User login
Swelling and pain 2 weeks after a dog bite
A 48-year-old man with gout, multiple sclerosis, and previously treated methicillin-resistant Staphylococcus aureus (MRSA) infection presented to the emergency room with pain and significant swelling at the site of a dog bite on his left forearm. He had been bitten 2 weeks earlier by a friend’s dog, and the bite had punctured the skin. He also had red streaking on the skin of the left arm from the wrist to the elbow, and he reported feeling “feverish” and having night sweats.
At first, the bite had seemed to improve, then swelling and pain had developed and increased. He reported this to his primary care physician, along with the information that he had previously had an anaphylactic reaction to penicillin and a cephalosporin. His physician, considering a penicillin allergy, started him on ciprofloxacin (Cipro) plus clindamycin (Cleocin). The patient took this for 5 days, but without improvement. The appearance of the red streaking on his left forearm prompted his presentation to our emergency room.
ORGANISMS IN DOG BITES
1. Which is the most common cause of infected dog bite?
- Pasteurella canis
- Streptococci and S aureus
- Erysipelothrix rhusiopathiae
- Capnocytophaga canimorsus
- Eikenella corrodens
Streptococci (50%) and S aureus (20% to 40%) are the organisms most commonly responsible for infected dog bites, as they are for other skin and soft-tissue infections.1P canis is unique to dog bite infections but accounts for only 18%.2E rhusiopathiae is an unusual isolate from cat and dog bites and is more commonly isolated from the mouths of fish and aquatic mammals. C canimorsus is a normal inhabitant of the oral cavity of dogs and cats but an unusual cause of wound infection from a dog bite. It is notable for sepsis and central nervous system infections uniquely associated with veterinarians, dog owners, kennel workers, and mail carriers.3E corrodens infection is more common with human bites.4
THE EVALUATION BEGINS
On examination, the patient had marked edema of the left forearm and pain in the joints of the left hand. His temperature was 100.2°F (37.9°C). Because of the duration and severity of symptoms, the examining physician was concerned about septic arthritis of the wrist, and the patient was admitted to the hospital.
In the hospital, our patient was thermodynamically stable without documented fever or chills. There was no open wound to culture, and blood cultures were negative. Marked edema and joint involvement raised suspicion of erysipeloid. This “cousin” of erysipelas often involves the underlying joint, is associated with edema, and produces systemic manifestations of fever and arthralgia.
Radiography of the left forearm and hand demonstrated multiple foci of demineralization within the carpal bones and proximal radius, attributed to disuse. Magnetic resonance imaging (MRI) the next day showed multiple bone infarcts in the carpal bones and the distal radius, with synovitis and fluid in the carpal joints and without adjacent osteomyelitis. Fluid was also seen in the soft tissues in the ulnar aspect of the left wrist, and tenosynovitis involving the flexor carpi radialis tendon was noted.
Arthrocentesis of his left radiocarpal joint produced synovial fluid negative for crystals and negative on Gram stain; the fluid was also sent for culture. The patient’s tetanus immunization was current, and the dog was known to have been immunized against rabies.
ANTIBIOTICS FOR INFECTED DOG BITES
2. Which antibiotic regimen would you choose for this patient?
- Oral amoxicillin and clavulanate
- Meropenem
- Vancomycin, clindamycin, aztreonam
- Clindamycin and levofloxacin
- Clindamycin and trimethoprim-sulfamethoxazole
Oral amoxicillin and clavulanate (Augmentin) is a judicious choice for prophylactic treatment of deep bites in the early stages of infection. However, our patient’s wound was no longer in the early stages of infection, and he had a history of an adverse reaction to penicillin.
Meropenem (Merrem IV) cross-reacts minimally with penicillin allergy and is reported to be safe in patients with a history of anaphylactic reactions to penicillin,5 but overuse of carbapenems has led to the development of carbapenem-resistant strains of Klebsiella, Stenotrophomonas, and Acinetobacter organisms.
Given the rise of MRSA infections and the common involvement of staphylococci, streptococci, and anaerobic bacteria in complicated dog bites, the combination of vancomycin and clindamycin is a good choice, and aztreonam (Azactam) would add empiric coverage of gram-negative enteric organisms.
