User login
Gorham-Stout disease (GSD) is a rare condition characterized by spontaneous idiopathic resorption of bone with lymphovascular proliferation and an absence of malignant features. It was originally described by Jackson1 in an 1838 report of a 36-year-old man whose “arm bone, between the shoulder and elbow” had completely vanished after 2 fractures. The disease was defined and its pathology characterized by Gorham and Stout2 in 1955 in a series of 24 patients. Despite about 200 reported cases in the literature,3 its etiology remains unclear. Any bone in the skeleton may be affected by GSD, although there is a predilection for the skull, humerus, clavicle, ribs, pelvis, and femur.4-6 It commonly manifests within the first 3 decades of life, but case reports range from as early as 2 months of age to the eighth decade.5,7
Gorham-Stout disease is a diagnosis of exclusion that requires careful consideration of the clinical context, radiographic findings, and histopathology. Typical histopathologic findings include benign lymphatic or vascular proliferation, involution of adipose tissue within the bone marrow, and thinning of bony trabeculae.6 Fibrous tissue may replace vascular tissue after the initial vasoproliferative, osteolytic phase.6 Some authors describe the disease as having 2 phases, the first with massive osteolysis followed by relative dormancy and the second without progression or re-ossification.8,9 Treatment remains controversial and is guided by management of the disease’s complications. Options range from careful observation and supportive management to aggressive surgical resection and reconstruction, with positive outcomes reported using many different modalities.10 Most treatment successes, however, hinge on halting bony resorption using medical and radiation therapy. Surgery is usually reserved as a salvage option for patients who have failed medical modalities and have residual symptoms or functional limitations.6
This case report describes the successful surgical management of a patient with pelvic GSD who had progressive pain and functional limitation despite exhaustive medical and radiation therapy. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A healthy 27-year-old man sought medical attention after a fall while mowing his lawn that resulted in difficulty ambulating. Radiographic studies showed discontinuous lytic lesions in the right periacetabular region and the right sacroiliac (SI) joint. Biopsy at an outside institution revealed an infiltration of thin-walled branching vascular channels involving intertrabecular marrow spaces and periosteal connective tissue. The vessels were devoid of a muscular coat and lined by flattened epithelium; these features were seen as consistent with GSD.
The patient was managed medically at the outside institution for approximately 2 years, with regimens consisting of zoledronate, denosumab, sorafenib, vincristine, sirolimus, and bevacizumab. Because there is no standard chemotherapy protocol for GSD, this broad regimen was likely an attempt by treating physicians to control disease progression before considering radiation or surgery. Zoledronate, a bisphosphonate, and denosumab, a monoclonal antibody against the receptor activator of nuclear factor κβ ligand (RANKL), both inhibit bone resorption, making them logical choices in treating an osteolytic disease. Sorafenib, vincristine, sirolimus, and bevacizumab may be of clinical benefit in GSD via inhibition of vascular proliferation, which is a key histologic feature in GSD. Sorafenib inhibits the vascular endothelial growth factor (VEGF) receptor, vincristine and sirolimus inhibit VEGF production, and bevacizumab is a monoclonal antibody targeting VEGF.
The patient’s disease continued to involve more of his right hemipelvis despite this extensive regimen of chemotherapy, and he experienced significant functional decline about 2 years after initial presentation, when he was no longer able to ambulate unassisted. Radiation therapy to the pelvis was attempted at the outside institution (6/15 MV photons, 5040 cGy, 28 fractions) without improvement. Three years after his initial injury, he presented to our clinic.
Now age 30 years, the patient ambulated only with crutches and endorsed minimal improvement in his pain over 3 years of treatment. Physical examination of the patient revealed that he was a tall, thin man in visible discomfort. Sensation was intact to light touch in the bilateral L1 to S1 nerve distributions. There was marked weakness of the right lower extremity, and his examination was limited by pain. He could not perform a straight leg raise on the right side. Right quadriceps strength was 4/5, and right hamstrings strength was 3/5. There was no weakness in the left leg. Reflexes were normal and symmetric bilaterally at the patellar and gastrocnemius soleus tendons. Distal circulatory status in both extremities was normal, and there were no deformities of the skin.
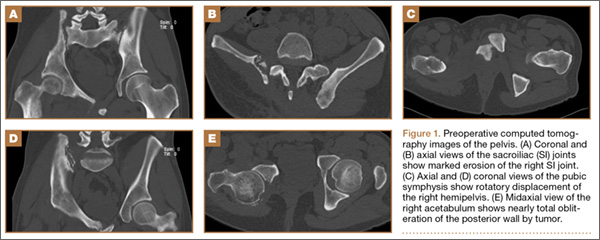
Figure 1 shows the patient’s computed tomography (CT) scan. Figures 1A and 1B reveal fragmentation of the posterior ilia and sacrum along both SI joints. Dislocation of the pubic symphysis is shown in Figures 1C and 1D, and discontinuous involvement of the ischium and posterior wall of the acetabulum is visible in Figure 1E.
Serum studies, including C-reactive protein, erythrocyte sedimentation rate, and a complete blood count, were within normal limits. A CT-guided core needle biopsy and aspiration of the right SI joint revealed no infection; pathology was nondiagnostic. Anesthetic injection of the hip joint resulted in no relief. As this man was severely functionally limited and had exhausted all medical and radiation treatment options, a collaborative decision was made to proceed with surgical management. Surgical options included spinopelvic fusion unilaterally or bilaterally, hip arthroplasty, or sacropelvic resection with or without reconstruction. The patient opted for intralesional surgery and spinopelvic fusion in place of more radical options.
