User login
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
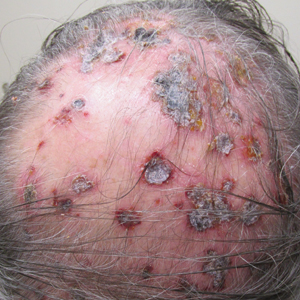
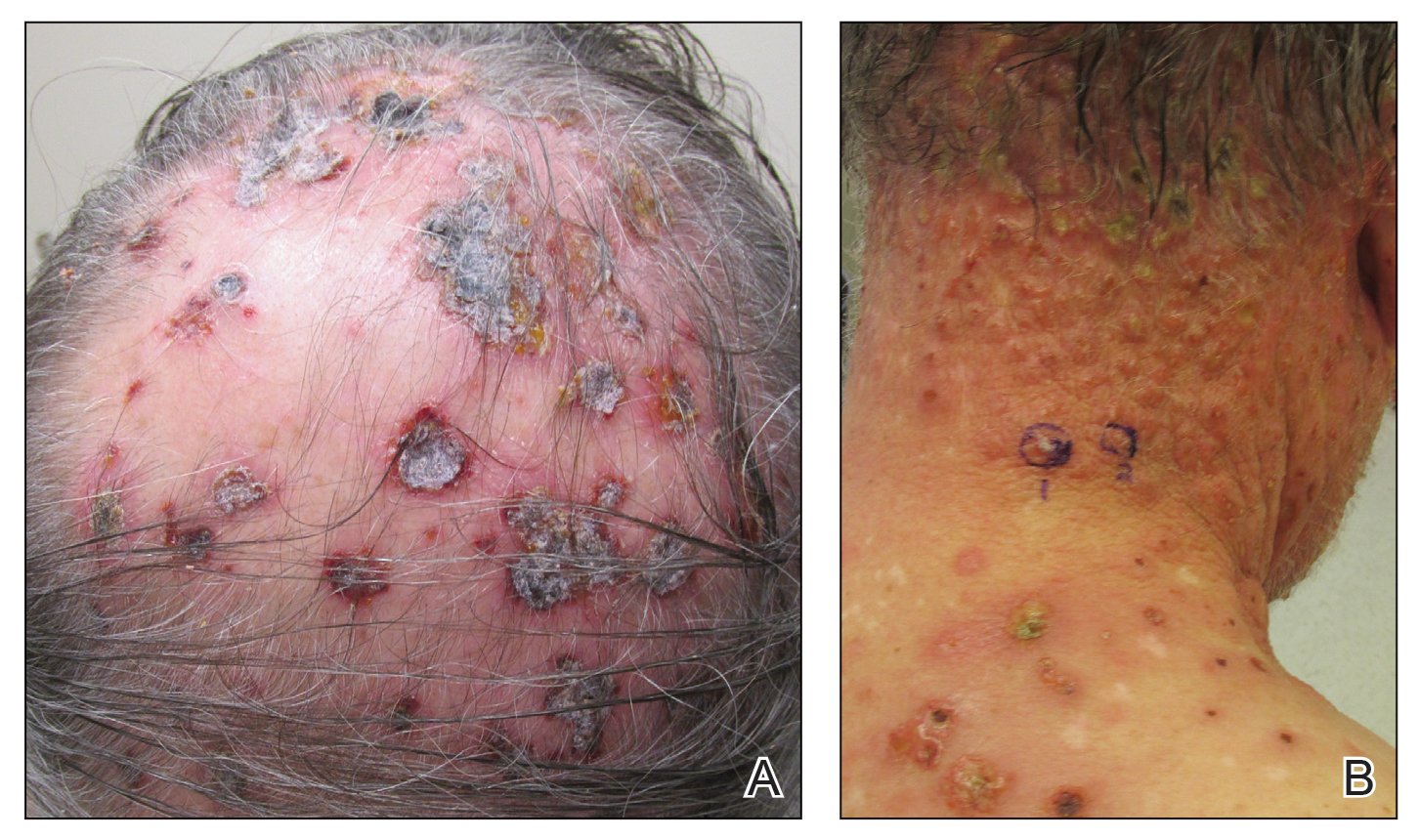
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
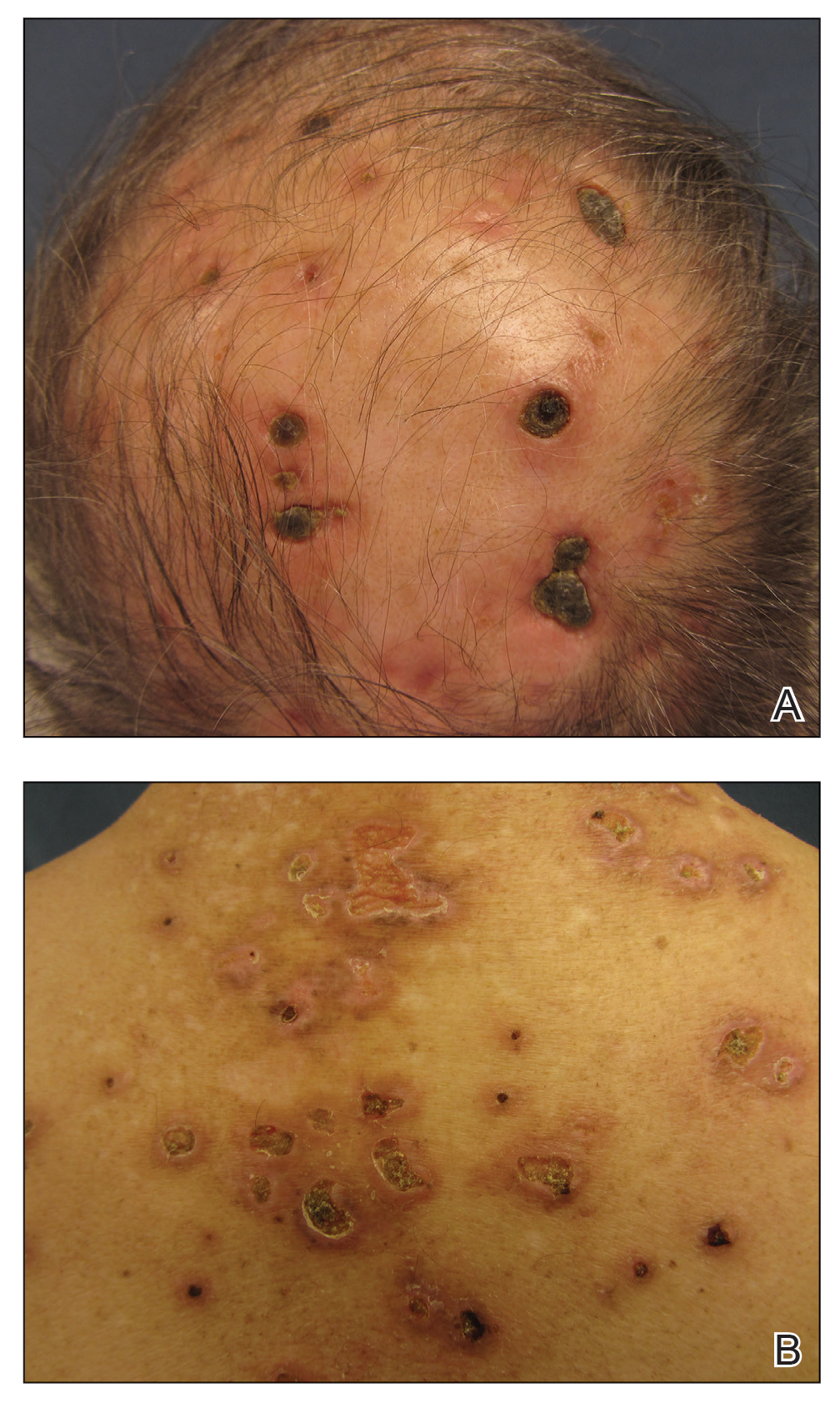
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
Practice Points
- Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder characterized by red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution.
- Histopathologically, LyP consists of a frequently CD30Mathematical Pi LT Std+ lymphocytic proliferation in multiple described patterns.
- Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy.