User login
You have a 54-year-old black patient with gout, diabetes mellitus, hypertension, and chronic kidney disease (CKD). He has an acute gout flare involving his right knee. In the past year he has had four attacks of gout in the ankles and knees, which you treated with intra-articular glucocorticoid injections. He has been on allopurinol (Zyloprim) 200 mg daily, but his last serum urate level was 9.4 mg/dL (reference range 3.0–8.0). His creatinine clearance is 45 mL/minute (reference range 85–125).
In view of his kidney disease, you are concerned about increasing his dose of allopurinol, but also about the need to treat his frequent attacks. How should you manage this patient?
GOUT IS CHALLENGING TO TREAT IN PATIENTS WITH KIDNEY DISEASE
A major challenge in treating patients with gout is to avoid therapeutic interactions with common comorbidities, including hypertension, insulin resistance, coronary artery disease, heart failure, and especially CKD.1
In this paper, we discuss approaches to and controversies in the management of gout and hyperuricemia in patients with CKD. Unfortunately, the evidence from clinical trials to guide treatment decisions is limited; therefore, decisions must often be based on experience and pathophysiologic principles.
GENERAL GOALS OF GOUT THERAPY
Depending on the patient and the stage of the disease, the goals in treating patients with gout are to:
- Terminate acute attacks as promptly and safely as possible
- Prevent recurrences of acute gout attacks
- Prevent or reverse complications resulting from deposition of monosodium urate in the joints, in the kidneys, or at other sites.
These goals are more difficult to achieve in patients with CKD because of the potential complications from many of the available drugs.
TERMINATING ACUTE GOUT FLARES
In patients with acute gout, treatment is aimed at quickly resolving pain and inflammation.
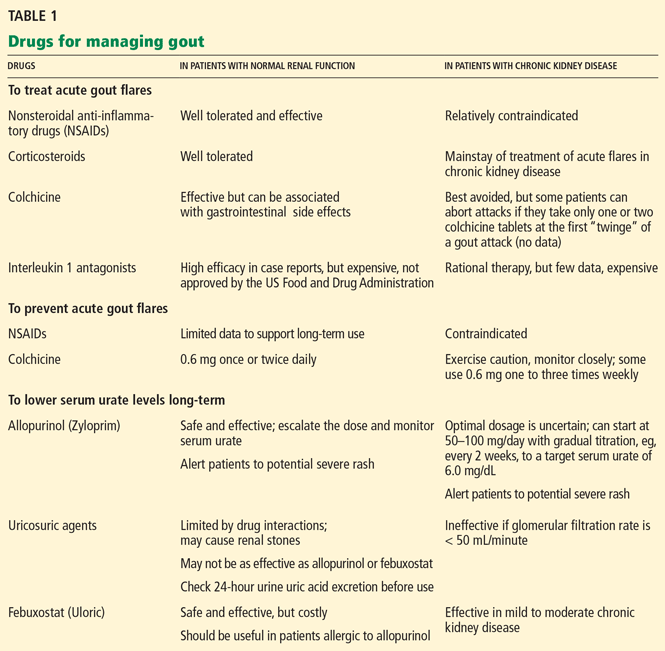
Several types of drugs can terminate acute gout flares. The choice in most situations is colchicine (Colcrys); a nonsteroidal anti-inflammatory drug (NSAID); a corticosteroid; or corticotropin (ACTH).
However, in patients with CKD, there are concerns about using colchicine or NSAIDs, and corticotropin is very expensive; thus, corticosteroids are often used.
Colchicine’s clearance is reduced in CKD
Colchicine is somewhat effective in treating acute gout attacks and probably more effective in preventing attacks.
Due to concerns about inappropriate dosing and reported deaths,2 the intravenous formulation is not available in many countries, including the United States.
After oral administration, colchicine is rapidly absorbed, with a bioavailability of up to 50%. It undergoes metabolism by the liver, and its metabolites are excreted by renal and biliary-intestinal routes. Up to 20% of the active drug is excreted by the kidneys.3
Colchicine’s clearance is significantly reduced in patients with renal or hepatic insufficiency, and the drug may accumulate in cells, with resultant toxicity.4 Colchicine-induced toxicity has been observed when the drug was used for acute treatment, as well as for chronic prophylaxis of gout in patients with CKD; thus, alternative agents for treating acute attacks should be considered.5,6 With prolonged use, reversible colchicine-induced axonal neuropathy, neutropenia, and vacuolar myopathy can develop in patients with CKD.7
In a trial in patients with normal renal function, nearly 100% who received an initial dose of 1 mg followed by 0.5 mg every 2 hours developed diarrhea at a median time of 24 hours.8 Emesis may also occur.
A lower dose of 1.8 mg (two 0.6-mg pills followed by one pill an hour later) was well tolerated but only moderately effective in treating acute gout, causing at least a 50% reduction in pain at 24 hours in only 38% of patients.9 This study does not clarify the dosage to use to completely resolve attacks. Using additional colchicine likely will increase the response rate, but will also increase side effects. Patients with CKD were not included.
Some patients, as shown in the above trial, can abort attacks by taking only one or two colchicine tablets when they feel the first “twinge” of an attack. This approach is likely to be safe in CKD, but it may be of value to only a few patients.
Nonsteroidal anti-inflammatory drugs can worsen chronic kidney disease
NSAIDs in high doses can effectively treat the pain and inflammation of acute gout. Indomethacin (Indocin) 50 mg three times daily has been standard NSAID therapy.
Other nonselective NSAIDs and NSAIDs that selectively inhibit cyclooxygenase 2 (COX-2) are effective, but all can cause acute renal toxicity or worsen CKD.10 Renal side effects include salt and water retention, acute tubular necrosis, acute interstitial nephritis, proteinuria, hypertension, hyperkalemia, and chronic renal injury.11
Even short-term use of high-dose NSAIDs should generally be avoided in patients with preexisting CKD, for whom there is no established safe threshold dose. When NSAIDs (including selective COX-2 inhibitors) are used, renal function should be monitored closely and the duration limited as much as possible.
Corticosteroids are often used to treat acute attacks
Due to the concerns about NSAIDs or colchicine to treat acute gout attacks in patients with CKD, corticosteroids are often used in this setting.
Intra-articular steroid injections are useful in treating acute gout limited to a single joint or bursa.12 However, one should first make sure that the joint is not infected: septic arthritis should ideally be excluded by arthrocentesis, particularly in immunosuppressed patients13 or those with end-stage renal disease, who are predisposed to bacteremia.
Oral, intramuscular, or intravenous steroids can provide complete relief from acute gout, although high doses (eg, prednisone 30–60 mg/day or the equivalent) are often needed. Common errors resulting in inefficacy include using too low a dose or not treating for a sufficient time before tapering or stopping. Groff and colleagues14 described 13 patients who received oral or intravenous steroids for acute gout. Nine patients received an initial single dose of prednisone ranging from 20 to 50 mg, with tapering over a mean of 10 days. Twelve of the 13 patients had improvement within 48 hours, and the signs and symptoms of acute gout resolved completely within 7 to 10 days.
We often give prednisone 40 mg daily until a day after the acute attack resolves and then taper over another 7 to 10 days. There are no data to guide steroid dosing in an evidence-based way, but we believe too short a course of therapy may result in return of symptoms.
Corticotropin and other agents: Effective but costly
Corticotropin is available for subcutaneous or intramuscular injection. A single intramuscular injection of corticotropin gel (H.P. Acthar, 25–80 IU) may terminate an acute gout attack.15 However, many patients need another injection after 24 to 72 hours, which would require another visit to the physician. This treatment has been touted by some as being more effective than corticosteroid therapy, possibly because of a unique peripheral mechanism of action in addition to stimulating cortisol release.16
We rarely use corticotropin, in view of its cost as well as concerns about excessive sodium and water retention due to the release of multiple hormones from the adrenal gland. This may be especially deleterious in patients with CKD or congestive heart failure.17
Parenteral anti-tumor necrosis factor agents or interleukin 1 antagonists can be dramatically effective but are also expensive.18,19 For example, anakinra (Kineret) 100 mg costs about $73, and multiple daily doses may be necessary.
Under unique conditions in which they can be safely used (eg, patients with CKD, diabetes mellitus, liver disease), they may be cost-effective if they can shorten the stay of a hospitalized patient with acute gout.
PROPHYLACTIC ANTI-INFLAMMATORY THERAPY FOR PATIENTS WITH GOUT
Between attacks, the goal is to prevent new attacks through prophylactic management, which may include anti-inflammatory and hypouricemic therapy along with dietary instruction (such as avoiding excessive beer, liquor, and fructose ingestion).
Colchicine can be used as prophylaxis, with caution and monitoring
Although colchicine is not 100% effective, it markedly reduces the flare rate when started in low doses at the time hypouricemic therapy is initiated.20,21 (Hypouricemic therapy is discussed below.) We generally try to continue this prophylactic therapy, if the patient tolerates it, for at least 6 months—longer if tophi are still present or if attacks continue to occur.
If renal function is intact, colchicine can be prescribed at a dosage of 0.6 mg orally once or twice daily.21 In CKD, since the clearance of colchicine is reduced,4 the dosage should be reduced. Patients on colchicine for prophylaxis must be carefully monitored if the glomerular filtration rate is less than 50 mL/minute, or colchicine should be avoided altogether.6 Laboratory testing for colchicine levels is not routinely available and may be of limited value in predicting adverse effects; thus, recommendations about dose adjustments in CKD are empiric.
Wallace et al22 recommended a dose of 0.6 mg once daily if the creatinine clearance is 35 to 49 mL/minute and 0.6 mg every 2 to 3 days if it is 10 to 34 mL/minute, but there are no published long-term safety or efficacy data validating these reasonable (based on available information) dosing regimens.
Even with dose adjustment, caution is needed. Low-dose daily colchicine may be associated with reversible neuromyopathy and bone marrow suppression.7,23 Patients with neuromyopathy may complain of myalgias, proximal muscle weakness, and numbness and may have areflexia and decreased sensation. Laboratory findings include elevated creatine kinase and aminotransferase levels. We regularly check for leukopenia or elevated creatine kinase and aspartate aminotransferase levels in patients with CKD who are receiving colchicine in any dose.
Prolonged colchicine therapy should probably be avoided in patients on hemodialysis, as this drug is not removed by dialysis or by exchange transfusion, and the risk of toxicity under these circumstances may be high.22 When there is no viable alternative and the drug is given, patients should be closely monitored for signs of toxicity.
Concurrent (even short-term) treatment with most macrolide antibiotics, particularly clarithromycin (Biaxin), most statin drugs, ketoconazole (Nizoral), cyclosporine, and likely other drugs predisposes to colchicine toxicity by altering its distribution and elimination, and can in rare cases cause morbidity or death.24–26
NSAIDs are not optimal as prophylaxis in patients with chronic kidney disease
Little information has been published about using NSAIDs chronically to prevent flares, but they are not the optimal drugs to use in patients with CKD, as discussed above. In patients with end-stage renal disease, there are also concerns about NSAID-induced gastric and intestinal bleeding.
Low-dose steroids may not be effective as prophylaxis
Lower doses of steroids may not be effective as prophylaxis against gout flares, consistent with the common observation that gout flares still occur in organ transplant recipients who are taking maintenance doses of prednisone.13
PREVENTING FLARES BY LOWERING SERUM URATE LEVELS
If tophi are present, if radiography shows evidence of damage, if attacks are frequent or disabling, or if there are relative contraindications to the drugs that would be needed to treat acute attacks, then hypouricemic therapy should be strongly considered to reduce the burden of urate in the body, resorb tophi, and ultimately reduce the frequency of gout flares.20
Although intermittent therapy for attacks or prolonged prophylactic use of colchicine may prevent recurrent episodes of gouty arthritis and may be reasonable for many patients, this approach does not prevent continued urate deposition, with the potential development of bony erosions, tophaceous deposits, and chronic arthritis.
The definitive therapy for gouty arthritis is to deplete the periarticular deposits of urate by maintaining a low serum urate level. Urate-lowering therapy, when indicated, is almost always lifelong.
Four strategies for lowering serum urate
The serum urate concentration can be lowered in four ways:
- Increasing renal uric acid excretion
- Altering the diet
- Decreasing urate synthesis
- Converting urate to a more soluble metabolite.
Increasing uric acid excretion is rarely effective if renal function is impaired
Probenecid, sulfinpyrazone (Anturane), and losartan (Cozaar) modestly increase uric acid secretion and reduce serum urate levels, but they are rarely effective if the creatinine clearance rate is less than 60 mL/minute, and they require significant fluid intake for maximal efficacy.
