User login
National Institute of Mental Health (NIMH): New Clinical Drug Evaluation Unit (NCDEU)
Lurasidone produces less daytime sleepiness than quetiapine XR
HOLLYWOOD, FLA. – Lurasidone resulted in significantly less daytime sleepiness and improved cognitive performance, compared with extended-release quetiapine in a long-term, double-blind, head-to-head comparative trial in patients with schizophrenia.
The 6-month, double-blind study involved 207 patients on lurasidone (Latuda) flexibly dosed at 40-160 mg once daily in the evening and 85 patients on extended-release quetiapine (Seroquel XR) at 200-800 mg/day.
One of the two major endpoints was change in daytime alertness as measured by the validated Epworth Sleepiness Scale. From a mean baseline ESS of 5.9, scores in the lurasidone group improved to a mean of 4.4. This was a significantly greater improvement in daytime sleepiness than with quetiapine, where scores went from 6.48 to 5.9, Philip D. Harvey, Ph.D., reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The other major endpoint was change over time in cognitive performance as assessed using the computerized CogState schizophrenia battery. From a mean baseline composite Z-score of –3.9, scores in the lurasidone group improved by roughly 1.5 points, a threefold greater gain than in patients on quetiapine, added Dr. Harvey, professor of psychiatry and behavioral sciences, and chief of the division of psychology at the University of Miami.
Although it seems logical that daytime sleepiness would impair cognition and interfere with everyday functional capacity, in a multivariate analysis, the superior cognitive performance at month 6 in the lurasidone group was independent of the antipsychotic agent’s effect upon sleepiness.
Lurasidone has greater affinity for serotonin 5HT7 and 5HT1A receptors than other second-generation antipsychotic agents. These receptors are abundant in areas of the brain involved in sleep, mood regulation, and memory. These pharmacologic attributes provide a biologic basis in support of the improvements in cognitive performance and daytime sleepiness seen in the head-to-head trial.
Dr. Harvey is a consultant to Sunovion Pharmaceuticals, which sponsored the comparative trial, as well as to a half-dozen other pharmaceutical companies.
HOLLYWOOD, FLA. – Lurasidone resulted in significantly less daytime sleepiness and improved cognitive performance, compared with extended-release quetiapine in a long-term, double-blind, head-to-head comparative trial in patients with schizophrenia.
The 6-month, double-blind study involved 207 patients on lurasidone (Latuda) flexibly dosed at 40-160 mg once daily in the evening and 85 patients on extended-release quetiapine (Seroquel XR) at 200-800 mg/day.
One of the two major endpoints was change in daytime alertness as measured by the validated Epworth Sleepiness Scale. From a mean baseline ESS of 5.9, scores in the lurasidone group improved to a mean of 4.4. This was a significantly greater improvement in daytime sleepiness than with quetiapine, where scores went from 6.48 to 5.9, Philip D. Harvey, Ph.D., reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The other major endpoint was change over time in cognitive performance as assessed using the computerized CogState schizophrenia battery. From a mean baseline composite Z-score of –3.9, scores in the lurasidone group improved by roughly 1.5 points, a threefold greater gain than in patients on quetiapine, added Dr. Harvey, professor of psychiatry and behavioral sciences, and chief of the division of psychology at the University of Miami.
Although it seems logical that daytime sleepiness would impair cognition and interfere with everyday functional capacity, in a multivariate analysis, the superior cognitive performance at month 6 in the lurasidone group was independent of the antipsychotic agent’s effect upon sleepiness.
Lurasidone has greater affinity for serotonin 5HT7 and 5HT1A receptors than other second-generation antipsychotic agents. These receptors are abundant in areas of the brain involved in sleep, mood regulation, and memory. These pharmacologic attributes provide a biologic basis in support of the improvements in cognitive performance and daytime sleepiness seen in the head-to-head trial.
Dr. Harvey is a consultant to Sunovion Pharmaceuticals, which sponsored the comparative trial, as well as to a half-dozen other pharmaceutical companies.
HOLLYWOOD, FLA. – Lurasidone resulted in significantly less daytime sleepiness and improved cognitive performance, compared with extended-release quetiapine in a long-term, double-blind, head-to-head comparative trial in patients with schizophrenia.
The 6-month, double-blind study involved 207 patients on lurasidone (Latuda) flexibly dosed at 40-160 mg once daily in the evening and 85 patients on extended-release quetiapine (Seroquel XR) at 200-800 mg/day.
One of the two major endpoints was change in daytime alertness as measured by the validated Epworth Sleepiness Scale. From a mean baseline ESS of 5.9, scores in the lurasidone group improved to a mean of 4.4. This was a significantly greater improvement in daytime sleepiness than with quetiapine, where scores went from 6.48 to 5.9, Philip D. Harvey, Ph.D., reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The other major endpoint was change over time in cognitive performance as assessed using the computerized CogState schizophrenia battery. From a mean baseline composite Z-score of –3.9, scores in the lurasidone group improved by roughly 1.5 points, a threefold greater gain than in patients on quetiapine, added Dr. Harvey, professor of psychiatry and behavioral sciences, and chief of the division of psychology at the University of Miami.
Although it seems logical that daytime sleepiness would impair cognition and interfere with everyday functional capacity, in a multivariate analysis, the superior cognitive performance at month 6 in the lurasidone group was independent of the antipsychotic agent’s effect upon sleepiness.
Lurasidone has greater affinity for serotonin 5HT7 and 5HT1A receptors than other second-generation antipsychotic agents. These receptors are abundant in areas of the brain involved in sleep, mood regulation, and memory. These pharmacologic attributes provide a biologic basis in support of the improvements in cognitive performance and daytime sleepiness seen in the head-to-head trial.
Dr. Harvey is a consultant to Sunovion Pharmaceuticals, which sponsored the comparative trial, as well as to a half-dozen other pharmaceutical companies.
AT THE NCDEU MEETING
Major finding: Patients with schizophrenia who were randomized to 6 months of double-blind lurasidone experienced significantly less daytime sleepiness than did those on extended-release quetiapine as reflected in their mean 1.5-point improvement over baseline on the Epworth Sleepiness Scale as compared with a 0.58-point improvement with quetiapine XR.
Data source: This was a randomized double-blind clinical trial involving 207 patients with schizophrenia assigned to lurasidone and 85 others placed on extended-release quetiapine.
Disclosures: The study was sponsored by Sunovion Pharmaceuticals, which markets lurasidone. The presenter is a consultant to the company.
Duloxetine proves beneficial in elderly GAD
HOLLYWOOD, FLA. – Duloxetine proved safe and effective for the treatment of generalized anxiety disorder in the elderly in a phase IV clinical trial restricted to patients aged 65 and up.
This study fills a major gap in the evidence base for duloxetine (Cymbalta). Generalized anxiety disorder (GAD) is one of the most common psychiatric disorders in the elderly, with a prevalence estimated at up to 7%. Yet prior studies of duloxetine for GAD largely excluded the elderly. Indeed, the product labeling states prominently that premarketing studies did not include enough patients over age 65 to determine whether they respond differently than younger subjects.
With the new evidence provided by the phase IV study, the answer to that question is now in: Duloxetine proved significantly more effective than placebo in seniors. Moreover, the discontinuation rate because of adverse events did not differ significantly between the duloxetine and placebo groups, Karla J. Alaka reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The study included 291 patients aged 65 or older (mean age, 72) who met DSM-IV-TR criteria for GAD. They were randomized to 10 weeks of double-blind treatment with duloxetine dosed at 30-120 mg/day or placebo. Throughout the study, roughly one-third of the duloxetine group remained on 30 mg/day, another third bumped up to 60 mg/day, one-quarter increased to 90 mg/day, and the remainder eventually received 120 mg/day.
Elderly patients with an additional Axis I diagnosis plus GAD were not eligible for the trial. However, patients with comorbid medical illnesses were not excluded from participation so long as those conditions were stable and not expected to result in hospitalization within the next 6 months. Fully 83% of subjects had one or more preexisting medical conditions for which they took concomitant medication during the study. Geriatricians and other physicians have become increasingly vocal about the absence of quality safety and efficacy data for many widely prescribed drugs in the elderly, where issues such as polypharmacy, drug-drug interactions, and slowed drug metabolism become key considerations.
The primary efficacy measure in the study was a change in the Hamilton Anxiety Scale (HAS) total score from baseline to week 10. From a mean baseline HAS score of 24.6, the duloxetine group averaged a 15.9-point decrease, significantly better than the 11.7-point drop with placebo, according to Ms. Alaka of Eli Lilly, Indianapolis.
The HAS response rate, defined by at least a 50% reduction in the HAS total score, was 75% in duloxetine-treated patients, compared with 56% in controls. The Hamilton Anxiety Scale remission rate, a more stringent endpoint requiring a week 10 total score of 10 or less, was achieved by 62% of the duloxetine group and 40% on placebo.
Duloxetine-treated patients also outperformed controls in terms of several other secondary endpoints. For example, the Sheehan Disability Scale global function impairment score in the duloxetine group improved by a mean of 7.6 points from a baseline score of 13.7, compared with a 4.3-point improvement with placebo.
Study discontinuation because of treatment-emergent adverse events occurred in 9.9% of elderly patients on duloxetine and 10.7% on placebo. The only adverse event that was significantly more common in the active treatment group was dry mouth, which occurred in 7% on duloxetine, compared with 1% of controls. Duloxetine was not associated with any clinically significant changes in laboratory findings, ECG parameters, or body weight.
The phase IV study was sponsored by Eli Lilly, which markets duloxetine. The presenter is a company employee.
HOLLYWOOD, FLA. – Duloxetine proved safe and effective for the treatment of generalized anxiety disorder in the elderly in a phase IV clinical trial restricted to patients aged 65 and up.
This study fills a major gap in the evidence base for duloxetine (Cymbalta). Generalized anxiety disorder (GAD) is one of the most common psychiatric disorders in the elderly, with a prevalence estimated at up to 7%. Yet prior studies of duloxetine for GAD largely excluded the elderly. Indeed, the product labeling states prominently that premarketing studies did not include enough patients over age 65 to determine whether they respond differently than younger subjects.
With the new evidence provided by the phase IV study, the answer to that question is now in: Duloxetine proved significantly more effective than placebo in seniors. Moreover, the discontinuation rate because of adverse events did not differ significantly between the duloxetine and placebo groups, Karla J. Alaka reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The study included 291 patients aged 65 or older (mean age, 72) who met DSM-IV-TR criteria for GAD. They were randomized to 10 weeks of double-blind treatment with duloxetine dosed at 30-120 mg/day or placebo. Throughout the study, roughly one-third of the duloxetine group remained on 30 mg/day, another third bumped up to 60 mg/day, one-quarter increased to 90 mg/day, and the remainder eventually received 120 mg/day.
Elderly patients with an additional Axis I diagnosis plus GAD were not eligible for the trial. However, patients with comorbid medical illnesses were not excluded from participation so long as those conditions were stable and not expected to result in hospitalization within the next 6 months. Fully 83% of subjects had one or more preexisting medical conditions for which they took concomitant medication during the study. Geriatricians and other physicians have become increasingly vocal about the absence of quality safety and efficacy data for many widely prescribed drugs in the elderly, where issues such as polypharmacy, drug-drug interactions, and slowed drug metabolism become key considerations.
The primary efficacy measure in the study was a change in the Hamilton Anxiety Scale (HAS) total score from baseline to week 10. From a mean baseline HAS score of 24.6, the duloxetine group averaged a 15.9-point decrease, significantly better than the 11.7-point drop with placebo, according to Ms. Alaka of Eli Lilly, Indianapolis.
The HAS response rate, defined by at least a 50% reduction in the HAS total score, was 75% in duloxetine-treated patients, compared with 56% in controls. The Hamilton Anxiety Scale remission rate, a more stringent endpoint requiring a week 10 total score of 10 or less, was achieved by 62% of the duloxetine group and 40% on placebo.
Duloxetine-treated patients also outperformed controls in terms of several other secondary endpoints. For example, the Sheehan Disability Scale global function impairment score in the duloxetine group improved by a mean of 7.6 points from a baseline score of 13.7, compared with a 4.3-point improvement with placebo.
Study discontinuation because of treatment-emergent adverse events occurred in 9.9% of elderly patients on duloxetine and 10.7% on placebo. The only adverse event that was significantly more common in the active treatment group was dry mouth, which occurred in 7% on duloxetine, compared with 1% of controls. Duloxetine was not associated with any clinically significant changes in laboratory findings, ECG parameters, or body weight.
The phase IV study was sponsored by Eli Lilly, which markets duloxetine. The presenter is a company employee.
HOLLYWOOD, FLA. – Duloxetine proved safe and effective for the treatment of generalized anxiety disorder in the elderly in a phase IV clinical trial restricted to patients aged 65 and up.
This study fills a major gap in the evidence base for duloxetine (Cymbalta). Generalized anxiety disorder (GAD) is one of the most common psychiatric disorders in the elderly, with a prevalence estimated at up to 7%. Yet prior studies of duloxetine for GAD largely excluded the elderly. Indeed, the product labeling states prominently that premarketing studies did not include enough patients over age 65 to determine whether they respond differently than younger subjects.
With the new evidence provided by the phase IV study, the answer to that question is now in: Duloxetine proved significantly more effective than placebo in seniors. Moreover, the discontinuation rate because of adverse events did not differ significantly between the duloxetine and placebo groups, Karla J. Alaka reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The study included 291 patients aged 65 or older (mean age, 72) who met DSM-IV-TR criteria for GAD. They were randomized to 10 weeks of double-blind treatment with duloxetine dosed at 30-120 mg/day or placebo. Throughout the study, roughly one-third of the duloxetine group remained on 30 mg/day, another third bumped up to 60 mg/day, one-quarter increased to 90 mg/day, and the remainder eventually received 120 mg/day.
Elderly patients with an additional Axis I diagnosis plus GAD were not eligible for the trial. However, patients with comorbid medical illnesses were not excluded from participation so long as those conditions were stable and not expected to result in hospitalization within the next 6 months. Fully 83% of subjects had one or more preexisting medical conditions for which they took concomitant medication during the study. Geriatricians and other physicians have become increasingly vocal about the absence of quality safety and efficacy data for many widely prescribed drugs in the elderly, where issues such as polypharmacy, drug-drug interactions, and slowed drug metabolism become key considerations.
The primary efficacy measure in the study was a change in the Hamilton Anxiety Scale (HAS) total score from baseline to week 10. From a mean baseline HAS score of 24.6, the duloxetine group averaged a 15.9-point decrease, significantly better than the 11.7-point drop with placebo, according to Ms. Alaka of Eli Lilly, Indianapolis.