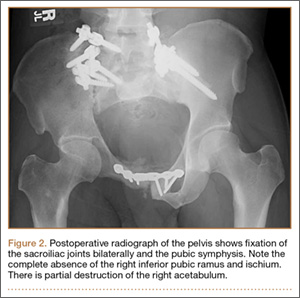
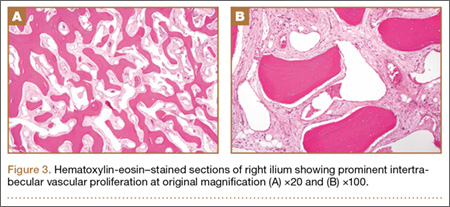
Thirty-seven months after his initial presentation, he underwent posterior spinal fusion L5 to S1, SI fusion, and anterior locking plate fixation of the pubic symphysis, as seen in Figure 2. Pathology from surgical specimens, seen at original magnification ×20 and ×100 in Figures 3A and 3B, respectively, showed prominent vascular proliferation in the right ilium, with reactive bone changes in the left ilium and right sacrum. A lytic lesion showed fibrous tissue with an embedded fragment of necrotic bone.
Six weeks after surgery, the patient had substantial improvement in his pain and was partially weight-bearing. He was able to ambulate with crutches and returned to work. The patient’s overall clinical status continued to improve throughout the postoperative course. He developed low back pain 7 months after surgery and was found to have a sacrococcygeal abscess and coccygeal fracture anterior to the sacrum. He underwent irrigation and débridement of the abscess and distal coccygectomy and was treated with 6 weeks of intravenous cefazolin and long-term suppression with levofloxacin and rifampin for methicillin-sensitive Staphylococcus aureus hardware infection and osteomyelitis. The patient’s clinical course subsequently improved. At latest follow-up 16 months after the index operation, pain was reported as manageable and mostly an annoyance. He was prescribed up to 40 mg of oxycodone daily for pain. The patient returned to work, ambulates with a cane (no other assistive devices), and reports being able to get around without any difficulty.
Discussion
Gorham-Stout disease is an exceedingly rare condition resulting in spontaneous osteolysis. Approximately 200 cases have been reported with no apparent gender, race, or familial predilection or systemic symptoms differentiating it from other etiologies of idiopathic osteolysis.6 These patients often seek medical attention after sustaining a pathologic fracture,6 when a broad differential diagnosis narrows to GSD only after biopsy excludes other possibilities and demonstrates characteristic angiomatosis without malignant features.2,4,6,8,10 Gorham-Stout disease appears more frequently at particular sites within the skeleton, and pelvic involvement is common—more than 20% of cases in 1 review.5,10 Limitations in the patient’s ability to ambulate invariably result from osteolysis of the pelvis, which is concerning considering the young age at which GSD typically presents. A variety of treatment modalities have been described for pelvic GSD, but surgery has been undertaken in relatively few cases.5
The diagnosis is one of exclusion after considering the clinical context and radiologic and pathologic findings. In this case, a pathologic fracture was discovered with osteolytic lesions throughout the hemipelvis. Biopsy excluded malignancy and demonstrated the key hemangiomatous vascular proliferation with thin-walled vessels that is classic for GSD. While our patient initially appeared to have 2 sites of disease, the surgical specimen revealed a primary site of vascular proliferation in the right ilium from which 2 apparent foci had spread, consistent with the typical monocentric presentation of GSD.11 A broad differential diagnosis must be considered at initial presentation, including osteomyelitis, metastatic disease, multiple myeloma, and primary bone sarcoma. Upon identifying a primary osteolytic process, several considerations besides GSD remain, such as Hajdu-Cheney syndrome, Winchester syndrome, multicentric osteolysis with nephropathy, familial osteolysis, Farber disease, and neurogenic osteolysis; most of these etiologies involve familial predispositions and/or systemic symptoms.
Treatment options for GSD include supportive care, medical therapy, radiation, and surgery. For pelvic GSD, numerous reports have demonstrated good outcomes with supportive management, since osteolysis often spontaneously arrests.8,9,12 Others have had success with medical treatments in attempts to halt bone resorption.6,13-15 Bisphosphonates are the cornerstone of medical therapy in GSD, as they appear to halt further osteoclastic bone breakdown. The levels of VEGF have been shown to be elevated in GSD,13 likely consistent with the vascular proliferation evident on pathology, and therapies such as bevacizumab and interferon α-2b have been used to target osteolysis via this pathway with good outcome.13,14,16 External beam-radiation therapy has been shown to prevent local progression of osteolysis in up to 80% of cases.4 However, even with arrest of bone resorption, damage to affected bone may have progressed to the point of significant functional limitation. This may be especially true in the pelvis.
