User login
The architecture of clinical research
I am flattered to present the inaugural talk in the biostatistics and clinical research design series on the architecture of clinical research. This content is based on the teachings of my mentor, Dr. Alvan Feinstein, who together with Dr. David Sackett, is credited with pioneering clinical epidemiology. Dr. Feinstein was a Sterling Professor at the Yale School of Medicine. His main opus of work is a book called, Clinical Epidemiology: The Architecture of Clinical Research.1 This paper is named in credit to Dr. Feinstein’s enormous contribution. I will review some important terms defined by Dr. Feinstein to provide the background necessary for the remainder of the talks in this series.
To start, I will frame this topic by asking the following question: Why do we do research? I’ll talk about the basic structure of research studies and provide a taxonomy, as Dr. Feinstein would say, a nomenclature with which to understand trial design and the sources of bias in those trials. Then, I will discuss these sources of bias in detail using the taxonomy that Dr. Feinstein described in his aforementioned book. Finally, I will share with you some examples of bias in clinical trials to help you better understand these concepts.
Now, the answer to the basic question posed above is: basically, we do cause-and-effect research to establish the causality of a risk factor or the efficacy of a therapy. Does cigarette smoking cause lung cancer? Does taking hydrochlorothiazide help systemic hypertension? Does air pollution worsen asthma? Does supplemental oxygen help patients with chronic obstructive pulmonary disease (COPD)?
Cause-and-effect research can be subsumed under 2 broad issues: causal risk factors and therapeutic efficacy. In his review of early false understandings in medicine that were based on anecdotal observation alone, Thomas cites many examples—“the undue longevity of useless and even harmful drugs can be laid at the door of authority,” ie, empiricism, lack of rigorous research.2 The field is full of these: yellow fever causality, the value of cupping, and even intermittent mandatory ventilation when it was described by John Downs in 1973 and touted as a superior mode for weaning patients from mechanical ventilation.3 Twenty-five years later, randomized controlled trials by Brochard et al4 indicated not only that intermittent mandatory ventilation was not the best mode to wean but was, in fact, the worst mode for weaning patients from mechanical ventilation compared with either pressure support or spontaneous breathing trials. Many more examples exist to demonstrate the false understandings that can be ascribed to lack of rigorous study or evidence in medicine.

Before systematically exploring the sources of bias in Feinstein’s construct, let us define some very basic terms from his book. Dr. Feinstein talks about the baseline state, which refers to the group of patients under study who are culled from a larger population to whom the results are intended to be applied (Figure 1).1 This baseline group is hopefully representative of this larger target population. As a nod to the later discussion, Dr. Feinstein would call bias introduced by unusual assembly of the study population from the larger intended population as “assembly bias.” So, if the group under study is not representative of either the patients you see or the world of patients with this condition or if there is something special or distinctively nonrepresentative about the study population, then the results may be subject to “assembly bias.” Assembly bias can compromise the so-called “external” validity of the study—its ability to be applied to populations beyond the study group.
Having assembled a baseline group for study, that group is classically allocated to 1 of 2 (or sometimes more than 2) compared therapies. In a controlled trial, patients can be allocated using a variety of strategies, including randomization. Using the paradigm diagram (Figure 1, which considers a 2-arm trial), patients are allocated to 1 of 2 compared groups—group A and group B. Then, in a treatment trial, 1 group receives the principal maneuver, which is the drug or intervention under study—for example, supplemental oxygen for patients with COPD. The comparative maneuver is allocated to group B, which also receives all the other treatments (called “co-maneuvers”) that are used to treat the condition under study. In a trial of supplemental oxygen for COPD evaluating lung function and exacerbation frequency as outcome measures, such co-maneuvers might include inhaled bronchodilators, inhaled corticosteroids, pulmonary rehabilitation, and Pneumovax vaccine. Ideally, these co-maneuvers are equally distributed between the compared groups (A and B).
So, in summary, we have a comparative maneuver, which is the nonadministration of supplemental oxygen in this proposed trial of supplemental oxygen in COPD, the principal maneuver—administration of oxygen—and all the co-maneuvers that are ideally equally distributed between both groups. This balanced distribution of co-maneuvers between the compared groups helps to ensure that any differences in the study outcome measures (ie, what is counted as the main impact of the intervention under study) can be solely attributed to the principal maneuver. When this condition—that the difference in outcomes can be reliably ascribed to the study intervention—is satisfied, the study is felt to be “internally” valid. As we will see, ensuring internal validity requires freedom from the many sources of what Dr. Feinstein calls “internal bias.”
Back to basic terms: “cohort” in Dr. Feinstein’s language is a group that shares common traits and is followed forward in a longitudinal study. The “outcome measure” is self-evident—it is what is being measured, with the “primary outcome” being the pre-defined measure that is considered the most important (and ideally most clinically relevant) impact of the study intervention. Later in this series of lectures, there will be discussions of power calculations and the so-called “effect size”—the magnitude of effect that the intervention is expected to produce and that is ideally deemed clinically important.
An important consideration in designing a trial is to define and declare the primary outcome measure carefully because defining the primary outcome measure has important implications for the study. I will provide an example from the alpha-1 antitrypsin deficiency literature. Some of you have probably read what has been called the RAPID trial.5 RAPID was a trial of augmentation therapy vs placebo in patients with severe alpha-1 antitrypsin deficiency. The primary outcome measure (which was pre-negotiated with the US Food and Drug Administration [FDA]) was computer tomography (CT) lung density determined at functional residual capacity (FRC) and total lung capacity (TLC). The trial failed to achieve statistical significance in regard to CT lung density, although the study authors argued that CT density measurements made at TLC were more reproducible than those made at FRC. When the results were analyzed by TLC alone, the results were statistically significant, but when they were analyzed with FRC and TLC combined, they were not. In the end, based on the pre-negotiated primary outcome measure of CT density based on both FRC and TLC, the FDA rejected the proposal for a label change to say that augmentation therapy slowed the loss of lung density even though the weight of evidence was clearly in its favor. This case exemplifies just how critical the choice of primary outcome measure can be.

The wash-out period refers to an interval in a subset of randomized trials called “crossover trials” in which the primary intervention is discontinued and the patient returns to his baseline state before the comparative maneuver is then implemented (Figure 2).6 In order to perform a crossover trial, it is important that the effects of the initial intervention can “wash out” or be fully extinguished. So, for example, in trials of radiation therapy vs surgery, it is impossible to do a crossover trial because the effects of radiation can never completely wash out nor can those of surgery, which are similarly permanent. For example, we cannot replace the colon once it is resected for cancer or replace the appendix once removed. Therefore, producing a wash-out requires some very specific pharmacokinetic and pharmacodynamic features in order for a crossover trial to be considered. Later talks in this series will discuss the enhanced statistical power of a crossover trial, where one is comparing every patient to him or herself rather than to another patient.
So, there is always an appetite to do a crossover trial as long as the criteria for wash-out can be met, namely again that the primary intervention can dissipate completely to the baseline state before the alternative intervention is implemented.
“Placebo” is a fairly self-evident and well-understood term; placebo refers to the administration of a maneuver in a way that is identical to the principal maneuver except that the placebo is not expected to exert any clinical effect.
“Blinding” is the unawareness of either the investigator or of the patient to which the intervention is being administered. “Single-blinding” refers to the condition in which either the study or the investigator (but not both) is unaware, and “double-blinding” refers to the condition in which both the subjects and the investigators are unaware. There can be some subtle issues that compromise whether the patient is aware of the intervention that he or she is receiving and that can potentially condition the patient’s response, particularly if there is any subjective component of the assessment of the outcome. So, blinding is important.
With these terms describing the elements of a clinical study now described, let us turn to the types of studies that comprise clinical research. The first group of study types is what Dr. Feinstein called descriptive studies—studies that simply describe phenomena without comparison to a control group. As an example of a descriptive study, Sehgal et al7 recently described the workup of a focal, segmental pneumonia in a patient taking pembrolizumab for lung cancer. In this paper, there were four other cases of focal pneumonia accompanying pembrolizumab use that were assembled from the literature, making this descriptive paper a so-called case series. A “case series” differs from a “single case report,” which reports a single patient experience. Though limited in their ability to establish cause and effect, case reports and case series can help researchers develop proof of principle, so I would not discount the value of case reports.8
I can cite a case report from of my own experience that demonstrates this point. In 1987, I saw a patient from Buffalo who had primary biliary cirrhosis and the hepatopulmonary syndrome (HPS). She was so debilitated by her HPS that she could not stand up without desaturating severely. Although she had normal liver synthetic function, she was severely debilitated by her HPS and the decision was made to offer her a liver transplant, which, at that time, was considered to be relatively contraindicated. Much to everyone’s amazement and satisfaction, her HPS completely resolved after the transplant surgery. Her oxygenation and alveolar-arterial oxygen gradient normalized, and her clubbing resolved. We reported this in a case report, which began to affect the way people thought about the feasibility of liver transplant for the HPS.8 The lesson is: do not underestimate the power of a thoughtful case report.
The second group of research study types is called “cohort studies,” in which one actually compares outcomes between 2 groups in the study. Cohort studies fall into the bucket of either “observational cohort studies,” in which allocation to the compared maneuvers is not performed by randomization but by any other strategy, and “randomized trials.” In observational studies, allocation could occur through physician choice, as when the physician prescribes a treatment to 1 group but not another, or by patient choice or circumstance. For example, an observational cohort study of the risk of cigarette smoking would compare outcomes between smokers and non-smokers where the patient choses to smoke under his/her own volition. Alternatively, the circumstances of an exposure could allocate someone to the principal maneuver, as when we are studying the effect of exposure to World Trade Center dust in the firefighters who responded or of exposure to nuclear radiation in Hiroshima survivors. These are examples of observational cohort studies that compare exposed individuals to unexposed individuals, where the exposure did not occur by randomization but by choice or unfortunate circumstance.
In contrast to observational studies, allocation in randomized trials occurs through a formal process. Randomization has the specific purpose of attempting to ensure that patients are allocated to 2 comparative groups from the baseline group with comparable risk for developing the outcome measure. When randomization is effective, differences in study outcomes can be reliably ascribed to the intervention rather than to differences in the baseline susceptibility of the compared groups.
While randomization is an excellent strategy to ensure baseline similarity between compared groups, randomization can fail, and its effectiveness must be checked. Specifically, in a randomized trial, it is customary to examine the compared groups at baseline on all features that can affect the likelihood of developing the outcome measure. If the groups turn out to be dissimilar at baseline in an important way, then the study is at risk for bias, which is specifically called “susceptibility bias” in Feinstein’s construct. Obviously, the larger number of baseline clinical and demographic features that can condition the likelihood of developing the outcome measure, the more difficult it is to achieve baseline similarity between compared groups and the more important it becomes to ensure that randomization has been effective. In this circumstance, larger numbers of participants in both compared groups are generally needed. More about susceptibility bias later.
There are generally 2 types of randomized trials: the so-called “parallel controlled trials” in which each group receives either the principal or the comparative maneuver and is followed and “crossover trials” in which each compared group receives both the principal maneuver and the co-maneuver at different times after an effective wash-out period. Wash-out was discussed above. Figure 2 shows an example of a crossover trial examining the effects of terbutaline on diaphragmatic function.6 The investigators administered terbutaline for a week, measured transdiaphragmatic pressures, gave the patient a terbutaline vacation (the “wash-out period”), and then crossed over those patients who were initially receiving terbutaline to placebo and initial placebo recipients to terbutaline, having remeasured diaphragmatic function after the wash-out period to assure that the patient’s diaphragmatic function prior to the second crossover was identical to his/her baseline state. If this return to baseline is accomplished, then the criteria from effective wash-out are satisfied.


As we begin to talk about sources of bias, consider a study in which we compare survival of patients allocated to surgery vs nonsurgical therapy for lung cancer (Figure 3).1 This study is subject to the first type of so-called “internal bias” in the Feinsteinian construct—so-called “selection bias.” For example, all patients treated surgically were considered healthy enough by their doctors to undergo surgery, whereas patients treated without surgery may have been deemed inoperable because of comorbidities, lung dysfunction, cardiac dysfunction, and so on. If the results of such a comparison show that the mortality rate among surgical patients in this study was lower, the question then becomes: is the improved survival in surgical candidates due to the superior efficacy of surgery vs other therapy or was the enhanced survival due to the surgical patients being healthier to begin with? You can intuitively sense that the answer to this question is that the enhanced survival may be due to the better health of patients treated surgically rather than to the surgery itself because of how the patients were selected to receive it. So, this is a simple example of what Dr. Feinstein would call “susceptibility bias.” Susceptibility bias occurs when the 2 baseline groups are not comparably at risk or susceptible to developing the outcome measure, leading the naïve investigator in this specific example to attribute the difference in outcomes to the superiority of surgery when in fact it may have nothing to do with the surgery vs. the other maneuver. When susceptibility bias is in play, the difference between the outcomes in the compared groups could be attributed to the baseline imbalance of the groups rather than to the principal maneuver itself.
Turning back to the taxonomy of bias, there are four types that can threaten internal validity—“susceptibility,” “performance,” “detection,” and “transfer” bias—and 1 type of bias (called “external bias”) that can affect the generalizability of the study called “assembly bias” (Table 1).

Figure 4 shows where these various sources of bias appear in the architecture of a clinical trial. As just discussed, susceptibility bias affects the baseline state and the comparability of the groups. Performance bias relates to how effective and how comparably the co-maneuvers are given and whether the primary intervention is potent enough to affect an outcome. Both transfer and detection bias operate in detecting the outcome, especially regarding the rigor and frequency with which they are investigated. Transfer bias has to do with selective loss to follow-up of those included in the trial. If there is a systematic reason for loss to follow-up that is related to the impact of the intervention, then the study is at risk for transfer bias. For example, in a randomized trial of drug A vs placebo for pneumonia, if drug A is effective but all the drug A recipients fail to follow-up because they feel too good to return for follow-up, then transfer bias could be causing the study to show nonefficacy even though the drug works. So, if those who respond favorably are systematically lost to follow-up, and if all the patients who felt lousy wanted to see the doctor and came back for follow-up, such transfer bias would bias towards nonefficacy. Specifically, only patients remaining in the trial would be those who failed to respond and that would dilute any difference between the 2 groups despite the active efficacy of drug A.
Hopefully, you are already beginning to get a sense that one has to be extremely disciplined in thinking about each of these sources of bias because they can have some very subtle nuances in randomized trials that can easily escape attention.
Returning to sources of bias, let’s consider the second type of bias, “performance bias.” Performance bias relates to the administration of the compared maneuvers—the primary or principal maneuver, compared with the comparative maneuver. Performance bias can occur when the main maneuver is not administered adequately or when the co-maneuvers are administered in an imbalanced way between the compared groups. Consider the example of the Long-Term Oxygen Treatment Trial (LOTT) trial, which compared use of supplemental oxygen with no supplemental oxygen in patients with stable COPD and resting or exercise-induced moderate desaturation.9 The principal outcome measure of LOTT was all-cause hospitalization or death. In such a study, many potential sources of performance bias exist. For example, performance bias might exist if none of the patients allocated to oxygen actually used supplemental oxygen. Alternately, to the extent that use of inhaled corticosteroids or antimuscarinic agents lessens the risk of COPD exacerbation, performance bias could occur if use of these co-maneuvers was imbalanced between the compared groups. As a specific extreme circumstance, if all patients in the nonoxygen group used these inhalers but none of the patients in the oxygen group did, then a lack of difference between exacerbation frequency could be related to this imbalance in co-maneuvers (a form of performance bias) rather than to the lack of efficacy of supplemental oxygen.
“Compliance bias” is a subset of performance bias which occurs when 2 conditions are satisfied: (1) the main maneuver is not administered adequately, and (2) the investigator is unaware of that nonreceipt so that this cannot be accounted for in interpreting the study results. For example, if a drug has efficacy but if no one in the treatment arm of the trial takes the drug, the absence of a difference in outcomes between the compared groups will be ascribed to nonefficacy, whereas “compliance bias” (ie, no one actually took the drug) could actually be the cause. Ideally, randomized studies should be evaluated on an “intention to treat” basis irrespective of compliance, but there is an analytic approach called “per protocol” analysis in which you can analyze the results according to whether the patient actually used the intervention in an effective way. “Per protocol” analysis is a secondary analysis of the primary results but it can nonetheless help determine whether the negative result is likely related to noncompliance or not.
A third type of internal bias, “detection bias,” is fairly straightforward. Detection bias is related to how avidly and how comparably the outcomes are measured between the 2 compared groups. Let’s say that you are conducting a trial of a new antibiotic and the primary outcome is colony counts on petri dishes of plated collected specimens. If the technicians who read the petri dish counts are unblinded, they may look at the colony counts with a biased eye, seeing fewer colonies on plates collected from patients receiving the antibiotic.
Overall, detection bias occurs when outcomes are ascertained or detected unequally between the compared groups, and detection bias can involve any of the following: is there comparable surveillance of the 2 groups for analysis of the outcome measure? Are the diagnostic tests comparably performed in both groups and is the interpretation comparably unbiased with equipoise? Investigators who know which patients are receiving an active drug and those who are not could experience subliminal bias that renders them more likely to find that the drug under study is efficacious.
Depending on the principal study maneuver, ensuring blinding can be challenging. To demonstrate this point, let’s consider the example of conducting a randomized control trial of Vicks VapoRub. Vicks VapoRub is an old product that smells like wintergreen and that mothers used to rub on the chests of their infants in the hope of speeding recovery from colds and bronchitis episodes. It was felt that the distinctive smell of the product was materially related to wintergreen, which gives rise to the odor. So, imagine a randomized trial of Vicks VaporRub. A trial is designed in which sick children receive Vicks VapoRub on their chest and others receive a placebo rub that lacks the distinctive wintergreen odor. But, the odor itself is felt to be related to how Vicks VapoRub actually works. Thus, it is the odor itself that creates the blinding challenge here.
The primary outcomes in this study are the duration of the child’s cold symptoms, as ascertained by pediatricians actually examining the children. So, pediatricians would come and listen to the infants’ chests: “Yeah, this chest is clear, but this other infant is still full of rhonchi,” and they would ascertain the outcome measure in this way. So, my blinding question to you is: how do you blind a trial of Vicks VapoRub given the conditions described? Namely, you put the VapoRub on the chest, it smells and the smell is the intervention—how do you blind such a trial?
The clever answer is that you should put Vicks VapoRub on the upper lips of all the examiners, so what they smell is Vicks VapoRub independent of whether the child they are examining also has the Vicks VapoRub or placebo on their chest. In this way, single blinding of the examiners is preserved and detection bias is averted. It is important to point out that double blinding could also be achieved by placing Vicks VapoRub on the child’s upper lip, but there is little reason to suspect that the infants being studied have a bias related to whether they smell the Vicks VapoRub.
The fourth potential source of internal bias is called “transfer bias.” Transfer bias is the selective loss to follow-up of patients from 1 of the 2 compared groups in the trial for a systematic reason. By systematic, I mean that that the drop-out is associated with the development of the outcome event or some impact of the intervention regarding the likelihood to develop the outcome event. As an example, if all patients respond favorably to a drug and everybody fails to follow up because they feel too good to come back, then that would bias the study towards nonefficacy even in the face of an efficacious intervention.
Finally, let’s consider a source of bias that can affect the “external validity,” or the generalizability of the study results to populations other than that included in the study itself. Dr. Feinstein calls this 5th type of bias “assembly bias” (Table 1).1 Assembly bias occurs when the results of the study cannot be reliably applied to populations outside the study itself.
For example, if I screen patients during a study of digoxin for heart rate control in atrial fibrillation, I could establish whether the subject was compliant or not by checking his/her serum digoxin levels. Serum levels of 0 indicate that the patient has not taken the digoxin. If I include a run-in period for the trial—an interval before the actual study when I am assessing potential subjects’ eligibility to participate—and check serum digoxin levels to include only patients who are shown to be taking the drug, then I am screening for study inclusion on compliance. In this way, I will have assembled a population that is highly compliant so that I can truly assess whether digoxin has efficacy in controlling the heart rate in patients with atrial fibrillation. At the same time, this study population is not highly representative of the population of patients with atrial fibrillation at large, because we know that rates of drug noncompliances may be as high as 30% to 40%. So, culling a population with run-in periods on demonstrated compliance criteria may be very important to assess efficacy (ie, whether the drug works), but this design will trade off on the effectiveness of the drug (ie, which asks the question “does the drug work in actual practice?”). This is because, in the yin-yang between assessing efficacy and assessing effectiveness, the focus on assessing efficacy naturally undermines the ability to assess whether the drug works in real-world conditions.
As another example of potential assembly bias, let’s say you are studying an antihypertensive drug at a Veterans Administration (VA) hospital, where most veterans are men. But you are treating women in your practice and wonder whether the drug, which works in a predominately male population, will work in your female patients. So, there could be assembly bias in applying the results of a VA study to a non-VA predominantly female population.
Having now described the design of clinical trials and the major sources of bias, let’s apply this thinking to the earliest clinical trial. James Lind, a British Naval officer, was credited with conducting the first clinical trial of citrus fruits for scurvy while sailing on the ship Salisbury in 1747.2 The question that Lind addressed was “does citrus fruit treat and prevent scurvy?” In describing this trial, Lind stated “I took 12 patients with scurvy, these patients were as similar as I could have them, had one diet common to all.” As you read this through your new Feinsteinian bias lens, Lind is addressing 2 potential sources of bias, namely, susceptibility bias and performance bias. In trying to make the “cases as similar as I could have them,” he is trying to avoid susceptibility bias and in “providing one diet common to all,” he is trying to avoid performance bias.
In terms of the intervention in this trial, these 12 patients were allocated in pairs to several interventions: a quart of cider a day, 25 drops of elixir of vitriol 3 times a day on an empty stomach, 2 spoonsful of vinegar 3 times a day on an empty stomach, ½ pint a day of sea water, 2 oranges and 1 lemon given every day, and a “bigness of nutmeg” 3 times per day. In describing the outcome of the trial, Lind states “the consequence was that the most sudden and visible good effects were perceived from the use of oranges and lemons; one of those who had taken them, being at the end of 6 days fit for duty. The spots were not indeed at that time quite off his body, nor his gums sound, but without any other medicine then a gargarism of elixir vitriol, he became quite healthy before we came into Plymouth which was on the 16th of June. The other was the best recovered of any in his condition; and being now deemed pretty well, was appointed nurse to the rest of the sick.”
In analyzing this trial, we could characterize it as a parallel controlled trial. Whether the allocation was done by randomization is not clear, but it was certainly an observational cohort study in that there were concurrent controls who were treated as similarly as possible except for the principal maneuver, which was the administration of citrus fruit. Already mentioned was the attention to averting susceptibility and performance bias. There was no evidence of compliance bias as the interventions were enforced, nor was there evidence of transfer bias because all subjects who were enrolled in the study completed the study because they were a captive group on a sailing ship. Finally, the likelihood of assembly bias seems small, as these sailors seemed to be representative of victims of scurvy in general, namely in being otherwise deprived of access to citrus fruits.
In terms of the statistical results of this study, subsequent analysis of the research showed that the impact of lemons and oranges was dramatic and showed a trend (P = .09) towards statistical significance. Notwithstanding the lack of a P < .05, Dr. Feinstein would likely say that this study satisfied the “intra-ocular test” in that the efficacy of the citrus fruit was so dramatic that it “hit you between the eyes.” He often argued that the widespread practice of prescribing penicillin for pneumococcal pneumonia was not based on the results of a convincing randomized controlled trial because the efficacy of penicillin in that setting was so dramatic that a randomized trial was not necessary (and potentially even unethical if the condition of “intra-ocular” efficacy was satisfied).
The final question to address in this lecture is whether randomized controlled trials, for all their rigor, always produce more reliable results than observational studies. This issue has been addressed by several authors.10–12 Sacks et al10 contended in 1983 that observational studies systematically overestimate the magnitude of association between exposure and outcome and therefore argued that randomized trials were more reliable than observational studies. Subsequent analyses tended to challenge this view.11,12 Specifically, Benson and Hartz11 compared the results of 136 reports regarding 19 different therapies that were studied between 1985 and 1998. In only 2 of the 19 analyses did the treatment effects in the observational studies fall outside the 95% confidence interval for the randomized controlled trial results. In this way, these authors argued that observational studies generally are concordant with the results of randomized trials. They stated that “our finding that observational studies and randomized controlled trials usually produce similar results differs from the conclusions of previous authors. The fundamental criticism of observational studies is that unrecognized confounding factors may distort the results. According to the conventional wisdom, this distortion is sufficiently common and unpredictable that observational studies are not liable and should not be funded. Our results suggested observational studies usually do provide valid information.”11
An additional analysis of this issue was performed by Concato et al,12 who identified 99 articles regarding 5 clinical topics. Again, the results from randomized trials were compared with those of observational cohort or case-controlled studies regarding the same intervention. The authors reported that “contrary to prevailing belief, the average results from well-designed observational studies did not systematically overestimate the magnitude of the associations between exposure and outcome as compared with the results of randomized, controlled trials on the same topic. Rather, the summary results of randomized, controlled trials and observational studies were remarkably similar.”12
On the basis of these studies, it appears that randomized control trials continue to serve as the gold standard in clinical research, but we must also recognize that circumstances often preclude the conduct of a randomized trial. As an example, consider a randomized trial of whether cigarette smoking is harmful, which, given the strong suspicion of harm, would be unethical in that patients cannot be randomized to smoke. Similarly, from the example before, a randomized trial of penicillin for pneumococcal pneumonia would be unethical because denying patients in the placebo group access to penicillin would exclude them from access to a drug that has “intra-ocular” efficacy. In circumstances like these, well-performed observational studies that are attentive to sources of bias can likely produce comparably reliable results to randomized trials.
In the end, of course, the interpretation of the study results requires the reader’s careful attention to potential sources of bias that can compromise study validity. The hope is that with Dr. Feinstein’s framework, you can be better equipped to think critically about study results that you review and to keenly ascertain whether there is any threat to internal or to external validity. Similarly, as you go on to design clinical trials yourselves, you can pay attention to these potential sources of bias that, if present, can compromise the reliability of the study conclusions internally or their applicability to patients outside of the study.
- Feinstein AR. Clinical Epidemiology: The Architecture of Clinical Research. Philadelphia, PA: WB Saunders; 1985.
- Thomas DP. Experiment versus authority: James Lind and Benjamin Rush. N Engl J Med 1969; 281:932–934.
- Downs JB, Klein EF Jr, Desautels D, Modell JH, Kirby RR. Intermittent mandatory ventilation: a new approach to weaning patients from mechanical ventilators. Chest 1973; 64:331–335.
- Brochard L, Rauss A, Benito S, et al. Comparison of three methods of gradual withdrawal from ventilatory support during weaning from mechanical ventilation. Am J Respir Crit Care Med 1994; 150:896–903.
- Chapman KR, Burdon JGW, Piitulainen E, et al; on behalf of the RAPID Trial Study Group. Intravenous augmentation treatment and lung density in severe 1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet 2015; 386:360–368.
- Stoller JK, Wiedemann HP, Loke J, Snyder P, Virgulto J, Matthay RA. Terbutaline and diaphragm function in chronic obstructive pulmonary disease: a double-blind randomized clinical trial. Br J Dis Chest 1988; 82:242–250.
- Sehgal S, Velcheti V, Mukhopadhyay S, Stoller JK. Focal lung infiltrate complicating PD-1 inhibitor use: a new pattern of drug-associated lung toxicity? Respir Med Case Rep 2016; 19:118–120.
- Stoller JK, Moodie D, Schiavone WA, et al. Reduction of intrapulmonary shunt and resolution of digital clubbing associated with primary biliary cirrhosis after liver transplantation. Hepatology 1990; 11:54–58.
- Albert RK, Au DH, Blackford AL, et al; for the Long-Term Oxygen Treatment Trial Group. A randomized trial of long-term oxygen for COPD with moderate desaturation. N Engl J Med 2016; 375:1617–1627.
- Sacks HS, Chalmers TC, Smith H Jr. Sensitivity and specificity of clinical trials: randomized v historical controls. Arch Intern Med 1983; 143:753–755.
- Benson K, Hartz AJ. A comparison of observational studies and randomized, controlled trials. N Engl J Med 2000; 342:1878–1886.
- Concato J, Shah N, Horwitz RI. Randomized, controlled trials, observational studies, and the hierarchy of research designs. N Engl J Med 2000; 342:1887–1892.
I am flattered to present the inaugural talk in the biostatistics and clinical research design series on the architecture of clinical research. This content is based on the teachings of my mentor, Dr. Alvan Feinstein, who together with Dr. David Sackett, is credited with pioneering clinical epidemiology. Dr. Feinstein was a Sterling Professor at the Yale School of Medicine. His main opus of work is a book called, Clinical Epidemiology: The Architecture of Clinical Research.1 This paper is named in credit to Dr. Feinstein’s enormous contribution. I will review some important terms defined by Dr. Feinstein to provide the background necessary for the remainder of the talks in this series.
To start, I will frame this topic by asking the following question: Why do we do research? I’ll talk about the basic structure of research studies and provide a taxonomy, as Dr. Feinstein would say, a nomenclature with which to understand trial design and the sources of bias in those trials. Then, I will discuss these sources of bias in detail using the taxonomy that Dr. Feinstein described in his aforementioned book. Finally, I will share with you some examples of bias in clinical trials to help you better understand these concepts.
Now, the answer to the basic question posed above is: basically, we do cause-and-effect research to establish the causality of a risk factor or the efficacy of a therapy. Does cigarette smoking cause lung cancer? Does taking hydrochlorothiazide help systemic hypertension? Does air pollution worsen asthma? Does supplemental oxygen help patients with chronic obstructive pulmonary disease (COPD)?
Cause-and-effect research can be subsumed under 2 broad issues: causal risk factors and therapeutic efficacy. In his review of early false understandings in medicine that were based on anecdotal observation alone, Thomas cites many examples—“the undue longevity of useless and even harmful drugs can be laid at the door of authority,” ie, empiricism, lack of rigorous research.2 The field is full of these: yellow fever causality, the value of cupping, and even intermittent mandatory ventilation when it was described by John Downs in 1973 and touted as a superior mode for weaning patients from mechanical ventilation.3 Twenty-five years later, randomized controlled trials by Brochard et al4 indicated not only that intermittent mandatory ventilation was not the best mode to wean but was, in fact, the worst mode for weaning patients from mechanical ventilation compared with either pressure support or spontaneous breathing trials. Many more examples exist to demonstrate the false understandings that can be ascribed to lack of rigorous study or evidence in medicine.
Before systematically exploring the sources of bias in Feinstein’s construct, let us define some very basic terms from his book. Dr. Feinstein talks about the baseline state, which refers to the group of patients under study who are culled from a larger population to whom the results are intended to be applied (Figure 1).1 This baseline group is hopefully representative of this larger target population. As a nod to the later discussion, Dr. Feinstein would call bias introduced by unusual assembly of the study population from the larger intended population as “assembly bias.” So, if the group under study is not representative of either the patients you see or the world of patients with this condition or if there is something special or distinctively nonrepresentative about the study population, then the results may be subject to “assembly bias.” Assembly bias can compromise the so-called “external” validity of the study—its ability to be applied to populations beyond the study group.
Having assembled a baseline group for study, that group is classically allocated to 1 of 2 (or sometimes more than 2) compared therapies. In a controlled trial, patients can be allocated using a variety of strategies, including randomization. Using the paradigm diagram (Figure 1, which considers a 2-arm trial), patients are allocated to 1 of 2 compared groups—group A and group B. Then, in a treatment trial, 1 group receives the principal maneuver, which is the drug or intervention under study—for example, supplemental oxygen for patients with COPD. The comparative maneuver is allocated to group B, which also receives all the other treatments (called “co-maneuvers”) that are used to treat the condition under study. In a trial of supplemental oxygen for COPD evaluating lung function and exacerbation frequency as outcome measures, such co-maneuvers might include inhaled bronchodilators, inhaled corticosteroids, pulmonary rehabilitation, and Pneumovax vaccine. Ideally, these co-maneuvers are equally distributed between the compared groups (A and B).
So, in summary, we have a comparative maneuver, which is the nonadministration of supplemental oxygen in this proposed trial of supplemental oxygen in COPD, the principal maneuver—administration of oxygen—and all the co-maneuvers that are ideally equally distributed between both groups. This balanced distribution of co-maneuvers between the compared groups helps to ensure that any differences in the study outcome measures (ie, what is counted as the main impact of the intervention under study) can be solely attributed to the principal maneuver. When this condition—that the difference in outcomes can be reliably ascribed to the study intervention—is satisfied, the study is felt to be “internally” valid. As we will see, ensuring internal validity requires freedom from the many sources of what Dr. Feinstein calls “internal bias.”
Back to basic terms: “cohort” in Dr. Feinstein’s language is a group that shares common traits and is followed forward in a longitudinal study. The “outcome measure” is self-evident—it is what is being measured, with the “primary outcome” being the pre-defined measure that is considered the most important (and ideally most clinically relevant) impact of the study intervention. Later in this series of lectures, there will be discussions of power calculations and the so-called “effect size”—the magnitude of effect that the intervention is expected to produce and that is ideally deemed clinically important.
An important consideration in designing a trial is to define and declare the primary outcome measure carefully because defining the primary outcome measure has important implications for the study. I will provide an example from the alpha-1 antitrypsin deficiency literature. Some of you have probably read what has been called the RAPID trial.5 RAPID was a trial of augmentation therapy vs placebo in patients with severe alpha-1 antitrypsin deficiency. The primary outcome measure (which was pre-negotiated with the US Food and Drug Administration [FDA]) was computer tomography (CT) lung density determined at functional residual capacity (FRC) and total lung capacity (TLC). The trial failed to achieve statistical significance in regard to CT lung density, although the study authors argued that CT density measurements made at TLC were more reproducible than those made at FRC. When the results were analyzed by TLC alone, the results were statistically significant, but when they were analyzed with FRC and TLC combined, they were not. In the end, based on the pre-negotiated primary outcome measure of CT density based on both FRC and TLC, the FDA rejected the proposal for a label change to say that augmentation therapy slowed the loss of lung density even though the weight of evidence was clearly in its favor. This case exemplifies just how critical the choice of primary outcome measure can be.
The wash-out period refers to an interval in a subset of randomized trials called “crossover trials” in which the primary intervention is discontinued and the patient returns to his baseline state before the comparative maneuver is then implemented (Figure 2).6 In order to perform a crossover trial, it is important that the effects of the initial intervention can “wash out” or be fully extinguished. So, for example, in trials of radiation therapy vs surgery, it is impossible to do a crossover trial because the effects of radiation can never completely wash out nor can those of surgery, which are similarly permanent. For example, we cannot replace the colon once it is resected for cancer or replace the appendix once removed. Therefore, producing a wash-out requires some very specific pharmacokinetic and pharmacodynamic features in order for a crossover trial to be considered. Later talks in this series will discuss the enhanced statistical power of a crossover trial, where one is comparing every patient to him or herself rather than to another patient.
So, there is always an appetite to do a crossover trial as long as the criteria for wash-out can be met, namely again that the primary intervention can dissipate completely to the baseline state before the alternative intervention is implemented.
“Placebo” is a fairly self-evident and well-understood term; placebo refers to the administration of a maneuver in a way that is identical to the principal maneuver except that the placebo is not expected to exert any clinical effect.
“Blinding” is the unawareness of either the investigator or of the patient to which the intervention is being administered. “Single-blinding” refers to the condition in which either the study or the investigator (but not both) is unaware, and “double-blinding” refers to the condition in which both the subjects and the investigators are unaware. There can be some subtle issues that compromise whether the patient is aware of the intervention that he or she is receiving and that can potentially condition the patient’s response, particularly if there is any subjective component of the assessment of the outcome. So, blinding is important.
With these terms describing the elements of a clinical study now described, let us turn to the types of studies that comprise clinical research. The first group of study types is what Dr. Feinstein called descriptive studies—studies that simply describe phenomena without comparison to a control group. As an example of a descriptive study, Sehgal et al7 recently described the workup of a focal, segmental pneumonia in a patient taking pembrolizumab for lung cancer. In this paper, there were four other cases of focal pneumonia accompanying pembrolizumab use that were assembled from the literature, making this descriptive paper a so-called case series. A “case series” differs from a “single case report,” which reports a single patient experience. Though limited in their ability to establish cause and effect, case reports and case series can help researchers develop proof of principle, so I would not discount the value of case reports.8
I can cite a case report from of my own experience that demonstrates this point. In 1987, I saw a patient from Buffalo who had primary biliary cirrhosis and the hepatopulmonary syndrome (HPS). She was so debilitated by her HPS that she could not stand up without desaturating severely. Although she had normal liver synthetic function, she was severely debilitated by her HPS and the decision was made to offer her a liver transplant, which, at that time, was considered to be relatively contraindicated. Much to everyone’s amazement and satisfaction, her HPS completely resolved after the transplant surgery. Her oxygenation and alveolar-arterial oxygen gradient normalized, and her clubbing resolved. We reported this in a case report, which began to affect the way people thought about the feasibility of liver transplant for the HPS.8 The lesson is: do not underestimate the power of a thoughtful case report.
The second group of research study types is called “cohort studies,” in which one actually compares outcomes between 2 groups in the study. Cohort studies fall into the bucket of either “observational cohort studies,” in which allocation to the compared maneuvers is not performed by randomization but by any other strategy, and “randomized trials.” In observational studies, allocation could occur through physician choice, as when the physician prescribes a treatment to 1 group but not another, or by patient choice or circumstance. For example, an observational cohort study of the risk of cigarette smoking would compare outcomes between smokers and non-smokers where the patient choses to smoke under his/her own volition. Alternatively, the circumstances of an exposure could allocate someone to the principal maneuver, as when we are studying the effect of exposure to World Trade Center dust in the firefighters who responded or of exposure to nuclear radiation in Hiroshima survivors. These are examples of observational cohort studies that compare exposed individuals to unexposed individuals, where the exposure did not occur by randomization but by choice or unfortunate circumstance.
In contrast to observational studies, allocation in randomized trials occurs through a formal process. Randomization has the specific purpose of attempting to ensure that patients are allocated to 2 comparative groups from the baseline group with comparable risk for developing the outcome measure. When randomization is effective, differences in study outcomes can be reliably ascribed to the intervention rather than to differences in the baseline susceptibility of the compared groups.
While randomization is an excellent strategy to ensure baseline similarity between compared groups, randomization can fail, and its effectiveness must be checked. Specifically, in a randomized trial, it is customary to examine the compared groups at baseline on all features that can affect the likelihood of developing the outcome measure. If the groups turn out to be dissimilar at baseline in an important way, then the study is at risk for bias, which is specifically called “susceptibility bias” in Feinstein’s construct. Obviously, the larger number of baseline clinical and demographic features that can condition the likelihood of developing the outcome measure, the more difficult it is to achieve baseline similarity between compared groups and the more important it becomes to ensure that randomization has been effective. In this circumstance, larger numbers of participants in both compared groups are generally needed. More about susceptibility bias later.
There are generally 2 types of randomized trials: the so-called “parallel controlled trials” in which each group receives either the principal or the comparative maneuver and is followed and “crossover trials” in which each compared group receives both the principal maneuver and the co-maneuver at different times after an effective wash-out period. Wash-out was discussed above. Figure 2 shows an example of a crossover trial examining the effects of terbutaline on diaphragmatic function.6 The investigators administered terbutaline for a week, measured transdiaphragmatic pressures, gave the patient a terbutaline vacation (the “wash-out period”), and then crossed over those patients who were initially receiving terbutaline to placebo and initial placebo recipients to terbutaline, having remeasured diaphragmatic function after the wash-out period to assure that the patient’s diaphragmatic function prior to the second crossover was identical to his/her baseline state. If this return to baseline is accomplished, then the criteria from effective wash-out are satisfied.
As we begin to talk about sources of bias, consider a study in which we compare survival of patients allocated to surgery vs nonsurgical therapy for lung cancer (Figure 3).1 This study is subject to the first type of so-called “internal bias” in the Feinsteinian construct—so-called “selection bias.” For example, all patients treated surgically were considered healthy enough by their doctors to undergo surgery, whereas patients treated without surgery may have been deemed inoperable because of comorbidities, lung dysfunction, cardiac dysfunction, and so on. If the results of such a comparison show that the mortality rate among surgical patients in this study was lower, the question then becomes: is the improved survival in surgical candidates due to the superior efficacy of surgery vs other therapy or was the enhanced survival due to the surgical patients being healthier to begin with? You can intuitively sense that the answer to this question is that the enhanced survival may be due to the better health of patients treated surgically rather than to the surgery itself because of how the patients were selected to receive it. So, this is a simple example of what Dr. Feinstein would call “susceptibility bias.” Susceptibility bias occurs when the 2 baseline groups are not comparably at risk or susceptible to developing the outcome measure, leading the naïve investigator in this specific example to attribute the difference in outcomes to the superiority of surgery when in fact it may have nothing to do with the surgery vs. the other maneuver. When susceptibility bias is in play, the difference between the outcomes in the compared groups could be attributed to the baseline imbalance of the groups rather than to the principal maneuver itself.
Turning back to the taxonomy of bias, there are four types that can threaten internal validity—“susceptibility,” “performance,” “detection,” and “transfer” bias—and 1 type of bias (called “external bias”) that can affect the generalizability of the study called “assembly bias” (Table 1).
