User login
The Diagnosis: Lichen Planus Pemphigoides
Lichen planus pemphigoides (LPP) is a rare autoimmune subepithelial blistering disorder with clinical, pathologic, and immunologic features of lichen planus (LP) and bullous pemphigoid (BP).1 It mainly arises in adults and usually is idiopathic but has been associated with certain infections,2 drugs such as angiotensin-converting enzyme inhibitors,3 phototherapy,4 and malignancy.5 Patients classically present with lichenoid lesions, tense vesiculobullae, and erosions.6 Vesiculobullae formation usually follows the development of lichenoid lesions, occurs on both lichenoid lesions and unaffected skin, and predominantly involves the lower extremities, as in our patient.1,6
The pathogenesis of LPP is not fully understood but likely represents a distinct entity rather than a subtype of BP or the simultaneous occurrence of LP and BP. Lichen planus pemphigoides generally has an earlier onset and better treatment response compared to BP.7 Further, autoantibodies in patients with LPP react to a novel epitope within the C-terminal portion of the BP-180 NC16A domain. Accordingly, it has been postulated that an inflammatory cutaneous process resulting from infection, phototherapy, or LP itself leads to damage of the epidermis and triggers a secondary blistering autoimmune dermatosis mediated by antibody formation against basement membrane (BM) antigens, such as BP-180.7
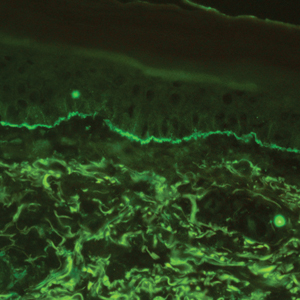
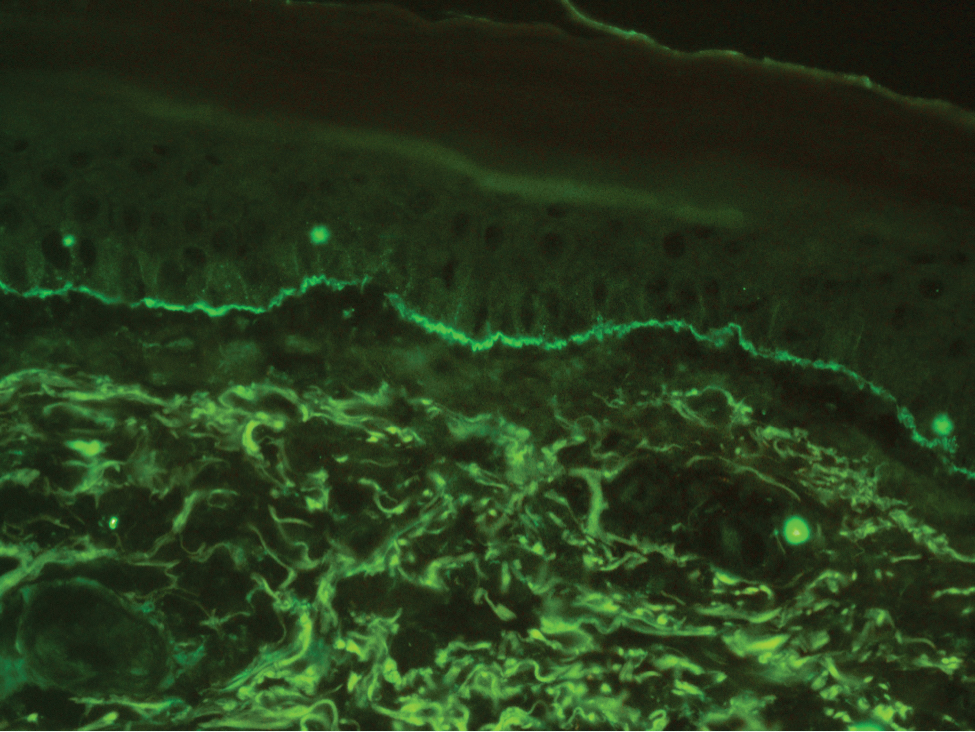
