inadequate knowledge, in a study by Taliercio, Chatburn, and this author,8 internal medicine residents scored only 63% correct on a 10-question quiz on diagnostic features of alpha-1 antitrypsin deficiency. There was no evidence of a training effect—senior residents scored no higher than interns.
Similarly, when Greulich et al7 surveyed German and Italian internists, general practitioners, and pulmonologists, one-fourth to one-half of them (depending on specialty and country) stated that they knew either very little or nothing at all about alpha-1 antitrypsin deficiency. In addition, 7% to 8% agreed with the statement, “There is no treatment available for this disease.”7
Nonadoption of clinical guidelines has been widely recognized in medicine and is evident in the failure to implement various recommended practices,9,10 such as low-stretch ventilation for acute respiratory distress syndrome and prophylaxis against deep vein thrombosis.
Finding the rest of the iceberg
Efforts to enhance compliance with guidelines on testing for alpha-1 antitrypsin deficiency have included using the electronic medical record to prompt physicians to test appropriate candidates.11–13
Jain et al13 examined the effect of installing such a prompting system to remind physicians to test for alpha-1 antitrypsin deficiency in patients with airflow obstruction that does not reverse with a bronchodilator—a recognized indication for testing for this disease according to standards endorsed by the American Thoracic Society and European Respiratory Society.14 At baseline, only 4.7% of appropriate candidates were being tested; after a prompt was installed in the electronic medical record, the rate rose to 15.1%, still a minority of candidates.
Another strategy is to empower respiratory therapists who perform pulmonary function tests to invite patients to be tested if their pulmonary function tests show postbronchodilator airflow obstruction. Rahaghi et al15 showed that using this strategy, 20 (0.63%) of 3,152 patients who were found to have fixed airflow obstruction when they underwent pulmonary function testing were newly diagnosed with severe deficiency of alpha-1 antitrypsin. Other targeted detection studies in patients with COPD estimated the prevalence of alpha-1 antitrypsin deficiency at up to 12%.3
PHYSIOLOGY AND PATHOPHYSIOLOGY OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Alpha-1 antitrypsin is a single-chain, 394-amino acid glycoprotein with three carbohydrate side chains found at asparagine residues along the primary structure.16
A major physiologic function of this molecule is to bind neutrophil elastase, which it does avidly. In a “mousetrap-like” mechanism,16 an active site on the alpha-1 antitrypsin molecule captures the neutrophil elastase and is cleaved, releasing steric energy in the molecule, catapulting the neutrophil elastase to the opposite side of the alpha-1 antitrypsin molecule, and inactivating it (Figure 1).
MM is normal, ZZ is not
Alpha-1 antitrypsin deficiency is inherited as an autosomal-codominant condition.17
The SERPINA1 gene, which codes for alpha-1 antitrypsin, is located on the long arm of the 14th chromosome, and more than 150 alleles of this gene have been identified to date. The normal allele is denoted M, and the allele most commonly associated with severe deficiency is denoted Z. People who are homozygous for the M allele (ie, normal) are called PI*MM (PI stands for “protease inhibitor”), and those who are homozygous for the Z allele are PI*ZZ. More than 90% of patients with severe alpha-1 antitrypsin deficiency are PI*ZZ.18
The Z allele has a single amino acid substitution (glutamic acid-to-lysine at position 342), which results in abnormal folding and formation of polymers of the Z molecule within hepatocytes.19,20 These polymers are recognized on liver biopsy as periodic acid-Schiff diastase-resistant eosinophilic inclusion bodies on histologic staining (Figure 2).
With alpha-1 antitrypsin trapped as Z-molecule polymers in the liver, the amount in the bloodstream falls, and there is a consequent decrease in the amount available in the lung to oppose the proteolytic burden of neutrophil elastase, especially in people who smoke or work in dusty environments.21
Tan et al22 have shown that some of the polymerized Z protein can escape the liver and circulate in the blood and that alveolar macrophages may also produce Z polymers. These Z polymers are chemotactic for neutrophils,23 so that their presence in the lung fuels the inflammatory cascade by recruiting more neutrophils to the lung, thereby increasing the proteolytic burden to the lung and increasing the risk of emphysema. Z monomers that do circulate can bind neutrophil elastase, but their binding avidity to neutrophil elastase is substantially lower than that of M-type alpha-1 antitrypsin.
CLINICAL MANIFESTATIONS
Alpha-1 antitrypsin deficiency of the PI*ZZ type is associated with two major clinical manifestations:
- Emphysema, resulting from the loss of proteolytic protection of the lung by alpha-1 antitrypsin (a toxic loss of function), and
- Liver diseases such as cirrhosis and chronic hepatitis, which result from abnormal accumulation of alpha-1 antitrypsin within hepatocytes (a toxic gain of function), and hepatoma.17
Other clinical manifestations of PI*ZZ alpha-1 antitrypsin deficiency include panniculitis and an association with cytoplasmic antineutrophil cytoplasmic antibody-positive vasculitis.17
Some uncertainty exists regarding the risk associated with the PI*MZ heterozygous state because there has been no systematic longitudinal study of people with this genotype. However, the weight of available experience suggests that PI*MZ individuals who have never smoked are not at increased risk of developing emphysema.24
Findings from a national registry: PI*ZZ COPD resembles ‘usual’ COPD
Distinguishing patients with alpha-1 antitrypsin deficiency from those with “usual” COPD (ie, without alpha-1 antitrypsin deficiency) can be difficult, as shown in data from the National Heart, Lung, and Blood Institute’s Alpha-1 Antitrypsin Deficiency Registry study.18 This multicenter, longitudinal, observational study contains the largest well-characterized cohort with severe deficiency of alpha-1 antitrypsin (PI*ZZ, PI*ZNull, etc), with 1,129 patients.