Figure 4 shows where these various sources of bias appear in the architecture of a clinical trial. As just discussed, susceptibility bias affects the baseline state and the comparability of the groups. Performance bias relates to how effective and how comparably the co-maneuvers are given and whether the primary intervention is potent enough to affect an outcome. Both transfer and detection bias operate in detecting the outcome, especially regarding the rigor and frequency with which they are investigated. Transfer bias has to do with selective loss to follow-up of those included in the trial. If there is a systematic reason for loss to follow-up that is related to the impact of the intervention, then the study is at risk for transfer bias. For example, in a randomized trial of drug A vs placebo for pneumonia, if drug A is effective but all the drug A recipients fail to follow-up because they feel too good to return for follow-up, then transfer bias could be causing the study to show nonefficacy even though the drug works. So, if those who respond favorably are systematically lost to follow-up, and if all the patients who felt lousy wanted to see the doctor and came back for follow-up, such transfer bias would bias towards nonefficacy. Specifically, only patients remaining in the trial would be those who failed to respond and that would dilute any difference between the 2 groups despite the active efficacy of drug A.
Hopefully, you are already beginning to get a sense that one has to be extremely disciplined in thinking about each of these sources of bias because they can have some very subtle nuances in randomized trials that can easily escape attention.
Returning to sources of bias, let’s consider the second type of bias, “performance bias.” Performance bias relates to the administration of the compared maneuvers—the primary or principal maneuver, compared with the comparative maneuver. Performance bias can occur when the main maneuver is not administered adequately or when the co-maneuvers are administered in an imbalanced way between the compared groups. Consider the example of the Long-Term Oxygen Treatment Trial (LOTT) trial, which compared use of supplemental oxygen with no supplemental oxygen in patients with stable COPD and resting or exercise-induced moderate desaturation.9 The principal outcome measure of LOTT was all-cause hospitalization or death. In such a study, many potential sources of performance bias exist. For example, performance bias might exist if none of the patients allocated to oxygen actually used supplemental oxygen. Alternately, to the extent that use of inhaled corticosteroids or antimuscarinic agents lessens the risk of COPD exacerbation, performance bias could occur if use of these co-maneuvers was imbalanced between the compared groups. As a specific extreme circumstance, if all patients in the nonoxygen group used these inhalers but none of the patients in the oxygen group did, then a lack of difference between exacerbation frequency could be related to this imbalance in co-maneuvers (a form of performance bias) rather than to the lack of efficacy of supplemental oxygen.
“Compliance bias” is a subset of performance bias which occurs when 2 conditions are satisfied: (1) the main maneuver is not administered adequately, and (2) the investigator is unaware of that nonreceipt so that this cannot be accounted for in interpreting the study results. For example, if a drug has efficacy but if no one in the treatment arm of the trial takes the drug, the absence of a difference in outcomes between the compared groups will be ascribed to nonefficacy, whereas “compliance bias” (ie, no one actually took the drug) could actually be the cause. Ideally, randomized studies should be evaluated on an “intention to treat” basis irrespective of compliance, but there is an analytic approach called “per protocol” analysis in which you can analyze the results according to whether the patient actually used the intervention in an effective way. “Per protocol” analysis is a secondary analysis of the primary results but it can nonetheless help determine whether the negative result is likely related to noncompliance or not.
A third type of internal bias, “detection bias,” is fairly straightforward. Detection bias is related to how avidly and how comparably the outcomes are measured between the 2 compared groups. Let’s say that you are conducting a trial of a new antibiotic and the primary outcome is colony counts on petri dishes of plated collected specimens. If the technicians who read the petri dish counts are unblinded, they may look at the colony counts with a biased eye, seeing fewer colonies on plates collected from patients receiving the antibiotic.
Overall, detection bias occurs when outcomes are ascertained or detected unequally between the compared groups, and detection bias can involve any of the following: is there comparable surveillance of the 2 groups for analysis of the outcome measure? Are the diagnostic tests comparably performed in both groups and is the interpretation comparably unbiased with equipoise? Investigators who know which patients are receiving an active drug and those who are not could experience subliminal bias that renders them more likely to find that the drug under study is efficacious.
Depending on the principal study maneuver, ensuring blinding can be challenging. To demonstrate this point, let’s consider the example of conducting a randomized control trial of Vicks VapoRub. Vicks VapoRub is an old product that smells like wintergreen and that mothers used to rub on the chests of their infants in the hope of speeding recovery from colds and bronchitis episodes. It was felt that the distinctive smell of the product was materially related to wintergreen, which gives rise to the odor. So, imagine a randomized trial of Vicks VaporRub. A trial is designed in which sick children receive Vicks VapoRub on their chest and others receive a placebo rub that lacks the distinctive wintergreen odor. But, the odor itself is felt to be related to how Vicks VapoRub actually works. Thus, it is the odor itself that creates the blinding challenge here.
The primary outcomes in this study are the duration of the child’s cold symptoms, as ascertained by pediatricians actually examining the children. So, pediatricians would come and listen to the infants’ chests: “Yeah, this chest is clear, but this other infant is still full of rhonchi,” and they would ascertain the outcome measure in this way. So, my blinding question to you is: how do you blind a trial of Vicks VapoRub given the conditions described? Namely, you put the VapoRub on the chest, it smells and the smell is the intervention—how do you blind such a trial?
The clever answer is that you should put Vicks VapoRub on the upper lips of all the examiners, so what they smell is Vicks VapoRub independent of whether the child they are examining also has the Vicks VapoRub or placebo on their chest. In this way, single blinding of the examiners is preserved and detection bias is averted. It is important to point out that double blinding could also be achieved by placing Vicks VapoRub on the child’s upper lip, but there is little reason to suspect that the infants being studied have a bias related to whether they smell the Vicks VapoRub.
The fourth potential source of internal bias is called “transfer bias.” Transfer bias is the selective loss to follow-up of patients from 1 of the 2 compared groups in the trial for a systematic reason. By systematic, I mean that that the drop-out is associated with the development of the outcome event or some impact of the intervention regarding the likelihood to develop the outcome event. As an example, if all patients respond favorably to a drug and everybody fails to follow up because they feel too good to come back, then that would bias the study towards nonefficacy even in the face of an efficacious intervention.
Finally, let’s consider a source of bias that can affect the “external validity,” or the generalizability of the study results to populations other than that included in the study itself. Dr. Feinstein calls this 5th type of bias “assembly bias” (Table 1).1 Assembly bias occurs when the results of the study cannot be reliably applied to populations outside the study itself.
For example, if I screen patients during a study of digoxin for heart rate control in atrial fibrillation, I could establish whether the subject was compliant or not by checking his/her serum digoxin levels. Serum levels of 0 indicate that the patient has not taken the digoxin. If I include a run-in period for the trial—an interval before the actual study when I am assessing potential subjects’ eligibility to participate—and check serum digoxin levels to include only patients who are shown to be taking the drug, then I am screening for study inclusion on compliance. In this way, I will have assembled a population that is highly compliant so that I can truly assess whether digoxin has efficacy in controlling the heart rate in patients with atrial fibrillation. At the same time, this study population is not highly representative of the population of patients with atrial fibrillation at large, because we know that rates of drug noncompliances may be as high as 30% to 40%. So, culling a population with run-in periods on demonstrated compliance criteria may be very important to assess efficacy (ie, whether the drug works), but this design will trade off on the effectiveness of the drug (ie, which asks the question “does the drug work in actual practice?”). This is because, in the yin-yang between assessing efficacy and assessing effectiveness, the focus on assessing efficacy naturally undermines the ability to assess whether the drug works in real-world conditions.
As another example of potential assembly bias, let’s say you are studying an antihypertensive drug at a Veterans Administration (VA) hospital, where most veterans are men. But you are treating women in your practice and wonder whether the drug, which works in a predominately male population, will work in your female patients. So, there could be assembly bias in applying the results of a VA study to a non-VA predominantly female population.
Having now described the design of clinical trials and the major sources of bias, let’s apply this thinking to the earliest clinical trial. James Lind, a British Naval officer, was credited with conducting the first clinical trial of citrus fruits for scurvy while sailing on the ship Salisbury in 1747.2 The question that Lind addressed was “does citrus fruit treat and prevent scurvy?” In describing this trial, Lind stated “I took 12 patients with scurvy, these patients were as similar as I could have them, had one diet common to all.” As you read this through your new Feinsteinian bias lens, Lind is addressing 2 potential sources of bias, namely, susceptibility bias and performance bias. In trying to make the “cases as similar as I could have them,” he is trying to avoid susceptibility bias and in “providing one diet common to all,” he is trying to avoid performance bias.
In terms of the intervention in this trial, these 12 patients were allocated in pairs to several interventions: a quart of cider a day, 25 drops of elixir of vitriol 3 times a day on an empty stomach, 2 spoonsful of vinegar 3 times a day on an empty stomach, ½ pint a day of sea water, 2 oranges and 1 lemon given every day, and a “bigness of nutmeg” 3 times per day. In describing the outcome of the trial, Lind states “the consequence was that the most sudden and visible good effects were perceived from the use of oranges and lemons; one of those who had taken them, being at the end of 6 days fit for duty. The spots were not indeed at that time quite off his body, nor his gums sound, but without any other medicine then a gargarism of elixir vitriol, he became quite healthy before we came into Plymouth which was on the 16th of June. The other was the best recovered of any in his condition; and being now deemed pretty well, was appointed nurse to the rest of the sick.”
In analyzing this trial, we could characterize it as a parallel controlled trial. Whether the allocation was done by randomization is not clear, but it was certainly an observational cohort study in that there were concurrent controls who were treated as similarly as possible except for the principal maneuver, which was the administration of citrus fruit. Already mentioned was the attention to averting susceptibility and performance bias. There was no evidence of compliance bias as the interventions were enforced, nor was there evidence of transfer bias because all subjects who were enrolled in the study completed the study because they were a captive group on a sailing ship. Finally, the likelihood of assembly bias seems small, as these sailors seemed to be representative of victims of scurvy in general, namely in being otherwise deprived of access to citrus fruits.
In terms of the statistical results of this study, subsequent analysis of the research showed that the impact of lemons and oranges was dramatic and showed a trend (P = .09) towards statistical significance. Notwithstanding the lack of a P < .05, Dr. Feinstein would likely say that this study satisfied the “intra-ocular test” in that the efficacy of the citrus fruit was so dramatic that it “hit you between the eyes.” He often argued that the widespread practice of prescribing penicillin for pneumococcal pneumonia was not based on the results of a convincing randomized controlled trial because the efficacy of penicillin in that setting was so dramatic that a randomized trial was not necessary (and potentially even unethical if the condition of “intra-ocular” efficacy was satisfied).
The final question to address in this lecture is whether randomized controlled trials, for all their rigor, always produce more reliable results than observational studies. This issue has been addressed by several authors.10–12 Sacks et al10 contended in 1983 that observational studies systematically overestimate the magnitude of association between exposure and outcome and therefore argued that randomized trials were more reliable than observational studies. Subsequent analyses tended to challenge this view.11,12 Specifically, Benson and Hartz11 compared the results of 136 reports regarding 19 different therapies that were studied between 1985 and 1998. In only 2 of the 19 analyses did the treatment effects in the observational studies fall outside the 95% confidence interval for the randomized controlled trial results. In this way, these authors argued that observational studies generally are concordant with the results of randomized trials. They stated that “our finding that observational studies and randomized controlled trials usually produce similar results differs from the conclusions of previous authors. The fundamental criticism of observational studies is that unrecognized confounding factors may distort the results. According to the conventional wisdom, this distortion is sufficiently common and unpredictable that observational studies are not liable and should not be funded. Our results suggested observational studies usually do provide valid information.”11
An additional analysis of this issue was performed by Concato et al,12 who identified 99 articles regarding 5 clinical topics. Again, the results from randomized trials were compared with those of observational cohort or case-controlled studies regarding the same intervention. The authors reported that “contrary to prevailing belief, the average results from well-designed observational studies did not systematically overestimate the magnitude of the associations between exposure and outcome as compared with the results of randomized, controlled trials on the same topic. Rather, the summary results of randomized, controlled trials and observational studies were remarkably similar.”12
On the basis of these studies, it appears that randomized control trials continue to serve as the gold standard in clinical research, but we must also recognize that circumstances often preclude the conduct of a randomized trial. As an example, consider a randomized trial of whether cigarette smoking is harmful, which, given the strong suspicion of harm, would be unethical in that patients cannot be randomized to smoke. Similarly, from the example before, a randomized trial of penicillin for pneumococcal pneumonia would be unethical because denying patients in the placebo group access to penicillin would exclude them from access to a drug that has “intra-ocular” efficacy. In circumstances like these, well-performed observational studies that are attentive to sources of bias can likely produce comparably reliable results to randomized trials.
In the end, of course, the interpretation of the study results requires the reader’s careful attention to potential sources of bias that can compromise study validity. The hope is that with Dr. Feinstein’s framework, you can be better equipped to think critically about study results that you review and to keenly ascertain whether there is any threat to internal or to external validity. Similarly, as you go on to design clinical trials yourselves, you can pay attention to these potential sources of bias that, if present, can compromise the reliability of the study conclusions internally or their applicability to patients outside of the study.
I am flattered to present the inaugural talk in the biostatistics and clinical research design series on the architecture of clinical research. This content is based on the teachings of my mentor, Dr. Alvan Feinstein, who together with Dr. David Sackett, is credited with pioneering clinical epidemiology. Dr. Feinstein was a Sterling Professor at the Yale School of Medicine. His main opus of work is a book called, Clinical Epidemiology: The Architecture of Clinical Research.1 This paper is named in credit to Dr. Feinstein’s enormous contribution. I will review some important terms defined by Dr. Feinstein to provide the background necessary for the remainder of the talks in this series.
To start, I will frame this topic by asking the following question: Why do we do research? I’ll talk about the basic structure of research studies and provide a taxonomy, as Dr. Feinstein would say, a nomenclature with which to understand trial design and the sources of bias in those trials. Then, I will discuss these sources of bias in detail using the taxonomy that Dr. Feinstein described in his aforementioned book. Finally, I will share with you some examples of bias in clinical trials to help you better understand these concepts.
Now, the answer to the basic question posed above is: basically, we do cause-and-effect research to establish the causality of a risk factor or the efficacy of a therapy. Does cigarette smoking cause lung cancer? Does taking hydrochlorothiazide help systemic hypertension? Does air pollution worsen asthma? Does supplemental oxygen help patients with chronic obstructive pulmonary disease (COPD)?
Cause-and-effect research can be subsumed under 2 broad issues: causal risk factors and therapeutic efficacy. In his review of early false understandings in medicine that were based on anecdotal observation alone, Thomas cites many examples—“the undue longevity of useless and even harmful drugs can be laid at the door of authority,” ie, empiricism, lack of rigorous research.2 The field is full of these: yellow fever causality, the value of cupping, and even intermittent mandatory ventilation when it was described by John Downs in 1973 and touted as a superior mode for weaning patients from mechanical ventilation.3 Twenty-five years later, randomized controlled trials by Brochard et al4 indicated not only that intermittent mandatory ventilation was not the best mode to wean but was, in fact, the worst mode for weaning patients from mechanical ventilation compared with either pressure support or spontaneous breathing trials. Many more examples exist to demonstrate the false understandings that can be ascribed to lack of rigorous study or evidence in medicine.
Before systematically exploring the sources of bias in Feinstein’s construct, let us define some very basic terms from his book. Dr. Feinstein talks about the baseline state, which refers to the group of patients under study who are culled from a larger population to whom the results are intended to be applied (Figure 1).1 This baseline group is hopefully representative of this larger target population. As a nod to the later discussion, Dr. Feinstein would call bias introduced by unusual assembly of the study population from the larger intended population as “assembly bias.” So, if the group under study is not representative of either the patients you see or the world of patients with this condition or if there is something special or distinctively nonrepresentative about the study population, then the results may be subject to “assembly bias.” Assembly bias can compromise the so-called “external” validity of the study—its ability to be applied to populations beyond the study group.
Having assembled a baseline group for study, that group is classically allocated to 1 of 2 (or sometimes more than 2) compared therapies. In a controlled trial, patients can be allocated using a variety of strategies, including randomization. Using the paradigm diagram (Figure 1, which considers a 2-arm trial), patients are allocated to 1 of 2 compared groups—group A and group B. Then, in a treatment trial, 1 group receives the principal maneuver, which is the drug or intervention under study—for example, supplemental oxygen for patients with COPD. The comparative maneuver is allocated to group B, which also receives all the other treatments (called “co-maneuvers”) that are used to treat the condition under study. In a trial of supplemental oxygen for COPD evaluating lung function and exacerbation frequency as outcome measures, such co-maneuvers might include inhaled bronchodilators, inhaled corticosteroids, pulmonary rehabilitation, and Pneumovax vaccine. Ideally, these co-maneuvers are equally distributed between the compared groups (A and B).
So, in summary, we have a comparative maneuver, which is the nonadministration of supplemental oxygen in this proposed trial of supplemental oxygen in COPD, the principal maneuver—administration of oxygen—and all the co-maneuvers that are ideally equally distributed between both groups. This balanced distribution of co-maneuvers between the compared groups helps to ensure that any differences in the study outcome measures (ie, what is counted as the main impact of the intervention under study) can be solely attributed to the principal maneuver. When this condition—that the difference in outcomes can be reliably ascribed to the study intervention—is satisfied, the study is felt to be “internally” valid. As we will see, ensuring internal validity requires freedom from the many sources of what Dr. Feinstein calls “internal bias.”
Back to basic terms: “cohort” in Dr. Feinstein’s language is a group that shares common traits and is followed forward in a longitudinal study. The “outcome measure” is self-evident—it is what is being measured, with the “primary outcome” being the pre-defined measure that is considered the most important (and ideally most clinically relevant) impact of the study intervention. Later in this series of lectures, there will be discussions of power calculations and the so-called “effect size”—the magnitude of effect that the intervention is expected to produce and that is ideally deemed clinically important.
An important consideration in designing a trial is to define and declare the primary outcome measure carefully because defining the primary outcome measure has important implications for the study. I will provide an example from the alpha-1 antitrypsin deficiency literature. Some of you have probably read what has been called the RAPID trial.5 RAPID was a trial of augmentation therapy vs placebo in patients with severe alpha-1 antitrypsin deficiency. The primary outcome measure (which was pre-negotiated with the US Food and Drug Administration [FDA]) was computer tomography (CT) lung density determined at functional residual capacity (FRC) and total lung capacity (TLC). The trial failed to achieve statistical significance in regard to CT lung density, although the study authors argued that CT density measurements made at TLC were more reproducible than those made at FRC. When the results were analyzed by TLC alone, the results were statistically significant, but when they were analyzed with FRC and TLC combined, they were not. In the end, based on the pre-negotiated primary outcome measure of CT density based on both FRC and TLC, the FDA rejected the proposal for a label change to say that augmentation therapy slowed the loss of lung density even though the weight of evidence was clearly in its favor. This case exemplifies just how critical the choice of primary outcome measure can be.
The wash-out period refers to an interval in a subset of randomized trials called “crossover trials” in which the primary intervention is discontinued and the patient returns to his baseline state before the comparative maneuver is then implemented (Figure 2).6 In order to perform a crossover trial, it is important that the effects of the initial intervention can “wash out” or be fully extinguished. So, for example, in trials of radiation therapy vs surgery, it is impossible to do a crossover trial because the effects of radiation can never completely wash out nor can those of surgery, which are similarly permanent. For example, we cannot replace the colon once it is resected for cancer or replace the appendix once removed. Therefore, producing a wash-out requires some very specific pharmacokinetic and pharmacodynamic features in order for a crossover trial to be considered. Later talks in this series will discuss the enhanced statistical power of a crossover trial, where one is comparing every patient to him or herself rather than to another patient.
So, there is always an appetite to do a crossover trial as long as the criteria for wash-out can be met, namely again that the primary intervention can dissipate completely to the baseline state before the alternative intervention is implemented.
“Placebo” is a fairly self-evident and well-understood term; placebo refers to the administration of a maneuver in a way that is identical to the principal maneuver except that the placebo is not expected to exert any clinical effect.
“Blinding” is the unawareness of either the investigator or of the patient to which the intervention is being administered. “Single-blinding” refers to the condition in which either the study or the investigator (but not both) is unaware, and “double-blinding” refers to the condition in which both the subjects and the investigators are unaware. There can be some subtle issues that compromise whether the patient is aware of the intervention that he or she is receiving and that can potentially condition the patient’s response, particularly if there is any subjective component of the assessment of the outcome. So, blinding is important.
With these terms describing the elements of a clinical study now described, let us turn to the types of studies that comprise clinical research. The first group of study types is what Dr. Feinstein called descriptive studies—studies that simply describe phenomena without comparison to a control group. As an example of a descriptive study, Sehgal et al7 recently described the workup of a focal, segmental pneumonia in a patient taking pembrolizumab for lung cancer. In this paper, there were four other cases of focal pneumonia accompanying pembrolizumab use that were assembled from the literature, making this descriptive paper a so-called case series. A “case series” differs from a “single case report,” which reports a single patient experience. Though limited in their ability to establish cause and effect, case reports and case series can help researchers develop proof of principle, so I would not discount the value of case reports.8
I can cite a case report from of my own experience that demonstrates this point. In 1987, I saw a patient from Buffalo who had primary biliary cirrhosis and the hepatopulmonary syndrome (HPS). She was so debilitated by her HPS that she could not stand up without desaturating severely. Although she had normal liver synthetic function, she was severely debilitated by her HPS and the decision was made to offer her a liver transplant, which, at that time, was considered to be relatively contraindicated. Much to everyone’s amazement and satisfaction, her HPS completely resolved after the transplant surgery. Her oxygenation and alveolar-arterial oxygen gradient normalized, and her clubbing resolved. We reported this in a case report, which began to affect the way people thought about the feasibility of liver transplant for the HPS.8 The lesson is: do not underestimate the power of a thoughtful case report.
The second group of research study types is called “cohort studies,” in which one actually compares outcomes between 2 groups in the study. Cohort studies fall into the bucket of either “observational cohort studies,” in which allocation to the compared maneuvers is not performed by randomization but by any other strategy, and “randomized trials.” In observational studies, allocation could occur through physician choice, as when the physician prescribes a treatment to 1 group but not another, or by patient choice or circumstance. For example, an observational cohort study of the risk of cigarette smoking would compare outcomes between smokers and non-smokers where the patient choses to smoke under his/her own volition. Alternatively, the circumstances of an exposure could allocate someone to the principal maneuver, as when we are studying the effect of exposure to World Trade Center dust in the firefighters who responded or of exposure to nuclear radiation in Hiroshima survivors. These are examples of observational cohort studies that compare exposed individuals to unexposed individuals, where the exposure did not occur by randomization but by choice or unfortunate circumstance.
In contrast to observational studies, allocation in randomized trials occurs through a formal process. Randomization has the specific purpose of attempting to ensure that patients are allocated to 2 comparative groups from the baseline group with comparable risk for developing the outcome measure. When randomization is effective, differences in study outcomes can be reliably ascribed to the intervention rather than to differences in the baseline susceptibility of the compared groups.
While randomization is an excellent strategy to ensure baseline similarity between compared groups, randomization can fail, and its effectiveness must be checked. Specifically, in a randomized trial, it is customary to examine the compared groups at baseline on all features that can affect the likelihood of developing the outcome measure. If the groups turn out to be dissimilar at baseline in an important way, then the study is at risk for bias, which is specifically called “susceptibility bias” in Feinstein’s construct. Obviously, the larger number of baseline clinical and demographic features that can condition the likelihood of developing the outcome measure, the more difficult it is to achieve baseline similarity between compared groups and the more important it becomes to ensure that randomization has been effective. In this circumstance, larger numbers of participants in both compared groups are generally needed. More about susceptibility bias later.
There are generally 2 types of randomized trials: the so-called “parallel controlled trials” in which each group receives either the principal or the comparative maneuver and is followed and “crossover trials” in which each compared group receives both the principal maneuver and the co-maneuver at different times after an effective wash-out period. Wash-out was discussed above. Figure 2 shows an example of a crossover trial examining the effects of terbutaline on diaphragmatic function.6 The investigators administered terbutaline for a week, measured transdiaphragmatic pressures, gave the patient a terbutaline vacation (the “wash-out period”), and then crossed over those patients who were initially receiving terbutaline to placebo and initial placebo recipients to terbutaline, having remeasured diaphragmatic function after the wash-out period to assure that the patient’s diaphragmatic function prior to the second crossover was identical to his/her baseline state. If this return to baseline is accomplished, then the criteria from effective wash-out are satisfied.
As we begin to talk about sources of bias, consider a study in which we compare survival of patients allocated to surgery vs nonsurgical therapy for lung cancer (Figure 3).1 This study is subject to the first type of so-called “internal bias” in the Feinsteinian construct—so-called “selection bias.” For example, all patients treated surgically were considered healthy enough by their doctors to undergo surgery, whereas patients treated without surgery may have been deemed inoperable because of comorbidities, lung dysfunction, cardiac dysfunction, and so on. If the results of such a comparison show that the mortality rate among surgical patients in this study was lower, the question then becomes: is the improved survival in surgical candidates due to the superior efficacy of surgery vs other therapy or was the enhanced survival due to the surgical patients being healthier to begin with? You can intuitively sense that the answer to this question is that the enhanced survival may be due to the better health of patients treated surgically rather than to the surgery itself because of how the patients were selected to receive it. So, this is a simple example of what Dr. Feinstein would call “susceptibility bias.” Susceptibility bias occurs when the 2 baseline groups are not comparably at risk or susceptible to developing the outcome measure, leading the naïve investigator in this specific example to attribute the difference in outcomes to the superiority of surgery when in fact it may have nothing to do with the surgery vs. the other maneuver. When susceptibility bias is in play, the difference between the outcomes in the compared groups could be attributed to the baseline imbalance of the groups rather than to the principal maneuver itself.
Turning back to the taxonomy of bias, there are four types that can threaten internal validity—“susceptibility,” “performance,” “detection,” and “transfer” bias—and 1 type of bias (called “external bias”) that can affect the generalizability of the study called “assembly bias” (Table 1).
Figure 4 shows where these various sources of bias appear in the architecture of a clinical trial. As just discussed, susceptibility bias affects the baseline state and the comparability of the groups. Performance bias relates to how effective and how comparably the co-maneuvers are given and whether the primary intervention is potent enough to affect an outcome. Both transfer and detection bias operate in detecting the outcome, especially regarding the rigor and frequency with which they are investigated. Transfer bias has to do with selective loss to follow-up of those included in the trial. If there is a systematic reason for loss to follow-up that is related to the impact of the intervention, then the study is at risk for transfer bias. For example, in a randomized trial of drug A vs placebo for pneumonia, if drug A is effective but all the drug A recipients fail to follow-up because they feel too good to return for follow-up, then transfer bias could be causing the study to show nonefficacy even though the drug works. So, if those who respond favorably are systematically lost to follow-up, and if all the patients who felt lousy wanted to see the doctor and came back for follow-up, such transfer bias would bias towards nonefficacy. Specifically, only patients remaining in the trial would be those who failed to respond and that would dilute any difference between the 2 groups despite the active efficacy of drug A.
Hopefully, you are already beginning to get a sense that one has to be extremely disciplined in thinking about each of these sources of bias because they can have some very subtle nuances in randomized trials that can easily escape attention.
Returning to sources of bias, let’s consider the second type of bias, “performance bias.” Performance bias relates to the administration of the compared maneuvers—the primary or principal maneuver, compared with the comparative maneuver. Performance bias can occur when the main maneuver is not administered adequately or when the co-maneuvers are administered in an imbalanced way between the compared groups. Consider the example of the Long-Term Oxygen Treatment Trial (LOTT) trial, which compared use of supplemental oxygen with no supplemental oxygen in patients with stable COPD and resting or exercise-induced moderate desaturation.9 The principal outcome measure of LOTT was all-cause hospitalization or death. In such a study, many potential sources of performance bias exist. For example, performance bias might exist if none of the patients allocated to oxygen actually used supplemental oxygen. Alternately, to the extent that use of inhaled corticosteroids or antimuscarinic agents lessens the risk of COPD exacerbation, performance bias could occur if use of these co-maneuvers was imbalanced between the compared groups. As a specific extreme circumstance, if all patients in the nonoxygen group used these inhalers but none of the patients in the oxygen group did, then a lack of difference between exacerbation frequency could be related to this imbalance in co-maneuvers (a form of performance bias) rather than to the lack of efficacy of supplemental oxygen.
“Compliance bias” is a subset of performance bias which occurs when 2 conditions are satisfied: (1) the main maneuver is not administered adequately, and (2) the investigator is unaware of that nonreceipt so that this cannot be accounted for in interpreting the study results. For example, if a drug has efficacy but if no one in the treatment arm of the trial takes the drug, the absence of a difference in outcomes between the compared groups will be ascribed to nonefficacy, whereas “compliance bias” (ie, no one actually took the drug) could actually be the cause. Ideally, randomized studies should be evaluated on an “intention to treat” basis irrespective of compliance, but there is an analytic approach called “per protocol” analysis in which you can analyze the results according to whether the patient actually used the intervention in an effective way. “Per protocol” analysis is a secondary analysis of the primary results but it can nonetheless help determine whether the negative result is likely related to noncompliance or not.
A third type of internal bias, “detection bias,” is fairly straightforward. Detection bias is related to how avidly and how comparably the outcomes are measured between the 2 compared groups. Let’s say that you are conducting a trial of a new antibiotic and the primary outcome is colony counts on petri dishes of plated collected specimens. If the technicians who read the petri dish counts are unblinded, they may look at the colony counts with a biased eye, seeing fewer colonies on plates collected from patients receiving the antibiotic.
Overall, detection bias occurs when outcomes are ascertained or detected unequally between the compared groups, and detection bias can involve any of the following: is there comparable surveillance of the 2 groups for analysis of the outcome measure? Are the diagnostic tests comparably performed in both groups and is the interpretation comparably unbiased with equipoise? Investigators who know which patients are receiving an active drug and those who are not could experience subliminal bias that renders them more likely to find that the drug under study is efficacious.
Depending on the principal study maneuver, ensuring blinding can be challenging. To demonstrate this point, let’s consider the example of conducting a randomized control trial of Vicks VapoRub. Vicks VapoRub is an old product that smells like wintergreen and that mothers used to rub on the chests of their infants in the hope of speeding recovery from colds and bronchitis episodes. It was felt that the distinctive smell of the product was materially related to wintergreen, which gives rise to the odor. So, imagine a randomized trial of Vicks VaporRub. A trial is designed in which sick children receive Vicks VapoRub on their chest and others receive a placebo rub that lacks the distinctive wintergreen odor. But, the odor itself is felt to be related to how Vicks VapoRub actually works. Thus, it is the odor itself that creates the blinding challenge here.
The primary outcomes in this study are the duration of the child’s cold symptoms, as ascertained by pediatricians actually examining the children. So, pediatricians would come and listen to the infants’ chests: “Yeah, this chest is clear, but this other infant is still full of rhonchi,” and they would ascertain the outcome measure in this way. So, my blinding question to you is: how do you blind a trial of Vicks VapoRub given the conditions described? Namely, you put the VapoRub on the chest, it smells and the smell is the intervention—how do you blind such a trial?
The clever answer is that you should put Vicks VapoRub on the upper lips of all the examiners, so what they smell is Vicks VapoRub independent of whether the child they are examining also has the Vicks VapoRub or placebo on their chest. In this way, single blinding of the examiners is preserved and detection bias is averted. It is important to point out that double blinding could also be achieved by placing Vicks VapoRub on the child’s upper lip, but there is little reason to suspect that the infants being studied have a bias related to whether they smell the Vicks VapoRub.
The fourth potential source of internal bias is called “transfer bias.” Transfer bias is the selective loss to follow-up of patients from 1 of the 2 compared groups in the trial for a systematic reason. By systematic, I mean that that the drop-out is associated with the development of the outcome event or some impact of the intervention regarding the likelihood to develop the outcome event. As an example, if all patients respond favorably to a drug and everybody fails to follow up because they feel too good to come back, then that would bias the study towards nonefficacy even in the face of an efficacious intervention.
Finally, let’s consider a source of bias that can affect the “external validity,” or the generalizability of the study results to populations other than that included in the study itself. Dr. Feinstein calls this 5th type of bias “assembly bias” (Table 1).1 Assembly bias occurs when the results of the study cannot be reliably applied to populations outside the study itself.
For example, if I screen patients during a study of digoxin for heart rate control in atrial fibrillation, I could establish whether the subject was compliant or not by checking his/her serum digoxin levels. Serum levels of 0 indicate that the patient has not taken the digoxin. If I include a run-in period for the trial—an interval before the actual study when I am assessing potential subjects’ eligibility to participate—and check serum digoxin levels to include only patients who are shown to be taking the drug, then I am screening for study inclusion on compliance. In this way, I will have assembled a population that is highly compliant so that I can truly assess whether digoxin has efficacy in controlling the heart rate in patients with atrial fibrillation. At the same time, this study population is not highly representative of the population of patients with atrial fibrillation at large, because we know that rates of drug noncompliances may be as high as 30% to 40%. So, culling a population with run-in periods on demonstrated compliance criteria may be very important to assess efficacy (ie, whether the drug works), but this design will trade off on the effectiveness of the drug (ie, which asks the question “does the drug work in actual practice?”). This is because, in the yin-yang between assessing efficacy and assessing effectiveness, the focus on assessing efficacy naturally undermines the ability to assess whether the drug works in real-world conditions.
As another example of potential assembly bias, let’s say you are studying an antihypertensive drug at a Veterans Administration (VA) hospital, where most veterans are men. But you are treating women in your practice and wonder whether the drug, which works in a predominately male population, will work in your female patients. So, there could be assembly bias in applying the results of a VA study to a non-VA predominantly female population.
Having now described the design of clinical trials and the major sources of bias, let’s apply this thinking to the earliest clinical trial. James Lind, a British Naval officer, was credited with conducting the first clinical trial of citrus fruits for scurvy while sailing on the ship Salisbury in 1747.2 The question that Lind addressed was “does citrus fruit treat and prevent scurvy?” In describing this trial, Lind stated “I took 12 patients with scurvy, these patients were as similar as I could have them, had one diet common to all.” As you read this through your new Feinsteinian bias lens, Lind is addressing 2 potential sources of bias, namely, susceptibility bias and performance bias. In trying to make the “cases as similar as I could have them,” he is trying to avoid susceptibility bias and in “providing one diet common to all,” he is trying to avoid performance bias.
In terms of the intervention in this trial, these 12 patients were allocated in pairs to several interventions: a quart of cider a day, 25 drops of elixir of vitriol 3 times a day on an empty stomach, 2 spoonsful of vinegar 3 times a day on an empty stomach, ½ pint a day of sea water, 2 oranges and 1 lemon given every day, and a “bigness of nutmeg” 3 times per day. In describing the outcome of the trial, Lind states “the consequence was that the most sudden and visible good effects were perceived from the use of oranges and lemons; one of those who had taken them, being at the end of 6 days fit for duty. The spots were not indeed at that time quite off his body, nor his gums sound, but without any other medicine then a gargarism of elixir vitriol, he became quite healthy before we came into Plymouth which was on the 16th of June. The other was the best recovered of any in his condition; and being now deemed pretty well, was appointed nurse to the rest of the sick.”
In analyzing this trial, we could characterize it as a parallel controlled trial. Whether the allocation was done by randomization is not clear, but it was certainly an observational cohort study in that there were concurrent controls who were treated as similarly as possible except for the principal maneuver, which was the administration of citrus fruit. Already mentioned was the attention to averting susceptibility and performance bias. There was no evidence of compliance bias as the interventions were enforced, nor was there evidence of transfer bias because all subjects who were enrolled in the study completed the study because they were a captive group on a sailing ship. Finally, the likelihood of assembly bias seems small, as these sailors seemed to be representative of victims of scurvy in general, namely in being otherwise deprived of access to citrus fruits.
In terms of the statistical results of this study, subsequent analysis of the research showed that the impact of lemons and oranges was dramatic and showed a trend (P = .09) towards statistical significance. Notwithstanding the lack of a P < .05, Dr. Feinstein would likely say that this study satisfied the “intra-ocular test” in that the efficacy of the citrus fruit was so dramatic that it “hit you between the eyes.” He often argued that the widespread practice of prescribing penicillin for pneumococcal pneumonia was not based on the results of a convincing randomized controlled trial because the efficacy of penicillin in that setting was so dramatic that a randomized trial was not necessary (and potentially even unethical if the condition of “intra-ocular” efficacy was satisfied).
The final question to address in this lecture is whether randomized controlled trials, for all their rigor, always produce more reliable results than observational studies. This issue has been addressed by several authors.10–12 Sacks et al10 contended in 1983 that observational studies systematically overestimate the magnitude of association between exposure and outcome and therefore argued that randomized trials were more reliable than observational studies. Subsequent analyses tended to challenge this view.11,12 Specifically, Benson and Hartz11 compared the results of 136 reports regarding 19 different therapies that were studied between 1985 and 1998. In only 2 of the 19 analyses did the treatment effects in the observational studies fall outside the 95% confidence interval for the randomized controlled trial results. In this way, these authors argued that observational studies generally are concordant with the results of randomized trials. They stated that “our finding that observational studies and randomized controlled trials usually produce similar results differs from the conclusions of previous authors. The fundamental criticism of observational studies is that unrecognized confounding factors may distort the results. According to the conventional wisdom, this distortion is sufficiently common and unpredictable that observational studies are not liable and should not be funded. Our results suggested observational studies usually do provide valid information.”11
An additional analysis of this issue was performed by Concato et al,12 who identified 99 articles regarding 5 clinical topics. Again, the results from randomized trials were compared with those of observational cohort or case-controlled studies regarding the same intervention. The authors reported that “contrary to prevailing belief, the average results from well-designed observational studies did not systematically overestimate the magnitude of the associations between exposure and outcome as compared with the results of randomized, controlled trials on the same topic. Rather, the summary results of randomized, controlled trials and observational studies were remarkably similar.”12
On the basis of these studies, it appears that randomized control trials continue to serve as the gold standard in clinical research, but we must also recognize that circumstances often preclude the conduct of a randomized trial. As an example, consider a randomized trial of whether cigarette smoking is harmful, which, given the strong suspicion of harm, would be unethical in that patients cannot be randomized to smoke. Similarly, from the example before, a randomized trial of penicillin for pneumococcal pneumonia would be unethical because denying patients in the placebo group access to penicillin would exclude them from access to a drug that has “intra-ocular” efficacy. In circumstances like these, well-performed observational studies that are attentive to sources of bias can likely produce comparably reliable results to randomized trials.
In the end, of course, the interpretation of the study results requires the reader’s careful attention to potential sources of bias that can compromise study validity. The hope is that with Dr. Feinstein’s framework, you can be better equipped to think critically about study results that you review and to keenly ascertain whether there is any threat to internal or to external validity. Similarly, as you go on to design clinical trials yourselves, you can pay attention to these potential sources of bias that, if present, can compromise the reliability of the study conclusions internally or their applicability to patients outside of the study.
- Feinstein AR. Clinical Epidemiology: The Architecture of Clinical Research. Philadelphia, PA: WB Saunders; 1985.
- Thomas DP. Experiment versus authority: James Lind and Benjamin Rush. N Engl J Med 1969; 281:932–934.
- Downs JB, Klein EF Jr, Desautels D, Modell JH, Kirby RR. Intermittent mandatory ventilation: a new approach to weaning patients from mechanical ventilators. Chest 1973; 64:331–335.
- Brochard L, Rauss A, Benito S, et al. Comparison of three methods of gradual withdrawal from ventilatory support during weaning from mechanical ventilation. Am J Respir Crit Care Med 1994; 150:896–903.
- Chapman KR, Burdon JGW, Piitulainen E, et al; on behalf of the RAPID Trial Study Group. Intravenous augmentation treatment and lung density in severe 1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet 2015; 386:360–368.
- Stoller JK, Wiedemann HP, Loke J, Snyder P, Virgulto J, Matthay RA. Terbutaline and diaphragm function in chronic obstructive pulmonary disease: a double-blind randomized clinical trial. Br J Dis Chest 1988; 82:242–250.
- Sehgal S, Velcheti V, Mukhopadhyay S, Stoller JK. Focal lung infiltrate complicating PD-1 inhibitor use: a new pattern of drug-associated lung toxicity? Respir Med Case Rep 2016; 19:118–120.
- Stoller JK, Moodie D, Schiavone WA, et al. Reduction of intrapulmonary shunt and resolution of digital clubbing associated with primary biliary cirrhosis after liver transplantation. Hepatology 1990; 11:54–58.
- Albert RK, Au DH, Blackford AL, et al; for the Long-Term Oxygen Treatment Trial Group. A randomized trial of long-term oxygen for COPD with moderate desaturation. N Engl J Med 2016; 375:1617–1627.
- Sacks HS, Chalmers TC, Smith H Jr. Sensitivity and specificity of clinical trials: randomized v historical controls. Arch Intern Med 1983; 143:753–755.
- Benson K, Hartz AJ. A comparison of observational studies and randomized, controlled trials. N Engl J Med 2000; 342:1878–1886.
