User login
The Diagnosis: Addison Disease in the Context of Polyglandular Autoimmune Syndrome Type 2
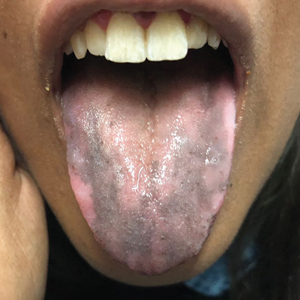
The patient’s hormone levels as well as distinct clinical features led to a diagnosis of Addison disease in the context of polyglandular autoimmune syndrome type 2 (PAS-2). Approximately 50% of PAS-2 cases are familiar, and different modes of inheritance—autosomal recessive, autosomal dominant, and polygenic—have been reported. Women are affected up to 3 times more often than men.1,2 The age of onset ranges from infancy to late adulthood, with most cases occurring in early adulthood. Primary adrenal insufficiency (Addison disease) is the principal manifestation of PAS-2. It appears in approximately 50% of patients, occurring simultaneously with autoimmune thyroid disease or diabetes mellitus in 20% of patients and following them in 30% of patients.1,2 Autoimmune thyroid diseases such as chronic autoimmune thyroiditis and occasionally Graves disease as well as type 1 diabetes mellitus also are common. Polyglandular autoimmune syndrome type 2 with primary adrenal insufficiency and autoimmune thyroid disease was formerly referred to as Schmidt syndrome.3 It must be differentiated from polyglandular autoimmune syndrome type 1, a rare condition that also is referred to as autoimmune polyendocrinopathycandidiasis-ectodermal dystrophy syndrome.1,3 As with any other cause of adrenal insufficiency, the treatment involves hormone replacement therapy up to normal levels and then tapering according to stress levels (ie, surgery or infections that require a dose increase). Our patient was diagnosed according to hormone levels and clinical features and was started on 30 mg daily of hydrocortisone and 50 μg daily of levothyroxine. No improvement in her condition was noted after 6 months of treatment. The patient is still under yearly follow-up, and the mucous hyperpigmentation faded approximately 6 months after hormonal homeostasis was achieved.
Peutz-Jeghers syndrome is inherited in an autosomal-dominant fashion. It is characterized by multiple hamartomatous polyps in the gastrointestinal tract, mucocutaneous pigmentation, and an increased risk for gastrointestinal and nongastrointestinal cancer. Mucocutaneous pigmented macules most commonly occur on the lips and perioral region, buccal mucosa, and the palms and soles. However, mucocutaneous pigmentation usually occurs during the first 1 to 2 years of life, increases in size and number over the ensuing years, and usually fades after puberty.4
Laugier-Hunziker syndrome is an acquired benign disorder presenting in adults with lentigines on the lips and buccal mucosa. It frequently is accompaniedby longitudinal melanonychia, macular pigmentation of the genitals, and involvement of the palms and soles. The diagnosis of Laugier-Hunziker syndrome is one of exclusion and is made after ruling out other causes of oral and labial hyperpigmentation, including physiologic pigmentation seen in darker-skinned individuals as well as inherited diseases associated with lentiginosis, requiring complete physical examination, endoscopy, and colonscopy.5
A wide variety of drugs and chemicals can lead to diffuse cutaneous hyperpigmentation. Increased production of melanin and/or the deposition of drug complexes or metals in the dermis is responsible for the skin discoloration. Drugs that most often cause hyperpigmentation on mucosal surfaces are hydroxychloroquine, minocycline, nicotine, silver, and some chemotherapy agents. The hyperpigmentation usually resolves with discontinuation of the offending agent, but the course may be prolonged over months to years.6
Changes in the skin and subcutaneous tissue occur in patients with Cushing syndrome. Hyperpigmentation is induced by increased secretion of adrenocorticotropic hormone, not cortisol, and occurs most often in patients with the ectopic adrenocorticotropic hormone syndrome. Hyperpigmentation may be generalized but is more intense in areas exposed to light (eg, face, neck, dorsal aspects of the hands) or to chronic mild trauma, friction, or pressure (eg, elbows, knees, spine, knuckles). Patchy pigmentation may occur on the inner surface of the lips and the buccal mucosa along the line of dental occlusion. Acanthosis nigricans also can be present in the axillae and around the neck.7
- Ferre EM, Rose SR, Rosenzweig SD, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathycandidiasis-ectodermal dystrophy. JCI Insight. 2016;1:E88782.
- Orlova EM, Sozaeva LS, Kareva MA, et al. Expanding the phenotypic and genotypic landscape of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. 2017;102:3546-3556.
