User login
Case
A 76-year-old man with Parkinson disease (PD) and hypertension presented to the ED with acute onset of severe tremulousness, blurred vision, salivation, lacrimation, diffuse muscle aches, and extremity weakness. His initial vital signs were: blood pressure, 175/74 mm Hg; heart rate, 62 beats/minute; respiratory rate, 16 breaths/minute; temperature, 37°C (98.6°F). Oxygen saturation was 100% on room air. On physical examination, the patient had excessive lacrimation and salivation, a coarse resting tremor, and 2/5 strength in both the upper and lower extremities. The remainder of the examination, including abdominal and pulmonary systems, was unremarkable compared with baseline findings.
How does the pathophysiology of PD explain how treatments are targeted?

What medications are used to treat PD? What are some associated complications?
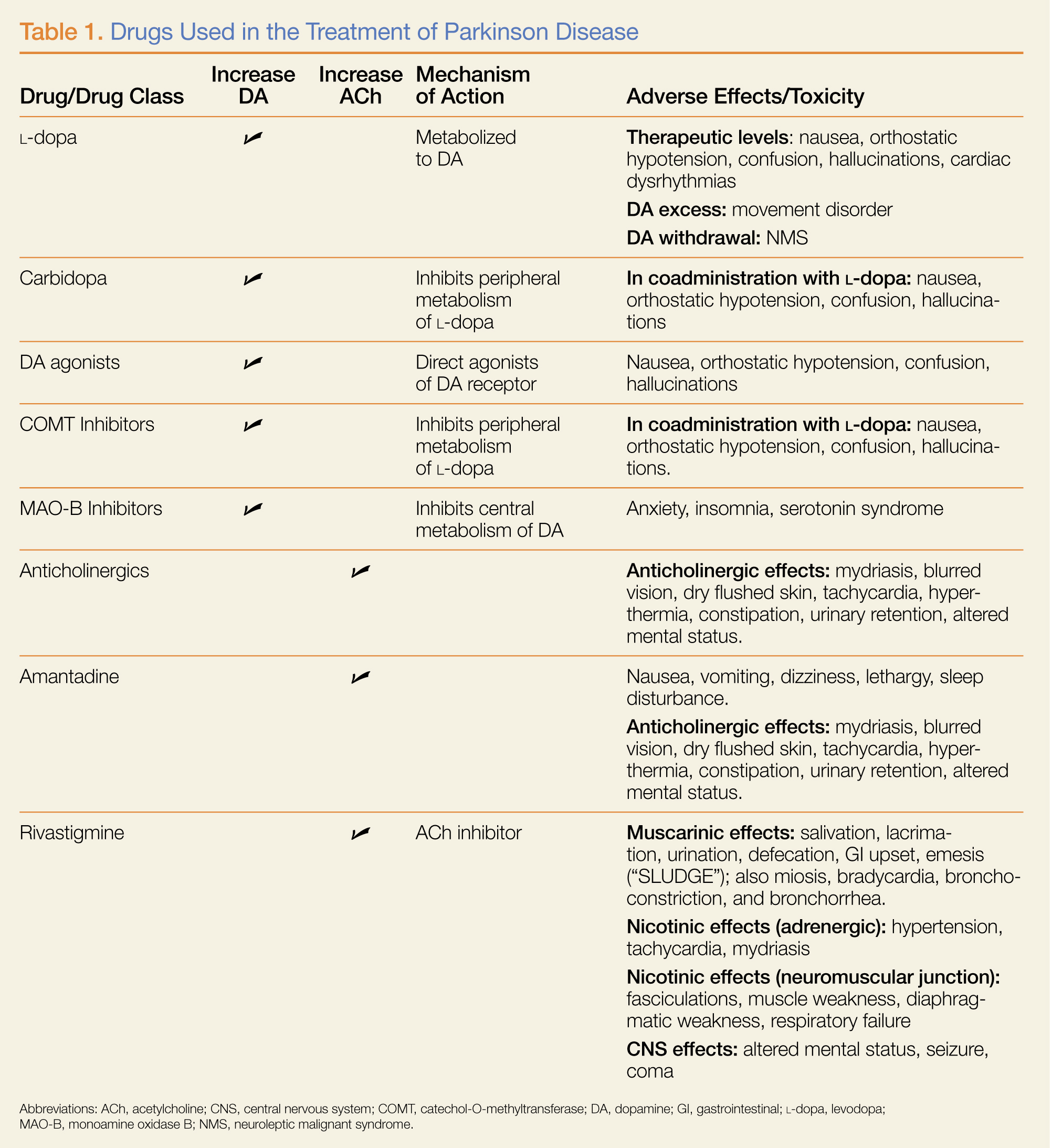
Dopamine Precursors and Agonists
(L-dopa) can be combined with the L-amino acid decarboxylase inhibitor carbidopa to prevent peripheral metabolism by this enzyme and thereby increase brain concentrations of DA following metabolism by DA decarboxylase in the central nervous system (CNS).1 Dopamine agonists, including bromocriptine, ropinirole, and pramipexole, do not depend on endogenous conversion to DA and have substantially longer durations of action, limiting the dose-related fluctuations in motor function common in some PD patients taking L-dopa.1 For these reasons, DA agonists have often replaced L-dopa as initial treatment, especially in younger patients. Catechol-O-methyltransferase inhibitors (tolcapone, entacapone) prevent peripheral breakdown of DA, allowing a higher fraction to reach the CNS.