- Concato J, Shah N, Horwitz RI. Randomized, controlled trials, observational studies, and the hierarchy of research designs. N Engl J Med 2000; 342:1887–1892.
- Feinstein AR. Clinical Epidemiology: The Architecture of Clinical Research. Philadelphia, PA: WB Saunders; 1985.
- Thomas DP. Experiment versus authority: James Lind and Benjamin Rush. N Engl J Med 1969; 281:932–934.
- Downs JB, Klein EF Jr, Desautels D, Modell JH, Kirby RR. Intermittent mandatory ventilation: a new approach to weaning patients from mechanical ventilators. Chest 1973; 64:331–335.
- Brochard L, Rauss A, Benito S, et al. Comparison of three methods of gradual withdrawal from ventilatory support during weaning from mechanical ventilation. Am J Respir Crit Care Med 1994; 150:896–903.
- Chapman KR, Burdon JGW, Piitulainen E, et al; on behalf of the RAPID Trial Study Group. Intravenous augmentation treatment and lung density in severe 1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet 2015; 386:360–368.
- Stoller JK, Wiedemann HP, Loke J, Snyder P, Virgulto J, Matthay RA. Terbutaline and diaphragm function in chronic obstructive pulmonary disease: a double-blind randomized clinical trial. Br J Dis Chest 1988; 82:242–250.
- Sehgal S, Velcheti V, Mukhopadhyay S, Stoller JK. Focal lung infiltrate complicating PD-1 inhibitor use: a new pattern of drug-associated lung toxicity? Respir Med Case Rep 2016; 19:118–120.
- Stoller JK, Moodie D, Schiavone WA, et al. Reduction of intrapulmonary shunt and resolution of digital clubbing associated with primary biliary cirrhosis after liver transplantation. Hepatology 1990; 11:54–58.
- Albert RK, Au DH, Blackford AL, et al; for the Long-Term Oxygen Treatment Trial Group. A randomized trial of long-term oxygen for COPD with moderate desaturation. N Engl J Med 2016; 375:1617–1627.
- Sacks HS, Chalmers TC, Smith H Jr. Sensitivity and specificity of clinical trials: randomized v historical controls. Arch Intern Med 1983; 143:753–755.
- Benson K, Hartz AJ. A comparison of observational studies and randomized, controlled trials. N Engl J Med 2000; 342:1878–1886.
- Concato J, Shah N, Horwitz RI. Randomized, controlled trials, observational studies, and the hierarchy of research designs. N Engl J Med 2000; 342:1887–1892.

Alpha-1 antitrypsin deficiency: An underrecognized, treatable cause of COPD
Alpha-1 antitrypsin deficiency is a common but underrecognized genetic condition that increases the risk of chronic obstructive pulmonary disease (COPD) and liver disease. Primary care providers can play a critical role in detecting it and managing patients who have it.
RECOGNIZED CASES ARE THE TIP OF THE ICEBERG
First described in 1963,1 alpha-1 antitrypsin deficiency is estimated to affect 100,000 Americans, fewer than 15,000 of whom have received a clinical diagnosis. As further evidence of its underrecognition,2–7 many patients experience long delays between their first symptoms and the diagnosis. Early studies indicated that the average diagnostic delay was 7.2 years,4 and the latest studies, as recent as 2013, indicate a similar diagnostic delay.7
Furthermore, many patients see multiple healthcare providers before receiving the correct diagnosis. A 1994 survey by this author4 found that 43.7% of patients who had severe deficiency of alpha-1 antitrypsin saw at least three physicians before the correct diagnosis was made.
Why is the disease underrecognized?
Several reasons may account for underrecognition of this disease. Many clinicians—including, unfortunately, many pulmonologists—do not know much about it,7,8 do not adhere to clinical guidelines,9,10 or harbor the misperception that there is no therapy available and, therefore, no compelling reason to make a diagnosis.7
Regarding inadequate knowledge, in a study by Taliercio, Chatburn, and this author,8 internal medicine residents scored only 63% correct on a 10-question quiz on diagnostic features of alpha-1 antitrypsin deficiency. There was no evidence of a training effect—senior residents scored no higher than interns.
Similarly, when Greulich et al7 surveyed German and Italian internists, general practitioners, and pulmonologists, one-fourth to one-half of them (depending on specialty and country) stated that they knew either very little or nothing at all about alpha-1 antitrypsin deficiency. In addition, 7% to 8% agreed with the statement, “There is no treatment available for this disease.”7
Nonadoption of clinical guidelines has been widely recognized in medicine and is evident in the failure to implement various recommended practices,9,10 such as low-stretch ventilation for acute respiratory distress syndrome and prophylaxis against deep vein thrombosis.
Finding the rest of the iceberg
Efforts to enhance compliance with guidelines on testing for alpha-1 antitrypsin deficiency have included using the electronic medical record to prompt physicians to test appropriate candidates.11–13
Jain et al13 examined the effect of installing such a prompting system to remind physicians to test for alpha-1 antitrypsin deficiency in patients with airflow obstruction that does not reverse with a bronchodilator—a recognized indication for testing for this disease according to standards endorsed by the American Thoracic Society and European Respiratory Society.14 At baseline, only 4.7% of appropriate candidates were being tested; after a prompt was installed in the electronic medical record, the rate rose to 15.1%, still a minority of candidates.
Another strategy is to empower respiratory therapists who perform pulmonary function tests to invite patients to be tested if their pulmonary function tests show postbronchodilator airflow obstruction. Rahaghi et al15 showed that using this strategy, 20 (0.63%) of 3,152 patients who were found to have fixed airflow obstruction when they underwent pulmonary function testing were newly diagnosed with severe deficiency of alpha-1 antitrypsin. Other targeted detection studies in patients with COPD estimated the prevalence of alpha-1 antitrypsin deficiency at up to 12%.3
PHYSIOLOGY AND PATHOPHYSIOLOGY OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Alpha-1 antitrypsin is a single-chain, 394-amino acid glycoprotein with three carbohydrate side chains found at asparagine residues along the primary structure.16
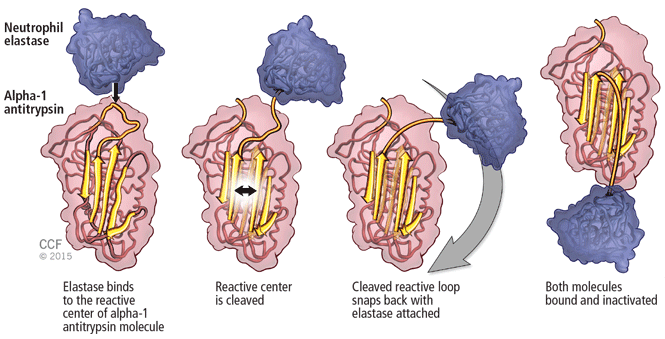
A major physiologic function of this molecule is to bind neutrophil elastase, which it does avidly. In a “mousetrap-like” mechanism,16 an active site on the alpha-1 antitrypsin molecule captures the neutrophil elastase and is cleaved, releasing steric energy in the molecule, catapulting the neutrophil elastase to the opposite side of the alpha-1 antitrypsin molecule, and inactivating it (Figure 1).
MM is normal, ZZ is not
Alpha-1 antitrypsin deficiency is inherited as an autosomal-codominant condition.17
The SERPINA1 gene, which codes for alpha-1 antitrypsin, is located on the long arm of the 14th chromosome, and more than 150 alleles of this gene have been identified to date. The normal allele is denoted M, and the allele most commonly associated with severe deficiency is denoted Z. People who are homozygous for the M allele (ie, normal) are called PI*MM (PI stands for “protease inhibitor”), and those who are homozygous for the Z allele are PI*ZZ. More than 90% of patients with severe alpha-1 antitrypsin deficiency are PI*ZZ.18
The Z allele has a single amino acid substitution (glutamic acid-to-lysine at position 342), which results in abnormal folding and formation of polymers of the Z molecule within hepatocytes.19,20 These polymers are recognized on liver biopsy as periodic acid-Schiff diastase-resistant eosinophilic inclusion bodies on histologic staining (Figure 2).
With alpha-1 antitrypsin trapped as Z-molecule polymers in the liver, the amount in the bloodstream falls, and there is a consequent decrease in the amount available in the lung to oppose the proteolytic burden of neutrophil elastase, especially in people who smoke or work in dusty environments.21
Tan et al22 have shown that some of the polymerized Z protein can escape the liver and circulate in the blood and that alveolar macrophages may also produce Z polymers. These Z polymers are chemotactic for neutrophils,23 so that their presence in the lung fuels the inflammatory cascade by recruiting more neutrophils to the lung, thereby increasing the proteolytic burden to the lung and increasing the risk of emphysema. Z monomers that do circulate can bind neutrophil elastase, but their binding avidity to neutrophil elastase is substantially lower than that of M-type alpha-1 antitrypsin.
CLINICAL MANIFESTATIONS
Alpha-1 antitrypsin deficiency of the PI*ZZ type is associated with two major clinical manifestations:
- Emphysema, resulting from the loss of proteolytic protection of the lung by alpha-1 antitrypsin (a toxic loss of function), and
- Liver diseases such as cirrhosis and chronic hepatitis, which result from abnormal accumulation of alpha-1 antitrypsin within hepatocytes (a toxic gain of function), and hepatoma.17
Other clinical manifestations of PI*ZZ alpha-1 antitrypsin deficiency include panniculitis and an association with cytoplasmic antineutrophil cytoplasmic antibody-positive vasculitis.17
Some uncertainty exists regarding the risk associated with the PI*MZ heterozygous state because there has been no systematic longitudinal study of people with this genotype. However, the weight of available experience suggests that PI*MZ individuals who have never smoked are not at increased risk of developing emphysema.24
Findings from a national registry: PI*ZZ COPD resembles ‘usual’ COPD
Distinguishing patients with alpha-1 antitrypsin deficiency from those with “usual” COPD (ie, without alpha-1 antitrypsin deficiency) can be difficult, as shown in data from the National Heart, Lung, and Blood Institute’s Alpha-1 Antitrypsin Deficiency Registry study.18 This multicenter, longitudinal, observational study contains the largest well-characterized cohort with severe deficiency of alpha-1 antitrypsin (PI*ZZ, PI*ZNull, etc), with 1,129 patients.
Pulmonary function test results were consistent with emphysema in most of the patients in the registry. Mean postbronchodilator pulmonary function values (± standard error of the mean) were:
- Forced expiratory volume in 1 second (FEV1) 46.7% of predicted (± 30%)
- Ratio of FEV1 to forced vital capacity 42.9% (± 20.4% )
- Mean diffusing capacity for carbon monoxide 50.3% of predicted (± 22.5%).
Like many patients with usual COPD, 60% of the registry patients demonstrated a component of airway reactivity, with significant reversal of airflow obstruction over three spirometries after receiving a dose of an inhaled bronchodilator (characterized by a 12% and 200-mL postbronchodilator rise in FEV1). Moreover, 78 patients had normal lung function.
Symptoms also resembled those in patients with usual emphysema, chronic bronchitis, or both. On enrollment in the registry, 83.9% of the patients had shortness of breath on exertion, 75.5% had wheezing with upper respiratory infections, 65.3% had wheezing without upper respiratory infection, 67.6% had recent debilitating chest illness, 42.4% had “usual” cough, and 49.6% had annual cough and phlegm episodes.
Imaging findings. Although the classic teaching is that emphysema due to alpha-1 antitrypsin deficiency produces lower-lobe hyperlucency on plain films, relying on this sign would lead to underrecognition, as 36% of PI*ZZ patients have apical-predominant emphysema on chest computed tomography,24 which resembles the usual centriacinar emphysema pattern. Figure 3 shows axial computed tomographic scans through the apices and the bases of the lungs of a patient with alpha-1 antitrypsin deficiency.

In view of these difficulties, guidelines from the American Thoracic Society and European Respiratory Society14 endorse testing for alpha-1 antitrypsin deficiency in all adults who have symptoms and fixed airflow obstruction (Table 1).
CONSEQUENCES OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Two large screening studies2,3,25,26 followed people who were identified at birth as having alpha-1 antitrypsin deficiency to examine the natural course of the disease.
The larger of the two studies27 tested 200,000 Swedish newborns. Follow-up of this cohort to age 35 indicated that 35-year-old never-smoking PI*ZZ individuals have normal lung function and no excess emphysema on computed tomography compared with normal peers matched for age and sex.27 In contrast, the few PI*ZZ ever-smokers demonstrated a lower level of transfer factor and significantly more emphysema on computed tomography than normal (PI*MM) never-smokers.
Faster decline in lung function
Data from the National Heart, Lung, and Blood Institute registry indicate that, on average, people with severe alpha-1 antitrypsin deficiency lose lung function faster than people without the disease.28 Specifically, in never-smokers in the registry, the average rate of FEV1 decline was 67 mL/year, and among ex-smokers, it was 54 mL/year. Both of these values exceed the general age-related rate of FEV1 decline of approximately 20 to 25 mL/year in never-smoking, normal adults. Among current smokers in the registry with severe alpha-1 antitrypsin deficiency, the rate of FEV1 decline was 109 mL/year.
Rates of FEV1 decline over time vary among groups with differing degrees of airflow obstruction. For example, PI*ZZ patients with moderate COPD (stage II of the four-stage Global Initiative for Chronic Obstructive Lung Disease classification system) lose lung function faster than patients with either milder or more severe degrees of airflow obstruction.29
As with COPD in general, exacerbations of COPD in people with severe deficiency of alpha-1 antitrypsin are associated with worsened clinical status. In one series,30 54% of 265 PI*ZZ patients experienced an exacerbation in the first year of follow-up, and 18% experienced at least three. Such exacerbations occurred in December and January in 32% of these individuals, likely due to a viral precipitant.
Increased mortality
Severe deficiency of alpha-1 antitrypsin is associated not only with severe morbidity but also death. In the national registry, the overall rate of death was 18.6% at 5 years of follow-up, or approximately 3% per year.28
A low FEV1 at entry was a bad sign. Patients entering the registry with FEV1 values below 15% of predicted had a 36% mortality rate at 3 years, compared with 2.6% in those whose baseline FEV1 exceeded 50% of predicted.
Underlying causes of death in registry participants included emphysema (accounting for 72% of deaths) and cirrhosis (10%),31 which were the only causes of death more frequent than in age- and sex-matched controls. In a series of never-smokers who had PI*ZZ alpha-1 antitrypsin deficiency,32 death was less frequently attributed to emphysema than in the national registry (46%) and more often attributed to cirrhosis (28%), indicating that never-smokers may more frequently escape the ravages of emphysema but experience a higher rate of developing cirrhosis later in life.33
DIAGNOSING ALPHA-1 ANTITRYPSIN DEFICIENCY
Available blood tests for alpha-1 antitrypsin deficiency include:
The serum alpha-1 antitrypsin level, most often done by nephelometry. Normal serum levels generally range from 100 to 220 mg/dL.
Phenotyping, usually performed by isoelectric focusing, which can identify different band patterns associated with different alleles.
Genotyping involves determining which alpha-1 antitrypsin alleles are present, most often using polymerase chain reaction testing targeting the S and Z alleles and occasionally set up to detect less common alleles such as F and I.17
Gene sequencing is occasionally necessary to achieve an accurate, definitive diagnosis.
Free, confidential testing is available
Clinical testing most often involves checking both a serum level and a phenotype or genotype. Such tests are often available in hospital laboratories and commercial laboratories, with testing also facilitated by the availability of free testing kits from several manufacturers of drugs for alpha-1 antitrypsin deficiency.
The Alpha-1 Foundation (www.alpha1.org)34 also offers a free, home-based confidential testing kit through a research protocol at the Medical University of South Carolina (alphaone@musc.edu) called the Alpha-1 Coded Testing (ACT) study. Patients can receive a kit and lancet at home, submit the dried blood-spot specimen, and receive in the mail a confidential serum level and genotype.
The availability of such home-based confidential testing allows patients to seek testing without a physician’s order and makes it easier for facilitated allied health providers, such as respiratory therapists, to recommend testing in appropriate clinical circumstances.15
TREATMENT OF ALPHA-1 ANTITRYPSIN DEFICIENCY
The treatment of patients with severe deficiency of alpha-1 antitrypsin and emphysema generally resembles that of patients with usual COPD. Specifically, smoking cessation, bronchodilators, occasionally inhaled steroids, supplemental oxygen, preventive vaccinations, and pulmonary rehabilitation are indicated as per usual clinical assessment.
Lung volume reduction surgery, which is beneficial in appropriate subsets of COPD patients, is generally less effective in those with severe alpha-1 antitrypsin deficiency,35 specifically because the magnitude of FEV1 increase and the duration of such a rise are lower than in usual COPD patients.
Augmentation therapy
Specific therapy for alpha-1 antitrypsin deficiency currently involves weekly intravenous infusions of purified, pooled human-plasma-derived alpha-1 antitrypsin, so-called augmentation therapy. Four drugs have been approved for use in the United States:
- Prolastin-C (Grifols, Barcelona, Spain)
- Aralast NP (Baxalta, Bonneckborn, IL)
- Zemaira (CSL Behring, King of Prussia, PA)
- Glassia (Baxalta, Bonneckborn, IL, and Kamada, Ness Ziona, Israel).
All of these were approved for use in the United States on the basis of biochemical efficacy. Specifically, infusion of these drugs has been shown to raise serum levels above a protective threshold value (generally considered 57 mg/dL, the value below which the risk of developing emphysema increases beyond normal).
Randomized controlled trials36,37 have addressed the efficacy of intravenous augmentation therapy, and although no single trial has been definitive, the weight of evidence shows that augmentation therapy can slow the progression of emphysema. For example, in a study by Dirksen et al,37 augmentation therapy was associated with a slower progression of emphysema as assessed by the rate of loss of lung density on computed tomography.
On the basis of the available evidence, the American Thoracic Society and European Respiratory Society14 have recommended augmentation therapy in individuals with “established airflow obstruction from alpha-1 antitrypsin deficiency.”14 Their guidelines go on to say that the evidence that augmentation therapy is beneficial “is stronger for individuals with moderate airflow obstruction (eg, FEV1 35%–60% of predicted) than for those with severe airflow obstruction. Augmentation therapy is not currently recommended for individuals without emphysema.”
The guidelines recognize that although augmentation therapy does not satisfy the usual criteria for cost-effectiveness (< $50,000 per quality-adjusted life year) due to its high cost (approximately $100,000 per year if paid for out of pocket),38 it is recommended for appropriate candidates because it is the only available specific therapy for severe deficiency of alpha-1 antitrypsin.
Novel therapies
In addition to current treatment approaches of augmentation therapy, a number of novel treatment strategies are being investigated, several of which hold much promise.
Gene therapy, using adeno-associated virus to transfect the normal human gene into individuals with severe deficiency of alpha-1 antitrypsin, has been undertaken and is currently under study. In addition, a variety of approaches to interdict production of abnormal Z protein from the liver are being examined, as well as inhaled hyaluronic acid to protect the lung.
- Laurell C, Eriksson A. The electrophoretic alpha-1 globulin pattern of serum in alpha-1 antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15:132–140.
- Aboussouan LS, Stoller JK. Detection of alpha-1 antitrypsin deficiency: a review. Respir Med 2009; 103:335–341.
- Stoller JK, Brantly M. The challenge of detecting alpha-1 antitrypsin deficiency. COPD 2013; 10(suppl 1):26–34.
- Stoller JK, Smith P, Yang P, Spray J. Physical and social impact of alpha 1-antitrypsin deficiency: results of a survey. Cleve Clin J Med 1994; 61:461–467.
- Stoller JK, Sandhaus RA, Turino G, Dickson R, Rodgers K, Strange C. Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest 2005; 128:1989–1994.
- Campos MA, Wanner A, Zhang G, Sandhaus RA. Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest 2005; 128:1179–1186.
- Greulich T, Ottaviani S, Bals R, et al. Alpha1-antitrypsin deficiency—diagnostic testing and disease awareness in Germany and Italy. Respir Med 2013; 107:1400–1408.
- Taliercio RM, Chatburn RL, Stoller JK. Knowledge of alpha-1 antitrypsin deficiency among internal medicine house officers and respiratory therapists: results of a survey. Respir Care 2010; 55:322–327.
- Rubenfeld GD, Cooper C, Carter G, Thompson BT, Hudson LD. Barriers to providing lung-protective ventilation to patients with acute lung injury. Crit Care Med 2004; 32:1289–1293.
- Cabana MD, Rand CS, Powe NR, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA 1999; 282:1458–1465.
- Rahaghi F, Ortega I, Rahaghi N, et al. Physician alert suggesting alpha-1 antitrypsin deficiency testing in pulmonary function test (PFT) results. COPD 2009; 6:26–30.
- Campos M, Hagenlocker B, Martinez N, et al. Impact of an electronic medical record clinical reminder to improve detection of COPD and alpha-1 antitrypsin deficiency in the Veterans Administration (VA) system (abstract). Am J Respir Crit Care Med 2011;183:A5356. www.atsjournals.org/doi/pdf/10.1164/ajrccm-conference.2011.183.1_MeetingAbstracts.A5356. Accessed May 24, 2016.
- Jain A, McCarthy K, Xu M, Stoller JK. Impact of a clinical decision support system in an electronic health record to enhance detection of alpha(1)-antitrypsin deficiency. Chest 2011;140:198–204.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Rahaghi FF, Sandhaus RA, Brantly ML, et al. The prevalence of alpha-1 antitrypsin deficiency among patients found to have airflow obstruction. COPD 2012; 9:352–358.
- Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency—a model for conformational diseases. N Engl J Med 2002; 346:45–53.
- Stoller JK, Aboussouan LS. A review of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2012; 185:246–259.
- McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of Alpha 1-Antitrypsin Deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Chest 1997; 111:394–403.
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992; 357:605–607.
- Lomas DA, Finch JT, Seyama K, Nukiwa T, Carrell RW. Alpha 1-antitrypsin Siiyama (Ser53-->Phe). Further evidence for intracellular loop-sheet polymerization. J Biol Chem 1993; 268:15333–15335.
- Mayer AS, Stoller JK, Bucher Bartelson B, James Ruttenber A, Sandhaus RA, Newman LS. Occupational exposure risks in individuals with PI*Z alpha(1)-antitrypsin deficiency. Am J Respir Crit Care Med 2000; 162:553–558.
- Tan L, Dickens JA, Demeo DL, et al. Circulating polymers in alpha1-antitrypsin deficiency. Eur Respir J 2014; 43:1501–1504.
- Parmar JS, Mahadeva R, Reed BJ, et al. Polymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysema. Am J Respir Cell Mol Biol 2002; 26:723–730.
- Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in alpha1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med 2014; 189:419–427.
- Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med 2004; 170:1172–1178.
- Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976; 294:1316–1321.
- O’Brien ML, Buist NR, Murphey WH. Neonatal screening for alpha1-antitrypsin deficiency. J Pediatr 1978; 92:1006–1010.
- Piitulainen E, Montero LC, Nystedt-Duzakin M, et al. Lung function and CT densitometry in subjects with alpha-1-antitrypsin deficiency and healthy controls at 35 years of age. COPD 2015; 12:162–167.
- The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Respir Crit Care Med 1998; 158:49–59.
- Dawkins PA, Dawkins CL, Wood AM, Nightingale PG, Stockley JA, Stockley RA. Rate of progression of lung function impairment in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1338–1344.
- Needham M, Stockley RA. Alpha 1-antitrypsin deficiency. 3: clinical manifestations and natural history. Thorax 2004; 59:441–445.
- Tomashefski JF Jr, Crystal RG, Wiedemann HP, Mascha E, Stoller JK. The bronchopulmonary pathology of alpha-1 antitrypsin (AAT) deficiency: findings of the Death Review Committee of the National Registry for Individuals with Severe Deficiency of Alpha-1 Antitrypsin. Hum Pathol 2004; 35:1452–1461.
- Tanash HA, Nilsson PM, Nilsson JA, Piitulainen E. Clinical course and prognosis of never-smokers with severe alpha-1-antitrypsin deficiency (PiZZ). Thorax 2008; 63:1091–1095.
- Walsh JW, Snider GL, Stoller JK. A review of the Alpha-1 Foundation: its formation, impact, and critical success factors. Respir Care 2006; 51:526–531.
- Rokadia HK, Stoller JK. Surgical and bronchoscopic lung volume reduction treatment for a-1 antitrypsin deficiency. Clin Pulm Med 2015; 22:279–285.
- Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1345–1353.
- Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160:1468–1472.
- Gildea TR, Shermock KM, Singer ME, Stoller JK. Cost-effectiveness analysis of augmentation therapy for severe alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2003; 167:1387–1392.
Alpha-1 antitrypsin deficiency is a common but underrecognized genetic condition that increases the risk of chronic obstructive pulmonary disease (COPD) and liver disease. Primary care providers can play a critical role in detecting it and managing patients who have it.
RECOGNIZED CASES ARE THE TIP OF THE ICEBERG
First described in 1963,1 alpha-1 antitrypsin deficiency is estimated to affect 100,000 Americans, fewer than 15,000 of whom have received a clinical diagnosis. As further evidence of its underrecognition,2–7 many patients experience long delays between their first symptoms and the diagnosis. Early studies indicated that the average diagnostic delay was 7.2 years,4 and the latest studies, as recent as 2013, indicate a similar diagnostic delay.7
Furthermore, many patients see multiple healthcare providers before receiving the correct diagnosis. A 1994 survey by this author4 found that 43.7% of patients who had severe deficiency of alpha-1 antitrypsin saw at least three physicians before the correct diagnosis was made.
Why is the disease underrecognized?
Several reasons may account for underrecognition of this disease. Many clinicians—including, unfortunately, many pulmonologists—do not know much about it,7,8 do not adhere to clinical guidelines,9,10 or harbor the misperception that there is no therapy available and, therefore, no compelling reason to make a diagnosis.7
Regarding inadequate knowledge, in a study by Taliercio, Chatburn, and this author,8 internal medicine residents scored only 63% correct on a 10-question quiz on diagnostic features of alpha-1 antitrypsin deficiency. There was no evidence of a training effect—senior residents scored no higher than interns.
Similarly, when Greulich et al7 surveyed German and Italian internists, general practitioners, and pulmonologists, one-fourth to one-half of them (depending on specialty and country) stated that they knew either very little or nothing at all about alpha-1 antitrypsin deficiency. In addition, 7% to 8% agreed with the statement, “There is no treatment available for this disease.”7
Nonadoption of clinical guidelines has been widely recognized in medicine and is evident in the failure to implement various recommended practices,9,10 such as low-stretch ventilation for acute respiratory distress syndrome and prophylaxis against deep vein thrombosis.
Finding the rest of the iceberg
Efforts to enhance compliance with guidelines on testing for alpha-1 antitrypsin deficiency have included using the electronic medical record to prompt physicians to test appropriate candidates.11–13
Jain et al13 examined the effect of installing such a prompting system to remind physicians to test for alpha-1 antitrypsin deficiency in patients with airflow obstruction that does not reverse with a bronchodilator—a recognized indication for testing for this disease according to standards endorsed by the American Thoracic Society and European Respiratory Society.14 At baseline, only 4.7% of appropriate candidates were being tested; after a prompt was installed in the electronic medical record, the rate rose to 15.1%, still a minority of candidates.
Another strategy is to empower respiratory therapists who perform pulmonary function tests to invite patients to be tested if their pulmonary function tests show postbronchodilator airflow obstruction. Rahaghi et al15 showed that using this strategy, 20 (0.63%) of 3,152 patients who were found to have fixed airflow obstruction when they underwent pulmonary function testing were newly diagnosed with severe deficiency of alpha-1 antitrypsin. Other targeted detection studies in patients with COPD estimated the prevalence of alpha-1 antitrypsin deficiency at up to 12%.3
PHYSIOLOGY AND PATHOPHYSIOLOGY OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Alpha-1 antitrypsin is a single-chain, 394-amino acid glycoprotein with three carbohydrate side chains found at asparagine residues along the primary structure.16
A major physiologic function of this molecule is to bind neutrophil elastase, which it does avidly. In a “mousetrap-like” mechanism,16 an active site on the alpha-1 antitrypsin molecule captures the neutrophil elastase and is cleaved, releasing steric energy in the molecule, catapulting the neutrophil elastase to the opposite side of the alpha-1 antitrypsin molecule, and inactivating it (Figure 1).
MM is normal, ZZ is not
Alpha-1 antitrypsin deficiency is inherited as an autosomal-codominant condition.17
The SERPINA1 gene, which codes for alpha-1 antitrypsin, is located on the long arm of the 14th chromosome, and more than 150 alleles of this gene have been identified to date. The normal allele is denoted M, and the allele most commonly associated with severe deficiency is denoted Z. People who are homozygous for the M allele (ie, normal) are called PI*MM (PI stands for “protease inhibitor”), and those who are homozygous for the Z allele are PI*ZZ. More than 90% of patients with severe alpha-1 antitrypsin deficiency are PI*ZZ.18
The Z allele has a single amino acid substitution (glutamic acid-to-lysine at position 342), which results in abnormal folding and formation of polymers of the Z molecule within hepatocytes.19,20 These polymers are recognized on liver biopsy as periodic acid-Schiff diastase-resistant eosinophilic inclusion bodies on histologic staining (Figure 2).
With alpha-1 antitrypsin trapped as Z-molecule polymers in the liver, the amount in the bloodstream falls, and there is a consequent decrease in the amount available in the lung to oppose the proteolytic burden of neutrophil elastase, especially in people who smoke or work in dusty environments.21
Tan et al22 have shown that some of the polymerized Z protein can escape the liver and circulate in the blood and that alveolar macrophages may also produce Z polymers. These Z polymers are chemotactic for neutrophils,23 so that their presence in the lung fuels the inflammatory cascade by recruiting more neutrophils to the lung, thereby increasing the proteolytic burden to the lung and increasing the risk of emphysema. Z monomers that do circulate can bind neutrophil elastase, but their binding avidity to neutrophil elastase is substantially lower than that of M-type alpha-1 antitrypsin.
CLINICAL MANIFESTATIONS
Alpha-1 antitrypsin deficiency of the PI*ZZ type is associated with two major clinical manifestations:
- Emphysema, resulting from the loss of proteolytic protection of the lung by alpha-1 antitrypsin (a toxic loss of function), and
- Liver diseases such as cirrhosis and chronic hepatitis, which result from abnormal accumulation of alpha-1 antitrypsin within hepatocytes (a toxic gain of function), and hepatoma.17
Other clinical manifestations of PI*ZZ alpha-1 antitrypsin deficiency include panniculitis and an association with cytoplasmic antineutrophil cytoplasmic antibody-positive vasculitis.17
Some uncertainty exists regarding the risk associated with the PI*MZ heterozygous state because there has been no systematic longitudinal study of people with this genotype. However, the weight of available experience suggests that PI*MZ individuals who have never smoked are not at increased risk of developing emphysema.24
Findings from a national registry: PI*ZZ COPD resembles ‘usual’ COPD
Distinguishing patients with alpha-1 antitrypsin deficiency from those with “usual” COPD (ie, without alpha-1 antitrypsin deficiency) can be difficult, as shown in data from the National Heart, Lung, and Blood Institute’s Alpha-1 Antitrypsin Deficiency Registry study.18 This multicenter, longitudinal, observational study contains the largest well-characterized cohort with severe deficiency of alpha-1 antitrypsin (PI*ZZ, PI*ZNull, etc), with 1,129 patients.
Pulmonary function test results were consistent with emphysema in most of the patients in the registry. Mean postbronchodilator pulmonary function values (± standard error of the mean) were:
- Forced expiratory volume in 1 second (FEV1) 46.7% of predicted (± 30%)
- Ratio of FEV1 to forced vital capacity 42.9% (± 20.4% )
- Mean diffusing capacity for carbon monoxide 50.3% of predicted (± 22.5%).
Like many patients with usual COPD, 60% of the registry patients demonstrated a component of airway reactivity, with significant reversal of airflow obstruction over three spirometries after receiving a dose of an inhaled bronchodilator (characterized by a 12% and 200-mL postbronchodilator rise in FEV1). Moreover, 78 patients had normal lung function.
Symptoms also resembled those in patients with usual emphysema, chronic bronchitis, or both. On enrollment in the registry, 83.9% of the patients had shortness of breath on exertion, 75.5% had wheezing with upper respiratory infections, 65.3% had wheezing without upper respiratory infection, 67.6% had recent debilitating chest illness, 42.4% had “usual” cough, and 49.6% had annual cough and phlegm episodes.
Imaging findings. Although the classic teaching is that emphysema due to alpha-1 antitrypsin deficiency produces lower-lobe hyperlucency on plain films, relying on this sign would lead to underrecognition, as 36% of PI*ZZ patients have apical-predominant emphysema on chest computed tomography,24 which resembles the usual centriacinar emphysema pattern. Figure 3 shows axial computed tomographic scans through the apices and the bases of the lungs of a patient with alpha-1 antitrypsin deficiency.
In view of these difficulties, guidelines from the American Thoracic Society and European Respiratory Society14 endorse testing for alpha-1 antitrypsin deficiency in all adults who have symptoms and fixed airflow obstruction (Table 1).
CONSEQUENCES OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Two large screening studies2,3,25,26 followed people who were identified at birth as having alpha-1 antitrypsin deficiency to examine the natural course of the disease.
The larger of the two studies27 tested 200,000 Swedish newborns. Follow-up of this cohort to age 35 indicated that 35-year-old never-smoking PI*ZZ individuals have normal lung function and no excess emphysema on computed tomography compared with normal peers matched for age and sex.27 In contrast, the few PI*ZZ ever-smokers demonstrated a lower level of transfer factor and significantly more emphysema on computed tomography than normal (PI*MM) never-smokers.
Faster decline in lung function
Data from the National Heart, Lung, and Blood Institute registry indicate that, on average, people with severe alpha-1 antitrypsin deficiency lose lung function faster than people without the disease.28 Specifically, in never-smokers in the registry, the average rate of FEV1 decline was 67 mL/year, and among ex-smokers, it was 54 mL/year. Both of these values exceed the general age-related rate of FEV1 decline of approximately 20 to 25 mL/year in never-smoking, normal adults. Among current smokers in the registry with severe alpha-1 antitrypsin deficiency, the rate of FEV1 decline was 109 mL/year.
Rates of FEV1 decline over time vary among groups with differing degrees of airflow obstruction. For example, PI*ZZ patients with moderate COPD (stage II of the four-stage Global Initiative for Chronic Obstructive Lung Disease classification system) lose lung function faster than patients with either milder or more severe degrees of airflow obstruction.29
As with COPD in general, exacerbations of COPD in people with severe deficiency of alpha-1 antitrypsin are associated with worsened clinical status. In one series,30 54% of 265 PI*ZZ patients experienced an exacerbation in the first year of follow-up, and 18% experienced at least three. Such exacerbations occurred in December and January in 32% of these individuals, likely due to a viral precipitant.
Increased mortality
Severe deficiency of alpha-1 antitrypsin is associated not only with severe morbidity but also death. In the national registry, the overall rate of death was 18.6% at 5 years of follow-up, or approximately 3% per year.28
A low FEV1 at entry was a bad sign. Patients entering the registry with FEV1 values below 15% of predicted had a 36% mortality rate at 3 years, compared with 2.6% in those whose baseline FEV1 exceeded 50% of predicted.
Underlying causes of death in registry participants included emphysema (accounting for 72% of deaths) and cirrhosis (10%),31 which were the only causes of death more frequent than in age- and sex-matched controls. In a series of never-smokers who had PI*ZZ alpha-1 antitrypsin deficiency,32 death was less frequently attributed to emphysema than in the national registry (46%) and more often attributed to cirrhosis (28%), indicating that never-smokers may more frequently escape the ravages of emphysema but experience a higher rate of developing cirrhosis later in life.33
DIAGNOSING ALPHA-1 ANTITRYPSIN DEFICIENCY
Available blood tests for alpha-1 antitrypsin deficiency include:
The serum alpha-1 antitrypsin level, most often done by nephelometry. Normal serum levels generally range from 100 to 220 mg/dL.
Phenotyping, usually performed by isoelectric focusing, which can identify different band patterns associated with different alleles.
Genotyping involves determining which alpha-1 antitrypsin alleles are present, most often using polymerase chain reaction testing targeting the S and Z alleles and occasionally set up to detect less common alleles such as F and I.17
Gene sequencing is occasionally necessary to achieve an accurate, definitive diagnosis.
Free, confidential testing is available
Clinical testing most often involves checking both a serum level and a phenotype or genotype. Such tests are often available in hospital laboratories and commercial laboratories, with testing also facilitated by the availability of free testing kits from several manufacturers of drugs for alpha-1 antitrypsin deficiency.
The Alpha-1 Foundation (www.alpha1.org)34 also offers a free, home-based confidential testing kit through a research protocol at the Medical University of South Carolina (alphaone@musc.edu) called the Alpha-1 Coded Testing (ACT) study. Patients can receive a kit and lancet at home, submit the dried blood-spot specimen, and receive in the mail a confidential serum level and genotype.
The availability of such home-based confidential testing allows patients to seek testing without a physician’s order and makes it easier for facilitated allied health providers, such as respiratory therapists, to recommend testing in appropriate clinical circumstances.15
TREATMENT OF ALPHA-1 ANTITRYPSIN DEFICIENCY
The treatment of patients with severe deficiency of alpha-1 antitrypsin and emphysema generally resembles that of patients with usual COPD. Specifically, smoking cessation, bronchodilators, occasionally inhaled steroids, supplemental oxygen, preventive vaccinations, and pulmonary rehabilitation are indicated as per usual clinical assessment.
Lung volume reduction surgery, which is beneficial in appropriate subsets of COPD patients, is generally less effective in those with severe alpha-1 antitrypsin deficiency,35 specifically because the magnitude of FEV1 increase and the duration of such a rise are lower than in usual COPD patients.
Augmentation therapy
Specific therapy for alpha-1 antitrypsin deficiency currently involves weekly intravenous infusions of purified, pooled human-plasma-derived alpha-1 antitrypsin, so-called augmentation therapy. Four drugs have been approved for use in the United States:
- Prolastin-C (Grifols, Barcelona, Spain)
- Aralast NP (Baxalta, Bonneckborn, IL)
- Zemaira (CSL Behring, King of Prussia, PA)
- Glassia (Baxalta, Bonneckborn, IL, and Kamada, Ness Ziona, Israel).
All of these were approved for use in the United States on the basis of biochemical efficacy. Specifically, infusion of these drugs has been shown to raise serum levels above a protective threshold value (generally considered 57 mg/dL, the value below which the risk of developing emphysema increases beyond normal).
Randomized controlled trials36,37 have addressed the efficacy of intravenous augmentation therapy, and although no single trial has been definitive, the weight of evidence shows that augmentation therapy can slow the progression of emphysema. For example, in a study by Dirksen et al,37 augmentation therapy was associated with a slower progression of emphysema as assessed by the rate of loss of lung density on computed tomography.
On the basis of the available evidence, the American Thoracic Society and European Respiratory Society14 have recommended augmentation therapy in individuals with “established airflow obstruction from alpha-1 antitrypsin deficiency.”14 Their guidelines go on to say that the evidence that augmentation therapy is beneficial “is stronger for individuals with moderate airflow obstruction (eg, FEV1 35%–60% of predicted) than for those with severe airflow obstruction. Augmentation therapy is not currently recommended for individuals without emphysema.”
The guidelines recognize that although augmentation therapy does not satisfy the usual criteria for cost-effectiveness (< $50,000 per quality-adjusted life year) due to its high cost (approximately $100,000 per year if paid for out of pocket),38 it is recommended for appropriate candidates because it is the only available specific therapy for severe deficiency of alpha-1 antitrypsin.
Novel therapies
In addition to current treatment approaches of augmentation therapy, a number of novel treatment strategies are being investigated, several of which hold much promise.
Gene therapy, using adeno-associated virus to transfect the normal human gene into individuals with severe deficiency of alpha-1 antitrypsin, has been undertaken and is currently under study. In addition, a variety of approaches to interdict production of abnormal Z protein from the liver are being examined, as well as inhaled hyaluronic acid to protect the lung.
Alpha-1 antitrypsin deficiency is a common but underrecognized genetic condition that increases the risk of chronic obstructive pulmonary disease (COPD) and liver disease. Primary care providers can play a critical role in detecting it and managing patients who have it.
RECOGNIZED CASES ARE THE TIP OF THE ICEBERG
First described in 1963,1 alpha-1 antitrypsin deficiency is estimated to affect 100,000 Americans, fewer than 15,000 of whom have received a clinical diagnosis. As further evidence of its underrecognition,2–7 many patients experience long delays between their first symptoms and the diagnosis. Early studies indicated that the average diagnostic delay was 7.2 years,4 and the latest studies, as recent as 2013, indicate a similar diagnostic delay.7
Furthermore, many patients see multiple healthcare providers before receiving the correct diagnosis. A 1994 survey by this author4 found that 43.7% of patients who had severe deficiency of alpha-1 antitrypsin saw at least three physicians before the correct diagnosis was made.
Why is the disease underrecognized?
Several reasons may account for underrecognition of this disease. Many clinicians—including, unfortunately, many pulmonologists—do not know much about it,7,8 do not adhere to clinical guidelines,9,10 or harbor the misperception that there is no therapy available and, therefore, no compelling reason to make a diagnosis.7
Regarding inadequate knowledge, in a study by Taliercio, Chatburn, and this author,8 internal medicine residents scored only 63% correct on a 10-question quiz on diagnostic features of alpha-1 antitrypsin deficiency. There was no evidence of a training effect—senior residents scored no higher than interns.