Pulmonary function test results were consistent with emphysema in most of the patients in the registry. Mean postbronchodilator pulmonary function values (± standard error of the mean) were:
- Forced expiratory volume in 1 second (FEV1) 46.7% of predicted (± 30%)
- Ratio of FEV1 to forced vital capacity 42.9% (± 20.4% )
- Mean diffusing capacity for carbon monoxide 50.3% of predicted (± 22.5%).
Like many patients with usual COPD, 60% of the registry patients demonstrated a component of airway reactivity, with significant reversal of airflow obstruction over three spirometries after receiving a dose of an inhaled bronchodilator (characterized by a 12% and 200-mL postbronchodilator rise in FEV1). Moreover, 78 patients had normal lung function.
Symptoms also resembled those in patients with usual emphysema, chronic bronchitis, or both. On enrollment in the registry, 83.9% of the patients had shortness of breath on exertion, 75.5% had wheezing with upper respiratory infections, 65.3% had wheezing without upper respiratory infection, 67.6% had recent debilitating chest illness, 42.4% had “usual” cough, and 49.6% had annual cough and phlegm episodes.
Imaging findings. Although the classic teaching is that emphysema due to alpha-1 antitrypsin deficiency produces lower-lobe hyperlucency on plain films, relying on this sign would lead to underrecognition, as 36% of PI*ZZ patients have apical-predominant emphysema on chest computed tomography,24 which resembles the usual centriacinar emphysema pattern. Figure 3 shows axial computed tomographic scans through the apices and the bases of the lungs of a patient with alpha-1 antitrypsin deficiency.
In view of these difficulties, guidelines from the American Thoracic Society and European Respiratory Society14 endorse testing for alpha-1 antitrypsin deficiency in all adults who have symptoms and fixed airflow obstruction (Table 1).
CONSEQUENCES OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Two large screening studies2,3,25,26 followed people who were identified at birth as having alpha-1 antitrypsin deficiency to examine the natural course of the disease.
The larger of the two studies27 tested 200,000 Swedish newborns. Follow-up of this cohort to age 35 indicated that 35-year-old never-smoking PI*ZZ individuals have normal lung function and no excess emphysema on computed tomography compared with normal peers matched for age and sex.27 In contrast, the few PI*ZZ ever-smokers demonstrated a lower level of transfer factor and significantly more emphysema on computed tomography than normal (PI*MM) never-smokers.
Faster decline in lung function
Data from the National Heart, Lung, and Blood Institute registry indicate that, on average, people with severe alpha-1 antitrypsin deficiency lose lung function faster than people without the disease.28 Specifically, in never-smokers in the registry, the average rate of FEV1 decline was 67 mL/year, and among ex-smokers, it was 54 mL/year. Both of these values exceed the general age-related rate of FEV1 decline of approximately 20 to 25 mL/year in never-smoking, normal adults. Among current smokers in the registry with severe alpha-1 antitrypsin deficiency, the rate of FEV1 decline was 109 mL/year.
Rates of FEV1 decline over time vary among groups with differing degrees of airflow obstruction. For example, PI*ZZ patients with moderate COPD (stage II of the four-stage Global Initiative for Chronic Obstructive Lung Disease classification system) lose lung function faster than patients with either milder or more severe degrees of airflow obstruction.29
As with COPD in general, exacerbations of COPD in people with severe deficiency of alpha-1 antitrypsin are associated with worsened clinical status. In one series,30 54% of 265 PI*ZZ patients experienced an exacerbation in the first year of follow-up, and 18% experienced at least three. Such exacerbations occurred in December and January in 32% of these individuals, likely due to a viral precipitant.
Increased mortality
Severe deficiency of alpha-1 antitrypsin is associated not only with severe morbidity but also death. In the national registry, the overall rate of death was 18.6% at 5 years of follow-up, or approximately 3% per year.28
A low FEV1 at entry was a bad sign. Patients entering the registry with FEV1 values below 15% of predicted had a 36% mortality rate at 3 years, compared with 2.6% in those whose baseline FEV1 exceeded 50% of predicted.
Underlying causes of death in registry participants included emphysema (accounting for 72% of deaths) and cirrhosis (10%),31 which were the only causes of death more frequent than in age- and sex-matched controls. In a series of never-smokers who had PI*ZZ alpha-1 antitrypsin deficiency,32 death was less frequently attributed to emphysema than in the national registry (46%) and more often attributed to cirrhosis (28%), indicating that never-smokers may more frequently escape the ravages of emphysema but experience a higher rate of developing cirrhosis later in life.33
DIAGNOSING ALPHA-1 ANTITRYPSIN DEFICIENCY
Available blood tests for alpha-1 antitrypsin deficiency include:
The serum alpha-1 antitrypsin level, most often done by nephelometry. Normal serum levels generally range from 100 to 220 mg/dL.
Phenotyping, usually performed by isoelectric focusing, which can identify different band patterns associated with different alleles.
Genotyping involves determining which alpha-1 antitrypsin alleles are present, most often using polymerase chain reaction testing targeting the S and Z alleles and occasionally set up to detect less common alleles such as F and I.17
Gene sequencing is occasionally necessary to achieve an accurate, definitive diagnosis.
