User login
Neurologic emergencies such as acute stroke, status epilepticus, subarachnoid hemorrhage, neuromuscular weakness, and spinal cord injury affect millions of Americans yearly.1,2 These conditions can be difficult to diagnose, and delays in recognition and treatment can have devastating results. Consequently, it is important for nonneurologists to be able to quickly recognize these conditions and initiate timely management, often while awaiting neurologic consultation.
Here, we review how to recognize and treat these common, serious conditions.
ACUTE ISCHEMIC STROKE: TIME IS OF THE ESSENCE
Stroke is the fourth leading cause of death in the United States and is one of the most common causes of disability worldwide.3–5 About 85% of strokes are ischemic, resulting from diminished vascular supply to the brain. Symptoms such as facial droop, unilateral weakness or numbness, aphasia, gaze deviation, and unsteadiness of gait may be seen. Time is of the essence, as all currently available interventions are safe and effective only within defined time windows.
Diagnosis and assessment
When acute ischemic stroke is suspected, the clinical history, time of onset, and basic neurologic examination should be obtained quickly.
The National Institutes of Health (NIH) stroke scale is an objective marker for assessing stroke severity as well as evolution of disease and should be obtained in all stroke patients. Scores range from 0 (best) to 42 (worst) (www.ninds.nih.gov/doctors/NIH_Stroke_Scale.pdf).
Time of onset of symptoms is essential to determine, since it guides eligibility for acute therapies. Clinicians should ascertain the last time the patient was seen to be neurologically well in order to estimate this time window as closely as possible.
Laboratory tests should include a fingerstick blood glucose measurement, coagulation studies, complete blood cell count, and basic metabolic profile.
Computed tomography (CT) of the head without contrast should be obtained immediately to exclude acute hemorrhage and any alternative diagnoses that could explain the patient’s symptoms. Acute brain ischemia is often not apparent on CT during the first few hours of injury. Therefore, a patient presenting with new focal neurologic deficits and an unremarkable result on CT of the head should be treated as having had an acute ischemic stroke, and interventional therapies should be considered.

Stroke mimics should be considered and treated, as appropriate (Table 1).
Acute management of ischemic stroke
Acute treatment should not be delayed by obtaining chest radiography, inserting a Foley catheter, or obtaining an electrocardiogram. The longer the time that elapses before treatment, the worse the functional outcome, underscoring the need for rapid decision-making.6–8
Lowering the head of the bed may provide benefit by promoting blood flow to ischemic brain tissue.9 However, this should not be done in patients with significantly elevated intracerebral pressure and concern for herniation.
Permissive hypertension (antihypertensive treatment only for blood pressure greater than 220/110 mm Hg) should be allowed per national guidelines to provide adequate perfusion to brain areas at risk of injury.10
Tissue plasminogen activator. Patients with ischemic stroke who present within 3 hours of symptom onset should be considered for intravenous administration of tissue plasminogen activator (tPA), a safe and effective therapy with nearly 2 decades of evidence to support its use.10 The treating physician should carefully review the risks and benefits of this therapy.
To receive tPA, the patient must have all of the following:
- Clinical diagnosis of ischemic stroke with measurable neurologic deficit
- Onset of symptoms within the past 3 hours
- Age 18 or older.
The patient must not have any of the following:
- Significant stroke within the past 3 months
- Severe traumatic head injury within the past 3 months
- History of significant intracerebral hemorrhage
- Previously ruptured arteriovenous malformation or intracranial aneurysm
- Central nervous system neoplasm
- Arterial puncture at a noncompressible site within the past 7 days
- Evidence of hemorrhage on CT of the head
- Evidence of ischemia in greater than 33% of the cerebral hemisphere on head CT
- History and symptoms strongly suggesting subarachnoid hemorrhage
- Persistent hypertension (systolic pressure ≥ 185 mm Hg or diastolic pressure ≥ 110 mm Hg)
- Evidence of acute significant bleeding (external or internal)
- Hypoglycemia—ie, serum glucose less than 50 mg/dL (< 2.8 mmol/L)
- Thrombocytopenia (platelet count < 100 × 109/L)
- Significant coagulopathy (international normalized ratio > 1.7, prothrombin time > 15 seconds, or abnormally elevated activated partial thromboplastin time)
- Current use of a factor Xa inhibitor or direct thrombin inhibitor.
Relative contraindications:
- Minor or rapidly resolving symptoms
- Major surgery or trauma within the past 14 days
- Gastrointestinal or urinary tract bleeding within the past 21 days
- Myocardial infarction in the past 3 months
- Unruptured intracranial aneurysm
- Seizure occurring at stroke onset
- Pregnancy.
If these criteria are satisfied, tPA should be given at a dose of 0.9 mg/kg intravenously over 60 minutes. Ten percent of the dose should be given as an initial bolus, followed by a constant infusion of the remaining 90% over 1 hour.
If tPA is given, the blood pressure must be kept lower than 185/110 mm Hg to minimize the risk of symptomatic intracerebral hemorrhage.
A subset of patients may benefit from receiving intravenous tPA between 3 and 4.5 hours after the onset of stroke symptoms. These include patients who are no more than 80 years old, who have not recently used oral anticoagulants, who do not have severe neurologic injury (ie, do not have NIH Stroke Scale scores > 25), and who do not have diabetes mellitus or a history of ischemic stroke.11 Although many hospitals have such a protocol for tPA up to 4.5 hours after the onset of stroke symptoms, this time window is not currently approved by the US Food and Drug Administration.
Intra-arterial therapy. Based on recent trials, some patients may benefit further from intra-arterial thrombolysis or mechanical thrombectomy, both delivered during catheter-based cerebral angiography, independent of intravenous tPA administration.12,13 These patients should be evaluated on a case-by-case basis by a neurologist and neurointerventional team. Time windows for these treatments generally extend to 6 hours from stroke onset and perhaps even longer in some situations (eg, basilar artery occlusion).
An antiplatelet agent should be started quickly in all stroke patients who do not receive tPA. Patients who receive tPA can begin receiving an antiplatelet agent 24 hours afterward.
Unfractionated heparin. There is no evidence to support the use of unfractionated heparin in most cases of acute ischemic stroke.10
Glucose control (in the range of 140–180 mg/dL) and fever control remain essential elements of post-acute stroke care to provide additional protection to the damaged brain.
For ischemic stroke due to atrial fibrillation
In ischemic stroke due to atrial fibrillation, early anticoagulation should be considered, based on the CHA2DS2-VASC risk of ischemic stroke vs the HAS-BLED risk of hemorrhage (calculators available at www.mdcalc.com).
In general, anticoagulation may be withheld during the first 72 hours while further stroke workup and evaluation of extent of injury are carried out, as there is an increased risk of hemorrhagic transformation of the ischemic stroke. Often, anticoagulation is resumed at a full dose between 72 hours and 2 weeks of the ischemic stroke.
ACUTE HEMORRHAGIC STROKE: BLOOD PRESSURE, COAGULATION

Approximately 15% of strokes are caused by intracerebral hemorrhage, which can be detected with noncontrast head CT with a sensitivity of 98.6% within 6 hours of the onset of bleeding.14 A common underlying cause of intracerebral hemorrhage is chronic poorly controlled hypertension, causing rupture of damaged (or “lipohyalinized”) vessels with resultant blood extravasation into the brain parenchyma. Other causes are less common (Table 2).
Treatment of acute hemorrhagic stroke
Acute treatment of intracerebral hemorrhage includes blood pressure control, reversal of underlying coagulopathy or anticoagulation, and sometimes intracranial pressure control. There is little role for surgery in most cases, based on findings of randomized trials.15
Blood pressure control. Many studies have investigated optimal blood pressure goals in acute intracerebral hemorrhage. Recent data suggest that early aggressive therapy, targeting a systolic blood pressure goal less than 140 mm Hg within the first hour, is safe and can lead to better functional outcomes than a more conservative blood-pressure-lowering target.16 Rapid-onset, short-acting antihypertensive agents in intravenous form, such as nicardipine and labetalol, are frequently used. Of note, this treatment strategy for hemorrhagic stroke is in direct contrast to the treatment of ischemic stroke, in which permissive hypertension (blood pressure goal < 220/110 mm Hg) is often pursued.
Reversal of any coagulation abnormalities should be done quickly in intracranial hemorrhage. Warfarin use has been shown to be a strong independent predictor of intracranial hemorrhage expansion, which increases the risk of death.17,18
Increasingly, agents other than vitamin K or fresh-frozen plasma are being used to rapidly reverse anticoagulation, including prothrombin complex concentrate (available in three- and four-factor preparations) and recombinant factor VIIa. While four-factor prothrombin complex concentrate and recombinant factor VIIa have been shown to be more efficacious than fresh-frozen plasma, there are limited data directly comparing these newer reversal agents against each other.19 The use of these medications is limited by availability and practitioner familiarity.20–22
Reversing anticoagulation due to target-specific oral anticoagulants. The acute management of intracranial hemorrhage in patients taking the new target-specific oral anticoagulants (eg, dabigatran, apixaban, rivaroxaban, edoxaban) remains challenging. Laboratory tests such as factor Xa levels are not readily available in many institutions and do not provide results in a timely fashion, and in the interim, acute hemorrhage and clinical deterioration may occur. Management strategies involve giving fresh-frozen plasma, prothrombin complex concentrate, and consideration of hemodialysis.23 Dabigatran reversal with idarucizumab has recently been shown to have efficacy.24
Vigilance for elevated intracranial pressure. Intracranial hemorrhage can occasionally cause elevated intracranial pressure, which should be treated rapidly. Any acute decline in mental status in a patient with intracranial hemorrhage requires emergency imaging to evaluate for expansion of hemorrhage.
SUBARACHNOID HEMORRHAGE
The sudden onset of a “thunderclap” headache (often described by patients as “the worst headache of my life”) suggests subarachnoid hemorrhage.
In contrast to intracranial hemorrhage, in subarachnoid hemorrhage blood collects mainly in the cerebral spinal fluid-containing spaces surrounding the brain, leading to a higher incidence of hydrocephalus from impaired drainage of cerebrospinal fluid. Nontraumatic subarachnoid hemorrhage is most often caused by rupture of an intracranial aneurysm, which can be a devastating event, with death rates approaching 50%.25
Diagnosis of subarachnoid hemorrhage
Noncontrast CT of the head is the main modality for diagnosing subarachnoid hemorrhage. Blood within the subarachnoid space is demonstrable in 92% of cases if CT is performed within the first 24 hours of hemorrhage, with an initial sensitivity of about 95% within the first 6 hours of onset.14,26,27 The longer CT is delayed, the lower the sensitivity.
Some studies suggest that a protocol of CT followed by CT angiography can safely exclude aneurysmal subarachnoid hemorrhage and obviate the need for lumbar puncture. However, further research is required to validate this approach.28
Lumbar puncture. If clinical suspicion of subarachnoid hemorrhage remains strong even though initial CT is negative, lumbar puncture must be performed for cerebrospinal fluid analysis.29 Xanthochromia (a yellowish pigmentation of the cerebrospinal fluid due to the degeneration of blood products that occurs within 8 to 12 hours of bleeding) should raise the alarm for subarachnoid hemorrhage; this sign may be present up to 4 weeks after the bleeding event.30
If lumbar puncture is contraindicated, then aneurysmal subarachnoid hemorrhage has not been ruled out, and further neurologic consultation should be pursued.
Management of subarachnoid hemorrhage
Early management of blood pressure for a ruptured intracranial aneurysm follows strategies similar to those for intracranial hemorrhage. Further investigation is rapidly directed toward an underlying vascular malformation, with intracranial vessel imaging such as CT angiography, magnetic resonance angiography, or the gold standard test—catheter-based cerebral angiography.
