User login
Atopic dermatitis (AD), or eczema, is the leading dermatologic diagnosis worldwide and is vexing to patients due to the itchiness of the rash. It is the leading cause of skin disease burden worldwide with a prevalence of 229,761,000 reported cases in 2010, presenting largely in preadolescence but also persisting through adulthood.1 Using the children’s life quality index, it has been demonstrated that AD has a greater impact on health-related quality of life than renal disease and cystic fibrosis.2 The overall burden of AD includes stress on the patient and his/her family as well as financial burdens that have been estimated to be similar to that of type 1 diabetes mellitus.3
Epidemiology of AD
The worldwide prevalence of AD varies by country and age group surveyed, with a higher prevalence in wealthy developed nations (eg, the United States) compared to poorer developing nations.4 Efforts to identify prevalence data for AD in the United States have been approached through a variety of strategies. A group in Oregon estimated the prevalence of AD in children aged 5 to 9 years to be 17.2% via a survey of parents (N=1465) and 11.8% with doctor-diagnosed eczema. In the same study, the question “Has a doctor ever said that your child has eczema?” was found to have a 91.3% predictive correlation.5 Analysis of the 2003 National Survey of Children’s Health demonstrated the overall US prevalence of pediatric AD to be 10.7% in 102,353 children 17 years or younger, with a range of 8.7% to 18.1% by region.6
In its evaluation of the worldwide prevalence of AD, the International Study of Asthma and Allergies in Childhood ranked the United States 17th.7,8 The prevalence of AD in developed countries such as the United States is fluid and is expected to increase if the trends from the last 20 years remain true. In an assessment of the National Health Interview Survey data from 1997 to 2011 based on responses to the question, “During the past 12 months, has your child had eczema or any kind of skin allergy?”, the Centers for Disease Control and Prevention identified an increase in the prevalence of AD in patients aged 0 to 17 years from 7.4% in 1997-1999 to 12.5% in 2009-2011.9 Rising prevalence seems to be paired with rising incidence in the total number of severe intractable cases, reduced clearance at the approach of grade school, or cases persisting into adulthood.
Racial Disparity in AD
Racial disparity worldwide and migration are thought to contribute to the prevalence of and therapeutic need for AD. For example, in the United Kingdom, the prevalence of AD in London-born Afro-Caribbean children versus white children (total cross-section, N=693 [junior school children]) was 16.3% and 8.7%, respectively.10 In the United States, black children were more likely to have AD than white children (odds ratio, 1.7).6 Asian and black children also were more likely to present to a physician for treatment of AD than white children.6,10-13
Definition and Diagnostic Considerations
According to Hanifin,14 “Eczema represents a family of inflammatory skin conditions characterized by pruritic, papulovesicular, sometimes weeping dermatitis. All demonstrate the histological hallmark of spongiosis, which helps to distinguish the eczemas from papulosquamous diseases such as psoriasis.”14 Atopic dermatitis is a variant of eczema; however, most laymen identify eczema and AD as being one and the same.
The Hanifin and Rajka15 criteria are the major diagnostic criteria for AD but are difficult to use in clinical practice. Three of the following 4 major criteria are needed for diagnosis: (1) pruritus, which is present universally; (2) typical morphology and distribution; (3) chronic or chronically relapsing dermatitis; and (4) personal and/or family history of atopy. Additionally, 3 of the following 23 minor criteria are needed for diagnosis: xerosis; ichthyosis vulgaris, palmar hyperlinearity, or keratosis pilaris; positive skin prick test; elevated serum IgE level; early age of onset; tendency toward cutaneous infections or impaired cell-mediated immunity; tendency toward nonspecific hand or foot dermatitis; nipple eczema; cheilitis; recurrent conjunctivitis; Dennie-Morgan fold (infraorbital fold); keratoconus; anterior subcapsular cataracts; orbital darkening; facial pallor or facial erythema; pityriasis alba; anterior neck folds; itching when sweating; intolerance to wool and lipid solvents; perifollicular accentuation; skin reactions from ingested foods or by food contact; environmental or emotional factors; and lesional/nonlesional white dermographism or delayed blanch.15-17
More pragmatic streamlined diagnostic criteria were established by Eichenfield et al.18 According to these guidelines, essential features for AD include pruritus and eczema. Important features seen in most cases and adding support to the diagnosis include early age of onset, atopy, and xerosis.18 In clinical practice, diagnosis is often made based on a pruritic relapsing condition in typical locations including the face, neck, and extensor surfaces in infants and children.
Age Considerations
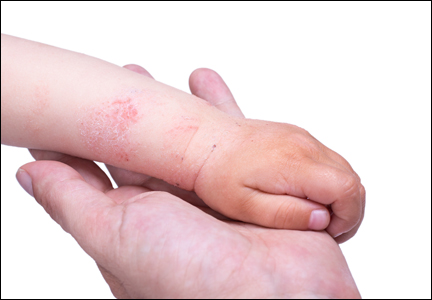
Diagnosis of AD is made by 5 years of age in 85% to 90% of children who will develop the disease and by age 1 year in 60% to 65%.6,19,20 Atopic dermatitis will persist into adulthood in up to one-third of children.21,22 Infantile AD is characterized by erythematous, oozing, excoriated plaques on the cheeks (sparing the nose), scalp, trunk, and extensor surfaces. Pruritus is always seen in AD and can be a source of morbidity.16-18 Seborrheic dermatitis may complicate or overlap with AD in infancy.22
By 2 years of age, most children who are going to develop AD begin to show disease signs of childhood AD characterized by flexural lesions and lesions on the neck and in the postauricular area with sparing of the diaper area.23 Adult AD often presents as eczema of the hands and/or feet. Hand eczema in adulthood is correlated with a prior history of childhood hand eczema and/or childhood AD as well as wet work and caring for small children.24 Children with skin of color may manifest with follicular eczema as their primary disease phenotype. Facial and eyelid dermatitis are more common in Asian females, infants, and teenagers.12,25 Other disease phenotypes that are common in patients with skin of color include lichenoid AD and postinflammatory hypopigmentation.12
Pathogenesis of AD
There are 2 theories on the pathogenesis of AD known as the inside-out and outside-in hypotheses.26 The inside-out hypothesis suggests that allergic triggering leads to a weakened skin barrier that furthers allergen introduction and presentation, while the outside-in hypothesis suggests that the skin barrier is weakened in AD and allows for the presentation of allergens. Both theories have validity and biologic basis, and both may in fact be true in certain individuals.26
The Skin Barrier: An Overview
The skin barrier is a complex set of factors present and functional at birth that seal the keratinocytes and the interkeratinocyte space so that the skin can perform key processes and functions including retention of fluid, exclusion of allergens, protection from UV light and solvents, and prevention of pathogen entry (eg, infections).27-29 The superficial stratum corneum or the cornified envelope consists of keratinocytes with intercellular stripes of hydrophobic and hydrophilic substances formed by various intercellular lipids, largely ceramides, cholesterol, and free fatty acids.30,31 Keratinocytes are the first responders to a variety of environmental insults with the production of IL-18, RANTES (regulated on activation, normal T-expressed, and presumably secreted), granulocyte-macrophage colony-stimulating factor, and thymic stromal lymphopoietin. These inflammatory substances produce acute and chronic inflammation, mast cell reactivity, and T-cell activation.14 Corneodesmosins link the keratinocytes. Peptidases released will cleave the corneodesmosins and allow normal desquamation or shedding of surface skin, which is replaced by division of stem cells in the basal layer.29
