User login
The Diagnosis: Plexiform Neurofibroma as a Manifestation of Neurofibromatosis Type I
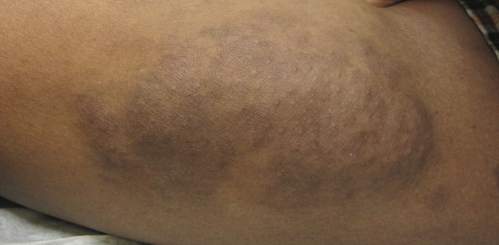
Physical examination revealed a large 10×8-cm subcutaneous nodule that was boggy and resembled a bag of worms on palpation. It was covered by slightly hyperpigmented skin. He also had numerous (>20) café au lait spots measuring 2 to 3 cm across the body and several others on the axillae. There were no gross eye findings. Otherwise the examination was unremarkable on the rest of the body. The patient’s paternal grandfather and aunt had similar macules and multiple nodules. The patient had mild to moderate learning difficulties. He was subsequently referred for genetic and ophthalmology evaluation.
Plexiform neurofibromas are usually benign nerve sheath tumors that are elongated and are multinodular, forming when the tumor involves either multiple trunks of a plexus or multiple fascicles of a large nerve such as the sciatic. Some plexiform neurofibromas resemble a bag of worms; others produce a massive ropy enlargement of the nerve.1,2 Plexiform neurofibromas are associated with cases of neurofibromatosis type I (NFI) and are themselves one of the diagnostic criteria for NFI.1
Plexiform neurofibromas are benign tumors that are the result of a genetic mutation in which loss of heterozygosity occurs, as is the case with the other predominant neoplasms of NFI, that results in unrestricted cell growth.3,4 Some patients have a loss of heterozygosity of this tumor suppression gene with overgrowth of neurofibromatosis on a Blaschko segment. One study in mouse models showed that stromal mast cells were involved in promoting inflammation and increasing tumor growth by mediation of mitogenic signals involved in vascular ingrowth, collagen deposition, and cellular proliferation.5 Plexiform neurofibromas are a presenting feature in 30% of NFI cases within the first year of life. They are extensive nerve sheath tumors with an unpredictable growth pattern that can involve multiple fascicles (ie, large nerves and their branches). Five percent become malignant and the transformation is often heralded by rapid growth and pain.6 If malignant transformation is suspected, biopsy is diagnostic. Magnetic resonance imaging with and without contrast can categorize them into 3 growth categories: superficial, displacing, and invasive.7 Because plexiform neurofibromas are rare tumors, it previously was common practice to delay surgical intervention until disfigurement or disability arose. Complete surgical resection at more advanced stages is nearly impossible given the networklike growth pattern that commonly encapsulates vital structures.8,9 Therefore, surgery has been used in the past for debulking the large growths that eventually will recur. A study of 9 small superficial plexiform neurofibromas in children aged 3 to 15 years documented treatment with early surgical resection, which showed complete resection and no relapse at 4 years. This study showed a promising strategy to prevent future extension of these fast-growing tumors into vital structures.8 There also are current clinical trials investigating sirolimus and peginterferon alfa-2b in patients with more invasive plexiform neurofibromas that are unable to undergo surgical resection due to encapsulation or proximity to essential anatomical structures (registered at www.clinicaltrials.gov with the identifiers NCT00652990 and NCT00678951, respectively).