The diagnosis of LPP ultimately is confirmed with immunohistologic analysis. Biopsy of LPP shows findings consistent with both LP and BP (quiz image [top]). In the lichenoid portion, biopsy reveals orthohyperkeratosis, hypergranulosis, and acanthosis of the epidermis; a bandlike infiltrate consisting primarily of lymphocytes in the upper dermis; and apoptotic keratinocytes (colloid bodies) and vacuolar degeneration at the dermoepidermal junction (DEJ).1 Biopsy of bullae reveals eosinophilic spongiosis, a subepithelial blister plane with eosinophils, and a mixed superficial inflammatory cell infiltrate. Direct immunofluorescence from perilesional skin reveals linear deposition of IgG and/or C3 at the DEJ (quiz image [bottom]).1 Measurement of anti-BM antibodies against BP-180 and BP-230 can be useful in suspected cases, as 50% to 60% of patients have circulating antibodies against these antigens.6 Remission usually is achieved with topical and systemic corticosteroids and/or steroid-sparing agents, with rare recurrence following lesion resolution.1 More recently, successful treatment with biologics such as ustekinumab has been reported.8
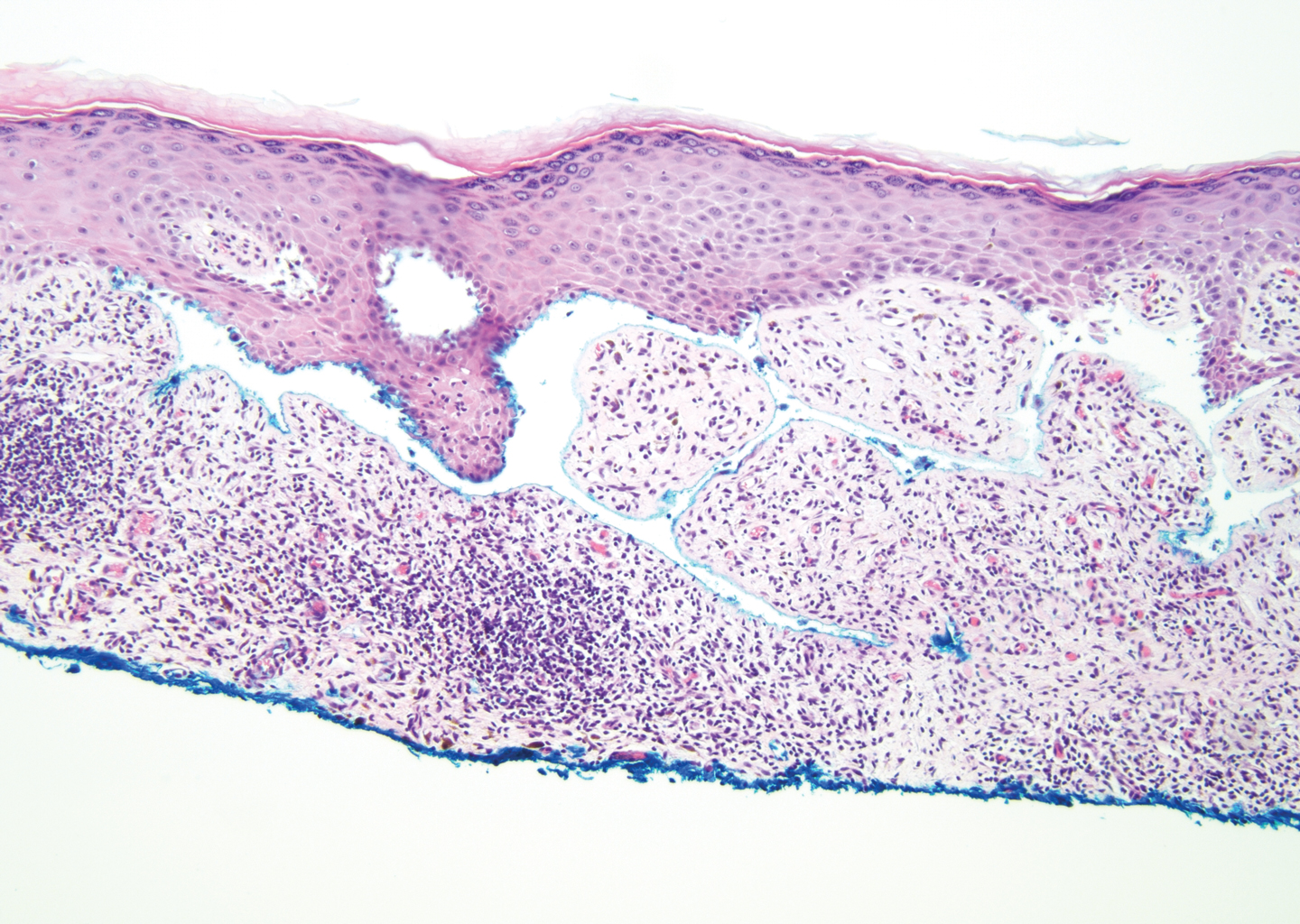
The predominant differential diagnosis for LPP is bullous LP, a variant of LP in which vesiculobullous disease occurs exclusively on preexisting LP lesions, commonly on the legs due to severe vacuolar degeneration at the DEJ. On histopathology, the characteristic features of LP (eg, orthohyperkeratosis, hypergranulosis, acanthosis, bandlike