The HAS response rate, defined by at least a 50% reduction in the HAS total score, was 75% in duloxetine-treated patients, compared with 56% in controls. The Hamilton Anxiety Scale remission rate, a more stringent endpoint requiring a week 10 total score of 10 or less, was achieved by 62% of the duloxetine group and 40% on placebo.
Duloxetine-treated patients also outperformed controls in terms of several other secondary endpoints. For example, the Sheehan Disability Scale global function impairment score in the duloxetine group improved by a mean of 7.6 points from a baseline score of 13.7, compared with a 4.3-point improvement with placebo.
Study discontinuation because of treatment-emergent adverse events occurred in 9.9% of elderly patients on duloxetine and 10.7% on placebo. The only adverse event that was significantly more common in the active treatment group was dry mouth, which occurred in 7% on duloxetine, compared with 1% of controls. Duloxetine was not associated with any clinically significant changes in laboratory findings, ECG parameters, or body weight.
The phase IV study was sponsored by Eli Lilly, which markets duloxetine. The presenter is a company employee.
AT THE NCDEU MEETING
Major finding: The first-ever randomized clinical trial of duloxetine for generalized anxiety disorder to include a substantial number of patients aged 65 and older showed a 62% remission rate with duloxetine, compared with 40% with placebo.
Data source: This 10-week, randomized, double-blind phase IV clinical trial included 291 patients with generalized anxiety disorder, all aged 65 or older.
Disclosures: The phase IV study was sponsored by Eli Lilly, which markets duloxetine. The presenter is a company employee.
Long-acting injectable antipsychotics provide better outcomes
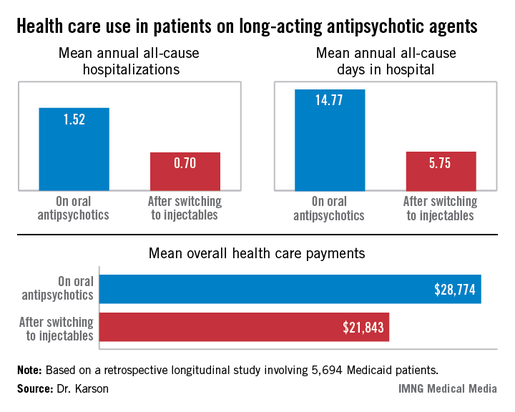
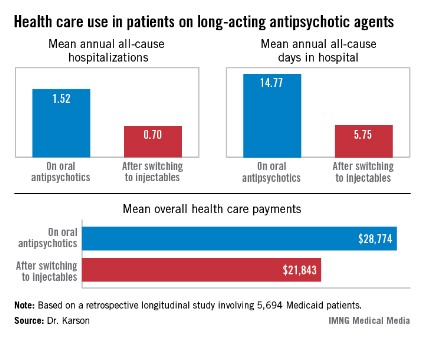
HOLLYWOOD, FLA. – Once patients were switched from oral antipsychotic agents to long-acting injectable ones, their annual all-cause hospitalization rate was cut by more than half in an observational study involving nearly 6,000 Medicaid patients with schizophrenia.
Moreover, the mean number of days per year spent in the hospital for any reason also dropped dramatically after the switch. Plus, the mean overall annual health care costs decreased by $6,901, Dr. Craig N. Karson reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
"We may have an opportunity here to improve the management of schizophrenia, and in particular the cost management," said Dr. Karson, a psychiatrist in Wayne, Pa.
He presented a retrospective longitudinal study of 5,694 Medicaid patients with schizophrenia who switched from oral to long-acting injectable antipsychotics during 2005-2010. The data came from the Thomson Reuters MarketScan Research Medicaid Database.

Forty-four percent of patients were placed on long-acting injectable risperidone, 41% on haloperidol, and the rest on fluphenazine, which were the three antipsychotic agents available in depot form during the study period.
Roughly 80% of the all-cause hospitalizations in this study were for schizophrenia.
The switch from oral to long-acting injectable antipsychotic agents was associated with a striking reduction in overall health care resource use, including a decrease in the mean annual number of all-cause hospitalizations from 1.52 to 0.70 (see box).
In order to see whether patients’ duration on long-acting injectable antipsychotics was related to health care resource use, Dr. Karson and his coinvestigators divided the study population into two groups: the 2,856 patients who were short-term users of long-acting injectable therapy, defined as less than 180 days of treatment; and the 2,838 users of long-acting injectables for at least 180 days. The short-term users averaged 0.79 all-cause hospitalizations annually; the long-term users, 0.61. The mean annualized total days in the hospital was 6.56 for the short-term users, compared with 4.93 in longer-term users.
Several speakers argued that the MarketScan database has significant methodologic shortcomings, including selection bias, rendering it better suited as a basis for hypothesis generation than for drawing firm conclusions. Dr. Karson acknowledged that the database is limited in that it provides a convenience sample rather than a random sample of the Medicaid population, but he argued that the shear size of the study population overcomes that limitation.
"Ask yourself, how many treatment studies do we have in psychiatry with almost 6,000 patients? Very few," he said. "When you get into these kinds of patient numbers, which provide enormous statistical power, and then use a very hard endpoint like annual all-cause hospitalizations, I think it changes the game a little. I would argue that these patients are representative of the Medicaid population with serious mental illness, regardless."
Discussant Dr. Stephen R. Marder, who was not involved in the study, agreed that the data are compelling."
"The differences seen in your study were large and persuasive. And I thought the analysis of short- versus long-term use showing a dose-response effect added to the confidence in the results. Being on depot medication longer makes a difference," concluded Dr. Marder, director of the section on psychosis at the University of California, Los Angeles, Neuropsychiatric Institute.
"The problem lies with us as providers: We see long-acting injectable antipsychotics as the last resort, said Dr. Marder, also professor of psychiatry and behavioral sciences at UCLA. "We’re probably reluctant to do it because we see the infrastructure for providing the therapy is very poor in many places."

At the VA medical center where he works, for example, patients formerly had access to depot medication on a 24/7 basis, so that if they had a job, they could obtain treatment without missing work. That service isn’t available, anymore, Dr. Marder noted.
His hope, he added, is that thought leaders in psychiatry will be able to influence implementation of the Affordable Care Act so as to create a much better infrastructure for providing long-acting injectable antipsychotic therapy. Ideally, access to long-acting injectable antipsychotics should be available in places where psychosocial and rehabilitation services are located. "That’s what seems to work the best," according to the psychiatrist.
Dr. Karson reported serving as a consultant to Otsuka America Pharmaceutical, which funded the study he presented, and to the Lieber Institute for Brain Development. Dr. Marder is a consultant to and/or has received research funding from Otsuka and 10 other companies.
HOLLYWOOD, FLA. – Once patients were switched from oral antipsychotic agents to long-acting injectable ones, their annual all-cause hospitalization rate was cut by more than half in an observational study involving nearly 6,000 Medicaid patients with schizophrenia.
Moreover, the mean number of days per year spent in the hospital for any reason also dropped dramatically after the switch. Plus, the mean overall annual health care costs decreased by $6,901, Dr. Craig N. Karson reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
"We may have an opportunity here to improve the management of schizophrenia, and in particular the cost management," said Dr. Karson, a psychiatrist in Wayne, Pa.
He presented a retrospective longitudinal study of 5,694 Medicaid patients with schizophrenia who switched from oral to long-acting injectable antipsychotics during 2005-2010. The data came from the Thomson Reuters MarketScan Research Medicaid Database.
Forty-four percent of patients were placed on long-acting injectable risperidone, 41% on haloperidol, and the rest on fluphenazine, which were the three antipsychotic agents available in depot form during the study period.
Roughly 80% of the all-cause hospitalizations in this study were for schizophrenia.
The switch from oral to long-acting injectable antipsychotic agents was associated with a striking reduction in overall health care resource use, including a decrease in the mean annual number of all-cause hospitalizations from 1.52 to 0.70 (see box).
In order to see whether patients’ duration on long-acting injectable antipsychotics was related to health care resource use, Dr. Karson and his coinvestigators divided the study population into two groups: the 2,856 patients who were short-term users of long-acting injectable therapy, defined as less than 180 days of treatment; and the 2,838 users of long-acting injectables for at least 180 days. The short-term users averaged 0.79 all-cause hospitalizations annually; the long-term users, 0.61. The mean annualized total days in the hospital was 6.56 for the short-term users, compared with 4.93 in longer-term users.
Several speakers argued that the MarketScan database has significant methodologic shortcomings, including selection bias, rendering it better suited as a basis for hypothesis generation than for drawing firm conclusions. Dr. Karson acknowledged that the database is limited in that it provides a convenience sample rather than a random sample of the Medicaid population, but he argued that the shear size of the study population overcomes that limitation.
"Ask yourself, how many treatment studies do we have in psychiatry with almost 6,000 patients? Very few," he said. "When you get into these kinds of patient numbers, which provide enormous statistical power, and then use a very hard endpoint like annual all-cause hospitalizations, I think it changes the game a little. I would argue that these patients are representative of the Medicaid population with serious mental illness, regardless."
Discussant Dr. Stephen R. Marder, who was not involved in the study, agreed that the data are compelling."
"The differences seen in your study were large and persuasive. And I thought the analysis of short- versus long-term use showing a dose-response effect added to the confidence in the results. Being on depot medication longer makes a difference," concluded Dr. Marder, director of the section on psychosis at the University of California, Los Angeles, Neuropsychiatric Institute.
"The problem lies with us as providers: We see long-acting injectable antipsychotics as the last resort, said Dr. Marder, also professor of psychiatry and behavioral sciences at UCLA. "We’re probably reluctant to do it because we see the infrastructure for providing the therapy is very poor in many places."
At the VA medical center where he works, for example, patients formerly had access to depot medication on a 24/7 basis, so that if they had a job, they could obtain treatment without missing work. That service isn’t available, anymore, Dr. Marder noted.
His hope, he added, is that thought leaders in psychiatry will be able to influence implementation of the Affordable Care Act so as to create a much better infrastructure for providing long-acting injectable antipsychotic therapy. Ideally, access to long-acting injectable antipsychotics should be available in places where psychosocial and rehabilitation services are located. "That’s what seems to work the best," according to the psychiatrist.
Dr. Karson reported serving as a consultant to Otsuka America Pharmaceutical, which funded the study he presented, and to the Lieber Institute for Brain Development. Dr. Marder is a consultant to and/or has received research funding from Otsuka and 10 other companies.
HOLLYWOOD, FLA. – Once patients were switched from oral antipsychotic agents to long-acting injectable ones, their annual all-cause hospitalization rate was cut by more than half in an observational study involving nearly 6,000 Medicaid patients with schizophrenia.
Moreover, the mean number of days per year spent in the hospital for any reason also dropped dramatically after the switch. Plus, the mean overall annual health care costs decreased by $6,901, Dr. Craig N. Karson reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
"We may have an opportunity here to improve the management of schizophrenia, and in particular the cost management," said Dr. Karson, a psychiatrist in Wayne, Pa.
He presented a retrospective longitudinal study of 5,694 Medicaid patients with schizophrenia who switched from oral to long-acting injectable antipsychotics during 2005-2010. The data came from the Thomson Reuters MarketScan Research Medicaid Database.
Forty-four percent of patients were placed on long-acting injectable risperidone, 41% on haloperidol, and the rest on fluphenazine, which were the three antipsychotic agents available in depot form during the study period.
Roughly 80% of the all-cause hospitalizations in this study were for schizophrenia.
The switch from oral to long-acting injectable antipsychotic agents was associated with a striking reduction in overall health care resource use, including a decrease in the mean annual number of all-cause hospitalizations from 1.52 to 0.70 (see box).
In order to see whether patients’ duration on long-acting injectable antipsychotics was related to health care resource use, Dr. Karson and his coinvestigators divided the study population into two groups: the 2,856 patients who were short-term users of long-acting injectable therapy, defined as less than 180 days of treatment; and the 2,838 users of long-acting injectables for at least 180 days. The short-term users averaged 0.79 all-cause hospitalizations annually; the long-term users, 0.61. The mean annualized total days in the hospital was 6.56 for the short-term users, compared with 4.93 in longer-term users.
Several speakers argued that the MarketScan database has significant methodologic shortcomings, including selection bias, rendering it better suited as a basis for hypothesis generation than for drawing firm conclusions. Dr. Karson acknowledged that the database is limited in that it provides a convenience sample rather than a random sample of the Medicaid population, but he argued that the shear size of the study population overcomes that limitation.
"Ask yourself, how many treatment studies do we have in psychiatry with almost 6,000 patients? Very few," he said. "When you get into these kinds of patient numbers, which provide enormous statistical power, and then use a very hard endpoint like annual all-cause hospitalizations, I think it changes the game a little. I would argue that these patients are representative of the Medicaid population with serious mental illness, regardless."
Discussant Dr. Stephen R. Marder, who was not involved in the study, agreed that the data are compelling."
"The differences seen in your study were large and persuasive. And I thought the analysis of short- versus long-term use showing a dose-response effect added to the confidence in the results. Being on depot medication longer makes a difference," concluded Dr. Marder, director of the section on psychosis at the University of California, Los Angeles, Neuropsychiatric Institute.
"The problem lies with us as providers: We see long-acting injectable antipsychotics as the last resort, said Dr. Marder, also professor of psychiatry and behavioral sciences at UCLA. "We’re probably reluctant to do it because we see the infrastructure for providing the therapy is very poor in many places."
At the VA medical center where he works, for example, patients formerly had access to depot medication on a 24/7 basis, so that if they had a job, they could obtain treatment without missing work. That service isn’t available, anymore, Dr. Marder noted.
His hope, he added, is that thought leaders in psychiatry will be able to influence implementation of the Affordable Care Act so as to create a much better infrastructure for providing long-acting injectable antipsychotic therapy. Ideally, access to long-acting injectable antipsychotics should be available in places where psychosocial and rehabilitation services are located. "That’s what seems to work the best," according to the psychiatrist.
Dr. Karson reported serving as a consultant to Otsuka America Pharmaceutical, which funded the study he presented, and to the Lieber Institute for Brain Development. Dr. Marder is a consultant to and/or has received research funding from Otsuka and 10 other companies.
AT THE NCDEU MEETING
Major finding: Medicaid patients with schizophrenia had a mean 1.52 hospitalizations per year for any cause while they were on oral antipsychotic medication, dropping to 0.7 all-cause hospitalizations per year after they switched to long-acting injectable therapy.
Data source: A retrospective observational study involving 5,694 Medicaid patients with schizophrenia drawn from the Thomson Reuters MarketScan Research Medicaid Database.
Disclosures: Dr. Karson reported serving as a consultant to Otsuka America Pharmaceutical, which funded the study he presented, and to the Lieber Institute for Brain Development. Dr. Marder is a consultant to and/or has received research funding from Otsuka and 10 other companies.
