User login
THE DIAGNOSIS:
Acquired Perforating Disorder of Renal Disease
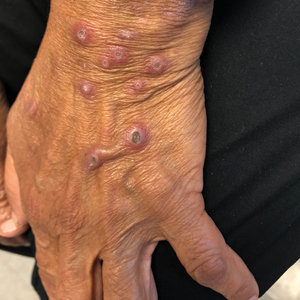
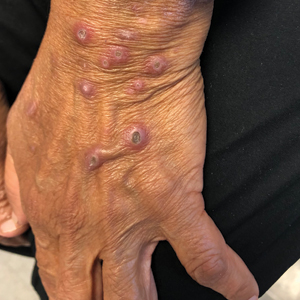
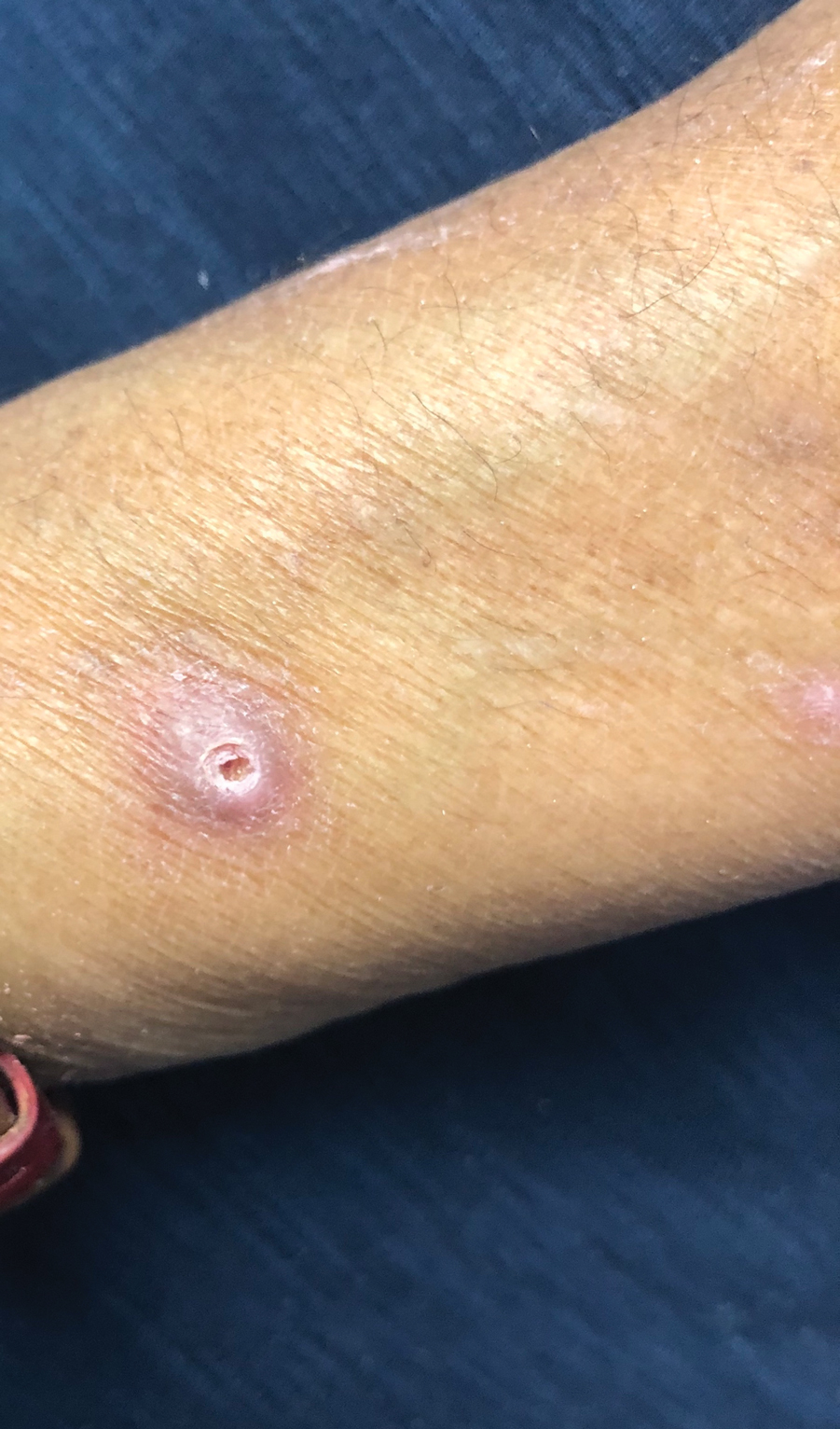
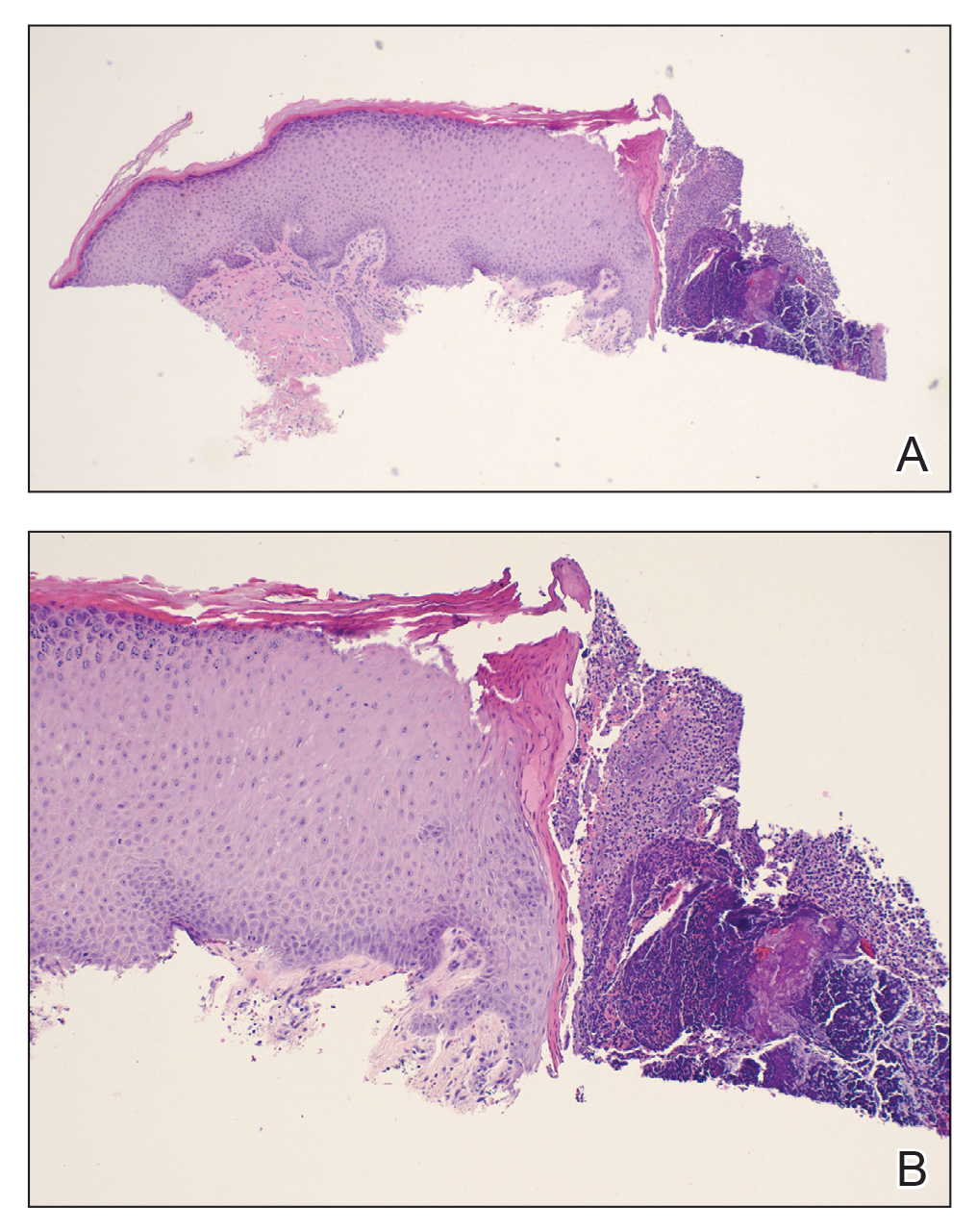
A papule with the central plug removed left a pitlike depression, representing Kyrle disease (Figure 1). A punch biopsy of the left forearm revealed epidermal hyperplasia (Figure 2A) surrounding a keratin plug that contained degenerated basophilic material (Figure 2B), confirming the diagnosis of acquired perforating disorder of renal disease (APDRD), classically described as Kyrle disease.
