User login
Duchenne muscular dystrophy (DMD) is a severe progressive inherited disease characterized by muscle wasting and ultimately culminating in death. It’s a common enough neuromuscular disorder that pediatricians and family practice physicians are likely to see at least a couple of patients with DMD over the course of their career,” John Brandsema, MD, Neuromuscular Section Head, Division of Neurology, Children’s Hospital of Philadelphia in Pennsylvania, said in an interview. Healthcare providers should therefore be familiar with the disorder so as to provide timely diagnosis and early intervention as well as practical and emotional support to the patient and family/caregivers as they traverse the challenging and often heartbreaking journey with this condition.
Pathophysiology and Disease Trajectory
DMD is caused by pathogenic variants in the X-linked DMD gene, leading to reduction in dystrophin, a protein that serves as a cytoskeletal integrator, stabilizing the plasma membrane of striated muscle cells. Dystrophin is critical for muscle membrane stability.2 In particular, mutations in the gene that encodes for dystrophin lead to dysfunction in Dp427m, which is the muscle isoform of dystrophin.3,4
DMD is one of several types of muscular dystrophies. All are progressive disorders. Over time, healthy muscle fibers disappear and are replaced by fibrotic tissue and fat, making the muscles “less able to generate force for everyday activity.”2 Ultimately, the skeletal muscle dysfunction affects not only the patient’s day-to-day mobility but other systems as well. Most patients with DMD eventually die of cardiac and/or respiratory failure between the ages of 20 and 40 years, with a median life expectancy of 22 years — although children born after 1990 have a somewhat higher median life expectancy (28 years), because of the improving standard of care.3,5
Typically, DMD first presents with developmental delays and weakness in skeletal leg muscles. As the disease goes through stages of progression, it starts involving upper extremities and other systems. (Table 1)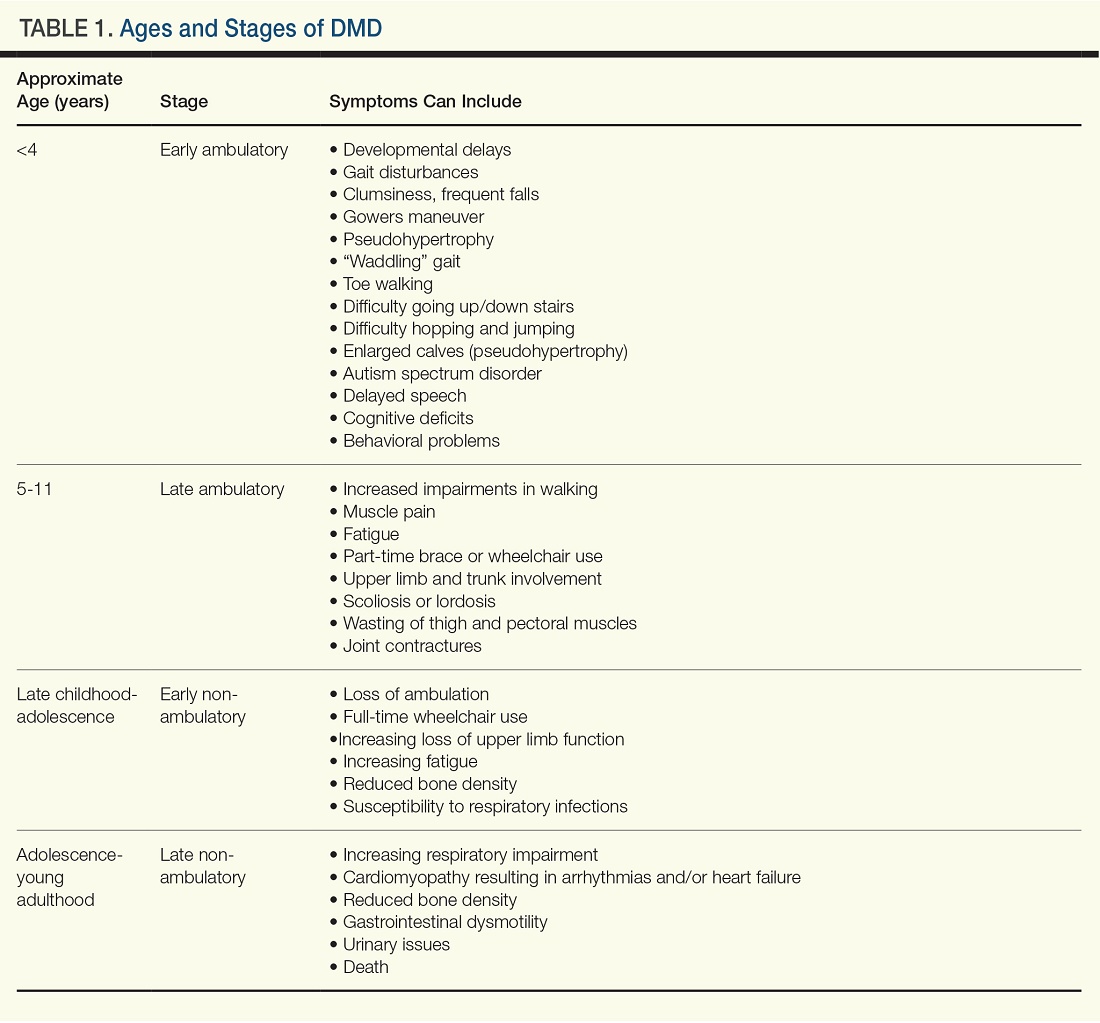
Genetic Causes of DMD
The DMD gene, located on the X chromosome, encodes for the production of dystrophin. Variants of this gene result in the lack of dystrophin protein, leading in turn to muscle fiber degeneration and the progressive symptoms of DMD. Because of the gene’s location on the X chromosome, males (who don’t have a second copy of the X chromosome) cannot compensate for the mutated gene, which is why the disease affects male children. Females with this mutation are carriers and typically do not develop the same severity of symptoms, although they might have milder muscle cramps, weakness, and cardiac issues.3
A female carrier with DMD (or any other X-linked disorder) has a 25% chance to have a carrier daughter, a 25% change of having a noncarrier daughter, a 25% chance of having an affected son, and a 25% chance of having a nonaffected son. A male with the disorder will pass the mutated gene on to his daughters who then become carriers. He cannot pass the disorder on to his sons because males inherit only the Y chromosome from their fathers.3
Diagnosing DMD
“It can take as long as 1-3 years for a child to be diagnosed with DMD,” Dr. Brandsema said. “Parents typically have concerns and know that something is ‘off’ about their child and they’re sent to various specialists, but it usually takes time for an accurate diagnosis to be made.” The mean age at diagnosis of DMD is between ages 4 and 5 years.6
Early identification of infants at risk for developing DMD can help move the needle toward earlier diagnosis. Newborn screening for DMD has been researched and piloted in several programs.6 In 2023, DMD was nominated for inclusion in the Recommended Universal Screening Panel (RUSP) for universal newborn screening. But in May 2024, the advisory committee on Heritable Disorders in Newborns and Children decided to postpone the vote to include DMD in the RUSP, requesting additional information to ensure an evidence-based decision.
In the absence of universal newborn screening for DMD, alternative approaches have been proposed to reduce the delay in clinical diagnosis and specialist referral, including increasing awareness among healthcare providers (eg, pediatricians, pediatric neurologists, and primary care physicians).6
The National Task Force for Early Identification of Childhood Neuromuscular Disorders delineates the steps necessary to identify pediatric muscle weakness and signs of neuromuscular disease. Primary care providers are encouraged to engage in regular developmental surveillance. A surveillance aid lays out the timetable for recommended visits, typical developmental milestones, and components of surveillance. Clinical evaluation includes a detailed patient history, family history, and physical examination.
If a neuromuscular condition is suspected, laboratory work should include creatinine phosphokinase (CK).6 Elevated serum CK points to leakage of CK through the muscle membrane, suggesting muscle damage. If CK is elevated, genetic testing should be performed; and, if negative, it should be followed by genetic sequencing that tests for small-scale mutations in the DMD gene. If that test is negative, a muscle biopsy should be performed to test for deep intronic mutations in the DMD gene.4
The diagnostic process and immediate steps after a confirmed DMD diagnosis is found in Figure 1.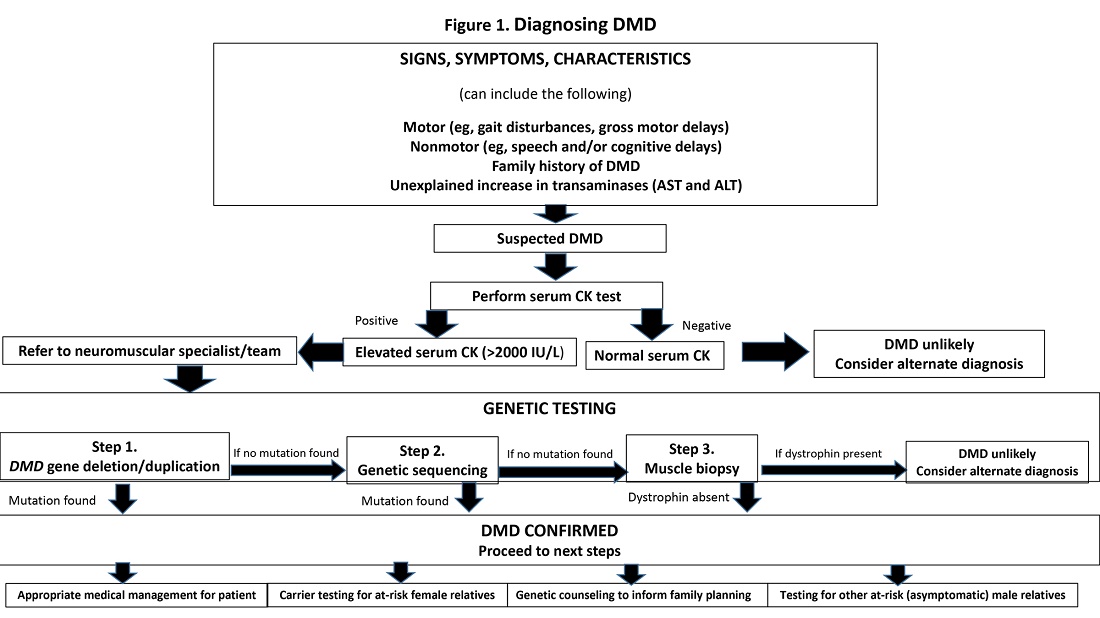
Targeting Inflammation in DMD
Traditionally, corticosteroids have been the only available medical treatment for DMD and they remain a cornerstone of DMD management. A meta-analysis found “moderate evidence” that corticosteroid therapy improves muscle strength and function in the short term (12 months), and strength up to 2 years.10
The two most common corticosteroids for DMD are prednisone and deflazacort. Deflazacort (Emflaza, PTC Therapeutics) was approved in 2017 to treat patients ages 5 years and older with DMD, subsequently expanded to 2 years and older. Deflazacort has been found to be more effective than prednisone in improving functional outcomes, delaying the onset of cardiomyopathy, and improving overall survival, with fewer adverse effects.11
In 2023, vamorolone (Agamree, Catalyst Pharmaceuticals) was approved by the Food and Drug Administration (FDA) to treat DMD patients (ages 2 years and older). Vamorolone is a dissociative steroidal anti-inflammatory that reduces bone morbidities and is regarded as a safer alternative than prednisone. A clinical trial comparing two doses of vamorolone with prednisone for 24 weeks found that vamorolone 6 mg/kg per day met the primary endpoint (time to stand velocity) and four sequential secondary motor function endpoints, with less bone morbidity, compared to prednisone.12 A more recent trial found improvements in motor outcomes at 48 weeks with a dose of 6 mg/kg per day of vamorolone. Bone morbidities of prednisone were reversed when the patient transitioned to vamorolone.13
“Steroid treatment has been proven to help, usually taken daily, although other schedules have been tried,” Dr. Brandsema said. However, all steroids are fraught with adverse effects and are suboptimal in the long term in reducing the disease burden.
The anti-inflammatory agent givinostat (Duvyzat, ITF Therapeutics), an oral histone deacetylase (HDAC) inhibitor, was approved in March 2024 for the treatment of DMD in patients 6 years of age and older. It is the first nonsteroidal drug to treat patients with all genetic variants of the disease, and it has a unique mechanism of action. Deficits in dystrophin can lead to increased HDAC activity in DMD, reducing the expression of genes involved in muscle regeneration. Givinostat therefore can help to counteract the pathogenic events downstream of dystrophin deficiency by inhibiting HDAC.14
Approval for givinostat was based on the phase 3 EPIDYS trial, which randomized 179 boys with DMD to receive either givinostat or placebo. Although results of a functional task worsened in both groups over the 12-month study period, the decline was significantly smaller with givinostat versus placebo. The most common adverse events were diarrhea and vomiting.14 Dr. Brandsema noted that monitoring of triglycerides and platelet count is required, as hypertriglyceridemia and thrombocytopenia can occur. This treatment was studied in tandem with corticosteroids as a combination approach to muscle stabilization.
New Pharmacotherapeutic Options: Exon-Skipping Agents
Today’s treatments have expanded beyond corticosteroids, with newer therapeutic options that include targeted exon-skipping therapies and, more recently, gene therapies. “These new treatment paradigms have changed the face of DMD treatment,” Dr. Brandsema said.

“Exon-skipping drugs in their current form have only a modest effect, but at least they’re a step in the right direction and a breakthrough, in terms of slowing disease progression,” Dr. Brandsema said.
Current exon-skipping agents use antisense phosphorodiamidate morpholino oligomers (PMOs) to restore a DMD open reading frame. Next-generation drugs called cell-penetrating peptide-conjugated PMOs (PPMOs) are being actively researched, Dr. Brandsema said. These agents have shown enhanced cellular uptake and more efficient dystrophin restoration, compared with unconjugated PMOs.15
There are currently four FDA-approved exon-skipping agents for DMD, all of which are administered via a weekly intravenous infusion: Casimersen (Amondys-45, SRP-4045), approved by the FDA in 2021; Eteplirsen (Exondys 51), approved in 2016; Golodirsen (Vyondys 53,SRP-4053), approved in 2019; and Vitolarsen (Viltepso), approved in 2020. They can be associated with multiple side effects, depending on the drug, including upper respiratory infection, fever, cough, rash, and gastrointestinal issues.16 These agents have the potential to help 30% of DMD patients, restoring low levels of dystrophin.16
Gene Transfer Therapies
Gene transfer therapies, a new class of agents, utilize a nonpathogenic viral vector (adeno-associated virus) to transfer specific genes to patients with DMD. Gene therapy involves overexpressing the micro-dystrophin gene to restore functional dystrophin expression.16
Multiple clinical trials of gene therapy are currently in progress. In 2023, delandistrogene moxeparvovec-rokl (Elevidys, Serepta) was granted accelerated FDA approval for ambulatory individuals with DMD between the ages of 4 and 5 years of age and a confirmed mutation in the DMD gene. It received expanded approval in June 2024 to include ambulatory and nonambulatory individuals aged 4 years and older with DMD and a confirmed mutation in the DMD gene (with the exception of exon 8 or 9 mutations).
The approval was based on preliminary data from two double-blind, placebo-controlled studies and two open-label studies, which enrolled a total of 218 male patients (including those who received placebo) with a confirmed disease-causing mutation in the DMD gene.
Delandistrogene moxeparvovec-rokl is delivered as a one-time infusion and has been associated with side effects and “a lot of potential issues,” Dr. Brandsema said. “We’ve seen cardiac effects, immune system effects, increased muscle inflammation and hepatic complications, and some people who became quite unwell were hospitalized for a long time.”
Fortunately, he added, “these seem to be rare but they do happen. Once the medication has been delivered, it’s permanently in the body, so you’re managing the side effects potentially on a long-term basis.”
It is critical to discuss the risks and benefits of this treatment with the family and caregivers and with the patient as well, if he old enough and able to participate in the decision-making progress. “We don’t want to give unrealistic expectations and we want people to be aware of the potential downside of this treatment,” he said. “This is a very complex discussion because the trajectory of the disease is so devastating and this treatment does hold out hope that other therapies don’t necessarily have.”
Nonpharmacologic Interventions
Physical therapy is a mainstay in DMD treatment, addressing protection of fragile muscles, preservation of strength, and prevention of muscle contractures.16 Given the respiratory impairments that occur with DMD progression, respiratory monitoring and therapy are essential; however, the number and type of evaluations and interventions vary with the stage of the disease, intensifying as the disease progresses.16 Similarly, cardiac monitoring should begin early, with patients screened for cardiac complications, and should intensify through the stages of disease progression.16
Bone health is compromised in patients with DMD, both as a result of corticosteroid treatment and as part of the disease itself. Fractures may be asymptomatic and may go unnoticed. Thus, bone health surveillance and maintenance are critical components of DMD management.16
Patients with DMD often experience gastrointestinal issues. They may experience weight gain because of lack of mobility and corticosteroid use in early stages, or weight loss as a result of diet or fluid imbalance, low bone density, or dysphagia in later stages. Patients should be closely followed by a nutritionist, a gastroenterologist as needed, and a physical therapist.16
Psychosocial support “should be developed and implemented across the lifespan in a manner that promotes thinking about the future and sets expectations that individuals will actively participate in their care and daily activities.”9 This includes psychological care, neuropsychological evaluations, and educational support.
Assisting Patients and Families Through the DMD Journey
DMD care is best delivered in a multidisciplinary setting, where physicians of relevant specialties, physical and occupational therapists, nutritionists, social workers, and genetic counselors collaborate. At Children’s Hospital of Philadelphia, DMD care is delivered through this collaborative model.
Unfortunately, Dr. Brandsema said, many patients don’t have this type of multidisciplinary resource available. “One specialist, such as a pulmonologist or neurologist, might have to be the sole source of care.” Or parents may have to ferry their child to multiple specialists in disparate locations, placing extra stress on an already-stressed family system.
“It’s helpful to connect the family with a comprehensive care center, if possible,” Dr. Brandsema advised. If that’s not available, then he suggests recommending educational opportunities and resources through national organizations such as the Muscular Dystrophy Association; Parent Project MD; NORD; Friends, Family and Duchenne; and Cure Duchenne. Families and caregivers, along with affected individuals, can get education and support from people who understand the day-to-day reality of living with this disease.
One of the major challenges that families face is navigating the high cost of treating DMD, especially the new medications, Dr. Brandsema said. “The authorization process can be intensive and long, and the family may need to take an active role, together with the provider team, in advocating for the patient to get access.”
Among her many responsibilities, Ms. Kaschak engages in care coordination tasks and management, helps patients and caregivers understand care plans, and provides psychosocial support and education about the disease process. She assists families in completing paperwork and navigating specialty authorizations, helping families understand and navigate the complex insurance process. “My role is to bridge gaps in care,” she said.
Dr. Brandsema noted that it’s important for couples to receive genetic counseling if they’re planning to have multiple children because there is a 50% chance that their next boy will be affected. About two thirds of mothers with children who have DMD are carriers, but many are not aware of it. Receiving counseling will enable them to understand their own risks of health complications, as well as the risk to future children.
Managing DMD Across the Lifespan
Another dimension of DMD care is providing resources and help to young people with DMD as they transition into adulthood. “In the past, we had limited treatment and mortality typically took place in the early 20s, so there weren’t a lot of patients who were adults. But as medication options have expanded and management of cardiac and respiratory failure has improved, we see a more significant proportion of adults who require adult-appropriate clinics — or, at the very least, specialists who are conversant in care or can provide care across the lifespan,” Dr. Brandsema said.
The DMD Care Considerations Working Group provides recommendations regarding care across the lifespan,9 as does the Adult North Star Network, of Muscular Dystrophy UK.17,18
Dr. Brandsema emphasized that, despite their disability, many adults with DMD “still engage with the community, and live life to its fullest.” It is to be hoped that, with ongoing research, earlier diagnosis, and improved treatment options, the future will look bright for people with DMD.
Dr. Brandsema has served as a consultant for Audentes, AveXis/Novartis, Biogen, Cytokinetics, Dyne, Edgewise, Fibrogen, Genentech, Marathon, Momenta/Janssen, NS Pharma, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Takeda, and WaVe. He is on the medical advisory council member for Cure SMA and is a site investigator for clinical trials with Alexion, Astellas, AveXis/Novartis, Biogen, Biohaven, Catabasis, CSL Behring, Cytokinetics, Dyne, Fibrogen, Genentech, Ionis, Lilly, ML Bio, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Summit, and WaVe. Ms. Kaschak has nothing to disclose.
References
1. Venugopal V and Pavlakis S. Duchenne Muscular Dystrophy. 2023 Jul 10. In: StatPearls [Internet]. Treasure Island, Florida: StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK482346/.
