User login
Acute Tender Papules on the Arms and Legs
The Diagnosis: Erythema Nodosum Leprosum
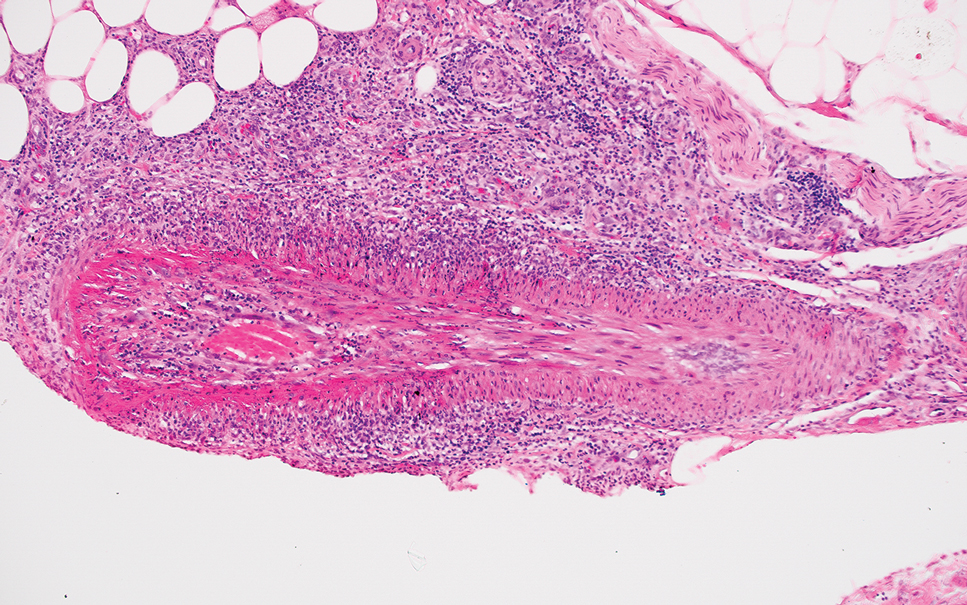
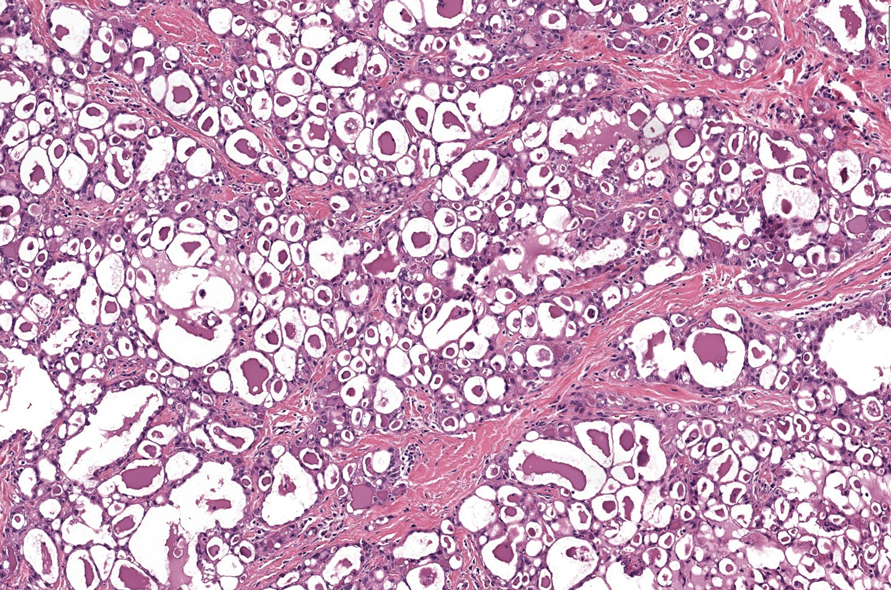
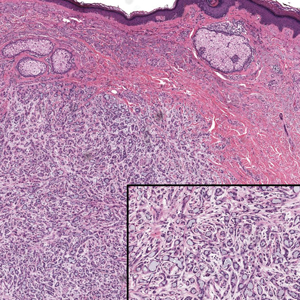
Erythema nodosum leprosum (ENL) is a type 2 reaction sometimes seen in patients infected with Mycobacterium leprae—primarily those with lepromatous or borderline lepromatous subtypes. Clinically, ENL manifests with abrupt onset of tender erythematous papules with associated fevers and general malaise. Studies have demonstrated a complex immune system reaction in ENL, but the detailed pathophysiology is not fully understood.1 Biopsies conducted within 24 hours of lesion formation are most elucidating. Foamy histiocytes admixed with neutrophils are seen in the subcutis, often causing a lobular panniculitis (quiz image).2 Neutrophils rarely are seen in other types of leprosy and thus are a useful diagnostic clue for ENL. Vasculitis of small- to medium-sized vessels can be seen but is not a necessary diagnostic criterion. Fite staining will highlight many acid-fast bacilli within the histiocytes (Figure 1).
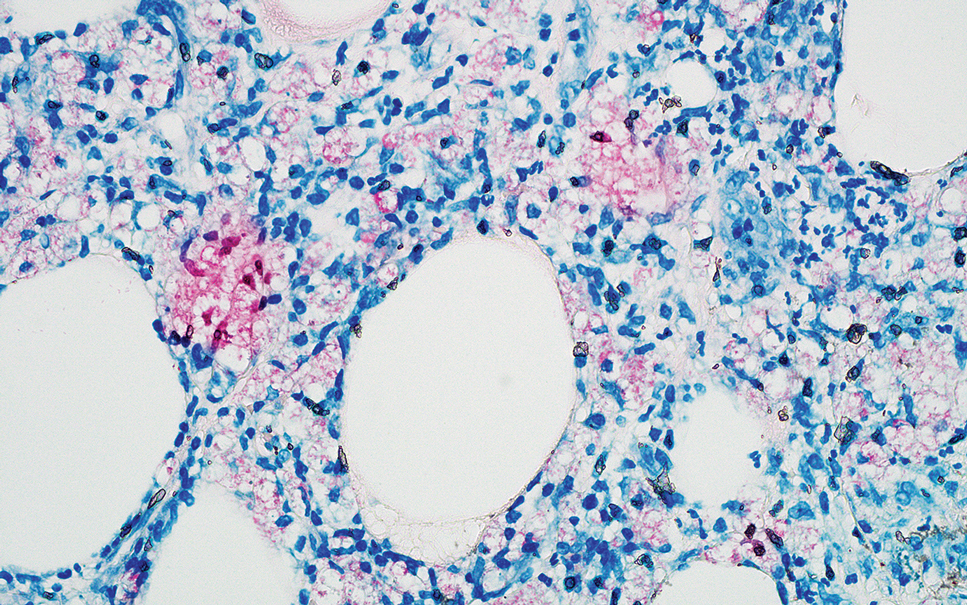
Erythema nodosum leprosum is treated with a combination of immunosuppressants such as prednisone and thalidomide. Our patient was taking triple-antibiotic therapy—dapsone, rifampin, and clofazimine—for lepromatous leprosy when the erythematous papules developed on the arms and legs. After a skin biopsy confirmed the diagnosis of ENL, he was started on prednisone 20 mg daily with plans for close follow-up. Unfortunately, the patient was subsequently lost to follow-up.
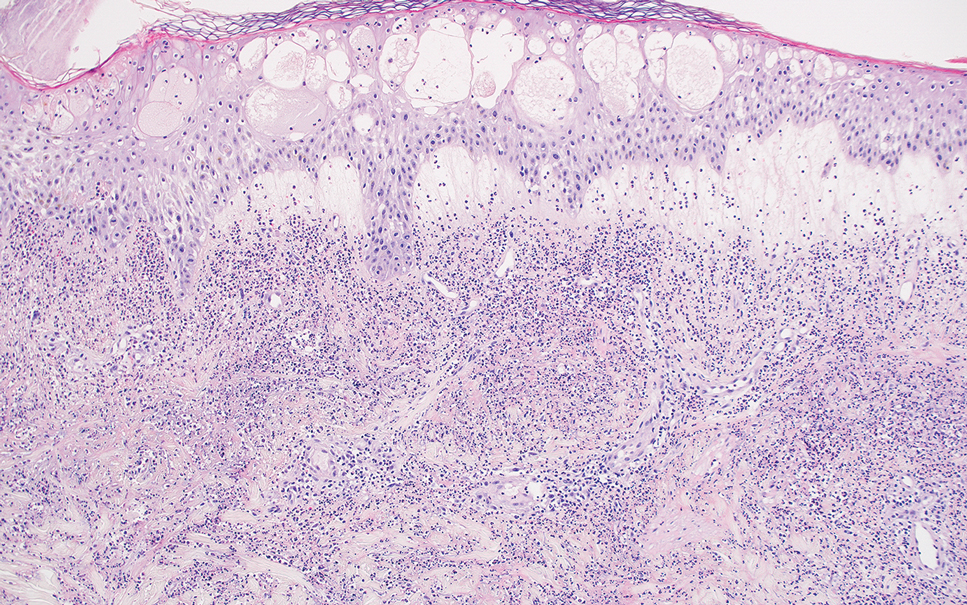
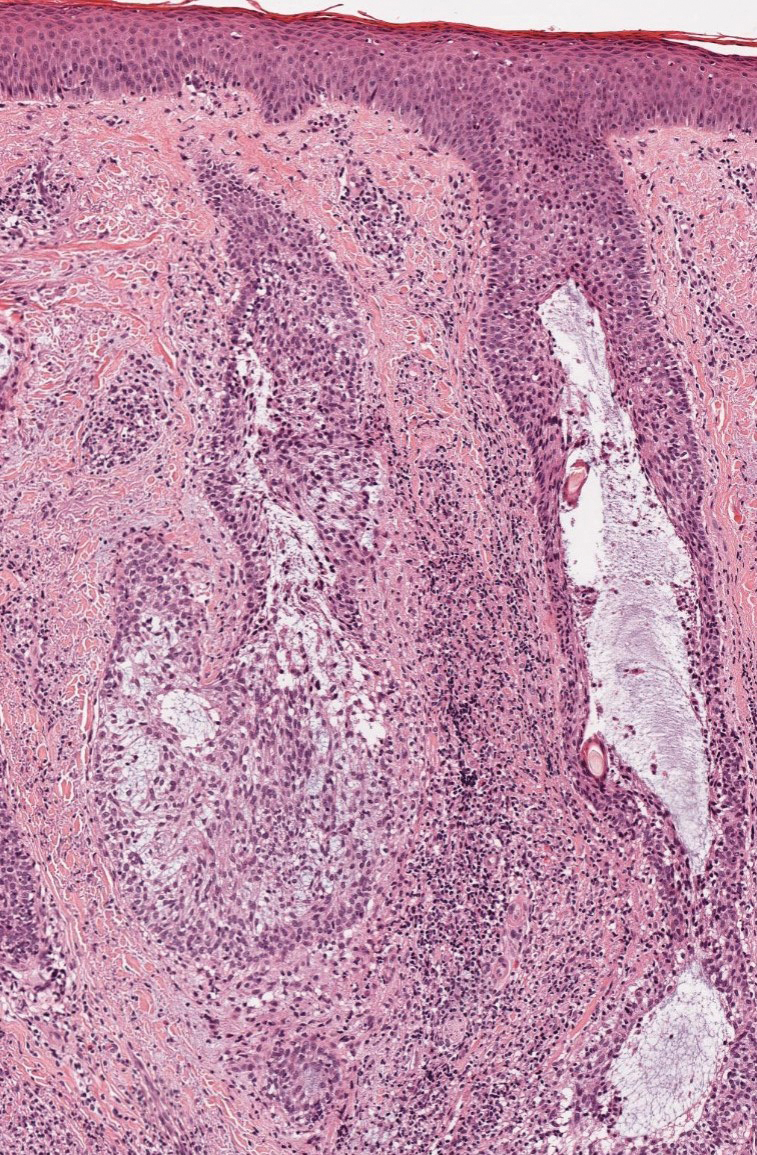
Acute febrile neutrophilic dermatosis (also known as Sweet syndrome) is an acute inflammatory disease characterized by abrupt onset of painful erythematous papules, plaques, or nodules on the skin. It often is seen in association with preceding infections (especially those in the upper respiratory or gastrointestinal tracts), hematologic malignancies, inflammatory bowel disease, or exposure to certain classes of medications (eg, granulocyte colony-stimulating factor, tyrosine kinase inhibitors, various antibiotics).3 Histologically, acute febrile neutrophilic dermatosis is characterized by dense neutrophilic infiltrates, often with notable dermal edema (Figure 2).4 Many cases also show leukocytoclastic vasculitis; however, foamy histiocytes are not a notable component of the inflammatory infiltrate, though a histiocytoid form of acute febrile neutrophilic dermatosis has been described.5 Infections must be rigorously ruled out prior to diagnosing a patient with acute febrile neutrophilic dermatosis, making it a diagnosis of exclusion.
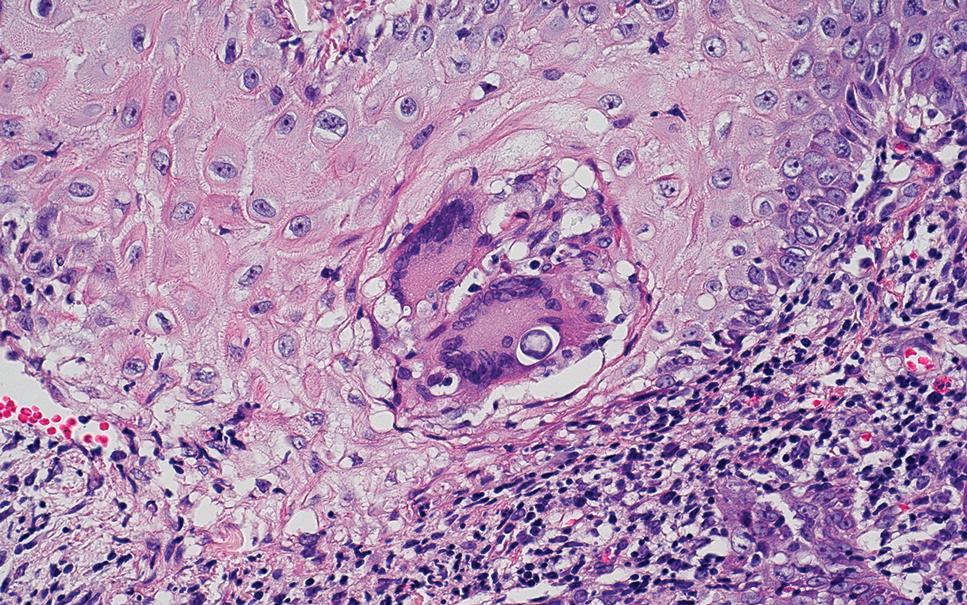
Cutaneous coccidioidomycosis is an infection caused by the dimorphic fungi Coccidioides immitis or Coccidioides posadasii. Cutaneous disease is rare but can occur from direct inoculation or dissemination from pulmonary disease in immunocompetent or immunocompromised patients. Papules, pustules, or plaques are seen clinically. Histologically, cutaneous coccidioidomycosis shows spherules that vary from 10 to 100 μm and are filled with multiple smaller endospores (Figure 3).6 Pseudoepitheliomatous hyperplasia with dense suppurative and granulomatous infiltrates also is seen.
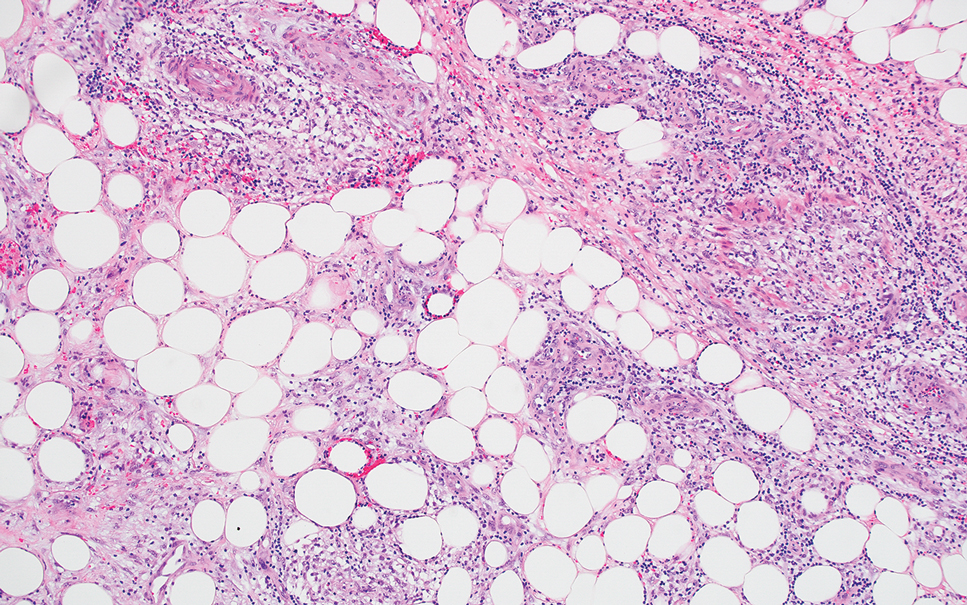
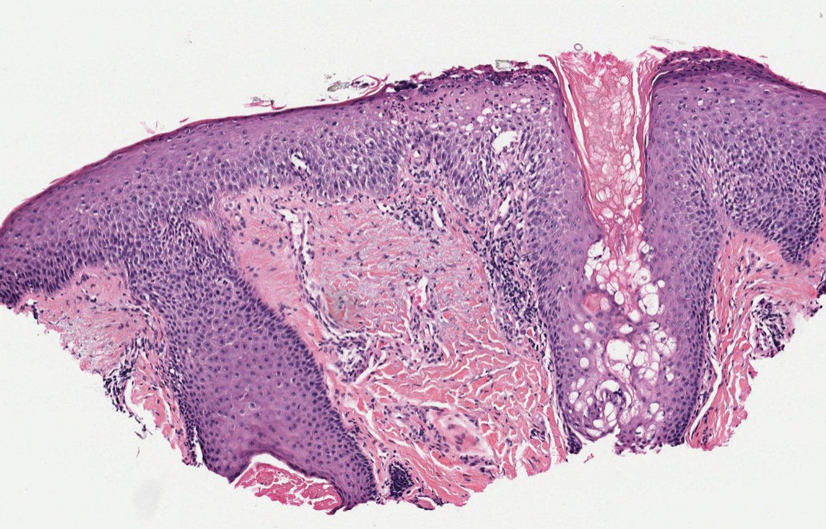
Erythema induratum is characterized by tender nodules on the lower extremities and has a substantial female predominance. Many cases are associated with Mycobacterium tuberculosis infection. The bacteria are not seen directly in the skin but are instead detectable through DNA polymerase chain reaction testing or investigation of other organ systems.7,8 Histologically, lesions show a lobular panniculitis with a mixed infiltrate. Vasculitis is seen in approximately 90% of erythema induratum cases vs approximately 25% of classic ENL cases (Figure 4),2,9 which has led some to use the term nodular vasculitis to describe this disease entity. Nodular vasculitis is considered by others to be a distinct disease entity in which there are clinical and histologic features similar to erythema induratum but no evidence of M tuberculosis infection.9
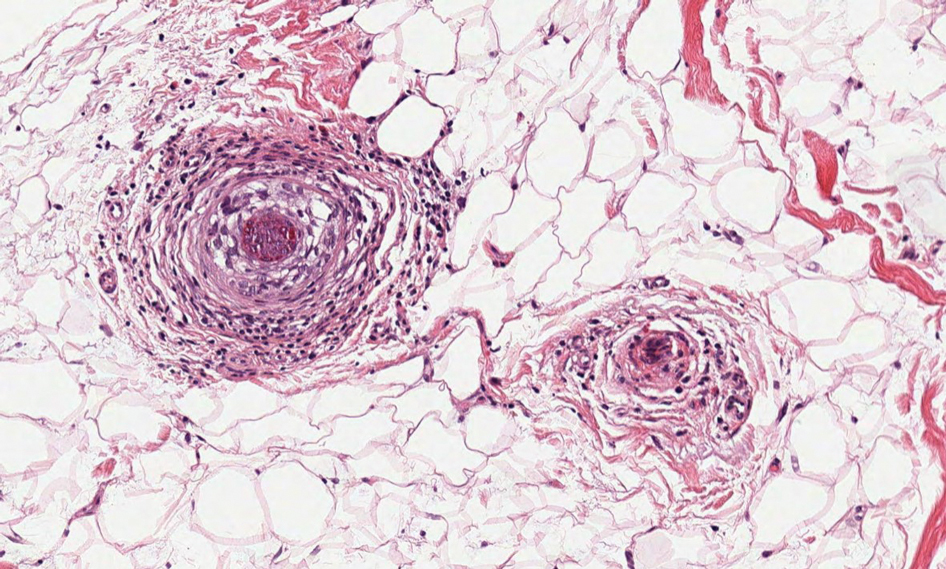
Polyarteritis nodosa is a vasculitis that affects medium- sized vessels of various organ systems. The presenting signs and symptoms vary based on the affected organ systems. Palpable to retiform purpura, livedo racemosa, subcutaneous nodules, or ulcers are seen when the skin is involved. The histologic hallmark is necrotizing vasculitis of medium-sized arterioles (Figure 5), although leukocytoclastic vasculitis of small-caliber vessels also can be seen in biopsies of affected skin.10 The vascular changes are said to be segmental, with uninvolved segments interspersed with involved segments. Antineutrophil cytoplasmic antibody (ANCA)– associated vasculitis also must be considered when one sees leukocytoclastic vasculitis of small-caliber vessels in the skin, as it can be distinguished most readily by detecting circulating antibodies specific for myeloperoxidase (MPO-ANCA) or proteinase 3 (PR3-ANCA).
- Polycarpou A, Walker SL, Lockwood DNJ. A systematic review of immunological studies of erythema nodosum leprosum. Front Immunol. 2017;8:233. doi:10.3389/fimmu.2017.00233
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45. doi:10.1016/j.clindermatol.2014.10.003
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:1-28. doi:10.1186/1750-1172-2-34
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133. doi:10.1097/01.dad.0000249887.59810.76
- Wilson TC, Stone MS, Swick BL. Histiocytoid Sweet syndrome with haloed myeloid cells masquerading as a cryptococcal infection. Am J Dermatopathology. 2014;36:264-269. doi:10.1097/DAD.0b013e31828b811b
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280. doi:10.1128/CMR.00053-10
- Schneider JW, Jordaan HF, Geiger DH, et al. Erythema induratum of Bazin: a clinicopathological study of 20 cases of Mycobacterium tuberculosis DNA in skin lesions by polymerase chain reaction. Am J Dermatopathol. 1995;17:350-356. doi:10.1097/00000372-199508000-00008
- Boonchai W, Suthipinittharm P, Mahaisavariya P. Panniculitis in tuberculosis: a clinicopathologic study of nodular panniculitis associated with tuberculosis. Int J Dermatol. 1998;37:361-363. doi:10.1046/j.1365-4362.1998.00299.x
- Segura S, Pujol RM, Trindade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851. doi:10.1016/j.jaad.2008.07.030
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathological analysis and a review of the published work. J Dermatol. 2010;37:85-93. doi:10.1111/j.1346-8138.2009.00752.x
The Diagnosis: Erythema Nodosum Leprosum
Erythema nodosum leprosum (ENL) is a type 2 reaction sometimes seen in patients infected with Mycobacterium leprae—primarily those with lepromatous or borderline lepromatous subtypes. Clinically, ENL manifests with abrupt onset of tender erythematous papules with associated fevers and general malaise. Studies have demonstrated a complex immune system reaction in ENL, but the detailed pathophysiology is not fully understood.1 Biopsies conducted within 24 hours of lesion formation are most elucidating. Foamy histiocytes admixed with neutrophils are seen in the subcutis, often causing a lobular panniculitis (quiz image).2 Neutrophils rarely are seen in other types of leprosy and thus are a useful diagnostic clue for ENL. Vasculitis of small- to medium-sized vessels can be seen but is not a necessary diagnostic criterion. Fite staining will highlight many acid-fast bacilli within the histiocytes (Figure 1).
Erythema nodosum leprosum is treated with a combination of immunosuppressants such as prednisone and thalidomide. Our patient was taking triple-antibiotic therapy—dapsone, rifampin, and clofazimine—for lepromatous leprosy when the erythematous papules developed on the arms and legs. After a skin biopsy confirmed the diagnosis of ENL, he was started on prednisone 20 mg daily with plans for close follow-up. Unfortunately, the patient was subsequently lost to follow-up.
Acute febrile neutrophilic dermatosis (also known as Sweet syndrome) is an acute inflammatory disease characterized by abrupt onset of painful erythematous papules, plaques, or nodules on the skin. It often is seen in association with preceding infections (especially those in the upper respiratory or gastrointestinal tracts), hematologic malignancies, inflammatory bowel disease, or exposure to certain classes of medications (eg, granulocyte colony-stimulating factor, tyrosine kinase inhibitors, various antibiotics).3 Histologically, acute febrile neutrophilic dermatosis is characterized by dense neutrophilic infiltrates, often with notable dermal edema (Figure 2).4 Many cases also show leukocytoclastic vasculitis; however, foamy histiocytes are not a notable component of the inflammatory infiltrate, though a histiocytoid form of acute febrile neutrophilic dermatosis has been described.5 Infections must be rigorously ruled out prior to diagnosing a patient with acute febrile neutrophilic dermatosis, making it a diagnosis of exclusion.
Cutaneous coccidioidomycosis is an infection caused by the dimorphic fungi Coccidioides immitis or Coccidioides posadasii. Cutaneous disease is rare but can occur from direct inoculation or dissemination from pulmonary disease in immunocompetent or immunocompromised patients. Papules, pustules, or plaques are seen clinically. Histologically, cutaneous coccidioidomycosis shows spherules that vary from 10 to 100 μm and are filled with multiple smaller endospores (Figure 3).6 Pseudoepitheliomatous hyperplasia with dense suppurative and granulomatous infiltrates also is seen.
Erythema induratum is characterized by tender nodules on the lower extremities and has a substantial female predominance. Many cases are associated with Mycobacterium tuberculosis infection. The bacteria are not seen directly in the skin but are instead detectable through DNA polymerase chain reaction testing or investigation of other organ systems.7,8 Histologically, lesions show a lobular panniculitis with a mixed infiltrate. Vasculitis is seen in approximately 90% of erythema induratum cases vs approximately 25% of classic ENL cases (Figure 4),2,9 which has led some to use the term nodular vasculitis to describe this disease entity. Nodular vasculitis is considered by others to be a distinct disease entity in which there are clinical and histologic features similar to erythema induratum but no evidence of M tuberculosis infection.9
Polyarteritis nodosa is a vasculitis that affects medium- sized vessels of various organ systems. The presenting signs and symptoms vary based on the affected organ systems. Palpable to retiform purpura, livedo racemosa, subcutaneous nodules, or ulcers are seen when the skin is involved. The histologic hallmark is necrotizing vasculitis of medium-sized arterioles (Figure 5), although leukocytoclastic vasculitis of small-caliber vessels also can be seen in biopsies of affected skin.10 The vascular changes are said to be segmental, with uninvolved segments interspersed with involved segments. Antineutrophil cytoplasmic antibody (ANCA)– associated vasculitis also must be considered when one sees leukocytoclastic vasculitis of small-caliber vessels in the skin, as it can be distinguished most readily by detecting circulating antibodies specific for myeloperoxidase (MPO-ANCA) or proteinase 3 (PR3-ANCA).
The Diagnosis: Erythema Nodosum Leprosum
Erythema nodosum leprosum (ENL) is a type 2 reaction sometimes seen in patients infected with Mycobacterium leprae—primarily those with lepromatous or borderline lepromatous subtypes. Clinically, ENL manifests with abrupt onset of tender erythematous papules with associated fevers and general malaise. Studies have demonstrated a complex immune system reaction in ENL, but the detailed pathophysiology is not fully understood.1 Biopsies conducted within 24 hours of lesion formation are most elucidating. Foamy histiocytes admixed with neutrophils are seen in the subcutis, often causing a lobular panniculitis (quiz image).2 Neutrophils rarely are seen in other types of leprosy and thus are a useful diagnostic clue for ENL. Vasculitis of small- to medium-sized vessels can be seen but is not a necessary diagnostic criterion. Fite staining will highlight many acid-fast bacilli within the histiocytes (Figure 1).
Erythema nodosum leprosum is treated with a combination of immunosuppressants such as prednisone and thalidomide. Our patient was taking triple-antibiotic therapy—dapsone, rifampin, and clofazimine—for lepromatous leprosy when the erythematous papules developed on the arms and legs. After a skin biopsy confirmed the diagnosis of ENL, he was started on prednisone 20 mg daily with plans for close follow-up. Unfortunately, the patient was subsequently lost to follow-up.
Acute febrile neutrophilic dermatosis (also known as Sweet syndrome) is an acute inflammatory disease characterized by abrupt onset of painful erythematous papules, plaques, or nodules on the skin. It often is seen in association with preceding infections (especially those in the upper respiratory or gastrointestinal tracts), hematologic malignancies, inflammatory bowel disease, or exposure to certain classes of medications (eg, granulocyte colony-stimulating factor, tyrosine kinase inhibitors, various antibiotics).3 Histologically, acute febrile neutrophilic dermatosis is characterized by dense neutrophilic infiltrates, often with notable dermal edema (Figure 2).4 Many cases also show leukocytoclastic vasculitis; however, foamy histiocytes are not a notable component of the inflammatory infiltrate, though a histiocytoid form of acute febrile neutrophilic dermatosis has been described.5 Infections must be rigorously ruled out prior to diagnosing a patient with acute febrile neutrophilic dermatosis, making it a diagnosis of exclusion.
Cutaneous coccidioidomycosis is an infection caused by the dimorphic fungi Coccidioides immitis or Coccidioides posadasii. Cutaneous disease is rare but can occur from direct inoculation or dissemination from pulmonary disease in immunocompetent or immunocompromised patients. Papules, pustules, or plaques are seen clinically. Histologically, cutaneous coccidioidomycosis shows spherules that vary from 10 to 100 μm and are filled with multiple smaller endospores (Figure 3).6 Pseudoepitheliomatous hyperplasia with dense suppurative and granulomatous infiltrates also is seen.
Erythema induratum is characterized by tender nodules on the lower extremities and has a substantial female predominance. Many cases are associated with Mycobacterium tuberculosis infection. The bacteria are not seen directly in the skin but are instead detectable through DNA polymerase chain reaction testing or investigation of other organ systems.7,8 Histologically, lesions show a lobular panniculitis with a mixed infiltrate. Vasculitis is seen in approximately 90% of erythema induratum cases vs approximately 25% of classic ENL cases (Figure 4),2,9 which has led some to use the term nodular vasculitis to describe this disease entity. Nodular vasculitis is considered by others to be a distinct disease entity in which there are clinical and histologic features similar to erythema induratum but no evidence of M tuberculosis infection.9
Polyarteritis nodosa is a vasculitis that affects medium- sized vessels of various organ systems. The presenting signs and symptoms vary based on the affected organ systems. Palpable to retiform purpura, livedo racemosa, subcutaneous nodules, or ulcers are seen when the skin is involved. The histologic hallmark is necrotizing vasculitis of medium-sized arterioles (Figure 5), although leukocytoclastic vasculitis of small-caliber vessels also can be seen in biopsies of affected skin.10 The vascular changes are said to be segmental, with uninvolved segments interspersed with involved segments. Antineutrophil cytoplasmic antibody (ANCA)– associated vasculitis also must be considered when one sees leukocytoclastic vasculitis of small-caliber vessels in the skin, as it can be distinguished most readily by detecting circulating antibodies specific for myeloperoxidase (MPO-ANCA) or proteinase 3 (PR3-ANCA).
- Polycarpou A, Walker SL, Lockwood DNJ. A systematic review of immunological studies of erythema nodosum leprosum. Front Immunol. 2017;8:233. doi:10.3389/fimmu.2017.00233
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45. doi:10.1016/j.clindermatol.2014.10.003
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:1-28. doi:10.1186/1750-1172-2-34
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133. doi:10.1097/01.dad.0000249887.59810.76
- Wilson TC, Stone MS, Swick BL. Histiocytoid Sweet syndrome with haloed myeloid cells masquerading as a cryptococcal infection. Am J Dermatopathology. 2014;36:264-269. doi:10.1097/DAD.0b013e31828b811b
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280. doi:10.1128/CMR.00053-10
- Schneider JW, Jordaan HF, Geiger DH, et al. Erythema induratum of Bazin: a clinicopathological study of 20 cases of Mycobacterium tuberculosis DNA in skin lesions by polymerase chain reaction. Am J Dermatopathol. 1995;17:350-356. doi:10.1097/00000372-199508000-00008
- Boonchai W, Suthipinittharm P, Mahaisavariya P. Panniculitis in tuberculosis: a clinicopathologic study of nodular panniculitis associated with tuberculosis. Int J Dermatol. 1998;37:361-363. doi:10.1046/j.1365-4362.1998.00299.x
- Segura S, Pujol RM, Trindade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851. doi:10.1016/j.jaad.2008.07.030
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathological analysis and a review of the published work. J Dermatol. 2010;37:85-93. doi:10.1111/j.1346-8138.2009.00752.x
- Polycarpou A, Walker SL, Lockwood DNJ. A systematic review of immunological studies of erythema nodosum leprosum. Front Immunol. 2017;8:233. doi:10.3389/fimmu.2017.00233
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45. doi:10.1016/j.clindermatol.2014.10.003
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:1-28. doi:10.1186/1750-1172-2-34
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133. doi:10.1097/01.dad.0000249887.59810.76
- Wilson TC, Stone MS, Swick BL. Histiocytoid Sweet syndrome with haloed myeloid cells masquerading as a cryptococcal infection. Am J Dermatopathology. 2014;36:264-269. doi:10.1097/DAD.0b013e31828b811b
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280. doi:10.1128/CMR.00053-10
- Schneider JW, Jordaan HF, Geiger DH, et al. Erythema induratum of Bazin: a clinicopathological study of 20 cases of Mycobacterium tuberculosis DNA in skin lesions by polymerase chain reaction. Am J Dermatopathol. 1995;17:350-356. doi:10.1097/00000372-199508000-00008
- Boonchai W, Suthipinittharm P, Mahaisavariya P. Panniculitis in tuberculosis: a clinicopathologic study of nodular panniculitis associated with tuberculosis. Int J Dermatol. 1998;37:361-363. doi:10.1046/j.1365-4362.1998.00299.x
- Segura S, Pujol RM, Trindade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851. doi:10.1016/j.jaad.2008.07.030
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathological analysis and a review of the published work. J Dermatol. 2010;37:85-93. doi:10.1111/j.1346-8138.2009.00752.x
A 66-year-old man presented with new tender erythematous papules scattered over the arms and legs. A biopsy of a lesion on the left thigh was performed.
Slowly Enlarging Nodule on the Neck
The Diagnosis: Microsecretory Adenocarcinoma
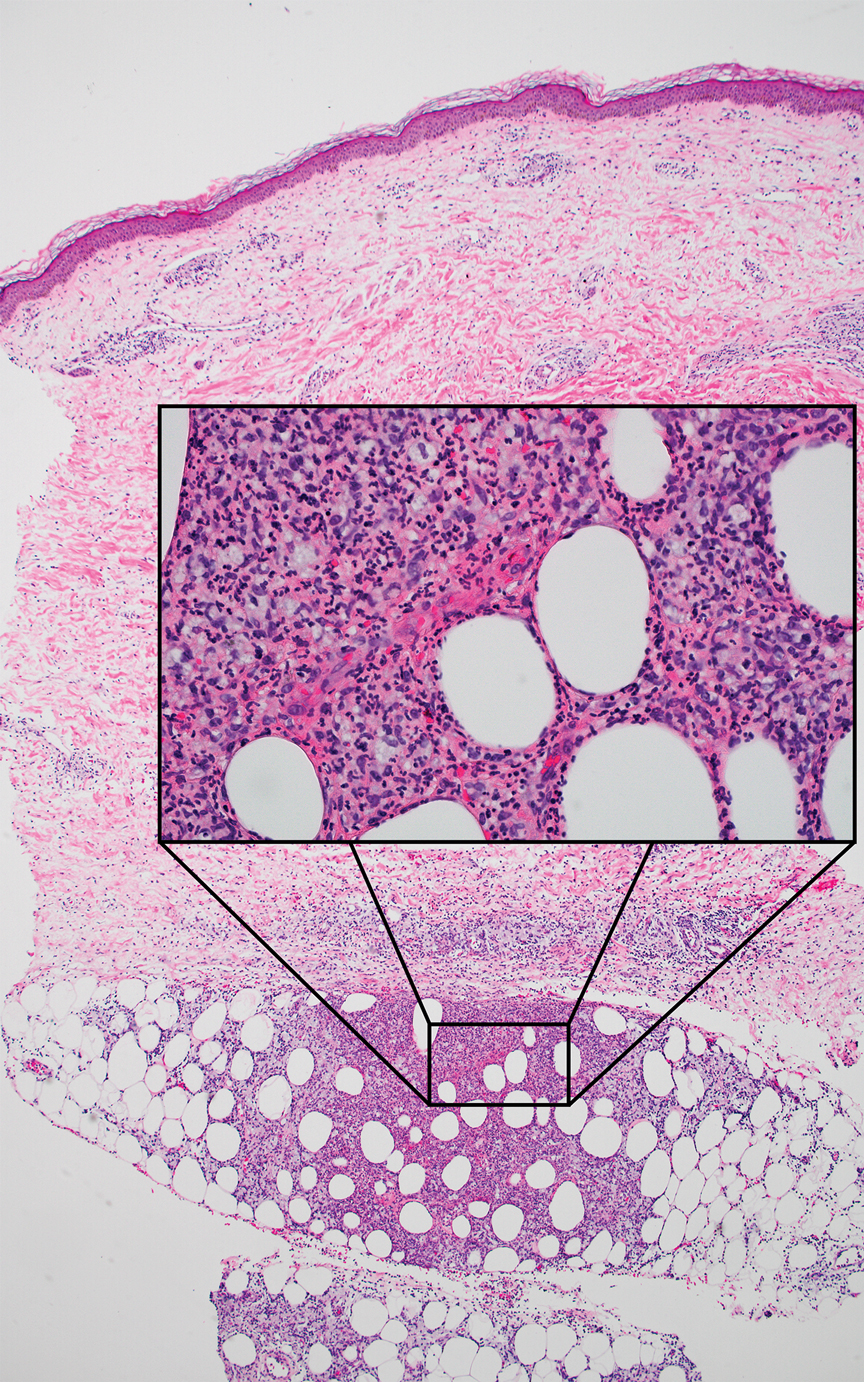
Microscopically, the tumor was relatively well circumscribed but had irregular borders. It consisted of microcysts and tubules lined by flattened to plump eosinophilic cells with mildly enlarged nuclei and intraluminal basophilic secretions. Peripheral lymphocytic aggregates also were seen in the mid and deep reticular dermis. Tumor necrosis, lymphovascular invasion, and notable mitotic activity were absent. Immunohistochemistry was diffusely positive for cytokeratin (CK) 7 and CK5/6. Occasional tumor cells showed variable expression of alpha smooth muscle actin, S-100 protein, and p40 and p63 antibodies. Immunohistochemistry was negative for CK20; GATA binding protein 3; MYB proto-oncogene, transcription factor; and insulinoma-associated protein 1. A dual-color, break-apart fluorescence in situ hybridization probe identified a rearrangement of the SS18 (SYT) gene locus on chromosome 18. The nodule was excised with clear surgical margins, and the patient had no evidence of recurrent disease or metastasis at 2-year follow-up.
In recent years, there has been a growing recognition of the pivotal role played by gene fusions in driving oncogenesis, encompassing a diverse range of benign and malignant cutaneous neoplasms. These investigations have shed light on previously unknown mechanisms and pathways contributing to the pathogenesis of these neoplastic conditions, offering invaluable insights into their underlying biology. As a result, our ability to classify and diagnose these cutaneous tumors has improved. A notable example of how our current understanding has evolved is the discovery of the new cutaneous adnexal tumor microsecretory adenocarcinoma (MSA). Initially described by Bishop et al1 in 2019 as predominantly occurring in the intraoral minor salivary glands, rare instances of primary cutaneous MSA involving the head and neck regions also have been reported.2 Microsecretory adenocarcinoma represents an important addition to the group of fusion-driven tumors with both salivary gland and cutaneous adnexal analogues, characterized by a MEF2C::SS18 gene fusion. This entity is now recognized as a group of cutaneous adnexal tumors with distinct gene fusions, including both relatively recently discovered entities (eg, secretory carcinoma with NTRK fusions) and previously known entities with newly identified gene fusions (eg, poroid neoplasms with NUTM1, YAP1, or WWTR1 fusions; hidradenomatous neoplasms with CRTC1::MAML2 fusions; and adenoid cystic carcinoma with MYB, MYBL1, and/or NFIB rearrangements).3
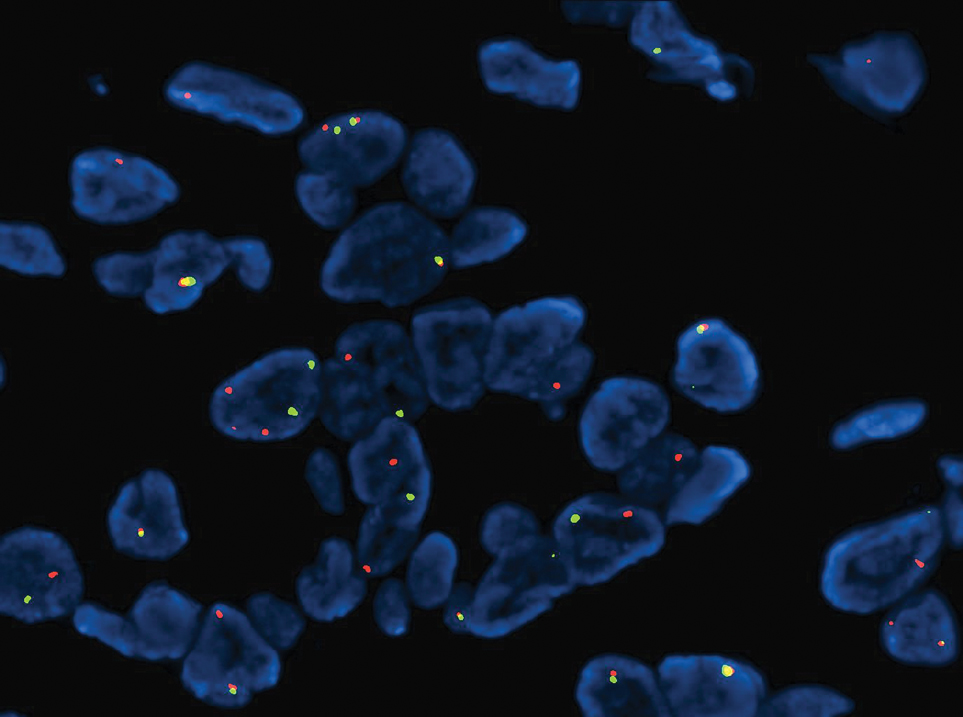
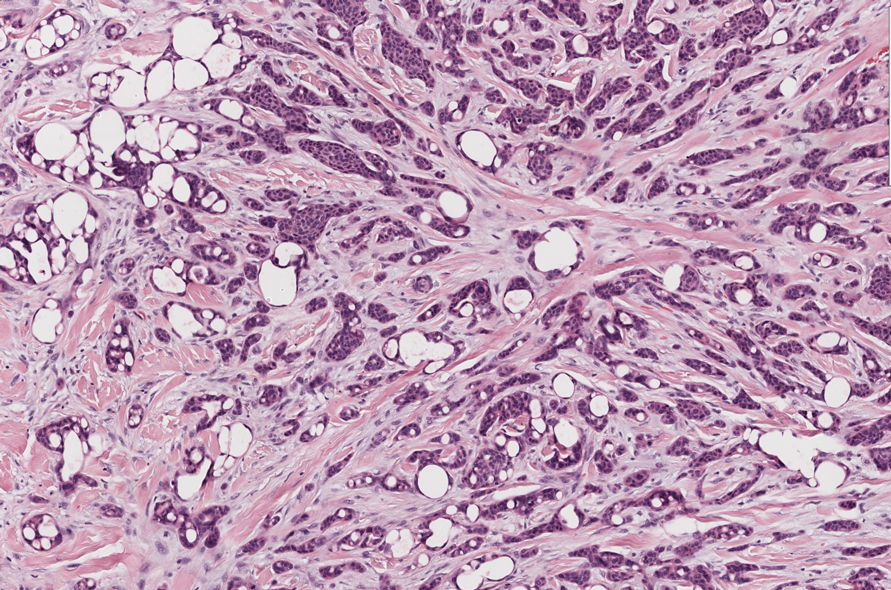
Microsecretory adenocarcinoma exhibits a high degree of morphologic consistency, characterized by a microcystic-predominant growth pattern, uniform intercalated ductlike tumor cells with attenuated eosinophilic to clear cytoplasm, monotonous oval hyperchromatic nuclei with indistinct nucleoli, abundant basophilic luminal secretions, and a variably cellular fibromyxoid stroma. It also shows rounded borders with subtle infiltrative growth. Occasionally, pseudoepitheliomatous hyperplasia, tumor-associated lymphoid proliferation, or metaplastic bone formation may accompany MSA. Perineural invasion is rare, necrosis is absent, and mitotic rates generally are low, contributing to its distinctive histopathologic features that aid in accurate diagnosis and differentiation from other entities. Immunohistochemistry reveals diffuse positivity for CK7 and patchy to diffuse expression of S-100 in tumor cells as well as variable expression of p40 and p63. Highly specific SS18 gene translocations at chromosome 18q are useful for diagnosing MSA when found alongside its characteristic appearance, and SS18 break-apart fluorescence in situ hybridization can serve reliably as an accurate diagnostic method (Figure 1).4 Our case illustrates how molecular analysis assists in distinguishing MSA from other cutaneous adnexal tumors, exemplifying the power of our evolving understanding in refining diagnostic accuracy and guiding targeted therapies in clinical practice.
The differential diagnosis of MSA includes tubular adenoma, secretory carcinoma, cribriform tumor (previously carcinoma), and metastatic adenocarcinoma. Tubular adenoma is a rare benign neoplasm that predominantly affects females and can manifest at any age in adulthood. It typically manifests as a slow-growing, occasionally pedunculated nodule, often measuring less than 2 cm. Although it most commonly manifests on the scalp, tubular adenoma also may arise in diverse sites such as the face, axillae, lower extremities, or genitalia.
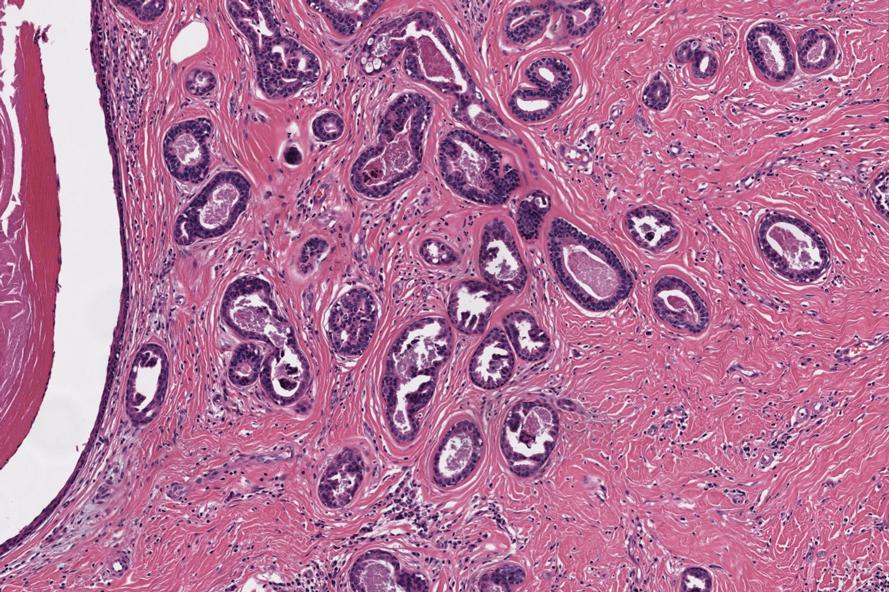
Notably, scalp lesions often are associated with nevus sebaceus of Jadassohn or syringocystadenoma papilliferum. Microscopically, tubular adenoma is well circumscribed within the dermis and may extend into the subcutis in some cases. Its distinctive appearance consists of variably sized tubules lined by a double or multilayered cuboidal to columnar epithelium, frequently displaying apocrine decapitation secretion (Figure 2). Cystic changes and intraluminal papillae devoid of true fibrovascular cores frequently are observed. Immunohistochemically, luminal epithelial cells express epithelial membrane antigen and carcinoembryonic antigen, while the myoepithelial layer expresses smooth muscle markers, p40, and S-100 protein. BRAF V600E mutation can be detected using immunohistochemistry, with excellent sensitivity and specificity using the anti-BRAF V600E antibody (clone VE1).5 Distinguishing tubular adenoma from MSA is achievable by observing its larger, more variable tubules, along with the consistent presence of a peripheral myoepithelial layer.
Secretory carcinoma is recognized as a low-grade gene fusion–driven carcinoma that primarily arises in salivary glands (both major and minor), with occasional occurrences in the breast and extremely rare instances in other locations such as the skin, thyroid gland, and lung.6 Although the axilla is the most common cutaneous site, diverse locations such as the neck, eyelids, extremities, and nipples also have been documented. Secretory carcinoma affects individuals across a wide age range (13–71 years).6 The hallmark tumors exhibit densely packed, sievelike microcystic glands and tubular spaces filled with abundant eosinophilic intraluminal secretions (Figure 3). Additionally, morphologic variants, such as predominantly papillary, papillary-cystic, macrocystic, solid, partially mucinous, and mixed-pattern neoplasms, have been described. Secretory carcinoma shares certain features with MSA; however, it is distinguished by the presence of pronounced eosinophilic secretions, plump and vacuolated cytoplasm, and a less conspicuous fibromyxoid stroma. Immunohistochemistry reveals tumor cells that are positive for CK7, SOX-10, S-100, mammaglobin, MUC4, and variably GATA-3. Genetically, secretory carcinoma exhibits distinct characteristics, commonly showing the ETV6::NTRK3 fusion, detectable through molecular techniques or pan-TRK immunohistochemistry, while RET fusions and other rare variants are less frequent.7
In 1998, Requena et al8 introduced the concept of primary cutaneous cribriform carcinoma. Despite initially being classified as a carcinoma, the malignant potential of this tumor remains uncertain. Consequently, the term cribriform tumor now has become the preferred terminology for denoting this rare entity.9 Primary cutaneous cribriform tumors are observed more commonly in women and typically affect individuals aged 20 to 55 years (mean, 44 years). Predominant locations include the upper and lower extremities, especially the thighs, knees, and legs, with additional cases occurring on the head and trunk. Microscopically, cribriform tumor is characterized by a partially circumscribed, unencapsulated dermal nodule composed of round or oval nuclei displaying hyperchromatism and mild pleomorphism. The defining aspect of its morphology revolves around interspersed small round cavities that give rise to the hallmark cribriform pattern (Figure 4). Although MSA occasionally may exhibit a cribriform architectural pattern, it typically lacks the distinctive feature of thin, threadlike, intraluminal bridging strands observed in cribriform tumors. Similarly, luminal cells within the cribriform tumor express CK7 and exhibit variable S-100 expression. It is recognized as an indolent neoplasm with uncertain malignant potential.
The histopathologic features of metastatic carcinomas can overlap with those of primary cutaneous tumors, particularly adnexal neoplasms.10 However, several key features can aid in the differentiation of cutaneous metastases, including a dermal-based growth pattern with or without subcutaneous involvement, the presence of multiple lesions, and the occurrence of lymphovascular invasion (Figure 5). Conversely, features that suggest a primary cutaneous adnexal neoplasm include the presence of superimposed in situ disease, carcinoma developing within a benign adnexal neoplasm, and notable stromal and/or vascular hyalinization within benign-appearing areas. In some cases, it can be difficult to determine the primary site of origin of a metastatic carcinoma to the skin based on morphologic features alone. In these cases, immunohistochemistry can be helpful. The most cost-effective and time-efficient approach to accurate diagnosis is to obtain a comprehensive clinical history. If there is a known history of cancer, a small panel of organ-specific immunohistochemical studies can be performed to confirm the diagnosis. If there is no known history, an algorithmic approach can be used to identify the primary site of origin. In all circumstances, it cannot be stressed enough that acquiring a thorough clinical history before conducting any diagnostic examinations is paramount.
- Bishop JA, Weinreb I, Swanson D, et al. Microsecretory adenocarcinoma: a novel salivary gland tumor characterized by a recurrent MEF2C-SS18 fusion. Am J Surg Pathol. 2019;43:1023-1032.
- Bishop JA, Williams EA, McLean AC, et al. Microsecretory adenocarcinoma of the skin harboring recurrent SS18 fusions: a cutaneous analog to a newly described salivary gland tumor. J Cutan Pathol. 2023;50:134-139.
- Macagno N, Sohier Pierre, Kervarrec T, et al. Recent advances on immunohistochemistry and molecular biology for the diagnosis of adnexal sweat gland tumors. Cancers (Basel). 2022;14:476.
- Bishop JA, Koduru P, Veremis BM, et al. SS18 break-apart fluorescence in situ hybridization is a practical and effective method for diagnosing microsecretory adenocarcinoma of salivary glands. Head Neck Pathol. 2021;15:723-726.
- Liau JY, Tsai JH, Huang WC, et al. BRAF and KRAS mutations in tubular apocrine adenoma and papillary eccrine adenoma of the skin. Hum Pathol. 2018;73:59-65.
- Chang MD, Arthur AK, Garcia JJ, et al. ETV6 rearrangement in a case of mammary analogue secretory carcinoma of the skin. J Cutan Pathol. 2016;43:1045-1049.
- Skalova A, Baneckova M, Thompson LDR, et al. Expanding the molecular spectrum of secretory carcinoma of salivary glands with a novel VIM-RET fusion. Am J Surg Pathol. 2020;44:1295-1307.
- Requena L, Kiryu H, Ackerman AB. Neoplasms With Apocrine Differentiation. Lippencott-Raven; 1998.
- Kazakov DV, Llamas-Velasco M, Fernandez-Flores A, et al. Cribriform tumour (previously carcinoma). In: WHO Classification of Tumours: Skin Tumours. 5th ed. International Agency for Research on Cancer; 2024.
- Habaermehl G, Ko J. Cutaneous metastases: a review and diagnostic approach to tumors of unknown origin. Arch Pathol Lab Med. 2019;143:943-957.
The Diagnosis: Microsecretory Adenocarcinoma
Microscopically, the tumor was relatively well circumscribed but had irregular borders. It consisted of microcysts and tubules lined by flattened to plump eosinophilic cells with mildly enlarged nuclei and intraluminal basophilic secretions. Peripheral lymphocytic aggregates also were seen in the mid and deep reticular dermis. Tumor necrosis, lymphovascular invasion, and notable mitotic activity were absent. Immunohistochemistry was diffusely positive for cytokeratin (CK) 7 and CK5/6. Occasional tumor cells showed variable expression of alpha smooth muscle actin, S-100 protein, and p40 and p63 antibodies. Immunohistochemistry was negative for CK20; GATA binding protein 3; MYB proto-oncogene, transcription factor; and insulinoma-associated protein 1. A dual-color, break-apart fluorescence in situ hybridization probe identified a rearrangement of the SS18 (SYT) gene locus on chromosome 18. The nodule was excised with clear surgical margins, and the patient had no evidence of recurrent disease or metastasis at 2-year follow-up.
In recent years, there has been a growing recognition of the pivotal role played by gene fusions in driving oncogenesis, encompassing a diverse range of benign and malignant cutaneous neoplasms. These investigations have shed light on previously unknown mechanisms and pathways contributing to the pathogenesis of these neoplastic conditions, offering invaluable insights into their underlying biology. As a result, our ability to classify and diagnose these cutaneous tumors has improved. A notable example of how our current understanding has evolved is the discovery of the new cutaneous adnexal tumor microsecretory adenocarcinoma (MSA). Initially described by Bishop et al1 in 2019 as predominantly occurring in the intraoral minor salivary glands, rare instances of primary cutaneous MSA involving the head and neck regions also have been reported.2 Microsecretory adenocarcinoma represents an important addition to the group of fusion-driven tumors with both salivary gland and cutaneous adnexal analogues, characterized by a MEF2C::SS18 gene fusion. This entity is now recognized as a group of cutaneous adnexal tumors with distinct gene fusions, including both relatively recently discovered entities (eg, secretory carcinoma with NTRK fusions) and previously known entities with newly identified gene fusions (eg, poroid neoplasms with NUTM1, YAP1, or WWTR1 fusions; hidradenomatous neoplasms with CRTC1::MAML2 fusions; and adenoid cystic carcinoma with MYB, MYBL1, and/or NFIB rearrangements).3
Microsecretory adenocarcinoma exhibits a high degree of morphologic consistency, characterized by a microcystic-predominant growth pattern, uniform intercalated ductlike tumor cells with attenuated eosinophilic to clear cytoplasm, monotonous oval hyperchromatic nuclei with indistinct nucleoli, abundant basophilic luminal secretions, and a variably cellular fibromyxoid stroma. It also shows rounded borders with subtle infiltrative growth. Occasionally, pseudoepitheliomatous hyperplasia, tumor-associated lymphoid proliferation, or metaplastic bone formation may accompany MSA. Perineural invasion is rare, necrosis is absent, and mitotic rates generally are low, contributing to its distinctive histopathologic features that aid in accurate diagnosis and differentiation from other entities. Immunohistochemistry reveals diffuse positivity for CK7 and patchy to diffuse expression of S-100 in tumor cells as well as variable expression of p40 and p63. Highly specific SS18 gene translocations at chromosome 18q are useful for diagnosing MSA when found alongside its characteristic appearance, and SS18 break-apart fluorescence in situ hybridization can serve reliably as an accurate diagnostic method (Figure 1).4 Our case illustrates how molecular analysis assists in distinguishing MSA from other cutaneous adnexal tumors, exemplifying the power of our evolving understanding in refining diagnostic accuracy and guiding targeted therapies in clinical practice.
The differential diagnosis of MSA includes tubular adenoma, secretory carcinoma, cribriform tumor (previously carcinoma), and metastatic adenocarcinoma. Tubular adenoma is a rare benign neoplasm that predominantly affects females and can manifest at any age in adulthood. It typically manifests as a slow-growing, occasionally pedunculated nodule, often measuring less than 2 cm. Although it most commonly manifests on the scalp, tubular adenoma also may arise in diverse sites such as the face, axillae, lower extremities, or genitalia.
Notably, scalp lesions often are associated with nevus sebaceus of Jadassohn or syringocystadenoma papilliferum. Microscopically, tubular adenoma is well circumscribed within the dermis and may extend into the subcutis in some cases. Its distinctive appearance consists of variably sized tubules lined by a double or multilayered cuboidal to columnar epithelium, frequently displaying apocrine decapitation secretion (Figure 2). Cystic changes and intraluminal papillae devoid of true fibrovascular cores frequently are observed. Immunohistochemically, luminal epithelial cells express epithelial membrane antigen and carcinoembryonic antigen, while the myoepithelial layer expresses smooth muscle markers, p40, and S-100 protein. BRAF V600E mutation can be detected using immunohistochemistry, with excellent sensitivity and specificity using the anti-BRAF V600E antibody (clone VE1).5 Distinguishing tubular adenoma from MSA is achievable by observing its larger, more variable tubules, along with the consistent presence of a peripheral myoepithelial layer.
Secretory carcinoma is recognized as a low-grade gene fusion–driven carcinoma that primarily arises in salivary glands (both major and minor), with occasional occurrences in the breast and extremely rare instances in other locations such as the skin, thyroid gland, and lung.6 Although the axilla is the most common cutaneous site, diverse locations such as the neck, eyelids, extremities, and nipples also have been documented. Secretory carcinoma affects individuals across a wide age range (13–71 years).6 The hallmark tumors exhibit densely packed, sievelike microcystic glands and tubular spaces filled with abundant eosinophilic intraluminal secretions (Figure 3). Additionally, morphologic variants, such as predominantly papillary, papillary-cystic, macrocystic, solid, partially mucinous, and mixed-pattern neoplasms, have been described. Secretory carcinoma shares certain features with MSA; however, it is distinguished by the presence of pronounced eosinophilic secretions, plump and vacuolated cytoplasm, and a less conspicuous fibromyxoid stroma. Immunohistochemistry reveals tumor cells that are positive for CK7, SOX-10, S-100, mammaglobin, MUC4, and variably GATA-3. Genetically, secretory carcinoma exhibits distinct characteristics, commonly showing the ETV6::NTRK3 fusion, detectable through molecular techniques or pan-TRK immunohistochemistry, while RET fusions and other rare variants are less frequent.7