Acquired perforating disorder of renal disease is an uncommon condition in the general population. It is associated with systemic disease, commonly diabetes mellitus and chronic renal failure, and is seen in up to 10% patients receiving hemodialysis.1 The underlying etiology and pathogenesis of APDRD remains unknown. It has been proposed to be a variant of prurigo nodularis, representing end-stage excoriated folliculitis.1 Given that most cases appear in patients with systemic disease and metabolic abnormalities, APDRD also has been classified under the spectrum of acquired perforating dermatoses, a group of disorders defined by transepithelial elimination of dermal connective tissue. Elevated levels of serum and tissue fibronectin, uremia, and hyperphosphatemia have been observed in patients with APDRD.1,2 Fibronectin stimulates epithelial migration and proliferation and may lead to expulsion of keratin. Furthermore, dermal deposition of excess urea and/or phosphate could initiate transepithelial elimination of material. Alternative hypotheses implicate abnormal keratinization or an imbalance between the rates of epidermal proliferation/ differentiation and keratin production, whereby keratin production outpaces the former. Keratin deposited within the dermis subsequently elicits an inflammatory response along with alterations in the local dermis and connective tissue. These components become intermixed and are extruded through the plug opening.3 Lastly, immune dysregulation resulting from systemic disease could contribute to APDRD through increased expression of IL-31, a cytokine thought to play a role in several pruritic inflammatory skin diseases.4
Although standardized treatment guidelines for APDRD have not been established, the mainstay of therapy is control of the underlying systemic disorder. Intense pruritus and repeated scratching may contribute to microtrauma and subsequent koebnerization of new lesions.3 Thus, ameliorating pruritus can provide both symptomatic relief and prevent the development of new lesions. Retinoids, UV light, oral antibiotics, antihistamines, corticosteroids, keratolytic agents, and immunosuppressants (eg, allopurinol, tacrolimus) have shown some benefit.4
The differential diagnoses for APDRD include arthropod hypersensitivity reactions, eruptive keratoacanthomas, keratosis pilaris, and prurigo nodularis. Arthropod hypersensitivity reactions are seen in patients with a history of a bite or sting from arthropods such as bees, fleas, mites, ticks, and spiders. These reactions cause symptoms of pain, burning, or pruritus and present heterogeneously. They can be edematous and appear as single or multiple papules, pustules, plaques, vesicles, and/or bullae. A central punctum or crusting also may be present. Eruptive keratoacanthomas are seen in Grzybowski syndrome and Ferguson-Smith disease. Grzybowski syndrome arises in the fifth to seventh decades of life and is characterized by the eruptive onset of hundreds to thousands of pruritic, dome-shaped, follicular papules with or without central keratin plugs. Ectropion, mucosal lesions, and masklike facies are other clinical characteristics of Grzybowski syndrome. Ferguson-Smith disease begins in the second decade of life. The eruption of multiple keratoacanthomas and/or squamous cell carcinomas occurs in crops, rapidly growing over 2 to 4 weeks, and then self-resolves. This disease is inherited in an autosomal-dominant manner and is associated with chromosome 9q22. Keratosis pilaris is a benign condition of follicular hyperkeratosis that can appear in any age group and usually is absent of symptoms. It is not associated with any systemic disease. Clinically, the condition appears as folliculocentric keratotic papules with varying degrees of perifollicular erythema located along the extensor surfaces. Keratosis pilaris and APDRD share features of a follicular hyperkeratosis and dilated infundibulum; however, perforation is absent in keratosis pilaris. Lastly, prurigo nodularis is another intensely pruritic dermatosis associated with renal disease that presents as papulonodules on the extensor surfaces of the arms and legs. A biopsy can help to distinguish prurigo nodularis from APDRD.
- Rice AS, Zedek D. Kyrle disease. StatPearls [internet]. StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK532886/
- McKinley-Grant L, Peebles J. Renal disease. In: Kelly A, Taylor SC, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. McGraw-Hill; 2016
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Forouzandeh M, Stratman S, Yosipovitch G. The treatment of Kyrle’s disease: a systematic review. J Eur Acad Dermatol Venereol. 2020;34:1457-1463. doi:10.1111/jdv.16182
THE DIAGNOSIS:
Acquired Perforating Disorder of Renal Disease
A papule with the central plug removed left a pitlike depression, representing Kyrle disease (Figure 1). A punch biopsy of the left forearm revealed epidermal hyperplasia (Figure 2A) surrounding a keratin plug that contained degenerated basophilic material (Figure 2B), confirming the diagnosis of acquired perforating disorder of renal disease (APDRD), classically described as Kyrle disease.