Cariprazine performs well for schizophrenia, bipolar mania
HOLLYWOOD, FLA. – The novel oral antipsychotic agent cariprazine showed efficacy across the broad spectrum of schizophrenia manifestations – including negative symptoms – in a multicenter phase III clinical trial.
This is welcome news. Current antipsychotic agents are only modestly effective in treating the negative symptoms, mood disturbances, and neurocognitive dysfunction that so often hamper recovery in patients with schizophrenia. In contrast, the investigational agent cariprazine demonstrated efficacy in all these domains, Dr. John M. Kane reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Cariprazine is a potent dopamine D3/D2 receptor partial agonist with preferential binding to D3 receptors. As such, it would be expected to have broad efficacy across the spectrum of schizophrenia symptoms. This has been borne out, first in animal studies and now in this phase III randomized trial, noted Dr. Kane, chairman of psychiatry at Zucker Hillside Hospital in Glen Oaks, N.Y., and professor of psychiatry, neurology, and neuroscience at the Albert Einstein College of Medicine in New York.

Forest Pharmaceuticals filed an application with the Food and Drug Administration last fall for marketing approval of cariprazine in the treatment of schizophrenia as well as for treatment of bipolar mania. The company expects a decision from the regulatory agency before year’s end.
Dr. Kane reported on 446 patients with acute exacerbation of schizophrenia placed on 6 weeks of double-blind placebo or cariprazine at either 3-6 mg/day or 6-9 mg/day. Sixty-one percent of subjects completed the study. The mean daily dosage of cariprazine in the lower-dose study arm was 4.2 mg/day, versus 6.6 mg/day in the higher-dose arm.
The primary study endpoint was improvement in Positive and Negative Syndrome Scale (PANSS) total score. From a baseline mean PANSS score of 96, indicative of marked illness, patients in the lower-dose cariprazine group showed a placebo-subtracted net 6.8-point improvement. Those on higher-dose therapy had a net 9.9-point improvement. Both dosing strategies were significantly more effective than was placebo. Patients on cariprazine at the more effective dosing regimen of 6-9 mg/day showed a significant advantage over placebo starting at week 1.
To more closely examine the drug’s efficacy in domains other than positive symptoms such as delusions and hallucinations, Dr. Kane and his coinvestigators conducted a post hoc analysis focusing on cariprazine’s impact on each of the five PANSS-derived Marder symptom factor groups. These five groupings are negative symptoms, positive symptoms, uncontrolled hostility/excitement, disorganized thought, and anxiety/depression.
Cariprazine at 6-9 mg/day proved superior to placebo in all five Marder factor groupings. Cariprazine at 3-6 mg/day outperformed placebo in all but the Marder negative symptom factor grouping, where a trend favoring the drug fell short of statistical significance.
Of the 30 individual items that make up the PANSS, higher-dose cariprazine was significantly more effective than was placebo in 21. These included most positive symptom factor items as well as others where conventional antipsychotic agents typically don’t fare very well, such as emotional withdrawal, active social avoidance, poor rapport, anxiety, depression, tension, poor impulse control, hostility, excitement, uncooperativeness, disturbance of volition, and conceptual disorganization.
Cariprazine at 3-6 mg/day outperformed placebo on 11 of the 30 individual PANSS items.
Long-term cariprazine in bipolar I
Also at the NCDEU meeting, Dr. Terence A. Ketter presented a phase III, open-label, 16-week, study of flexibly dosed cariprazine in 402 patients with bipolar I disorder.
Dosing of cariprazine was permitted at 3-12 mg/day. The actual mean dose used in the study was 6.2 mg/day. The primary study endpoints involved safety and tolerability, since the drug already had demonstrated efficacy in three separate 3-week-long studies in acute mania.
This 16-week study was undertaken in recognition that the real challenge in bipolar I disorder is long-term management. Nearly half of all patients who respond to initial conventional therapies relapse within 2 years, mainly because of high rates of nonadherence, explained Dr. Ketter, professor of psychiatry and behavioral sciences and chief of the bipolar disorders clinic at Stanford (Calif.) University.
Just 33% of participants completed the 16-week study. Sixteen percent of subjects withdrew because of adverse events, roughly the same number as for protocol violations.
The major tolerability issue in the phase III trial was treatment-emergent akathisia, which occurred in 37% of participants. In addition, 7% experienced treatment-emergent extrapyramidal symptoms as defined by a Simpson-Angus Scale score of 3 or less at baseline rising to greater than 3 on treatment.
Ninety-eight percent of these cases of treatment-emergent akathisia or extrapyramidal symptoms were rated as mild to moderate in intensity. Five percent of subjects dropped out of the trial because of treatment-emergent akathisia, making this the No. 1 reason for discontinuation because of adverse events, followed by depression at 2%. Only 0.7% of subjects dropped out because of extrapyramidal symptoms, the same small number as quit because of worsening mania.
No clinically meaningful changes occurred in lipids, liver enzymes, ECG parameters, or prolactin levels. Systolic and diastolic blood pressure each rose modestly by less than 2 mm Hg. Weight gain was minimal: a mean increase of 0.9 kg over 16 weeks, with an accompanying mean 1.0-cm increase in waist circumference.
While this was primarily a phase III safety and tolerability study, efficacy was also measured. At week 16, 63% of subjects met study criteria for disease remission as defined by a Young Mania Rating Scale (YMRS) total score of 12 or less, starting from a mean baseline YMRS score of 26.1. Most of the symptomatic improvement was attained within the first 3 weeks of treatment, with a mean 13.6-point reduction in the YMRS, modestly improved upon to a mean 15.2-point reduction by week 16.
These schizophrenia and bipolar I disorder studies were sponsored by Forest Pharmaceuticals. Dr. Kane reported serving as a consultant and/or having received honoraria for lectures from Forest and 17 other pharmaceutical companies. Dr. Ketter reported serving as a consultant to Forest and four other pharmaceutical companies as well as receiving lecture honoraria from GlaxoSmithKline and Otsuka.
HOLLYWOOD, FLA. – The novel oral antipsychotic agent cariprazine showed efficacy across the broad spectrum of schizophrenia manifestations – including negative symptoms – in a multicenter phase III clinical trial.
This is welcome news. Current antipsychotic agents are only modestly effective in treating the negative symptoms, mood disturbances, and neurocognitive dysfunction that so often hamper recovery in patients with schizophrenia. In contrast, the investigational agent cariprazine demonstrated efficacy in all these domains, Dr. John M. Kane reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Cariprazine is a potent dopamine D3/D2 receptor partial agonist with preferential binding to D3 receptors. As such, it would be expected to have broad efficacy across the spectrum of schizophrenia symptoms. This has been borne out, first in animal studies and now in this phase III randomized trial, noted Dr. Kane, chairman of psychiatry at Zucker Hillside Hospital in Glen Oaks, N.Y., and professor of psychiatry, neurology, and neuroscience at the Albert Einstein College of Medicine in New York.
Forest Pharmaceuticals filed an application with the Food and Drug Administration last fall for marketing approval of cariprazine in the treatment of schizophrenia as well as for treatment of bipolar mania. The company expects a decision from the regulatory agency before year’s end.
Dr. Kane reported on 446 patients with acute exacerbation of schizophrenia placed on 6 weeks of double-blind placebo or cariprazine at either 3-6 mg/day or 6-9 mg/day. Sixty-one percent of subjects completed the study. The mean daily dosage of cariprazine in the lower-dose study arm was 4.2 mg/day, versus 6.6 mg/day in the higher-dose arm.
The primary study endpoint was improvement in Positive and Negative Syndrome Scale (PANSS) total score. From a baseline mean PANSS score of 96, indicative of marked illness, patients in the lower-dose cariprazine group showed a placebo-subtracted net 6.8-point improvement. Those on higher-dose therapy had a net 9.9-point improvement. Both dosing strategies were significantly more effective than was placebo. Patients on cariprazine at the more effective dosing regimen of 6-9 mg/day showed a significant advantage over placebo starting at week 1.
To more closely examine the drug’s efficacy in domains other than positive symptoms such as delusions and hallucinations, Dr. Kane and his coinvestigators conducted a post hoc analysis focusing on cariprazine’s impact on each of the five PANSS-derived Marder symptom factor groups. These five groupings are negative symptoms, positive symptoms, uncontrolled hostility/excitement, disorganized thought, and anxiety/depression.
Cariprazine at 6-9 mg/day proved superior to placebo in all five Marder factor groupings. Cariprazine at 3-6 mg/day outperformed placebo in all but the Marder negative symptom factor grouping, where a trend favoring the drug fell short of statistical significance.
Of the 30 individual items that make up the PANSS, higher-dose cariprazine was significantly more effective than was placebo in 21. These included most positive symptom factor items as well as others where conventional antipsychotic agents typically don’t fare very well, such as emotional withdrawal, active social avoidance, poor rapport, anxiety, depression, tension, poor impulse control, hostility, excitement, uncooperativeness, disturbance of volition, and conceptual disorganization.
Cariprazine at 3-6 mg/day outperformed placebo on 11 of the 30 individual PANSS items.
Long-term cariprazine in bipolar I
Also at the NCDEU meeting, Dr. Terence A. Ketter presented a phase III, open-label, 16-week, study of flexibly dosed cariprazine in 402 patients with bipolar I disorder.
Dosing of cariprazine was permitted at 3-12 mg/day. The actual mean dose used in the study was 6.2 mg/day. The primary study endpoints involved safety and tolerability, since the drug already had demonstrated efficacy in three separate 3-week-long studies in acute mania.
This 16-week study was undertaken in recognition that the real challenge in bipolar I disorder is long-term management. Nearly half of all patients who respond to initial conventional therapies relapse within 2 years, mainly because of high rates of nonadherence, explained Dr. Ketter, professor of psychiatry and behavioral sciences and chief of the bipolar disorders clinic at Stanford (Calif.) University.
Just 33% of participants completed the 16-week study. Sixteen percent of subjects withdrew because of adverse events, roughly the same number as for protocol violations.
The major tolerability issue in the phase III trial was treatment-emergent akathisia, which occurred in 37% of participants. In addition, 7% experienced treatment-emergent extrapyramidal symptoms as defined by a Simpson-Angus Scale score of 3 or less at baseline rising to greater than 3 on treatment.
Ninety-eight percent of these cases of treatment-emergent akathisia or extrapyramidal symptoms were rated as mild to moderate in intensity. Five percent of subjects dropped out of the trial because of treatment-emergent akathisia, making this the No. 1 reason for discontinuation because of adverse events, followed by depression at 2%. Only 0.7% of subjects dropped out because of extrapyramidal symptoms, the same small number as quit because of worsening mania.
No clinically meaningful changes occurred in lipids, liver enzymes, ECG parameters, or prolactin levels. Systolic and diastolic blood pressure each rose modestly by less than 2 mm Hg. Weight gain was minimal: a mean increase of 0.9 kg over 16 weeks, with an accompanying mean 1.0-cm increase in waist circumference.
While this was primarily a phase III safety and tolerability study, efficacy was also measured. At week 16, 63% of subjects met study criteria for disease remission as defined by a Young Mania Rating Scale (YMRS) total score of 12 or less, starting from a mean baseline YMRS score of 26.1. Most of the symptomatic improvement was attained within the first 3 weeks of treatment, with a mean 13.6-point reduction in the YMRS, modestly improved upon to a mean 15.2-point reduction by week 16.
These schizophrenia and bipolar I disorder studies were sponsored by Forest Pharmaceuticals. Dr. Kane reported serving as a consultant and/or having received honoraria for lectures from Forest and 17 other pharmaceutical companies. Dr. Ketter reported serving as a consultant to Forest and four other pharmaceutical companies as well as receiving lecture honoraria from GlaxoSmithKline and Otsuka.
HOLLYWOOD, FLA. – The novel oral antipsychotic agent cariprazine showed efficacy across the broad spectrum of schizophrenia manifestations – including negative symptoms – in a multicenter phase III clinical trial.
This is welcome news. Current antipsychotic agents are only modestly effective in treating the negative symptoms, mood disturbances, and neurocognitive dysfunction that so often hamper recovery in patients with schizophrenia. In contrast, the investigational agent cariprazine demonstrated efficacy in all these domains, Dr. John M. Kane reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Cariprazine is a potent dopamine D3/D2 receptor partial agonist with preferential binding to D3 receptors. As such, it would be expected to have broad efficacy across the spectrum of schizophrenia symptoms. This has been borne out, first in animal studies and now in this phase III randomized trial, noted Dr. Kane, chairman of psychiatry at Zucker Hillside Hospital in Glen Oaks, N.Y., and professor of psychiatry, neurology, and neuroscience at the Albert Einstein College of Medicine in New York.
Forest Pharmaceuticals filed an application with the Food and Drug Administration last fall for marketing approval of cariprazine in the treatment of schizophrenia as well as for treatment of bipolar mania. The company expects a decision from the regulatory agency before year’s end.
Dr. Kane reported on 446 patients with acute exacerbation of schizophrenia placed on 6 weeks of double-blind placebo or cariprazine at either 3-6 mg/day or 6-9 mg/day. Sixty-one percent of subjects completed the study. The mean daily dosage of cariprazine in the lower-dose study arm was 4.2 mg/day, versus 6.6 mg/day in the higher-dose arm.
The primary study endpoint was improvement in Positive and Negative Syndrome Scale (PANSS) total score. From a baseline mean PANSS score of 96, indicative of marked illness, patients in the lower-dose cariprazine group showed a placebo-subtracted net 6.8-point improvement. Those on higher-dose therapy had a net 9.9-point improvement. Both dosing strategies were significantly more effective than was placebo. Patients on cariprazine at the more effective dosing regimen of 6-9 mg/day showed a significant advantage over placebo starting at week 1.
To more closely examine the drug’s efficacy in domains other than positive symptoms such as delusions and hallucinations, Dr. Kane and his coinvestigators conducted a post hoc analysis focusing on cariprazine’s impact on each of the five PANSS-derived Marder symptom factor groups. These five groupings are negative symptoms, positive symptoms, uncontrolled hostility/excitement, disorganized thought, and anxiety/depression.
Cariprazine at 6-9 mg/day proved superior to placebo in all five Marder factor groupings. Cariprazine at 3-6 mg/day outperformed placebo in all but the Marder negative symptom factor grouping, where a trend favoring the drug fell short of statistical significance.
Of the 30 individual items that make up the PANSS, higher-dose cariprazine was significantly more effective than was placebo in 21. These included most positive symptom factor items as well as others where conventional antipsychotic agents typically don’t fare very well, such as emotional withdrawal, active social avoidance, poor rapport, anxiety, depression, tension, poor impulse control, hostility, excitement, uncooperativeness, disturbance of volition, and conceptual disorganization.
Cariprazine at 3-6 mg/day outperformed placebo on 11 of the 30 individual PANSS items.