In 1998, Requena et al8 introduced the concept of primary cutaneous cribriform carcinoma. Despite initially being classified as a carcinoma, the malignant potential of this tumor remains uncertain. Consequently, the term cribriform tumor now has become the preferred terminology for denoting this rare entity.9 Primary cutaneous cribriform tumors are observed more commonly in women and typically affect individuals aged 20 to 55 years (mean, 44 years). Predominant locations include the upper and lower extremities, especially the thighs, knees, and legs, with additional cases occurring on the head and trunk. Microscopically, cribriform tumor is characterized by a partially circumscribed, unencapsulated dermal nodule composed of round or oval nuclei displaying hyperchromatism and mild pleomorphism. The defining aspect of its morphology revolves around interspersed small round cavities that give rise to the hallmark cribriform pattern (Figure 4). Although MSA occasionally may exhibit a cribriform architectural pattern, it typically lacks the distinctive feature of thin, threadlike, intraluminal bridging strands observed in cribriform tumors. Similarly, luminal cells within the cribriform tumor express CK7 and exhibit variable S-100 expression. It is recognized as an indolent neoplasm with uncertain malignant potential.
The histopathologic features of metastatic carcinomas can overlap with those of primary cutaneous tumors, particularly adnexal neoplasms.10 However, several key features can aid in the differentiation of cutaneous metastases, including a dermal-based growth pattern with or without subcutaneous involvement, the presence of multiple lesions, and the occurrence of lymphovascular invasion (Figure 5). Conversely, features that suggest a primary cutaneous adnexal neoplasm include the presence of superimposed in situ disease, carcinoma developing within a benign adnexal neoplasm, and notable stromal and/or vascular hyalinization within benign-appearing areas. In some cases, it can be difficult to determine the primary site of origin of a metastatic carcinoma to the skin based on morphologic features alone. In these cases, immunohistochemistry can be helpful. The most cost-effective and time-efficient approach to accurate diagnosis is to obtain a comprehensive clinical history. If there is a known history of cancer, a small panel of organ-specific immunohistochemical studies can be performed to confirm the diagnosis. If there is no known history, an algorithmic approach can be used to identify the primary site of origin. In all circumstances, it cannot be stressed enough that acquiring a thorough clinical history before conducting any diagnostic examinations is paramount.
The Diagnosis: Microsecretory Adenocarcinoma
Microscopically, the tumor was relatively well circumscribed but had irregular borders. It consisted of microcysts and tubules lined by flattened to plump eosinophilic cells with mildly enlarged nuclei and intraluminal basophilic secretions. Peripheral lymphocytic aggregates also were seen in the mid and deep reticular dermis. Tumor necrosis, lymphovascular invasion, and notable mitotic activity were absent. Immunohistochemistry was diffusely positive for cytokeratin (CK) 7 and CK5/6. Occasional tumor cells showed variable expression of alpha smooth muscle actin, S-100 protein, and p40 and p63 antibodies. Immunohistochemistry was negative for CK20; GATA binding protein 3; MYB proto-oncogene, transcription factor; and insulinoma-associated protein 1. A dual-color, break-apart fluorescence in situ hybridization probe identified a rearrangement of the SS18 (SYT) gene locus on chromosome 18. The nodule was excised with clear surgical margins, and the patient had no evidence of recurrent disease or metastasis at 2-year follow-up.
In recent years, there has been a growing recognition of the pivotal role played by gene fusions in driving oncogenesis, encompassing a diverse range of benign and malignant cutaneous neoplasms. These investigations have shed light on previously unknown mechanisms and pathways contributing to the pathogenesis of these neoplastic conditions, offering invaluable insights into their underlying biology. As a result, our ability to classify and diagnose these cutaneous tumors has improved. A notable example of how our current understanding has evolved is the discovery of the new cutaneous adnexal tumor microsecretory adenocarcinoma (MSA). Initially described by Bishop et al1 in 2019 as predominantly occurring in the intraoral minor salivary glands, rare instances of primary cutaneous MSA involving the head and neck regions also have been reported.2 Microsecretory adenocarcinoma represents an important addition to the group of fusion-driven tumors with both salivary gland and cutaneous adnexal analogues, characterized by a MEF2C::SS18 gene fusion. This entity is now recognized as a group of cutaneous adnexal tumors with distinct gene fusions, including both relatively recently discovered entities (eg, secretory carcinoma with NTRK fusions) and previously known entities with newly identified gene fusions (eg, poroid neoplasms with NUTM1, YAP1, or WWTR1 fusions; hidradenomatous neoplasms with CRTC1::MAML2 fusions; and adenoid cystic carcinoma with MYB, MYBL1, and/or NFIB rearrangements).3
Microsecretory adenocarcinoma exhibits a high degree of morphologic consistency, characterized by a microcystic-predominant growth pattern, uniform intercalated ductlike tumor cells with attenuated eosinophilic to clear cytoplasm, monotonous oval hyperchromatic nuclei with indistinct nucleoli, abundant basophilic luminal secretions, and a variably cellular fibromyxoid stroma. It also shows rounded borders with subtle infiltrative growth. Occasionally, pseudoepitheliomatous hyperplasia, tumor-associated lymphoid proliferation, or metaplastic bone formation may accompany MSA. Perineural invasion is rare, necrosis is absent, and mitotic rates generally are low, contributing to its distinctive histopathologic features that aid in accurate diagnosis and differentiation from other entities. Immunohistochemistry reveals diffuse positivity for CK7 and patchy to diffuse expression of S-100 in tumor cells as well as variable expression of p40 and p63. Highly specific SS18 gene translocations at chromosome 18q are useful for diagnosing MSA when found alongside its characteristic appearance, and SS18 break-apart fluorescence in situ hybridization can serve reliably as an accurate diagnostic method (Figure 1).4 Our case illustrates how molecular analysis assists in distinguishing MSA from other cutaneous adnexal tumors, exemplifying the power of our evolving understanding in refining diagnostic accuracy and guiding targeted therapies in clinical practice.
The differential diagnosis of MSA includes tubular adenoma, secretory carcinoma, cribriform tumor (previously carcinoma), and metastatic adenocarcinoma. Tubular adenoma is a rare benign neoplasm that predominantly affects females and can manifest at any age in adulthood. It typically manifests as a slow-growing, occasionally pedunculated nodule, often measuring less than 2 cm. Although it most commonly manifests on the scalp, tubular adenoma also may arise in diverse sites such as the face, axillae, lower extremities, or genitalia.
Notably, scalp lesions often are associated with nevus sebaceus of Jadassohn or syringocystadenoma papilliferum. Microscopically, tubular adenoma is well circumscribed within the dermis and may extend into the subcutis in some cases. Its distinctive appearance consists of variably sized tubules lined by a double or multilayered cuboidal to columnar epithelium, frequently displaying apocrine decapitation secretion (Figure 2). Cystic changes and intraluminal papillae devoid of true fibrovascular cores frequently are observed. Immunohistochemically, luminal epithelial cells express epithelial membrane antigen and carcinoembryonic antigen, while the myoepithelial layer expresses smooth muscle markers, p40, and S-100 protein. BRAF V600E mutation can be detected using immunohistochemistry, with excellent sensitivity and specificity using the anti-BRAF V600E antibody (clone VE1).5 Distinguishing tubular adenoma from MSA is achievable by observing its larger, more variable tubules, along with the consistent presence of a peripheral myoepithelial layer.
Secretory carcinoma is recognized as a low-grade gene fusion–driven carcinoma that primarily arises in salivary glands (both major and minor), with occasional occurrences in the breast and extremely rare instances in other locations such as the skin, thyroid gland, and lung.6 Although the axilla is the most common cutaneous site, diverse locations such as the neck, eyelids, extremities, and nipples also have been documented. Secretory carcinoma affects individuals across a wide age range (13–71 years).6 The hallmark tumors exhibit densely packed, sievelike microcystic glands and tubular spaces filled with abundant eosinophilic intraluminal secretions (Figure 3). Additionally, morphologic variants, such as predominantly papillary, papillary-cystic, macrocystic, solid, partially mucinous, and mixed-pattern neoplasms, have been described. Secretory carcinoma shares certain features with MSA; however, it is distinguished by the presence of pronounced eosinophilic secretions, plump and vacuolated cytoplasm, and a less conspicuous fibromyxoid stroma. Immunohistochemistry reveals tumor cells that are positive for CK7, SOX-10, S-100, mammaglobin, MUC4, and variably GATA-3. Genetically, secretory carcinoma exhibits distinct characteristics, commonly showing the ETV6::NTRK3 fusion, detectable through molecular techniques or pan-TRK immunohistochemistry, while RET fusions and other rare variants are less frequent.7
In 1998, Requena et al8 introduced the concept of primary cutaneous cribriform carcinoma. Despite initially being classified as a carcinoma, the malignant potential of this tumor remains uncertain. Consequently, the term cribriform tumor now has become the preferred terminology for denoting this rare entity.9 Primary cutaneous cribriform tumors are observed more commonly in women and typically affect individuals aged 20 to 55 years (mean, 44 years). Predominant locations include the upper and lower extremities, especially the thighs, knees, and legs, with additional cases occurring on the head and trunk. Microscopically, cribriform tumor is characterized by a partially circumscribed, unencapsulated dermal nodule composed of round or oval nuclei displaying hyperchromatism and mild pleomorphism. The defining aspect of its morphology revolves around interspersed small round cavities that give rise to the hallmark cribriform pattern (Figure 4). Although MSA occasionally may exhibit a cribriform architectural pattern, it typically lacks the distinctive feature of thin, threadlike, intraluminal bridging strands observed in cribriform tumors. Similarly, luminal cells within the cribriform tumor express CK7 and exhibit variable S-100 expression. It is recognized as an indolent neoplasm with uncertain malignant potential.
The histopathologic features of metastatic carcinomas can overlap with those of primary cutaneous tumors, particularly adnexal neoplasms.10 However, several key features can aid in the differentiation of cutaneous metastases, including a dermal-based growth pattern with or without subcutaneous involvement, the presence of multiple lesions, and the occurrence of lymphovascular invasion (Figure 5). Conversely, features that suggest a primary cutaneous adnexal neoplasm include the presence of superimposed in situ disease, carcinoma developing within a benign adnexal neoplasm, and notable stromal and/or vascular hyalinization within benign-appearing areas. In some cases, it can be difficult to determine the primary site of origin of a metastatic carcinoma to the skin based on morphologic features alone. In these cases, immunohistochemistry can be helpful. The most cost-effective and time-efficient approach to accurate diagnosis is to obtain a comprehensive clinical history. If there is a known history of cancer, a small panel of organ-specific immunohistochemical studies can be performed to confirm the diagnosis. If there is no known history, an algorithmic approach can be used to identify the primary site of origin. In all circumstances, it cannot be stressed enough that acquiring a thorough clinical history before conducting any diagnostic examinations is paramount.
- Bishop JA, Weinreb I, Swanson D, et al. Microsecretory adenocarcinoma: a novel salivary gland tumor characterized by a recurrent MEF2C-SS18 fusion. Am J Surg Pathol. 2019;43:1023-1032.
- Bishop JA, Williams EA, McLean AC, et al. Microsecretory adenocarcinoma of the skin harboring recurrent SS18 fusions: a cutaneous analog to a newly described salivary gland tumor. J Cutan Pathol. 2023;50:134-139.
- Macagno N, Sohier Pierre, Kervarrec T, et al. Recent advances on immunohistochemistry and molecular biology for the diagnosis of adnexal sweat gland tumors. Cancers (Basel). 2022;14:476.
- Bishop JA, Koduru P, Veremis BM, et al. SS18 break-apart fluorescence in situ hybridization is a practical and effective method for diagnosing microsecretory adenocarcinoma of salivary glands. Head Neck Pathol. 2021;15:723-726.
- Liau JY, Tsai JH, Huang WC, et al. BRAF and KRAS mutations in tubular apocrine adenoma and papillary eccrine adenoma of the skin. Hum Pathol. 2018;73:59-65.
- Chang MD, Arthur AK, Garcia JJ, et al. ETV6 rearrangement in a case of mammary analogue secretory carcinoma of the skin. J Cutan Pathol. 2016;43:1045-1049.
- Skalova A, Baneckova M, Thompson LDR, et al. Expanding the molecular spectrum of secretory carcinoma of salivary glands with a novel VIM-RET fusion. Am J Surg Pathol. 2020;44:1295-1307.
- Requena L, Kiryu H, Ackerman AB. Neoplasms With Apocrine Differentiation. Lippencott-Raven; 1998.
- Kazakov DV, Llamas-Velasco M, Fernandez-Flores A, et al. Cribriform tumour (previously carcinoma). In: WHO Classification of Tumours: Skin Tumours. 5th ed. International Agency for Research on Cancer; 2024.
- Habaermehl G, Ko J. Cutaneous metastases: a review and diagnostic approach to tumors of unknown origin. Arch Pathol Lab Med. 2019;143:943-957.
- Bishop JA, Weinreb I, Swanson D, et al. Microsecretory adenocarcinoma: a novel salivary gland tumor characterized by a recurrent MEF2C-SS18 fusion. Am J Surg Pathol. 2019;43:1023-1032.
- Bishop JA, Williams EA, McLean AC, et al. Microsecretory adenocarcinoma of the skin harboring recurrent SS18 fusions: a cutaneous analog to a newly described salivary gland tumor. J Cutan Pathol. 2023;50:134-139.
- Macagno N, Sohier Pierre, Kervarrec T, et al. Recent advances on immunohistochemistry and molecular biology for the diagnosis of adnexal sweat gland tumors. Cancers (Basel). 2022;14:476.
- Bishop JA, Koduru P, Veremis BM, et al. SS18 break-apart fluorescence in situ hybridization is a practical and effective method for diagnosing microsecretory adenocarcinoma of salivary glands. Head Neck Pathol. 2021;15:723-726.
- Liau JY, Tsai JH, Huang WC, et al. BRAF and KRAS mutations in tubular apocrine adenoma and papillary eccrine adenoma of the skin. Hum Pathol. 2018;73:59-65.
- Chang MD, Arthur AK, Garcia JJ, et al. ETV6 rearrangement in a case of mammary analogue secretory carcinoma of the skin. J Cutan Pathol. 2016;43:1045-1049.
- Skalova A, Baneckova M, Thompson LDR, et al. Expanding the molecular spectrum of secretory carcinoma of salivary glands with a novel VIM-RET fusion. Am J Surg Pathol. 2020;44:1295-1307.
- Requena L, Kiryu H, Ackerman AB. Neoplasms With Apocrine Differentiation. Lippencott-Raven; 1998.
- Kazakov DV, Llamas-Velasco M, Fernandez-Flores A, et al. Cribriform tumour (previously carcinoma). In: WHO Classification of Tumours: Skin Tumours. 5th ed. International Agency for Research on Cancer; 2024.
- Habaermehl G, Ko J. Cutaneous metastases: a review and diagnostic approach to tumors of unknown origin. Arch Pathol Lab Med. 2019;143:943-957.
A 74-year-old man presented with an asymptomatic nodule on the left neck measuring approximately 2 cm. An excisional biopsy was obtained for histopathologic evaluation.
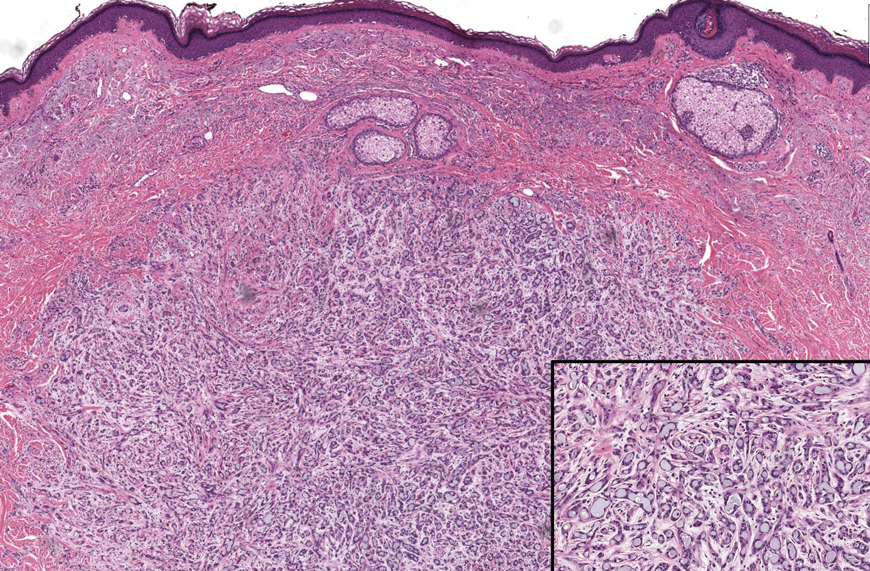
Progressive Eyelash Loss and Scale of the Right Eyelid
The Diagnosis: Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) characterized by folliculotropism and follicular-based lesions. The clinical manifestation of FMF can vary and includes patches, plaques, or tumors resembling nonfolliculotropic MF; acneform lesions including comedones and pustules; or areas of alopecia. Lesions commonly involve the head and neck but also can be seen on the trunk or extremities. Folliculotropic mycosis fungoides can be accompanied by pruritus or superimposed secondary infection.
Histologic features of FMF include follicular (perifollicular or intrafollicular) infiltration by atypical T cells showing cerebriform nuclei.1 In early lesions, there may be only mild superficial perivascular inflammation without notable lymphocyte atypia, making diagnosis challenging. 2,3 Mucinous degeneration of the follicles—termed follicular mucinosis—is a common histologic finding in FMF.1,2 Follicular mucinosis is not exclusive to FMF; it can be primary/idiopathic or secondary to underlying inflammatory or neoplastic disorders such as FMF. On immunohistochemistry, FMF most commonly demonstrates a helper T cell phenotype that is positive for CD3 and CD4 and negative for CD8, with aberrant loss of CD7 and variably CD5, which is similar to classic MF. Occasionally, larger CD30+ cells also can be present in the dermis. T-cell gene rearrangement studies will demonstrate T-cell receptor clonality in most cases.2
Many large retrospective cohort studies have suggested that patients with FMF have a worse prognosis than classic MF, with a 5-year survival rate of 62% to 87% for early-stage FMF vs more than 90% for classic patchand plaque-stage MF.4-7 However, a 2016 study suggested histologic evaluation may be able to further differentiate clinically identical cases into indolent and aggressive forms of FMF with considerably different outcomes based on the density of the perifollicular infiltrate.5 The presence of follicular mucinosis has no impact on prognosis compared to cases without follicular mucinosis.1,2
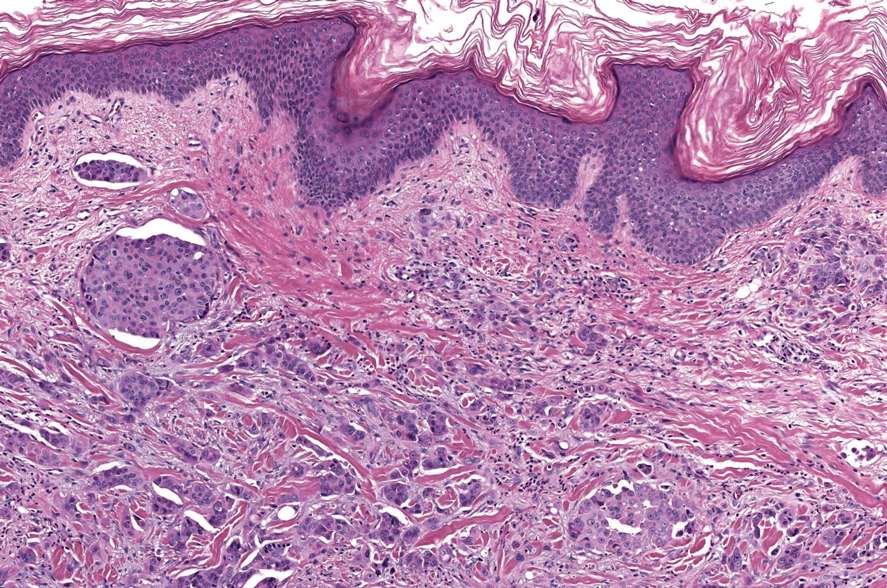
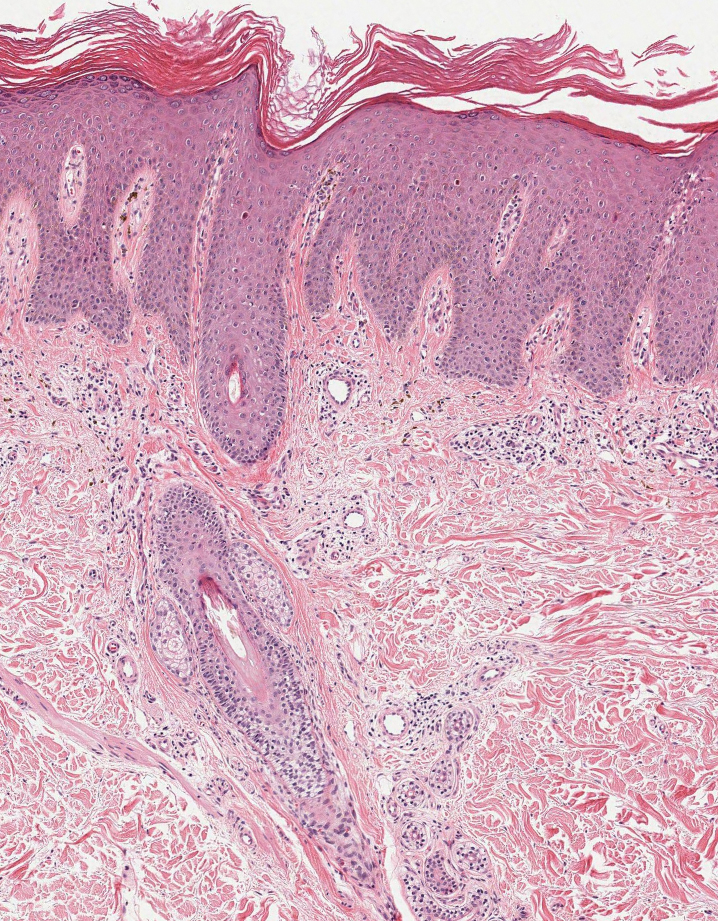
Alopecia mucinosa is characterized by infiltrating, erythematous, scaling plaques localized to the head and neck.8 It is diagnosed clinically, and histopathology shows follicular mucinosis. The terms alopecia mucinosa and follicular mucinosis often are used interchangeably. Over the past few decades, 3 variants have been categorized: primary acute, primary chronic, and secondary. The primary acute form manifests in children and young adults as solitary lesions, which often resolve spontaneously. In contrast, the primary chronic form manifests in older adults as multiple disseminated lesions with a chronic relapsing course.8,9 The secondary form can occur in the setting of other disorders, including lupus erythematosus, hypertrophic lichen planus, alopecia areata, and neoplasms such as MF or Hodgkin lymphoma.9 The histopathologic findings are similar for all types of alopecia mucinosa, with cystic pools of mucin deposition in the sebaceous glands and external root sheath of the follicles as well as associated inflammation composed of lymphocytes and eosinophils (Figure 1).9,10 The inflammatory infiltrate rarely extends into the epidermis or upper portion of the hair follicle. Although histopathology alone cannot reliably distinguish between primary and secondary forms of alopecia mucinosa, MF (including follicular MF) or another underlying cutaneous T-cell lymphoma should be considered if inflammation extends into the upper dermis, epidermis, or follicles or is in a dense bandlike distribution.11 On immunohistochemistry, lymphocytes should show positivity for CD3, CD4, and CD8. The CD4:CD8 ratio often is 1:1 in alopecia mucinosa, while in FMF it is approximately 3:1.10 CD7 commonly is negative but can be present in a small percentage of cases.12 T-cell receptor gene rearrangement studies have detected clonality in both primary and secondary alopecia mucinosa and thus cannot be used alone to distinguish between the two.10 Given the overlap in histopathologic and immunohistochemical features of primary and secondary alopecia mucinosa, definitive diagnosis cannot be made with any single modality and should be based on correlating clinical presentation, histopathology, immunohistochemistry, and molecular analyses.
Inflammatory dermatoses including seborrheic dermatitis also are in the differential diagnosis for FMF. Seborrheic dermatitis is a common chronic inflammatory skin disorder affecting 1% to 3% of the general population. 13 Patients usually present with scaly and greasy plaques and papules localized to areas with increased sebaceous glands and high sebum production such as the face, scalp, and intertriginous regions. The distribution often is symmetrical, and the severity of disease can vary substantially.13 Sebopsoriasis is an entity with overlapping features of seborrheic dermatitis and psoriasis, including thicker, more erythematous plaques that are more elevated. Histopathology of seborrheic dermatitis reveals spongiotic inflammation in the epidermis characterized by rounding of the keratinocytes, widening of the intercellular spaces, and accumulation of intracellular edema, causing the formation of clear spaces in the epidermis (Figure 2). Focal parakeratosis, usually in the follicular ostia, and mounds of scaly crust often are present. 14 A periodic acid–Schiff stain should be performed to rule out infectious dermatophytes, which can show similar clinical and histologic features. More chronic cases of seborrheic dermatitis often can take on histologic features of psoriasis, namely epidermal hyperplasia with thinning over dermal papillae, though the hyperplasia in psoriasis is more regular.
Alopecia areata is an immune-mediated disorder characterized by nonscarring hair loss; it affects approximately 0.1% to 0.2% of the general population.15 The pathogenesis involves the premature transition of hair follicles in the anagen (growth) phase to the catagen ( nonproliferative/involution) and telogen (resting) phases, resulting in sudden hair shedding and decreased regrowth. Clinically, it is characterized by asymptomatic hair loss that occurs most frequently on the scalp and other areas of the head, including eyelashes, eyebrows, and facial hair, but also can occur on the extremities. There are several variants; the most common is patchy alopecia, which features smooth circular areas of hair loss that progress over several weeks. Some patients can progress to loss of all scalp hairs (alopecia totalis) or all hairs throughout the body (alopecia universalis). 15 Patients typically will have spontaneous regrowth of hair, with up to 50% of those with limited hair loss recovering within a year.16 The disease has a chronic/ relapsing course, and patients often will have multiple episodes of hair loss. Histopathologic features can vary depending on the stage of disease. In acute cases, a peribulbar lymphocytic infiltrate preferentially involving anagen-stage hair follicles is seen, with associated necrosis, edema, and pigment incontinence (Figure 3).16 In chronic alopecia areata, the inflammation may be less brisk, and follicular miniaturization often is seen. Additionally, increased proportions of catagen- or telogen-stage follicles are present.16,17 On immunohistochemistry, lymphocytes express both CD4 and CD8, with a slightly increased CD4:CD8 ratio in active disease.18
Psoriatic alopecia describes hair loss that occurs in patients with psoriasis. Patients present with scaly, erythematous, psoriasiform plaques or patches, as well as decreased hair density, finer hairs, and increased dystrophic hair bulbs within the psoriatic plaques.19 It often is nonscarring and resolves with therapy, though scarring may occur with secondary infection. Psoriatic alopecia may occur in the setting of classic psoriasis and also may occur in psoriasiform drug eruptions, including those caused by tumor necrosis factor inhibitors.20,21 Histologic features include atrophy of sebaceous glands, epidermal changes with hypogranulosis and psoriasiform hyperplasia, decreased hair follicle density, and neutrophils in the stratum spinosum (Figure 4). There often is associated perifollicular lymphocytic inflammation with small lymphocytes that do not have notable morphologic abnormalities.
- Willemze R, Cerroni L, Kempf W, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood. 2019;133:1703-1714. doi:10.1182/blood-2018-11-881268
- Malveira MIB, Pascoal G, Gamonal SBL, et al. Folliculotropic mycosis fungoides: challenging clinical, histopathological and immunohistochemical diagnosis. An Bras Dermatol. 2017;92(5 suppl 1):73-75. doi:10.1590/abd1806-4841.20175634
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525- 530. doi:10.1034/j.1600-0560.2001.281006.x
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides: a distinct disease entity with or without associated follicular mucinosis: a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198. doi:10.1001/archderm.138.2.191
- van Santen S, Roach REJ, van Doorn R, et al. Clinical staging and prognostic factors in folliculotropic mycosis fungoides. JAMA Dermatol. 2016;152:992-1000. doi:10.1001/jamadermatol.2016.1597
- Lehman JS, Cook-Norris RH, Weed BR, et al. Folliculotropic mycosis fungoides: single-center study and systematic review. Arch Dermatol. 2010;146:607-613. doi:10.1001/archdermatol.2010.101
- Gerami P, Rosen S, Kuzel T, et al. Folliculotropic mycosis fungoides: an aggressive variant of cutaneous T-cell lymphoma. Arch Dermatol. 2008;144:738-746. doi:10.1001/archderm.144.6.738
- Büchner SA, Meier M, Rufli TH. Follicular mucinosis associated with mycosis fungoides. Dermatology. 1991;183:66-67. doi:10.1159/000247639
- Akinsanya AO, Tschen JA. Follicular mucinosis: a case report. Cureus. 2019;11:E4746. doi:10.7759/cureus.4746
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19. doi:10.1111/j.1600-0560.2009.01338.x
- Khalil J, Kurban M, Abbas O. Follicular mucinosis: a review. Int J Dermatol. 2021;60:159-165. doi:10.1111/ijd.15165
- Zvulunov A, Shkalim V, Ben-Amitai D, et al. Clinical and histopathologic spectrum of alopecia mucinosa/follicular mucinosis and its natural history in children. J Am Acad Dermatol. 2012;67:1174-1181. doi:10.1016/j.jaad.2012.04.015
- Dessinioti C, Katsambas A. Seborrheic dermatitis: etiology, risk factors, and treatments: facts and controversies. Clin Dermatol. 2013;31:343-351. doi:10.1016/j.clindermatol.2013.01.001
- Gupta AK, Bluhm R. Seborrheic dermatitis. J Eur Acad Dermatol Venereol. 2004;18:13-26; quiz 19-20. doi:10.1111/j .1468-3083.2004.00693.x
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: disease characteristics, clinical evaluation, and new perspectives on pathogenesis. J Am Acad Dermatol. 2018;78:1-12. doi:10.1016/j .jaad.2017.04.1141
- Alkhalifah A, Alsantali A, Wang E, et al. Alopecia areata update: part I. clinical picture, histopathology, and pathogenesis. J Am Acad Dermatol. 2010;62:177-88, quiz 189-90. doi:10.1016/j.jaad.2009.10.032
- Whiting DA. Histopathologic features of alopecia areata: a new look. Arch Dermatol. 2003;139:1555-1559. doi:10.1001/archderm .139.12.1555
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11(2 pt 1):216-223. doi:10.1016 /s0190-9622(84)70152-6
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Afaasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-539. doi:10.1111/cup.12932
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? A clinicohistologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
The Diagnosis: Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) characterized by folliculotropism and follicular-based lesions. The clinical manifestation of FMF can vary and includes patches, plaques, or tumors resembling nonfolliculotropic MF; acneform lesions including comedones and pustules; or areas of alopecia. Lesions commonly involve the head and neck but also can be seen on the trunk or extremities. Folliculotropic mycosis fungoides can be accompanied by pruritus or superimposed secondary infection.
Histologic features of FMF include follicular (perifollicular or intrafollicular) infiltration by atypical T cells showing cerebriform nuclei.1 In early lesions, there may be only mild superficial perivascular inflammation without notable lymphocyte atypia, making diagnosis challenging. 2,3 Mucinous degeneration of the follicles—termed follicular mucinosis—is a common histologic finding in FMF.1,2 Follicular mucinosis is not exclusive to FMF; it can be primary/idiopathic or secondary to underlying inflammatory or neoplastic disorders such as FMF. On immunohistochemistry, FMF most commonly demonstrates a helper T cell phenotype that is positive for CD3 and CD4 and negative for CD8, with aberrant loss of CD7 and variably CD5, which is similar to classic MF. Occasionally, larger CD30+ cells also can be present in the dermis. T-cell gene rearrangement studies will demonstrate T-cell receptor clonality in most cases.2
Many large retrospective cohort studies have suggested that patients with FMF have a worse prognosis than classic MF, with a 5-year survival rate of 62% to 87% for early-stage FMF vs more than 90% for classic patchand plaque-stage MF.4-7 However, a 2016 study suggested histologic evaluation may be able to further differentiate clinically identical cases into indolent and aggressive forms of FMF with considerably different outcomes based on the density of the perifollicular infiltrate.5 The presence of follicular mucinosis has no impact on prognosis compared to cases without follicular mucinosis.1,2
Alopecia mucinosa is characterized by infiltrating, erythematous, scaling plaques localized to the head and neck.8 It is diagnosed clinically, and histopathology shows follicular mucinosis. The terms alopecia mucinosa and follicular mucinosis often are used interchangeably. Over the past few decades, 3 variants have been categorized: primary acute, primary chronic, and secondary. The primary acute form manifests in children and young adults as solitary lesions, which often resolve spontaneously. In contrast, the primary chronic form manifests in older adults as multiple disseminated lesions with a chronic relapsing course.8,9 The secondary form can occur in the setting of other disorders, including lupus erythematosus, hypertrophic lichen planus, alopecia areata, and neoplasms such as MF or Hodgkin lymphoma.9 The histopathologic findings are similar for all types of alopecia mucinosa, with cystic pools of mucin deposition in the sebaceous glands and external root sheath of the follicles as well as associated inflammation composed of lymphocytes and eosinophils (Figure 1).9,10 The inflammatory infiltrate rarely extends into the epidermis or upper portion of the hair follicle. Although histopathology alone cannot reliably distinguish between primary and secondary forms of alopecia mucinosa, MF (including follicular MF) or another underlying cutaneous T-cell lymphoma should be considered if inflammation extends into the upper dermis, epidermis, or follicles or is in a dense bandlike distribution.11 On immunohistochemistry, lymphocytes should show positivity for CD3, CD4, and CD8. The CD4:CD8 ratio often is 1:1 in alopecia mucinosa, while in FMF it is approximately 3:1.10 CD7 commonly is negative but can be present in a small percentage of cases.12 T-cell receptor gene rearrangement studies have detected clonality in both primary and secondary alopecia mucinosa and thus cannot be used alone to distinguish between the two.10 Given the overlap in histopathologic and immunohistochemical features of primary and secondary alopecia mucinosa, definitive diagnosis cannot be made with any single modality and should be based on correlating clinical presentation, histopathology, immunohistochemistry, and molecular analyses.
Inflammatory dermatoses including seborrheic dermatitis also are in the differential diagnosis for FMF. Seborrheic dermatitis is a common chronic inflammatory skin disorder affecting 1% to 3% of the general population. 13 Patients usually present with scaly and greasy plaques and papules localized to areas with increased sebaceous glands and high sebum production such as the face, scalp, and intertriginous regions. The distribution often is symmetrical, and the severity of disease can vary substantially.13 Sebopsoriasis is an entity with overlapping features of seborrheic dermatitis and psoriasis, including thicker, more erythematous plaques that are more elevated. Histopathology of seborrheic dermatitis reveals spongiotic inflammation in the epidermis characterized by rounding of the keratinocytes, widening of the intercellular spaces, and accumulation of intracellular edema, causing the formation of clear spaces in the epidermis (Figure 2). Focal parakeratosis, usually in the follicular ostia, and mounds of scaly crust often are present. 14 A periodic acid–Schiff stain should be performed to rule out infectious dermatophytes, which can show similar clinical and histologic features. More chronic cases of seborrheic dermatitis often can take on histologic features of psoriasis, namely epidermal hyperplasia with thinning over dermal papillae, though the hyperplasia in psoriasis is more regular.
Alopecia areata is an immune-mediated disorder characterized by nonscarring hair loss; it affects approximately 0.1% to 0.2% of the general population.15 The pathogenesis involves the premature transition of hair follicles in the anagen (growth) phase to the catagen ( nonproliferative/involution) and telogen (resting) phases, resulting in sudden hair shedding and decreased regrowth. Clinically, it is characterized by asymptomatic hair loss that occurs most frequently on the scalp and other areas of the head, including eyelashes, eyebrows, and facial hair, but also can occur on the extremities. There are several variants; the most common is patchy alopecia, which features smooth circular areas of hair loss that progress over several weeks. Some patients can progress to loss of all scalp hairs (alopecia totalis) or all hairs throughout the body (alopecia universalis). 15 Patients typically will have spontaneous regrowth of hair, with up to 50% of those with limited hair loss recovering within a year.16 The disease has a chronic/ relapsing course, and patients often will have multiple episodes of hair loss. Histopathologic features can vary depending on the stage of disease. In acute cases, a peribulbar lymphocytic infiltrate preferentially involving anagen-stage hair follicles is seen, with associated necrosis, edema, and pigment incontinence (Figure 3).16 In chronic alopecia areata, the inflammation may be less brisk, and follicular miniaturization often is seen. Additionally, increased proportions of catagen- or telogen-stage follicles are present.16,17 On immunohistochemistry, lymphocytes express both CD4 and CD8, with a slightly increased CD4:CD8 ratio in active disease.18
Psoriatic alopecia describes hair loss that occurs in patients with psoriasis. Patients present with scaly, erythematous, psoriasiform plaques or patches, as well as decreased hair density, finer hairs, and increased dystrophic hair bulbs within the psoriatic plaques.19 It often is nonscarring and resolves with therapy, though scarring may occur with secondary infection. Psoriatic alopecia may occur in the setting of classic psoriasis and also may occur in psoriasiform drug eruptions, including those caused by tumor necrosis factor inhibitors.20,21 Histologic features include atrophy of sebaceous glands, epidermal changes with hypogranulosis and psoriasiform hyperplasia, decreased hair follicle density, and neutrophils in the stratum spinosum (Figure 4). There often is associated perifollicular lymphocytic inflammation with small lymphocytes that do not have notable morphologic abnormalities.
The Diagnosis: Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) characterized by folliculotropism and follicular-based lesions. The clinical manifestation of FMF can vary and includes patches, plaques, or tumors resembling nonfolliculotropic MF; acneform lesions including comedones and pustules; or areas of alopecia. Lesions commonly involve the head and neck but also can be seen on the trunk or extremities. Folliculotropic mycosis fungoides can be accompanied by pruritus or superimposed secondary infection.
Histologic features of FMF include follicular (perifollicular or intrafollicular) infiltration by atypical T cells showing cerebriform nuclei.1 In early lesions, there may be only mild superficial perivascular inflammation without notable lymphocyte atypia, making diagnosis challenging. 2,3 Mucinous degeneration of the follicles—termed follicular mucinosis—is a common histologic finding in FMF.1,2 Follicular mucinosis is not exclusive to FMF; it can be primary/idiopathic or secondary to underlying inflammatory or neoplastic disorders such as FMF. On immunohistochemistry, FMF most commonly demonstrates a helper T cell phenotype that is positive for CD3 and CD4 and negative for CD8, with aberrant loss of CD7 and variably CD5, which is similar to classic MF. Occasionally, larger CD30+ cells also can be present in the dermis. T-cell gene rearrangement studies will demonstrate T-cell receptor clonality in most cases.2