Pain, development of a neurologic deficit, or enlargement of a preexisting plexiform neurofibroma may signal a malignant peripheral nerve sheath tumor (MPNST) and require immediate evaluation.10 Examination by magnetic resonance imaging and positron emission tomography is useful in distinguishing benign and MPNSTs,8,11,12 but definitive differentiation can only be made by histologic examination of the tumor. Complete surgical excision, when possible, is the only treatment that offers the possibility of cure of MPNSTs. Adjuvant chemotherapy or radiotherapy also is sometimes used, though benefit has not been clearly established.8,9,13,14
Death certificate and population-based studies have shown that approximately 10% of patients with NFI have a reduced life expectancy due to MPNSTs; indeed, these tumors arising from plexiform neurofibromas are the main cause of death in adults with NFI. In 2003, Mautner et al7 studied 50 individuals with NFI. The objective was to establish magnetic resonance imaging criteria for MPNST and to test their usefulness in detecting early malignant change in plexiform neurofibromas. This study found that MPNST in patients with NFI frequently showed inhomogeneous contrast enhancement. This inhomogeneity was due to necrosis and hemorrhage, as shown by macroscopic and histologic analysis of amputated limbs in 2 patients within the study. The investigators found it to be possible to detect malignant transformation at an early stage in patients with no overt clinical signs of progression.7 Careful follow-up will determine how frequently early malignancy can be detected and if it is worthwhile carrying out magnetic resonance imaging at defined intervals.2,7,10,15,16
- Friedman JM. Neurofibromatosis 1. In: Pagon RA, ed. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1109/. Updated September 4, 2014. Accessed April 6, 2015.
- Friedman JM, Riccardi VM. Clinical epidemiological features. In: Friedman JM, Gutmann DH, MacCollin M, et al, eds. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. Baltimore, MD: Johns Hopkins University Press; 1999:29-86.
- Bausch B, Borozdin W, Mautner VF, et al; European-American Phaeochromocytoma Registry Study Group. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with phaeochromocytoma and neurofibromatosis type 1. J Clin Endocrinol Metab. 2007;92:2784-2792.
- Bottillo I, Ahlquist T, Brekke H, et al. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J Pathol. 2009;217:693-701.
- Staser K, Yang FC, Clapp DW. Pathogenesis of plexiform neurofibroma: tumor-stromal/hematopoietic interactions in tumor progression. Ann Rev Pathol. 2012;7:469-495.
- Murphey MD, Smith WS, Smith SE, et al. From the archives of the AFIP: imaging of musculoskeletal neurogenic tumors: radiologic-pathologic correlation. Radiographics. 1999;19:1253-1280.
- Mautner VF, Friedrich RE, von Deimling A, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis type 1: MRI supports the diagnosis of malignant plexiform neurofibroma. Neuroradiology. 2003;45:618-625.
- Friedrich RE, Schmelzle R, Hartmann M, et al. Resection of small plexiform neurofibromas in neurofibromatosis type 1 children. World J Surg Oncol. 2005;3:6.
- Gottfried ON, Viskochil DH, Fults DW, et al. Molecular, genetic, and cellular pathogenesis of neurofibromas and surgical implications. Neurosurgery. 2006;58:1-16.
- Valeyrie-Allanore L, Ismaïli N, Bastuji-Garin S, et al. Symptoms associated with malignancy of peripheral nerve sheath tumours: a retrospective study of 69 patients with neurofibromatosis 1. Br J Dermatol. 2005;153:79-82.
- Bensaid B, Giammarile F, Mognetti T, et al. Utility of 18 FDG positron emission tomography in detection of sarcomatous transformation in neurofibromatosis type 1. Ann Dermatol Venereol. 2007;134:735-741.
- Ferner RE, Lucas JD, O’Doherty MJ, et al. Evaluation of (18)fluorodeoxyglucose positron emission tomography ((18)FDG PET) in the detection of malignant peripheral nerve sheath tumours arising from within plexiform neurofibromas in neurofibromatosis 1. J Neurol Neurosurg Psychiatry. 2000;68:3353-3357.
- Baujat B, Krastinova-Lolov D, Blumen M, et al. Radiofrequency in the treatment of craniofacial plexiform neurofibromatosis: a pilot study. Plast Reconstr Surg. 2006;117:1261-1268.
- Hummel T, Anyane-Yeboa A, Mo J, et al. Response of NF1-related plexiform neurofibroma to high-dose carboplatin. Pediatr Blood Cancer. 2011;56:488-490.
- Feldmann R, Schuierer G, Wessel A, et al. Development of MRI T2 hyperintensities and cognitive functioning in patients with neurofibromatosis type 1. Acta Paediatr. 2010;99:1657-1660.
- Blazo MA, Lewis RA, Chintagumpala MM, et al. Outcomes of systematic screening for optic pathway tumors in children with neurofibromatosis type 1. Am J Med Genet A. 2004;127A:224-229.