Uricosuric drugs probably should be avoided in patients who excrete more than 1,000 mg of uric acid per day on a normal diet, since urinary uric acid stones may form. In practice, however, patients are given losartan to treat hypertension without attention to uric acid excretion.
More-potent urocosuric drugs are being tested in clinical trials.
Altering the diet: Traditional advice confirmed
The Health Professionals Follow-up Study27,28 prospectively examined the relation between diet and gout over 12 years in 47,150 men. The study confirmed some long-standing beliefs, such as that consuming meat, seafood, beer, and liquor increases the risk. Other risk factors were consumption of sugar-sweetened soft drinks and fructose, adiposity, weight gain, hypertension, and diuretic use. On the other hand, protein, wine, and purine-rich vegetables were not associated with gout flares. Low-fat dairy products may have a protective effect. Weight loss was found to be protective.
Low-purine diets are not very palatable, are difficult to adhere to, and are at best only minimally effective, lowering serum urate by 1 to 2 mg/dL. Low-protein diets designed to slow progression of CKD will likely also have only a slight effect on serum urate. Dietary change alone is not likely to dramatically lower serum urate levels.
Metabolizing urate with exogenous uricase
Rasburicase (Elitek) effectively converts urate to allantoin, which is more soluble, but rasburicase is fraught with allergic reactions and cannot be used as chronic therapy.
A pegylated intravenous uricase29 has just been approved by the US Food and Drug Administration (FDA); the retail cost is not yet known. It is dramatically effective in those patients able to use it chronically, but it has not been fully evaluated in patients with CKD.
Decreasing urate synthesis with allopurinol
Allopurinol acts by competitively inhibiting xanthine oxidase, the enzyme that converts hypoxanthine to xanthine and xanthine to uric acid. The drug, a structural analogue of hypoxanthine, is converted by xanthine oxidase to oxypurinol, which is an even more effective inhibitor of xanthine oxidase than allopurinol.
Allopurinol is metabolized in the liver and has a half-life of 1 to 3 hours, but oxypurinol, which is excreted in the urine, has a half-life of 12 to 17 hours. Because of these pharmacokinetic properties, allopurinol can usually be given once daily, and the dosage required to reduce serum urate levels should in theory be lower in patients with lower glomerular filtration rates.
Allopurinol (100- and 300-mg tablets) is approved by the FDA in doses of up to 800 mg/day to treat hyperuricemia in patients with gout,30 while guidelines from the British Society of Rheumatology advocate a maximum dose of 900 mg/day.31 These maximum doses are based on the limited amount of data with higher doses, not on documented toxicity.
Practice survey data in the United States indicate that most physicians prescribe no greater than 300 mg daily, although this dosage is likely to reduce the serum urate to less than 6 mg/dL—the goal level—in fewer than 50% of patients.20,32 Patients with normal renal function occasionally require more than 1,000 mg daily to reduce the serum urate level to less than 6 mg/dL.
How low should the serum urate level be?
Ideally, therapy should keep the serum urate level significantly below 6.7 mg/dL, the approximate saturation point of urate in physiologic fluids.
Lowering the serum urate level from 10 mg/dL to 7 mg/dL may seem encouraging, and the urate level may be in the laboratory “normal” range; however, urate may continue to precipitate in tissues if the concentration is greater than 6.7 mg/dL. A target of 6 mg/dL, used in clinical studies, is far enough below the saturation level to provide some margin for fluctuations in serum levels. A serum level of 6.0 mg/dL has thus been arbitrarily proposed as a reasonable therapeutic target.
The lower the serum urate level achieved during hypouricemic therapy, the faster the reduction in tophaceous deposits. With adequate urate lowering, tophi can be visibly reduced in less than a year of hypouricemic therapy.33,34
We have as yet no convincing evidence that lowering the serum urate level to less than 6.0 mg/dL is harmful, despite theoretical concerns that urate is a beneficial circulating antioxidant and epidemiologic observations that urate levels have been inversely correlated with progression of Parkinson disease.
Start low, go slow to avoid a flare
Rapid reduction of the serum urate level in a patient with chronic hyperuricemia and gout is likely to induce an acute flare.20 We have traditionally used a “start low and increase slowly” approach to escalating hypouricemic therapy in hopes of reducing the likelihood of causing a gout flare.
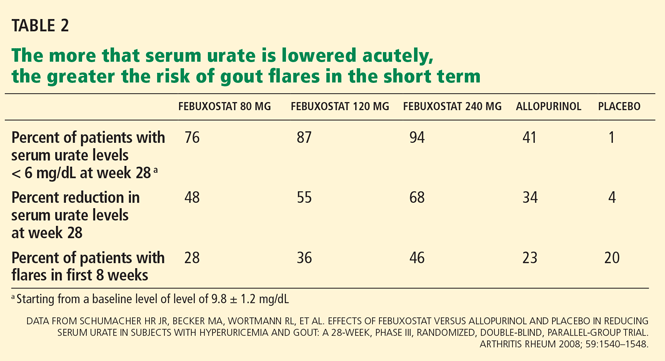
Without anti-inflammatory prophylaxis, acute flares associated with urate-lowering are extremely likely. In a 28-week trial of allopurinol, febuxostat, and placebo by Schumacher et al,33 during the first 8 weeks, when prophylaxis against gout flare was provided with either colchicine 0.6 mg once daily or naproxen (Naprosyn) 250 mg twice daily, the proportion of patients requiring treatment of gout flares was still 23% to 46%. When prophylaxis was stopped, the flare rate increased further.33
IS IT NECESSARY TO ADJUST THE ALLOPURINOL DOSE IN CHRONIC KIDNEY DISEASE?
In 1984, Hande et al35 proposed that allopurinol doses be lower in patients with renal insufficiency, with a dosage scale based on creatinine clearance.
Their thoughtful proposal was based on data from six of their own patients and 72 others with severe allopurinol toxicity, mainly allopurinol hypersensitivity syndrome, reported in the literature.
Perez-Ruiz et al36 noted that patients who had experienced adverse effects from allopurinol in their series were likely to have had received “higher” doses of allopurinol, if the dosage was corrected for reduced oxypurinol elimination based on their estimated creatinine clearance.
However, most of these reactions occurred soon after initiating therapy, a temporal pattern more typical of non-dose-dependent allergic reactions. Additionally, allopurinol hypersensitivity has been linked to T-cell-mediated immune reactions to oxypurinol,37 a mechanism not likely linked to drug levels.
Arguments against dose adjustment
Despite the compelling information that allopurinol reactions are more common in CKD, adjusting the dosage of allopurinol has not been clearly shown to reduce the frequency of these reactions.
In a small retrospective analysis, Vázquez-Mellado et al38 reported that adjusting the allopurinol dosage according to creatinine clearance did not decrease the incidence of allopurinol hypersensitivity.
In a study in 250 patients, Dalbeth et al39 showed that the overall incidence of hypersensitivity reaction was 1.6%, and the incidence of allergic reactions did not decrease when allopurinol was given according to the dosing guidelines proposed by Hande et al.35 However, it is worth noting that, of the patients who received the recommended lower doses, only 19% achieved the target serum urate level of 6 mg/dL.39
Silverberg et al40 found that of 15 patients who developed hypersensitivity reactions to allopurinol, 10 had received doses that were low or appropriate according to the guidelines of Hande et al.35
More recently, Stamp et al41 found that gradually increasing the allopurinol dose above the proposed creatinine clearance-based dose was safe and effective. Thirty-one (89%) of the 35 patients who completed the study achieved the target serum urate level of 6 mg/dL, while only 3 of 45 who started the study developed rashes, which were not serious.
The small number of patients in these studies limits any strong conclusion, but at present there is no interventional study showing that allopurinol dosing adjustment based on glomerular filtration rate is effective or safer than dosing based on the serum urate level.
Our view on allopurinol dosing adjustment
We believe the initial observations of Hande et al35 and the subsequent meticulous data from Perez-Ruiz et al36 suggest a relationship between CKD and the occurrence of severe allopurinol reactions. However, these observations do not prove that dose adjustment will prevent these reactions.
In patients with normal kidney function, the FDA30 and the European League Against Rheumatism (EULAR)42 recommend slow upward titration, starting with 100 to 200 mg/day, which we agree should decrease the frequency of acute gout flares. The dose is increased by increments of 100 mg/day at intervals of 1 week (FDA recommendation) or 2 to 4 weeks (EULAR recommendation) until the serum urate level is lower than 6 mg/dL.
We believe the optimal approach to allopurinol dosing in patients with CKD remains uncertain. We generally escalate the dose slowly, with ongoing frequent laboratory and clinical monitoring, and we do not limit the maximal dose as suggested by Hande et al.35
An alternative strategy is to use the newer, far more expensive xanthine oxidase inhibitor febuxostat in patients with CKD, since it is not excreted by the kidney. We usually first try escalating doses of allopurinol.
FEBUXOSTAT, AN ALTERNATIVE TO ALLOPURINOL
Febuxostat is an oral nonpurine inhibitor of xanthine oxidase.43 Approved by the FDA in 2009, it is available in 40- and 80-mg tablets.
Unlike allopurinol, febuxostat is metabolized primarily by hepatic glucuronide formation and oxidation and then excreted in stool and urine,44 making it in theory an attractive agent in patients with renal insufficiency, bypassing the controversial dose-adjustment issue with allopurinol.
In the Febuxostat Versus Allopurinol Controlled Trial (FACT),20 a 52-week randomized, double-blind study in hyperuricemic patients with gout, serum urate levels were reduced to less than 6.0 mg/dL in over 50% of patients receiving febuxostat 80 mg or 120 mg once daily, while only 21% of patients receiving 300 mg of allopurinol achieved this goal. This does not imply that allopurinol at higher doses, as should be used in clinical practice,45 would not be equally effective. Patients with CKD were not included in this trial.
In the study by Schumacher et al,33 febuxostat 80, 120, or 240 mg once daily reduced serum urate. A small subset (35 patients) had mild to moderate renal insufficiency (serum creatinine 1.5–2 mg/dL).33 The number of patients with renal insufficiency who achieved the primary end point of a serum urate level lower than 6 mg/dL was 4 (44%) of 9 in the febuxostat 80-mg group, 5 (46%) of 11 in the 120-mg group, and 3 (60%) of 5 in the 240-mg group, while none of the 10 patients in the dose-adjusted allopurinol group achieved the primary end point (P < .05). Of note, 41% of the patients with normal renal function who received allopurinol achieved the primary end point.33 As proposed above, if the allopurinol dose had been slowly increased in the patients with renal insufficiency, it might have been equally effective.
Febuxostat has not been thoroughly evaluated in patients with severe CKD or in patients on hemodialysis.
A presumed niche indication of febuxostat is in patients allergic to allopurinol, since the drugs are not similar in chemical structure. However, at present, experience with this use is limited. Allopurinol-allergic patients were excluded from the clinical trials; thus, if there is any allergic overlap, it would not likely have been recognized in those studies. The FDA has received reports of patients who were allergic to allopurinol also having reactions to febuxostat, and it is currently evaluating these reports (personal communication).
Concern was raised over cardiovascular adverse events in patients treated with febuxostat during clinical trials. In the FACT trial, two patients died of cardiac causes.20 In the study by Schumacher et al,33 11 of 670 patients experienced cardiac adverse events in the febuxostat group vs 3 of 268 in the allopurinol group. Events included atrial fibrillation, chest pain, coronary artery disease, and myocardial infarction. However, this difference was not statistically significant.
Febuxostat costs much more than allopurinol. Currently, patients pay $153.88 for 1 month of febuxostat 40 or 80 mg from Cleveland Clinic pharmacy; 1 month of allopurinol costs $17.45 (300 mg) or $14.00 (100 mg). We believe febuxostat should be reserved for patients with documented intolerance to allopurinol in effective doses.
Monitoring serum urate levels is important in all patients on hypouricemic therapy so that dosage adjustments can be made until the target serum urate concentration is reached. In patients failing to meet target serum urate levels, patient adherence with the prescribed dosing should be specifically addressed because as many as 50% of patients do not adhere to their prescribed regimen.
DOES URATE-LOWERING THERAPY HAVE BENEFITS BEYOND GOUT?
Despite experimental animal data and a strong epidemiologic association between hyperuri-cemia and hypertension,46 metabolic syndrome, and rates of cardiovascular and all-cause mortality,47 the evidence from interventional trials so far does not support the routine use of hypo-uricemic therapy to prevent these outcomes.
