User login
A 52-year-old woman presented with a 4-day history of progressive dyspnea, nonproductive cough, pleuritic chest pain, and subjective fevers. She described dyspnea at rest, which worsened with exertion. She reported no chills, night sweats, weight change, wheezing, hemoptysis, orthopnea, lower extremity edema, or nasal congestion. She also denied myalgia, arthralgia, or joint swelling. She reported no rash, itching, or peripheral lymphadenopathy. She had no seasonal allergies. She was treated for presumed bronchitis with azithromycin by her primary care provider 4 days prior to presentation but experienced progressive dyspnea.
The constellation of dry cough, fever, and dyspnea is often infectious in origin, with the nonproductive, dry cough more suggestive of a viral than bacterial syndrome. Atypical organisms such as Mycoplasma pneumoniae, Legionella pneumophila, and Chlamydia pneumoniae may also present with these symptoms. Noninfectious etiologies should also be considered, including pulmonary embolism, systemic lupus erythematosus, asbestosis, hypersensitivity pneumonitis, sarcoidosis, and lung cancer. The dyspnea at rest stands out as a worrisome feature, as it implies hypoxia; therefore, an oxygen saturation is necessary to quickly determine her peripheral oxygen saturation.
Her past medical history was notable for lung adenocarcinoma, for which she had undergone right upper lobectomy, without chemotherapy or radiation, 13 years ago without recurrence. She had no history of chronic obstructive pulmonary disease, asthma, or pneumonia, nor a family history of chronic obstructive pulmonary disease, asthma, pneumonia, or lung cancer. Her only medication was azithromycin. She drank alcohol on occasion and denied illicit drug use. Three weeks prior to admission, she began smoking 4 to 5 cigarettes per day after 13 years of abstinence. Her smoking history prior to abstinence was 1 pack per day for 20 years. She worked as a department store remodeler; she had no exposure to asbestos, mold, or water-damaged wood. She reported no recent travel, sick contacts, or exposure to animals.
A primary lung neoplasm with a pleural effusion could cause her shortness of breath and pleuritic chest pain. Her history of lung cancer at age 39 raises the possibility of recurrence. For cigarette smokers, a second lung cancer may occur many years after the first diagnosis and treatment, even if they have quit smoking. A review of her original cancer records is essential to confirm the diagnosis of pulmonary adenocarcinoma. What is now being described as pulmonary adenocarcinoma may have been a metastatic lesion arising from outside the lung. Although unlikely, a primary adenocarcinoma may remain active.
Infectious etiologies continue to merit consideration. A parapneumonic effusion from a pneumonia or an empyema are consistent with her symptoms. Systemic lupus erythematosus can cause lung disease with pleural effusions. She does exhibit dyspnea and pleurisy, which are consistent with autoimmune disease, but does not exhibit some of the more typical autoimmune symptoms such as arthralgias, joint swelling, and rash. Pneumothorax could also produce her symptoms; however, pneumothorax usually occurs spontaneously in younger patients or after trauma or a procedure. Remote right upper lobectomy would not be a cause of pneumothorax now. Her reported history makes lung disease or pneumoconiosis due to occupational exposure to mold or aspergillosis a possibility. Legionellosis, histoplasmosis, or coccidioidomycosis should be considered if she lives in or has visited a high-risk area. Pulmonary embolism remains a concern for all patients with new-onset shortness of breath. Decision support tools, such as the Wells criteria, are valuable, but the gestalt of the physician does not lag far behind in accuracy.
Cardiac disease is also in the differential. Bibasilar crackles, third heart sound gallop, and jugular vein distension would suggest heart failure. A pericardial friction rub would be highly suggestive of pericarditis. A paradoxical pulse would raise concern for pericardial tamponade. Pleurisy may be associated with a pericardial effusion, making viral pericarditis and myocarditis possibilities.
She was in moderate distress with tachypnea and increased work of breathing. Her temperature was 36.7°C, heart rate 104 beats per minute, respiratory rate 24 breaths per minute, oxygen saturation was 88% on room air, 94% on 3 liters of oxygen, and blood pressure was 147/61 mmHg. Auscultation of the lungs revealed bibasilar crackles and decreased breath sounds at the bases. She was tachycardic, with a regular rhythm and no appreciable murmurs, rubs, or gallops. There was no jugular venous distention or lower extremity edema. Her thyroid was palpable, without appreciation of nodules. Skin and musculoskeletal examinations were normal.
Unless she is immunocompromised, infection has become lower in the differential, as she is afebrile. Decreased breath sounds at the bases and bibasilar crackles may be due to pleural effusions. Congestive heart failure is a possibility, especially given her dyspnea and bibasilar crackles. Volume overload from renal failure is possible, but she does not have other signs of volume overload such as lower extremity edema or jugular venous distension. It is important to note that crackles may be due to other etiologies, including atelectasis, fibrosis, or pneumonia. Pulmonary embolism may cause hypoxia, tachycardia, and pleural effusions. Additional diseases may present similarly, including human immunodeficiency virus with Pneumocystis jirovecii, causing dyspnea, tachypnea, and tachycardia; hematologic malignancy with anemia, causing dyspnea and tachycardia; and thyrotoxic states with thyromegaly, causing dyspnea and tachycardia. Thyroid storm patients appear in distress, are tachycardic, and may have thyromegaly.
Moderate distress, increased work of breathing, tachycardia, tachypnea, and hypoxia are all worrisome signs. Her temperature is subnormal, although this may not be accurate, as oral temperatures may register lower in patients with increased respiratory rates because of increased air flow across the thermometer. Bibasilar crackles with decreased bibasilar sounds require further investigation. A complete blood count, complete metabolic profile, troponin, arterial blood gas (ABG), electrocardiogram (ECG), and chest radiograph are warranted.
Laboratory studies revealed a white blood cell count of 8600 per mm3 with 11% bands and 7.3% eosinophils, and a hemoglobin count of 15 gm/dL. Basic metabolic panel, liver function tests, coagulation panel, and urinalysis were within normal limits, including serum creatinine 0.7 mg/dL, sodium 143 mmoL/L, chloride 104 mmoL/L, bicarbonate 30 mEq/L, anion gap 9 mmoL/L, and blood urea nitrogen 12 mg/dL. Chest radiograph disclosed diffusely increased interstitial markings and a small left pleural effusion (Figure 1).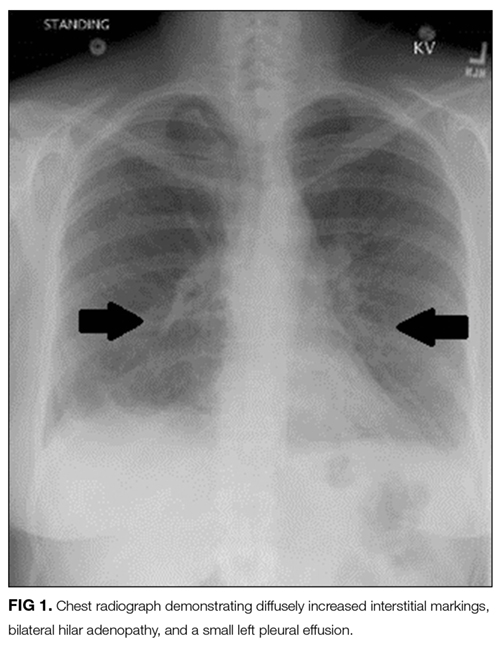
Her bandemia suggests infection. Stress can cause a leukocytosis by demargination of mature white blood cells; however, stress does not often cause immature cells such as bands to appear. Her chest radiograph with diffuse interstitial markings is consistent with a community-acquired pneumonia. Empiric antibiotic therapy should be initiated because of the possibility of community-acquired pneumonia. Recent studies demonstrate that steroids decrease mortality, the need for mechanical ventilation, and the length of stay for patients hospitalized with community-acquired pneumonia; therefore, this patient should also be treated with steroids.
