User login
In many patients, certain drugs do not work as well as expected, whereas in other patients they cause toxic effects, even at lower doses. For some patients, the reason may be genetic.
Sizeable minorities of the population carry genetic variants—polymorphisms— that affect their response to various drugs. Thanks to genetic research, our understanding of the variability of drug response has advanced markedly in the last decade. Many relevant polymorphisms have been identified, and tests for some of them are available.
Armed with the knowledge of their patients’ genetic status, physicians could predict their response to certain drugs, leading to better efficacy, fewer adverse drug reactions, and a better cost-benefit ratio.
The possible impact is substantial, since more than half of the drugs most commonly involved in adverse drug reactions are metabolized by polymorphic enzymes.1 Adverse drug reactions remain a significant detriment to public health, having a substantial impact on rates of morbidity and death and on healthcare costs.2–8 In the United States, adverse drug reactions are a leading cause of death in hospitalized patients4 and are annually responsible for hundreds of thousands of deaths and hundreds of billions of dollars in added costs.2,4,6–8

In the meantime, physicians can educate their patients and promote efforts to incorporate genomic information into standard clinical decision-making.
This article offers an overview of pharmacogenomic testing, discussing implications and limitations of a few validated tests. Specifically, we will discuss testing that is relevant when using warfarin (Coumadin), clopidogrel (Plavix), statins, tamoxifen (Nolvadex), codeine, and psychotropic medications, as well as the future role of pharmacogenomic testing in medicine.
WHAT IS PHARMACOGENOMICS?
Pharmacogenomics is the study of how genetic factors relate to interindividual variability of drug response.
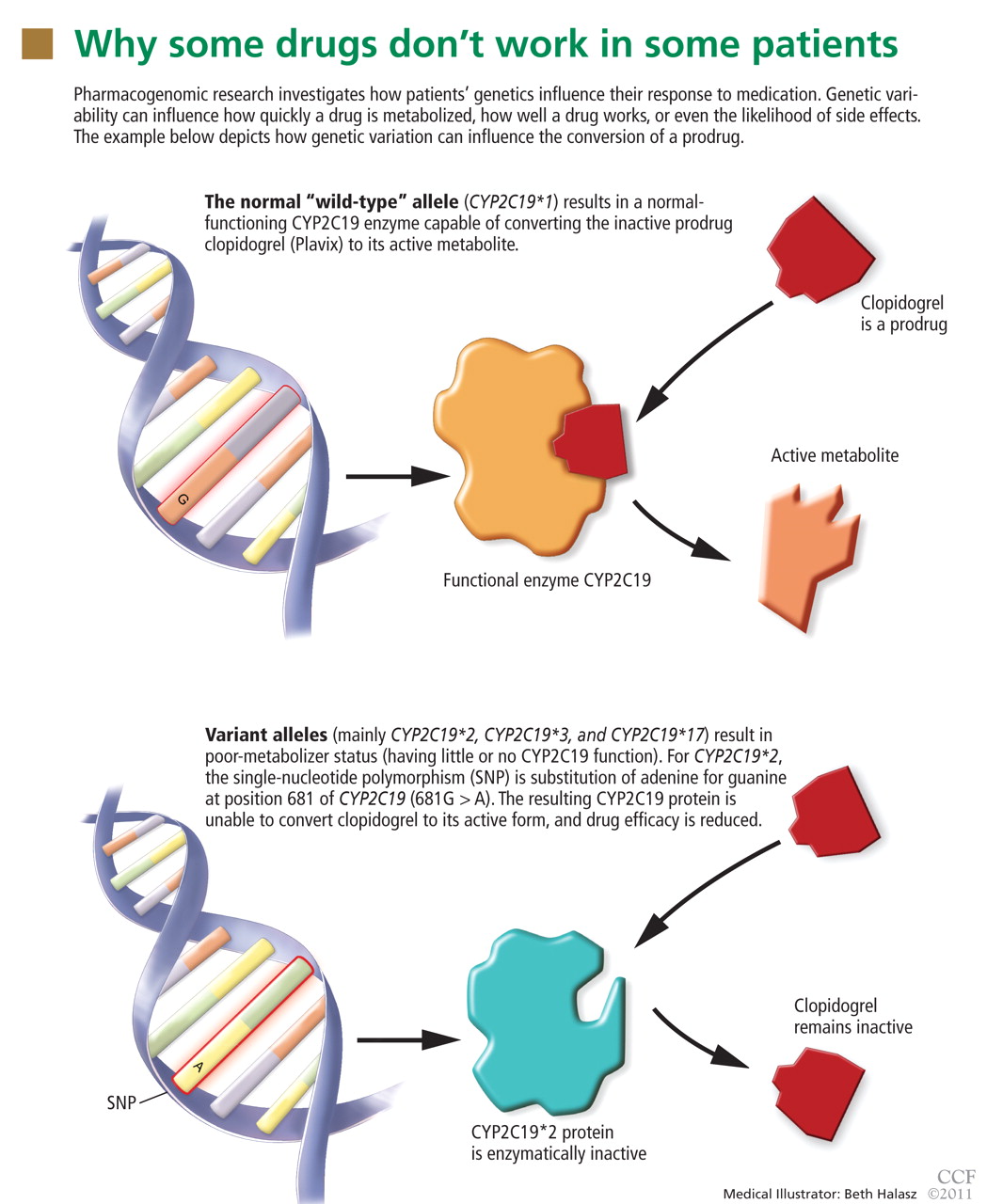
Many clinicians may not be familiar with the background and terminology used in the pharmacogenomic literature. Below, a brief review of the terminology is followed by a schematic describing the various stages of research involved in pharmacogenomics and the advancement of a test into standard practice.
The review and schematic may be helpful for evaluating the clinical significance of pharmacogenomics-related articles.
From genotype to phenotype
Genotype refers to the coding sequence of DNA base pairs for a particular gene, and phenotype (eg, disease or drug response) refers to a trait resulting from the protein product encoded by the gene. The name of a gene often refers to its protein product and is italicized (eg, the CYP3A4 gene encodes for the CYP3A4 enzyme).
Two alleles per autosomal gene (one paternal and one maternal) form the genotype. Heterozygotes possess two different alleles, and homozygotes possess two of the same alleles. The most common allele in a population is referred to as the wild type, and allele frequencies can vary greatly in different populations.9
Most sequence variations are single nucleotide polymorphisms (SNPs, pronounced “snips”), a single DNA base pair substitution that may result in a different gene product. SNPs can be classified as structural RNA polymorphisms (srSNPs), regulatory polymorphisms (rSNPs), or polymorphisms in coding regions (cSNPs)10: srSNPs alter mRNA processing and translation, rSNPs alter transcription, and cSNPs alter protein sequence and function.
Recently, genetic associations with a phenotype have been done on a large scale, with millions of SNPs measured in each of many subjects. This approach, called a genomewide association study or GWAS, has revealed countless candidate genes for clinical traits, but only a few have resulted in a practical clinical application. SNPs may by themselves exert a pharmacokinetic effect (ie, how the body processes the drug), a pharmacodynamic effect (ie, how the drug affects the body), or both, or they may act in concert with other genetic factors. Pharmacodynamic effects can result from a pharmacokinetic effect or can result from variations in a pharmacologic target.
Establishing a genotype-phenotype association can involve clinical studies, animal transgenic studies, or molecular and cellular functional assays.
Clinical applications are emerging

WARFARIN: IMPORTANCE OF CYP2C9, VKORC1
Warfarin is used for the long-term treatment and prevention of thromboembolic events.
This drug has a narrow therapeutic window and shows substantial interpatient dose variability. The start of warfarin therapy is associated with one of the highest rates of adverse events and emergency room visits of any single drug.12 More than 2 million patients start warfarin each year in the United States alone,13 and about 20% of them are hospitalized within the first 6 months because of bleeding due to overanticoagulation.14
The findings from a recent study suggest that pharmacogenomic testing may eventually allow more patients to safely benefit from warfarin therapy. In this large, nationwide, prospective study, hospitalization rates were 30% lower when pharmacogenomic testing was used.14 However, no reduction was seen in the time needed to reach the target international normalized ratio (INR) or in the need for INR checks at 6 months. Furthermore, this study used historical control data, and some or all of the reduction in hospitalization rates may be attributed to more frequent INR checks in the patients who underwent testing than in the historical control group.
A relationship between warfarin dose requirements and the genetic status of CYP2C9, which encodes a major drug-metabolizing enzyme, has been demonstrated in retrospective and prospective studies.15–17
S-warfarin is metabolized by CYP2C9, which is polymorphic
Warfarin contains equal amounts of two isomers, designated S and R. S-warfarin, which is more potent, is metabolized principally by CYP2C9, while R-warfarin is metabolized by CYP1A2, CYP2C19, and CYP3A4.
People who possess two copies of the wild type CYP2C9 gene CYP2C9*1 metabolize warfarin very well and so are called “extensive warfarin metabolizers.” Carriers of the allelic variants CYP2C9*2 and CYP2C9*3 (which have point mutations in exons 3 and 7 of CYP2C9, respectively), have less capacity. Compared with those who are homozygous for the wild-type gene, homozygous carriers of CYP2C9* 3 clear S-warfarin at a rate that is 90% lower, and those with the CYP2C9*1/*3, CYP2C9* 1/*2, CYP2C9*2/*2, or CYP2C9*2/*3 genotypes clear it at a rate 50% to 75% lower. A meta-analysis of 12 studies found that the CYP2C9 genotype accounted for 12% of the interindividual variability of warfarin dose requirements.18
About 8% of whites carry at least one copy of CYP2C9*2, as do 1% of African Americans; the allele is rare in Asian populations. The frequency of CYP2C9*3 is 6% in whites, 1% in African Americans, and 3% in Asians.19,20 People with CYP2C9*4 or CYP2C9*5 have a diminished capacity to clear warfarin; however, these variants occur so infrequently that their clinical relevance may be minimal.
Warfarin’s target, VKOR, is also polymorphic
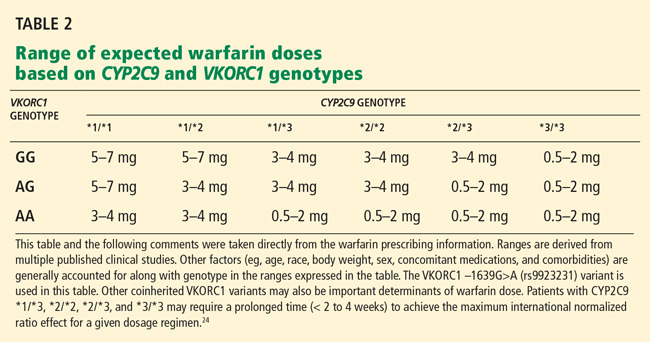
Genetic variation in warfarin’s pharmacologic target, vitamin K 2,3-epoxide reductase (VKOR), also influences dose requirements. Warfarin decreases the synthesis of vitamin-K-dependent clotting factors by inhibiting VKOR. This inhibition depends on the patient’s C1 subunit gene, VKORC1. Patients with a guanine-to-adenine SNP 1,639 bases upstream of VKORC1 (−1639G>A) need lower warfarin doses—an average of 25% lower in those with the GA genotype (ie, one allele has guanine in the −1639 position and the other allele has adenine in that position) and 50% lower in those with the AA genotype compared with the wild-type genotype GG.21 This promoter SNP, positioned upstream (ie, before the gene-coding region), greatly influences VKORC1 expression.
A meta-analysis of 10 studies found that the VKORC1 polymorphism accounts for 25% of the interindividual variation in warfarin dose.18 In one study, the frequency of the AA genotype in a white population was 14%, AG 47%, and GG 39%; in a Chinese population the frequency of AA was 82%, AG 18%, and GG 0.35%.22
CYP4F2 and GGCX also affect warfarin’s dose requirements
Genetic variations in the enzymes CYP4F2 and gamma-glutamyl carboxylase (GGCX) also influence warfarin dose requirements. Although the data are limited and the effects are smaller than those of CYP2C9 and VKORC1, people with a SNP in CYP4F2 need 8% higher doses of warfarin, while those with a SNP in GGCX need 6% lower doses.23
CYP2C9 and VKORC1 testing is available
Currently, the clinical pharmacogenetic tests relevant for warfarin use are for CYP2C9 and VKORC1.10
The FDA has approved four warfarin pharmacogenetic test kits, but most third-party payers are reluctant to reimburse for testing because it is not currently considered a standard of care. Testing typically costs a few hundred dollars, but it should become less expensive as it becomes more commonplace. The current FDA-approved product label for warfarin does not recommend routine pharmacogenomic testing for determining initial or maintenance doses, but it does acknowledge that dose requirements are influenced by CYP2C9 and VKORC1 and states that genotype information, when available, can assist in selecting the starting dose.24
Clinical trials of warfarin pharmacogenomic testing are under way
Although genetic status can greatly influence an individual patient’s warfarin dosing requirement, routine prospective pharmacogenomic testing is not endorsed by the FDA or by other expert panels26 because there is currently insufficient evidence to recommend for or against it.
Several large prospective trials are under way. For example, the National Heart, Lung, and Blood Institute began a prospective trial in about 1,200 patients to evaluate the use of clinical plus genetic information to guide the initiation of warfarin therapy and to improve anticoagulation control for patients.27 The results, expected in September 2011, and those of other large prospective trials should provide adequate evidence for making recommendations about the clinical utility of routine pharmacogenetic testing for guiding warfarin therapy.
Several recent cost-utility and cost-effectiveness studies have attempted to quantify the value of pharmacogenomic testing for warfarin therapy,28–30 but their analyses are largely limited because the benefit (clinical utility) is yet to be sufficiently characterized.
The relevance of such analyses may soon be drastically diminished, as several non-vitamin-K-dependent blood thinners such as rivaroxaban (Xarelto), dabigatran (Pradaxa), and apixaban are poised to enter clinical practice.31
CLOPIDOGREL IS ACTIVATED BY CYP2C19
Clopidogrel, taken by about 40 million patients worldwide, is used to prevent atherothrombotic events and cardiac stent thrombosis when given along with aspirin.
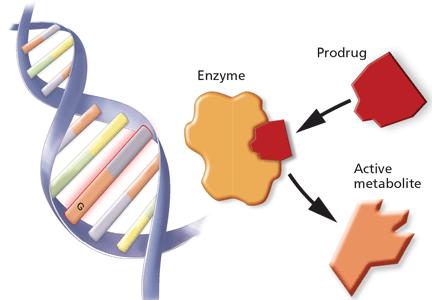
Studies of clopidogrel pharmacogenomics
A recent genome-wide association study conducted in a cohort of 429 healthy Amish persons revealed a SNP in CYP2C19 to be associated with a diminished response to clopidogrel and to account for 12% of the variation in drug response.33 Traditional factors (the patient’s age, body-mass index, and cholesterol level) combined accounted for less than 10% of the variation.