The stratum granulosum is the layer beneath the stratum corneum that co-contributes to barrier activity. The stratum granulosum is absent or reduced histologically in ichthyosis vulgaris,32 a form of skin dryness linked to filaggrin mutations and AD. Filaggrin breakdown creates natural moisturizing factor, a series of hygroscopic compounds that attract water into the skin.33 Histidine, a filaggrin breakdown product, is used by urocanic acid to process UV light insults.34 Filaggrin also contributes to other barrier functions including pH and stratum corneum cohesion as well as paracellular permeability of the stratum corneum. Tight junctions in the stratum granulosum include claudin-1 and claudin-6 and provide another barrier feature.29
The skin barrier is composed of lipids and keratinocytes. Ceramides, which represent one type of lipids, are reduced in AD, causing alteration in the lamellar pattern35 and increased transepidermal water loss. Furthermore, the stratum corneum is thickened in AD, possibly in response to trauma, and hydration is reduced.36 Filaggrin (chromosome arm 1q21.3) is formed from the 400-kDa+ precursor profilaggrin through dephosphorylation and cleavage, and it performs an essential function in the skin barrier through its differential cleavage and breakdown as well as release of natural moisturizing factor and other compounds.37 Filaggrin mutations are linked to AD and ichthyosis vulgaris; however, barrier defects as evidenced by transepidermal water loss in the absence of filaggrin mutation are sufficient to allow for sensitization to allergens through the skin.29 Filaggrin mutations have been associated with AD development and vary in prevalence worldwide. In the United Kingdom, a prevalence study of filaggrin mutations in patients aged 7 to 9 years (N=792) demonstrated an 18.4% carrier rate in AD patients versus 12.9% in controls.34 A similar study in Sweden (N=3301) showed carrier rates of 13.5% versus 6.5%, respectively.38 Although filaggrin mutations are lower in black patients,39 ceramide content may be reduced in this population, demonstrating that a variety of skin barrier defects can result in AD. Carriers of filaggrin mutations are more likely to have eczema on skin exposed to environmental factors (eg, face, hands).40
Barrier Defects Contributing to AD
The breakdown of the stratum corneum allows for antigen presentation to Langerhans cells, the dendritic antigen-presenting cells of the skin. Breaks in the stratum corneum may occur from scratching. These macroscopic breaks are large, whereas the breaks that otherwise occur due to barrier breakdown may be more microscopic in nature. Scratching causes aggravation of the helper T cell (TH2) response.29 For example, it allows the dendritic ends of Langerhans cells to be exposed to antigens. The dendritic ends capture allergens through IgE (may be elevated in AD29), which is bound to the high-affinity FCER1 receptors on Langerhans cells. Rather than causing a type I hypersensitivity reaction, these Langerhans cells are activated and move to the lymph nodes where they present antigen and initiate a cascade of proinflammatory activity. This TH2 cascade includes release of cytokines such as IL-2, IL-4, IL-8, IL-10, tumor necrosis factor α, and IFN-γ.26,29
Transepidermal water loss and barrier dysfunction contribute to disease activity and facilitate food/environmental allergen sensitization by allowing increased penetration of allergens through the skin to be presented by Langerhans cells to TH1 cells (sensitization phase). The Langerhans cells can reach their dendritic ends through tight junctions and into the stratum corneum, allowing them to reach surface allergens when the barrier is impaired. Ultimate expansion to systemic allergy (effector phase) occurs when dendritic cells move to draining lymph nodes, causing antigen presentation to CD4 and/or CD8 cells. Langerhans cells and dendritic cell sensitization through the weakened skin is believed to be the basis or role of barrier disruption as a trigger of atopic diseases, including AD and food and environmental allergies.
Many different forms of barrier disruption can cause a TH2 response in AD. The TH2 response triggers a constellation of proinflammatory activities including release of IL-4, associated with eosinophilia and elevated IgE levels, the latter being minor criterion in the diagnosis of AD.15 One mechanism by which the TH2 response is elicited may be the release of molecules such as danger-associated molecule patterns that may elicit recruitment of other inflammatory cells. Helper T cell (TH2) activity also can worsen barrier defects through IL-4 and IL-13 release, which can reduce filaggrin expression,29,41 and can aggravate barrier dysfunction in AD.
Inflammatory activation in AD also may involve inflammatory dendritic epidermal cells (IDECs). The IDECs can be tolerogenic or immunogenic mature phenotypes. The IDECs activate helper T cells (TH1), which may contribute to long-term AD activity.
Conclusion
Atopic dermatitis is a common skin condition worldwide and is characterized by the hallmark of pruritus and features that include a typical pattern, history of atopy (personal or family), and usually xerosis and early disease onset. Barrier dysfunction and immune dysregulation are prominent in AD, both of which aggravate the other and may encourage increased development of allergies and other forms of atopy over time.
1. Hay RJ, Johns NE, Williams HC, et al. The global burden of skin disease in 2010: an analysis of the prevalence and impact of skin conditions. J Invest Dermatol. 2014;134:1527-1534.
2. Beattie PE, Lewis-Jones MS. A comparative study of impairment of quality of life in children with skin disease and children with other chronic childhood diseases. Br J Dermatol. 2006;155:145-151.
3. Su JC, Kemp AS, Varigos GA, et al. Atopic eczema: its impact on the family and financial cost. Arch Dis Child. 1997;76:159-162.
4. Garg N, Silverberg JI. Epidemiology of childhood atopic dermatitis. Clin Dermatol. 2015;33:281-288.
5. Laughter D, Istvan JA, Tofte SJ, et al. The prevalence of atopic dermatitis in Oregon schoolchildren. J Am Acad Dermatol. 2000;43:649-655.
6. Shaw TE, Currie GP, Koudelka CW, et al. Eczema prevalence in the United States: data from the 2003 National Survey of Children’s Health. J Invest Dermatol. 2011;131:67-73.
7. Odhiambo JA, Williams HC, Clayton TO, et al. Global variations in prevalence of eczema symptoms in children from ISAAC Phase Three. J Allergy Clin Immunol. 2009;124:1251-1258.
8. Worldwide variation in prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and atopic eczema: ISAAC. The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee. Lancet. 1998;351:1225-1232.
9. Hansen TE, Evjenth B, Holt J. Increasing prevalence of asthma, allergic rhinoconjunctivitis and eczema among schoolchildren: three surveys during the period 1985-2008. Acta Paediatr. 2013;102:47-52.
10. Williams HC, Pembroke AC, Forsdyke H, et al. London-born black Caribbean children are at increased risk of atopic dermatitis. J Am Acad Dermatol. 1995;32:212-217.
11. Horii KA, Simon SD, Liu DY, et al. Atopic dermatitis in children in the United States, 1997-2004: visit trends, patient and provider characteristics, and prescribing patterns. Pediatrics. 2007;120:e527-e534.
12. Silverberg NB. Eczematous diseases. In: Silverberg NB. Atlas of Pediatric Cutaneous Biodiversity. New York, NY: Springer; 2012:69-88.
13. Gupta J, Grube E, Ericksen MB, et al. Intrinsically defective skin barrier function in children with atopic dermatitis correlates with disease severity. J Allergy Clin Immunol. 2008;121:725-730.
14. Hanifin JM. Evolving concepts of pathogenesis in atopic dermatitis and other eczemas. J Invest Dermatol. 2009;129:320-322.
15. Hanifin JM, Rajka G. Diagnostic features of atopic dermatitis. Acta Derm Venereol Suppl (Stockh). 1980;92:44-47.
16. Queille-Roussel C, Raynaud F, Saurat JH. A prospective computerized study of 500 cases of atopic dermatitis in childhood. I. Initial analysis of 250 parameters. Acta Derm Venereol Suppl (Stockh). 1985;114:87-92.