We present a case of a patient who continued to deteriorate after maximal medical and radiation therapy. Many reported cases of pelvic GSD have had good outcomes with some combination of conservative management, medical therapy, and radiation. However, in our patient, the pelvis and lumbosacral spine were unstable as a result of significant bone loss and fracture, and his clinical deterioration was dramatic. We considered reasonable surgical approaches, including local intralesional débridement and massive en bloc resection with structural allograft. We chose the less radical procedure given the patient’s age, minimal surgical history, and personal preference. Although structural pelvic allograft has been successful in a few cases, there remains a high risk of complications, such as fracture, resorption, or infection.17 We considered the addition of hip arthroplasty with either scenario, but we elected not to perform this component given his young age and lack of symptomatic improvement with diagnostic anesthetic hip injection. The key to this patient’s surgical reconstruction, aside from eliminating gross disease, was the stabilization of the spinopelvic junction and pelvic ring. His functional improvement as early as 6 weeks after surgery demonstrates that surgery can have an important role for patients with pelvic GSD who fail medical and radiation therapy.
1. Jackson JBS. A boneless arm. Boston Med Surg J. 1838;18:368-369.
2. Gorham LW, Stout AP. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone): its relation to hemangiomatosis. J Bone Joint Surg Am. 1955;37(5):985-1004.
3. Lehmann G, Pfeil A, Böttcher J, et al. Benefit of a 17-year long-term bisphosphonate therapy in a patient with Gorham-Stout syndrome. Arch Orthop Trauma Surg. 2009;129(7):967-972.
4. Heyd R, Micke O, Surholt C, et al; German Cooperative Group on Radiotherapy for Benign Diseases (GCG-BD). Radiation therapy for Gorham-Stout syndrome: results of a national patterns-of-care study and literature review. Int J Radiat Oncol Biol Phys. 2011;81(3):e179-e185.
5. Kulenkampff HA, Richter GM, Hasse WE, Adler CP. Massive pelvic osteolysis in the Gorham-Stout syndrome. Int Orthop. 1990;14(4):361-366.
6. Ruggieri P, Montalti M, Angelini A, Alberghini M, Mercuri M. Gorham-Stout disease: the experience of the Rizzoli Institute and review of the literature. Skeletal Radiol. 2011;40(11):1391-1397.
7. Vinée P, Tanyü MO, Hauenstein KH, Sigmund G, Stöver B, Adler CP. CT and MRI of Gorham syndrome. J Comput Assist Tomogr. 1994;18(6):985-989.
8. Boyer P, Bourgeois P, Boyer O, Catonné Y, Saillant G. Massive Gorham-Stout syndrome of the pelvis. Clin Rheumatol. 2005;24(5):551-555.
9. Malde R, Agrawal HM, Ghosh SL, Dinshaw KA. Vanishing bone disease involving the pelvis. J Cancer Res Ther. 2005;1(4):227-228.
10. Kuriyama DK, McElligott SC, Glaser DW, Thompson KS. Treatment of Gorham-Stout disease with zoledronic acid and interferon-α: a case report and literature review. J Pediatr Hematol Oncol. 2010;32(8):579-584.
11. Tie ML, Poland GA, Rosenow EC III. Chylothorax in Gorham’s syndrome. A common complication of a rare disease. Chest. 1994;105(1):208-213.
12. Möller G, Priemel M, Amling M, Werner M, Kuhlmey AS, Delling G. The Gorham-Stout syndrome (Gorham’s massive osteolysis). A report of six cases with histopathological findings. J Bone Joint Surg Br. 1999;81(3):501-506.
13. Dupond JL, Bermont L, Runge M, de Billy M. Plasma VEGF determination in disseminated lymphangiomatosis—Gorham-Stout syndrome: a marker of activity? A case report with a 5-year follow-up. Bone. 2010;46(3):873-876.
14. Wang JD, Chang TK, Cheng YY, et al. A child with dyspnea and unstable gait. Pediatr Hemat Oncol. 2007;24(4):321-324.
15. Zheng MW, Yang M, Qiu JX, et al. Gorham-Stout syndrome presenting in a 5-year-old girl with a successful bisphosphonate therapeutic effect. Exp Ther Med. 2012;4(3):449-451.
16. Timke C, Krause MF, Oppermann HC, Leuschner I, Claviez A. Interferon alpha 2b treatment in an eleven-year-old boy with disseminated lymphangiomatosis. Pediatr Blood Cancer. 2007;48(1):108-111.
17. Stöve J, Reichelt A. Massive osteolysis of the pelvis, femur and sacral bone with a Gorham-Stout syndrome. Arch Orthop Trauma Surg. 1995;114(4):207-210.