With respect to side effects, all of the dopaminergic medications can cause nausea, hallucinations, confusion, and orthostatic hypotension.
Anticholinergic Drugs
Although the precise mechanism by which anticholinergic drugs improve PD is not fully understood, agents such as trihexyphenidyl, benztropine mesylate, and diphenhydramine hydrochloride were prescribed even before the discovery of L-dopa and continue to be used today.1 Adverse effects are a function of the antimuscarinic (anticholinergic) properties of the drugs and may include mydriasis and blurred vision, dry flushed skin, tachycardia, hyperthermia, constipation, urinary retention, and altered mental status.
Amantadine
In addition to the anticholinergics, amantadine is also used to treat PD. This antiviral agent alters DA release in the brain, produces anticholinergic effects, and blocks N-methyl-D-aspartate glutamate receptors.1 Common adverse drug effects include anticholinergic signs as well as nausea, vomiting, dizziness, lethargy, and sleep disturbance, all of which are usually mild and reversible.
Case Continuation
A review of the patient’s medication history revealed he has been taking L-dopa/carbidopa. In addition to L-dopa/carbidopa, he was recently prescribed transdermal rivastigmine patches (13.3 mg/24 h). At bedtime the evening prior to presentation, the patient applied more than 20 rivastigmine patches. Approximately 5 hours later, he awoke with the previously described findings whereupon his wife removed the patches and brought him to the ED.
What is rivastigmine and what is its role in PD
Rivastigmine is a carbamate-type cholinesterase inhibitor (CEI) indicated for the treatment of mild-to-moderate dementia associated with PD and Alzheimer disease.2 Tacrine, a medicinal noncarbamate CEI, is also prescribed for this use.2 Both drugs increase ACh concentrations in relevant brain regions and foster the formation of new memory.
Cholinesterase inhibitors are mechanistically analogous to the insecticidal carbamates (eg, aldicarb) and the organophosphates (OPs) (eg, malathion). They inhibit the metabolism of ACh by acetylcholinesterase (AChE) in the various cholinergic synapses, increasing the intrasynaptic concentration of ACh.
Additional AChEs include physostigmine, a carbamate commonly used in the ED to treat anticholinergic toxicity. Physostigmine raises the local synaptic concentration of ACh to compete for the muscarinic ACh receptor with drugs such as diphenhydramine or atropine. Other CEIs (eg, neostigmine, pyridostigmine, edrophonium) are used to raise intrasynaptic ACh concentrations and overcome antibody blockade of nicotinic ACh receptors at the neuromuscular junction in patients with myasthenia gravis.
What is the toxidrome associated with carbamate overdose
Carbamate toxicity, as manifested by the cholinergic toxidrome, largely resembles OP toxicity but with an important difference: Both OPs and carbamates function by binding to and inhibiting AChE; however, the carbamate-AChE bond undergoes spontaneous hydrolysis, thereby reactivating the enzyme. Consequently, the clinical effects of carbamate toxicity, though potentially severe, are self-limited and usually only last 24 hours or less.4
How should this patient be managed?