Eosinophilia may be seen in drug reactions, allergies, pulmonary emboli, pleural effusions, and occasionally in malignancy. Eosinophilic pneumonia typically has the “reverse pulmonary edema” picture, with infiltrates in the periphery and not centrally, as in congestive heart failure.
A serum bicarbonate of 30 mEq/L suggests a metabolic compensation for a chronic respiratory acidosis as renal compensation, and rise in bicarbonate generally takes 3 days. She may have been hypoxic longer than her symptoms suggest.
An ABG should be ordered to determine the degree of hypoxia and whether a higher level of care is indicated. The abnormal chest radiograph, along with her hypoxia, merits a closer look at her lung parenchyma with chest computed tomography (CT). A D-dimer would be beneficial to rule out pulmonary embolism. If the D-dimer is positive, chest CT with contrast is indicated to determine if a pulmonary embolism is present. A brain natriuretic peptide would assist in the diagnosis of congestive heart failure. A sputum culture and Gram stain and respiratory viral panel may establish a pathogen for pneumonia. An ECG and troponin to rule out myocardial infarction should be performed as well.
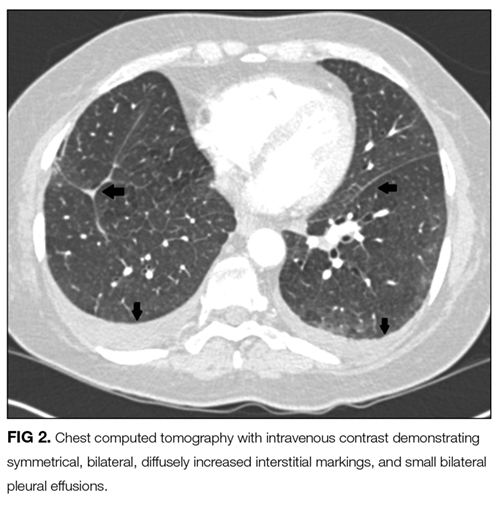
The presence of hilar and subcarinal lymph nodes expands the differential. Stage IV pulmonary sarcoid may present with diffuse infiltrates and nodes, although the acuity in this case makes it less likely. A very aggressive malignancy such as Burkitt lymphoma may have these findings. Acute viral and atypical pneumonias remain possible. Right middle lobe syndrome may cause partial collapse of the right middle lobe. Tuberculosis can be associated with right middle lobe syndrome; however, in this day and age an obstructing mass is more likely the cause. Pulmonary disease, such as cryptogenic organizing pneumonia, idiopathic pulmonary fibrosis, and interstitial lung disease, should be considered in patients with pneumonia unresponsive to antibiotics. Lung biopsy and bronchoalveolar lavage (BAL) would help make the diagnosis and should be the next step, unless her degree of hypoxia is prohibitive. Similarly, thoracentesis with analysis of the pleural fluid for cell count, Gram stain, and culture may help make the diagnosis. Thoracentesis should be done with fluoroscopic guidance, given the risk of pneumothorax, which would further compromise her tenuous respiratory status.
Thoracentesis was attempted, but the pleural effusion was too small to provide a sample. Subsequent serum blood counts with differential showed an increased eosinophilia to 20% and resolved bandemia. Upon further questioning, she recalled several months of extensive, daily, fine-dust exposure from demolition during the remodeling of a new building.
Hypereosinophilia and pulmonary infiltrates narrow the differential considerably to include asthma; parasitic infection, such as the pulmonary phase of ascariasis; exposure, such as to dust, cigarettes, or asbestosis; or hypereosinophilic syndromes characterized by peripheral eosinophilia, along with a tissue eosinophilia, causing organ dysfunction. Idiopathic hypereosinophilic syndrome, a hypereosinophilic syndrome of unknown etiology despite extensive diagnostic testing, is rare, and eosinophilic leukemia even rarer. Her history strongly suggests exposure. Many eosinophilic diseases respond rapidly to steroids, and response to treatment would help narrow the diagnosis. If she does not respond to steroids, a lung and/or bone marrow biopsy would be the next step.
A BAL of the right middle lobe revealed 51% eosinophils, 3% neutrophils, 15% macrophages, and 28% lymphocytes. Gram stain, as well as cultures for bacteria, acid fast bacilli, fungus, herpes simplex virus, and cytomegalovirus cultures, were negative. Transbronchial lung biopsy revealed focal interstitial fibrosis and inflammation, without evidence of infection.
Eosinophils are primarily located in tissues; therefore, peripheral blood eosinophil counts often underestimate the degree of infiltration into end organs such as the lung. With 50% eosinophils, her BAL reflects this. Mold, fungus, chemical, and particle exposure could produce an eosinophilic BAL. She does not appear to be at risk for parasitic exposure. Eosinophilic granulomatosis (previously known as Churg-Strauss) is a consideration, but the lack of signs of vasculitis and wheezing make this less likely. A negative antineutrophil cytoplasmic antibody may provide reassurance. “Fine dust exposure” is consistent with environmental exposure but not a specific antigen. Steroids provide a brisk eosinophil reduction and are appropriate for this patient. There is the possibility of missing infectious or parasitic etiologies; therefore, a culture of BAL fluid should be sent.
Eosinophilic infiltration may lead to fibrosis, as was found on the lung biopsy. She should be counseled to avoid “fine dust exposure” in the future. Follow-up lung imaging and pulmonary function tests (PFTs) should be performed once her acute illness resolves. She should be strongly urged not to smoke tobacco. Interestingly, there are reports that ex-smokers who restart smoking have an increased risk of eosinophilic pneumonia, but in this case dust exposure is the more likely etiology.
She was diagnosed with acute eosinophilic pneumonia (AEP). Antibiotics were discontinued, and oral prednisone was initiated at 40 mg daily, with a brisk response and resolution of her dyspnea. She was discharged with a 6-week prednisone taper. She had no cough, dyspnea, chest pain, or fevers at her follow-up 14 days after discharge. On a 6-week, postdischarge phone call, she continued to report no symptoms, and she maintained abstinence from cigarette smoking.
This case highlights that the very best test in any medical situation is a thorough, detailed history and physical examination. A comprehensive history with physical examination is noninvasive, safe, and cheap. Had the history of fine-dust exposure been known, it is likely that a great deal of testing and money would have been saved. The patient would have been diagnosed and treated earlier, and suffered less.