Long-term cariprazine in bipolar I
Also at the NCDEU meeting, Dr. Terence A. Ketter presented a phase III, open-label, 16-week, study of flexibly dosed cariprazine in 402 patients with bipolar I disorder.
Dosing of cariprazine was permitted at 3-12 mg/day. The actual mean dose used in the study was 6.2 mg/day. The primary study endpoints involved safety and tolerability, since the drug already had demonstrated efficacy in three separate 3-week-long studies in acute mania.
This 16-week study was undertaken in recognition that the real challenge in bipolar I disorder is long-term management. Nearly half of all patients who respond to initial conventional therapies relapse within 2 years, mainly because of high rates of nonadherence, explained Dr. Ketter, professor of psychiatry and behavioral sciences and chief of the bipolar disorders clinic at Stanford (Calif.) University.
Just 33% of participants completed the 16-week study. Sixteen percent of subjects withdrew because of adverse events, roughly the same number as for protocol violations.
The major tolerability issue in the phase III trial was treatment-emergent akathisia, which occurred in 37% of participants. In addition, 7% experienced treatment-emergent extrapyramidal symptoms as defined by a Simpson-Angus Scale score of 3 or less at baseline rising to greater than 3 on treatment.
Ninety-eight percent of these cases of treatment-emergent akathisia or extrapyramidal symptoms were rated as mild to moderate in intensity. Five percent of subjects dropped out of the trial because of treatment-emergent akathisia, making this the No. 1 reason for discontinuation because of adverse events, followed by depression at 2%. Only 0.7% of subjects dropped out because of extrapyramidal symptoms, the same small number as quit because of worsening mania.
No clinically meaningful changes occurred in lipids, liver enzymes, ECG parameters, or prolactin levels. Systolic and diastolic blood pressure each rose modestly by less than 2 mm Hg. Weight gain was minimal: a mean increase of 0.9 kg over 16 weeks, with an accompanying mean 1.0-cm increase in waist circumference.
While this was primarily a phase III safety and tolerability study, efficacy was also measured. At week 16, 63% of subjects met study criteria for disease remission as defined by a Young Mania Rating Scale (YMRS) total score of 12 or less, starting from a mean baseline YMRS score of 26.1. Most of the symptomatic improvement was attained within the first 3 weeks of treatment, with a mean 13.6-point reduction in the YMRS, modestly improved upon to a mean 15.2-point reduction by week 16.
These schizophrenia and bipolar I disorder studies were sponsored by Forest Pharmaceuticals. Dr. Kane reported serving as a consultant and/or having received honoraria for lectures from Forest and 17 other pharmaceutical companies. Dr. Ketter reported serving as a consultant to Forest and four other pharmaceutical companies as well as receiving lecture honoraria from GlaxoSmithKline and Otsuka.
AT THE NCDEU MEETING
Major finding: Schizophrenia study: Patients in the higher-dose cariprazine group showed a net 9.9-point improvement in Positive and Negative Syndrome Scale total score. Bipolar I study: Among those who took cariprazine, 63% met study criteria for disease remission as defined by a Young Mania Rating Scale total score of 12 or less.
Data source: Multicenter phase III trial of 446 patients with acute exacerbation of schizophrenia and a phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I.
Disclosures: Studies were funded by Forest Pharmaceuticals. Dr. Kane and Dr. Ketter reported serving as consultants to Forest.

Memantine delays driving impairment in mild Alzheimer's
HOLLYWOOD, FLA. – Treatment with memantine appears to delay progressive driving impairment in patients with mild Alzheimer’s disease, according to a year-long, double-blind, albeit small, randomized controlled trial.
"For many patients with Alzheimer’s disease, driving cessation is a major life-changing event that negatively impacts them and their caregivers. Until highly effective disease-altering treatments are available, there is a need to develop therapies that can prolong independent functioning," Dr. Peter J. Holland said in explaining the rationale for the driving skills study he presented at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.

He chose memantine for the study for a couple of reasons. Preclinical evidence suggests the drug might have neuroprotective properties. And, in his experience, it’s a very well-tolerated drug.
There are a number of large controlled studies in which the dropout rate from adverse events was higher in the placebo group than with memantine, observed Dr. Holland, a psychiatrist and research professor of biomedical science at Florida Atlantic University, Boca Raton.
He reported on 60 otherwise healthy subjects with mild Alzheimer’s disease who were randomized to 12 months of double-blind memantine titrated to a target of 20 mg/day or placebo. All participants were still driving. The primary study endpoint was their score on the DriveABLE On Road Evaluation at 12 months. This structured, behind-the-wheel road test measures driving-related neurocognitive performance.
All 13 subjects on memantine either improved or maintained their baseline driving ability at 12 months, as did only 9 of 12 on placebo, a statistically significant difference (P = .05).
Asked about the high dropout rate, Dr. Holland noted that it’s a real challenge to keep older patients with Alzheimer’s disease in a study lasting a full year.
"The P value is there. I’d like to see the study replicated. If this holds true, it’s such a benign medication – it’s so well tolerated – that if it has a chance of being helpful, I think it’s worth further study," Dr. Holland added.
Memantine’s approved indication is in treating patients with moderate to-severe Alzheimer’s disease, so this application of the drug to delay driving impairment in patients with mild Alzheimer’s disease is off-label use.
In addition to the DriveABLE On Road test, study participants completed a battery of neurocognitive assessments at baseline, 6, and 12 months. These assessments were chosen because they test skills necessary for safe driving, including executive function, selective attention, and visuospatial abilities.
Thus far, Dr. Holland has determined that the memantine-treated group performed significantly better than controls at 12 months on the Rey-Osterrieth Complex Figure Test. In contrast, no significant difference between the two groups was noted on the Trail Making Part B test. As the study was only recently completed, the investigator is still analyzing the results on the Useful Field of View, Motor Free Visual Perception Test, and Alzheimer's Disease Assessment Scale-Cognitive test.
An estimated 5.2 million Americans aged 65 years or older have Alzheimer’s disease. And according to the American Automobile Association, in another dozen years, fully one-quarter of all U.S. drivers will be 65 years or older. A medication that slows driving impairment would have a large clinical impact, Dr. Holland noted.
His investigator-initiated study was funded by a research grant from Forest Pharmaceuticals.
HOLLYWOOD, FLA. – Treatment with memantine appears to delay progressive driving impairment in patients with mild Alzheimer’s disease, according to a year-long, double-blind, albeit small, randomized controlled trial.
"For many patients with Alzheimer’s disease, driving cessation is a major life-changing event that negatively impacts them and their caregivers. Until highly effective disease-altering treatments are available, there is a need to develop therapies that can prolong independent functioning," Dr. Peter J. Holland said in explaining the rationale for the driving skills study he presented at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
He chose memantine for the study for a couple of reasons. Preclinical evidence suggests the drug might have neuroprotective properties. And, in his experience, it’s a very well-tolerated drug.
There are a number of large controlled studies in which the dropout rate from adverse events was higher in the placebo group than with memantine, observed Dr. Holland, a psychiatrist and research professor of biomedical science at Florida Atlantic University, Boca Raton.
He reported on 60 otherwise healthy subjects with mild Alzheimer’s disease who were randomized to 12 months of double-blind memantine titrated to a target of 20 mg/day or placebo. All participants were still driving. The primary study endpoint was their score on the DriveABLE On Road Evaluation at 12 months. This structured, behind-the-wheel road test measures driving-related neurocognitive performance.
All 13 subjects on memantine either improved or maintained their baseline driving ability at 12 months, as did only 9 of 12 on placebo, a statistically significant difference (P = .05).
Asked about the high dropout rate, Dr. Holland noted that it’s a real challenge to keep older patients with Alzheimer’s disease in a study lasting a full year.
"The P value is there. I’d like to see the study replicated. If this holds true, it’s such a benign medication – it’s so well tolerated – that if it has a chance of being helpful, I think it’s worth further study," Dr. Holland added.
Memantine’s approved indication is in treating patients with moderate to-severe Alzheimer’s disease, so this application of the drug to delay driving impairment in patients with mild Alzheimer’s disease is off-label use.
In addition to the DriveABLE On Road test, study participants completed a battery of neurocognitive assessments at baseline, 6, and 12 months. These assessments were chosen because they test skills necessary for safe driving, including executive function, selective attention, and visuospatial abilities.
Thus far, Dr. Holland has determined that the memantine-treated group performed significantly better than controls at 12 months on the Rey-Osterrieth Complex Figure Test. In contrast, no significant difference between the two groups was noted on the Trail Making Part B test. As the study was only recently completed, the investigator is still analyzing the results on the Useful Field of View, Motor Free Visual Perception Test, and Alzheimer's Disease Assessment Scale-Cognitive test.
An estimated 5.2 million Americans aged 65 years or older have Alzheimer’s disease. And according to the American Automobile Association, in another dozen years, fully one-quarter of all U.S. drivers will be 65 years or older. A medication that slows driving impairment would have a large clinical impact, Dr. Holland noted.
His investigator-initiated study was funded by a research grant from Forest Pharmaceuticals.
HOLLYWOOD, FLA. – Treatment with memantine appears to delay progressive driving impairment in patients with mild Alzheimer’s disease, according to a year-long, double-blind, albeit small, randomized controlled trial.
"For many patients with Alzheimer’s disease, driving cessation is a major life-changing event that negatively impacts them and their caregivers. Until highly effective disease-altering treatments are available, there is a need to develop therapies that can prolong independent functioning," Dr. Peter J. Holland said in explaining the rationale for the driving skills study he presented at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
He chose memantine for the study for a couple of reasons. Preclinical evidence suggests the drug might have neuroprotective properties. And, in his experience, it’s a very well-tolerated drug.
There are a number of large controlled studies in which the dropout rate from adverse events was higher in the placebo group than with memantine, observed Dr. Holland, a psychiatrist and research professor of biomedical science at Florida Atlantic University, Boca Raton.
He reported on 60 otherwise healthy subjects with mild Alzheimer’s disease who were randomized to 12 months of double-blind memantine titrated to a target of 20 mg/day or placebo. All participants were still driving. The primary study endpoint was their score on the DriveABLE On Road Evaluation at 12 months. This structured, behind-the-wheel road test measures driving-related neurocognitive performance.
All 13 subjects on memantine either improved or maintained their baseline driving ability at 12 months, as did only 9 of 12 on placebo, a statistically significant difference (P = .05).
Asked about the high dropout rate, Dr. Holland noted that it’s a real challenge to keep older patients with Alzheimer’s disease in a study lasting a full year.
"The P value is there. I’d like to see the study replicated. If this holds true, it’s such a benign medication – it’s so well tolerated – that if it has a chance of being helpful, I think it’s worth further study," Dr. Holland added.
Memantine’s approved indication is in treating patients with moderate to-severe Alzheimer’s disease, so this application of the drug to delay driving impairment in patients with mild Alzheimer’s disease is off-label use.
In addition to the DriveABLE On Road test, study participants completed a battery of neurocognitive assessments at baseline, 6, and 12 months. These assessments were chosen because they test skills necessary for safe driving, including executive function, selective attention, and visuospatial abilities.
Thus far, Dr. Holland has determined that the memantine-treated group performed significantly better than controls at 12 months on the Rey-Osterrieth Complex Figure Test. In contrast, no significant difference between the two groups was noted on the Trail Making Part B test. As the study was only recently completed, the investigator is still analyzing the results on the Useful Field of View, Motor Free Visual Perception Test, and Alzheimer's Disease Assessment Scale-Cognitive test.
An estimated 5.2 million Americans aged 65 years or older have Alzheimer’s disease. And according to the American Automobile Association, in another dozen years, fully one-quarter of all U.S. drivers will be 65 years or older. A medication that slows driving impairment would have a large clinical impact, Dr. Holland noted.
His investigator-initiated study was funded by a research grant from Forest Pharmaceuticals.
AT THE NCDEU MEETING
Major finding: One hundred percent of patients with mild Alzheimer’s disease passed a structured on-the-road driving test after 12 months on memantine, compared to 75% on placebo in a small double-blind randomized trial.
Data source: A randomized, double-blind, prospective, 12-month study involving 60 patients with mild Alzheimer’s disease.
Disclosures: The study was funded by an investigator-initiated research grant from Forest Pharmaceuticals.

Treating adult ADHD improves parenting performance
HOLLYWOOD, FLA. – Pharmacologic treatment of parental attention-deficit/hyperactivity disorder might provide a novel means of improving parenting skills while reducing inappropriate behaviors in their unmedicated children with the disorder, according to Dr. James G. Waxmonsky.
In a structured study conducted by formally trained evaluators in a university family behavioral sciences center, treatment of parental ADHD with lisdexamfetamine dimesylate (Vyvanse) not only resulted in the expected reduction in parental ADHD symptoms, but was also associated with improved parenting performance and more harmonious child behavior in the laboratory setting, he reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Parents on ADHD medication gradually increased their use of praise over time by fivefold, while those on placebo did not change their use of praise. The parents on lisdexamfetamine dimesylate (LDX) also were significantly more verbally responsive to their child. There was less negative talk. Parents issued fewer commands, as well.

Even before the treated adults’ parenting performance began to show objective improvement, their children displayed a reduction in inappropriate and oppositional behaviors during joint homework assignments and other observed interactions.
"Most effects emerged several weeks into treatment with the optimal dose. The strength of effects on parenting behaviors paralleled that seen in trials of established parent training programs, while the degree of improvements in children’s oppositional behaviors matched that seen with stimulant medication," according to Dr. Waxmonsky, a child and adolescent psychiatrist serving as medical director of the center for children and families at Florida International University, Miami.
He presented a study involving 38 parents who met full DSM-IV criteria for ADHD and who also had a 5- to 15-year-old child with ADHD. The parents were started on LDX at 30 mg/day, titrated over the next several weeks to a maximum of 70 mg/day or until occurrence of at least a 30% reduction in scores on the ADHD Rating Scale. The mean optimized drug dose was 50 mg/day, with an average 58% reduction in ADHD Rating Scale score severity.
Once the medication was optimized, the parent and child were assessed using the Dyadic Parent-Child Interaction Coding System in the behavior laboratory on two occasions 1 week apart. In these sessions, the parent and child collaborated on a simulated homework task as well as an age-appropriate nonacademic task such as joint play or planning a family activity. In the first session, the blinded parent was on medication; for the second session, on placebo. The child with ADHD remained unmedicated throughout the study.
Significant improvement in parental ADHD symptoms was noted after just 1 week on the optimal medication dose. But in the initial pair of evaluations, the first of which was conducted when parents had been on their optimal drug dose for just 1-2 weeks, there was no significant difference between parenting behaviors when the parents were on and off medication.