The general approach to a patient with medical carbamate toxicity is similar to that of a patient with OP poisoning. Dermal exposure, as is the case with this patient, should prompt skin decontamination to minimize ongoing exposure. Patch removal is necessary but is not sufficient to prevent ongoing absorption, since a depot of medication typically forms in the dermal tissue. In the presence of significant or life-threatening muscarinic effects (eg, bronchorrhea, bronchospasm, seizure), an antimuscarinic agent such as atropine is indicated. Various dosing schemes of atropine exist; at our institution, we recommend an initial dose of 1 to 3 mg intravenously (IV), with escalating doses every 5 minutes until reversal of bronchorrhea and bronchospasm occur.4 This is followed by initiation of an atropine infusion at a rate of 10% to 20% of the total loading dose per hour (to a maximum of 2 mg/h).4
Pralidoxime (2-PAM) and other oximes, accelerate the reactivation of carbamate-inhibited AChE and have effects at both the nicotinic and muscarinic synapses. Reactivation results in the enhanced metabolism of intrasynaptic ACh and decreased clinical cholinergic effects. Since atropine is only effective at muscarinic receptors, oximes were administered in this case to reverse neuromuscular weakness.
Although early administration of 2-PAM is indicated in the setting of significant OP poisoning (due to irreversible inhibition of AChE), its use for medical carbamate toxicity is controversial. Early animal studies of carbamate toxicity suggested that treatment with oximes worsened outcomes; however, this has not been demonstrated in more recent studies.5,6 Therefore, although 2-PAM may be beneficial in treating cases of clinically significant carbamate poisoning (which can be prolonged and severe), these benefits should be weighed against the potential risks.
Case Conclusion
Upon arrival to the ED, the patient’s skin was cleansed thoroughly. As he did not exhibit muscarinic findings of bradycardia, bronchoconstriction, or bronchorrhea, atropine was not indicated. He was treated conservatively with IV fluid hydration and admitted to the medicine floor. Since he continued to exhibit profound extremity weakness with no improvement 12 hours from the onset of symptoms, pralidoxime 1 g IV was administered over a 30-minute period. Shortly thereafter, patient’s motor strength improved from 2/5 to 4/5 in both upper and lower extremities. No complications were noted, and the patient‘s weakness and tremulousness continued to resolve. He was transferred to a skilled nursing facility on hospital day 6.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Standaert DG, Roberson ED. Treatment of central nervous system degenerative disorders. In: Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill; 2011:609-628
- Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318(7184):633-638.
- Exelon Patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2013.
- Eddleston M, Clark RF. Insecticides: organic phosphorus compounds and carbamates. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:1450-1466.
- Natoff IL, Reiff B. Effect of oximes on the acute toxicity of anticholinesterase carbamates. Toxicol Appl Pharmacol. 1973;25(4):569-575.
- Mercurio-Zappala M, Hack JB, Salvador A, Hoffman RS. Pralidoxime in carbaryl poisoning: an animal model. Hum Exp Toxicol. 2007;26(2)125-129.
Case
A 76-year-old man with Parkinson disease (PD) and hypertension presented to the ED with acute onset of severe tremulousness, blurred vision, salivation, lacrimation, diffuse muscle aches, and extremity weakness. His initial vital signs were: blood pressure, 175/74 mm Hg; heart rate, 62 beats/minute; respiratory rate, 16 breaths/minute; temperature, 37°C (98.6°F). Oxygen saturation was 100% on room air. On physical examination, the patient had excessive lacrimation and salivation, a coarse resting tremor, and 2/5 strength in both the upper and lower extremities. The remainder of the examination, including abdominal and pulmonary systems, was unremarkable compared with baseline findings.
How does the pathophysiology of PD explain how treatments are targeted?
What medications are used to treat PD? What are some associated complications?
Dopamine Precursors and Agonists
(L-dopa) can be combined with the L-amino acid decarboxylase inhibitor carbidopa to prevent peripheral metabolism by this enzyme and thereby increase brain concentrations of DA following metabolism by DA decarboxylase in the central nervous system (CNS).1 Dopamine agonists, including bromocriptine, ropinirole, and pramipexole, do not depend on endogenous conversion to DA and have substantially longer durations of action, limiting the dose-related fluctuations in motor function common in some PD patients taking L-dopa.1 For these reasons, DA agonists have often replaced L-dopa as initial treatment, especially in younger patients. Catechol-O-methyltransferase inhibitors (tolcapone, entacapone) prevent peripheral breakdown of DA, allowing a higher fraction to reach the CNS.
With respect to side effects, all of the dopaminergic medications can cause nausea, hallucinations, confusion, and orthostatic hypotension.