Aneurysms are treated (or “secured”) either by surgical clipping or by endovascular coiling. Endovascular coiling is preferable in cases in which both can be safely attempted.31 If the facility lacks the resources to do these procedures, the patient should be referred to a nearby tertiary care center.
INTRACRANIAL HYPERTENSION: DANGER OF BRAIN HERNIATION
A number of conditions can cause an acute intracranial pressure elevation. The danger of brain herniation requires that therapies be implemented rapidly to prevent catastrophic neurologic injury. In many situations, nonneurologists are the first responders and therefore should be familiar with basic intracranial pressure management.
Initial symptoms of acute rise in intracranial pressure
As intracranial pressure rises, pressure is typically equally distributed throughout the cranial vault, leading to dysfunction of the ascending reticular activating system, which clinically manifests as the inability to stay alert despite varying degrees of noxious stimulation. Progressive cranial neuropathies (often starting with pupillary abnormalities) and coma are often seen in this setting as the upper brainstem begins to be compressed.
Initial assessment and treatment of elevated intracranial pressure
Noncontrast CT of the head is often obtained immediately when acutely elevated intracranial pressure is suspected. If clinical examination and radiographic findings are consistent with intracranial hypertension, prompt measures can be started at the bedside.
Elevate the head of the bed to 30 degrees to promote venous drainage and reduce intracranial pressure. (In contrast, most other hemodynamically unstable patients are placed flat or in the Trendelenburg position.)
Intubation should be done quickly in cases of airway compromise, and hyperventilation should be started with a goal Paco2 of 30 to 35 mm Hg. This hypocarbic strategy promotes cerebral vasoconstriction and a transient decrease in intracranial pressure.
Hyperosmolar therapy allows for transient intracranial volume decompression and is the mainstay of emergency medical treatment of intracranial hypertension. Mannitol is a hyperosmolar polysaccharide that promotes osmotic diuresis and removes excessive cerebral water. In the acute setting, it can be given as an intravenous bolus of 1 to 2 g/kg through a peripheral intravenous line, followed by a bolus every 4 to 6 hours. Hypotension can occur after diuresis, and renal function should be closely monitored since frequent mannitol use can promote acute tubular necrosis. In patients who are anuric, the medication is typically not used.
Hypertonic saline (typically 3% sodium chloride, though different concentrations are available) is an alternative that helps draw interstitial fluid into the intravascular space, decreasing cerebral edema and maintaining hemodynamic stability. Relative contraindications include congestive heart failure or renal failure leading to pulmonary edema from volume overload. Hypertonic saline can be given as a bolus or a constant infusion. Some institutions have rapid access to 23.4% saline, which can be given as a 30-mL bolus but typically requires a central venous catheter for rapid infusion.
Comatose patients with radiographic findings of hydrocephalus, epidural or subdural hematoma, or mass effect with midline shift warrant prompt neurosurgical consultation for further surgical measures of intracranial pressure control and monitoring.
The ‘blown’ pupil

The physician should be concerned about elevated intracranial pressure if a patient has mydriasis, ie, an abnormally dilated (“blown”) pupil, which is a worrisome sign in the setting of true intracranial hypertension. However, many different processes can cause mydriasis and should be kept in mind when evaluating this finding (Table 3).32 If radiographic findings do not suggest elevated intracranial pressure, further workup into these other processes should be pursued.
STATUS EPILEPTICUS: SEIZURE CONTROL IS IMPORTANT
A continuous unremitting seizure lasting longer than 5 minutes or recurrent seizure activity in a patient who does not regain consciousness between seizures should be treated as status epilepticus. All seizure types carry the risk of progressing to status epilepticus, and responsiveness to antiepileptic drug therapy is inversely related to the duration of seizures. It is imperative that seizure activity be treated early and aggressively to prevent recalcitrant seizure activity, neuronal damage, and progression to status epilepticus.33

Once the ABCs of emergency stabilization have been performed (ie, airway, breathing, circulation), antiepileptic drug therapy should start immediately using established algorithms (Figure 1).34–36 During the course of treatment, the reliability of the neurologic examination may be limited due to medication effects or continued status epilepticus, making continuous video electroencephalographic monitoring often necessary to guide further therapy in patients who are not rapidly recovering.34–38
Once status epilepticus has resolved, further investigation into the underlying cause should be pursued quickly, especially in patients without a previous diagnosis of epilepsy. Head CT with contrast or magnetic resonance imaging can be used to look for any structural abnormality that may explain seizures. Basic laboratory tests including toxicology screening can identify a common trigger such as hypoglycemia or stimulant use. Fever or other possible signs of meningitis should be investigated further with cerebrospinal fluid analysis.
SPINAL CORD INJURY
Acute spinal cord injury can lead to substantial long-term neurologic impairment and should be suspected in any patient presenting with focal motor loss, sensory loss, or both with sparing of the cranial nerves and mental status. Causes of injury include compression (traumatic or nontraumatic) and inflammatory and noninflammatory myelopathies.
The location of the injury can be inferred by analyzing the symptoms, which can point to the cord level and indicate whether the anterior or posterior of the cord is involved. Anterior cord injury tends to affect the descending corticospinal and pyramidal tracts, resulting in motor deficits and weakness. Posterior cord injury involves the dorsal columns, leading to deficits of vibration sensation and proprioception. High cervical cord injuries tend to involve varying degrees of quadriparesis, sensory loss, and sometimes respiratory compromise. A clinical history of bilateral lower-extremity weakness, a “band-like” sensory complaint around the lower chest or abdomen, or both, can suggest thoracic cord involvement. Symptoms isolated to one or both lower extremities along with lower back pain and bowel or bladder involvement may point to injury of the lumbosacral cord.
Basic management of spinal cord injury includes decompression of the bladder and initial protection against further injury with a stabilizing collar or brace.
Magnetic resonance imaging with and without contrast is the ideal study to evaluate injuries to the spinal cord itself. While CT is helpful in identifying bony disease of the spinal column (eg, evaluating traumatic fractures), it is not helpful in viewing intrinsic cord pathology.
Traumatic myelopathy
Traumatic spinal cord injury is usually suggested by the clinical history and confirmed with CT. In this setting, early consultation with a neurosurgeon is required to prevent permanent cord injury.
Guidelines suggest maintaining a mean arterial pressure greater than 85 to 90 mm Hg for the first 7 days after traumatic spinal cord injury, a particular problem in the setting of hemodynamic instability, which can accompany lesions above the midthoracic level.39,40
Patients with vertebral body misalignment should be placed in an appropriate stabilizing collar or brace until a medically trained professional deems it appropriate to discontinue the device, or until surgical stabilization is performed.
Methylprednisone is a controversial intervention for acute spinal cord trauma, lacking clear benefit in meta-analyses.41
Nontraumatic compressive myelopathy
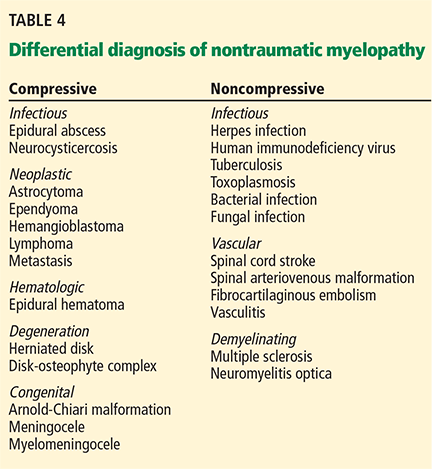
Patients with nontraumatic compressive myelopathy tend to present with varying degrees of back pain and worsening sensorimotor function. The differential diagnosis includes epidural abscesses, hematoma, metastatic neoplasm, and osteophyte compression (Table 4). The clinical history helps to guide therapy and should involve assessment for previous spinal column injury, immunocompromised state, travel history (which provides information on risks of exposure to a variety of diseases, including infections), and constitutional symptoms such as fever and weight loss.
Epidural abscess can have devastating results if missed. Red flags such as recent illness, intravenous drug use, focal back pain, fever, worsening numbness or weakness, and bowel or bladder incontinence should raise suspicion of this disorder. Emergency magnetic resonance imaging is required to diagnose this condition, and treatment involves urgent administration of antibiotics and consideration of surgical drainage.
Noncompressive myelopathies
There are numerous causes of noncompressive spinal cord injury (Table 4), and the etiology may be inflammatory (eg, “myelitis”) or noninflammatory. The diagnostic workup may require both magnetic resonance imaging and cerebrospinal fluid analysis. Acute disease-targeted therapy is rarely indicated and can be deferred until a full diagnostic workup has been completed.
NEUROMUSCULAR DISEASE: IS VENTILATION NEEDED?

Diseases involving the motor components of the peripheral nervous system (Table 5) share the common risk of causing ventilatory failure due to weakness of the diaphragm, intercostal muscles, and upper-airway muscles. Clinicians need to be aware of this risk and view these disorders as neurologic emergencies.
Determining when these patients require mechanical intubation is a challenge. Serial measurements of maximum inspiratory force and vital capacity are important and can be accomplished quickly at the bedside by a respiratory therapist. A maximum inspiratory force less than –30 cm H2O or a vital capacity less than 20 mL/kg, or both, are worrisome markers that raise concern for impending ventilatory failure. Serial measurements can detect changes in these values that might indicate the need for elective intubation. In any patient presenting with weakness of the limbs, these measurements are an important step in the initial evaluation.
Myasthenic crisis
Myasthenia gravis is caused by autoantibodies directed against postsynaptic acetylcholine receptors. Patients demonstrate muscle weakness, usually in a proximal pattern, with fatigue, respiratory distress, nasal speech, ophthalmoparesis, and dysphagia. Exacerbations can occur as a response to recent infection, surgery, or medications such as neuromuscular blocking agents or aminoglycosides.
Myasthenic crisis, while uncommon, is a life-threatening emergency characterized by bulbar or respiratory failure secondary to muscle weakness. It can occur in patients already diagnosed with myasthenia gravis or may be the initial manifestation of the disease.42–49 Intubation and mechanical ventilation are frequently required. Postoperative myasthenic patients in whom extubation has been delayed more than 24 hours should be considered in crisis.45
The diagnosis of myasthenia gravis can be made by serum autoantibody testing, electromyography, and nerve conduction studies (with repetitive stimulation) or administration of edrophonium in patients with obvious ptosis.
The mainstay of therapy for myasthenic crisis is either intravenous immunoglobulin at a dose of 2 g/kg over 2 to 5 days or plasmapheresis (5–7 exchanges over 7–14 days). Corticosteroids are not recommended in myasthenic crisis in patients who are not intubated, as they can potentiate an initial worsening of crisis. Once the patient begins to show clinical improvement, outpatient pyridostigmine and immunosuppressive medications can be resumed at a low dose and titrated as tolerated.
Acute inflammatory demyelinating polyneuropathy (Guillain-Barré syndrome)
Acute inflammatory demyelinating polyneuropathy is an autoimmune disorder involving autoantibodies against axons or myelin in the peripheral nervous system.
This disease should be suspected in a patient who is developing worsening muscle weakness (usually with areflexia) over the course of days to weeks. Occasionally, a recent diarrheal or other systemic infectious trigger can be identified. Blood pressure instability and cardiac arrhythmia can also be seen due to autonomic nerve involvement. Although classically described as an “ascending paralysis,” other variants of this disease have distinct clinical presentations (eg, the descending paralysis, ataxia, areflexia, ophthalmoparesis of the Miller Fisher syndrome).
Acute inflammatory demyelinating polyneuropathy is diagnosed by electromyography and nerve conduction studies. A cerebrospinal fluid profile demonstrating elevated protein and few white blood cells is typical.