Similarly, hyperuricemia has long been associated with renal disease, and there has been debate as to whether hyperuricemia is a result of kidney dysfunction or a contributing factor.46,48–51 A few studies have documented improvement of renal function after initiation of hypouricemic therapy.52 However, treating asymptomatic hyperuricemia to preserve kidney function remains controversial.
A recent study indicates that lowering the serum urate level with allopurinol can lower the blood pressure in hyperuricemic adolescents who have newly diagnosed primary hypertension.53 This does not indicate, however, that initiating hypouricemic therapy in patients with preexisting, long-standing hypertension will be successful.
RECOMMENDED FOR OUR PATIENT
As for our diabetic patient with an acute gout flare and creatinine clearance rate of 45 mL/minute, we would recommend:
- Aspirating the knee, sending the fluid for bacterial culture, and then treating it with a local glucocorticoid injection
- Starting colchicine 0.6 mg every day, with frequent monitoring for signs of toxicity (muscle pain, weakness, leukopenia, and elevations of creatine kinase and aspartate aminotransferase)
- Increasing his allopurinol dose by 100 mg every 2 to 4 weeks until the target serum urate level of less than 6.0 mg/dL is reached
- If he cannot tolerate allopurinol or if the target serum urate level is not achieved despite adequate doses of allopurinol (about 800 mg), we would switch to febuxostat 40 mg and increase the dose as needed to achieve the desired urate level.
- Vázquez-Mellado J, García CG, Vázquez SG, et al. Metabolic syndrome and ischemic heart disease in gout. J Clin Rheumatol 2004; 10:105–109.
- Bonnel RA, Villalba ML, Karwoski CB, Beitz J. Deaths associated with inappropriate intravenous colchicine administration. J Emerg Med 2002; 22:385–387.
- Achtert G, Scherrmann JM, Christen MO. Pharmacokinetics/bioavailability of colchicine in healthy male volunteers. Eur J Drug Metab Pharmacokinet 1989; 14:317–322.
- Ben-Chetrit E, Scherrmann JM, Zylber-Katz E, Levy M. Colchicine disposition in patients with familial Mediterranean fever with renal impairment. J Rheumatol 1994; 21:710–713.
- Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum 1991; 21:143–155.
- Aronoff G, Brater DC, Schrier R, Bennett WM. Use of drugs in patients with renal insufficiency. Workshop report. Blood Purif 1994; 12:14–19.
- Kuncl RW, Duncan G, Watson D, Alderson K, Rogawski MA, Peper M. Colchicine myopathy and neuropathy. N Engl J Med 1987; 316:1562–1568.
- Ahern MJ, Reid C, Gordon TP, McCredie M, Brooks PM, Jones M. Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med 1987; 17:301–304.
- Terkeltaub R, Furst D, Bennett K, Kook K, Crockett RS, Davis WM. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum 2010, 62:1060–1068.
- Whelton A. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: physiologic foundations and clinical implications. Am J Med 1999; 106:13S–24S.
- Wali RK, Henrich WL. Recent developments in toxic nephropathy. Curr Opin Nephrol Hypertens 2002; 11:155–163.
- Fernández C, Noguera R, González JA, Pascual E. Treatment of acute attacks of gout with a small dose of intraarticular triamcinolone acetonide. J Rheumatol 1999; 26:2285–2286.
- Clive DM. Renal transplant-associated hyperuricemia and gout. J Am Soc Nephrol 2000; 11:974–979.
- Groff GD, Franck WA, Raddatz DA. Systemic steroid therapy for acute gout: a clinical trial and review of the literature. Semin Arthritis Rheum 1990; 19:329–336.
- Ritter J, Kerr LD, Valeriano-Marcet J, Spiera H. ACTH revisited: effective treatment for acute crystal induced synovitis in patients with multiple medical problems. J Rheumatol 1994; 21:696–699.
- Getting SJ, Christian HC, Flower RJ, Perretti M. Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis Rheum 2002; 46:2765–2775.
- Connell JM, Whitworth JA, Davies DL, Lever AF, Richards AM, Fraser R. Effects of ACTH and cortisol administration on blood pressure, electrolyte metabolism, atrial natriuretic peptide and renal function in normal man. J Hypertens 1987; 5:425–433.
- Tausche AK, Richter K, Grässler A, Hänsel S, Roch B, Schröder HE. Severe gouty arthritis refractory to anti-inflammatory drugs: treatment with anti-tumour necrosis factor alpha as a new therapeutic option. Ann Rheum Dis 2004; 63:1351–1352.
- So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther 2007; 9:R28.
- Becker MA, Schumacher HR, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med 2005; 353:2450–2461.
- Borstad GC, Bryant LR, Abel MP, Scroggie DA, Harris MD, Alloway JA. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. J Rheumatol 2004; 31:2429–2432.
- Wallace SL, Singer JZ, Duncan GJ, Wigley FM, Kuncl RW. Renal function predicts colchicine toxicity: guidelines for the prophylactic use of colchicine in gout. J Rheumatol 1991; 18:264–269.
- Wilbur K, Makowsky M. Colchicine myotoxicity: case reports and literature review. Pharmacotherapy 2004; 24:1784–1792.
- Hung IF, Wu AK, Cheng VC, et al. Fatal interaction between clarithromycin and colchicine in patients with renal insufficiency: a retrospective study. Clin Infect Dis 2005; 41:291–300.
- Alayli G, Cengiz K, Cantürk F, Durmus D, Akyol Y, Menekse EB. Acute myopathy in a patient with concomitant use of pravastatin and colchicine. Ann Pharmacother 2005; 39:1358–1361.
- Ducloux D, Schuller V, Bresson-Vautrin C, Chalopin JM. Colchicine myopathy in renal transplant recipients on cyclosporin. Nephrol Dial Transplant 1997; 12:2389–2392.
- Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med 2004; 350:1093–1103.
- Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ 2008; 336:309–312.
- Sundy JS, Becker MA, Baraf HS, et al; Pegloticase Phase 2 Study Investigators. Reduction of plasma urate levels following treatment with multiple doses of pegloticase (polyethylene glycol-conjugated uricase) in patients with treatment-failure gout: results of a phase II randomized study. Arthritis Rheum 2008; 58:2882–2891.
- US National Library of Medicine. About DailyMed. FDA information: allopurinol tablet. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?id=5047. Accessed August 27, 2010.
- Jordan KM, Cameron JS, Snaith M, et al; British Society for Rheumatology and British Health Professionals in Rheumatology Standards, Guidelines and Audit Working Group (SGAWG). British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology (Oxford) 2007; 46:1372–1374.
- Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, Herrero-Beites A, García-Erauskin G, Ruiz-Lucea E. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis 1998; 57:545–549.
- Schumacher HR, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum 2008; 59:1540–1548.
- Perez-Ruiz F, Calabozo M, Pijoan JI, Herrero-Beites AM, Ruibal A. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum 2002; 47:356–360.
- Hande KR, Noone RM, Stone WJ. Severe allopurinol toxicity. Description and guidelines for prevention in patients with renal insufficiency. Am J Med 1984; 76:47–56.
- Perez-Ruiz F, Hernando I, Villar I, Nolla JM. Correction of allopurinol dosing should be based on clearance of creatinine, but not plasma creatinine levels: another insight to allopurinol-related toxicity. J Clin Rheumatol 2005; 11:129–133.
- Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A 2005; 102:4134–4139.
- Vázquez-Mellado J, Morales EM, Pacheco-Tena C, Burgos-Vargas R. Relation between adverse events associated with allopurinol and renal function in patients with gout. Ann Rheum Dis 2001; 60:981–983.
- Dalbeth N, Kumar S, Stamp L, Gow P. Dose adjustment of allopurinol according to creatinine clearance does not provide adequate control of hyperuricemia in patients with gout. J Rheumatol 2006; 33:1646–1650.
- Silverberg MS, Mallela R, Lesse AJ, Bonner MR, Baer AN, Li C. Allopurinol hypersensitivity reactions: a case-control study of the role of renal dosing (abstract). Arthritis Rheum 2009; 60(suppl 10):1106.
- Stamp LK, O'Donnell JL, Zhang M, et al. Using allopurinol above the dose based on creatinine clearance is effective and safe in chronic gout, including in those with renal impairment. Arthritis Rhem 2010; doi:10.1002/art.30119. E-pub ahead of print. Accessed 10/29/2010.
- Zhang W, Doherty M, Bardin T, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis 2006; 65:1312–1324.
- Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem 2003; 278:1848–1855.
- Khosravan R, Grabowski BA, Wu JT, Joseph-Ridge N, Vernillet L. Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin Pharmacokinet 2006; 45:821–841.
- Reinders MK, Haagsma C, Jansen TL, et al. A randomised controlled trial on the efficacy and tolerability with dose escalation of allopurinol 300–600 mg/day versus benzbromarone 100–200 mg/day in patients with gout. Ann Rheum Dis 2009; 68:892–897.
- Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001; 38:1101–1106.
- Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med 2008; 359:1811–1821.
- Beck LH. Requiem for gouty nephropathy. Kidney Int 1986; 30:280–287.
- Tomita M, Mizuno S, Yamanaka H, et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol 2000; 10:403–409.
- Kang DH, Nakagawa T. Uric acid and chronic renal disease: possible implication of hyperuricemia on progression of renal disease. Semin Nephrol 2005; 25:43–49.
- Iseki K, Oshiro S, Tozawa M, Iseki C, Ikemiya Y, Takishita S. Significance of hyperuricemia on the early detection of renal failure in a cohort of screened subjects. Hypertens Res 2001; 24:691–697.
- Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med 1987; 82:421–426.
- Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis 2006; 47:51–59.
- Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA 2008; 300:924–932.
You have a 54-year-old black patient with gout, diabetes mellitus, hypertension, and chronic kidney disease (CKD). He has an acute gout flare involving his right knee. In the past year he has had four attacks of gout in the ankles and knees, which you treated with intra-articular glucocorticoid injections. He has been on allopurinol (Zyloprim) 200 mg daily, but his last serum urate level was 9.4 mg/dL (reference range 3.0–8.0). His creatinine clearance is 45 mL/minute (reference range 85–125).
In view of his kidney disease, you are concerned about increasing his dose of allopurinol, but also about the need to treat his frequent attacks. How should you manage this patient?
GOUT IS CHALLENGING TO TREAT IN PATIENTS WITH KIDNEY DISEASE
A major challenge in treating patients with gout is to avoid therapeutic interactions with common comorbidities, including hypertension, insulin resistance, coronary artery disease, heart failure, and especially CKD.1
In this paper, we discuss approaches to and controversies in the management of gout and hyperuricemia in patients with CKD. Unfortunately, the evidence from clinical trials to guide treatment decisions is limited; therefore, decisions must often be based on experience and pathophysiologic principles.
GENERAL GOALS OF GOUT THERAPY
Depending on the patient and the stage of the disease, the goals in treating patients with gout are to:
- Terminate acute attacks as promptly and safely as possible
- Prevent recurrences of acute gout attacks
- Prevent or reverse complications resulting from deposition of monosodium urate in the joints, in the kidneys, or at other sites.
These goals are more difficult to achieve in patients with CKD because of the potential complications from many of the available drugs.
TERMINATING ACUTE GOUT FLARES
In patients with acute gout, treatment is aimed at quickly resolving pain and inflammation.
Several types of drugs can terminate acute gout flares. The choice in most situations is colchicine (Colcrys); a nonsteroidal anti-inflammatory drug (NSAID); a corticosteroid; or corticotropin (ACTH).
However, in patients with CKD, there are concerns about using colchicine or NSAIDs, and corticotropin is very expensive; thus, corticosteroids are often used.
Colchicine’s clearance is reduced in CKD
Colchicine is somewhat effective in treating acute gout attacks and probably more effective in preventing attacks.
Due to concerns about inappropriate dosing and reported deaths,2 the intravenous formulation is not available in many countries, including the United States.
After oral administration, colchicine is rapidly absorbed, with a bioavailability of up to 50%. It undergoes metabolism by the liver, and its metabolites are excreted by renal and biliary-intestinal routes. Up to 20% of the active drug is excreted by the kidneys.3
Colchicine’s clearance is significantly reduced in patients with renal or hepatic insufficiency, and the drug may accumulate in cells, with resultant toxicity.4 Colchicine-induced toxicity has been observed when the drug was used for acute treatment, as well as for chronic prophylaxis of gout in patients with CKD; thus, alternative agents for treating acute attacks should be considered.5,6 With prolonged use, reversible colchicine-induced axonal neuropathy, neutropenia, and vacuolar myopathy can develop in patients with CKD.7
In a trial in patients with normal renal function, nearly 100% who received an initial dose of 1 mg followed by 0.5 mg every 2 hours developed diarrhea at a median time of 24 hours.8 Emesis may also occur.
A lower dose of 1.8 mg (two 0.6-mg pills followed by one pill an hour later) was well tolerated but only moderately effective in treating acute gout, causing at least a 50% reduction in pain at 24 hours in only 38% of patients.9 This study does not clarify the dosage to use to completely resolve attacks. Using additional colchicine likely will increase the response rate, but will also increase side effects. Patients with CKD were not included.
Some patients, as shown in the above trial, can abort attacks by taking only one or two colchicine tablets when they feel the first “twinge” of an attack. This approach is likely to be safe in CKD, but it may be of value to only a few patients.
Nonsteroidal anti-inflammatory drugs can worsen chronic kidney disease
NSAIDs in high doses can effectively treat the pain and inflammation of acute gout. Indomethacin (Indocin) 50 mg three times daily has been standard NSAID therapy.
Other nonselective NSAIDs and NSAIDs that selectively inhibit cyclooxygenase 2 (COX-2) are effective, but all can cause acute renal toxicity or worsen CKD.10 Renal side effects include salt and water retention, acute tubular necrosis, acute interstitial nephritis, proteinuria, hypertension, hyperkalemia, and chronic renal injury.11
Even short-term use of high-dose NSAIDs should generally be avoided in patients with preexisting CKD, for whom there is no established safe threshold dose. When NSAIDs (including selective COX-2 inhibitors) are used, renal function should be monitored closely and the duration limited as much as possible.
Corticosteroids are often used to treat acute attacks
Due to the concerns about NSAIDs or colchicine to treat acute gout attacks in patients with CKD, corticosteroids are often used in this setting.
Intra-articular steroid injections are useful in treating acute gout limited to a single joint or bursa.12 However, one should first make sure that the joint is not infected: septic arthritis should ideally be excluded by arthrocentesis, particularly in immunosuppressed patients13 or those with end-stage renal disease, who are predisposed to bacteremia.
Oral, intramuscular, or intravenous steroids can provide complete relief from acute gout, although high doses (eg, prednisone 30–60 mg/day or the equivalent) are often needed. Common errors resulting in inefficacy include using too low a dose or not treating for a sufficient time before tapering or stopping. Groff and colleagues14 described 13 patients who received oral or intravenous steroids for acute gout. Nine patients received an initial single dose of prednisone ranging from 20 to 50 mg, with tapering over a mean of 10 days. Twelve of the 13 patients had improvement within 48 hours, and the signs and symptoms of acute gout resolved completely within 7 to 10 days.
We often give prednisone 40 mg daily until a day after the acute attack resolves and then taper over another 7 to 10 days. There are no data to guide steroid dosing in an evidence-based way, but we believe too short a course of therapy may result in return of symptoms.
Corticotropin and other agents: Effective but costly
Corticotropin is available for subcutaneous or intramuscular injection. A single intramuscular injection of corticotropin gel (H.P. Acthar, 25–80 IU) may terminate an acute gout attack.15 However, many patients need another injection after 24 to 72 hours, which would require another visit to the physician. This treatment has been touted by some as being more effective than corticosteroid therapy, possibly because of a unique peripheral mechanism of action in addition to stimulating cortisol release.16
We rarely use corticotropin, in view of its cost as well as concerns about excessive sodium and water retention due to the release of multiple hormones from the adrenal gland. This may be especially deleterious in patients with CKD or congestive heart failure.17
Parenteral anti-tumor necrosis factor agents or interleukin 1 antagonists can be dramatically effective but are also expensive.18,19 For example, anakinra (Kineret) 100 mg costs about $73, and multiple daily doses may be necessary.
Under unique conditions in which they can be safely used (eg, patients with CKD, diabetes mellitus, liver disease), they may be cost-effective if they can shorten the stay of a hospitalized patient with acute gout.
PROPHYLACTIC ANTI-INFLAMMATORY THERAPY FOR PATIENTS WITH GOUT
Between attacks, the goal is to prevent new attacks through prophylactic management, which may include anti-inflammatory and hypouricemic therapy along with dietary instruction (such as avoiding excessive beer, liquor, and fructose ingestion).
Colchicine can be used as prophylaxis, with caution and monitoring
Although colchicine is not 100% effective, it markedly reduces the flare rate when started in low doses at the time hypouricemic therapy is initiated.20,21 (Hypouricemic therapy is discussed below.) We generally try to continue this prophylactic therapy, if the patient tolerates it, for at least 6 months—longer if tophi are still present or if attacks continue to occur.
If renal function is intact, colchicine can be prescribed at a dosage of 0.6 mg orally once or twice daily.21 In CKD, since the clearance of colchicine is reduced,4 the dosage should be reduced. Patients on colchicine for prophylaxis must be carefully monitored if the glomerular filtration rate is less than 50 mL/minute, or colchicine should be avoided altogether.6 Laboratory testing for colchicine levels is not routinely available and may be of limited value in predicting adverse effects; thus, recommendations about dose adjustments in CKD are empiric.
Wallace et al22 recommended a dose of 0.6 mg once daily if the creatinine clearance is 35 to 49 mL/minute and 0.6 mg every 2 to 3 days if it is 10 to 34 mL/minute, but there are no published long-term safety or efficacy data validating these reasonable (based on available information) dosing regimens.
Even with dose adjustment, caution is needed. Low-dose daily colchicine may be associated with reversible neuromyopathy and bone marrow suppression.7,23 Patients with neuromyopathy may complain of myalgias, proximal muscle weakness, and numbness and may have areflexia and decreased sensation. Laboratory findings include elevated creatine kinase and aminotransferase levels. We regularly check for leukopenia or elevated creatine kinase and aspartate aminotransferase levels in patients with CKD who are receiving colchicine in any dose.
Prolonged colchicine therapy should probably be avoided in patients on hemodialysis, as this drug is not removed by dialysis or by exchange transfusion, and the risk of toxicity under these circumstances may be high.22 When there is no viable alternative and the drug is given, patients should be closely monitored for signs of toxicity.
Concurrent (even short-term) treatment with most macrolide antibiotics, particularly clarithromycin (Biaxin), most statin drugs, ketoconazole (Nizoral), cyclosporine, and likely other drugs predisposes to colchicine toxicity by altering its distribution and elimination, and can in rare cases cause morbidity or death.24–26
NSAIDs are not optimal as prophylaxis in patients with chronic kidney disease
Little information has been published about using NSAIDs chronically to prevent flares, but they are not the optimal drugs to use in patients with CKD, as discussed above. In patients with end-stage renal disease, there are also concerns about NSAID-induced gastric and intestinal bleeding.
Low-dose steroids may not be effective as prophylaxis
Lower doses of steroids may not be effective as prophylaxis against gout flares, consistent with the common observation that gout flares still occur in organ transplant recipients who are taking maintenance doses of prednisone.13
PREVENTING FLARES BY LOWERING SERUM URATE LEVELS
If tophi are present, if radiography shows evidence of damage, if attacks are frequent or disabling, or if there are relative contraindications to the drugs that would be needed to treat acute attacks, then hypouricemic therapy should be strongly considered to reduce the burden of urate in the body, resorb tophi, and ultimately reduce the frequency of gout flares.20
Although intermittent therapy for attacks or prolonged prophylactic use of colchicine may prevent recurrent episodes of gouty arthritis and may be reasonable for many patients, this approach does not prevent continued urate deposition, with the potential development of bony erosions, tophaceous deposits, and chronic arthritis.
The definitive therapy for gouty arthritis is to deplete the periarticular deposits of urate by maintaining a low serum urate level. Urate-lowering therapy, when indicated, is almost always lifelong.
Four strategies for lowering serum urate
The serum urate concentration can be lowered in four ways:
- Increasing renal uric acid excretion
- Altering the diet
- Decreasing urate synthesis
- Converting urate to a more soluble metabolite.
Increasing uric acid excretion is rarely effective if renal function is impaired
Probenecid, sulfinpyrazone (Anturane), and losartan (Cozaar) modestly increase uric acid secretion and reduce serum urate levels, but they are rarely effective if the creatinine clearance rate is less than 60 mL/minute, and they require significant fluid intake for maximal efficacy.
Uricosuric drugs probably should be avoided in patients who excrete more than 1,000 mg of uric acid per day on a normal diet, since urinary uric acid stones may form. In practice, however, patients are given losartan to treat hypertension without attention to uric acid excretion.
More-potent urocosuric drugs are being tested in clinical trials.
Altering the diet: Traditional advice confirmed
The Health Professionals Follow-up Study27,28 prospectively examined the relation between diet and gout over 12 years in 47,150 men. The study confirmed some long-standing beliefs, such as that consuming meat, seafood, beer, and liquor increases the risk. Other risk factors were consumption of sugar-sweetened soft drinks and fructose, adiposity, weight gain, hypertension, and diuretic use. On the other hand, protein, wine, and purine-rich vegetables were not associated with gout flares. Low-fat dairy products may have a protective effect. Weight loss was found to be protective.
Low-purine diets are not very palatable, are difficult to adhere to, and are at best only minimally effective, lowering serum urate by 1 to 2 mg/dL. Low-protein diets designed to slow progression of CKD will likely also have only a slight effect on serum urate. Dietary change alone is not likely to dramatically lower serum urate levels.
Metabolizing urate with exogenous uricase
Rasburicase (Elitek) effectively converts urate to allantoin, which is more soluble, but rasburicase is fraught with allergic reactions and cannot be used as chronic therapy.
A pegylated intravenous uricase29 has just been approved by the US Food and Drug Administration (FDA); the retail cost is not yet known. It is dramatically effective in those patients able to use it chronically, but it has not been fully evaluated in patients with CKD.
Decreasing urate synthesis with allopurinol
Allopurinol acts by competitively inhibiting xanthine oxidase, the enzyme that converts hypoxanthine to xanthine and xanthine to uric acid. The drug, a structural analogue of hypoxanthine, is converted by xanthine oxidase to oxypurinol, which is an even more effective inhibitor of xanthine oxidase than allopurinol.
Allopurinol is metabolized in the liver and has a half-life of 1 to 3 hours, but oxypurinol, which is excreted in the urine, has a half-life of 12 to 17 hours. Because of these pharmacokinetic properties, allopurinol can usually be given once daily, and the dosage required to reduce serum urate levels should in theory be lower in patients with lower glomerular filtration rates.
Allopurinol (100- and 300-mg tablets) is approved by the FDA in doses of up to 800 mg/day to treat hyperuricemia in patients with gout,30 while guidelines from the British Society of Rheumatology advocate a maximum dose of 900 mg/day.31 These maximum doses are based on the limited amount of data with higher doses, not on documented toxicity.
Practice survey data in the United States indicate that most physicians prescribe no greater than 300 mg daily, although this dosage is likely to reduce the serum urate to less than 6 mg/dL—the goal level—in fewer than 50% of patients.20,32 Patients with normal renal function occasionally require more than 1,000 mg daily to reduce the serum urate level to less than 6 mg/dL.
How low should the serum urate level be?
Ideally, therapy should keep the serum urate level significantly below 6.7 mg/dL, the approximate saturation point of urate in physiologic fluids.
Lowering the serum urate level from 10 mg/dL to 7 mg/dL may seem encouraging, and the urate level may be in the laboratory “normal” range; however, urate may continue to precipitate in tissues if the concentration is greater than 6.7 mg/dL. A target of 6 mg/dL, used in clinical studies, is far enough below the saturation level to provide some margin for fluctuations in serum levels. A serum level of 6.0 mg/dL has thus been arbitrarily proposed as a reasonable therapeutic target.
The lower the serum urate level achieved during hypouricemic therapy, the faster the reduction in tophaceous deposits. With adequate urate lowering, tophi can be visibly reduced in less than a year of hypouricemic therapy.33,34
We have as yet no convincing evidence that lowering the serum urate level to less than 6.0 mg/dL is harmful, despite theoretical concerns that urate is a beneficial circulating antioxidant and epidemiologic observations that urate levels have been inversely correlated with progression of Parkinson disease.
Start low, go slow to avoid a flare
Rapid reduction of the serum urate level in a patient with chronic hyperuricemia and gout is likely to induce an acute flare.20 We have traditionally used a “start low and increase slowly” approach to escalating hypouricemic therapy in hopes of reducing the likelihood of causing a gout flare.
Without anti-inflammatory prophylaxis, acute flares associated with urate-lowering are extremely likely. In a 28-week trial of allopurinol, febuxostat, and placebo by Schumacher et al,33 during the first 8 weeks, when prophylaxis against gout flare was provided with either colchicine 0.6 mg once daily or naproxen (Naprosyn) 250 mg twice daily, the proportion of patients requiring treatment of gout flares was still 23% to 46%. When prophylaxis was stopped, the flare rate increased further.33
IS IT NECESSARY TO ADJUST THE ALLOPURINOL DOSE IN CHRONIC KIDNEY DISEASE?
In 1984, Hande et al35 proposed that allopurinol doses be lower in patients with renal insufficiency, with a dosage scale based on creatinine clearance.
Their thoughtful proposal was based on data from six of their own patients and 72 others with severe allopurinol toxicity, mainly allopurinol hypersensitivity syndrome, reported in the literature.
Perez-Ruiz et al36 noted that patients who had experienced adverse effects from allopurinol in their series were likely to have had received “higher” doses of allopurinol, if the dosage was corrected for reduced oxypurinol elimination based on their estimated creatinine clearance.
However, most of these reactions occurred soon after initiating therapy, a temporal pattern more typical of non-dose-dependent allergic reactions. Additionally, allopurinol hypersensitivity has been linked to T-cell-mediated immune reactions to oxypurinol,37 a mechanism not likely linked to drug levels.
Arguments against dose adjustment
Despite the compelling information that allopurinol reactions are more common in CKD, adjusting the dosage of allopurinol has not been clearly shown to reduce the frequency of these reactions.
In a small retrospective analysis, Vázquez-Mellado et al38 reported that adjusting the allopurinol dosage according to creatinine clearance did not decrease the incidence of allopurinol hypersensitivity.
In a study in 250 patients, Dalbeth et al39 showed that the overall incidence of hypersensitivity reaction was 1.6%, and the incidence of allergic reactions did not decrease when allopurinol was given according to the dosing guidelines proposed by Hande et al.35 However, it is worth noting that, of the patients who received the recommended lower doses, only 19% achieved the target serum urate level of 6 mg/dL.39
Silverberg et al40 found that of 15 patients who developed hypersensitivity reactions to allopurinol, 10 had received doses that were low or appropriate according to the guidelines of Hande et al.35
More recently, Stamp et al41 found that gradually increasing the allopurinol dose above the proposed creatinine clearance-based dose was safe and effective. Thirty-one (89%) of the 35 patients who completed the study achieved the target serum urate level of 6 mg/dL, while only 3 of 45 who started the study developed rashes, which were not serious.
The small number of patients in these studies limits any strong conclusion, but at present there is no interventional study showing that allopurinol dosing adjustment based on glomerular filtration rate is effective or safer than dosing based on the serum urate level.
Our view on allopurinol dosing adjustment
We believe the initial observations of Hande et al35 and the subsequent meticulous data from Perez-Ruiz et al36 suggest a relationship between CKD and the occurrence of severe allopurinol reactions. However, these observations do not prove that dose adjustment will prevent these reactions.
In patients with normal kidney function, the FDA30 and the European League Against Rheumatism (EULAR)42 recommend slow upward titration, starting with 100 to 200 mg/day, which we agree should decrease the frequency of acute gout flares. The dose is increased by increments of 100 mg/day at intervals of 1 week (FDA recommendation) or 2 to 4 weeks (EULAR recommendation) until the serum urate level is lower than 6 mg/dL.
We believe the optimal approach to allopurinol dosing in patients with CKD remains uncertain. We generally escalate the dose slowly, with ongoing frequent laboratory and clinical monitoring, and we do not limit the maximal dose as suggested by Hande et al.35
An alternative strategy is to use the newer, far more expensive xanthine oxidase inhibitor febuxostat in patients with CKD, since it is not excreted by the kidney. We usually first try escalating doses of allopurinol.
FEBUXOSTAT, AN ALTERNATIVE TO ALLOPURINOL
Febuxostat is an oral nonpurine inhibitor of xanthine oxidase.43 Approved by the FDA in 2009, it is available in 40- and 80-mg tablets.
Unlike allopurinol, febuxostat is metabolized primarily by hepatic glucuronide formation and oxidation and then excreted in stool and urine,44 making it in theory an attractive agent in patients with renal insufficiency, bypassing the controversial dose-adjustment issue with allopurinol.
In the Febuxostat Versus Allopurinol Controlled Trial (FACT),20 a 52-week randomized, double-blind study in hyperuricemic patients with gout, serum urate levels were reduced to less than 6.0 mg/dL in over 50% of patients receiving febuxostat 80 mg or 120 mg once daily, while only 21% of patients receiving 300 mg of allopurinol achieved this goal. This does not imply that allopurinol at higher doses, as should be used in clinical practice,45 would not be equally effective. Patients with CKD were not included in this trial.
In the study by Schumacher et al,33 febuxostat 80, 120, or 240 mg once daily reduced serum urate. A small subset (35 patients) had mild to moderate renal insufficiency (serum creatinine 1.5–2 mg/dL).33 The number of patients with renal insufficiency who achieved the primary end point of a serum urate level lower than 6 mg/dL was 4 (44%) of 9 in the febuxostat 80-mg group, 5 (46%) of 11 in the 120-mg group, and 3 (60%) of 5 in the 240-mg group, while none of the 10 patients in the dose-adjusted allopurinol group achieved the primary end point (P < .05). Of note, 41% of the patients with normal renal function who received allopurinol achieved the primary end point.33 As proposed above, if the allopurinol dose had been slowly increased in the patients with renal insufficiency, it might have been equally effective.
Febuxostat has not been thoroughly evaluated in patients with severe CKD or in patients on hemodialysis.
A presumed niche indication of febuxostat is in patients allergic to allopurinol, since the drugs are not similar in chemical structure. However, at present, experience with this use is limited. Allopurinol-allergic patients were excluded from the clinical trials; thus, if there is any allergic overlap, it would not likely have been recognized in those studies. The FDA has received reports of patients who were allergic to allopurinol also having reactions to febuxostat, and it is currently evaluating these reports (personal communication).
Concern was raised over cardiovascular adverse events in patients treated with febuxostat during clinical trials. In the FACT trial, two patients died of cardiac causes.20 In the study by Schumacher et al,33 11 of 670 patients experienced cardiac adverse events in the febuxostat group vs 3 of 268 in the allopurinol group. Events included atrial fibrillation, chest pain, coronary artery disease, and myocardial infarction. However, this difference was not statistically significant.
Febuxostat costs much more than allopurinol. Currently, patients pay $153.88 for 1 month of febuxostat 40 or 80 mg from Cleveland Clinic pharmacy; 1 month of allopurinol costs $17.45 (300 mg) or $14.00 (100 mg). We believe febuxostat should be reserved for patients with documented intolerance to allopurinol in effective doses.
Monitoring serum urate levels is important in all patients on hypouricemic therapy so that dosage adjustments can be made until the target serum urate concentration is reached. In patients failing to meet target serum urate levels, patient adherence with the prescribed dosing should be specifically addressed because as many as 50% of patients do not adhere to their prescribed regimen.
DOES URATE-LOWERING THERAPY HAVE BENEFITS BEYOND GOUT?
Despite experimental animal data and a strong epidemiologic association between hyperuri-cemia and hypertension,46 metabolic syndrome, and rates of cardiovascular and all-cause mortality,47 the evidence from interventional trials so far does not support the routine use of hypo-uricemic therapy to prevent these outcomes.
Similarly, hyperuricemia has long been associated with renal disease, and there has been debate as to whether hyperuricemia is a result of kidney dysfunction or a contributing factor.46,48–51 A few studies have documented improvement of renal function after initiation of hypouricemic therapy.52 However, treating asymptomatic hyperuricemia to preserve kidney function remains controversial.
A recent study indicates that lowering the serum urate level with allopurinol can lower the blood pressure in hyperuricemic adolescents who have newly diagnosed primary hypertension.53 This does not indicate, however, that initiating hypouricemic therapy in patients with preexisting, long-standing hypertension will be successful.
RECOMMENDED FOR OUR PATIENT
As for our diabetic patient with an acute gout flare and creatinine clearance rate of 45 mL/minute, we would recommend:
- Aspirating the knee, sending the fluid for bacterial culture, and then treating it with a local glucocorticoid injection
- Starting colchicine 0.6 mg every day, with frequent monitoring for signs of toxicity (muscle pain, weakness, leukopenia, and elevations of creatine kinase and aspartate aminotransferase)
- Increasing his allopurinol dose by 100 mg every 2 to 4 weeks until the target serum urate level of less than 6.0 mg/dL is reached
- If he cannot tolerate allopurinol or if the target serum urate level is not achieved despite adequate doses of allopurinol (about 800 mg), we would switch to febuxostat 40 mg and increase the dose as needed to achieve the desired urate level.
You have a 54-year-old black patient with gout, diabetes mellitus, hypertension, and chronic kidney disease (CKD). He has an acute gout flare involving his right knee. In the past year he has had four attacks of gout in the ankles and knees, which you treated with intra-articular glucocorticoid injections. He has been on allopurinol (Zyloprim) 200 mg daily, but his last serum urate level was 9.4 mg/dL (reference range 3.0–8.0). His creatinine clearance is 45 mL/minute (reference range 85–125).
In view of his kidney disease, you are concerned about increasing his dose of allopurinol, but also about the need to treat his frequent attacks. How should you manage this patient?
GOUT IS CHALLENGING TO TREAT IN PATIENTS WITH KIDNEY DISEASE
A major challenge in treating patients with gout is to avoid therapeutic interactions with common comorbidities, including hypertension, insulin resistance, coronary artery disease, heart failure, and especially CKD.1
In this paper, we discuss approaches to and controversies in the management of gout and hyperuricemia in patients with CKD. Unfortunately, the evidence from clinical trials to guide treatment decisions is limited; therefore, decisions must often be based on experience and pathophysiologic principles.
GENERAL GOALS OF GOUT THERAPY
Depending on the patient and the stage of the disease, the goals in treating patients with gout are to:
- Terminate acute attacks as promptly and safely as possible
- Prevent recurrences of acute gout attacks
- Prevent or reverse complications resulting from deposition of monosodium urate in the joints, in the kidneys, or at other sites.
These goals are more difficult to achieve in patients with CKD because of the potential complications from many of the available drugs.
TERMINATING ACUTE GOUT FLARES
In patients with acute gout, treatment is aimed at quickly resolving pain and inflammation.
Several types of drugs can terminate acute gout flares. The choice in most situations is colchicine (Colcrys); a nonsteroidal anti-inflammatory drug (NSAID); a corticosteroid; or corticotropin (ACTH).
However, in patients with CKD, there are concerns about using colchicine or NSAIDs, and corticotropin is very expensive; thus, corticosteroids are often used.
Colchicine’s clearance is reduced in CKD
Colchicine is somewhat effective in treating acute gout attacks and probably more effective in preventing attacks.
Due to concerns about inappropriate dosing and reported deaths,2 the intravenous formulation is not available in many countries, including the United States.
After oral administration, colchicine is rapidly absorbed, with a bioavailability of up to 50%. It undergoes metabolism by the liver, and its metabolites are excreted by renal and biliary-intestinal routes. Up to 20% of the active drug is excreted by the kidneys.3
Colchicine’s clearance is significantly reduced in patients with renal or hepatic insufficiency, and the drug may accumulate in cells, with resultant toxicity.4 Colchicine-induced toxicity has been observed when the drug was used for acute treatment, as well as for chronic prophylaxis of gout in patients with CKD; thus, alternative agents for treating acute attacks should be considered.5,6 With prolonged use, reversible colchicine-induced axonal neuropathy, neutropenia, and vacuolar myopathy can develop in patients with CKD.7
In a trial in patients with normal renal function, nearly 100% who received an initial dose of 1 mg followed by 0.5 mg every 2 hours developed diarrhea at a median time of 24 hours.8 Emesis may also occur.
A lower dose of 1.8 mg (two 0.6-mg pills followed by one pill an hour later) was well tolerated but only moderately effective in treating acute gout, causing at least a 50% reduction in pain at 24 hours in only 38% of patients.9 This study does not clarify the dosage to use to completely resolve attacks. Using additional colchicine likely will increase the response rate, but will also increase side effects. Patients with CKD were not included.
Some patients, as shown in the above trial, can abort attacks by taking only one or two colchicine tablets when they feel the first “twinge” of an attack. This approach is likely to be safe in CKD, but it may be of value to only a few patients.
Nonsteroidal anti-inflammatory drugs can worsen chronic kidney disease
NSAIDs in high doses can effectively treat the pain and inflammation of acute gout. Indomethacin (Indocin) 50 mg three times daily has been standard NSAID therapy.