Levofloxacin (Levaquin) also covers gramnegative enteric organisms, but Fusobacterium canifelinum, a common anaerobe in the oral flora of dogs and cats, is intrinsically resistant to fluoroquinolones.
Clindamycin and levofloxacin would be a good step-down oral regimen. Pasteurella multocida has variable sensitivity to the commonly used agents dicloxacillin (Dynapen), cephalexin (Keflex), macrolides, and clindamycin, but it is a less likely pathogen at this late stage and could be covered with levofloxacin alone.
C canimorsus is resistant to trimethoprim-sulfamethoxazole (Bactrim) and cephalexin, but is well covered by clindamycin.6
CASE CONTINUED
Our patient was started on intravenous vancomycin, clindamycin, and aztreonam for coverage of dog-mouth flora. Blood cultures and cultures of synovial fluid of the left wrist were negative. Vancomycin was discontinued after 48 hours when blood cultures did not grow staphylococcal organisms, but clindamycin and aztreonam were continued for a total of 8 days to treat possible infection with anaerobic and gram-negative enteric pathogens.
To test for autonomic dysfunction, a plastic pen case drawn lightly across each forearm revealed a loss of tactile adherence (ie, areas where moist, sweaty skin impeded the movement of the pen case) on the affected forearm, a sign of underlying nerve injury. The affected forearm was sensitive to light touch, with pain out of proportion to the stimulus.
ARRIVING AT THE DIAGNOSIS
Based on the wide distribution of inflammation, autonomic dysfunction (shown by differences in temperature and sweating), radiographic evidence of demineralization, hyperesthesia, and lack of improvement in pain and swelling after two courses of antibiotics, the patient’s clinical course was determined to be consistent with complex regional pain syndrome type 1, previously referred to as reflex sympathetic dystrophy.
Symptoms of complex regional pain syndrome traditionally include pain, regional edema, joint stiffness, muscular atrophy, vasomotor disturbances (causing temperature variability and erythema), regional diaphoresis, and localized skeletal demineralization on radiography.
Complex regional pain syndrome type 1 occurs as regional pain and inflammation as an excessive sympathetic reaction to an often minor insult, without nerve injury. When the syndrome occurs in a patient with obvious partial nerve injury, it is categorized as type 2 (formerly known as causalgia). The two types are clinically indistinguishable and are not uncommon. About 10% of all patients with complex regional pain syndrome have obvious nerve injury (complex regional pain syndrome type 2). In a study of 109 patients with Colles fracture, 25% developed symptoms of complex regional pain syndrome.7
Complex regional pain syndrome is difficult to diagnose, as it resembles many other ailments, such as gout, infection, bone tumor, stress fracture, and arthritis. Its pathophysiology is poorly understood, but it is believed to result from a “short circuit” in the reflex arc between somatic afferent sensory fibers and autonomic sympathetic efferent fibers, and this is thought to explain the increased sympathetic stimulation.
Although the pathophysiology is likely the same in type 1 and type 2, electromyography with a nerve conduction study is a reliable way to detect nerve damage and thus distinguish between the two types of complex regional pain syndrome.8
Our understanding of this syndrome is evolving. A recent study using sensory testing showed that 33% of patients with type 1 had combinations of increased and decreased thresholds for the detection of thermal, vibratory, and mechanical stimuli in the distribution of discrete peripheral nerves, suggesting that the patients actually had type 2.9
CONFIRMING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
3. Which of the following is the best way to confirm complex regional pain syndrome type 1?
- Erythrocyte sedimentation rate, C-reactive protein, and complete blood cell count
- Plain radiography of the hand and forearm
- Three-phase technetium bone scan
- The Budapest diagnostic criteria
- MRI
- Autonomic testing

Complex regional pain syndrome type 1 is a clinical diagnosis. Diagnostic studies lack sensitivity and specificity but may confirm complex regional pain syndrome type 1 or rule out other diagnoses. The Budapest diagnostic criteria10 (Table 1) may be the best way to confirm this diagnosis. The criteria are as follows: continuing pain disproportionate to an inciting event, coupled with three of four symptoms plus at least one sign from sensory, vasomotor, sudomotor, and motor-trophic categories.