Treatment, as in myasthenic crisis, involves intravenous immunoglobulin or plasmapheresis. Corticosteroids are ineffective. Anticipation of ventilatory failure and expectant intubation is essential, given the progressive nature of the disorder.50
- Pitts SR, Niska RW, Xu J, Burt CW. National hospital ambulatory medical care survey: 2006 emergency department summary. Natl Health Stat Report 2008; 7:1–38.
- McMullan JT, Knight WA, Clark JF, Beyette FR, Pancioli A. Time-critical neurological emergencies: the unfulfilled role for point-of-care testing. Int J Emerg Med 2010; 3:127–131.
- Centers for Disease Control and Prevention (CDC). Prevalence of stroke: United States, 2006–2010. MMWR Morb Mortal Wkly Rep 2012; 61:379–382.
- Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet 2012; 380:2095–2128.
- Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1,160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet 2012; 380:2163–2196.
- Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA stroke study group. N Engl J Med 1995; 333:1581–1587.
- Hacke W, Donnan G, Fieschi C, et al; ATLANTIS Trials Investigators; ECASS Trials Investigators; NINDS rt-PA Study Group Investigators. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet 2004; 363:768–774.
- Saver JL, Fonarrow GC, Smith EE, et al. Time to treatment with intravenous tissue plasminogen activator and outcome from acute ischemic stroke. JAMA 2013; 309:2480–2488.
- Wojner-Alexander AW, Garami Z, Chernyshev OY, Alexandrov AV. Heads down: flat positioning improves blood flow velocity in acute ischemic stroke. Neurology 2005; 64:1354–1357.
- Jauch EC, Saver JL, Adams HP Jr, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013; 44:870–947.
- Hacke W, Kaste M, Bluhmki E, et al; ECASS Investigators. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 2008; 359:1317–1329.
- Berkhemer OA, Fransen PSS, Beumer D, et al; MR CLEAN Investigators. A randomized trial of intraarterial treatment for acute ischemic stroke. N Eng J Med 2015; 372:11–20.
- Campbell BC, Mitchell PJ, Kleinig TJ, et al; EXTEND-IA Investigators. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N Engl J Med 2015; 372:1009–1018.
- Backes D, Rinkel GJ, Kemperman H, Linn FH, Vergouwen MD. Time-dependent test characteristics of head computed tomography in patients suspected of nontraumatic subarachnoid hemorrhage. Stroke 2012; 43:2115–2119.
- Mendelow AD, Gregson BA, Fernandes HM, et al; STICH investigators. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet 2005; 365: 387–397.
- Anderson CS, Helley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Flibotte JJ, Hagan N, O'Donnell J, Greenberg SM, Rosand J. Warfarin, hematoma expansion, and outcome of intracerebral hemorrhage. Neurology 2004; 63:1059–1064.
- Davis SM, Broderick J, Hennerici M, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Hematoma growth is a determinant of mortality and poor outcome after intracerebral hemorrhage. Neurology 2006; 66:1175–1181.
- Woo CH, Patel N, Conell C, et al. Rapid warfarin reversal in the setting of intracranial hemorrhage: a comparison of plasma, recombinant activated factor VII, and prothrombin complex concentrate. World Neurosurg 2014; 81:110–115.
- Broderick J, Connolly S, Feldmann E, et al; American Heart Association; American Stroke Association Stroke Council; High Blood Pressure Research Council; Quality of Care and Outcomes in Research Interdisciplinary Working Group. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Stroke 2007; 38:2001–2023.
- Goldstein JN, Thomas SH, Frontiero V, et al. Timing of fresh frozen plasma administration and rapid correction of coagulopathy in warfarin-related intracerebral hemorrhage. Stroke 2006, 37:151–155.
- Chapman SA, Irwin ED, Beal AL, Kulinski NM, Hutson KE, Thorson MA. Prothrombin complex concentrate versus standard therapies for INR reversal in trauma patients receiving warfarin. Ann Pharmacother 2011; 45:869–875.
- Fawole A, Daw HA, Crowther MA. Practical management of bleeding due to the anticoagulants dabigatran, rivaroxaban, and apixaban. Cleve Clin J Med 2013; 80:443–451.
- Pollack CV Jr, Reilly PA, Eikelboom J, et al. Idarucizumab for dabigatran reversal. N Engl J Med 2015; 373:511-520.
- Broderick JP, Brott TG, Duldner JE, Tomsick T, Leach A. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke 1994; 25:1342–1347.
- Kassell NF, Torner JC, Haley EC Jr, Jane JA, Adams HP, Kongable GL. The international cooperative study on the timing of aneurysm surgery. Part 1: overall management results. J Neurosurg 1990; 73:18–36.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Vermuelen M, Hasan D, Blijenberg BG, Hijdra A, van Gijn J. Xanthochromia after subarachnoid haemorrhage needs no revisitation. J Neurol Neurosurg Psychiatry 1989; 52:826–828.
- Molyneaux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid hemorrhage trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Caglayan HZ, Colpak IA, Kansu T. A diagnostic challenge: dilated pupil. Curr Opin Ophthalmol 2013; 24:550–557.
- Brophy GM, Bell R, Claassen J, et al; Neurocritical Care Society Status Epilepticus Guideline Writing Committee. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care 2012; 17:3–23.
- Chang CW, Bleck TP. Status epilepticus. Neurol Clin 1995; 13:529–548.
- Treiman DM. Generalized convulsive status epilepticus in the adult. Epilepsia 1993; 34(suppl 1):S2–S11.
- Leppick IE. Status epilepticus: the next decade. Neurology 1990; 40(suppl 2):4–9.
- Aranda A, Foucart G, Ducassé JL, Grolleau S, McGonigal A, Valton L. Generalized convulsive status epilepticus management in adults: a cohort study with evaluation of professional practice. Epilepsia 2010; 51:2159–2167.
- DeLorenzo RJ, Waterhouse EJ, Towne AR, et al. Persistent nonconvulsive status epilepticus after the control of convulsive status epilepticus. Epilepsia 1998; 39:833–840.
- Casha S, Christie S. A systematic review of intensive cardiopulmonary management after spinal cord injury. J Neurotrauma 2011; 28:1479–1495.
- Walters BC, Hadley MN, Hurlbert RJ, et al; American Association of Neurological Surgeons; Congress of Neurological Surgeons. Guidelines for the management of acute cervical spine and spinal cord injuries: 2013 update. Neurosurgery 2013; 60(suppl 1):82–91.
- Hurlbert RJ, Hadley MN, Walters BC, et al. Pharmacological therapy for acute spinal cord injury. Neurosurgery 2013; 72(suppl 2):93–105.
- Cohen MS, Younger D. Aspects of the natural history of myasthenia gravis: crisis and death. Ann NY Acad Sci 1981; 377:670–677.
- Belack RS, Sanders DB. On the concept of myasthenic crisis. J Clin Neuromuscul Dis 2002; 4:40–42.
- Chaudhuri A, Behan PO. Myasthenic crisis. QJM 2009; 102:97–107.
- Mayer SA. Intensive care of the myasthenic patient. Neurology 1997; 48(suppl 5):70S–75S.
- Jani-Acsadi A, Lisak RP. Myasthenic crisis: guidelines for prevention and treatment. J Neurol Sci 2007; 261:127–133.
- Bershad EM, Feen ES, Suarez JI. Myasthenia gravis crisis. South Med J 2008; 101:63–69.
- Ahmed S, Kirmani JF, Janjua N, et al. An update on myasthenic crisis. Curr Treat Options Neurol 2005; 7:129–141.
- Godoy DA, Vaz de Mello LJ, Masotti L, Napoli MD. The myasthenic patient in crisis: an update of the management in neurointensive care unit. Arq Neuropsiquiatr 2013; 71:627–639.
- Hughes RA, Wijdicks EF, Benson E, et al; Multidisciplinary Consensus Group. Supportive care for patients with Guillain-Barré syndrome: Arch Neurol 2005; 62:1194–1198.
Neurologic emergencies such as acute stroke, status epilepticus, subarachnoid hemorrhage, neuromuscular weakness, and spinal cord injury affect millions of Americans yearly.1,2 These conditions can be difficult to diagnose, and delays in recognition and treatment can have devastating results. Consequently, it is important for nonneurologists to be able to quickly recognize these conditions and initiate timely management, often while awaiting neurologic consultation.
Here, we review how to recognize and treat these common, serious conditions.
ACUTE ISCHEMIC STROKE: TIME IS OF THE ESSENCE
Stroke is the fourth leading cause of death in the United States and is one of the most common causes of disability worldwide.3–5 About 85% of strokes are ischemic, resulting from diminished vascular supply to the brain. Symptoms such as facial droop, unilateral weakness or numbness, aphasia, gaze deviation, and unsteadiness of gait may be seen. Time is of the essence, as all currently available interventions are safe and effective only within defined time windows.
Diagnosis and assessment
When acute ischemic stroke is suspected, the clinical history, time of onset, and basic neurologic examination should be obtained quickly.
The National Institutes of Health (NIH) stroke scale is an objective marker for assessing stroke severity as well as evolution of disease and should be obtained in all stroke patients. Scores range from 0 (best) to 42 (worst) (www.ninds.nih.gov/doctors/NIH_Stroke_Scale.pdf).
Time of onset of symptoms is essential to determine, since it guides eligibility for acute therapies. Clinicians should ascertain the last time the patient was seen to be neurologically well in order to estimate this time window as closely as possible.
Laboratory tests should include a fingerstick blood glucose measurement, coagulation studies, complete blood cell count, and basic metabolic profile.
Computed tomography (CT) of the head without contrast should be obtained immediately to exclude acute hemorrhage and any alternative diagnoses that could explain the patient’s symptoms. Acute brain ischemia is often not apparent on CT during the first few hours of injury. Therefore, a patient presenting with new focal neurologic deficits and an unremarkable result on CT of the head should be treated as having had an acute ischemic stroke, and interventional therapies should be considered.
Stroke mimics should be considered and treated, as appropriate (Table 1).
Acute management of ischemic stroke
Acute treatment should not be delayed by obtaining chest radiography, inserting a Foley catheter, or obtaining an electrocardiogram. The longer the time that elapses before treatment, the worse the functional outcome, underscoring the need for rapid decision-making.6–8
Lowering the head of the bed may provide benefit by promoting blood flow to ischemic brain tissue.9 However, this should not be done in patients with significantly elevated intracerebral pressure and concern for herniation.
Permissive hypertension (antihypertensive treatment only for blood pressure greater than 220/110 mm Hg) should be allowed per national guidelines to provide adequate perfusion to brain areas at risk of injury.10
Tissue plasminogen activator. Patients with ischemic stroke who present within 3 hours of symptom onset should be considered for intravenous administration of tissue plasminogen activator (tPA), a safe and effective therapy with nearly 2 decades of evidence to support its use.10 The treating physician should carefully review the risks and benefits of this therapy.
To receive tPA, the patient must have all of the following:
- Clinical diagnosis of ischemic stroke with measurable neurologic deficit
- Onset of symptoms within the past 3 hours
- Age 18 or older.