Gorham-Stout disease (GSD) is a rare condition characterized by spontaneous idiopathic resorption of bone with lymphovascular proliferation and an absence of malignant features. It was originally described by Jackson1 in an 1838 report of a 36-year-old man whose “arm bone, between the shoulder and elbow” had completely vanished after 2 fractures. The disease was defined and its pathology characterized by Gorham and Stout2 in 1955 in a series of 24 patients. Despite about 200 reported cases in the literature,3 its etiology remains unclear. Any bone in the skeleton may be affected by GSD, although there is a predilection for the skull, humerus, clavicle, ribs, pelvis, and femur.4-6 It commonly manifests within the first 3 decades of life, but case reports range from as early as 2 months of age to the eighth decade.5,7
Gorham-Stout disease is a diagnosis of exclusion that requires careful consideration of the clinical context, radiographic findings, and histopathology. Typical histopathologic findings include benign lymphatic or vascular proliferation, involution of adipose tissue within the bone marrow, and thinning of bony trabeculae.6 Fibrous tissue may replace vascular tissue after the initial vasoproliferative, osteolytic phase.6 Some authors describe the disease as having 2 phases, the first with massive osteolysis followed by relative dormancy and the second without progression or re-ossification.8,9 Treatment remains controversial and is guided by management of the disease’s complications. Options range from careful observation and supportive management to aggressive surgical resection and reconstruction, with positive outcomes reported using many different modalities.10 Most treatment successes, however, hinge on halting bony resorption using medical and radiation therapy. Surgery is usually reserved as a salvage option for patients who have failed medical modalities and have residual symptoms or functional limitations.6
This case report describes the successful surgical management of a patient with pelvic GSD who had progressive pain and functional limitation despite exhaustive medical and radiation therapy. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A healthy 27-year-old man sought medical attention after a fall while mowing his lawn that resulted in difficulty ambulating. Radiographic studies showed discontinuous lytic lesions in the right periacetabular region and the right sacroiliac (SI) joint. Biopsy at an outside institution revealed an infiltration of thin-walled branching vascular channels involving intertrabecular marrow spaces and periosteal connective tissue. The vessels were devoid of a muscular coat and lined by flattened epithelium; these features were seen as consistent with GSD.
The patient was managed medically at the outside institution for approximately 2 years, with regimens consisting of zoledronate, denosumab, sorafenib, vincristine, sirolimus, and bevacizumab. Because there is no standard chemotherapy protocol for GSD, this broad regimen was likely an attempt by treating physicians to control disease progression before considering radiation or surgery. Zoledronate, a bisphosphonate, and denosumab, a monoclonal antibody against the receptor activator of nuclear factor κβ ligand (RANKL), both inhibit bone resorption, making them logical choices in treating an osteolytic disease. Sorafenib, vincristine, sirolimus, and bevacizumab may be of clinical benefit in GSD via inhibition of vascular proliferation, which is a key histologic feature in GSD. Sorafenib inhibits the vascular endothelial growth factor (VEGF) receptor, vincristine and sirolimus inhibit VEGF production, and bevacizumab is a monoclonal antibody targeting VEGF.
The patient’s disease continued to involve more of his right hemipelvis despite this extensive regimen of chemotherapy, and he experienced significant functional decline about 2 years after initial presentation, when he was no longer able to ambulate unassisted. Radiation therapy to the pelvis was attempted at the outside institution (6/15 MV photons, 5040 cGy, 28 fractions) without improvement. Three years after his initial injury, he presented to our clinic.
Now age 30 years, the patient ambulated only with crutches and endorsed minimal improvement in his pain over 3 years of treatment. Physical examination of the patient revealed that he was a tall, thin man in visible discomfort. Sensation was intact to light touch in the bilateral L1 to S1 nerve distributions. There was marked weakness of the right lower extremity, and his examination was limited by pain. He could not perform a straight leg raise on the right side. Right quadriceps strength was 4/5, and right hamstrings strength was 3/5. There was no weakness in the left leg. Reflexes were normal and symmetric bilaterally at the patellar and gastrocnemius soleus tendons. Distal circulatory status in both extremities was normal, and there were no deformities of the skin.
Figure 1 shows the patient’s computed tomography (CT) scan. Figures 1A and 1B reveal fragmentation of the posterior ilia and sacrum along both SI joints. Dislocation of the pubic symphysis is shown in Figures 1C and 1D, and discontinuous involvement of the ischium and posterior wall of the acetabulum is visible in Figure 1E.
Serum studies, including C-reactive protein, erythrocyte sedimentation rate, and a complete blood count, were within normal limits. A CT-guided core needle biopsy and aspiration of the right SI joint revealed no infection; pathology was nondiagnostic. Anesthetic injection of the hip joint resulted in no relief. As this man was severely functionally limited and had exhausted all medical and radiation treatment options, a collaborative decision was made to proceed with surgical management. Surgical options included spinopelvic fusion unilaterally or bilaterally, hip arthroplasty, or sacropelvic resection with or without reconstruction. The patient opted for intralesional surgery and spinopelvic fusion in place of more radical options.
Thirty-seven months after his initial presentation, he underwent posterior spinal fusion L5 to S1, SI fusion, and anterior locking plate fixation of the pubic symphysis, as seen in Figure 2. Pathology from surgical specimens, seen at original magnification ×20 and ×100 in Figures 3A and 3B, respectively, showed prominent vascular proliferation in the right ilium, with reactive bone changes in the left ilium and right sacrum. A lytic lesion showed fibrous tissue with an embedded fragment of necrotic bone.
Six weeks after surgery, the patient had substantial improvement in his pain and was partially weight-bearing. He was able to ambulate with crutches and returned to work. The patient’s overall clinical status continued to improve throughout the postoperative course. He developed low back pain 7 months after surgery and was found to have a sacrococcygeal abscess and coccygeal fracture anterior to the sacrum. He underwent irrigation and débridement of the abscess and distal coccygectomy and was treated with 6 weeks of intravenous cefazolin and long-term suppression with levofloxacin and rifampin for methicillin-sensitive Staphylococcus aureus hardware infection and osteomyelitis. The patient’s clinical course subsequently improved. At latest follow-up 16 months after the index operation, pain was reported as manageable and mostly an annoyance. He was prescribed up to 40 mg of oxycodone daily for pain. The patient returned to work, ambulates with a cane (no other assistive devices), and reports being able to get around without any difficulty.