- Ahonen P, Myllärniemi S, Sipilä I, et al. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829-1836.
- Utsunomiya J, Gocho H, Miyanaga T, et al. Peutz-Jeghers syndrome: its natural course and management. Johns Hopkins Med J. 1975;136:71-82.
- Nayak RS, Kotrashetti VS, Hosmani JV. Laugier-Hunziker syndrome. J Oral Maxillofac Pathol. 2012;16:245-250.
- Krause W. Drug-induced hyperpigmentation: a systematic review. J Dtsch Dermatol Ges. 2013;11:644-651.
- Newell-Price J, Trainer P, Besser M, et al. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr Rev. 1998;19:647-672.
The Diagnosis: Addison Disease in the Context of Polyglandular Autoimmune Syndrome Type 2
The patient’s hormone levels as well as distinct clinical features led to a diagnosis of Addison disease in the context of polyglandular autoimmune syndrome type 2 (PAS-2). Approximately 50% of PAS-2 cases are familiar, and different modes of inheritance—autosomal recessive, autosomal dominant, and polygenic—have been reported. Women are affected up to 3 times more often than men.1,2 The age of onset ranges from infancy to late adulthood, with most cases occurring in early adulthood. Primary adrenal insufficiency (Addison disease) is the principal manifestation of PAS-2. It appears in approximately 50% of patients, occurring simultaneously with autoimmune thyroid disease or diabetes mellitus in 20% of patients and following them in 30% of patients.1,2 Autoimmune thyroid diseases such as chronic autoimmune thyroiditis and occasionally Graves disease as well as type 1 diabetes mellitus also are common. Polyglandular autoimmune syndrome type 2 with primary adrenal insufficiency and autoimmune thyroid disease was formerly referred to as Schmidt syndrome.3 It must be differentiated from polyglandular autoimmune syndrome type 1, a rare condition that also is referred to as autoimmune polyendocrinopathycandidiasis-ectodermal dystrophy syndrome.1,3 As with any other cause of adrenal insufficiency, the treatment involves hormone replacement therapy up to normal levels and then tapering according to stress levels (ie, surgery or infections that require a dose increase). Our patient was diagnosed according to hormone levels and clinical features and was started on 30 mg daily of hydrocortisone and 50 μg daily of levothyroxine. No improvement in her condition was noted after 6 months of treatment. The patient is still under yearly follow-up, and the mucous hyperpigmentation faded approximately 6 months after hormonal homeostasis was achieved.
Peutz-Jeghers syndrome is inherited in an autosomal-dominant fashion. It is characterized by multiple hamartomatous polyps in the gastrointestinal tract, mucocutaneous pigmentation, and an increased risk for gastrointestinal and nongastrointestinal cancer. Mucocutaneous pigmented macules most commonly occur on the lips and perioral region, buccal mucosa, and the palms and soles. However, mucocutaneous pigmentation usually occurs during the first 1 to 2 years of life, increases in size and number over the ensuing years, and usually fades after puberty.4
Laugier-Hunziker syndrome is an acquired benign disorder presenting in adults with lentigines on the lips and buccal mucosa. It frequently is accompaniedby longitudinal melanonychia, macular pigmentation of the genitals, and involvement of the palms and soles. The diagnosis of Laugier-Hunziker syndrome is one of exclusion and is made after ruling out other causes of oral and labial hyperpigmentation, including physiologic pigmentation seen in darker-skinned individuals as well as inherited diseases associated with lentiginosis, requiring complete physical examination, endoscopy, and colonscopy.5
A wide variety of drugs and chemicals can lead to diffuse cutaneous hyperpigmentation. Increased production of melanin and/or the deposition of drug complexes or metals in the dermis is responsible for the skin discoloration. Drugs that most often cause hyperpigmentation on mucosal surfaces are hydroxychloroquine, minocycline, nicotine, silver, and some chemotherapy agents. The hyperpigmentation usually resolves with discontinuation of the offending agent, but the course may be prolonged over months to years.6
Changes in the skin and subcutaneous tissue occur in patients with Cushing syndrome. Hyperpigmentation is induced by increased secretion of adrenocorticotropic hormone, not cortisol, and occurs most often in patients with the ectopic adrenocorticotropic hormone syndrome. Hyperpigmentation may be generalized but is more intense in areas exposed to light (eg, face, neck, dorsal aspects of the hands) or to chronic mild trauma, friction, or pressure (eg, elbows, knees, spine, knuckles). Patchy pigmentation may occur on the inner surface of the lips and the buccal mucosa along the line of dental occlusion. Acanthosis nigricans also can be present in the axillae and around the neck.7