lymphocytic infiltrate, colloid bodies) along with subepidermal clefting will be seen. However, in bullous LP (Figure 1) there is an absence of linear IgG and/or C3 deposition at the DEJ on direct immunofluorescence. Furthermore, patients lack circulating antibodies against BP-180 and BP-230.9
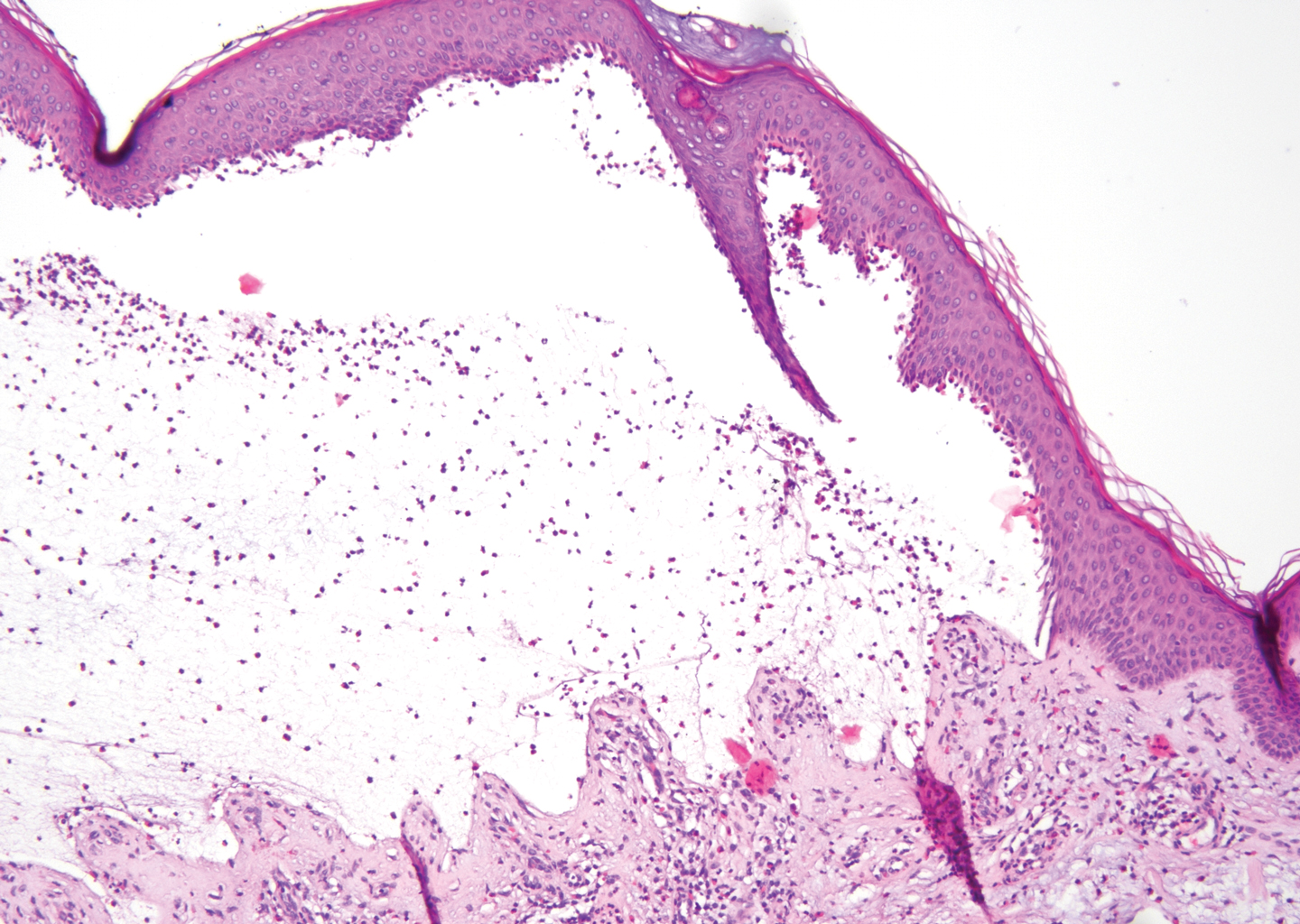
Lichen planus pemphigoides also can be confused with BP. Bullous pemphigoid is the most common autoimmune blistering disorder; typically arises in older adults; and is caused by autoantibody formation against hemidesmosomal proteins, particularly BP-180 and BP-230. Patients classically present with tense bullae and erosions on an erythematous, urticarial, or normal base. These lesions often are pruritic and concentrated on the trunk, axillary and inguinal folds, and extremity flexures. Histopathologic examination of a bulla edge reveals the classic findings seen in BP (eg, eosinophilic spongiosis, subepithelial blister plane with eosinophils)(Figure 2). Direct immunofluorescence of perilesional skin reveals linear IgG and/or C3 deposition along the DEJ. A large subset of patients also has circulating antibodies against BP-180 and BP-230. In contrast to LPP, however, patients with BP do not develop lichenoid lesions clinically or a lichenoid tissue reaction histopathologically.10
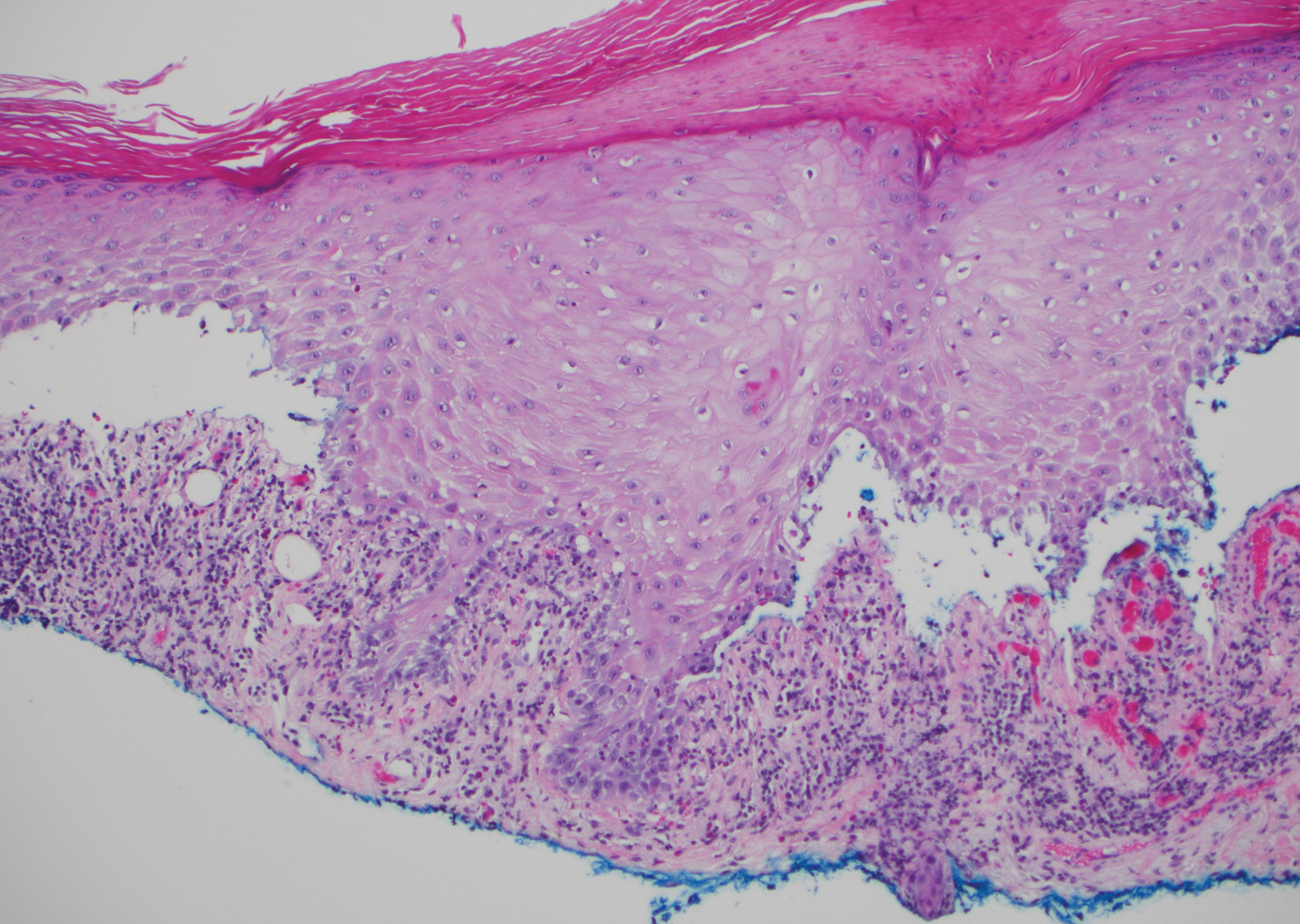