The Diagnosis: Plexiform Neurofibroma as a Manifestation of Neurofibromatosis Type I
Physical examination revealed a large 10×8-cm subcutaneous nodule that was boggy and resembled a bag of worms on palpation. It was covered by slightly hyperpigmented skin. He also had numerous (>20) café au lait spots measuring 2 to 3 cm across the body and several others on the axillae. There were no gross eye findings. Otherwise the examination was unremarkable on the rest of the body. The patient’s paternal grandfather and aunt had similar macules and multiple nodules. The patient had mild to moderate learning difficulties. He was subsequently referred for genetic and ophthalmology evaluation.
Plexiform neurofibromas are usually benign nerve sheath tumors that are elongated and are multinodular, forming when the tumor involves either multiple trunks of a plexus or multiple fascicles of a large nerve such as the sciatic. Some plexiform neurofibromas resemble a bag of worms; others produce a massive ropy enlargement of the nerve.1,2 Plexiform neurofibromas are associated with cases of neurofibromatosis type I (NFI) and are themselves one of the diagnostic criteria for NFI.1
Plexiform neurofibromas are benign tumors that are the result of a genetic mutation in which loss of heterozygosity occurs, as is the case with the other predominant neoplasms of NFI, that results in unrestricted cell growth.3,4 Some patients have a loss of heterozygosity of this tumor suppression gene with overgrowth of neurofibromatosis on a Blaschko segment. One study in mouse models showed that stromal mast cells were involved in promoting inflammation and increasing tumor growth by mediation of mitogenic signals involved in vascular ingrowth, collagen deposition, and cellular proliferation.5 Plexiform neurofibromas are a presenting feature in 30% of NFI cases within the first year of life. They are extensive nerve sheath tumors with an unpredictable growth pattern that can involve multiple fascicles (ie, large nerves and their branches). Five percent become malignant and the transformation is often heralded by rapid growth and pain.6 If malignant transformation is suspected, biopsy is diagnostic. Magnetic resonance imaging with and without contrast can categorize them into 3 growth categories: superficial, displacing, and invasive.7 Because plexiform neurofibromas are rare tumors, it previously was common practice to delay surgical intervention until disfigurement or disability arose. Complete surgical resection at more advanced stages is nearly impossible given the networklike growth pattern that commonly encapsulates vital structures.8,9 Therefore, surgery has been used in the past for debulking the large growths that eventually will recur. A study of 9 small superficial plexiform neurofibromas in children aged 3 to 15 years documented treatment with early surgical resection, which showed complete resection and no relapse at 4 years. This study showed a promising strategy to prevent future extension of these fast-growing tumors into vital structures.8 There also are current clinical trials investigating sirolimus and peginterferon alfa-2b in patients with more invasive plexiform neurofibromas that are unable to undergo surgical resection due to encapsulation or proximity to essential anatomical structures (registered at www.clinicaltrials.gov with the identifiers NCT00652990 and NCT00678951, respectively).
Pain, development of a neurologic deficit, or enlargement of a preexisting plexiform neurofibroma may signal a malignant peripheral nerve sheath tumor (MPNST) and require immediate evaluation.10 Examination by magnetic resonance imaging and positron emission tomography is useful in distinguishing benign and MPNSTs,8,11,12 but definitive differentiation can only be made by histologic examination of the tumor. Complete surgical excision, when possible, is the only treatment that offers the possibility of cure of MPNSTs. Adjuvant chemotherapy or radiotherapy also is sometimes used, though benefit has not been clearly established.8,9,13,14
Death certificate and population-based studies have shown that approximately 10% of patients with NFI have a reduced life expectancy due to MPNSTs; indeed, these tumors arising from plexiform neurofibromas are the main cause of death in adults with NFI. In 2003, Mautner et al7 studied 50 individuals with NFI. The objective was to establish magnetic resonance imaging criteria for MPNST and to test their usefulness in detecting early malignant change in plexiform neurofibromas. This study found that MPNST in patients with NFI frequently showed inhomogeneous contrast enhancement. This inhomogeneity was due to necrosis and hemorrhage, as shown by macroscopic and histologic analysis of amputated limbs in 2 patients within the study. The investigators found it to be possible to detect malignant transformation at an early stage in patients with no overt clinical signs of progression.7 Careful follow-up will determine how frequently early malignancy can be detected and if it is worthwhile carrying out magnetic resonance imaging at defined intervals.2,7,10,15,16
The Diagnosis: Plexiform Neurofibroma as a Manifestation of Neurofibromatosis Type I
Physical examination revealed a large 10×8-cm subcutaneous nodule that was boggy and resembled a bag of worms on palpation. It was covered by slightly hyperpigmented skin. He also had numerous (>20) café au lait spots measuring 2 to 3 cm across the body and several others on the axillae. There were no gross eye findings. Otherwise the examination was unremarkable on the rest of the body. The patient’s paternal grandfather and aunt had similar macules and multiple nodules. The patient had mild to moderate learning difficulties. He was subsequently referred for genetic and ophthalmology evaluation.