Discussion
Gorham-Stout disease is an exceedingly rare condition resulting in spontaneous osteolysis. Approximately 200 cases have been reported with no apparent gender, race, or familial predilection or systemic symptoms differentiating it from other etiologies of idiopathic osteolysis.6 These patients often seek medical attention after sustaining a pathologic fracture,6 when a broad differential diagnosis narrows to GSD only after biopsy excludes other possibilities and demonstrates characteristic angiomatosis without malignant features.2,4,6,8,10 Gorham-Stout disease appears more frequently at particular sites within the skeleton, and pelvic involvement is common—more than 20% of cases in 1 review.5,10 Limitations in the patient’s ability to ambulate invariably result from osteolysis of the pelvis, which is concerning considering the young age at which GSD typically presents. A variety of treatment modalities have been described for pelvic GSD, but surgery has been undertaken in relatively few cases.5
The diagnosis is one of exclusion after considering the clinical context and radiologic and pathologic findings. In this case, a pathologic fracture was discovered with osteolytic lesions throughout the hemipelvis. Biopsy excluded malignancy and demonstrated the key hemangiomatous vascular proliferation with thin-walled vessels that is classic for GSD. While our patient initially appeared to have 2 sites of disease, the surgical specimen revealed a primary site of vascular proliferation in the right ilium from which 2 apparent foci had spread, consistent with the typical monocentric presentation of GSD.11 A broad differential diagnosis must be considered at initial presentation, including osteomyelitis, metastatic disease, multiple myeloma, and primary bone sarcoma. Upon identifying a primary osteolytic process, several considerations besides GSD remain, such as Hajdu-Cheney syndrome, Winchester syndrome, multicentric osteolysis with nephropathy, familial osteolysis, Farber disease, and neurogenic osteolysis; most of these etiologies involve familial predispositions and/or systemic symptoms.
Treatment options for GSD include supportive care, medical therapy, radiation, and surgery. For pelvic GSD, numerous reports have demonstrated good outcomes with supportive management, since osteolysis often spontaneously arrests.8,9,12 Others have had success with medical treatments in attempts to halt bone resorption.6,13-15 Bisphosphonates are the cornerstone of medical therapy in GSD, as they appear to halt further osteoclastic bone breakdown. The levels of VEGF have been shown to be elevated in GSD,13 likely consistent with the vascular proliferation evident on pathology, and therapies such as bevacizumab and interferon α-2b have been used to target osteolysis via this pathway with good outcome.13,14,16 External beam-radiation therapy has been shown to prevent local progression of osteolysis in up to 80% of cases.4 However, even with arrest of bone resorption, damage to affected bone may have progressed to the point of significant functional limitation. This may be especially true in the pelvis.
We present a case of a patient who continued to deteriorate after maximal medical and radiation therapy. Many reported cases of pelvic GSD have had good outcomes with some combination of conservative management, medical therapy, and radiation. However, in our patient, the pelvis and lumbosacral spine were unstable as a result of significant bone loss and fracture, and his clinical deterioration was dramatic. We considered reasonable surgical approaches, including local intralesional débridement and massive en bloc resection with structural allograft. We chose the less radical procedure given the patient’s age, minimal surgical history, and personal preference. Although structural pelvic allograft has been successful in a few cases, there remains a high risk of complications, such as fracture, resorption, or infection.17 We considered the addition of hip arthroplasty with either scenario, but we elected not to perform this component given his young age and lack of symptomatic improvement with diagnostic anesthetic hip injection. The key to this patient’s surgical reconstruction, aside from eliminating gross disease, was the stabilization of the spinopelvic junction and pelvic ring. His functional improvement as early as 6 weeks after surgery demonstrates that surgery can have an important role for patients with pelvic GSD who fail medical and radiation therapy.
Gorham-Stout disease (GSD) is a rare condition characterized by spontaneous idiopathic resorption of bone with lymphovascular proliferation and an absence of malignant features. It was originally described by Jackson1 in an 1838 report of a 36-year-old man whose “arm bone, between the shoulder and elbow” had completely vanished after 2 fractures. The disease was defined and its pathology characterized by Gorham and Stout2 in 1955 in a series of 24 patients. Despite about 200 reported cases in the literature,3 its etiology remains unclear. Any bone in the skeleton may be affected by GSD, although there is a predilection for the skull, humerus, clavicle, ribs, pelvis, and femur.4-6 It commonly manifests within the first 3 decades of life, but case reports range from as early as 2 months of age to the eighth decade.5,7
Gorham-Stout disease is a diagnosis of exclusion that requires careful consideration of the clinical context, radiographic findings, and histopathology. Typical histopathologic findings include benign lymphatic or vascular proliferation, involution of adipose tissue within the bone marrow, and thinning of bony trabeculae.6 Fibrous tissue may replace vascular tissue after the initial vasoproliferative, osteolytic phase.6 Some authors describe the disease as having 2 phases, the first with massive osteolysis followed by relative dormancy and the second without progression or re-ossification.8,9 Treatment remains controversial and is guided by management of the disease’s complications. Options range from careful observation and supportive management to aggressive surgical resection and reconstruction, with positive outcomes reported using many different modalities.10 Most treatment successes, however, hinge on halting bony resorption using medical and radiation therapy. Surgery is usually reserved as a salvage option for patients who have failed medical modalities and have residual symptoms or functional limitations.6
This case report describes the successful surgical management of a patient with pelvic GSD who had progressive pain and functional limitation despite exhaustive medical and radiation therapy. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A healthy 27-year-old man sought medical attention after a fall while mowing his lawn that resulted in difficulty ambulating. Radiographic studies showed discontinuous lytic lesions in the right periacetabular region and the right sacroiliac (SI) joint. Biopsy at an outside institution revealed an infiltration of thin-walled branching vascular channels involving intertrabecular marrow spaces and periosteal connective tissue. The vessels were devoid of a muscular coat and lined by flattened epithelium; these features were seen as consistent with GSD.