COMMENTARY
First described in 1989,1,2 AEP is an uncommon cause of acute respiratory failure. Cases have been reported throughout the world, including in the United States, Belgium, Japan, and Iraq.2,3 AEP is an acute febrile illness with cough, chest pain, and dyspnea for fewer than 7 days, diffuse pulmonary infiltrates on chest radiograph, hypoxemia, no history of asthma or atopic disease, no infection, and greater than 25% eosinophils on a BAL.1,3 Physical examination typically reveals fever, tachypnea, and crackles on auscultation.1 Peripheral blood eosinophilia is inconsistently seen at presentation but generally observed as the disease progresses.1 Peripheral eosinophilia at presentation is positively correlated with a milder course of AEP, including higher oxygen saturation and fewer intensive care admissions.4 Acute respiratory failure in AEP progresses rapidly, often within hours.1 Delayed recognition of AEP may lead to respiratory failure, requiring intubation, and even to death.1
Reticular markings with Kerley-B lines, mixed reticular and alveolar infiltrates, and pleural effusions are usually found on chest radiography.1 Bilateral areas of ground-glass attenuation, interlobular septal thickening, bronchovascular bundle thickening, and pleural effusions are seen on chest CT.5 Marked eosinophilic infiltration of the interstitium and alveolar spaces, as well as diffuse alveolar damage with hyaline membrane fibroblast proliferation and inflammatory cells, are present on lung biopsy.1 Restriction with impaired diffusion capacity is found on PFTs. However, PFTs return to normal after recovery.1
AEP is distinguished from other pulmonary diseases by BAL, lung biopsy, symptoms, symptom course, and/or radiographically. AEP is often misdiagnosed as severe community-acquired pneumonia and/or acute respiratory distress syndrome, as AEP tends to occur in previously healthy individuals who have diffuse infiltrates on chest radiograph, fevers, and acute, often severe, respiratory symptoms.1-3 Other eosinophilic lung diseases to rule out include simple pulmonary eosinophilia, chronic eosinophilic pneumonia, eosinophilic granulomatosis with polyangitis (Churg-Strauss), idiopathic hypereosinophilic syndrome, infection, and drug reactions.1,3,5 Simple eosinophilic pneumonia is characterized by no symptoms or very mild pulmonary symptoms and transient patchy infiltrates on radiography.3,5 Patients with simple pulmonary eosinophilia do not have interlobular septal thickening, thickening of the bronchovascular bundles, or pleural effusions radiographically, as seen with AEP.5 Chronic eosinophilic pneumonia is subacute, with respiratory symptoms of more than 3 months in duration, in contrast with the 7 days of respiratory symptoms for AEP, and is also not associated with interlobular septal thickening, thickening of the bronchovascular bundles, or pleural effusions on radiography.3,5 Unlike AEP, chronic eosinophilic pneumonia often recurs after the course of steroids has ended.3 In contrast with AEP, eosinophilic granulomatosis with polyangitis is associated with concomitant asthma and the involvement of nonpulmonary organs.3 Idiopathic hypereosinophilic syndrome is characterized by extremely high peripheral eosinophilia and by eosinophilic involvement of multiple organs, and it requires chronic steroid use.3 Patients with allergic bronchopulmonary aspergillosis (ABPA), in contrast with AEP, typically have steroid-dependent asthma and chronic respiratory symptoms.3 ABPA also differs from AEP in that radiographic infiltrates are localized and transient, and the syndrome may relapse after steroid treatment.3 Other infectious etiologies that may present similarly to AEP include invasive pulmonary aspergillosis, pulmonary coccidiodomycosis, Pneumocystis jioveri pneumonia, pulmonary toxocariasis, pulmonary filariasis, paragonimiasis, and Loeffler syndrome (pneumonia due to Strongyloides, Ascaris, or hookworms), highlighting the importance of a thorough travel and exposure history.1,3 Several drugs may cause eosinophilic lung disease, including nitrofurantoin, tetracyclines, phenytoin, L-tryptophan, acetaminophen, ampicillin, heroin, and cocaine, which necessitates a thorough review of medication and illegal drug use.3
Steroids and supportive care are the treatment of choice for AEP, although spontaneous resolution has been seen.1,3 Significant clinical improvement occurs within 24 to 48 hours of steroid initiation.1,3 Optimal dose and duration of therapy have not been determined; however, methylprednisolone 125 mg intravenously every 6 hours until improvement is an often-used option.1 Tapers vary from 2 to 12 weeks with no difference in outcome.1-3 AEP does not recur after appropriate treatment with steroids.1,3
Little is known about the etiology of AEP. It usually occurs in young, healthy individuals and is presumed to be an unusual, acute hypersensitivity reaction to an inhaled allergen.1 A report of 18 US soldiers deployed in or near Iraq proposed dust exposure and cigarette or cigar smoking as a cause of AEP.2 Similar to our patient’s fine-dust exposure and recent onset of cigarette smoking, the soldiers were exposed to the dusty, arid environment for at least 1 day and had been smoking for at least 1 month.2 The authors proposed that small dust particles irritate alveoli, stimulating eosinophils, which are exacerbated by the onset of smoking. Alternatively, cigarette smoke may prime the lung such that dust triggers an inflammatory cascade, resulting in AEP.
TEACHING POINTS
- With the potential for the rapid progression of respiratory failure, it is imperative that the diagnosis of AEP be considered for a patient with diffuse infiltrates on a chest radiograph and acute respiratory failure of unknown cause.
- A thorough history of exposure is key to including AEP in the differential of acute pulmonary disease, with recent-onset cigarette smoking and dust exposure.
- The rapid initiation of steroids leads to a full recovery without recurrence and may be life-saving in AEP.
Disclosure
The authors report no conflicts of interest.
1. Allen J. Acute eosinophilic pneumonia. Semin Respir Crit Care Med. 2006;27:142-147. PubMed
2. Shorr AF, Scoville SL, Cersovsky SB, et al. Acute eosinophilic pneumonia among US military personnel deployed in or near Iraq. JAMA. 2004;292:2997-3005. PubMed
3. Pope-Harman AL, Davis WB, Allen ED, Christoforidis AJ, Allen JN. Acute eosinophilic pneumonia. A summary of 15 cases and review of the literature. Medicine (Baltimore). 1996;75(6):334-342. PubMed
4. Jhun BW, Kim SJ, Kim K, Lee JE. Clinical implications of initial peripheral eosinophilia in acute eosinophilic pneumonia. Respirology. 2014;19:1059-1065. PubMed
5. Daimon T, Johkoh T, Sumikawa H, et al. Acute eosinophilic pneumonia: Thin-section CT findings in 29 patients. Eur J Radiol. 2008;65:462-467. PubMed
A 52-year-old woman presented with a 4-day history of progressive dyspnea, nonproductive cough, pleuritic chest pain, and subjective fevers. She described dyspnea at rest, which worsened with exertion. She reported no chills, night sweats, weight change, wheezing, hemoptysis, orthopnea, lower extremity edema, or nasal congestion. She also denied myalgia, arthralgia, or joint swelling. She reported no rash, itching, or peripheral lymphadenopathy. She had no seasonal allergies. She was treated for presumed bronchitis with azithromycin by her primary care provider 4 days prior to presentation but experienced progressive dyspnea.
The constellation of dry cough, fever, and dyspnea is often infectious in origin, with the nonproductive, dry cough more suggestive of a viral than bacterial syndrome. Atypical organisms such as Mycoplasma pneumoniae, Legionella pneumophila, and Chlamydia pneumoniae may also present with these symptoms. Noninfectious etiologies should also be considered, including pulmonary embolism, systemic lupus erythematosus, asbestosis, hypersensitivity pneumonitis, sarcoidosis, and lung cancer. The dyspnea at rest stands out as a worrisome feature, as it implies hypoxia; therefore, an oxygen saturation is necessary to quickly determine her peripheral oxygen saturation.