17. Böhme M, Svensson A, Kull I, et al. Hanifin’s and Rajka’s minor criteria for atopic dermatitis: which do 2-year-olds exhibit? J Am Acad Dermatol. 2000;43:785-792.
18. Eichenfield LF, Hanifin JM, Luger TA, et al. Consensus conference on pediatric atopic dermatitis. J Am Acad Dermatol. 2003;49:1088-1095.
19. Kay J, Gawkrodger DJ, Mortimer MJ, et al. The prevalence of childhood atopic eczema in a general population. J Am Acad Dermatol. 1994;30:35-39.
20. Perkin MR, Strachan DP, Williams HC, et al. Natural history of atopic dermatitis and its relationship to serum total immunoglobulin E in a population-based birth cohort study. Pediatr Allergy Immunol. 2004;15:221-229.
21. Ellis CN, Mancini AJ, Paller AS, et al. Understanding and managing atopic dermatitis in adult patients. Semin Cutan Med Surg. 2012;31(suppl 2):S18-S22.
22. Elish D, Silverberg NB. Infantile seborrheic dermatitis. Cutis. 2006;77:297-300.
23. Meding B, Wrangsjö K, Järvholm B. Hand eczema extent and morphology—association and influence on long-term prognosis. J Invest Dermatol. 2007;127:2147-2151.
24. Mortz CG, Bindslev-Jensen C, Andersen KE. Hand eczema in The Odense Adolescence Cohort Study on Atopic Diseases and Dermatitis (TOACS): prevalence, incidence and risk factors from adolescence to adulthood [published online August 7, 2014]. Br J Dermatol. 2014;171:313-323.
25. Kiken DA, Silverberg NB. Atopic dermatitis in children, part 1: epidemiology, clinical features, and complications. Cutis. 2006;78:241-247.
26. Silverberg NB, Silverberg JI. Inside out or outside in: does atopic dermatitis disrupt barrier function or does disruption of barrier function trigger atopic dermatitis? Cutis. 2015;96:359-361.
27. Visscher MO, Adam R, Brink S, et al. Newborn infant skin: physiology, development, and care [published online December 8, 2014]. Clin Dermatol. 2015;33:271-280.
28. Miyagaki T, Sugaya M. Recent advances in atopic dermatitis and psoriasis: genetic background, barrier function, and therapeutic targets. J Dermatol Sci. 2015;78:89-94.
29. De Benedetto A, Kubo A, Beck LA. Skin barrier disruption: a requirement for allergen sensitization? J Invest Dermatol. 2012;132:949-963.
30. Elias PM, Schmuth M. Abnormal skin barrier in the etiopathogenesis of atopic dermatitis. Curr Opin Allergy Clin Immunol. 2009;9:437-446.
31. Janssens M, van Smeden J, Gooris GS, et al. Lamellar lipid organization and ceramide composition in the stratum corneum of patients with atopic eczema. J Invest Dermatol. 2011;131:2136-2138.
32. Fitch N, Segool R, Ferenczy A, et al. Dominant ichthyosis vulgaris with an ultrastructurally normal granular layer. Clin Genet. 1976;9:71-76.
33. Chandar P, Nole G, Johnson AW. Understanding natural moisturizing mechanisms: implications for moisturizer technology. Cutis. 2009;84(suppl 1):2-15.
34. Brown SJ, Relton CL, Liao H, et al. Filaggrin null mutations and childhood atopic eczema: a population-based case-control study. J Allergy Clin Immunol. 2008;121:940-946.
35. Marenholz I, Rivera VA, Esparza-Gordillo J, et al. Association screening in the Epidermal Differentiation Complex (EDC) identifies an SPRR3 repeat number variant as a risk factor for eczema. J Invest Dermatol. 2011;131:1644-1649.
36. Nemoto-Hasebe I, Akiyama M, Nomura T, et al. Clinical severity correlates with impaired barrier in filaggrin-related eczema. J Invest Dermatol. 2009;129:682-689.
37. Hoste E, Kemperman P, Devos M, et al. Caspase-14 is required for filaggrin degradation to natural moisturizing factors in the skin. J Invest Dermatol. 2011;131:2233-2241.
38. Ballardini N, Kull I, Söderhäll C, et al. Eczema severity in preadolescent children and its relation to sex, filaggrin mutations, asthma, rhinitis, aggravating factors and topical treatment: a report from the BAMSE birth cohort. Br J Dermatol. 2013;168:588-594.
39. Margolis DJ, Apter AJ, Gupta J, et al. The persistence of atopic dermatitis and filaggrin (FLG) mutations in a US longitudinal cohort. J Allergy Clin Immunol. 2012;130:912-917.
40. Carson CG, Rasmussen MA, Thyssen JP, et al. Clinical presentation of atopic dermatitis by filaggrin gene mutation status during the first 7 years of life in a prospective cohort study. PLoS One. 2012;7:e48678.
41. Paller AS. Latest approaches to treating atopic dermatitis. Chem Immunol Allergy. 2012;96:132-140.
Atopic dermatitis (AD), or eczema, is the leading dermatologic diagnosis worldwide and is vexing to patients due to the itchiness of the rash. It is the leading cause of skin disease burden worldwide with a prevalence of 229,761,000 reported cases in 2010, presenting largely in preadolescence but also persisting through adulthood.1 Using the children’s life quality index, it has been demonstrated that AD has a greater impact on health-related quality of life than renal disease and cystic fibrosis.2 The overall burden of AD includes stress on the patient and his/her family as well as financial burdens that have been estimated to be similar to that of type 1 diabetes mellitus.3
Epidemiology of AD
The worldwide prevalence of AD varies by country and age group surveyed, with a higher prevalence in wealthy developed nations (eg, the United States) compared to poorer developing nations.4 Efforts to identify prevalence data for AD in the United States have been approached through a variety of strategies. A group in Oregon estimated the prevalence of AD in children aged 5 to 9 years to be 17.2% via a survey of parents (N=1465) and 11.8% with doctor-diagnosed eczema. In the same study, the question “Has a doctor ever said that your child has eczema?” was found to have a 91.3% predictive correlation.5 Analysis of the 2003 National Survey of Children’s Health demonstrated the overall US prevalence of pediatric AD to be 10.7% in 102,353 children 17 years or younger, with a range of 8.7% to 18.1% by region.6
In its evaluation of the worldwide prevalence of AD, the International Study of Asthma and Allergies in Childhood ranked the United States 17th.7,8 The prevalence of AD in developed countries such as the United States is fluid and is expected to increase if the trends from the last 20 years remain true. In an assessment of the National Health Interview Survey data from 1997 to 2011 based on responses to the question, “During the past 12 months, has your child had eczema or any kind of skin allergy?”, the Centers for Disease Control and Prevention identified an increase in the prevalence of AD in patients aged 0 to 17 years from 7.4% in 1997-1999 to 12.5% in 2009-2011.9 Rising prevalence seems to be paired with rising incidence in the total number of severe intractable cases, reduced clearance at the approach of grade school, or cases persisting into adulthood.