The patient was managed medically at the outside institution for approximately 2 years, with regimens consisting of zoledronate, denosumab, sorafenib, vincristine, sirolimus, and bevacizumab. Because there is no standard chemotherapy protocol for GSD, this broad regimen was likely an attempt by treating physicians to control disease progression before considering radiation or surgery. Zoledronate, a bisphosphonate, and denosumab, a monoclonal antibody against the receptor activator of nuclear factor κβ ligand (RANKL), both inhibit bone resorption, making them logical choices in treating an osteolytic disease. Sorafenib, vincristine, sirolimus, and bevacizumab may be of clinical benefit in GSD via inhibition of vascular proliferation, which is a key histologic feature in GSD. Sorafenib inhibits the vascular endothelial growth factor (VEGF) receptor, vincristine and sirolimus inhibit VEGF production, and bevacizumab is a monoclonal antibody targeting VEGF.
The patient’s disease continued to involve more of his right hemipelvis despite this extensive regimen of chemotherapy, and he experienced significant functional decline about 2 years after initial presentation, when he was no longer able to ambulate unassisted. Radiation therapy to the pelvis was attempted at the outside institution (6/15 MV photons, 5040 cGy, 28 fractions) without improvement. Three years after his initial injury, he presented to our clinic.
Now age 30 years, the patient ambulated only with crutches and endorsed minimal improvement in his pain over 3 years of treatment. Physical examination of the patient revealed that he was a tall, thin man in visible discomfort. Sensation was intact to light touch in the bilateral L1 to S1 nerve distributions. There was marked weakness of the right lower extremity, and his examination was limited by pain. He could not perform a straight leg raise on the right side. Right quadriceps strength was 4/5, and right hamstrings strength was 3/5. There was no weakness in the left leg. Reflexes were normal and symmetric bilaterally at the patellar and gastrocnemius soleus tendons. Distal circulatory status in both extremities was normal, and there were no deformities of the skin.
Figure 1 shows the patient’s computed tomography (CT) scan. Figures 1A and 1B reveal fragmentation of the posterior ilia and sacrum along both SI joints. Dislocation of the pubic symphysis is shown in Figures 1C and 1D, and discontinuous involvement of the ischium and posterior wall of the acetabulum is visible in Figure 1E.
Serum studies, including C-reactive protein, erythrocyte sedimentation rate, and a complete blood count, were within normal limits. A CT-guided core needle biopsy and aspiration of the right SI joint revealed no infection; pathology was nondiagnostic. Anesthetic injection of the hip joint resulted in no relief. As this man was severely functionally limited and had exhausted all medical and radiation treatment options, a collaborative decision was made to proceed with surgical management. Surgical options included spinopelvic fusion unilaterally or bilaterally, hip arthroplasty, or sacropelvic resection with or without reconstruction. The patient opted for intralesional surgery and spinopelvic fusion in place of more radical options.
Thirty-seven months after his initial presentation, he underwent posterior spinal fusion L5 to S1, SI fusion, and anterior locking plate fixation of the pubic symphysis, as seen in Figure 2. Pathology from surgical specimens, seen at original magnification ×20 and ×100 in Figures 3A and 3B, respectively, showed prominent vascular proliferation in the right ilium, with reactive bone changes in the left ilium and right sacrum. A lytic lesion showed fibrous tissue with an embedded fragment of necrotic bone.
Six weeks after surgery, the patient had substantial improvement in his pain and was partially weight-bearing. He was able to ambulate with crutches and returned to work. The patient’s overall clinical status continued to improve throughout the postoperative course. He developed low back pain 7 months after surgery and was found to have a sacrococcygeal abscess and coccygeal fracture anterior to the sacrum. He underwent irrigation and débridement of the abscess and distal coccygectomy and was treated with 6 weeks of intravenous cefazolin and long-term suppression with levofloxacin and rifampin for methicillin-sensitive Staphylococcus aureus hardware infection and osteomyelitis. The patient’s clinical course subsequently improved. At latest follow-up 16 months after the index operation, pain was reported as manageable and mostly an annoyance. He was prescribed up to 40 mg of oxycodone daily for pain. The patient returned to work, ambulates with a cane (no other assistive devices), and reports being able to get around without any difficulty.