Her past medical history was notable for lung adenocarcinoma, for which she had undergone right upper lobectomy, without chemotherapy or radiation, 13 years ago without recurrence. She had no history of chronic obstructive pulmonary disease, asthma, or pneumonia, nor a family history of chronic obstructive pulmonary disease, asthma, pneumonia, or lung cancer. Her only medication was azithromycin. She drank alcohol on occasion and denied illicit drug use. Three weeks prior to admission, she began smoking 4 to 5 cigarettes per day after 13 years of abstinence. Her smoking history prior to abstinence was 1 pack per day for 20 years. She worked as a department store remodeler; she had no exposure to asbestos, mold, or water-damaged wood. She reported no recent travel, sick contacts, or exposure to animals.
A primary lung neoplasm with a pleural effusion could cause her shortness of breath and pleuritic chest pain. Her history of lung cancer at age 39 raises the possibility of recurrence. For cigarette smokers, a second lung cancer may occur many years after the first diagnosis and treatment, even if they have quit smoking. A review of her original cancer records is essential to confirm the diagnosis of pulmonary adenocarcinoma. What is now being described as pulmonary adenocarcinoma may have been a metastatic lesion arising from outside the lung. Although unlikely, a primary adenocarcinoma may remain active.
Infectious etiologies continue to merit consideration. A parapneumonic effusion from a pneumonia or an empyema are consistent with her symptoms. Systemic lupus erythematosus can cause lung disease with pleural effusions. She does exhibit dyspnea and pleurisy, which are consistent with autoimmune disease, but does not exhibit some of the more typical autoimmune symptoms such as arthralgias, joint swelling, and rash. Pneumothorax could also produce her symptoms; however, pneumothorax usually occurs spontaneously in younger patients or after trauma or a procedure. Remote right upper lobectomy would not be a cause of pneumothorax now. Her reported history makes lung disease or pneumoconiosis due to occupational exposure to mold or aspergillosis a possibility. Legionellosis, histoplasmosis, or coccidioidomycosis should be considered if she lives in or has visited a high-risk area. Pulmonary embolism remains a concern for all patients with new-onset shortness of breath. Decision support tools, such as the Wells criteria, are valuable, but the gestalt of the physician does not lag far behind in accuracy.
Cardiac disease is also in the differential. Bibasilar crackles, third heart sound gallop, and jugular vein distension would suggest heart failure. A pericardial friction rub would be highly suggestive of pericarditis. A paradoxical pulse would raise concern for pericardial tamponade. Pleurisy may be associated with a pericardial effusion, making viral pericarditis and myocarditis possibilities.
She was in moderate distress with tachypnea and increased work of breathing. Her temperature was 36.7°C, heart rate 104 beats per minute, respiratory rate 24 breaths per minute, oxygen saturation was 88% on room air, 94% on 3 liters of oxygen, and blood pressure was 147/61 mmHg. Auscultation of the lungs revealed bibasilar crackles and decreased breath sounds at the bases. She was tachycardic, with a regular rhythm and no appreciable murmurs, rubs, or gallops. There was no jugular venous distention or lower extremity edema. Her thyroid was palpable, without appreciation of nodules. Skin and musculoskeletal examinations were normal.
Unless she is immunocompromised, infection has become lower in the differential, as she is afebrile. Decreased breath sounds at the bases and bibasilar crackles may be due to pleural effusions. Congestive heart failure is a possibility, especially given her dyspnea and bibasilar crackles. Volume overload from renal failure is possible, but she does not have other signs of volume overload such as lower extremity edema or jugular venous distension. It is important to note that crackles may be due to other etiologies, including atelectasis, fibrosis, or pneumonia. Pulmonary embolism may cause hypoxia, tachycardia, and pleural effusions. Additional diseases may present similarly, including human immunodeficiency virus with Pneumocystis jirovecii, causing dyspnea, tachypnea, and tachycardia; hematologic malignancy with anemia, causing dyspnea and tachycardia; and thyrotoxic states with thyromegaly, causing dyspnea and tachycardia. Thyroid storm patients appear in distress, are tachycardic, and may have thyromegaly.
Moderate distress, increased work of breathing, tachycardia, tachypnea, and hypoxia are all worrisome signs. Her temperature is subnormal, although this may not be accurate, as oral temperatures may register lower in patients with increased respiratory rates because of increased air flow across the thermometer. Bibasilar crackles with decreased bibasilar sounds require further investigation. A complete blood count, complete metabolic profile, troponin, arterial blood gas (ABG), electrocardiogram (ECG), and chest radiograph are warranted.
Laboratory studies revealed a white blood cell count of 8600 per mm3 with 11% bands and 7.3% eosinophils, and a hemoglobin count of 15 gm/dL. Basic metabolic panel, liver function tests, coagulation panel, and urinalysis were within normal limits, including serum creatinine 0.7 mg/dL, sodium 143 mmoL/L, chloride 104 mmoL/L, bicarbonate 30 mEq/L, anion gap 9 mmoL/L, and blood urea nitrogen 12 mg/dL. Chest radiograph disclosed diffusely increased interstitial markings and a small left pleural effusion (Figure 1).
Her bandemia suggests infection. Stress can cause a leukocytosis by demargination of mature white blood cells; however, stress does not often cause immature cells such as bands to appear. Her chest radiograph with diffuse interstitial markings is consistent with a community-acquired pneumonia. Empiric antibiotic therapy should be initiated because of the possibility of community-acquired pneumonia. Recent studies demonstrate that steroids decrease mortality, the need for mechanical ventilation, and the length of stay for patients hospitalized with community-acquired pneumonia; therefore, this patient should also be treated with steroids.
Eosinophilia may be seen in drug reactions, allergies, pulmonary emboli, pleural effusions, and occasionally in malignancy. Eosinophilic pneumonia typically has the “reverse pulmonary edema” picture, with infiltrates in the periphery and not centrally, as in congestive heart failure.
A serum bicarbonate of 30 mEq/L suggests a metabolic compensation for a chronic respiratory acidosis as renal compensation, and rise in bicarbonate generally takes 3 days. She may have been hypoxic longer than her symptoms suggest.
An ABG should be ordered to determine the degree of hypoxia and whether a higher level of care is indicated. The abnormal chest radiograph, along with her hypoxia, merits a closer look at her lung parenchyma with chest computed tomography (CT). A D-dimer would be beneficial to rule out pulmonary embolism. If the D-dimer is positive, chest CT with contrast is indicated to determine if a pulmonary embolism is present. A brain natriuretic peptide would assist in the diagnosis of congestive heart failure. A sputum culture and Gram stain and respiratory viral panel may establish a pathogen for pneumonia. An ECG and troponin to rule out myocardial infarction should be performed as well.
The presence of hilar and subcarinal lymph nodes expands the differential. Stage IV pulmonary sarcoid may present with diffuse infiltrates and nodes, although the acuity in this case makes it less likely. A very aggressive malignancy such as Burkitt lymphoma may have these findings. Acute viral and atypical pneumonias remain possible. Right middle lobe syndrome may cause partial collapse of the right middle lobe. Tuberculosis can be associated with right middle lobe syndrome; however, in this day and age an obstructing mass is more likely the cause. Pulmonary disease, such as cryptogenic organizing pneumonia, idiopathic pulmonary fibrosis, and interstitial lung disease, should be considered in patients with pneumonia unresponsive to antibiotics. Lung biopsy and bronchoalveolar lavage (BAL) would help make the diagnosis and should be the next step, unless her degree of hypoxia is prohibitive. Similarly, thoracentesis with analysis of the pleural fluid for cell count, Gram stain, and culture may help make the diagnosis. Thoracentesis should be done with fluoroscopic guidance, given the risk of pneumothorax, which would further compromise her tenuous respiratory status.