The patient must not have any of the following:
- Significant stroke within the past 3 months
- Severe traumatic head injury within the past 3 months
- History of significant intracerebral hemorrhage
- Previously ruptured arteriovenous malformation or intracranial aneurysm
- Central nervous system neoplasm
- Arterial puncture at a noncompressible site within the past 7 days
- Evidence of hemorrhage on CT of the head
- Evidence of ischemia in greater than 33% of the cerebral hemisphere on head CT
- History and symptoms strongly suggesting subarachnoid hemorrhage
- Persistent hypertension (systolic pressure ≥ 185 mm Hg or diastolic pressure ≥ 110 mm Hg)
- Evidence of acute significant bleeding (external or internal)
- Hypoglycemia—ie, serum glucose less than 50 mg/dL (< 2.8 mmol/L)
- Thrombocytopenia (platelet count < 100 × 109/L)
- Significant coagulopathy (international normalized ratio > 1.7, prothrombin time > 15 seconds, or abnormally elevated activated partial thromboplastin time)
- Current use of a factor Xa inhibitor or direct thrombin inhibitor.
Relative contraindications:
- Minor or rapidly resolving symptoms
- Major surgery or trauma within the past 14 days
- Gastrointestinal or urinary tract bleeding within the past 21 days
- Myocardial infarction in the past 3 months
- Unruptured intracranial aneurysm
- Seizure occurring at stroke onset
- Pregnancy.
If these criteria are satisfied, tPA should be given at a dose of 0.9 mg/kg intravenously over 60 minutes. Ten percent of the dose should be given as an initial bolus, followed by a constant infusion of the remaining 90% over 1 hour.
If tPA is given, the blood pressure must be kept lower than 185/110 mm Hg to minimize the risk of symptomatic intracerebral hemorrhage.
A subset of patients may benefit from receiving intravenous tPA between 3 and 4.5 hours after the onset of stroke symptoms. These include patients who are no more than 80 years old, who have not recently used oral anticoagulants, who do not have severe neurologic injury (ie, do not have NIH Stroke Scale scores > 25), and who do not have diabetes mellitus or a history of ischemic stroke.11 Although many hospitals have such a protocol for tPA up to 4.5 hours after the onset of stroke symptoms, this time window is not currently approved by the US Food and Drug Administration.
Intra-arterial therapy. Based on recent trials, some patients may benefit further from intra-arterial thrombolysis or mechanical thrombectomy, both delivered during catheter-based cerebral angiography, independent of intravenous tPA administration.12,13 These patients should be evaluated on a case-by-case basis by a neurologist and neurointerventional team. Time windows for these treatments generally extend to 6 hours from stroke onset and perhaps even longer in some situations (eg, basilar artery occlusion).
An antiplatelet agent should be started quickly in all stroke patients who do not receive tPA. Patients who receive tPA can begin receiving an antiplatelet agent 24 hours afterward.
Unfractionated heparin. There is no evidence to support the use of unfractionated heparin in most cases of acute ischemic stroke.10
Glucose control (in the range of 140–180 mg/dL) and fever control remain essential elements of post-acute stroke care to provide additional protection to the damaged brain.
For ischemic stroke due to atrial fibrillation
In ischemic stroke due to atrial fibrillation, early anticoagulation should be considered, based on the CHA2DS2-VASC risk of ischemic stroke vs the HAS-BLED risk of hemorrhage (calculators available at www.mdcalc.com).
In general, anticoagulation may be withheld during the first 72 hours while further stroke workup and evaluation of extent of injury are carried out, as there is an increased risk of hemorrhagic transformation of the ischemic stroke. Often, anticoagulation is resumed at a full dose between 72 hours and 2 weeks of the ischemic stroke.
ACUTE HEMORRHAGIC STROKE: BLOOD PRESSURE, COAGULATION
Approximately 15% of strokes are caused by intracerebral hemorrhage, which can be detected with noncontrast head CT with a sensitivity of 98.6% within 6 hours of the onset of bleeding.14 A common underlying cause of intracerebral hemorrhage is chronic poorly controlled hypertension, causing rupture of damaged (or “lipohyalinized”) vessels with resultant blood extravasation into the brain parenchyma. Other causes are less common (Table 2).
Treatment of acute hemorrhagic stroke
Acute treatment of intracerebral hemorrhage includes blood pressure control, reversal of underlying coagulopathy or anticoagulation, and sometimes intracranial pressure control. There is little role for surgery in most cases, based on findings of randomized trials.15
Blood pressure control. Many studies have investigated optimal blood pressure goals in acute intracerebral hemorrhage. Recent data suggest that early aggressive therapy, targeting a systolic blood pressure goal less than 140 mm Hg within the first hour, is safe and can lead to better functional outcomes than a more conservative blood-pressure-lowering target.16 Rapid-onset, short-acting antihypertensive agents in intravenous form, such as nicardipine and labetalol, are frequently used. Of note, this treatment strategy for hemorrhagic stroke is in direct contrast to the treatment of ischemic stroke, in which permissive hypertension (blood pressure goal < 220/110 mm Hg) is often pursued.
Reversal of any coagulation abnormalities should be done quickly in intracranial hemorrhage. Warfarin use has been shown to be a strong independent predictor of intracranial hemorrhage expansion, which increases the risk of death.17,18
Increasingly, agents other than vitamin K or fresh-frozen plasma are being used to rapidly reverse anticoagulation, including prothrombin complex concentrate (available in three- and four-factor preparations) and recombinant factor VIIa. While four-factor prothrombin complex concentrate and recombinant factor VIIa have been shown to be more efficacious than fresh-frozen plasma, there are limited data directly comparing these newer reversal agents against each other.19 The use of these medications is limited by availability and practitioner familiarity.20–22
Reversing anticoagulation due to target-specific oral anticoagulants. The acute management of intracranial hemorrhage in patients taking the new target-specific oral anticoagulants (eg, dabigatran, apixaban, rivaroxaban, edoxaban) remains challenging. Laboratory tests such as factor Xa levels are not readily available in many institutions and do not provide results in a timely fashion, and in the interim, acute hemorrhage and clinical deterioration may occur. Management strategies involve giving fresh-frozen plasma, prothrombin complex concentrate, and consideration of hemodialysis.23 Dabigatran reversal with idarucizumab has recently been shown to have efficacy.24
Vigilance for elevated intracranial pressure. Intracranial hemorrhage can occasionally cause elevated intracranial pressure, which should be treated rapidly. Any acute decline in mental status in a patient with intracranial hemorrhage requires emergency imaging to evaluate for expansion of hemorrhage.
SUBARACHNOID HEMORRHAGE
The sudden onset of a “thunderclap” headache (often described by patients as “the worst headache of my life”) suggests subarachnoid hemorrhage.
In contrast to intracranial hemorrhage, in subarachnoid hemorrhage blood collects mainly in the cerebral spinal fluid-containing spaces surrounding the brain, leading to a higher incidence of hydrocephalus from impaired drainage of cerebrospinal fluid. Nontraumatic subarachnoid hemorrhage is most often caused by rupture of an intracranial aneurysm, which can be a devastating event, with death rates approaching 50%.25
Diagnosis of subarachnoid hemorrhage
Noncontrast CT of the head is the main modality for diagnosing subarachnoid hemorrhage. Blood within the subarachnoid space is demonstrable in 92% of cases if CT is performed within the first 24 hours of hemorrhage, with an initial sensitivity of about 95% within the first 6 hours of onset.14,26,27 The longer CT is delayed, the lower the sensitivity.
Some studies suggest that a protocol of CT followed by CT angiography can safely exclude aneurysmal subarachnoid hemorrhage and obviate the need for lumbar puncture. However, further research is required to validate this approach.28
Lumbar puncture. If clinical suspicion of subarachnoid hemorrhage remains strong even though initial CT is negative, lumbar puncture must be performed for cerebrospinal fluid analysis.29 Xanthochromia (a yellowish pigmentation of the cerebrospinal fluid due to the degeneration of blood products that occurs within 8 to 12 hours of bleeding) should raise the alarm for subarachnoid hemorrhage; this sign may be present up to 4 weeks after the bleeding event.30
If lumbar puncture is contraindicated, then aneurysmal subarachnoid hemorrhage has not been ruled out, and further neurologic consultation should be pursued.
Management of subarachnoid hemorrhage
Early management of blood pressure for a ruptured intracranial aneurysm follows strategies similar to those for intracranial hemorrhage. Further investigation is rapidly directed toward an underlying vascular malformation, with intracranial vessel imaging such as CT angiography, magnetic resonance angiography, or the gold standard test—catheter-based cerebral angiography.
Aneurysms are treated (or “secured”) either by surgical clipping or by endovascular coiling. Endovascular coiling is preferable in cases in which both can be safely attempted.31 If the facility lacks the resources to do these procedures, the patient should be referred to a nearby tertiary care center.
INTRACRANIAL HYPERTENSION: DANGER OF BRAIN HERNIATION
A number of conditions can cause an acute intracranial pressure elevation. The danger of brain herniation requires that therapies be implemented rapidly to prevent catastrophic neurologic injury. In many situations, nonneurologists are the first responders and therefore should be familiar with basic intracranial pressure management.
Initial symptoms of acute rise in intracranial pressure
As intracranial pressure rises, pressure is typically equally distributed throughout the cranial vault, leading to dysfunction of the ascending reticular activating system, which clinically manifests as the inability to stay alert despite varying degrees of noxious stimulation. Progressive cranial neuropathies (often starting with pupillary abnormalities) and coma are often seen in this setting as the upper brainstem begins to be compressed.
Initial assessment and treatment of elevated intracranial pressure
Noncontrast CT of the head is often obtained immediately when acutely elevated intracranial pressure is suspected. If clinical examination and radiographic findings are consistent with intracranial hypertension, prompt measures can be started at the bedside.
Elevate the head of the bed to 30 degrees to promote venous drainage and reduce intracranial pressure. (In contrast, most other hemodynamically unstable patients are placed flat or in the Trendelenburg position.)
Intubation should be done quickly in cases of airway compromise, and hyperventilation should be started with a goal Paco2 of 30 to 35 mm Hg. This hypocarbic strategy promotes cerebral vasoconstriction and a transient decrease in intracranial pressure.
Hyperosmolar therapy allows for transient intracranial volume decompression and is the mainstay of emergency medical treatment of intracranial hypertension. Mannitol is a hyperosmolar polysaccharide that promotes osmotic diuresis and removes excessive cerebral water. In the acute setting, it can be given as an intravenous bolus of 1 to 2 g/kg through a peripheral intravenous line, followed by a bolus every 4 to 6 hours. Hypotension can occur after diuresis, and renal function should be closely monitored since frequent mannitol use can promote acute tubular necrosis. In patients who are anuric, the medication is typically not used.
Hypertonic saline (typically 3% sodium chloride, though different concentrations are available) is an alternative that helps draw interstitial fluid into the intravascular space, decreasing cerebral edema and maintaining hemodynamic stability. Relative contraindications include congestive heart failure or renal failure leading to pulmonary edema from volume overload. Hypertonic saline can be given as a bolus or a constant infusion. Some institutions have rapid access to 23.4% saline, which can be given as a 30-mL bolus but typically requires a central venous catheter for rapid infusion.
Comatose patients with radiographic findings of hydrocephalus, epidural or subdural hematoma, or mass effect with midline shift warrant prompt neurosurgical consultation for further surgical measures of intracranial pressure control and monitoring.
The ‘blown’ pupil
The physician should be concerned about elevated intracranial pressure if a patient has mydriasis, ie, an abnormally dilated (“blown”) pupil, which is a worrisome sign in the setting of true intracranial hypertension. However, many different processes can cause mydriasis and should be kept in mind when evaluating this finding (Table 3).32 If radiographic findings do not suggest elevated intracranial pressure, further workup into these other processes should be pursued.