In contrast to the lack of change in parenting behaviors during this initial study phase, significant reductions in the children’s inappropriate behavior during the homework task was documented when the parent was on LDX versus placebo. One plausible explanation for this observation is that a reduction in parental ADHD symptoms triggers improved child behavior. Then, as parents note improvements in their child’s behavior, they might respond by engaging in more positive parenting behaviors, in turn fostering further improvement over time in their child’s behavior. That would account for what was seen later in the study, Dr. Waxmonsky said.
In phase II of the study, the blinded parents were randomized to 8 weeks of optimized medication or placebo, followed by another evaluation of parent-child interaction in the behavior lab. This time, striking improvements in parenting performance were evident in the adults on extended duration of optimized medication compared with those on placebo. Moreover, the improvements in the behavior of the children whose parents were on LDX compared with that of the kids whose parents were on placebo were even larger than those seen in the first phase of the study. For example, children whose parents had been on optimized LDX for 8 weeks showed a fourfold greater reduction in inappropriate and oppositional behaviors than did children of parents on placebo.
The chief limitation of this in-depth study, Dr. Waxmonsky, is its small size. Almost none of the participating parents had ever been on anti-ADHD medication before; 10 of the 38 dropped out because of medication side effects, most prominently appetite loss, insomnia, and headaches.
The study was funded by a research grant from Shire Pharmaceuticals.
HOLLYWOOD, FLA. – Pharmacologic treatment of parental attention-deficit/hyperactivity disorder might provide a novel means of improving parenting skills while reducing inappropriate behaviors in their unmedicated children with the disorder, according to Dr. James G. Waxmonsky.
In a structured study conducted by formally trained evaluators in a university family behavioral sciences center, treatment of parental ADHD with lisdexamfetamine dimesylate (Vyvanse) not only resulted in the expected reduction in parental ADHD symptoms, but was also associated with improved parenting performance and more harmonious child behavior in the laboratory setting, he reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Parents on ADHD medication gradually increased their use of praise over time by fivefold, while those on placebo did not change their use of praise. The parents on lisdexamfetamine dimesylate (LDX) also were significantly more verbally responsive to their child. There was less negative talk. Parents issued fewer commands, as well.
Even before the treated adults’ parenting performance began to show objective improvement, their children displayed a reduction in inappropriate and oppositional behaviors during joint homework assignments and other observed interactions.
"Most effects emerged several weeks into treatment with the optimal dose. The strength of effects on parenting behaviors paralleled that seen in trials of established parent training programs, while the degree of improvements in children’s oppositional behaviors matched that seen with stimulant medication," according to Dr. Waxmonsky, a child and adolescent psychiatrist serving as medical director of the center for children and families at Florida International University, Miami.
He presented a study involving 38 parents who met full DSM-IV criteria for ADHD and who also had a 5- to 15-year-old child with ADHD. The parents were started on LDX at 30 mg/day, titrated over the next several weeks to a maximum of 70 mg/day or until occurrence of at least a 30% reduction in scores on the ADHD Rating Scale. The mean optimized drug dose was 50 mg/day, with an average 58% reduction in ADHD Rating Scale score severity.
Once the medication was optimized, the parent and child were assessed using the Dyadic Parent-Child Interaction Coding System in the behavior laboratory on two occasions 1 week apart. In these sessions, the parent and child collaborated on a simulated homework task as well as an age-appropriate nonacademic task such as joint play or planning a family activity. In the first session, the blinded parent was on medication; for the second session, on placebo. The child with ADHD remained unmedicated throughout the study.
Significant improvement in parental ADHD symptoms was noted after just 1 week on the optimal medication dose. But in the initial pair of evaluations, the first of which was conducted when parents had been on their optimal drug dose for just 1-2 weeks, there was no significant difference between parenting behaviors when the parents were on and off medication.
In contrast to the lack of change in parenting behaviors during this initial study phase, significant reductions in the children’s inappropriate behavior during the homework task was documented when the parent was on LDX versus placebo. One plausible explanation for this observation is that a reduction in parental ADHD symptoms triggers improved child behavior. Then, as parents note improvements in their child’s behavior, they might respond by engaging in more positive parenting behaviors, in turn fostering further improvement over time in their child’s behavior. That would account for what was seen later in the study, Dr. Waxmonsky said.
In phase II of the study, the blinded parents were randomized to 8 weeks of optimized medication or placebo, followed by another evaluation of parent-child interaction in the behavior lab. This time, striking improvements in parenting performance were evident in the adults on extended duration of optimized medication compared with those on placebo. Moreover, the improvements in the behavior of the children whose parents were on LDX compared with that of the kids whose parents were on placebo were even larger than those seen in the first phase of the study. For example, children whose parents had been on optimized LDX for 8 weeks showed a fourfold greater reduction in inappropriate and oppositional behaviors than did children of parents on placebo.
The chief limitation of this in-depth study, Dr. Waxmonsky, is its small size. Almost none of the participating parents had ever been on anti-ADHD medication before; 10 of the 38 dropped out because of medication side effects, most prominently appetite loss, insomnia, and headaches.
The study was funded by a research grant from Shire Pharmaceuticals.
HOLLYWOOD, FLA. – Pharmacologic treatment of parental attention-deficit/hyperactivity disorder might provide a novel means of improving parenting skills while reducing inappropriate behaviors in their unmedicated children with the disorder, according to Dr. James G. Waxmonsky.
In a structured study conducted by formally trained evaluators in a university family behavioral sciences center, treatment of parental ADHD with lisdexamfetamine dimesylate (Vyvanse) not only resulted in the expected reduction in parental ADHD symptoms, but was also associated with improved parenting performance and more harmonious child behavior in the laboratory setting, he reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Parents on ADHD medication gradually increased their use of praise over time by fivefold, while those on placebo did not change their use of praise. The parents on lisdexamfetamine dimesylate (LDX) also were significantly more verbally responsive to their child. There was less negative talk. Parents issued fewer commands, as well.
Even before the treated adults’ parenting performance began to show objective improvement, their children displayed a reduction in inappropriate and oppositional behaviors during joint homework assignments and other observed interactions.
"Most effects emerged several weeks into treatment with the optimal dose. The strength of effects on parenting behaviors paralleled that seen in trials of established parent training programs, while the degree of improvements in children’s oppositional behaviors matched that seen with stimulant medication," according to Dr. Waxmonsky, a child and adolescent psychiatrist serving as medical director of the center for children and families at Florida International University, Miami.
He presented a study involving 38 parents who met full DSM-IV criteria for ADHD and who also had a 5- to 15-year-old child with ADHD. The parents were started on LDX at 30 mg/day, titrated over the next several weeks to a maximum of 70 mg/day or until occurrence of at least a 30% reduction in scores on the ADHD Rating Scale. The mean optimized drug dose was 50 mg/day, with an average 58% reduction in ADHD Rating Scale score severity.
Once the medication was optimized, the parent and child were assessed using the Dyadic Parent-Child Interaction Coding System in the behavior laboratory on two occasions 1 week apart. In these sessions, the parent and child collaborated on a simulated homework task as well as an age-appropriate nonacademic task such as joint play or planning a family activity. In the first session, the blinded parent was on medication; for the second session, on placebo. The child with ADHD remained unmedicated throughout the study.
Significant improvement in parental ADHD symptoms was noted after just 1 week on the optimal medication dose. But in the initial pair of evaluations, the first of which was conducted when parents had been on their optimal drug dose for just 1-2 weeks, there was no significant difference between parenting behaviors when the parents were on and off medication.
In contrast to the lack of change in parenting behaviors during this initial study phase, significant reductions in the children’s inappropriate behavior during the homework task was documented when the parent was on LDX versus placebo. One plausible explanation for this observation is that a reduction in parental ADHD symptoms triggers improved child behavior. Then, as parents note improvements in their child’s behavior, they might respond by engaging in more positive parenting behaviors, in turn fostering further improvement over time in their child’s behavior. That would account for what was seen later in the study, Dr. Waxmonsky said.
In phase II of the study, the blinded parents were randomized to 8 weeks of optimized medication or placebo, followed by another evaluation of parent-child interaction in the behavior lab. This time, striking improvements in parenting performance were evident in the adults on extended duration of optimized medication compared with those on placebo. Moreover, the improvements in the behavior of the children whose parents were on LDX compared with that of the kids whose parents were on placebo were even larger than those seen in the first phase of the study. For example, children whose parents had been on optimized LDX for 8 weeks showed a fourfold greater reduction in inappropriate and oppositional behaviors than did children of parents on placebo.
The chief limitation of this in-depth study, Dr. Waxmonsky, is its small size. Almost none of the participating parents had ever been on anti-ADHD medication before; 10 of the 38 dropped out because of medication side effects, most prominently appetite loss, insomnia, and headaches.
The study was funded by a research grant from Shire Pharmaceuticals.
AT THE NCDEU MEETING
Major finding: Parenting performance improved when adults with ADHD underwent pharmacologic treatment. Marked improvements also were documented in the behavior of their children, who also had ADHD but remained unmedicated.
Data source: A study conducted in a university behavior lab involving 38 parents with ADHD, each with a 5- to 15-year-old child who also had ADHD.
Disclosures: The study was funded by a research grant from Shire Pharmaceuticals.
Vortioxetine effective in treatment-resistant depression
HOLLYWOOD, FLA. – The investigational antidepressant vortioxetine achieved a 55% remission rate in a large, double-blind study of patients with major depressive disorder switched to the drug after an inadequate response to one of a half-dozen approved selective serotonin reuptake inhibitors or selective norepinephrine reuptake inhibitors.
"I think it’s quite impressive to see a remission rate on the order of 55% in this difficult-to-treat population," Dr. Marianne Dragheim observed in presenting the study findings at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.

After all, in the landmark NIMH-sponsored sequential treatment alternatives to relieve depression, or STAR*D trial, in which citalopram nonresponders were switched to second-line treatment with sustained-release bupropion, extended-release venlafaxine, or sertraline, the mean remission rate after 12-14 weeks on the backup medication was just 31% (N. Engl. J. Med. 2006;354:1231-42), noted Dr. Dragheim of H. Lundbeck A/S, Valby, Denmark.
Vortioxetine is a first of its kind multimodal antidepressant. It is an inhibitor of the 5-hydroxytryptamine (5-HT) or serotonin transporter. In addition, vortioxetine is a 5-HT3, 5-HT7, and 5-HT10 receptor antagonist, a 5-HT1A receptor agonist, and a 5-HT1B receptor partial agonist.
Vortioxetine is currently under review at the Food and Drug Administration for possible marketing approval. The evidence submitted to the FDA includes seven positive studies, including four phase III randomized trials presented in May at the annual meeting of the American Psychiatric Association in San Francisco. The switching study Dr. Dragheim presented at the NCDEU meeting was completed so recently that it was not included in the FDA’s data package.
She reported on 501 European patients with major depressive disorder who responded inadequately to at least 6 weeks of monotherapy with citalopram, escitalopram, sertraline, paroxetine, venlafaxine, or duloxetine at approved doses. The study participants, all of whom wanted to change their antidepressant because of inadequate response, were randomized to 12 weeks of double-blind treatment with flexibly dosed vortioxetine at 10-20 mg/day or agomelatine at 25-50 mg/day. Agomelatine is approved as an antidepressant in the European Union and elsewhere in the world, but not in the United States.
The primary study endpoint was the change in scores on the Montgomery-Åsberg Depression Rating Scale (MADRS) between baseline and week 8. From a baseline MADRS total score of 29, the vortioxetine group improved by a mean of 2.2 more points than the agomelatine group, a statistically significant difference.
In addition, the vortioxetine-treated patients fared significantly better in terms of numerous secondary endpoints. They had a mean 1.9-point greater improvement than did the agomelatine group at 8 weeks in the Hamilton Anxiety Rating Scale total score, a 0.3-point greater improvement on the Clinical Global Impression-Severity, and a 2.2-point bigger improvement on the Sheehan Disability Scale.
The week-8 response rate as reflected in at least a 50% improvement from baseline in MADRS score was 61.5% in the vortioxetine group and 47.3% with agomelatine.
Remission as defined by a MADRS score of 10 or less occurred in 40.5% of the vortioxetine group and 29.5% of the agomelatine group at week 8, and in 55.2% vs. 39.4%, respectively, at week 12 (P = .0002).
Study withdrawal because of adverse events occurred in 5.9% of the vortioxetine group, compared with 9.5% of patients on agomelatine. Nausea, reported by 16% of patients on vortioxetine, was the only side effect more common in patients on that drug; however, less than 1% of subjects on vortioxetine left the study because of nausea.
Vortioxetine is being developed jointly by H. Lundbeck and Takeda Pharmaceutical Co.Dr. Dragheim is a company employee.
HOLLYWOOD, FLA. – The investigational antidepressant vortioxetine achieved a 55% remission rate in a large, double-blind study of patients with major depressive disorder switched to the drug after an inadequate response to one of a half-dozen approved selective serotonin reuptake inhibitors or selective norepinephrine reuptake inhibitors.
"I think it’s quite impressive to see a remission rate on the order of 55% in this difficult-to-treat population," Dr. Marianne Dragheim observed in presenting the study findings at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
After all, in the landmark NIMH-sponsored sequential treatment alternatives to relieve depression, or STAR*D trial, in which citalopram nonresponders were switched to second-line treatment with sustained-release bupropion, extended-release venlafaxine, or sertraline, the mean remission rate after 12-14 weeks on the backup medication was just 31% (N. Engl. J. Med. 2006;354:1231-42), noted Dr. Dragheim of H. Lundbeck A/S, Valby, Denmark.
Vortioxetine is a first of its kind multimodal antidepressant. It is an inhibitor of the 5-hydroxytryptamine (5-HT) or serotonin transporter. In addition, vortioxetine is a 5-HT3, 5-HT7, and 5-HT10 receptor antagonist, a 5-HT1A receptor agonist, and a 5-HT1B receptor partial agonist.
Vortioxetine is currently under review at the Food and Drug Administration for possible marketing approval. The evidence submitted to the FDA includes seven positive studies, including four phase III randomized trials presented in May at the annual meeting of the American Psychiatric Association in San Francisco. The switching study Dr. Dragheim presented at the NCDEU meeting was completed so recently that it was not included in the FDA’s data package.
She reported on 501 European patients with major depressive disorder who responded inadequately to at least 6 weeks of monotherapy with citalopram, escitalopram, sertraline, paroxetine, venlafaxine, or duloxetine at approved doses. The study participants, all of whom wanted to change their antidepressant because of inadequate response, were randomized to 12 weeks of double-blind treatment with flexibly dosed vortioxetine at 10-20 mg/day or agomelatine at 25-50 mg/day. Agomelatine is approved as an antidepressant in the European Union and elsewhere in the world, but not in the United States.
The primary study endpoint was the change in scores on the Montgomery-Åsberg Depression Rating Scale (MADRS) between baseline and week 8. From a baseline MADRS total score of 29, the vortioxetine group improved by a mean of 2.2 more points than the agomelatine group, a statistically significant difference.