Plexiform neurofibromas are usually benign nerve sheath tumors that are elongated and are multinodular, forming when the tumor involves either multiple trunks of a plexus or multiple fascicles of a large nerve such as the sciatic. Some plexiform neurofibromas resemble a bag of worms; others produce a massive ropy enlargement of the nerve.1,2 Plexiform neurofibromas are associated with cases of neurofibromatosis type I (NFI) and are themselves one of the diagnostic criteria for NFI.1
Plexiform neurofibromas are benign tumors that are the result of a genetic mutation in which loss of heterozygosity occurs, as is the case with the other predominant neoplasms of NFI, that results in unrestricted cell growth.3,4 Some patients have a loss of heterozygosity of this tumor suppression gene with overgrowth of neurofibromatosis on a Blaschko segment. One study in mouse models showed that stromal mast cells were involved in promoting inflammation and increasing tumor growth by mediation of mitogenic signals involved in vascular ingrowth, collagen deposition, and cellular proliferation.5 Plexiform neurofibromas are a presenting feature in 30% of NFI cases within the first year of life. They are extensive nerve sheath tumors with an unpredictable growth pattern that can involve multiple fascicles (ie, large nerves and their branches). Five percent become malignant and the transformation is often heralded by rapid growth and pain.6 If malignant transformation is suspected, biopsy is diagnostic. Magnetic resonance imaging with and without contrast can categorize them into 3 growth categories: superficial, displacing, and invasive.7 Because plexiform neurofibromas are rare tumors, it previously was common practice to delay surgical intervention until disfigurement or disability arose. Complete surgical resection at more advanced stages is nearly impossible given the networklike growth pattern that commonly encapsulates vital structures.8,9 Therefore, surgery has been used in the past for debulking the large growths that eventually will recur. A study of 9 small superficial plexiform neurofibromas in children aged 3 to 15 years documented treatment with early surgical resection, which showed complete resection and no relapse at 4 years. This study showed a promising strategy to prevent future extension of these fast-growing tumors into vital structures.8 There also are current clinical trials investigating sirolimus and peginterferon alfa-2b in patients with more invasive plexiform neurofibromas that are unable to undergo surgical resection due to encapsulation or proximity to essential anatomical structures (registered at www.clinicaltrials.gov with the identifiers NCT00652990 and NCT00678951, respectively).
Pain, development of a neurologic deficit, or enlargement of a preexisting plexiform neurofibroma may signal a malignant peripheral nerve sheath tumor (MPNST) and require immediate evaluation.10 Examination by magnetic resonance imaging and positron emission tomography is useful in distinguishing benign and MPNSTs,8,11,12 but definitive differentiation can only be made by histologic examination of the tumor. Complete surgical excision, when possible, is the only treatment that offers the possibility of cure of MPNSTs. Adjuvant chemotherapy or radiotherapy also is sometimes used, though benefit has not been clearly established.8,9,13,14
Death certificate and population-based studies have shown that approximately 10% of patients with NFI have a reduced life expectancy due to MPNSTs; indeed, these tumors arising from plexiform neurofibromas are the main cause of death in adults with NFI. In 2003, Mautner et al7 studied 50 individuals with NFI. The objective was to establish magnetic resonance imaging criteria for MPNST and to test their usefulness in detecting early malignant change in plexiform neurofibromas. This study found that MPNST in patients with NFI frequently showed inhomogeneous contrast enhancement. This inhomogeneity was due to necrosis and hemorrhage, as shown by macroscopic and histologic analysis of amputated limbs in 2 patients within the study. The investigators found it to be possible to detect malignant transformation at an early stage in patients with no overt clinical signs of progression.7 Careful follow-up will determine how frequently early malignancy can be detected and if it is worthwhile carrying out magnetic resonance imaging at defined intervals.2,7,10,15,16
- Friedman JM. Neurofibromatosis 1. In: Pagon RA, ed. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1109/. Updated September 4, 2014. Accessed April 6, 2015.