Other nonselective NSAIDs and NSAIDs that selectively inhibit cyclooxygenase 2 (COX-2) are effective, but all can cause acute renal toxicity or worsen CKD.10 Renal side effects include salt and water retention, acute tubular necrosis, acute interstitial nephritis, proteinuria, hypertension, hyperkalemia, and chronic renal injury.11
Even short-term use of high-dose NSAIDs should generally be avoided in patients with preexisting CKD, for whom there is no established safe threshold dose. When NSAIDs (including selective COX-2 inhibitors) are used, renal function should be monitored closely and the duration limited as much as possible.
Corticosteroids are often used to treat acute attacks
Due to the concerns about NSAIDs or colchicine to treat acute gout attacks in patients with CKD, corticosteroids are often used in this setting.
Intra-articular steroid injections are useful in treating acute gout limited to a single joint or bursa.12 However, one should first make sure that the joint is not infected: septic arthritis should ideally be excluded by arthrocentesis, particularly in immunosuppressed patients13 or those with end-stage renal disease, who are predisposed to bacteremia.
Oral, intramuscular, or intravenous steroids can provide complete relief from acute gout, although high doses (eg, prednisone 30–60 mg/day or the equivalent) are often needed. Common errors resulting in inefficacy include using too low a dose or not treating for a sufficient time before tapering or stopping. Groff and colleagues14 described 13 patients who received oral or intravenous steroids for acute gout. Nine patients received an initial single dose of prednisone ranging from 20 to 50 mg, with tapering over a mean of 10 days. Twelve of the 13 patients had improvement within 48 hours, and the signs and symptoms of acute gout resolved completely within 7 to 10 days.
We often give prednisone 40 mg daily until a day after the acute attack resolves and then taper over another 7 to 10 days. There are no data to guide steroid dosing in an evidence-based way, but we believe too short a course of therapy may result in return of symptoms.
Corticotropin and other agents: Effective but costly
Corticotropin is available for subcutaneous or intramuscular injection. A single intramuscular injection of corticotropin gel (H.P. Acthar, 25–80 IU) may terminate an acute gout attack.15 However, many patients need another injection after 24 to 72 hours, which would require another visit to the physician. This treatment has been touted by some as being more effective than corticosteroid therapy, possibly because of a unique peripheral mechanism of action in addition to stimulating cortisol release.16
We rarely use corticotropin, in view of its cost as well as concerns about excessive sodium and water retention due to the release of multiple hormones from the adrenal gland. This may be especially deleterious in patients with CKD or congestive heart failure.17
Parenteral anti-tumor necrosis factor agents or interleukin 1 antagonists can be dramatically effective but are also expensive.18,19 For example, anakinra (Kineret) 100 mg costs about $73, and multiple daily doses may be necessary.
Under unique conditions in which they can be safely used (eg, patients with CKD, diabetes mellitus, liver disease), they may be cost-effective if they can shorten the stay of a hospitalized patient with acute gout.
PROPHYLACTIC ANTI-INFLAMMATORY THERAPY FOR PATIENTS WITH GOUT
Between attacks, the goal is to prevent new attacks through prophylactic management, which may include anti-inflammatory and hypouricemic therapy along with dietary instruction (such as avoiding excessive beer, liquor, and fructose ingestion).
Colchicine can be used as prophylaxis, with caution and monitoring
Although colchicine is not 100% effective, it markedly reduces the flare rate when started in low doses at the time hypouricemic therapy is initiated.20,21 (Hypouricemic therapy is discussed below.) We generally try to continue this prophylactic therapy, if the patient tolerates it, for at least 6 months—longer if tophi are still present or if attacks continue to occur.
If renal function is intact, colchicine can be prescribed at a dosage of 0.6 mg orally once or twice daily.21 In CKD, since the clearance of colchicine is reduced,4 the dosage should be reduced. Patients on colchicine for prophylaxis must be carefully monitored if the glomerular filtration rate is less than 50 mL/minute, or colchicine should be avoided altogether.6 Laboratory testing for colchicine levels is not routinely available and may be of limited value in predicting adverse effects; thus, recommendations about dose adjustments in CKD are empiric.
Wallace et al22 recommended a dose of 0.6 mg once daily if the creatinine clearance is 35 to 49 mL/minute and 0.6 mg every 2 to 3 days if it is 10 to 34 mL/minute, but there are no published long-term safety or efficacy data validating these reasonable (based on available information) dosing regimens.
Even with dose adjustment, caution is needed. Low-dose daily colchicine may be associated with reversible neuromyopathy and bone marrow suppression.7,23 Patients with neuromyopathy may complain of myalgias, proximal muscle weakness, and numbness and may have areflexia and decreased sensation. Laboratory findings include elevated creatine kinase and aminotransferase levels. We regularly check for leukopenia or elevated creatine kinase and aspartate aminotransferase levels in patients with CKD who are receiving colchicine in any dose.
Prolonged colchicine therapy should probably be avoided in patients on hemodialysis, as this drug is not removed by dialysis or by exchange transfusion, and the risk of toxicity under these circumstances may be high.22 When there is no viable alternative and the drug is given, patients should be closely monitored for signs of toxicity.
Concurrent (even short-term) treatment with most macrolide antibiotics, particularly clarithromycin (Biaxin), most statin drugs, ketoconazole (Nizoral), cyclosporine, and likely other drugs predisposes to colchicine toxicity by altering its distribution and elimination, and can in rare cases cause morbidity or death.24–26
NSAIDs are not optimal as prophylaxis in patients with chronic kidney disease
Little information has been published about using NSAIDs chronically to prevent flares, but they are not the optimal drugs to use in patients with CKD, as discussed above. In patients with end-stage renal disease, there are also concerns about NSAID-induced gastric and intestinal bleeding.
Low-dose steroids may not be effective as prophylaxis
Lower doses of steroids may not be effective as prophylaxis against gout flares, consistent with the common observation that gout flares still occur in organ transplant recipients who are taking maintenance doses of prednisone.13
PREVENTING FLARES BY LOWERING SERUM URATE LEVELS
If tophi are present, if radiography shows evidence of damage, if attacks are frequent or disabling, or if there are relative contraindications to the drugs that would be needed to treat acute attacks, then hypouricemic therapy should be strongly considered to reduce the burden of urate in the body, resorb tophi, and ultimately reduce the frequency of gout flares.20
Although intermittent therapy for attacks or prolonged prophylactic use of colchicine may prevent recurrent episodes of gouty arthritis and may be reasonable for many patients, this approach does not prevent continued urate deposition, with the potential development of bony erosions, tophaceous deposits, and chronic arthritis.
The definitive therapy for gouty arthritis is to deplete the periarticular deposits of urate by maintaining a low serum urate level. Urate-lowering therapy, when indicated, is almost always lifelong.
Four strategies for lowering serum urate
The serum urate concentration can be lowered in four ways:
- Increasing renal uric acid excretion
- Altering the diet
- Decreasing urate synthesis
- Converting urate to a more soluble metabolite.
Increasing uric acid excretion is rarely effective if renal function is impaired
Probenecid, sulfinpyrazone (Anturane), and losartan (Cozaar) modestly increase uric acid secretion and reduce serum urate levels, but they are rarely effective if the creatinine clearance rate is less than 60 mL/minute, and they require significant fluid intake for maximal efficacy.
Uricosuric drugs probably should be avoided in patients who excrete more than 1,000 mg of uric acid per day on a normal diet, since urinary uric acid stones may form. In practice, however, patients are given losartan to treat hypertension without attention to uric acid excretion.
More-potent urocosuric drugs are being tested in clinical trials.
Altering the diet: Traditional advice confirmed
The Health Professionals Follow-up Study27,28 prospectively examined the relation between diet and gout over 12 years in 47,150 men. The study confirmed some long-standing beliefs, such as that consuming meat, seafood, beer, and liquor increases the risk. Other risk factors were consumption of sugar-sweetened soft drinks and fructose, adiposity, weight gain, hypertension, and diuretic use. On the other hand, protein, wine, and purine-rich vegetables were not associated with gout flares. Low-fat dairy products may have a protective effect. Weight loss was found to be protective.
Low-purine diets are not very palatable, are difficult to adhere to, and are at best only minimally effective, lowering serum urate by 1 to 2 mg/dL. Low-protein diets designed to slow progression of CKD will likely also have only a slight effect on serum urate. Dietary change alone is not likely to dramatically lower serum urate levels.
Metabolizing urate with exogenous uricase
Rasburicase (Elitek) effectively converts urate to allantoin, which is more soluble, but rasburicase is fraught with allergic reactions and cannot be used as chronic therapy.
A pegylated intravenous uricase29 has just been approved by the US Food and Drug Administration (FDA); the retail cost is not yet known. It is dramatically effective in those patients able to use it chronically, but it has not been fully evaluated in patients with CKD.
Decreasing urate synthesis with allopurinol
Allopurinol acts by competitively inhibiting xanthine oxidase, the enzyme that converts hypoxanthine to xanthine and xanthine to uric acid. The drug, a structural analogue of hypoxanthine, is converted by xanthine oxidase to oxypurinol, which is an even more effective inhibitor of xanthine oxidase than allopurinol.
Allopurinol is metabolized in the liver and has a half-life of 1 to 3 hours, but oxypurinol, which is excreted in the urine, has a half-life of 12 to 17 hours. Because of these pharmacokinetic properties, allopurinol can usually be given once daily, and the dosage required to reduce serum urate levels should in theory be lower in patients with lower glomerular filtration rates.
Allopurinol (100- and 300-mg tablets) is approved by the FDA in doses of up to 800 mg/day to treat hyperuricemia in patients with gout,30 while guidelines from the British Society of Rheumatology advocate a maximum dose of 900 mg/day.31 These maximum doses are based on the limited amount of data with higher doses, not on documented toxicity.
Practice survey data in the United States indicate that most physicians prescribe no greater than 300 mg daily, although this dosage is likely to reduce the serum urate to less than 6 mg/dL—the goal level—in fewer than 50% of patients.20,32 Patients with normal renal function occasionally require more than 1,000 mg daily to reduce the serum urate level to less than 6 mg/dL.
How low should the serum urate level be?
Ideally, therapy should keep the serum urate level significantly below 6.7 mg/dL, the approximate saturation point of urate in physiologic fluids.
Lowering the serum urate level from 10 mg/dL to 7 mg/dL may seem encouraging, and the urate level may be in the laboratory “normal” range; however, urate may continue to precipitate in tissues if the concentration is greater than 6.7 mg/dL. A target of 6 mg/dL, used in clinical studies, is far enough below the saturation level to provide some margin for fluctuations in serum levels. A serum level of 6.0 mg/dL has thus been arbitrarily proposed as a reasonable therapeutic target.
The lower the serum urate level achieved during hypouricemic therapy, the faster the reduction in tophaceous deposits. With adequate urate lowering, tophi can be visibly reduced in less than a year of hypouricemic therapy.33,34
We have as yet no convincing evidence that lowering the serum urate level to less than 6.0 mg/dL is harmful, despite theoretical concerns that urate is a beneficial circulating antioxidant and epidemiologic observations that urate levels have been inversely correlated with progression of Parkinson disease.
Start low, go slow to avoid a flare
Rapid reduction of the serum urate level in a patient with chronic hyperuricemia and gout is likely to induce an acute flare.20 We have traditionally used a “start low and increase slowly” approach to escalating hypouricemic therapy in hopes of reducing the likelihood of causing a gout flare.
Without anti-inflammatory prophylaxis, acute flares associated with urate-lowering are extremely likely. In a 28-week trial of allopurinol, febuxostat, and placebo by Schumacher et al,33 during the first 8 weeks, when prophylaxis against gout flare was provided with either colchicine 0.6 mg once daily or naproxen (Naprosyn) 250 mg twice daily, the proportion of patients requiring treatment of gout flares was still 23% to 46%. When prophylaxis was stopped, the flare rate increased further.33
IS IT NECESSARY TO ADJUST THE ALLOPURINOL DOSE IN CHRONIC KIDNEY DISEASE?
In 1984, Hande et al35 proposed that allopurinol doses be lower in patients with renal insufficiency, with a dosage scale based on creatinine clearance.
Their thoughtful proposal was based on data from six of their own patients and 72 others with severe allopurinol toxicity, mainly allopurinol hypersensitivity syndrome, reported in the literature.
Perez-Ruiz et al36 noted that patients who had experienced adverse effects from allopurinol in their series were likely to have had received “higher” doses of allopurinol, if the dosage was corrected for reduced oxypurinol elimination based on their estimated creatinine clearance.