Thoracentesis was attempted, but the pleural effusion was too small to provide a sample. Subsequent serum blood counts with differential showed an increased eosinophilia to 20% and resolved bandemia. Upon further questioning, she recalled several months of extensive, daily, fine-dust exposure from demolition during the remodeling of a new building.
Hypereosinophilia and pulmonary infiltrates narrow the differential considerably to include asthma; parasitic infection, such as the pulmonary phase of ascariasis; exposure, such as to dust, cigarettes, or asbestosis; or hypereosinophilic syndromes characterized by peripheral eosinophilia, along with a tissue eosinophilia, causing organ dysfunction. Idiopathic hypereosinophilic syndrome, a hypereosinophilic syndrome of unknown etiology despite extensive diagnostic testing, is rare, and eosinophilic leukemia even rarer. Her history strongly suggests exposure. Many eosinophilic diseases respond rapidly to steroids, and response to treatment would help narrow the diagnosis. If she does not respond to steroids, a lung and/or bone marrow biopsy would be the next step.
A BAL of the right middle lobe revealed 51% eosinophils, 3% neutrophils, 15% macrophages, and 28% lymphocytes. Gram stain, as well as cultures for bacteria, acid fast bacilli, fungus, herpes simplex virus, and cytomegalovirus cultures, were negative. Transbronchial lung biopsy revealed focal interstitial fibrosis and inflammation, without evidence of infection.
Eosinophils are primarily located in tissues; therefore, peripheral blood eosinophil counts often underestimate the degree of infiltration into end organs such as the lung. With 50% eosinophils, her BAL reflects this. Mold, fungus, chemical, and particle exposure could produce an eosinophilic BAL. She does not appear to be at risk for parasitic exposure. Eosinophilic granulomatosis (previously known as Churg-Strauss) is a consideration, but the lack of signs of vasculitis and wheezing make this less likely. A negative antineutrophil cytoplasmic antibody may provide reassurance. “Fine dust exposure” is consistent with environmental exposure but not a specific antigen. Steroids provide a brisk eosinophil reduction and are appropriate for this patient. There is the possibility of missing infectious or parasitic etiologies; therefore, a culture of BAL fluid should be sent.
Eosinophilic infiltration may lead to fibrosis, as was found on the lung biopsy. She should be counseled to avoid “fine dust exposure” in the future. Follow-up lung imaging and pulmonary function tests (PFTs) should be performed once her acute illness resolves. She should be strongly urged not to smoke tobacco. Interestingly, there are reports that ex-smokers who restart smoking have an increased risk of eosinophilic pneumonia, but in this case dust exposure is the more likely etiology.
She was diagnosed with acute eosinophilic pneumonia (AEP). Antibiotics were discontinued, and oral prednisone was initiated at 40 mg daily, with a brisk response and resolution of her dyspnea. She was discharged with a 6-week prednisone taper. She had no cough, dyspnea, chest pain, or fevers at her follow-up 14 days after discharge. On a 6-week, postdischarge phone call, she continued to report no symptoms, and she maintained abstinence from cigarette smoking.
This case highlights that the very best test in any medical situation is a thorough, detailed history and physical examination. A comprehensive history with physical examination is noninvasive, safe, and cheap. Had the history of fine-dust exposure been known, it is likely that a great deal of testing and money would have been saved. The patient would have been diagnosed and treated earlier, and suffered less.
COMMENTARY
First described in 1989,1,2 AEP is an uncommon cause of acute respiratory failure. Cases have been reported throughout the world, including in the United States, Belgium, Japan, and Iraq.2,3 AEP is an acute febrile illness with cough, chest pain, and dyspnea for fewer than 7 days, diffuse pulmonary infiltrates on chest radiograph, hypoxemia, no history of asthma or atopic disease, no infection, and greater than 25% eosinophils on a BAL.1,3 Physical examination typically reveals fever, tachypnea, and crackles on auscultation.1 Peripheral blood eosinophilia is inconsistently seen at presentation but generally observed as the disease progresses.1 Peripheral eosinophilia at presentation is positively correlated with a milder course of AEP, including higher oxygen saturation and fewer intensive care admissions.4 Acute respiratory failure in AEP progresses rapidly, often within hours.1 Delayed recognition of AEP may lead to respiratory failure, requiring intubation, and even to death.1
Reticular markings with Kerley-B lines, mixed reticular and alveolar infiltrates, and pleural effusions are usually found on chest radiography.1 Bilateral areas of ground-glass attenuation, interlobular septal thickening, bronchovascular bundle thickening, and pleural effusions are seen on chest CT.5 Marked eosinophilic infiltration of the interstitium and alveolar spaces, as well as diffuse alveolar damage with hyaline membrane fibroblast proliferation and inflammatory cells, are present on lung biopsy.1 Restriction with impaired diffusion capacity is found on PFTs. However, PFTs return to normal after recovery.1
AEP is distinguished from other pulmonary diseases by BAL, lung biopsy, symptoms, symptom course, and/or radiographically. AEP is often misdiagnosed as severe community-acquired pneumonia and/or acute respiratory distress syndrome, as AEP tends to occur in previously healthy individuals who have diffuse infiltrates on chest radiograph, fevers, and acute, often severe, respiratory symptoms.1-3 Other eosinophilic lung diseases to rule out include simple pulmonary eosinophilia, chronic eosinophilic pneumonia, eosinophilic granulomatosis with polyangitis (Churg-Strauss), idiopathic hypereosinophilic syndrome, infection, and drug reactions.1,3,5 Simple eosinophilic pneumonia is characterized by no symptoms or very mild pulmonary symptoms and transient patchy infiltrates on radiography.3,5 Patients with simple pulmonary eosinophilia do not have interlobular septal thickening, thickening of the bronchovascular bundles, or pleural effusions radiographically, as seen with AEP.5 Chronic eosinophilic pneumonia is subacute, with respiratory symptoms of more than 3 months in duration, in contrast with the 7 days of respiratory symptoms for AEP, and is also not associated with interlobular septal thickening, thickening of the bronchovascular bundles, or pleural effusions on radiography.3,5 Unlike AEP, chronic eosinophilic pneumonia often recurs after the course of steroids has ended.3 In contrast with AEP, eosinophilic granulomatosis with polyangitis is associated with concomitant asthma and the involvement of nonpulmonary organs.3 Idiopathic hypereosinophilic syndrome is characterized by extremely high peripheral eosinophilia and by eosinophilic involvement of multiple organs, and it requires chronic steroid use.3 Patients with allergic bronchopulmonary aspergillosis (ABPA), in contrast with AEP, typically have steroid-dependent asthma and chronic respiratory symptoms.3 ABPA also differs from AEP in that radiographic infiltrates are localized and transient, and the syndrome may relapse after steroid treatment.3 Other infectious etiologies that may present similarly to AEP include invasive pulmonary aspergillosis, pulmonary coccidiodomycosis, Pneumocystis jioveri pneumonia, pulmonary toxocariasis, pulmonary filariasis, paragonimiasis, and Loeffler syndrome (pneumonia due to Strongyloides, Ascaris, or hookworms), highlighting the importance of a thorough travel and exposure history.1,3 Several drugs may cause eosinophilic lung disease, including nitrofurantoin, tetracyclines, phenytoin, L-tryptophan, acetaminophen, ampicillin, heroin, and cocaine, which necessitates a thorough review of medication and illegal drug use.3
Steroids and supportive care are the treatment of choice for AEP, although spontaneous resolution has been seen.1,3 Significant clinical improvement occurs within 24 to 48 hours of steroid initiation.1,3 Optimal dose and duration of therapy have not been determined; however, methylprednisolone 125 mg intravenously every 6 hours until improvement is an often-used option.1 Tapers vary from 2 to 12 weeks with no difference in outcome.1-3 AEP does not recur after appropriate treatment with steroids.1,3
Little is known about the etiology of AEP. It usually occurs in young, healthy individuals and is presumed to be an unusual, acute hypersensitivity reaction to an inhaled allergen.1 A report of 18 US soldiers deployed in or near Iraq proposed dust exposure and cigarette or cigar smoking as a cause of AEP.2 Similar to our patient’s fine-dust exposure and recent onset of cigarette smoking, the soldiers were exposed to the dusty, arid environment for at least 1 day and had been smoking for at least 1 month.2 The authors proposed that small dust particles irritate alveoli, stimulating eosinophils, which are exacerbated by the onset of smoking. Alternatively, cigarette smoke may prime the lung such that dust triggers an inflammatory cascade, resulting in AEP.