In addition, the vortioxetine-treated patients fared significantly better in terms of numerous secondary endpoints. They had a mean 1.9-point greater improvement than did the agomelatine group at 8 weeks in the Hamilton Anxiety Rating Scale total score, a 0.3-point greater improvement on the Clinical Global Impression-Severity, and a 2.2-point bigger improvement on the Sheehan Disability Scale.
The week-8 response rate as reflected in at least a 50% improvement from baseline in MADRS score was 61.5% in the vortioxetine group and 47.3% with agomelatine.
Remission as defined by a MADRS score of 10 or less occurred in 40.5% of the vortioxetine group and 29.5% of the agomelatine group at week 8, and in 55.2% vs. 39.4%, respectively, at week 12 (P = .0002).
Study withdrawal because of adverse events occurred in 5.9% of the vortioxetine group, compared with 9.5% of patients on agomelatine. Nausea, reported by 16% of patients on vortioxetine, was the only side effect more common in patients on that drug; however, less than 1% of subjects on vortioxetine left the study because of nausea.
Vortioxetine is being developed jointly by H. Lundbeck and Takeda Pharmaceutical Co.Dr. Dragheim is a company employee.
HOLLYWOOD, FLA. – The investigational antidepressant vortioxetine achieved a 55% remission rate in a large, double-blind study of patients with major depressive disorder switched to the drug after an inadequate response to one of a half-dozen approved selective serotonin reuptake inhibitors or selective norepinephrine reuptake inhibitors.
"I think it’s quite impressive to see a remission rate on the order of 55% in this difficult-to-treat population," Dr. Marianne Dragheim observed in presenting the study findings at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
After all, in the landmark NIMH-sponsored sequential treatment alternatives to relieve depression, or STAR*D trial, in which citalopram nonresponders were switched to second-line treatment with sustained-release bupropion, extended-release venlafaxine, or sertraline, the mean remission rate after 12-14 weeks on the backup medication was just 31% (N. Engl. J. Med. 2006;354:1231-42), noted Dr. Dragheim of H. Lundbeck A/S, Valby, Denmark.
Vortioxetine is a first of its kind multimodal antidepressant. It is an inhibitor of the 5-hydroxytryptamine (5-HT) or serotonin transporter. In addition, vortioxetine is a 5-HT3, 5-HT7, and 5-HT10 receptor antagonist, a 5-HT1A receptor agonist, and a 5-HT1B receptor partial agonist.
Vortioxetine is currently under review at the Food and Drug Administration for possible marketing approval. The evidence submitted to the FDA includes seven positive studies, including four phase III randomized trials presented in May at the annual meeting of the American Psychiatric Association in San Francisco. The switching study Dr. Dragheim presented at the NCDEU meeting was completed so recently that it was not included in the FDA’s data package.
She reported on 501 European patients with major depressive disorder who responded inadequately to at least 6 weeks of monotherapy with citalopram, escitalopram, sertraline, paroxetine, venlafaxine, or duloxetine at approved doses. The study participants, all of whom wanted to change their antidepressant because of inadequate response, were randomized to 12 weeks of double-blind treatment with flexibly dosed vortioxetine at 10-20 mg/day or agomelatine at 25-50 mg/day. Agomelatine is approved as an antidepressant in the European Union and elsewhere in the world, but not in the United States.
The primary study endpoint was the change in scores on the Montgomery-Åsberg Depression Rating Scale (MADRS) between baseline and week 8. From a baseline MADRS total score of 29, the vortioxetine group improved by a mean of 2.2 more points than the agomelatine group, a statistically significant difference.
In addition, the vortioxetine-treated patients fared significantly better in terms of numerous secondary endpoints. They had a mean 1.9-point greater improvement than did the agomelatine group at 8 weeks in the Hamilton Anxiety Rating Scale total score, a 0.3-point greater improvement on the Clinical Global Impression-Severity, and a 2.2-point bigger improvement on the Sheehan Disability Scale.
The week-8 response rate as reflected in at least a 50% improvement from baseline in MADRS score was 61.5% in the vortioxetine group and 47.3% with agomelatine.
Remission as defined by a MADRS score of 10 or less occurred in 40.5% of the vortioxetine group and 29.5% of the agomelatine group at week 8, and in 55.2% vs. 39.4%, respectively, at week 12 (P = .0002).
Study withdrawal because of adverse events occurred in 5.9% of the vortioxetine group, compared with 9.5% of patients on agomelatine. Nausea, reported by 16% of patients on vortioxetine, was the only side effect more common in patients on that drug; however, less than 1% of subjects on vortioxetine left the study because of nausea.
Vortioxetine is being developed jointly by H. Lundbeck and Takeda Pharmaceutical Co.Dr. Dragheim is a company employee.
AT THE NCDEU MEETING
Major finding: Patients with major depressive disorder who did not respond adequately to a course of selective serotonin reuptake inhibitor or selective norepinephrine reuptake inhibitor therapy and who were then switched to double-blind treatment with the novel antidepressant vortioxetine had a 55% remission rate after 8 weeks of therapy.
Data source: A 501-patient, multicenter, randomized comparator European study.
Disclosures: The study was sponsored by H. Lundbeck A/S. Dr. Dragheim is a company employee.

Valacyclovir improves cognition in bipolar patients
HOLLYWOOD, FLA. – A 4-month course of the oral antiviral agent valacyclovir boosted cognition in herpes simplex virus-1–seropositive patients with bipolar disorder and cognitive impairment in a randomized, double-blind placebo-controlled clinical trial.
In this 60-patient study, 53% of participants assigned to valacyclovir (Valtrex) exhibited a clinically meaningful improvement in cognitive function, defined as at least a 10-point gain over baseline on the Repeatable Battery for the Assessment of Neurological Status (RBANS), compared with 14% in the placebo arm, Dr. Jennifer L. Payne reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.

The impressive improvement in response to valacyclovir documented in this study lends support to the viral hypothesis of mental illness, she said. This hypothesis posits that infection with one of the herpes viruses can in genetically vulnerable individuals lead to major mental illness, including schizophrenia, bipolar disorder, and perhaps Alzheimer’s disease.
"The idea behind this is that episodic reactivation of the virus in the [central nervous system] by stress or other stimuli could trigger mood, cognitive, or psychotic symptoms. If you treat patients you see this all the time: A patient comes under some kind of stress and becomes psychiatrically ill. One of the thoughts is that [herpes simplex virus] could underlie some of that psychopathology," according to Dr. Payne, a psychiatrist and director of the women’s mood disorders center at Johns Hopkins University, Baltimore.
She credited Faith B. Dickerson, Ph.D., of the Sheppard Pratt Institute in Baltimore with earlier groundbreaking work identifying a link between herpes simplex virus type 1 (HSV-1) and cognitive dysfunction in bipolar disorder. Dr. Dickerson and her colleagues reported a roughly 20-fold increased risk for cognitive impairment on the RBANS in HSV-1–seropositive, compared with seronegative patients with bipolar disorder (Biol. Psychiatry 2004;55:588-93).
The RBANS is a paper and pencil test that takes roughly 25 minutes. The mean score in the general population is 100. One standard deviation below the mean is a score of 85. Upon retaking the test, 16% of the general population are able to improve their score by 10 points or more, a rate close to the placebo group’s performance in Dr. Payne’s bipolar disorder study. The RBANS has individual sections for attention, immediate memory, delayed memory, language, and visuospatial construction.
HSV-1 typically causes oral herpes lesions. It’s an extremely common infection. By middle age, roughly 70% of Americans have serologic antibody titers to HSV-1. The virus infects the CNS and often remains in a latent state for many years, punctuated by symptomatic recurrences.
Dr. Payne screened 106 bipolar disorder patients; 84 proved HSV-1 seropositive. The mean baseline RBANS score in the seropositive patients was 75, compared with 92 in the seronegative cohort. The observed association between HSV-1 serologic status and RBANS score remained significant in a multivariate logistic regression analysis after investigators controlled for education level.
The 60 study participants were bipolar disorder outpatients, with an average age of 43. Thirty-seven percent met diagnostic criteria for bipolar I disorder; the rest, for bipolar II disorder. All were required to have baseline cognitive impairment as reflected by an RBANS score of 85 or less. Their average baseline Montgomery-Asberg Depression Rating Scale (MADRS) score was 24, with a Young Mania Rating Scale (YMRS) score of 8. Patients remained on their usual psychiatric medications during the study.
Participants in the 4-month trial were evaluated every 2 weeks for a change in mood symptoms using the MADRS and YMRS. The results came as a surprise.
"Our hypothesis had been that cognitive improvement with valacyclovir would be associated with improvement in depression, but the MADRS scores didn’t change over time," according to Dr. Payne.
As is typical in months-long clinical trials conducted in patients with bipolar disorder, there was a high dropout rate. Mean RBANS scores in the 19 patients in the valacyclovir group who completed the study improved from a baseline of 67.6 to 77.7 at 4 months. The 22 study completers in the control group showed no significant change in scores over time.
Dr. Payne said that if these results are confirmed in another clinical trial – and she plans to conduct one including seropositive bipolar disorder patients who are not cognitively impaired – it would be practice changing.
"If these findings hold up, it would indicate that as clinicians, we need to be testing for HSV-1 and treating it in our patients," she said.
Her future plans also include studying HSV-1 antibody status, cognition, and the possible impact of antiviral therapy in patients with major depressive disorder.
Patients with bipolar disorder often complain and exhibit symptoms of cognitive dysfunction, particularly in the domains of attention, memory, and executive function. The dysfunction typically worsens during manic or depressive episodes, but it’s often still present when bipolar disorder patients are affectively neutral.
The randomized trial was funded by the Stanley Medical Research Institute. Dr. Payne reported serving as a consultant to Pfizer and AstraZeneca.
HOLLYWOOD, FLA. – A 4-month course of the oral antiviral agent valacyclovir boosted cognition in herpes simplex virus-1–seropositive patients with bipolar disorder and cognitive impairment in a randomized, double-blind placebo-controlled clinical trial.
In this 60-patient study, 53% of participants assigned to valacyclovir (Valtrex) exhibited a clinically meaningful improvement in cognitive function, defined as at least a 10-point gain over baseline on the Repeatable Battery for the Assessment of Neurological Status (RBANS), compared with 14% in the placebo arm, Dr. Jennifer L. Payne reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The impressive improvement in response to valacyclovir documented in this study lends support to the viral hypothesis of mental illness, she said. This hypothesis posits that infection with one of the herpes viruses can in genetically vulnerable individuals lead to major mental illness, including schizophrenia, bipolar disorder, and perhaps Alzheimer’s disease.
"The idea behind this is that episodic reactivation of the virus in the [central nervous system] by stress or other stimuli could trigger mood, cognitive, or psychotic symptoms. If you treat patients you see this all the time: A patient comes under some kind of stress and becomes psychiatrically ill. One of the thoughts is that [herpes simplex virus] could underlie some of that psychopathology," according to Dr. Payne, a psychiatrist and director of the women’s mood disorders center at Johns Hopkins University, Baltimore.
She credited Faith B. Dickerson, Ph.D., of the Sheppard Pratt Institute in Baltimore with earlier groundbreaking work identifying a link between herpes simplex virus type 1 (HSV-1) and cognitive dysfunction in bipolar disorder. Dr. Dickerson and her colleagues reported a roughly 20-fold increased risk for cognitive impairment on the RBANS in HSV-1–seropositive, compared with seronegative patients with bipolar disorder (Biol. Psychiatry 2004;55:588-93).
The RBANS is a paper and pencil test that takes roughly 25 minutes. The mean score in the general population is 100. One standard deviation below the mean is a score of 85. Upon retaking the test, 16% of the general population are able to improve their score by 10 points or more, a rate close to the placebo group’s performance in Dr. Payne’s bipolar disorder study. The RBANS has individual sections for attention, immediate memory, delayed memory, language, and visuospatial construction.
HSV-1 typically causes oral herpes lesions. It’s an extremely common infection. By middle age, roughly 70% of Americans have serologic antibody titers to HSV-1. The virus infects the CNS and often remains in a latent state for many years, punctuated by symptomatic recurrences.
Dr. Payne screened 106 bipolar disorder patients; 84 proved HSV-1 seropositive. The mean baseline RBANS score in the seropositive patients was 75, compared with 92 in the seronegative cohort. The observed association between HSV-1 serologic status and RBANS score remained significant in a multivariate logistic regression analysis after investigators controlled for education level.
The 60 study participants were bipolar disorder outpatients, with an average age of 43. Thirty-seven percent met diagnostic criteria for bipolar I disorder; the rest, for bipolar II disorder. All were required to have baseline cognitive impairment as reflected by an RBANS score of 85 or less. Their average baseline Montgomery-Asberg Depression Rating Scale (MADRS) score was 24, with a Young Mania Rating Scale (YMRS) score of 8. Patients remained on their usual psychiatric medications during the study.
Participants in the 4-month trial were evaluated every 2 weeks for a change in mood symptoms using the MADRS and YMRS. The results came as a surprise.
"Our hypothesis had been that cognitive improvement with valacyclovir would be associated with improvement in depression, but the MADRS scores didn’t change over time," according to Dr. Payne.
As is typical in months-long clinical trials conducted in patients with bipolar disorder, there was a high dropout rate. Mean RBANS scores in the 19 patients in the valacyclovir group who completed the study improved from a baseline of 67.6 to 77.7 at 4 months. The 22 study completers in the control group showed no significant change in scores over time.
Dr. Payne said that if these results are confirmed in another clinical trial – and she plans to conduct one including seropositive bipolar disorder patients who are not cognitively impaired – it would be practice changing.
"If these findings hold up, it would indicate that as clinicians, we need to be testing for HSV-1 and treating it in our patients," she said.
Her future plans also include studying HSV-1 antibody status, cognition, and the possible impact of antiviral therapy in patients with major depressive disorder.
Patients with bipolar disorder often complain and exhibit symptoms of cognitive dysfunction, particularly in the domains of attention, memory, and executive function. The dysfunction typically worsens during manic or depressive episodes, but it’s often still present when bipolar disorder patients are affectively neutral.
The randomized trial was funded by the Stanley Medical Research Institute. Dr. Payne reported serving as a consultant to Pfizer and AstraZeneca.
HOLLYWOOD, FLA. – A 4-month course of the oral antiviral agent valacyclovir boosted cognition in herpes simplex virus-1–seropositive patients with bipolar disorder and cognitive impairment in a randomized, double-blind placebo-controlled clinical trial.