Findings were similar in a subsequent investigation in 227 cardiac patients receiving clopidogrel: 21% of those with the variant had a cardiovascular ischemic event or died during a 1-year follow-up period compared with 10% of those without the variant (hazard ratio 2.42, P = .02).33
A 12-year prospective study investigating clopidogrel efficacy in 300 cardiac patients under the age of 45 used cardiovascular death, nonfatal myocardial infarction, and urgent coronary revascularization as end points. It concluded that the only independent predictor of these events was the patient’s CYP2C19 status.34
A study in 2,200 patients with recent myocardial infarction examined whether any of the known allelic variations that modulate clopidogrel’s absorption (ABCB1), metabolic activation (CYP3A4/5 and CYP2C19), or biologic activity (P2RY12 and ITGB3) was associated with a higher rate of the combined end point of all-cause mortality, nonfatal myocardial infarction, or stroke. None of the SNPs in CYP3A4/5, P2RY12, or ITGB3 that were evaluated was associated with a higher risk at 1 year. However, the allelic variations modulating clopidogrel’s absorption (ABCB1) and metabolism (CYP2C19) were associated with higher event rates. Patients with two variant ABCB1 alleles had a higher adjusted hazard ratio (95% confidence interval [CI] 1.2–2.47) than those with the wild-type allele. Patients who had one or two CYP2C19 loss-of-function alleles had a higher event rate than those with two wild-type alleles (95% CI 1.10–3.58 and 1.71–7.51, respectively).35
Conversely, researchers from the Population Health Research Institute found no association between poor-metabolizer status and treatment outcomes when CYP2C19 analysis was retrospectively added to the findings of two large clinical trials (combined N > 5,000). However, patients with acute coronary syndrome benefited more from clopidogrel treatment if they were ultra-rapid metabolizers (possessing the gain-of-function allele CYP2C19*17).36
Current status of clopidogrel testing: Uncertain
A current FDA boxed warning states that poor CYP2C19 metabolizers may not benefit from clopidogrel and recommends that prescribers consider alternative treatment for patients in this category.37 However, routine CYP2C19 testing is not recommended, and no firm recommendations have been established regarding dose adjustments for CYP2C19 status.

In 2010, the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association collectively pronounced the current evidence base insufficient for recommending routine pharmacogenomic testing.39
Needed are large-scale studies examining the cost-effectiveness and clinical utility of genotype-guided clopidogrel therapy compared with other therapy options such as prasugrel (Effient), an analogue not metabolized by CYP2C19. One such study, sponsored by Medco Health Solutions, plans to enroll 14,600 cardiac patients and has an estimated completion date in June 2011.40 The expectation that clopidogrel will be available in generic form in 2012 adds to the uncertainty regarding the cost-effectiveness of CYP2C19 testing for clopidogrel therapy.
STATINS: SLC01B1*5 INCREASES MYOPATHY RISK
Statins lower the concentration of low-density lipoprotein cholesterol (LDL-C), resulting in a relative-risk reduction of about 20% for each 1 mmol/L (39 mg/dL) decrement in LDL-C.41 They are one of the most commonly prescribed classes of drugs, but their side effects can limit their appeal: statin-induced myopathy occurs in about 1:1,000 to 1:10,000 patients and is difficult to predict.
SLC01B1. The Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH), a genome-wide association study, recently found a SNP (SLCO1B1* 5) in the SLC01B1 gene to be associated with a higher risk of statin-induced myopathy in cardiac patients receiving simvastatin (Zocor) 40 or 80 mg daily.42 The SLC01B1 gene, located on chromosome 12, influences the extent of the drug’s hepatic uptake and its serum concentration. Only the SLC01B1*5 SNP emerged as a predictor of statin-induced myopathy across the entire genome.42
The authors believe the findings are likely to apply to other statins. The mechanisms leading to statin-induced myopathy and the impact of statin pharmacogenomics are still unclear.43
CYP3A4. Other genetic variants may play a vital role in determining response to statin therapy. Carriers of a newly identified CYP3A4 polymorphism (intron 6 SNP, rs35599367, C>T) required significantly lower statin doses (0.2–0.6 times less) for optimal lipid control. The analyses included atorvastatin (Lipitor), simvastatin, and lovastatin (Mevacor), and the association was robust (P = .019).44
Statin pharmacogenomic testing is not routinely recommended
Routine pharmacogenomic testing for statin therapy is not recommended. Additional studies are needed to determine the clinical utility and cost-effectiveness of pharmacogenomic testing (involving a combination of several polymorphisms) in various patient populations delineated by type of statin, dose, and concomitant use of other drugs.
TAMOXIFEN IS ACTIVATED BY CYP2D6
Tamoxifen is prescribed to prevent the recurrence of estrogen-receptor-positive breast cancer, to treat metastatic breast cancer, to prevent cancer in high-risk populations, and to treat ductal carcinoma in situ.
Tamoxifen is metabolized to form endoxifen, which has much higher potency and higher systemic levels than tamoxifen.45 Both CYP2D6 and CYP3A4/5 are required to produce endoxifen via two intermediates, but CYP2D6 catalyzes the critical step leading to metabolic activation.
The CYP2D6 gene is highly polymorphic, with more than 75 allelic variants identified. Extensive literature is available describing the influence of CYP2D6 polymorphisms on tamoxifen metabolism and therapy outcomes.46–52 Several CYP2D6 variants result in reduced or no enzyme activity, and people who have more than two normally functioning alleles have exaggerated enzyme activity (gene amplification).
Classification of CYP2D6 status
Several systems have been developed to categorize the phenotypic activity of CYP2D6 based on genotype.
A genetic basis for the observed diversity in the metabolism of cytochrome P450 substrates was recognized more than 30 years ago. People were categorized as either extensive or poor metabolizers, reflecting normal vs impaired ability to metabolize the CYP2D6 substrates sparteine and debrisoquine. Later work expanded this system to include categories for intermediate (between poor and extensive) and ultra-rapid (better than extensive) metabolizers.
The genetic basis for these categories includes homozygosity for dysfunctional variants (the poor-metabolizer group) or extra copies of normal functioning variants (the ultra-rapid-metabolizer group).
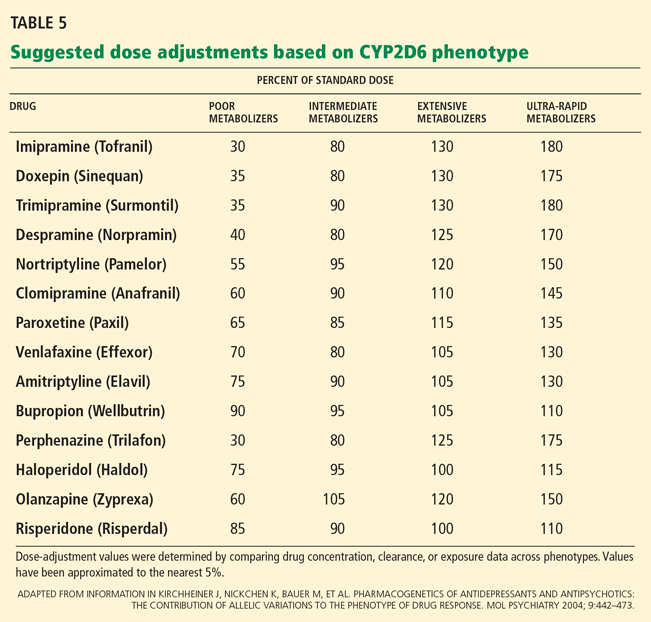
Newer systems have been described for characterizing the CYP2D6 activity phenotype whereby CYP2D6 variants are assigned activity scores.53–56 The various scoring systems have been reviewed by Kirchheiner.57

A recent version of the activity scoring system also takes into consideration the many drugs that inhibit CYP2D6, such as amiodarone (Cordarone) and fluoxetine (Prozac) that can reduce the action of tamoxifen if given with it (Table 4).58 For example, the tamoxifen exposure (as predicted by the CYP2D6-activity score) experienced by a CYP2D6 extensive metabolizer taking a CYP2D6-inhibiting drug may be similar to the exposure experienced by a CYP2D6 poor metabolizer receiving the same tamoxifen dose but not taking a CYP2D6-inhibiting drug.
Likewise, the activity score of a CYP2D6 intermediate metabolizer taking a CYP2D6-inducing drug may be similar to that of a CYP2D6 ultra-rapid metabolizer not taking a CYP2D6-inducing drug. Examples of CYP2D6 inducers are dexamethasone, rifampin, and hyperforin (St. John’s wort).
While the newer systems are reported to provide better correlations between genotype and phenotype scores, the older scoring systems and the categorical names are still widely used (eg, in the FDA-approved AmpliChip CYP450 test from Roche,59 which includes genotype data for CYP2D6 and CYP2C19).
No firm recommendations for CYP2D6 testing in tamoxifen users
The different genotypes and phenotypes vary in prevalence in different ethnic groups, and significantly different activity levels for endoxifen formation are observed. Tamoxifen lacks efficacy in those who are poor CYP2D6 metabolizers—ie, about 7% of the white population.
However, the FDA has not made firm recommendations about CYP2D6 testing for prescribing tamoxifen because the evidence of benefit, although suggestive, has been considered insufficient.
Clinicians should be aware that tamoxifen’s efficacy is greatly reduced by concomitant therapy with CYP2D6-inhibiting drugs (Table 4).
Other genes affecting tamoxifen: CYP3A4/5, SULT1A1, and UGT2B15
Some investigators propose that polymorphisms in additional genes encoding enzymes in the tamoxifen metabolic and elimination pathways (eg, CYP3A4/5, SULT1A1, and UGT2B15) also need to be considered to account adequately for interindividual variation in drug response.
For example, CYP3A4 and CYP3A5 are also polymorphic, and large interindividual variation exists in their enzyme activities. These enzymes have overlapping substrate specificities, represent the most abundant drug-metabolizing enzymes in the human liver, and are involved in the biotransformation of a broad range of endogenous substrates and most drugs.60
Clinical studies evaluating the impact of CYP3A4/5 polymorphisms have been inconsistent in their conclusions, which is generally attributed to the relatively low functional impact or the low prevalence of the SNPs evaluated. Many of the nearly 100 CYP3A4/5 polymorphisms identified have not yet been characterized regarding their functional impact on enzyme expression or activity. CYP-3A4/5 enzyme activity is highly variable between individuals and warrants further study of its role in outcomes of tamoxifen therapy. Ongoing and future prospective clinical trials evaluating CYP2D6, CYP3A4/5, and other relevant polymorphisms are necessary to define their clinical relevance before routine genetic testing for tamoxifen can be justified.
CODEINE IS ALSO ACTIVATED BY CYP2D6
Codeine also depends on the CYP2D6 gene, as it must be activated to its more potent opioid metabolites, including morphine. Poor CYP2D6 metabolizers do not benefit from codeine therapy.
The pharmacogenomics of codeine has become a hot topic, especially regarding breast-feeding mothers. The debate was ignited with the publication in 2006 of a case report of an infant’s death, apparently the result of metabolic polymorphisms.61 The evolution of this debate and the outcome of the case may be noteworthy to clinicians, as they illustrate the gravity of public and patient interest in pharmacogenomic testing. In this case, the breast-feeding mother had taken codeine regularly for about 14 days when her 13-day-old infant died from toxic levels of morphine. Unknown to her and the prescriber, both the mother and infant were ultra-rapid CYP2D6 metabolizers, resulting in a more rapid and extensive conversion of codeine to morphine.
A logical strategy for preventing similar deaths would be routine CYP2D6 genotyping when prescribing codeine to breast-feeding mothers. However, after several investigations examined the metabolic and excretion pathways of codeine in their entirety, the FDA did not recommend routine CYP2D6 testing when prescribing codeine to breastfeeding mothers because several other factors, including rare genetic variations of other enzymes, proved necessary for reaching the opioid toxicity leading to the infant’s death.62
PHARMACOGENOMICS OF PSYCHOTROPIC DRUGS
Pharmacogenomic testing has clinical utility for some psychotropic drugs.
HLA-B and carbamazepine
Considered a standard of care, HLA-B genotyping is appropriate before prescribing carbamazepine (Tegretol, Equetro) to patients in populations in which HLAB*1502 is likely to be present, such as Asians. Carriers of HLAB* 1502 are at higher risk of life-threatening skin reactions such as Stevens-Johnson syndrome.11
Several other pharmacogenomic applications for psychotropic medications have been suggested, but routine testing has not been recommended by the FDA or endorsed by any expert panel because sufficient clinical utility and cost-effectiveness have not been demonstrated. A brief summary of study findings and a few practical suggestions follow.
Polymorphisms in metabolizing enzymes have been investigated in patients receiving psychotropic drugs.
CYP2D6 and antidepressants
Many antidepressants show significant differences in plasma drug levels with CYP2D6 polymorphisms (in descending order of influence)55:
- Imipramine (Tofranil)
- Doxepin (Adapin, Silenor, Sinequan)
- Maprotiline (Deprilept, Ludiomil, Psymion)
- Trimipramine (Surmontil)
- Desipramine (Noraprim)
- Nortriptyline (Aventyl, Pamelor)
- Clomipramine (Anafranil)
- Paroxetine (Paxil)
- Venlafaxine (Effexor)
- Amitriptyline (Elavil)
- Mianserin
- Trazadone (Desyrel)
- Bupropion (Wellbutrin)
- Nefazodone (Serzone)
- Citalopram (Celexa)
- Sertraline (Zoloft).
CYP2D6 and antipsychotics
Several antipsychotics are also influenced by CYP2D6 polymorphisms (also in descending order of influence)55:
- Perphenazine (Trilafon)
- Thioridazine (Mellaril)
- Olanzapine (Zyprexa)
- Zuclopenthixol (Cisordinol, Clopixol, Acuphase)
- Aripiprazole (Abilify)
- Flupentixol (Depixol, Fluanxol)
- Haloperidol (Haldol)
- Perazine (Taxilan)
- Risperidone (Risperdal)
- Pimozide (Orap).
CYP2C19 and antidepressants
CYP2C19 polymorphisms are likewise associated with differences in drug metabolism for many antidepressants, such as (in descending order of CYP2C19-mediated influence)55:
- Trimipramine
- Doxepin
- Amitriptyline
- Imipramine
- Citalopram (Celexa)
- Clomipramine
- Moclobemide (Aurorix, Manerix)
- Sertraline
- Fluvoxamine (Luvox).
Clinical relevance of CYP2D6 and CYP2C19
Several studies have demonstrated that poor and intermediate CYP2D6 metabolizers have a higher incidence of adverse effects when taking CYP2D6-dependent antidepressants63–68; however, an almost equal number of studies did not find statistically significant associations.69–72 Likewise, several studies have found an association between ultra-rapid CYP2D6 metabolizer status and diminished response to antidepressants,65,73,74 but no association was found in a larger retrospective study.75
Routine CYP2D6 and CYP2C19 screening is not recommended when prescribing psychotropic drugs. However, reviews of the pharmacokinetic data have suggested a few practical applications when genetic status is already known. In general, clinicians can consider reducing the dose of tricyclic antidepressants by about 50% when prescribing to CYP2D6-poor-metabolizers.55,76–78
Genes that affect serotonin metabolism
Several genes in the serotonin pathway have been investigated to determine whether they influence patients’ susceptibility to depression and adverse effects and response to psychotropic medications.
SLC6A4. Polymorphisms in the promoter region of the serotonin transporter gene SLC6A4 appear to influence the treatment response and side-effect profiles of selective serotonin reuptake inhibitors (SSRIs). Carriers of the SLC6A4 5-HTTLPR L alleles have fewer side effects79 and better response to SSRI treatment, and carriers of the S allele have a higher incidence of antidepressant-induced mania80 and poorer response to SSRI treatment.81
5-HT. Polymorphisms in serotonin receptors (2A and 2C subtypes) appear to influence SSRI response and side effects. Carriers of 5-HT 2A C alleles had more severe adverse effects from paroxetine,71 but another 5-HT 2A polymorphism common to Asians is associated with better response to antidepressant therapy.82 A 5-HT 2C polymorphism was associated with a lower incidence of antipsychotic-induced weight gain.83
Although the understanding of these relationships is incomplete and routine pharmacogenomic testing is not currently recommended, reviews of the pharmacodynamic data have suggested a few practical applications when a patient’s genetic status is already known. One should consider:
- Selecting treatments other than SSRIs for depressed patients known to possess the SLC6A4 variant
- Selecting citalopram for depressed patients known to carry the 5-HT 2A polymorphism
- Avoiding treatment with antipsychotic drugs for patients known to possess the 5-HT 2C polymorphism.