TEACHING POINTS
- With the potential for the rapid progression of respiratory failure, it is imperative that the diagnosis of AEP be considered for a patient with diffuse infiltrates on a chest radiograph and acute respiratory failure of unknown cause.
- A thorough history of exposure is key to including AEP in the differential of acute pulmonary disease, with recent-onset cigarette smoking and dust exposure.
- The rapid initiation of steroids leads to a full recovery without recurrence and may be life-saving in AEP.
Disclosure
The authors report no conflicts of interest.
A 52-year-old woman presented with a 4-day history of progressive dyspnea, nonproductive cough, pleuritic chest pain, and subjective fevers. She described dyspnea at rest, which worsened with exertion. She reported no chills, night sweats, weight change, wheezing, hemoptysis, orthopnea, lower extremity edema, or nasal congestion. She also denied myalgia, arthralgia, or joint swelling. She reported no rash, itching, or peripheral lymphadenopathy. She had no seasonal allergies. She was treated for presumed bronchitis with azithromycin by her primary care provider 4 days prior to presentation but experienced progressive dyspnea.
The constellation of dry cough, fever, and dyspnea is often infectious in origin, with the nonproductive, dry cough more suggestive of a viral than bacterial syndrome. Atypical organisms such as Mycoplasma pneumoniae, Legionella pneumophila, and Chlamydia pneumoniae may also present with these symptoms. Noninfectious etiologies should also be considered, including pulmonary embolism, systemic lupus erythematosus, asbestosis, hypersensitivity pneumonitis, sarcoidosis, and lung cancer. The dyspnea at rest stands out as a worrisome feature, as it implies hypoxia; therefore, an oxygen saturation is necessary to quickly determine her peripheral oxygen saturation.
Her past medical history was notable for lung adenocarcinoma, for which she had undergone right upper lobectomy, without chemotherapy or radiation, 13 years ago without recurrence. She had no history of chronic obstructive pulmonary disease, asthma, or pneumonia, nor a family history of chronic obstructive pulmonary disease, asthma, pneumonia, or lung cancer. Her only medication was azithromycin. She drank alcohol on occasion and denied illicit drug use. Three weeks prior to admission, she began smoking 4 to 5 cigarettes per day after 13 years of abstinence. Her smoking history prior to abstinence was 1 pack per day for 20 years. She worked as a department store remodeler; she had no exposure to asbestos, mold, or water-damaged wood. She reported no recent travel, sick contacts, or exposure to animals.
A primary lung neoplasm with a pleural effusion could cause her shortness of breath and pleuritic chest pain. Her history of lung cancer at age 39 raises the possibility of recurrence. For cigarette smokers, a second lung cancer may occur many years after the first diagnosis and treatment, even if they have quit smoking. A review of her original cancer records is essential to confirm the diagnosis of pulmonary adenocarcinoma. What is now being described as pulmonary adenocarcinoma may have been a metastatic lesion arising from outside the lung. Although unlikely, a primary adenocarcinoma may remain active.
Infectious etiologies continue to merit consideration. A parapneumonic effusion from a pneumonia or an empyema are consistent with her symptoms. Systemic lupus erythematosus can cause lung disease with pleural effusions. She does exhibit dyspnea and pleurisy, which are consistent with autoimmune disease, but does not exhibit some of the more typical autoimmune symptoms such as arthralgias, joint swelling, and rash. Pneumothorax could also produce her symptoms; however, pneumothorax usually occurs spontaneously in younger patients or after trauma or a procedure. Remote right upper lobectomy would not be a cause of pneumothorax now. Her reported history makes lung disease or pneumoconiosis due to occupational exposure to mold or aspergillosis a possibility. Legionellosis, histoplasmosis, or coccidioidomycosis should be considered if she lives in or has visited a high-risk area. Pulmonary embolism remains a concern for all patients with new-onset shortness of breath. Decision support tools, such as the Wells criteria, are valuable, but the gestalt of the physician does not lag far behind in accuracy.
Cardiac disease is also in the differential. Bibasilar crackles, third heart sound gallop, and jugular vein distension would suggest heart failure. A pericardial friction rub would be highly suggestive of pericarditis. A paradoxical pulse would raise concern for pericardial tamponade. Pleurisy may be associated with a pericardial effusion, making viral pericarditis and myocarditis possibilities.
She was in moderate distress with tachypnea and increased work of breathing. Her temperature was 36.7°C, heart rate 104 beats per minute, respiratory rate 24 breaths per minute, oxygen saturation was 88% on room air, 94% on 3 liters of oxygen, and blood pressure was 147/61 mmHg. Auscultation of the lungs revealed bibasilar crackles and decreased breath sounds at the bases. She was tachycardic, with a regular rhythm and no appreciable murmurs, rubs, or gallops. There was no jugular venous distention or lower extremity edema. Her thyroid was palpable, without appreciation of nodules. Skin and musculoskeletal examinations were normal.
Unless she is immunocompromised, infection has become lower in the differential, as she is afebrile. Decreased breath sounds at the bases and bibasilar crackles may be due to pleural effusions. Congestive heart failure is a possibility, especially given her dyspnea and bibasilar crackles. Volume overload from renal failure is possible, but she does not have other signs of volume overload such as lower extremity edema or jugular venous distension. It is important to note that crackles may be due to other etiologies, including atelectasis, fibrosis, or pneumonia. Pulmonary embolism may cause hypoxia, tachycardia, and pleural effusions. Additional diseases may present similarly, including human immunodeficiency virus with Pneumocystis jirovecii, causing dyspnea, tachypnea, and tachycardia; hematologic malignancy with anemia, causing dyspnea and tachycardia; and thyrotoxic states with thyromegaly, causing dyspnea and tachycardia. Thyroid storm patients appear in distress, are tachycardic, and may have thyromegaly.