The Diagnosis: Addison Disease in the Context of Polyglandular Autoimmune Syndrome Type 2
The patient’s hormone levels as well as distinct clinical features led to a diagnosis of Addison disease in the context of polyglandular autoimmune syndrome type 2 (PAS-2). Approximately 50% of PAS-2 cases are familiar, and different modes of inheritance—autosomal recessive, autosomal dominant, and polygenic—have been reported. Women are affected up to 3 times more often than men.1,2 The age of onset ranges from infancy to late adulthood, with most cases occurring in early adulthood. Primary adrenal insufficiency (Addison disease) is the principal manifestation of PAS-2. It appears in approximately 50% of patients, occurring simultaneously with autoimmune thyroid disease or diabetes mellitus in 20% of patients and following them in 30% of patients.1,2 Autoimmune thyroid diseases such as chronic autoimmune thyroiditis and occasionally Graves disease as well as type 1 diabetes mellitus also are common. Polyglandular autoimmune syndrome type 2 with primary adrenal insufficiency and autoimmune thyroid disease was formerly referred to as Schmidt syndrome.3 It must be differentiated from polyglandular autoimmune syndrome type 1, a rare condition that also is referred to as autoimmune polyendocrinopathycandidiasis-ectodermal dystrophy syndrome.1,3 As with any other cause of adrenal insufficiency, the treatment involves hormone replacement therapy up to normal levels and then tapering according to stress levels (ie, surgery or infections that require a dose increase). Our patient was diagnosed according to hormone levels and clinical features and was started on 30 mg daily of hydrocortisone and 50 μg daily of levothyroxine. No improvement in her condition was noted after 6 months of treatment. The patient is still under yearly follow-up, and the mucous hyperpigmentation faded approximately 6 months after hormonal homeostasis was achieved.
Peutz-Jeghers syndrome is inherited in an autosomal-dominant fashion. It is characterized by multiple hamartomatous polyps in the gastrointestinal tract, mucocutaneous pigmentation, and an increased risk for gastrointestinal and nongastrointestinal cancer. Mucocutaneous pigmented macules most commonly occur on the lips and perioral region, buccal mucosa, and the palms and soles. However, mucocutaneous pigmentation usually occurs during the first 1 to 2 years of life, increases in size and number over the ensuing years, and usually fades after puberty.4
Laugier-Hunziker syndrome is an acquired benign disorder presenting in adults with lentigines on the lips and buccal mucosa. It frequently is accompaniedby longitudinal melanonychia, macular pigmentation of the genitals, and involvement of the palms and soles. The diagnosis of Laugier-Hunziker syndrome is one of exclusion and is made after ruling out other causes of oral and labial hyperpigmentation, including physiologic pigmentation seen in darker-skinned individuals as well as inherited diseases associated with lentiginosis, requiring complete physical examination, endoscopy, and colonscopy.5
A wide variety of drugs and chemicals can lead to diffuse cutaneous hyperpigmentation. Increased production of melanin and/or the deposition of drug complexes or metals in the dermis is responsible for the skin discoloration. Drugs that most often cause hyperpigmentation on mucosal surfaces are hydroxychloroquine, minocycline, nicotine, silver, and some chemotherapy agents. The hyperpigmentation usually resolves with discontinuation of the offending agent, but the course may be prolonged over months to years.6
Changes in the skin and subcutaneous tissue occur in patients with Cushing syndrome. Hyperpigmentation is induced by increased secretion of adrenocorticotropic hormone, not cortisol, and occurs most often in patients with the ectopic adrenocorticotropic hormone syndrome. Hyperpigmentation may be generalized but is more intense in areas exposed to light (eg, face, neck, dorsal aspects of the hands) or to chronic mild trauma, friction, or pressure (eg, elbows, knees, spine, knuckles). Patchy pigmentation may occur on the inner surface of the lips and the buccal mucosa along the line of dental occlusion. Acanthosis nigricans also can be present in the axillae and around the neck.7
- Ferre EM, Rose SR, Rosenzweig SD, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathycandidiasis-ectodermal dystrophy. JCI Insight. 2016;1:E88782.
- Orlova EM, Sozaeva LS, Kareva MA, et al. Expanding the phenotypic and genotypic landscape of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. 2017;102:3546-3556.
- Ahonen P, Myllärniemi S, Sipilä I, et al. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829-1836.
- Utsunomiya J, Gocho H, Miyanaga T, et al. Peutz-Jeghers syndrome: its natural course and management. Johns Hopkins Med J. 1975;136:71-82.