Anticholinergic Drugs
Although the precise mechanism by which anticholinergic drugs improve PD is not fully understood, agents such as trihexyphenidyl, benztropine mesylate, and diphenhydramine hydrochloride were prescribed even before the discovery of L-dopa and continue to be used today.1 Adverse effects are a function of the antimuscarinic (anticholinergic) properties of the drugs and may include mydriasis and blurred vision, dry flushed skin, tachycardia, hyperthermia, constipation, urinary retention, and altered mental status.
Amantadine
In addition to the anticholinergics, amantadine is also used to treat PD. This antiviral agent alters DA release in the brain, produces anticholinergic effects, and blocks N-methyl-D-aspartate glutamate receptors.1 Common adverse drug effects include anticholinergic signs as well as nausea, vomiting, dizziness, lethargy, and sleep disturbance, all of which are usually mild and reversible.
Case Continuation
A review of the patient’s medication history revealed he has been taking L-dopa/carbidopa. In addition to L-dopa/carbidopa, he was recently prescribed transdermal rivastigmine patches (13.3 mg/24 h). At bedtime the evening prior to presentation, the patient applied more than 20 rivastigmine patches. Approximately 5 hours later, he awoke with the previously described findings whereupon his wife removed the patches and brought him to the ED.
What is rivastigmine and what is its role in PD
Rivastigmine is a carbamate-type cholinesterase inhibitor (CEI) indicated for the treatment of mild-to-moderate dementia associated with PD and Alzheimer disease.2 Tacrine, a medicinal noncarbamate CEI, is also prescribed for this use.2 Both drugs increase ACh concentrations in relevant brain regions and foster the formation of new memory.
Cholinesterase inhibitors are mechanistically analogous to the insecticidal carbamates (eg, aldicarb) and the organophosphates (OPs) (eg, malathion). They inhibit the metabolism of ACh by acetylcholinesterase (AChE) in the various cholinergic synapses, increasing the intrasynaptic concentration of ACh.
Additional AChEs include physostigmine, a carbamate commonly used in the ED to treat anticholinergic toxicity. Physostigmine raises the local synaptic concentration of ACh to compete for the muscarinic ACh receptor with drugs such as diphenhydramine or atropine. Other CEIs (eg, neostigmine, pyridostigmine, edrophonium) are used to raise intrasynaptic ACh concentrations and overcome antibody blockade of nicotinic ACh receptors at the neuromuscular junction in patients with myasthenia gravis.
What is the toxidrome associated with carbamate overdose
Carbamate toxicity, as manifested by the cholinergic toxidrome, largely resembles OP toxicity but with an important difference: Both OPs and carbamates function by binding to and inhibiting AChE; however, the carbamate-AChE bond undergoes spontaneous hydrolysis, thereby reactivating the enzyme. Consequently, the clinical effects of carbamate toxicity, though potentially severe, are self-limited and usually only last 24 hours or less.4
How should this patient be managed?
The general approach to a patient with medical carbamate toxicity is similar to that of a patient with OP poisoning. Dermal exposure, as is the case with this patient, should prompt skin decontamination to minimize ongoing exposure. Patch removal is necessary but is not sufficient to prevent ongoing absorption, since a depot of medication typically forms in the dermal tissue. In the presence of significant or life-threatening muscarinic effects (eg, bronchorrhea, bronchospasm, seizure), an antimuscarinic agent such as atropine is indicated. Various dosing schemes of atropine exist; at our institution, we recommend an initial dose of 1 to 3 mg intravenously (IV), with escalating doses every 5 minutes until reversal of bronchorrhea and bronchospasm occur.4 This is followed by initiation of an atropine infusion at a rate of 10% to 20% of the total loading dose per hour (to a maximum of 2 mg/h).4
Pralidoxime (2-PAM) and other oximes, accelerate the reactivation of carbamate-inhibited AChE and have effects at both the nicotinic and muscarinic synapses. Reactivation results in the enhanced metabolism of intrasynaptic ACh and decreased clinical cholinergic effects. Since atropine is only effective at muscarinic receptors, oximes were administered in this case to reverse neuromuscular weakness.
Although early administration of 2-PAM is indicated in the setting of significant OP poisoning (due to irreversible inhibition of AChE), its use for medical carbamate toxicity is controversial. Early animal studies of carbamate toxicity suggested that treatment with oximes worsened outcomes; however, this has not been demonstrated in more recent studies.5,6 Therefore, although 2-PAM may be beneficial in treating cases of clinically significant carbamate poisoning (which can be prolonged and severe), these benefits should be weighed against the potential risks.