Moderate distress, increased work of breathing, tachycardia, tachypnea, and hypoxia are all worrisome signs. Her temperature is subnormal, although this may not be accurate, as oral temperatures may register lower in patients with increased respiratory rates because of increased air flow across the thermometer. Bibasilar crackles with decreased bibasilar sounds require further investigation. A complete blood count, complete metabolic profile, troponin, arterial blood gas (ABG), electrocardiogram (ECG), and chest radiograph are warranted.
Laboratory studies revealed a white blood cell count of 8600 per mm3 with 11% bands and 7.3% eosinophils, and a hemoglobin count of 15 gm/dL. Basic metabolic panel, liver function tests, coagulation panel, and urinalysis were within normal limits, including serum creatinine 0.7 mg/dL, sodium 143 mmoL/L, chloride 104 mmoL/L, bicarbonate 30 mEq/L, anion gap 9 mmoL/L, and blood urea nitrogen 12 mg/dL. Chest radiograph disclosed diffusely increased interstitial markings and a small left pleural effusion (Figure 1).
Her bandemia suggests infection. Stress can cause a leukocytosis by demargination of mature white blood cells; however, stress does not often cause immature cells such as bands to appear. Her chest radiograph with diffuse interstitial markings is consistent with a community-acquired pneumonia. Empiric antibiotic therapy should be initiated because of the possibility of community-acquired pneumonia. Recent studies demonstrate that steroids decrease mortality, the need for mechanical ventilation, and the length of stay for patients hospitalized with community-acquired pneumonia; therefore, this patient should also be treated with steroids.
Eosinophilia may be seen in drug reactions, allergies, pulmonary emboli, pleural effusions, and occasionally in malignancy. Eosinophilic pneumonia typically has the “reverse pulmonary edema” picture, with infiltrates in the periphery and not centrally, as in congestive heart failure.
A serum bicarbonate of 30 mEq/L suggests a metabolic compensation for a chronic respiratory acidosis as renal compensation, and rise in bicarbonate generally takes 3 days. She may have been hypoxic longer than her symptoms suggest.
An ABG should be ordered to determine the degree of hypoxia and whether a higher level of care is indicated. The abnormal chest radiograph, along with her hypoxia, merits a closer look at her lung parenchyma with chest computed tomography (CT). A D-dimer would be beneficial to rule out pulmonary embolism. If the D-dimer is positive, chest CT with contrast is indicated to determine if a pulmonary embolism is present. A brain natriuretic peptide would assist in the diagnosis of congestive heart failure. A sputum culture and Gram stain and respiratory viral panel may establish a pathogen for pneumonia. An ECG and troponin to rule out myocardial infarction should be performed as well.
The presence of hilar and subcarinal lymph nodes expands the differential. Stage IV pulmonary sarcoid may present with diffuse infiltrates and nodes, although the acuity in this case makes it less likely. A very aggressive malignancy such as Burkitt lymphoma may have these findings. Acute viral and atypical pneumonias remain possible. Right middle lobe syndrome may cause partial collapse of the right middle lobe. Tuberculosis can be associated with right middle lobe syndrome; however, in this day and age an obstructing mass is more likely the cause. Pulmonary disease, such as cryptogenic organizing pneumonia, idiopathic pulmonary fibrosis, and interstitial lung disease, should be considered in patients with pneumonia unresponsive to antibiotics. Lung biopsy and bronchoalveolar lavage (BAL) would help make the diagnosis and should be the next step, unless her degree of hypoxia is prohibitive. Similarly, thoracentesis with analysis of the pleural fluid for cell count, Gram stain, and culture may help make the diagnosis. Thoracentesis should be done with fluoroscopic guidance, given the risk of pneumothorax, which would further compromise her tenuous respiratory status.
Thoracentesis was attempted, but the pleural effusion was too small to provide a sample. Subsequent serum blood counts with differential showed an increased eosinophilia to 20% and resolved bandemia. Upon further questioning, she recalled several months of extensive, daily, fine-dust exposure from demolition during the remodeling of a new building.
Hypereosinophilia and pulmonary infiltrates narrow the differential considerably to include asthma; parasitic infection, such as the pulmonary phase of ascariasis; exposure, such as to dust, cigarettes, or asbestosis; or hypereosinophilic syndromes characterized by peripheral eosinophilia, along with a tissue eosinophilia, causing organ dysfunction. Idiopathic hypereosinophilic syndrome, a hypereosinophilic syndrome of unknown etiology despite extensive diagnostic testing, is rare, and eosinophilic leukemia even rarer. Her history strongly suggests exposure. Many eosinophilic diseases respond rapidly to steroids, and response to treatment would help narrow the diagnosis. If she does not respond to steroids, a lung and/or bone marrow biopsy would be the next step.
A BAL of the right middle lobe revealed 51% eosinophils, 3% neutrophils, 15% macrophages, and 28% lymphocytes. Gram stain, as well as cultures for bacteria, acid fast bacilli, fungus, herpes simplex virus, and cytomegalovirus cultures, were negative. Transbronchial lung biopsy revealed focal interstitial fibrosis and inflammation, without evidence of infection.
Eosinophils are primarily located in tissues; therefore, peripheral blood eosinophil counts often underestimate the degree of infiltration into end organs such as the lung. With 50% eosinophils, her BAL reflects this. Mold, fungus, chemical, and particle exposure could produce an eosinophilic BAL. She does not appear to be at risk for parasitic exposure. Eosinophilic granulomatosis (previously known as Churg-Strauss) is a consideration, but the lack of signs of vasculitis and wheezing make this less likely. A negative antineutrophil cytoplasmic antibody may provide reassurance. “Fine dust exposure” is consistent with environmental exposure but not a specific antigen. Steroids provide a brisk eosinophil reduction and are appropriate for this patient. There is the possibility of missing infectious or parasitic etiologies; therefore, a culture of BAL fluid should be sent.
Eosinophilic infiltration may lead to fibrosis, as was found on the lung biopsy. She should be counseled to avoid “fine dust exposure” in the future. Follow-up lung imaging and pulmonary function tests (PFTs) should be performed once her acute illness resolves. She should be strongly urged not to smoke tobacco. Interestingly, there are reports that ex-smokers who restart smoking have an increased risk of eosinophilic pneumonia, but in this case dust exposure is the more likely etiology.
She was diagnosed with acute eosinophilic pneumonia (AEP). Antibiotics were discontinued, and oral prednisone was initiated at 40 mg daily, with a brisk response and resolution of her dyspnea. She was discharged with a 6-week prednisone taper. She had no cough, dyspnea, chest pain, or fevers at her follow-up 14 days after discharge. On a 6-week, postdischarge phone call, she continued to report no symptoms, and she maintained abstinence from cigarette smoking.
This case highlights that the very best test in any medical situation is a thorough, detailed history and physical examination. A comprehensive history with physical examination is noninvasive, safe, and cheap. Had the history of fine-dust exposure been known, it is likely that a great deal of testing and money would have been saved. The patient would have been diagnosed and treated earlier, and suffered less.