STATUS EPILEPTICUS: SEIZURE CONTROL IS IMPORTANT
A continuous unremitting seizure lasting longer than 5 minutes or recurrent seizure activity in a patient who does not regain consciousness between seizures should be treated as status epilepticus. All seizure types carry the risk of progressing to status epilepticus, and responsiveness to antiepileptic drug therapy is inversely related to the duration of seizures. It is imperative that seizure activity be treated early and aggressively to prevent recalcitrant seizure activity, neuronal damage, and progression to status epilepticus.33
Once the ABCs of emergency stabilization have been performed (ie, airway, breathing, circulation), antiepileptic drug therapy should start immediately using established algorithms (Figure 1).34–36 During the course of treatment, the reliability of the neurologic examination may be limited due to medication effects or continued status epilepticus, making continuous video electroencephalographic monitoring often necessary to guide further therapy in patients who are not rapidly recovering.34–38
Once status epilepticus has resolved, further investigation into the underlying cause should be pursued quickly, especially in patients without a previous diagnosis of epilepsy. Head CT with contrast or magnetic resonance imaging can be used to look for any structural abnormality that may explain seizures. Basic laboratory tests including toxicology screening can identify a common trigger such as hypoglycemia or stimulant use. Fever or other possible signs of meningitis should be investigated further with cerebrospinal fluid analysis.
SPINAL CORD INJURY
Acute spinal cord injury can lead to substantial long-term neurologic impairment and should be suspected in any patient presenting with focal motor loss, sensory loss, or both with sparing of the cranial nerves and mental status. Causes of injury include compression (traumatic or nontraumatic) and inflammatory and noninflammatory myelopathies.
The location of the injury can be inferred by analyzing the symptoms, which can point to the cord level and indicate whether the anterior or posterior of the cord is involved. Anterior cord injury tends to affect the descending corticospinal and pyramidal tracts, resulting in motor deficits and weakness. Posterior cord injury involves the dorsal columns, leading to deficits of vibration sensation and proprioception. High cervical cord injuries tend to involve varying degrees of quadriparesis, sensory loss, and sometimes respiratory compromise. A clinical history of bilateral lower-extremity weakness, a “band-like” sensory complaint around the lower chest or abdomen, or both, can suggest thoracic cord involvement. Symptoms isolated to one or both lower extremities along with lower back pain and bowel or bladder involvement may point to injury of the lumbosacral cord.
Basic management of spinal cord injury includes decompression of the bladder and initial protection against further injury with a stabilizing collar or brace.
Magnetic resonance imaging with and without contrast is the ideal study to evaluate injuries to the spinal cord itself. While CT is helpful in identifying bony disease of the spinal column (eg, evaluating traumatic fractures), it is not helpful in viewing intrinsic cord pathology.
Traumatic myelopathy
Traumatic spinal cord injury is usually suggested by the clinical history and confirmed with CT. In this setting, early consultation with a neurosurgeon is required to prevent permanent cord injury.
Guidelines suggest maintaining a mean arterial pressure greater than 85 to 90 mm Hg for the first 7 days after traumatic spinal cord injury, a particular problem in the setting of hemodynamic instability, which can accompany lesions above the midthoracic level.39,40
Patients with vertebral body misalignment should be placed in an appropriate stabilizing collar or brace until a medically trained professional deems it appropriate to discontinue the device, or until surgical stabilization is performed.
Methylprednisone is a controversial intervention for acute spinal cord trauma, lacking clear benefit in meta-analyses.41
Nontraumatic compressive myelopathy
Patients with nontraumatic compressive myelopathy tend to present with varying degrees of back pain and worsening sensorimotor function. The differential diagnosis includes epidural abscesses, hematoma, metastatic neoplasm, and osteophyte compression (Table 4). The clinical history helps to guide therapy and should involve assessment for previous spinal column injury, immunocompromised state, travel history (which provides information on risks of exposure to a variety of diseases, including infections), and constitutional symptoms such as fever and weight loss.
Epidural abscess can have devastating results if missed. Red flags such as recent illness, intravenous drug use, focal back pain, fever, worsening numbness or weakness, and bowel or bladder incontinence should raise suspicion of this disorder. Emergency magnetic resonance imaging is required to diagnose this condition, and treatment involves urgent administration of antibiotics and consideration of surgical drainage.
Noncompressive myelopathies
There are numerous causes of noncompressive spinal cord injury (Table 4), and the etiology may be inflammatory (eg, “myelitis”) or noninflammatory. The diagnostic workup may require both magnetic resonance imaging and cerebrospinal fluid analysis. Acute disease-targeted therapy is rarely indicated and can be deferred until a full diagnostic workup has been completed.
NEUROMUSCULAR DISEASE: IS VENTILATION NEEDED?
Diseases involving the motor components of the peripheral nervous system (Table 5) share the common risk of causing ventilatory failure due to weakness of the diaphragm, intercostal muscles, and upper-airway muscles. Clinicians need to be aware of this risk and view these disorders as neurologic emergencies.
Determining when these patients require mechanical intubation is a challenge. Serial measurements of maximum inspiratory force and vital capacity are important and can be accomplished quickly at the bedside by a respiratory therapist. A maximum inspiratory force less than –30 cm H2O or a vital capacity less than 20 mL/kg, or both, are worrisome markers that raise concern for impending ventilatory failure. Serial measurements can detect changes in these values that might indicate the need for elective intubation. In any patient presenting with weakness of the limbs, these measurements are an important step in the initial evaluation.
Myasthenic crisis
Myasthenia gravis is caused by autoantibodies directed against postsynaptic acetylcholine receptors. Patients demonstrate muscle weakness, usually in a proximal pattern, with fatigue, respiratory distress, nasal speech, ophthalmoparesis, and dysphagia. Exacerbations can occur as a response to recent infection, surgery, or medications such as neuromuscular blocking agents or aminoglycosides.
Myasthenic crisis, while uncommon, is a life-threatening emergency characterized by bulbar or respiratory failure secondary to muscle weakness. It can occur in patients already diagnosed with myasthenia gravis or may be the initial manifestation of the disease.42–49 Intubation and mechanical ventilation are frequently required. Postoperative myasthenic patients in whom extubation has been delayed more than 24 hours should be considered in crisis.45
The diagnosis of myasthenia gravis can be made by serum autoantibody testing, electromyography, and nerve conduction studies (with repetitive stimulation) or administration of edrophonium in patients with obvious ptosis.
The mainstay of therapy for myasthenic crisis is either intravenous immunoglobulin at a dose of 2 g/kg over 2 to 5 days or plasmapheresis (5–7 exchanges over 7–14 days). Corticosteroids are not recommended in myasthenic crisis in patients who are not intubated, as they can potentiate an initial worsening of crisis. Once the patient begins to show clinical improvement, outpatient pyridostigmine and immunosuppressive medications can be resumed at a low dose and titrated as tolerated.
Acute inflammatory demyelinating polyneuropathy (Guillain-Barré syndrome)
Acute inflammatory demyelinating polyneuropathy is an autoimmune disorder involving autoantibodies against axons or myelin in the peripheral nervous system.
This disease should be suspected in a patient who is developing worsening muscle weakness (usually with areflexia) over the course of days to weeks. Occasionally, a recent diarrheal or other systemic infectious trigger can be identified. Blood pressure instability and cardiac arrhythmia can also be seen due to autonomic nerve involvement. Although classically described as an “ascending paralysis,” other variants of this disease have distinct clinical presentations (eg, the descending paralysis, ataxia, areflexia, ophthalmoparesis of the Miller Fisher syndrome).
Acute inflammatory demyelinating polyneuropathy is diagnosed by electromyography and nerve conduction studies. A cerebrospinal fluid profile demonstrating elevated protein and few white blood cells is typical.
Treatment, as in myasthenic crisis, involves intravenous immunoglobulin or plasmapheresis. Corticosteroids are ineffective. Anticipation of ventilatory failure and expectant intubation is essential, given the progressive nature of the disorder.50
Neurologic emergencies such as acute stroke, status epilepticus, subarachnoid hemorrhage, neuromuscular weakness, and spinal cord injury affect millions of Americans yearly.1,2 These conditions can be difficult to diagnose, and delays in recognition and treatment can have devastating results. Consequently, it is important for nonneurologists to be able to quickly recognize these conditions and initiate timely management, often while awaiting neurologic consultation.
Here, we review how to recognize and treat these common, serious conditions.
ACUTE ISCHEMIC STROKE: TIME IS OF THE ESSENCE
Stroke is the fourth leading cause of death in the United States and is one of the most common causes of disability worldwide.3–5 About 85% of strokes are ischemic, resulting from diminished vascular supply to the brain. Symptoms such as facial droop, unilateral weakness or numbness, aphasia, gaze deviation, and unsteadiness of gait may be seen. Time is of the essence, as all currently available interventions are safe and effective only within defined time windows.
Diagnosis and assessment
When acute ischemic stroke is suspected, the clinical history, time of onset, and basic neurologic examination should be obtained quickly.
The National Institutes of Health (NIH) stroke scale is an objective marker for assessing stroke severity as well as evolution of disease and should be obtained in all stroke patients. Scores range from 0 (best) to 42 (worst) (www.ninds.nih.gov/doctors/NIH_Stroke_Scale.pdf).
Time of onset of symptoms is essential to determine, since it guides eligibility for acute therapies. Clinicians should ascertain the last time the patient was seen to be neurologically well in order to estimate this time window as closely as possible.
Laboratory tests should include a fingerstick blood glucose measurement, coagulation studies, complete blood cell count, and basic metabolic profile.
Computed tomography (CT) of the head without contrast should be obtained immediately to exclude acute hemorrhage and any alternative diagnoses that could explain the patient’s symptoms. Acute brain ischemia is often not apparent on CT during the first few hours of injury. Therefore, a patient presenting with new focal neurologic deficits and an unremarkable result on CT of the head should be treated as having had an acute ischemic stroke, and interventional therapies should be considered.
Stroke mimics should be considered and treated, as appropriate (Table 1).
Acute management of ischemic stroke
Acute treatment should not be delayed by obtaining chest radiography, inserting a Foley catheter, or obtaining an electrocardiogram. The longer the time that elapses before treatment, the worse the functional outcome, underscoring the need for rapid decision-making.6–8
Lowering the head of the bed may provide benefit by promoting blood flow to ischemic brain tissue.9 However, this should not be done in patients with significantly elevated intracerebral pressure and concern for herniation.
Permissive hypertension (antihypertensive treatment only for blood pressure greater than 220/110 mm Hg) should be allowed per national guidelines to provide adequate perfusion to brain areas at risk of injury.10
Tissue plasminogen activator. Patients with ischemic stroke who present within 3 hours of symptom onset should be considered for intravenous administration of tissue plasminogen activator (tPA), a safe and effective therapy with nearly 2 decades of evidence to support its use.10 The treating physician should carefully review the risks and benefits of this therapy.
To receive tPA, the patient must have all of the following:
- Clinical diagnosis of ischemic stroke with measurable neurologic deficit
- Onset of symptoms within the past 3 hours
- Age 18 or older.
The patient must not have any of the following:
- Significant stroke within the past 3 months
- Severe traumatic head injury within the past 3 months
- History of significant intracerebral hemorrhage
- Previously ruptured arteriovenous malformation or intracranial aneurysm
- Central nervous system neoplasm
- Arterial puncture at a noncompressible site within the past 7 days
- Evidence of hemorrhage on CT of the head
- Evidence of ischemia in greater than 33% of the cerebral hemisphere on head CT
- History and symptoms strongly suggesting subarachnoid hemorrhage
- Persistent hypertension (systolic pressure ≥ 185 mm Hg or diastolic pressure ≥ 110 mm Hg)
- Evidence of acute significant bleeding (external or internal)
- Hypoglycemia—ie, serum glucose less than 50 mg/dL (< 2.8 mmol/L)
- Thrombocytopenia (platelet count < 100 × 109/L)
- Significant coagulopathy (international normalized ratio > 1.7, prothrombin time > 15 seconds, or abnormally elevated activated partial thromboplastin time)
- Current use of a factor Xa inhibitor or direct thrombin inhibitor.
Relative contraindications:
- Minor or rapidly resolving symptoms
- Major surgery or trauma within the past 14 days
- Gastrointestinal or urinary tract bleeding within the past 21 days
- Myocardial infarction in the past 3 months
- Unruptured intracranial aneurysm
- Seizure occurring at stroke onset
- Pregnancy.
If these criteria are satisfied, tPA should be given at a dose of 0.9 mg/kg intravenously over 60 minutes. Ten percent of the dose should be given as an initial bolus, followed by a constant infusion of the remaining 90% over 1 hour.
If tPA is given, the blood pressure must be kept lower than 185/110 mm Hg to minimize the risk of symptomatic intracerebral hemorrhage.
A subset of patients may benefit from receiving intravenous tPA between 3 and 4.5 hours after the onset of stroke symptoms. These include patients who are no more than 80 years old, who have not recently used oral anticoagulants, who do not have severe neurologic injury (ie, do not have NIH Stroke Scale scores > 25), and who do not have diabetes mellitus or a history of ischemic stroke.11 Although many hospitals have such a protocol for tPA up to 4.5 hours after the onset of stroke symptoms, this time window is not currently approved by the US Food and Drug Administration.
Intra-arterial therapy. Based on recent trials, some patients may benefit further from intra-arterial thrombolysis or mechanical thrombectomy, both delivered during catheter-based cerebral angiography, independent of intravenous tPA administration.12,13 These patients should be evaluated on a case-by-case basis by a neurologist and neurointerventional team. Time windows for these treatments generally extend to 6 hours from stroke onset and perhaps even longer in some situations (eg, basilar artery occlusion).
An antiplatelet agent should be started quickly in all stroke patients who do not receive tPA. Patients who receive tPA can begin receiving an antiplatelet agent 24 hours afterward.
Unfractionated heparin. There is no evidence to support the use of unfractionated heparin in most cases of acute ischemic stroke.10
Glucose control (in the range of 140–180 mg/dL) and fever control remain essential elements of post-acute stroke care to provide additional protection to the damaged brain.
For ischemic stroke due to atrial fibrillation
In ischemic stroke due to atrial fibrillation, early anticoagulation should be considered, based on the CHA2DS2-VASC risk of ischemic stroke vs the HAS-BLED risk of hemorrhage (calculators available at www.mdcalc.com).
In general, anticoagulation may be withheld during the first 72 hours while further stroke workup and evaluation of extent of injury are carried out, as there is an increased risk of hemorrhagic transformation of the ischemic stroke. Often, anticoagulation is resumed at a full dose between 72 hours and 2 weeks of the ischemic stroke.
ACUTE HEMORRHAGIC STROKE: BLOOD PRESSURE, COAGULATION
Approximately 15% of strokes are caused by intracerebral hemorrhage, which can be detected with noncontrast head CT with a sensitivity of 98.6% within 6 hours of the onset of bleeding.14 A common underlying cause of intracerebral hemorrhage is chronic poorly controlled hypertension, causing rupture of damaged (or “lipohyalinized”) vessels with resultant blood extravasation into the brain parenchyma. Other causes are less common (Table 2).
Treatment of acute hemorrhagic stroke
Acute treatment of intracerebral hemorrhage includes blood pressure control, reversal of underlying coagulopathy or anticoagulation, and sometimes intracranial pressure control. There is little role for surgery in most cases, based on findings of randomized trials.15
Blood pressure control. Many studies have investigated optimal blood pressure goals in acute intracerebral hemorrhage. Recent data suggest that early aggressive therapy, targeting a systolic blood pressure goal less than 140 mm Hg within the first hour, is safe and can lead to better functional outcomes than a more conservative blood-pressure-lowering target.16 Rapid-onset, short-acting antihypertensive agents in intravenous form, such as nicardipine and labetalol, are frequently used. Of note, this treatment strategy for hemorrhagic stroke is in direct contrast to the treatment of ischemic stroke, in which permissive hypertension (blood pressure goal < 220/110 mm Hg) is often pursued.
Reversal of any coagulation abnormalities should be done quickly in intracranial hemorrhage. Warfarin use has been shown to be a strong independent predictor of intracranial hemorrhage expansion, which increases the risk of death.17,18
Increasingly, agents other than vitamin K or fresh-frozen plasma are being used to rapidly reverse anticoagulation, including prothrombin complex concentrate (available in three- and four-factor preparations) and recombinant factor VIIa. While four-factor prothrombin complex concentrate and recombinant factor VIIa have been shown to be more efficacious than fresh-frozen plasma, there are limited data directly comparing these newer reversal agents against each other.19 The use of these medications is limited by availability and practitioner familiarity.20–22
Reversing anticoagulation due to target-specific oral anticoagulants. The acute management of intracranial hemorrhage in patients taking the new target-specific oral anticoagulants (eg, dabigatran, apixaban, rivaroxaban, edoxaban) remains challenging. Laboratory tests such as factor Xa levels are not readily available in many institutions and do not provide results in a timely fashion, and in the interim, acute hemorrhage and clinical deterioration may occur. Management strategies involve giving fresh-frozen plasma, prothrombin complex concentrate, and consideration of hemodialysis.23 Dabigatran reversal with idarucizumab has recently been shown to have efficacy.24
Vigilance for elevated intracranial pressure. Intracranial hemorrhage can occasionally cause elevated intracranial pressure, which should be treated rapidly. Any acute decline in mental status in a patient with intracranial hemorrhage requires emergency imaging to evaluate for expansion of hemorrhage.
SUBARACHNOID HEMORRHAGE
The sudden onset of a “thunderclap” headache (often described by patients as “the worst headache of my life”) suggests subarachnoid hemorrhage.
In contrast to intracranial hemorrhage, in subarachnoid hemorrhage blood collects mainly in the cerebral spinal fluid-containing spaces surrounding the brain, leading to a higher incidence of hydrocephalus from impaired drainage of cerebrospinal fluid. Nontraumatic subarachnoid hemorrhage is most often caused by rupture of an intracranial aneurysm, which can be a devastating event, with death rates approaching 50%.25
Diagnosis of subarachnoid hemorrhage
Noncontrast CT of the head is the main modality for diagnosing subarachnoid hemorrhage. Blood within the subarachnoid space is demonstrable in 92% of cases if CT is performed within the first 24 hours of hemorrhage, with an initial sensitivity of about 95% within the first 6 hours of onset.14,26,27 The longer CT is delayed, the lower the sensitivity.
Some studies suggest that a protocol of CT followed by CT angiography can safely exclude aneurysmal subarachnoid hemorrhage and obviate the need for lumbar puncture. However, further research is required to validate this approach.28
Lumbar puncture. If clinical suspicion of subarachnoid hemorrhage remains strong even though initial CT is negative, lumbar puncture must be performed for cerebrospinal fluid analysis.29 Xanthochromia (a yellowish pigmentation of the cerebrospinal fluid due to the degeneration of blood products that occurs within 8 to 12 hours of bleeding) should raise the alarm for subarachnoid hemorrhage; this sign may be present up to 4 weeks after the bleeding event.30
If lumbar puncture is contraindicated, then aneurysmal subarachnoid hemorrhage has not been ruled out, and further neurologic consultation should be pursued.
Management of subarachnoid hemorrhage
Early management of blood pressure for a ruptured intracranial aneurysm follows strategies similar to those for intracranial hemorrhage. Further investigation is rapidly directed toward an underlying vascular malformation, with intracranial vessel imaging such as CT angiography, magnetic resonance angiography, or the gold standard test—catheter-based cerebral angiography.
Aneurysms are treated (or “secured”) either by surgical clipping or by endovascular coiling. Endovascular coiling is preferable in cases in which both can be safely attempted.31 If the facility lacks the resources to do these procedures, the patient should be referred to a nearby tertiary care center.
INTRACRANIAL HYPERTENSION: DANGER OF BRAIN HERNIATION
A number of conditions can cause an acute intracranial pressure elevation. The danger of brain herniation requires that therapies be implemented rapidly to prevent catastrophic neurologic injury. In many situations, nonneurologists are the first responders and therefore should be familiar with basic intracranial pressure management.
Initial symptoms of acute rise in intracranial pressure
As intracranial pressure rises, pressure is typically equally distributed throughout the cranial vault, leading to dysfunction of the ascending reticular activating system, which clinically manifests as the inability to stay alert despite varying degrees of noxious stimulation. Progressive cranial neuropathies (often starting with pupillary abnormalities) and coma are often seen in this setting as the upper brainstem begins to be compressed.
Initial assessment and treatment of elevated intracranial pressure
Noncontrast CT of the head is often obtained immediately when acutely elevated intracranial pressure is suspected. If clinical examination and radiographic findings are consistent with intracranial hypertension, prompt measures can be started at the bedside.
Elevate the head of the bed to 30 degrees to promote venous drainage and reduce intracranial pressure. (In contrast, most other hemodynamically unstable patients are placed flat or in the Trendelenburg position.)
Intubation should be done quickly in cases of airway compromise, and hyperventilation should be started with a goal Paco2 of 30 to 35 mm Hg. This hypocarbic strategy promotes cerebral vasoconstriction and a transient decrease in intracranial pressure.
Hyperosmolar therapy allows for transient intracranial volume decompression and is the mainstay of emergency medical treatment of intracranial hypertension. Mannitol is a hyperosmolar polysaccharide that promotes osmotic diuresis and removes excessive cerebral water. In the acute setting, it can be given as an intravenous bolus of 1 to 2 g/kg through a peripheral intravenous line, followed by a bolus every 4 to 6 hours. Hypotension can occur after diuresis, and renal function should be closely monitored since frequent mannitol use can promote acute tubular necrosis. In patients who are anuric, the medication is typically not used.
Hypertonic saline (typically 3% sodium chloride, though different concentrations are available) is an alternative that helps draw interstitial fluid into the intravascular space, decreasing cerebral edema and maintaining hemodynamic stability. Relative contraindications include congestive heart failure or renal failure leading to pulmonary edema from volume overload. Hypertonic saline can be given as a bolus or a constant infusion. Some institutions have rapid access to 23.4% saline, which can be given as a 30-mL bolus but typically requires a central venous catheter for rapid infusion.
Comatose patients with radiographic findings of hydrocephalus, epidural or subdural hematoma, or mass effect with midline shift warrant prompt neurosurgical consultation for further surgical measures of intracranial pressure control and monitoring.
The ‘blown’ pupil
The physician should be concerned about elevated intracranial pressure if a patient has mydriasis, ie, an abnormally dilated (“blown”) pupil, which is a worrisome sign in the setting of true intracranial hypertension. However, many different processes can cause mydriasis and should be kept in mind when evaluating this finding (Table 3).32 If radiographic findings do not suggest elevated intracranial pressure, further workup into these other processes should be pursued.
STATUS EPILEPTICUS: SEIZURE CONTROL IS IMPORTANT
A continuous unremitting seizure lasting longer than 5 minutes or recurrent seizure activity in a patient who does not regain consciousness between seizures should be treated as status epilepticus. All seizure types carry the risk of progressing to status epilepticus, and responsiveness to antiepileptic drug therapy is inversely related to the duration of seizures. It is imperative that seizure activity be treated early and aggressively to prevent recalcitrant seizure activity, neuronal damage, and progression to status epilepticus.33
Once the ABCs of emergency stabilization have been performed (ie, airway, breathing, circulation), antiepileptic drug therapy should start immediately using established algorithms (Figure 1).34–36 During the course of treatment, the reliability of the neurologic examination may be limited due to medication effects or continued status epilepticus, making continuous video electroencephalographic monitoring often necessary to guide further therapy in patients who are not rapidly recovering.34–38
Once status epilepticus has resolved, further investigation into the underlying cause should be pursued quickly, especially in patients without a previous diagnosis of epilepsy. Head CT with contrast or magnetic resonance imaging can be used to look for any structural abnormality that may explain seizures. Basic laboratory tests including toxicology screening can identify a common trigger such as hypoglycemia or stimulant use. Fever or other possible signs of meningitis should be investigated further with cerebrospinal fluid analysis.
SPINAL CORD INJURY
Acute spinal cord injury can lead to substantial long-term neurologic impairment and should be suspected in any patient presenting with focal motor loss, sensory loss, or both with sparing of the cranial nerves and mental status. Causes of injury include compression (traumatic or nontraumatic) and inflammatory and noninflammatory myelopathies.
The location of the injury can be inferred by analyzing the symptoms, which can point to the cord level and indicate whether the anterior or posterior of the cord is involved. Anterior cord injury tends to affect the descending corticospinal and pyramidal tracts, resulting in motor deficits and weakness. Posterior cord injury involves the dorsal columns, leading to deficits of vibration sensation and proprioception. High cervical cord injuries tend to involve varying degrees of quadriparesis, sensory loss, and sometimes respiratory compromise. A clinical history of bilateral lower-extremity weakness, a “band-like” sensory complaint around the lower chest or abdomen, or both, can suggest thoracic cord involvement. Symptoms isolated to one or both lower extremities along with lower back pain and bowel or bladder involvement may point to injury of the lumbosacral cord.
Basic management of spinal cord injury includes decompression of the bladder and initial protection against further injury with a stabilizing collar or brace.
Magnetic resonance imaging with and without contrast is the ideal study to evaluate injuries to the spinal cord itself. While CT is helpful in identifying bony disease of the spinal column (eg, evaluating traumatic fractures), it is not helpful in viewing intrinsic cord pathology.
Traumatic myelopathy
Traumatic spinal cord injury is usually suggested by the clinical history and confirmed with CT. In this setting, early consultation with a neurosurgeon is required to prevent permanent cord injury.
Guidelines suggest maintaining a mean arterial pressure greater than 85 to 90 mm Hg for the first 7 days after traumatic spinal cord injury, a particular problem in the setting of hemodynamic instability, which can accompany lesions above the midthoracic level.39,40
Patients with vertebral body misalignment should be placed in an appropriate stabilizing collar or brace until a medically trained professional deems it appropriate to discontinue the device, or until surgical stabilization is performed.
Methylprednisone is a controversial intervention for acute spinal cord trauma, lacking clear benefit in meta-analyses.41
Nontraumatic compressive myelopathy
Patients with nontraumatic compressive myelopathy tend to present with varying degrees of back pain and worsening sensorimotor function. The differential diagnosis includes epidural abscesses, hematoma, metastatic neoplasm, and osteophyte compression (Table 4). The clinical history helps to guide therapy and should involve assessment for previous spinal column injury, immunocompromised state, travel history (which provides information on risks of exposure to a variety of diseases, including infections), and constitutional symptoms such as fever and weight loss.
Epidural abscess can have devastating results if missed. Red flags such as recent illness, intravenous drug use, focal back pain, fever, worsening numbness or weakness, and bowel or bladder incontinence should raise suspicion of this disorder. Emergency magnetic resonance imaging is required to diagnose this condition, and treatment involves urgent administration of antibiotics and consideration of surgical drainage.
Noncompressive myelopathies
There are numerous causes of noncompressive spinal cord injury (Table 4), and the etiology may be inflammatory (eg, “myelitis”) or noninflammatory. The diagnostic workup may require both magnetic resonance imaging and cerebrospinal fluid analysis. Acute disease-targeted therapy is rarely indicated and can be deferred until a full diagnostic workup has been completed.
NEUROMUSCULAR DISEASE: IS VENTILATION NEEDED?
Diseases involving the motor components of the peripheral nervous system (Table 5) share the common risk of causing ventilatory failure due to weakness of the diaphragm, intercostal muscles, and upper-airway muscles. Clinicians need to be aware of this risk and view these disorders as neurologic emergencies.
Determining when these patients require mechanical intubation is a challenge. Serial measurements of maximum inspiratory force and vital capacity are important and can be accomplished quickly at the bedside by a respiratory therapist. A maximum inspiratory force less than –30 cm H2O or a vital capacity less than 20 mL/kg, or both, are worrisome markers that raise concern for impending ventilatory failure. Serial measurements can detect changes in these values that might indicate the need for elective intubation. In any patient presenting with weakness of the limbs, these measurements are an important step in the initial evaluation.
Myasthenic crisis
Myasthenia gravis is caused by autoantibodies directed against postsynaptic acetylcholine receptors. Patients demonstrate muscle weakness, usually in a proximal pattern, with fatigue, respiratory distress, nasal speech, ophthalmoparesis, and dysphagia. Exacerbations can occur as a response to recent infection, surgery, or medications such as neuromuscular blocking agents or aminoglycosides.
Myasthenic crisis, while uncommon, is a life-threatening emergency characterized by bulbar or respiratory failure secondary to muscle weakness. It can occur in patients already diagnosed with myasthenia gravis or may be the initial manifestation of the disease.42–49 Intubation and mechanical ventilation are frequently required. Postoperative myasthenic patients in whom extubation has been delayed more than 24 hours should be considered in crisis.45
The diagnosis of myasthenia gravis can be made by serum autoantibody testing, electromyography, and nerve conduction studies (with repetitive stimulation) or administration of edrophonium in patients with obvious ptosis.
The mainstay of therapy for myasthenic crisis is either intravenous immunoglobulin at a dose of 2 g/kg over 2 to 5 days or plasmapheresis (5–7 exchanges over 7–14 days). Corticosteroids are not recommended in myasthenic crisis in patients who are not intubated, as they can potentiate an initial worsening of crisis. Once the patient begins to show clinical improvement, outpatient pyridostigmine and immunosuppressive medications can be resumed at a low dose and titrated as tolerated.
Acute inflammatory demyelinating polyneuropathy (Guillain-Barré syndrome)
Acute inflammatory demyelinating polyneuropathy is an autoimmune disorder involving autoantibodies against axons or myelin in the peripheral nervous system.
This disease should be suspected in a patient who is developing worsening muscle weakness (usually with areflexia) over the course of days to weeks. Occasionally, a recent diarrheal or other systemic infectious trigger can be identified. Blood pressure instability and cardiac arrhythmia can also be seen due to autonomic nerve involvement. Although classically described as an “ascending paralysis,” other variants of this disease have distinct clinical presentations (eg, the descending paralysis, ataxia, areflexia, ophthalmoparesis of the Miller Fisher syndrome).
Acute inflammatory demyelinating polyneuropathy is diagnosed by electromyography and nerve conduction studies. A cerebrospinal fluid profile demonstrating elevated protein and few white blood cells is typical.
Treatment, as in myasthenic crisis, involves intravenous immunoglobulin or plasmapheresis. Corticosteroids are ineffective. Anticipation of ventilatory failure and expectant intubation is essential, given the progressive nature of the disorder.50
- Pitts SR, Niska RW, Xu J, Burt CW. National hospital ambulatory medical care survey: 2006 emergency department summary. Natl Health Stat Report 2008; 7:1–38.
- McMullan JT, Knight WA, Clark JF, Beyette FR, Pancioli A. Time-critical neurological emergencies: the unfulfilled role for point-of-care testing. Int J Emerg Med 2010; 3:127–131.
- Centers for Disease Control and Prevention (CDC). Prevalence of stroke: United States, 2006–2010. MMWR Morb Mortal Wkly Rep 2012; 61:379–382.
- Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet 2012; 380:2095–2128.
- Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1,160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet 2012; 380:2163–2196.
- Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA stroke study group. N Engl J Med 1995; 333:1581–1587.
- Hacke W, Donnan G, Fieschi C, et al; ATLANTIS Trials Investigators; ECASS Trials Investigators; NINDS rt-PA Study Group Investigators. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet 2004; 363:768–774.
- Saver JL, Fonarrow GC, Smith EE, et al. Time to treatment with intravenous tissue plasminogen activator and outcome from acute ischemic stroke. JAMA 2013; 309:2480–2488.
- Wojner-Alexander AW, Garami Z, Chernyshev OY, Alexandrov AV. Heads down: flat positioning improves blood flow velocity in acute ischemic stroke. Neurology 2005; 64:1354–1357.
- Jauch EC, Saver JL, Adams HP Jr, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013; 44:870–947.
- Hacke W, Kaste M, Bluhmki E, et al; ECASS Investigators. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 2008; 359:1317–1329.
- Berkhemer OA, Fransen PSS, Beumer D, et al; MR CLEAN Investigators. A randomized trial of intraarterial treatment for acute ischemic stroke. N Eng J Med 2015; 372:11–20.
- Campbell BC, Mitchell PJ, Kleinig TJ, et al; EXTEND-IA Investigators. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N Engl J Med 2015; 372:1009–1018.
- Backes D, Rinkel GJ, Kemperman H, Linn FH, Vergouwen MD. Time-dependent test characteristics of head computed tomography in patients suspected of nontraumatic subarachnoid hemorrhage. Stroke 2012; 43:2115–2119.
- Mendelow AD, Gregson BA, Fernandes HM, et al; STICH investigators. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet 2005; 365: 387–397.
- Anderson CS, Helley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Flibotte JJ, Hagan N, O'Donnell J, Greenberg SM, Rosand J. Warfarin, hematoma expansion, and outcome of intracerebral hemorrhage. Neurology 2004; 63:1059–1064.
- Davis SM, Broderick J, Hennerici M, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Hematoma growth is a determinant of mortality and poor outcome after intracerebral hemorrhage. Neurology 2006; 66:1175–1181.
- Woo CH, Patel N, Conell C, et al. Rapid warfarin reversal in the setting of intracranial hemorrhage: a comparison of plasma, recombinant activated factor VII, and prothrombin complex concentrate. World Neurosurg 2014; 81:110–115.
- Broderick J, Connolly S, Feldmann E, et al; American Heart Association; American Stroke Association Stroke Council; High Blood Pressure Research Council; Quality of Care and Outcomes in Research Interdisciplinary Working Group. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Stroke 2007; 38:2001–2023.
- Goldstein JN, Thomas SH, Frontiero V, et al. Timing of fresh frozen plasma administration and rapid correction of coagulopathy in warfarin-related intracerebral hemorrhage. Stroke 2006, 37:151–155.
- Chapman SA, Irwin ED, Beal AL, Kulinski NM, Hutson KE, Thorson MA. Prothrombin complex concentrate versus standard therapies for INR reversal in trauma patients receiving warfarin. Ann Pharmacother 2011; 45:869–875.
- Fawole A, Daw HA, Crowther MA. Practical management of bleeding due to the anticoagulants dabigatran, rivaroxaban, and apixaban. Cleve Clin J Med 2013; 80:443–451.
- Pollack CV Jr, Reilly PA, Eikelboom J, et al. Idarucizumab for dabigatran reversal. N Engl J Med 2015; 373:511-520.
- Broderick JP, Brott TG, Duldner JE, Tomsick T, Leach A. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke 1994; 25:1342–1347.
- Kassell NF, Torner JC, Haley EC Jr, Jane JA, Adams HP, Kongable GL. The international cooperative study on the timing of aneurysm surgery. Part 1: overall management results. J Neurosurg 1990; 73:18–36.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Vermuelen M, Hasan D, Blijenberg BG, Hijdra A, van Gijn J. Xanthochromia after subarachnoid haemorrhage needs no revisitation. J Neurol Neurosurg Psychiatry 1989; 52:826–828.