THE FUTURE OF PHARMACOGENOMIC TESTING
The examples discussed in this article provide some insight about how pharmacogenomic testing is maturing and slowly being integrated into the practice of medicine. They also illustrate the complexity of the multiple stages of research that pharmacogenomic applications must go through in order to be adopted as standard practice.
In the future, pharmacogenomic data will continue to accumulate, and the clinical utility of many other pharmacogenomic tests may be uncovered. The FDA provides information on emerging pharmacogenomic tests at its Web site, www.fda.gov.11 Its up-to-date “Table of Valid Genomic Biomarkers in the Context of Approved Drug Labels” includes boxed warnings, recommendations, research outcomes, and relevant population genetics.
If the FDA continues its current policy, prospective randomized trials that show improvement in patient outcomes will remain the gold standard for determining the clinical significance of a pharmacogenomic test. Furthermore, cost-benefit analyses are likely to continue dictating policy regarding pharmacogenomic testing, and cost-benefit profiles should improve as technology advances and as information gathered from a single test becomes applicable to multiple medications and clinical scenarios.
In the meantime, physicians should become familiar with the terms used in medical genetics and pharmacogenomics and begin to understand genetic contributions to the outcomes of drug therapy. For example, understanding the consequences of metabolizer status and the frequency of variants in a given population can be tremendously helpful when advising our patients about anticipating potential problems when taking specific medications and about making informed decisions about pharmacogenomic testing.
This exchange of information alone may go a long way in improving therapy outcomes even when prospective pharmacogenomic testing is not routinely performed. Furthermore, an increasing number of patients will already have genotyping information available when they come to us, and clinicians need to be aware of the many pharmacogenomic applications recommended by the FDA when genetic status is known.10
- King HC, Sinha AA. Gene expression profile analysis by DNA micro-arrays: promise and pitfalls. JAMA 2001; 286:2280–2288.
- Nuckols TK, Paddock SM, Bower AG, et al. Costs of intravenous adverse drug events in academic and nonacademic intensive care units. Med Care 2008; 46:17–24.
- Gurwitz JH, Field TS, Judge J, et al. The incidence of adverse events in two long-term care facilities. Am J Med 2005; 118:251–258.
- Vargas E, Terleira A, Hernando F, et al. Effects of adverse drug reactions on length of stay in surgical intensive care units. Crit Care Med 2003; 31:694–698.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events in older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Classen DC, Pestotnik SL, Evans RS, Lloyd JF, Burke JP. Adverse drug events in hospitalized patients. Excess length of stay, extra costs, and attributable mortality. JAMA 1997; 277:301–306.
- Ernst FR, Grizzle AJ. Drug-related morbidity and mortality: updating the cost-of-illness model. J Am Pharm Assoc (Wash) 2001; 41:192–199.
- Bradford LD. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 2002; 3:229–243.
- Sadee W. Measuring cis-acting regulatory variants genome-wide: new insights into expression genetics and disease susceptibility. Genome Med 2009; 1:116.
- US Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labels. http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm. Accessed 1/18/2011.
- Budnitz DS, Shehab N, Kegler SR, Richards CL. Medication use leading to emergency department visits for adverse drug events in older adults. Ann Intern Med 2007; 147:755–765.
- Elias DJ, Topol EJ. Warfarin pharmacogenomics: a big step forward for individualized medicine: enlightened dosing of warfarin. Eur J Hum Genet 2008; 16:532–534.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Rettie AE, Wienkers LC, Gonzalez FJ, Trager WF, Korzekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Au N, Rettie AE. Pharmacogenomics of 4-hydroxycoumarin anticoagulants. Drug Metab Rev 2008; 40:355–375.
- García-Martín E, Martínez C, Ladero JM, Agúndez JA. Interethnic and intraethnic variability of CYP2C8 and CYP2C9 polymorphisms in healthy individuals. Mol Diagn Ther 2006; 10:29–40.
- Sullivan-Klose TH, Ghanayem BI, Bell DA, et al. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics 1996; 6:341–349.
- Wen MS, Lee M, Chen JJ, et al. Prospective study of warfarin dosage requirements based on CYP2C9 and VKORC1 genotypes. Clin Pharmacol Ther 2008; 84:83–89.
- Larramendy-Gozalo C, Yang JQ, Verstuyft C, et al. Genetic polymorphism of vitamin K epoxide reductase (VKORC1) 1173C>T in a Chinese and a Caucasian population. Basic Clin Pharmacol Toxicol 2006; 98:611–613.
- Caldwell MD, Awad T, Johnson JA, et al. CYP4F2 genetic variant alters required warfarin dose. Blood 2008; 111:4106–4112.
- Bristol-Myers Squibb. Coumadin (warfarin sodium) Prescribing Information. January 2010.
- Barnes-Jewish Hospital at Washington University Medical Center. Warfarin dosing. http://warfarindosing.orgAccessed 1/20/2011.
- Flockhart DA, O’Kane D, Williams MS, et al. Pharmacogenetic testing of CYP2C9 and VKORC1 alleles for warfarin. Genet Med 2008; 10:139–150.
- National Institutes of Health. ClinicalTrials.gov. http://clinicaltrials.gov. Accessed January 20, 2011.
- Eckman MH, Rosand J, Greenberg SM, Gage BF. Cost-effectiveness of using pharmacogenetic information in warfarin dosing for patients with nonvalvular atrial fibrillation. Ann Intern Med 2009; 150:73–83.
- Meckley LM, Gudgeon JM, Anderson JL, Williams MS, Veenstra DL. A policy model to evaluate the benefits, risks, and costs of warfarin pharmacogenomic testing. Pharmacoeconomics 2010; 28:61–74.
- Patrick AR, Avron J, Choudhry NK. Cost-effectiveness of genotype-guided warfarin dosing for patients with atrial fibrillation. Circ Cardiovasc Qual Outcomes 2009; 2:429–436.
- Haas S. New oral Xa and IIa inhibitors: updates on clinical trial results. J Thromb Thrombolysis 2008; 25:52–60.
- Bhatt DL. Tailoring antiplatelet therapy based on pharmacogenomics: how well do the data fit? JAMA 2009; 302:896–897.
- Shuldiner AR, O’Connell JR, Bliden KP, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009; 302:849–857.
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Paré G, Mehta SR, Yusuf S, et al. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med 2010; 363:1704–1714.
- Bristol-Myers Squibb/Sanofi Pharmaceutical Partnership. Plavix (clopidogrel bisulfate) prescribing information. August 2010.
- P450 Drug Interaction Table. Indiana University School of Medicine. http://medicine.iupui.edu/clinpharm/ddis/table.asp. Accessed 1/21/2011.
- Society for Cardiovascular Angiography and Interventions; Holmes DR, Dehmer GJ, Kaul S, Leifer D, O’Gara PT, Stein CM. ACCF/AHA clopidogrel clinical alert: approaches to the FDA “boxed warning”: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association. Circulation 2010; 122:537–557.
- National Institutes of Health. Genotype Guided Comparison of Clopidogrel and Prasugrel Outcomes Study. http://clinicaltrialsfeeds.org/clinical-trials/show/NCT00995514. Accessed 1/20/2011.
- Amarenco P, Labreuche J. Lipid management in the prevention of stroke: Review and updated meta-analysis of statins for stroke prevention. Lancet Neurol 2009; 8:453–463.
- SEARCH Collaborative Group, Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Romaine SP, Bailey KM, Hall AS, Balmforth AJ. The influence of SLC01B1 (OATP1B1) gene polymorphisms on response to statin therapy. Pharmacogenomics J 2010; 10:1–11.
- Wang D, Guo Y, Wrighton SA, Cooke GE, Sadee W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J 2010; Apr 13 [Epub ahead of print].
- Goetz MP, Rae JM, Suman VJ, et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 2005; 23:9312–9318.
- Beverage JN, Sissung TM, Sion AM, Danesi R, Figg WD. CYP2D6 polymorphisms and the impact on tamoxifen therapy. J Pharm Sci 2007; 96:2224–2231.
- Flockhart D. CYP2D6 genotyping and the pharmacogenetics of tamoxifen. Clin Adv Hematol Oncol 2008; 6:493–494.
- Goetz MP, Kamal A, Ames MM. Tamoxifen pharmacogenomics: the role of CYP2D6 as a predictor of drug response. Clin Pharmacol Ther 2008; 83:160–166.
- Stearns V, Rae JM. Pharmacogenetics and breast cancer endocrine therapy: CYP2D6 as a predictive factor for tamoxifen metabolism and drug response? Expert Rev Mol Med 2008; 10:e34.
- Dezentjé VO, Guchelaar HJ, Nortier JW, van del Velde CJ, Gelderblom H. Clinical implications of CYP2D6 genotyping in tamoxifen treatment for breast cancer. Clin Cancer Res 2009; 15:15–21.
- Higgins MJ, Rae JM, Flockhart DA, Hayes DF, Stearns V. Pharmacogenetics of tamoxifen: who should undergo CYP2D6 genetic testing? J Natl Compr Canc Netw 2009; 7:203–213.
- Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer 2009; 9:576–586.
- Steimer W, Zöpf K, von Amelunxen S, et al. Allele-specific change of concentration and functional gene dose for the prediction of steady-state serum concentrations of amitriptyline and nortriptyline in CYP2C19 and CYP2D6 extensive and intermediate metabolizers. Clinical Cancer 2004; 50:1623–1633.
- Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharm Ther 2008; 83:234–242.
- Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004; 9:442–473.
- Kirchheiner J, Schmidt H, Tzvetkov M, et al. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J 2007; 7:257–265.
- Kirchheiner J. CYP2D6 phenotype prediction from genotype: which system is the best? Clin Pharmacol Ther 2008; 83:225–227.
- Borges S, Desta Z, Jin Y, et al. Composite functional genetic and comedication CYP2D6 activity score in predicting tamoxifen drug exposure among breast cancer patients. J Clin Pharmacol 2010; 50:450–458.
- Hoffmann-La Roche Ltd. AmpliChip CYP450 Test. http://www.roche.com/assays/Pages/AmpliChipCYP450Test.aspx. Accessed 1/21/2011.
- Anzenbacher P, Anzenbacherová E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci 2001; 58:737–747.
- Koren G, Cairns J, Chitayat D, Gaedigk A, Leeder SJ. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet 2006; 368:704.
- Willmann S, Edginton AN, Coboeken K, Ahr G, Lippert J. Risk to the breast-fed neonate from codeine treatment to the mother: a quantitative mechanistic modeling study. Clin Pharm Ther 2009; 86:634–643.
- Grzesiak M, Beszłej A, Lebioda A, Jonkisz A, Dobosz T, Kienja A. [Retrospective assessment of the antidepressants tolerance in the group of patients with diagnosis of depression and different CYP2D6 genotype.] [In Polish] Psychiatr Pol 2003; 37:433–444.
- Laika B, Leucht S, Heres S, Steimer W. Intermediate metabolizer: increased side effects in psychoactive drug therapy. The key to cost-effectiveness of pretreatment CYP2D6 screening? Pharmacogenomics J 2009; 9:395–403.
- Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: Impact on adverse effects and nonresponse during treatment with antidepressants—a pilot study. Clin Pharm Ther 2004; 75:386–393.
- McAlpine DE, O’Kane DJ, Black JL, Mrazek DA. Cytochrome P450 2D6 genotype variation and venlafaxine dosage. Mayo Clin Proc 2007; 82:1065–1068.
- Chen S, Chou WH, Blouin RA, et al. The cytochrome P450 2D6 (CYP2D6) enzyme polymorphism: screening costs and influence on clinical outcomes in psychiatry. Clin Pharmacol Ther 1996; 60:522–534.
- Shams ME, Arneth B, Hiemke C, et al. CYP2D6 polymorphism and clinical effect of the antidepressant venlafaxine. J Clin Pharm Ther 2006; 31:493–502.
- Whyte EM, Romkes M, Mulsant BH, et al. CYP2D6 genotype and venlafaxine-XR concentrations in depressed elderly. Int J Geriatr Psychiatry 2006; 21:542–549.
- Roberts RL, Mulder RT, Joyce PR, Luty SE, Kennedy MA. No evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2004; 19:17–23.
- Murphy GM, Kremer C, Rodrigues HE, Schatzberg AF. Pharmacogenetics of antidepressant medication intolerance. Am J Psychiatry 2003; 160:1830–1835.
- Gillman PK. Re: no evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2005; 20:61–62.
- Gex-Fabry M, Eap CB, Oneda B, et al. CYP2D6 and ABCB1 genetic variability: influence on paroxetine plasma level and therapeutic response. Ther Drug Monit 2008; 30:474–482.
- Kawanishi C, Lundgren S, Agren H, Bertilsson L. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol 2004; 59:803–807.
- Serretti A, Calati R, Massat I, et al. Cytochrome P450 CYP1A2, CYP2C9, CYP2C19 and CYP2D6 genes are not associated with response and remission in a sample of depressive patients. Int Clin Psychopharmacol 2009; 24:250–256.
- de Leon J, Armstrong SC, Cozza KL. Clinical guidelines for psychiatrists for the use of pharmacogenetic testing for CYP450 2D6 and CYP450 2C19. Psychosomatics 2006; 47:75–85.
- de Leon J, Susce MT, Johnson M, et al. DNA microarray technology in the clinical environment: the AmpliChip CYP450 test for CYP2D6 and CYP2C19 genotyping. CNS Spectr 2009; 14:19–34.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Horstmann S, Binder EB. Pharmacogenomics of antidepressant drugs. Pharmacol Ther 2009; 124:57–73.
- Ferreira Ade A, Neves FS, da Rocha FF, et al. The role of 5-HTTLPR polymorphism in antidepressant-associated mania in bipolar disorder. J Affect Disord 2009; 112:267–272.
- Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry 2007; 12:247–257.
- Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry 2010; 15:473–500.
- Reynolds GP, Zhang Z, Zhang X. Polymorphism of the promoter region of the serotonin 5-HT(2C) receptor gene and clozapine-induced weight gain. Am J Psychiatry 2003; 160:677–679.
In many patients, certain drugs do not work as well as expected, whereas in other patients they cause toxic effects, even at lower doses. For some patients, the reason may be genetic.
Sizeable minorities of the population carry genetic variants—polymorphisms— that affect their response to various drugs. Thanks to genetic research, our understanding of the variability of drug response has advanced markedly in the last decade. Many relevant polymorphisms have been identified, and tests for some of them are available.
Armed with the knowledge of their patients’ genetic status, physicians could predict their response to certain drugs, leading to better efficacy, fewer adverse drug reactions, and a better cost-benefit ratio.
The possible impact is substantial, since more than half of the drugs most commonly involved in adverse drug reactions are metabolized by polymorphic enzymes.1 Adverse drug reactions remain a significant detriment to public health, having a substantial impact on rates of morbidity and death and on healthcare costs.2–8 In the United States, adverse drug reactions are a leading cause of death in hospitalized patients4 and are annually responsible for hundreds of thousands of deaths and hundreds of billions of dollars in added costs.2,4,6–8
In the meantime, physicians can educate their patients and promote efforts to incorporate genomic information into standard clinical decision-making.
This article offers an overview of pharmacogenomic testing, discussing implications and limitations of a few validated tests. Specifically, we will discuss testing that is relevant when using warfarin (Coumadin), clopidogrel (Plavix), statins, tamoxifen (Nolvadex), codeine, and psychotropic medications, as well as the future role of pharmacogenomic testing in medicine.
WHAT IS PHARMACOGENOMICS?
Pharmacogenomics is the study of how genetic factors relate to interindividual variability of drug response.
Many clinicians may not be familiar with the background and terminology used in the pharmacogenomic literature. Below, a brief review of the terminology is followed by a schematic describing the various stages of research involved in pharmacogenomics and the advancement of a test into standard practice.
The review and schematic may be helpful for evaluating the clinical significance of pharmacogenomics-related articles.
From genotype to phenotype
Genotype refers to the coding sequence of DNA base pairs for a particular gene, and phenotype (eg, disease or drug response) refers to a trait resulting from the protein product encoded by the gene. The name of a gene often refers to its protein product and is italicized (eg, the CYP3A4 gene encodes for the CYP3A4 enzyme).
Two alleles per autosomal gene (one paternal and one maternal) form the genotype. Heterozygotes possess two different alleles, and homozygotes possess two of the same alleles. The most common allele in a population is referred to as the wild type, and allele frequencies can vary greatly in different populations.9
Most sequence variations are single nucleotide polymorphisms (SNPs, pronounced “snips”), a single DNA base pair substitution that may result in a different gene product. SNPs can be classified as structural RNA polymorphisms (srSNPs), regulatory polymorphisms (rSNPs), or polymorphisms in coding regions (cSNPs)10: srSNPs alter mRNA processing and translation, rSNPs alter transcription, and cSNPs alter protein sequence and function.
Recently, genetic associations with a phenotype have been done on a large scale, with millions of SNPs measured in each of many subjects. This approach, called a genomewide association study or GWAS, has revealed countless candidate genes for clinical traits, but only a few have resulted in a practical clinical application. SNPs may by themselves exert a pharmacokinetic effect (ie, how the body processes the drug), a pharmacodynamic effect (ie, how the drug affects the body), or both, or they may act in concert with other genetic factors. Pharmacodynamic effects can result from a pharmacokinetic effect or can result from variations in a pharmacologic target.
Establishing a genotype-phenotype association can involve clinical studies, animal transgenic studies, or molecular and cellular functional assays.
Clinical applications are emerging
WARFARIN: IMPORTANCE OF CYP2C9, VKORC1
Warfarin is used for the long-term treatment and prevention of thromboembolic events.
This drug has a narrow therapeutic window and shows substantial interpatient dose variability. The start of warfarin therapy is associated with one of the highest rates of adverse events and emergency room visits of any single drug.12 More than 2 million patients start warfarin each year in the United States alone,13 and about 20% of them are hospitalized within the first 6 months because of bleeding due to overanticoagulation.14
The findings from a recent study suggest that pharmacogenomic testing may eventually allow more patients to safely benefit from warfarin therapy. In this large, nationwide, prospective study, hospitalization rates were 30% lower when pharmacogenomic testing was used.14 However, no reduction was seen in the time needed to reach the target international normalized ratio (INR) or in the need for INR checks at 6 months. Furthermore, this study used historical control data, and some or all of the reduction in hospitalization rates may be attributed to more frequent INR checks in the patients who underwent testing than in the historical control group.
A relationship between warfarin dose requirements and the genetic status of CYP2C9, which encodes a major drug-metabolizing enzyme, has been demonstrated in retrospective and prospective studies.15–17
S-warfarin is metabolized by CYP2C9, which is polymorphic
Warfarin contains equal amounts of two isomers, designated S and R. S-warfarin, which is more potent, is metabolized principally by CYP2C9, while R-warfarin is metabolized by CYP1A2, CYP2C19, and CYP3A4.
People who possess two copies of the wild type CYP2C9 gene CYP2C9*1 metabolize warfarin very well and so are called “extensive warfarin metabolizers.” Carriers of the allelic variants CYP2C9*2 and CYP2C9*3 (which have point mutations in exons 3 and 7 of CYP2C9, respectively), have less capacity. Compared with those who are homozygous for the wild-type gene, homozygous carriers of CYP2C9* 3 clear S-warfarin at a rate that is 90% lower, and those with the CYP2C9*1/*3, CYP2C9* 1/*2, CYP2C9*2/*2, or CYP2C9*2/*3 genotypes clear it at a rate 50% to 75% lower. A meta-analysis of 12 studies found that the CYP2C9 genotype accounted for 12% of the interindividual variability of warfarin dose requirements.18
About 8% of whites carry at least one copy of CYP2C9*2, as do 1% of African Americans; the allele is rare in Asian populations. The frequency of CYP2C9*3 is 6% in whites, 1% in African Americans, and 3% in Asians.19,20 People with CYP2C9*4 or CYP2C9*5 have a diminished capacity to clear warfarin; however, these variants occur so infrequently that their clinical relevance may be minimal.
Warfarin’s target, VKOR, is also polymorphic
Genetic variation in warfarin’s pharmacologic target, vitamin K 2,3-epoxide reductase (VKOR), also influences dose requirements. Warfarin decreases the synthesis of vitamin-K-dependent clotting factors by inhibiting VKOR. This inhibition depends on the patient’s C1 subunit gene, VKORC1. Patients with a guanine-to-adenine SNP 1,639 bases upstream of VKORC1 (−1639G>A) need lower warfarin doses—an average of 25% lower in those with the GA genotype (ie, one allele has guanine in the −1639 position and the other allele has adenine in that position) and 50% lower in those with the AA genotype compared with the wild-type genotype GG.21 This promoter SNP, positioned upstream (ie, before the gene-coding region), greatly influences VKORC1 expression.
A meta-analysis of 10 studies found that the VKORC1 polymorphism accounts for 25% of the interindividual variation in warfarin dose.18 In one study, the frequency of the AA genotype in a white population was 14%, AG 47%, and GG 39%; in a Chinese population the frequency of AA was 82%, AG 18%, and GG 0.35%.22
CYP4F2 and GGCX also affect warfarin’s dose requirements
Genetic variations in the enzymes CYP4F2 and gamma-glutamyl carboxylase (GGCX) also influence warfarin dose requirements. Although the data are limited and the effects are smaller than those of CYP2C9 and VKORC1, people with a SNP in CYP4F2 need 8% higher doses of warfarin, while those with a SNP in GGCX need 6% lower doses.23
CYP2C9 and VKORC1 testing is available
Currently, the clinical pharmacogenetic tests relevant for warfarin use are for CYP2C9 and VKORC1.10
The FDA has approved four warfarin pharmacogenetic test kits, but most third-party payers are reluctant to reimburse for testing because it is not currently considered a standard of care. Testing typically costs a few hundred dollars, but it should become less expensive as it becomes more commonplace. The current FDA-approved product label for warfarin does not recommend routine pharmacogenomic testing for determining initial or maintenance doses, but it does acknowledge that dose requirements are influenced by CYP2C9 and VKORC1 and states that genotype information, when available, can assist in selecting the starting dose.24
Clinical trials of warfarin pharmacogenomic testing are under way
Although genetic status can greatly influence an individual patient’s warfarin dosing requirement, routine prospective pharmacogenomic testing is not endorsed by the FDA or by other expert panels26 because there is currently insufficient evidence to recommend for or against it.
Several large prospective trials are under way. For example, the National Heart, Lung, and Blood Institute began a prospective trial in about 1,200 patients to evaluate the use of clinical plus genetic information to guide the initiation of warfarin therapy and to improve anticoagulation control for patients.27 The results, expected in September 2011, and those of other large prospective trials should provide adequate evidence for making recommendations about the clinical utility of routine pharmacogenetic testing for guiding warfarin therapy.
Several recent cost-utility and cost-effectiveness studies have attempted to quantify the value of pharmacogenomic testing for warfarin therapy,28–30 but their analyses are largely limited because the benefit (clinical utility) is yet to be sufficiently characterized.
The relevance of such analyses may soon be drastically diminished, as several non-vitamin-K-dependent blood thinners such as rivaroxaban (Xarelto), dabigatran (Pradaxa), and apixaban are poised to enter clinical practice.31
CLOPIDOGREL IS ACTIVATED BY CYP2C19
Clopidogrel, taken by about 40 million patients worldwide, is used to prevent atherothrombotic events and cardiac stent thrombosis when given along with aspirin.
Studies of clopidogrel pharmacogenomics
A recent genome-wide association study conducted in a cohort of 429 healthy Amish persons revealed a SNP in CYP2C19 to be associated with a diminished response to clopidogrel and to account for 12% of the variation in drug response.33 Traditional factors (the patient’s age, body-mass index, and cholesterol level) combined accounted for less than 10% of the variation.
Findings were similar in a subsequent investigation in 227 cardiac patients receiving clopidogrel: 21% of those with the variant had a cardiovascular ischemic event or died during a 1-year follow-up period compared with 10% of those without the variant (hazard ratio 2.42, P = .02).33
A 12-year prospective study investigating clopidogrel efficacy in 300 cardiac patients under the age of 45 used cardiovascular death, nonfatal myocardial infarction, and urgent coronary revascularization as end points. It concluded that the only independent predictor of these events was the patient’s CYP2C19 status.34
A study in 2,200 patients with recent myocardial infarction examined whether any of the known allelic variations that modulate clopidogrel’s absorption (ABCB1), metabolic activation (CYP3A4/5 and CYP2C19), or biologic activity (P2RY12 and ITGB3) was associated with a higher rate of the combined end point of all-cause mortality, nonfatal myocardial infarction, or stroke. None of the SNPs in CYP3A4/5, P2RY12, or ITGB3 that were evaluated was associated with a higher risk at 1 year. However, the allelic variations modulating clopidogrel’s absorption (ABCB1) and metabolism (CYP2C19) were associated with higher event rates. Patients with two variant ABCB1 alleles had a higher adjusted hazard ratio (95% confidence interval [CI] 1.2–2.47) than those with the wild-type allele. Patients who had one or two CYP2C19 loss-of-function alleles had a higher event rate than those with two wild-type alleles (95% CI 1.10–3.58 and 1.71–7.51, respectively).35
Conversely, researchers from the Population Health Research Institute found no association between poor-metabolizer status and treatment outcomes when CYP2C19 analysis was retrospectively added to the findings of two large clinical trials (combined N > 5,000). However, patients with acute coronary syndrome benefited more from clopidogrel treatment if they were ultra-rapid metabolizers (possessing the gain-of-function allele CYP2C19*17).36
Current status of clopidogrel testing: Uncertain
A current FDA boxed warning states that poor CYP2C19 metabolizers may not benefit from clopidogrel and recommends that prescribers consider alternative treatment for patients in this category.37 However, routine CYP2C19 testing is not recommended, and no firm recommendations have been established regarding dose adjustments for CYP2C19 status.
In 2010, the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association collectively pronounced the current evidence base insufficient for recommending routine pharmacogenomic testing.39
Needed are large-scale studies examining the cost-effectiveness and clinical utility of genotype-guided clopidogrel therapy compared with other therapy options such as prasugrel (Effient), an analogue not metabolized by CYP2C19. One such study, sponsored by Medco Health Solutions, plans to enroll 14,600 cardiac patients and has an estimated completion date in June 2011.40 The expectation that clopidogrel will be available in generic form in 2012 adds to the uncertainty regarding the cost-effectiveness of CYP2C19 testing for clopidogrel therapy.
STATINS: SLC01B1*5 INCREASES MYOPATHY RISK
Statins lower the concentration of low-density lipoprotein cholesterol (LDL-C), resulting in a relative-risk reduction of about 20% for each 1 mmol/L (39 mg/dL) decrement in LDL-C.41 They are one of the most commonly prescribed classes of drugs, but their side effects can limit their appeal: statin-induced myopathy occurs in about 1:1,000 to 1:10,000 patients and is difficult to predict.
SLC01B1. The Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH), a genome-wide association study, recently found a SNP (SLCO1B1* 5) in the SLC01B1 gene to be associated with a higher risk of statin-induced myopathy in cardiac patients receiving simvastatin (Zocor) 40 or 80 mg daily.42 The SLC01B1 gene, located on chromosome 12, influences the extent of the drug’s hepatic uptake and its serum concentration. Only the SLC01B1*5 SNP emerged as a predictor of statin-induced myopathy across the entire genome.42
The authors believe the findings are likely to apply to other statins. The mechanisms leading to statin-induced myopathy and the impact of statin pharmacogenomics are still unclear.43
CYP3A4. Other genetic variants may play a vital role in determining response to statin therapy. Carriers of a newly identified CYP3A4 polymorphism (intron 6 SNP, rs35599367, C>T) required significantly lower statin doses (0.2–0.6 times less) for optimal lipid control. The analyses included atorvastatin (Lipitor), simvastatin, and lovastatin (Mevacor), and the association was robust (P = .019).44
Statin pharmacogenomic testing is not routinely recommended
Routine pharmacogenomic testing for statin therapy is not recommended. Additional studies are needed to determine the clinical utility and cost-effectiveness of pharmacogenomic testing (involving a combination of several polymorphisms) in various patient populations delineated by type of statin, dose, and concomitant use of other drugs.
TAMOXIFEN IS ACTIVATED BY CYP2D6
Tamoxifen is prescribed to prevent the recurrence of estrogen-receptor-positive breast cancer, to treat metastatic breast cancer, to prevent cancer in high-risk populations, and to treat ductal carcinoma in situ.
Tamoxifen is metabolized to form endoxifen, which has much higher potency and higher systemic levels than tamoxifen.45 Both CYP2D6 and CYP3A4/5 are required to produce endoxifen via two intermediates, but CYP2D6 catalyzes the critical step leading to metabolic activation.
The CYP2D6 gene is highly polymorphic, with more than 75 allelic variants identified. Extensive literature is available describing the influence of CYP2D6 polymorphisms on tamoxifen metabolism and therapy outcomes.46–52 Several CYP2D6 variants result in reduced or no enzyme activity, and people who have more than two normally functioning alleles have exaggerated enzyme activity (gene amplification).
Classification of CYP2D6 status
Several systems have been developed to categorize the phenotypic activity of CYP2D6 based on genotype.
A genetic basis for the observed diversity in the metabolism of cytochrome P450 substrates was recognized more than 30 years ago. People were categorized as either extensive or poor metabolizers, reflecting normal vs impaired ability to metabolize the CYP2D6 substrates sparteine and debrisoquine. Later work expanded this system to include categories for intermediate (between poor and extensive) and ultra-rapid (better than extensive) metabolizers.
The genetic basis for these categories includes homozygosity for dysfunctional variants (the poor-metabolizer group) or extra copies of normal functioning variants (the ultra-rapid-metabolizer group).
Newer systems have been described for characterizing the CYP2D6 activity phenotype whereby CYP2D6 variants are assigned activity scores.53–56 The various scoring systems have been reviewed by Kirchheiner.57
A recent version of the activity scoring system also takes into consideration the many drugs that inhibit CYP2D6, such as amiodarone (Cordarone) and fluoxetine (Prozac) that can reduce the action of tamoxifen if given with it (Table 4).58 For example, the tamoxifen exposure (as predicted by the CYP2D6-activity score) experienced by a CYP2D6 extensive metabolizer taking a CYP2D6-inhibiting drug may be similar to the exposure experienced by a CYP2D6 poor metabolizer receiving the same tamoxifen dose but not taking a CYP2D6-inhibiting drug.
Likewise, the activity score of a CYP2D6 intermediate metabolizer taking a CYP2D6-inducing drug may be similar to that of a CYP2D6 ultra-rapid metabolizer not taking a CYP2D6-inducing drug. Examples of CYP2D6 inducers are dexamethasone, rifampin, and hyperforin (St. John’s wort).
While the newer systems are reported to provide better correlations between genotype and phenotype scores, the older scoring systems and the categorical names are still widely used (eg, in the FDA-approved AmpliChip CYP450 test from Roche,59 which includes genotype data for CYP2D6 and CYP2C19).
No firm recommendations for CYP2D6 testing in tamoxifen users
The different genotypes and phenotypes vary in prevalence in different ethnic groups, and significantly different activity levels for endoxifen formation are observed. Tamoxifen lacks efficacy in those who are poor CYP2D6 metabolizers—ie, about 7% of the white population.
However, the FDA has not made firm recommendations about CYP2D6 testing for prescribing tamoxifen because the evidence of benefit, although suggestive, has been considered insufficient.
Clinicians should be aware that tamoxifen’s efficacy is greatly reduced by concomitant therapy with CYP2D6-inhibiting drugs (Table 4).
Other genes affecting tamoxifen: CYP3A4/5, SULT1A1, and UGT2B15
Some investigators propose that polymorphisms in additional genes encoding enzymes in the tamoxifen metabolic and elimination pathways (eg, CYP3A4/5, SULT1A1, and UGT2B15) also need to be considered to account adequately for interindividual variation in drug response.
For example, CYP3A4 and CYP3A5 are also polymorphic, and large interindividual variation exists in their enzyme activities. These enzymes have overlapping substrate specificities, represent the most abundant drug-metabolizing enzymes in the human liver, and are involved in the biotransformation of a broad range of endogenous substrates and most drugs.60
Clinical studies evaluating the impact of CYP3A4/5 polymorphisms have been inconsistent in their conclusions, which is generally attributed to the relatively low functional impact or the low prevalence of the SNPs evaluated. Many of the nearly 100 CYP3A4/5 polymorphisms identified have not yet been characterized regarding their functional impact on enzyme expression or activity. CYP-3A4/5 enzyme activity is highly variable between individuals and warrants further study of its role in outcomes of tamoxifen therapy. Ongoing and future prospective clinical trials evaluating CYP2D6, CYP3A4/5, and other relevant polymorphisms are necessary to define their clinical relevance before routine genetic testing for tamoxifen can be justified.
CODEINE IS ALSO ACTIVATED BY CYP2D6
Codeine also depends on the CYP2D6 gene, as it must be activated to its more potent opioid metabolites, including morphine. Poor CYP2D6 metabolizers do not benefit from codeine therapy.
The pharmacogenomics of codeine has become a hot topic, especially regarding breast-feeding mothers. The debate was ignited with the publication in 2006 of a case report of an infant’s death, apparently the result of metabolic polymorphisms.61 The evolution of this debate and the outcome of the case may be noteworthy to clinicians, as they illustrate the gravity of public and patient interest in pharmacogenomic testing. In this case, the breast-feeding mother had taken codeine regularly for about 14 days when her 13-day-old infant died from toxic levels of morphine. Unknown to her and the prescriber, both the mother and infant were ultra-rapid CYP2D6 metabolizers, resulting in a more rapid and extensive conversion of codeine to morphine.
A logical strategy for preventing similar deaths would be routine CYP2D6 genotyping when prescribing codeine to breast-feeding mothers. However, after several investigations examined the metabolic and excretion pathways of codeine in their entirety, the FDA did not recommend routine CYP2D6 testing when prescribing codeine to breastfeeding mothers because several other factors, including rare genetic variations of other enzymes, proved necessary for reaching the opioid toxicity leading to the infant’s death.62
PHARMACOGENOMICS OF PSYCHOTROPIC DRUGS
Pharmacogenomic testing has clinical utility for some psychotropic drugs.
HLA-B and carbamazepine
Considered a standard of care, HLA-B genotyping is appropriate before prescribing carbamazepine (Tegretol, Equetro) to patients in populations in which HLAB*1502 is likely to be present, such as Asians. Carriers of HLAB* 1502 are at higher risk of life-threatening skin reactions such as Stevens-Johnson syndrome.11
Several other pharmacogenomic applications for psychotropic medications have been suggested, but routine testing has not been recommended by the FDA or endorsed by any expert panel because sufficient clinical utility and cost-effectiveness have not been demonstrated. A brief summary of study findings and a few practical suggestions follow.
Polymorphisms in metabolizing enzymes have been investigated in patients receiving psychotropic drugs.
CYP2D6 and antidepressants
Many antidepressants show significant differences in plasma drug levels with CYP2D6 polymorphisms (in descending order of influence)55:
- Imipramine (Tofranil)
- Doxepin (Adapin, Silenor, Sinequan)
- Maprotiline (Deprilept, Ludiomil, Psymion)
- Trimipramine (Surmontil)
- Desipramine (Noraprim)
- Nortriptyline (Aventyl, Pamelor)
- Clomipramine (Anafranil)
- Paroxetine (Paxil)
- Venlafaxine (Effexor)
- Amitriptyline (Elavil)
- Mianserin
- Trazadone (Desyrel)
- Bupropion (Wellbutrin)
- Nefazodone (Serzone)
- Citalopram (Celexa)
- Sertraline (Zoloft).
CYP2D6 and antipsychotics
Several antipsychotics are also influenced by CYP2D6 polymorphisms (also in descending order of influence)55:
- Perphenazine (Trilafon)
- Thioridazine (Mellaril)
- Olanzapine (Zyprexa)
- Zuclopenthixol (Cisordinol, Clopixol, Acuphase)
- Aripiprazole (Abilify)
- Flupentixol (Depixol, Fluanxol)
- Haloperidol (Haldol)
- Perazine (Taxilan)
- Risperidone (Risperdal)
- Pimozide (Orap).
CYP2C19 and antidepressants
CYP2C19 polymorphisms are likewise associated with differences in drug metabolism for many antidepressants, such as (in descending order of CYP2C19-mediated influence)55:
- Trimipramine
- Doxepin
- Amitriptyline
- Imipramine
- Citalopram (Celexa)
- Clomipramine
- Moclobemide (Aurorix, Manerix)
- Sertraline
- Fluvoxamine (Luvox).
Clinical relevance of CYP2D6 and CYP2C19
Several studies have demonstrated that poor and intermediate CYP2D6 metabolizers have a higher incidence of adverse effects when taking CYP2D6-dependent antidepressants63–68; however, an almost equal number of studies did not find statistically significant associations.69–72 Likewise, several studies have found an association between ultra-rapid CYP2D6 metabolizer status and diminished response to antidepressants,65,73,74 but no association was found in a larger retrospective study.75
Routine CYP2D6 and CYP2C19 screening is not recommended when prescribing psychotropic drugs. However, reviews of the pharmacokinetic data have suggested a few practical applications when genetic status is already known. In general, clinicians can consider reducing the dose of tricyclic antidepressants by about 50% when prescribing to CYP2D6-poor-metabolizers.55,76–78
Genes that affect serotonin metabolism
Several genes in the serotonin pathway have been investigated to determine whether they influence patients’ susceptibility to depression and adverse effects and response to psychotropic medications.
SLC6A4. Polymorphisms in the promoter region of the serotonin transporter gene SLC6A4 appear to influence the treatment response and side-effect profiles of selective serotonin reuptake inhibitors (SSRIs). Carriers of the SLC6A4 5-HTTLPR L alleles have fewer side effects79 and better response to SSRI treatment, and carriers of the S allele have a higher incidence of antidepressant-induced mania80 and poorer response to SSRI treatment.81
5-HT. Polymorphisms in serotonin receptors (2A and 2C subtypes) appear to influence SSRI response and side effects. Carriers of 5-HT 2A C alleles had more severe adverse effects from paroxetine,71 but another 5-HT 2A polymorphism common to Asians is associated with better response to antidepressant therapy.82 A 5-HT 2C polymorphism was associated with a lower incidence of antipsychotic-induced weight gain.83
Although the understanding of these relationships is incomplete and routine pharmacogenomic testing is not currently recommended, reviews of the pharmacodynamic data have suggested a few practical applications when a patient’s genetic status is already known. One should consider:
- Selecting treatments other than SSRIs for depressed patients known to possess the SLC6A4 variant
- Selecting citalopram for depressed patients known to carry the 5-HT 2A polymorphism
- Avoiding treatment with antipsychotic drugs for patients known to possess the 5-HT 2C polymorphism.
THE FUTURE OF PHARMACOGENOMIC TESTING
The examples discussed in this article provide some insight about how pharmacogenomic testing is maturing and slowly being integrated into the practice of medicine. They also illustrate the complexity of the multiple stages of research that pharmacogenomic applications must go through in order to be adopted as standard practice.
In the future, pharmacogenomic data will continue to accumulate, and the clinical utility of many other pharmacogenomic tests may be uncovered. The FDA provides information on emerging pharmacogenomic tests at its Web site, www.fda.gov.11 Its up-to-date “Table of Valid Genomic Biomarkers in the Context of Approved Drug Labels” includes boxed warnings, recommendations, research outcomes, and relevant population genetics.
If the FDA continues its current policy, prospective randomized trials that show improvement in patient outcomes will remain the gold standard for determining the clinical significance of a pharmacogenomic test. Furthermore, cost-benefit analyses are likely to continue dictating policy regarding pharmacogenomic testing, and cost-benefit profiles should improve as technology advances and as information gathered from a single test becomes applicable to multiple medications and clinical scenarios.
In the meantime, physicians should become familiar with the terms used in medical genetics and pharmacogenomics and begin to understand genetic contributions to the outcomes of drug therapy. For example, understanding the consequences of metabolizer status and the frequency of variants in a given population can be tremendously helpful when advising our patients about anticipating potential problems when taking specific medications and about making informed decisions about pharmacogenomic testing.
This exchange of information alone may go a long way in improving therapy outcomes even when prospective pharmacogenomic testing is not routinely performed. Furthermore, an increasing number of patients will already have genotyping information available when they come to us, and clinicians need to be aware of the many pharmacogenomic applications recommended by the FDA when genetic status is known.10
In many patients, certain drugs do not work as well as expected, whereas in other patients they cause toxic effects, even at lower doses. For some patients, the reason may be genetic.
Sizeable minorities of the population carry genetic variants—polymorphisms— that affect their response to various drugs. Thanks to genetic research, our understanding of the variability of drug response has advanced markedly in the last decade. Many relevant polymorphisms have been identified, and tests for some of them are available.
Armed with the knowledge of their patients’ genetic status, physicians could predict their response to certain drugs, leading to better efficacy, fewer adverse drug reactions, and a better cost-benefit ratio.
The possible impact is substantial, since more than half of the drugs most commonly involved in adverse drug reactions are metabolized by polymorphic enzymes.1 Adverse drug reactions remain a significant detriment to public health, having a substantial impact on rates of morbidity and death and on healthcare costs.2–8 In the United States, adverse drug reactions are a leading cause of death in hospitalized patients4 and are annually responsible for hundreds of thousands of deaths and hundreds of billions of dollars in added costs.2,4,6–8
In the meantime, physicians can educate their patients and promote efforts to incorporate genomic information into standard clinical decision-making.
This article offers an overview of pharmacogenomic testing, discussing implications and limitations of a few validated tests. Specifically, we will discuss testing that is relevant when using warfarin (Coumadin), clopidogrel (Plavix), statins, tamoxifen (Nolvadex), codeine, and psychotropic medications, as well as the future role of pharmacogenomic testing in medicine.
WHAT IS PHARMACOGENOMICS?
Pharmacogenomics is the study of how genetic factors relate to interindividual variability of drug response.
Many clinicians may not be familiar with the background and terminology used in the pharmacogenomic literature. Below, a brief review of the terminology is followed by a schematic describing the various stages of research involved in pharmacogenomics and the advancement of a test into standard practice.
The review and schematic may be helpful for evaluating the clinical significance of pharmacogenomics-related articles.
From genotype to phenotype
Genotype refers to the coding sequence of DNA base pairs for a particular gene, and phenotype (eg, disease or drug response) refers to a trait resulting from the protein product encoded by the gene. The name of a gene often refers to its protein product and is italicized (eg, the CYP3A4 gene encodes for the CYP3A4 enzyme).
Two alleles per autosomal gene (one paternal and one maternal) form the genotype. Heterozygotes possess two different alleles, and homozygotes possess two of the same alleles. The most common allele in a population is referred to as the wild type, and allele frequencies can vary greatly in different populations.9
Most sequence variations are single nucleotide polymorphisms (SNPs, pronounced “snips”), a single DNA base pair substitution that may result in a different gene product. SNPs can be classified as structural RNA polymorphisms (srSNPs), regulatory polymorphisms (rSNPs), or polymorphisms in coding regions (cSNPs)10: srSNPs alter mRNA processing and translation, rSNPs alter transcription, and cSNPs alter protein sequence and function.
Recently, genetic associations with a phenotype have been done on a large scale, with millions of SNPs measured in each of many subjects. This approach, called a genomewide association study or GWAS, has revealed countless candidate genes for clinical traits, but only a few have resulted in a practical clinical application. SNPs may by themselves exert a pharmacokinetic effect (ie, how the body processes the drug), a pharmacodynamic effect (ie, how the drug affects the body), or both, or they may act in concert with other genetic factors. Pharmacodynamic effects can result from a pharmacokinetic effect or can result from variations in a pharmacologic target.
Establishing a genotype-phenotype association can involve clinical studies, animal transgenic studies, or molecular and cellular functional assays.
Clinical applications are emerging
WARFARIN: IMPORTANCE OF CYP2C9, VKORC1
Warfarin is used for the long-term treatment and prevention of thromboembolic events.
This drug has a narrow therapeutic window and shows substantial interpatient dose variability. The start of warfarin therapy is associated with one of the highest rates of adverse events and emergency room visits of any single drug.12 More than 2 million patients start warfarin each year in the United States alone,13 and about 20% of them are hospitalized within the first 6 months because of bleeding due to overanticoagulation.14
The findings from a recent study suggest that pharmacogenomic testing may eventually allow more patients to safely benefit from warfarin therapy. In this large, nationwide, prospective study, hospitalization rates were 30% lower when pharmacogenomic testing was used.14 However, no reduction was seen in the time needed to reach the target international normalized ratio (INR) or in the need for INR checks at 6 months. Furthermore, this study used historical control data, and some or all of the reduction in hospitalization rates may be attributed to more frequent INR checks in the patients who underwent testing than in the historical control group.
A relationship between warfarin dose requirements and the genetic status of CYP2C9, which encodes a major drug-metabolizing enzyme, has been demonstrated in retrospective and prospective studies.15–17
S-warfarin is metabolized by CYP2C9, which is polymorphic
Warfarin contains equal amounts of two isomers, designated S and R. S-warfarin, which is more potent, is metabolized principally by CYP2C9, while R-warfarin is metabolized by CYP1A2, CYP2C19, and CYP3A4.
People who possess two copies of the wild type CYP2C9 gene CYP2C9*1 metabolize warfarin very well and so are called “extensive warfarin metabolizers.” Carriers of the allelic variants CYP2C9*2 and CYP2C9*3 (which have point mutations in exons 3 and 7 of CYP2C9, respectively), have less capacity. Compared with those who are homozygous for the wild-type gene, homozygous carriers of CYP2C9* 3 clear S-warfarin at a rate that is 90% lower, and those with the CYP2C9*1/*3, CYP2C9* 1/*2, CYP2C9*2/*2, or CYP2C9*2/*3 genotypes clear it at a rate 50% to 75% lower. A meta-analysis of 12 studies found that the CYP2C9 genotype accounted for 12% of the interindividual variability of warfarin dose requirements.18
About 8% of whites carry at least one copy of CYP2C9*2, as do 1% of African Americans; the allele is rare in Asian populations. The frequency of CYP2C9*3 is 6% in whites, 1% in African Americans, and 3% in Asians.19,20 People with CYP2C9*4 or CYP2C9*5 have a diminished capacity to clear warfarin; however, these variants occur so infrequently that their clinical relevance may be minimal.
Warfarin’s target, VKOR, is also polymorphic
Genetic variation in warfarin’s pharmacologic target, vitamin K 2,3-epoxide reductase (VKOR), also influences dose requirements. Warfarin decreases the synthesis of vitamin-K-dependent clotting factors by inhibiting VKOR. This inhibition depends on the patient’s C1 subunit gene, VKORC1. Patients with a guanine-to-adenine SNP 1,639 bases upstream of VKORC1 (−1639G>A) need lower warfarin doses—an average of 25% lower in those with the GA genotype (ie, one allele has guanine in the −1639 position and the other allele has adenine in that position) and 50% lower in those with the AA genotype compared with the wild-type genotype GG.21 This promoter SNP, positioned upstream (ie, before the gene-coding region), greatly influences VKORC1 expression.
A meta-analysis of 10 studies found that the VKORC1 polymorphism accounts for 25% of the interindividual variation in warfarin dose.18 In one study, the frequency of the AA genotype in a white population was 14%, AG 47%, and GG 39%; in a Chinese population the frequency of AA was 82%, AG 18%, and GG 0.35%.22
CYP4F2 and GGCX also affect warfarin’s dose requirements
Genetic variations in the enzymes CYP4F2 and gamma-glutamyl carboxylase (GGCX) also influence warfarin dose requirements. Although the data are limited and the effects are smaller than those of CYP2C9 and VKORC1, people with a SNP in CYP4F2 need 8% higher doses of warfarin, while those with a SNP in GGCX need 6% lower doses.23
CYP2C9 and VKORC1 testing is available
Currently, the clinical pharmacogenetic tests relevant for warfarin use are for CYP2C9 and VKORC1.10
The FDA has approved four warfarin pharmacogenetic test kits, but most third-party payers are reluctant to reimburse for testing because it is not currently considered a standard of care. Testing typically costs a few hundred dollars, but it should become less expensive as it becomes more commonplace. The current FDA-approved product label for warfarin does not recommend routine pharmacogenomic testing for determining initial or maintenance doses, but it does acknowledge that dose requirements are influenced by CYP2C9 and VKORC1 and states that genotype information, when available, can assist in selecting the starting dose.24
Clinical trials of warfarin pharmacogenomic testing are under way
Although genetic status can greatly influence an individual patient’s warfarin dosing requirement, routine prospective pharmacogenomic testing is not endorsed by the FDA or by other expert panels26 because there is currently insufficient evidence to recommend for or against it.
Several large prospective trials are under way. For example, the National Heart, Lung, and Blood Institute began a prospective trial in about 1,200 patients to evaluate the use of clinical plus genetic information to guide the initiation of warfarin therapy and to improve anticoagulation control for patients.27 The results, expected in September 2011, and those of other large prospective trials should provide adequate evidence for making recommendations about the clinical utility of routine pharmacogenetic testing for guiding warfarin therapy.
Several recent cost-utility and cost-effectiveness studies have attempted to quantify the value of pharmacogenomic testing for warfarin therapy,28–30 but their analyses are largely limited because the benefit (clinical utility) is yet to be sufficiently characterized.
The relevance of such analyses may soon be drastically diminished, as several non-vitamin-K-dependent blood thinners such as rivaroxaban (Xarelto), dabigatran (Pradaxa), and apixaban are poised to enter clinical practice.31
CLOPIDOGREL IS ACTIVATED BY CYP2C19
Clopidogrel, taken by about 40 million patients worldwide, is used to prevent atherothrombotic events and cardiac stent thrombosis when given along with aspirin.
Studies of clopidogrel pharmacogenomics
A recent genome-wide association study conducted in a cohort of 429 healthy Amish persons revealed a SNP in CYP2C19 to be associated with a diminished response to clopidogrel and to account for 12% of the variation in drug response.33 Traditional factors (the patient’s age, body-mass index, and cholesterol level) combined accounted for less than 10% of the variation.
Findings were similar in a subsequent investigation in 227 cardiac patients receiving clopidogrel: 21% of those with the variant had a cardiovascular ischemic event or died during a 1-year follow-up period compared with 10% of those without the variant (hazard ratio 2.42, P = .02).33
A 12-year prospective study investigating clopidogrel efficacy in 300 cardiac patients under the age of 45 used cardiovascular death, nonfatal myocardial infarction, and urgent coronary revascularization as end points. It concluded that the only independent predictor of these events was the patient’s CYP2C19 status.34
A study in 2,200 patients with recent myocardial infarction examined whether any of the known allelic variations that modulate clopidogrel’s absorption (ABCB1), metabolic activation (CYP3A4/5 and CYP2C19), or biologic activity (P2RY12 and ITGB3) was associated with a higher rate of the combined end point of all-cause mortality, nonfatal myocardial infarction, or stroke. None of the SNPs in CYP3A4/5, P2RY12, or ITGB3 that were evaluated was associated with a higher risk at 1 year. However, the allelic variations modulating clopidogrel’s absorption (ABCB1) and metabolism (CYP2C19) were associated with higher event rates. Patients with two variant ABCB1 alleles had a higher adjusted hazard ratio (95% confidence interval [CI] 1.2–2.47) than those with the wild-type allele. Patients who had one or two CYP2C19 loss-of-function alleles had a higher event rate than those with two wild-type alleles (95% CI 1.10–3.58 and 1.71–7.51, respectively).35
Conversely, researchers from the Population Health Research Institute found no association between poor-metabolizer status and treatment outcomes when CYP2C19 analysis was retrospectively added to the findings of two large clinical trials (combined N > 5,000). However, patients with acute coronary syndrome benefited more from clopidogrel treatment if they were ultra-rapid metabolizers (possessing the gain-of-function allele CYP2C19*17).36
Current status of clopidogrel testing: Uncertain
A current FDA boxed warning states that poor CYP2C19 metabolizers may not benefit from clopidogrel and recommends that prescribers consider alternative treatment for patients in this category.37 However, routine CYP2C19 testing is not recommended, and no firm recommendations have been established regarding dose adjustments for CYP2C19 status.
In 2010, the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association collectively pronounced the current evidence base insufficient for recommending routine pharmacogenomic testing.39
Needed are large-scale studies examining the cost-effectiveness and clinical utility of genotype-guided clopidogrel therapy compared with other therapy options such as prasugrel (Effient), an analogue not metabolized by CYP2C19. One such study, sponsored by Medco Health Solutions, plans to enroll 14,600 cardiac patients and has an estimated completion date in June 2011.40 The expectation that clopidogrel will be available in generic form in 2012 adds to the uncertainty regarding the cost-effectiveness of CYP2C19 testing for clopidogrel therapy.
STATINS: SLC01B1*5 INCREASES MYOPATHY RISK
Statins lower the concentration of low-density lipoprotein cholesterol (LDL-C), resulting in a relative-risk reduction of about 20% for each 1 mmol/L (39 mg/dL) decrement in LDL-C.41 They are one of the most commonly prescribed classes of drugs, but their side effects can limit their appeal: statin-induced myopathy occurs in about 1:1,000 to 1:10,000 patients and is difficult to predict.
SLC01B1. The Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH), a genome-wide association study, recently found a SNP (SLCO1B1* 5) in the SLC01B1 gene to be associated with a higher risk of statin-induced myopathy in cardiac patients receiving simvastatin (Zocor) 40 or 80 mg daily.42 The SLC01B1 gene, located on chromosome 12, influences the extent of the drug’s hepatic uptake and its serum concentration. Only the SLC01B1*5 SNP emerged as a predictor of statin-induced myopathy across the entire genome.42
The authors believe the findings are likely to apply to other statins. The mechanisms leading to statin-induced myopathy and the impact of statin pharmacogenomics are still unclear.43
CYP3A4. Other genetic variants may play a vital role in determining response to statin therapy. Carriers of a newly identified CYP3A4 polymorphism (intron 6 SNP, rs35599367, C>T) required significantly lower statin doses (0.2–0.6 times less) for optimal lipid control. The analyses included atorvastatin (Lipitor), simvastatin, and lovastatin (Mevacor), and the association was robust (P = .019).44
Statin pharmacogenomic testing is not routinely recommended
Routine pharmacogenomic testing for statin therapy is not recommended. Additional studies are needed to determine the clinical utility and cost-effectiveness of pharmacogenomic testing (involving a combination of several polymorphisms) in various patient populations delineated by type of statin, dose, and concomitant use of other drugs.
TAMOXIFEN IS ACTIVATED BY CYP2D6
Tamoxifen is prescribed to prevent the recurrence of estrogen-receptor-positive breast cancer, to treat metastatic breast cancer, to prevent cancer in high-risk populations, and to treat ductal carcinoma in situ.
Tamoxifen is metabolized to form endoxifen, which has much higher potency and higher systemic levels than tamoxifen.45 Both CYP2D6 and CYP3A4/5 are required to produce endoxifen via two intermediates, but CYP2D6 catalyzes the critical step leading to metabolic activation.
The CYP2D6 gene is highly polymorphic, with more than 75 allelic variants identified. Extensive literature is available describing the influence of CYP2D6 polymorphisms on tamoxifen metabolism and therapy outcomes.46–52 Several CYP2D6 variants result in reduced or no enzyme activity, and people who have more than two normally functioning alleles have exaggerated enzyme activity (gene amplification).
Classification of CYP2D6 status
Several systems have been developed to categorize the phenotypic activity of CYP2D6 based on genotype.
A genetic basis for the observed diversity in the metabolism of cytochrome P450 substrates was recognized more than 30 years ago. People were categorized as either extensive or poor metabolizers, reflecting normal vs impaired ability to metabolize the CYP2D6 substrates sparteine and debrisoquine. Later work expanded this system to include categories for intermediate (between poor and extensive) and ultra-rapid (better than extensive) metabolizers.
The genetic basis for these categories includes homozygosity for dysfunctional variants (the poor-metabolizer group) or extra copies of normal functioning variants (the ultra-rapid-metabolizer group).
Newer systems have been described for characterizing the CYP2D6 activity phenotype whereby CYP2D6 variants are assigned activity scores.53–56 The various scoring systems have been reviewed by Kirchheiner.57
A recent version of the activity scoring system also takes into consideration the many drugs that inhibit CYP2D6, such as amiodarone (Cordarone) and fluoxetine (Prozac) that can reduce the action of tamoxifen if given with it (Table 4).58 For example, the tamoxifen exposure (as predicted by the CYP2D6-activity score) experienced by a CYP2D6 extensive metabolizer taking a CYP2D6-inhibiting drug may be similar to the exposure experienced by a CYP2D6 poor metabolizer receiving the same tamoxifen dose but not taking a CYP2D6-inhibiting drug.
Likewise, the activity score of a CYP2D6 intermediate metabolizer taking a CYP2D6-inducing drug may be similar to that of a CYP2D6 ultra-rapid metabolizer not taking a CYP2D6-inducing drug. Examples of CYP2D6 inducers are dexamethasone, rifampin, and hyperforin (St. John’s wort).
While the newer systems are reported to provide better correlations between genotype and phenotype scores, the older scoring systems and the categorical names are still widely used (eg, in the FDA-approved AmpliChip CYP450 test from Roche,59 which includes genotype data for CYP2D6 and CYP2C19).
No firm recommendations for CYP2D6 testing in tamoxifen users
The different genotypes and phenotypes vary in prevalence in different ethnic groups, and significantly different activity levels for endoxifen formation are observed. Tamoxifen lacks efficacy in those who are poor CYP2D6 metabolizers—ie, about 7% of the white population.
However, the FDA has not made firm recommendations about CYP2D6 testing for prescribing tamoxifen because the evidence of benefit, although suggestive, has been considered insufficient.
Clinicians should be aware that tamoxifen’s efficacy is greatly reduced by concomitant therapy with CYP2D6-inhibiting drugs (Table 4).
Other genes affecting tamoxifen: CYP3A4/5, SULT1A1, and UGT2B15
Some investigators propose that polymorphisms in additional genes encoding enzymes in the tamoxifen metabolic and elimination pathways (eg, CYP3A4/5, SULT1A1, and UGT2B15) also need to be considered to account adequately for interindividual variation in drug response.
For example, CYP3A4 and CYP3A5 are also polymorphic, and large interindividual variation exists in their enzyme activities. These enzymes have overlapping substrate specificities, represent the most abundant drug-metabolizing enzymes in the human liver, and are involved in the biotransformation of a broad range of endogenous substrates and most drugs.60
Clinical studies evaluating the impact of CYP3A4/5 polymorphisms have been inconsistent in their conclusions, which is generally attributed to the relatively low functional impact or the low prevalence of the SNPs evaluated. Many of the nearly 100 CYP3A4/5 polymorphisms identified have not yet been characterized regarding their functional impact on enzyme expression or activity. CYP-3A4/5 enzyme activity is highly variable between individuals and warrants further study of its role in outcomes of tamoxifen therapy. Ongoing and future prospective clinical trials evaluating CYP2D6, CYP3A4/5, and other relevant polymorphisms are necessary to define their clinical relevance before routine genetic testing for tamoxifen can be justified.
CODEINE IS ALSO ACTIVATED BY CYP2D6
Codeine also depends on the CYP2D6 gene, as it must be activated to its more potent opioid metabolites, including morphine. Poor CYP2D6 metabolizers do not benefit from codeine therapy.
The pharmacogenomics of codeine has become a hot topic, especially regarding breast-feeding mothers. The debate was ignited with the publication in 2006 of a case report of an infant’s death, apparently the result of metabolic polymorphisms.61 The evolution of this debate and the outcome of the case may be noteworthy to clinicians, as they illustrate the gravity of public and patient interest in pharmacogenomic testing. In this case, the breast-feeding mother had taken codeine regularly for about 14 days when her 13-day-old infant died from toxic levels of morphine. Unknown to her and the prescriber, both the mother and infant were ultra-rapid CYP2D6 metabolizers, resulting in a more rapid and extensive conversion of codeine to morphine.
A logical strategy for preventing similar deaths would be routine CYP2D6 genotyping when prescribing codeine to breast-feeding mothers. However, after several investigations examined the metabolic and excretion pathways of codeine in their entirety, the FDA did not recommend routine CYP2D6 testing when prescribing codeine to breastfeeding mothers because several other factors, including rare genetic variations of other enzymes, proved necessary for reaching the opioid toxicity leading to the infant’s death.62
PHARMACOGENOMICS OF PSYCHOTROPIC DRUGS
Pharmacogenomic testing has clinical utility for some psychotropic drugs.
HLA-B and carbamazepine
Considered a standard of care, HLA-B genotyping is appropriate before prescribing carbamazepine (Tegretol, Equetro) to patients in populations in which HLAB*1502 is likely to be present, such as Asians. Carriers of HLAB* 1502 are at higher risk of life-threatening skin reactions such as Stevens-Johnson syndrome.11
Several other pharmacogenomic applications for psychotropic medications have been suggested, but routine testing has not been recommended by the FDA or endorsed by any expert panel because sufficient clinical utility and cost-effectiveness have not been demonstrated. A brief summary of study findings and a few practical suggestions follow.