- Nayak RS, Kotrashetti VS, Hosmani JV. Laugier-Hunziker syndrome. J Oral Maxillofac Pathol. 2012;16:245-250.
- Krause W. Drug-induced hyperpigmentation: a systematic review. J Dtsch Dermatol Ges. 2013;11:644-651.
- Newell-Price J, Trainer P, Besser M, et al. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr Rev. 1998;19:647-672.
- Ferre EM, Rose SR, Rosenzweig SD, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathycandidiasis-ectodermal dystrophy. JCI Insight. 2016;1:E88782.
- Orlova EM, Sozaeva LS, Kareva MA, et al. Expanding the phenotypic and genotypic landscape of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. 2017;102:3546-3556.
- Ahonen P, Myllärniemi S, Sipilä I, et al. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829-1836.
- Utsunomiya J, Gocho H, Miyanaga T, et al. Peutz-Jeghers syndrome: its natural course and management. Johns Hopkins Med J. 1975;136:71-82.
- Nayak RS, Kotrashetti VS, Hosmani JV. Laugier-Hunziker syndrome. J Oral Maxillofac Pathol. 2012;16:245-250.
- Krause W. Drug-induced hyperpigmentation: a systematic review. J Dtsch Dermatol Ges. 2013;11:644-651.
- Newell-Price J, Trainer P, Besser M, et al. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr Rev. 1998;19:647-672.
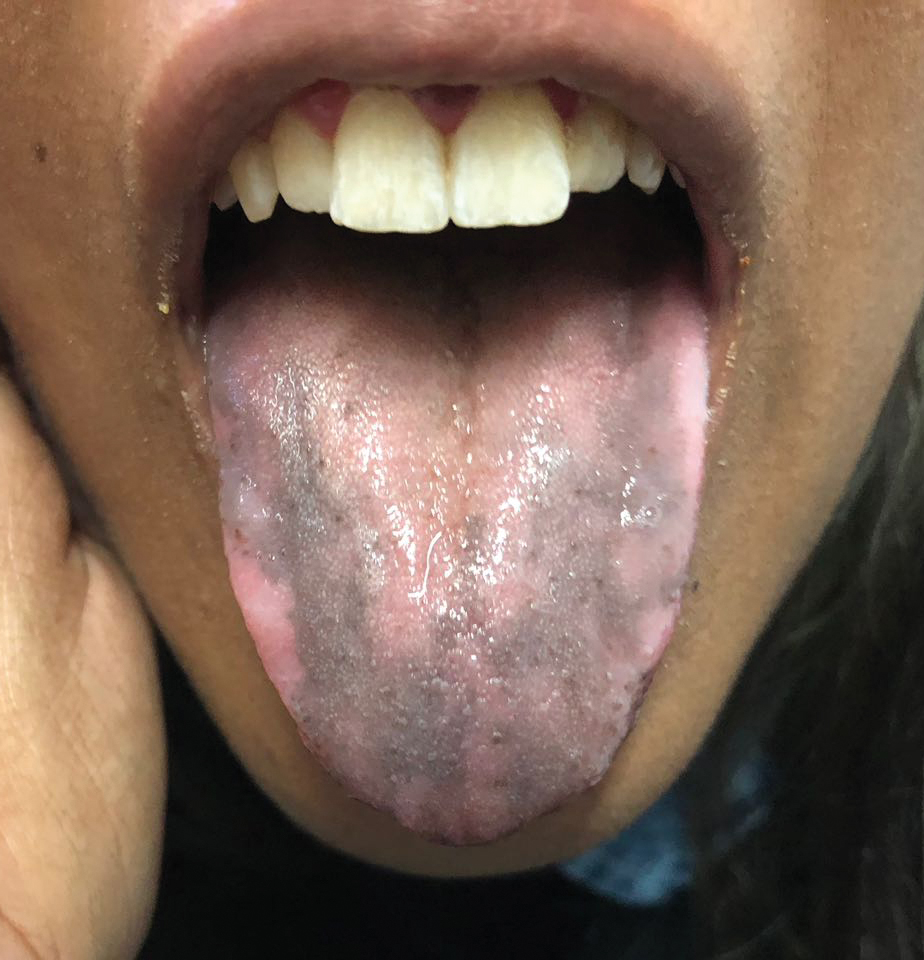
An otherwise healthy 17-year-old adolescent girl from Spain presented with hyperpigmentation on the tongue of several weeks’ duration. She denied licking graphite pencils or pens. Physical examination revealed pigmentation in the palmar creases and a slight generalized tan. The patient denied sun exposure. Neither melanonychia nor genital hyperpigmented lesions were noted. Blood tests showed overt hypothyroidism.