- Molyneaux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid hemorrhage trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Caglayan HZ, Colpak IA, Kansu T. A diagnostic challenge: dilated pupil. Curr Opin Ophthalmol 2013; 24:550–557.
- Brophy GM, Bell R, Claassen J, et al; Neurocritical Care Society Status Epilepticus Guideline Writing Committee. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care 2012; 17:3–23.
- Chang CW, Bleck TP. Status epilepticus. Neurol Clin 1995; 13:529–548.
- Treiman DM. Generalized convulsive status epilepticus in the adult. Epilepsia 1993; 34(suppl 1):S2–S11.
- Leppick IE. Status epilepticus: the next decade. Neurology 1990; 40(suppl 2):4–9.
- Aranda A, Foucart G, Ducassé JL, Grolleau S, McGonigal A, Valton L. Generalized convulsive status epilepticus management in adults: a cohort study with evaluation of professional practice. Epilepsia 2010; 51:2159–2167.
- DeLorenzo RJ, Waterhouse EJ, Towne AR, et al. Persistent nonconvulsive status epilepticus after the control of convulsive status epilepticus. Epilepsia 1998; 39:833–840.
- Casha S, Christie S. A systematic review of intensive cardiopulmonary management after spinal cord injury. J Neurotrauma 2011; 28:1479–1495.
- Walters BC, Hadley MN, Hurlbert RJ, et al; American Association of Neurological Surgeons; Congress of Neurological Surgeons. Guidelines for the management of acute cervical spine and spinal cord injuries: 2013 update. Neurosurgery 2013; 60(suppl 1):82–91.
- Hurlbert RJ, Hadley MN, Walters BC, et al. Pharmacological therapy for acute spinal cord injury. Neurosurgery 2013; 72(suppl 2):93–105.
- Cohen MS, Younger D. Aspects of the natural history of myasthenia gravis: crisis and death. Ann NY Acad Sci 1981; 377:670–677.
- Belack RS, Sanders DB. On the concept of myasthenic crisis. J Clin Neuromuscul Dis 2002; 4:40–42.
- Chaudhuri A, Behan PO. Myasthenic crisis. QJM 2009; 102:97–107.
- Mayer SA. Intensive care of the myasthenic patient. Neurology 1997; 48(suppl 5):70S–75S.
- Jani-Acsadi A, Lisak RP. Myasthenic crisis: guidelines for prevention and treatment. J Neurol Sci 2007; 261:127–133.
- Bershad EM, Feen ES, Suarez JI. Myasthenia gravis crisis. South Med J 2008; 101:63–69.
- Ahmed S, Kirmani JF, Janjua N, et al. An update on myasthenic crisis. Curr Treat Options Neurol 2005; 7:129–141.
- Godoy DA, Vaz de Mello LJ, Masotti L, Napoli MD. The myasthenic patient in crisis: an update of the management in neurointensive care unit. Arq Neuropsiquiatr 2013; 71:627–639.
- Hughes RA, Wijdicks EF, Benson E, et al; Multidisciplinary Consensus Group. Supportive care for patients with Guillain-Barré syndrome: Arch Neurol 2005; 62:1194–1198.
- Pitts SR, Niska RW, Xu J, Burt CW. National hospital ambulatory medical care survey: 2006 emergency department summary. Natl Health Stat Report 2008; 7:1–38.
- McMullan JT, Knight WA, Clark JF, Beyette FR, Pancioli A. Time-critical neurological emergencies: the unfulfilled role for point-of-care testing. Int J Emerg Med 2010; 3:127–131.
- Centers for Disease Control and Prevention (CDC). Prevalence of stroke: United States, 2006–2010. MMWR Morb Mortal Wkly Rep 2012; 61:379–382.
- Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet 2012; 380:2095–2128.
- Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1,160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet 2012; 380:2163–2196.
- Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA stroke study group. N Engl J Med 1995; 333:1581–1587.
- Hacke W, Donnan G, Fieschi C, et al; ATLANTIS Trials Investigators; ECASS Trials Investigators; NINDS rt-PA Study Group Investigators. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet 2004; 363:768–774.
- Saver JL, Fonarrow GC, Smith EE, et al. Time to treatment with intravenous tissue plasminogen activator and outcome from acute ischemic stroke. JAMA 2013; 309:2480–2488.
- Wojner-Alexander AW, Garami Z, Chernyshev OY, Alexandrov AV. Heads down: flat positioning improves blood flow velocity in acute ischemic stroke. Neurology 2005; 64:1354–1357.
- Jauch EC, Saver JL, Adams HP Jr, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013; 44:870–947.
- Hacke W, Kaste M, Bluhmki E, et al; ECASS Investigators. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 2008; 359:1317–1329.
- Berkhemer OA, Fransen PSS, Beumer D, et al; MR CLEAN Investigators. A randomized trial of intraarterial treatment for acute ischemic stroke. N Eng J Med 2015; 372:11–20.
- Campbell BC, Mitchell PJ, Kleinig TJ, et al; EXTEND-IA Investigators. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N Engl J Med 2015; 372:1009–1018.
- Backes D, Rinkel GJ, Kemperman H, Linn FH, Vergouwen MD. Time-dependent test characteristics of head computed tomography in patients suspected of nontraumatic subarachnoid hemorrhage. Stroke 2012; 43:2115–2119.
- Mendelow AD, Gregson BA, Fernandes HM, et al; STICH investigators. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet 2005; 365: 387–397.
- Anderson CS, Helley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Flibotte JJ, Hagan N, O'Donnell J, Greenberg SM, Rosand J. Warfarin, hematoma expansion, and outcome of intracerebral hemorrhage. Neurology 2004; 63:1059–1064.
- Davis SM, Broderick J, Hennerici M, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Hematoma growth is a determinant of mortality and poor outcome after intracerebral hemorrhage. Neurology 2006; 66:1175–1181.
- Woo CH, Patel N, Conell C, et al. Rapid warfarin reversal in the setting of intracranial hemorrhage: a comparison of plasma, recombinant activated factor VII, and prothrombin complex concentrate. World Neurosurg 2014; 81:110–115.
- Broderick J, Connolly S, Feldmann E, et al; American Heart Association; American Stroke Association Stroke Council; High Blood Pressure Research Council; Quality of Care and Outcomes in Research Interdisciplinary Working Group. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Stroke 2007; 38:2001–2023.
- Goldstein JN, Thomas SH, Frontiero V, et al. Timing of fresh frozen plasma administration and rapid correction of coagulopathy in warfarin-related intracerebral hemorrhage. Stroke 2006, 37:151–155.
- Chapman SA, Irwin ED, Beal AL, Kulinski NM, Hutson KE, Thorson MA. Prothrombin complex concentrate versus standard therapies for INR reversal in trauma patients receiving warfarin. Ann Pharmacother 2011; 45:869–875.
- Fawole A, Daw HA, Crowther MA. Practical management of bleeding due to the anticoagulants dabigatran, rivaroxaban, and apixaban. Cleve Clin J Med 2013; 80:443–451.
- Pollack CV Jr, Reilly PA, Eikelboom J, et al. Idarucizumab for dabigatran reversal. N Engl J Med 2015; 373:511-520.
- Broderick JP, Brott TG, Duldner JE, Tomsick T, Leach A. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke 1994; 25:1342–1347.
- Kassell NF, Torner JC, Haley EC Jr, Jane JA, Adams HP, Kongable GL. The international cooperative study on the timing of aneurysm surgery. Part 1: overall management results. J Neurosurg 1990; 73:18–36.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Vermuelen M, Hasan D, Blijenberg BG, Hijdra A, van Gijn J. Xanthochromia after subarachnoid haemorrhage needs no revisitation. J Neurol Neurosurg Psychiatry 1989; 52:826–828.
- Molyneaux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid hemorrhage trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Caglayan HZ, Colpak IA, Kansu T. A diagnostic challenge: dilated pupil. Curr Opin Ophthalmol 2013; 24:550–557.
- Brophy GM, Bell R, Claassen J, et al; Neurocritical Care Society Status Epilepticus Guideline Writing Committee. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care 2012; 17:3–23.
- Chang CW, Bleck TP. Status epilepticus. Neurol Clin 1995; 13:529–548.
- Treiman DM. Generalized convulsive status epilepticus in the adult. Epilepsia 1993; 34(suppl 1):S2–S11.
- Leppick IE. Status epilepticus: the next decade. Neurology 1990; 40(suppl 2):4–9.
- Aranda A, Foucart G, Ducassé JL, Grolleau S, McGonigal A, Valton L. Generalized convulsive status epilepticus management in adults: a cohort study with evaluation of professional practice. Epilepsia 2010; 51:2159–2167.
- DeLorenzo RJ, Waterhouse EJ, Towne AR, et al. Persistent nonconvulsive status epilepticus after the control of convulsive status epilepticus. Epilepsia 1998; 39:833–840.
- Casha S, Christie S. A systematic review of intensive cardiopulmonary management after spinal cord injury. J Neurotrauma 2011; 28:1479–1495.
- Walters BC, Hadley MN, Hurlbert RJ, et al; American Association of Neurological Surgeons; Congress of Neurological Surgeons. Guidelines for the management of acute cervical spine and spinal cord injuries: 2013 update. Neurosurgery 2013; 60(suppl 1):82–91.
- Hurlbert RJ, Hadley MN, Walters BC, et al. Pharmacological therapy for acute spinal cord injury. Neurosurgery 2013; 72(suppl 2):93–105.
- Cohen MS, Younger D. Aspects of the natural history of myasthenia gravis: crisis and death. Ann NY Acad Sci 1981; 377:670–677.
- Belack RS, Sanders DB. On the concept of myasthenic crisis. J Clin Neuromuscul Dis 2002; 4:40–42.
- Chaudhuri A, Behan PO. Myasthenic crisis. QJM 2009; 102:97–107.
- Mayer SA. Intensive care of the myasthenic patient. Neurology 1997; 48(suppl 5):70S–75S.
- Jani-Acsadi A, Lisak RP. Myasthenic crisis: guidelines for prevention and treatment. J Neurol Sci 2007; 261:127–133.
- Bershad EM, Feen ES, Suarez JI. Myasthenia gravis crisis. South Med J 2008; 101:63–69.
- Ahmed S, Kirmani JF, Janjua N, et al. An update on myasthenic crisis. Curr Treat Options Neurol 2005; 7:129–141.
- Godoy DA, Vaz de Mello LJ, Masotti L, Napoli MD. The myasthenic patient in crisis: an update of the management in neurointensive care unit. Arq Neuropsiquiatr 2013; 71:627–639.
- Hughes RA, Wijdicks EF, Benson E, et al; Multidisciplinary Consensus Group. Supportive care for patients with Guillain-Barré syndrome: Arch Neurol 2005; 62:1194–1198.
KEY POINTS
- Patients with possible acute ischemic stroke should be assessed quickly to see if they should receive tissue plasminogen activator, which should be started within 3 hours of stroke onset. Computed tomography (CT) of the head without contrast should be done immediately to rule out acute hemorrhagic stroke.
- Acute treatment of intracerebral hemorrhage includes blood pressure control, reversal of underlying coagulopathy, and sometimes intracranial pressure control.
- If the clinical suspicion of subarachnoid hemorrhage remains strong even though initial CT was negative, lumbar puncture is mandatory.
- Hyperosmolar therapy is the mainstay of emergency medical treatment of intracranial hypertension.
- Seizure activity must be treated aggressively to prevent recalcitrant seizure activity, neuronal damage, and progression to status epilepticus.