Bullous systemic lupus erythematosus (SLE), a rare cutaneous manifestation of SLE, typically arises in young women of African descent and is due to autoantibody formation against type VII collagen and other BM-zone antigens. Patients generally present with acute onset of tense vesiculobullae on a normal or erythematous base, which often are transient and heal without milia or scarring. Common sites of involvement include the trunk, arms, neck, face, and vermilion border, as well as the oral mucosa. The diagnosis of bullous SLE requires that patients fulfill the criteria for SLE and is confirmed by immunohistologic analysis. Biopsy of a bulla edge reveals a subepidermal blister containing neutrophils and increased mucin within the reticular dermis (Figure 3). Direct immunofluorescence of perilesional skin most commonly reveals linear and/or granular deposition of IgG, IgA, C3, and IgM at the DEJ.11

Bullous tinea is a manifestation of cutaneous dermatophytosis that usually occurs in the setting of tinea pedis. Common causative dermatophytes include Trichophyton mentagrophytes, Trichophyton rubrum, and Epidermophyton floccosum. Diagnosis is made by demonstration of fungal hyphae on potassium hydroxide preparation of the blister roof, biopsy with periodic acid-Schiff stain, or fungal culture. If routine histopathologic analysis is performed, epidermal spongiosis with varying degrees of papillary dermal edema is seen, along with abundant fungal elements in the stratum corneum (Figure 4). Direct immunofluorescence of perilesional skin usually is negative, but C3 deposition in a linear and/or granular pattern along the DEJ has been reported.12