Free, confidential testing is available
Clinical testing most often involves checking both a serum level and a phenotype or genotype. Such tests are often available in hospital laboratories and commercial laboratories, with testing also facilitated by the availability of free testing kits from several manufacturers of drugs for alpha-1 antitrypsin deficiency.
The Alpha-1 Foundation (www.alpha1.org)34 also offers a free, home-based confidential testing kit through a research protocol at the Medical University of South Carolina (alphaone@musc.edu) called the Alpha-1 Coded Testing (ACT) study. Patients can receive a kit and lancet at home, submit the dried blood-spot specimen, and receive in the mail a confidential serum level and genotype.
The availability of such home-based confidential testing allows patients to seek testing without a physician’s order and makes it easier for facilitated allied health providers, such as respiratory therapists, to recommend testing in appropriate clinical circumstances.15
TREATMENT OF ALPHA-1 ANTITRYPSIN DEFICIENCY
The treatment of patients with severe deficiency of alpha-1 antitrypsin and emphysema generally resembles that of patients with usual COPD. Specifically, smoking cessation, bronchodilators, occasionally inhaled steroids, supplemental oxygen, preventive vaccinations, and pulmonary rehabilitation are indicated as per usual clinical assessment.
Lung volume reduction surgery, which is beneficial in appropriate subsets of COPD patients, is generally less effective in those with severe alpha-1 antitrypsin deficiency,35 specifically because the magnitude of FEV1 increase and the duration of such a rise are lower than in usual COPD patients.
Augmentation therapy
Specific therapy for alpha-1 antitrypsin deficiency currently involves weekly intravenous infusions of purified, pooled human-plasma-derived alpha-1 antitrypsin, so-called augmentation therapy. Four drugs have been approved for use in the United States:
- Prolastin-C (Grifols, Barcelona, Spain)
- Aralast NP (Baxalta, Bonneckborn, IL)
- Zemaira (CSL Behring, King of Prussia, PA)
- Glassia (Baxalta, Bonneckborn, IL, and Kamada, Ness Ziona, Israel).
All of these were approved for use in the United States on the basis of biochemical efficacy. Specifically, infusion of these drugs has been shown to raise serum levels above a protective threshold value (generally considered 57 mg/dL, the value below which the risk of developing emphysema increases beyond normal).
Randomized controlled trials36,37 have addressed the efficacy of intravenous augmentation therapy, and although no single trial has been definitive, the weight of evidence shows that augmentation therapy can slow the progression of emphysema. For example, in a study by Dirksen et al,37 augmentation therapy was associated with a slower progression of emphysema as assessed by the rate of loss of lung density on computed tomography.
On the basis of the available evidence, the American Thoracic Society and European Respiratory Society14 have recommended augmentation therapy in individuals with “established airflow obstruction from alpha-1 antitrypsin deficiency.”14 Their guidelines go on to say that the evidence that augmentation therapy is beneficial “is stronger for individuals with moderate airflow obstruction (eg, FEV1 35%–60% of predicted) than for those with severe airflow obstruction. Augmentation therapy is not currently recommended for individuals without emphysema.”
The guidelines recognize that although augmentation therapy does not satisfy the usual criteria for cost-effectiveness (< $50,000 per quality-adjusted life year) due to its high cost (approximately $100,000 per year if paid for out of pocket),38 it is recommended for appropriate candidates because it is the only available specific therapy for severe deficiency of alpha-1 antitrypsin.
Novel therapies
In addition to current treatment approaches of augmentation therapy, a number of novel treatment strategies are being investigated, several of which hold much promise.
Gene therapy, using adeno-associated virus to transfect the normal human gene into individuals with severe deficiency of alpha-1 antitrypsin, has been undertaken and is currently under study. In addition, a variety of approaches to interdict production of abnormal Z protein from the liver are being examined, as well as inhaled hyaluronic acid to protect the lung.
Alpha-1 antitrypsin deficiency is a common but underrecognized genetic condition that increases the risk of chronic obstructive pulmonary disease (COPD) and liver disease. Primary care providers can play a critical role in detecting it and managing patients who have it.
RECOGNIZED CASES ARE THE TIP OF THE ICEBERG
First described in 1963,1 alpha-1 antitrypsin deficiency is estimated to affect 100,000 Americans, fewer than 15,000 of whom have received a clinical diagnosis. As further evidence of its underrecognition,2–7 many patients experience long delays between their first symptoms and the diagnosis. Early studies indicated that the average diagnostic delay was 7.2 years,4 and the latest studies, as recent as 2013, indicate a similar diagnostic delay.7
Furthermore, many patients see multiple healthcare providers before receiving the correct diagnosis. A 1994 survey by this author4 found that 43.7% of patients who had severe deficiency of alpha-1 antitrypsin saw at least three physicians before the correct diagnosis was made.
Why is the disease underrecognized?
Several reasons may account for underrecognition of this disease. Many clinicians—including, unfortunately, many pulmonologists—do not know much about it,7,8 do not adhere to clinical guidelines,9,10 or harbor the misperception that there is no therapy available and, therefore, no compelling reason to make a diagnosis.7
Regarding inadequate knowledge, in a study by Taliercio, Chatburn, and this author,8 internal medicine residents scored only 63% correct on a 10-question quiz on diagnostic features of alpha-1 antitrypsin deficiency. There was no evidence of a training effect—senior residents scored no higher than interns.
