User login
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.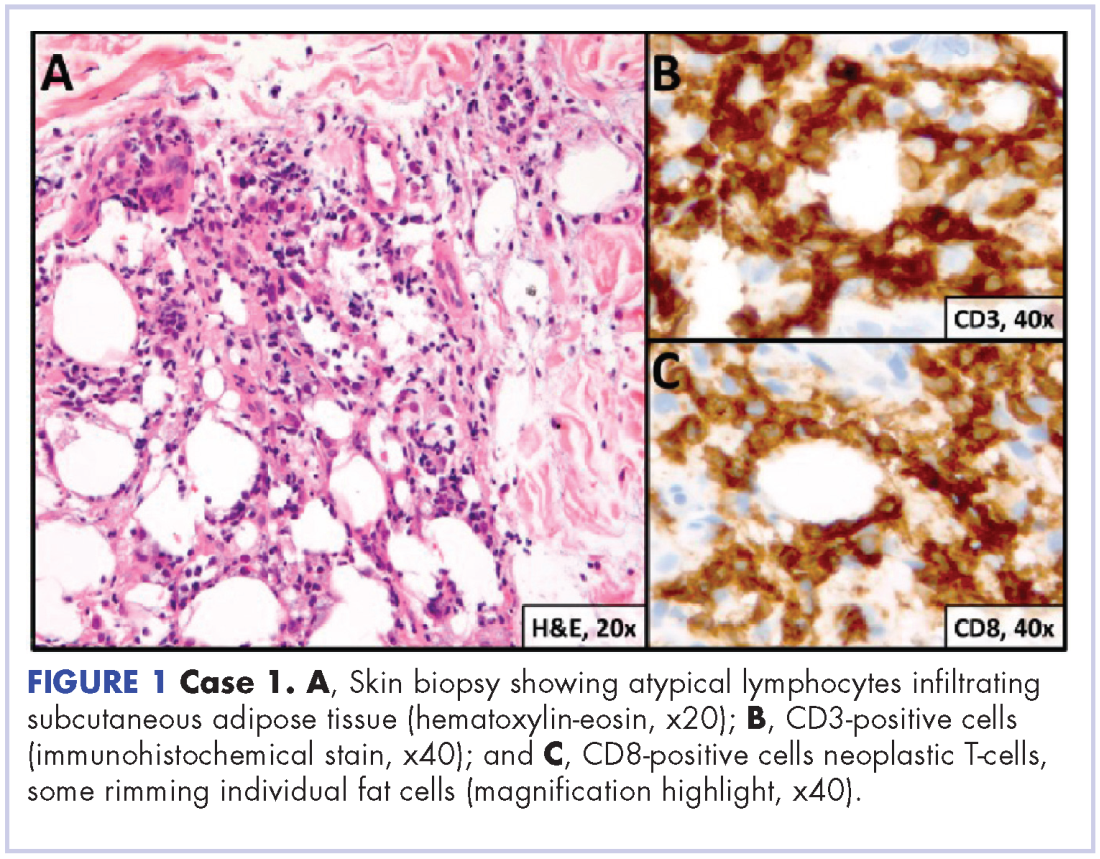
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.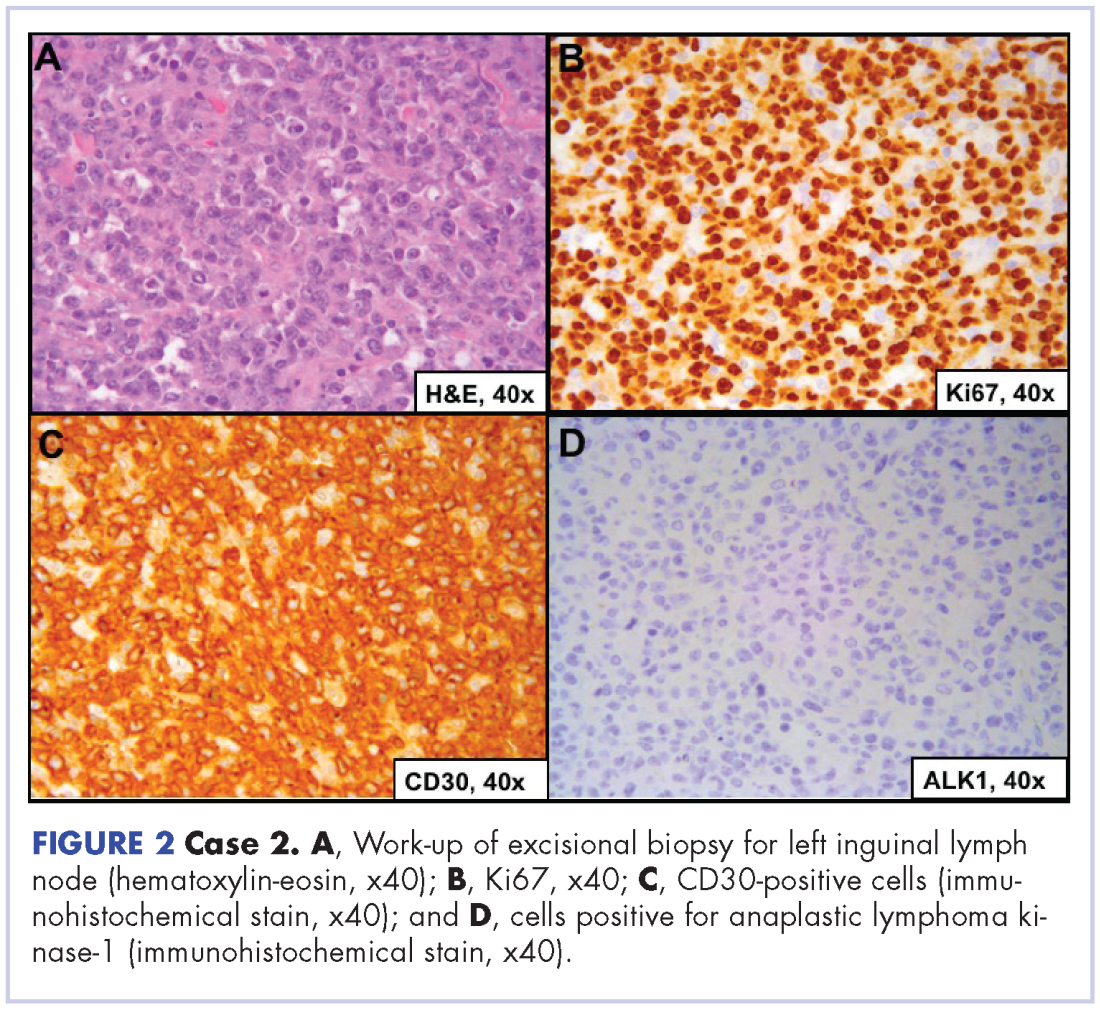