Case Conclusion
Upon arrival to the ED, the patient’s skin was cleansed thoroughly. As he did not exhibit muscarinic findings of bradycardia, bronchoconstriction, or bronchorrhea, atropine was not indicated. He was treated conservatively with IV fluid hydration and admitted to the medicine floor. Since he continued to exhibit profound extremity weakness with no improvement 12 hours from the onset of symptoms, pralidoxime 1 g IV was administered over a 30-minute period. Shortly thereafter, patient’s motor strength improved from 2/5 to 4/5 in both upper and lower extremities. No complications were noted, and the patient‘s weakness and tremulousness continued to resolve. He was transferred to a skilled nursing facility on hospital day 6.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 76-year-old man with Parkinson disease (PD) and hypertension presented to the ED with acute onset of severe tremulousness, blurred vision, salivation, lacrimation, diffuse muscle aches, and extremity weakness. His initial vital signs were: blood pressure, 175/74 mm Hg; heart rate, 62 beats/minute; respiratory rate, 16 breaths/minute; temperature, 37°C (98.6°F). Oxygen saturation was 100% on room air. On physical examination, the patient had excessive lacrimation and salivation, a coarse resting tremor, and 2/5 strength in both the upper and lower extremities. The remainder of the examination, including abdominal and pulmonary systems, was unremarkable compared with baseline findings.
How does the pathophysiology of PD explain how treatments are targeted?
What medications are used to treat PD? What are some associated complications?
Dopamine Precursors and Agonists
(L-dopa) can be combined with the L-amino acid decarboxylase inhibitor carbidopa to prevent peripheral metabolism by this enzyme and thereby increase brain concentrations of DA following metabolism by DA decarboxylase in the central nervous system (CNS).1 Dopamine agonists, including bromocriptine, ropinirole, and pramipexole, do not depend on endogenous conversion to DA and have substantially longer durations of action, limiting the dose-related fluctuations in motor function common in some PD patients taking L-dopa.1 For these reasons, DA agonists have often replaced L-dopa as initial treatment, especially in younger patients. Catechol-O-methyltransferase inhibitors (tolcapone, entacapone) prevent peripheral breakdown of DA, allowing a higher fraction to reach the CNS.
With respect to side effects, all of the dopaminergic medications can cause nausea, hallucinations, confusion, and orthostatic hypotension.
Anticholinergic Drugs
Although the precise mechanism by which anticholinergic drugs improve PD is not fully understood, agents such as trihexyphenidyl, benztropine mesylate, and diphenhydramine hydrochloride were prescribed even before the discovery of L-dopa and continue to be used today.1 Adverse effects are a function of the antimuscarinic (anticholinergic) properties of the drugs and may include mydriasis and blurred vision, dry flushed skin, tachycardia, hyperthermia, constipation, urinary retention, and altered mental status.
Amantadine
In addition to the anticholinergics, amantadine is also used to treat PD. This antiviral agent alters DA release in the brain, produces anticholinergic effects, and blocks N-methyl-D-aspartate glutamate receptors.1 Common adverse drug effects include anticholinergic signs as well as nausea, vomiting, dizziness, lethargy, and sleep disturbance, all of which are usually mild and reversible.
Case Continuation
A review of the patient’s medication history revealed he has been taking L-dopa/carbidopa. In addition to L-dopa/carbidopa, he was recently prescribed transdermal rivastigmine patches (13.3 mg/24 h). At bedtime the evening prior to presentation, the patient applied more than 20 rivastigmine patches. Approximately 5 hours later, he awoke with the previously described findings whereupon his wife removed the patches and brought him to the ED.
What is rivastigmine and what is its role in PD
Rivastigmine is a carbamate-type cholinesterase inhibitor (CEI) indicated for the treatment of mild-to-moderate dementia associated with PD and Alzheimer disease.2 Tacrine, a medicinal noncarbamate CEI, is also prescribed for this use.2 Both drugs increase ACh concentrations in relevant brain regions and foster the formation of new memory.
Cholinesterase inhibitors are mechanistically analogous to the insecticidal carbamates (eg, aldicarb) and the organophosphates (OPs) (eg, malathion). They inhibit the metabolism of ACh by acetylcholinesterase (AChE) in the various cholinergic synapses, increasing the intrasynaptic concentration of ACh.