Many large retrospective cohort studies have suggested that patients with FMF have a worse prognosis than classic MF, with a 5-year survival rate of 62% to 87% for early-stage FMF vs more than 90% for classic patchand plaque-stage MF.4-7 However, a 2016 study suggested histologic evaluation may be able to further differentiate clinically identical cases into indolent and aggressive forms of FMF with considerably different outcomes based on the density of the perifollicular infiltrate.5 The presence of follicular mucinosis has no impact on prognosis compared to cases without follicular mucinosis.1,2
Alopecia mucinosa is characterized by infiltrating, erythematous, scaling plaques localized to the head and neck.8 It is diagnosed clinically, and histopathology shows follicular mucinosis. The terms alopecia mucinosa and follicular mucinosis often are used interchangeably. Over the past few decades, 3 variants have been categorized: primary acute, primary chronic, and secondary. The primary acute form manifests in children and young adults as solitary lesions, which often resolve spontaneously. In contrast, the primary chronic form manifests in older adults as multiple disseminated lesions with a chronic relapsing course.8,9 The secondary form can occur in the setting of other disorders, including lupus erythematosus, hypertrophic lichen planus, alopecia areata, and neoplasms such as MF or Hodgkin lymphoma.9 The histopathologic findings are similar for all types of alopecia mucinosa, with cystic pools of mucin deposition in the sebaceous glands and external root sheath of the follicles as well as associated inflammation composed of lymphocytes and eosinophils (Figure 1).9,10 The inflammatory infiltrate rarely extends into the epidermis or upper portion of the hair follicle. Although histopathology alone cannot reliably distinguish between primary and secondary forms of alopecia mucinosa, MF (including follicular MF) or another underlying cutaneous T-cell lymphoma should be considered if inflammation extends into the upper dermis, epidermis, or follicles or is in a dense bandlike distribution.11 On immunohistochemistry, lymphocytes should show positivity for CD3, CD4, and CD8. The CD4:CD8 ratio often is 1:1 in alopecia mucinosa, while in FMF it is approximately 3:1.10 CD7 commonly is negative but can be present in a small percentage of cases.12 T-cell receptor gene rearrangement studies have detected clonality in both primary and secondary alopecia mucinosa and thus cannot be used alone to distinguish between the two.10 Given the overlap in histopathologic and immunohistochemical features of primary and secondary alopecia mucinosa, definitive diagnosis cannot be made with any single modality and should be based on correlating clinical presentation, histopathology, immunohistochemistry, and molecular analyses.
Inflammatory dermatoses including seborrheic dermatitis also are in the differential diagnosis for FMF. Seborrheic dermatitis is a common chronic inflammatory skin disorder affecting 1% to 3% of the general population. 13 Patients usually present with scaly and greasy plaques and papules localized to areas with increased sebaceous glands and high sebum production such as the face, scalp, and intertriginous regions. The distribution often is symmetrical, and the severity of disease can vary substantially.13 Sebopsoriasis is an entity with overlapping features of seborrheic dermatitis and psoriasis, including thicker, more erythematous plaques that are more elevated. Histopathology of seborrheic dermatitis reveals spongiotic inflammation in the epidermis characterized by rounding of the keratinocytes, widening of the intercellular spaces, and accumulation of intracellular edema, causing the formation of clear spaces in the epidermis (Figure 2). Focal parakeratosis, usually in the follicular ostia, and mounds of scaly crust often are present. 14 A periodic acid–Schiff stain should be performed to rule out infectious dermatophytes, which can show similar clinical and histologic features. More chronic cases of seborrheic dermatitis often can take on histologic features of psoriasis, namely epidermal hyperplasia with thinning over dermal papillae, though the hyperplasia in psoriasis is more regular.
Alopecia areata is an immune-mediated disorder characterized by nonscarring hair loss; it affects approximately 0.1% to 0.2% of the general population.15 The pathogenesis involves the premature transition of hair follicles in the anagen (growth) phase to the catagen ( nonproliferative/involution) and telogen (resting) phases, resulting in sudden hair shedding and decreased regrowth. Clinically, it is characterized by asymptomatic hair loss that occurs most frequently on the scalp and other areas of the head, including eyelashes, eyebrows, and facial hair, but also can occur on the extremities. There are several variants; the most common is patchy alopecia, which features smooth circular areas of hair loss that progress over several weeks. Some patients can progress to loss of all scalp hairs (alopecia totalis) or all hairs throughout the body (alopecia universalis). 15 Patients typically will have spontaneous regrowth of hair, with up to 50% of those with limited hair loss recovering within a year.16 The disease has a chronic/ relapsing course, and patients often will have multiple episodes of hair loss. Histopathologic features can vary depending on the stage of disease. In acute cases, a peribulbar lymphocytic infiltrate preferentially involving anagen-stage hair follicles is seen, with associated necrosis, edema, and pigment incontinence (Figure 3).16 In chronic alopecia areata, the inflammation may be less brisk, and follicular miniaturization often is seen. Additionally, increased proportions of catagen- or telogen-stage follicles are present.16,17 On immunohistochemistry, lymphocytes express both CD4 and CD8, with a slightly increased CD4:CD8 ratio in active disease.18
Psoriatic alopecia describes hair loss that occurs in patients with psoriasis. Patients present with scaly, erythematous, psoriasiform plaques or patches, as well as decreased hair density, finer hairs, and increased dystrophic hair bulbs within the psoriatic plaques.19 It often is nonscarring and resolves with therapy, though scarring may occur with secondary infection. Psoriatic alopecia may occur in the setting of classic psoriasis and also may occur in psoriasiform drug eruptions, including those caused by tumor necrosis factor inhibitors.20,21 Histologic features include atrophy of sebaceous glands, epidermal changes with hypogranulosis and psoriasiform hyperplasia, decreased hair follicle density, and neutrophils in the stratum spinosum (Figure 4). There often is associated perifollicular lymphocytic inflammation with small lymphocytes that do not have notable morphologic abnormalities.
- Willemze R, Cerroni L, Kempf W, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood. 2019;133:1703-1714. doi:10.1182/blood-2018-11-881268
- Malveira MIB, Pascoal G, Gamonal SBL, et al. Folliculotropic mycosis fungoides: challenging clinical, histopathological and immunohistochemical diagnosis. An Bras Dermatol. 2017;92(5 suppl 1):73-75. doi:10.1590/abd1806-4841.20175634
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525- 530. doi:10.1034/j.1600-0560.2001.281006.x
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides: a distinct disease entity with or without associated follicular mucinosis: a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198. doi:10.1001/archderm.138.2.191
- van Santen S, Roach REJ, van Doorn R, et al. Clinical staging and prognostic factors in folliculotropic mycosis fungoides. JAMA Dermatol. 2016;152:992-1000. doi:10.1001/jamadermatol.2016.1597
- Lehman JS, Cook-Norris RH, Weed BR, et al. Folliculotropic mycosis fungoides: single-center study and systematic review. Arch Dermatol. 2010;146:607-613. doi:10.1001/archdermatol.2010.101
- Gerami P, Rosen S, Kuzel T, et al. Folliculotropic mycosis fungoides: an aggressive variant of cutaneous T-cell lymphoma. Arch Dermatol. 2008;144:738-746. doi:10.1001/archderm.144.6.738
- Büchner SA, Meier M, Rufli TH. Follicular mucinosis associated with mycosis fungoides. Dermatology. 1991;183:66-67. doi:10.1159/000247639
- Akinsanya AO, Tschen JA. Follicular mucinosis: a case report. Cureus. 2019;11:E4746. doi:10.7759/cureus.4746
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19. doi:10.1111/j.1600-0560.2009.01338.x
- Khalil J, Kurban M, Abbas O. Follicular mucinosis: a review. Int J Dermatol. 2021;60:159-165. doi:10.1111/ijd.15165
- Zvulunov A, Shkalim V, Ben-Amitai D, et al. Clinical and histopathologic spectrum of alopecia mucinosa/follicular mucinosis and its natural history in children. J Am Acad Dermatol. 2012;67:1174-1181. doi:10.1016/j.jaad.2012.04.015
- Dessinioti C, Katsambas A. Seborrheic dermatitis: etiology, risk factors, and treatments: facts and controversies. Clin Dermatol. 2013;31:343-351. doi:10.1016/j.clindermatol.2013.01.001
- Gupta AK, Bluhm R. Seborrheic dermatitis. J Eur Acad Dermatol Venereol. 2004;18:13-26; quiz 19-20. doi:10.1111/j .1468-3083.2004.00693.x
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: disease characteristics, clinical evaluation, and new perspectives on pathogenesis. J Am Acad Dermatol. 2018;78:1-12. doi:10.1016/j .jaad.2017.04.1141
- Alkhalifah A, Alsantali A, Wang E, et al. Alopecia areata update: part I. clinical picture, histopathology, and pathogenesis. J Am Acad Dermatol. 2010;62:177-88, quiz 189-90. doi:10.1016/j.jaad.2009.10.032
- Whiting DA. Histopathologic features of alopecia areata: a new look. Arch Dermatol. 2003;139:1555-1559. doi:10.1001/archderm .139.12.1555
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11(2 pt 1):216-223. doi:10.1016 /s0190-9622(84)70152-6
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Afaasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-539. doi:10.1111/cup.12932
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? A clinicohistologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
- Willemze R, Cerroni L, Kempf W, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood. 2019;133:1703-1714. doi:10.1182/blood-2018-11-881268
- Malveira MIB, Pascoal G, Gamonal SBL, et al. Folliculotropic mycosis fungoides: challenging clinical, histopathological and immunohistochemical diagnosis. An Bras Dermatol. 2017;92(5 suppl 1):73-75. doi:10.1590/abd1806-4841.20175634
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525- 530. doi:10.1034/j.1600-0560.2001.281006.x
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides: a distinct disease entity with or without associated follicular mucinosis: a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198. doi:10.1001/archderm.138.2.191
- van Santen S, Roach REJ, van Doorn R, et al. Clinical staging and prognostic factors in folliculotropic mycosis fungoides. JAMA Dermatol. 2016;152:992-1000. doi:10.1001/jamadermatol.2016.1597
- Lehman JS, Cook-Norris RH, Weed BR, et al. Folliculotropic mycosis fungoides: single-center study and systematic review. Arch Dermatol. 2010;146:607-613. doi:10.1001/archdermatol.2010.101
- Gerami P, Rosen S, Kuzel T, et al. Folliculotropic mycosis fungoides: an aggressive variant of cutaneous T-cell lymphoma. Arch Dermatol. 2008;144:738-746. doi:10.1001/archderm.144.6.738
- Büchner SA, Meier M, Rufli TH. Follicular mucinosis associated with mycosis fungoides. Dermatology. 1991;183:66-67. doi:10.1159/000247639
- Akinsanya AO, Tschen JA. Follicular mucinosis: a case report. Cureus. 2019;11:E4746. doi:10.7759/cureus.4746
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19. doi:10.1111/j.1600-0560.2009.01338.x
- Khalil J, Kurban M, Abbas O. Follicular mucinosis: a review. Int J Dermatol. 2021;60:159-165. doi:10.1111/ijd.15165
- Zvulunov A, Shkalim V, Ben-Amitai D, et al. Clinical and histopathologic spectrum of alopecia mucinosa/follicular mucinosis and its natural history in children. J Am Acad Dermatol. 2012;67:1174-1181. doi:10.1016/j.jaad.2012.04.015
- Dessinioti C, Katsambas A. Seborrheic dermatitis: etiology, risk factors, and treatments: facts and controversies. Clin Dermatol. 2013;31:343-351. doi:10.1016/j.clindermatol.2013.01.001
- Gupta AK, Bluhm R. Seborrheic dermatitis. J Eur Acad Dermatol Venereol. 2004;18:13-26; quiz 19-20. doi:10.1111/j .1468-3083.2004.00693.x
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: disease characteristics, clinical evaluation, and new perspectives on pathogenesis. J Am Acad Dermatol. 2018;78:1-12. doi:10.1016/j .jaad.2017.04.1141
- Alkhalifah A, Alsantali A, Wang E, et al. Alopecia areata update: part I. clinical picture, histopathology, and pathogenesis. J Am Acad Dermatol. 2010;62:177-88, quiz 189-90. doi:10.1016/j.jaad.2009.10.032
- Whiting DA. Histopathologic features of alopecia areata: a new look. Arch Dermatol. 2003;139:1555-1559. doi:10.1001/archderm .139.12.1555
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11(2 pt 1):216-223. doi:10.1016 /s0190-9622(84)70152-6
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Afaasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-539. doi:10.1111/cup.12932
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? A clinicohistologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
An 88-year-old man presented with progressive eyelash loss and scale involving the right eyelids (top). Dermatopathologic examination was performed (bottom).
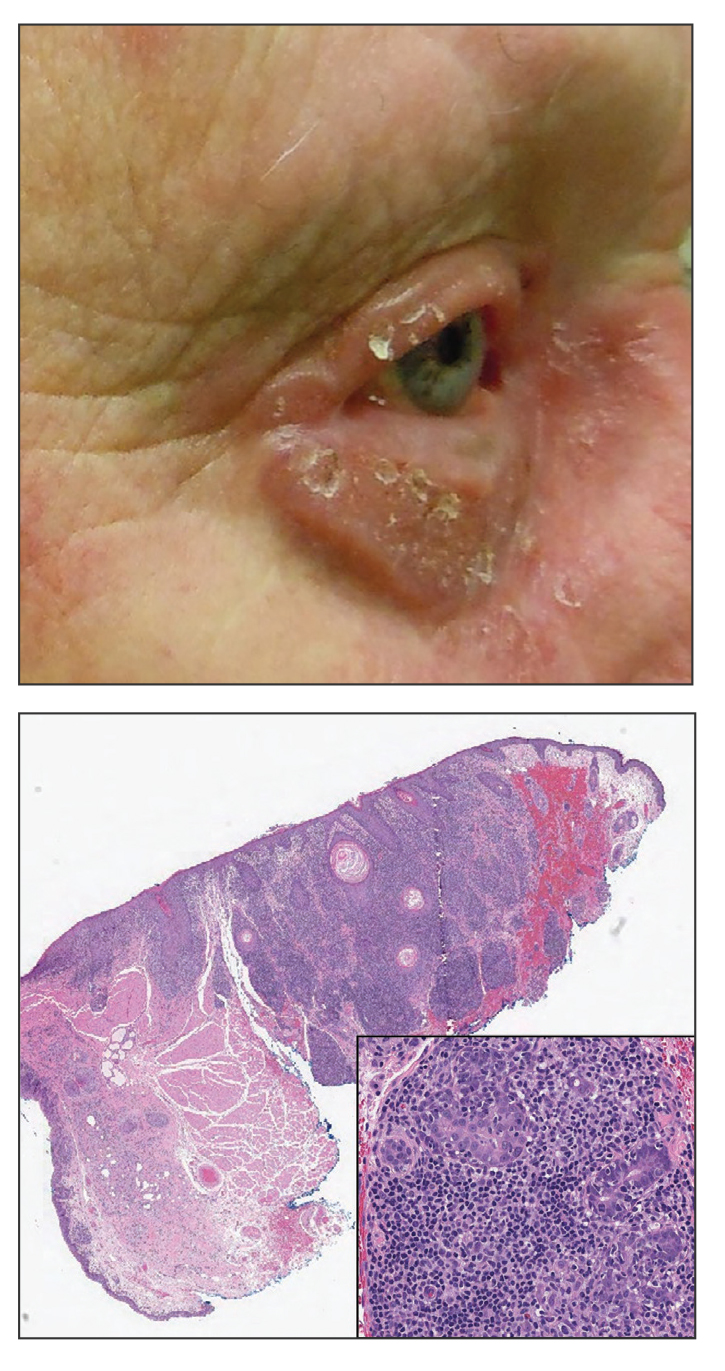
Erythematous Flaky Rash on the Toe
The Diagnosis: Necrolytic Migratory Erythema
Necrolytic migratory erythema (NME) is a waxing and waning rash associated with rare pancreatic neuroendocrine tumors called glucagonomas. It is characterized by pruritic and painful, well-demarcated, erythematous plaques that manifest in the intertriginous areas and on the perineum and buttocks.1 Due to the evolving nature of the rash, the histopathologic findings in NME vary depending on the stage of the cutaneous lesions at the time of biopsy.2 Multiple dyskeratotic keratinocytes spanning all epidermal layers may be a diagnostic clue in early lesions of NME.3 Typical features of longstanding lesions include confluent parakeratosis, psoriasiform hyperplasia with mild or absent spongiosis, and upper epidermal necrosis with keratinocyte vacuolization and pallor.4 Morphologic features that are present prior to the development of epidermal vacuolation and necrosis frequently are misattributed to psoriasis or eczema. Long-standing lesions also may develop a neutrophilic infiltrate with subcorneal and intraepidermal pustules.2 Other common features include a discrete perivascular lymphocytic infiltrate and an erosive or encrusted epidermis.5 Although direct immunofluorescence typically is negative, nonspecific findings can be seen, including apoptotic keratinocytes labeling with fibrinogen and C3, as well as scattered, clumped, IgM-positive cytoid bodies present at the dermal-epidermal junction (DEJ).6 Biopsies also have shown scattered, clumped, IgM-positive cytoid bodies present at the DEJ.5
Psoriasis is a chronic relapsing papulosquamous disorder characterized by scaly erythematous plaques often overlying the extensor surfaces of the extremities. Histopathology shows a psoriasiform pattern of inflammation with thinning of the suprapapillary plates and elongation of the rete ridges. Further diagnostic clues of psoriasis include regular acanthosis, characteristic Munro microabscesses with neutrophils in a hyperkeratotic stratum corneum (Figure 1), hypogranulosis, and neutrophilic spongiform pustules of Kogoj in the stratum spinosum. Generally, there is a lack of the epidermal necrosis seen with NME.7,8
Lichen simplex chronicus manifests as pruritic, often hyperpigmented, well-defined, lichenified plaques with excoriation following repetitive mechanical trauma, commonly on the lower lateral legs, posterior neck, and flexural areas.9 The histologic landscape is marked by well-developed lesions evolving to show compact orthokeratosis, hypergranulosis, irregularly elongated rete ridges (ie, irregular acanthosis), and papillary dermal fibrosis with vertical streaking of collagen (Figure 2).9,10
Subacute cutaneous lupus erythematosus (SCLE) is recognized clinically by scaly/psoriasiform and annular lesions with mild or absent systemic involvement. Common histopathologic findings include epidermal atrophy, vacuolar interface dermatitis with hydropic degeneration of the basal layer, a subepidermal lymphocytic infiltrate, and a periadnexal and perivascular infiltrate (Figure 3).11 Upper dermal edema, spotty necrosis of individual cells in the epidermis, dermal-epidermal separation caused by prominent basal cell degeneration, and accumulation of acid mucopolysaccharides (mucin) are other histologic features associated with SCLE.12,13
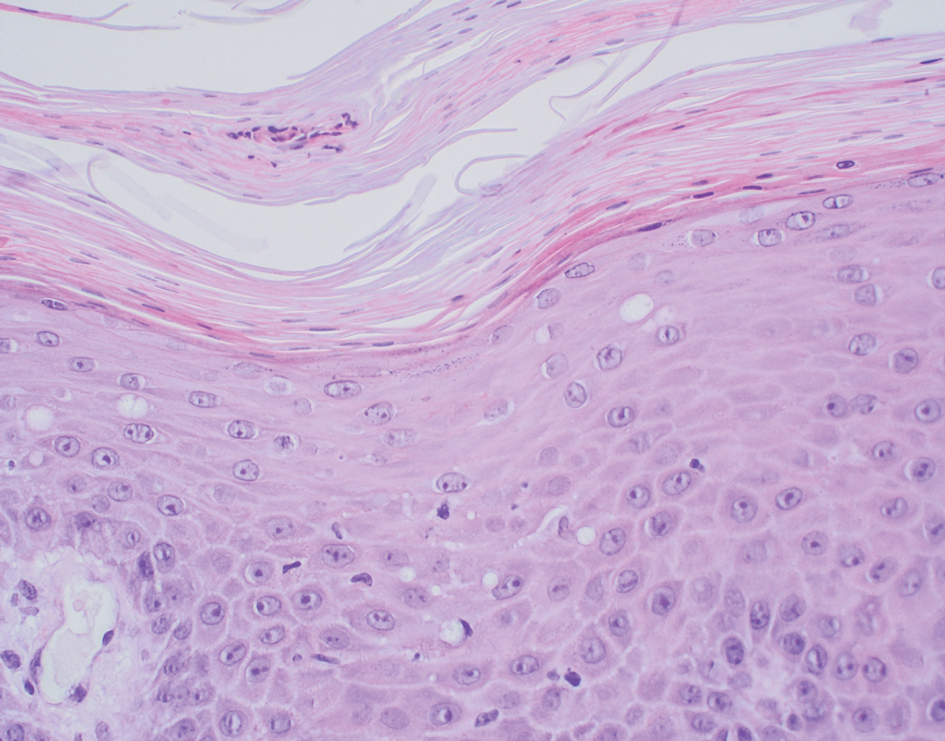
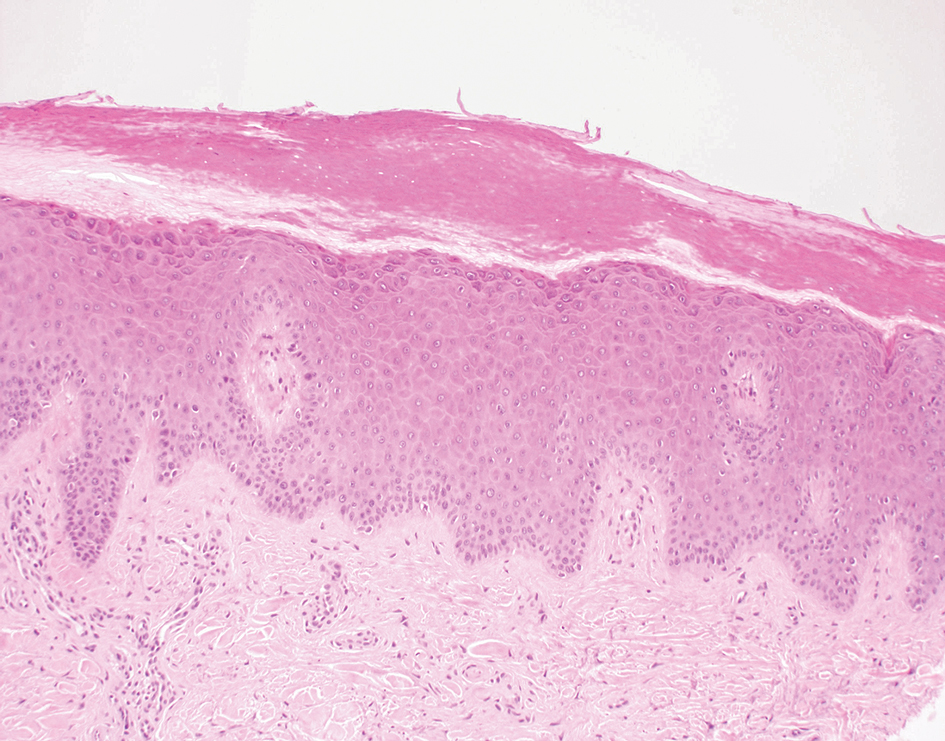
The immunofluorescence pattern in SCLE features dustlike particles of IgG deposition in the epidermis, subepidermal region, and dermal cellular infiltrate. Lesions also may have granular deposition of immunoreactions at the DEJ.11,13
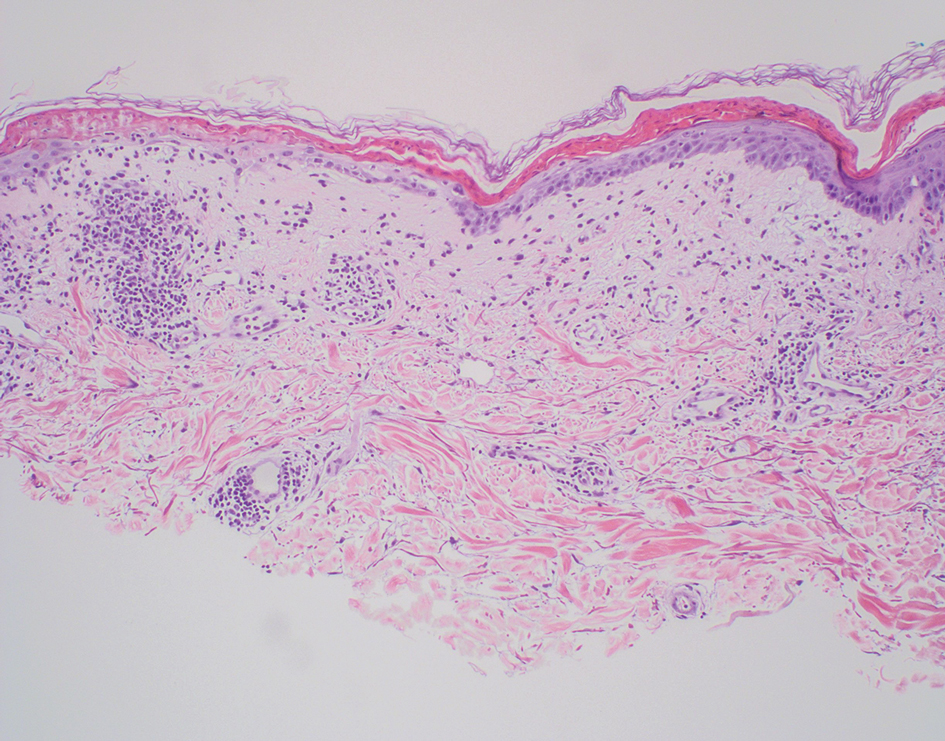
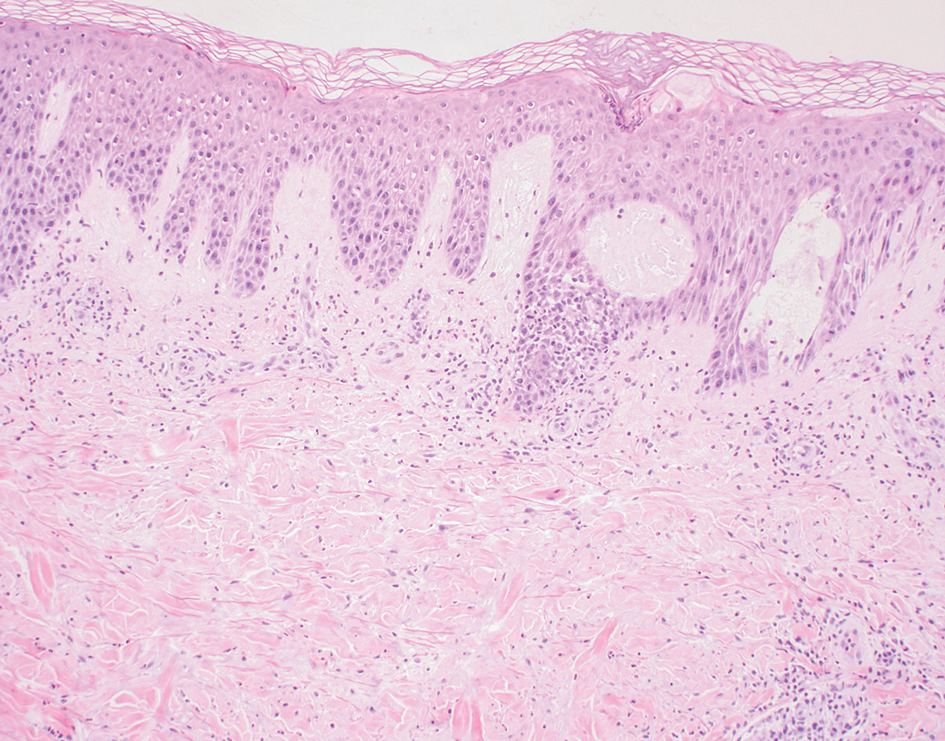
The manifestation of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome (also known as drug-induced hypersensitivity syndrome) is variable, with a morbilliform rash that spreads from the face to the entire body, urticaria, atypical target lesions, purpuriform lesions, lymphadenopathy, and exfoliative dermatitis.14 The nonspecific morphologic features of DRESS syndrome lesions are associated with variable histologic features, which include focal interface changes with vacuolar alteration of the basal layer; atypical lymphocytes with hyperchromic nuclei; and a superficial, inconsistently dense, perivascular lymphocytic infiltrate. Other relatively common histopathologic patterns include an upper dermis with dilated blood vessels, spongiosis with exocytosis of lymphocytes (Figure 4), and necrotic keratinocytes. Although peripheral eosinophilia is an important diagnostic criterion and is observed consistently, eosinophils are variably present on skin biopsy.15,16 Given the histopathologic variability and nonspecific findings, clinical correlation is required when diagnosing DRESS syndrome.
- Halvorson SA, Gilbert E, Hopkins RS, et al. Putting the pieces together: necrolytic migratory erythema and the glucagonoma syndrome. J Gen Intern Med. 2013;28:1525-1529. doi:10.1007 /s11606-013-2490-5
- Toberer F, Hartschuh W, Wiedemeyer K. Glucagonoma-associated necrolytic migratory erythema: the broad spectrum of the clinical and histopathological findings and clues to the diagnosis. Am J Dermatopathol. 2019;41:E29-E32. doi:10.1097DAD .0000000000001219
- Hunt SJ, Narus VT, Abell E. Necrolytic migratory erythema: dyskeratotic dermatitis, a clue to early diagnosis. J Am Acad Dermatol. 1991; 24:473-477. doi:10.1016/0190-9622(91)70076-e
- van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol. 2004;151:531-537. doi:10.1530/eje.0.1510531
- Pujol RM, Wang C-Y E, el-Azhary RA, et al. Necrolytic migratory erythema: clinicopathologic study of 13 cases. Int J Dermatol. 2004;43:12- 18. doi:10.1111/j.1365-4632.2004.01844.x
- Johnson SM, Smoller BR, Lamps LW, et al. Necrolytic migratory erythema as the only presenting sign of a glucagonoma. J Am Acad Dermatol. 2003;49:325-328. doi:10.1067/s0190-9622(02)61774-8
- De Rosa G, Mignogna C. The histopathology of psoriasis. Reumatismo. 2007;59(suppl 1):46-48. doi:10.4081/reumatismo.2007.1s.46
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Bhutani T, Liao W, Nakamura M, eds. Evidence-Based Psoriasis. Springer; 2018:1-16. doi:10.1007/978-3-319-90107-7_1
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68. doi:10.22551/2019.24.0603.10155
- O’Keefe RJ, Scurry JP, Dennerstein G, et al. Audit of 114 nonneoplastic vulvar biopsies. Br J Obstet Gynaecol. 1995;102:780-786. doi:10.1111/j.1471-0528.1995.tb10842.x
- Parodi A, Caproni M, Cardinali C, et al P. Clinical, histological and immunopathological features of 58 patients with subacute cutaneous lupus erythematosus. Dermatology. 2000;200:6-10. doi:10.1159/000018307
- Lyon CC, Blewitt R, Harrison PV. Subacute cutaneous lupus erythematosus: two cases of delayed diagnosis. Acta Derm Venereol. 1998;78:57-59. doi:10.1080/00015559850135869
- David-Bajar KM. Subacute cutaneous lupus erythematosus. J Invest Dermatol. 1993;100:2S-8S. doi:10.1111/1523-1747.ep12355164
- Paulmann M, Mockenhaupt M. Severe drug-induced skin reactions: clinical features, diagnosis, etiology, and therapy. J Dtsch Dermatol Ges. 2015;13:625-643. doi:10.1111/ddg.12747
- Borroni G, Torti S, Pezzini C, et al. Histopathologic spectrum of drug reaction with eosinophilia and systemic symptoms (DRESS): a diagnosis that needs clinico-pathological correlation. G Ital Dermatol Venereol. 2014;149:291-300.
- Ortonne N, Valeyrie-Allanore L, Bastuji-Garin S, et al. Histopathology of drug rash with eosinophilia and systemic symptoms syndrome: a morphological and phenotypical study. Br J Dermatol. 2015;173:50-58. doi:10.1111/bjd.13683
The Diagnosis: Necrolytic Migratory Erythema
Necrolytic migratory erythema (NME) is a waxing and waning rash associated with rare pancreatic neuroendocrine tumors called glucagonomas. It is characterized by pruritic and painful, well-demarcated, erythematous plaques that manifest in the intertriginous areas and on the perineum and buttocks.1 Due to the evolving nature of the rash, the histopathologic findings in NME vary depending on the stage of the cutaneous lesions at the time of biopsy.2 Multiple dyskeratotic keratinocytes spanning all epidermal layers may be a diagnostic clue in early lesions of NME.3 Typical features of longstanding lesions include confluent parakeratosis, psoriasiform hyperplasia with mild or absent spongiosis, and upper epidermal necrosis with keratinocyte vacuolization and pallor.4 Morphologic features that are present prior to the development of epidermal vacuolation and necrosis frequently are misattributed to psoriasis or eczema. Long-standing lesions also may develop a neutrophilic infiltrate with subcorneal and intraepidermal pustules.2 Other common features include a discrete perivascular lymphocytic infiltrate and an erosive or encrusted epidermis.5 Although direct immunofluorescence typically is negative, nonspecific findings can be seen, including apoptotic keratinocytes labeling with fibrinogen and C3, as well as scattered, clumped, IgM-positive cytoid bodies present at the dermal-epidermal junction (DEJ).6 Biopsies also have shown scattered, clumped, IgM-positive cytoid bodies present at the DEJ.5
Psoriasis is a chronic relapsing papulosquamous disorder characterized by scaly erythematous plaques often overlying the extensor surfaces of the extremities. Histopathology shows a psoriasiform pattern of inflammation with thinning of the suprapapillary plates and elongation of the rete ridges. Further diagnostic clues of psoriasis include regular acanthosis, characteristic Munro microabscesses with neutrophils in a hyperkeratotic stratum corneum (Figure 1), hypogranulosis, and neutrophilic spongiform pustules of Kogoj in the stratum spinosum. Generally, there is a lack of the epidermal necrosis seen with NME.7,8
Lichen simplex chronicus manifests as pruritic, often hyperpigmented, well-defined, lichenified plaques with excoriation following repetitive mechanical trauma, commonly on the lower lateral legs, posterior neck, and flexural areas.9 The histologic landscape is marked by well-developed lesions evolving to show compact orthokeratosis, hypergranulosis, irregularly elongated rete ridges (ie, irregular acanthosis), and papillary dermal fibrosis with vertical streaking of collagen (Figure 2).9,10
Subacute cutaneous lupus erythematosus (SCLE) is recognized clinically by scaly/psoriasiform and annular lesions with mild or absent systemic involvement. Common histopathologic findings include epidermal atrophy, vacuolar interface dermatitis with hydropic degeneration of the basal layer, a subepidermal lymphocytic infiltrate, and a periadnexal and perivascular infiltrate (Figure 3).11 Upper dermal edema, spotty necrosis of individual cells in the epidermis, dermal-epidermal separation caused by prominent basal cell degeneration, and accumulation of acid mucopolysaccharides (mucin) are other histologic features associated with SCLE.12,13
The immunofluorescence pattern in SCLE features dustlike particles of IgG deposition in the epidermis, subepidermal region, and dermal cellular infiltrate. Lesions also may have granular deposition of immunoreactions at the DEJ.11,13
The manifestation of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome (also known as drug-induced hypersensitivity syndrome) is variable, with a morbilliform rash that spreads from the face to the entire body, urticaria, atypical target lesions, purpuriform lesions, lymphadenopathy, and exfoliative dermatitis.14 The nonspecific morphologic features of DRESS syndrome lesions are associated with variable histologic features, which include focal interface changes with vacuolar alteration of the basal layer; atypical lymphocytes with hyperchromic nuclei; and a superficial, inconsistently dense, perivascular lymphocytic infiltrate. Other relatively common histopathologic patterns include an upper dermis with dilated blood vessels, spongiosis with exocytosis of lymphocytes (Figure 4), and necrotic keratinocytes. Although peripheral eosinophilia is an important diagnostic criterion and is observed consistently, eosinophils are variably present on skin biopsy.15,16 Given the histopathologic variability and nonspecific findings, clinical correlation is required when diagnosing DRESS syndrome.
The Diagnosis: Necrolytic Migratory Erythema
Necrolytic migratory erythema (NME) is a waxing and waning rash associated with rare pancreatic neuroendocrine tumors called glucagonomas. It is characterized by pruritic and painful, well-demarcated, erythematous plaques that manifest in the intertriginous areas and on the perineum and buttocks.1 Due to the evolving nature of the rash, the histopathologic findings in NME vary depending on the stage of the cutaneous lesions at the time of biopsy.2 Multiple dyskeratotic keratinocytes spanning all epidermal layers may be a diagnostic clue in early lesions of NME.3 Typical features of longstanding lesions include confluent parakeratosis, psoriasiform hyperplasia with mild or absent spongiosis, and upper epidermal necrosis with keratinocyte vacuolization and pallor.4 Morphologic features that are present prior to the development of epidermal vacuolation and necrosis frequently are misattributed to psoriasis or eczema. Long-standing lesions also may develop a neutrophilic infiltrate with subcorneal and intraepidermal pustules.2 Other common features include a discrete perivascular lymphocytic infiltrate and an erosive or encrusted epidermis.5 Although direct immunofluorescence typically is negative, nonspecific findings can be seen, including apoptotic keratinocytes labeling with fibrinogen and C3, as well as scattered, clumped, IgM-positive cytoid bodies present at the dermal-epidermal junction (DEJ).6 Biopsies also have shown scattered, clumped, IgM-positive cytoid bodies present at the DEJ.5
Psoriasis is a chronic relapsing papulosquamous disorder characterized by scaly erythematous plaques often overlying the extensor surfaces of the extremities. Histopathology shows a psoriasiform pattern of inflammation with thinning of the suprapapillary plates and elongation of the rete ridges. Further diagnostic clues of psoriasis include regular acanthosis, characteristic Munro microabscesses with neutrophils in a hyperkeratotic stratum corneum (Figure 1), hypogranulosis, and neutrophilic spongiform pustules of Kogoj in the stratum spinosum. Generally, there is a lack of the epidermal necrosis seen with NME.7,8
Lichen simplex chronicus manifests as pruritic, often hyperpigmented, well-defined, lichenified plaques with excoriation following repetitive mechanical trauma, commonly on the lower lateral legs, posterior neck, and flexural areas.9 The histologic landscape is marked by well-developed lesions evolving to show compact orthokeratosis, hypergranulosis, irregularly elongated rete ridges (ie, irregular acanthosis), and papillary dermal fibrosis with vertical streaking of collagen (Figure 2).9,10
Subacute cutaneous lupus erythematosus (SCLE) is recognized clinically by scaly/psoriasiform and annular lesions with mild or absent systemic involvement. Common histopathologic findings include epidermal atrophy, vacuolar interface dermatitis with hydropic degeneration of the basal layer, a subepidermal lymphocytic infiltrate, and a periadnexal and perivascular infiltrate (Figure 3).11 Upper dermal edema, spotty necrosis of individual cells in the epidermis, dermal-epidermal separation caused by prominent basal cell degeneration, and accumulation of acid mucopolysaccharides (mucin) are other histologic features associated with SCLE.12,13
The immunofluorescence pattern in SCLE features dustlike particles of IgG deposition in the epidermis, subepidermal region, and dermal cellular infiltrate. Lesions also may have granular deposition of immunoreactions at the DEJ.11,13
The manifestation of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome (also known as drug-induced hypersensitivity syndrome) is variable, with a morbilliform rash that spreads from the face to the entire body, urticaria, atypical target lesions, purpuriform lesions, lymphadenopathy, and exfoliative dermatitis.14 The nonspecific morphologic features of DRESS syndrome lesions are associated with variable histologic features, which include focal interface changes with vacuolar alteration of the basal layer; atypical lymphocytes with hyperchromic nuclei; and a superficial, inconsistently dense, perivascular lymphocytic infiltrate. Other relatively common histopathologic patterns include an upper dermis with dilated blood vessels, spongiosis with exocytosis of lymphocytes (Figure 4), and necrotic keratinocytes. Although peripheral eosinophilia is an important diagnostic criterion and is observed consistently, eosinophils are variably present on skin biopsy.15,16 Given the histopathologic variability and nonspecific findings, clinical correlation is required when diagnosing DRESS syndrome.
- Halvorson SA, Gilbert E, Hopkins RS, et al. Putting the pieces together: necrolytic migratory erythema and the glucagonoma syndrome. J Gen Intern Med. 2013;28:1525-1529. doi:10.1007 /s11606-013-2490-5
- Toberer F, Hartschuh W, Wiedemeyer K. Glucagonoma-associated necrolytic migratory erythema: the broad spectrum of the clinical and histopathological findings and clues to the diagnosis. Am J Dermatopathol. 2019;41:E29-E32. doi:10.1097DAD .0000000000001219
- Hunt SJ, Narus VT, Abell E. Necrolytic migratory erythema: dyskeratotic dermatitis, a clue to early diagnosis. J Am Acad Dermatol. 1991; 24:473-477. doi:10.1016/0190-9622(91)70076-e
- van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol. 2004;151:531-537. doi:10.1530/eje.0.1510531
- Pujol RM, Wang C-Y E, el-Azhary RA, et al. Necrolytic migratory erythema: clinicopathologic study of 13 cases. Int J Dermatol. 2004;43:12- 18. doi:10.1111/j.1365-4632.2004.01844.x
- Johnson SM, Smoller BR, Lamps LW, et al. Necrolytic migratory erythema as the only presenting sign of a glucagonoma. J Am Acad Dermatol. 2003;49:325-328. doi:10.1067/s0190-9622(02)61774-8
- De Rosa G, Mignogna C. The histopathology of psoriasis. Reumatismo. 2007;59(suppl 1):46-48. doi:10.4081/reumatismo.2007.1s.46
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Bhutani T, Liao W, Nakamura M, eds. Evidence-Based Psoriasis. Springer; 2018:1-16. doi:10.1007/978-3-319-90107-7_1
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68. doi:10.22551/2019.24.0603.10155
- O’Keefe RJ, Scurry JP, Dennerstein G, et al. Audit of 114 nonneoplastic vulvar biopsies. Br J Obstet Gynaecol. 1995;102:780-786. doi:10.1111/j.1471-0528.1995.tb10842.x
- Parodi A, Caproni M, Cardinali C, et al P. Clinical, histological and immunopathological features of 58 patients with subacute cutaneous lupus erythematosus. Dermatology. 2000;200:6-10. doi:10.1159/000018307
- Lyon CC, Blewitt R, Harrison PV. Subacute cutaneous lupus erythematosus: two cases of delayed diagnosis. Acta Derm Venereol. 1998;78:57-59. doi:10.1080/00015559850135869
- David-Bajar KM. Subacute cutaneous lupus erythematosus. J Invest Dermatol. 1993;100:2S-8S. doi:10.1111/1523-1747.ep12355164
- Paulmann M, Mockenhaupt M. Severe drug-induced skin reactions: clinical features, diagnosis, etiology, and therapy. J Dtsch Dermatol Ges. 2015;13:625-643. doi:10.1111/ddg.12747
- Borroni G, Torti S, Pezzini C, et al. Histopathologic spectrum of drug reaction with eosinophilia and systemic symptoms (DRESS): a diagnosis that needs clinico-pathological correlation. G Ital Dermatol Venereol. 2014;149:291-300.
- Ortonne N, Valeyrie-Allanore L, Bastuji-Garin S, et al. Histopathology of drug rash with eosinophilia and systemic symptoms syndrome: a morphological and phenotypical study. Br J Dermatol. 2015;173:50-58. doi:10.1111/bjd.13683
- Halvorson SA, Gilbert E, Hopkins RS, et al. Putting the pieces together: necrolytic migratory erythema and the glucagonoma syndrome. J Gen Intern Med. 2013;28:1525-1529. doi:10.1007 /s11606-013-2490-5
- Toberer F, Hartschuh W, Wiedemeyer K. Glucagonoma-associated necrolytic migratory erythema: the broad spectrum of the clinical and histopathological findings and clues to the diagnosis. Am J Dermatopathol. 2019;41:E29-E32. doi:10.1097DAD .0000000000001219
- Hunt SJ, Narus VT, Abell E. Necrolytic migratory erythema: dyskeratotic dermatitis, a clue to early diagnosis. J Am Acad Dermatol. 1991; 24:473-477. doi:10.1016/0190-9622(91)70076-e
- van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol. 2004;151:531-537. doi:10.1530/eje.0.1510531
- Pujol RM, Wang C-Y E, el-Azhary RA, et al. Necrolytic migratory erythema: clinicopathologic study of 13 cases. Int J Dermatol. 2004;43:12- 18. doi:10.1111/j.1365-4632.2004.01844.x
- Johnson SM, Smoller BR, Lamps LW, et al. Necrolytic migratory erythema as the only presenting sign of a glucagonoma. J Am Acad Dermatol. 2003;49:325-328. doi:10.1067/s0190-9622(02)61774-8
- De Rosa G, Mignogna C. The histopathology of psoriasis. Reumatismo. 2007;59(suppl 1):46-48. doi:10.4081/reumatismo.2007.1s.46
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Bhutani T, Liao W, Nakamura M, eds. Evidence-Based Psoriasis. Springer; 2018:1-16. doi:10.1007/978-3-319-90107-7_1
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68. doi:10.22551/2019.24.0603.10155
- O’Keefe RJ, Scurry JP, Dennerstein G, et al. Audit of 114 nonneoplastic vulvar biopsies. Br J Obstet Gynaecol. 1995;102:780-786. doi:10.1111/j.1471-0528.1995.tb10842.x
- Parodi A, Caproni M, Cardinali C, et al P. Clinical, histological and immunopathological features of 58 patients with subacute cutaneous lupus erythematosus. Dermatology. 2000;200:6-10. doi:10.1159/000018307
- Lyon CC, Blewitt R, Harrison PV. Subacute cutaneous lupus erythematosus: two cases of delayed diagnosis. Acta Derm Venereol. 1998;78:57-59. doi:10.1080/00015559850135869
- David-Bajar KM. Subacute cutaneous lupus erythematosus. J Invest Dermatol. 1993;100:2S-8S. doi:10.1111/1523-1747.ep12355164
- Paulmann M, Mockenhaupt M. Severe drug-induced skin reactions: clinical features, diagnosis, etiology, and therapy. J Dtsch Dermatol Ges. 2015;13:625-643. doi:10.1111/ddg.12747
- Borroni G, Torti S, Pezzini C, et al. Histopathologic spectrum of drug reaction with eosinophilia and systemic symptoms (DRESS): a diagnosis that needs clinico-pathological correlation. G Ital Dermatol Venereol. 2014;149:291-300.
- Ortonne N, Valeyrie-Allanore L, Bastuji-Garin S, et al. Histopathology of drug rash with eosinophilia and systemic symptoms syndrome: a morphological and phenotypical study. Br J Dermatol. 2015;173:50-58. doi:10.1111/bjd.13683
A 62-year-old man presented with an erythematous flaky rash associated with burning pain on the right medial second toe that persisted for several months. Prior treatment with econazole, ciclopirox, and oral amoxicillin had failed. A shave biopsy was performed.
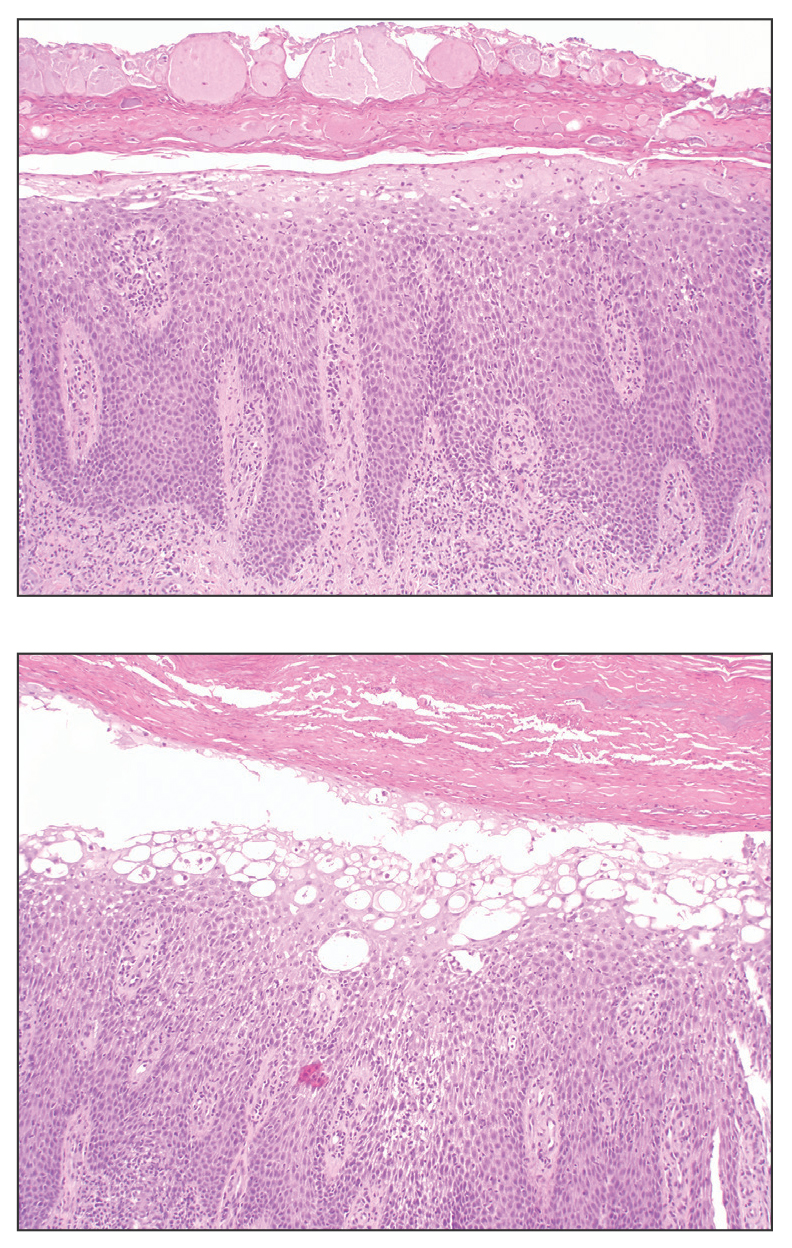
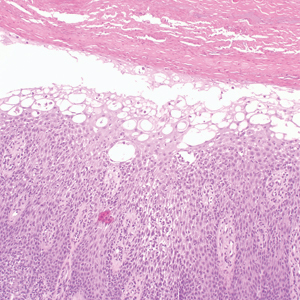
Multiple Asymptomatic Dome-Shaped Papules on the Scalp
The Diagnosis: Spiradenocylindroma
Shave biopsies of our patient’s lesions showed wellcircumscribed dermal nodules resembling a spiradenoma with 3 cell populations: those with lighter nuclei, darker nuclei, and scattered lymphocytes. However, the conspicuous globules of basement membrane material were reminiscent of a cylindroma. These overlapping features and the patient’s history of cylindroma were suggestive of a diagnosis of spiradenocylindroma.
Spiradenocylindroma is an uncommon dermal tumor with features that overlap with spiradenoma and cylindroma.1 It may manifest as a solitary lesion or multiple lesions and can occur sporadically or in the context of a family history. Histologically, it must be distinguished from other intradermal basaloid neoplasms including conventional cylindroma and spiradenoma, dermal duct tumor, hidradenoma, and trichoblastoma.
When patients present with multiple cylindromas, spiradenomas, or spiradenocylindromas, physicians should consider genetic testing and review of the family history to assess for cylindromatosis gene mutations or Brooke-Spiegler syndrome. Biopsy and histologic examination are important because malignant tumors can evolve from pre-existing spiradenocylindromas, cylindromas, and spiradenomas,2 with an increased risk in patients with Brooke-Spiegler syndrome.1 Our patient declined further genetic workup but continues to follow up with dermatology for monitoring of lesions.
Dermal duct tumors are morphologic variants of poromas that are derived from sweat gland lineage and usually manifest as solitary dome-shaped papules, plaques, or nodules most often seen on acral surfaces as well as the head and neck.3 Clinically, they may be indistinguishable from spiradenocylindromas and require biopsy for histologic evaluation. They can be distinguished from spiradenocylindroma by the presence of small dermal nodules composed of cuboidal cells with ample pink cytoplasm and cuticle-lined ducts (Figure 1).