However, most of these reactions occurred soon after initiating therapy, a temporal pattern more typical of non-dose-dependent allergic reactions. Additionally, allopurinol hypersensitivity has been linked to T-cell-mediated immune reactions to oxypurinol,37 a mechanism not likely linked to drug levels.
Arguments against dose adjustment
Despite the compelling information that allopurinol reactions are more common in CKD, adjusting the dosage of allopurinol has not been clearly shown to reduce the frequency of these reactions.
In a small retrospective analysis, Vázquez-Mellado et al38 reported that adjusting the allopurinol dosage according to creatinine clearance did not decrease the incidence of allopurinol hypersensitivity.
In a study in 250 patients, Dalbeth et al39 showed that the overall incidence of hypersensitivity reaction was 1.6%, and the incidence of allergic reactions did not decrease when allopurinol was given according to the dosing guidelines proposed by Hande et al.35 However, it is worth noting that, of the patients who received the recommended lower doses, only 19% achieved the target serum urate level of 6 mg/dL.39
Silverberg et al40 found that of 15 patients who developed hypersensitivity reactions to allopurinol, 10 had received doses that were low or appropriate according to the guidelines of Hande et al.35
More recently, Stamp et al41 found that gradually increasing the allopurinol dose above the proposed creatinine clearance-based dose was safe and effective. Thirty-one (89%) of the 35 patients who completed the study achieved the target serum urate level of 6 mg/dL, while only 3 of 45 who started the study developed rashes, which were not serious.
The small number of patients in these studies limits any strong conclusion, but at present there is no interventional study showing that allopurinol dosing adjustment based on glomerular filtration rate is effective or safer than dosing based on the serum urate level.
Our view on allopurinol dosing adjustment
We believe the initial observations of Hande et al35 and the subsequent meticulous data from Perez-Ruiz et al36 suggest a relationship between CKD and the occurrence of severe allopurinol reactions. However, these observations do not prove that dose adjustment will prevent these reactions.
In patients with normal kidney function, the FDA30 and the European League Against Rheumatism (EULAR)42 recommend slow upward titration, starting with 100 to 200 mg/day, which we agree should decrease the frequency of acute gout flares. The dose is increased by increments of 100 mg/day at intervals of 1 week (FDA recommendation) or 2 to 4 weeks (EULAR recommendation) until the serum urate level is lower than 6 mg/dL.
We believe the optimal approach to allopurinol dosing in patients with CKD remains uncertain. We generally escalate the dose slowly, with ongoing frequent laboratory and clinical monitoring, and we do not limit the maximal dose as suggested by Hande et al.35
An alternative strategy is to use the newer, far more expensive xanthine oxidase inhibitor febuxostat in patients with CKD, since it is not excreted by the kidney. We usually first try escalating doses of allopurinol.
FEBUXOSTAT, AN ALTERNATIVE TO ALLOPURINOL
Febuxostat is an oral nonpurine inhibitor of xanthine oxidase.43 Approved by the FDA in 2009, it is available in 40- and 80-mg tablets.
Unlike allopurinol, febuxostat is metabolized primarily by hepatic glucuronide formation and oxidation and then excreted in stool and urine,44 making it in theory an attractive agent in patients with renal insufficiency, bypassing the controversial dose-adjustment issue with allopurinol.
In the Febuxostat Versus Allopurinol Controlled Trial (FACT),20 a 52-week randomized, double-blind study in hyperuricemic patients with gout, serum urate levels were reduced to less than 6.0 mg/dL in over 50% of patients receiving febuxostat 80 mg or 120 mg once daily, while only 21% of patients receiving 300 mg of allopurinol achieved this goal. This does not imply that allopurinol at higher doses, as should be used in clinical practice,45 would not be equally effective. Patients with CKD were not included in this trial.
In the study by Schumacher et al,33 febuxostat 80, 120, or 240 mg once daily reduced serum urate. A small subset (35 patients) had mild to moderate renal insufficiency (serum creatinine 1.5–2 mg/dL).33 The number of patients with renal insufficiency who achieved the primary end point of a serum urate level lower than 6 mg/dL was 4 (44%) of 9 in the febuxostat 80-mg group, 5 (46%) of 11 in the 120-mg group, and 3 (60%) of 5 in the 240-mg group, while none of the 10 patients in the dose-adjusted allopurinol group achieved the primary end point (P < .05). Of note, 41% of the patients with normal renal function who received allopurinol achieved the primary end point.33 As proposed above, if the allopurinol dose had been slowly increased in the patients with renal insufficiency, it might have been equally effective.
Febuxostat has not been thoroughly evaluated in patients with severe CKD or in patients on hemodialysis.
A presumed niche indication of febuxostat is in patients allergic to allopurinol, since the drugs are not similar in chemical structure. However, at present, experience with this use is limited. Allopurinol-allergic patients were excluded from the clinical trials; thus, if there is any allergic overlap, it would not likely have been recognized in those studies. The FDA has received reports of patients who were allergic to allopurinol also having reactions to febuxostat, and it is currently evaluating these reports (personal communication).
Concern was raised over cardiovascular adverse events in patients treated with febuxostat during clinical trials. In the FACT trial, two patients died of cardiac causes.20 In the study by Schumacher et al,33 11 of 670 patients experienced cardiac adverse events in the febuxostat group vs 3 of 268 in the allopurinol group. Events included atrial fibrillation, chest pain, coronary artery disease, and myocardial infarction. However, this difference was not statistically significant.
Febuxostat costs much more than allopurinol. Currently, patients pay $153.88 for 1 month of febuxostat 40 or 80 mg from Cleveland Clinic pharmacy; 1 month of allopurinol costs $17.45 (300 mg) or $14.00 (100 mg). We believe febuxostat should be reserved for patients with documented intolerance to allopurinol in effective doses.
Monitoring serum urate levels is important in all patients on hypouricemic therapy so that dosage adjustments can be made until the target serum urate concentration is reached. In patients failing to meet target serum urate levels, patient adherence with the prescribed dosing should be specifically addressed because as many as 50% of patients do not adhere to their prescribed regimen.
DOES URATE-LOWERING THERAPY HAVE BENEFITS BEYOND GOUT?
Despite experimental animal data and a strong epidemiologic association between hyperuri-cemia and hypertension,46 metabolic syndrome, and rates of cardiovascular and all-cause mortality,47 the evidence from interventional trials so far does not support the routine use of hypo-uricemic therapy to prevent these outcomes.
Similarly, hyperuricemia has long been associated with renal disease, and there has been debate as to whether hyperuricemia is a result of kidney dysfunction or a contributing factor.46,48–51 A few studies have documented improvement of renal function after initiation of hypouricemic therapy.52 However, treating asymptomatic hyperuricemia to preserve kidney function remains controversial.
A recent study indicates that lowering the serum urate level with allopurinol can lower the blood pressure in hyperuricemic adolescents who have newly diagnosed primary hypertension.53 This does not indicate, however, that initiating hypouricemic therapy in patients with preexisting, long-standing hypertension will be successful.
RECOMMENDED FOR OUR PATIENT
As for our diabetic patient with an acute gout flare and creatinine clearance rate of 45 mL/minute, we would recommend:
- Aspirating the knee, sending the fluid for bacterial culture, and then treating it with a local glucocorticoid injection
- Starting colchicine 0.6 mg every day, with frequent monitoring for signs of toxicity (muscle pain, weakness, leukopenia, and elevations of creatine kinase and aspartate aminotransferase)
- Increasing his allopurinol dose by 100 mg every 2 to 4 weeks until the target serum urate level of less than 6.0 mg/dL is reached
- If he cannot tolerate allopurinol or if the target serum urate level is not achieved despite adequate doses of allopurinol (about 800 mg), we would switch to febuxostat 40 mg and increase the dose as needed to achieve the desired urate level.
- Vázquez-Mellado J, García CG, Vázquez SG, et al. Metabolic syndrome and ischemic heart disease in gout. J Clin Rheumatol 2004; 10:105–109.
- Bonnel RA, Villalba ML, Karwoski CB, Beitz J. Deaths associated with inappropriate intravenous colchicine administration. J Emerg Med 2002; 22:385–387.
- Achtert G, Scherrmann JM, Christen MO. Pharmacokinetics/bioavailability of colchicine in healthy male volunteers. Eur J Drug Metab Pharmacokinet 1989; 14:317–322.
- Ben-Chetrit E, Scherrmann JM, Zylber-Katz E, Levy M. Colchicine disposition in patients with familial Mediterranean fever with renal impairment. J Rheumatol 1994; 21:710–713.
- Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum 1991; 21:143–155.
- Aronoff G, Brater DC, Schrier R, Bennett WM. Use of drugs in patients with renal insufficiency. Workshop report. Blood Purif 1994; 12:14–19.
- Kuncl RW, Duncan G, Watson D, Alderson K, Rogawski MA, Peper M. Colchicine myopathy and neuropathy. N Engl J Med 1987; 316:1562–1568.
- Ahern MJ, Reid C, Gordon TP, McCredie M, Brooks PM, Jones M. Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med 1987; 17:301–304.
- Terkeltaub R, Furst D, Bennett K, Kook K, Crockett RS, Davis WM. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum 2010, 62:1060–1068.
- Whelton A. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: physiologic foundations and clinical implications. Am J Med 1999; 106:13S–24S.
- Wali RK, Henrich WL. Recent developments in toxic nephropathy. Curr Opin Nephrol Hypertens 2002; 11:155–163.
- Fernández C, Noguera R, González JA, Pascual E. Treatment of acute attacks of gout with a small dose of intraarticular triamcinolone acetonide. J Rheumatol 1999; 26:2285–2286.
- Clive DM. Renal transplant-associated hyperuricemia and gout. J Am Soc Nephrol 2000; 11:974–979.
- Groff GD, Franck WA, Raddatz DA. Systemic steroid therapy for acute gout: a clinical trial and review of the literature. Semin Arthritis Rheum 1990; 19:329–336.
- Ritter J, Kerr LD, Valeriano-Marcet J, Spiera H. ACTH revisited: effective treatment for acute crystal induced synovitis in patients with multiple medical problems. J Rheumatol 1994; 21:696–699.
- Getting SJ, Christian HC, Flower RJ, Perretti M. Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis Rheum 2002; 46:2765–2775.
- Connell JM, Whitworth JA, Davies DL, Lever AF, Richards AM, Fraser R. Effects of ACTH and cortisol administration on blood pressure, electrolyte metabolism, atrial natriuretic peptide and renal function in normal man. J Hypertens 1987; 5:425–433.
- Tausche AK, Richter K, Grässler A, Hänsel S, Roch B, Schröder HE. Severe gouty arthritis refractory to anti-inflammatory drugs: treatment with anti-tumour necrosis factor alpha as a new therapeutic option. Ann Rheum Dis 2004; 63:1351–1352.
- So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther 2007; 9:R28.
- Becker MA, Schumacher HR, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med 2005; 353:2450–2461.
- Borstad GC, Bryant LR, Abel MP, Scroggie DA, Harris MD, Alloway JA. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. J Rheumatol 2004; 31:2429–2432.
- Wallace SL, Singer JZ, Duncan GJ, Wigley FM, Kuncl RW. Renal function predicts colchicine toxicity: guidelines for the prophylactic use of colchicine in gout. J Rheumatol 1991; 18:264–269.
- Wilbur K, Makowsky M. Colchicine myotoxicity: case reports and literature review. Pharmacotherapy 2004; 24:1784–1792.
- Hung IF, Wu AK, Cheng VC, et al. Fatal interaction between clarithromycin and colchicine in patients with renal insufficiency: a retrospective study. Clin Infect Dis 2005; 41:291–300.
- Alayli G, Cengiz K, Cantürk F, Durmus D, Akyol Y, Menekse EB. Acute myopathy in a patient with concomitant use of pravastatin and colchicine. Ann Pharmacother 2005; 39:1358–1361.
- Ducloux D, Schuller V, Bresson-Vautrin C, Chalopin JM. Colchicine myopathy in renal transplant recipients on cyclosporin. Nephrol Dial Transplant 1997; 12:2389–2392.
- Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med 2004; 350:1093–1103.
- Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ 2008; 336:309–312.
- Sundy JS, Becker MA, Baraf HS, et al; Pegloticase Phase 2 Study Investigators. Reduction of plasma urate levels following treatment with multiple doses of pegloticase (polyethylene glycol-conjugated uricase) in patients with treatment-failure gout: results of a phase II randomized study. Arthritis Rheum 2008; 58:2882–2891.