Lichen planus pemphigoides is a rare disease entity and often presents a diagnostic challenge to clinicians. The differential for LPP includes bullous LP as well as other bullous disorders. Ultimately, the diagnosis is confirmed through immunohistologic analysis. Timely diagnosis of LPP is crucial, as most patients can achieve long-term remission with appropriate treatment.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Mohanarao TS, Kumar GA, Chennamsetty K, et al. Childhood lichen planus pemphigoides triggered by chickenpox. Indian Dermatol Online J. 2014;5:S98-S100.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Shimada H, Shono T, Sakai T, et al. Lichen planus pemphigoides concomitant with rectal adenocarcinoma: fortuitous or a true association? Eur J Dermatol. 2015;25:501-503.
- Matos-Pires E, Campos S, Lencastre A, et al. Lichen planus pemphigoides. J Dtsch Dermatol Ges. 2018;16:335-337.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Knisley RR, Petropolis AA, Mackey VT. Lichen planus pemphigoides treated with ustekinumab. Cutis. 2017;100:415-418.
- Wagner G, Rose C, Sachse MM. Clinical variants of lichen planus. J Dtsch Dermatol Ges. 2013;11:309-319.
- Bagci IS, Horvath ON, Ruzicka T, et al. Bullous pemphigoid. Autoimmun Rev. 2017;16:445-455.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Miller DD, Bhawan J. Bullous tinea pedis with direct immunofluorescence positivity: when is a positive result not autoimmune bullous disease? Am J Dermatopathol. 2013;35:587-594.