- Friedman JM, Riccardi VM. Clinical epidemiological features. In: Friedman JM, Gutmann DH, MacCollin M, et al, eds. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. Baltimore, MD: Johns Hopkins University Press; 1999:29-86.
- Bausch B, Borozdin W, Mautner VF, et al; European-American Phaeochromocytoma Registry Study Group. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with phaeochromocytoma and neurofibromatosis type 1. J Clin Endocrinol Metab. 2007;92:2784-2792.
- Bottillo I, Ahlquist T, Brekke H, et al. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J Pathol. 2009;217:693-701.
- Staser K, Yang FC, Clapp DW. Pathogenesis of plexiform neurofibroma: tumor-stromal/hematopoietic interactions in tumor progression. Ann Rev Pathol. 2012;7:469-495.
- Murphey MD, Smith WS, Smith SE, et al. From the archives of the AFIP: imaging of musculoskeletal neurogenic tumors: radiologic-pathologic correlation. Radiographics. 1999;19:1253-1280.
- Mautner VF, Friedrich RE, von Deimling A, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis type 1: MRI supports the diagnosis of malignant plexiform neurofibroma. Neuroradiology. 2003;45:618-625.
- Friedrich RE, Schmelzle R, Hartmann M, et al. Resection of small plexiform neurofibromas in neurofibromatosis type 1 children. World J Surg Oncol. 2005;3:6.
- Gottfried ON, Viskochil DH, Fults DW, et al. Molecular, genetic, and cellular pathogenesis of neurofibromas and surgical implications. Neurosurgery. 2006;58:1-16.
- Valeyrie-Allanore L, Ismaïli N, Bastuji-Garin S, et al. Symptoms associated with malignancy of peripheral nerve sheath tumours: a retrospective study of 69 patients with neurofibromatosis 1. Br J Dermatol. 2005;153:79-82.
- Bensaid B, Giammarile F, Mognetti T, et al. Utility of 18 FDG positron emission tomography in detection of sarcomatous transformation in neurofibromatosis type 1. Ann Dermatol Venereol. 2007;134:735-741.
- Ferner RE, Lucas JD, O’Doherty MJ, et al. Evaluation of (18)fluorodeoxyglucose positron emission tomography ((18)FDG PET) in the detection of malignant peripheral nerve sheath tumours arising from within plexiform neurofibromas in neurofibromatosis 1. J Neurol Neurosurg Psychiatry. 2000;68:3353-3357.
- Baujat B, Krastinova-Lolov D, Blumen M, et al. Radiofrequency in the treatment of craniofacial plexiform neurofibromatosis: a pilot study. Plast Reconstr Surg. 2006;117:1261-1268.
- Hummel T, Anyane-Yeboa A, Mo J, et al. Response of NF1-related plexiform neurofibroma to high-dose carboplatin. Pediatr Blood Cancer. 2011;56:488-490.
- Feldmann R, Schuierer G, Wessel A, et al. Development of MRI T2 hyperintensities and cognitive functioning in patients with neurofibromatosis type 1. Acta Paediatr. 2010;99:1657-1660.
- Blazo MA, Lewis RA, Chintagumpala MM, et al. Outcomes of systematic screening for optic pathway tumors in children with neurofibromatosis type 1. Am J Med Genet A. 2004;127A:224-229.