Racial Disparity in AD
Racial disparity worldwide and migration are thought to contribute to the prevalence of and therapeutic need for AD. For example, in the United Kingdom, the prevalence of AD in London-born Afro-Caribbean children versus white children (total cross-section, N=693 [junior school children]) was 16.3% and 8.7%, respectively.10 In the United States, black children were more likely to have AD than white children (odds ratio, 1.7).6 Asian and black children also were more likely to present to a physician for treatment of AD than white children.6,10-13
Definition and Diagnostic Considerations
According to Hanifin,14 “Eczema represents a family of inflammatory skin conditions characterized by pruritic, papulovesicular, sometimes weeping dermatitis. All demonstrate the histological hallmark of spongiosis, which helps to distinguish the eczemas from papulosquamous diseases such as psoriasis.”14 Atopic dermatitis is a variant of eczema; however, most laymen identify eczema and AD as being one and the same.
The Hanifin and Rajka15 criteria are the major diagnostic criteria for AD but are difficult to use in clinical practice. Three of the following 4 major criteria are needed for diagnosis: (1) pruritus, which is present universally; (2) typical morphology and distribution; (3) chronic or chronically relapsing dermatitis; and (4) personal and/or family history of atopy. Additionally, 3 of the following 23 minor criteria are needed for diagnosis: xerosis; ichthyosis vulgaris, palmar hyperlinearity, or keratosis pilaris; positive skin prick test; elevated serum IgE level; early age of onset; tendency toward cutaneous infections or impaired cell-mediated immunity; tendency toward nonspecific hand or foot dermatitis; nipple eczema; cheilitis; recurrent conjunctivitis; Dennie-Morgan fold (infraorbital fold); keratoconus; anterior subcapsular cataracts; orbital darkening; facial pallor or facial erythema; pityriasis alba; anterior neck folds; itching when sweating; intolerance to wool and lipid solvents; perifollicular accentuation; skin reactions from ingested foods or by food contact; environmental or emotional factors; and lesional/nonlesional white dermographism or delayed blanch.15-17
More pragmatic streamlined diagnostic criteria were established by Eichenfield et al.18 According to these guidelines, essential features for AD include pruritus and eczema. Important features seen in most cases and adding support to the diagnosis include early age of onset, atopy, and xerosis.18 In clinical practice, diagnosis is often made based on a pruritic relapsing condition in typical locations including the face, neck, and extensor surfaces in infants and children.
Age Considerations
Diagnosis of AD is made by 5 years of age in 85% to 90% of children who will develop the disease and by age 1 year in 60% to 65%.6,19,20 Atopic dermatitis will persist into adulthood in up to one-third of children.21,22 Infantile AD is characterized by erythematous, oozing, excoriated plaques on the cheeks (sparing the nose), scalp, trunk, and extensor surfaces. Pruritus is always seen in AD and can be a source of morbidity.16-18 Seborrheic dermatitis may complicate or overlap with AD in infancy.22
By 2 years of age, most children who are going to develop AD begin to show disease signs of childhood AD characterized by flexural lesions and lesions on the neck and in the postauricular area with sparing of the diaper area.23 Adult AD often presents as eczema of the hands and/or feet. Hand eczema in adulthood is correlated with a prior history of childhood hand eczema and/or childhood AD as well as wet work and caring for small children.24 Children with skin of color may manifest with follicular eczema as their primary disease phenotype. Facial and eyelid dermatitis are more common in Asian females, infants, and teenagers.12,25 Other disease phenotypes that are common in patients with skin of color include lichenoid AD and postinflammatory hypopigmentation.12
Pathogenesis of AD
There are 2 theories on the pathogenesis of AD known as the inside-out and outside-in hypotheses.26 The inside-out hypothesis suggests that allergic triggering leads to a weakened skin barrier that furthers allergen introduction and presentation, while the outside-in hypothesis suggests that the skin barrier is weakened in AD and allows for the presentation of allergens. Both theories have validity and biologic basis, and both may in fact be true in certain individuals.26
The Skin Barrier: An Overview
The skin barrier is a complex set of factors present and functional at birth that seal the keratinocytes and the interkeratinocyte space so that the skin can perform key processes and functions including retention of fluid, exclusion of allergens, protection from UV light and solvents, and prevention of pathogen entry (eg, infections).27-29 The superficial stratum corneum or the cornified envelope consists of keratinocytes with intercellular stripes of hydrophobic and hydrophilic substances formed by various intercellular lipids, largely ceramides, cholesterol, and free fatty acids.30,31 Keratinocytes are the first responders to a variety of environmental insults with the production of IL-18, RANTES (regulated on activation, normal T-expressed, and presumably secreted), granulocyte-macrophage colony-stimulating factor, and thymic stromal lymphopoietin. These inflammatory substances produce acute and chronic inflammation, mast cell reactivity, and T-cell activation.14 Corneodesmosins link the keratinocytes. Peptidases released will cleave the corneodesmosins and allow normal desquamation or shedding of surface skin, which is replaced by division of stem cells in the basal layer.29
The stratum granulosum is the layer beneath the stratum corneum that co-contributes to barrier activity. The stratum granulosum is absent or reduced histologically in ichthyosis vulgaris,32 a form of skin dryness linked to filaggrin mutations and AD. Filaggrin breakdown creates natural moisturizing factor, a series of hygroscopic compounds that attract water into the skin.33 Histidine, a filaggrin breakdown product, is used by urocanic acid to process UV light insults.34 Filaggrin also contributes to other barrier functions including pH and stratum corneum cohesion as well as paracellular permeability of the stratum corneum. Tight junctions in the stratum granulosum include claudin-1 and claudin-6 and provide another barrier feature.29
The skin barrier is composed of lipids and keratinocytes. Ceramides, which represent one type of lipids, are reduced in AD, causing alteration in the lamellar pattern35 and increased transepidermal water loss. Furthermore, the stratum corneum is thickened in AD, possibly in response to trauma, and hydration is reduced.36 Filaggrin (chromosome arm 1q21.3) is formed from the 400-kDa+ precursor profilaggrin through dephosphorylation and cleavage, and it performs an essential function in the skin barrier through its differential cleavage and breakdown as well as release of natural moisturizing factor and other compounds.37 Filaggrin mutations are linked to AD and ichthyosis vulgaris; however, barrier defects as evidenced by transepidermal water loss in the absence of filaggrin mutation are sufficient to allow for sensitization to allergens through the skin.29 Filaggrin mutations have been associated with AD development and vary in prevalence worldwide. In the United Kingdom, a prevalence study of filaggrin mutations in patients aged 7 to 9 years (N=792) demonstrated an 18.4% carrier rate in AD patients versus 12.9% in controls.34 A similar study in Sweden (N=3301) showed carrier rates of 13.5% versus 6.5%, respectively.38 Although filaggrin mutations are lower in black patients,39 ceramide content may be reduced in this population, demonstrating that a variety of skin barrier defects can result in AD. Carriers of filaggrin mutations are more likely to have eczema on skin exposed to environmental factors (eg, face, hands).40
Barrier Defects Contributing to AD
The breakdown of the stratum corneum allows for antigen presentation to Langerhans cells, the dendritic antigen-presenting cells of the skin. Breaks in the stratum corneum may occur from scratching. These macroscopic breaks are large, whereas the breaks that otherwise occur due to barrier breakdown may be more microscopic in nature. Scratching causes aggravation of the helper T cell (TH2) response.29 For example, it allows the dendritic ends of Langerhans cells to be exposed to antigens. The dendritic ends capture allergens through IgE (may be elevated in AD29), which is bound to the high-affinity FCER1 receptors on Langerhans cells. Rather than causing a type I hypersensitivity reaction, these Langerhans cells are activated and move to the lymph nodes where they present antigen and initiate a cascade of proinflammatory activity. This TH2 cascade includes release of cytokines such as IL-2, IL-4, IL-8, IL-10, tumor necrosis factor α, and IFN-γ.26,29
Transepidermal water loss and barrier dysfunction contribute to disease activity and facilitate food/environmental allergen sensitization by allowing increased penetration of allergens through the skin to be presented by Langerhans cells to TH1 cells (sensitization phase). The Langerhans cells can reach their dendritic ends through tight junctions and into the stratum corneum, allowing them to reach surface allergens when the barrier is impaired. Ultimate expansion to systemic allergy (effector phase) occurs when dendritic cells move to draining lymph nodes, causing antigen presentation to CD4 and/or CD8 cells. Langerhans cells and dendritic cell sensitization through the weakened skin is believed to be the basis or role of barrier disruption as a trigger of atopic diseases, including AD and food and environmental allergies.