Similarly, when Greulich et al7 surveyed German and Italian internists, general practitioners, and pulmonologists, one-fourth to one-half of them (depending on specialty and country) stated that they knew either very little or nothing at all about alpha-1 antitrypsin deficiency. In addition, 7% to 8% agreed with the statement, “There is no treatment available for this disease.”7
Nonadoption of clinical guidelines has been widely recognized in medicine and is evident in the failure to implement various recommended practices,9,10 such as low-stretch ventilation for acute respiratory distress syndrome and prophylaxis against deep vein thrombosis.
Finding the rest of the iceberg
Efforts to enhance compliance with guidelines on testing for alpha-1 antitrypsin deficiency have included using the electronic medical record to prompt physicians to test appropriate candidates.11–13
Jain et al13 examined the effect of installing such a prompting system to remind physicians to test for alpha-1 antitrypsin deficiency in patients with airflow obstruction that does not reverse with a bronchodilator—a recognized indication for testing for this disease according to standards endorsed by the American Thoracic Society and European Respiratory Society.14 At baseline, only 4.7% of appropriate candidates were being tested; after a prompt was installed in the electronic medical record, the rate rose to 15.1%, still a minority of candidates.
Another strategy is to empower respiratory therapists who perform pulmonary function tests to invite patients to be tested if their pulmonary function tests show postbronchodilator airflow obstruction. Rahaghi et al15 showed that using this strategy, 20 (0.63%) of 3,152 patients who were found to have fixed airflow obstruction when they underwent pulmonary function testing were newly diagnosed with severe deficiency of alpha-1 antitrypsin. Other targeted detection studies in patients with COPD estimated the prevalence of alpha-1 antitrypsin deficiency at up to 12%.3
PHYSIOLOGY AND PATHOPHYSIOLOGY OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Alpha-1 antitrypsin is a single-chain, 394-amino acid glycoprotein with three carbohydrate side chains found at asparagine residues along the primary structure.16
A major physiologic function of this molecule is to bind neutrophil elastase, which it does avidly. In a “mousetrap-like” mechanism,16 an active site on the alpha-1 antitrypsin molecule captures the neutrophil elastase and is cleaved, releasing steric energy in the molecule, catapulting the neutrophil elastase to the opposite side of the alpha-1 antitrypsin molecule, and inactivating it (Figure 1).
MM is normal, ZZ is not
Alpha-1 antitrypsin deficiency is inherited as an autosomal-codominant condition.17
The SERPINA1 gene, which codes for alpha-1 antitrypsin, is located on the long arm of the 14th chromosome, and more than 150 alleles of this gene have been identified to date. The normal allele is denoted M, and the allele most commonly associated with severe deficiency is denoted Z. People who are homozygous for the M allele (ie, normal) are called PI*MM (PI stands for “protease inhibitor”), and those who are homozygous for the Z allele are PI*ZZ. More than 90% of patients with severe alpha-1 antitrypsin deficiency are PI*ZZ.18
The Z allele has a single amino acid substitution (glutamic acid-to-lysine at position 342), which results in abnormal folding and formation of polymers of the Z molecule within hepatocytes.19,20 These polymers are recognized on liver biopsy as periodic acid-Schiff diastase-resistant eosinophilic inclusion bodies on histologic staining (Figure 2).
With alpha-1 antitrypsin trapped as Z-molecule polymers in the liver, the amount in the bloodstream falls, and there is a consequent decrease in the amount available in the lung to oppose the proteolytic burden of neutrophil elastase, especially in people who smoke or work in dusty environments.21
Tan et al22 have shown that some of the polymerized Z protein can escape the liver and circulate in the blood and that alveolar macrophages may also produce Z polymers. These Z polymers are chemotactic for neutrophils,23 so that their presence in the lung fuels the inflammatory cascade by recruiting more neutrophils to the lung, thereby increasing the proteolytic burden to the lung and increasing the risk of emphysema. Z monomers that do circulate can bind neutrophil elastase, but their binding avidity to neutrophil elastase is substantially lower than that of M-type alpha-1 antitrypsin.
CLINICAL MANIFESTATIONS
Alpha-1 antitrypsin deficiency of the PI*ZZ type is associated with two major clinical manifestations:
- Emphysema, resulting from the loss of proteolytic protection of the lung by alpha-1 antitrypsin (a toxic loss of function), and
- Liver diseases such as cirrhosis and chronic hepatitis, which result from abnormal accumulation of alpha-1 antitrypsin within hepatocytes (a toxic gain of function), and hepatoma.17
Other clinical manifestations of PI*ZZ alpha-1 antitrypsin deficiency include panniculitis and an association with cytoplasmic antineutrophil cytoplasmic antibody-positive vasculitis.17
Some uncertainty exists regarding the risk associated with the PI*MZ heterozygous state because there has been no systematic longitudinal study of people with this genotype. However, the weight of available experience suggests that PI*MZ individuals who have never smoked are not at increased risk of developing emphysema.24
Findings from a national registry: PI*ZZ COPD resembles ‘usual’ COPD
Distinguishing patients with alpha-1 antitrypsin deficiency from those with “usual” COPD (ie, without alpha-1 antitrypsin deficiency) can be difficult, as shown in data from the National Heart, Lung, and Blood Institute’s Alpha-1 Antitrypsin Deficiency Registry study.18 This multicenter, longitudinal, observational study contains the largest well-characterized cohort with severe deficiency of alpha-1 antitrypsin (PI*ZZ, PI*ZNull, etc), with 1,129 patients.
Pulmonary function test results were consistent with emphysema in most of the patients in the registry. Mean postbronchodilator pulmonary function values (± standard error of the mean) were:
- Forced expiratory volume in 1 second (FEV1) 46.7% of predicted (± 30%)
- Ratio of FEV1 to forced vital capacity 42.9% (± 20.4% )
- Mean diffusing capacity for carbon monoxide 50.3% of predicted (± 22.5%).
Like many patients with usual COPD, 60% of the registry patients demonstrated a component of airway reactivity, with significant reversal of airflow obstruction over three spirometries after receiving a dose of an inhaled bronchodilator (characterized by a 12% and 200-mL postbronchodilator rise in FEV1). Moreover, 78 patients had normal lung function.
Symptoms also resembled those in patients with usual emphysema, chronic bronchitis, or both. On enrollment in the registry, 83.9% of the patients had shortness of breath on exertion, 75.5% had wheezing with upper respiratory infections, 65.3% had wheezing without upper respiratory infection, 67.6% had recent debilitating chest illness, 42.4% had “usual” cough, and 49.6% had annual cough and phlegm episodes.
Imaging findings. Although the classic teaching is that emphysema due to alpha-1 antitrypsin deficiency produces lower-lobe hyperlucency on plain films, relying on this sign would lead to underrecognition, as 36% of PI*ZZ patients have apical-predominant emphysema on chest computed tomography,24 which resembles the usual centriacinar emphysema pattern. Figure 3 shows axial computed tomographic scans through the apices and the bases of the lungs of a patient with alpha-1 antitrypsin deficiency.
In view of these difficulties, guidelines from the American Thoracic Society and European Respiratory Society14 endorse testing for alpha-1 antitrypsin deficiency in all adults who have symptoms and fixed airflow obstruction (Table 1).
CONSEQUENCES OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Two large screening studies2,3,25,26 followed people who were identified at birth as having alpha-1 antitrypsin deficiency to examine the natural course of the disease.
The larger of the two studies27 tested 200,000 Swedish newborns. Follow-up of this cohort to age 35 indicated that 35-year-old never-smoking PI*ZZ individuals have normal lung function and no excess emphysema on computed tomography compared with normal peers matched for age and sex.27 In contrast, the few PI*ZZ ever-smokers demonstrated a lower level of transfer factor and significantly more emphysema on computed tomography than normal (PI*MM) never-smokers.
Faster decline in lung function
Data from the National Heart, Lung, and Blood Institute registry indicate that, on average, people with severe alpha-1 antitrypsin deficiency lose lung function faster than people without the disease.28 Specifically, in never-smokers in the registry, the average rate of FEV1 decline was 67 mL/year, and among ex-smokers, it was 54 mL/year. Both of these values exceed the general age-related rate of FEV1 decline of approximately 20 to 25 mL/year in never-smoking, normal adults. Among current smokers in the registry with severe alpha-1 antitrypsin deficiency, the rate of FEV1 decline was 109 mL/year.
Rates of FEV1 decline over time vary among groups with differing degrees of airflow obstruction. For example, PI*ZZ patients with moderate COPD (stage II of the four-stage Global Initiative for Chronic Obstructive Lung Disease classification system) lose lung function faster than patients with either milder or more severe degrees of airflow obstruction.29
As with COPD in general, exacerbations of COPD in people with severe deficiency of alpha-1 antitrypsin are associated with worsened clinical status. In one series,30 54% of 265 PI*ZZ patients experienced an exacerbation in the first year of follow-up, and 18% experienced at least three. Such exacerbations occurred in December and January in 32% of these individuals, likely due to a viral precipitant.
Increased mortality
Severe deficiency of alpha-1 antitrypsin is associated not only with severe morbidity but also death. In the national registry, the overall rate of death was 18.6% at 5 years of follow-up, or approximately 3% per year.28
A low FEV1 at entry was a bad sign. Patients entering the registry with FEV1 values below 15% of predicted had a 36% mortality rate at 3 years, compared with 2.6% in those whose baseline FEV1 exceeded 50% of predicted.
Underlying causes of death in registry participants included emphysema (accounting for 72% of deaths) and cirrhosis (10%),31 which were the only causes of death more frequent than in age- and sex-matched controls. In a series of never-smokers who had PI*ZZ alpha-1 antitrypsin deficiency,32 death was less frequently attributed to emphysema than in the national registry (46%) and more often attributed to cirrhosis (28%), indicating that never-smokers may more frequently escape the ravages of emphysema but experience a higher rate of developing cirrhosis later in life.33
DIAGNOSING ALPHA-1 ANTITRYPSIN DEFICIENCY
Available blood tests for alpha-1 antitrypsin deficiency include:
The serum alpha-1 antitrypsin level, most often done by nephelometry. Normal serum levels generally range from 100 to 220 mg/dL.
Phenotyping, usually performed by isoelectric focusing, which can identify different band patterns associated with different alleles.
Genotyping involves determining which alpha-1 antitrypsin alleles are present, most often using polymerase chain reaction testing targeting the S and Z alleles and occasionally set up to detect less common alleles such as F and I.17
Gene sequencing is occasionally necessary to achieve an accurate, definitive diagnosis.
Free, confidential testing is available
Clinical testing most often involves checking both a serum level and a phenotype or genotype. Such tests are often available in hospital laboratories and commercial laboratories, with testing also facilitated by the availability of free testing kits from several manufacturers of drugs for alpha-1 antitrypsin deficiency.
The Alpha-1 Foundation (www.alpha1.org)34 also offers a free, home-based confidential testing kit through a research protocol at the Medical University of South Carolina (alphaone@musc.edu) called the Alpha-1 Coded Testing (ACT) study. Patients can receive a kit and lancet at home, submit the dried blood-spot specimen, and receive in the mail a confidential serum level and genotype.
The availability of such home-based confidential testing allows patients to seek testing without a physician’s order and makes it easier for facilitated allied health providers, such as respiratory therapists, to recommend testing in appropriate clinical circumstances.15
TREATMENT OF ALPHA-1 ANTITRYPSIN DEFICIENCY
The treatment of patients with severe deficiency of alpha-1 antitrypsin and emphysema generally resembles that of patients with usual COPD. Specifically, smoking cessation, bronchodilators, occasionally inhaled steroids, supplemental oxygen, preventive vaccinations, and pulmonary rehabilitation are indicated as per usual clinical assessment.
Lung volume reduction surgery, which is beneficial in appropriate subsets of COPD patients, is generally less effective in those with severe alpha-1 antitrypsin deficiency,35 specifically because the magnitude of FEV1 increase and the duration of such a rise are lower than in usual COPD patients.
Augmentation therapy
Specific therapy for alpha-1 antitrypsin deficiency currently involves weekly intravenous infusions of purified, pooled human-plasma-derived alpha-1 antitrypsin, so-called augmentation therapy. Four drugs have been approved for use in the United States:
- Prolastin-C (Grifols, Barcelona, Spain)
- Aralast NP (Baxalta, Bonneckborn, IL)
- Zemaira (CSL Behring, King of Prussia, PA)
- Glassia (Baxalta, Bonneckborn, IL, and Kamada, Ness Ziona, Israel).
All of these were approved for use in the United States on the basis of biochemical efficacy. Specifically, infusion of these drugs has been shown to raise serum levels above a protective threshold value (generally considered 57 mg/dL, the value below which the risk of developing emphysema increases beyond normal).
Randomized controlled trials36,37 have addressed the efficacy of intravenous augmentation therapy, and although no single trial has been definitive, the weight of evidence shows that augmentation therapy can slow the progression of emphysema. For example, in a study by Dirksen et al,37 augmentation therapy was associated with a slower progression of emphysema as assessed by the rate of loss of lung density on computed tomography.
On the basis of the available evidence, the American Thoracic Society and European Respiratory Society14 have recommended augmentation therapy in individuals with “established airflow obstruction from alpha-1 antitrypsin deficiency.”14 Their guidelines go on to say that the evidence that augmentation therapy is beneficial “is stronger for individuals with moderate airflow obstruction (eg, FEV1 35%–60% of predicted) than for those with severe airflow obstruction. Augmentation therapy is not currently recommended for individuals without emphysema.”
The guidelines recognize that although augmentation therapy does not satisfy the usual criteria for cost-effectiveness (< $50,000 per quality-adjusted life year) due to its high cost (approximately $100,000 per year if paid for out of pocket),38 it is recommended for appropriate candidates because it is the only available specific therapy for severe deficiency of alpha-1 antitrypsin.
Novel therapies
In addition to current treatment approaches of augmentation therapy, a number of novel treatment strategies are being investigated, several of which hold much promise.
Gene therapy, using adeno-associated virus to transfect the normal human gene into individuals with severe deficiency of alpha-1 antitrypsin, has been undertaken and is currently under study. In addition, a variety of approaches to interdict production of abnormal Z protein from the liver are being examined, as well as inhaled hyaluronic acid to protect the lung.
- Laurell C, Eriksson A. The electrophoretic alpha-1 globulin pattern of serum in alpha-1 antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15:132–140.
- Aboussouan LS, Stoller JK. Detection of alpha-1 antitrypsin deficiency: a review. Respir Med 2009; 103:335–341.
- Stoller JK, Brantly M. The challenge of detecting alpha-1 antitrypsin deficiency. COPD 2013; 10(suppl 1):26–34.
- Stoller JK, Smith P, Yang P, Spray J. Physical and social impact of alpha 1-antitrypsin deficiency: results of a survey. Cleve Clin J Med 1994; 61:461–467.
- Stoller JK, Sandhaus RA, Turino G, Dickson R, Rodgers K, Strange C. Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest 2005; 128:1989–1994.
- Campos MA, Wanner A, Zhang G, Sandhaus RA. Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest 2005; 128:1179–1186.
- Greulich T, Ottaviani S, Bals R, et al. Alpha1-antitrypsin deficiency—diagnostic testing and disease awareness in Germany and Italy. Respir Med 2013; 107:1400–1408.
- Taliercio RM, Chatburn RL, Stoller JK. Knowledge of alpha-1 antitrypsin deficiency among internal medicine house officers and respiratory therapists: results of a survey. Respir Care 2010; 55:322–327.
- Rubenfeld GD, Cooper C, Carter G, Thompson BT, Hudson LD. Barriers to providing lung-protective ventilation to patients with acute lung injury. Crit Care Med 2004; 32:1289–1293.
- Cabana MD, Rand CS, Powe NR, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA 1999; 282:1458–1465.
- Rahaghi F, Ortega I, Rahaghi N, et al. Physician alert suggesting alpha-1 antitrypsin deficiency testing in pulmonary function test (PFT) results. COPD 2009; 6:26–30.
- Campos M, Hagenlocker B, Martinez N, et al. Impact of an electronic medical record clinical reminder to improve detection of COPD and alpha-1 antitrypsin deficiency in the Veterans Administration (VA) system (abstract). Am J Respir Crit Care Med 2011;183:A5356. www.atsjournals.org/doi/pdf/10.1164/ajrccm-conference.2011.183.1_MeetingAbstracts.A5356. Accessed May 24, 2016.
- Jain A, McCarthy K, Xu M, Stoller JK. Impact of a clinical decision support system in an electronic health record to enhance detection of alpha(1)-antitrypsin deficiency. Chest 2011;140:198–204.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Rahaghi FF, Sandhaus RA, Brantly ML, et al. The prevalence of alpha-1 antitrypsin deficiency among patients found to have airflow obstruction. COPD 2012; 9:352–358.
- Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency—a model for conformational diseases. N Engl J Med 2002; 346:45–53.
- Stoller JK, Aboussouan LS. A review of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2012; 185:246–259.
- McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of Alpha 1-Antitrypsin Deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Chest 1997; 111:394–403.
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992; 357:605–607.
- Lomas DA, Finch JT, Seyama K, Nukiwa T, Carrell RW. Alpha 1-antitrypsin Siiyama (Ser53-->Phe). Further evidence for intracellular loop-sheet polymerization. J Biol Chem 1993; 268:15333–15335.
- Mayer AS, Stoller JK, Bucher Bartelson B, James Ruttenber A, Sandhaus RA, Newman LS. Occupational exposure risks in individuals with PI*Z alpha(1)-antitrypsin deficiency. Am J Respir Crit Care Med 2000; 162:553–558.
- Tan L, Dickens JA, Demeo DL, et al. Circulating polymers in alpha1-antitrypsin deficiency. Eur Respir J 2014; 43:1501–1504.
- Parmar JS, Mahadeva R, Reed BJ, et al. Polymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysema. Am J Respir Cell Mol Biol 2002; 26:723–730.
- Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in alpha1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med 2014; 189:419–427.
- Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med 2004; 170:1172–1178.
- Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976; 294:1316–1321.
- O’Brien ML, Buist NR, Murphey WH. Neonatal screening for alpha1-antitrypsin deficiency. J Pediatr 1978; 92:1006–1010.
- Piitulainen E, Montero LC, Nystedt-Duzakin M, et al. Lung function and CT densitometry in subjects with alpha-1-antitrypsin deficiency and healthy controls at 35 years of age. COPD 2015; 12:162–167.
- The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Respir Crit Care Med 1998; 158:49–59.
- Dawkins PA, Dawkins CL, Wood AM, Nightingale PG, Stockley JA, Stockley RA. Rate of progression of lung function impairment in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1338–1344.
- Needham M, Stockley RA. Alpha 1-antitrypsin deficiency. 3: clinical manifestations and natural history. Thorax 2004; 59:441–445.
- Tomashefski JF Jr, Crystal RG, Wiedemann HP, Mascha E, Stoller JK. The bronchopulmonary pathology of alpha-1 antitrypsin (AAT) deficiency: findings of the Death Review Committee of the National Registry for Individuals with Severe Deficiency of Alpha-1 Antitrypsin. Hum Pathol 2004; 35:1452–1461.
- Tanash HA, Nilsson PM, Nilsson JA, Piitulainen E. Clinical course and prognosis of never-smokers with severe alpha-1-antitrypsin deficiency (PiZZ). Thorax 2008; 63:1091–1095.
- Walsh JW, Snider GL, Stoller JK. A review of the Alpha-1 Foundation: its formation, impact, and critical success factors. Respir Care 2006; 51:526–531.
- Rokadia HK, Stoller JK. Surgical and bronchoscopic lung volume reduction treatment for a-1 antitrypsin deficiency. Clin Pulm Med 2015; 22:279–285.
- Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1345–1353.
- Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160:1468–1472.
- Gildea TR, Shermock KM, Singer ME, Stoller JK. Cost-effectiveness analysis of augmentation therapy for severe alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2003; 167:1387–1392.
- Laurell C, Eriksson A. The electrophoretic alpha-1 globulin pattern of serum in alpha-1 antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15:132–140.
- Aboussouan LS, Stoller JK. Detection of alpha-1 antitrypsin deficiency: a review. Respir Med 2009; 103:335–341.
- Stoller JK, Brantly M. The challenge of detecting alpha-1 antitrypsin deficiency. COPD 2013; 10(suppl 1):26–34.
- Stoller JK, Smith P, Yang P, Spray J. Physical and social impact of alpha 1-antitrypsin deficiency: results of a survey. Cleve Clin J Med 1994; 61:461–467.
- Stoller JK, Sandhaus RA, Turino G, Dickson R, Rodgers K, Strange C. Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest 2005; 128:1989–1994.
- Campos MA, Wanner A, Zhang G, Sandhaus RA. Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest 2005; 128:1179–1186.
- Greulich T, Ottaviani S, Bals R, et al. Alpha1-antitrypsin deficiency—diagnostic testing and disease awareness in Germany and Italy. Respir Med 2013; 107:1400–1408.
- Taliercio RM, Chatburn RL, Stoller JK. Knowledge of alpha-1 antitrypsin deficiency among internal medicine house officers and respiratory therapists: results of a survey. Respir Care 2010; 55:322–327.
- Rubenfeld GD, Cooper C, Carter G, Thompson BT, Hudson LD. Barriers to providing lung-protective ventilation to patients with acute lung injury. Crit Care Med 2004; 32:1289–1293.
- Cabana MD, Rand CS, Powe NR, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA 1999; 282:1458–1465.
- Rahaghi F, Ortega I, Rahaghi N, et al. Physician alert suggesting alpha-1 antitrypsin deficiency testing in pulmonary function test (PFT) results. COPD 2009; 6:26–30.
- Campos M, Hagenlocker B, Martinez N, et al. Impact of an electronic medical record clinical reminder to improve detection of COPD and alpha-1 antitrypsin deficiency in the Veterans Administration (VA) system (abstract). Am J Respir Crit Care Med 2011;183:A5356. www.atsjournals.org/doi/pdf/10.1164/ajrccm-conference.2011.183.1_MeetingAbstracts.A5356. Accessed May 24, 2016.
- Jain A, McCarthy K, Xu M, Stoller JK. Impact of a clinical decision support system in an electronic health record to enhance detection of alpha(1)-antitrypsin deficiency. Chest 2011;140:198–204.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Rahaghi FF, Sandhaus RA, Brantly ML, et al. The prevalence of alpha-1 antitrypsin deficiency among patients found to have airflow obstruction. COPD 2012; 9:352–358.
- Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency—a model for conformational diseases. N Engl J Med 2002; 346:45–53.
- Stoller JK, Aboussouan LS. A review of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2012; 185:246–259.
- McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of Alpha 1-Antitrypsin Deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Chest 1997; 111:394–403.
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992; 357:605–607.
- Lomas DA, Finch JT, Seyama K, Nukiwa T, Carrell RW. Alpha 1-antitrypsin Siiyama (Ser53-->Phe). Further evidence for intracellular loop-sheet polymerization. J Biol Chem 1993; 268:15333–15335.
- Mayer AS, Stoller JK, Bucher Bartelson B, James Ruttenber A, Sandhaus RA, Newman LS. Occupational exposure risks in individuals with PI*Z alpha(1)-antitrypsin deficiency. Am J Respir Crit Care Med 2000; 162:553–558.
- Tan L, Dickens JA, Demeo DL, et al. Circulating polymers in alpha1-antitrypsin deficiency. Eur Respir J 2014; 43:1501–1504.
- Parmar JS, Mahadeva R, Reed BJ, et al. Polymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysema. Am J Respir Cell Mol Biol 2002; 26:723–730.
- Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in alpha1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med 2014; 189:419–427.
- Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med 2004; 170:1172–1178.
- Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976; 294:1316–1321.
- O’Brien ML, Buist NR, Murphey WH. Neonatal screening for alpha1-antitrypsin deficiency. J Pediatr 1978; 92:1006–1010.
- Piitulainen E, Montero LC, Nystedt-Duzakin M, et al. Lung function and CT densitometry in subjects with alpha-1-antitrypsin deficiency and healthy controls at 35 years of age. COPD 2015; 12:162–167.
- The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Respir Crit Care Med 1998; 158:49–59.
- Dawkins PA, Dawkins CL, Wood AM, Nightingale PG, Stockley JA, Stockley RA. Rate of progression of lung function impairment in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1338–1344.
- Needham M, Stockley RA. Alpha 1-antitrypsin deficiency. 3: clinical manifestations and natural history. Thorax 2004; 59:441–445.
- Tomashefski JF Jr, Crystal RG, Wiedemann HP, Mascha E, Stoller JK. The bronchopulmonary pathology of alpha-1 antitrypsin (AAT) deficiency: findings of the Death Review Committee of the National Registry for Individuals with Severe Deficiency of Alpha-1 Antitrypsin. Hum Pathol 2004; 35:1452–1461.
- Tanash HA, Nilsson PM, Nilsson JA, Piitulainen E. Clinical course and prognosis of never-smokers with severe alpha-1-antitrypsin deficiency (PiZZ). Thorax 2008; 63:1091–1095.
- Walsh JW, Snider GL, Stoller JK. A review of the Alpha-1 Foundation: its formation, impact, and critical success factors. Respir Care 2006; 51:526–531.
- Rokadia HK, Stoller JK. Surgical and bronchoscopic lung volume reduction treatment for a-1 antitrypsin deficiency. Clin Pulm Med 2015; 22:279–285.
- Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1345–1353.
- Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160:1468–1472.
- Gildea TR, Shermock KM, Singer ME, Stoller JK. Cost-effectiveness analysis of augmentation therapy for severe alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2003; 167:1387–1392.
KEY POINTS
- Only about 15% of people who have alpha-1 antitrypsin disease have received a diagnosis of it.
- The disease is genetic. People who are homozygous for the Z allele of the gene that codes for alpha-1 antitrypsin are at increased risk of lung and liver disease.
- Chronic obstructive pulmonary disease (COPD) due to alpha-1 antitrypsin deficiency is difficult to distinguish from “usual” COPD on a clinical basis, but blood tests are available.
- The basic care of a patient with COPD due to alpha-1 antitrypsin disease is the same as for any patient with COPD, ie, with bronchodilators, inhaled steroids, supplemental oxygen, preventive vaccinations, and pulmonary rehabilitation as indicated. Specific treatment consists of weekly infusions of alpha-1 antitrypsin (augmentation therapy).
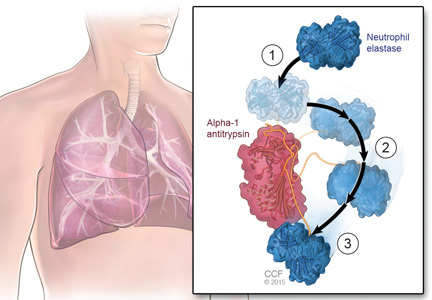
Electronic siloing: An unintended consequence of the electronic health record
For all the purported benefits of the electronic health record (EHR), an unintended adverse effect is “electronic siloing.”
I define electronic siloing as the isolating effect of the EHR on clinical workflow that drives caregivers to work in silos, ie, alone at their workstations, thereby discouraging spontaneous interaction. To the extent that increasing evidence supports the importance of interaction among clinical colleagues and of teamwork to optimize clinical outcomes, electronic siloing threatens optimal practice and quality.
Mindfulness that the EHR can foster siloing will help mitigate the risk, as can novel solutions such as using “viewbox watering holes”1 and embedding secure social messaging functions within the EHR, thereby allowing clinicians to reach out to colleagues with clinical challenges in the moment.
THE EHR BRINGS CHANGES, GOOD AND BAD
The EHR represents a major change in health care, with reported benefits that include standardized ordering, reduced medical errors, embedded protocols for guideline-based care, data access to analyze clinical practice patterns and outcomes, and enhanced communication among colleagues who are geographically separated (eg, virtual consults2). On the basis of these benefits and the federal Medicare and Medicaid financial incentives associated with “meaningful use,” the EHR is being increasingly adopted.3–5
Yet for all these benefits and the promise that technology can enhance interaction among health care providers, unintended risks of the EHR paradoxically threaten optimal clinical care.6 Recognized risks include the threat to care should the EHR fail,6 the time and inefficiency costs of typing and multiple log-ons, and the perpetuation of errors in the medical record caused by the cutting and pasting of clinical notes.
Indeed, a substantial body of literature on sociotechnical interactions—how technology affects human patterns of practice—informs analyses of the impact of changing from a paper medical chart to an EHR.6,8–12 For example, in a review of the impact of computerized physician order entry on inpatient clinical workflow, Niazkhani et al11 noted that computerized ordering can change communication channels and collaboration mechanisms. More specifically, they point out that these systems can “replace interpersonal contacts that may result in fewer opportunities for team-wide negotiations.”11
Similarly, Ash et al8 cited the unintended consequences of patient care information systems, especially increased overreliance on the system to communicate, which can undermine direct communication between healthcare providers.
Finally, Dykstra10 described the “reciprocal impact” of computerized physician order entry systems on communication between physicians and nurses. One observer stated, “[You] start doing physician order entry and direct entry of notes and you move that away from the ward into a room and now you eliminate the sense of team, and the kind of human communication that really was essential… You create physician separation.”10 Taken together, these observations suggest that the EHR and computerized order entry in particular can disrupt interaction between physicians and other health care providers, such as nurses and pharmacists.
BENEFITS OF TEAMWORK
A growing body of evidence indicates that teamwork and collaboration among health care providers—which involve frequent, critical face-to-face interaction—has clinical benefit. Demonstrated benefits of teamwork in health care11 include lower surgical and intensive care unit mortality rates, fewer errors in emergency room management, better neonatal resuscitation, and enhanced diagnostic accuracy in interpreting images and biopsies.12,13
As a specific example of the benefits of face-to-face conversation for interpreting chest images, O’Donovan et al14 showed that the diagnostic accuracy of a pulmonologist and thoracic radiologist in assessing rounded atelectasis was better when they reviewed chest CT scans together than when they interpreted the images solo.
Similarly, Flaherty et al15 showed that the level of agreement among pulmonologists, chest radiologists, and lung pathologists progressively increased as interaction and conversation increased when assessing the etiology of patients’ interstitial lung diseases.
As yet another demonstrable benefit of teamwork that should command interest in the current reimbursement-attentive era, analyses by Press Ganey16 and by Gallup have shown that the single best correlate of high patient satisfaction scores regarding hospitalization (including Hospital Consumer Assessment of Healthcare Providers and Systems ratings) is patients’ perception that their caregivers functioned as a team serving their needs.
The current perspective extends this observation about the unintended adverse effects of the EHR by suggesting that the EHR can inadvertently lessen spontaneous interaction between physicians as they care for outpatients. I have proposed the term electronic siloing to reflect the isolating impact of the EHR on clinical workflow that drives caregivers to work alone at their workstations, thereby discouraging spontaneous interaction between colleagues (eg, between primary care physicians and subspecialists, and between subspecialists in different disciplines). Because spontaneous face-to-face encounters and conversations among clinicians can encourage clinical insights that benefit patient care, electronic siloing can undermine optimal care. My thesis here is that the EHR predisposes to electronic siloing and that the solution is to first recognize and then to design care to prevent this effect.
DECLINE OF THE ‘CURBSIDE’ CONSULT
How does the subtle but sinister effect of electronic siloing really manifest itself at the bedside? I’ll offer an example from my personal clinical experience and then review similar examples from other clinical settings.
First, consider the following real change in clinical workflow that was caused by implementing the EHR in a pulmonary outpatient clinic and its impact on clinical hallway discussions among pulmonologists caring for their outpatients (Figure 1).
The pre-EHR scene was a straight corridor of examination rooms with a long desk outside the rooms and a bank of x-ray viewboxes where clinicians would review films, gather their thoughts, and write notes before re-entering the patient’s room to discuss recommendations. This scene was undoubtedly common in outpatient clinics of all types around the world.
In the bygone era of paper charting and printed x-ray films, the pulmonologists seeing their patients in examination rooms along this corridor and seated next to one another while they wrote their notes would frequently turn to a colleague seated next to them and request a “curbside” consult, ie, an opinion on the films and the case. Typically, a brief, spontaneous conversation would follow, either confirming the requester’s impressions or raising some new, unconsidered approaches. The effect of these brief, spontaneous conversations was either a new diagnostic or treatment consideration or enhanced clinician confidence in the current plan of care. Each outcome has great merit.
Now consider the same scenario in the EHR era. Printed films and viewboxes are gone (which has the benefits of lower production cost and better film retrieval), and images are now reviewed digitally on computer workstations. Workstations are characteristically spread out along the corridor at distances or may be mounted on mobile platforms. Often, physicians now retreat to their nearby offices to write notes, allowing easier access to workstations or to use voice transcription software to record notes. The net effect of this physical separation and of the subtle but powerful change in workflow is that spontaneous curbside consults over a chest film are less likely to occur and, to the extent that such interactions enhance diagnostic accuracy, beneficial face-to-face clinical discussions are less likely. This is the risk of electronic siloing realized.
Defenders of the EHR will point out that the EHR does not preclude such face-to-face encounters. While technically this is correct, it is also equally true that such encounters are less likely because they no longer flow naturally from the workflow of writing a note side-by-side with colleagues with the films displayed nearby. Pressured for time, clinicians learn efficiency of motion and are simply less likely to leave their workstations to seek another colleague who, in turn, may be tethered to a workstation and absorbed in keyboarding and monitor-watching. The net effect is that such spontaneous face-to-face encounters are clearly less common in the EHR era.
Electronic siloing undoubtedly occurs in many other outpatient and inpatient settings in other specialties. For example, consults between orthopedic surgeons seeing outpatients must be similarly affected, as might be discussions between pathologists reviewing tissue slides on a multiheaded microscope vs individually at their own microscopes or work stations. Indeed, observations that computerized order entry isolates physicians from nurses and that the EHR undermines communication between inpatient health care providers6,8–11 represent other manifestations of electronic siloing.
Another variant of siloing occurs when there are not enough computers to go around. When clinicians seek but cannot find available workstations on the hospital ward, they move from the ward to their offices or other locations, separating them from the nurses and other physicians caring for those patients and, thereby, creating isolation and another form of siloing. A related theme is the importance of architecture in driving desirable interactions in the workplace in general and in hospitals in particular,17,18 where interchanges between health care providers are critical to enhancing quality of care.
OUT OF THE SILO, INTO THE FIELD
So, given the many clear benefits of the EHR and its current wave of adoption in health care, how can we maximize the benefits of the EHR while minimizing the adverse effects of electronic siloing?
The key point is that we must realize, appreciate, and prioritize the value of face-toface interaction among providers as we try to offer optimal care to patients with ever more complex clinical problems.
In doing so, clinical workspaces and the number and placement of workstations must be designed with an explicit intent and priority to encourage interchange between providers and to avoid electronic siloing. As an example related to reviewing images, imaging suites and clinics should be designed with the concept of a viewbox watering hole1 in which clinicians arrayed in a common space could review images on their individual computers but could easily prompt colleagues and send an image to a large, centrally visible monitor for the group’s review and comment. Furthermore, the EHR workflows themselves should drive caregivers to the patient rather than requiring their attention to the keyboard and the monitor. One could also imagine embedding secure social messaging within the EHR to encourage interactions among clinicians about pressing clinical challenges they are facing in the moment.
Overall, only through mindfulness of electronic siloing and of its subtle but adverse effects will we break out of the silos and emerge onto the fields of optimal health care.
- Saunder BF. CT Suite: The Work of Diagnosis in the Age of Noninvasive Cutting. Durham, NC: Duke University Press; 2008.
- Palen TE, Price D, Shetterly S, Wallace KB. Comparing virtual consults to traditional consults using an electronic health record: an observational case-control study. BMC Med Inform Decis Mak 2012; 12:65.
- Black AD, Car J, Pagliari C, et al. The impact of eHealth on the quality and safety of health care: a systematic overview. PLoS Med 2011; 8:e1000387.
- Goldzweig CL, Towfigh A, Maglione M, Shekelle PG. Costs and benefits of health information technology: new trends from the literature. Health Aff (Millwood) 2009; 28:w282–w293.
- Police RL, Foster T, Wong KS. Adoption and use of health information technology in physician practice organisations: systematic review. Inform Prim Care 2010; 18:245–258.
- Holroyd-Leduc JM, Lorenzetti D, Straus SE, Sykes L, Quan H. The impact of the electronic medical record on structure, process, and outcomes within primary care: a systematic review of the evidence. J Am Med Inform Assoc 2011; 18:732–737.
- Bohmer RM, McFarlan FW, Adler-Milstein JR. Information technology and clinical operations at Beth Israel Deaconess Medical Center. Harvard Business School 2007; Case 607-150.
- Ash JS, Berg M, Coiera E. Some unintended consequences of information technology in health care: the nature of patient care information system-related errors. J Am Med Inform Assoc 2004; 11:104–112.
- Berg M, Toussaint P. The mantra of modeling and the forgotten powers of paper: a sociotechnical view on the development of process-oriented ICT in health care. Int J Med Inform 2003; 69:223–234.
- Dykstra R. Computerized physician order entry and communication: reciprocal impacts. Proc AMIA Symp 2002:230–234.
- Niazkhani Z, Pirnejad H, Berg M, Aarts J. The impact of computerized provider order entry systems on inpatient clinical workflow: a literature review. J Am Med Inform Assoc 2009; 16:539–549.
- Carayon P. Human factors of complex sociotechnical systems. Appl Ergon 2006; 37:525–535.
- Wheeler D, Stoller JK. Teamwork, teambuilding and leadership in respiratory and health care. Can J Resp Ther 2011; 47. 1:6–11.
- O’Donovan PB, Schenk M, Lim K, Obuchowski N, Stoller JK. Evaluation of the reliability of computed tomographic criteria used in the diagnosis of round atelectasis. J Thorac Imaging 1997; 12:54–58.
- Flaherty KR, King TE, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170:904–910.
- Press Ganey Associates, Inc. Press Ganey mean score correlations to HCAHPS “Rate Hospital 0-10.” 2010. http://www.pressganey.com/ourSolutions/hospitalSettings/satisfactionPerformanceSuite/HCAHPS_Insights.aspx. Accessed May 30, 2013.
- Stoller JK. A physician’s view of hospital design. The impact of verticality on interaction. Architecture 1988; 77:121–122.
- Becker FD, Steele F, editors. Workplace by Design: Mapping the High-Performance Workplace. San Francisco, CA: Jossey-Bass; 1995.
For all the purported benefits of the electronic health record (EHR), an unintended adverse effect is “electronic siloing.”
I define electronic siloing as the isolating effect of the EHR on clinical workflow that drives caregivers to work in silos, ie, alone at their workstations, thereby discouraging spontaneous interaction. To the extent that increasing evidence supports the importance of interaction among clinical colleagues and of teamwork to optimize clinical outcomes, electronic siloing threatens optimal practice and quality.
Mindfulness that the EHR can foster siloing will help mitigate the risk, as can novel solutions such as using “viewbox watering holes”1 and embedding secure social messaging functions within the EHR, thereby allowing clinicians to reach out to colleagues with clinical challenges in the moment.
THE EHR BRINGS CHANGES, GOOD AND BAD
The EHR represents a major change in health care, with reported benefits that include standardized ordering, reduced medical errors, embedded protocols for guideline-based care, data access to analyze clinical practice patterns and outcomes, and enhanced communication among colleagues who are geographically separated (eg, virtual consults2). On the basis of these benefits and the federal Medicare and Medicaid financial incentives associated with “meaningful use,” the EHR is being increasingly adopted.3–5
Yet for all these benefits and the promise that technology can enhance interaction among health care providers, unintended risks of the EHR paradoxically threaten optimal clinical care.6 Recognized risks include the threat to care should the EHR fail,6 the time and inefficiency costs of typing and multiple log-ons, and the perpetuation of errors in the medical record caused by the cutting and pasting of clinical notes.
Indeed, a substantial body of literature on sociotechnical interactions—how technology affects human patterns of practice—informs analyses of the impact of changing from a paper medical chart to an EHR.6,8–12 For example, in a review of the impact of computerized physician order entry on inpatient clinical workflow, Niazkhani et al11 noted that computerized ordering can change communication channels and collaboration mechanisms. More specifically, they point out that these systems can “replace interpersonal contacts that may result in fewer opportunities for team-wide negotiations.”11
Similarly, Ash et al8 cited the unintended consequences of patient care information systems, especially increased overreliance on the system to communicate, which can undermine direct communication between healthcare providers.
Finally, Dykstra10 described the “reciprocal impact” of computerized physician order entry systems on communication between physicians and nurses. One observer stated, “[You] start doing physician order entry and direct entry of notes and you move that away from the ward into a room and now you eliminate the sense of team, and the kind of human communication that really was essential… You create physician separation.”10 Taken together, these observations suggest that the EHR and computerized order entry in particular can disrupt interaction between physicians and other health care providers, such as nurses and pharmacists.