Polymorphisms in metabolizing enzymes have been investigated in patients receiving psychotropic drugs.
CYP2D6 and antidepressants
Many antidepressants show significant differences in plasma drug levels with CYP2D6 polymorphisms (in descending order of influence)55:
- Imipramine (Tofranil)
- Doxepin (Adapin, Silenor, Sinequan)
- Maprotiline (Deprilept, Ludiomil, Psymion)
- Trimipramine (Surmontil)
- Desipramine (Noraprim)
- Nortriptyline (Aventyl, Pamelor)
- Clomipramine (Anafranil)
- Paroxetine (Paxil)
- Venlafaxine (Effexor)
- Amitriptyline (Elavil)
- Mianserin
- Trazadone (Desyrel)
- Bupropion (Wellbutrin)
- Nefazodone (Serzone)
- Citalopram (Celexa)
- Sertraline (Zoloft).
CYP2D6 and antipsychotics
Several antipsychotics are also influenced by CYP2D6 polymorphisms (also in descending order of influence)55:
- Perphenazine (Trilafon)
- Thioridazine (Mellaril)
- Olanzapine (Zyprexa)
- Zuclopenthixol (Cisordinol, Clopixol, Acuphase)
- Aripiprazole (Abilify)
- Flupentixol (Depixol, Fluanxol)
- Haloperidol (Haldol)
- Perazine (Taxilan)
- Risperidone (Risperdal)
- Pimozide (Orap).
CYP2C19 and antidepressants
CYP2C19 polymorphisms are likewise associated with differences in drug metabolism for many antidepressants, such as (in descending order of CYP2C19-mediated influence)55:
- Trimipramine
- Doxepin
- Amitriptyline
- Imipramine
- Citalopram (Celexa)
- Clomipramine
- Moclobemide (Aurorix, Manerix)
- Sertraline
- Fluvoxamine (Luvox).
Clinical relevance of CYP2D6 and CYP2C19
Several studies have demonstrated that poor and intermediate CYP2D6 metabolizers have a higher incidence of adverse effects when taking CYP2D6-dependent antidepressants63–68; however, an almost equal number of studies did not find statistically significant associations.69–72 Likewise, several studies have found an association between ultra-rapid CYP2D6 metabolizer status and diminished response to antidepressants,65,73,74 but no association was found in a larger retrospective study.75
Routine CYP2D6 and CYP2C19 screening is not recommended when prescribing psychotropic drugs. However, reviews of the pharmacokinetic data have suggested a few practical applications when genetic status is already known. In general, clinicians can consider reducing the dose of tricyclic antidepressants by about 50% when prescribing to CYP2D6-poor-metabolizers.55,76–78
Genes that affect serotonin metabolism
Several genes in the serotonin pathway have been investigated to determine whether they influence patients’ susceptibility to depression and adverse effects and response to psychotropic medications.
SLC6A4. Polymorphisms in the promoter region of the serotonin transporter gene SLC6A4 appear to influence the treatment response and side-effect profiles of selective serotonin reuptake inhibitors (SSRIs). Carriers of the SLC6A4 5-HTTLPR L alleles have fewer side effects79 and better response to SSRI treatment, and carriers of the S allele have a higher incidence of antidepressant-induced mania80 and poorer response to SSRI treatment.81
5-HT. Polymorphisms in serotonin receptors (2A and 2C subtypes) appear to influence SSRI response and side effects. Carriers of 5-HT 2A C alleles had more severe adverse effects from paroxetine,71 but another 5-HT 2A polymorphism common to Asians is associated with better response to antidepressant therapy.82 A 5-HT 2C polymorphism was associated with a lower incidence of antipsychotic-induced weight gain.83
Although the understanding of these relationships is incomplete and routine pharmacogenomic testing is not currently recommended, reviews of the pharmacodynamic data have suggested a few practical applications when a patient’s genetic status is already known. One should consider:
- Selecting treatments other than SSRIs for depressed patients known to possess the SLC6A4 variant
- Selecting citalopram for depressed patients known to carry the 5-HT 2A polymorphism
- Avoiding treatment with antipsychotic drugs for patients known to possess the 5-HT 2C polymorphism.
THE FUTURE OF PHARMACOGENOMIC TESTING
The examples discussed in this article provide some insight about how pharmacogenomic testing is maturing and slowly being integrated into the practice of medicine. They also illustrate the complexity of the multiple stages of research that pharmacogenomic applications must go through in order to be adopted as standard practice.
In the future, pharmacogenomic data will continue to accumulate, and the clinical utility of many other pharmacogenomic tests may be uncovered. The FDA provides information on emerging pharmacogenomic tests at its Web site, www.fda.gov.11 Its up-to-date “Table of Valid Genomic Biomarkers in the Context of Approved Drug Labels” includes boxed warnings, recommendations, research outcomes, and relevant population genetics.
If the FDA continues its current policy, prospective randomized trials that show improvement in patient outcomes will remain the gold standard for determining the clinical significance of a pharmacogenomic test. Furthermore, cost-benefit analyses are likely to continue dictating policy regarding pharmacogenomic testing, and cost-benefit profiles should improve as technology advances and as information gathered from a single test becomes applicable to multiple medications and clinical scenarios.
In the meantime, physicians should become familiar with the terms used in medical genetics and pharmacogenomics and begin to understand genetic contributions to the outcomes of drug therapy. For example, understanding the consequences of metabolizer status and the frequency of variants in a given population can be tremendously helpful when advising our patients about anticipating potential problems when taking specific medications and about making informed decisions about pharmacogenomic testing.
This exchange of information alone may go a long way in improving therapy outcomes even when prospective pharmacogenomic testing is not routinely performed. Furthermore, an increasing number of patients will already have genotyping information available when they come to us, and clinicians need to be aware of the many pharmacogenomic applications recommended by the FDA when genetic status is known.10
- King HC, Sinha AA. Gene expression profile analysis by DNA micro-arrays: promise and pitfalls. JAMA 2001; 286:2280–2288.
- Nuckols TK, Paddock SM, Bower AG, et al. Costs of intravenous adverse drug events in academic and nonacademic intensive care units. Med Care 2008; 46:17–24.
- Gurwitz JH, Field TS, Judge J, et al. The incidence of adverse events in two long-term care facilities. Am J Med 2005; 118:251–258.
- Vargas E, Terleira A, Hernando F, et al. Effects of adverse drug reactions on length of stay in surgical intensive care units. Crit Care Med 2003; 31:694–698.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events in older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Classen DC, Pestotnik SL, Evans RS, Lloyd JF, Burke JP. Adverse drug events in hospitalized patients. Excess length of stay, extra costs, and attributable mortality. JAMA 1997; 277:301–306.
- Ernst FR, Grizzle AJ. Drug-related morbidity and mortality: updating the cost-of-illness model. J Am Pharm Assoc (Wash) 2001; 41:192–199.
- Bradford LD. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 2002; 3:229–243.
- Sadee W. Measuring cis-acting regulatory variants genome-wide: new insights into expression genetics and disease susceptibility. Genome Med 2009; 1:116.
- US Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labels. http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm. Accessed 1/18/2011.
- Budnitz DS, Shehab N, Kegler SR, Richards CL. Medication use leading to emergency department visits for adverse drug events in older adults. Ann Intern Med 2007; 147:755–765.
- Elias DJ, Topol EJ. Warfarin pharmacogenomics: a big step forward for individualized medicine: enlightened dosing of warfarin. Eur J Hum Genet 2008; 16:532–534.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Rettie AE, Wienkers LC, Gonzalez FJ, Trager WF, Korzekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Au N, Rettie AE. Pharmacogenomics of 4-hydroxycoumarin anticoagulants. Drug Metab Rev 2008; 40:355–375.
- García-Martín E, Martínez C, Ladero JM, Agúndez JA. Interethnic and intraethnic variability of CYP2C8 and CYP2C9 polymorphisms in healthy individuals. Mol Diagn Ther 2006; 10:29–40.
- Sullivan-Klose TH, Ghanayem BI, Bell DA, et al. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics 1996; 6:341–349.
- Wen MS, Lee M, Chen JJ, et al. Prospective study of warfarin dosage requirements based on CYP2C9 and VKORC1 genotypes. Clin Pharmacol Ther 2008; 84:83–89.
- Larramendy-Gozalo C, Yang JQ, Verstuyft C, et al. Genetic polymorphism of vitamin K epoxide reductase (VKORC1) 1173C>T in a Chinese and a Caucasian population. Basic Clin Pharmacol Toxicol 2006; 98:611–613.
- Caldwell MD, Awad T, Johnson JA, et al. CYP4F2 genetic variant alters required warfarin dose. Blood 2008; 111:4106–4112.
- Bristol-Myers Squibb. Coumadin (warfarin sodium) Prescribing Information. January 2010.
- Barnes-Jewish Hospital at Washington University Medical Center. Warfarin dosing. http://warfarindosing.orgAccessed 1/20/2011.
- Flockhart DA, O’Kane D, Williams MS, et al. Pharmacogenetic testing of CYP2C9 and VKORC1 alleles for warfarin. Genet Med 2008; 10:139–150.
- National Institutes of Health. ClinicalTrials.gov. http://clinicaltrials.gov. Accessed January 20, 2011.
- Eckman MH, Rosand J, Greenberg SM, Gage BF. Cost-effectiveness of using pharmacogenetic information in warfarin dosing for patients with nonvalvular atrial fibrillation. Ann Intern Med 2009; 150:73–83.
- Meckley LM, Gudgeon JM, Anderson JL, Williams MS, Veenstra DL. A policy model to evaluate the benefits, risks, and costs of warfarin pharmacogenomic testing. Pharmacoeconomics 2010; 28:61–74.
- Patrick AR, Avron J, Choudhry NK. Cost-effectiveness of genotype-guided warfarin dosing for patients with atrial fibrillation. Circ Cardiovasc Qual Outcomes 2009; 2:429–436.
- Haas S. New oral Xa and IIa inhibitors: updates on clinical trial results. J Thromb Thrombolysis 2008; 25:52–60.
- Bhatt DL. Tailoring antiplatelet therapy based on pharmacogenomics: how well do the data fit? JAMA 2009; 302:896–897.
- Shuldiner AR, O’Connell JR, Bliden KP, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009; 302:849–857.
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Paré G, Mehta SR, Yusuf S, et al. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med 2010; 363:1704–1714.
- Bristol-Myers Squibb/Sanofi Pharmaceutical Partnership. Plavix (clopidogrel bisulfate) prescribing information. August 2010.
- P450 Drug Interaction Table. Indiana University School of Medicine. http://medicine.iupui.edu/clinpharm/ddis/table.asp. Accessed 1/21/2011.
- Society for Cardiovascular Angiography and Interventions; Holmes DR, Dehmer GJ, Kaul S, Leifer D, O’Gara PT, Stein CM. ACCF/AHA clopidogrel clinical alert: approaches to the FDA “boxed warning”: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association. Circulation 2010; 122:537–557.
- National Institutes of Health. Genotype Guided Comparison of Clopidogrel and Prasugrel Outcomes Study. http://clinicaltrialsfeeds.org/clinical-trials/show/NCT00995514. Accessed 1/20/2011.
- Amarenco P, Labreuche J. Lipid management in the prevention of stroke: Review and updated meta-analysis of statins for stroke prevention. Lancet Neurol 2009; 8:453–463.
- SEARCH Collaborative Group, Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Romaine SP, Bailey KM, Hall AS, Balmforth AJ. The influence of SLC01B1 (OATP1B1) gene polymorphisms on response to statin therapy. Pharmacogenomics J 2010; 10:1–11.
- Wang D, Guo Y, Wrighton SA, Cooke GE, Sadee W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J 2010; Apr 13 [Epub ahead of print].
- Goetz MP, Rae JM, Suman VJ, et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 2005; 23:9312–9318.
- Beverage JN, Sissung TM, Sion AM, Danesi R, Figg WD. CYP2D6 polymorphisms and the impact on tamoxifen therapy. J Pharm Sci 2007; 96:2224–2231.
- Flockhart D. CYP2D6 genotyping and the pharmacogenetics of tamoxifen. Clin Adv Hematol Oncol 2008; 6:493–494.
- Goetz MP, Kamal A, Ames MM. Tamoxifen pharmacogenomics: the role of CYP2D6 as a predictor of drug response. Clin Pharmacol Ther 2008; 83:160–166.
- Stearns V, Rae JM. Pharmacogenetics and breast cancer endocrine therapy: CYP2D6 as a predictive factor for tamoxifen metabolism and drug response? Expert Rev Mol Med 2008; 10:e34.
- Dezentjé VO, Guchelaar HJ, Nortier JW, van del Velde CJ, Gelderblom H. Clinical implications of CYP2D6 genotyping in tamoxifen treatment for breast cancer. Clin Cancer Res 2009; 15:15–21.
- Higgins MJ, Rae JM, Flockhart DA, Hayes DF, Stearns V. Pharmacogenetics of tamoxifen: who should undergo CYP2D6 genetic testing? J Natl Compr Canc Netw 2009; 7:203–213.
- Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer 2009; 9:576–586.
- Steimer W, Zöpf K, von Amelunxen S, et al. Allele-specific change of concentration and functional gene dose for the prediction of steady-state serum concentrations of amitriptyline and nortriptyline in CYP2C19 and CYP2D6 extensive and intermediate metabolizers. Clinical Cancer 2004; 50:1623–1633.
- Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharm Ther 2008; 83:234–242.
- Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004; 9:442–473.
- Kirchheiner J, Schmidt H, Tzvetkov M, et al. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J 2007; 7:257–265.
- Kirchheiner J. CYP2D6 phenotype prediction from genotype: which system is the best? Clin Pharmacol Ther 2008; 83:225–227.
- Borges S, Desta Z, Jin Y, et al. Composite functional genetic and comedication CYP2D6 activity score in predicting tamoxifen drug exposure among breast cancer patients. J Clin Pharmacol 2010; 50:450–458.
- Hoffmann-La Roche Ltd. AmpliChip CYP450 Test. http://www.roche.com/assays/Pages/AmpliChipCYP450Test.aspx. Accessed 1/21/2011.
- Anzenbacher P, Anzenbacherová E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci 2001; 58:737–747.
- Koren G, Cairns J, Chitayat D, Gaedigk A, Leeder SJ. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet 2006; 368:704.
- Willmann S, Edginton AN, Coboeken K, Ahr G, Lippert J. Risk to the breast-fed neonate from codeine treatment to the mother: a quantitative mechanistic modeling study. Clin Pharm Ther 2009; 86:634–643.
- Grzesiak M, Beszłej A, Lebioda A, Jonkisz A, Dobosz T, Kienja A. [Retrospective assessment of the antidepressants tolerance in the group of patients with diagnosis of depression and different CYP2D6 genotype.] [In Polish] Psychiatr Pol 2003; 37:433–444.
- Laika B, Leucht S, Heres S, Steimer W. Intermediate metabolizer: increased side effects in psychoactive drug therapy. The key to cost-effectiveness of pretreatment CYP2D6 screening? Pharmacogenomics J 2009; 9:395–403.
- Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: Impact on adverse effects and nonresponse during treatment with antidepressants—a pilot study. Clin Pharm Ther 2004; 75:386–393.
- McAlpine DE, O’Kane DJ, Black JL, Mrazek DA. Cytochrome P450 2D6 genotype variation and venlafaxine dosage. Mayo Clin Proc 2007; 82:1065–1068.