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).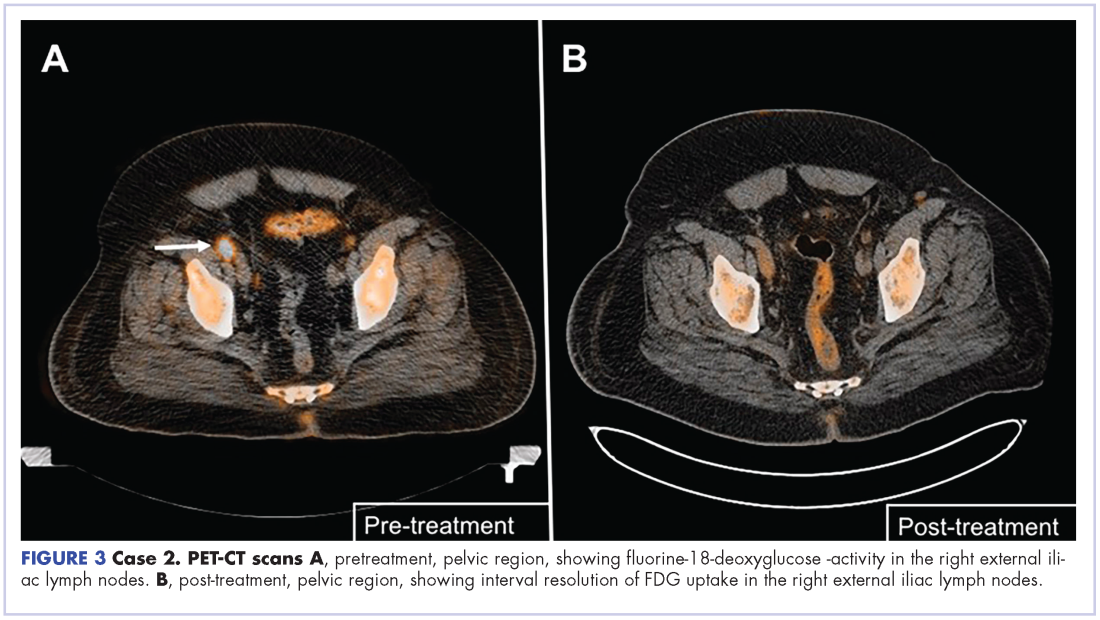
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.