Acquired perforating disorder of renal disease is an uncommon condition in the general population. It is associated with systemic disease, commonly diabetes mellitus and chronic renal failure, and is seen in up to 10% patients receiving hemodialysis.1 The underlying etiology and pathogenesis of APDRD remains unknown. It has been proposed to be a variant of prurigo nodularis, representing end-stage excoriated folliculitis.1 Given that most cases appear in patients with systemic disease and metabolic abnormalities, APDRD also has been classified under the spectrum of acquired perforating dermatoses, a group of disorders defined by transepithelial elimination of dermal connective tissue. Elevated levels of serum and tissue fibronectin, uremia, and hyperphosphatemia have been observed in patients with APDRD.1,2 Fibronectin stimulates epithelial migration and proliferation and may lead to expulsion of keratin. Furthermore, dermal deposition of excess urea and/or phosphate could initiate transepithelial elimination of material. Alternative hypotheses implicate abnormal keratinization or an imbalance between the rates of epidermal proliferation/ differentiation and keratin production, whereby keratin production outpaces the former. Keratin deposited within the dermis subsequently elicits an inflammatory response along with alterations in the local dermis and connective tissue. These components become intermixed and are extruded through the plug opening.3 Lastly, immune dysregulation resulting from systemic disease could contribute to APDRD through increased expression of IL-31, a cytokine thought to play a role in several pruritic inflammatory skin diseases.4
Although standardized treatment guidelines for APDRD have not been established, the mainstay of therapy is control of the underlying systemic disorder. Intense pruritus and repeated scratching may contribute to microtrauma and subsequent koebnerization of new lesions.3 Thus, ameliorating pruritus can provide both symptomatic relief and prevent the development of new lesions. Retinoids, UV light, oral antibiotics, antihistamines, corticosteroids, keratolytic agents, and immunosuppressants (eg, allopurinol, tacrolimus) have shown some benefit.4
The differential diagnoses for APDRD include arthropod hypersensitivity reactions, eruptive keratoacanthomas, keratosis pilaris, and prurigo nodularis. Arthropod hypersensitivity reactions are seen in patients with a history of a bite or sting from arthropods such as bees, fleas, mites, ticks, and spiders. These reactions cause symptoms of pain, burning, or pruritus and present heterogeneously. They can be edematous and appear as single or multiple papules, pustules, plaques, vesicles, and/or bullae. A central punctum or crusting also may be present. Eruptive keratoacanthomas are seen in Grzybowski syndrome and Ferguson-Smith disease. Grzybowski syndrome arises in the fifth to seventh decades of life and is characterized by the eruptive onset of hundreds to thousands of pruritic, dome-shaped, follicular papules with or without central keratin plugs. Ectropion, mucosal lesions, and masklike facies are other clinical characteristics of Grzybowski syndrome. Ferguson-Smith disease begins in the second decade of life. The eruption of multiple keratoacanthomas and/or squamous cell carcinomas occurs in crops, rapidly growing over 2 to 4 weeks, and then self-resolves. This disease is inherited in an autosomal-dominant manner and is associated with chromosome 9q22. Keratosis pilaris is a benign condition of follicular hyperkeratosis that can appear in any age group and usually is absent of symptoms. It is not associated with any systemic disease. Clinically, the condition appears as folliculocentric keratotic papules with varying degrees of perifollicular erythema located along the extensor surfaces. Keratosis pilaris and APDRD share features of a follicular hyperkeratosis and dilated infundibulum; however, perforation is absent in keratosis pilaris. Lastly, prurigo nodularis is another intensely pruritic dermatosis associated with renal disease that presents as papulonodules on the extensor surfaces of the arms and legs. A biopsy can help to distinguish prurigo nodularis from APDRD.
THE DIAGNOSIS:
Acquired Perforating Disorder of Renal Disease
A papule with the central plug removed left a pitlike depression, representing Kyrle disease (Figure 1). A punch biopsy of the left forearm revealed epidermal hyperplasia (Figure 2A) surrounding a keratin plug that contained degenerated basophilic material (Figure 2B), confirming the diagnosis of acquired perforating disorder of renal disease (APDRD), classically described as Kyrle disease.