Laboratory tests are not helpful because acute-phase reactants and blood counts remain normal in these patients.
Plain radiography is not sensitive in early diagnosis, but at 2 weeks it may show patchy areas of osteopenia in adjacent bones throughout the region, as well as subsequent diffuse demineralization.
Three-phase bone scanning is more sensitive than plain radiography, with 75% of patients showing regional disparities in blood flow in early sequences and increased bone uptake in the later sequences.
MRI is a sensitive early test, as it better defines focal areas of bone loss and increased T2 bone signal in adjacent bone, as well as early soft-tissue changes. Computed tomography does not show early specific changes in muscle, tendon, or bone and so is not recommended.
THE EVALUATION CONTINUES
The admitting diagnosis was septic arthritis, and our patient underwent computed tomography, which showed focal demineralization that could have represented bone infarcts or infection, confounding the diagnosis of complex regional pain syndrome.
Autonomic nerve testing can help distinguish complex regional pain syndrome from other disorders. Complex regional pain syndrome is characterized by increased sympathetic activity and results in increased sweat output. Autonomic testing includes resting sweat output, resting skin temperature, and quantitative sudomotor axon reflex testing. In one study, an increase in resting sweat output used in conjunction with quantitative sudomotor axon reflex testing predicted the diagnosis of complex regional pain syndrome with a specificity of 98%.11 However, autonomic testing is limited to academic centers and is not readily available.
TREATING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
4. Which is the best first-line therapy for complex regional pain syndrome type 1?
- Stellate ganglion nerve block
- Occupational therapy to splint the wrist and forearm
- Oral corticosteroids
- Physical therapy to prevent loss of joint motion
- Tricyclic antidepressant drugs (eg, amitriptyline), pregabalin, and bisphosphonates
Physical therapy should be started early in all patients, with range-of-motion exercises to prevent contracture and enhance mobility.
Stellate ganglion nerve block has been used to counter severe sympathetic hyperactivity, but it also may aggravate symptoms of complex regional pain syndrome and so remains a controversial treatment.
Immobilization and splinting should be avoided, as this will augment edema, pain, and contracture of joints.
Corticosteroids do not shorten the course or assuage symptoms and may increase edema.
Amitriptyline (Elavil) and pregabalin (Lyrica) have been used successfully to counter extended courses of allodynia and hyperalgesia. Bisphosphonates may decrease bone loss and pain and may be needed should the course be complicated by myositis ossificans, a form of dystrophic bone formation in juxtaposed tendon and muscle related to neuroactivation of fibroblasts and osteoblasts.
THE COURSE OF COMPLEX REGIONAL PAIN SYNDROME
Traditionally, type 1 was divided into three stages—an early inflammatory stage, a dystrophic stage, and a late atrophic stage.12 Although there is no evidence to support a consistent three-stage evolution, the severity of symptoms may help determine the best approach to management.13
Patients initially exhibit burning or throbbing pain, diffuse aching, sensitivity to touch or cold (allodynia), localized edema, and vasomotor disturbances of variable intensity that may produce altered color and temperature. Topical capsaicin cream; a tricyclic antidepressant; an anticonvulsant such as gabapentin (Neurontin), pregabalin, or lamotrigine (Lamictal); or a nonsteroidal anti-inflammatory drug should be tried first. Some of these treatments are poorly tolerated in elderly patients. If pain persists, nasal calcitonin may be added. Trigger-point injections with an anesthetic or glucocorticoid may be tried.
The management of early complex regional pain syndrome is sometimes supplemented with systemic corticosteroids, but reviews of randomized controlled trials have failed to show efficacy.14
Later in the course, patients may suffer persistent soft-tissue edema, accompanied by thickening of the skin and periarticular soft tissues, muscle wasting, and the skin changes of brawny edema. Regional blockade of sympathetic ganglions, epidural administration of clonidine, implantable peripheral nerve stimulators, and spinal cord stimulators have all been applied by experts in pain management and may provide benefit. Progression of the syndrome may include cyanosis, mottling, increased sweating, abnormal hair growth, and diffuse swelling in nonarticular tissue.