BENEFITS OF TEAMWORK
A growing body of evidence indicates that teamwork and collaboration among health care providers—which involve frequent, critical face-to-face interaction—has clinical benefit. Demonstrated benefits of teamwork in health care11 include lower surgical and intensive care unit mortality rates, fewer errors in emergency room management, better neonatal resuscitation, and enhanced diagnostic accuracy in interpreting images and biopsies.12,13
As a specific example of the benefits of face-to-face conversation for interpreting chest images, O’Donovan et al14 showed that the diagnostic accuracy of a pulmonologist and thoracic radiologist in assessing rounded atelectasis was better when they reviewed chest CT scans together than when they interpreted the images solo.
Similarly, Flaherty et al15 showed that the level of agreement among pulmonologists, chest radiologists, and lung pathologists progressively increased as interaction and conversation increased when assessing the etiology of patients’ interstitial lung diseases.
As yet another demonstrable benefit of teamwork that should command interest in the current reimbursement-attentive era, analyses by Press Ganey16 and by Gallup have shown that the single best correlate of high patient satisfaction scores regarding hospitalization (including Hospital Consumer Assessment of Healthcare Providers and Systems ratings) is patients’ perception that their caregivers functioned as a team serving their needs.
The current perspective extends this observation about the unintended adverse effects of the EHR by suggesting that the EHR can inadvertently lessen spontaneous interaction between physicians as they care for outpatients. I have proposed the term electronic siloing to reflect the isolating impact of the EHR on clinical workflow that drives caregivers to work alone at their workstations, thereby discouraging spontaneous interaction between colleagues (eg, between primary care physicians and subspecialists, and between subspecialists in different disciplines). Because spontaneous face-to-face encounters and conversations among clinicians can encourage clinical insights that benefit patient care, electronic siloing can undermine optimal care. My thesis here is that the EHR predisposes to electronic siloing and that the solution is to first recognize and then to design care to prevent this effect.
DECLINE OF THE ‘CURBSIDE’ CONSULT
How does the subtle but sinister effect of electronic siloing really manifest itself at the bedside? I’ll offer an example from my personal clinical experience and then review similar examples from other clinical settings.
First, consider the following real change in clinical workflow that was caused by implementing the EHR in a pulmonary outpatient clinic and its impact on clinical hallway discussions among pulmonologists caring for their outpatients (Figure 1).
The pre-EHR scene was a straight corridor of examination rooms with a long desk outside the rooms and a bank of x-ray viewboxes where clinicians would review films, gather their thoughts, and write notes before re-entering the patient’s room to discuss recommendations. This scene was undoubtedly common in outpatient clinics of all types around the world.
In the bygone era of paper charting and printed x-ray films, the pulmonologists seeing their patients in examination rooms along this corridor and seated next to one another while they wrote their notes would frequently turn to a colleague seated next to them and request a “curbside” consult, ie, an opinion on the films and the case. Typically, a brief, spontaneous conversation would follow, either confirming the requester’s impressions or raising some new, unconsidered approaches. The effect of these brief, spontaneous conversations was either a new diagnostic or treatment consideration or enhanced clinician confidence in the current plan of care. Each outcome has great merit.
Now consider the same scenario in the EHR era. Printed films and viewboxes are gone (which has the benefits of lower production cost and better film retrieval), and images are now reviewed digitally on computer workstations. Workstations are characteristically spread out along the corridor at distances or may be mounted on mobile platforms. Often, physicians now retreat to their nearby offices to write notes, allowing easier access to workstations or to use voice transcription software to record notes. The net effect of this physical separation and of the subtle but powerful change in workflow is that spontaneous curbside consults over a chest film are less likely to occur and, to the extent that such interactions enhance diagnostic accuracy, beneficial face-to-face clinical discussions are less likely. This is the risk of electronic siloing realized.
Defenders of the EHR will point out that the EHR does not preclude such face-to-face encounters. While technically this is correct, it is also equally true that such encounters are less likely because they no longer flow naturally from the workflow of writing a note side-by-side with colleagues with the films displayed nearby. Pressured for time, clinicians learn efficiency of motion and are simply less likely to leave their workstations to seek another colleague who, in turn, may be tethered to a workstation and absorbed in keyboarding and monitor-watching. The net effect is that such spontaneous face-to-face encounters are clearly less common in the EHR era.
Electronic siloing undoubtedly occurs in many other outpatient and inpatient settings in other specialties. For example, consults between orthopedic surgeons seeing outpatients must be similarly affected, as might be discussions between pathologists reviewing tissue slides on a multiheaded microscope vs individually at their own microscopes or work stations. Indeed, observations that computerized order entry isolates physicians from nurses and that the EHR undermines communication between inpatient health care providers6,8–11 represent other manifestations of electronic siloing.
Another variant of siloing occurs when there are not enough computers to go around. When clinicians seek but cannot find available workstations on the hospital ward, they move from the ward to their offices or other locations, separating them from the nurses and other physicians caring for those patients and, thereby, creating isolation and another form of siloing. A related theme is the importance of architecture in driving desirable interactions in the workplace in general and in hospitals in particular,17,18 where interchanges between health care providers are critical to enhancing quality of care.
OUT OF THE SILO, INTO THE FIELD
So, given the many clear benefits of the EHR and its current wave of adoption in health care, how can we maximize the benefits of the EHR while minimizing the adverse effects of electronic siloing?
The key point is that we must realize, appreciate, and prioritize the value of face-toface interaction among providers as we try to offer optimal care to patients with ever more complex clinical problems.
In doing so, clinical workspaces and the number and placement of workstations must be designed with an explicit intent and priority to encourage interchange between providers and to avoid electronic siloing. As an example related to reviewing images, imaging suites and clinics should be designed with the concept of a viewbox watering hole1 in which clinicians arrayed in a common space could review images on their individual computers but could easily prompt colleagues and send an image to a large, centrally visible monitor for the group’s review and comment. Furthermore, the EHR workflows themselves should drive caregivers to the patient rather than requiring their attention to the keyboard and the monitor. One could also imagine embedding secure social messaging within the EHR to encourage interactions among clinicians about pressing clinical challenges they are facing in the moment.
Overall, only through mindfulness of electronic siloing and of its subtle but adverse effects will we break out of the silos and emerge onto the fields of optimal health care.
For all the purported benefits of the electronic health record (EHR), an unintended adverse effect is “electronic siloing.”
I define electronic siloing as the isolating effect of the EHR on clinical workflow that drives caregivers to work in silos, ie, alone at their workstations, thereby discouraging spontaneous interaction. To the extent that increasing evidence supports the importance of interaction among clinical colleagues and of teamwork to optimize clinical outcomes, electronic siloing threatens optimal practice and quality.
Mindfulness that the EHR can foster siloing will help mitigate the risk, as can novel solutions such as using “viewbox watering holes”1 and embedding secure social messaging functions within the EHR, thereby allowing clinicians to reach out to colleagues with clinical challenges in the moment.
THE EHR BRINGS CHANGES, GOOD AND BAD
The EHR represents a major change in health care, with reported benefits that include standardized ordering, reduced medical errors, embedded protocols for guideline-based care, data access to analyze clinical practice patterns and outcomes, and enhanced communication among colleagues who are geographically separated (eg, virtual consults2). On the basis of these benefits and the federal Medicare and Medicaid financial incentives associated with “meaningful use,” the EHR is being increasingly adopted.3–5
Yet for all these benefits and the promise that technology can enhance interaction among health care providers, unintended risks of the EHR paradoxically threaten optimal clinical care.6 Recognized risks include the threat to care should the EHR fail,6 the time and inefficiency costs of typing and multiple log-ons, and the perpetuation of errors in the medical record caused by the cutting and pasting of clinical notes.
Indeed, a substantial body of literature on sociotechnical interactions—how technology affects human patterns of practice—informs analyses of the impact of changing from a paper medical chart to an EHR.6,8–12 For example, in a review of the impact of computerized physician order entry on inpatient clinical workflow, Niazkhani et al11 noted that computerized ordering can change communication channels and collaboration mechanisms. More specifically, they point out that these systems can “replace interpersonal contacts that may result in fewer opportunities for team-wide negotiations.”11
Similarly, Ash et al8 cited the unintended consequences of patient care information systems, especially increased overreliance on the system to communicate, which can undermine direct communication between healthcare providers.
Finally, Dykstra10 described the “reciprocal impact” of computerized physician order entry systems on communication between physicians and nurses. One observer stated, “[You] start doing physician order entry and direct entry of notes and you move that away from the ward into a room and now you eliminate the sense of team, and the kind of human communication that really was essential… You create physician separation.”10 Taken together, these observations suggest that the EHR and computerized order entry in particular can disrupt interaction between physicians and other health care providers, such as nurses and pharmacists.
BENEFITS OF TEAMWORK
A growing body of evidence indicates that teamwork and collaboration among health care providers—which involve frequent, critical face-to-face interaction—has clinical benefit. Demonstrated benefits of teamwork in health care11 include lower surgical and intensive care unit mortality rates, fewer errors in emergency room management, better neonatal resuscitation, and enhanced diagnostic accuracy in interpreting images and biopsies.12,13
As a specific example of the benefits of face-to-face conversation for interpreting chest images, O’Donovan et al14 showed that the diagnostic accuracy of a pulmonologist and thoracic radiologist in assessing rounded atelectasis was better when they reviewed chest CT scans together than when they interpreted the images solo.
Similarly, Flaherty et al15 showed that the level of agreement among pulmonologists, chest radiologists, and lung pathologists progressively increased as interaction and conversation increased when assessing the etiology of patients’ interstitial lung diseases.
As yet another demonstrable benefit of teamwork that should command interest in the current reimbursement-attentive era, analyses by Press Ganey16 and by Gallup have shown that the single best correlate of high patient satisfaction scores regarding hospitalization (including Hospital Consumer Assessment of Healthcare Providers and Systems ratings) is patients’ perception that their caregivers functioned as a team serving their needs.
The current perspective extends this observation about the unintended adverse effects of the EHR by suggesting that the EHR can inadvertently lessen spontaneous interaction between physicians as they care for outpatients. I have proposed the term electronic siloing to reflect the isolating impact of the EHR on clinical workflow that drives caregivers to work alone at their workstations, thereby discouraging spontaneous interaction between colleagues (eg, between primary care physicians and subspecialists, and between subspecialists in different disciplines). Because spontaneous face-to-face encounters and conversations among clinicians can encourage clinical insights that benefit patient care, electronic siloing can undermine optimal care. My thesis here is that the EHR predisposes to electronic siloing and that the solution is to first recognize and then to design care to prevent this effect.
DECLINE OF THE ‘CURBSIDE’ CONSULT
How does the subtle but sinister effect of electronic siloing really manifest itself at the bedside? I’ll offer an example from my personal clinical experience and then review similar examples from other clinical settings.
First, consider the following real change in clinical workflow that was caused by implementing the EHR in a pulmonary outpatient clinic and its impact on clinical hallway discussions among pulmonologists caring for their outpatients (Figure 1).
The pre-EHR scene was a straight corridor of examination rooms with a long desk outside the rooms and a bank of x-ray viewboxes where clinicians would review films, gather their thoughts, and write notes before re-entering the patient’s room to discuss recommendations. This scene was undoubtedly common in outpatient clinics of all types around the world.
In the bygone era of paper charting and printed x-ray films, the pulmonologists seeing their patients in examination rooms along this corridor and seated next to one another while they wrote their notes would frequently turn to a colleague seated next to them and request a “curbside” consult, ie, an opinion on the films and the case. Typically, a brief, spontaneous conversation would follow, either confirming the requester’s impressions or raising some new, unconsidered approaches. The effect of these brief, spontaneous conversations was either a new diagnostic or treatment consideration or enhanced clinician confidence in the current plan of care. Each outcome has great merit.
Now consider the same scenario in the EHR era. Printed films and viewboxes are gone (which has the benefits of lower production cost and better film retrieval), and images are now reviewed digitally on computer workstations. Workstations are characteristically spread out along the corridor at distances or may be mounted on mobile platforms. Often, physicians now retreat to their nearby offices to write notes, allowing easier access to workstations or to use voice transcription software to record notes. The net effect of this physical separation and of the subtle but powerful change in workflow is that spontaneous curbside consults over a chest film are less likely to occur and, to the extent that such interactions enhance diagnostic accuracy, beneficial face-to-face clinical discussions are less likely. This is the risk of electronic siloing realized.
Defenders of the EHR will point out that the EHR does not preclude such face-to-face encounters. While technically this is correct, it is also equally true that such encounters are less likely because they no longer flow naturally from the workflow of writing a note side-by-side with colleagues with the films displayed nearby. Pressured for time, clinicians learn efficiency of motion and are simply less likely to leave their workstations to seek another colleague who, in turn, may be tethered to a workstation and absorbed in keyboarding and monitor-watching. The net effect is that such spontaneous face-to-face encounters are clearly less common in the EHR era.
Electronic siloing undoubtedly occurs in many other outpatient and inpatient settings in other specialties. For example, consults between orthopedic surgeons seeing outpatients must be similarly affected, as might be discussions between pathologists reviewing tissue slides on a multiheaded microscope vs individually at their own microscopes or work stations. Indeed, observations that computerized order entry isolates physicians from nurses and that the EHR undermines communication between inpatient health care providers6,8–11 represent other manifestations of electronic siloing.
Another variant of siloing occurs when there are not enough computers to go around. When clinicians seek but cannot find available workstations on the hospital ward, they move from the ward to their offices or other locations, separating them from the nurses and other physicians caring for those patients and, thereby, creating isolation and another form of siloing. A related theme is the importance of architecture in driving desirable interactions in the workplace in general and in hospitals in particular,17,18 where interchanges between health care providers are critical to enhancing quality of care.
OUT OF THE SILO, INTO THE FIELD
So, given the many clear benefits of the EHR and its current wave of adoption in health care, how can we maximize the benefits of the EHR while minimizing the adverse effects of electronic siloing?
The key point is that we must realize, appreciate, and prioritize the value of face-toface interaction among providers as we try to offer optimal care to patients with ever more complex clinical problems.
In doing so, clinical workspaces and the number and placement of workstations must be designed with an explicit intent and priority to encourage interchange between providers and to avoid electronic siloing. As an example related to reviewing images, imaging suites and clinics should be designed with the concept of a viewbox watering hole1 in which clinicians arrayed in a common space could review images on their individual computers but could easily prompt colleagues and send an image to a large, centrally visible monitor for the group’s review and comment. Furthermore, the EHR workflows themselves should drive caregivers to the patient rather than requiring their attention to the keyboard and the monitor. One could also imagine embedding secure social messaging within the EHR to encourage interactions among clinicians about pressing clinical challenges they are facing in the moment.
Overall, only through mindfulness of electronic siloing and of its subtle but adverse effects will we break out of the silos and emerge onto the fields of optimal health care.
- Saunder BF. CT Suite: The Work of Diagnosis in the Age of Noninvasive Cutting. Durham, NC: Duke University Press; 2008.
- Palen TE, Price D, Shetterly S, Wallace KB. Comparing virtual consults to traditional consults using an electronic health record: an observational case-control study. BMC Med Inform Decis Mak 2012; 12:65.
- Black AD, Car J, Pagliari C, et al. The impact of eHealth on the quality and safety of health care: a systematic overview. PLoS Med 2011; 8:e1000387.
- Goldzweig CL, Towfigh A, Maglione M, Shekelle PG. Costs and benefits of health information technology: new trends from the literature. Health Aff (Millwood) 2009; 28:w282–w293.
- Police RL, Foster T, Wong KS. Adoption and use of health information technology in physician practice organisations: systematic review. Inform Prim Care 2010; 18:245–258.
- Holroyd-Leduc JM, Lorenzetti D, Straus SE, Sykes L, Quan H. The impact of the electronic medical record on structure, process, and outcomes within primary care: a systematic review of the evidence. J Am Med Inform Assoc 2011; 18:732–737.
- Bohmer RM, McFarlan FW, Adler-Milstein JR. Information technology and clinical operations at Beth Israel Deaconess Medical Center. Harvard Business School 2007; Case 607-150.
- Ash JS, Berg M, Coiera E. Some unintended consequences of information technology in health care: the nature of patient care information system-related errors. J Am Med Inform Assoc 2004; 11:104–112.
- Berg M, Toussaint P. The mantra of modeling and the forgotten powers of paper: a sociotechnical view on the development of process-oriented ICT in health care. Int J Med Inform 2003; 69:223–234.
- Dykstra R. Computerized physician order entry and communication: reciprocal impacts. Proc AMIA Symp 2002:230–234.
- Niazkhani Z, Pirnejad H, Berg M, Aarts J. The impact of computerized provider order entry systems on inpatient clinical workflow: a literature review. J Am Med Inform Assoc 2009; 16:539–549.
- Carayon P. Human factors of complex sociotechnical systems. Appl Ergon 2006; 37:525–535.
- Wheeler D, Stoller JK. Teamwork, teambuilding and leadership in respiratory and health care. Can J Resp Ther 2011; 47. 1:6–11.
- O’Donovan PB, Schenk M, Lim K, Obuchowski N, Stoller JK. Evaluation of the reliability of computed tomographic criteria used in the diagnosis of round atelectasis. J Thorac Imaging 1997; 12:54–58.
- Flaherty KR, King TE, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170:904–910.
- Press Ganey Associates, Inc. Press Ganey mean score correlations to HCAHPS “Rate Hospital 0-10.” 2010. http://www.pressganey.com/ourSolutions/hospitalSettings/satisfactionPerformanceSuite/HCAHPS_Insights.aspx. Accessed May 30, 2013.
- Stoller JK. A physician’s view of hospital design. The impact of verticality on interaction. Architecture 1988; 77:121–122.
- Becker FD, Steele F, editors. Workplace by Design: Mapping the High-Performance Workplace. San Francisco, CA: Jossey-Bass; 1995.
- Saunder BF. CT Suite: The Work of Diagnosis in the Age of Noninvasive Cutting. Durham, NC: Duke University Press; 2008.
- Palen TE, Price D, Shetterly S, Wallace KB. Comparing virtual consults to traditional consults using an electronic health record: an observational case-control study. BMC Med Inform Decis Mak 2012; 12:65.
- Black AD, Car J, Pagliari C, et al. The impact of eHealth on the quality and safety of health care: a systematic overview. PLoS Med 2011; 8:e1000387.
- Goldzweig CL, Towfigh A, Maglione M, Shekelle PG. Costs and benefits of health information technology: new trends from the literature. Health Aff (Millwood) 2009; 28:w282–w293.
- Police RL, Foster T, Wong KS. Adoption and use of health information technology in physician practice organisations: systematic review. Inform Prim Care 2010; 18:245–258.
- Holroyd-Leduc JM, Lorenzetti D, Straus SE, Sykes L, Quan H. The impact of the electronic medical record on structure, process, and outcomes within primary care: a systematic review of the evidence. J Am Med Inform Assoc 2011; 18:732–737.
- Bohmer RM, McFarlan FW, Adler-Milstein JR. Information technology and clinical operations at Beth Israel Deaconess Medical Center. Harvard Business School 2007; Case 607-150.
- Ash JS, Berg M, Coiera E. Some unintended consequences of information technology in health care: the nature of patient care information system-related errors. J Am Med Inform Assoc 2004; 11:104–112.
- Berg M, Toussaint P. The mantra of modeling and the forgotten powers of paper: a sociotechnical view on the development of process-oriented ICT in health care. Int J Med Inform 2003; 69:223–234.
- Dykstra R. Computerized physician order entry and communication: reciprocal impacts. Proc AMIA Symp 2002:230–234.
- Niazkhani Z, Pirnejad H, Berg M, Aarts J. The impact of computerized provider order entry systems on inpatient clinical workflow: a literature review. J Am Med Inform Assoc 2009; 16:539–549.
- Carayon P. Human factors of complex sociotechnical systems. Appl Ergon 2006; 37:525–535.
- Wheeler D, Stoller JK. Teamwork, teambuilding and leadership in respiratory and health care. Can J Resp Ther 2011; 47. 1:6–11.
- O’Donovan PB, Schenk M, Lim K, Obuchowski N, Stoller JK. Evaluation of the reliability of computed tomographic criteria used in the diagnosis of round atelectasis. J Thorac Imaging 1997; 12:54–58.
- Flaherty KR, King TE, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170:904–910.
- Press Ganey Associates, Inc. Press Ganey mean score correlations to HCAHPS “Rate Hospital 0-10.” 2010. http://www.pressganey.com/ourSolutions/hospitalSettings/satisfactionPerformanceSuite/HCAHPS_Insights.aspx. Accessed May 30, 2013.
- Stoller JK. A physician’s view of hospital design. The impact of verticality on interaction. Architecture 1988; 77:121–122.
- Becker FD, Steele F, editors. Workplace by Design: Mapping the High-Performance Workplace. San Francisco, CA: Jossey-Bass; 1995.
Caring for VIPs: Nine principles
Medical tourism is on the rise,1 and since medical tourists are often very important persons (VIPs), hospital-based physicians may be more likely to care for celebrities, royalty, and political leaders. But even in hospitals that do not see medical tourists, physicians will often care for VIP patients such as hospital trustees and board members, prominent physicians, and community leaders.2–4
However, caring for VIPs raises special issues and challenges. In a situation often referred to as the “VIP syndrome,”5–9 a patient’s special social or political status—or our perceptions of it—induces changes in behaviors and clinical practice that create a “vicious circle of VIP pressure and staff withdrawal”9 that can lead to poor outcomes.
Based on their experience caring for three American presidents, Mariano and McLeod7 offered three directives for caring for VIPs:
- Vow to value your medical skills and judgment
- Intend to command the medical aspects of the situation
- Practice medicine the same way for all your patients.7
In this paper, we hope to extend the sparse literature on the VIP syndrome by proposing nine principles of caring for VIPs, with recommendations specific to the type of VIP where applicable.
PRINCIPLE 1: DON’T BEND THE RULES
Caring for VIPs creates pressures to change usual clinical wisdom and practices. But it is essential to resist changing time-honored, effective clinical judgment and practices.
To preserve usual clinical practice, clinicians must be constantly vigilant as to whether their judgment is being clouded by the circumstances. As Smith and Shesser noted in 1988, “Since the standard operating procedures […] are designed for the efficient delivery of high-quality care, any deviation from these procedures increases the possibility that care may be compromised.”5 In other words, suspending usual practice when caring for a VIP patient can imperil the patient.2–5,10,11 When caring for VIP physicians, for example, circumventing usual medical and administrative routines and the difficulties that caring for colleagues poses for nurses and physicians have led to poor medical care and outcomes, as well as to hostility.2–4
A striking example of the potential effects of VIP syndrome is the death of Eleanor Roosevelt from miliary tuberculosis acutissima: she was misdiagnosed with aplastic anemia on the basis of only the results of a bone marrow aspirate study, and she was treated with steroids. The desire to spare this VIP patient the discomfort of a bone marrow biopsy, on which tuberculous granulomata were more likely to have been seen, caused the true diagnosis to be missed and resulted in the administration of a hazardous medication.11 The hard lesson here is that we must resist the pressure to simplify or change customary medical care to avoid causing a VIP patient discomfort or putting the patient through a complex procedure.
We recommend discussing these issues explicitly with the VIP patient and family at the outset so that everyone can appreciate the importance of usual care. An early conversation can communicate the clinician’s experience in the care of such patients and can be reassuring. As Smith and Shesser noted, “Usually, the VIP is relieved if the physician states explicitly, ‘I am going to treat you as I would any other patient.’ ”5
PRINCIPLE 2: WORK AS A TEAM, NOT IN ‘SILOS’
Teamwork is essential for good clinical outcomes, 12–14 especially when the clinical problem is complex, as is often the case when people travel long distances to receive care. All consultants involved in the patient’s care must not only attend to their own clinical issues but also communicate amply with their colleagues.
At the same time, we must recognize that medical practice “is not a committee process; it must be clear at all times which physician is responsible for directing clinical care.”5 One physician must be in charge of the overall care. Seeking the input of other physicians must not be allowed to diffuse responsibility. The primary attending physician must speak with the consultants, summarize their views, and then communicate the findings and the plan of care to the patient and family.
Paradoxically, teamwork can be challenged when circumstances lead consultants to defer communicating directly with the family in favor of the primary physician’s doing so. Similarly, consultants must avoid any temptation to simply “do their thing” and not communicate with one another, thereby potentially offering “siloed,” discoordinated care.
We propose designating a primary physician to take charge of the care and the communication. This physician must have the time to talk with each team member about how best to communicate the individual findings to the patient and family. At times, the primary physician may also ask the consultants to communicate directly with the patient and family when needed.
PRINCIPLE 3: COMMUNICATE, COMMUNICATE, COMMUNICATE
As a corollary of principle 2, heightened communication is essential when caring for VIP patients. Communication should include the patient, the family, visiting physicians who accompany the patient, and the physicians providing care. Communicating with the media and with other uninvolved individuals is addressed in principle 4.
The logistic and security challenges of transporting VIP patients through the hospital for tests or therapy demand increased communication. Scheduling a computed tomographic scan may involve arranging an off-hours appointment in the radiology department (to minimize security risks and disruption to other patients’ schedules), assuring the off-hours availability of allied health providers to accompany the patient, alerting hospital security, and discussing the appointment with the patient and the patient’s entourage.
PRINCIPLE 4: CAREFULLY MANAGE COMMUNICATION WITH THE MEDIA
Although the news media and the public may demand medical information about patients who are celebrities, political luminaries, or royalty, the confidentiality of the physician-patient relationship must be protected. The release of health information is at the sole discretion of the patient or a designated surrogate.
The care of President Ronald Reagan after the 1981 assassination attempt is a benchmark of how to release information to the public.10 A single physician held regularly scheduled press conferences, and these were intentionally held away from the site of the President’s care.
Designating a senior hospital physician to communicate with the media is desirable, and the physician-spokesperson can call on specialists from the patient care team (eg, a critical care physician), when appropriate, to provide further information.
Early implementation of an explicit and structured media communication plan is advisable, especially when the VIP patient is a political or royal figure for whom public clamor for information will be vigorous. A successful communication strategy balances the public’s demand for information with the need to protect the patient’s confidentiality.
PRINCIPLE 5: RESIST ‘CHAIRPERSON’S SYNDROME’
“Chairperson’s syndrome”5 is pressure for the VIP patient to be cared for by the department chairperson. The pressure may come from the patient, family, or attendants, who may assume that the chairperson is the best doctor for the clinical circumstance. The pressure may also come from the chairperson, who feels the need to “take command” in a situation with high visibility. Nevertheless, designation of a chairperson to care for a VIP patient is appropriate only when the chairperson is indeed the clinician who has the most expertise in the patient’s clinical issues.
As in principle 1, in academic medical centers, we encourage the participation of trainees in the care of VIP patients because excluding them could disrupt the usual flow of care, and because trainees offer a currency and facility with the nuances of hospital practice and routine that are advantageous to the patient’s care.
PRINCIPLE 6: CARE SHOULD OCCUR WHERE IT IS MOST APPROPRIATE
Decisions about where to place the VIP patient during the medical visit can fall victim to the VIP syndrome if the expectations of the patient or family conflict with usual clinical practice and judgment about the optimal care venue.
For example, caring for the patient in a setting away from the mainstream clinical environment may offer the appeal of privacy or enhanced security but can under some circumstances impede optimal care, including prolonging the response time during emergencies and disrupting the optimal care routine and teamwork of allied health providers.
Critical care services and monitoring are best provided in the intensive care unit, and attempts to relocate the patient away from the intensive care unit should be resisted. We recommend a candid discussion of the importance of keeping the patient in the intensive care unit to ensure optimal care by a seasoned clinical team with short response times if urgencies should arise.
At the same time, a request to transfer a VIP patient to a special setting designed for private care with special amenities (eg, appealing room decor, adjacent sleeping rooms for family members, enhanced security) available in some hospitals15–16 can be honored as soon as the patient’s condition permits. The benefits of such amenities are often greatly appreciated and can reduce stress and thereby promote recovery. The benefits of enhanced security in sequestered venues may especially drive the decision to move when clinically prudent (see principle 7).
PRINCIPLE 7: PROTECT THE PATIENT’S SECURITY
Providing security is another essential part of caring for VIPs, especially celebrities, political figures, and royalty. Protecting the patient from bodily harm requires special attention to the patient’s location, caregiver access, and other logistic matters.
As indicated in principle 6, the patient’s clinical needs are paramount in determining where the patient receives care. If the patient requires care in a mainstream hospital location such as the intensive care unit, modifications of the unit may be needed to alter access, to accommodate security personnel, and to restrict caregivers’ access to the patient. Modifications include structural changes to windows, special credentials (eg, badges) for essential providers, arranging transports within the hospital for elective procedures during off-hours, and providing around-the-clock security personnel near the patient.
As important as it is to protect VIP patients from bodily harm during the visit, it is equally important to protect them from attacks on confidentiality via unauthorized access to the electronic medical record, and this is perhaps the more difficult challenge, as examples of breaches abound.10,17–19 Although the duty to protect against these breaches rests with the hospital, the use of “pop-ups” in the electronic medical record can flash a warning that only employees with legitimate clinical reasons should access the record. These warnings should also cite the penalties for unauthorized review of the record, which is supported by the Health Insurance Portability and Accountability Act (HIPAA). Access to celebrities’ health records could be restricted to a few predetermined health care providers.
PRINCIPLE 8: BE CAREFUL ABOUT ACCEPTING OR DECLINING GIFTS
VIP patients often present gifts to physicians, and giving gifts to doctors is a common and long-standing practice.20,21 Patients offer gifts out of gratitude, affection, desperation, or the desire to garner special treatment or indebtedness. VIP patients from gifting cultures may be especially likely to offer gifts to their providers, and the gifts can be lavish.
The “ethical calculus”21 of whether to accept or decline a gift depends on the circumstances and on what motivates the offer, and the physician needs to consider the patient’s reasons for giving the gift.
In general, gifts should be accepted only with caution during the acute episode of care. The acceptance of a gift from a VIP patient or family member may be interpreted by the gift-giver as a sort of unspoken promise, and this misunderstanding may strain the physician-patient relationship, especially if the clinical course deteriorates.
Rather than accept a gift during an episode of acute care, we suggest that the physician graciously decline the gift and offer to accept the gift at the end of the episode of acute care—that is, if the offerer still feels so inclined and remembers. Explaining the reason for deferring the gift can decrease the risk of misunderstandings or of unmet expectations by the gift-giver. Also, deferring the acceptance of a gift allows the caregiver to affirm the commitment to excellent care that is free of gifts, thereby ensuring that the patient will be confident of a similar level of care by providers who have not been offered gifts.
On the other hand, declining a gift may cause more damage than accepting it, particularly if the VIP patient is from a culture in which refusing a gift is impolite.22 A sensible compromise may be to adopt the recommendations of the American Academy of Pediatrics23—ie, attempt to appreciate appropriate gifts and graciously refuse those that are not.
PRINCIPLE 9: WORKING WITH THE PATIENT’S PERSONAL PHYSICIANS
VIP patients, perhaps especially royalty, may be accompanied by their own physicians and may also wish to bring in consultants from other institutions. Though this outside involvement poses challenges (eg, providing access to medical records, arranging briefings, attending bedside rounds), we believe it should be encouraged when the issue is raised. Furthermore, institutions and caregivers should anticipate these requests and identify potential outside consultants whose names can be volunteered if the issue arises.
Again, if VIP patients wish to involve physicians from outside the institution where they are receiving care, this should not be viewed as an expression of doubt about the care being received. Rather, we prefer to view it as an opportunity to validate current management or to entertain alternative approaches. Most often, when an outside consultant confirms the current medical care, this can have the beneficial effect of increasing confidence and facilitating management.
In a similar way, when VIP patients bring their own physician, whose judgment and care they trust, this represents an opportunity to engage the patient’s trusted physician-advisor in clinical decision-making and thus optimize communication with the patient. Collegial interactions with these physician-colleagues can facilitate communication and decision-making for the patient.
- Ehrbeck T, Guevara C, Mango PD. Mapping the market for medical travel. Health Care: Strategy & Analysis. McKinsey Quarterly 2008 May;1–11.
- Stoudemire A, Rhoads JM. When the doctor needs a doctor: special considerations for the physician-patient. Ann Intern Med 1983; 98:654–659.
- Schneck SA. “Doctoring” doctors and their families. JAMA 1998; 280:2039–2042.
- Adshead G. Healing ourselves: ethical issues in the care of sick doctors. Adv Psychiatr Treat 2005; 11:330–337.
- Smith MS, Shesser RF. The emergency care of the VIP patient. N Engl J Med 1988; 319:1421–1423.
- Block AJ. Beware of the VIP syndrome. Chest 1993; 104:989.
- Mariano EC, McLeod JA. Emergency care for the VIP patient. Intensive Care Medicine 2007. http://dx.doi.org/10.1007/978-0-387-49518-7_88. Accessed December 27, 2010.
- Schenkenberg T, Kochenour NK, Botkin JR. Ethical considerations in clinical care of the “VIP”. J Clin Ethics 2007; 18:56–63.
- Weintraub W. “The VIP syndrome”: a clinical study in hospital psychiatry. J Nerv Ment Dis 1964; 138:181–193.
- Weiss YG, Mor-Yosef S, Sprung CL, Weissman C, Weiss Y. Caring for a major government official: challenges and lessons learned. Crit Care Med 2007; 35:1769–1772.
- Lerner BH. Revisiting the death of Eleanor Roosevelt: was the diagnosis of tuberculosis missed? Int J Tuberc Lung Dis 2001; 5:1080–1085.
- Lee TH. Turning doctors into leaders. Harv Bus Rev 2010; 88:50–58.
- Clemmer TP, Spuhler VJ, Berwick DM, Nolan TW. Cooperation: the foundation of improvement. Ann Intern Med 1998; 128:1004–1009.
- Morey JC, Simon R, Jay GD, et al. Error reduction and performance improvement in the emergency department through formal teamwork training: evaluation results of the MedTeams project. Health Serv Res 2002; 37:1553–1581.
- VIP ward at Walter Reed gets scrutiny. USA Today. http://www.usatoday.com/news/washington/2007-03-15-walter-reed-vip_N.htm. Accessed December 27, 2010.
- Robins RS, Post JM. When Illness Strikes the Leader. The Dilemma of the Captive King. New Haven: Yale University Press; 1995.
- Carr J. Breach of Britney Spears patient data reported. SC Magazine, March 19, 2008. http://www.scmagazineus.com/breach-of-britney-spears-patient-data-reported/article/108141/. Accessed December 27, 2010.
- Collins T. Sir Bobby Robson’s electronic health records viewed illicitly by NHS staff. ComputerWeekly.com, September 24, 2007. http://www.computerweekly.com/blogs/tony_collins/2007/09/bobby-robsons-medical-records-1.html. Accessed December 27, 2010.
- Ornstein C. Kaiser hospital fined $250,000 for privacy breach in octuplet case. Propublica.org, May 15, 2009. http://www.propublica.org/article/kaiser-hospital-fined-250000-for-privacy-breach-in-octuplet-case-515. Accessed December 27, 2010.
- Levene MI, Sireling L. Gift giving to hospital doctors—in the mouth of the gift horse. Br Med J 1980; 281:1685.
- Lyckholm LJ. Should physicians accept gifts from patients? JAMA 1998; 280:1944–1946.
- Takayama JI. Giving and receiving gifts: one perspective. West J Med 2001; 175:138–139.
- Committee on Bioethics. From the American Academy of Pediatrics: policy statements—pediatrician-family-patient relationships: managing the boundaries. Pediatrics 2009; 124:1685–1688.
Medical tourism is on the rise,1 and since medical tourists are often very important persons (VIPs), hospital-based physicians may be more likely to care for celebrities, royalty, and political leaders. But even in hospitals that do not see medical tourists, physicians will often care for VIP patients such as hospital trustees and board members, prominent physicians, and community leaders.2–4
However, caring for VIPs raises special issues and challenges. In a situation often referred to as the “VIP syndrome,”5–9 a patient’s special social or political status—or our perceptions of it—induces changes in behaviors and clinical practice that create a “vicious circle of VIP pressure and staff withdrawal”9 that can lead to poor outcomes.
Based on their experience caring for three American presidents, Mariano and McLeod7 offered three directives for caring for VIPs:
- Vow to value your medical skills and judgment
- Intend to command the medical aspects of the situation
- Practice medicine the same way for all your patients.7
In this paper, we hope to extend the sparse literature on the VIP syndrome by proposing nine principles of caring for VIPs, with recommendations specific to the type of VIP where applicable.
PRINCIPLE 1: DON’T BEND THE RULES
Caring for VIPs creates pressures to change usual clinical wisdom and practices. But it is essential to resist changing time-honored, effective clinical judgment and practices.
To preserve usual clinical practice, clinicians must be constantly vigilant as to whether their judgment is being clouded by the circumstances. As Smith and Shesser noted in 1988, “Since the standard operating procedures […] are designed for the efficient delivery of high-quality care, any deviation from these procedures increases the possibility that care may be compromised.”5 In other words, suspending usual practice when caring for a VIP patient can imperil the patient.2–5,10,11 When caring for VIP physicians, for example, circumventing usual medical and administrative routines and the difficulties that caring for colleagues poses for nurses and physicians have led to poor medical care and outcomes, as well as to hostility.2–4
A striking example of the potential effects of VIP syndrome is the death of Eleanor Roosevelt from miliary tuberculosis acutissima: she was misdiagnosed with aplastic anemia on the basis of only the results of a bone marrow aspirate study, and she was treated with steroids. The desire to spare this VIP patient the discomfort of a bone marrow biopsy, on which tuberculous granulomata were more likely to have been seen, caused the true diagnosis to be missed and resulted in the administration of a hazardous medication.11 The hard lesson here is that we must resist the pressure to simplify or change customary medical care to avoid causing a VIP patient discomfort or putting the patient through a complex procedure.
We recommend discussing these issues explicitly with the VIP patient and family at the outset so that everyone can appreciate the importance of usual care. An early conversation can communicate the clinician’s experience in the care of such patients and can be reassuring. As Smith and Shesser noted, “Usually, the VIP is relieved if the physician states explicitly, ‘I am going to treat you as I would any other patient.’ ”5
PRINCIPLE 2: WORK AS A TEAM, NOT IN ‘SILOS’
Teamwork is essential for good clinical outcomes, 12–14 especially when the clinical problem is complex, as is often the case when people travel long distances to receive care. All consultants involved in the patient’s care must not only attend to their own clinical issues but also communicate amply with their colleagues.
At the same time, we must recognize that medical practice “is not a committee process; it must be clear at all times which physician is responsible for directing clinical care.”5 One physician must be in charge of the overall care. Seeking the input of other physicians must not be allowed to diffuse responsibility. The primary attending physician must speak with the consultants, summarize their views, and then communicate the findings and the plan of care to the patient and family.
Paradoxically, teamwork can be challenged when circumstances lead consultants to defer communicating directly with the family in favor of the primary physician’s doing so. Similarly, consultants must avoid any temptation to simply “do their thing” and not communicate with one another, thereby potentially offering “siloed,” discoordinated care.
We propose designating a primary physician to take charge of the care and the communication. This physician must have the time to talk with each team member about how best to communicate the individual findings to the patient and family. At times, the primary physician may also ask the consultants to communicate directly with the patient and family when needed.
PRINCIPLE 3: COMMUNICATE, COMMUNICATE, COMMUNICATE
As a corollary of principle 2, heightened communication is essential when caring for VIP patients. Communication should include the patient, the family, visiting physicians who accompany the patient, and the physicians providing care. Communicating with the media and with other uninvolved individuals is addressed in principle 4.
The logistic and security challenges of transporting VIP patients through the hospital for tests or therapy demand increased communication. Scheduling a computed tomographic scan may involve arranging an off-hours appointment in the radiology department (to minimize security risks and disruption to other patients’ schedules), assuring the off-hours availability of allied health providers to accompany the patient, alerting hospital security, and discussing the appointment with the patient and the patient’s entourage.
PRINCIPLE 4: CAREFULLY MANAGE COMMUNICATION WITH THE MEDIA
Although the news media and the public may demand medical information about patients who are celebrities, political luminaries, or royalty, the confidentiality of the physician-patient relationship must be protected. The release of health information is at the sole discretion of the patient or a designated surrogate.
The care of President Ronald Reagan after the 1981 assassination attempt is a benchmark of how to release information to the public.10 A single physician held regularly scheduled press conferences, and these were intentionally held away from the site of the President’s care.
Designating a senior hospital physician to communicate with the media is desirable, and the physician-spokesperson can call on specialists from the patient care team (eg, a critical care physician), when appropriate, to provide further information.
Early implementation of an explicit and structured media communication plan is advisable, especially when the VIP patient is a political or royal figure for whom public clamor for information will be vigorous. A successful communication strategy balances the public’s demand for information with the need to protect the patient’s confidentiality.
PRINCIPLE 5: RESIST ‘CHAIRPERSON’S SYNDROME’
“Chairperson’s syndrome”5 is pressure for the VIP patient to be cared for by the department chairperson. The pressure may come from the patient, family, or attendants, who may assume that the chairperson is the best doctor for the clinical circumstance. The pressure may also come from the chairperson, who feels the need to “take command” in a situation with high visibility. Nevertheless, designation of a chairperson to care for a VIP patient is appropriate only when the chairperson is indeed the clinician who has the most expertise in the patient’s clinical issues.
As in principle 1, in academic medical centers, we encourage the participation of trainees in the care of VIP patients because excluding them could disrupt the usual flow of care, and because trainees offer a currency and facility with the nuances of hospital practice and routine that are advantageous to the patient’s care.