- Friedman JM. Neurofibromatosis 1. In: Pagon RA, ed. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1109/. Updated September 4, 2014. Accessed April 6, 2015.
- Friedman JM, Riccardi VM. Clinical epidemiological features. In: Friedman JM, Gutmann DH, MacCollin M, et al, eds. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. Baltimore, MD: Johns Hopkins University Press; 1999:29-86.
- Bausch B, Borozdin W, Mautner VF, et al; European-American Phaeochromocytoma Registry Study Group. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with phaeochromocytoma and neurofibromatosis type 1. J Clin Endocrinol Metab. 2007;92:2784-2792.
- Bottillo I, Ahlquist T, Brekke H, et al. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J Pathol. 2009;217:693-701.
- Staser K, Yang FC, Clapp DW. Pathogenesis of plexiform neurofibroma: tumor-stromal/hematopoietic interactions in tumor progression. Ann Rev Pathol. 2012;7:469-495.
- Murphey MD, Smith WS, Smith SE, et al. From the archives of the AFIP: imaging of musculoskeletal neurogenic tumors: radiologic-pathologic correlation. Radiographics. 1999;19:1253-1280.
- Mautner VF, Friedrich RE, von Deimling A, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis type 1: MRI supports the diagnosis of malignant plexiform neurofibroma. Neuroradiology. 2003;45:618-625.
- Friedrich RE, Schmelzle R, Hartmann M, et al. Resection of small plexiform neurofibromas in neurofibromatosis type 1 children. World J Surg Oncol. 2005;3:6.
- Gottfried ON, Viskochil DH, Fults DW, et al. Molecular, genetic, and cellular pathogenesis of neurofibromas and surgical implications. Neurosurgery. 2006;58:1-16.
- Valeyrie-Allanore L, Ismaïli N, Bastuji-Garin S, et al. Symptoms associated with malignancy of peripheral nerve sheath tumours: a retrospective study of 69 patients with neurofibromatosis 1. Br J Dermatol. 2005;153:79-82.
- Bensaid B, Giammarile F, Mognetti T, et al. Utility of 18 FDG positron emission tomography in detection of sarcomatous transformation in neurofibromatosis type 1. Ann Dermatol Venereol. 2007;134:735-741.
- Ferner RE, Lucas JD, O’Doherty MJ, et al. Evaluation of (18)fluorodeoxyglucose positron emission tomography ((18)FDG PET) in the detection of malignant peripheral nerve sheath tumours arising from within plexiform neurofibromas in neurofibromatosis 1. J Neurol Neurosurg Psychiatry. 2000;68:3353-3357.
- Baujat B, Krastinova-Lolov D, Blumen M, et al. Radiofrequency in the treatment of craniofacial plexiform neurofibromatosis: a pilot study. Plast Reconstr Surg. 2006;117:1261-1268.
- Hummel T, Anyane-Yeboa A, Mo J, et al. Response of NF1-related plexiform neurofibroma to high-dose carboplatin. Pediatr Blood Cancer. 2011;56:488-490.
- Feldmann R, Schuierer G, Wessel A, et al. Development of MRI T2 hyperintensities and cognitive functioning in patients with neurofibromatosis type 1. Acta Paediatr. 2010;99:1657-1660.
- Blazo MA, Lewis RA, Chintagumpala MM, et al. Outcomes of systematic screening for optic pathway tumors in children with neurofibromatosis type 1. Am J Med Genet A. 2004;127A:224-229.
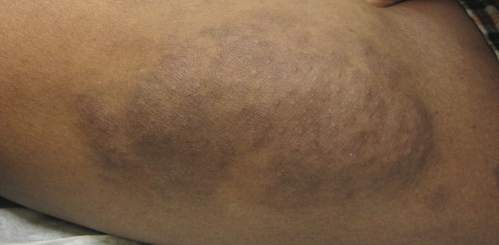
An 11-year-old boy presented for evaluation of a 10×8-cm tortuous lesion on the right posterior leg. Although it had been present since birth, the patient’s mother reported recent growth of the lesion. The lesion was noted to occasionally become irritated and pruritic. The patient’s history was remarkable for asthma.