Similarly, when Greulich et al7 surveyed German and Italian internists, general practitioners, and pulmonologists, one-fourth to one-half of them (depending on specialty and country) stated that they knew either very little or nothing at all about alpha-1 antitrypsin deficiency. In addition, 7% to 8% agreed with the statement, “There is no treatment available for this disease.”7
Nonadoption of clinical guidelines has been widely recognized in medicine and is evident in the failure to implement various recommended practices,9,10 such as low-stretch ventilation for acute respiratory distress syndrome and prophylaxis against deep vein thrombosis.
Finding the rest of the iceberg
Efforts to enhance compliance with guidelines on testing for alpha-1 antitrypsin deficiency have included using the electronic medical record to prompt physicians to test appropriate candidates.11–13
Jain et al13 examined the effect of installing such a prompting system to remind physicians to test for alpha-1 antitrypsin deficiency in patients with airflow obstruction that does not reverse with a bronchodilator—a recognized indication for testing for this disease according to standards endorsed by the American Thoracic Society and European Respiratory Society.14 At baseline, only 4.7% of appropriate candidates were being tested; after a prompt was installed in the electronic medical record, the rate rose to 15.1%, still a minority of candidates.
Another strategy is to empower respiratory therapists who perform pulmonary function tests to invite patients to be tested if their pulmonary function tests show postbronchodilator airflow obstruction. Rahaghi et al15 showed that using this strategy, 20 (0.63%) of 3,152 patients who were found to have fixed airflow obstruction when they underwent pulmonary function testing were newly diagnosed with severe deficiency of alpha-1 antitrypsin. Other targeted detection studies in patients with COPD estimated the prevalence of alpha-1 antitrypsin deficiency at up to 12%.3
PHYSIOLOGY AND PATHOPHYSIOLOGY OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Alpha-1 antitrypsin is a single-chain, 394-amino acid glycoprotein with three carbohydrate side chains found at asparagine residues along the primary structure.16
A major physiologic function of this molecule is to bind neutrophil elastase, which it does avidly. In a “mousetrap-like” mechanism,16 an active site on the alpha-1 antitrypsin molecule captures the neutrophil elastase and is cleaved, releasing steric energy in the molecule, catapulting the neutrophil elastase to the opposite side of the alpha-1 antitrypsin molecule, and inactivating it (Figure 1).
MM is normal, ZZ is not
Alpha-1 antitrypsin deficiency is inherited as an autosomal-codominant condition.17
The SERPINA1 gene, which codes for alpha-1 antitrypsin, is located on the long arm of the 14th chromosome, and more than 150 alleles of this gene have been identified to date. The normal allele is denoted M, and the allele most commonly associated with severe deficiency is denoted Z. People who are homozygous for the M allele (ie, normal) are called PI*MM (PI stands for “protease inhibitor”), and those who are homozygous for the Z allele are PI*ZZ. More than 90% of patients with severe alpha-1 antitrypsin deficiency are PI*ZZ.18
The Z allele has a single amino acid substitution (glutamic acid-to-lysine at position 342), which results in abnormal folding and formation of polymers of the Z molecule within hepatocytes.19,20 These polymers are recognized on liver biopsy as periodic acid-Schiff diastase-resistant eosinophilic inclusion bodies on histologic staining (Figure 2).
With alpha-1 antitrypsin trapped as Z-molecule polymers in the liver, the amount in the bloodstream falls, and there is a consequent decrease in the amount available in the lung to oppose the proteolytic burden of neutrophil elastase, especially in people who smoke or work in dusty environments.21
Tan et al22 have shown that some of the polymerized Z protein can escape the liver and circulate in the blood and that alveolar macrophages may also produce Z polymers. These Z polymers are chemotactic for neutrophils,23 so that their presence in the lung fuels the inflammatory cascade by recruiting more neutrophils to the lung, thereby increasing the proteolytic burden to the lung and increasing the risk of emphysema. Z monomers that do circulate can bind neutrophil elastase, but their binding avidity to neutrophil elastase is substantially lower than that of M-type alpha-1 antitrypsin.
CLINICAL MANIFESTATIONS
Alpha-1 antitrypsin deficiency of the PI*ZZ type is associated with two major clinical manifestations:
- Emphysema, resulting from the loss of proteolytic protection of the lung by alpha-1 antitrypsin (a toxic loss of function), and
- Liver diseases such as cirrhosis and chronic hepatitis, which result from abnormal accumulation of alpha-1 antitrypsin within hepatocytes (a toxic gain of function), and hepatoma.17
Other clinical manifestations of PI*ZZ alpha-1 antitrypsin deficiency include panniculitis and an association with cytoplasmic antineutrophil cytoplasmic antibody-positive vasculitis.17
Some uncertainty exists regarding the risk associated with the PI*MZ heterozygous state because there has been no systematic longitudinal study of people with this genotype. However, the weight of available experience suggests that PI*MZ individuals who have never smoked are not at increased risk of developing emphysema.24
Findings from a national registry: PI*ZZ COPD resembles ‘usual’ COPD
Distinguishing patients with alpha-1 antitrypsin deficiency from those with “usual” COPD (ie, without alpha-1 antitrypsin deficiency) can be difficult, as shown in data from the National Heart, Lung, and Blood Institute’s Alpha-1 Antitrypsin Deficiency Registry study.18 This multicenter, longitudinal, observational study contains the largest well-characterized cohort with severe deficiency of alpha-1 antitrypsin (PI*ZZ, PI*ZNull, etc), with 1,129 patients.