Additional AChEs include physostigmine, a carbamate commonly used in the ED to treat anticholinergic toxicity. Physostigmine raises the local synaptic concentration of ACh to compete for the muscarinic ACh receptor with drugs such as diphenhydramine or atropine. Other CEIs (eg, neostigmine, pyridostigmine, edrophonium) are used to raise intrasynaptic ACh concentrations and overcome antibody blockade of nicotinic ACh receptors at the neuromuscular junction in patients with myasthenia gravis.
What is the toxidrome associated with carbamate overdose
Carbamate toxicity, as manifested by the cholinergic toxidrome, largely resembles OP toxicity but with an important difference: Both OPs and carbamates function by binding to and inhibiting AChE; however, the carbamate-AChE bond undergoes spontaneous hydrolysis, thereby reactivating the enzyme. Consequently, the clinical effects of carbamate toxicity, though potentially severe, are self-limited and usually only last 24 hours or less.4
How should this patient be managed?
The general approach to a patient with medical carbamate toxicity is similar to that of a patient with OP poisoning. Dermal exposure, as is the case with this patient, should prompt skin decontamination to minimize ongoing exposure. Patch removal is necessary but is not sufficient to prevent ongoing absorption, since a depot of medication typically forms in the dermal tissue. In the presence of significant or life-threatening muscarinic effects (eg, bronchorrhea, bronchospasm, seizure), an antimuscarinic agent such as atropine is indicated. Various dosing schemes of atropine exist; at our institution, we recommend an initial dose of 1 to 3 mg intravenously (IV), with escalating doses every 5 minutes until reversal of bronchorrhea and bronchospasm occur.4 This is followed by initiation of an atropine infusion at a rate of 10% to 20% of the total loading dose per hour (to a maximum of 2 mg/h).4
Pralidoxime (2-PAM) and other oximes, accelerate the reactivation of carbamate-inhibited AChE and have effects at both the nicotinic and muscarinic synapses. Reactivation results in the enhanced metabolism of intrasynaptic ACh and decreased clinical cholinergic effects. Since atropine is only effective at muscarinic receptors, oximes were administered in this case to reverse neuromuscular weakness.
Although early administration of 2-PAM is indicated in the setting of significant OP poisoning (due to irreversible inhibition of AChE), its use for medical carbamate toxicity is controversial. Early animal studies of carbamate toxicity suggested that treatment with oximes worsened outcomes; however, this has not been demonstrated in more recent studies.5,6 Therefore, although 2-PAM may be beneficial in treating cases of clinically significant carbamate poisoning (which can be prolonged and severe), these benefits should be weighed against the potential risks.
Case Conclusion
Upon arrival to the ED, the patient’s skin was cleansed thoroughly. As he did not exhibit muscarinic findings of bradycardia, bronchoconstriction, or bronchorrhea, atropine was not indicated. He was treated conservatively with IV fluid hydration and admitted to the medicine floor. Since he continued to exhibit profound extremity weakness with no improvement 12 hours from the onset of symptoms, pralidoxime 1 g IV was administered over a 30-minute period. Shortly thereafter, patient’s motor strength improved from 2/5 to 4/5 in both upper and lower extremities. No complications were noted, and the patient‘s weakness and tremulousness continued to resolve. He was transferred to a skilled nursing facility on hospital day 6.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Standaert DG, Roberson ED. Treatment of central nervous system degenerative disorders. In: Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill; 2011:609-628
- Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318(7184):633-638.
- Exelon Patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2013.
- Eddleston M, Clark RF. Insecticides: organic phosphorus compounds and carbamates. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:1450-1466.
- Natoff IL, Reiff B. Effect of oximes on the acute toxicity of anticholinesterase carbamates. Toxicol Appl Pharmacol. 1973;25(4):569-575.
- Mercurio-Zappala M, Hack JB, Salvador A, Hoffman RS. Pralidoxime in carbaryl poisoning: an animal model. Hum Exp Toxicol. 2007;26(2)125-129.
- Standaert DG, Roberson ED. Treatment of central nervous system degenerative disorders. In: Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill; 2011:609-628
- Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318(7184):633-638.
- Exelon Patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2013.
- Eddleston M, Clark RF. Insecticides: organic phosphorus compounds and carbamates. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:1450-1466.
- Natoff IL, Reiff B. Effect of oximes on the acute toxicity of anticholinesterase carbamates. Toxicol Appl Pharmacol. 1973;25(4):569-575.
- Mercurio-Zappala M, Hack JB, Salvador A, Hoffman RS. Pralidoxime in carbaryl poisoning: an animal model. Hum Exp Toxicol. 2007;26(2)125-129.