In this 60-patient study, 53% of participants assigned to valacyclovir (Valtrex) exhibited a clinically meaningful improvement in cognitive function, defined as at least a 10-point gain over baseline on the Repeatable Battery for the Assessment of Neurological Status (RBANS), compared with 14% in the placebo arm, Dr. Jennifer L. Payne reported at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
The impressive improvement in response to valacyclovir documented in this study lends support to the viral hypothesis of mental illness, she said. This hypothesis posits that infection with one of the herpes viruses can in genetically vulnerable individuals lead to major mental illness, including schizophrenia, bipolar disorder, and perhaps Alzheimer’s disease.
"The idea behind this is that episodic reactivation of the virus in the [central nervous system] by stress or other stimuli could trigger mood, cognitive, or psychotic symptoms. If you treat patients you see this all the time: A patient comes under some kind of stress and becomes psychiatrically ill. One of the thoughts is that [herpes simplex virus] could underlie some of that psychopathology," according to Dr. Payne, a psychiatrist and director of the women’s mood disorders center at Johns Hopkins University, Baltimore.
She credited Faith B. Dickerson, Ph.D., of the Sheppard Pratt Institute in Baltimore with earlier groundbreaking work identifying a link between herpes simplex virus type 1 (HSV-1) and cognitive dysfunction in bipolar disorder. Dr. Dickerson and her colleagues reported a roughly 20-fold increased risk for cognitive impairment on the RBANS in HSV-1–seropositive, compared with seronegative patients with bipolar disorder (Biol. Psychiatry 2004;55:588-93).
The RBANS is a paper and pencil test that takes roughly 25 minutes. The mean score in the general population is 100. One standard deviation below the mean is a score of 85. Upon retaking the test, 16% of the general population are able to improve their score by 10 points or more, a rate close to the placebo group’s performance in Dr. Payne’s bipolar disorder study. The RBANS has individual sections for attention, immediate memory, delayed memory, language, and visuospatial construction.
HSV-1 typically causes oral herpes lesions. It’s an extremely common infection. By middle age, roughly 70% of Americans have serologic antibody titers to HSV-1. The virus infects the CNS and often remains in a latent state for many years, punctuated by symptomatic recurrences.
Dr. Payne screened 106 bipolar disorder patients; 84 proved HSV-1 seropositive. The mean baseline RBANS score in the seropositive patients was 75, compared with 92 in the seronegative cohort. The observed association between HSV-1 serologic status and RBANS score remained significant in a multivariate logistic regression analysis after investigators controlled for education level.
The 60 study participants were bipolar disorder outpatients, with an average age of 43. Thirty-seven percent met diagnostic criteria for bipolar I disorder; the rest, for bipolar II disorder. All were required to have baseline cognitive impairment as reflected by an RBANS score of 85 or less. Their average baseline Montgomery-Asberg Depression Rating Scale (MADRS) score was 24, with a Young Mania Rating Scale (YMRS) score of 8. Patients remained on their usual psychiatric medications during the study.
Participants in the 4-month trial were evaluated every 2 weeks for a change in mood symptoms using the MADRS and YMRS. The results came as a surprise.
"Our hypothesis had been that cognitive improvement with valacyclovir would be associated with improvement in depression, but the MADRS scores didn’t change over time," according to Dr. Payne.
As is typical in months-long clinical trials conducted in patients with bipolar disorder, there was a high dropout rate. Mean RBANS scores in the 19 patients in the valacyclovir group who completed the study improved from a baseline of 67.6 to 77.7 at 4 months. The 22 study completers in the control group showed no significant change in scores over time.
Dr. Payne said that if these results are confirmed in another clinical trial – and she plans to conduct one including seropositive bipolar disorder patients who are not cognitively impaired – it would be practice changing.
"If these findings hold up, it would indicate that as clinicians, we need to be testing for HSV-1 and treating it in our patients," she said.
Her future plans also include studying HSV-1 antibody status, cognition, and the possible impact of antiviral therapy in patients with major depressive disorder.
Patients with bipolar disorder often complain and exhibit symptoms of cognitive dysfunction, particularly in the domains of attention, memory, and executive function. The dysfunction typically worsens during manic or depressive episodes, but it’s often still present when bipolar disorder patients are affectively neutral.
The randomized trial was funded by the Stanley Medical Research Institute. Dr. Payne reported serving as a consultant to Pfizer and AstraZeneca.
AT THE NCDEU MEETING
Major finding: Fifty-three percent of herpes simplex virus-1–seropositive patients with bipolar disorder and cognitive impairment exhibited objective cognitive improvement after 4 months on valacyclovir, as did a mere 14% on placebo.
Data source: A 60-patient randomized, double-blind, placebo-controlled clinical trial.
Disclosures: The study was sponsored by the Stanley Medical Research Institute. Dr. Payne reported serving as a consultant to Pfizer and AstraZeneca.

Bremelanotide improves female sexual dysfunction
HOLLYWOOD, FLA. – Subcutaneous bremelanotide self-administered at home by premenopausal women with sexual dysfunction significantly boosted sexual arousal and desire and their number of satisfying sexual events, based on data from a phase IIb clinical trial.
The novel therapy proved effective both in women with hypoactive sexual desire disorder and in those with combined hypoactive sexual desire disorder/female sexual arousal disorder, among the most common forms of female sexual dysfunction, Dr. Anita H. Clayton noted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Female sexual dysfunction is distressing, very common, and multifactorial, and there is at present no approved pharmacotherapy for these disorders, according to Dr. Clayton, professor of psychiatry and neurobehavioral sciences at the University of Virginia, Charlottesville. Thus, this represents an area of significant unmet medical need, she said.
Bremelanotide is a cyclic 7-amino-acid melanocortin peptide. It is a synthetic analog of the hormone alpha-melanocyte–stimulating hormone (alpha-MSH). It functions as a melanocortin-4 receptor agonist. Bremelanotide, like alpha-MSH, is thought to modulate brain pathways involved in sexual response, Dr. Clayton explained.
She reported data from a phase IIb randomized, double-blind, multicenter trial involving 327 women who met diagnostic criteria for hypoactive sexual desire disorder or combined hypoactive sexual desire disorder/female sexual arousal disorder. After receiving instruction in self-administration of subcutaneous injections, all participants underwent 4 weeks of single-blind placebo self-dosing at home on an as-needed basis. Then they were randomized to 12 weeks of double-blind home treatment with placebo or bremelanotide at 0.75 mg, 1.25 mg, or 1.75 mg in prefilled syringes. Participants were instructed to inject themselves approximately 45 minutes prior to sexual activity. They were not to exceed 1 dose per day, or 16 doses in a 4-week period.
The primary endpoint was change between the numbers of satisfying sexual events during the 28-day baseline period on placebo and during the final 28 days of the 12-week double-blind study period, using the Female Sexual Encounter Profile-Revised
The mean increase was 0.2 events in placebo-treated controls. Women randomized to 0.75 of bremelanotide didn’t fare significantly better than that.
However, women using bremelanotide at 1.25 mg had a mean 0.7-event increase from baseline, and those on 1.75 mg averaged a 0.8-event increase, both of which were significantly better than placebo.
Secondary endpoints were also positive, in dose-dependent fashion. The mean change over time in the Female Sexual Function Index total score, a validated measure of overall sexual functioning, was 1.88 for placebo, 3.6 in the pooled analysis of patients on bremelanotide at 1.25 or 1.75 mg, and 4.4 in those on 1.75 mg.
Similarly, the mean improvement on the FSDS-DAO (Female Sexual Distress Scale-Desire/Arousal/Orgasm) total score, an indicator of sexual dysfunction–related distress, was –6.8 for placebo, –11.1 for the pooled group on 1.25 or 1.75 mg of bremelanotide, and –13.1 for women on 1.75 mg.
These are clinically meaningful improvements, according to Dr. Clayton. Of note, the mean total score improvements, compared with baseline on these outcome measures, were still growing during the third and final month of double-blind treatment.
Also encouraging was the large percentage of women on bremelanotide whose scores reached thresholds indicative of normal levels of sexual function, she continued. For example, a Female Sexual Function Index total score greater than 26.5 was achieved in 42.7% of women on bremelanotide at 0.75 mg, 45.5% of those on 1.25 mg, and 49% on 1.75 mg, compared with 36.5% of placebo-treated controls. Moreover, a FSDS-DAO total score less than 18 was attained by 28.5% of women on placebo, 40.5% of those on bremelanotide at 0.75 mg, 45.6% on 1.25 mg, and 47.5% of those on 1.75 mg.
The drug therapy was safe and generally well tolerated. The most common bremelanotide-associated side effects were nausea, facial flushing, and headache, which affected 9%-24% of patients in dose-dependent fashion and were typically mild to moderate in nature.
Bremelanotide-treated patients averaged an increase in blood pressure of approximately 2 mm Hg, largely restricted to the first 4 hours after dosing. The number of patients forced to withdraw from the study based on predefined blood pressure change criteria was evenly distributed among the placebo and treatment groups, which was reassuring, Dr. Clayton said. Approximately 5 years ago, development of an intranasal formulation of bremelanotide for treatment of male erectile dysfunction as well as female sexual dysfunction was discontinued because of concerns about significant drug-induced hypertension. The current study, as well as other data, indicates that hypertension isn’t an issue with subcutaneous administration, she noted.
A definitive phase III clinical trial of at-home, self-administered subcutaneous bremelanotide for treatment of female sexual dysfunction is anticipated to start later this year.
Dr. Clayton reported receiving research support and consulting fees from Palatin Technologies, which is developing bremelanotide, as well as from other pharmaceutical companies.
HOLLYWOOD, FLA. – Subcutaneous bremelanotide self-administered at home by premenopausal women with sexual dysfunction significantly boosted sexual arousal and desire and their number of satisfying sexual events, based on data from a phase IIb clinical trial.
The novel therapy proved effective both in women with hypoactive sexual desire disorder and in those with combined hypoactive sexual desire disorder/female sexual arousal disorder, among the most common forms of female sexual dysfunction, Dr. Anita H. Clayton noted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Female sexual dysfunction is distressing, very common, and multifactorial, and there is at present no approved pharmacotherapy for these disorders, according to Dr. Clayton, professor of psychiatry and neurobehavioral sciences at the University of Virginia, Charlottesville. Thus, this represents an area of significant unmet medical need, she said.
Bremelanotide is a cyclic 7-amino-acid melanocortin peptide. It is a synthetic analog of the hormone alpha-melanocyte–stimulating hormone (alpha-MSH). It functions as a melanocortin-4 receptor agonist. Bremelanotide, like alpha-MSH, is thought to modulate brain pathways involved in sexual response, Dr. Clayton explained.
She reported data from a phase IIb randomized, double-blind, multicenter trial involving 327 women who met diagnostic criteria for hypoactive sexual desire disorder or combined hypoactive sexual desire disorder/female sexual arousal disorder. After receiving instruction in self-administration of subcutaneous injections, all participants underwent 4 weeks of single-blind placebo self-dosing at home on an as-needed basis. Then they were randomized to 12 weeks of double-blind home treatment with placebo or bremelanotide at 0.75 mg, 1.25 mg, or 1.75 mg in prefilled syringes. Participants were instructed to inject themselves approximately 45 minutes prior to sexual activity. They were not to exceed 1 dose per day, or 16 doses in a 4-week period.
The primary endpoint was change between the numbers of satisfying sexual events during the 28-day baseline period on placebo and during the final 28 days of the 12-week double-blind study period, using the Female Sexual Encounter Profile-Revised
The mean increase was 0.2 events in placebo-treated controls. Women randomized to 0.75 of bremelanotide didn’t fare significantly better than that.
However, women using bremelanotide at 1.25 mg had a mean 0.7-event increase from baseline, and those on 1.75 mg averaged a 0.8-event increase, both of which were significantly better than placebo.
Secondary endpoints were also positive, in dose-dependent fashion. The mean change over time in the Female Sexual Function Index total score, a validated measure of overall sexual functioning, was 1.88 for placebo, 3.6 in the pooled analysis of patients on bremelanotide at 1.25 or 1.75 mg, and 4.4 in those on 1.75 mg.
Similarly, the mean improvement on the FSDS-DAO (Female Sexual Distress Scale-Desire/Arousal/Orgasm) total score, an indicator of sexual dysfunction–related distress, was –6.8 for placebo, –11.1 for the pooled group on 1.25 or 1.75 mg of bremelanotide, and –13.1 for women on 1.75 mg.
These are clinically meaningful improvements, according to Dr. Clayton. Of note, the mean total score improvements, compared with baseline on these outcome measures, were still growing during the third and final month of double-blind treatment.
Also encouraging was the large percentage of women on bremelanotide whose scores reached thresholds indicative of normal levels of sexual function, she continued. For example, a Female Sexual Function Index total score greater than 26.5 was achieved in 42.7% of women on bremelanotide at 0.75 mg, 45.5% of those on 1.25 mg, and 49% on 1.75 mg, compared with 36.5% of placebo-treated controls. Moreover, a FSDS-DAO total score less than 18 was attained by 28.5% of women on placebo, 40.5% of those on bremelanotide at 0.75 mg, 45.6% on 1.25 mg, and 47.5% of those on 1.75 mg.
The drug therapy was safe and generally well tolerated. The most common bremelanotide-associated side effects were nausea, facial flushing, and headache, which affected 9%-24% of patients in dose-dependent fashion and were typically mild to moderate in nature.
Bremelanotide-treated patients averaged an increase in blood pressure of approximately 2 mm Hg, largely restricted to the first 4 hours after dosing. The number of patients forced to withdraw from the study based on predefined blood pressure change criteria was evenly distributed among the placebo and treatment groups, which was reassuring, Dr. Clayton said. Approximately 5 years ago, development of an intranasal formulation of bremelanotide for treatment of male erectile dysfunction as well as female sexual dysfunction was discontinued because of concerns about significant drug-induced hypertension. The current study, as well as other data, indicates that hypertension isn’t an issue with subcutaneous administration, she noted.
A definitive phase III clinical trial of at-home, self-administered subcutaneous bremelanotide for treatment of female sexual dysfunction is anticipated to start later this year.
Dr. Clayton reported receiving research support and consulting fees from Palatin Technologies, which is developing bremelanotide, as well as from other pharmaceutical companies.
HOLLYWOOD, FLA. – Subcutaneous bremelanotide self-administered at home by premenopausal women with sexual dysfunction significantly boosted sexual arousal and desire and their number of satisfying sexual events, based on data from a phase IIb clinical trial.
The novel therapy proved effective both in women with hypoactive sexual desire disorder and in those with combined hypoactive sexual desire disorder/female sexual arousal disorder, among the most common forms of female sexual dysfunction, Dr. Anita H. Clayton noted at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
Female sexual dysfunction is distressing, very common, and multifactorial, and there is at present no approved pharmacotherapy for these disorders, according to Dr. Clayton, professor of psychiatry and neurobehavioral sciences at the University of Virginia, Charlottesville. Thus, this represents an area of significant unmet medical need, she said.