2. Gao QQ and McNally EM. Compr Physiol. 2015 Jul 1;5(3):1223-39. doi: 10.1002/cphy.c140048.
3. Duan D et al. Nat Rev Dis Primers. 2021 Feb 18;7(1):13. doi: 10.1038/s41572-021-00248-3.
4. Aartsma-Rus A et al. J Pediatr. 2019 Jan:204:305-313.e14. doi: 10.1016/j.jpeds.2018.10.043.
5. Broomfield J et al. Neurology. 2021 Dec 7;97(23):e2304-e2314. doi: 10.1212/WNL.0000000000012910.
6. Mercuri E et al. Front Pediatr. 2023 Nov 10:11:1276144. doi: 10.1212/WNL.0000000000012910.
7. Birnkrant DJ et al. Lancet Neurol. 2018 Mar;17(3):251-267. doi: 10.1016/S1474-4422(18)30024-3.
8. Birnkrant DJ et al. Lancet Neurol. 2018 Apr;17(4):347-361. doi: 10.1016/S1474-4422(18)30025-5.
9. Birnkrant DJ et al. Lancet Neurol. 2018 May;17(5):445-455. doi: 10.1016/S1474-4422(18)30026-7.
10. Matthews E et al. Cochrane Database Syst Rev. 2016 May 5;2016(5):CD003725. doi: 10.1002/14651858.CD003725.pub4.
11. Bylo M et al. Ann Pharmacother. 2020 Aug;54(8):788-794. doi: 10.1177/1060028019900500.
12. Guglieri M et al. JAMA Neurol. 2022 Oct 1;79(10):1005-1014. doi: 10.1001/jamaneurol.2022.2480.
13. Dang UJ et al. Neurology. 2024 Mar 12;102(5):e208112. doi: 10.1212/WNL.0000000000208112.
14. Mercuri E et al. Lancet Neurol. 2024 Apr;23(4):393-403. doi: 10.1016/S1474-4422(24)00036-X.
15. Gushchina LV et al. Mol Ther Nucleic Acids. 2022 Nov 9:30:479-492. doi: 10.1016/j.omtn.2022.10.025.
16. Patterson G et al. Eur J Pharmacol. 2023 May 15:947:175675. doi: 10.1016/j.ejphar.2023.175675.
17. Quinlivan R et al. J Neuromuscul Dis. 2021;8(6):899-926. doi: 10.3233/JND-200609.
18. Narayan S et al. J Neuromuscul Dis. 2022;9(3):365-381. doi: 10.3233/JND-210707.
Duchenne muscular dystrophy (DMD) is a severe progressive inherited disease characterized by muscle wasting and ultimately culminating in death. It’s a common enough neuromuscular disorder that pediatricians and family practice physicians are likely to see at least a couple of patients with DMD over the course of their career,” John Brandsema, MD, Neuromuscular Section Head, Division of Neurology, Children’s Hospital of Philadelphia in Pennsylvania, said in an interview. Healthcare providers should therefore be familiar with the disorder so as to provide timely diagnosis and early intervention as well as practical and emotional support to the patient and family/caregivers as they traverse the challenging and often heartbreaking journey with this condition.
Pathophysiology and Disease Trajectory
DMD is caused by pathogenic variants in the X-linked DMD gene, leading to reduction in dystrophin, a protein that serves as a cytoskeletal integrator, stabilizing the plasma membrane of striated muscle cells. Dystrophin is critical for muscle membrane stability.2 In particular, mutations in the gene that encodes for dystrophin lead to dysfunction in Dp427m, which is the muscle isoform of dystrophin.3,4
DMD is one of several types of muscular dystrophies. All are progressive disorders. Over time, healthy muscle fibers disappear and are replaced by fibrotic tissue and fat, making the muscles “less able to generate force for everyday activity.”2 Ultimately, the skeletal muscle dysfunction affects not only the patient’s day-to-day mobility but other systems as well. Most patients with DMD eventually die of cardiac and/or respiratory failure between the ages of 20 and 40 years, with a median life expectancy of 22 years — although children born after 1990 have a somewhat higher median life expectancy (28 years), because of the improving standard of care.3,5
Typically, DMD first presents with developmental delays and weakness in skeletal leg muscles. As the disease goes through stages of progression, it starts involving upper extremities and other systems. (Table 1)
Genetic Causes of DMD
The DMD gene, located on the X chromosome, encodes for the production of dystrophin. Variants of this gene result in the lack of dystrophin protein, leading in turn to muscle fiber degeneration and the progressive symptoms of DMD. Because of the gene’s location on the X chromosome, males (who don’t have a second copy of the X chromosome) cannot compensate for the mutated gene, which is why the disease affects male children. Females with this mutation are carriers and typically do not develop the same severity of symptoms, although they might have milder muscle cramps, weakness, and cardiac issues.3
A female carrier with DMD (or any other X-linked disorder) has a 25% chance to have a carrier daughter, a 25% change of having a noncarrier daughter, a 25% chance of having an affected son, and a 25% chance of having a nonaffected son. A male with the disorder will pass the mutated gene on to his daughters who then become carriers. He cannot pass the disorder on to his sons because males inherit only the Y chromosome from their fathers.3
Diagnosing DMD
“It can take as long as 1-3 years for a child to be diagnosed with DMD,” Dr. Brandsema said. “Parents typically have concerns and know that something is ‘off’ about their child and they’re sent to various specialists, but it usually takes time for an accurate diagnosis to be made.” The mean age at diagnosis of DMD is between ages 4 and 5 years.6
Early identification of infants at risk for developing DMD can help move the needle toward earlier diagnosis. Newborn screening for DMD has been researched and piloted in several programs.6 In 2023, DMD was nominated for inclusion in the Recommended Universal Screening Panel (RUSP) for universal newborn screening. But in May 2024, the advisory committee on Heritable Disorders in Newborns and Children decided to postpone the vote to include DMD in the RUSP, requesting additional information to ensure an evidence-based decision.
In the absence of universal newborn screening for DMD, alternative approaches have been proposed to reduce the delay in clinical diagnosis and specialist referral, including increasing awareness among healthcare providers (eg, pediatricians, pediatric neurologists, and primary care physicians).6
The National Task Force for Early Identification of Childhood Neuromuscular Disorders delineates the steps necessary to identify pediatric muscle weakness and signs of neuromuscular disease. Primary care providers are encouraged to engage in regular developmental surveillance. A surveillance aid lays out the timetable for recommended visits, typical developmental milestones, and components of surveillance. Clinical evaluation includes a detailed patient history, family history, and physical examination.
If a neuromuscular condition is suspected, laboratory work should include creatinine phosphokinase (CK).6 Elevated serum CK points to leakage of CK through the muscle membrane, suggesting muscle damage. If CK is elevated, genetic testing should be performed; and, if negative, it should be followed by genetic sequencing that tests for small-scale mutations in the DMD gene. If that test is negative, a muscle biopsy should be performed to test for deep intronic mutations in the DMD gene.4
The diagnostic process and immediate steps after a confirmed DMD diagnosis is found in Figure 1.
Targeting Inflammation in DMD
Traditionally, corticosteroids have been the only available medical treatment for DMD and they remain a cornerstone of DMD management. A meta-analysis found “moderate evidence” that corticosteroid therapy improves muscle strength and function in the short term (12 months), and strength up to 2 years.10
The two most common corticosteroids for DMD are prednisone and deflazacort. Deflazacort (Emflaza, PTC Therapeutics) was approved in 2017 to treat patients ages 5 years and older with DMD, subsequently expanded to 2 years and older. Deflazacort has been found to be more effective than prednisone in improving functional outcomes, delaying the onset of cardiomyopathy, and improving overall survival, with fewer adverse effects.11
In 2023, vamorolone (Agamree, Catalyst Pharmaceuticals) was approved by the Food and Drug Administration (FDA) to treat DMD patients (ages 2 years and older). Vamorolone is a dissociative steroidal anti-inflammatory that reduces bone morbidities and is regarded as a safer alternative than prednisone. A clinical trial comparing two doses of vamorolone with prednisone for 24 weeks found that vamorolone 6 mg/kg per day met the primary endpoint (time to stand velocity) and four sequential secondary motor function endpoints, with less bone morbidity, compared to prednisone.12 A more recent trial found improvements in motor outcomes at 48 weeks with a dose of 6 mg/kg per day of vamorolone. Bone morbidities of prednisone were reversed when the patient transitioned to vamorolone.13
“Steroid treatment has been proven to help, usually taken daily, although other schedules have been tried,” Dr. Brandsema said. However, all steroids are fraught with adverse effects and are suboptimal in the long term in reducing the disease burden.