Pulmonary function test results were consistent with emphysema in most of the patients in the registry. Mean postbronchodilator pulmonary function values (± standard error of the mean) were:
- Forced expiratory volume in 1 second (FEV1) 46.7% of predicted (± 30%)
- Ratio of FEV1 to forced vital capacity 42.9% (± 20.4% )
- Mean diffusing capacity for carbon monoxide 50.3% of predicted (± 22.5%).
Like many patients with usual COPD, 60% of the registry patients demonstrated a component of airway reactivity, with significant reversal of airflow obstruction over three spirometries after receiving a dose of an inhaled bronchodilator (characterized by a 12% and 200-mL postbronchodilator rise in FEV1). Moreover, 78 patients had normal lung function.
Symptoms also resembled those in patients with usual emphysema, chronic bronchitis, or both. On enrollment in the registry, 83.9% of the patients had shortness of breath on exertion, 75.5% had wheezing with upper respiratory infections, 65.3% had wheezing without upper respiratory infection, 67.6% had recent debilitating chest illness, 42.4% had “usual” cough, and 49.6% had annual cough and phlegm episodes.
Imaging findings. Although the classic teaching is that emphysema due to alpha-1 antitrypsin deficiency produces lower-lobe hyperlucency on plain films, relying on this sign would lead to underrecognition, as 36% of PI*ZZ patients have apical-predominant emphysema on chest computed tomography,24 which resembles the usual centriacinar emphysema pattern. Figure 3 shows axial computed tomographic scans through the apices and the bases of the lungs of a patient with alpha-1 antitrypsin deficiency.
In view of these difficulties, guidelines from the American Thoracic Society and European Respiratory Society14 endorse testing for alpha-1 antitrypsin deficiency in all adults who have symptoms and fixed airflow obstruction (Table 1).
CONSEQUENCES OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Two large screening studies2,3,25,26 followed people who were identified at birth as having alpha-1 antitrypsin deficiency to examine the natural course of the disease.
The larger of the two studies27 tested 200,000 Swedish newborns. Follow-up of this cohort to age 35 indicated that 35-year-old never-smoking PI*ZZ individuals have normal lung function and no excess emphysema on computed tomography compared with normal peers matched for age and sex.27 In contrast, the few PI*ZZ ever-smokers demonstrated a lower level of transfer factor and significantly more emphysema on computed tomography than normal (PI*MM) never-smokers.
Faster decline in lung function
Data from the National Heart, Lung, and Blood Institute registry indicate that, on average, people with severe alpha-1 antitrypsin deficiency lose lung function faster than people without the disease.28 Specifically, in never-smokers in the registry, the average rate of FEV1 decline was 67 mL/year, and among ex-smokers, it was 54 mL/year. Both of these values exceed the general age-related rate of FEV1 decline of approximately 20 to 25 mL/year in never-smoking, normal adults. Among current smokers in the registry with severe alpha-1 antitrypsin deficiency, the rate of FEV1 decline was 109 mL/year.
Rates of FEV1 decline over time vary among groups with differing degrees of airflow obstruction. For example, PI*ZZ patients with moderate COPD (stage II of the four-stage Global Initiative for Chronic Obstructive Lung Disease classification system) lose lung function faster than patients with either milder or more severe degrees of airflow obstruction.29
As with COPD in general, exacerbations of COPD in people with severe deficiency of alpha-1 antitrypsin are associated with worsened clinical status. In one series,30 54% of 265 PI*ZZ patients experienced an exacerbation in the first year of follow-up, and 18% experienced at least three. Such exacerbations occurred in December and January in 32% of these individuals, likely due to a viral precipitant.
Increased mortality
Severe deficiency of alpha-1 antitrypsin is associated not only with severe morbidity but also death. In the national registry, the overall rate of death was 18.6% at 5 years of follow-up, or approximately 3% per year.28
A low FEV1 at entry was a bad sign. Patients entering the registry with FEV1 values below 15% of predicted had a 36% mortality rate at 3 years, compared with 2.6% in those whose baseline FEV1 exceeded 50% of predicted.
Underlying causes of death in registry participants included emphysema (accounting for 72% of deaths) and cirrhosis (10%),31 which were the only causes of death more frequent than in age- and sex-matched controls. In a series of never-smokers who had PI*ZZ alpha-1 antitrypsin deficiency,32 death was less frequently attributed to emphysema than in the national registry (46%) and more often attributed to cirrhosis (28%), indicating that never-smokers may more frequently escape the ravages of emphysema but experience a higher rate of developing cirrhosis later in life.33
DIAGNOSING ALPHA-1 ANTITRYPSIN DEFICIENCY
Available blood tests for alpha-1 antitrypsin deficiency include:
The serum alpha-1 antitrypsin level, most often done by nephelometry. Normal serum levels generally range from 100 to 220 mg/dL.
Phenotyping, usually performed by isoelectric focusing, which can identify different band patterns associated with different alleles.
Genotyping involves determining which alpha-1 antitrypsin alleles are present, most often using polymerase chain reaction testing targeting the S and Z alleles and occasionally set up to detect less common alleles such as F and I.17
Gene sequencing is occasionally necessary to achieve an accurate, definitive diagnosis.