Trichoblastomas typically are deep-seated basaloid follicular neoplasms on the scalp with papillary mesenchyme resembling the normal fibrous sheath of the hair follicle, often replete with papillary mesenchymal bodies (Figure 2). There generally are no retraction spaces between its basaloid nests and the surrounding stroma, which is unlikely to contain mucin relative to basal cell carcinoma (BCC).4,5

Adenoid cystic carcinoma is a rare salivary gland tumor that can metastasize to the skin and rarely arises as a primary skin adnexal tumor. It manifests as a slowgrowing mass that can be tender to palpation.6 Histologic examination shows dermal islands with cribriform blue and pink spaces. Compared to BCC, adenoid cystic carcinoma cells are enlarged and epithelioid with relatively scarce cytoplasm (Figure 3).6,7 Adenoid cystic carcinoma can show variable growth patterns including infiltrative nests and trabeculae. Perineural invasion is common, and there is a high risk for local recurrence.7 First-line therapy usually is surgical, and postoperative radiotherapy may be required.6,7

Nodular BCC commonly manifests as an enlarging nonhealing lesion on sun-exposed skin and has many subtypes, typically with arborizing telangiectases on dermoscopy. Histopathologic examination of nodular BCC reveals a nest of basaloid follicular germinative cells in the dermis with peripheral palisading and a fibromyxoid stroma (Figure 4).8 Patients with Brooke-Spiegler syndrome are at increased risk for nodular BCC, which may be clinically indistinguishable from spiradenoma, cylindroma, and spiradenocylindroma, necessitating histologic assessment.