The anti-inflammatory agent givinostat (Duvyzat, ITF Therapeutics), an oral histone deacetylase (HDAC) inhibitor, was approved in March 2024 for the treatment of DMD in patients 6 years of age and older. It is the first nonsteroidal drug to treat patients with all genetic variants of the disease, and it has a unique mechanism of action. Deficits in dystrophin can lead to increased HDAC activity in DMD, reducing the expression of genes involved in muscle regeneration. Givinostat therefore can help to counteract the pathogenic events downstream of dystrophin deficiency by inhibiting HDAC.14
Approval for givinostat was based on the phase 3 EPIDYS trial, which randomized 179 boys with DMD to receive either givinostat or placebo. Although results of a functional task worsened in both groups over the 12-month study period, the decline was significantly smaller with givinostat versus placebo. The most common adverse events were diarrhea and vomiting.14 Dr. Brandsema noted that monitoring of triglycerides and platelet count is required, as hypertriglyceridemia and thrombocytopenia can occur. This treatment was studied in tandem with corticosteroids as a combination approach to muscle stabilization.
New Pharmacotherapeutic Options: Exon-Skipping Agents
Today’s treatments have expanded beyond corticosteroids, with newer therapeutic options that include targeted exon-skipping therapies and, more recently, gene therapies. “These new treatment paradigms have changed the face of DMD treatment,” Dr. Brandsema said.
“Exon-skipping drugs in their current form have only a modest effect, but at least they’re a step in the right direction and a breakthrough, in terms of slowing disease progression,” Dr. Brandsema said.
Current exon-skipping agents use antisense phosphorodiamidate morpholino oligomers (PMOs) to restore a DMD open reading frame. Next-generation drugs called cell-penetrating peptide-conjugated PMOs (PPMOs) are being actively researched, Dr. Brandsema said. These agents have shown enhanced cellular uptake and more efficient dystrophin restoration, compared with unconjugated PMOs.15
There are currently four FDA-approved exon-skipping agents for DMD, all of which are administered via a weekly intravenous infusion: Casimersen (Amondys-45, SRP-4045), approved by the FDA in 2021; Eteplirsen (Exondys 51), approved in 2016; Golodirsen (Vyondys 53,SRP-4053), approved in 2019; and Vitolarsen (Viltepso), approved in 2020. They can be associated with multiple side effects, depending on the drug, including upper respiratory infection, fever, cough, rash, and gastrointestinal issues.16 These agents have the potential to help 30% of DMD patients, restoring low levels of dystrophin.16
Gene Transfer Therapies
Gene transfer therapies, a new class of agents, utilize a nonpathogenic viral vector (adeno-associated virus) to transfer specific genes to patients with DMD. Gene therapy involves overexpressing the micro-dystrophin gene to restore functional dystrophin expression.16
Multiple clinical trials of gene therapy are currently in progress. In 2023, delandistrogene moxeparvovec-rokl (Elevidys, Serepta) was granted accelerated FDA approval for ambulatory individuals with DMD between the ages of 4 and 5 years of age and a confirmed mutation in the DMD gene. It received expanded approval in June 2024 to include ambulatory and nonambulatory individuals aged 4 years and older with DMD and a confirmed mutation in the DMD gene (with the exception of exon 8 or 9 mutations).
The approval was based on preliminary data from two double-blind, placebo-controlled studies and two open-label studies, which enrolled a total of 218 male patients (including those who received placebo) with a confirmed disease-causing mutation in the DMD gene.
Delandistrogene moxeparvovec-rokl is delivered as a one-time infusion and has been associated with side effects and “a lot of potential issues,” Dr. Brandsema said. “We’ve seen cardiac effects, immune system effects, increased muscle inflammation and hepatic complications, and some people who became quite unwell were hospitalized for a long time.”
Fortunately, he added, “these seem to be rare but they do happen. Once the medication has been delivered, it’s permanently in the body, so you’re managing the side effects potentially on a long-term basis.”
It is critical to discuss the risks and benefits of this treatment with the family and caregivers and with the patient as well, if he old enough and able to participate in the decision-making progress. “We don’t want to give unrealistic expectations and we want people to be aware of the potential downside of this treatment,” he said. “This is a very complex discussion because the trajectory of the disease is so devastating and this treatment does hold out hope that other therapies don’t necessarily have.”
Nonpharmacologic Interventions
Physical therapy is a mainstay in DMD treatment, addressing protection of fragile muscles, preservation of strength, and prevention of muscle contractures.16 Given the respiratory impairments that occur with DMD progression, respiratory monitoring and therapy are essential; however, the number and type of evaluations and interventions vary with the stage of the disease, intensifying as the disease progresses.16 Similarly, cardiac monitoring should begin early, with patients screened for cardiac complications, and should intensify through the stages of disease progression.16
Bone health is compromised in patients with DMD, both as a result of corticosteroid treatment and as part of the disease itself. Fractures may be asymptomatic and may go unnoticed. Thus, bone health surveillance and maintenance are critical components of DMD management.16
Patients with DMD often experience gastrointestinal issues. They may experience weight gain because of lack of mobility and corticosteroid use in early stages, or weight loss as a result of diet or fluid imbalance, low bone density, or dysphagia in later stages. Patients should be closely followed by a nutritionist, a gastroenterologist as needed, and a physical therapist.16
Psychosocial support “should be developed and implemented across the lifespan in a manner that promotes thinking about the future and sets expectations that individuals will actively participate in their care and daily activities.”9 This includes psychological care, neuropsychological evaluations, and educational support.
Assisting Patients and Families Through the DMD Journey
DMD care is best delivered in a multidisciplinary setting, where physicians of relevant specialties, physical and occupational therapists, nutritionists, social workers, and genetic counselors collaborate. At Children’s Hospital of Philadelphia, DMD care is delivered through this collaborative model.
Unfortunately, Dr. Brandsema said, many patients don’t have this type of multidisciplinary resource available. “One specialist, such as a pulmonologist or neurologist, might have to be the sole source of care.” Or parents may have to ferry their child to multiple specialists in disparate locations, placing extra stress on an already-stressed family system.
“It’s helpful to connect the family with a comprehensive care center, if possible,” Dr. Brandsema advised. If that’s not available, then he suggests recommending educational opportunities and resources through national organizations such as the Muscular Dystrophy Association; Parent Project MD; NORD; Friends, Family and Duchenne; and Cure Duchenne. Families and caregivers, along with affected individuals, can get education and support from people who understand the day-to-day reality of living with this disease.
One of the major challenges that families face is navigating the high cost of treating DMD, especially the new medications, Dr. Brandsema said. “The authorization process can be intensive and long, and the family may need to take an active role, together with the provider team, in advocating for the patient to get access.”
Among her many responsibilities, Ms. Kaschak engages in care coordination tasks and management, helps patients and caregivers understand care plans, and provides psychosocial support and education about the disease process. She assists families in completing paperwork and navigating specialty authorizations, helping families understand and navigate the complex insurance process. “My role is to bridge gaps in care,” she said.