Acquired perforating disorder of renal disease is an uncommon condition in the general population. It is associated with systemic disease, commonly diabetes mellitus and chronic renal failure, and is seen in up to 10% patients receiving hemodialysis.1 The underlying etiology and pathogenesis of APDRD remains unknown. It has been proposed to be a variant of prurigo nodularis, representing end-stage excoriated folliculitis.1 Given that most cases appear in patients with systemic disease and metabolic abnormalities, APDRD also has been classified under the spectrum of acquired perforating dermatoses, a group of disorders defined by transepithelial elimination of dermal connective tissue. Elevated levels of serum and tissue fibronectin, uremia, and hyperphosphatemia have been observed in patients with APDRD.1,2 Fibronectin stimulates epithelial migration and proliferation and may lead to expulsion of keratin. Furthermore, dermal deposition of excess urea and/or phosphate could initiate transepithelial elimination of material. Alternative hypotheses implicate abnormal keratinization or an imbalance between the rates of epidermal proliferation/ differentiation and keratin production, whereby keratin production outpaces the former. Keratin deposited within the dermis subsequently elicits an inflammatory response along with alterations in the local dermis and connective tissue. These components become intermixed and are extruded through the plug opening.3 Lastly, immune dysregulation resulting from systemic disease could contribute to APDRD through increased expression of IL-31, a cytokine thought to play a role in several pruritic inflammatory skin diseases.4
Although standardized treatment guidelines for APDRD have not been established, the mainstay of therapy is control of the underlying systemic disorder. Intense pruritus and repeated scratching may contribute to microtrauma and subsequent koebnerization of new lesions.3 Thus, ameliorating pruritus can provide both symptomatic relief and prevent the development of new lesions. Retinoids, UV light, oral antibiotics, antihistamines, corticosteroids, keratolytic agents, and immunosuppressants (eg, allopurinol, tacrolimus) have shown some benefit.4
The differential diagnoses for APDRD include arthropod hypersensitivity reactions, eruptive keratoacanthomas, keratosis pilaris, and prurigo nodularis. Arthropod hypersensitivity reactions are seen in patients with a history of a bite or sting from arthropods such as bees, fleas, mites, ticks, and spiders. These reactions cause symptoms of pain, burning, or pruritus and present heterogeneously. They can be edematous and appear as single or multiple papules, pustules, plaques, vesicles, and/or bullae. A central punctum or crusting also may be present. Eruptive keratoacanthomas are seen in Grzybowski syndrome and Ferguson-Smith disease. Grzybowski syndrome arises in the fifth to seventh decades of life and is characterized by the eruptive onset of hundreds to thousands of pruritic, dome-shaped, follicular papules with or without central keratin plugs. Ectropion, mucosal lesions, and masklike facies are other clinical characteristics of Grzybowski syndrome. Ferguson-Smith disease begins in the second decade of life. The eruption of multiple keratoacanthomas and/or squamous cell carcinomas occurs in crops, rapidly growing over 2 to 4 weeks, and then self-resolves. This disease is inherited in an autosomal-dominant manner and is associated with chromosome 9q22. Keratosis pilaris is a benign condition of follicular hyperkeratosis that can appear in any age group and usually is absent of symptoms. It is not associated with any systemic disease. Clinically, the condition appears as folliculocentric keratotic papules with varying degrees of perifollicular erythema located along the extensor surfaces. Keratosis pilaris and APDRD share features of a follicular hyperkeratosis and dilated infundibulum; however, perforation is absent in keratosis pilaris. Lastly, prurigo nodularis is another intensely pruritic dermatosis associated with renal disease that presents as papulonodules on the extensor surfaces of the arms and legs. A biopsy can help to distinguish prurigo nodularis from APDRD.
- Rice AS, Zedek D. Kyrle disease. StatPearls [internet]. StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK532886/
- McKinley-Grant L, Peebles J. Renal disease. In: Kelly A, Taylor SC, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. McGraw-Hill; 2016
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Forouzandeh M, Stratman S, Yosipovitch G. The treatment of Kyrle’s disease: a systematic review. J Eur Acad Dermatol Venereol. 2020;34:1457-1463. doi:10.1111/jdv.16182
- Rice AS, Zedek D. Kyrle disease. StatPearls [internet]. StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK532886/
- McKinley-Grant L, Peebles J. Renal disease. In: Kelly A, Taylor SC, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. McGraw-Hill; 2016
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Forouzandeh M, Stratman S, Yosipovitch G. The treatment of Kyrle’s disease: a systematic review. J Eur Acad Dermatol Venereol. 2020;34:1457-1463. doi:10.1111/jdv.16182
A 74-year-old woman with a 30-year history of type 2 diabetes mellitus presented to our dermatology clinic with a pruritic eruption on the trunk, arms, and legs of 2 months’ duration. Several over-the-counter moisturizers had been used without improvement, and the pruritus was notably impacting her sleep. Physical examination revealed discrete, hyperkeratotic, predominantly follicular, eruptive papules with hyperkeratotic plugs diffusely distributed on the trunk, arms, and legs.