- Chen S, Chou WH, Blouin RA, et al. The cytochrome P450 2D6 (CYP2D6) enzyme polymorphism: screening costs and influence on clinical outcomes in psychiatry. Clin Pharmacol Ther 1996; 60:522–534.
- Shams ME, Arneth B, Hiemke C, et al. CYP2D6 polymorphism and clinical effect of the antidepressant venlafaxine. J Clin Pharm Ther 2006; 31:493–502.
- Whyte EM, Romkes M, Mulsant BH, et al. CYP2D6 genotype and venlafaxine-XR concentrations in depressed elderly. Int J Geriatr Psychiatry 2006; 21:542–549.
- Roberts RL, Mulder RT, Joyce PR, Luty SE, Kennedy MA. No evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2004; 19:17–23.
- Murphy GM, Kremer C, Rodrigues HE, Schatzberg AF. Pharmacogenetics of antidepressant medication intolerance. Am J Psychiatry 2003; 160:1830–1835.
- Gillman PK. Re: no evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2005; 20:61–62.
- Gex-Fabry M, Eap CB, Oneda B, et al. CYP2D6 and ABCB1 genetic variability: influence on paroxetine plasma level and therapeutic response. Ther Drug Monit 2008; 30:474–482.
- Kawanishi C, Lundgren S, Agren H, Bertilsson L. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol 2004; 59:803–807.
- Serretti A, Calati R, Massat I, et al. Cytochrome P450 CYP1A2, CYP2C9, CYP2C19 and CYP2D6 genes are not associated with response and remission in a sample of depressive patients. Int Clin Psychopharmacol 2009; 24:250–256.
- de Leon J, Armstrong SC, Cozza KL. Clinical guidelines for psychiatrists for the use of pharmacogenetic testing for CYP450 2D6 and CYP450 2C19. Psychosomatics 2006; 47:75–85.
- de Leon J, Susce MT, Johnson M, et al. DNA microarray technology in the clinical environment: the AmpliChip CYP450 test for CYP2D6 and CYP2C19 genotyping. CNS Spectr 2009; 14:19–34.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Horstmann S, Binder EB. Pharmacogenomics of antidepressant drugs. Pharmacol Ther 2009; 124:57–73.
- Ferreira Ade A, Neves FS, da Rocha FF, et al. The role of 5-HTTLPR polymorphism in antidepressant-associated mania in bipolar disorder. J Affect Disord 2009; 112:267–272.
- Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry 2007; 12:247–257.
- Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry 2010; 15:473–500.
- Reynolds GP, Zhang Z, Zhang X. Polymorphism of the promoter region of the serotonin 5-HT(2C) receptor gene and clozapine-induced weight gain. Am J Psychiatry 2003; 160:677–679.
- King HC, Sinha AA. Gene expression profile analysis by DNA micro-arrays: promise and pitfalls. JAMA 2001; 286:2280–2288.
- Nuckols TK, Paddock SM, Bower AG, et al. Costs of intravenous adverse drug events in academic and nonacademic intensive care units. Med Care 2008; 46:17–24.
- Gurwitz JH, Field TS, Judge J, et al. The incidence of adverse events in two long-term care facilities. Am J Med 2005; 118:251–258.
- Vargas E, Terleira A, Hernando F, et al. Effects of adverse drug reactions on length of stay in surgical intensive care units. Crit Care Med 2003; 31:694–698.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events in older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Classen DC, Pestotnik SL, Evans RS, Lloyd JF, Burke JP. Adverse drug events in hospitalized patients. Excess length of stay, extra costs, and attributable mortality. JAMA 1997; 277:301–306.
- Ernst FR, Grizzle AJ. Drug-related morbidity and mortality: updating the cost-of-illness model. J Am Pharm Assoc (Wash) 2001; 41:192–199.
- Bradford LD. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 2002; 3:229–243.
- Sadee W. Measuring cis-acting regulatory variants genome-wide: new insights into expression genetics and disease susceptibility. Genome Med 2009; 1:116.
- US Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labels. http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm. Accessed 1/18/2011.
- Budnitz DS, Shehab N, Kegler SR, Richards CL. Medication use leading to emergency department visits for adverse drug events in older adults. Ann Intern Med 2007; 147:755–765.
- Elias DJ, Topol EJ. Warfarin pharmacogenomics: a big step forward for individualized medicine: enlightened dosing of warfarin. Eur J Hum Genet 2008; 16:532–534.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Rettie AE, Wienkers LC, Gonzalez FJ, Trager WF, Korzekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Au N, Rettie AE. Pharmacogenomics of 4-hydroxycoumarin anticoagulants. Drug Metab Rev 2008; 40:355–375.
- García-Martín E, Martínez C, Ladero JM, Agúndez JA. Interethnic and intraethnic variability of CYP2C8 and CYP2C9 polymorphisms in healthy individuals. Mol Diagn Ther 2006; 10:29–40.
- Sullivan-Klose TH, Ghanayem BI, Bell DA, et al. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics 1996; 6:341–349.
- Wen MS, Lee M, Chen JJ, et al. Prospective study of warfarin dosage requirements based on CYP2C9 and VKORC1 genotypes. Clin Pharmacol Ther 2008; 84:83–89.
- Larramendy-Gozalo C, Yang JQ, Verstuyft C, et al. Genetic polymorphism of vitamin K epoxide reductase (VKORC1) 1173C>T in a Chinese and a Caucasian population. Basic Clin Pharmacol Toxicol 2006; 98:611–613.
- Caldwell MD, Awad T, Johnson JA, et al. CYP4F2 genetic variant alters required warfarin dose. Blood 2008; 111:4106–4112.
- Bristol-Myers Squibb. Coumadin (warfarin sodium) Prescribing Information. January 2010.
- Barnes-Jewish Hospital at Washington University Medical Center. Warfarin dosing. http://warfarindosing.orgAccessed 1/20/2011.
- Flockhart DA, O’Kane D, Williams MS, et al. Pharmacogenetic testing of CYP2C9 and VKORC1 alleles for warfarin. Genet Med 2008; 10:139–150.
- National Institutes of Health. ClinicalTrials.gov. http://clinicaltrials.gov. Accessed January 20, 2011.
- Eckman MH, Rosand J, Greenberg SM, Gage BF. Cost-effectiveness of using pharmacogenetic information in warfarin dosing for patients with nonvalvular atrial fibrillation. Ann Intern Med 2009; 150:73–83.
- Meckley LM, Gudgeon JM, Anderson JL, Williams MS, Veenstra DL. A policy model to evaluate the benefits, risks, and costs of warfarin pharmacogenomic testing. Pharmacoeconomics 2010; 28:61–74.
- Patrick AR, Avron J, Choudhry NK. Cost-effectiveness of genotype-guided warfarin dosing for patients with atrial fibrillation. Circ Cardiovasc Qual Outcomes 2009; 2:429–436.
- Haas S. New oral Xa and IIa inhibitors: updates on clinical trial results. J Thromb Thrombolysis 2008; 25:52–60.
- Bhatt DL. Tailoring antiplatelet therapy based on pharmacogenomics: how well do the data fit? JAMA 2009; 302:896–897.
- Shuldiner AR, O’Connell JR, Bliden KP, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009; 302:849–857.
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Paré G, Mehta SR, Yusuf S, et al. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med 2010; 363:1704–1714.
- Bristol-Myers Squibb/Sanofi Pharmaceutical Partnership. Plavix (clopidogrel bisulfate) prescribing information. August 2010.
- P450 Drug Interaction Table. Indiana University School of Medicine. http://medicine.iupui.edu/clinpharm/ddis/table.asp. Accessed 1/21/2011.
- Society for Cardiovascular Angiography and Interventions; Holmes DR, Dehmer GJ, Kaul S, Leifer D, O’Gara PT, Stein CM. ACCF/AHA clopidogrel clinical alert: approaches to the FDA “boxed warning”: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association. Circulation 2010; 122:537–557.
- National Institutes of Health. Genotype Guided Comparison of Clopidogrel and Prasugrel Outcomes Study. http://clinicaltrialsfeeds.org/clinical-trials/show/NCT00995514. Accessed 1/20/2011.
- Amarenco P, Labreuche J. Lipid management in the prevention of stroke: Review and updated meta-analysis of statins for stroke prevention. Lancet Neurol 2009; 8:453–463.
- SEARCH Collaborative Group, Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Romaine SP, Bailey KM, Hall AS, Balmforth AJ. The influence of SLC01B1 (OATP1B1) gene polymorphisms on response to statin therapy. Pharmacogenomics J 2010; 10:1–11.
- Wang D, Guo Y, Wrighton SA, Cooke GE, Sadee W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J 2010; Apr 13 [Epub ahead of print].
- Goetz MP, Rae JM, Suman VJ, et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 2005; 23:9312–9318.
- Beverage JN, Sissung TM, Sion AM, Danesi R, Figg WD. CYP2D6 polymorphisms and the impact on tamoxifen therapy. J Pharm Sci 2007; 96:2224–2231.
- Flockhart D. CYP2D6 genotyping and the pharmacogenetics of tamoxifen. Clin Adv Hematol Oncol 2008; 6:493–494.
- Goetz MP, Kamal A, Ames MM. Tamoxifen pharmacogenomics: the role of CYP2D6 as a predictor of drug response. Clin Pharmacol Ther 2008; 83:160–166.
- Stearns V, Rae JM. Pharmacogenetics and breast cancer endocrine therapy: CYP2D6 as a predictive factor for tamoxifen metabolism and drug response? Expert Rev Mol Med 2008; 10:e34.
- Dezentjé VO, Guchelaar HJ, Nortier JW, van del Velde CJ, Gelderblom H. Clinical implications of CYP2D6 genotyping in tamoxifen treatment for breast cancer. Clin Cancer Res 2009; 15:15–21.
- Higgins MJ, Rae JM, Flockhart DA, Hayes DF, Stearns V. Pharmacogenetics of tamoxifen: who should undergo CYP2D6 genetic testing? J Natl Compr Canc Netw 2009; 7:203–213.
- Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer 2009; 9:576–586.
- Steimer W, Zöpf K, von Amelunxen S, et al. Allele-specific change of concentration and functional gene dose for the prediction of steady-state serum concentrations of amitriptyline and nortriptyline in CYP2C19 and CYP2D6 extensive and intermediate metabolizers. Clinical Cancer 2004; 50:1623–1633.
- Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharm Ther 2008; 83:234–242.
- Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004; 9:442–473.
- Kirchheiner J, Schmidt H, Tzvetkov M, et al. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J 2007; 7:257–265.
- Kirchheiner J. CYP2D6 phenotype prediction from genotype: which system is the best? Clin Pharmacol Ther 2008; 83:225–227.
- Borges S, Desta Z, Jin Y, et al. Composite functional genetic and comedication CYP2D6 activity score in predicting tamoxifen drug exposure among breast cancer patients. J Clin Pharmacol 2010; 50:450–458.
- Hoffmann-La Roche Ltd. AmpliChip CYP450 Test. http://www.roche.com/assays/Pages/AmpliChipCYP450Test.aspx. Accessed 1/21/2011.
- Anzenbacher P, Anzenbacherová E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci 2001; 58:737–747.
- Koren G, Cairns J, Chitayat D, Gaedigk A, Leeder SJ. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet 2006; 368:704.
- Willmann S, Edginton AN, Coboeken K, Ahr G, Lippert J. Risk to the breast-fed neonate from codeine treatment to the mother: a quantitative mechanistic modeling study. Clin Pharm Ther 2009; 86:634–643.
- Grzesiak M, Beszłej A, Lebioda A, Jonkisz A, Dobosz T, Kienja A. [Retrospective assessment of the antidepressants tolerance in the group of patients with diagnosis of depression and different CYP2D6 genotype.] [In Polish] Psychiatr Pol 2003; 37:433–444.
- Laika B, Leucht S, Heres S, Steimer W. Intermediate metabolizer: increased side effects in psychoactive drug therapy. The key to cost-effectiveness of pretreatment CYP2D6 screening? Pharmacogenomics J 2009; 9:395–403.
- Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: Impact on adverse effects and nonresponse during treatment with antidepressants—a pilot study. Clin Pharm Ther 2004; 75:386–393.
- McAlpine DE, O’Kane DJ, Black JL, Mrazek DA. Cytochrome P450 2D6 genotype variation and venlafaxine dosage. Mayo Clin Proc 2007; 82:1065–1068.
- Chen S, Chou WH, Blouin RA, et al. The cytochrome P450 2D6 (CYP2D6) enzyme polymorphism: screening costs and influence on clinical outcomes in psychiatry. Clin Pharmacol Ther 1996; 60:522–534.
- Shams ME, Arneth B, Hiemke C, et al. CYP2D6 polymorphism and clinical effect of the antidepressant venlafaxine. J Clin Pharm Ther 2006; 31:493–502.
- Whyte EM, Romkes M, Mulsant BH, et al. CYP2D6 genotype and venlafaxine-XR concentrations in depressed elderly. Int J Geriatr Psychiatry 2006; 21:542–549.
- Roberts RL, Mulder RT, Joyce PR, Luty SE, Kennedy MA. No evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2004; 19:17–23.
- Murphy GM, Kremer C, Rodrigues HE, Schatzberg AF. Pharmacogenetics of antidepressant medication intolerance. Am J Psychiatry 2003; 160:1830–1835.
- Gillman PK. Re: no evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2005; 20:61–62.
- Gex-Fabry M, Eap CB, Oneda B, et al. CYP2D6 and ABCB1 genetic variability: influence on paroxetine plasma level and therapeutic response. Ther Drug Monit 2008; 30:474–482.
- Kawanishi C, Lundgren S, Agren H, Bertilsson L. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol 2004; 59:803–807.
- Serretti A, Calati R, Massat I, et al. Cytochrome P450 CYP1A2, CYP2C9, CYP2C19 and CYP2D6 genes are not associated with response and remission in a sample of depressive patients. Int Clin Psychopharmacol 2009; 24:250–256.
- de Leon J, Armstrong SC, Cozza KL. Clinical guidelines for psychiatrists for the use of pharmacogenetic testing for CYP450 2D6 and CYP450 2C19. Psychosomatics 2006; 47:75–85.
- de Leon J, Susce MT, Johnson M, et al. DNA microarray technology in the clinical environment: the AmpliChip CYP450 test for CYP2D6 and CYP2C19 genotyping. CNS Spectr 2009; 14:19–34.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Horstmann S, Binder EB. Pharmacogenomics of antidepressant drugs. Pharmacol Ther 2009; 124:57–73.
- Ferreira Ade A, Neves FS, da Rocha FF, et al. The role of 5-HTTLPR polymorphism in antidepressant-associated mania in bipolar disorder. J Affect Disord 2009; 112:267–272.
- Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry 2007; 12:247–257.
- Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry 2010; 15:473–500.
- Reynolds GP, Zhang Z, Zhang X. Polymorphism of the promoter region of the serotonin 5-HT(2C) receptor gene and clozapine-induced weight gain. Am J Psychiatry 2003; 160:677–679.
KEY POINTS
- Polymorphisms that affect the pharmacokinetics and pharmacodynamics of specific drugs are common.
- Testing for certain polymorphisms before prescribing certain drugs could help avoid adverse drug effects and improve efficacy.
- Pharmacogenomic testing has only recently begun to enter clinical practice, and routine testing is currently limited to a few clinical scenarios. However, additional applications may be just around the corner.
- Many pharmacogenomic tests are available, but testing has not yet been recommended for most drugs. Needed are large-scale trials to show that routine testing improves patient outcomes.