Discussion
Gorham-Stout disease is an exceedingly rare condition resulting in spontaneous osteolysis. Approximately 200 cases have been reported with no apparent gender, race, or familial predilection or systemic symptoms differentiating it from other etiologies of idiopathic osteolysis.6 These patients often seek medical attention after sustaining a pathologic fracture,6 when a broad differential diagnosis narrows to GSD only after biopsy excludes other possibilities and demonstrates characteristic angiomatosis without malignant features.2,4,6,8,10 Gorham-Stout disease appears more frequently at particular sites within the skeleton, and pelvic involvement is common—more than 20% of cases in 1 review.5,10 Limitations in the patient’s ability to ambulate invariably result from osteolysis of the pelvis, which is concerning considering the young age at which GSD typically presents. A variety of treatment modalities have been described for pelvic GSD, but surgery has been undertaken in relatively few cases.5
The diagnosis is one of exclusion after considering the clinical context and radiologic and pathologic findings. In this case, a pathologic fracture was discovered with osteolytic lesions throughout the hemipelvis. Biopsy excluded malignancy and demonstrated the key hemangiomatous vascular proliferation with thin-walled vessels that is classic for GSD. While our patient initially appeared to have 2 sites of disease, the surgical specimen revealed a primary site of vascular proliferation in the right ilium from which 2 apparent foci had spread, consistent with the typical monocentric presentation of GSD.11 A broad differential diagnosis must be considered at initial presentation, including osteomyelitis, metastatic disease, multiple myeloma, and primary bone sarcoma. Upon identifying a primary osteolytic process, several considerations besides GSD remain, such as Hajdu-Cheney syndrome, Winchester syndrome, multicentric osteolysis with nephropathy, familial osteolysis, Farber disease, and neurogenic osteolysis; most of these etiologies involve familial predispositions and/or systemic symptoms.
Treatment options for GSD include supportive care, medical therapy, radiation, and surgery. For pelvic GSD, numerous reports have demonstrated good outcomes with supportive management, since osteolysis often spontaneously arrests.8,9,12 Others have had success with medical treatments in attempts to halt bone resorption.6,13-15 Bisphosphonates are the cornerstone of medical therapy in GSD, as they appear to halt further osteoclastic bone breakdown. The levels of VEGF have been shown to be elevated in GSD,13 likely consistent with the vascular proliferation evident on pathology, and therapies such as bevacizumab and interferon α-2b have been used to target osteolysis via this pathway with good outcome.13,14,16 External beam-radiation therapy has been shown to prevent local progression of osteolysis in up to 80% of cases.4 However, even with arrest of bone resorption, damage to affected bone may have progressed to the point of significant functional limitation. This may be especially true in the pelvis.
We present a case of a patient who continued to deteriorate after maximal medical and radiation therapy. Many reported cases of pelvic GSD have had good outcomes with some combination of conservative management, medical therapy, and radiation. However, in our patient, the pelvis and lumbosacral spine were unstable as a result of significant bone loss and fracture, and his clinical deterioration was dramatic. We considered reasonable surgical approaches, including local intralesional débridement and massive en bloc resection with structural allograft. We chose the less radical procedure given the patient’s age, minimal surgical history, and personal preference. Although structural pelvic allograft has been successful in a few cases, there remains a high risk of complications, such as fracture, resorption, or infection.17 We considered the addition of hip arthroplasty with either scenario, but we elected not to perform this component given his young age and lack of symptomatic improvement with diagnostic anesthetic hip injection. The key to this patient’s surgical reconstruction, aside from eliminating gross disease, was the stabilization of the spinopelvic junction and pelvic ring. His functional improvement as early as 6 weeks after surgery demonstrates that surgery can have an important role for patients with pelvic GSD who fail medical and radiation therapy.
1. Jackson JBS. A boneless arm. Boston Med Surg J. 1838;18:368-369.
2. Gorham LW, Stout AP. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone): its relation to hemangiomatosis. J Bone Joint Surg Am. 1955;37(5):985-1004.
3. Lehmann G, Pfeil A, Böttcher J, et al. Benefit of a 17-year long-term bisphosphonate therapy in a patient with Gorham-Stout syndrome. Arch Orthop Trauma Surg. 2009;129(7):967-972.
4. Heyd R, Micke O, Surholt C, et al; German Cooperative Group on Radiotherapy for Benign Diseases (GCG-BD). Radiation therapy for Gorham-Stout syndrome: results of a national patterns-of-care study and literature review. Int J Radiat Oncol Biol Phys. 2011;81(3):e179-e185.
5. Kulenkampff HA, Richter GM, Hasse WE, Adler CP. Massive pelvic osteolysis in the Gorham-Stout syndrome. Int Orthop. 1990;14(4):361-366.
6. Ruggieri P, Montalti M, Angelini A, Alberghini M, Mercuri M. Gorham-Stout disease: the experience of the Rizzoli Institute and review of the literature. Skeletal Radiol. 2011;40(11):1391-1397.
7. Vinée P, Tanyü MO, Hauenstein KH, Sigmund G, Stöver B, Adler CP. CT and MRI of Gorham syndrome. J Comput Assist Tomogr. 1994;18(6):985-989.
8. Boyer P, Bourgeois P, Boyer O, Catonné Y, Saillant G. Massive Gorham-Stout syndrome of the pelvis. Clin Rheumatol. 2005;24(5):551-555.
9. Malde R, Agrawal HM, Ghosh SL, Dinshaw KA. Vanishing bone disease involving the pelvis. J Cancer Res Ther. 2005;1(4):227-228.