Dr. Brandsema noted that it’s important for couples to receive genetic counseling if they’re planning to have multiple children because there is a 50% chance that their next boy will be affected. About two thirds of mothers with children who have DMD are carriers, but many are not aware of it. Receiving counseling will enable them to understand their own risks of health complications, as well as the risk to future children.
Managing DMD Across the Lifespan
Another dimension of DMD care is providing resources and help to young people with DMD as they transition into adulthood. “In the past, we had limited treatment and mortality typically took place in the early 20s, so there weren’t a lot of patients who were adults. But as medication options have expanded and management of cardiac and respiratory failure has improved, we see a more significant proportion of adults who require adult-appropriate clinics — or, at the very least, specialists who are conversant in care or can provide care across the lifespan,” Dr. Brandsema said.
The DMD Care Considerations Working Group provides recommendations regarding care across the lifespan,9 as does the Adult North Star Network, of Muscular Dystrophy UK.17,18
Dr. Brandsema emphasized that, despite their disability, many adults with DMD “still engage with the community, and live life to its fullest.” It is to be hoped that, with ongoing research, earlier diagnosis, and improved treatment options, the future will look bright for people with DMD.
Dr. Brandsema has served as a consultant for Audentes, AveXis/Novartis, Biogen, Cytokinetics, Dyne, Edgewise, Fibrogen, Genentech, Marathon, Momenta/Janssen, NS Pharma, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Takeda, and WaVe. He is on the medical advisory council member for Cure SMA and is a site investigator for clinical trials with Alexion, Astellas, AveXis/Novartis, Biogen, Biohaven, Catabasis, CSL Behring, Cytokinetics, Dyne, Fibrogen, Genentech, Ionis, Lilly, ML Bio, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Summit, and WaVe. Ms. Kaschak has nothing to disclose.
References
1. Venugopal V and Pavlakis S. Duchenne Muscular Dystrophy. 2023 Jul 10. In: StatPearls [Internet]. Treasure Island, Florida: StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK482346/.
2. Gao QQ and McNally EM. Compr Physiol. 2015 Jul 1;5(3):1223-39. doi: 10.1002/cphy.c140048.
3. Duan D et al. Nat Rev Dis Primers. 2021 Feb 18;7(1):13. doi: 10.1038/s41572-021-00248-3.
4. Aartsma-Rus A et al. J Pediatr. 2019 Jan:204:305-313.e14. doi: 10.1016/j.jpeds.2018.10.043.
5. Broomfield J et al. Neurology. 2021 Dec 7;97(23):e2304-e2314. doi: 10.1212/WNL.0000000000012910.
6. Mercuri E et al. Front Pediatr. 2023 Nov 10:11:1276144. doi: 10.1212/WNL.0000000000012910.
7. Birnkrant DJ et al. Lancet Neurol. 2018 Mar;17(3):251-267. doi: 10.1016/S1474-4422(18)30024-3.
8. Birnkrant DJ et al. Lancet Neurol. 2018 Apr;17(4):347-361. doi: 10.1016/S1474-4422(18)30025-5.
9. Birnkrant DJ et al. Lancet Neurol. 2018 May;17(5):445-455. doi: 10.1016/S1474-4422(18)30026-7.
10. Matthews E et al. Cochrane Database Syst Rev. 2016 May 5;2016(5):CD003725. doi: 10.1002/14651858.CD003725.pub4.
11. Bylo M et al. Ann Pharmacother. 2020 Aug;54(8):788-794. doi: 10.1177/1060028019900500.
12. Guglieri M et al. JAMA Neurol. 2022 Oct 1;79(10):1005-1014. doi: 10.1001/jamaneurol.2022.2480.
13. Dang UJ et al. Neurology. 2024 Mar 12;102(5):e208112. doi: 10.1212/WNL.0000000000208112.
14. Mercuri E et al. Lancet Neurol. 2024 Apr;23(4):393-403. doi: 10.1016/S1474-4422(24)00036-X.
15. Gushchina LV et al. Mol Ther Nucleic Acids. 2022 Nov 9:30:479-492. doi: 10.1016/j.omtn.2022.10.025.
16. Patterson G et al. Eur J Pharmacol. 2023 May 15:947:175675. doi: 10.1016/j.ejphar.2023.175675.
17. Quinlivan R et al. J Neuromuscul Dis. 2021;8(6):899-926. doi: 10.3233/JND-200609.
18. Narayan S et al. J Neuromuscul Dis. 2022;9(3):365-381. doi: 10.3233/JND-210707.
Duchenne muscular dystrophy (DMD) is a severe progressive inherited disease characterized by muscle wasting and ultimately culminating in death. It’s a common enough neuromuscular disorder that pediatricians and family practice physicians are likely to see at least a couple of patients with DMD over the course of their career,” John Brandsema, MD, Neuromuscular Section Head, Division of Neurology, Children’s Hospital of Philadelphia in Pennsylvania, said in an interview. Healthcare providers should therefore be familiar with the disorder so as to provide timely diagnosis and early intervention as well as practical and emotional support to the patient and family/caregivers as they traverse the challenging and often heartbreaking journey with this condition.
Pathophysiology and Disease Trajectory
DMD is caused by pathogenic variants in the X-linked DMD gene, leading to reduction in dystrophin, a protein that serves as a cytoskeletal integrator, stabilizing the plasma membrane of striated muscle cells. Dystrophin is critical for muscle membrane stability.2 In particular, mutations in the gene that encodes for dystrophin lead to dysfunction in Dp427m, which is the muscle isoform of dystrophin.3,4
DMD is one of several types of muscular dystrophies. All are progressive disorders. Over time, healthy muscle fibers disappear and are replaced by fibrotic tissue and fat, making the muscles “less able to generate force for everyday activity.”2 Ultimately, the skeletal muscle dysfunction affects not only the patient’s day-to-day mobility but other systems as well. Most patients with DMD eventually die of cardiac and/or respiratory failure between the ages of 20 and 40 years, with a median life expectancy of 22 years — although children born after 1990 have a somewhat higher median life expectancy (28 years), because of the improving standard of care.3,5
Typically, DMD first presents with developmental delays and weakness in skeletal leg muscles. As the disease goes through stages of progression, it starts involving upper extremities and other systems. (Table 1)
Genetic Causes of DMD
The DMD gene, located on the X chromosome, encodes for the production of dystrophin. Variants of this gene result in the lack of dystrophin protein, leading in turn to muscle fiber degeneration and the progressive symptoms of DMD. Because of the gene’s location on the X chromosome, males (who don’t have a second copy of the X chromosome) cannot compensate for the mutated gene, which is why the disease affects male children. Females with this mutation are carriers and typically do not develop the same severity of symptoms, although they might have milder muscle cramps, weakness, and cardiac issues.3
A female carrier with DMD (or any other X-linked disorder) has a 25% chance to have a carrier daughter, a 25% change of having a noncarrier daughter, a 25% chance of having an affected son, and a 25% chance of having a nonaffected son. A male with the disorder will pass the mutated gene on to his daughters who then become carriers. He cannot pass the disorder on to his sons because males inherit only the Y chromosome from their fathers.3
Diagnosing DMD
“It can take as long as 1-3 years for a child to be diagnosed with DMD,” Dr. Brandsema said. “Parents typically have concerns and know that something is ‘off’ about their child and they’re sent to various specialists, but it usually takes time for an accurate diagnosis to be made.” The mean age at diagnosis of DMD is between ages 4 and 5 years.6
Early identification of infants at risk for developing DMD can help move the needle toward earlier diagnosis. Newborn screening for DMD has been researched and piloted in several programs.6 In 2023, DMD was nominated for inclusion in the Recommended Universal Screening Panel (RUSP) for universal newborn screening. But in May 2024, the advisory committee on Heritable Disorders in Newborns and Children decided to postpone the vote to include DMD in the RUSP, requesting additional information to ensure an evidence-based decision.