The Diagnosis: Lichen Planus Pemphigoides
Lichen planus pemphigoides (LPP) is a rare autoimmune subepithelial blistering disorder with clinical, pathologic, and immunologic features of lichen planus (LP) and bullous pemphigoid (BP).1 It mainly arises in adults and usually is idiopathic but has been associated with certain infections,2 drugs such as angiotensin-converting enzyme inhibitors,3 phototherapy,4 and malignancy.5 Patients classically present with lichenoid lesions, tense vesiculobullae, and erosions.6 Vesiculobullae formation usually follows the development of lichenoid lesions, occurs on both lichenoid lesions and unaffected skin, and predominantly involves the lower extremities, as in our patient.1,6
The pathogenesis of LPP is not fully understood but likely represents a distinct entity rather than a subtype of BP or the simultaneous occurrence of LP and BP. Lichen planus pemphigoides generally has an earlier onset and better treatment response compared to BP.7 Further, autoantibodies in patients with LPP react to a novel epitope within the C-terminal portion of the BP-180 NC16A domain. Accordingly, it has been postulated that an inflammatory cutaneous process resulting from infection, phototherapy, or LP itself leads to damage of the epidermis and triggers a secondary blistering autoimmune dermatosis mediated by antibody formation against basement membrane (BM) antigens, such as BP-180.7
The diagnosis of LPP ultimately is confirmed with immunohistologic analysis. Biopsy of LPP shows findings consistent with both LP and BP (quiz image [top]). In the lichenoid portion, biopsy reveals orthohyperkeratosis, hypergranulosis, and acanthosis of the epidermis; a bandlike infiltrate consisting primarily of lymphocytes in the upper dermis; and apoptotic keratinocytes (colloid bodies) and vacuolar degeneration at the dermoepidermal junction (DEJ).1 Biopsy of bullae reveals eosinophilic spongiosis, a subepithelial blister plane with eosinophils, and a mixed superficial inflammatory cell infiltrate. Direct immunofluorescence from perilesional skin reveals linear deposition of IgG and/or C3 at the DEJ (quiz image [bottom]).1 Measurement of anti-BM antibodies against BP-180 and BP-230 can be useful in suspected cases, as 50% to 60% of patients have circulating antibodies against these antigens.6 Remission usually is achieved with topical and systemic corticosteroids and/or steroid-sparing agents, with rare recurrence following lesion resolution.1 More recently, successful treatment with biologics such as ustekinumab has been reported.8
The predominant differential diagnosis for LPP is bullous LP, a variant of LP in which vesiculobullous disease occurs exclusively on preexisting LP lesions, commonly on the legs due to severe vacuolar degeneration at the DEJ. On histopathology, the characteristic features of LP (eg, orthohyperkeratosis, hypergranulosis, acanthosis, bandlike lymphocytic infiltrate, colloid bodies) along with subepidermal clefting will be seen. However, in bullous LP (Figure 1) there is an absence of linear IgG and/or C3 deposition at the DEJ on direct immunofluorescence. Furthermore, patients lack circulating antibodies against BP-180 and BP-230.9
Lichen planus pemphigoides also can be confused with BP. Bullous pemphigoid is the most common autoimmune blistering disorder; typically arises in older adults; and is caused by autoantibody formation against hemidesmosomal proteins, particularly BP-180 and BP-230. Patients classically present with tense bullae and erosions on an erythematous, urticarial, or normal base. These lesions often are pruritic and concentrated on the trunk, axillary and inguinal folds, and extremity flexures. Histopathologic examination of a bulla edge reveals the classic findings seen in BP (eg, eosinophilic spongiosis, subepithelial blister plane with eosinophils)(Figure 2). Direct immunofluorescence of perilesional skin reveals linear IgG and/or C3 deposition along the DEJ. A large subset of patients also has circulating antibodies against BP-180 and BP-230. In contrast to LPP, however, patients with BP do not develop lichenoid lesions clinically or a lichenoid tissue reaction histopathologically.10
Bullous systemic lupus erythematosus (SLE), a rare cutaneous manifestation of SLE, typically arises in young women of African descent and is due to autoantibody formation against type VII collagen and other BM-zone antigens. Patients generally present with acute onset of tense vesiculobullae on a normal or erythematous base, which often are transient and heal without milia or scarring. Common sites of involvement include the trunk, arms, neck, face, and vermilion border, as well as the oral mucosa. The diagnosis of bullous SLE requires that patients fulfill the criteria for SLE and is confirmed by immunohistologic analysis. Biopsy of a bulla edge reveals a subepidermal blister containing neutrophils and increased mucin within the reticular dermis (Figure 3). Direct immunofluorescence of perilesional skin most commonly reveals linear and/or granular deposition of IgG, IgA, C3, and IgM at the DEJ.11
Bullous tinea is a manifestation of cutaneous dermatophytosis that usually occurs in the setting of tinea pedis. Common causative dermatophytes include Trichophyton mentagrophytes, Trichophyton rubrum, and Epidermophyton floccosum. Diagnosis is made by demonstration of fungal hyphae on potassium hydroxide preparation of the blister roof, biopsy with periodic acid-Schiff stain, or fungal culture. If routine histopathologic analysis is performed, epidermal spongiosis with varying degrees of papillary dermal edema is seen, along with abundant fungal elements in the stratum corneum (Figure 4). Direct immunofluorescence of perilesional skin usually is negative, but C3 deposition in a linear and/or granular pattern along the DEJ has been reported.12
Lichen planus pemphigoides is a rare disease entity and often presents a diagnostic challenge to clinicians. The differential for LPP includes bullous LP as well as other bullous disorders. Ultimately, the diagnosis is confirmed through immunohistologic analysis. Timely diagnosis of LPP is crucial, as most patients can achieve long-term remission with appropriate treatment.