COMMENTARY
First described in 1989,1,2 AEP is an uncommon cause of acute respiratory failure. Cases have been reported throughout the world, including in the United States, Belgium, Japan, and Iraq.2,3 AEP is an acute febrile illness with cough, chest pain, and dyspnea for fewer than 7 days, diffuse pulmonary infiltrates on chest radiograph, hypoxemia, no history of asthma or atopic disease, no infection, and greater than 25% eosinophils on a BAL.1,3 Physical examination typically reveals fever, tachypnea, and crackles on auscultation.1 Peripheral blood eosinophilia is inconsistently seen at presentation but generally observed as the disease progresses.1 Peripheral eosinophilia at presentation is positively correlated with a milder course of AEP, including higher oxygen saturation and fewer intensive care admissions.4 Acute respiratory failure in AEP progresses rapidly, often within hours.1 Delayed recognition of AEP may lead to respiratory failure, requiring intubation, and even to death.1
Reticular markings with Kerley-B lines, mixed reticular and alveolar infiltrates, and pleural effusions are usually found on chest radiography.1 Bilateral areas of ground-glass attenuation, interlobular septal thickening, bronchovascular bundle thickening, and pleural effusions are seen on chest CT.5 Marked eosinophilic infiltration of the interstitium and alveolar spaces, as well as diffuse alveolar damage with hyaline membrane fibroblast proliferation and inflammatory cells, are present on lung biopsy.1 Restriction with impaired diffusion capacity is found on PFTs. However, PFTs return to normal after recovery.1
AEP is distinguished from other pulmonary diseases by BAL, lung biopsy, symptoms, symptom course, and/or radiographically. AEP is often misdiagnosed as severe community-acquired pneumonia and/or acute respiratory distress syndrome, as AEP tends to occur in previously healthy individuals who have diffuse infiltrates on chest radiograph, fevers, and acute, often severe, respiratory symptoms.1-3 Other eosinophilic lung diseases to rule out include simple pulmonary eosinophilia, chronic eosinophilic pneumonia, eosinophilic granulomatosis with polyangitis (Churg-Strauss), idiopathic hypereosinophilic syndrome, infection, and drug reactions.1,3,5 Simple eosinophilic pneumonia is characterized by no symptoms or very mild pulmonary symptoms and transient patchy infiltrates on radiography.3,5 Patients with simple pulmonary eosinophilia do not have interlobular septal thickening, thickening of the bronchovascular bundles, or pleural effusions radiographically, as seen with AEP.5 Chronic eosinophilic pneumonia is subacute, with respiratory symptoms of more than 3 months in duration, in contrast with the 7 days of respiratory symptoms for AEP, and is also not associated with interlobular septal thickening, thickening of the bronchovascular bundles, or pleural effusions on radiography.3,5 Unlike AEP, chronic eosinophilic pneumonia often recurs after the course of steroids has ended.3 In contrast with AEP, eosinophilic granulomatosis with polyangitis is associated with concomitant asthma and the involvement of nonpulmonary organs.3 Idiopathic hypereosinophilic syndrome is characterized by extremely high peripheral eosinophilia and by eosinophilic involvement of multiple organs, and it requires chronic steroid use.3 Patients with allergic bronchopulmonary aspergillosis (ABPA), in contrast with AEP, typically have steroid-dependent asthma and chronic respiratory symptoms.3 ABPA also differs from AEP in that radiographic infiltrates are localized and transient, and the syndrome may relapse after steroid treatment.3 Other infectious etiologies that may present similarly to AEP include invasive pulmonary aspergillosis, pulmonary coccidiodomycosis, Pneumocystis jioveri pneumonia, pulmonary toxocariasis, pulmonary filariasis, paragonimiasis, and Loeffler syndrome (pneumonia due to Strongyloides, Ascaris, or hookworms), highlighting the importance of a thorough travel and exposure history.1,3 Several drugs may cause eosinophilic lung disease, including nitrofurantoin, tetracyclines, phenytoin, L-tryptophan, acetaminophen, ampicillin, heroin, and cocaine, which necessitates a thorough review of medication and illegal drug use.3
Steroids and supportive care are the treatment of choice for AEP, although spontaneous resolution has been seen.1,3 Significant clinical improvement occurs within 24 to 48 hours of steroid initiation.1,3 Optimal dose and duration of therapy have not been determined; however, methylprednisolone 125 mg intravenously every 6 hours until improvement is an often-used option.1 Tapers vary from 2 to 12 weeks with no difference in outcome.1-3 AEP does not recur after appropriate treatment with steroids.1,3
Little is known about the etiology of AEP. It usually occurs in young, healthy individuals and is presumed to be an unusual, acute hypersensitivity reaction to an inhaled allergen.1 A report of 18 US soldiers deployed in or near Iraq proposed dust exposure and cigarette or cigar smoking as a cause of AEP.2 Similar to our patient’s fine-dust exposure and recent onset of cigarette smoking, the soldiers were exposed to the dusty, arid environment for at least 1 day and had been smoking for at least 1 month.2 The authors proposed that small dust particles irritate alveoli, stimulating eosinophils, which are exacerbated by the onset of smoking. Alternatively, cigarette smoke may prime the lung such that dust triggers an inflammatory cascade, resulting in AEP.
TEACHING POINTS
- With the potential for the rapid progression of respiratory failure, it is imperative that the diagnosis of AEP be considered for a patient with diffuse infiltrates on a chest radiograph and acute respiratory failure of unknown cause.
- A thorough history of exposure is key to including AEP in the differential of acute pulmonary disease, with recent-onset cigarette smoking and dust exposure.
- The rapid initiation of steroids leads to a full recovery without recurrence and may be life-saving in AEP.
Disclosure
The authors report no conflicts of interest.
1. Allen J. Acute eosinophilic pneumonia. Semin Respir Crit Care Med. 2006;27:142-147. PubMed
2. Shorr AF, Scoville SL, Cersovsky SB, et al. Acute eosinophilic pneumonia among US military personnel deployed in or near Iraq. JAMA. 2004;292:2997-3005. PubMed
3. Pope-Harman AL, Davis WB, Allen ED, Christoforidis AJ, Allen JN. Acute eosinophilic pneumonia. A summary of 15 cases and review of the literature. Medicine (Baltimore). 1996;75(6):334-342. PubMed
4. Jhun BW, Kim SJ, Kim K, Lee JE. Clinical implications of initial peripheral eosinophilia in acute eosinophilic pneumonia. Respirology. 2014;19:1059-1065. PubMed
5. Daimon T, Johkoh T, Sumikawa H, et al. Acute eosinophilic pneumonia: Thin-section CT findings in 29 patients. Eur J Radiol. 2008;65:462-467. PubMed
1. Allen J. Acute eosinophilic pneumonia. Semin Respir Crit Care Med. 2006;27:142-147. PubMed
2. Shorr AF, Scoville SL, Cersovsky SB, et al. Acute eosinophilic pneumonia among US military personnel deployed in or near Iraq. JAMA. 2004;292:2997-3005. PubMed
3. Pope-Harman AL, Davis WB, Allen ED, Christoforidis AJ, Allen JN. Acute eosinophilic pneumonia. A summary of 15 cases and review of the literature. Medicine (Baltimore). 1996;75(6):334-342. PubMed
4. Jhun BW, Kim SJ, Kim K, Lee JE. Clinical implications of initial peripheral eosinophilia in acute eosinophilic pneumonia. Respirology. 2014;19:1059-1065. PubMed
5. Daimon T, Johkoh T, Sumikawa H, et al. Acute eosinophilic pneumonia: Thin-section CT findings in 29 patients. Eur J Radiol. 2008;65:462-467. PubMed