- Facchini V, Colangeli W, Bozza F, et al. A rare histopathological spiradenocylindroma: a case report. Clin Ter. 2022;173:292-294. doi:10.7417/ CT.2022.2433
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update [published online March 14, 2016]. Head Neck Pathol. 2016;10:125-30. doi:10.1007/s12105-016-0705-x
- Miller AC, Adjei S, Temiz LA, et al. Dermal duct tumor: a diagnostic dilemma. Dermatopathology (Basel). 2022;9:36-47. doi:10.3390/dermatopathology9010007
- Elston DM. Pilar and sebaceous neoplasms. In: Elston DM, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Elsevier; 2018:71-85.
- McCalmont TH, Pincus LB. Adnexal neoplasms. In: Bolognia J, Schaffer J, Cerroni, L. Dermatology. 4th ed. Elsevier; 2017:1930-1953.
- Coca-Pelaz A, Rodrigo JP, Bradley PJ, et al. Adenoid cystic carcinoma of the head and neck—an update [published online May 2, 2015]. Oral Oncol. 2015;51:652-661. doi:10.1016/j.oraloncology.2015.04.005
- Tonev ID, Pirgova YS, Conev NV. Primary adenoid cystic carcinoma of the skin with multiple local recurrences. Case Rep Oncol. 2015;8:251- 255. doi:10.1159/000431082
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations [published online May 18, 2018]. J Am Acad Dermatol. 2019;80:303-317. doi:10.1016/j.jaad.2018.03.060
The Diagnosis: Spiradenocylindroma
Shave biopsies of our patient’s lesions showed wellcircumscribed dermal nodules resembling a spiradenoma with 3 cell populations: those with lighter nuclei, darker nuclei, and scattered lymphocytes. However, the conspicuous globules of basement membrane material were reminiscent of a cylindroma. These overlapping features and the patient’s history of cylindroma were suggestive of a diagnosis of spiradenocylindroma.
Spiradenocylindroma is an uncommon dermal tumor with features that overlap with spiradenoma and cylindroma.1 It may manifest as a solitary lesion or multiple lesions and can occur sporadically or in the context of a family history. Histologically, it must be distinguished from other intradermal basaloid neoplasms including conventional cylindroma and spiradenoma, dermal duct tumor, hidradenoma, and trichoblastoma.
When patients present with multiple cylindromas, spiradenomas, or spiradenocylindromas, physicians should consider genetic testing and review of the family history to assess for cylindromatosis gene mutations or Brooke-Spiegler syndrome. Biopsy and histologic examination are important because malignant tumors can evolve from pre-existing spiradenocylindromas, cylindromas, and spiradenomas,2 with an increased risk in patients with Brooke-Spiegler syndrome.1 Our patient declined further genetic workup but continues to follow up with dermatology for monitoring of lesions.
Dermal duct tumors are morphologic variants of poromas that are derived from sweat gland lineage and usually manifest as solitary dome-shaped papules, plaques, or nodules most often seen on acral surfaces as well as the head and neck.3 Clinically, they may be indistinguishable from spiradenocylindromas and require biopsy for histologic evaluation. They can be distinguished from spiradenocylindroma by the presence of small dermal nodules composed of cuboidal cells with ample pink cytoplasm and cuticle-lined ducts (Figure 1).
Trichoblastomas typically are deep-seated basaloid follicular neoplasms on the scalp with papillary mesenchyme resembling the normal fibrous sheath of the hair follicle, often replete with papillary mesenchymal bodies (Figure 2). There generally are no retraction spaces between its basaloid nests and the surrounding stroma, which is unlikely to contain mucin relative to basal cell carcinoma (BCC).4,5
Adenoid cystic carcinoma is a rare salivary gland tumor that can metastasize to the skin and rarely arises as a primary skin adnexal tumor. It manifests as a slowgrowing mass that can be tender to palpation.6 Histologic examination shows dermal islands with cribriform blue and pink spaces. Compared to BCC, adenoid cystic carcinoma cells are enlarged and epithelioid with relatively scarce cytoplasm (Figure 3).6,7 Adenoid cystic carcinoma can show variable growth patterns including infiltrative nests and trabeculae. Perineural invasion is common, and there is a high risk for local recurrence.7 First-line therapy usually is surgical, and postoperative radiotherapy may be required.6,7
Nodular BCC commonly manifests as an enlarging nonhealing lesion on sun-exposed skin and has many subtypes, typically with arborizing telangiectases on dermoscopy. Histopathologic examination of nodular BCC reveals a nest of basaloid follicular germinative cells in the dermis with peripheral palisading and a fibromyxoid stroma (Figure 4).8 Patients with Brooke-Spiegler syndrome are at increased risk for nodular BCC, which may be clinically indistinguishable from spiradenoma, cylindroma, and spiradenocylindroma, necessitating histologic assessment.
The Diagnosis: Spiradenocylindroma
Shave biopsies of our patient’s lesions showed wellcircumscribed dermal nodules resembling a spiradenoma with 3 cell populations: those with lighter nuclei, darker nuclei, and scattered lymphocytes. However, the conspicuous globules of basement membrane material were reminiscent of a cylindroma. These overlapping features and the patient’s history of cylindroma were suggestive of a diagnosis of spiradenocylindroma.
Spiradenocylindroma is an uncommon dermal tumor with features that overlap with spiradenoma and cylindroma.1 It may manifest as a solitary lesion or multiple lesions and can occur sporadically or in the context of a family history. Histologically, it must be distinguished from other intradermal basaloid neoplasms including conventional cylindroma and spiradenoma, dermal duct tumor, hidradenoma, and trichoblastoma.
When patients present with multiple cylindromas, spiradenomas, or spiradenocylindromas, physicians should consider genetic testing and review of the family history to assess for cylindromatosis gene mutations or Brooke-Spiegler syndrome. Biopsy and histologic examination are important because malignant tumors can evolve from pre-existing spiradenocylindromas, cylindromas, and spiradenomas,2 with an increased risk in patients with Brooke-Spiegler syndrome.1 Our patient declined further genetic workup but continues to follow up with dermatology for monitoring of lesions.
Dermal duct tumors are morphologic variants of poromas that are derived from sweat gland lineage and usually manifest as solitary dome-shaped papules, plaques, or nodules most often seen on acral surfaces as well as the head and neck.3 Clinically, they may be indistinguishable from spiradenocylindromas and require biopsy for histologic evaluation. They can be distinguished from spiradenocylindroma by the presence of small dermal nodules composed of cuboidal cells with ample pink cytoplasm and cuticle-lined ducts (Figure 1).
Trichoblastomas typically are deep-seated basaloid follicular neoplasms on the scalp with papillary mesenchyme resembling the normal fibrous sheath of the hair follicle, often replete with papillary mesenchymal bodies (Figure 2). There generally are no retraction spaces between its basaloid nests and the surrounding stroma, which is unlikely to contain mucin relative to basal cell carcinoma (BCC).4,5
Adenoid cystic carcinoma is a rare salivary gland tumor that can metastasize to the skin and rarely arises as a primary skin adnexal tumor. It manifests as a slowgrowing mass that can be tender to palpation.6 Histologic examination shows dermal islands with cribriform blue and pink spaces. Compared to BCC, adenoid cystic carcinoma cells are enlarged and epithelioid with relatively scarce cytoplasm (Figure 3).6,7 Adenoid cystic carcinoma can show variable growth patterns including infiltrative nests and trabeculae. Perineural invasion is common, and there is a high risk for local recurrence.7 First-line therapy usually is surgical, and postoperative radiotherapy may be required.6,7
Nodular BCC commonly manifests as an enlarging nonhealing lesion on sun-exposed skin and has many subtypes, typically with arborizing telangiectases on dermoscopy. Histopathologic examination of nodular BCC reveals a nest of basaloid follicular germinative cells in the dermis with peripheral palisading and a fibromyxoid stroma (Figure 4).8 Patients with Brooke-Spiegler syndrome are at increased risk for nodular BCC, which may be clinically indistinguishable from spiradenoma, cylindroma, and spiradenocylindroma, necessitating histologic assessment.
- Facchini V, Colangeli W, Bozza F, et al. A rare histopathological spiradenocylindroma: a case report. Clin Ter. 2022;173:292-294. doi:10.7417/ CT.2022.2433
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update [published online March 14, 2016]. Head Neck Pathol. 2016;10:125-30. doi:10.1007/s12105-016-0705-x
- Miller AC, Adjei S, Temiz LA, et al. Dermal duct tumor: a diagnostic dilemma. Dermatopathology (Basel). 2022;9:36-47. doi:10.3390/dermatopathology9010007
- Elston DM. Pilar and sebaceous neoplasms. In: Elston DM, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Elsevier; 2018:71-85.
- McCalmont TH, Pincus LB. Adnexal neoplasms. In: Bolognia J, Schaffer J, Cerroni, L. Dermatology. 4th ed. Elsevier; 2017:1930-1953.
- Coca-Pelaz A, Rodrigo JP, Bradley PJ, et al. Adenoid cystic carcinoma of the head and neck—an update [published online May 2, 2015]. Oral Oncol. 2015;51:652-661. doi:10.1016/j.oraloncology.2015.04.005
- Tonev ID, Pirgova YS, Conev NV. Primary adenoid cystic carcinoma of the skin with multiple local recurrences. Case Rep Oncol. 2015;8:251- 255. doi:10.1159/000431082
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations [published online May 18, 2018]. J Am Acad Dermatol. 2019;80:303-317. doi:10.1016/j.jaad.2018.03.060
- Facchini V, Colangeli W, Bozza F, et al. A rare histopathological spiradenocylindroma: a case report. Clin Ter. 2022;173:292-294. doi:10.7417/ CT.2022.2433
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update [published online March 14, 2016]. Head Neck Pathol. 2016;10:125-30. doi:10.1007/s12105-016-0705-x
- Miller AC, Adjei S, Temiz LA, et al. Dermal duct tumor: a diagnostic dilemma. Dermatopathology (Basel). 2022;9:36-47. doi:10.3390/dermatopathology9010007
- Elston DM. Pilar and sebaceous neoplasms. In: Elston DM, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Elsevier; 2018:71-85.
- McCalmont TH, Pincus LB. Adnexal neoplasms. In: Bolognia J, Schaffer J, Cerroni, L. Dermatology. 4th ed. Elsevier; 2017:1930-1953.
- Coca-Pelaz A, Rodrigo JP, Bradley PJ, et al. Adenoid cystic carcinoma of the head and neck—an update [published online May 2, 2015]. Oral Oncol. 2015;51:652-661. doi:10.1016/j.oraloncology.2015.04.005
- Tonev ID, Pirgova YS, Conev NV. Primary adenoid cystic carcinoma of the skin with multiple local recurrences. Case Rep Oncol. 2015;8:251- 255. doi:10.1159/000431082
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations [published online May 18, 2018]. J Am Acad Dermatol. 2019;80:303-317. doi:10.1016/j.jaad.2018.03.060
A 62-year-old man with a history of cylindromas presented to our clinic with multiple asymptomatic, 3- to 4-mm, nonmobile, dome-shaped, telangiectatic, pink papules over the parietal and vertex scalp that had been present for more than 10 years without change. Several family members had similar lesions that had not been evaluated by a physician, and there had been no genetic evaluation. Shave biopsies of several lesions were performed.