Free, confidential testing is available
Clinical testing most often involves checking both a serum level and a phenotype or genotype. Such tests are often available in hospital laboratories and commercial laboratories, with testing also facilitated by the availability of free testing kits from several manufacturers of drugs for alpha-1 antitrypsin deficiency.
The Alpha-1 Foundation (www.alpha1.org)34 also offers a free, home-based confidential testing kit through a research protocol at the Medical University of South Carolina (alphaone@musc.edu) called the Alpha-1 Coded Testing (ACT) study. Patients can receive a kit and lancet at home, submit the dried blood-spot specimen, and receive in the mail a confidential serum level and genotype.
The availability of such home-based confidential testing allows patients to seek testing without a physician’s order and makes it easier for facilitated allied health providers, such as respiratory therapists, to recommend testing in appropriate clinical circumstances.15
TREATMENT OF ALPHA-1 ANTITRYPSIN DEFICIENCY
The treatment of patients with severe deficiency of alpha-1 antitrypsin and emphysema generally resembles that of patients with usual COPD. Specifically, smoking cessation, bronchodilators, occasionally inhaled steroids, supplemental oxygen, preventive vaccinations, and pulmonary rehabilitation are indicated as per usual clinical assessment.
Lung volume reduction surgery, which is beneficial in appropriate subsets of COPD patients, is generally less effective in those with severe alpha-1 antitrypsin deficiency,35 specifically because the magnitude of FEV1 increase and the duration of such a rise are lower than in usual COPD patients.
Augmentation therapy
Specific therapy for alpha-1 antitrypsin deficiency currently involves weekly intravenous infusions of purified, pooled human-plasma-derived alpha-1 antitrypsin, so-called augmentation therapy. Four drugs have been approved for use in the United States:
- Prolastin-C (Grifols, Barcelona, Spain)
- Aralast NP (Baxalta, Bonneckborn, IL)
- Zemaira (CSL Behring, King of Prussia, PA)
- Glassia (Baxalta, Bonneckborn, IL, and Kamada, Ness Ziona, Israel).
All of these were approved for use in the United States on the basis of biochemical efficacy. Specifically, infusion of these drugs has been shown to raise serum levels above a protective threshold value (generally considered 57 mg/dL, the value below which the risk of developing emphysema increases beyond normal).
Randomized controlled trials36,37 have addressed the efficacy of intravenous augmentation therapy, and although no single trial has been definitive, the weight of evidence shows that augmentation therapy can slow the progression of emphysema. For example, in a study by Dirksen et al,37 augmentation therapy was associated with a slower progression of emphysema as assessed by the rate of loss of lung density on computed tomography.
On the basis of the available evidence, the American Thoracic Society and European Respiratory Society14 have recommended augmentation therapy in individuals with “established airflow obstruction from alpha-1 antitrypsin deficiency.”14 Their guidelines go on to say that the evidence that augmentation therapy is beneficial “is stronger for individuals with moderate airflow obstruction (eg, FEV1 35%–60% of predicted) than for those with severe airflow obstruction. Augmentation therapy is not currently recommended for individuals without emphysema.”
The guidelines recognize that although augmentation therapy does not satisfy the usual criteria for cost-effectiveness (< $50,000 per quality-adjusted life year) due to its high cost (approximately $100,000 per year if paid for out of pocket),38 it is recommended for appropriate candidates because it is the only available specific therapy for severe deficiency of alpha-1 antitrypsin.
Novel therapies
In addition to current treatment approaches of augmentation therapy, a number of novel treatment strategies are being investigated, several of which hold much promise.
Gene therapy, using adeno-associated virus to transfect the normal human gene into individuals with severe deficiency of alpha-1 antitrypsin, has been undertaken and is currently under study. In addition, a variety of approaches to interdict production of abnormal Z protein from the liver are being examined, as well as inhaled hyaluronic acid to protect the lung.
- Laurell C, Eriksson A. The electrophoretic alpha-1 globulin pattern of serum in alpha-1 antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15:132–140.
- Aboussouan LS, Stoller JK. Detection of alpha-1 antitrypsin deficiency: a review. Respir Med 2009; 103:335–341.
- Stoller JK, Brantly M. The challenge of detecting alpha-1 antitrypsin deficiency. COPD 2013; 10(suppl 1):26–34.
- Stoller JK, Smith P, Yang P, Spray J. Physical and social impact of alpha 1-antitrypsin deficiency: results of a survey. Cleve Clin J Med 1994; 61:461–467.
- Stoller JK, Sandhaus RA, Turino G, Dickson R, Rodgers K, Strange C. Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest 2005; 128:1989–1994.
- Campos MA, Wanner A, Zhang G, Sandhaus RA. Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest 2005; 128:1179–1186.
- Greulich T, Ottaviani S, Bals R, et al. Alpha1-antitrypsin deficiency—diagnostic testing and disease awareness in Germany and Italy. Respir Med 2013; 107:1400–1408.
- Taliercio RM, Chatburn RL, Stoller JK. Knowledge of alpha-1 antitrypsin deficiency among internal medicine house officers and respiratory therapists: results of a survey. Respir Care 2010; 55:322–327.
- Rubenfeld GD, Cooper C, Carter G, Thompson BT, Hudson LD. Barriers to providing lung-protective ventilation to patients with acute lung injury. Crit Care Med 2004; 32:1289–1293.
- Cabana MD, Rand CS, Powe NR, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA 1999; 282:1458–1465.