In the absence of universal newborn screening for DMD, alternative approaches have been proposed to reduce the delay in clinical diagnosis and specialist referral, including increasing awareness among healthcare providers (eg, pediatricians, pediatric neurologists, and primary care physicians).6
The National Task Force for Early Identification of Childhood Neuromuscular Disorders delineates the steps necessary to identify pediatric muscle weakness and signs of neuromuscular disease. Primary care providers are encouraged to engage in regular developmental surveillance. A surveillance aid lays out the timetable for recommended visits, typical developmental milestones, and components of surveillance. Clinical evaluation includes a detailed patient history, family history, and physical examination.
If a neuromuscular condition is suspected, laboratory work should include creatinine phosphokinase (CK).6 Elevated serum CK points to leakage of CK through the muscle membrane, suggesting muscle damage. If CK is elevated, genetic testing should be performed; and, if negative, it should be followed by genetic sequencing that tests for small-scale mutations in the DMD gene. If that test is negative, a muscle biopsy should be performed to test for deep intronic mutations in the DMD gene.4
The diagnostic process and immediate steps after a confirmed DMD diagnosis is found in Figure 1.
Targeting Inflammation in DMD
Traditionally, corticosteroids have been the only available medical treatment for DMD and they remain a cornerstone of DMD management. A meta-analysis found “moderate evidence” that corticosteroid therapy improves muscle strength and function in the short term (12 months), and strength up to 2 years.10
The two most common corticosteroids for DMD are prednisone and deflazacort. Deflazacort (Emflaza, PTC Therapeutics) was approved in 2017 to treat patients ages 5 years and older with DMD, subsequently expanded to 2 years and older. Deflazacort has been found to be more effective than prednisone in improving functional outcomes, delaying the onset of cardiomyopathy, and improving overall survival, with fewer adverse effects.11
In 2023, vamorolone (Agamree, Catalyst Pharmaceuticals) was approved by the Food and Drug Administration (FDA) to treat DMD patients (ages 2 years and older). Vamorolone is a dissociative steroidal anti-inflammatory that reduces bone morbidities and is regarded as a safer alternative than prednisone. A clinical trial comparing two doses of vamorolone with prednisone for 24 weeks found that vamorolone 6 mg/kg per day met the primary endpoint (time to stand velocity) and four sequential secondary motor function endpoints, with less bone morbidity, compared to prednisone.12 A more recent trial found improvements in motor outcomes at 48 weeks with a dose of 6 mg/kg per day of vamorolone. Bone morbidities of prednisone were reversed when the patient transitioned to vamorolone.13
“Steroid treatment has been proven to help, usually taken daily, although other schedules have been tried,” Dr. Brandsema said. However, all steroids are fraught with adverse effects and are suboptimal in the long term in reducing the disease burden.
The anti-inflammatory agent givinostat (Duvyzat, ITF Therapeutics), an oral histone deacetylase (HDAC) inhibitor, was approved in March 2024 for the treatment of DMD in patients 6 years of age and older. It is the first nonsteroidal drug to treat patients with all genetic variants of the disease, and it has a unique mechanism of action. Deficits in dystrophin can lead to increased HDAC activity in DMD, reducing the expression of genes involved in muscle regeneration. Givinostat therefore can help to counteract the pathogenic events downstream of dystrophin deficiency by inhibiting HDAC.14
Approval for givinostat was based on the phase 3 EPIDYS trial, which randomized 179 boys with DMD to receive either givinostat or placebo. Although results of a functional task worsened in both groups over the 12-month study period, the decline was significantly smaller with givinostat versus placebo. The most common adverse events were diarrhea and vomiting.14 Dr. Brandsema noted that monitoring of triglycerides and platelet count is required, as hypertriglyceridemia and thrombocytopenia can occur. This treatment was studied in tandem with corticosteroids as a combination approach to muscle stabilization.
New Pharmacotherapeutic Options: Exon-Skipping Agents
Today’s treatments have expanded beyond corticosteroids, with newer therapeutic options that include targeted exon-skipping therapies and, more recently, gene therapies. “These new treatment paradigms have changed the face of DMD treatment,” Dr. Brandsema said.
“Exon-skipping drugs in their current form have only a modest effect, but at least they’re a step in the right direction and a breakthrough, in terms of slowing disease progression,” Dr. Brandsema said.
Current exon-skipping agents use antisense phosphorodiamidate morpholino oligomers (PMOs) to restore a DMD open reading frame. Next-generation drugs called cell-penetrating peptide-conjugated PMOs (PPMOs) are being actively researched, Dr. Brandsema said. These agents have shown enhanced cellular uptake and more efficient dystrophin restoration, compared with unconjugated PMOs.15
There are currently four FDA-approved exon-skipping agents for DMD, all of which are administered via a weekly intravenous infusion: Casimersen (Amondys-45, SRP-4045), approved by the FDA in 2021; Eteplirsen (Exondys 51), approved in 2016; Golodirsen (Vyondys 53,SRP-4053), approved in 2019; and Vitolarsen (Viltepso), approved in 2020. They can be associated with multiple side effects, depending on the drug, including upper respiratory infection, fever, cough, rash, and gastrointestinal issues.16 These agents have the potential to help 30% of DMD patients, restoring low levels of dystrophin.16
Gene Transfer Therapies
Gene transfer therapies, a new class of agents, utilize a nonpathogenic viral vector (adeno-associated virus) to transfer specific genes to patients with DMD. Gene therapy involves overexpressing the micro-dystrophin gene to restore functional dystrophin expression.16
Multiple clinical trials of gene therapy are currently in progress. In 2023, delandistrogene moxeparvovec-rokl (Elevidys, Serepta) was granted accelerated FDA approval for ambulatory individuals with DMD between the ages of 4 and 5 years of age and a confirmed mutation in the DMD gene. It received expanded approval in June 2024 to include ambulatory and nonambulatory individuals aged 4 years and older with DMD and a confirmed mutation in the DMD gene (with the exception of exon 8 or 9 mutations).
The approval was based on preliminary data from two double-blind, placebo-controlled studies and two open-label studies, which enrolled a total of 218 male patients (including those who received placebo) with a confirmed disease-causing mutation in the DMD gene.
Delandistrogene moxeparvovec-rokl is delivered as a one-time infusion and has been associated with side effects and “a lot of potential issues,” Dr. Brandsema said. “We’ve seen cardiac effects, immune system effects, increased muscle inflammation and hepatic complications, and some people who became quite unwell were hospitalized for a long time.”
Fortunately, he added, “these seem to be rare but they do happen. Once the medication has been delivered, it’s permanently in the body, so you’re managing the side effects potentially on a long-term basis.”
It is critical to discuss the risks and benefits of this treatment with the family and caregivers and with the patient as well, if he old enough and able to participate in the decision-making progress. “We don’t want to give unrealistic expectations and we want people to be aware of the potential downside of this treatment,” he said. “This is a very complex discussion because the trajectory of the disease is so devastating and this treatment does hold out hope that other therapies don’t necessarily have.”