PRINCIPLE 6: CARE SHOULD OCCUR WHERE IT IS MOST APPROPRIATE
Decisions about where to place the VIP patient during the medical visit can fall victim to the VIP syndrome if the expectations of the patient or family conflict with usual clinical practice and judgment about the optimal care venue.
For example, caring for the patient in a setting away from the mainstream clinical environment may offer the appeal of privacy or enhanced security but can under some circumstances impede optimal care, including prolonging the response time during emergencies and disrupting the optimal care routine and teamwork of allied health providers.
Critical care services and monitoring are best provided in the intensive care unit, and attempts to relocate the patient away from the intensive care unit should be resisted. We recommend a candid discussion of the importance of keeping the patient in the intensive care unit to ensure optimal care by a seasoned clinical team with short response times if urgencies should arise.
At the same time, a request to transfer a VIP patient to a special setting designed for private care with special amenities (eg, appealing room decor, adjacent sleeping rooms for family members, enhanced security) available in some hospitals15–16 can be honored as soon as the patient’s condition permits. The benefits of such amenities are often greatly appreciated and can reduce stress and thereby promote recovery. The benefits of enhanced security in sequestered venues may especially drive the decision to move when clinically prudent (see principle 7).
PRINCIPLE 7: PROTECT THE PATIENT’S SECURITY
Providing security is another essential part of caring for VIPs, especially celebrities, political figures, and royalty. Protecting the patient from bodily harm requires special attention to the patient’s location, caregiver access, and other logistic matters.
As indicated in principle 6, the patient’s clinical needs are paramount in determining where the patient receives care. If the patient requires care in a mainstream hospital location such as the intensive care unit, modifications of the unit may be needed to alter access, to accommodate security personnel, and to restrict caregivers’ access to the patient. Modifications include structural changes to windows, special credentials (eg, badges) for essential providers, arranging transports within the hospital for elective procedures during off-hours, and providing around-the-clock security personnel near the patient.
As important as it is to protect VIP patients from bodily harm during the visit, it is equally important to protect them from attacks on confidentiality via unauthorized access to the electronic medical record, and this is perhaps the more difficult challenge, as examples of breaches abound.10,17–19 Although the duty to protect against these breaches rests with the hospital, the use of “pop-ups” in the electronic medical record can flash a warning that only employees with legitimate clinical reasons should access the record. These warnings should also cite the penalties for unauthorized review of the record, which is supported by the Health Insurance Portability and Accountability Act (HIPAA). Access to celebrities’ health records could be restricted to a few predetermined health care providers.
PRINCIPLE 8: BE CAREFUL ABOUT ACCEPTING OR DECLINING GIFTS
VIP patients often present gifts to physicians, and giving gifts to doctors is a common and long-standing practice.20,21 Patients offer gifts out of gratitude, affection, desperation, or the desire to garner special treatment or indebtedness. VIP patients from gifting cultures may be especially likely to offer gifts to their providers, and the gifts can be lavish.
The “ethical calculus”21 of whether to accept or decline a gift depends on the circumstances and on what motivates the offer, and the physician needs to consider the patient’s reasons for giving the gift.
In general, gifts should be accepted only with caution during the acute episode of care. The acceptance of a gift from a VIP patient or family member may be interpreted by the gift-giver as a sort of unspoken promise, and this misunderstanding may strain the physician-patient relationship, especially if the clinical course deteriorates.
Rather than accept a gift during an episode of acute care, we suggest that the physician graciously decline the gift and offer to accept the gift at the end of the episode of acute care—that is, if the offerer still feels so inclined and remembers. Explaining the reason for deferring the gift can decrease the risk of misunderstandings or of unmet expectations by the gift-giver. Also, deferring the acceptance of a gift allows the caregiver to affirm the commitment to excellent care that is free of gifts, thereby ensuring that the patient will be confident of a similar level of care by providers who have not been offered gifts.
On the other hand, declining a gift may cause more damage than accepting it, particularly if the VIP patient is from a culture in which refusing a gift is impolite.22 A sensible compromise may be to adopt the recommendations of the American Academy of Pediatrics23—ie, attempt to appreciate appropriate gifts and graciously refuse those that are not.
PRINCIPLE 9: WORKING WITH THE PATIENT’S PERSONAL PHYSICIANS
VIP patients, perhaps especially royalty, may be accompanied by their own physicians and may also wish to bring in consultants from other institutions. Though this outside involvement poses challenges (eg, providing access to medical records, arranging briefings, attending bedside rounds), we believe it should be encouraged when the issue is raised. Furthermore, institutions and caregivers should anticipate these requests and identify potential outside consultants whose names can be volunteered if the issue arises.
Again, if VIP patients wish to involve physicians from outside the institution where they are receiving care, this should not be viewed as an expression of doubt about the care being received. Rather, we prefer to view it as an opportunity to validate current management or to entertain alternative approaches. Most often, when an outside consultant confirms the current medical care, this can have the beneficial effect of increasing confidence and facilitating management.
In a similar way, when VIP patients bring their own physician, whose judgment and care they trust, this represents an opportunity to engage the patient’s trusted physician-advisor in clinical decision-making and thus optimize communication with the patient. Collegial interactions with these physician-colleagues can facilitate communication and decision-making for the patient.
Medical tourism is on the rise,1 and since medical tourists are often very important persons (VIPs), hospital-based physicians may be more likely to care for celebrities, royalty, and political leaders. But even in hospitals that do not see medical tourists, physicians will often care for VIP patients such as hospital trustees and board members, prominent physicians, and community leaders.2–4
However, caring for VIPs raises special issues and challenges. In a situation often referred to as the “VIP syndrome,”5–9 a patient’s special social or political status—or our perceptions of it—induces changes in behaviors and clinical practice that create a “vicious circle of VIP pressure and staff withdrawal”9 that can lead to poor outcomes.
Based on their experience caring for three American presidents, Mariano and McLeod7 offered three directives for caring for VIPs:
- Vow to value your medical skills and judgment
- Intend to command the medical aspects of the situation
- Practice medicine the same way for all your patients.7
In this paper, we hope to extend the sparse literature on the VIP syndrome by proposing nine principles of caring for VIPs, with recommendations specific to the type of VIP where applicable.
PRINCIPLE 1: DON’T BEND THE RULES
Caring for VIPs creates pressures to change usual clinical wisdom and practices. But it is essential to resist changing time-honored, effective clinical judgment and practices.
To preserve usual clinical practice, clinicians must be constantly vigilant as to whether their judgment is being clouded by the circumstances. As Smith and Shesser noted in 1988, “Since the standard operating procedures […] are designed for the efficient delivery of high-quality care, any deviation from these procedures increases the possibility that care may be compromised.”5 In other words, suspending usual practice when caring for a VIP patient can imperil the patient.2–5,10,11 When caring for VIP physicians, for example, circumventing usual medical and administrative routines and the difficulties that caring for colleagues poses for nurses and physicians have led to poor medical care and outcomes, as well as to hostility.2–4
A striking example of the potential effects of VIP syndrome is the death of Eleanor Roosevelt from miliary tuberculosis acutissima: she was misdiagnosed with aplastic anemia on the basis of only the results of a bone marrow aspirate study, and she was treated with steroids. The desire to spare this VIP patient the discomfort of a bone marrow biopsy, on which tuberculous granulomata were more likely to have been seen, caused the true diagnosis to be missed and resulted in the administration of a hazardous medication.11 The hard lesson here is that we must resist the pressure to simplify or change customary medical care to avoid causing a VIP patient discomfort or putting the patient through a complex procedure.
We recommend discussing these issues explicitly with the VIP patient and family at the outset so that everyone can appreciate the importance of usual care. An early conversation can communicate the clinician’s experience in the care of such patients and can be reassuring. As Smith and Shesser noted, “Usually, the VIP is relieved if the physician states explicitly, ‘I am going to treat you as I would any other patient.’ ”5
PRINCIPLE 2: WORK AS A TEAM, NOT IN ‘SILOS’
Teamwork is essential for good clinical outcomes, 12–14 especially when the clinical problem is complex, as is often the case when people travel long distances to receive care. All consultants involved in the patient’s care must not only attend to their own clinical issues but also communicate amply with their colleagues.
At the same time, we must recognize that medical practice “is not a committee process; it must be clear at all times which physician is responsible for directing clinical care.”5 One physician must be in charge of the overall care. Seeking the input of other physicians must not be allowed to diffuse responsibility. The primary attending physician must speak with the consultants, summarize their views, and then communicate the findings and the plan of care to the patient and family.
Paradoxically, teamwork can be challenged when circumstances lead consultants to defer communicating directly with the family in favor of the primary physician’s doing so. Similarly, consultants must avoid any temptation to simply “do their thing” and not communicate with one another, thereby potentially offering “siloed,” discoordinated care.
We propose designating a primary physician to take charge of the care and the communication. This physician must have the time to talk with each team member about how best to communicate the individual findings to the patient and family. At times, the primary physician may also ask the consultants to communicate directly with the patient and family when needed.
PRINCIPLE 3: COMMUNICATE, COMMUNICATE, COMMUNICATE
As a corollary of principle 2, heightened communication is essential when caring for VIP patients. Communication should include the patient, the family, visiting physicians who accompany the patient, and the physicians providing care. Communicating with the media and with other uninvolved individuals is addressed in principle 4.
The logistic and security challenges of transporting VIP patients through the hospital for tests or therapy demand increased communication. Scheduling a computed tomographic scan may involve arranging an off-hours appointment in the radiology department (to minimize security risks and disruption to other patients’ schedules), assuring the off-hours availability of allied health providers to accompany the patient, alerting hospital security, and discussing the appointment with the patient and the patient’s entourage.
PRINCIPLE 4: CAREFULLY MANAGE COMMUNICATION WITH THE MEDIA
Although the news media and the public may demand medical information about patients who are celebrities, political luminaries, or royalty, the confidentiality of the physician-patient relationship must be protected. The release of health information is at the sole discretion of the patient or a designated surrogate.
The care of President Ronald Reagan after the 1981 assassination attempt is a benchmark of how to release information to the public.10 A single physician held regularly scheduled press conferences, and these were intentionally held away from the site of the President’s care.
Designating a senior hospital physician to communicate with the media is desirable, and the physician-spokesperson can call on specialists from the patient care team (eg, a critical care physician), when appropriate, to provide further information.
Early implementation of an explicit and structured media communication plan is advisable, especially when the VIP patient is a political or royal figure for whom public clamor for information will be vigorous. A successful communication strategy balances the public’s demand for information with the need to protect the patient’s confidentiality.
PRINCIPLE 5: RESIST ‘CHAIRPERSON’S SYNDROME’
“Chairperson’s syndrome”5 is pressure for the VIP patient to be cared for by the department chairperson. The pressure may come from the patient, family, or attendants, who may assume that the chairperson is the best doctor for the clinical circumstance. The pressure may also come from the chairperson, who feels the need to “take command” in a situation with high visibility. Nevertheless, designation of a chairperson to care for a VIP patient is appropriate only when the chairperson is indeed the clinician who has the most expertise in the patient’s clinical issues.
As in principle 1, in academic medical centers, we encourage the participation of trainees in the care of VIP patients because excluding them could disrupt the usual flow of care, and because trainees offer a currency and facility with the nuances of hospital practice and routine that are advantageous to the patient’s care.
PRINCIPLE 6: CARE SHOULD OCCUR WHERE IT IS MOST APPROPRIATE
Decisions about where to place the VIP patient during the medical visit can fall victim to the VIP syndrome if the expectations of the patient or family conflict with usual clinical practice and judgment about the optimal care venue.
For example, caring for the patient in a setting away from the mainstream clinical environment may offer the appeal of privacy or enhanced security but can under some circumstances impede optimal care, including prolonging the response time during emergencies and disrupting the optimal care routine and teamwork of allied health providers.
Critical care services and monitoring are best provided in the intensive care unit, and attempts to relocate the patient away from the intensive care unit should be resisted. We recommend a candid discussion of the importance of keeping the patient in the intensive care unit to ensure optimal care by a seasoned clinical team with short response times if urgencies should arise.
At the same time, a request to transfer a VIP patient to a special setting designed for private care with special amenities (eg, appealing room decor, adjacent sleeping rooms for family members, enhanced security) available in some hospitals15–16 can be honored as soon as the patient’s condition permits. The benefits of such amenities are often greatly appreciated and can reduce stress and thereby promote recovery. The benefits of enhanced security in sequestered venues may especially drive the decision to move when clinically prudent (see principle 7).
PRINCIPLE 7: PROTECT THE PATIENT’S SECURITY
Providing security is another essential part of caring for VIPs, especially celebrities, political figures, and royalty. Protecting the patient from bodily harm requires special attention to the patient’s location, caregiver access, and other logistic matters.
As indicated in principle 6, the patient’s clinical needs are paramount in determining where the patient receives care. If the patient requires care in a mainstream hospital location such as the intensive care unit, modifications of the unit may be needed to alter access, to accommodate security personnel, and to restrict caregivers’ access to the patient. Modifications include structural changes to windows, special credentials (eg, badges) for essential providers, arranging transports within the hospital for elective procedures during off-hours, and providing around-the-clock security personnel near the patient.
As important as it is to protect VIP patients from bodily harm during the visit, it is equally important to protect them from attacks on confidentiality via unauthorized access to the electronic medical record, and this is perhaps the more difficult challenge, as examples of breaches abound.10,17–19 Although the duty to protect against these breaches rests with the hospital, the use of “pop-ups” in the electronic medical record can flash a warning that only employees with legitimate clinical reasons should access the record. These warnings should also cite the penalties for unauthorized review of the record, which is supported by the Health Insurance Portability and Accountability Act (HIPAA). Access to celebrities’ health records could be restricted to a few predetermined health care providers.
PRINCIPLE 8: BE CAREFUL ABOUT ACCEPTING OR DECLINING GIFTS
VIP patients often present gifts to physicians, and giving gifts to doctors is a common and long-standing practice.20,21 Patients offer gifts out of gratitude, affection, desperation, or the desire to garner special treatment or indebtedness. VIP patients from gifting cultures may be especially likely to offer gifts to their providers, and the gifts can be lavish.
The “ethical calculus”21 of whether to accept or decline a gift depends on the circumstances and on what motivates the offer, and the physician needs to consider the patient’s reasons for giving the gift.
In general, gifts should be accepted only with caution during the acute episode of care. The acceptance of a gift from a VIP patient or family member may be interpreted by the gift-giver as a sort of unspoken promise, and this misunderstanding may strain the physician-patient relationship, especially if the clinical course deteriorates.
Rather than accept a gift during an episode of acute care, we suggest that the physician graciously decline the gift and offer to accept the gift at the end of the episode of acute care—that is, if the offerer still feels so inclined and remembers. Explaining the reason for deferring the gift can decrease the risk of misunderstandings or of unmet expectations by the gift-giver. Also, deferring the acceptance of a gift allows the caregiver to affirm the commitment to excellent care that is free of gifts, thereby ensuring that the patient will be confident of a similar level of care by providers who have not been offered gifts.
On the other hand, declining a gift may cause more damage than accepting it, particularly if the VIP patient is from a culture in which refusing a gift is impolite.22 A sensible compromise may be to adopt the recommendations of the American Academy of Pediatrics23—ie, attempt to appreciate appropriate gifts and graciously refuse those that are not.
PRINCIPLE 9: WORKING WITH THE PATIENT’S PERSONAL PHYSICIANS
VIP patients, perhaps especially royalty, may be accompanied by their own physicians and may also wish to bring in consultants from other institutions. Though this outside involvement poses challenges (eg, providing access to medical records, arranging briefings, attending bedside rounds), we believe it should be encouraged when the issue is raised. Furthermore, institutions and caregivers should anticipate these requests and identify potential outside consultants whose names can be volunteered if the issue arises.
Again, if VIP patients wish to involve physicians from outside the institution where they are receiving care, this should not be viewed as an expression of doubt about the care being received. Rather, we prefer to view it as an opportunity to validate current management or to entertain alternative approaches. Most often, when an outside consultant confirms the current medical care, this can have the beneficial effect of increasing confidence and facilitating management.
In a similar way, when VIP patients bring their own physician, whose judgment and care they trust, this represents an opportunity to engage the patient’s trusted physician-advisor in clinical decision-making and thus optimize communication with the patient. Collegial interactions with these physician-colleagues can facilitate communication and decision-making for the patient.
- Ehrbeck T, Guevara C, Mango PD. Mapping the market for medical travel. Health Care: Strategy & Analysis. McKinsey Quarterly 2008 May;1–11.
- Stoudemire A, Rhoads JM. When the doctor needs a doctor: special considerations for the physician-patient. Ann Intern Med 1983; 98:654–659.
- Schneck SA. “Doctoring” doctors and their families. JAMA 1998; 280:2039–2042.
- Adshead G. Healing ourselves: ethical issues in the care of sick doctors. Adv Psychiatr Treat 2005; 11:330–337.
- Smith MS, Shesser RF. The emergency care of the VIP patient. N Engl J Med 1988; 319:1421–1423.
- Block AJ. Beware of the VIP syndrome. Chest 1993; 104:989.
- Mariano EC, McLeod JA. Emergency care for the VIP patient. Intensive Care Medicine 2007. http://dx.doi.org/10.1007/978-0-387-49518-7_88. Accessed December 27, 2010.
- Schenkenberg T, Kochenour NK, Botkin JR. Ethical considerations in clinical care of the “VIP”. J Clin Ethics 2007; 18:56–63.
- Weintraub W. “The VIP syndrome”: a clinical study in hospital psychiatry. J Nerv Ment Dis 1964; 138:181–193.
- Weiss YG, Mor-Yosef S, Sprung CL, Weissman C, Weiss Y. Caring for a major government official: challenges and lessons learned. Crit Care Med 2007; 35:1769–1772.
- Lerner BH. Revisiting the death of Eleanor Roosevelt: was the diagnosis of tuberculosis missed? Int J Tuberc Lung Dis 2001; 5:1080–1085.
- Lee TH. Turning doctors into leaders. Harv Bus Rev 2010; 88:50–58.
- Clemmer TP, Spuhler VJ, Berwick DM, Nolan TW. Cooperation: the foundation of improvement. Ann Intern Med 1998; 128:1004–1009.
- Morey JC, Simon R, Jay GD, et al. Error reduction and performance improvement in the emergency department through formal teamwork training: evaluation results of the MedTeams project. Health Serv Res 2002; 37:1553–1581.
- VIP ward at Walter Reed gets scrutiny. USA Today. http://www.usatoday.com/news/washington/2007-03-15-walter-reed-vip_N.htm. Accessed December 27, 2010.
- Robins RS, Post JM. When Illness Strikes the Leader. The Dilemma of the Captive King. New Haven: Yale University Press; 1995.
- Carr J. Breach of Britney Spears patient data reported. SC Magazine, March 19, 2008. http://www.scmagazineus.com/breach-of-britney-spears-patient-data-reported/article/108141/. Accessed December 27, 2010.
- Collins T. Sir Bobby Robson’s electronic health records viewed illicitly by NHS staff. ComputerWeekly.com, September 24, 2007. http://www.computerweekly.com/blogs/tony_collins/2007/09/bobby-robsons-medical-records-1.html. Accessed December 27, 2010.
- Ornstein C. Kaiser hospital fined $250,000 for privacy breach in octuplet case. Propublica.org, May 15, 2009. http://www.propublica.org/article/kaiser-hospital-fined-250000-for-privacy-breach-in-octuplet-case-515. Accessed December 27, 2010.
- Levene MI, Sireling L. Gift giving to hospital doctors—in the mouth of the gift horse. Br Med J 1980; 281:1685.
- Lyckholm LJ. Should physicians accept gifts from patients? JAMA 1998; 280:1944–1946.
- Takayama JI. Giving and receiving gifts: one perspective. West J Med 2001; 175:138–139.
- Committee on Bioethics. From the American Academy of Pediatrics: policy statements—pediatrician-family-patient relationships: managing the boundaries. Pediatrics 2009; 124:1685–1688.
- Ehrbeck T, Guevara C, Mango PD. Mapping the market for medical travel. Health Care: Strategy & Analysis. McKinsey Quarterly 2008 May;1–11.
- Stoudemire A, Rhoads JM. When the doctor needs a doctor: special considerations for the physician-patient. Ann Intern Med 1983; 98:654–659.
- Schneck SA. “Doctoring” doctors and their families. JAMA 1998; 280:2039–2042.
- Adshead G. Healing ourselves: ethical issues in the care of sick doctors. Adv Psychiatr Treat 2005; 11:330–337.
- Smith MS, Shesser RF. The emergency care of the VIP patient. N Engl J Med 1988; 319:1421–1423.
- Block AJ. Beware of the VIP syndrome. Chest 1993; 104:989.
- Mariano EC, McLeod JA. Emergency care for the VIP patient. Intensive Care Medicine 2007. http://dx.doi.org/10.1007/978-0-387-49518-7_88. Accessed December 27, 2010.
- Schenkenberg T, Kochenour NK, Botkin JR. Ethical considerations in clinical care of the “VIP”. J Clin Ethics 2007; 18:56–63.
- Weintraub W. “The VIP syndrome”: a clinical study in hospital psychiatry. J Nerv Ment Dis 1964; 138:181–193.
- Weiss YG, Mor-Yosef S, Sprung CL, Weissman C, Weiss Y. Caring for a major government official: challenges and lessons learned. Crit Care Med 2007; 35:1769–1772.
- Lerner BH. Revisiting the death of Eleanor Roosevelt: was the diagnosis of tuberculosis missed? Int J Tuberc Lung Dis 2001; 5:1080–1085.
- Lee TH. Turning doctors into leaders. Harv Bus Rev 2010; 88:50–58.
- Clemmer TP, Spuhler VJ, Berwick DM, Nolan TW. Cooperation: the foundation of improvement. Ann Intern Med 1998; 128:1004–1009.
- Morey JC, Simon R, Jay GD, et al. Error reduction and performance improvement in the emergency department through formal teamwork training: evaluation results of the MedTeams project. Health Serv Res 2002; 37:1553–1581.
- VIP ward at Walter Reed gets scrutiny. USA Today. http://www.usatoday.com/news/washington/2007-03-15-walter-reed-vip_N.htm. Accessed December 27, 2010.
- Robins RS, Post JM. When Illness Strikes the Leader. The Dilemma of the Captive King. New Haven: Yale University Press; 1995.
- Carr J. Breach of Britney Spears patient data reported. SC Magazine, March 19, 2008. http://www.scmagazineus.com/breach-of-britney-spears-patient-data-reported/article/108141/. Accessed December 27, 2010.
- Collins T. Sir Bobby Robson’s electronic health records viewed illicitly by NHS staff. ComputerWeekly.com, September 24, 2007. http://www.computerweekly.com/blogs/tony_collins/2007/09/bobby-robsons-medical-records-1.html. Accessed December 27, 2010.
- Ornstein C. Kaiser hospital fined $250,000 for privacy breach in octuplet case. Propublica.org, May 15, 2009. http://www.propublica.org/article/kaiser-hospital-fined-250000-for-privacy-breach-in-octuplet-case-515. Accessed December 27, 2010.
- Levene MI, Sireling L. Gift giving to hospital doctors—in the mouth of the gift horse. Br Med J 1980; 281:1685.
- Lyckholm LJ. Should physicians accept gifts from patients? JAMA 1998; 280:1944–1946.
- Takayama JI. Giving and receiving gifts: one perspective. West J Med 2001; 175:138–139.
- Committee on Bioethics. From the American Academy of Pediatrics: policy statements—pediatrician-family-patient relationships: managing the boundaries. Pediatrics 2009; 124:1685–1688.
KEY POINTS
- Caring for VIPs creates pressures to change usual clinical wisdom and practices. But it is essential to resist changing time-honored, effective clinical practices and overriding one’s clinical judgment.
- Designating a chairperson to head the care of a VIP patient is appropriate only if the chairperson is the best clinician for the case.
- Although in some cases placing a VIP patient in a more private and remote setting may be appropriate, the patient is generally best served by receiving critical care services in the intensive care unit.
A woman with ulcerating, painful skin lesions
Q: On the basis of the skin findings, which test should be ordered to establish a diagnosis?
- Serum anti-nuclear antibody (ANA)
- Alpha-1 antitrypsin serum level
- Angiotensin-converting enzyme (ACE)
- Serum amylase
A: The lesions of a lobular, neutrophilic panniculitis should raise the possibility of alpha-1 antitrypsin deficiency. Hence, measuring the alpha-1 antitrypsin serum level is the best answer.
Many other conditions can give rise to panniculitis, including pancreatitis, lupus, and sarcoidosis, each of which is suggested by the various other test choices above. However, although the ANA level might be elevated in lupus, the ANA is quite nonspecific. The serum ACE level is frequently ordered as a screening test for sarcoidosis, although it has very little utility in its diagnosis. Panniculitis due to pancreatitis with an elevated serum amylase level would be relatively unlikely in the absence of pancreatic symptoms (eg, abdominal pain).
Panniculitis may be the only clinical manifestation of alpha-1 antitrypsin deficiency, which can also be accompanied (depending on the phenotype) by chronic obstructive pulmonary disease and cirrhosis. Since alpha-1 antitrypsin deficiency is underrecognized in general, suspecting it when patients present with panniculitis will likely enhance its detection. Similarly, national guidelines recommend testing for alpha-1 antitrypsin deficiency in patients with either symptomatic, fixed airflow obstruction or cirrhosis that is otherwise unexplained, as well as in patients with panniculitis.1
PANNICULITIS DUE TO ALPHA-1 ANTITRYPSIN DEFICIENCY

Likely due to proteolytic damage
Though incompletely understood, the panniculitis in alpha-1 antitrypsin deficiency is likely the result of unopposed proteolytic damage in the subcutaneous fat by membrane-bound serine proteases, akin to the pathogenesis of pulmonary emphysema in people with severe deficiency of alpha-1 antitrypsin. Supporting evidence for the inflammatory, proteolytic pathogenesis of panniculitis in alpha-1 anti-trypsin deficiency includes the presence of inflammatory exudates in the subcutaneous tissues, as well as the rapid improvement seen with the infusion of purified pooled human alpha-1 proteinase inhibitor.2–4
Red, painful nodules
Panniculitis due to alpha-1 antitrypsin deficiency classically presents as red, painful nodules that may break down and ooze an oily discharge.5–10 As in the patient presented here, common sites of occurrence are areas of trauma, eg, on the thighs and buttocks, abdomen, and upper extremities (Figure 1). Indeed, in a review of the 41 reported cases of panniculitis related to alpha-1 antitrypsin deficiency, Geraminejad et al11 reported that the erythe-matous plaques and nodules occurred on the thighs, hips, buttocks, or groin in 44% of cases in which the location was cited. Factors predisposing to panniculitis include trauma (cited in 35% of instances), cryosurgery, and, in the case of alpha-1 antitrypsin deficiency, extravasation of intravenous clarithromycin (Biaxin).11–13
Clinical features that distinguish the panniculitis associated with alpha-1 antitrypsin deficiency from other types of panniculitis include ulceration and an oily discharge, both of which were present in the patient discussed here.
Neutrophils, necrosis, scarring, fibrosis
Several distinctive phases and features characterize the histology of panniculitis associated with alpha-1 antitrypsin deficiency.5,7 Initially, neutrophils briskly infiltrate the reticular dermis, splaying the collagen bundles. In the subcutaneous fat, the neutrophilic infiltrate is in a lobular pattern, affecting individual adipocytes. Rarely, a septal pattern or a mixed lobular and septal pattern can be seen. This phase is followed by dissolution of the dermal collagen, with liquefactive necrosis of the subcutaneous fat (clinically appearing as ulceration and leading to oily drainage). In the late stage, there is scarring and fibrosis with little or no inflammation.
Various treatments tried
Various therapies for panniculitis associated with alpha-1 antitrypsin deficiency have been tried, including corticosteroids, doxycycline (Vibramycin), dapsone, plasma exchange, liver transplantation, and intravenous pooled human plasma alpha-1 proteinase inhibitor (so-called augmentation therapy). Because panniculitis associated with alpha-1 antitrypsin deficiency is rare, neither controlled, blinded studies nor even large observational series have been reported. However, the limited reported experience with augmentation therapy suggests that it can confer rapid and dramatic improvement in panniculitis in patients with alpha-1 antitrypsin deficiency.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Smith KC, Pittelkow MR, Su WP. Panniculitis associated with severe alpha-1 antitrypsin deficiency. Treatment and review of the literature. Arch Dermatol 1987; 123:1655–1661.
- Furey NL, Golden RS, Potts SR. Treatment of alpha-1 antitrypsin deficiency, massive edema, and panniculitis with alpha-1 protease inhibitor [letter]. Ann Intern Med 1996; 125:699.
- O’Riordan K, Blei A, Rao MS, Abecassis M. Alpha-1 antitrypsin deficiency-associated panniculitis: resolution with intravenous alpha-1 antitrypsin administration and liver transplantation. Transplantation 1997; 63:480–482.
- Stoller JK, Piliang M. Panniculitis in alpha-1 antitrypsin deficiency: a review. Clin Pulm Med 2008; 15:113–117.
- Hendrick SJ, Silverman AK, Solomon AR, Headington JT. Alpha-1 antitrypsin deficiency associated with panniculitis. J Am Acad Derm 1988; 18:684–692.
- Loche F, Tremeau-Martinage C, Laplanche G, Massip P, Bazex J. Panniculitis revealing qualitative alpha-1 antitrypsine deficiency (MS variant). Eur J Dermatol 1999; 9:565–567.
- McBean J, Sable A, Maude J, Robinson-Bostom L. Alpha 1-antitrypsin deficiency panniculitis. Cutis 2003; 71:205–209.
- Pittelkow MR, Smith KC, Su WP. Alpha-1 antitrypsin deficiency and panniculitis. Perspectives on disease relationship and replacement therapy. Am J Med 1988; 84:80–86.
- Requena L, Sánchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001; 45:325–361.
- Geraminejad P, DeBloom JR, Walling HW, Sontheimer RD, VanBeek M. Alpha-1-antitrypsin associated panniculitis: the MS variant. J Am Acad Dermatol 2004; 51:645–655.
- Linares-Barrios M, Conejo-Mir IS, Artola Igarza JL, Navarrete M. Panniculitis due to alpha-1 antitrypsin deficiency induced by cryosurgery [letter]. Br J Dermatol 1998; 138:552–553.
- Parr DG, Stewart DG, Hero I, Stockley RA. Panniculitis secondary to extravasation of clarithromycin in a patient with alpha 1-antitrypsin deficiency (phenotype PiZ). Br J Dermatol 2003; 149:410–413.
Q: On the basis of the skin findings, which test should be ordered to establish a diagnosis?
- Serum anti-nuclear antibody (ANA)
- Alpha-1 antitrypsin serum level
- Angiotensin-converting enzyme (ACE)
- Serum amylase
A: The lesions of a lobular, neutrophilic panniculitis should raise the possibility of alpha-1 antitrypsin deficiency. Hence, measuring the alpha-1 antitrypsin serum level is the best answer.
Many other conditions can give rise to panniculitis, including pancreatitis, lupus, and sarcoidosis, each of which is suggested by the various other test choices above. However, although the ANA level might be elevated in lupus, the ANA is quite nonspecific. The serum ACE level is frequently ordered as a screening test for sarcoidosis, although it has very little utility in its diagnosis. Panniculitis due to pancreatitis with an elevated serum amylase level would be relatively unlikely in the absence of pancreatic symptoms (eg, abdominal pain).
Panniculitis may be the only clinical manifestation of alpha-1 antitrypsin deficiency, which can also be accompanied (depending on the phenotype) by chronic obstructive pulmonary disease and cirrhosis. Since alpha-1 antitrypsin deficiency is underrecognized in general, suspecting it when patients present with panniculitis will likely enhance its detection. Similarly, national guidelines recommend testing for alpha-1 antitrypsin deficiency in patients with either symptomatic, fixed airflow obstruction or cirrhosis that is otherwise unexplained, as well as in patients with panniculitis.1
PANNICULITIS DUE TO ALPHA-1 ANTITRYPSIN DEFICIENCY
Likely due to proteolytic damage
Though incompletely understood, the panniculitis in alpha-1 antitrypsin deficiency is likely the result of unopposed proteolytic damage in the subcutaneous fat by membrane-bound serine proteases, akin to the pathogenesis of pulmonary emphysema in people with severe deficiency of alpha-1 antitrypsin. Supporting evidence for the inflammatory, proteolytic pathogenesis of panniculitis in alpha-1 anti-trypsin deficiency includes the presence of inflammatory exudates in the subcutaneous tissues, as well as the rapid improvement seen with the infusion of purified pooled human alpha-1 proteinase inhibitor.2–4
Red, painful nodules
Panniculitis due to alpha-1 antitrypsin deficiency classically presents as red, painful nodules that may break down and ooze an oily discharge.5–10 As in the patient presented here, common sites of occurrence are areas of trauma, eg, on the thighs and buttocks, abdomen, and upper extremities (Figure 1). Indeed, in a review of the 41 reported cases of panniculitis related to alpha-1 antitrypsin deficiency, Geraminejad et al11 reported that the erythe-matous plaques and nodules occurred on the thighs, hips, buttocks, or groin in 44% of cases in which the location was cited. Factors predisposing to panniculitis include trauma (cited in 35% of instances), cryosurgery, and, in the case of alpha-1 antitrypsin deficiency, extravasation of intravenous clarithromycin (Biaxin).11–13
Clinical features that distinguish the panniculitis associated with alpha-1 antitrypsin deficiency from other types of panniculitis include ulceration and an oily discharge, both of which were present in the patient discussed here.
Neutrophils, necrosis, scarring, fibrosis
Several distinctive phases and features characterize the histology of panniculitis associated with alpha-1 antitrypsin deficiency.5,7 Initially, neutrophils briskly infiltrate the reticular dermis, splaying the collagen bundles. In the subcutaneous fat, the neutrophilic infiltrate is in a lobular pattern, affecting individual adipocytes. Rarely, a septal pattern or a mixed lobular and septal pattern can be seen. This phase is followed by dissolution of the dermal collagen, with liquefactive necrosis of the subcutaneous fat (clinically appearing as ulceration and leading to oily drainage). In the late stage, there is scarring and fibrosis with little or no inflammation.
Various treatments tried
Various therapies for panniculitis associated with alpha-1 antitrypsin deficiency have been tried, including corticosteroids, doxycycline (Vibramycin), dapsone, plasma exchange, liver transplantation, and intravenous pooled human plasma alpha-1 proteinase inhibitor (so-called augmentation therapy). Because panniculitis associated with alpha-1 antitrypsin deficiency is rare, neither controlled, blinded studies nor even large observational series have been reported. However, the limited reported experience with augmentation therapy suggests that it can confer rapid and dramatic improvement in panniculitis in patients with alpha-1 antitrypsin deficiency.
Q: On the basis of the skin findings, which test should be ordered to establish a diagnosis?
- Serum anti-nuclear antibody (ANA)
- Alpha-1 antitrypsin serum level
- Angiotensin-converting enzyme (ACE)
- Serum amylase
A: The lesions of a lobular, neutrophilic panniculitis should raise the possibility of alpha-1 antitrypsin deficiency. Hence, measuring the alpha-1 antitrypsin serum level is the best answer.
Many other conditions can give rise to panniculitis, including pancreatitis, lupus, and sarcoidosis, each of which is suggested by the various other test choices above. However, although the ANA level might be elevated in lupus, the ANA is quite nonspecific. The serum ACE level is frequently ordered as a screening test for sarcoidosis, although it has very little utility in its diagnosis. Panniculitis due to pancreatitis with an elevated serum amylase level would be relatively unlikely in the absence of pancreatic symptoms (eg, abdominal pain).
Panniculitis may be the only clinical manifestation of alpha-1 antitrypsin deficiency, which can also be accompanied (depending on the phenotype) by chronic obstructive pulmonary disease and cirrhosis. Since alpha-1 antitrypsin deficiency is underrecognized in general, suspecting it when patients present with panniculitis will likely enhance its detection. Similarly, national guidelines recommend testing for alpha-1 antitrypsin deficiency in patients with either symptomatic, fixed airflow obstruction or cirrhosis that is otherwise unexplained, as well as in patients with panniculitis.1
PANNICULITIS DUE TO ALPHA-1 ANTITRYPSIN DEFICIENCY
Likely due to proteolytic damage
Though incompletely understood, the panniculitis in alpha-1 antitrypsin deficiency is likely the result of unopposed proteolytic damage in the subcutaneous fat by membrane-bound serine proteases, akin to the pathogenesis of pulmonary emphysema in people with severe deficiency of alpha-1 antitrypsin. Supporting evidence for the inflammatory, proteolytic pathogenesis of panniculitis in alpha-1 anti-trypsin deficiency includes the presence of inflammatory exudates in the subcutaneous tissues, as well as the rapid improvement seen with the infusion of purified pooled human alpha-1 proteinase inhibitor.2–4
Red, painful nodules
Panniculitis due to alpha-1 antitrypsin deficiency classically presents as red, painful nodules that may break down and ooze an oily discharge.5–10 As in the patient presented here, common sites of occurrence are areas of trauma, eg, on the thighs and buttocks, abdomen, and upper extremities (Figure 1). Indeed, in a review of the 41 reported cases of panniculitis related to alpha-1 antitrypsin deficiency, Geraminejad et al11 reported that the erythe-matous plaques and nodules occurred on the thighs, hips, buttocks, or groin in 44% of cases in which the location was cited. Factors predisposing to panniculitis include trauma (cited in 35% of instances), cryosurgery, and, in the case of alpha-1 antitrypsin deficiency, extravasation of intravenous clarithromycin (Biaxin).11–13
Clinical features that distinguish the panniculitis associated with alpha-1 antitrypsin deficiency from other types of panniculitis include ulceration and an oily discharge, both of which were present in the patient discussed here.
Neutrophils, necrosis, scarring, fibrosis
Several distinctive phases and features characterize the histology of panniculitis associated with alpha-1 antitrypsin deficiency.5,7 Initially, neutrophils briskly infiltrate the reticular dermis, splaying the collagen bundles. In the subcutaneous fat, the neutrophilic infiltrate is in a lobular pattern, affecting individual adipocytes. Rarely, a septal pattern or a mixed lobular and septal pattern can be seen. This phase is followed by dissolution of the dermal collagen, with liquefactive necrosis of the subcutaneous fat (clinically appearing as ulceration and leading to oily drainage). In the late stage, there is scarring and fibrosis with little or no inflammation.
Various treatments tried
Various therapies for panniculitis associated with alpha-1 antitrypsin deficiency have been tried, including corticosteroids, doxycycline (Vibramycin), dapsone, plasma exchange, liver transplantation, and intravenous pooled human plasma alpha-1 proteinase inhibitor (so-called augmentation therapy). Because panniculitis associated with alpha-1 antitrypsin deficiency is rare, neither controlled, blinded studies nor even large observational series have been reported. However, the limited reported experience with augmentation therapy suggests that it can confer rapid and dramatic improvement in panniculitis in patients with alpha-1 antitrypsin deficiency.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Smith KC, Pittelkow MR, Su WP. Panniculitis associated with severe alpha-1 antitrypsin deficiency. Treatment and review of the literature. Arch Dermatol 1987; 123:1655–1661.
- Furey NL, Golden RS, Potts SR. Treatment of alpha-1 antitrypsin deficiency, massive edema, and panniculitis with alpha-1 protease inhibitor [letter]. Ann Intern Med 1996; 125:699.
- O’Riordan K, Blei A, Rao MS, Abecassis M. Alpha-1 antitrypsin deficiency-associated panniculitis: resolution with intravenous alpha-1 antitrypsin administration and liver transplantation. Transplantation 1997; 63:480–482.
- Stoller JK, Piliang M. Panniculitis in alpha-1 antitrypsin deficiency: a review. Clin Pulm Med 2008; 15:113–117.
- Hendrick SJ, Silverman AK, Solomon AR, Headington JT. Alpha-1 antitrypsin deficiency associated with panniculitis. J Am Acad Derm 1988; 18:684–692.
- Loche F, Tremeau-Martinage C, Laplanche G, Massip P, Bazex J. Panniculitis revealing qualitative alpha-1 antitrypsine deficiency (MS variant). Eur J Dermatol 1999; 9:565–567.
- McBean J, Sable A, Maude J, Robinson-Bostom L. Alpha 1-antitrypsin deficiency panniculitis. Cutis 2003; 71:205–209.
- Pittelkow MR, Smith KC, Su WP. Alpha-1 antitrypsin deficiency and panniculitis. Perspectives on disease relationship and replacement therapy. Am J Med 1988; 84:80–86.
- Requena L, Sánchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001; 45:325–361.
- Geraminejad P, DeBloom JR, Walling HW, Sontheimer RD, VanBeek M. Alpha-1-antitrypsin associated panniculitis: the MS variant. J Am Acad Dermatol 2004; 51:645–655.
- Linares-Barrios M, Conejo-Mir IS, Artola Igarza JL, Navarrete M. Panniculitis due to alpha-1 antitrypsin deficiency induced by cryosurgery [letter]. Br J Dermatol 1998; 138:552–553.