Bremelanotide is a cyclic 7-amino-acid melanocortin peptide. It is a synthetic analog of the hormone alpha-melanocyte–stimulating hormone (alpha-MSH). It functions as a melanocortin-4 receptor agonist. Bremelanotide, like alpha-MSH, is thought to modulate brain pathways involved in sexual response, Dr. Clayton explained.
She reported data from a phase IIb randomized, double-blind, multicenter trial involving 327 women who met diagnostic criteria for hypoactive sexual desire disorder or combined hypoactive sexual desire disorder/female sexual arousal disorder. After receiving instruction in self-administration of subcutaneous injections, all participants underwent 4 weeks of single-blind placebo self-dosing at home on an as-needed basis. Then they were randomized to 12 weeks of double-blind home treatment with placebo or bremelanotide at 0.75 mg, 1.25 mg, or 1.75 mg in prefilled syringes. Participants were instructed to inject themselves approximately 45 minutes prior to sexual activity. They were not to exceed 1 dose per day, or 16 doses in a 4-week period.
The primary endpoint was change between the numbers of satisfying sexual events during the 28-day baseline period on placebo and during the final 28 days of the 12-week double-blind study period, using the Female Sexual Encounter Profile-Revised
The mean increase was 0.2 events in placebo-treated controls. Women randomized to 0.75 of bremelanotide didn’t fare significantly better than that.
However, women using bremelanotide at 1.25 mg had a mean 0.7-event increase from baseline, and those on 1.75 mg averaged a 0.8-event increase, both of which were significantly better than placebo.
Secondary endpoints were also positive, in dose-dependent fashion. The mean change over time in the Female Sexual Function Index total score, a validated measure of overall sexual functioning, was 1.88 for placebo, 3.6 in the pooled analysis of patients on bremelanotide at 1.25 or 1.75 mg, and 4.4 in those on 1.75 mg.
Similarly, the mean improvement on the FSDS-DAO (Female Sexual Distress Scale-Desire/Arousal/Orgasm) total score, an indicator of sexual dysfunction–related distress, was –6.8 for placebo, –11.1 for the pooled group on 1.25 or 1.75 mg of bremelanotide, and –13.1 for women on 1.75 mg.
These are clinically meaningful improvements, according to Dr. Clayton. Of note, the mean total score improvements, compared with baseline on these outcome measures, were still growing during the third and final month of double-blind treatment.
Also encouraging was the large percentage of women on bremelanotide whose scores reached thresholds indicative of normal levels of sexual function, she continued. For example, a Female Sexual Function Index total score greater than 26.5 was achieved in 42.7% of women on bremelanotide at 0.75 mg, 45.5% of those on 1.25 mg, and 49% on 1.75 mg, compared with 36.5% of placebo-treated controls. Moreover, a FSDS-DAO total score less than 18 was attained by 28.5% of women on placebo, 40.5% of those on bremelanotide at 0.75 mg, 45.6% on 1.25 mg, and 47.5% of those on 1.75 mg.
The drug therapy was safe and generally well tolerated. The most common bremelanotide-associated side effects were nausea, facial flushing, and headache, which affected 9%-24% of patients in dose-dependent fashion and were typically mild to moderate in nature.
Bremelanotide-treated patients averaged an increase in blood pressure of approximately 2 mm Hg, largely restricted to the first 4 hours after dosing. The number of patients forced to withdraw from the study based on predefined blood pressure change criteria was evenly distributed among the placebo and treatment groups, which was reassuring, Dr. Clayton said. Approximately 5 years ago, development of an intranasal formulation of bremelanotide for treatment of male erectile dysfunction as well as female sexual dysfunction was discontinued because of concerns about significant drug-induced hypertension. The current study, as well as other data, indicates that hypertension isn’t an issue with subcutaneous administration, she noted.
A definitive phase III clinical trial of at-home, self-administered subcutaneous bremelanotide for treatment of female sexual dysfunction is anticipated to start later this year.
Dr. Clayton reported receiving research support and consulting fees from Palatin Technologies, which is developing bremelanotide, as well as from other pharmaceutical companies.
AT THE NCDEU MEETING
Major finding: Self-administered subcutaneous injections of the novel agent bremelanotide at the top dose tested resulted in quadruple the number of satisfying sexual events, compared with placebo, in premenopausal women with female sexual dysfunction.
Data source: A multicenter, double-blind, phase IIb randomized trial involving 397 women with female sexual dysfunction, 327 of whom were evaluable.
Disclosures: The phase IIb study was sponsored by Palatin Technologies. The presenter has received research funding and consulting fees from this and other pharmaceutical companies.
Vyvanse shows promise for binge-eating disorder
HOLLYWOOD, FLA. – Not a single medication is approved for treatment of binge-eating disorder, but that could change if the favorable results of an ongoing phase III randomized trial of lisdexamfetamine mirror those of a recently completed phase II study.
Binge-eating disorder (BED) is the most common eating disorder. It is characterized by excessive food intake accompanied by psychological distress, but without the purging or fasting that are the hallmarks of bulimia nervosa and anorexia nervosa, respectively. Patients with BED are often severely obese, depressed, and have metabolic disorders.

BED is "gaining increasing recognition as a very significant public health problem, but it remains underdiagnosed," Dr. Susan L. McElroy observed at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
She presented the results of a 31-site, randomized, double-blind, placebo-controlled clinical trial of lisdexamfetamine (Vyvanse) in 213 patients with moderate or severe BED as defined by DSM-IV-TR. The study, in which participants recorded their binge-eating episodes in a daily diary, entailed a 2-week baseline period, 11 weeks of double-blind therapy, and a 1-week follow-up phase.
Patients were assigned to placebo or to lisdexamfetamine at 30, 50, or 70 mg/day. This was a forced-dose titration study. Everyone assigned to lisdexamfetamine started at 30 mg/day. After week 1, patients assigned to 50 or 70 mg/day increased their daily dose by 20 mg/wk until reaching their target.
Subjects assigned to lisdexamfetamine at 50 mg/day had a mean baseline of 4.54 binge-eating days/wk. By week 11 on the drug, this had improved to 0.31 days/wk.
This was a significantly better result than in placebo-treated controls, who went from 4.29 binge-eating days per week at baseline to 1.13 per week after 11 weeks. Patients assigned to lisdexamfetamine at 70 mg/day did best of all, improving from 4.47 binge-eating days/wk at baseline to 0.11 after 11 weeks.
In addition, 67% of patients in the 70-mg/day group had been free of any binge-eating episodes for 1 week at week 11, compared with 56% of subjects on lisdexamfetamine at 50 mg/day and 34% on placebo. These differences were statistically significant.
This was a dose-ranging study, and indeed a linear dose-response relationship was found. The 30-mg/day dose was not more effective than placebo. Using statistical analysis, the minimum effective dose of lisdexamfetamine for treatment of BED was estimated at 34 mg/day, said Dr. McElroy, chief research officer at the Lindner Center of HOPE in Mason, Ohio, and professor of psychiatry and behavioral neuroscience at the University of Cincinnati.
Lisdexamfetamine is a d-amphetamine prodrug that inhibits dopamine reuptake and stimulates release of monoamine neurotransmitters. The therapeutic rationale for its use in BED lies in the observation that BED is associated with abnormal signaling by the dopamine and norepinephrine neurotransmitter systems, she explained.
The drug’s approved indication is for treatment of attention-deficit/hyperactivity disorder, for which the recommended starting dose is 30 mg/day, with adjustments up to 70 mg/day permitted.
The side effects noted in the phase II BED study were typical of those seen when lisdexamfetamine is prescribed for ADHD.
This study was sponsored by Shire. Dr. McElroy is a consultant to or a member of the scientific advisory boards of Shire and a half-dozen other pharmaceutical companies.
Dr. Susan L. McElroy, Vyvanse
HOLLYWOOD, FLA. – Not a single medication is approved for treatment of binge-eating disorder, but that could change if the favorable results of an ongoing phase III randomized trial of lisdexamfetamine mirror those of a recently completed phase II study.
Binge-eating disorder (BED) is the most common eating disorder. It is characterized by excessive food intake accompanied by psychological distress, but without the purging or fasting that are the hallmarks of bulimia nervosa and anorexia nervosa, respectively. Patients with BED are often severely obese, depressed, and have metabolic disorders.
BED is "gaining increasing recognition as a very significant public health problem, but it remains underdiagnosed," Dr. Susan L. McElroy observed at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
She presented the results of a 31-site, randomized, double-blind, placebo-controlled clinical trial of lisdexamfetamine (Vyvanse) in 213 patients with moderate or severe BED as defined by DSM-IV-TR. The study, in which participants recorded their binge-eating episodes in a daily diary, entailed a 2-week baseline period, 11 weeks of double-blind therapy, and a 1-week follow-up phase.
Patients were assigned to placebo or to lisdexamfetamine at 30, 50, or 70 mg/day. This was a forced-dose titration study. Everyone assigned to lisdexamfetamine started at 30 mg/day. After week 1, patients assigned to 50 or 70 mg/day increased their daily dose by 20 mg/wk until reaching their target.
Subjects assigned to lisdexamfetamine at 50 mg/day had a mean baseline of 4.54 binge-eating days/wk. By week 11 on the drug, this had improved to 0.31 days/wk.
This was a significantly better result than in placebo-treated controls, who went from 4.29 binge-eating days per week at baseline to 1.13 per week after 11 weeks. Patients assigned to lisdexamfetamine at 70 mg/day did best of all, improving from 4.47 binge-eating days/wk at baseline to 0.11 after 11 weeks.
In addition, 67% of patients in the 70-mg/day group had been free of any binge-eating episodes for 1 week at week 11, compared with 56% of subjects on lisdexamfetamine at 50 mg/day and 34% on placebo. These differences were statistically significant.
This was a dose-ranging study, and indeed a linear dose-response relationship was found. The 30-mg/day dose was not more effective than placebo. Using statistical analysis, the minimum effective dose of lisdexamfetamine for treatment of BED was estimated at 34 mg/day, said Dr. McElroy, chief research officer at the Lindner Center of HOPE in Mason, Ohio, and professor of psychiatry and behavioral neuroscience at the University of Cincinnati.
Lisdexamfetamine is a d-amphetamine prodrug that inhibits dopamine reuptake and stimulates release of monoamine neurotransmitters. The therapeutic rationale for its use in BED lies in the observation that BED is associated with abnormal signaling by the dopamine and norepinephrine neurotransmitter systems, she explained.
The drug’s approved indication is for treatment of attention-deficit/hyperactivity disorder, for which the recommended starting dose is 30 mg/day, with adjustments up to 70 mg/day permitted.
The side effects noted in the phase II BED study were typical of those seen when lisdexamfetamine is prescribed for ADHD.
This study was sponsored by Shire. Dr. McElroy is a consultant to or a member of the scientific advisory boards of Shire and a half-dozen other pharmaceutical companies.
HOLLYWOOD, FLA. – Not a single medication is approved for treatment of binge-eating disorder, but that could change if the favorable results of an ongoing phase III randomized trial of lisdexamfetamine mirror those of a recently completed phase II study.
Binge-eating disorder (BED) is the most common eating disorder. It is characterized by excessive food intake accompanied by psychological distress, but without the purging or fasting that are the hallmarks of bulimia nervosa and anorexia nervosa, respectively. Patients with BED are often severely obese, depressed, and have metabolic disorders.
BED is "gaining increasing recognition as a very significant public health problem, but it remains underdiagnosed," Dr. Susan L. McElroy observed at a meeting of the New Clinical Drug Evaluation Unit sponsored by the National Institute of Mental Health.
She presented the results of a 31-site, randomized, double-blind, placebo-controlled clinical trial of lisdexamfetamine (Vyvanse) in 213 patients with moderate or severe BED as defined by DSM-IV-TR. The study, in which participants recorded their binge-eating episodes in a daily diary, entailed a 2-week baseline period, 11 weeks of double-blind therapy, and a 1-week follow-up phase.
Patients were assigned to placebo or to lisdexamfetamine at 30, 50, or 70 mg/day. This was a forced-dose titration study. Everyone assigned to lisdexamfetamine started at 30 mg/day. After week 1, patients assigned to 50 or 70 mg/day increased their daily dose by 20 mg/wk until reaching their target.
Subjects assigned to lisdexamfetamine at 50 mg/day had a mean baseline of 4.54 binge-eating days/wk. By week 11 on the drug, this had improved to 0.31 days/wk.
This was a significantly better result than in placebo-treated controls, who went from 4.29 binge-eating days per week at baseline to 1.13 per week after 11 weeks. Patients assigned to lisdexamfetamine at 70 mg/day did best of all, improving from 4.47 binge-eating days/wk at baseline to 0.11 after 11 weeks.
In addition, 67% of patients in the 70-mg/day group had been free of any binge-eating episodes for 1 week at week 11, compared with 56% of subjects on lisdexamfetamine at 50 mg/day and 34% on placebo. These differences were statistically significant.
This was a dose-ranging study, and indeed a linear dose-response relationship was found. The 30-mg/day dose was not more effective than placebo. Using statistical analysis, the minimum effective dose of lisdexamfetamine for treatment of BED was estimated at 34 mg/day, said Dr. McElroy, chief research officer at the Lindner Center of HOPE in Mason, Ohio, and professor of psychiatry and behavioral neuroscience at the University of Cincinnati.
Lisdexamfetamine is a d-amphetamine prodrug that inhibits dopamine reuptake and stimulates release of monoamine neurotransmitters. The therapeutic rationale for its use in BED lies in the observation that BED is associated with abnormal signaling by the dopamine and norepinephrine neurotransmitter systems, she explained.
The drug’s approved indication is for treatment of attention-deficit/hyperactivity disorder, for which the recommended starting dose is 30 mg/day, with adjustments up to 70 mg/day permitted.
The side effects noted in the phase II BED study were typical of those seen when lisdexamfetamine is prescribed for ADHD.
This study was sponsored by Shire. Dr. McElroy is a consultant to or a member of the scientific advisory boards of Shire and a half-dozen other pharmaceutical companies.
Dr. Susan L. McElroy, Vyvanse
Dr. Susan L. McElroy, Vyvanse
AT THE NCDEU MEETING
Major finding: Patients with moderate or severe binge-eating disorder showed a dose-dependent reduction in binge-eating episodes in response to lisdexamfetamine dimesylate. Binge-eating days per week in those assigned to the top dose of 70 mg/day dropped from a mean of 4.47 at baseline to 0.11 after 11 weeks.
Data source: Randomized, double-blind, placebo-controlled, multicenter phase II study involving 213 patients.
Disclosures: The study was sponsored by Shire, which markets lisdexamfetamine for treatment of ADHD. Dr. McElroy is a consultant to the company.