The Diagnosis: Lichen Planus Pemphigoides
Lichen planus pemphigoides (LPP) is a rare autoimmune subepithelial blistering disorder with clinical, pathologic, and immunologic features of lichen planus (LP) and bullous pemphigoid (BP).1 It mainly arises in adults and usually is idiopathic but has been associated with certain infections,2 drugs such as angiotensin-converting enzyme inhibitors,3 phototherapy,4 and malignancy.5 Patients classically present with lichenoid lesions, tense vesiculobullae, and erosions.6 Vesiculobullae formation usually follows the development of lichenoid lesions, occurs on both lichenoid lesions and unaffected skin, and predominantly involves the lower extremities, as in our patient.1,6
The pathogenesis of LPP is not fully understood but likely represents a distinct entity rather than a subtype of BP or the simultaneous occurrence of LP and BP. Lichen planus pemphigoides generally has an earlier onset and better treatment response compared to BP.7 Further, autoantibodies in patients with LPP react to a novel epitope within the C-terminal portion of the BP-180 NC16A domain. Accordingly, it has been postulated that an inflammatory cutaneous process resulting from infection, phototherapy, or LP itself leads to damage of the epidermis and triggers a secondary blistering autoimmune dermatosis mediated by antibody formation against basement membrane (BM) antigens, such as BP-180.7
The diagnosis of LPP ultimately is confirmed with immunohistologic analysis. Biopsy of LPP shows findings consistent with both LP and BP (quiz image [top]). In the lichenoid portion, biopsy reveals orthohyperkeratosis, hypergranulosis, and acanthosis of the epidermis; a bandlike infiltrate consisting primarily of lymphocytes in the upper dermis; and apoptotic keratinocytes (colloid bodies) and vacuolar degeneration at the dermoepidermal junction (DEJ).1 Biopsy of bullae reveals eosinophilic spongiosis, a subepithelial blister plane with eosinophils, and a mixed superficial inflammatory cell infiltrate. Direct immunofluorescence from perilesional skin reveals linear deposition of IgG and/or C3 at the DEJ (quiz image [bottom]).1 Measurement of anti-BM antibodies against BP-180 and BP-230 can be useful in suspected cases, as 50% to 60% of patients have circulating antibodies against these antigens.6 Remission usually is achieved with topical and systemic corticosteroids and/or steroid-sparing agents, with rare recurrence following lesion resolution.1 More recently, successful treatment with biologics such as ustekinumab has been reported.8
The predominant differential diagnosis for LPP is bullous LP, a variant of LP in which vesiculobullous disease occurs exclusively on preexisting LP lesions, commonly on the legs due to severe vacuolar degeneration at the DEJ. On histopathology, the characteristic features of LP (eg, orthohyperkeratosis, hypergranulosis, acanthosis, bandlike lymphocytic infiltrate, colloid bodies) along with subepidermal clefting will be seen. However, in bullous LP (Figure 1) there is an absence of linear IgG and/or C3 deposition at the DEJ on direct immunofluorescence. Furthermore, patients lack circulating antibodies against BP-180 and BP-230.9
Lichen planus pemphigoides also can be confused with BP. Bullous pemphigoid is the most common autoimmune blistering disorder; typically arises in older adults; and is caused by autoantibody formation against hemidesmosomal proteins, particularly BP-180 and BP-230. Patients classically present with tense bullae and erosions on an erythematous, urticarial, or normal base. These lesions often are pruritic and concentrated on the trunk, axillary and inguinal folds, and extremity flexures. Histopathologic examination of a bulla edge reveals the classic findings seen in BP (eg, eosinophilic spongiosis, subepithelial blister plane with eosinophils)(Figure 2). Direct immunofluorescence of perilesional skin reveals linear IgG and/or C3 deposition along the DEJ. A large subset of patients also has circulating antibodies against BP-180 and BP-230. In contrast to LPP, however, patients with BP do not develop lichenoid lesions clinically or a lichenoid tissue reaction histopathologically.10
Bullous systemic lupus erythematosus (SLE), a rare cutaneous manifestation of SLE, typically arises in young women of African descent and is due to autoantibody formation against type VII collagen and other BM-zone antigens. Patients generally present with acute onset of tense vesiculobullae on a normal or erythematous base, which often are transient and heal without milia or scarring. Common sites of involvement include the trunk, arms, neck, face, and vermilion border, as well as the oral mucosa. The diagnosis of bullous SLE requires that patients fulfill the criteria for SLE and is confirmed by immunohistologic analysis. Biopsy of a bulla edge reveals a subepidermal blister containing neutrophils and increased mucin within the reticular dermis (Figure 3). Direct immunofluorescence of perilesional skin most commonly reveals linear and/or granular deposition of IgG, IgA, C3, and IgM at the DEJ.11