- Parr DG, Stewart DG, Hero I, Stockley RA. Panniculitis secondary to extravasation of clarithromycin in a patient with alpha 1-antitrypsin deficiency (phenotype PiZ). Br J Dermatol 2003; 149:410–413.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Smith KC, Pittelkow MR, Su WP. Panniculitis associated with severe alpha-1 antitrypsin deficiency. Treatment and review of the literature. Arch Dermatol 1987; 123:1655–1661.
- Furey NL, Golden RS, Potts SR. Treatment of alpha-1 antitrypsin deficiency, massive edema, and panniculitis with alpha-1 protease inhibitor [letter]. Ann Intern Med 1996; 125:699.
- O’Riordan K, Blei A, Rao MS, Abecassis M. Alpha-1 antitrypsin deficiency-associated panniculitis: resolution with intravenous alpha-1 antitrypsin administration and liver transplantation. Transplantation 1997; 63:480–482.
- Stoller JK, Piliang M. Panniculitis in alpha-1 antitrypsin deficiency: a review. Clin Pulm Med 2008; 15:113–117.
- Hendrick SJ, Silverman AK, Solomon AR, Headington JT. Alpha-1 antitrypsin deficiency associated with panniculitis. J Am Acad Derm 1988; 18:684–692.
- Loche F, Tremeau-Martinage C, Laplanche G, Massip P, Bazex J. Panniculitis revealing qualitative alpha-1 antitrypsine deficiency (MS variant). Eur J Dermatol 1999; 9:565–567.
- McBean J, Sable A, Maude J, Robinson-Bostom L. Alpha 1-antitrypsin deficiency panniculitis. Cutis 2003; 71:205–209.
- Pittelkow MR, Smith KC, Su WP. Alpha-1 antitrypsin deficiency and panniculitis. Perspectives on disease relationship and replacement therapy. Am J Med 1988; 84:80–86.
- Requena L, Sánchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001; 45:325–361.
- Geraminejad P, DeBloom JR, Walling HW, Sontheimer RD, VanBeek M. Alpha-1-antitrypsin associated panniculitis: the MS variant. J Am Acad Dermatol 2004; 51:645–655.
- Linares-Barrios M, Conejo-Mir IS, Artola Igarza JL, Navarrete M. Panniculitis due to alpha-1 antitrypsin deficiency induced by cryosurgery [letter]. Br J Dermatol 1998; 138:552–553.
- Parr DG, Stewart DG, Hero I, Stockley RA. Panniculitis secondary to extravasation of clarithromycin in a patient with alpha 1-antitrypsin deficiency (phenotype PiZ). Br J Dermatol 2003; 149:410–413.
Diffuse alveolar hemorrhage: Diagnosing it and finding the cause
Diffuse alveolar hemorrhage can complicate a large number of clinical conditions. It may present in different ways and may be life-threatening, and it poses an important challenge for the clinician.1
Diffuse alveolar hemorrhage is an uncommon condition in which blood floods the alveoli, usually at multiple sites. It is also known as intrapulmonary hemorrhage, diffuse pulmonary hemorrhage, pulmonary alveolar hemorrhage, pulmonary capillary hemorrhage, alveolar bleeding, or microvascular pulmonary hemorrhage.
In this article we review the causes, clinical features, diagnostic criteria, treatment, and prognosis of diffuse alveolar hemorrhage.
CAUSES OF DIFFUSE ALVEOLAR HEMORRHAGE

THREE CHARACTERISTIC PATTERNS
In general, diffuse alveolar hemorrhage can occur in three characteristic patterns, which reflect the nature of the underlying vascular injury1:
Diffuse alveolar hemorrhage associated with vasculitis or capillaritis. As described by Spencer4 50 years ago, pulmonary capillaritis is the most frequent underlying histologic lesion described in diffuse alveolar hemorrhage. Neutrophils infiltrate the interalveolar and peri-bronchiolar septal vessels (pulmonary interstitium),5 leading to anatomic disruption of the capillaries (ie, impairment of the alveolocapillary barrier) and to extravasation of red blood cells into the alveoli and interstitium. Neutrophil apoptosis and fragmentation, with subsequent release of the intracellular proteolytic enzymes and reactive oxygen species, beget more inflammation, intra-alveolar neutrophilic nuclear dust, fibrin and inflammatory exudate, and fibrinoid necrosis of the interstitium.6,7
‘Bland’ pulmonary hemorrhage (ie, without capillaritis or vasculitis). In this pattern, red blood cells leak into the alveoli without any evidence of inflammation or destruction of the alveolar capillaries, venules, and arterioles. The epithelial lesions are usually microscopic and are scattered geographically.
Diffuse alveolar hemorrhage associated with another process or condition (eg, diffuse alveolar damage, lymphangioleiomyomatosis, drug-induced lung injury, metastatic tumor to the lungs, mitral stenosis). Diffuse alveolar damage is the main underlying lesion of the acute respiratory distress syndrome and is characterized by formation of an intra-alveolar hyaline membrane, by interstitial edema with minimal inflammation, and, at times, by “secondary” diffuse alveolar hemorrhage. In this third category of diffuse alveolar hemorrhage, the underlying process causes alveolar hemorrhage by processes other than pulmonary vascular inflammation or direct extravasation of red cells.
THE CLINICAL PRESENTATION
The clinical presentation of diffuse alveolar hemorrhage may reflect either alveolar bleeding alone or features of the underlying cause (eg, hematuria in Wegener granulomatosis, arthritis in systemic lupus erythematosus). Hence, its recognition requires a high degree of suspicion.
Some patients present with severe acute respiratory distress requiring mechanical ventilation. However, dyspnea, cough, and fever are the common initial symptoms and are most often acute or subacute (ie, present for less than a week). The fever is usually due to the underlying cause, such as lupus.
Hemoptysis may be absent at the time of presentation in up to a third of patients because the total alveolar volume is large and can absorb large amounts of blood, without extending more proximally into the airways. Apparent hemoptysis, if present, must be differentiated from hematemesis or pseudohemoptysis (alveolar flooding with fluid that resembles blood, as in Serratia marcescens pneumonia, in which the reddish hue of the infecting organism can create the impression of alveolar bleeding).
DIAGNOSTIC EVALUATION
Generally speaking, dyspnea, cough, hemoptysis, and new alveolar infiltrates in conjunction with bloody bronchoalveolar lavage specimens (with numerous erythrocytes and siderophages) establish the diagnosis of diffuse alveolar hemorrhage. Surgical biopsy from the lung or another organ involved by an underlying condition is often necessary.
Physical examination
The physical findings are nonspecific and may reflect the underlying systemic vasculitis or collagen vascular disorder (eg, with accompanying rash, purpura, eye lesions, hepatosplenomegaly, or clubbing).
Imaging studies
Radiography may show new or old or both new and old patchy or diffuse alveolar opacities. Recurrent episodes of hemorrhage may lead to reticular interstitial opacities due to pulmonary fibrosis, usually with minimal (if any) honeycombing. Kerley B lines suggest mitral valve disease or pulmonary veno-occlusive disease as the cause of the hemorrhage.
Computed tomography may show areas of consolidation interspersed with areas of ground-glass attenuation and preserved, normal areas.
Currently, nuclear imaging such as gallium or tagged red blood cell studies have little role in evaluating diffuse alveolar hemorrhage. Other nuclear studies, geared to reveal breakdown of the microcirculatory integrity and extravasation of red blood cells out of the vessels, have also not been proven useful.
Evaluating pulmonary function
Diffuse alveolar hemorrhage may cause impairment of oxygen transfer and hypoxemia. In addition, it can cause several other abnormalities of pulmonary function.
Increased diffusing capacity. Because blood in the lungs can absorb inhaled carbon monoxide, the diffusing capacity for carbon monoxide (DLCO) may be distinctively increased. Serial increases in the DLCO may indicate progressive alveolar hemorrhage. However, the clinical instability of patients experiencing active alveolar bleeding precludes performing the DLCO measurement maneuvers, rendering the DLCO test relatively impractical.
Restrictive changes. Because recurrent episodes of diffuse alveolar hemorrhage can lead to interstitial fibrosis, restrictive changes—ie, decreased total lung capacity, decreased forced vital capacity (FVC), and preserved ratio of the forced expiratory volume in 1 second (FEV1) to the FVC—may characterize diffuse alveolar hemorrhage.
Obstructive changes (less common). Less commonly, patients with diffuse alveolar hemorrhage may have spirometric changes indicating airflow obstruction—ie, decreased FEV1 and decreased ratio of FEV1 to FVC—possibly because neutrophilic infiltration from blood extravasation into the alveolar sacs causes release of reactive oxygen species and proteolytic enzymes, which in turn may cause small airway and parenchymal damage such as bronchiolitis and emphysema. A pattern of obstructive lung disease associated with recurrent diffuse alveolar hemorrhage should prompt consideration of an underlying condition that can cause airflow obstruction, such as sarcoidosis, microscopic polyangiitis, or Wegener granulomatosis, or, less commonly, lymphangioleiomyomatosis, histiocytosis X, pulmonary capillaritis, or sometimes idiopathic pulmonary hemosiderosis.
As an example of an unusual circumstance, we have described elsewhere a case of a woman with idiopathic pulmonary hemosiderosis with multiple episodes of diffuse alveolar hemorrhage and resultant emphysema.8 Radiographic images showed several very large cysts, one of which herniated through the incision site of an open lung biopsy.
Decreased exhaled nitric oxide. Though currently unavailable in most clinical pulmonary function laboratories, evaluation of exhaled gas or condensate may have value in diagnosing diffuse alveolar hemorrhage.9 Specifically, because increased intra-alveolar hemoglobin binds nitric oxide, as it does carbon monoxide, levels of exhaled nitric oxide may be decreased in diffuse alveolar hemorrhage. In contrast to the difficulty of measuring DLCO in patients with active alveolar bleeding or hemoptysis, analysis of exhaled gas is clinically feasible, making this a promising diagnostic test.
Laboratory evaluation
Hematologic assessment in patients with diffuse alveolar hemorrhage generally reveals:
- Acute or chronic anemia
- Leukocytosis
- Elevated erythrocyte sedimentation rate
- Elevated C-reactive protein level (particularly in patients whose alveolar hemorrhage is due to systemic disease or vasculitis, or both).
Renal abnormalities such as elevated blood urea nitrogen and serum creatinine or abnormal findings on urinalysis (with hematuria, proteinuria, and red blood cell casts indicating glomerulonephritis) can also occur, as diffuse alveolar hemorrhage may complicate several pulmonary-renal syndromes such as Goodpasture syndrome and Wegener granulomatosis.
Bronchoscopy
The diagnostic evaluation in diffuse alveolar hemorrhage usually includes bronchoscopic examination,10 which serves two purposes:
- To document alveolar hemorrhage by bronchoalveolar lavage and to exclude airway sources of bleeding by visual inspection
- To exclude an associated infection.
Based on experience with nonmassive hemoptysis of all causes (but not exclusively diffuse alveolar hemorrhage), the diagnostic yield of bronchoscopy is higher if the procedure is performed within the first 48 hours of symptoms rather than later. Evidence supporting diffuse alveolar hemorrhage is persistent (or even increasing) blood on three sequential lavage aliquots from a single affected area of the lung.
FINDING THE UNDERLYING CAUSE
Once the diagnosis of diffuse alveolar hemorrhage is established, the clinician must ascertain whether an underlying cause is present. Serologic studies may prove important, although the results are generally not available in a manner timely enough to guide immediate management.
When a pulmonary-renal syndrome is suggested by accompanying hematuria or renal dysfunction, antiglomerular basement membrane antibody and antineutrophil cytoplasmic antibody (ANCA) levels should be checked. Tests for complement fractions C3 and C4, anti-double-stranded DNA, and antiphospholipid antibodies should be ordered if an underlying condition such as lupus or antiphospholipid antibody syndrome is suspected (Table 2).11
If the underlying cause remains elusive after a thorough clinical evaluation that includes imaging studies, serologic studies, and bronchoscopy, then surgical biopsy should be considered.1 Which organ to biopsy (eg, lung, sinus, kidney) depends on the level of suspicion for a specific cause. For example, suspicion of Wegener granulomato-sis with hematuria or renal dysfunction might prompt renal biopsy. However, lung biopsy often needs to be performed with video-assisted thoracoscopy, especially when disease is confined to the lung (as in idiopathic pulmonary hemosiderosis or pauci-immune pulmonary capillaritis). Renal biopsy specimens should also undergo immunofluores-cence staining, which may reveal linear deposition of immunoglobulins and immune complexes along the basement membrane in patients with Goodpasture syndrome, or of granular deposits in patients with systemic lupus erythematosus.
TWO GENERAL CLINICAL SCENARIOS
In general, the clinician will be confronted by one of two scenarios: a patient with diffuse alveolar hemorrhage and associated systemic findings, or a patient with hemorrhage and no associated systemic findings.
Hemorrhage with associated systemic findings
Certain clues from the history raise suspicion of diffuse alveolar hemorrhage:
- Recent infection suggests Henoch-Schönlein purpura or cryoglobulinemic vasculitis
- Use of a possibly offending drug such as an anticoagulant, D-penicillamine (Cuprimine, Depen), nitrofurantoin (Furadantin, Macrobid, Macrodantin), amiodarone (Cordarone), propylthiouracil, cocaine, or sirolimus (Rapamune, Rapamycin)
- Exposure to toxic agents such as trimellitic anhydride, insecticides, and pesticides
- A known comorbid condition such as vasculitis, connective tissue disease, mitral valve disease, or solid organ or stem cell transplantation.
If asthma, eosinophilia, pulmonary infiltrates, and diffuse alveolar hemorrhage coexist, consideration should be given to Churg-Strauss syndrome. If sinus disease, skin manifestations, pulmonary parenchymal nodules, and cavitary lesions coexist with positivity for antiproteinase 3 c-ANCA and biopsy-proven granulomata, then Wegener granulomatosis should be considered. Similarly, diffuse alveolar hemorrhage with glomerulonephritis and skin manifestations, positivity for p-ANCA, and necrotizing nongranulomatous lesions on end-organ biopsy may lead to a diagnosis of microscopic polyangiitis. In a young smoker with glomeru-lonephritis and diffuse alveolar hemorrhage presenting as either bland alveolar hemorrhage or pulmonary capillaritis, Goodpasture syndrome or antiglomerular basement membrane antibody disease should be considered.
Hemorrhage with no associated systemic findings
When the above conditions have been considered but no suggestive findings are found, the following four conditions should be considered:
- Antiglomerular basement membrane antibody disease in limited pulmonary form or onset: positivity to the antibody with linear deposits in the lungs would be diagnostic in such a case
- Pulmonary-limited microscopic polyangiitis positive for p-ANCA (a positive anti-myeloperoxidase p-ANCA test makes the diagnosis)
- Pauci-immune isolated pulmonary capillaritis, when the biopsy shows evidence of neutrophilic pulmonary capillaritis
- Idiopathic pulmonary hemosiderosis, a diagnosis of exclusion, when the biopsy shows evidence of acute, subacute, and chronic bland diffuse alveolar hemorrhage and no evidence of vasculitis.
TREATMENT OF DIFFUSE ALVEOLAR HEMORRHAGE
Therapy for diffuse alveolar hemorrhage consists of treating both the autoimmune destruction of the alveolar capillary membrane and the underlying condition. Corticosteroids and immunosuppressive agents remain the gold standard for most patients. Recombinant-activated human factor VII seems to be a promising new therapy, although further evaluation is needed.
Immunosuppressive agents are the mainstay of therapy for diffuse alveolar hemorrhage, especially if associated with systemic or pulmonary vasculitis, Goodpasture syndrome, and connective tissue disorders. Most experts recommend intravenous methylprednisolone (Solu-Medrol) (up to 500 mg every 6 hours, although lower doses seem to have similar efficacy) for 4 or 5 days, followed by a gradual taper to maintenance doses of oral steroids.
In patients with pulmonary-renal syndrome, therapy should be started as soon as possible to prevent irreversible renal failure.
Besides corticosteroids, other immunosuppressive drugs such as cyclophosphamide (Cytoxan), azathioprine (Imuran), mycophenolate mofetil (CellCept), and etanercept (Enbrel) may be used in diffuse alveolar hemorrhage, especially when the condition is severe, when first-line therapy with corticosteroids has proven ineffective (generally not advised, unless the condition is mild) or when specific underlying causes are present (eg, Wegener granulomatosis, Goodpasture syndrome, systemic lupus erythematosus). Intravenous cyclophosphamide (2 mg/kg/day, adjusted to renal function) is generally the preferred adjunctive immunosuppressive drug and may be continued for several weeks or until adverse effects occur, such as blood marrow suppression, infection, or hematuria. Thereafter, most clinicians switch to consolidative or maintenance therapy with methotrexate or another agent.
Plasmapheresis is indicated for diffuse alveolar hemorrhage associated with Good-pasture syndrome or with other vasculitic processes in which the titers of pathogenetic immunoglobulins and immune complexes are very high: for example, ANCA-associated vasculitis with overwhelming endothelial injury and a hypercoagulable state. However, the merits of plasmapharesis in diffuse alveolar hemorrhage associated with conditions other than Goodpasture syndrome has not been evaluated in prospective studies.
It remains unclear whether intravenous immunoglobulin therapy adds to the treatment of diffuse alveolar hemorrhage due to vasculitis or other connective tissue disease.
Several case reports have reported successful use of recombinant activated human factor VII in treating alveolar hemorrhage due to allogeneic hematopoietic stem cell transplantation, ANCA-associated vasculitis, systemic lupus erythematosus, or antiphospholipid syndrome. If borne out by larger experience, recombinant activated human factor VII may gain more widespread use in diffuse alveolar hemorrhage.
Other possible management measures include supplemental oxygen, bronchodilators, reversal of any coagulopathy, intubation with bronchial tamponade, protective strategies for the less involved lung, and mechanical ventilation.
PROGNOSIS
The prognosis for diffuse alveolar hemorrhage depends on the underlying cause (Table 3).
Recurrent episodes may lead to various degrees of interstitial fibrosis, especially in patients with underlying Wegener granulo-matosis, mitral stenosis, long-standing and severe mitral regurgitation, and idiopathic pulmonary hemosiderosis. Obstructive lung disease may also complicate microscopic polyangiitis and idiopathic pulmonary hemosiderosis.
Acknowledgment: We acknowledge and appreciate the assistance of Dr. Carol Farver, who provided the pathologic specimens.
- Ioachimescu OCLaurent GL, Shapiro SD. Alveolar hemorrhage. Encyclopedia of Respiratory Medicine. Amsterdam: Academic Press, 2006:92–100.
- Travis WD, Colby TV, Lombard C, Carpenter HA. A clinicopathologic study of 34 cases of diffuse pulmonary hemorrhage with lung biopsy confirmation. Am J Surg Pathol 1990; 14:1112–1125.
- Jennings CA, King TE, Tuder R, Cherniak RM, Schwarz MI. Diffuse alveolar hemorrhage with underlying isolated, pauciimmune pulmonary capillaritis. Am J Respir Crit Care Med 1997; 155:1101–1109.
- Spencer H. Pulmonary lesions in polyarteritis nodosa. Br J Tuberc Dis Chest 1957; 51:123–130.
- Travis WD. Pathology of pulmonary vasculitis. Semin Respir Crit Care Med 2004; 25:475–482.
- Schwarz MI, Brown KK. Small vessel vasculitis of the lung. Thorax 2000; 55:502–510.
- Collard HR, Schwarz MI. Diffuse alveolar hemorrhage. Clin Chest Med 2004; 25:583–592.
- Ioachimescu OC, Jennings C. Intercostal lung cyst hernia in idiopathic pulmonary hemosiderosis (cyst necessitans). Mayo Clin Proc 2006; 81:692.
- Rolla G, Heffler E, Guida G, Bergia R, Bucca C. Exhaled NO in diffuse alveolar haemorrhage. Thorax 2005; 60:614–615.
- Dweik RA, Stoller JK. Role of bronchoscopy in massive hemoptysis. Clin Chest Med 1999; 20:89–105.
- Ioachimescu OCLaurent GL, Shapiro SD. Autoantibodies. Encyclopedia of Respiratory Medicine. Amsterdam: Academic Press, 2006:219–227.
- Watts RA, Carruthers DM, Scott DG. Epidemiology of systemic vasculitis: changing incidence or definition? Semin Arthritis Rheum 1995; 25:28–34.
- Watts RA, Lane SE, Bentham G, Scott DG. Epidemiology of systemic vasculitis: a ten-year study in the United Kingdom. Arthritis Rheum 2000; 43:414–419.
- Watts RA, Jolliffe VA, Carruthers DM, Lockwood M, Scott DG. Effect of classification on the incidence of polyarteritis nodosa and microscopic polyangiitis. Arthritis Rheum 1996; 39:1208–1212.
- Ioachimescu OC, Kotch A, Stoller JK. Idiopathic pulmonary hemosiderosis in adults. Clin Pulm Med 2005; 12:16–25.
- Reinhold-Keller E, Herlyn K, Wagner-Bastmeyer R, et al. No difference in the incidences of vasculitides between north and south Germany: first results of the German vasculitis register. Rheumatology (Oxford) 2002; 41:540–549.
- Mahr A, Guillevin L, Poissonnet M, Ayme S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener’s granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum 2004; 51:92–99.
- Koldingsnes W, Nossent H. Epidemiology of Wegener’s granulomatosis in northern Norway. Arthritis Rheum 2000; 43:2481–2487.
- Kelly PT, Haponik EF. Goodpasture syndrome: molecular and clinical advances. Medicine (Baltimore) 1994; 73:171–185.
- Travis WD, Leslie KOLeslie KO, Wick MR. Pulmonary vasculitis and pulmonary hemorhage. Practical Pulmonary Pathology – a Diagnostic Approach. Philadelphia: Churchill Livingstone-Elsevier, 2005;335–378.
- Jennette JC, Thomas DB, Falk RJ. Microscopic polyangiitis (microscopic polyarteritis). Semin Diagn Pathol 2001; 18:3–13.
- Katzenstein AKatzenstein A, Askin F. Alveolar hemorrhage syndromes. Surgical Pathology of Non-neoplastic Lung Disease. Philadelphia: WB Saunders, 1997:153–159.
- Schwarz MI, Cherniack RM, King TEMurray JF, Nadel J. Diffuse alveolar hemorrhage and other rare infiltrative disorders. Textbook of Respiratory Medicine. Philadelphia: WB Saunders, 2000:1733–1755.
- Lynch JP, Leatherman JWFishman A. Alveolar hemorrhage syndromes. Fishman’s Pulmonary Diseases and Disorders. New York: McGraw-Hill, 1998:1193–1210.
- Cordier JF, Valeyre D, Guillevin L, Loire R, Brechot JM. Pulmonary Wegener’s granulomatosis. A clinical and imaging study of 77 cases. Chest 1990; 97:906–912.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener’s granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med 1983; 98:76–85.
- Reinhold-Keller E, Beuge N, Latza U, et al. An interdisciplinary approach to the care of patients with Wegener’s granulomatosis: long-term outcome in 155 patients. Arthritis Rheum 2000; 43:1021–1032.
- Langford CA, Hoffman GS. Rare diseases 3: Wegener’s granulomatosis. Thorax 1999; 54:629–637.
- Mark EJ, Matsubara O, Tan-Liu NS, Fienberg R. The pulmonary biopsy in the early diagnosis of Wegener’s (pathergic) granulomatosis: a study based on 35 open lung biopsies. Hum Pathol 1988; 19:1065–1071.
- Sheehan RE, Flint JD, Muller NL. Computed tomography features of the thoracic manifestations of Wegener granulomatosis. J Thorac Imaging 2003: 18:34–41.
- Specks USchwarz MI, King TE. Pulmonary vasculitis. Interstitial Lung Disease. Decker BC. Hamilton, Ontario, Canada: Decker, 2003:599–631.
- Ten Berge IJ, Wilmink JM, Meyer CJ, et al. Clinical and immunological follow-up of patients with severe renal disease in Wegener’s granulo-matosis. Am J Nephrol 1985; 5:21–29.
- Brandwein S, Esdaile J, Danoff D, Tannenbaum H. Wegener’s granulo-matosis. Clinical features and outcome in 13 patients. Arch Intern Med 1983; 143:476–479.
- Pinching AJ, Lockwood CM, Pussell BA, et al. Wegener’s granulomatosis: observations on 18 patients with severe renal disease. Q J Med 1983; 52:435–460.
- Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med 1997; 337:1512–1523.
- Lauque D, Cadranel J, Lazor R, et al. Microscopic polyangiitis with alveolar hemorrhage. A study of 29 cases and review of the literature. Groupe d’Études et de Recherche sur les Maladies “Orphelines” Pulmonaires. Medicine (Baltimore) 2000; 79:222–233.
- Johnson JP, Moore J, Austin HA, Balow JE, Antonovych TT, Wilson CB. Therapy of anti-glomerular basement membrane antibody disease: analysis of prognostic significance of clinical, pathologic and treatment factors. Medicine (Baltimore) 1985; 64:219–227.
- Savage CO, Winearls CG, Evans DJ, Rees AJ, Lockwood CM. Microscopic polyarteritis: presentation, pathology, and prognosis. Q J Med 1985; 56:467–483.
- Haworth SJ, Savage CO, Carr D. Pulmonary hemorrhage complicating Wegener’s granulomatosis and microscopic polyarteritis. Br Med J 1985; 290:1175–1178.
- Smyth L, Gaskin G, Pusey CD. Microscopic polyangiitis. Semin Respir Crit Care Med 2004; 25:523–533.
- Lanham JG, Elkon KB, Pusey CD, Hughes GR. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine (Baltimore) 1984; 63:65–81.
- Leatherman JW. Autoimmune diffuse alveolar hemorrhage. Clin Pulm Med 1994; 1:356–364.
- Boyce NW, Holdsworth SR. Pulmonary manifestations of the clinical syndrome of acute glomerulonephritis and lung hemorrhage. Am J Kidney Dis 1986; 8:31–36.
- Emlen W. Systemic lupus erythematosus and mixed connective tissue disease. Immunol Allergy Clin North Am 1979; 105:291–311.
- Hunninghake GW, Fauci AS. Pulmonary involvement in the collagen vascular diseases. Am Rev Respir Dis 1979; 119:471–503.
- Keane MP, Lynch JP. Pleuropulmonary manifestations of systemic lupus erythematosus. Thorax 2000; 55:159–166.
- Zamora MR, Warner ML, Tuder R, Schwarz MI. Diffuse alveolar hemorrhage and systemic lupus erythematosus. Clinical presentation, histology, survival, and outcome. Medicine (Baltimore) 1997; 76:192–202.
- Lee CK, Koh JH, Cha HS, et al. Pulmonary alveolar hemorrhage in patients with rheumatic diseases in Korea. Scand J Rheumatol 2000; 29:288–294.
- Vazquez-Del Mercado M, Mendoza-Topete A, Best-Aguilera CR, Garcia-De La Torre I. Diffuse alveolar hemorrhage in limited cutaneous systemic sclerosis with positive perinuclear antineutrophil cytoplasmic antibodies. J Rheumatol 1996; 23:1821–1823.
- Fenlon HM, Doran M, Sant SM, Breatnach E. High-resolution chest CT in systemic lupus erythematosus. AJR Am J Roentgenol 1996; 166:301–307.
- Ioachimescu OC. Idiopathic pulmonary hemosiderosis in adults. Pneumologia 2003; 52:38–43.
- Ioachimescu OC, Sieber S, Kotch A. Idiopathic pulmonary haemosiderosis revisited. Eur Respir J 2004; 24:162–170.
- Franks TJ, Koss MN. Pulmonary capillaritis. Curr Opin Pulm Med 2000; 6:430–435.
- Travis WD, Hoffman GS, Leavitt RY, Pass HI, Fauci AS. Surgical pathology of the lung in Wegener’s granulomatosis. Review of 87 open lung biopsies from 67 patients. Am J Surg Pathol 1991; 15:315–333.
- Zashin S, Fattor R, Fortin D. Microscopic polyarteritis: a forgotten aetiology of haemoptysis and rapidly progressive glomerulonephritis. Ann Rheum Dis 1990; 49:53–56.
- Yoshikawa Y, Watanabe T. Pulmonary lesions in Wegener’s granulo-matosis: a clinicopathologic study of 22 autopsy cases. Hum Pathol 1986; 17:401–410.
- Teague CA, Doak PB, Simpson IJ, Rainer SP, Herdson PB. Goodpasture’s syndrome: an analysis of 29 cases. Kidney Int 1978; 13:492–504.
- Abu-Shakra M, Smythe H, Lewtas J, Badley E, Weber D, Keystone E. Outcome of polyarteritis nodosa and Churg-Strauss syndrome. An analysis of twenty-five patients. Arthritis Rheum 1994; 37:1798–1803.
- Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore) 1999; 78:26–37.
- Schwab EP, Schumacher HR, Freundlich B, Callegari PE. Pulmonary alveolar hemorrhage in systemic lupus erythematosus. Semin Arthritis Rheum 1993; 23:8–15.
- Koh WH, Thumboo J, Boey ML. Pulmonary haemorrhage in Oriental patients with systemic lupus erythematosus. Lupus 1997; 6:713–716.
Diffuse alveolar hemorrhage can complicate a large number of clinical conditions. It may present in different ways and may be life-threatening, and it poses an important challenge for the clinician.1
Diffuse alveolar hemorrhage is an uncommon condition in which blood floods the alveoli, usually at multiple sites. It is also known as intrapulmonary hemorrhage, diffuse pulmonary hemorrhage, pulmonary alveolar hemorrhage, pulmonary capillary hemorrhage, alveolar bleeding, or microvascular pulmonary hemorrhage.
In this article we review the causes, clinical features, diagnostic criteria, treatment, and prognosis of diffuse alveolar hemorrhage.
CAUSES OF DIFFUSE ALVEOLAR HEMORRHAGE
THREE CHARACTERISTIC PATTERNS
In general, diffuse alveolar hemorrhage can occur in three characteristic patterns, which reflect the nature of the underlying vascular injury1:
Diffuse alveolar hemorrhage associated with vasculitis or capillaritis. As described by Spencer4 50 years ago, pulmonary capillaritis is the most frequent underlying histologic lesion described in diffuse alveolar hemorrhage. Neutrophils infiltrate the interalveolar and peri-bronchiolar septal vessels (pulmonary interstitium),5 leading to anatomic disruption of the capillaries (ie, impairment of the alveolocapillary barrier) and to extravasation of red blood cells into the alveoli and interstitium. Neutrophil apoptosis and fragmentation, with subsequent release of the intracellular proteolytic enzymes and reactive oxygen species, beget more inflammation, intra-alveolar neutrophilic nuclear dust, fibrin and inflammatory exudate, and fibrinoid necrosis of the interstitium.6,7
‘Bland’ pulmonary hemorrhage (ie, without capillaritis or vasculitis). In this pattern, red blood cells leak into the alveoli without any evidence of inflammation or destruction of the alveolar capillaries, venules, and arterioles. The epithelial lesions are usually microscopic and are scattered geographically.
Diffuse alveolar hemorrhage associated with another process or condition (eg, diffuse alveolar damage, lymphangioleiomyomatosis, drug-induced lung injury, metastatic tumor to the lungs, mitral stenosis). Diffuse alveolar damage is the main underlying lesion of the acute respiratory distress syndrome and is characterized by formation of an intra-alveolar hyaline membrane, by interstitial edema with minimal inflammation, and, at times, by “secondary” diffuse alveolar hemorrhage. In this third category of diffuse alveolar hemorrhage, the underlying process causes alveolar hemorrhage by processes other than pulmonary vascular inflammation or direct extravasation of red cells.
THE CLINICAL PRESENTATION
The clinical presentation of diffuse alveolar hemorrhage may reflect either alveolar bleeding alone or features of the underlying cause (eg, hematuria in Wegener granulomatosis, arthritis in systemic lupus erythematosus). Hence, its recognition requires a high degree of suspicion.
Some patients present with severe acute respiratory distress requiring mechanical ventilation. However, dyspnea, cough, and fever are the common initial symptoms and are most often acute or subacute (ie, present for less than a week). The fever is usually due to the underlying cause, such as lupus.
Hemoptysis may be absent at the time of presentation in up to a third of patients because the total alveolar volume is large and can absorb large amounts of blood, without extending more proximally into the airways. Apparent hemoptysis, if present, must be differentiated from hematemesis or pseudohemoptysis (alveolar flooding with fluid that resembles blood, as in Serratia marcescens pneumonia, in which the reddish hue of the infecting organism can create the impression of alveolar bleeding).
DIAGNOSTIC EVALUATION
Generally speaking, dyspnea, cough, hemoptysis, and new alveolar infiltrates in conjunction with bloody bronchoalveolar lavage specimens (with numerous erythrocytes and siderophages) establish the diagnosis of diffuse alveolar hemorrhage. Surgical biopsy from the lung or another organ involved by an underlying condition is often necessary.
Physical examination
The physical findings are nonspecific and may reflect the underlying systemic vasculitis or collagen vascular disorder (eg, with accompanying rash, purpura, eye lesions, hepatosplenomegaly, or clubbing).
Imaging studies
Radiography may show new or old or both new and old patchy or diffuse alveolar opacities. Recurrent episodes of hemorrhage may lead to reticular interstitial opacities due to pulmonary fibrosis, usually with minimal (if any) honeycombing. Kerley B lines suggest mitral valve disease or pulmonary veno-occlusive disease as the cause of the hemorrhage.
Computed tomography may show areas of consolidation interspersed with areas of ground-glass attenuation and preserved, normal areas.
Currently, nuclear imaging such as gallium or tagged red blood cell studies have little role in evaluating diffuse alveolar hemorrhage. Other nuclear studies, geared to reveal breakdown of the microcirculatory integrity and extravasation of red blood cells out of the vessels, have also not been proven useful.
Evaluating pulmonary function
Diffuse alveolar hemorrhage may cause impairment of oxygen transfer and hypoxemia. In addition, it can cause several other abnormalities of pulmonary function.
Increased diffusing capacity. Because blood in the lungs can absorb inhaled carbon monoxide, the diffusing capacity for carbon monoxide (DLCO) may be distinctively increased. Serial increases in the DLCO may indicate progressive alveolar hemorrhage. However, the clinical instability of patients experiencing active alveolar bleeding precludes performing the DLCO measurement maneuvers, rendering the DLCO test relatively impractical.
Restrictive changes. Because recurrent episodes of diffuse alveolar hemorrhage can lead to interstitial fibrosis, restrictive changes—ie, decreased total lung capacity, decreased forced vital capacity (FVC), and preserved ratio of the forced expiratory volume in 1 second (FEV1) to the FVC—may characterize diffuse alveolar hemorrhage.
Obstructive changes (less common). Less commonly, patients with diffuse alveolar hemorrhage may have spirometric changes indicating airflow obstruction—ie, decreased FEV1 and decreased ratio of FEV1 to FVC—possibly because neutrophilic infiltration from blood extravasation into the alveolar sacs causes release of reactive oxygen species and proteolytic enzymes, which in turn may cause small airway and parenchymal damage such as bronchiolitis and emphysema. A pattern of obstructive lung disease associated with recurrent diffuse alveolar hemorrhage should prompt consideration of an underlying condition that can cause airflow obstruction, such as sarcoidosis, microscopic polyangiitis, or Wegener granulomatosis, or, less commonly, lymphangioleiomyomatosis, histiocytosis X, pulmonary capillaritis, or sometimes idiopathic pulmonary hemosiderosis.
As an example of an unusual circumstance, we have described elsewhere a case of a woman with idiopathic pulmonary hemosiderosis with multiple episodes of diffuse alveolar hemorrhage and resultant emphysema.8 Radiographic images showed several very large cysts, one of which herniated through the incision site of an open lung biopsy.
Decreased exhaled nitric oxide. Though currently unavailable in most clinical pulmonary function laboratories, evaluation of exhaled gas or condensate may have value in diagnosing diffuse alveolar hemorrhage.9 Specifically, because increased intra-alveolar hemoglobin binds nitric oxide, as it does carbon monoxide, levels of exhaled nitric oxide may be decreased in diffuse alveolar hemorrhage. In contrast to the difficulty of measuring DLCO in patients with active alveolar bleeding or hemoptysis, analysis of exhaled gas is clinically feasible, making this a promising diagnostic test.
Laboratory evaluation
Hematologic assessment in patients with diffuse alveolar hemorrhage generally reveals:
- Acute or chronic anemia
- Leukocytosis
- Elevated erythrocyte sedimentation rate
- Elevated C-reactive protein level (particularly in patients whose alveolar hemorrhage is due to systemic disease or vasculitis, or both).
Renal abnormalities such as elevated blood urea nitrogen and serum creatinine or abnormal findings on urinalysis (with hematuria, proteinuria, and red blood cell casts indicating glomerulonephritis) can also occur, as diffuse alveolar hemorrhage may complicate several pulmonary-renal syndromes such as Goodpasture syndrome and Wegener granulomatosis.
Bronchoscopy
The diagnostic evaluation in diffuse alveolar hemorrhage usually includes bronchoscopic examination,10 which serves two purposes:
- To document alveolar hemorrhage by bronchoalveolar lavage and to exclude airway sources of bleeding by visual inspection
- To exclude an associated infection.
Based on experience with nonmassive hemoptysis of all causes (but not exclusively diffuse alveolar hemorrhage), the diagnostic yield of bronchoscopy is higher if the procedure is performed within the first 48 hours of symptoms rather than later. Evidence supporting diffuse alveolar hemorrhage is persistent (or even increasing) blood on three sequential lavage aliquots from a single affected area of the lung.
FINDING THE UNDERLYING CAUSE
Once the diagnosis of diffuse alveolar hemorrhage is established, the clinician must ascertain whether an underlying cause is present. Serologic studies may prove important, although the results are generally not available in a manner timely enough to guide immediate management.
When a pulmonary-renal syndrome is suggested by accompanying hematuria or renal dysfunction, antiglomerular basement membrane antibody and antineutrophil cytoplasmic antibody (ANCA) levels should be checked. Tests for complement fractions C3 and C4, anti-double-stranded DNA, and antiphospholipid antibodies should be ordered if an underlying condition such as lupus or antiphospholipid antibody syndrome is suspected (Table 2).11
If the underlying cause remains elusive after a thorough clinical evaluation that includes imaging studies, serologic studies, and bronchoscopy, then surgical biopsy should be considered.1 Which organ to biopsy (eg, lung, sinus, kidney) depends on the level of suspicion for a specific cause. For example, suspicion of Wegener granulomato-sis with hematuria or renal dysfunction might prompt renal biopsy. However, lung biopsy often needs to be performed with video-assisted thoracoscopy, especially when disease is confined to the lung (as in idiopathic pulmonary hemosiderosis or pauci-immune pulmonary capillaritis). Renal biopsy specimens should also undergo immunofluores-cence staining, which may reveal linear deposition of immunoglobulins and immune complexes along the basement membrane in patients with Goodpasture syndrome, or of granular deposits in patients with systemic lupus erythematosus.
TWO GENERAL CLINICAL SCENARIOS
In general, the clinician will be confronted by one of two scenarios: a patient with diffuse alveolar hemorrhage and associated systemic findings, or a patient with hemorrhage and no associated systemic findings.
Hemorrhage with associated systemic findings
Certain clues from the history raise suspicion of diffuse alveolar hemorrhage:
- Recent infection suggests Henoch-Schönlein purpura or cryoglobulinemic vasculitis
- Use of a possibly offending drug such as an anticoagulant, D-penicillamine (Cuprimine, Depen), nitrofurantoin (Furadantin, Macrobid, Macrodantin), amiodarone (Cordarone), propylthiouracil, cocaine, or sirolimus (Rapamune, Rapamycin)
- Exposure to toxic agents such as trimellitic anhydride, insecticides, and pesticides
- A known comorbid condition such as vasculitis, connective tissue disease, mitral valve disease, or solid organ or stem cell transplantation.
If asthma, eosinophilia, pulmonary infiltrates, and diffuse alveolar hemorrhage coexist, consideration should be given to Churg-Strauss syndrome. If sinus disease, skin manifestations, pulmonary parenchymal nodules, and cavitary lesions coexist with positivity for antiproteinase 3 c-ANCA and biopsy-proven granulomata, then Wegener granulomatosis should be considered. Similarly, diffuse alveolar hemorrhage with glomerulonephritis and skin manifestations, positivity for p-ANCA, and necrotizing nongranulomatous lesions on end-organ biopsy may lead to a diagnosis of microscopic polyangiitis. In a young smoker with glomeru-lonephritis and diffuse alveolar hemorrhage presenting as either bland alveolar hemorrhage or pulmonary capillaritis, Goodpasture syndrome or antiglomerular basement membrane antibody disease should be considered.
Hemorrhage with no associated systemic findings
When the above conditions have been considered but no suggestive findings are found, the following four conditions should be considered:
- Antiglomerular basement membrane antibody disease in limited pulmonary form or onset: positivity to the antibody with linear deposits in the lungs would be diagnostic in such a case
- Pulmonary-limited microscopic polyangiitis positive for p-ANCA (a positive anti-myeloperoxidase p-ANCA test makes the diagnosis)
- Pauci-immune isolated pulmonary capillaritis, when the biopsy shows evidence of neutrophilic pulmonary capillaritis
- Idiopathic pulmonary hemosiderosis, a diagnosis of exclusion, when the biopsy shows evidence of acute, subacute, and chronic bland diffuse alveolar hemorrhage and no evidence of vasculitis.