Many different forms of barrier disruption can cause a TH2 response in AD. The TH2 response triggers a constellation of proinflammatory activities including release of IL-4, associated with eosinophilia and elevated IgE levels, the latter being minor criterion in the diagnosis of AD.15 One mechanism by which the TH2 response is elicited may be the release of molecules such as danger-associated molecule patterns that may elicit recruitment of other inflammatory cells. Helper T cell (TH2) activity also can worsen barrier defects through IL-4 and IL-13 release, which can reduce filaggrin expression,29,41 and can aggravate barrier dysfunction in AD.
Inflammatory activation in AD also may involve inflammatory dendritic epidermal cells (IDECs). The IDECs can be tolerogenic or immunogenic mature phenotypes. The IDECs activate helper T cells (TH1), which may contribute to long-term AD activity.
Conclusion
Atopic dermatitis is a common skin condition worldwide and is characterized by the hallmark of pruritus and features that include a typical pattern, history of atopy (personal or family), and usually xerosis and early disease onset. Barrier dysfunction and immune dysregulation are prominent in AD, both of which aggravate the other and may encourage increased development of allergies and other forms of atopy over time.
Atopic dermatitis (AD), or eczema, is the leading dermatologic diagnosis worldwide and is vexing to patients due to the itchiness of the rash. It is the leading cause of skin disease burden worldwide with a prevalence of 229,761,000 reported cases in 2010, presenting largely in preadolescence but also persisting through adulthood.1 Using the children’s life quality index, it has been demonstrated that AD has a greater impact on health-related quality of life than renal disease and cystic fibrosis.2 The overall burden of AD includes stress on the patient and his/her family as well as financial burdens that have been estimated to be similar to that of type 1 diabetes mellitus.3
Epidemiology of AD
The worldwide prevalence of AD varies by country and age group surveyed, with a higher prevalence in wealthy developed nations (eg, the United States) compared to poorer developing nations.4 Efforts to identify prevalence data for AD in the United States have been approached through a variety of strategies. A group in Oregon estimated the prevalence of AD in children aged 5 to 9 years to be 17.2% via a survey of parents (N=1465) and 11.8% with doctor-diagnosed eczema. In the same study, the question “Has a doctor ever said that your child has eczema?” was found to have a 91.3% predictive correlation.5 Analysis of the 2003 National Survey of Children’s Health demonstrated the overall US prevalence of pediatric AD to be 10.7% in 102,353 children 17 years or younger, with a range of 8.7% to 18.1% by region.6
In its evaluation of the worldwide prevalence of AD, the International Study of Asthma and Allergies in Childhood ranked the United States 17th.7,8 The prevalence of AD in developed countries such as the United States is fluid and is expected to increase if the trends from the last 20 years remain true. In an assessment of the National Health Interview Survey data from 1997 to 2011 based on responses to the question, “During the past 12 months, has your child had eczema or any kind of skin allergy?”, the Centers for Disease Control and Prevention identified an increase in the prevalence of AD in patients aged 0 to 17 years from 7.4% in 1997-1999 to 12.5% in 2009-2011.9 Rising prevalence seems to be paired with rising incidence in the total number of severe intractable cases, reduced clearance at the approach of grade school, or cases persisting into adulthood.
Racial Disparity in AD
Racial disparity worldwide and migration are thought to contribute to the prevalence of and therapeutic need for AD. For example, in the United Kingdom, the prevalence of AD in London-born Afro-Caribbean children versus white children (total cross-section, N=693 [junior school children]) was 16.3% and 8.7%, respectively.10 In the United States, black children were more likely to have AD than white children (odds ratio, 1.7).6 Asian and black children also were more likely to present to a physician for treatment of AD than white children.6,10-13
Definition and Diagnostic Considerations
According to Hanifin,14 “Eczema represents a family of inflammatory skin conditions characterized by pruritic, papulovesicular, sometimes weeping dermatitis. All demonstrate the histological hallmark of spongiosis, which helps to distinguish the eczemas from papulosquamous diseases such as psoriasis.”14 Atopic dermatitis is a variant of eczema; however, most laymen identify eczema and AD as being one and the same.
The Hanifin and Rajka15 criteria are the major diagnostic criteria for AD but are difficult to use in clinical practice. Three of the following 4 major criteria are needed for diagnosis: (1) pruritus, which is present universally; (2) typical morphology and distribution; (3) chronic or chronically relapsing dermatitis; and (4) personal and/or family history of atopy. Additionally, 3 of the following 23 minor criteria are needed for diagnosis: xerosis; ichthyosis vulgaris, palmar hyperlinearity, or keratosis pilaris; positive skin prick test; elevated serum IgE level; early age of onset; tendency toward cutaneous infections or impaired cell-mediated immunity; tendency toward nonspecific hand or foot dermatitis; nipple eczema; cheilitis; recurrent conjunctivitis; Dennie-Morgan fold (infraorbital fold); keratoconus; anterior subcapsular cataracts; orbital darkening; facial pallor or facial erythema; pityriasis alba; anterior neck folds; itching when sweating; intolerance to wool and lipid solvents; perifollicular accentuation; skin reactions from ingested foods or by food contact; environmental or emotional factors; and lesional/nonlesional white dermographism or delayed blanch.15-17
More pragmatic streamlined diagnostic criteria were established by Eichenfield et al.18 According to these guidelines, essential features for AD include pruritus and eczema. Important features seen in most cases and adding support to the diagnosis include early age of onset, atopy, and xerosis.18 In clinical practice, diagnosis is often made based on a pruritic relapsing condition in typical locations including the face, neck, and extensor surfaces in infants and children.
Age Considerations
Diagnosis of AD is made by 5 years of age in 85% to 90% of children who will develop the disease and by age 1 year in 60% to 65%.6,19,20 Atopic dermatitis will persist into adulthood in up to one-third of children.21,22 Infantile AD is characterized by erythematous, oozing, excoriated plaques on the cheeks (sparing the nose), scalp, trunk, and extensor surfaces. Pruritus is always seen in AD and can be a source of morbidity.16-18 Seborrheic dermatitis may complicate or overlap with AD in infancy.22
By 2 years of age, most children who are going to develop AD begin to show disease signs of childhood AD characterized by flexural lesions and lesions on the neck and in the postauricular area with sparing of the diaper area.23 Adult AD often presents as eczema of the hands and/or feet. Hand eczema in adulthood is correlated with a prior history of childhood hand eczema and/or childhood AD as well as wet work and caring for small children.24 Children with skin of color may manifest with follicular eczema as their primary disease phenotype. Facial and eyelid dermatitis are more common in Asian females, infants, and teenagers.12,25 Other disease phenotypes that are common in patients with skin of color include lichenoid AD and postinflammatory hypopigmentation.12
Pathogenesis of AD
There are 2 theories on the pathogenesis of AD known as the inside-out and outside-in hypotheses.26 The inside-out hypothesis suggests that allergic triggering leads to a weakened skin barrier that furthers allergen introduction and presentation, while the outside-in hypothesis suggests that the skin barrier is weakened in AD and allows for the presentation of allergens. Both theories have validity and biologic basis, and both may in fact be true in certain individuals.26
The Skin Barrier: An Overview
The skin barrier is a complex set of factors present and functional at birth that seal the keratinocytes and the interkeratinocyte space so that the skin can perform key processes and functions including retention of fluid, exclusion of allergens, protection from UV light and solvents, and prevention of pathogen entry (eg, infections).27-29 The superficial stratum corneum or the cornified envelope consists of keratinocytes with intercellular stripes of hydrophobic and hydrophilic substances formed by various intercellular lipids, largely ceramides, cholesterol, and free fatty acids.30,31 Keratinocytes are the first responders to a variety of environmental insults with the production of IL-18, RANTES (regulated on activation, normal T-expressed, and presumably secreted), granulocyte-macrophage colony-stimulating factor, and thymic stromal lymphopoietin. These inflammatory substances produce acute and chronic inflammation, mast cell reactivity, and T-cell activation.14 Corneodesmosins link the keratinocytes. Peptidases released will cleave the corneodesmosins and allow normal desquamation or shedding of surface skin, which is replaced by division of stem cells in the basal layer.29
The stratum granulosum is the layer beneath the stratum corneum that co-contributes to barrier activity. The stratum granulosum is absent or reduced histologically in ichthyosis vulgaris,32 a form of skin dryness linked to filaggrin mutations and AD. Filaggrin breakdown creates natural moisturizing factor, a series of hygroscopic compounds that attract water into the skin.33 Histidine, a filaggrin breakdown product, is used by urocanic acid to process UV light insults.34 Filaggrin also contributes to other barrier functions including pH and stratum corneum cohesion as well as paracellular permeability of the stratum corneum. Tight junctions in the stratum granulosum include claudin-1 and claudin-6 and provide another barrier feature.29
The skin barrier is composed of lipids and keratinocytes. Ceramides, which represent one type of lipids, are reduced in AD, causing alteration in the lamellar pattern35 and increased transepidermal water loss. Furthermore, the stratum corneum is thickened in AD, possibly in response to trauma, and hydration is reduced.36 Filaggrin (chromosome arm 1q21.3) is formed from the 400-kDa+ precursor profilaggrin through dephosphorylation and cleavage, and it performs an essential function in the skin barrier through its differential cleavage and breakdown as well as release of natural moisturizing factor and other compounds.37 Filaggrin mutations are linked to AD and ichthyosis vulgaris; however, barrier defects as evidenced by transepidermal water loss in the absence of filaggrin mutation are sufficient to allow for sensitization to allergens through the skin.29 Filaggrin mutations have been associated with AD development and vary in prevalence worldwide. In the United Kingdom, a prevalence study of filaggrin mutations in patients aged 7 to 9 years (N=792) demonstrated an 18.4% carrier rate in AD patients versus 12.9% in controls.34 A similar study in Sweden (N=3301) showed carrier rates of 13.5% versus 6.5%, respectively.38 Although filaggrin mutations are lower in black patients,39 ceramide content may be reduced in this population, demonstrating that a variety of skin barrier defects can result in AD. Carriers of filaggrin mutations are more likely to have eczema on skin exposed to environmental factors (eg, face, hands).40
Barrier Defects Contributing to AD
The breakdown of the stratum corneum allows for antigen presentation to Langerhans cells, the dendritic antigen-presenting cells of the skin. Breaks in the stratum corneum may occur from scratching. These macroscopic breaks are large, whereas the breaks that otherwise occur due to barrier breakdown may be more microscopic in nature. Scratching causes aggravation of the helper T cell (TH2) response.29 For example, it allows the dendritic ends of Langerhans cells to be exposed to antigens. The dendritic ends capture allergens through IgE (may be elevated in AD29), which is bound to the high-affinity FCER1 receptors on Langerhans cells. Rather than causing a type I hypersensitivity reaction, these Langerhans cells are activated and move to the lymph nodes where they present antigen and initiate a cascade of proinflammatory activity. This TH2 cascade includes release of cytokines such as IL-2, IL-4, IL-8, IL-10, tumor necrosis factor α, and IFN-γ.26,29
Transepidermal water loss and barrier dysfunction contribute to disease activity and facilitate food/environmental allergen sensitization by allowing increased penetration of allergens through the skin to be presented by Langerhans cells to TH1 cells (sensitization phase). The Langerhans cells can reach their dendritic ends through tight junctions and into the stratum corneum, allowing them to reach surface allergens when the barrier is impaired. Ultimate expansion to systemic allergy (effector phase) occurs when dendritic cells move to draining lymph nodes, causing antigen presentation to CD4 and/or CD8 cells. Langerhans cells and dendritic cell sensitization through the weakened skin is believed to be the basis or role of barrier disruption as a trigger of atopic diseases, including AD and food and environmental allergies.
Many different forms of barrier disruption can cause a TH2 response in AD. The TH2 response triggers a constellation of proinflammatory activities including release of IL-4, associated with eosinophilia and elevated IgE levels, the latter being minor criterion in the diagnosis of AD.15 One mechanism by which the TH2 response is elicited may be the release of molecules such as danger-associated molecule patterns that may elicit recruitment of other inflammatory cells. Helper T cell (TH2) activity also can worsen barrier defects through IL-4 and IL-13 release, which can reduce filaggrin expression,29,41 and can aggravate barrier dysfunction in AD.
Inflammatory activation in AD also may involve inflammatory dendritic epidermal cells (IDECs). The IDECs can be tolerogenic or immunogenic mature phenotypes. The IDECs activate helper T cells (TH1), which may contribute to long-term AD activity.
Conclusion
Atopic dermatitis is a common skin condition worldwide and is characterized by the hallmark of pruritus and features that include a typical pattern, history of atopy (personal or family), and usually xerosis and early disease onset. Barrier dysfunction and immune dysregulation are prominent in AD, both of which aggravate the other and may encourage increased development of allergies and other forms of atopy over time.
1. Hay RJ, Johns NE, Williams HC, et al. The global burden of skin disease in 2010: an analysis of the prevalence and impact of skin conditions. J Invest Dermatol. 2014;134:1527-1534.
2. Beattie PE, Lewis-Jones MS. A comparative study of impairment of quality of life in children with skin disease and children with other chronic childhood diseases. Br J Dermatol. 2006;155:145-151.
3. Su JC, Kemp AS, Varigos GA, et al. Atopic eczema: its impact on the family and financial cost. Arch Dis Child. 1997;76:159-162.
4. Garg N, Silverberg JI. Epidemiology of childhood atopic dermatitis. Clin Dermatol. 2015;33:281-288.
5. Laughter D, Istvan JA, Tofte SJ, et al. The prevalence of atopic dermatitis in Oregon schoolchildren. J Am Acad Dermatol. 2000;43:649-655.
6. Shaw TE, Currie GP, Koudelka CW, et al. Eczema prevalence in the United States: data from the 2003 National Survey of Children’s Health. J Invest Dermatol. 2011;131:67-73.
7. Odhiambo JA, Williams HC, Clayton TO, et al. Global variations in prevalence of eczema symptoms in children from ISAAC Phase Three. J Allergy Clin Immunol. 2009;124:1251-1258.