- US National Library of Medicine. About DailyMed. FDA information: allopurinol tablet. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?id=5047. Accessed August 27, 2010.
- Jordan KM, Cameron JS, Snaith M, et al; British Society for Rheumatology and British Health Professionals in Rheumatology Standards, Guidelines and Audit Working Group (SGAWG). British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology (Oxford) 2007; 46:1372–1374.
- Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, Herrero-Beites A, García-Erauskin G, Ruiz-Lucea E. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis 1998; 57:545–549.
- Schumacher HR, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum 2008; 59:1540–1548.
- Perez-Ruiz F, Calabozo M, Pijoan JI, Herrero-Beites AM, Ruibal A. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum 2002; 47:356–360.
- Hande KR, Noone RM, Stone WJ. Severe allopurinol toxicity. Description and guidelines for prevention in patients with renal insufficiency. Am J Med 1984; 76:47–56.
- Perez-Ruiz F, Hernando I, Villar I, Nolla JM. Correction of allopurinol dosing should be based on clearance of creatinine, but not plasma creatinine levels: another insight to allopurinol-related toxicity. J Clin Rheumatol 2005; 11:129–133.
- Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A 2005; 102:4134–4139.
- Vázquez-Mellado J, Morales EM, Pacheco-Tena C, Burgos-Vargas R. Relation between adverse events associated with allopurinol and renal function in patients with gout. Ann Rheum Dis 2001; 60:981–983.
- Dalbeth N, Kumar S, Stamp L, Gow P. Dose adjustment of allopurinol according to creatinine clearance does not provide adequate control of hyperuricemia in patients with gout. J Rheumatol 2006; 33:1646–1650.
- Silverberg MS, Mallela R, Lesse AJ, Bonner MR, Baer AN, Li C. Allopurinol hypersensitivity reactions: a case-control study of the role of renal dosing (abstract). Arthritis Rheum 2009; 60(suppl 10):1106.
- Stamp LK, O'Donnell JL, Zhang M, et al. Using allopurinol above the dose based on creatinine clearance is effective and safe in chronic gout, including in those with renal impairment. Arthritis Rhem 2010; doi:10.1002/art.30119. E-pub ahead of print. Accessed 10/29/2010.
- Zhang W, Doherty M, Bardin T, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis 2006; 65:1312–1324.
- Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem 2003; 278:1848–1855.
- Khosravan R, Grabowski BA, Wu JT, Joseph-Ridge N, Vernillet L. Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin Pharmacokinet 2006; 45:821–841.
- Reinders MK, Haagsma C, Jansen TL, et al. A randomised controlled trial on the efficacy and tolerability with dose escalation of allopurinol 300–600 mg/day versus benzbromarone 100–200 mg/day in patients with gout. Ann Rheum Dis 2009; 68:892–897.
- Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001; 38:1101–1106.
- Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med 2008; 359:1811–1821.
- Beck LH. Requiem for gouty nephropathy. Kidney Int 1986; 30:280–287.
- Tomita M, Mizuno S, Yamanaka H, et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol 2000; 10:403–409.
- Kang DH, Nakagawa T. Uric acid and chronic renal disease: possible implication of hyperuricemia on progression of renal disease. Semin Nephrol 2005; 25:43–49.
- Iseki K, Oshiro S, Tozawa M, Iseki C, Ikemiya Y, Takishita S. Significance of hyperuricemia on the early detection of renal failure in a cohort of screened subjects. Hypertens Res 2001; 24:691–697.
- Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med 1987; 82:421–426.
- Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis 2006; 47:51–59.
- Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA 2008; 300:924–932.
- Vázquez-Mellado J, García CG, Vázquez SG, et al. Metabolic syndrome and ischemic heart disease in gout. J Clin Rheumatol 2004; 10:105–109.
- Bonnel RA, Villalba ML, Karwoski CB, Beitz J. Deaths associated with inappropriate intravenous colchicine administration. J Emerg Med 2002; 22:385–387.
- Achtert G, Scherrmann JM, Christen MO. Pharmacokinetics/bioavailability of colchicine in healthy male volunteers. Eur J Drug Metab Pharmacokinet 1989; 14:317–322.
- Ben-Chetrit E, Scherrmann JM, Zylber-Katz E, Levy M. Colchicine disposition in patients with familial Mediterranean fever with renal impairment. J Rheumatol 1994; 21:710–713.
- Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum 1991; 21:143–155.
- Aronoff G, Brater DC, Schrier R, Bennett WM. Use of drugs in patients with renal insufficiency. Workshop report. Blood Purif 1994; 12:14–19.
- Kuncl RW, Duncan G, Watson D, Alderson K, Rogawski MA, Peper M. Colchicine myopathy and neuropathy. N Engl J Med 1987; 316:1562–1568.
- Ahern MJ, Reid C, Gordon TP, McCredie M, Brooks PM, Jones M. Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med 1987; 17:301–304.
- Terkeltaub R, Furst D, Bennett K, Kook K, Crockett RS, Davis WM. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum 2010, 62:1060–1068.
- Whelton A. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: physiologic foundations and clinical implications. Am J Med 1999; 106:13S–24S.
- Wali RK, Henrich WL. Recent developments in toxic nephropathy. Curr Opin Nephrol Hypertens 2002; 11:155–163.
- Fernández C, Noguera R, González JA, Pascual E. Treatment of acute attacks of gout with a small dose of intraarticular triamcinolone acetonide. J Rheumatol 1999; 26:2285–2286.
- Clive DM. Renal transplant-associated hyperuricemia and gout. J Am Soc Nephrol 2000; 11:974–979.
- Groff GD, Franck WA, Raddatz DA. Systemic steroid therapy for acute gout: a clinical trial and review of the literature. Semin Arthritis Rheum 1990; 19:329–336.
- Ritter J, Kerr LD, Valeriano-Marcet J, Spiera H. ACTH revisited: effective treatment for acute crystal induced synovitis in patients with multiple medical problems. J Rheumatol 1994; 21:696–699.
- Getting SJ, Christian HC, Flower RJ, Perretti M. Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis Rheum 2002; 46:2765–2775.
- Connell JM, Whitworth JA, Davies DL, Lever AF, Richards AM, Fraser R. Effects of ACTH and cortisol administration on blood pressure, electrolyte metabolism, atrial natriuretic peptide and renal function in normal man. J Hypertens 1987; 5:425–433.
- Tausche AK, Richter K, Grässler A, Hänsel S, Roch B, Schröder HE. Severe gouty arthritis refractory to anti-inflammatory drugs: treatment with anti-tumour necrosis factor alpha as a new therapeutic option. Ann Rheum Dis 2004; 63:1351–1352.
- So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther 2007; 9:R28.
- Becker MA, Schumacher HR, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med 2005; 353:2450–2461.
- Borstad GC, Bryant LR, Abel MP, Scroggie DA, Harris MD, Alloway JA. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. J Rheumatol 2004; 31:2429–2432.
- Wallace SL, Singer JZ, Duncan GJ, Wigley FM, Kuncl RW. Renal function predicts colchicine toxicity: guidelines for the prophylactic use of colchicine in gout. J Rheumatol 1991; 18:264–269.
- Wilbur K, Makowsky M. Colchicine myotoxicity: case reports and literature review. Pharmacotherapy 2004; 24:1784–1792.
- Hung IF, Wu AK, Cheng VC, et al. Fatal interaction between clarithromycin and colchicine in patients with renal insufficiency: a retrospective study. Clin Infect Dis 2005; 41:291–300.
- Alayli G, Cengiz K, Cantürk F, Durmus D, Akyol Y, Menekse EB. Acute myopathy in a patient with concomitant use of pravastatin and colchicine. Ann Pharmacother 2005; 39:1358–1361.
- Ducloux D, Schuller V, Bresson-Vautrin C, Chalopin JM. Colchicine myopathy in renal transplant recipients on cyclosporin. Nephrol Dial Transplant 1997; 12:2389–2392.
- Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med 2004; 350:1093–1103.
- Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ 2008; 336:309–312.
- Sundy JS, Becker MA, Baraf HS, et al; Pegloticase Phase 2 Study Investigators. Reduction of plasma urate levels following treatment with multiple doses of pegloticase (polyethylene glycol-conjugated uricase) in patients with treatment-failure gout: results of a phase II randomized study. Arthritis Rheum 2008; 58:2882–2891.
- US National Library of Medicine. About DailyMed. FDA information: allopurinol tablet. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?id=5047. Accessed August 27, 2010.
- Jordan KM, Cameron JS, Snaith M, et al; British Society for Rheumatology and British Health Professionals in Rheumatology Standards, Guidelines and Audit Working Group (SGAWG). British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology (Oxford) 2007; 46:1372–1374.
- Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, Herrero-Beites A, García-Erauskin G, Ruiz-Lucea E. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis 1998; 57:545–549.
- Schumacher HR, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum 2008; 59:1540–1548.
- Perez-Ruiz F, Calabozo M, Pijoan JI, Herrero-Beites AM, Ruibal A. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum 2002; 47:356–360.
- Hande KR, Noone RM, Stone WJ. Severe allopurinol toxicity. Description and guidelines for prevention in patients with renal insufficiency. Am J Med 1984; 76:47–56.
- Perez-Ruiz F, Hernando I, Villar I, Nolla JM. Correction of allopurinol dosing should be based on clearance of creatinine, but not plasma creatinine levels: another insight to allopurinol-related toxicity. J Clin Rheumatol 2005; 11:129–133.
- Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A 2005; 102:4134–4139.
- Vázquez-Mellado J, Morales EM, Pacheco-Tena C, Burgos-Vargas R. Relation between adverse events associated with allopurinol and renal function in patients with gout. Ann Rheum Dis 2001; 60:981–983.
- Dalbeth N, Kumar S, Stamp L, Gow P. Dose adjustment of allopurinol according to creatinine clearance does not provide adequate control of hyperuricemia in patients with gout. J Rheumatol 2006; 33:1646–1650.
- Silverberg MS, Mallela R, Lesse AJ, Bonner MR, Baer AN, Li C. Allopurinol hypersensitivity reactions: a case-control study of the role of renal dosing (abstract). Arthritis Rheum 2009; 60(suppl 10):1106.
- Stamp LK, O'Donnell JL, Zhang M, et al. Using allopurinol above the dose based on creatinine clearance is effective and safe in chronic gout, including in those with renal impairment. Arthritis Rhem 2010; doi:10.1002/art.30119. E-pub ahead of print. Accessed 10/29/2010.
- Zhang W, Doherty M, Bardin T, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis 2006; 65:1312–1324.
- Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem 2003; 278:1848–1855.
- Khosravan R, Grabowski BA, Wu JT, Joseph-Ridge N, Vernillet L. Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin Pharmacokinet 2006; 45:821–841.
- Reinders MK, Haagsma C, Jansen TL, et al. A randomised controlled trial on the efficacy and tolerability with dose escalation of allopurinol 300–600 mg/day versus benzbromarone 100–200 mg/day in patients with gout. Ann Rheum Dis 2009; 68:892–897.
- Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001; 38:1101–1106.
- Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med 2008; 359:1811–1821.
- Beck LH. Requiem for gouty nephropathy. Kidney Int 1986; 30:280–287.
- Tomita M, Mizuno S, Yamanaka H, et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol 2000; 10:403–409.
- Kang DH, Nakagawa T. Uric acid and chronic renal disease: possible implication of hyperuricemia on progression of renal disease. Semin Nephrol 2005; 25:43–49.
- Iseki K, Oshiro S, Tozawa M, Iseki C, Ikemiya Y, Takishita S. Significance of hyperuricemia on the early detection of renal failure in a cohort of screened subjects. Hypertens Res 2001; 24:691–697.
- Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med 1987; 82:421–426.
- Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis 2006; 47:51–59.
- Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA 2008; 300:924–932.
KEY POINTS
- Owing to concerns about using colchicine and nonsteroidal anti-inflammatory drugs (NSAIDs) in patients with CKD, glucocorticoids (local injections or systemic therapy) are often used to treat acute attacks. Corticotropin (Acthar), anti-tumor necrosis factor agents, and interleukin 1 antagonists are effective but expensive.
- Colchicine can be used in low doses as prophylaxis, with caution and appropriate monitoring. NSAIDs should be avoided, and glucocorticoids may not be effective for this purpose.
- Whether the dosage of allopurinol should be lower in patients with CKD remains controversial. We start with a low dose and slowly increase it, with a goal serum urate level of less than 6.0 mg/dL.
- Febuxostat (Uloric), like allopurinol, is a xanthine oxidase inhibitor, but the elimination of the active drug is not by the kidney. Nevertheless, we try allopurinol in escalating doses first, due to major cost differences.