10. Kuriyama DK, McElligott SC, Glaser DW, Thompson KS. Treatment of Gorham-Stout disease with zoledronic acid and interferon-α: a case report and literature review. J Pediatr Hematol Oncol. 2010;32(8):579-584.
11. Tie ML, Poland GA, Rosenow EC III. Chylothorax in Gorham’s syndrome. A common complication of a rare disease. Chest. 1994;105(1):208-213.
12. Möller G, Priemel M, Amling M, Werner M, Kuhlmey AS, Delling G. The Gorham-Stout syndrome (Gorham’s massive osteolysis). A report of six cases with histopathological findings. J Bone Joint Surg Br. 1999;81(3):501-506.
13. Dupond JL, Bermont L, Runge M, de Billy M. Plasma VEGF determination in disseminated lymphangiomatosis—Gorham-Stout syndrome: a marker of activity? A case report with a 5-year follow-up. Bone. 2010;46(3):873-876.
14. Wang JD, Chang TK, Cheng YY, et al. A child with dyspnea and unstable gait. Pediatr Hemat Oncol. 2007;24(4):321-324.
15. Zheng MW, Yang M, Qiu JX, et al. Gorham-Stout syndrome presenting in a 5-year-old girl with a successful bisphosphonate therapeutic effect. Exp Ther Med. 2012;4(3):449-451.
16. Timke C, Krause MF, Oppermann HC, Leuschner I, Claviez A. Interferon alpha 2b treatment in an eleven-year-old boy with disseminated lymphangiomatosis. Pediatr Blood Cancer. 2007;48(1):108-111.
17. Stöve J, Reichelt A. Massive osteolysis of the pelvis, femur and sacral bone with a Gorham-Stout syndrome. Arch Orthop Trauma Surg. 1995;114(4):207-210.
1. Jackson JBS. A boneless arm. Boston Med Surg J. 1838;18:368-369.
2. Gorham LW, Stout AP. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone): its relation to hemangiomatosis. J Bone Joint Surg Am. 1955;37(5):985-1004.
3. Lehmann G, Pfeil A, Böttcher J, et al. Benefit of a 17-year long-term bisphosphonate therapy in a patient with Gorham-Stout syndrome. Arch Orthop Trauma Surg. 2009;129(7):967-972.
4. Heyd R, Micke O, Surholt C, et al; German Cooperative Group on Radiotherapy for Benign Diseases (GCG-BD). Radiation therapy for Gorham-Stout syndrome: results of a national patterns-of-care study and literature review. Int J Radiat Oncol Biol Phys. 2011;81(3):e179-e185.
5. Kulenkampff HA, Richter GM, Hasse WE, Adler CP. Massive pelvic osteolysis in the Gorham-Stout syndrome. Int Orthop. 1990;14(4):361-366.
6. Ruggieri P, Montalti M, Angelini A, Alberghini M, Mercuri M. Gorham-Stout disease: the experience of the Rizzoli Institute and review of the literature. Skeletal Radiol. 2011;40(11):1391-1397.
7. Vinée P, Tanyü MO, Hauenstein KH, Sigmund G, Stöver B, Adler CP. CT and MRI of Gorham syndrome. J Comput Assist Tomogr. 1994;18(6):985-989.
8. Boyer P, Bourgeois P, Boyer O, Catonné Y, Saillant G. Massive Gorham-Stout syndrome of the pelvis. Clin Rheumatol. 2005;24(5):551-555.
9. Malde R, Agrawal HM, Ghosh SL, Dinshaw KA. Vanishing bone disease involving the pelvis. J Cancer Res Ther. 2005;1(4):227-228.
10. Kuriyama DK, McElligott SC, Glaser DW, Thompson KS. Treatment of Gorham-Stout disease with zoledronic acid and interferon-α: a case report and literature review. J Pediatr Hematol Oncol. 2010;32(8):579-584.
11. Tie ML, Poland GA, Rosenow EC III. Chylothorax in Gorham’s syndrome. A common complication of a rare disease. Chest. 1994;105(1):208-213.
12. Möller G, Priemel M, Amling M, Werner M, Kuhlmey AS, Delling G. The Gorham-Stout syndrome (Gorham’s massive osteolysis). A report of six cases with histopathological findings. J Bone Joint Surg Br. 1999;81(3):501-506.
13. Dupond JL, Bermont L, Runge M, de Billy M. Plasma VEGF determination in disseminated lymphangiomatosis—Gorham-Stout syndrome: a marker of activity? A case report with a 5-year follow-up. Bone. 2010;46(3):873-876.
14. Wang JD, Chang TK, Cheng YY, et al. A child with dyspnea and unstable gait. Pediatr Hemat Oncol. 2007;24(4):321-324.
15. Zheng MW, Yang M, Qiu JX, et al. Gorham-Stout syndrome presenting in a 5-year-old girl with a successful bisphosphonate therapeutic effect. Exp Ther Med. 2012;4(3):449-451.
16. Timke C, Krause MF, Oppermann HC, Leuschner I, Claviez A. Interferon alpha 2b treatment in an eleven-year-old boy with disseminated lymphangiomatosis. Pediatr Blood Cancer. 2007;48(1):108-111.
17. Stöve J, Reichelt A. Massive osteolysis of the pelvis, femur and sacral bone with a Gorham-Stout syndrome. Arch Orthop Trauma Surg. 1995;114(4):207-210.