8. Worldwide variation in prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and atopic eczema: ISAAC. The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee. Lancet. 1998;351:1225-1232.
9. Hansen TE, Evjenth B, Holt J. Increasing prevalence of asthma, allergic rhinoconjunctivitis and eczema among schoolchildren: three surveys during the period 1985-2008. Acta Paediatr. 2013;102:47-52.
10. Williams HC, Pembroke AC, Forsdyke H, et al. London-born black Caribbean children are at increased risk of atopic dermatitis. J Am Acad Dermatol. 1995;32:212-217.
11. Horii KA, Simon SD, Liu DY, et al. Atopic dermatitis in children in the United States, 1997-2004: visit trends, patient and provider characteristics, and prescribing patterns. Pediatrics. 2007;120:e527-e534.
12. Silverberg NB. Eczematous diseases. In: Silverberg NB. Atlas of Pediatric Cutaneous Biodiversity. New York, NY: Springer; 2012:69-88.
13. Gupta J, Grube E, Ericksen MB, et al. Intrinsically defective skin barrier function in children with atopic dermatitis correlates with disease severity. J Allergy Clin Immunol. 2008;121:725-730.
14. Hanifin JM. Evolving concepts of pathogenesis in atopic dermatitis and other eczemas. J Invest Dermatol. 2009;129:320-322.
15. Hanifin JM, Rajka G. Diagnostic features of atopic dermatitis. Acta Derm Venereol Suppl (Stockh). 1980;92:44-47.
16. Queille-Roussel C, Raynaud F, Saurat JH. A prospective computerized study of 500 cases of atopic dermatitis in childhood. I. Initial analysis of 250 parameters. Acta Derm Venereol Suppl (Stockh). 1985;114:87-92.
17. Böhme M, Svensson A, Kull I, et al. Hanifin’s and Rajka’s minor criteria for atopic dermatitis: which do 2-year-olds exhibit? J Am Acad Dermatol. 2000;43:785-792.
18. Eichenfield LF, Hanifin JM, Luger TA, et al. Consensus conference on pediatric atopic dermatitis. J Am Acad Dermatol. 2003;49:1088-1095.
19. Kay J, Gawkrodger DJ, Mortimer MJ, et al. The prevalence of childhood atopic eczema in a general population. J Am Acad Dermatol. 1994;30:35-39.
20. Perkin MR, Strachan DP, Williams HC, et al. Natural history of atopic dermatitis and its relationship to serum total immunoglobulin E in a population-based birth cohort study. Pediatr Allergy Immunol. 2004;15:221-229.
21. Ellis CN, Mancini AJ, Paller AS, et al. Understanding and managing atopic dermatitis in adult patients. Semin Cutan Med Surg. 2012;31(suppl 2):S18-S22.
22. Elish D, Silverberg NB. Infantile seborrheic dermatitis. Cutis. 2006;77:297-300.
23. Meding B, Wrangsjö K, Järvholm B. Hand eczema extent and morphology—association and influence on long-term prognosis. J Invest Dermatol. 2007;127:2147-2151.
24. Mortz CG, Bindslev-Jensen C, Andersen KE. Hand eczema in The Odense Adolescence Cohort Study on Atopic Diseases and Dermatitis (TOACS): prevalence, incidence and risk factors from adolescence to adulthood [published online August 7, 2014]. Br J Dermatol. 2014;171:313-323.
25. Kiken DA, Silverberg NB. Atopic dermatitis in children, part 1: epidemiology, clinical features, and complications. Cutis. 2006;78:241-247.
26. Silverberg NB, Silverberg JI. Inside out or outside in: does atopic dermatitis disrupt barrier function or does disruption of barrier function trigger atopic dermatitis? Cutis. 2015;96:359-361.
27. Visscher MO, Adam R, Brink S, et al. Newborn infant skin: physiology, development, and care [published online December 8, 2014]. Clin Dermatol. 2015;33:271-280.
28. Miyagaki T, Sugaya M. Recent advances in atopic dermatitis and psoriasis: genetic background, barrier function, and therapeutic targets. J Dermatol Sci. 2015;78:89-94.
29. De Benedetto A, Kubo A, Beck LA. Skin barrier disruption: a requirement for allergen sensitization? J Invest Dermatol. 2012;132:949-963.
30. Elias PM, Schmuth M. Abnormal skin barrier in the etiopathogenesis of atopic dermatitis. Curr Opin Allergy Clin Immunol. 2009;9:437-446.
31. Janssens M, van Smeden J, Gooris GS, et al. Lamellar lipid organization and ceramide composition in the stratum corneum of patients with atopic eczema. J Invest Dermatol. 2011;131:2136-2138.
32. Fitch N, Segool R, Ferenczy A, et al. Dominant ichthyosis vulgaris with an ultrastructurally normal granular layer. Clin Genet. 1976;9:71-76.
33. Chandar P, Nole G, Johnson AW. Understanding natural moisturizing mechanisms: implications for moisturizer technology. Cutis. 2009;84(suppl 1):2-15.
34. Brown SJ, Relton CL, Liao H, et al. Filaggrin null mutations and childhood atopic eczema: a population-based case-control study. J Allergy Clin Immunol. 2008;121:940-946.
35. Marenholz I, Rivera VA, Esparza-Gordillo J, et al. Association screening in the Epidermal Differentiation Complex (EDC) identifies an SPRR3 repeat number variant as a risk factor for eczema. J Invest Dermatol. 2011;131:1644-1649.
36. Nemoto-Hasebe I, Akiyama M, Nomura T, et al. Clinical severity correlates with impaired barrier in filaggrin-related eczema. J Invest Dermatol. 2009;129:682-689.
37. Hoste E, Kemperman P, Devos M, et al. Caspase-14 is required for filaggrin degradation to natural moisturizing factors in the skin. J Invest Dermatol. 2011;131:2233-2241.
38. Ballardini N, Kull I, Söderhäll C, et al. Eczema severity in preadolescent children and its relation to sex, filaggrin mutations, asthma, rhinitis, aggravating factors and topical treatment: a report from the BAMSE birth cohort. Br J Dermatol. 2013;168:588-594.
39. Margolis DJ, Apter AJ, Gupta J, et al. The persistence of atopic dermatitis and filaggrin (FLG) mutations in a US longitudinal cohort. J Allergy Clin Immunol. 2012;130:912-917.
40. Carson CG, Rasmussen MA, Thyssen JP, et al. Clinical presentation of atopic dermatitis by filaggrin gene mutation status during the first 7 years of life in a prospective cohort study. PLoS One. 2012;7:e48678.
41. Paller AS. Latest approaches to treating atopic dermatitis. Chem Immunol Allergy. 2012;96:132-140.
1. Hay RJ, Johns NE, Williams HC, et al. The global burden of skin disease in 2010: an analysis of the prevalence and impact of skin conditions. J Invest Dermatol. 2014;134:1527-1534.
2. Beattie PE, Lewis-Jones MS. A comparative study of impairment of quality of life in children with skin disease and children with other chronic childhood diseases. Br J Dermatol. 2006;155:145-151.
3. Su JC, Kemp AS, Varigos GA, et al. Atopic eczema: its impact on the family and financial cost. Arch Dis Child. 1997;76:159-162.
4. Garg N, Silverberg JI. Epidemiology of childhood atopic dermatitis. Clin Dermatol. 2015;33:281-288.
5. Laughter D, Istvan JA, Tofte SJ, et al. The prevalence of atopic dermatitis in Oregon schoolchildren. J Am Acad Dermatol. 2000;43:649-655.