- Rahaghi F, Ortega I, Rahaghi N, et al. Physician alert suggesting alpha-1 antitrypsin deficiency testing in pulmonary function test (PFT) results. COPD 2009; 6:26–30.
- Campos M, Hagenlocker B, Martinez N, et al. Impact of an electronic medical record clinical reminder to improve detection of COPD and alpha-1 antitrypsin deficiency in the Veterans Administration (VA) system (abstract). Am J Respir Crit Care Med 2011;183:A5356. www.atsjournals.org/doi/pdf/10.1164/ajrccm-conference.2011.183.1_MeetingAbstracts.A5356. Accessed May 24, 2016.
- Jain A, McCarthy K, Xu M, Stoller JK. Impact of a clinical decision support system in an electronic health record to enhance detection of alpha(1)-antitrypsin deficiency. Chest 2011;140:198–204.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Rahaghi FF, Sandhaus RA, Brantly ML, et al. The prevalence of alpha-1 antitrypsin deficiency among patients found to have airflow obstruction. COPD 2012; 9:352–358.
- Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency—a model for conformational diseases. N Engl J Med 2002; 346:45–53.
- Stoller JK, Aboussouan LS. A review of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2012; 185:246–259.
- McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of Alpha 1-Antitrypsin Deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Chest 1997; 111:394–403.
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992; 357:605–607.
- Lomas DA, Finch JT, Seyama K, Nukiwa T, Carrell RW. Alpha 1-antitrypsin Siiyama (Ser53-->Phe). Further evidence for intracellular loop-sheet polymerization. J Biol Chem 1993; 268:15333–15335.
- Mayer AS, Stoller JK, Bucher Bartelson B, James Ruttenber A, Sandhaus RA, Newman LS. Occupational exposure risks in individuals with PI*Z alpha(1)-antitrypsin deficiency. Am J Respir Crit Care Med 2000; 162:553–558.
- Tan L, Dickens JA, Demeo DL, et al. Circulating polymers in alpha1-antitrypsin deficiency. Eur Respir J 2014; 43:1501–1504.
- Parmar JS, Mahadeva R, Reed BJ, et al. Polymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysema. Am J Respir Cell Mol Biol 2002; 26:723–730.
- Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in alpha1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med 2014; 189:419–427.
- Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med 2004; 170:1172–1178.
- Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976; 294:1316–1321.
- O’Brien ML, Buist NR, Murphey WH. Neonatal screening for alpha1-antitrypsin deficiency. J Pediatr 1978; 92:1006–1010.
- Piitulainen E, Montero LC, Nystedt-Duzakin M, et al. Lung function and CT densitometry in subjects with alpha-1-antitrypsin deficiency and healthy controls at 35 years of age. COPD 2015; 12:162–167.
- The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Respir Crit Care Med 1998; 158:49–59.
- Dawkins PA, Dawkins CL, Wood AM, Nightingale PG, Stockley JA, Stockley RA. Rate of progression of lung function impairment in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1338–1344.
- Needham M, Stockley RA. Alpha 1-antitrypsin deficiency. 3: clinical manifestations and natural history. Thorax 2004; 59:441–445.
- Tomashefski JF Jr, Crystal RG, Wiedemann HP, Mascha E, Stoller JK. The bronchopulmonary pathology of alpha-1 antitrypsin (AAT) deficiency: findings of the Death Review Committee of the National Registry for Individuals with Severe Deficiency of Alpha-1 Antitrypsin. Hum Pathol 2004; 35:1452–1461.
- Tanash HA, Nilsson PM, Nilsson JA, Piitulainen E. Clinical course and prognosis of never-smokers with severe alpha-1-antitrypsin deficiency (PiZZ). Thorax 2008; 63:1091–1095.
- Walsh JW, Snider GL, Stoller JK. A review of the Alpha-1 Foundation: its formation, impact, and critical success factors. Respir Care 2006; 51:526–531.
- Rokadia HK, Stoller JK. Surgical and bronchoscopic lung volume reduction treatment for a-1 antitrypsin deficiency. Clin Pulm Med 2015; 22:279–285.
- Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1345–1353.
- Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160:1468–1472.
- Gildea TR, Shermock KM, Singer ME, Stoller JK. Cost-effectiveness analysis of augmentation therapy for severe alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2003; 167:1387–1392.
- Laurell C, Eriksson A. The electrophoretic alpha-1 globulin pattern of serum in alpha-1 antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15:132–140.
- Aboussouan LS, Stoller JK. Detection of alpha-1 antitrypsin deficiency: a review. Respir Med 2009; 103:335–341.
- Stoller JK, Brantly M. The challenge of detecting alpha-1 antitrypsin deficiency. COPD 2013; 10(suppl 1):26–34.
- Stoller JK, Smith P, Yang P, Spray J. Physical and social impact of alpha 1-antitrypsin deficiency: results of a survey. Cleve Clin J Med 1994; 61:461–467.
- Stoller JK, Sandhaus RA, Turino G, Dickson R, Rodgers K, Strange C. Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest 2005; 128:1989–1994.
- Campos MA, Wanner A, Zhang G, Sandhaus RA. Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest 2005; 128:1179–1186.
- Greulich T, Ottaviani S, Bals R, et al. Alpha1-antitrypsin deficiency—diagnostic testing and disease awareness in Germany and Italy. Respir Med 2013; 107:1400–1408.
- Taliercio RM, Chatburn RL, Stoller JK. Knowledge of alpha-1 antitrypsin deficiency among internal medicine house officers and respiratory therapists: results of a survey. Respir Care 2010; 55:322–327.