Tender Dermal Nodule on the Temple
The Diagnosis: Lymphoepithelioma-like Carcinoma
Lymphoepithelioma-like carcinoma (LELC) is a rare, poorly differentiated, primary cutaneous neoplasm that occurs on sun-exposed skin, particularly on the head and neck of elderly individuals. It often manifests as an asymptomatic, slow-growing, flesh-colored or erythematous dermal nodule, though ulceration and tenderness have been reported.1 Histopathologically, these neoplasms often are poorly circumscribed and can infiltrate surrounding subcutaneous and soft tissue. As a biphasic tumor, LELC is characterized by islands, nests, or trabeculae of epithelioid cells within the mid dermis surrounded by a dense lymphocytic infiltrate with plasma cells (Figure 1).1 The epithelial component rarely communicates with the overlying epidermis and is composed of atypical polygonal cells with eosinophilic cytoplasm, vesicular nuclei, prominent nucleoli, and frequent mitosis.2 These epithelial nests can be highlighted by pancytokeratin AE1/AE3 or other epithelial differentiation markers (eg, CAM 5.2, CK5/6, epithelial membrane antigen, high-molecular-weight cytokeratin), while the surrounding lymphocytic infiltrate consists of an admixture of T cells and B cells. Lymphoepithelioma-like carcinomas also can demonstrate sebaceous, eccrine, or follicular differentiations.3 The epithelial nests of LELC also are positive for p63 and epithelial membrane antigen.2
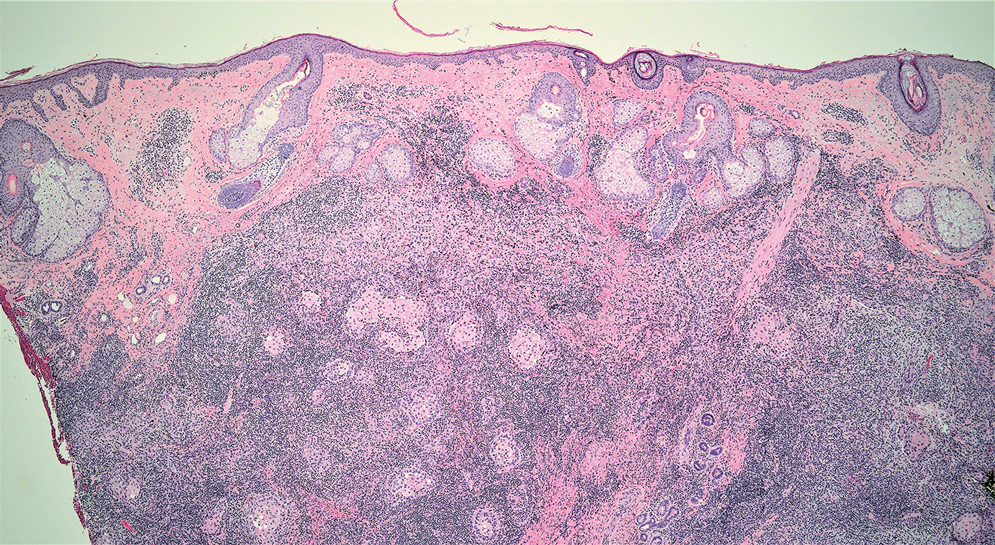
The usual treatment of LELC is wide local excision or Mohs micrographic surgery.1 Despite the poorly differentiated morphology of the tumor, LELC has a generally good prognosis with low metastatic potential and few reports of local recurrence after incomplete excision.3 Patients who are not candidates for surgery as well as recalcitrant cases are managed with radiotherapy.1
Cutaneous lymphadenoma (CL) is a benign adnexal neoplasm that manifests as a small, solitary, fleshcolored nodule usually in the head and neck region.4 Histologically, CL consists of well-circumscribed epithelial nests within the dermis that are peripherally outlined by palisading basaloid cells and filled with clear to eosinophilic epithelioid cells (Figure 2).5 The fibrotic tumor stroma often is infiltrated by numerous intralobular dendritic cells and lymphocytes that occasionally can be arranged in germinal center–like nodules.4 The lymphoepithelial nature of CL can be challenging to distinguish morphologically from LELC, and immunohistochemistry stains may be required. In CL, both the basaloid and epithelioid cells stain positive for pancytokeratin AE1/ AE3, but the peripheral palisaded basaloid cells also stain positive for BerEP4. Additionally, the fibrotic stroma can be highlighted by CD34 and the intralobular dendritic cells by S-100.4
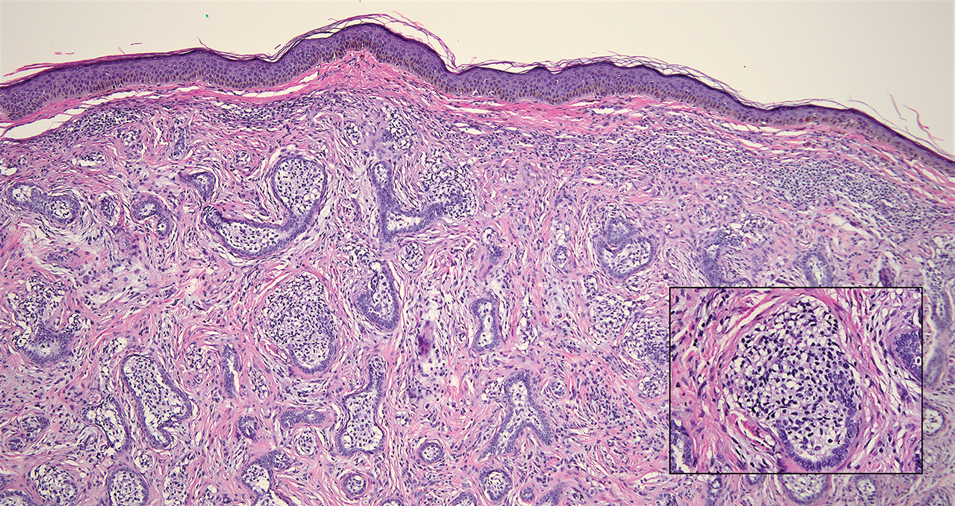
Nasopharyngeal carcinoma (NPC), formerly known as lymphoepithelioma, refers to carcinoma arising within the epithelium of the nasopharynx.6 Endemic to China, NPC manifests as an enlarging nasopharyngeal mass, causing clinical symptoms such as nasal obstruction and epistaxis.7 Histologically, nonkeratinizing NPC exhibits a biphasic morphology consisting of epithelioid neoplastic cells and background lymphocytic infiltrates (Figure 3). The epithelial component consists of round to oval neoplastic cells with amphophilic to eosinophilic cytoplasm, vesicular nuclei, and prominent nucleoli.6 Nasopharyngeal carcinoma is associated strongly with the Epstein-Barr virus while LELC is not; thus, Epstein- Barr encoding region in situ hybridization can reliably distinguish these entities. Metastatic NPC is rare but has been reported; therefore, it is highly recommended to perform an otolaryngologic examination in addition to testing for Epstein-Barr virus reactivity as part of a complete evaluation.8
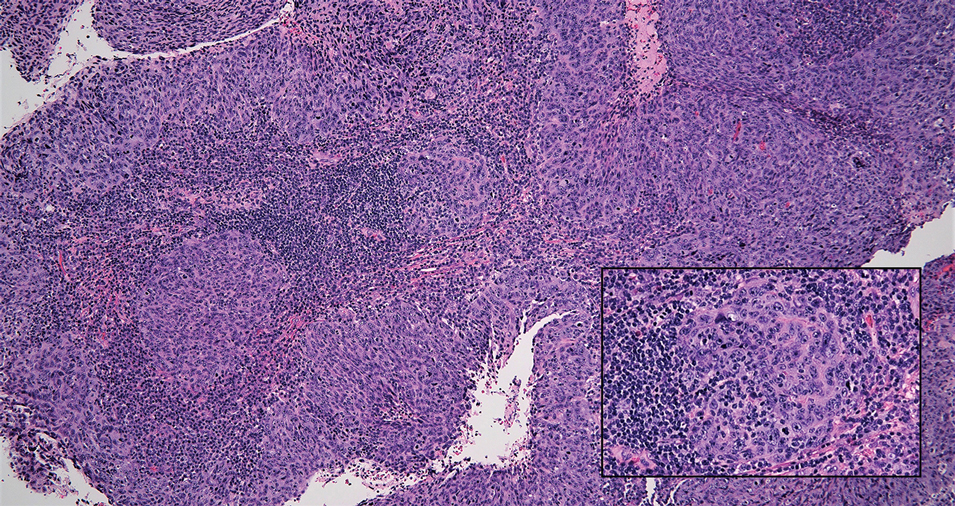
Cutaneous squamous cell carcinoma (SCC) is a common epidermal malignancy with multiple subtypes and variable morphology. The clinical presentation of SCC is similar to LELC—an enlarging hyperkeratotic papule or nodule on sun-exposed skin that often is ulcerated and tender.9 Histologically, poorly differentiated nonkeratinizing SCC can form nests and trabeculae of epithelioid cells that are stained by epithelial differentiation markers, resembling the epithelioid nests of LELC. Distinguishing between LELC and poorly differentiated SCC with robust inflammatory infiltrate can be challenging (Figure 4). In fact, some experts support LELC as an SCC variant rather than a separate entity.9 However, in contrast to LELC, the dermal nests of SCC usually maintain an epidermal connection and often are associated with an overlying area of SCC in situ or welldifferentiated SCC.3
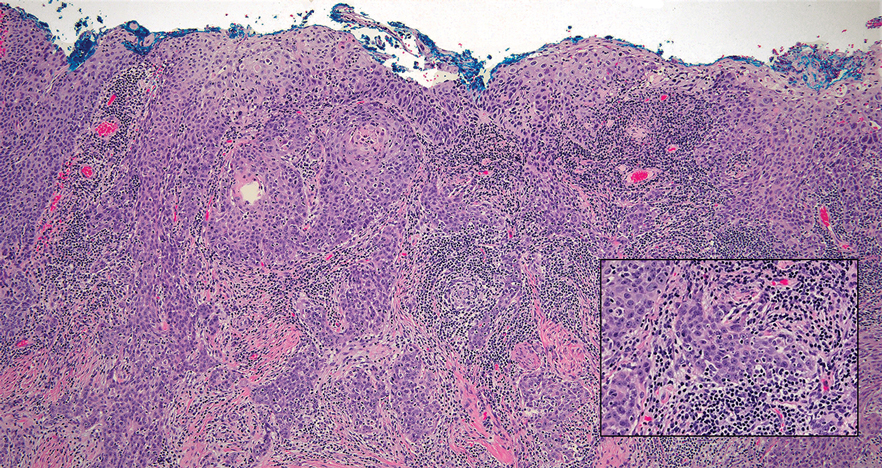
Mycosis fungoides (MF) is a primary cutaneous T-cell lymphoma. It is the most common type of cutaneous lymphoma, accounting for almost 50% of all reported cases.10 Classic MF has an indolent course and progresses through several clinical stages. Patches and plaques characterize early stages; lymphadenopathy indicates progression to later stages in which erythroderma may develop with coalescence of patches, plaques, and tumors; and MF present in blood or lymph nodes characterizes the late stage. Each stage of MF is different histologically—from a superficial lichenoid infiltrate with exocytosis of malignant T cells in the patch stage, to more robust epidermotropism and dermal infiltrate in the plaque stage, and finally a dense dermal infiltrate in the late stage.11 The rare syringotropic variant of MF clinically manifests as solitary or multiple erythematous lesions, often with overlying alopecia. Syringotropic MF uniquely exhibits folliculotropism and syringotropism along with syringometaplasia on histologic evaluation (Figure 5).12 The syringometaplasia can be difficult to distinguish from the epithelial nests of LELC, particularly with the lymphocytic background. Immunohistochemical panels for T-cell markers can highlight aberrant T cells in syringotropic MF through their usual loss of CD5 and CD7, in comparison to normal T cells in LELC.11 An elevated CD4:CD8 ratio of 4:1 and molecular analysis for T-cell receptor gene clonal rearrangements also can support the diagnosis of MF.12
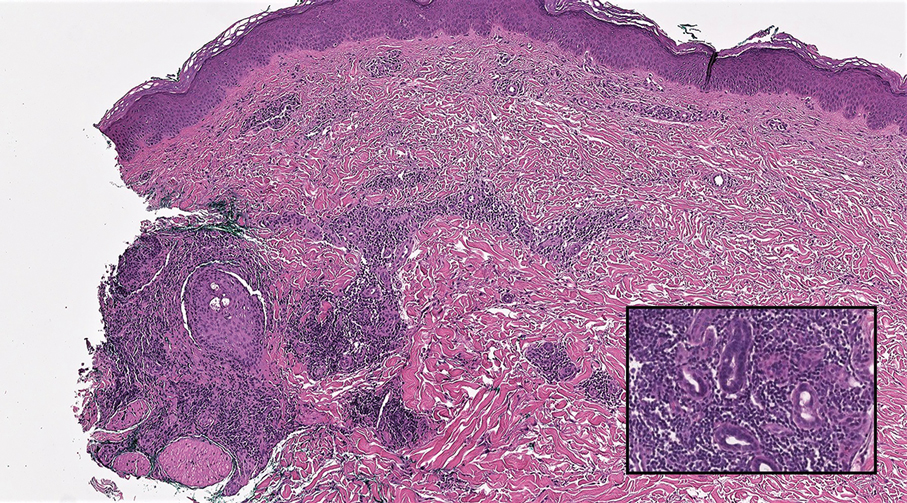
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Fisher JC, White RM, Hurd DS. Lymphoepithelioma-like carcinoma of the skin: a case of one patient presenting with two primary cutaneous neoplasms. J Am Osteopath Coll Dermatol. 2015;33:40-41.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Yu R, Salama S, Alowami S. Cutaneous lymphadenoma: a rare case and brief review of a diagnostic pitfall. Rare Tumors. 2014;6:5358.
- Monteagudo C, Fúnez R, Sánchez-Sendra B, et al. Cutaneous lymphadenoma is a distinct trichoblastoma-like lymphoepithelial tumor with diffuse androgen receptor immunoreactivity, Notch1 ligand in Reed-Sternberg-like Cells, and common EGFR somatic mutations. Am J Surg Pathol. 2021;45:1382-1390.
- Stelow EB, Wenig BM. Update from the 4th edition of the World Health Organization classification of head and neck tumours: nasopharynx. Head Neck Pathol. 2017;11:16-22.
- Almomani MH, Zulfiqar H, Nagalli S. Nasopharyngeal carcinoma (NPC, lymphoepithelioma). StatPearls Publishing; 2022.
- Lassen CB, Lock-Andersen J. Lymphoepithelioma-like carcinoma of the skin: a case with perineural invasion. Plast Reconstr Surg Glob Open. 2014;2:E252.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer Staging Guidelines, Prognostic Factors, and Histopathologic Variants. Adv Anat Pathol. 2017;24:171-194.
- Pileri A, Facchetti F, Rütten A, et al. Syringotropic mycosis fungoides: a rare variant of the disease with peculiar clinicopathologic features. Am J Surg Pathol. 2011;35:100-109.
- Ryu HJ, Kim SI, Jang HO, et al. Evaluation of the International Society for Cutaneous Lymphoma Algorithm for the Diagnosis of Early Mycosis Fungoides [published October 15, 2021]. Cells. 2021;10:2758. doi:10.3390/cells10102758
- Lehmer LM, Amber KT, de Feraudy SM. Syringotropic mycosis fungoides: a rare form of cutaneous T-cell lymphoma enabling a histopathologic “sigh of relief.” Am J Dermatopathol. 2017;39:920-923.
The Diagnosis: Lymphoepithelioma-like Carcinoma
Lymphoepithelioma-like carcinoma (LELC) is a rare, poorly differentiated, primary cutaneous neoplasm that occurs on sun-exposed skin, particularly on the head and neck of elderly individuals. It often manifests as an asymptomatic, slow-growing, flesh-colored or erythematous dermal nodule, though ulceration and tenderness have been reported.1 Histopathologically, these neoplasms often are poorly circumscribed and can infiltrate surrounding subcutaneous and soft tissue. As a biphasic tumor, LELC is characterized by islands, nests, or trabeculae of epithelioid cells within the mid dermis surrounded by a dense lymphocytic infiltrate with plasma cells (Figure 1).1 The epithelial component rarely communicates with the overlying epidermis and is composed of atypical polygonal cells with eosinophilic cytoplasm, vesicular nuclei, prominent nucleoli, and frequent mitosis.2 These epithelial nests can be highlighted by pancytokeratin AE1/AE3 or other epithelial differentiation markers (eg, CAM 5.2, CK5/6, epithelial membrane antigen, high-molecular-weight cytokeratin), while the surrounding lymphocytic infiltrate consists of an admixture of T cells and B cells. Lymphoepithelioma-like carcinomas also can demonstrate sebaceous, eccrine, or follicular differentiations.3 The epithelial nests of LELC also are positive for p63 and epithelial membrane antigen.2
The usual treatment of LELC is wide local excision or Mohs micrographic surgery.1 Despite the poorly differentiated morphology of the tumor, LELC has a generally good prognosis with low metastatic potential and few reports of local recurrence after incomplete excision.3 Patients who are not candidates for surgery as well as recalcitrant cases are managed with radiotherapy.1
Cutaneous lymphadenoma (CL) is a benign adnexal neoplasm that manifests as a small, solitary, fleshcolored nodule usually in the head and neck region.4 Histologically, CL consists of well-circumscribed epithelial nests within the dermis that are peripherally outlined by palisading basaloid cells and filled with clear to eosinophilic epithelioid cells (Figure 2).5 The fibrotic tumor stroma often is infiltrated by numerous intralobular dendritic cells and lymphocytes that occasionally can be arranged in germinal center–like nodules.4 The lymphoepithelial nature of CL can be challenging to distinguish morphologically from LELC, and immunohistochemistry stains may be required. In CL, both the basaloid and epithelioid cells stain positive for pancytokeratin AE1/ AE3, but the peripheral palisaded basaloid cells also stain positive for BerEP4. Additionally, the fibrotic stroma can be highlighted by CD34 and the intralobular dendritic cells by S-100.4
Nasopharyngeal carcinoma (NPC), formerly known as lymphoepithelioma, refers to carcinoma arising within the epithelium of the nasopharynx.6 Endemic to China, NPC manifests as an enlarging nasopharyngeal mass, causing clinical symptoms such as nasal obstruction and epistaxis.7 Histologically, nonkeratinizing NPC exhibits a biphasic morphology consisting of epithelioid neoplastic cells and background lymphocytic infiltrates (Figure 3). The epithelial component consists of round to oval neoplastic cells with amphophilic to eosinophilic cytoplasm, vesicular nuclei, and prominent nucleoli.6 Nasopharyngeal carcinoma is associated strongly with the Epstein-Barr virus while LELC is not; thus, Epstein- Barr encoding region in situ hybridization can reliably distinguish these entities. Metastatic NPC is rare but has been reported; therefore, it is highly recommended to perform an otolaryngologic examination in addition to testing for Epstein-Barr virus reactivity as part of a complete evaluation.8
Cutaneous squamous cell carcinoma (SCC) is a common epidermal malignancy with multiple subtypes and variable morphology. The clinical presentation of SCC is similar to LELC—an enlarging hyperkeratotic papule or nodule on sun-exposed skin that often is ulcerated and tender.9 Histologically, poorly differentiated nonkeratinizing SCC can form nests and trabeculae of epithelioid cells that are stained by epithelial differentiation markers, resembling the epithelioid nests of LELC. Distinguishing between LELC and poorly differentiated SCC with robust inflammatory infiltrate can be challenging (Figure 4). In fact, some experts support LELC as an SCC variant rather than a separate entity.9 However, in contrast to LELC, the dermal nests of SCC usually maintain an epidermal connection and often are associated with an overlying area of SCC in situ or welldifferentiated SCC.3
Mycosis fungoides (MF) is a primary cutaneous T-cell lymphoma. It is the most common type of cutaneous lymphoma, accounting for almost 50% of all reported cases.10 Classic MF has an indolent course and progresses through several clinical stages. Patches and plaques characterize early stages; lymphadenopathy indicates progression to later stages in which erythroderma may develop with coalescence of patches, plaques, and tumors; and MF present in blood or lymph nodes characterizes the late stage. Each stage of MF is different histologically—from a superficial lichenoid infiltrate with exocytosis of malignant T cells in the patch stage, to more robust epidermotropism and dermal infiltrate in the plaque stage, and finally a dense dermal infiltrate in the late stage.11 The rare syringotropic variant of MF clinically manifests as solitary or multiple erythematous lesions, often with overlying alopecia. Syringotropic MF uniquely exhibits folliculotropism and syringotropism along with syringometaplasia on histologic evaluation (Figure 5).12 The syringometaplasia can be difficult to distinguish from the epithelial nests of LELC, particularly with the lymphocytic background. Immunohistochemical panels for T-cell markers can highlight aberrant T cells in syringotropic MF through their usual loss of CD5 and CD7, in comparison to normal T cells in LELC.11 An elevated CD4:CD8 ratio of 4:1 and molecular analysis for T-cell receptor gene clonal rearrangements also can support the diagnosis of MF.12
The Diagnosis: Lymphoepithelioma-like Carcinoma
Lymphoepithelioma-like carcinoma (LELC) is a rare, poorly differentiated, primary cutaneous neoplasm that occurs on sun-exposed skin, particularly on the head and neck of elderly individuals. It often manifests as an asymptomatic, slow-growing, flesh-colored or erythematous dermal nodule, though ulceration and tenderness have been reported.1 Histopathologically, these neoplasms often are poorly circumscribed and can infiltrate surrounding subcutaneous and soft tissue. As a biphasic tumor, LELC is characterized by islands, nests, or trabeculae of epithelioid cells within the mid dermis surrounded by a dense lymphocytic infiltrate with plasma cells (Figure 1).1 The epithelial component rarely communicates with the overlying epidermis and is composed of atypical polygonal cells with eosinophilic cytoplasm, vesicular nuclei, prominent nucleoli, and frequent mitosis.2 These epithelial nests can be highlighted by pancytokeratin AE1/AE3 or other epithelial differentiation markers (eg, CAM 5.2, CK5/6, epithelial membrane antigen, high-molecular-weight cytokeratin), while the surrounding lymphocytic infiltrate consists of an admixture of T cells and B cells. Lymphoepithelioma-like carcinomas also can demonstrate sebaceous, eccrine, or follicular differentiations.3 The epithelial nests of LELC also are positive for p63 and epithelial membrane antigen.2
The usual treatment of LELC is wide local excision or Mohs micrographic surgery.1 Despite the poorly differentiated morphology of the tumor, LELC has a generally good prognosis with low metastatic potential and few reports of local recurrence after incomplete excision.3 Patients who are not candidates for surgery as well as recalcitrant cases are managed with radiotherapy.1
Cutaneous lymphadenoma (CL) is a benign adnexal neoplasm that manifests as a small, solitary, fleshcolored nodule usually in the head and neck region.4 Histologically, CL consists of well-circumscribed epithelial nests within the dermis that are peripherally outlined by palisading basaloid cells and filled with clear to eosinophilic epithelioid cells (Figure 2).5 The fibrotic tumor stroma often is infiltrated by numerous intralobular dendritic cells and lymphocytes that occasionally can be arranged in germinal center–like nodules.4 The lymphoepithelial nature of CL can be challenging to distinguish morphologically from LELC, and immunohistochemistry stains may be required. In CL, both the basaloid and epithelioid cells stain positive for pancytokeratin AE1/ AE3, but the peripheral palisaded basaloid cells also stain positive for BerEP4. Additionally, the fibrotic stroma can be highlighted by CD34 and the intralobular dendritic cells by S-100.4
Nasopharyngeal carcinoma (NPC), formerly known as lymphoepithelioma, refers to carcinoma arising within the epithelium of the nasopharynx.6 Endemic to China, NPC manifests as an enlarging nasopharyngeal mass, causing clinical symptoms such as nasal obstruction and epistaxis.7 Histologically, nonkeratinizing NPC exhibits a biphasic morphology consisting of epithelioid neoplastic cells and background lymphocytic infiltrates (Figure 3). The epithelial component consists of round to oval neoplastic cells with amphophilic to eosinophilic cytoplasm, vesicular nuclei, and prominent nucleoli.6 Nasopharyngeal carcinoma is associated strongly with the Epstein-Barr virus while LELC is not; thus, Epstein- Barr encoding region in situ hybridization can reliably distinguish these entities. Metastatic NPC is rare but has been reported; therefore, it is highly recommended to perform an otolaryngologic examination in addition to testing for Epstein-Barr virus reactivity as part of a complete evaluation.8
Cutaneous squamous cell carcinoma (SCC) is a common epidermal malignancy with multiple subtypes and variable morphology. The clinical presentation of SCC is similar to LELC—an enlarging hyperkeratotic papule or nodule on sun-exposed skin that often is ulcerated and tender.9 Histologically, poorly differentiated nonkeratinizing SCC can form nests and trabeculae of epithelioid cells that are stained by epithelial differentiation markers, resembling the epithelioid nests of LELC. Distinguishing between LELC and poorly differentiated SCC with robust inflammatory infiltrate can be challenging (Figure 4). In fact, some experts support LELC as an SCC variant rather than a separate entity.9 However, in contrast to LELC, the dermal nests of SCC usually maintain an epidermal connection and often are associated with an overlying area of SCC in situ or welldifferentiated SCC.3
Mycosis fungoides (MF) is a primary cutaneous T-cell lymphoma. It is the most common type of cutaneous lymphoma, accounting for almost 50% of all reported cases.10 Classic MF has an indolent course and progresses through several clinical stages. Patches and plaques characterize early stages; lymphadenopathy indicates progression to later stages in which erythroderma may develop with coalescence of patches, plaques, and tumors; and MF present in blood or lymph nodes characterizes the late stage. Each stage of MF is different histologically—from a superficial lichenoid infiltrate with exocytosis of malignant T cells in the patch stage, to more robust epidermotropism and dermal infiltrate in the plaque stage, and finally a dense dermal infiltrate in the late stage.11 The rare syringotropic variant of MF clinically manifests as solitary or multiple erythematous lesions, often with overlying alopecia. Syringotropic MF uniquely exhibits folliculotropism and syringotropism along with syringometaplasia on histologic evaluation (Figure 5).12 The syringometaplasia can be difficult to distinguish from the epithelial nests of LELC, particularly with the lymphocytic background. Immunohistochemical panels for T-cell markers can highlight aberrant T cells in syringotropic MF through their usual loss of CD5 and CD7, in comparison to normal T cells in LELC.11 An elevated CD4:CD8 ratio of 4:1 and molecular analysis for T-cell receptor gene clonal rearrangements also can support the diagnosis of MF.12
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Fisher JC, White RM, Hurd DS. Lymphoepithelioma-like carcinoma of the skin: a case of one patient presenting with two primary cutaneous neoplasms. J Am Osteopath Coll Dermatol. 2015;33:40-41.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Yu R, Salama S, Alowami S. Cutaneous lymphadenoma: a rare case and brief review of a diagnostic pitfall. Rare Tumors. 2014;6:5358.
- Monteagudo C, Fúnez R, Sánchez-Sendra B, et al. Cutaneous lymphadenoma is a distinct trichoblastoma-like lymphoepithelial tumor with diffuse androgen receptor immunoreactivity, Notch1 ligand in Reed-Sternberg-like Cells, and common EGFR somatic mutations. Am J Surg Pathol. 2021;45:1382-1390.
- Stelow EB, Wenig BM. Update from the 4th edition of the World Health Organization classification of head and neck tumours: nasopharynx. Head Neck Pathol. 2017;11:16-22.
- Almomani MH, Zulfiqar H, Nagalli S. Nasopharyngeal carcinoma (NPC, lymphoepithelioma). StatPearls Publishing; 2022.
- Lassen CB, Lock-Andersen J. Lymphoepithelioma-like carcinoma of the skin: a case with perineural invasion. Plast Reconstr Surg Glob Open. 2014;2:E252.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer Staging Guidelines, Prognostic Factors, and Histopathologic Variants. Adv Anat Pathol. 2017;24:171-194.
- Pileri A, Facchetti F, Rütten A, et al. Syringotropic mycosis fungoides: a rare variant of the disease with peculiar clinicopathologic features. Am J Surg Pathol. 2011;35:100-109.
- Ryu HJ, Kim SI, Jang HO, et al. Evaluation of the International Society for Cutaneous Lymphoma Algorithm for the Diagnosis of Early Mycosis Fungoides [published October 15, 2021]. Cells. 2021;10:2758. doi:10.3390/cells10102758
- Lehmer LM, Amber KT, de Feraudy SM. Syringotropic mycosis fungoides: a rare form of cutaneous T-cell lymphoma enabling a histopathologic “sigh of relief.” Am J Dermatopathol. 2017;39:920-923.
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Fisher JC, White RM, Hurd DS. Lymphoepithelioma-like carcinoma of the skin: a case of one patient presenting with two primary cutaneous neoplasms. J Am Osteopath Coll Dermatol. 2015;33:40-41.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Yu R, Salama S, Alowami S. Cutaneous lymphadenoma: a rare case and brief review of a diagnostic pitfall. Rare Tumors. 2014;6:5358.
- Monteagudo C, Fúnez R, Sánchez-Sendra B, et al. Cutaneous lymphadenoma is a distinct trichoblastoma-like lymphoepithelial tumor with diffuse androgen receptor immunoreactivity, Notch1 ligand in Reed-Sternberg-like Cells, and common EGFR somatic mutations. Am J Surg Pathol. 2021;45:1382-1390.
- Stelow EB, Wenig BM. Update from the 4th edition of the World Health Organization classification of head and neck tumours: nasopharynx. Head Neck Pathol. 2017;11:16-22.
- Almomani MH, Zulfiqar H, Nagalli S. Nasopharyngeal carcinoma (NPC, lymphoepithelioma). StatPearls Publishing; 2022.
- Lassen CB, Lock-Andersen J. Lymphoepithelioma-like carcinoma of the skin: a case with perineural invasion. Plast Reconstr Surg Glob Open. 2014;2:E252.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer Staging Guidelines, Prognostic Factors, and Histopathologic Variants. Adv Anat Pathol. 2017;24:171-194.
- Pileri A, Facchetti F, Rütten A, et al. Syringotropic mycosis fungoides: a rare variant of the disease with peculiar clinicopathologic features. Am J Surg Pathol. 2011;35:100-109.
- Ryu HJ, Kim SI, Jang HO, et al. Evaluation of the International Society for Cutaneous Lymphoma Algorithm for the Diagnosis of Early Mycosis Fungoides [published October 15, 2021]. Cells. 2021;10:2758. doi:10.3390/cells10102758
- Lehmer LM, Amber KT, de Feraudy SM. Syringotropic mycosis fungoides: a rare form of cutaneous T-cell lymphoma enabling a histopathologic “sigh of relief.” Am J Dermatopathol. 2017;39:920-923.
A 77-year-old man presented with a 1.2-cm dermal nodule on the left temple of 1 year’s duration. The lesion had become tender and darker in color. An excision was performed and submitted for histologic examination. Additional immunohistochemistry staining for Epstein-Barr virus was negative.
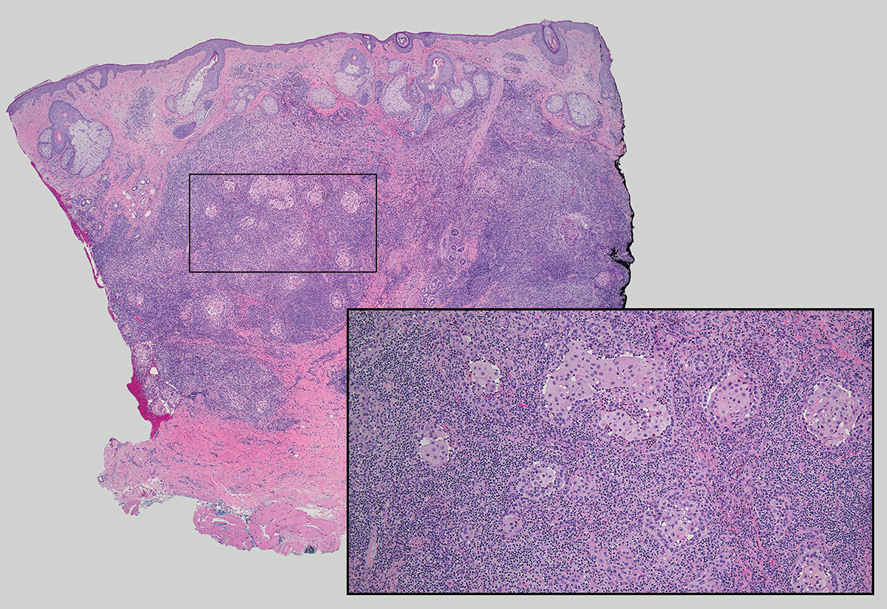
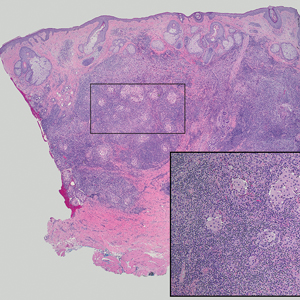
Hyperkeratotic Nodule on the Knee in a Patient With KID Syndrome
The Diagnosis: Proliferating Pilar Cyst
Histopathology revealed an extensive lobulated epithelial proliferation in a characteristic “rolls and scrolls” pattern (Figure 1). This finding along with the patient’s prior diagnosis of keratitis-ichthyosisdeafness (KID) syndrome supported the diagnosis of a proliferating pilar cyst.
.")
Pilar (or trichilemmal) cysts are common dermal cysts typically found on the outer root sheath of hair follicles. They clinically manifest as multiple yellow dome-shaped nodules without central puncta. They are slow growing and histologically are characterized as cysts with a stratified squamous epithelium demonstrating lack of a granular layer (trichilemmal keratinization) with bright red keratin contents and central focal calcification (Figure 2). Pilar cysts are more common in adult women and may be inherited through an autosomal-dominant pattern.1
.")
Proliferating pilar cysts represent less than 3% of all pilar cysts.2 In addition to the characteristic features of a pilar cyst, proliferating pilar cysts generally are larger (can be >6-cm wide) and are more ulcerative.3 Histopathology of proliferating pilar cysts reveals a more extensive epithelial proliferation, yielding a rolls and scrolls appearance, and may demonstrate nuclear atypia.4 Proliferating pilar cysts classically manifest as large, raised, smooth and/or ulcerated nodules on the scalp accompanied by areas of excessive hair growth in older women. They generally arise from pre-existing pilar cysts but also may occur sporadically.4
The development of multiple proliferating pilar cysts has been observed in patients with KID syndrome, a rare congenital ectodermal disorder characterized by a triad of vascularizing keratitis, hyperkeratosis, and sensorineural deafness.5,6 It is caused by a missense mutation of the GJB2 gene encoding for connexin 26, a gap junction that facilitates intercellular signaling and is expressed in a variety of structures including the cochlea, cornea, sweat glands, and inner and outer root sheaths of hair follicles.7
The differential diagnosis for proliferating pilar cysts includes pilomatrixomas, squamous cell carcinomas, and malignant proliferating pilar tumors. Pilomatrixomas (or calcifying epitheliomas of Malherbe) are the most common adnexal skin tumors in the pediatric population and most commonly present on the head, neck, and arms.8 They also can manifest in adults. Pilomatrixomas are benign dermal-subcutaneous tumors encapsulated by connective tissue that are found on the hair matrix and are histologically characterized by basaloid cells, shadow (or ghost) cells, dystrophic calcifications, and giant cells.9 The amount of basaloid cells and shadow cells can vary. Tumor progression results in the enucleation of the basaloid cells to form eosinophilic shadow cells in which calcification can occur. Giant cell granulomas may form contiguous with the calcifications. Both proliferating pilar cysts and pilomatrixomas have a rolls and scrolls appearance on low-power microscopy, but the latter are differentiated by their shadow cells and basaloid areas (Figure 3).
.")
Squamous cell carcinoma (SCC) is the second most common nonmelanoma skin cancer and more commonly affects men. Risk factors for SCC include immunosuppression and exposure to UV radiation. Histopathology of well-differentiated SCCs reveals invasive squamous cells with larger nuclei and a glassy appearance in addition to possible mitotic figures and keratin pearls (Figure 4). They typically manifest in sun-exposed areas such as the scalp, face, forearms, dorsal aspects of the hands, and lower legs.10 Proliferating pilar tumors often lack the nuclear atypia and invasive architecture of a well-differentiated SCC.
 and full-thickness atypia (H&E, original magnification ×20).")
Features of malignant proliferating pilar tumors overlap with proliferating pilar cysts. In addition to the proliferative epithelium with abrupt trichilemmal keratinization that is typical of a proliferating pilar cyst, a malignant proliferating pilar tumor will demonstrate invasion into the surrounding tissue and lymph nodes, mitotic and architectural atypia, and necrosis (Figure 5).11 Malignant proliferating pilar tumors grow rapidly, ranging in size from 1 to 10 cm, and may develop from pre-existing or proliferating pilar cysts or de novo.
.")
The development of multiple proliferating pilar cysts and thus increased risk for progression to malignant proliferating pilar tumors has been observed in patients with KID syndrome.6 Our case highlights the importance of early screening and recognition of proliferating pilar tumors in patients with this condition.
- Poiares Baptista A, Garcia E Silva L, Born MC. Proliferating trichilemmal cyst. J Cutan Pathol. 1983;10:178-187.
- Al Aboud DM, Yarrarapu SNS, Patel BC. Pilar cyst. StatPearls. StatPearls Publishing; 2023.
- Kim UG, Kook DB, Kim TH, et al. Trichilemmal carcinoma from proliferating trichilemmal cyst on the posterior neck [published online March 25, 2017]. Arch Craniofac Surg. 2017;18:50-53. doi:10.7181/acfs.2017.18.1.50
- Folpe AL, Reisenauer AK, Mentzel T, et al. Proliferating trichilemmal tumors: clinicopathologic evaluation is a guide to biologic behavior. J Cutan Pathol. 2003;30:492-498.
- Alsabbagh M. Keratitis-ichthyosis-deafness syndrome: a comprehensive review of cutaneous and systemic manifestations. Pediatr Dermatol. 2023;40:19-27.
- Nyquist GG, Mumm C, Grau R, et al. Malignant proliferating pilar tumors arising in KID syndrome: a report of two patients. Am J Med Genet A. 2007;143A:734-741.
- Richard G, Rouan F, Willoughby CE, et al. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am J Hum Genet. 2002;70: 1341-1348.
- Lee SI, Choi JH, Sung KY, et al. Proliferating pilar tumor of the cheek misdiagnosed as squamous cell carcinoma. Indian J Dermatol. 2022;67:207.
- Thompson LD. Pilomatricoma. Ear Nose Throat J. 2012;91:18-20.
- Waldman A, Schmults C. Cutaneous squamous cell carcinoma. Hematol Oncol Clin North Am. 2019;33:1-12.
- Cavanagh G, Negbenebor NA, Robinson-Bostom L, et al. Two cases of malignant proliferating trichilemmal tumor (MPTT) and review of literature. R I Med J (2013). 2022;105:12-16.
The Diagnosis: Proliferating Pilar Cyst
Histopathology revealed an extensive lobulated epithelial proliferation in a characteristic “rolls and scrolls” pattern (Figure 1). This finding along with the patient’s prior diagnosis of keratitis-ichthyosisdeafness (KID) syndrome supported the diagnosis of a proliferating pilar cyst.
Pilar (or trichilemmal) cysts are common dermal cysts typically found on the outer root sheath of hair follicles. They clinically manifest as multiple yellow dome-shaped nodules without central puncta. They are slow growing and histologically are characterized as cysts with a stratified squamous epithelium demonstrating lack of a granular layer (trichilemmal keratinization) with bright red keratin contents and central focal calcification (Figure 2). Pilar cysts are more common in adult women and may be inherited through an autosomal-dominant pattern.1
Proliferating pilar cysts represent less than 3% of all pilar cysts.2 In addition to the characteristic features of a pilar cyst, proliferating pilar cysts generally are larger (can be >6-cm wide) and are more ulcerative.3 Histopathology of proliferating pilar cysts reveals a more extensive epithelial proliferation, yielding a rolls and scrolls appearance, and may demonstrate nuclear atypia.4 Proliferating pilar cysts classically manifest as large, raised, smooth and/or ulcerated nodules on the scalp accompanied by areas of excessive hair growth in older women. They generally arise from pre-existing pilar cysts but also may occur sporadically.4
The development of multiple proliferating pilar cysts has been observed in patients with KID syndrome, a rare congenital ectodermal disorder characterized by a triad of vascularizing keratitis, hyperkeratosis, and sensorineural deafness.5,6 It is caused by a missense mutation of the GJB2 gene encoding for connexin 26, a gap junction that facilitates intercellular signaling and is expressed in a variety of structures including the cochlea, cornea, sweat glands, and inner and outer root sheaths of hair follicles.7
The differential diagnosis for proliferating pilar cysts includes pilomatrixomas, squamous cell carcinomas, and malignant proliferating pilar tumors. Pilomatrixomas (or calcifying epitheliomas of Malherbe) are the most common adnexal skin tumors in the pediatric population and most commonly present on the head, neck, and arms.8 They also can manifest in adults. Pilomatrixomas are benign dermal-subcutaneous tumors encapsulated by connective tissue that are found on the hair matrix and are histologically characterized by basaloid cells, shadow (or ghost) cells, dystrophic calcifications, and giant cells.9 The amount of basaloid cells and shadow cells can vary. Tumor progression results in the enucleation of the basaloid cells to form eosinophilic shadow cells in which calcification can occur. Giant cell granulomas may form contiguous with the calcifications. Both proliferating pilar cysts and pilomatrixomas have a rolls and scrolls appearance on low-power microscopy, but the latter are differentiated by their shadow cells and basaloid areas (Figure 3).
Squamous cell carcinoma (SCC) is the second most common nonmelanoma skin cancer and more commonly affects men. Risk factors for SCC include immunosuppression and exposure to UV radiation. Histopathology of well-differentiated SCCs reveals invasive squamous cells with larger nuclei and a glassy appearance in addition to possible mitotic figures and keratin pearls (Figure 4). They typically manifest in sun-exposed areas such as the scalp, face, forearms, dorsal aspects of the hands, and lower legs.10 Proliferating pilar tumors often lack the nuclear atypia and invasive architecture of a well-differentiated SCC.
Features of malignant proliferating pilar tumors overlap with proliferating pilar cysts. In addition to the proliferative epithelium with abrupt trichilemmal keratinization that is typical of a proliferating pilar cyst, a malignant proliferating pilar tumor will demonstrate invasion into the surrounding tissue and lymph nodes, mitotic and architectural atypia, and necrosis (Figure 5).11 Malignant proliferating pilar tumors grow rapidly, ranging in size from 1 to 10 cm, and may develop from pre-existing or proliferating pilar cysts or de novo.
The development of multiple proliferating pilar cysts and thus increased risk for progression to malignant proliferating pilar tumors has been observed in patients with KID syndrome.6 Our case highlights the importance of early screening and recognition of proliferating pilar tumors in patients with this condition.
The Diagnosis: Proliferating Pilar Cyst
Histopathology revealed an extensive lobulated epithelial proliferation in a characteristic “rolls and scrolls” pattern (Figure 1). This finding along with the patient’s prior diagnosis of keratitis-ichthyosisdeafness (KID) syndrome supported the diagnosis of a proliferating pilar cyst.
Pilar (or trichilemmal) cysts are common dermal cysts typically found on the outer root sheath of hair follicles. They clinically manifest as multiple yellow dome-shaped nodules without central puncta. They are slow growing and histologically are characterized as cysts with a stratified squamous epithelium demonstrating lack of a granular layer (trichilemmal keratinization) with bright red keratin contents and central focal calcification (Figure 2). Pilar cysts are more common in adult women and may be inherited through an autosomal-dominant pattern.1
Proliferating pilar cysts represent less than 3% of all pilar cysts.2 In addition to the characteristic features of a pilar cyst, proliferating pilar cysts generally are larger (can be >6-cm wide) and are more ulcerative.3 Histopathology of proliferating pilar cysts reveals a more extensive epithelial proliferation, yielding a rolls and scrolls appearance, and may demonstrate nuclear atypia.4 Proliferating pilar cysts classically manifest as large, raised, smooth and/or ulcerated nodules on the scalp accompanied by areas of excessive hair growth in older women. They generally arise from pre-existing pilar cysts but also may occur sporadically.4
The development of multiple proliferating pilar cysts has been observed in patients with KID syndrome, a rare congenital ectodermal disorder characterized by a triad of vascularizing keratitis, hyperkeratosis, and sensorineural deafness.5,6 It is caused by a missense mutation of the GJB2 gene encoding for connexin 26, a gap junction that facilitates intercellular signaling and is expressed in a variety of structures including the cochlea, cornea, sweat glands, and inner and outer root sheaths of hair follicles.7
The differential diagnosis for proliferating pilar cysts includes pilomatrixomas, squamous cell carcinomas, and malignant proliferating pilar tumors. Pilomatrixomas (or calcifying epitheliomas of Malherbe) are the most common adnexal skin tumors in the pediatric population and most commonly present on the head, neck, and arms.8 They also can manifest in adults. Pilomatrixomas are benign dermal-subcutaneous tumors encapsulated by connective tissue that are found on the hair matrix and are histologically characterized by basaloid cells, shadow (or ghost) cells, dystrophic calcifications, and giant cells.9 The amount of basaloid cells and shadow cells can vary. Tumor progression results in the enucleation of the basaloid cells to form eosinophilic shadow cells in which calcification can occur. Giant cell granulomas may form contiguous with the calcifications. Both proliferating pilar cysts and pilomatrixomas have a rolls and scrolls appearance on low-power microscopy, but the latter are differentiated by their shadow cells and basaloid areas (Figure 3).
Squamous cell carcinoma (SCC) is the second most common nonmelanoma skin cancer and more commonly affects men. Risk factors for SCC include immunosuppression and exposure to UV radiation. Histopathology of well-differentiated SCCs reveals invasive squamous cells with larger nuclei and a glassy appearance in addition to possible mitotic figures and keratin pearls (Figure 4). They typically manifest in sun-exposed areas such as the scalp, face, forearms, dorsal aspects of the hands, and lower legs.10 Proliferating pilar tumors often lack the nuclear atypia and invasive architecture of a well-differentiated SCC.
Features of malignant proliferating pilar tumors overlap with proliferating pilar cysts. In addition to the proliferative epithelium with abrupt trichilemmal keratinization that is typical of a proliferating pilar cyst, a malignant proliferating pilar tumor will demonstrate invasion into the surrounding tissue and lymph nodes, mitotic and architectural atypia, and necrosis (Figure 5).11 Malignant proliferating pilar tumors grow rapidly, ranging in size from 1 to 10 cm, and may develop from pre-existing or proliferating pilar cysts or de novo.
The development of multiple proliferating pilar cysts and thus increased risk for progression to malignant proliferating pilar tumors has been observed in patients with KID syndrome.6 Our case highlights the importance of early screening and recognition of proliferating pilar tumors in patients with this condition.
- Poiares Baptista A, Garcia E Silva L, Born MC. Proliferating trichilemmal cyst. J Cutan Pathol. 1983;10:178-187.
- Al Aboud DM, Yarrarapu SNS, Patel BC. Pilar cyst. StatPearls. StatPearls Publishing; 2023.
- Kim UG, Kook DB, Kim TH, et al. Trichilemmal carcinoma from proliferating trichilemmal cyst on the posterior neck [published online March 25, 2017]. Arch Craniofac Surg. 2017;18:50-53. doi:10.7181/acfs.2017.18.1.50
- Folpe AL, Reisenauer AK, Mentzel T, et al. Proliferating trichilemmal tumors: clinicopathologic evaluation is a guide to biologic behavior. J Cutan Pathol. 2003;30:492-498.
- Alsabbagh M. Keratitis-ichthyosis-deafness syndrome: a comprehensive review of cutaneous and systemic manifestations. Pediatr Dermatol. 2023;40:19-27.
- Nyquist GG, Mumm C, Grau R, et al. Malignant proliferating pilar tumors arising in KID syndrome: a report of two patients. Am J Med Genet A. 2007;143A:734-741.
- Richard G, Rouan F, Willoughby CE, et al. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am J Hum Genet. 2002;70: 1341-1348.
- Lee SI, Choi JH, Sung KY, et al. Proliferating pilar tumor of the cheek misdiagnosed as squamous cell carcinoma. Indian J Dermatol. 2022;67:207.
- Thompson LD. Pilomatricoma. Ear Nose Throat J. 2012;91:18-20.
- Waldman A, Schmults C. Cutaneous squamous cell carcinoma. Hematol Oncol Clin North Am. 2019;33:1-12.
- Cavanagh G, Negbenebor NA, Robinson-Bostom L, et al. Two cases of malignant proliferating trichilemmal tumor (MPTT) and review of literature. R I Med J (2013). 2022;105:12-16.
- Poiares Baptista A, Garcia E Silva L, Born MC. Proliferating trichilemmal cyst. J Cutan Pathol. 1983;10:178-187.
- Al Aboud DM, Yarrarapu SNS, Patel BC. Pilar cyst. StatPearls. StatPearls Publishing; 2023.
- Kim UG, Kook DB, Kim TH, et al. Trichilemmal carcinoma from proliferating trichilemmal cyst on the posterior neck [published online March 25, 2017]. Arch Craniofac Surg. 2017;18:50-53. doi:10.7181/acfs.2017.18.1.50
- Folpe AL, Reisenauer AK, Mentzel T, et al. Proliferating trichilemmal tumors: clinicopathologic evaluation is a guide to biologic behavior. J Cutan Pathol. 2003;30:492-498.
- Alsabbagh M. Keratitis-ichthyosis-deafness syndrome: a comprehensive review of cutaneous and systemic manifestations. Pediatr Dermatol. 2023;40:19-27.
- Nyquist GG, Mumm C, Grau R, et al. Malignant proliferating pilar tumors arising in KID syndrome: a report of two patients. Am J Med Genet A. 2007;143A:734-741.
- Richard G, Rouan F, Willoughby CE, et al. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am J Hum Genet. 2002;70: 1341-1348.
- Lee SI, Choi JH, Sung KY, et al. Proliferating pilar tumor of the cheek misdiagnosed as squamous cell carcinoma. Indian J Dermatol. 2022;67:207.
- Thompson LD. Pilomatricoma. Ear Nose Throat J. 2012;91:18-20.
- Waldman A, Schmults C. Cutaneous squamous cell carcinoma. Hematol Oncol Clin North Am. 2019;33:1-12.
- Cavanagh G, Negbenebor NA, Robinson-Bostom L, et al. Two cases of malignant proliferating trichilemmal tumor (MPTT) and review of literature. R I Med J (2013). 2022;105:12-16.
A 28-year-old man presented with an 8-mm, tender, mildly hyperkeratotic nodule on the right knee (top) of unknown duration. He had a history of mild keratitis-ichthyosis-deafness (KID) syndrome that was diagnosed based on the presence of congenital erythrokeratoderma, hearing issues identified at 2 years of age, palmoplantar keratoderma, keratitis, photophobia, chronic fungal nail infections, and alopecia and later was confirmed with a chromosome microarray for the GJB2 gene, which is associated with a connexin 26 mutation. A shave biopsy of the nodule was performed (bottom).


Annular Erythematous Plaques on the Back
The Diagnosis: Granuloma Annulare
The biopsies revealed palisading granulomatous dermatitis consistent with granuloma annulare (GA). This diagnosis was supported by the clinical presentation and histopathologic findings. Although the pathogenesis of GA is unclear, it is a benign, self-limiting condition. Primarily affected sites include the trunk and forearms. Generalized GA (or GA with ≥10 lesions) may warrant workup for malignancy, as it may represent a paraneoplastic process.1 Histopathology reveals granulomas comprising a dermal lymphohistiocytic infiltrate as well as central mucin and nuclear debris. There are a few histologic subtypes of GA, including palisading and interstitial, which refer to the distribution of the histiocytic infiltrate.2,3 This case—with palisading histiocytes lining the collection of necrobiosis and mucin (bottom quiz image)—features palisading GA. Notably, GA exhibits central rather than diffuse mucin.4
Erythema gyratum repens is a paraneoplastic arcuate erythema that manifests as erythematous figurate, gyrate, or annular plaques exhibiting a trailing scale. Clinically, erythema gyratum repens spreads rapidly—as quickly as 1 cm/d—and can be extensive (as in this case). Histopathology ruled out this diagnosis in our patient. Nonspecific findings of acanthosis, parakeratosis, and superficial spongiosis can be found in erythema gyratum repens. A superficial and deep perivascular lymphohistiocytic infiltrate may be seen in figurate erythemas (Figure 1).5 Unlike GA, this infiltrate does not form granulomas, is more superficial, and does not contain mucin.
.")
Histopathology also can help establish the diagnosis of leprosy and its specific subtype, as leprosy exists on a spectrum from tuberculoid to lepromatous, with a great deal of overlap in between.6 Lepromatous leprosy has many cutaneous clinical presentations but typically manifests as erythematous papules or nodules. It is multibacillary, and these mycobacteria form clumps known as globi that can be seen on Fite stain.7 In lepromatous leprosy, there is a characteristic dense lymphohistiocytic infiltrate (Figure 2) above which a Grenz zone can be seen.4,8 There are no well-formed granulomas in lepromatous leprosy, unlike in tuberculoid leprosy, which is paucibacillary and creates a granulomatous response surrounding nerves and adnexal structures.6
.")
Mycosis fungoides (MF) is the most common cutaneous lymphoma. There are patch, plaque, and tumor stages of MF, each of which exhibits various histopathologic findings.9 In early patch-stage MF, lymphocytes have perinuclear clearing, and the degree of lymphocytic infiltrate is out of proportion to the spongiosis present. Epidermotropism and Pautrier microabscesses often are present in the epidermis (Figure 3). In the plaque stage, there is a denser lymphoid infiltrate in a lichenoid pattern with epidermotropism and Pautrier microabscesses. The tumor stage shows a dense dermal lymphoid infiltrate with more atypia and typically a lack of epidermotropism. Rarely, MF can exhibit a granulomatous variant in which epithelioid histiocytes collect to form granulomas along with atypical lymphocytes.10
.")
The diagnosis of cutaneous sarcoidosis requires clinicopathologic corroboration. Histopathology demonstrates epithelioid histiocytes forming noncaseating granulomas with little to no lymphocytic infiltrate (Figure 4). There typically is no necrosis or necrobiosis as there is in GA. The diagnosis of sarcoidosis can be challenging histopathologically, and stains should be used to rule out infectious processes.4 Asteroid bodies— star-shaped eosinophilic inclusions within giant cells—may be present but are nonspecific for sarcoidosis.11 Schaumann bodies—inclusions of calcifications within giant cells—also may be present and can aid in diagnosis.12
.")
- Kovich O, Burgin S. Generalized granuloma annulare [published online December 30, 2005]. Dermatol Online J. 2005;11:23.
- Al Ameer MA, Al-Natour SH, Alsahaf HAA, et al. Eruptive granuloma annulare in an elderly man with diabetes [published online January 14, 2022]. Cureus. 2022;14:E21242. doi:10.7759/cureus.21242
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, et al, eds. Dermatology. Mosby; 2003:1455.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 3rd ed. Elsevier; 2018.
- Gore M, Winters ME. Erythema gyratum repens: a rare paraneoplastic rash. West J Emerg Med. 2011;12:556-558. doi:10.5811/westjem.2010.11.2090
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020;83:1-14. doi:10.1016/j.jaad.2019.12.080
- Pedley JC, Harman DJ, Waudby H, et al. Leprosy in peripheral nerves: histopathological findings in 119 untreated patients in Nepal. J Neurol Neurosurg Psychiatry. 1980;43:198-204. doi:10.1136/jnnp.43.3.198
- Booth AV, Kovich OI. Lepromatous leprosy [published online January 27, 2007]. Dermatol Online J. 2007;13:9.
- Robson A. The pathology of cutaneous T-cell lymphoma. Oncology (Williston Park). 2007;21(2 suppl 1):9-12.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001/archdermatol.2008.46
- Azar HA, Lunardelli C. Collagen nature of asteroid bodies of giant cells in sarcoidosis. Am J Pathol. 1969;57:81-92.
- Sreeja C, Priyadarshini A, Premika, et al. Sarcoidosis—a review article. J Oral Maxillofac Pathol. 2022;26:242-253. doi:10.4103 /jomfp.jomfp_373_21
The Diagnosis: Granuloma Annulare
The biopsies revealed palisading granulomatous dermatitis consistent with granuloma annulare (GA). This diagnosis was supported by the clinical presentation and histopathologic findings. Although the pathogenesis of GA is unclear, it is a benign, self-limiting condition. Primarily affected sites include the trunk and forearms. Generalized GA (or GA with ≥10 lesions) may warrant workup for malignancy, as it may represent a paraneoplastic process.1 Histopathology reveals granulomas comprising a dermal lymphohistiocytic infiltrate as well as central mucin and nuclear debris. There are a few histologic subtypes of GA, including palisading and interstitial, which refer to the distribution of the histiocytic infiltrate.2,3 This case—with palisading histiocytes lining the collection of necrobiosis and mucin (bottom quiz image)—features palisading GA. Notably, GA exhibits central rather than diffuse mucin.4
Erythema gyratum repens is a paraneoplastic arcuate erythema that manifests as erythematous figurate, gyrate, or annular plaques exhibiting a trailing scale. Clinically, erythema gyratum repens spreads rapidly—as quickly as 1 cm/d—and can be extensive (as in this case). Histopathology ruled out this diagnosis in our patient. Nonspecific findings of acanthosis, parakeratosis, and superficial spongiosis can be found in erythema gyratum repens. A superficial and deep perivascular lymphohistiocytic infiltrate may be seen in figurate erythemas (Figure 1).5 Unlike GA, this infiltrate does not form granulomas, is more superficial, and does not contain mucin.
Histopathology also can help establish the diagnosis of leprosy and its specific subtype, as leprosy exists on a spectrum from tuberculoid to lepromatous, with a great deal of overlap in between.6 Lepromatous leprosy has many cutaneous clinical presentations but typically manifests as erythematous papules or nodules. It is multibacillary, and these mycobacteria form clumps known as globi that can be seen on Fite stain.7 In lepromatous leprosy, there is a characteristic dense lymphohistiocytic infiltrate (Figure 2) above which a Grenz zone can be seen.4,8 There are no well-formed granulomas in lepromatous leprosy, unlike in tuberculoid leprosy, which is paucibacillary and creates a granulomatous response surrounding nerves and adnexal structures.6
Mycosis fungoides (MF) is the most common cutaneous lymphoma. There are patch, plaque, and tumor stages of MF, each of which exhibits various histopathologic findings.9 In early patch-stage MF, lymphocytes have perinuclear clearing, and the degree of lymphocytic infiltrate is out of proportion to the spongiosis present. Epidermotropism and Pautrier microabscesses often are present in the epidermis (Figure 3). In the plaque stage, there is a denser lymphoid infiltrate in a lichenoid pattern with epidermotropism and Pautrier microabscesses. The tumor stage shows a dense dermal lymphoid infiltrate with more atypia and typically a lack of epidermotropism. Rarely, MF can exhibit a granulomatous variant in which epithelioid histiocytes collect to form granulomas along with atypical lymphocytes.10
The diagnosis of cutaneous sarcoidosis requires clinicopathologic corroboration. Histopathology demonstrates epithelioid histiocytes forming noncaseating granulomas with little to no lymphocytic infiltrate (Figure 4). There typically is no necrosis or necrobiosis as there is in GA. The diagnosis of sarcoidosis can be challenging histopathologically, and stains should be used to rule out infectious processes.4 Asteroid bodies— star-shaped eosinophilic inclusions within giant cells—may be present but are nonspecific for sarcoidosis.11 Schaumann bodies—inclusions of calcifications within giant cells—also may be present and can aid in diagnosis.12
The Diagnosis: Granuloma Annulare
The biopsies revealed palisading granulomatous dermatitis consistent with granuloma annulare (GA). This diagnosis was supported by the clinical presentation and histopathologic findings. Although the pathogenesis of GA is unclear, it is a benign, self-limiting condition. Primarily affected sites include the trunk and forearms. Generalized GA (or GA with ≥10 lesions) may warrant workup for malignancy, as it may represent a paraneoplastic process.1 Histopathology reveals granulomas comprising a dermal lymphohistiocytic infiltrate as well as central mucin and nuclear debris. There are a few histologic subtypes of GA, including palisading and interstitial, which refer to the distribution of the histiocytic infiltrate.2,3 This case—with palisading histiocytes lining the collection of necrobiosis and mucin (bottom quiz image)—features palisading GA. Notably, GA exhibits central rather than diffuse mucin.4
Erythema gyratum repens is a paraneoplastic arcuate erythema that manifests as erythematous figurate, gyrate, or annular plaques exhibiting a trailing scale. Clinically, erythema gyratum repens spreads rapidly—as quickly as 1 cm/d—and can be extensive (as in this case). Histopathology ruled out this diagnosis in our patient. Nonspecific findings of acanthosis, parakeratosis, and superficial spongiosis can be found in erythema gyratum repens. A superficial and deep perivascular lymphohistiocytic infiltrate may be seen in figurate erythemas (Figure 1).5 Unlike GA, this infiltrate does not form granulomas, is more superficial, and does not contain mucin.
Histopathology also can help establish the diagnosis of leprosy and its specific subtype, as leprosy exists on a spectrum from tuberculoid to lepromatous, with a great deal of overlap in between.6 Lepromatous leprosy has many cutaneous clinical presentations but typically manifests as erythematous papules or nodules. It is multibacillary, and these mycobacteria form clumps known as globi that can be seen on Fite stain.7 In lepromatous leprosy, there is a characteristic dense lymphohistiocytic infiltrate (Figure 2) above which a Grenz zone can be seen.4,8 There are no well-formed granulomas in lepromatous leprosy, unlike in tuberculoid leprosy, which is paucibacillary and creates a granulomatous response surrounding nerves and adnexal structures.6
Mycosis fungoides (MF) is the most common cutaneous lymphoma. There are patch, plaque, and tumor stages of MF, each of which exhibits various histopathologic findings.9 In early patch-stage MF, lymphocytes have perinuclear clearing, and the degree of lymphocytic infiltrate is out of proportion to the spongiosis present. Epidermotropism and Pautrier microabscesses often are present in the epidermis (Figure 3). In the plaque stage, there is a denser lymphoid infiltrate in a lichenoid pattern with epidermotropism and Pautrier microabscesses. The tumor stage shows a dense dermal lymphoid infiltrate with more atypia and typically a lack of epidermotropism. Rarely, MF can exhibit a granulomatous variant in which epithelioid histiocytes collect to form granulomas along with atypical lymphocytes.10
The diagnosis of cutaneous sarcoidosis requires clinicopathologic corroboration. Histopathology demonstrates epithelioid histiocytes forming noncaseating granulomas with little to no lymphocytic infiltrate (Figure 4). There typically is no necrosis or necrobiosis as there is in GA. The diagnosis of sarcoidosis can be challenging histopathologically, and stains should be used to rule out infectious processes.4 Asteroid bodies— star-shaped eosinophilic inclusions within giant cells—may be present but are nonspecific for sarcoidosis.11 Schaumann bodies—inclusions of calcifications within giant cells—also may be present and can aid in diagnosis.12
- Kovich O, Burgin S. Generalized granuloma annulare [published online December 30, 2005]. Dermatol Online J. 2005;11:23.
- Al Ameer MA, Al-Natour SH, Alsahaf HAA, et al. Eruptive granuloma annulare in an elderly man with diabetes [published online January 14, 2022]. Cureus. 2022;14:E21242. doi:10.7759/cureus.21242
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, et al, eds. Dermatology. Mosby; 2003:1455.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 3rd ed. Elsevier; 2018.
- Gore M, Winters ME. Erythema gyratum repens: a rare paraneoplastic rash. West J Emerg Med. 2011;12:556-558. doi:10.5811/westjem.2010.11.2090
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020;83:1-14. doi:10.1016/j.jaad.2019.12.080
- Pedley JC, Harman DJ, Waudby H, et al. Leprosy in peripheral nerves: histopathological findings in 119 untreated patients in Nepal. J Neurol Neurosurg Psychiatry. 1980;43:198-204. doi:10.1136/jnnp.43.3.198
- Booth AV, Kovich OI. Lepromatous leprosy [published online January 27, 2007]. Dermatol Online J. 2007;13:9.
- Robson A. The pathology of cutaneous T-cell lymphoma. Oncology (Williston Park). 2007;21(2 suppl 1):9-12.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001/archdermatol.2008.46
- Azar HA, Lunardelli C. Collagen nature of asteroid bodies of giant cells in sarcoidosis. Am J Pathol. 1969;57:81-92.
- Sreeja C, Priyadarshini A, Premika, et al. Sarcoidosis—a review article. J Oral Maxillofac Pathol. 2022;26:242-253. doi:10.4103 /jomfp.jomfp_373_21
- Kovich O, Burgin S. Generalized granuloma annulare [published online December 30, 2005]. Dermatol Online J. 2005;11:23.
- Al Ameer MA, Al-Natour SH, Alsahaf HAA, et al. Eruptive granuloma annulare in an elderly man with diabetes [published online January 14, 2022]. Cureus. 2022;14:E21242. doi:10.7759/cureus.21242
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, et al, eds. Dermatology. Mosby; 2003:1455.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 3rd ed. Elsevier; 2018.
- Gore M, Winters ME. Erythema gyratum repens: a rare paraneoplastic rash. West J Emerg Med. 2011;12:556-558. doi:10.5811/westjem.2010.11.2090
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020;83:1-14. doi:10.1016/j.jaad.2019.12.080
- Pedley JC, Harman DJ, Waudby H, et al. Leprosy in peripheral nerves: histopathological findings in 119 untreated patients in Nepal. J Neurol Neurosurg Psychiatry. 1980;43:198-204. doi:10.1136/jnnp.43.3.198
- Booth AV, Kovich OI. Lepromatous leprosy [published online January 27, 2007]. Dermatol Online J. 2007;13:9.
- Robson A. The pathology of cutaneous T-cell lymphoma. Oncology (Williston Park). 2007;21(2 suppl 1):9-12.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001/archdermatol.2008.46
- Azar HA, Lunardelli C. Collagen nature of asteroid bodies of giant cells in sarcoidosis. Am J Pathol. 1969;57:81-92.
- Sreeja C, Priyadarshini A, Premika, et al. Sarcoidosis—a review article. J Oral Maxillofac Pathol. 2022;26:242-253. doi:10.4103 /jomfp.jomfp_373_21
An 84-year-old man presented to the clinic for evaluation of a pruritic rash on the back of 6 months’ duration that spread to the neck and chest over the past 2 months and then to the abdomen and thighs more recently. His primary care provider prescribed a 1-week course of oral steroids and steroid cream. The oral medication did not help, but the cream alleviated the pruritus. He had a medical history of coronary artery disease, hypertension, and diabetes mellitus. He also had a rash on the forearms that had waxed and waned for many years but was not associated with pruritus. He had not sought medical care for the rash and had never treated it. Physical examination revealed pink to violaceous annular plaques with central clearing and raised borders that coalesced into larger plaques on the trunk (top). Dusky, scaly, pink plaques were present on the dorsal forearms. Three punch biopsies—2 from the upper back (bottom) and 1 from the left forearm—all demonstrated consistent findings.


Pink Papules on the Cheek
The Diagnosis: Cutaneous Rosai-Dorfman Disease
Rosai-Dorfman disease is a rare benign non- Langerhans cell histiocytopathy that can manifest initially with lymph node involvement—classically, massive painless cervical lymphadenopathy.1 Cutaneous Rosai-Dorfman disease (CRDD) is a variant that can be associated with lymph node and internal involvement, but more than 80% of cases lack extracutaneous involvement.2,3 In cases with extracutaneous involvement, lymph node disease is most frequent.3 Cutaneous Rosai-Dorfman disease unassociated with extracutaneous disease is a benign self-limiting histiocytopathy that manifests as painless red-brown, yellow, or fleshcolored nodules, plaques, or papules that may become tender or ulcerated.4
Cutaneous Rosai-Dorfman disease represents a benign histiocytopathy of resident dendritic cell derivation.3 A characteristic immunohistochemical finding is S-100 positivity, which might suggest a Langerhans cell transdifferentiation phenotype, but other markers corroborative of a Langerhans cell phenotype—namely CD1a and langerin—will be negative. Biopsies typically show a mid to deep dermal histiocytic infiltration in a variably dense polymorphous inflammatory cell background comprised of a mixture of lymphocytes, plasma cells, and neutrophils.3 At times the extent of lymphocytic infiltration can be to a magnitude that resembles a lymphoma on histopathology. In our patient, lymphoma was excluded based on clinical presentation, as this patient lacked the typical symptoms of lymphadenopathy or B symptoms that come with B-cell lymphoma.5
The histiocytes in CRDD are characteristically large mononuclear cells exhibiting a low nuclear to cytoplasmic ratio reflective of the voluminous, nonvacuolated, watery cytoplasm. They have ill-defined cytoplasmic membranes resulting in a seemingly syncytial growth pattern. A hallmark of the histiocytes is emperipolesis characterized by intracytoplasmic localization of intact inflammatory cells including neutrophils, lymphocytes, and plasma cells.3
The differential diagnosis of CRDD includes Langerhans cell histiocytosis (LCH), indeterminate cell histiocytosis, xanthogranuloma, and reticulohistiocytoma. All of these conditions can be differentiated by their unique histopathologic and phenotypic characteristics.
Langerhans cell histiocytosis is a distinct clonal histiocytopathy that has a varied presentation ranging from cutaneous confined cases manifesting as a solitary lesion to one of disseminated cutaneous disease with the potential for multiorgan involvement. Regardless of the variant of LCH, the hallmark cell is one showing an eccentrically disposed, reniform nucleus with an open chromatin and abundant eosinophilic cytoplasm (Figure 1).6 Both LCH and CRDD stain positive for S-100. However, unlike the histiocytes in CRDD, those seen in LCH stain positive for CD1a and langerin and would not express factor XIIIA. Additionally, the neoplastic cells would not exhibit the same extent of CD68 positivity seen in CRDD.6 Treatment of LCH depends on the extent of disease, especially for the presence or absence of extracutaneous disease.7

A variant of LCH is indeterminate cell histiocytosis, which can be seen in neonates or adults. It represents a neoplastic proliferation of Langerhans cells that are devoid of Birbeck granules, reflective of an immature early phase of differentiation in the skin prior to the cells acquiring the Birbeck granule (as would be seen in neonates) or a later phase of differentiation after the mature Langerhans cell has encountered antigen and is en route to the lymph node (typically seen in adults).8 The phenotypic profile is identical to conventional LCH except the cells do not express langerin. Microscopically, the infiltrates are composed of Langerhans cells that are morphologically indistinguishable from classic LCH but without epidermotropism and exhibit a dominant localization in the dermis typically unassociated with other inflammatory cell elements (Figure 2).9

Xanthogranuloma is seen in young children (juvenile xanthogranuloma) as a solitary lesion, though a multifocal cutaneous variant and extracutaneous presentations have been described. Similar lesions can be seen in adults.10 These lesions are evolutionary in their morphology. In the inception of a juvenile xanthogranuloma, the lesions are highly cellular, and the histiocytes typically are poorly lipidized; there may be a dearth of other inflammatory cell elements. As the lesions mature, the histiocytes become lipidized, and characteristic Touton giant cells that exhibit a wreath of nuclei with peripheral lipidization may develop (Figure 3). In the later stages, there is considerable hyalinizing fibrosis, and the cells can acquire a spindled appearance. The absence of emperipolesis and the presence of notable lipidization are useful light microscopy features differentiating xanthogranuloma from CRDD.11 Treatment of xanthogranuloma can range from a conservative monitoring approach to an aggressive approach combining various antineoplastic therapies with immunosuppressive agents.12

Solitary and multicentric reticulohistiocytoma is another form of resident dendritic cell histiocytopathy that can resemble Rosai-Dorfman disease. It is a dermal histiocytic infiltrate accompanied by a polymorphous inflammatory cell infiltrate (Figure 4) and can show variable fibrosis.13 One of the hallmarks is the distinct amphophilic cytoplasms, possibly attributable to nuclear DNA released into the cytoplasm from effete nuclei.13 The solitary form is unassociated with systemic disease, whereas the multicentric variant can be a paraneoplastic syndrome in the setting of solid and hematologic malignancies.14 In addition, in the multicentric variant, similar lesions can affect any organ but there can be a proclivity to involve the hand and knee joints, leading to a crippling arthritis.15 We presented a case of CRDD, a benign resident dendritic cell histiocytopathy that can manifest as a cutaneous confined process in the skin where the clinical course is characteristically benign. It potentially can be confused with LCH, indeterminate cell histiocytosis, xanthogranuloma, and reticulohistiocytoma. For a solitary lesion, intralesional triamcinolone injection and excision are options. Multifocal cutaneous disease or CRDD with notable extracutaneous disease may require systemic treatment.16 In our patient, one intralesional triamcinolone injection was performed with notable resolution.

- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24:385.
- Ahmed A, Crowson N, Magro CM. A comprehensive assessment of cutaneous Rosai-Dorfman disease. Ann Diagn Pathol. 2019;40:166-173.
- Frater JL, Maddox JS, Obadiah JM, et al. Cutaneous Rosai-Dorfman disease: comprehensive review of cases reported in the medical literature since 1990 and presentation of an illustrative case. J Cutan Med Surg. 2006;10:281-290.
- Friedberg JW, Fisher RI. Diffuse large B-cell lymphoma. Hematol Oncol Clin North Am. 2008;22:941-952. Doi:10.1016/j.hoc.2008.07.002
- Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med. 2018;379:856-868.
- Board PPTE. Langerhans cell histiocytosis treatment (PDQ®). In: PDQ Cancer Information Summaries [Internet]. National Cancer Institute (US); 2009.
- Chu A, Eisinger M, Lee JS, et al. Immunoelectron microscopic identification of Langerhans cells using a new antigenic marker. J Invest Dermatol. 1982;78:177-180. doi:10.1111/1523-1747.ep12506352
- Berti E, Gianotti R, Alessi E. Unusual cutaneous histiocytosis expressing an intermediate immunophenotype between Langerhans’ cells and dermal macrophages. Arch Dermatol. 1988;124:1250-1253. doi:10.1001/archderm.1988.01670080062020
- Cypel TKS, Zuker RM. Juvenile xanthogranuloma: case report and review of the literature. Can J Plast Surg. 2008;16:175-177.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. In: StatPearls [Internet]. StatPearls Publishing; 2022.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492. doi:10.1016/j. clindermatol.2006.07.010
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521.
- Gold RH, Metzger AL, Mirra JM, et al. Multicentric reticulohistiocytosis (lipoid dermato-arthritis). An erosive polyarthritis with distinctive clinical, roentgenographic and pathologic features. Am J Roentgenol Radium Ther Nucl Med. 1975;124:610-624. doi:10.2214/ajr.124.4.610
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327. doi:10.1177/107327481402100408
The Diagnosis: Cutaneous Rosai-Dorfman Disease
Rosai-Dorfman disease is a rare benign non- Langerhans cell histiocytopathy that can manifest initially with lymph node involvement—classically, massive painless cervical lymphadenopathy.1 Cutaneous Rosai-Dorfman disease (CRDD) is a variant that can be associated with lymph node and internal involvement, but more than 80% of cases lack extracutaneous involvement.2,3 In cases with extracutaneous involvement, lymph node disease is most frequent.3 Cutaneous Rosai-Dorfman disease unassociated with extracutaneous disease is a benign self-limiting histiocytopathy that manifests as painless red-brown, yellow, or fleshcolored nodules, plaques, or papules that may become tender or ulcerated.4
Cutaneous Rosai-Dorfman disease represents a benign histiocytopathy of resident dendritic cell derivation.3 A characteristic immunohistochemical finding is S-100 positivity, which might suggest a Langerhans cell transdifferentiation phenotype, but other markers corroborative of a Langerhans cell phenotype—namely CD1a and langerin—will be negative. Biopsies typically show a mid to deep dermal histiocytic infiltration in a variably dense polymorphous inflammatory cell background comprised of a mixture of lymphocytes, plasma cells, and neutrophils.3 At times the extent of lymphocytic infiltration can be to a magnitude that resembles a lymphoma on histopathology. In our patient, lymphoma was excluded based on clinical presentation, as this patient lacked the typical symptoms of lymphadenopathy or B symptoms that come with B-cell lymphoma.5
The histiocytes in CRDD are characteristically large mononuclear cells exhibiting a low nuclear to cytoplasmic ratio reflective of the voluminous, nonvacuolated, watery cytoplasm. They have ill-defined cytoplasmic membranes resulting in a seemingly syncytial growth pattern. A hallmark of the histiocytes is emperipolesis characterized by intracytoplasmic localization of intact inflammatory cells including neutrophils, lymphocytes, and plasma cells.3
The differential diagnosis of CRDD includes Langerhans cell histiocytosis (LCH), indeterminate cell histiocytosis, xanthogranuloma, and reticulohistiocytoma. All of these conditions can be differentiated by their unique histopathologic and phenotypic characteristics.
Langerhans cell histiocytosis is a distinct clonal histiocytopathy that has a varied presentation ranging from cutaneous confined cases manifesting as a solitary lesion to one of disseminated cutaneous disease with the potential for multiorgan involvement. Regardless of the variant of LCH, the hallmark cell is one showing an eccentrically disposed, reniform nucleus with an open chromatin and abundant eosinophilic cytoplasm (Figure 1).6 Both LCH and CRDD stain positive for S-100. However, unlike the histiocytes in CRDD, those seen in LCH stain positive for CD1a and langerin and would not express factor XIIIA. Additionally, the neoplastic cells would not exhibit the same extent of CD68 positivity seen in CRDD.6 Treatment of LCH depends on the extent of disease, especially for the presence or absence of extracutaneous disease.7
A variant of LCH is indeterminate cell histiocytosis, which can be seen in neonates or adults. It represents a neoplastic proliferation of Langerhans cells that are devoid of Birbeck granules, reflective of an immature early phase of differentiation in the skin prior to the cells acquiring the Birbeck granule (as would be seen in neonates) or a later phase of differentiation after the mature Langerhans cell has encountered antigen and is en route to the lymph node (typically seen in adults).8 The phenotypic profile is identical to conventional LCH except the cells do not express langerin. Microscopically, the infiltrates are composed of Langerhans cells that are morphologically indistinguishable from classic LCH but without epidermotropism and exhibit a dominant localization in the dermis typically unassociated with other inflammatory cell elements (Figure 2).9
Xanthogranuloma is seen in young children (juvenile xanthogranuloma) as a solitary lesion, though a multifocal cutaneous variant and extracutaneous presentations have been described. Similar lesions can be seen in adults.10 These lesions are evolutionary in their morphology. In the inception of a juvenile xanthogranuloma, the lesions are highly cellular, and the histiocytes typically are poorly lipidized; there may be a dearth of other inflammatory cell elements. As the lesions mature, the histiocytes become lipidized, and characteristic Touton giant cells that exhibit a wreath of nuclei with peripheral lipidization may develop (Figure 3). In the later stages, there is considerable hyalinizing fibrosis, and the cells can acquire a spindled appearance. The absence of emperipolesis and the presence of notable lipidization are useful light microscopy features differentiating xanthogranuloma from CRDD.11 Treatment of xanthogranuloma can range from a conservative monitoring approach to an aggressive approach combining various antineoplastic therapies with immunosuppressive agents.12
Solitary and multicentric reticulohistiocytoma is another form of resident dendritic cell histiocytopathy that can resemble Rosai-Dorfman disease. It is a dermal histiocytic infiltrate accompanied by a polymorphous inflammatory cell infiltrate (Figure 4) and can show variable fibrosis.13 One of the hallmarks is the distinct amphophilic cytoplasms, possibly attributable to nuclear DNA released into the cytoplasm from effete nuclei.13 The solitary form is unassociated with systemic disease, whereas the multicentric variant can be a paraneoplastic syndrome in the setting of solid and hematologic malignancies.14 In addition, in the multicentric variant, similar lesions can affect any organ but there can be a proclivity to involve the hand and knee joints, leading to a crippling arthritis.15 We presented a case of CRDD, a benign resident dendritic cell histiocytopathy that can manifest as a cutaneous confined process in the skin where the clinical course is characteristically benign. It potentially can be confused with LCH, indeterminate cell histiocytosis, xanthogranuloma, and reticulohistiocytoma. For a solitary lesion, intralesional triamcinolone injection and excision are options. Multifocal cutaneous disease or CRDD with notable extracutaneous disease may require systemic treatment.16 In our patient, one intralesional triamcinolone injection was performed with notable resolution.
The Diagnosis: Cutaneous Rosai-Dorfman Disease
Rosai-Dorfman disease is a rare benign non- Langerhans cell histiocytopathy that can manifest initially with lymph node involvement—classically, massive painless cervical lymphadenopathy.1 Cutaneous Rosai-Dorfman disease (CRDD) is a variant that can be associated with lymph node and internal involvement, but more than 80% of cases lack extracutaneous involvement.2,3 In cases with extracutaneous involvement, lymph node disease is most frequent.3 Cutaneous Rosai-Dorfman disease unassociated with extracutaneous disease is a benign self-limiting histiocytopathy that manifests as painless red-brown, yellow, or fleshcolored nodules, plaques, or papules that may become tender or ulcerated.4
Cutaneous Rosai-Dorfman disease represents a benign histiocytopathy of resident dendritic cell derivation.3 A characteristic immunohistochemical finding is S-100 positivity, which might suggest a Langerhans cell transdifferentiation phenotype, but other markers corroborative of a Langerhans cell phenotype—namely CD1a and langerin—will be negative. Biopsies typically show a mid to deep dermal histiocytic infiltration in a variably dense polymorphous inflammatory cell background comprised of a mixture of lymphocytes, plasma cells, and neutrophils.3 At times the extent of lymphocytic infiltration can be to a magnitude that resembles a lymphoma on histopathology. In our patient, lymphoma was excluded based on clinical presentation, as this patient lacked the typical symptoms of lymphadenopathy or B symptoms that come with B-cell lymphoma.5
The histiocytes in CRDD are characteristically large mononuclear cells exhibiting a low nuclear to cytoplasmic ratio reflective of the voluminous, nonvacuolated, watery cytoplasm. They have ill-defined cytoplasmic membranes resulting in a seemingly syncytial growth pattern. A hallmark of the histiocytes is emperipolesis characterized by intracytoplasmic localization of intact inflammatory cells including neutrophils, lymphocytes, and plasma cells.3
The differential diagnosis of CRDD includes Langerhans cell histiocytosis (LCH), indeterminate cell histiocytosis, xanthogranuloma, and reticulohistiocytoma. All of these conditions can be differentiated by their unique histopathologic and phenotypic characteristics.
Langerhans cell histiocytosis is a distinct clonal histiocytopathy that has a varied presentation ranging from cutaneous confined cases manifesting as a solitary lesion to one of disseminated cutaneous disease with the potential for multiorgan involvement. Regardless of the variant of LCH, the hallmark cell is one showing an eccentrically disposed, reniform nucleus with an open chromatin and abundant eosinophilic cytoplasm (Figure 1).6 Both LCH and CRDD stain positive for S-100. However, unlike the histiocytes in CRDD, those seen in LCH stain positive for CD1a and langerin and would not express factor XIIIA. Additionally, the neoplastic cells would not exhibit the same extent of CD68 positivity seen in CRDD.6 Treatment of LCH depends on the extent of disease, especially for the presence or absence of extracutaneous disease.7
A variant of LCH is indeterminate cell histiocytosis, which can be seen in neonates or adults. It represents a neoplastic proliferation of Langerhans cells that are devoid of Birbeck granules, reflective of an immature early phase of differentiation in the skin prior to the cells acquiring the Birbeck granule (as would be seen in neonates) or a later phase of differentiation after the mature Langerhans cell has encountered antigen and is en route to the lymph node (typically seen in adults).8 The phenotypic profile is identical to conventional LCH except the cells do not express langerin. Microscopically, the infiltrates are composed of Langerhans cells that are morphologically indistinguishable from classic LCH but without epidermotropism and exhibit a dominant localization in the dermis typically unassociated with other inflammatory cell elements (Figure 2).9
Xanthogranuloma is seen in young children (juvenile xanthogranuloma) as a solitary lesion, though a multifocal cutaneous variant and extracutaneous presentations have been described. Similar lesions can be seen in adults.10 These lesions are evolutionary in their morphology. In the inception of a juvenile xanthogranuloma, the lesions are highly cellular, and the histiocytes typically are poorly lipidized; there may be a dearth of other inflammatory cell elements. As the lesions mature, the histiocytes become lipidized, and characteristic Touton giant cells that exhibit a wreath of nuclei with peripheral lipidization may develop (Figure 3). In the later stages, there is considerable hyalinizing fibrosis, and the cells can acquire a spindled appearance. The absence of emperipolesis and the presence of notable lipidization are useful light microscopy features differentiating xanthogranuloma from CRDD.11 Treatment of xanthogranuloma can range from a conservative monitoring approach to an aggressive approach combining various antineoplastic therapies with immunosuppressive agents.12
Solitary and multicentric reticulohistiocytoma is another form of resident dendritic cell histiocytopathy that can resemble Rosai-Dorfman disease. It is a dermal histiocytic infiltrate accompanied by a polymorphous inflammatory cell infiltrate (Figure 4) and can show variable fibrosis.13 One of the hallmarks is the distinct amphophilic cytoplasms, possibly attributable to nuclear DNA released into the cytoplasm from effete nuclei.13 The solitary form is unassociated with systemic disease, whereas the multicentric variant can be a paraneoplastic syndrome in the setting of solid and hematologic malignancies.14 In addition, in the multicentric variant, similar lesions can affect any organ but there can be a proclivity to involve the hand and knee joints, leading to a crippling arthritis.15 We presented a case of CRDD, a benign resident dendritic cell histiocytopathy that can manifest as a cutaneous confined process in the skin where the clinical course is characteristically benign. It potentially can be confused with LCH, indeterminate cell histiocytosis, xanthogranuloma, and reticulohistiocytoma. For a solitary lesion, intralesional triamcinolone injection and excision are options. Multifocal cutaneous disease or CRDD with notable extracutaneous disease may require systemic treatment.16 In our patient, one intralesional triamcinolone injection was performed with notable resolution.
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24:385.
- Ahmed A, Crowson N, Magro CM. A comprehensive assessment of cutaneous Rosai-Dorfman disease. Ann Diagn Pathol. 2019;40:166-173.
- Frater JL, Maddox JS, Obadiah JM, et al. Cutaneous Rosai-Dorfman disease: comprehensive review of cases reported in the medical literature since 1990 and presentation of an illustrative case. J Cutan Med Surg. 2006;10:281-290.
- Friedberg JW, Fisher RI. Diffuse large B-cell lymphoma. Hematol Oncol Clin North Am. 2008;22:941-952. Doi:10.1016/j.hoc.2008.07.002
- Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med. 2018;379:856-868.
- Board PPTE. Langerhans cell histiocytosis treatment (PDQ®). In: PDQ Cancer Information Summaries [Internet]. National Cancer Institute (US); 2009.
- Chu A, Eisinger M, Lee JS, et al. Immunoelectron microscopic identification of Langerhans cells using a new antigenic marker. J Invest Dermatol. 1982;78:177-180. doi:10.1111/1523-1747.ep12506352
- Berti E, Gianotti R, Alessi E. Unusual cutaneous histiocytosis expressing an intermediate immunophenotype between Langerhans’ cells and dermal macrophages. Arch Dermatol. 1988;124:1250-1253. doi:10.1001/archderm.1988.01670080062020
- Cypel TKS, Zuker RM. Juvenile xanthogranuloma: case report and review of the literature. Can J Plast Surg. 2008;16:175-177.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. In: StatPearls [Internet]. StatPearls Publishing; 2022.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492. doi:10.1016/j. clindermatol.2006.07.010
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521.
- Gold RH, Metzger AL, Mirra JM, et al. Multicentric reticulohistiocytosis (lipoid dermato-arthritis). An erosive polyarthritis with distinctive clinical, roentgenographic and pathologic features. Am J Roentgenol Radium Ther Nucl Med. 1975;124:610-624. doi:10.2214/ajr.124.4.610
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327. doi:10.1177/107327481402100408
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24:385.
- Ahmed A, Crowson N, Magro CM. A comprehensive assessment of cutaneous Rosai-Dorfman disease. Ann Diagn Pathol. 2019;40:166-173.
- Frater JL, Maddox JS, Obadiah JM, et al. Cutaneous Rosai-Dorfman disease: comprehensive review of cases reported in the medical literature since 1990 and presentation of an illustrative case. J Cutan Med Surg. 2006;10:281-290.
- Friedberg JW, Fisher RI. Diffuse large B-cell lymphoma. Hematol Oncol Clin North Am. 2008;22:941-952. Doi:10.1016/j.hoc.2008.07.002
- Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med. 2018;379:856-868.
- Board PPTE. Langerhans cell histiocytosis treatment (PDQ®). In: PDQ Cancer Information Summaries [Internet]. National Cancer Institute (US); 2009.
- Chu A, Eisinger M, Lee JS, et al. Immunoelectron microscopic identification of Langerhans cells using a new antigenic marker. J Invest Dermatol. 1982;78:177-180. doi:10.1111/1523-1747.ep12506352
- Berti E, Gianotti R, Alessi E. Unusual cutaneous histiocytosis expressing an intermediate immunophenotype between Langerhans’ cells and dermal macrophages. Arch Dermatol. 1988;124:1250-1253. doi:10.1001/archderm.1988.01670080062020
- Cypel TKS, Zuker RM. Juvenile xanthogranuloma: case report and review of the literature. Can J Plast Surg. 2008;16:175-177.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. In: StatPearls [Internet]. StatPearls Publishing; 2022.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492. doi:10.1016/j. clindermatol.2006.07.010
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521.
- Gold RH, Metzger AL, Mirra JM, et al. Multicentric reticulohistiocytosis (lipoid dermato-arthritis). An erosive polyarthritis with distinctive clinical, roentgenographic and pathologic features. Am J Roentgenol Radium Ther Nucl Med. 1975;124:610-624. doi:10.2214/ajr.124.4.610
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327. doi:10.1177/107327481402100408
A 31-year-old woman presented with a slow-growing, tender, pruritic lesion on the right cheek of 4 to 5 months’ duration. She had been applying petroleum jelly and hydrocortisone cream 2.5% without any improvement. Physical examination revealed a 1×5-mm, pearly pink, erythematous, crusted papule with arborizing vessels surrounded by scattered pink papules with white dots within. No cervical lymphadenopathy was appreciated on physical examination, and the patient denied any other systemic symptoms. Shave and punch biopsies of the lesion were performed; stains for microorganisms were negative. The biopsy showed a dense reticular mixed inflammatory cell infiltrate comprised of a mixture of histiocytes (top), lymphocytes, neutrophils, and plasma cells that assumed a diffuse growth pattern within the dermis. The histiocytes exhibited abundant watery cytoplasms with ill-defined cytoplasmic membranes; intact leukocytes were found within the cytoplasms. The histiocytes demonstrated a unique phenotype characterized by S-100 (bottom) and CD68 positivity.

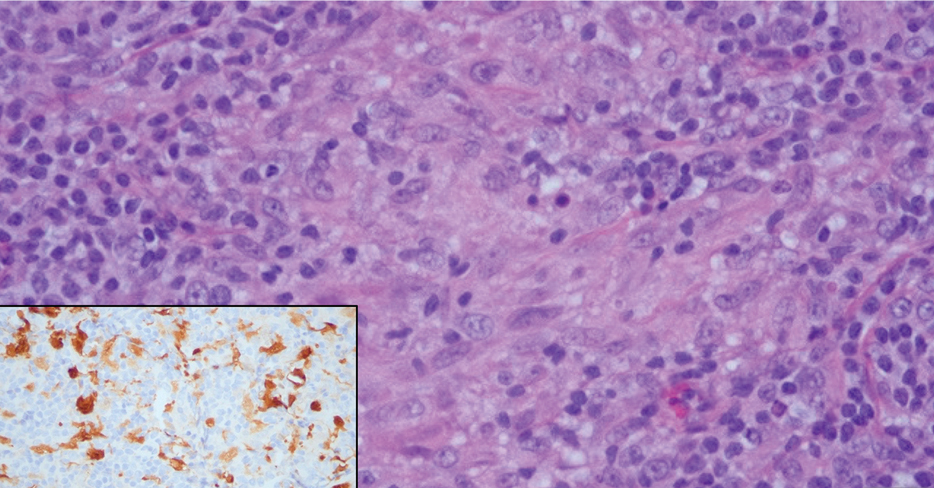

Painful Growing Nodule on the Right Calf
The Diagnosis: Merkel Cell Carcinoma
Multiple diagnoses should be considered for a small, round, blue cell neoplasm of the skin, including both primary and metastatic entities. In our patient, histopathology revealed sheets and nests of infiltrative neoplastic cells with dispersed chromatin, minimal cytoplasm, and multiple mitoses (quiz image 1).1 The lesional cells were in the dermis and superficial subcutaneous tissue but did not appear to be arising from the epidermis. Lymphovascular invasion also was evident on additional sections. Metastatic disease was identified in 3 sentinel lymph nodes from the right inguinal and right iliac regions. These features were compatible with a diagnosis of Merkel cell carcinoma (MCC).
Merkel cell carcinoma is a rare malignant neuroendocrine cutaneous tumor with a worldwide incidence of 0.1 to 1.6 cases per 100,000 individuals annually.2 The typical patient is older than 75 years with fair skin and a history of extensive sun exposure. Immunocompromised individuals are predisposed and more susceptible to infection with the Merkel cell polyomavirus, which promotes oncogenesis in the majority of MCCs. Our patient’s history of combined variable immunodeficiency likely explains her presentation at a younger age.
The prognosis in patients with MCC is poor, with 5-year survival rates of 51% for local disease, 35% for nodal disease, and 14% for systemic metastases. Survival also is reduced in cases with head/ neck primary tumors and polyomavirus-negative tumors, as well as in immunocompromised patients.2 Treatment of resectable MCC consists of Mohs micrographic surgery or wide local excision depending on the patient’s cosmetic concerns. Radiation therapy is recommended for cases with increased risk for recurrence or positive surgical margins, as well as when additional resection is impossible. A study investigating immunotherapy with nivolumab demonstrated complete pathologic response and radiographic tumor regression in nearly half of patients when given 4 weeks prior to surgery.3
Immunohistochemistry is essential in discerning MCC from other small blue cell tumors. Most MCC cases show positive expression of neuroendocrine markers such as synaptophysin, chromogranin, and insulinomaassociated protein 1. Perinuclear dotlike staining with cytokeratin (CK) 20 (quiz image 2) commonly is seen, but up to 15% of cases may be CK20 negative. Many of these CK20-negative cases also express CK7. This tumor also may stain with paired box 5 (PAX-5), CD99, terminal deoxynucleotidyl transferase, Ber-EP4, and CD1171,4; melanoma stains (ie, human melanoma black [HMB] 45, SRYrelated HMB-box 10 [SOX-10], S-100, melanoma antigen recognized by T-cells 1 [MART-1]) should be negative. However, PAX-5 expression may be a potential pitfall given that B-cell lymphomas also would express that marker and could mimic MCC histologically. Therefore, other universal lymphoid markers such as CD45 should be ordered to rule out this entity. Even with one or a few aberrant stains, a diagnosis of MCC still can be rendered using the histomorphology and the overall staining profile.4 Of prognostic significance, p63 expression is associated with more aggressive tumors, while Bcl-2 expression is favorable, as it offers an additional targeted treatment option.5,6
Basal cell carcinoma (BCC) is linked to excessive sun exposure and is the most common skin cancer. Similar to MCC, it typically is mitotically active and hyperchromatic; however, lymphovascular invasion or metastasis almost never is observed in BCC, whereas approximately one-third of MCC cases have metastasized by the time of diagnosis. Additionally, BCC lacks the perinuclear dotlike staining seen with CK20.2,7 Features present in BCC that are unusual for MCC include peripheral nuclear palisading, mucin, and retraction artifact on paraffin-embedded sections (Figure 1).7

Leukemia cutis (or cutaneous infiltrates of leukemia) commonly displays a perivascular and periadnexal pattern in the dermis and subcutis. These infiltrates of neoplastic leukocytes can congregate into sheets, sometimes with an overlying Grenz zone, or form single-file infiltrates (Figure 2).1,4 The neoplastic cells can be monomorphic or atypical and commonly are susceptible to crush artifact.4 Although the immunohistochemical profile varies depending on the etiology of the underlying leukemia, broad hematologic markers such as CD43 and CD45 are helpful to discern these malignancies from MCC.4

Being neuroendocrine in origin, metastatic small cell carcinoma (Figure 3) strongly mimics MCC histologically and usually stains with synaptophysin, chromogranin, and insulinoma-associated protein 1. Both tumor cells typically exhibit nuclear molding and high mitotic rates. Although small cell carcinoma is more likely to stain with high-molecular-weight cytokeratins (ie, CK7), it is not uncommon for these tumors to express lowmolecular- weight cytokeratins such as CK20. Because most cases originate from the lungs, these lesions should be positive for thyroid transcription factor 1 and negative for PAX-5, whereas MCC would show the reverse for those stains.1 Ultimately, however, clinical correlation with imaging results is the single best methodology for differentiation.

Small cell melanoma, a variant of nevoid melanoma, can strongly resemble an MCC or a lymphoma. Usually located on the scalp or arising from a congenital nevus, small cell melanomas are aggressive and confer an unfavorable prognosis. Histologically, they consist of nests to sheets of atypical cells within the epidermis and dermis. These cells typically exhibit hyperchromatic nuclei, minimal cytoplasm, and frequent mitoses (Figure 4). Furthermore, the cells do not display maturation based on depth.8 These tumors usually are positive for HMB45, S-100, MART-1, SOX-10, and tyrosinase, all of which are extremely unlikely to stain an MCC.1

- Patterson JW, Hosler GA. Weedon’s Skin Pathology. 4th ed. Churchill Livingstone/Elsevier; 2016.
- Walsh NM, Cerroni L. Merkel cell carcinoma: a review. J Cutan Pathol. 2021;48:411-421.
- Topalian SL, Bhatia S, Amin A, et al. Neoadjuvant nivolumab for patients with resectable Merkel cell carcinoma in the CheckMate 358 Trial. J Clin Oncol. 2020;38:2476-2488.
- Rapini RP. Practical Dermatopathology. 3rd ed. Elsevier; 2021.
- Asioli S, Righi A, Volante M, et al. p63 expression as a new prognostic marker in Merkel cell carcinoma. Cancer. 2007;110:640-647.
- Verhaegen ME, Mangelberger D, Weick JW, et al. Merkel cell carcinoma dependence on Bcl-2 family members for survival. J Invest Dermatol. 2014;134:2241-2250.
- Le MD, O’Steen LH, Cassarino DS. A rare case of CK20/CK7 double negative Merkel cell carcinoma. Am J Dermatopathol. 2017;39:208-211.
- North JP, Bastian BC, Lazar AJ. Melanoma. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin With Clinical Correlations. 5th ed. Elsevier; 2020.
The Diagnosis: Merkel Cell Carcinoma
Multiple diagnoses should be considered for a small, round, blue cell neoplasm of the skin, including both primary and metastatic entities. In our patient, histopathology revealed sheets and nests of infiltrative neoplastic cells with dispersed chromatin, minimal cytoplasm, and multiple mitoses (quiz image 1).1 The lesional cells were in the dermis and superficial subcutaneous tissue but did not appear to be arising from the epidermis. Lymphovascular invasion also was evident on additional sections. Metastatic disease was identified in 3 sentinel lymph nodes from the right inguinal and right iliac regions. These features were compatible with a diagnosis of Merkel cell carcinoma (MCC).
Merkel cell carcinoma is a rare malignant neuroendocrine cutaneous tumor with a worldwide incidence of 0.1 to 1.6 cases per 100,000 individuals annually.2 The typical patient is older than 75 years with fair skin and a history of extensive sun exposure. Immunocompromised individuals are predisposed and more susceptible to infection with the Merkel cell polyomavirus, which promotes oncogenesis in the majority of MCCs. Our patient’s history of combined variable immunodeficiency likely explains her presentation at a younger age.
The prognosis in patients with MCC is poor, with 5-year survival rates of 51% for local disease, 35% for nodal disease, and 14% for systemic metastases. Survival also is reduced in cases with head/ neck primary tumors and polyomavirus-negative tumors, as well as in immunocompromised patients.2 Treatment of resectable MCC consists of Mohs micrographic surgery or wide local excision depending on the patient’s cosmetic concerns. Radiation therapy is recommended for cases with increased risk for recurrence or positive surgical margins, as well as when additional resection is impossible. A study investigating immunotherapy with nivolumab demonstrated complete pathologic response and radiographic tumor regression in nearly half of patients when given 4 weeks prior to surgery.3
Immunohistochemistry is essential in discerning MCC from other small blue cell tumors. Most MCC cases show positive expression of neuroendocrine markers such as synaptophysin, chromogranin, and insulinomaassociated protein 1. Perinuclear dotlike staining with cytokeratin (CK) 20 (quiz image 2) commonly is seen, but up to 15% of cases may be CK20 negative. Many of these CK20-negative cases also express CK7. This tumor also may stain with paired box 5 (PAX-5), CD99, terminal deoxynucleotidyl transferase, Ber-EP4, and CD1171,4; melanoma stains (ie, human melanoma black [HMB] 45, SRYrelated HMB-box 10 [SOX-10], S-100, melanoma antigen recognized by T-cells 1 [MART-1]) should be negative. However, PAX-5 expression may be a potential pitfall given that B-cell lymphomas also would express that marker and could mimic MCC histologically. Therefore, other universal lymphoid markers such as CD45 should be ordered to rule out this entity. Even with one or a few aberrant stains, a diagnosis of MCC still can be rendered using the histomorphology and the overall staining profile.4 Of prognostic significance, p63 expression is associated with more aggressive tumors, while Bcl-2 expression is favorable, as it offers an additional targeted treatment option.5,6
Basal cell carcinoma (BCC) is linked to excessive sun exposure and is the most common skin cancer. Similar to MCC, it typically is mitotically active and hyperchromatic; however, lymphovascular invasion or metastasis almost never is observed in BCC, whereas approximately one-third of MCC cases have metastasized by the time of diagnosis. Additionally, BCC lacks the perinuclear dotlike staining seen with CK20.2,7 Features present in BCC that are unusual for MCC include peripheral nuclear palisading, mucin, and retraction artifact on paraffin-embedded sections (Figure 1).7
Leukemia cutis (or cutaneous infiltrates of leukemia) commonly displays a perivascular and periadnexal pattern in the dermis and subcutis. These infiltrates of neoplastic leukocytes can congregate into sheets, sometimes with an overlying Grenz zone, or form single-file infiltrates (Figure 2).1,4 The neoplastic cells can be monomorphic or atypical and commonly are susceptible to crush artifact.4 Although the immunohistochemical profile varies depending on the etiology of the underlying leukemia, broad hematologic markers such as CD43 and CD45 are helpful to discern these malignancies from MCC.4
Being neuroendocrine in origin, metastatic small cell carcinoma (Figure 3) strongly mimics MCC histologically and usually stains with synaptophysin, chromogranin, and insulinoma-associated protein 1. Both tumor cells typically exhibit nuclear molding and high mitotic rates. Although small cell carcinoma is more likely to stain with high-molecular-weight cytokeratins (ie, CK7), it is not uncommon for these tumors to express lowmolecular- weight cytokeratins such as CK20. Because most cases originate from the lungs, these lesions should be positive for thyroid transcription factor 1 and negative for PAX-5, whereas MCC would show the reverse for those stains.1 Ultimately, however, clinical correlation with imaging results is the single best methodology for differentiation.
Small cell melanoma, a variant of nevoid melanoma, can strongly resemble an MCC or a lymphoma. Usually located on the scalp or arising from a congenital nevus, small cell melanomas are aggressive and confer an unfavorable prognosis. Histologically, they consist of nests to sheets of atypical cells within the epidermis and dermis. These cells typically exhibit hyperchromatic nuclei, minimal cytoplasm, and frequent mitoses (Figure 4). Furthermore, the cells do not display maturation based on depth.8 These tumors usually are positive for HMB45, S-100, MART-1, SOX-10, and tyrosinase, all of which are extremely unlikely to stain an MCC.1
The Diagnosis: Merkel Cell Carcinoma
Multiple diagnoses should be considered for a small, round, blue cell neoplasm of the skin, including both primary and metastatic entities. In our patient, histopathology revealed sheets and nests of infiltrative neoplastic cells with dispersed chromatin, minimal cytoplasm, and multiple mitoses (quiz image 1).1 The lesional cells were in the dermis and superficial subcutaneous tissue but did not appear to be arising from the epidermis. Lymphovascular invasion also was evident on additional sections. Metastatic disease was identified in 3 sentinel lymph nodes from the right inguinal and right iliac regions. These features were compatible with a diagnosis of Merkel cell carcinoma (MCC).
Merkel cell carcinoma is a rare malignant neuroendocrine cutaneous tumor with a worldwide incidence of 0.1 to 1.6 cases per 100,000 individuals annually.2 The typical patient is older than 75 years with fair skin and a history of extensive sun exposure. Immunocompromised individuals are predisposed and more susceptible to infection with the Merkel cell polyomavirus, which promotes oncogenesis in the majority of MCCs. Our patient’s history of combined variable immunodeficiency likely explains her presentation at a younger age.
The prognosis in patients with MCC is poor, with 5-year survival rates of 51% for local disease, 35% for nodal disease, and 14% for systemic metastases. Survival also is reduced in cases with head/ neck primary tumors and polyomavirus-negative tumors, as well as in immunocompromised patients.2 Treatment of resectable MCC consists of Mohs micrographic surgery or wide local excision depending on the patient’s cosmetic concerns. Radiation therapy is recommended for cases with increased risk for recurrence or positive surgical margins, as well as when additional resection is impossible. A study investigating immunotherapy with nivolumab demonstrated complete pathologic response and radiographic tumor regression in nearly half of patients when given 4 weeks prior to surgery.3
Immunohistochemistry is essential in discerning MCC from other small blue cell tumors. Most MCC cases show positive expression of neuroendocrine markers such as synaptophysin, chromogranin, and insulinomaassociated protein 1. Perinuclear dotlike staining with cytokeratin (CK) 20 (quiz image 2) commonly is seen, but up to 15% of cases may be CK20 negative. Many of these CK20-negative cases also express CK7. This tumor also may stain with paired box 5 (PAX-5), CD99, terminal deoxynucleotidyl transferase, Ber-EP4, and CD1171,4; melanoma stains (ie, human melanoma black [HMB] 45, SRYrelated HMB-box 10 [SOX-10], S-100, melanoma antigen recognized by T-cells 1 [MART-1]) should be negative. However, PAX-5 expression may be a potential pitfall given that B-cell lymphomas also would express that marker and could mimic MCC histologically. Therefore, other universal lymphoid markers such as CD45 should be ordered to rule out this entity. Even with one or a few aberrant stains, a diagnosis of MCC still can be rendered using the histomorphology and the overall staining profile.4 Of prognostic significance, p63 expression is associated with more aggressive tumors, while Bcl-2 expression is favorable, as it offers an additional targeted treatment option.5,6
Basal cell carcinoma (BCC) is linked to excessive sun exposure and is the most common skin cancer. Similar to MCC, it typically is mitotically active and hyperchromatic; however, lymphovascular invasion or metastasis almost never is observed in BCC, whereas approximately one-third of MCC cases have metastasized by the time of diagnosis. Additionally, BCC lacks the perinuclear dotlike staining seen with CK20.2,7 Features present in BCC that are unusual for MCC include peripheral nuclear palisading, mucin, and retraction artifact on paraffin-embedded sections (Figure 1).7
Leukemia cutis (or cutaneous infiltrates of leukemia) commonly displays a perivascular and periadnexal pattern in the dermis and subcutis. These infiltrates of neoplastic leukocytes can congregate into sheets, sometimes with an overlying Grenz zone, or form single-file infiltrates (Figure 2).1,4 The neoplastic cells can be monomorphic or atypical and commonly are susceptible to crush artifact.4 Although the immunohistochemical profile varies depending on the etiology of the underlying leukemia, broad hematologic markers such as CD43 and CD45 are helpful to discern these malignancies from MCC.4
Being neuroendocrine in origin, metastatic small cell carcinoma (Figure 3) strongly mimics MCC histologically and usually stains with synaptophysin, chromogranin, and insulinoma-associated protein 1. Both tumor cells typically exhibit nuclear molding and high mitotic rates. Although small cell carcinoma is more likely to stain with high-molecular-weight cytokeratins (ie, CK7), it is not uncommon for these tumors to express lowmolecular- weight cytokeratins such as CK20. Because most cases originate from the lungs, these lesions should be positive for thyroid transcription factor 1 and negative for PAX-5, whereas MCC would show the reverse for those stains.1 Ultimately, however, clinical correlation with imaging results is the single best methodology for differentiation.
Small cell melanoma, a variant of nevoid melanoma, can strongly resemble an MCC or a lymphoma. Usually located on the scalp or arising from a congenital nevus, small cell melanomas are aggressive and confer an unfavorable prognosis. Histologically, they consist of nests to sheets of atypical cells within the epidermis and dermis. These cells typically exhibit hyperchromatic nuclei, minimal cytoplasm, and frequent mitoses (Figure 4). Furthermore, the cells do not display maturation based on depth.8 These tumors usually are positive for HMB45, S-100, MART-1, SOX-10, and tyrosinase, all of which are extremely unlikely to stain an MCC.1
- Patterson JW, Hosler GA. Weedon’s Skin Pathology. 4th ed. Churchill Livingstone/Elsevier; 2016.
- Walsh NM, Cerroni L. Merkel cell carcinoma: a review. J Cutan Pathol. 2021;48:411-421.
- Topalian SL, Bhatia S, Amin A, et al. Neoadjuvant nivolumab for patients with resectable Merkel cell carcinoma in the CheckMate 358 Trial. J Clin Oncol. 2020;38:2476-2488.
- Rapini RP. Practical Dermatopathology. 3rd ed. Elsevier; 2021.
- Asioli S, Righi A, Volante M, et al. p63 expression as a new prognostic marker in Merkel cell carcinoma. Cancer. 2007;110:640-647.
- Verhaegen ME, Mangelberger D, Weick JW, et al. Merkel cell carcinoma dependence on Bcl-2 family members for survival. J Invest Dermatol. 2014;134:2241-2250.
- Le MD, O’Steen LH, Cassarino DS. A rare case of CK20/CK7 double negative Merkel cell carcinoma. Am J Dermatopathol. 2017;39:208-211.
- North JP, Bastian BC, Lazar AJ. Melanoma. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin With Clinical Correlations. 5th ed. Elsevier; 2020.
- Patterson JW, Hosler GA. Weedon’s Skin Pathology. 4th ed. Churchill Livingstone/Elsevier; 2016.
- Walsh NM, Cerroni L. Merkel cell carcinoma: a review. J Cutan Pathol. 2021;48:411-421.
- Topalian SL, Bhatia S, Amin A, et al. Neoadjuvant nivolumab for patients with resectable Merkel cell carcinoma in the CheckMate 358 Trial. J Clin Oncol. 2020;38:2476-2488.
- Rapini RP. Practical Dermatopathology. 3rd ed. Elsevier; 2021.
- Asioli S, Righi A, Volante M, et al. p63 expression as a new prognostic marker in Merkel cell carcinoma. Cancer. 2007;110:640-647.
- Verhaegen ME, Mangelberger D, Weick JW, et al. Merkel cell carcinoma dependence on Bcl-2 family members for survival. J Invest Dermatol. 2014;134:2241-2250.
- Le MD, O’Steen LH, Cassarino DS. A rare case of CK20/CK7 double negative Merkel cell carcinoma. Am J Dermatopathol. 2017;39:208-211.
- North JP, Bastian BC, Lazar AJ. Melanoma. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin With Clinical Correlations. 5th ed. Elsevier; 2020.
A 47-year-old woman with a history of combined variable immunodeficiency presented with a 2.6×2.4-cm nodule on the lateral aspect of the right calf that was first noticed 2 years prior as a smaller nodule. It increased in size and became painful to touch over the last 3 to 4 months. Following diagnostic biopsy, the nodule was removed by wide local excision and was tan-brown on gross dissection. The lesion showed dotlike perinuclear positivity with cytokeratin 20 immunostaining. Positron emission tomography–computed tomography showed no evidence of lung lesions. A complete blood cell count was within reference range.

.")