6. Shaw TE, Currie GP, Koudelka CW, et al. Eczema prevalence in the United States: data from the 2003 National Survey of Children’s Health. J Invest Dermatol. 2011;131:67-73.
7. Odhiambo JA, Williams HC, Clayton TO, et al. Global variations in prevalence of eczema symptoms in children from ISAAC Phase Three. J Allergy Clin Immunol. 2009;124:1251-1258.
8. Worldwide variation in prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and atopic eczema: ISAAC. The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee. Lancet. 1998;351:1225-1232.
9. Hansen TE, Evjenth B, Holt J. Increasing prevalence of asthma, allergic rhinoconjunctivitis and eczema among schoolchildren: three surveys during the period 1985-2008. Acta Paediatr. 2013;102:47-52.
10. Williams HC, Pembroke AC, Forsdyke H, et al. London-born black Caribbean children are at increased risk of atopic dermatitis. J Am Acad Dermatol. 1995;32:212-217.
11. Horii KA, Simon SD, Liu DY, et al. Atopic dermatitis in children in the United States, 1997-2004: visit trends, patient and provider characteristics, and prescribing patterns. Pediatrics. 2007;120:e527-e534.
12. Silverberg NB. Eczematous diseases. In: Silverberg NB. Atlas of Pediatric Cutaneous Biodiversity. New York, NY: Springer; 2012:69-88.
13. Gupta J, Grube E, Ericksen MB, et al. Intrinsically defective skin barrier function in children with atopic dermatitis correlates with disease severity. J Allergy Clin Immunol. 2008;121:725-730.
14. Hanifin JM. Evolving concepts of pathogenesis in atopic dermatitis and other eczemas. J Invest Dermatol. 2009;129:320-322.
15. Hanifin JM, Rajka G. Diagnostic features of atopic dermatitis. Acta Derm Venereol Suppl (Stockh). 1980;92:44-47.
16. Queille-Roussel C, Raynaud F, Saurat JH. A prospective computerized study of 500 cases of atopic dermatitis in childhood. I. Initial analysis of 250 parameters. Acta Derm Venereol Suppl (Stockh). 1985;114:87-92.
17. Böhme M, Svensson A, Kull I, et al. Hanifin’s and Rajka’s minor criteria for atopic dermatitis: which do 2-year-olds exhibit? J Am Acad Dermatol. 2000;43:785-792.
18. Eichenfield LF, Hanifin JM, Luger TA, et al. Consensus conference on pediatric atopic dermatitis. J Am Acad Dermatol. 2003;49:1088-1095.
19. Kay J, Gawkrodger DJ, Mortimer MJ, et al. The prevalence of childhood atopic eczema in a general population. J Am Acad Dermatol. 1994;30:35-39.
20. Perkin MR, Strachan DP, Williams HC, et al. Natural history of atopic dermatitis and its relationship to serum total immunoglobulin E in a population-based birth cohort study. Pediatr Allergy Immunol. 2004;15:221-229.
21. Ellis CN, Mancini AJ, Paller AS, et al. Understanding and managing atopic dermatitis in adult patients. Semin Cutan Med Surg. 2012;31(suppl 2):S18-S22.
22. Elish D, Silverberg NB. Infantile seborrheic dermatitis. Cutis. 2006;77:297-300.
23. Meding B, Wrangsjö K, Järvholm B. Hand eczema extent and morphology—association and influence on long-term prognosis. J Invest Dermatol. 2007;127:2147-2151.
24. Mortz CG, Bindslev-Jensen C, Andersen KE. Hand eczema in The Odense Adolescence Cohort Study on Atopic Diseases and Dermatitis (TOACS): prevalence, incidence and risk factors from adolescence to adulthood [published online August 7, 2014]. Br J Dermatol. 2014;171:313-323.
25. Kiken DA, Silverberg NB. Atopic dermatitis in children, part 1: epidemiology, clinical features, and complications. Cutis. 2006;78:241-247.
26. Silverberg NB, Silverberg JI. Inside out or outside in: does atopic dermatitis disrupt barrier function or does disruption of barrier function trigger atopic dermatitis? Cutis. 2015;96:359-361.
27. Visscher MO, Adam R, Brink S, et al. Newborn infant skin: physiology, development, and care [published online December 8, 2014]. Clin Dermatol. 2015;33:271-280.
28. Miyagaki T, Sugaya M. Recent advances in atopic dermatitis and psoriasis: genetic background, barrier function, and therapeutic targets. J Dermatol Sci. 2015;78:89-94.
29. De Benedetto A, Kubo A, Beck LA. Skin barrier disruption: a requirement for allergen sensitization? J Invest Dermatol. 2012;132:949-963.
30. Elias PM, Schmuth M. Abnormal skin barrier in the etiopathogenesis of atopic dermatitis. Curr Opin Allergy Clin Immunol. 2009;9:437-446.
31. Janssens M, van Smeden J, Gooris GS, et al. Lamellar lipid organization and ceramide composition in the stratum corneum of patients with atopic eczema. J Invest Dermatol. 2011;131:2136-2138.
32. Fitch N, Segool R, Ferenczy A, et al. Dominant ichthyosis vulgaris with an ultrastructurally normal granular layer. Clin Genet. 1976;9:71-76.
33. Chandar P, Nole G, Johnson AW. Understanding natural moisturizing mechanisms: implications for moisturizer technology. Cutis. 2009;84(suppl 1):2-15.
34. Brown SJ, Relton CL, Liao H, et al. Filaggrin null mutations and childhood atopic eczema: a population-based case-control study. J Allergy Clin Immunol. 2008;121:940-946.
35. Marenholz I, Rivera VA, Esparza-Gordillo J, et al. Association screening in the Epidermal Differentiation Complex (EDC) identifies an SPRR3 repeat number variant as a risk factor for eczema. J Invest Dermatol. 2011;131:1644-1649.
36. Nemoto-Hasebe I, Akiyama M, Nomura T, et al. Clinical severity correlates with impaired barrier in filaggrin-related eczema. J Invest Dermatol. 2009;129:682-689.
37. Hoste E, Kemperman P, Devos M, et al. Caspase-14 is required for filaggrin degradation to natural moisturizing factors in the skin. J Invest Dermatol. 2011;131:2233-2241.
38. Ballardini N, Kull I, Söderhäll C, et al. Eczema severity in preadolescent children and its relation to sex, filaggrin mutations, asthma, rhinitis, aggravating factors and topical treatment: a report from the BAMSE birth cohort. Br J Dermatol. 2013;168:588-594.
39. Margolis DJ, Apter AJ, Gupta J, et al. The persistence of atopic dermatitis and filaggrin (FLG) mutations in a US longitudinal cohort. J Allergy Clin Immunol. 2012;130:912-917.
40. Carson CG, Rasmussen MA, Thyssen JP, et al. Clinical presentation of atopic dermatitis by filaggrin gene mutation status during the first 7 years of life in a prospective cohort study. PLoS One. 2012;7:e48678.
41. Paller AS. Latest approaches to treating atopic dermatitis. Chem Immunol Allergy. 2012;96:132-140.
Practice Points
- The impact of atopic dermatitis (AD) on health-related quality of life mimics that of chronic childhood illnesses such as cystic fibrosis.
- The prevalence of pediatric AD in the United States is estimated at more than 10% of children, with a 1.7 increased odds ratio in black children.
- Diagnosis generally is made based on the presence of a pruritic eczematous eruption with typical morphology and a personal and/or family history of atopy.
- Atopic dermatitis is caused by a complex interplay of skin barrier dysfunction and immune tendency toward allergy development.