- Rubenfeld GD, Cooper C, Carter G, Thompson BT, Hudson LD. Barriers to providing lung-protective ventilation to patients with acute lung injury. Crit Care Med 2004; 32:1289–1293.
- Cabana MD, Rand CS, Powe NR, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA 1999; 282:1458–1465.
- Rahaghi F, Ortega I, Rahaghi N, et al. Physician alert suggesting alpha-1 antitrypsin deficiency testing in pulmonary function test (PFT) results. COPD 2009; 6:26–30.
- Campos M, Hagenlocker B, Martinez N, et al. Impact of an electronic medical record clinical reminder to improve detection of COPD and alpha-1 antitrypsin deficiency in the Veterans Administration (VA) system (abstract). Am J Respir Crit Care Med 2011;183:A5356. www.atsjournals.org/doi/pdf/10.1164/ajrccm-conference.2011.183.1_MeetingAbstracts.A5356. Accessed May 24, 2016.
- Jain A, McCarthy K, Xu M, Stoller JK. Impact of a clinical decision support system in an electronic health record to enhance detection of alpha(1)-antitrypsin deficiency. Chest 2011;140:198–204.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Rahaghi FF, Sandhaus RA, Brantly ML, et al. The prevalence of alpha-1 antitrypsin deficiency among patients found to have airflow obstruction. COPD 2012; 9:352–358.
- Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency—a model for conformational diseases. N Engl J Med 2002; 346:45–53.
- Stoller JK, Aboussouan LS. A review of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2012; 185:246–259.
- McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of Alpha 1-Antitrypsin Deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Chest 1997; 111:394–403.
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992; 357:605–607.
- Lomas DA, Finch JT, Seyama K, Nukiwa T, Carrell RW. Alpha 1-antitrypsin Siiyama (Ser53-->Phe). Further evidence for intracellular loop-sheet polymerization. J Biol Chem 1993; 268:15333–15335.
- Mayer AS, Stoller JK, Bucher Bartelson B, James Ruttenber A, Sandhaus RA, Newman LS. Occupational exposure risks in individuals with PI*Z alpha(1)-antitrypsin deficiency. Am J Respir Crit Care Med 2000; 162:553–558.
- Tan L, Dickens JA, Demeo DL, et al. Circulating polymers in alpha1-antitrypsin deficiency. Eur Respir J 2014; 43:1501–1504.
- Parmar JS, Mahadeva R, Reed BJ, et al. Polymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysema. Am J Respir Cell Mol Biol 2002; 26:723–730.
- Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in alpha1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med 2014; 189:419–427.
- Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med 2004; 170:1172–1178.
- Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976; 294:1316–1321.
- O’Brien ML, Buist NR, Murphey WH. Neonatal screening for alpha1-antitrypsin deficiency. J Pediatr 1978; 92:1006–1010.
- Piitulainen E, Montero LC, Nystedt-Duzakin M, et al. Lung function and CT densitometry in subjects with alpha-1-antitrypsin deficiency and healthy controls at 35 years of age. COPD 2015; 12:162–167.
- The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Respir Crit Care Med 1998; 158:49–59.
- Dawkins PA, Dawkins CL, Wood AM, Nightingale PG, Stockley JA, Stockley RA. Rate of progression of lung function impairment in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1338–1344.
- Needham M, Stockley RA. Alpha 1-antitrypsin deficiency. 3: clinical manifestations and natural history. Thorax 2004; 59:441–445.
- Tomashefski JF Jr, Crystal RG, Wiedemann HP, Mascha E, Stoller JK. The bronchopulmonary pathology of alpha-1 antitrypsin (AAT) deficiency: findings of the Death Review Committee of the National Registry for Individuals with Severe Deficiency of Alpha-1 Antitrypsin. Hum Pathol 2004; 35:1452–1461.
- Tanash HA, Nilsson PM, Nilsson JA, Piitulainen E. Clinical course and prognosis of never-smokers with severe alpha-1-antitrypsin deficiency (PiZZ). Thorax 2008; 63:1091–1095.
- Walsh JW, Snider GL, Stoller JK. A review of the Alpha-1 Foundation: its formation, impact, and critical success factors. Respir Care 2006; 51:526–531.
- Rokadia HK, Stoller JK. Surgical and bronchoscopic lung volume reduction treatment for a-1 antitrypsin deficiency. Clin Pulm Med 2015; 22:279–285.
- Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1345–1353.
- Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160:1468–1472.
- Gildea TR, Shermock KM, Singer ME, Stoller JK. Cost-effectiveness analysis of augmentation therapy for severe alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2003; 167:1387–1392.
KEY POINTS
- Only about 15% of people who have alpha-1 antitrypsin disease have received a diagnosis of it.
- The disease is genetic. People who are homozygous for the Z allele of the gene that codes for alpha-1 antitrypsin are at increased risk of lung and liver disease.
- Chronic obstructive pulmonary disease (COPD) due to alpha-1 antitrypsin deficiency is difficult to distinguish from “usual” COPD on a clinical basis, but blood tests are available.
- The basic care of a patient with COPD due to alpha-1 antitrypsin disease is the same as for any patient with COPD, ie, with bronchodilators, inhaled steroids, supplemental oxygen, preventive vaccinations, and pulmonary rehabilitation as indicated. Specific treatment consists of weekly infusions of alpha-1 antitrypsin (augmentation therapy).