TREATMENT OF DIFFUSE ALVEOLAR HEMORRHAGE
Therapy for diffuse alveolar hemorrhage consists of treating both the autoimmune destruction of the alveolar capillary membrane and the underlying condition. Corticosteroids and immunosuppressive agents remain the gold standard for most patients. Recombinant-activated human factor VII seems to be a promising new therapy, although further evaluation is needed.
Immunosuppressive agents are the mainstay of therapy for diffuse alveolar hemorrhage, especially if associated with systemic or pulmonary vasculitis, Goodpasture syndrome, and connective tissue disorders. Most experts recommend intravenous methylprednisolone (Solu-Medrol) (up to 500 mg every 6 hours, although lower doses seem to have similar efficacy) for 4 or 5 days, followed by a gradual taper to maintenance doses of oral steroids.
In patients with pulmonary-renal syndrome, therapy should be started as soon as possible to prevent irreversible renal failure.
Besides corticosteroids, other immunosuppressive drugs such as cyclophosphamide (Cytoxan), azathioprine (Imuran), mycophenolate mofetil (CellCept), and etanercept (Enbrel) may be used in diffuse alveolar hemorrhage, especially when the condition is severe, when first-line therapy with corticosteroids has proven ineffective (generally not advised, unless the condition is mild) or when specific underlying causes are present (eg, Wegener granulomatosis, Goodpasture syndrome, systemic lupus erythematosus). Intravenous cyclophosphamide (2 mg/kg/day, adjusted to renal function) is generally the preferred adjunctive immunosuppressive drug and may be continued for several weeks or until adverse effects occur, such as blood marrow suppression, infection, or hematuria. Thereafter, most clinicians switch to consolidative or maintenance therapy with methotrexate or another agent.
Plasmapheresis is indicated for diffuse alveolar hemorrhage associated with Good-pasture syndrome or with other vasculitic processes in which the titers of pathogenetic immunoglobulins and immune complexes are very high: for example, ANCA-associated vasculitis with overwhelming endothelial injury and a hypercoagulable state. However, the merits of plasmapharesis in diffuse alveolar hemorrhage associated with conditions other than Goodpasture syndrome has not been evaluated in prospective studies.
It remains unclear whether intravenous immunoglobulin therapy adds to the treatment of diffuse alveolar hemorrhage due to vasculitis or other connective tissue disease.
Several case reports have reported successful use of recombinant activated human factor VII in treating alveolar hemorrhage due to allogeneic hematopoietic stem cell transplantation, ANCA-associated vasculitis, systemic lupus erythematosus, or antiphospholipid syndrome. If borne out by larger experience, recombinant activated human factor VII may gain more widespread use in diffuse alveolar hemorrhage.
Other possible management measures include supplemental oxygen, bronchodilators, reversal of any coagulopathy, intubation with bronchial tamponade, protective strategies for the less involved lung, and mechanical ventilation.
PROGNOSIS
The prognosis for diffuse alveolar hemorrhage depends on the underlying cause (Table 3).
Recurrent episodes may lead to various degrees of interstitial fibrosis, especially in patients with underlying Wegener granulo-matosis, mitral stenosis, long-standing and severe mitral regurgitation, and idiopathic pulmonary hemosiderosis. Obstructive lung disease may also complicate microscopic polyangiitis and idiopathic pulmonary hemosiderosis.
Acknowledgment: We acknowledge and appreciate the assistance of Dr. Carol Farver, who provided the pathologic specimens.
Diffuse alveolar hemorrhage can complicate a large number of clinical conditions. It may present in different ways and may be life-threatening, and it poses an important challenge for the clinician.1
Diffuse alveolar hemorrhage is an uncommon condition in which blood floods the alveoli, usually at multiple sites. It is also known as intrapulmonary hemorrhage, diffuse pulmonary hemorrhage, pulmonary alveolar hemorrhage, pulmonary capillary hemorrhage, alveolar bleeding, or microvascular pulmonary hemorrhage.
In this article we review the causes, clinical features, diagnostic criteria, treatment, and prognosis of diffuse alveolar hemorrhage.
CAUSES OF DIFFUSE ALVEOLAR HEMORRHAGE
THREE CHARACTERISTIC PATTERNS
In general, diffuse alveolar hemorrhage can occur in three characteristic patterns, which reflect the nature of the underlying vascular injury1:
Diffuse alveolar hemorrhage associated with vasculitis or capillaritis. As described by Spencer4 50 years ago, pulmonary capillaritis is the most frequent underlying histologic lesion described in diffuse alveolar hemorrhage. Neutrophils infiltrate the interalveolar and peri-bronchiolar septal vessels (pulmonary interstitium),5 leading to anatomic disruption of the capillaries (ie, impairment of the alveolocapillary barrier) and to extravasation of red blood cells into the alveoli and interstitium. Neutrophil apoptosis and fragmentation, with subsequent release of the intracellular proteolytic enzymes and reactive oxygen species, beget more inflammation, intra-alveolar neutrophilic nuclear dust, fibrin and inflammatory exudate, and fibrinoid necrosis of the interstitium.6,7
‘Bland’ pulmonary hemorrhage (ie, without capillaritis or vasculitis). In this pattern, red blood cells leak into the alveoli without any evidence of inflammation or destruction of the alveolar capillaries, venules, and arterioles. The epithelial lesions are usually microscopic and are scattered geographically.
Diffuse alveolar hemorrhage associated with another process or condition (eg, diffuse alveolar damage, lymphangioleiomyomatosis, drug-induced lung injury, metastatic tumor to the lungs, mitral stenosis). Diffuse alveolar damage is the main underlying lesion of the acute respiratory distress syndrome and is characterized by formation of an intra-alveolar hyaline membrane, by interstitial edema with minimal inflammation, and, at times, by “secondary” diffuse alveolar hemorrhage. In this third category of diffuse alveolar hemorrhage, the underlying process causes alveolar hemorrhage by processes other than pulmonary vascular inflammation or direct extravasation of red cells.
THE CLINICAL PRESENTATION
The clinical presentation of diffuse alveolar hemorrhage may reflect either alveolar bleeding alone or features of the underlying cause (eg, hematuria in Wegener granulomatosis, arthritis in systemic lupus erythematosus). Hence, its recognition requires a high degree of suspicion.
Some patients present with severe acute respiratory distress requiring mechanical ventilation. However, dyspnea, cough, and fever are the common initial symptoms and are most often acute or subacute (ie, present for less than a week). The fever is usually due to the underlying cause, such as lupus.
Hemoptysis may be absent at the time of presentation in up to a third of patients because the total alveolar volume is large and can absorb large amounts of blood, without extending more proximally into the airways. Apparent hemoptysis, if present, must be differentiated from hematemesis or pseudohemoptysis (alveolar flooding with fluid that resembles blood, as in Serratia marcescens pneumonia, in which the reddish hue of the infecting organism can create the impression of alveolar bleeding).
DIAGNOSTIC EVALUATION
Generally speaking, dyspnea, cough, hemoptysis, and new alveolar infiltrates in conjunction with bloody bronchoalveolar lavage specimens (with numerous erythrocytes and siderophages) establish the diagnosis of diffuse alveolar hemorrhage. Surgical biopsy from the lung or another organ involved by an underlying condition is often necessary.
Physical examination
The physical findings are nonspecific and may reflect the underlying systemic vasculitis or collagen vascular disorder (eg, with accompanying rash, purpura, eye lesions, hepatosplenomegaly, or clubbing).
Imaging studies
Radiography may show new or old or both new and old patchy or diffuse alveolar opacities. Recurrent episodes of hemorrhage may lead to reticular interstitial opacities due to pulmonary fibrosis, usually with minimal (if any) honeycombing. Kerley B lines suggest mitral valve disease or pulmonary veno-occlusive disease as the cause of the hemorrhage.
Computed tomography may show areas of consolidation interspersed with areas of ground-glass attenuation and preserved, normal areas.
Currently, nuclear imaging such as gallium or tagged red blood cell studies have little role in evaluating diffuse alveolar hemorrhage. Other nuclear studies, geared to reveal breakdown of the microcirculatory integrity and extravasation of red blood cells out of the vessels, have also not been proven useful.
Evaluating pulmonary function
Diffuse alveolar hemorrhage may cause impairment of oxygen transfer and hypoxemia. In addition, it can cause several other abnormalities of pulmonary function.
Increased diffusing capacity. Because blood in the lungs can absorb inhaled carbon monoxide, the diffusing capacity for carbon monoxide (DLCO) may be distinctively increased. Serial increases in the DLCO may indicate progressive alveolar hemorrhage. However, the clinical instability of patients experiencing active alveolar bleeding precludes performing the DLCO measurement maneuvers, rendering the DLCO test relatively impractical.
Restrictive changes. Because recurrent episodes of diffuse alveolar hemorrhage can lead to interstitial fibrosis, restrictive changes—ie, decreased total lung capacity, decreased forced vital capacity (FVC), and preserved ratio of the forced expiratory volume in 1 second (FEV1) to the FVC—may characterize diffuse alveolar hemorrhage.
Obstructive changes (less common). Less commonly, patients with diffuse alveolar hemorrhage may have spirometric changes indicating airflow obstruction—ie, decreased FEV1 and decreased ratio of FEV1 to FVC—possibly because neutrophilic infiltration from blood extravasation into the alveolar sacs causes release of reactive oxygen species and proteolytic enzymes, which in turn may cause small airway and parenchymal damage such as bronchiolitis and emphysema. A pattern of obstructive lung disease associated with recurrent diffuse alveolar hemorrhage should prompt consideration of an underlying condition that can cause airflow obstruction, such as sarcoidosis, microscopic polyangiitis, or Wegener granulomatosis, or, less commonly, lymphangioleiomyomatosis, histiocytosis X, pulmonary capillaritis, or sometimes idiopathic pulmonary hemosiderosis.
As an example of an unusual circumstance, we have described elsewhere a case of a woman with idiopathic pulmonary hemosiderosis with multiple episodes of diffuse alveolar hemorrhage and resultant emphysema.8 Radiographic images showed several very large cysts, one of which herniated through the incision site of an open lung biopsy.
Decreased exhaled nitric oxide. Though currently unavailable in most clinical pulmonary function laboratories, evaluation of exhaled gas or condensate may have value in diagnosing diffuse alveolar hemorrhage.9 Specifically, because increased intra-alveolar hemoglobin binds nitric oxide, as it does carbon monoxide, levels of exhaled nitric oxide may be decreased in diffuse alveolar hemorrhage. In contrast to the difficulty of measuring DLCO in patients with active alveolar bleeding or hemoptysis, analysis of exhaled gas is clinically feasible, making this a promising diagnostic test.
Laboratory evaluation
Hematologic assessment in patients with diffuse alveolar hemorrhage generally reveals:
- Acute or chronic anemia
- Leukocytosis
- Elevated erythrocyte sedimentation rate
- Elevated C-reactive protein level (particularly in patients whose alveolar hemorrhage is due to systemic disease or vasculitis, or both).
Renal abnormalities such as elevated blood urea nitrogen and serum creatinine or abnormal findings on urinalysis (with hematuria, proteinuria, and red blood cell casts indicating glomerulonephritis) can also occur, as diffuse alveolar hemorrhage may complicate several pulmonary-renal syndromes such as Goodpasture syndrome and Wegener granulomatosis.
Bronchoscopy
The diagnostic evaluation in diffuse alveolar hemorrhage usually includes bronchoscopic examination,10 which serves two purposes:
- To document alveolar hemorrhage by bronchoalveolar lavage and to exclude airway sources of bleeding by visual inspection
- To exclude an associated infection.
Based on experience with nonmassive hemoptysis of all causes (but not exclusively diffuse alveolar hemorrhage), the diagnostic yield of bronchoscopy is higher if the procedure is performed within the first 48 hours of symptoms rather than later. Evidence supporting diffuse alveolar hemorrhage is persistent (or even increasing) blood on three sequential lavage aliquots from a single affected area of the lung.
FINDING THE UNDERLYING CAUSE
Once the diagnosis of diffuse alveolar hemorrhage is established, the clinician must ascertain whether an underlying cause is present. Serologic studies may prove important, although the results are generally not available in a manner timely enough to guide immediate management.
When a pulmonary-renal syndrome is suggested by accompanying hematuria or renal dysfunction, antiglomerular basement membrane antibody and antineutrophil cytoplasmic antibody (ANCA) levels should be checked. Tests for complement fractions C3 and C4, anti-double-stranded DNA, and antiphospholipid antibodies should be ordered if an underlying condition such as lupus or antiphospholipid antibody syndrome is suspected (Table 2).11
If the underlying cause remains elusive after a thorough clinical evaluation that includes imaging studies, serologic studies, and bronchoscopy, then surgical biopsy should be considered.1 Which organ to biopsy (eg, lung, sinus, kidney) depends on the level of suspicion for a specific cause. For example, suspicion of Wegener granulomato-sis with hematuria or renal dysfunction might prompt renal biopsy. However, lung biopsy often needs to be performed with video-assisted thoracoscopy, especially when disease is confined to the lung (as in idiopathic pulmonary hemosiderosis or pauci-immune pulmonary capillaritis). Renal biopsy specimens should also undergo immunofluores-cence staining, which may reveal linear deposition of immunoglobulins and immune complexes along the basement membrane in patients with Goodpasture syndrome, or of granular deposits in patients with systemic lupus erythematosus.
TWO GENERAL CLINICAL SCENARIOS
In general, the clinician will be confronted by one of two scenarios: a patient with diffuse alveolar hemorrhage and associated systemic findings, or a patient with hemorrhage and no associated systemic findings.
Hemorrhage with associated systemic findings
Certain clues from the history raise suspicion of diffuse alveolar hemorrhage:
- Recent infection suggests Henoch-Schönlein purpura or cryoglobulinemic vasculitis
- Use of a possibly offending drug such as an anticoagulant, D-penicillamine (Cuprimine, Depen), nitrofurantoin (Furadantin, Macrobid, Macrodantin), amiodarone (Cordarone), propylthiouracil, cocaine, or sirolimus (Rapamune, Rapamycin)
- Exposure to toxic agents such as trimellitic anhydride, insecticides, and pesticides
- A known comorbid condition such as vasculitis, connective tissue disease, mitral valve disease, or solid organ or stem cell transplantation.
If asthma, eosinophilia, pulmonary infiltrates, and diffuse alveolar hemorrhage coexist, consideration should be given to Churg-Strauss syndrome. If sinus disease, skin manifestations, pulmonary parenchymal nodules, and cavitary lesions coexist with positivity for antiproteinase 3 c-ANCA and biopsy-proven granulomata, then Wegener granulomatosis should be considered. Similarly, diffuse alveolar hemorrhage with glomerulonephritis and skin manifestations, positivity for p-ANCA, and necrotizing nongranulomatous lesions on end-organ biopsy may lead to a diagnosis of microscopic polyangiitis. In a young smoker with glomeru-lonephritis and diffuse alveolar hemorrhage presenting as either bland alveolar hemorrhage or pulmonary capillaritis, Goodpasture syndrome or antiglomerular basement membrane antibody disease should be considered.
Hemorrhage with no associated systemic findings
When the above conditions have been considered but no suggestive findings are found, the following four conditions should be considered:
- Antiglomerular basement membrane antibody disease in limited pulmonary form or onset: positivity to the antibody with linear deposits in the lungs would be diagnostic in such a case
- Pulmonary-limited microscopic polyangiitis positive for p-ANCA (a positive anti-myeloperoxidase p-ANCA test makes the diagnosis)
- Pauci-immune isolated pulmonary capillaritis, when the biopsy shows evidence of neutrophilic pulmonary capillaritis
- Idiopathic pulmonary hemosiderosis, a diagnosis of exclusion, when the biopsy shows evidence of acute, subacute, and chronic bland diffuse alveolar hemorrhage and no evidence of vasculitis.
TREATMENT OF DIFFUSE ALVEOLAR HEMORRHAGE
Therapy for diffuse alveolar hemorrhage consists of treating both the autoimmune destruction of the alveolar capillary membrane and the underlying condition. Corticosteroids and immunosuppressive agents remain the gold standard for most patients. Recombinant-activated human factor VII seems to be a promising new therapy, although further evaluation is needed.
Immunosuppressive agents are the mainstay of therapy for diffuse alveolar hemorrhage, especially if associated with systemic or pulmonary vasculitis, Goodpasture syndrome, and connective tissue disorders. Most experts recommend intravenous methylprednisolone (Solu-Medrol) (up to 500 mg every 6 hours, although lower doses seem to have similar efficacy) for 4 or 5 days, followed by a gradual taper to maintenance doses of oral steroids.
In patients with pulmonary-renal syndrome, therapy should be started as soon as possible to prevent irreversible renal failure.
Besides corticosteroids, other immunosuppressive drugs such as cyclophosphamide (Cytoxan), azathioprine (Imuran), mycophenolate mofetil (CellCept), and etanercept (Enbrel) may be used in diffuse alveolar hemorrhage, especially when the condition is severe, when first-line therapy with corticosteroids has proven ineffective (generally not advised, unless the condition is mild) or when specific underlying causes are present (eg, Wegener granulomatosis, Goodpasture syndrome, systemic lupus erythematosus). Intravenous cyclophosphamide (2 mg/kg/day, adjusted to renal function) is generally the preferred adjunctive immunosuppressive drug and may be continued for several weeks or until adverse effects occur, such as blood marrow suppression, infection, or hematuria. Thereafter, most clinicians switch to consolidative or maintenance therapy with methotrexate or another agent.
Plasmapheresis is indicated for diffuse alveolar hemorrhage associated with Good-pasture syndrome or with other vasculitic processes in which the titers of pathogenetic immunoglobulins and immune complexes are very high: for example, ANCA-associated vasculitis with overwhelming endothelial injury and a hypercoagulable state. However, the merits of plasmapharesis in diffuse alveolar hemorrhage associated with conditions other than Goodpasture syndrome has not been evaluated in prospective studies.
It remains unclear whether intravenous immunoglobulin therapy adds to the treatment of diffuse alveolar hemorrhage due to vasculitis or other connective tissue disease.
Several case reports have reported successful use of recombinant activated human factor VII in treating alveolar hemorrhage due to allogeneic hematopoietic stem cell transplantation, ANCA-associated vasculitis, systemic lupus erythematosus, or antiphospholipid syndrome. If borne out by larger experience, recombinant activated human factor VII may gain more widespread use in diffuse alveolar hemorrhage.
Other possible management measures include supplemental oxygen, bronchodilators, reversal of any coagulopathy, intubation with bronchial tamponade, protective strategies for the less involved lung, and mechanical ventilation.
PROGNOSIS
The prognosis for diffuse alveolar hemorrhage depends on the underlying cause (Table 3).
Recurrent episodes may lead to various degrees of interstitial fibrosis, especially in patients with underlying Wegener granulo-matosis, mitral stenosis, long-standing and severe mitral regurgitation, and idiopathic pulmonary hemosiderosis. Obstructive lung disease may also complicate microscopic polyangiitis and idiopathic pulmonary hemosiderosis.
Acknowledgment: We acknowledge and appreciate the assistance of Dr. Carol Farver, who provided the pathologic specimens.
- Ioachimescu OCLaurent GL, Shapiro SD. Alveolar hemorrhage. Encyclopedia of Respiratory Medicine. Amsterdam: Academic Press, 2006:92–100.
- Travis WD, Colby TV, Lombard C, Carpenter HA. A clinicopathologic study of 34 cases of diffuse pulmonary hemorrhage with lung biopsy confirmation. Am J Surg Pathol 1990; 14:1112–1125.
- Jennings CA, King TE, Tuder R, Cherniak RM, Schwarz MI. Diffuse alveolar hemorrhage with underlying isolated, pauciimmune pulmonary capillaritis. Am J Respir Crit Care Med 1997; 155:1101–1109.
- Spencer H. Pulmonary lesions in polyarteritis nodosa. Br J Tuberc Dis Chest 1957; 51:123–130.
- Travis WD. Pathology of pulmonary vasculitis. Semin Respir Crit Care Med 2004; 25:475–482.
- Schwarz MI, Brown KK. Small vessel vasculitis of the lung. Thorax 2000; 55:502–510.
- Collard HR, Schwarz MI. Diffuse alveolar hemorrhage. Clin Chest Med 2004; 25:583–592.
- Ioachimescu OC, Jennings C. Intercostal lung cyst hernia in idiopathic pulmonary hemosiderosis (cyst necessitans). Mayo Clin Proc 2006; 81:692.
- Rolla G, Heffler E, Guida G, Bergia R, Bucca C. Exhaled NO in diffuse alveolar haemorrhage. Thorax 2005; 60:614–615.
- Dweik RA, Stoller JK. Role of bronchoscopy in massive hemoptysis. Clin Chest Med 1999; 20:89–105.
- Ioachimescu OCLaurent GL, Shapiro SD. Autoantibodies. Encyclopedia of Respiratory Medicine. Amsterdam: Academic Press, 2006:219–227.
- Watts RA, Carruthers DM, Scott DG. Epidemiology of systemic vasculitis: changing incidence or definition? Semin Arthritis Rheum 1995; 25:28–34.
- Watts RA, Lane SE, Bentham G, Scott DG. Epidemiology of systemic vasculitis: a ten-year study in the United Kingdom. Arthritis Rheum 2000; 43:414–419.
- Watts RA, Jolliffe VA, Carruthers DM, Lockwood M, Scott DG. Effect of classification on the incidence of polyarteritis nodosa and microscopic polyangiitis. Arthritis Rheum 1996; 39:1208–1212.
- Ioachimescu OC, Kotch A, Stoller JK. Idiopathic pulmonary hemosiderosis in adults. Clin Pulm Med 2005; 12:16–25.
- Reinhold-Keller E, Herlyn K, Wagner-Bastmeyer R, et al. No difference in the incidences of vasculitides between north and south Germany: first results of the German vasculitis register. Rheumatology (Oxford) 2002; 41:540–549.
- Mahr A, Guillevin L, Poissonnet M, Ayme S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener’s granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum 2004; 51:92–99.
- Koldingsnes W, Nossent H. Epidemiology of Wegener’s granulomatosis in northern Norway. Arthritis Rheum 2000; 43:2481–2487.
- Kelly PT, Haponik EF. Goodpasture syndrome: molecular and clinical advances. Medicine (Baltimore) 1994; 73:171–185.
- Travis WD, Leslie KOLeslie KO, Wick MR. Pulmonary vasculitis and pulmonary hemorhage. Practical Pulmonary Pathology – a Diagnostic Approach. Philadelphia: Churchill Livingstone-Elsevier, 2005;335–378.
- Jennette JC, Thomas DB, Falk RJ. Microscopic polyangiitis (microscopic polyarteritis). Semin Diagn Pathol 2001; 18:3–13.
- Katzenstein AKatzenstein A, Askin F. Alveolar hemorrhage syndromes. Surgical Pathology of Non-neoplastic Lung Disease. Philadelphia: WB Saunders, 1997:153–159.
- Schwarz MI, Cherniack RM, King TEMurray JF, Nadel J. Diffuse alveolar hemorrhage and other rare infiltrative disorders. Textbook of Respiratory Medicine. Philadelphia: WB Saunders, 2000:1733–1755.
- Lynch JP, Leatherman JWFishman A. Alveolar hemorrhage syndromes. Fishman’s Pulmonary Diseases and Disorders. New York: McGraw-Hill, 1998:1193–1210.
- Cordier JF, Valeyre D, Guillevin L, Loire R, Brechot JM. Pulmonary Wegener’s granulomatosis. A clinical and imaging study of 77 cases. Chest 1990; 97:906–912.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener’s granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med 1983; 98:76–85.
- Reinhold-Keller E, Beuge N, Latza U, et al. An interdisciplinary approach to the care of patients with Wegener’s granulomatosis: long-term outcome in 155 patients. Arthritis Rheum 2000; 43:1021–1032.
- Langford CA, Hoffman GS. Rare diseases 3: Wegener’s granulomatosis. Thorax 1999; 54:629–637.
- Mark EJ, Matsubara O, Tan-Liu NS, Fienberg R. The pulmonary biopsy in the early diagnosis of Wegener’s (pathergic) granulomatosis: a study based on 35 open lung biopsies. Hum Pathol 1988; 19:1065–1071.
- Sheehan RE, Flint JD, Muller NL. Computed tomography features of the thoracic manifestations of Wegener granulomatosis. J Thorac Imaging 2003: 18:34–41.
- Specks USchwarz MI, King TE. Pulmonary vasculitis. Interstitial Lung Disease. Decker BC. Hamilton, Ontario, Canada: Decker, 2003:599–631.
- Ten Berge IJ, Wilmink JM, Meyer CJ, et al. Clinical and immunological follow-up of patients with severe renal disease in Wegener’s granulo-matosis. Am J Nephrol 1985; 5:21–29.
- Brandwein S, Esdaile J, Danoff D, Tannenbaum H. Wegener’s granulo-matosis. Clinical features and outcome in 13 patients. Arch Intern Med 1983; 143:476–479.
- Pinching AJ, Lockwood CM, Pussell BA, et al. Wegener’s granulomatosis: observations on 18 patients with severe renal disease. Q J Med 1983; 52:435–460.
- Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med 1997; 337:1512–1523.
- Lauque D, Cadranel J, Lazor R, et al. Microscopic polyangiitis with alveolar hemorrhage. A study of 29 cases and review of the literature. Groupe d’Études et de Recherche sur les Maladies “Orphelines” Pulmonaires. Medicine (Baltimore) 2000; 79:222–233.
- Johnson JP, Moore J, Austin HA, Balow JE, Antonovych TT, Wilson CB. Therapy of anti-glomerular basement membrane antibody disease: analysis of prognostic significance of clinical, pathologic and treatment factors. Medicine (Baltimore) 1985; 64:219–227.
- Savage CO, Winearls CG, Evans DJ, Rees AJ, Lockwood CM. Microscopic polyarteritis: presentation, pathology, and prognosis. Q J Med 1985; 56:467–483.
- Haworth SJ, Savage CO, Carr D. Pulmonary hemorrhage complicating Wegener’s granulomatosis and microscopic polyarteritis. Br Med J 1985; 290:1175–1178.
- Smyth L, Gaskin G, Pusey CD. Microscopic polyangiitis. Semin Respir Crit Care Med 2004; 25:523–533.
- Lanham JG, Elkon KB, Pusey CD, Hughes GR. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine (Baltimore) 1984; 63:65–81.
- Leatherman JW. Autoimmune diffuse alveolar hemorrhage. Clin Pulm Med 1994; 1:356–364.
- Boyce NW, Holdsworth SR. Pulmonary manifestations of the clinical syndrome of acute glomerulonephritis and lung hemorrhage. Am J Kidney Dis 1986; 8:31–36.
- Emlen W. Systemic lupus erythematosus and mixed connective tissue disease. Immunol Allergy Clin North Am 1979; 105:291–311.
- Hunninghake GW, Fauci AS. Pulmonary involvement in the collagen vascular diseases. Am Rev Respir Dis 1979; 119:471–503.
- Keane MP, Lynch JP. Pleuropulmonary manifestations of systemic lupus erythematosus. Thorax 2000; 55:159–166.
- Zamora MR, Warner ML, Tuder R, Schwarz MI. Diffuse alveolar hemorrhage and systemic lupus erythematosus. Clinical presentation, histology, survival, and outcome. Medicine (Baltimore) 1997; 76:192–202.
- Lee CK, Koh JH, Cha HS, et al. Pulmonary alveolar hemorrhage in patients with rheumatic diseases in Korea. Scand J Rheumatol 2000; 29:288–294.
- Vazquez-Del Mercado M, Mendoza-Topete A, Best-Aguilera CR, Garcia-De La Torre I. Diffuse alveolar hemorrhage in limited cutaneous systemic sclerosis with positive perinuclear antineutrophil cytoplasmic antibodies. J Rheumatol 1996; 23:1821–1823.
- Fenlon HM, Doran M, Sant SM, Breatnach E. High-resolution chest CT in systemic lupus erythematosus. AJR Am J Roentgenol 1996; 166:301–307.
- Ioachimescu OC. Idiopathic pulmonary hemosiderosis in adults. Pneumologia 2003; 52:38–43.
- Ioachimescu OC, Sieber S, Kotch A. Idiopathic pulmonary haemosiderosis revisited. Eur Respir J 2004; 24:162–170.
- Franks TJ, Koss MN. Pulmonary capillaritis. Curr Opin Pulm Med 2000; 6:430–435.
- Travis WD, Hoffman GS, Leavitt RY, Pass HI, Fauci AS. Surgical pathology of the lung in Wegener’s granulomatosis. Review of 87 open lung biopsies from 67 patients. Am J Surg Pathol 1991; 15:315–333.
- Zashin S, Fattor R, Fortin D. Microscopic polyarteritis: a forgotten aetiology of haemoptysis and rapidly progressive glomerulonephritis. Ann Rheum Dis 1990; 49:53–56.
- Yoshikawa Y, Watanabe T. Pulmonary lesions in Wegener’s granulo-matosis: a clinicopathologic study of 22 autopsy cases. Hum Pathol 1986; 17:401–410.
- Teague CA, Doak PB, Simpson IJ, Rainer SP, Herdson PB. Goodpasture’s syndrome: an analysis of 29 cases. Kidney Int 1978; 13:492–504.
- Abu-Shakra M, Smythe H, Lewtas J, Badley E, Weber D, Keystone E. Outcome of polyarteritis nodosa and Churg-Strauss syndrome. An analysis of twenty-five patients. Arthritis Rheum 1994; 37:1798–1803.
- Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore) 1999; 78:26–37.
- Schwab EP, Schumacher HR, Freundlich B, Callegari PE. Pulmonary alveolar hemorrhage in systemic lupus erythematosus. Semin Arthritis Rheum 1993; 23:8–15.
- Koh WH, Thumboo J, Boey ML. Pulmonary haemorrhage in Oriental patients with systemic lupus erythematosus. Lupus 1997; 6:713–716.
- Ioachimescu OCLaurent GL, Shapiro SD. Alveolar hemorrhage. Encyclopedia of Respiratory Medicine. Amsterdam: Academic Press, 2006:92–100.
- Travis WD, Colby TV, Lombard C, Carpenter HA. A clinicopathologic study of 34 cases of diffuse pulmonary hemorrhage with lung biopsy confirmation. Am J Surg Pathol 1990; 14:1112–1125.
- Jennings CA, King TE, Tuder R, Cherniak RM, Schwarz MI. Diffuse alveolar hemorrhage with underlying isolated, pauciimmune pulmonary capillaritis. Am J Respir Crit Care Med 1997; 155:1101–1109.
- Spencer H. Pulmonary lesions in polyarteritis nodosa. Br J Tuberc Dis Chest 1957; 51:123–130.
- Travis WD. Pathology of pulmonary vasculitis. Semin Respir Crit Care Med 2004; 25:475–482.
- Schwarz MI, Brown KK. Small vessel vasculitis of the lung. Thorax 2000; 55:502–510.
- Collard HR, Schwarz MI. Diffuse alveolar hemorrhage. Clin Chest Med 2004; 25:583–592.
- Ioachimescu OC, Jennings C. Intercostal lung cyst hernia in idiopathic pulmonary hemosiderosis (cyst necessitans). Mayo Clin Proc 2006; 81:692.
- Rolla G, Heffler E, Guida G, Bergia R, Bucca C. Exhaled NO in diffuse alveolar haemorrhage. Thorax 2005; 60:614–615.
- Dweik RA, Stoller JK. Role of bronchoscopy in massive hemoptysis. Clin Chest Med 1999; 20:89–105.
- Ioachimescu OCLaurent GL, Shapiro SD. Autoantibodies. Encyclopedia of Respiratory Medicine. Amsterdam: Academic Press, 2006:219–227.
- Watts RA, Carruthers DM, Scott DG. Epidemiology of systemic vasculitis: changing incidence or definition? Semin Arthritis Rheum 1995; 25:28–34.
- Watts RA, Lane SE, Bentham G, Scott DG. Epidemiology of systemic vasculitis: a ten-year study in the United Kingdom. Arthritis Rheum 2000; 43:414–419.
- Watts RA, Jolliffe VA, Carruthers DM, Lockwood M, Scott DG. Effect of classification on the incidence of polyarteritis nodosa and microscopic polyangiitis. Arthritis Rheum 1996; 39:1208–1212.
- Ioachimescu OC, Kotch A, Stoller JK. Idiopathic pulmonary hemosiderosis in adults. Clin Pulm Med 2005; 12:16–25.
- Reinhold-Keller E, Herlyn K, Wagner-Bastmeyer R, et al. No difference in the incidences of vasculitides between north and south Germany: first results of the German vasculitis register. Rheumatology (Oxford) 2002; 41:540–549.
- Mahr A, Guillevin L, Poissonnet M, Ayme S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener’s granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum 2004; 51:92–99.
- Koldingsnes W, Nossent H. Epidemiology of Wegener’s granulomatosis in northern Norway. Arthritis Rheum 2000; 43:2481–2487.
- Kelly PT, Haponik EF. Goodpasture syndrome: molecular and clinical advances. Medicine (Baltimore) 1994; 73:171–185.
- Travis WD, Leslie KOLeslie KO, Wick MR. Pulmonary vasculitis and pulmonary hemorhage. Practical Pulmonary Pathology – a Diagnostic Approach. Philadelphia: Churchill Livingstone-Elsevier, 2005;335–378.
- Jennette JC, Thomas DB, Falk RJ. Microscopic polyangiitis (microscopic polyarteritis). Semin Diagn Pathol 2001; 18:3–13.
- Katzenstein AKatzenstein A, Askin F. Alveolar hemorrhage syndromes. Surgical Pathology of Non-neoplastic Lung Disease. Philadelphia: WB Saunders, 1997:153–159.
- Schwarz MI, Cherniack RM, King TEMurray JF, Nadel J. Diffuse alveolar hemorrhage and other rare infiltrative disorders. Textbook of Respiratory Medicine. Philadelphia: WB Saunders, 2000:1733–1755.
- Lynch JP, Leatherman JWFishman A. Alveolar hemorrhage syndromes. Fishman’s Pulmonary Diseases and Disorders. New York: McGraw-Hill, 1998:1193–1210.
- Cordier JF, Valeyre D, Guillevin L, Loire R, Brechot JM. Pulmonary Wegener’s granulomatosis. A clinical and imaging study of 77 cases. Chest 1990; 97:906–912.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener’s granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med 1983; 98:76–85.
- Reinhold-Keller E, Beuge N, Latza U, et al. An interdisciplinary approach to the care of patients with Wegener’s granulomatosis: long-term outcome in 155 patients. Arthritis Rheum 2000; 43:1021–1032.
- Langford CA, Hoffman GS. Rare diseases 3: Wegener’s granulomatosis. Thorax 1999; 54:629–637.
- Mark EJ, Matsubara O, Tan-Liu NS, Fienberg R. The pulmonary biopsy in the early diagnosis of Wegener’s (pathergic) granulomatosis: a study based on 35 open lung biopsies. Hum Pathol 1988; 19:1065–1071.
- Sheehan RE, Flint JD, Muller NL. Computed tomography features of the thoracic manifestations of Wegener granulomatosis. J Thorac Imaging 2003: 18:34–41.
- Specks USchwarz MI, King TE. Pulmonary vasculitis. Interstitial Lung Disease. Decker BC. Hamilton, Ontario, Canada: Decker, 2003:599–631.
- Ten Berge IJ, Wilmink JM, Meyer CJ, et al. Clinical and immunological follow-up of patients with severe renal disease in Wegener’s granulo-matosis. Am J Nephrol 1985; 5:21–29.
- Brandwein S, Esdaile J, Danoff D, Tannenbaum H. Wegener’s granulo-matosis. Clinical features and outcome in 13 patients. Arch Intern Med 1983; 143:476–479.
- Pinching AJ, Lockwood CM, Pussell BA, et al. Wegener’s granulomatosis: observations on 18 patients with severe renal disease. Q J Med 1983; 52:435–460.
- Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med 1997; 337:1512–1523.
- Lauque D, Cadranel J, Lazor R, et al. Microscopic polyangiitis with alveolar hemorrhage. A study of 29 cases and review of the literature. Groupe d’Études et de Recherche sur les Maladies “Orphelines” Pulmonaires. Medicine (Baltimore) 2000; 79:222–233.
- Johnson JP, Moore J, Austin HA, Balow JE, Antonovych TT, Wilson CB. Therapy of anti-glomerular basement membrane antibody disease: analysis of prognostic significance of clinical, pathologic and treatment factors. Medicine (Baltimore) 1985; 64:219–227.
- Savage CO, Winearls CG, Evans DJ, Rees AJ, Lockwood CM. Microscopic polyarteritis: presentation, pathology, and prognosis. Q J Med 1985; 56:467–483.
- Haworth SJ, Savage CO, Carr D. Pulmonary hemorrhage complicating Wegener’s granulomatosis and microscopic polyarteritis. Br Med J 1985; 290:1175–1178.
- Smyth L, Gaskin G, Pusey CD. Microscopic polyangiitis. Semin Respir Crit Care Med 2004; 25:523–533.
- Lanham JG, Elkon KB, Pusey CD, Hughes GR. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine (Baltimore) 1984; 63:65–81.
- Leatherman JW. Autoimmune diffuse alveolar hemorrhage. Clin Pulm Med 1994; 1:356–364.
- Boyce NW, Holdsworth SR. Pulmonary manifestations of the clinical syndrome of acute glomerulonephritis and lung hemorrhage. Am J Kidney Dis 1986; 8:31–36.
- Emlen W. Systemic lupus erythematosus and mixed connective tissue disease. Immunol Allergy Clin North Am 1979; 105:291–311.
- Hunninghake GW, Fauci AS. Pulmonary involvement in the collagen vascular diseases. Am Rev Respir Dis 1979; 119:471–503.
- Keane MP, Lynch JP. Pleuropulmonary manifestations of systemic lupus erythematosus. Thorax 2000; 55:159–166.
- Zamora MR, Warner ML, Tuder R, Schwarz MI. Diffuse alveolar hemorrhage and systemic lupus erythematosus. Clinical presentation, histology, survival, and outcome. Medicine (Baltimore) 1997; 76:192–202.
- Lee CK, Koh JH, Cha HS, et al. Pulmonary alveolar hemorrhage in patients with rheumatic diseases in Korea. Scand J Rheumatol 2000; 29:288–294.
- Vazquez-Del Mercado M, Mendoza-Topete A, Best-Aguilera CR, Garcia-De La Torre I. Diffuse alveolar hemorrhage in limited cutaneous systemic sclerosis with positive perinuclear antineutrophil cytoplasmic antibodies. J Rheumatol 1996; 23:1821–1823.
- Fenlon HM, Doran M, Sant SM, Breatnach E. High-resolution chest CT in systemic lupus erythematosus. AJR Am J Roentgenol 1996; 166:301–307.
- Ioachimescu OC. Idiopathic pulmonary hemosiderosis in adults. Pneumologia 2003; 52:38–43.
- Ioachimescu OC, Sieber S, Kotch A. Idiopathic pulmonary haemosiderosis revisited. Eur Respir J 2004; 24:162–170.
- Franks TJ, Koss MN. Pulmonary capillaritis. Curr Opin Pulm Med 2000; 6:430–435.
- Travis WD, Hoffman GS, Leavitt RY, Pass HI, Fauci AS. Surgical pathology of the lung in Wegener’s granulomatosis. Review of 87 open lung biopsies from 67 patients. Am J Surg Pathol 1991; 15:315–333.
- Zashin S, Fattor R, Fortin D. Microscopic polyarteritis: a forgotten aetiology of haemoptysis and rapidly progressive glomerulonephritis. Ann Rheum Dis 1990; 49:53–56.
- Yoshikawa Y, Watanabe T. Pulmonary lesions in Wegener’s granulo-matosis: a clinicopathologic study of 22 autopsy cases. Hum Pathol 1986; 17:401–410.
- Teague CA, Doak PB, Simpson IJ, Rainer SP, Herdson PB. Goodpasture’s syndrome: an analysis of 29 cases. Kidney Int 1978; 13:492–504.
- Abu-Shakra M, Smythe H, Lewtas J, Badley E, Weber D, Keystone E. Outcome of polyarteritis nodosa and Churg-Strauss syndrome. An analysis of twenty-five patients. Arthritis Rheum 1994; 37:1798–1803.
- Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore) 1999; 78:26–37.
- Schwab EP, Schumacher HR, Freundlich B, Callegari PE. Pulmonary alveolar hemorrhage in systemic lupus erythematosus. Semin Arthritis Rheum 1993; 23:8–15.
- Koh WH, Thumboo J, Boey ML. Pulmonary haemorrhage in Oriental patients with systemic lupus erythematosus. Lupus 1997; 6:713–716.
KEY POINTS
- Most patients present with dyspnea, cough, hemoptysis, and new alveolar infiltrates. Early bronchoscopy with bronchoalveolar lavage is generally required to confirm the diagnosis; blood in the lavage specimens (with numerous erythrocytes and siderophages) establishes the diagnosis.
- Therapy targets both the autoimmune destruction of the alveolar capillary membrane and the underlying condition. Corticosteroids and immunosuppressive agents remain the gold standard.
- In patients with diffuse alveolar hemorrhage and renal impairment (pulmonary-renal syndrome), kidney biopsy can be considered to identify the cause and to direct therapy.