Bullous tinea is a manifestation of cutaneous dermatophytosis that usually occurs in the setting of tinea pedis. Common causative dermatophytes include Trichophyton mentagrophytes, Trichophyton rubrum, and Epidermophyton floccosum. Diagnosis is made by demonstration of fungal hyphae on potassium hydroxide preparation of the blister roof, biopsy with periodic acid-Schiff stain, or fungal culture. If routine histopathologic analysis is performed, epidermal spongiosis with varying degrees of papillary dermal edema is seen, along with abundant fungal elements in the stratum corneum (Figure 4). Direct immunofluorescence of perilesional skin usually is negative, but C3 deposition in a linear and/or granular pattern along the DEJ has been reported.12
Lichen planus pemphigoides is a rare disease entity and often presents a diagnostic challenge to clinicians. The differential for LPP includes bullous LP as well as other bullous disorders. Ultimately, the diagnosis is confirmed through immunohistologic analysis. Timely diagnosis of LPP is crucial, as most patients can achieve long-term remission with appropriate treatment.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Mohanarao TS, Kumar GA, Chennamsetty K, et al. Childhood lichen planus pemphigoides triggered by chickenpox. Indian Dermatol Online J. 2014;5:S98-S100.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Shimada H, Shono T, Sakai T, et al. Lichen planus pemphigoides concomitant with rectal adenocarcinoma: fortuitous or a true association? Eur J Dermatol. 2015;25:501-503.
- Matos-Pires E, Campos S, Lencastre A, et al. Lichen planus pemphigoides. J Dtsch Dermatol Ges. 2018;16:335-337.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Knisley RR, Petropolis AA, Mackey VT. Lichen planus pemphigoides treated with ustekinumab. Cutis. 2017;100:415-418.
- Wagner G, Rose C, Sachse MM. Clinical variants of lichen planus. J Dtsch Dermatol Ges. 2013;11:309-319.
- Bagci IS, Horvath ON, Ruzicka T, et al. Bullous pemphigoid. Autoimmun Rev. 2017;16:445-455.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Miller DD, Bhawan J. Bullous tinea pedis with direct immunofluorescence positivity: when is a positive result not autoimmune bullous disease? Am J Dermatopathol. 2013;35:587-594.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Mohanarao TS, Kumar GA, Chennamsetty K, et al. Childhood lichen planus pemphigoides triggered by chickenpox. Indian Dermatol Online J. 2014;5:S98-S100.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Shimada H, Shono T, Sakai T, et al. Lichen planus pemphigoides concomitant with rectal adenocarcinoma: fortuitous or a true association? Eur J Dermatol. 2015;25:501-503.
- Matos-Pires E, Campos S, Lencastre A, et al. Lichen planus pemphigoides. J Dtsch Dermatol Ges. 2018;16:335-337.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Knisley RR, Petropolis AA, Mackey VT. Lichen planus pemphigoides treated with ustekinumab. Cutis. 2017;100:415-418.
- Wagner G, Rose C, Sachse MM. Clinical variants of lichen planus. J Dtsch Dermatol Ges. 2013;11:309-319.
- Bagci IS, Horvath ON, Ruzicka T, et al. Bullous pemphigoid. Autoimmun Rev. 2017;16:445-455.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Miller DD, Bhawan J. Bullous tinea pedis with direct immunofluorescence positivity: when is a positive result not autoimmune bullous disease? Am J Dermatopathol. 2013;35:587-594.
A 72-year-old woman presented to our dermatology clinic with a rash of several months' duration that began as itchy bumps on the wrists and spread to involve the legs. Approximately 2 months prior to presentation, she noted blisters on the feet and legs. She initially went to her primary care physician, who prescribed levofloxacin, cephalexin, and a 5-day course of prednisone. The prednisone initially helped; however the rash worsened on discontinuation. In our clinic, the patient had scattered tense bullae and numerous erosions with crust on the dorsum of the feet and legs, some of which were in conjunction with violaceous papules and plaques. There also was hypertrophic scale on the soles of the feet. A potassium hydroxide preparation of skin scrapings from the feet was negative for fungal elements. Two shave biopsies of a violaceous plaque and bulla as well as a perilesional punch biopsy from the leg were obtained.