Nonpharmacologic Interventions
Physical therapy is a mainstay in DMD treatment, addressing protection of fragile muscles, preservation of strength, and prevention of muscle contractures.16 Given the respiratory impairments that occur with DMD progression, respiratory monitoring and therapy are essential; however, the number and type of evaluations and interventions vary with the stage of the disease, intensifying as the disease progresses.16 Similarly, cardiac monitoring should begin early, with patients screened for cardiac complications, and should intensify through the stages of disease progression.16
Bone health is compromised in patients with DMD, both as a result of corticosteroid treatment and as part of the disease itself. Fractures may be asymptomatic and may go unnoticed. Thus, bone health surveillance and maintenance are critical components of DMD management.16
Patients with DMD often experience gastrointestinal issues. They may experience weight gain because of lack of mobility and corticosteroid use in early stages, or weight loss as a result of diet or fluid imbalance, low bone density, or dysphagia in later stages. Patients should be closely followed by a nutritionist, a gastroenterologist as needed, and a physical therapist.16
Psychosocial support “should be developed and implemented across the lifespan in a manner that promotes thinking about the future and sets expectations that individuals will actively participate in their care and daily activities.”9 This includes psychological care, neuropsychological evaluations, and educational support.
Assisting Patients and Families Through the DMD Journey
DMD care is best delivered in a multidisciplinary setting, where physicians of relevant specialties, physical and occupational therapists, nutritionists, social workers, and genetic counselors collaborate. At Children’s Hospital of Philadelphia, DMD care is delivered through this collaborative model.
Unfortunately, Dr. Brandsema said, many patients don’t have this type of multidisciplinary resource available. “One specialist, such as a pulmonologist or neurologist, might have to be the sole source of care.” Or parents may have to ferry their child to multiple specialists in disparate locations, placing extra stress on an already-stressed family system.
“It’s helpful to connect the family with a comprehensive care center, if possible,” Dr. Brandsema advised. If that’s not available, then he suggests recommending educational opportunities and resources through national organizations such as the Muscular Dystrophy Association; Parent Project MD; NORD; Friends, Family and Duchenne; and Cure Duchenne. Families and caregivers, along with affected individuals, can get education and support from people who understand the day-to-day reality of living with this disease.
One of the major challenges that families face is navigating the high cost of treating DMD, especially the new medications, Dr. Brandsema said. “The authorization process can be intensive and long, and the family may need to take an active role, together with the provider team, in advocating for the patient to get access.”
Among her many responsibilities, Ms. Kaschak engages in care coordination tasks and management, helps patients and caregivers understand care plans, and provides psychosocial support and education about the disease process. She assists families in completing paperwork and navigating specialty authorizations, helping families understand and navigate the complex insurance process. “My role is to bridge gaps in care,” she said.
Dr. Brandsema noted that it’s important for couples to receive genetic counseling if they’re planning to have multiple children because there is a 50% chance that their next boy will be affected. About two thirds of mothers with children who have DMD are carriers, but many are not aware of it. Receiving counseling will enable them to understand their own risks of health complications, as well as the risk to future children.
Managing DMD Across the Lifespan
Another dimension of DMD care is providing resources and help to young people with DMD as they transition into adulthood. “In the past, we had limited treatment and mortality typically took place in the early 20s, so there weren’t a lot of patients who were adults. But as medication options have expanded and management of cardiac and respiratory failure has improved, we see a more significant proportion of adults who require adult-appropriate clinics — or, at the very least, specialists who are conversant in care or can provide care across the lifespan,” Dr. Brandsema said.
The DMD Care Considerations Working Group provides recommendations regarding care across the lifespan,9 as does the Adult North Star Network, of Muscular Dystrophy UK.17,18
Dr. Brandsema emphasized that, despite their disability, many adults with DMD “still engage with the community, and live life to its fullest.” It is to be hoped that, with ongoing research, earlier diagnosis, and improved treatment options, the future will look bright for people with DMD.
Dr. Brandsema has served as a consultant for Audentes, AveXis/Novartis, Biogen, Cytokinetics, Dyne, Edgewise, Fibrogen, Genentech, Marathon, Momenta/Janssen, NS Pharma, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Takeda, and WaVe. He is on the medical advisory council member for Cure SMA and is a site investigator for clinical trials with Alexion, Astellas, AveXis/Novartis, Biogen, Biohaven, Catabasis, CSL Behring, Cytokinetics, Dyne, Fibrogen, Genentech, Ionis, Lilly, ML Bio, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Summit, and WaVe. Ms. Kaschak has nothing to disclose.
References
1. Venugopal V and Pavlakis S. Duchenne Muscular Dystrophy. 2023 Jul 10. In: StatPearls [Internet]. Treasure Island, Florida: StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK482346/.
2. Gao QQ and McNally EM. Compr Physiol. 2015 Jul 1;5(3):1223-39. doi: 10.1002/cphy.c140048.
3. Duan D et al. Nat Rev Dis Primers. 2021 Feb 18;7(1):13. doi: 10.1038/s41572-021-00248-3.
4. Aartsma-Rus A et al. J Pediatr. 2019 Jan:204:305-313.e14. doi: 10.1016/j.jpeds.2018.10.043.
5. Broomfield J et al. Neurology. 2021 Dec 7;97(23):e2304-e2314. doi: 10.1212/WNL.0000000000012910.
6. Mercuri E et al. Front Pediatr. 2023 Nov 10:11:1276144. doi: 10.1212/WNL.0000000000012910.
7. Birnkrant DJ et al. Lancet Neurol. 2018 Mar;17(3):251-267. doi: 10.1016/S1474-4422(18)30024-3.
8. Birnkrant DJ et al. Lancet Neurol. 2018 Apr;17(4):347-361. doi: 10.1016/S1474-4422(18)30025-5.
9. Birnkrant DJ et al. Lancet Neurol. 2018 May;17(5):445-455. doi: 10.1016/S1474-4422(18)30026-7.
10. Matthews E et al. Cochrane Database Syst Rev. 2016 May 5;2016(5):CD003725. doi: 10.1002/14651858.CD003725.pub4.
11. Bylo M et al. Ann Pharmacother. 2020 Aug;54(8):788-794. doi: 10.1177/1060028019900500.
12. Guglieri M et al. JAMA Neurol. 2022 Oct 1;79(10):1005-1014. doi: 10.1001/jamaneurol.2022.2480.
13. Dang UJ et al. Neurology. 2024 Mar 12;102(5):e208112. doi: 10.1212/WNL.0000000000208112.
14. Mercuri E et al. Lancet Neurol. 2024 Apr;23(4):393-403. doi: 10.1016/S1474-4422(24)00036-X.
15. Gushchina LV et al. Mol Ther Nucleic Acids. 2022 Nov 9:30:479-492. doi: 10.1016/j.omtn.2022.10.025.
16. Patterson G et al. Eur J Pharmacol. 2023 May 15:947:175675. doi: 10.1016/j.ejphar.2023.175675.
17. Quinlivan R et al. J Neuromuscul Dis. 2021;8(6):899-926. doi: 10.3233/JND-200609.
18. Narayan S et al. J Neuromuscul Dis. 2022;9(3):365-381. doi: 10.3233/JND-210707.