It is always acceptable to refer to an experienced pain management specialist, and a multidisciplinary approach is recommended at the outset.12
OUR PATIENT’S CARE CONTINUED
Our patient’s forearm and wrist were placed in a sling to keep his left arm elevated when active. This helped control sympathetic vascular edema and throbbing pain. Physical therapy with range-of-motion exercises prevented contracture.
He was discharged home on limited oxycodone as needed, with close follow-up by his primary care physician to monitor his pain symptoms. The pain and swelling slowly improved over the next 2 months, but he suffered a fall, twisting his left wrist. This minor injury was followed by more intense pain and swelling of the forearm, hand, and wrist.
COMORBIDITIES
5. Which of the following statements about conditions associated with complex regional pain syndrome most likely applies to our patient?
- Gout is likely following minor trauma
- Minor trauma or surgical bone biopsy may reactivate complex regional pain syndrome
- Septic hip arthritis due to MRSA may have reemerged and seeded the wrist
- Patients with multiple sclerosis have a propensity for complex regional pain syndrome
- Complex regional pain syndrome type 1 begets type 2
Gout does follow minor injury, but our patient’s uric acid was well controlled on allopurinol (Zyloprim), and gout is unlikely to be causing polyarticular swelling of the hand, wrist, and forearm.
Minor trauma, sometimes inconsequential enough to have been completely forgotten, may either initiate complex regional pain syndrome or, as seen here, reactivate it. Bone changes seen on MRI sometimes trigger surgical bone biopsy, only to reactivate the dysesthesia and sympathetic vascular reaction. Surgery should be avoided. Trauma and surgery are causative rather than associative comorbidities.
Sepsis due to MRSA after total hip arthroplasty may be reactivated, especially in the setting of immunosuppressive treatment. But the diffuse bone changes seen in multiple carpal, radial, and ulnar bones suggest generalized vascular and sympathetic disarray, most consistent with complex regional pain syndrome type 1.
AN ASSOCIATION WITH MULTIPLE SCLEROSIS?
Multiple sclerosis and other central neuropathic conditions such as stroke are associated with complex regional pain syndrome type 1.15,16
A hypothetical cause for the higher prevalence of complex regional pain syndrome in patients with multiple sclerosis may be demyelination resulting in aberrant signaling and overreaction to distal pain receptors. Demyelination of neurons within the autonomic or spinothalamic tracts potentially increases susceptibility to development of the pain syndrome.
Our patient had an apparent stimulus for the development of the syndrome, ie, the initial dog bite, and the wrist injury later may have caused peripheral nerve injury. Such injury may lead to release of vasodilatory neuropeptides including substance P from stimulated cutaneous nerves with cell bodies in the dorsal root ganglia. Excessive vasodilation and increased vascular permeability result in the affected limb becoming edematous and causing cutaneous nerves to be further activated. Stimulated cutaneous neurons normally have an inhibitory influence on sympathetic activity at the level of entry of the dorsal root ganglia in the cord. In complex regional pain syndrome, this inhibition is lost, resulting in a hyperactive somatosympathetic reflex.17 Underlying multiple sclerosis may have contributed to the loss of inhibition by the cutaneous nerves on the sympathetic system.
CASE CONCLUDED
We continued to closely follow this patient, who was on a self-directed program of physical therapy. One year after the original dog bite, the complex regional pain syndrome had completely resolved.
- Talan DA, Citron DM, Abrahamian FM, Moran GJ, Goldstein EJ. Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group. N Engl J Med 1999; 340:85–92.
- Holst E, Rollof J, Larsson L, Nielsen JP. Characterization and distribution of Pasteurella species recovered from infected humans. J Clin Microbiol 1992; 30:2984–2987.
- Jolivet-Gougeon A, Sixou JL, Tamanai-Shacoori Z, Bonnaure-Mallet M. Antimicrobial treatment of Capnocytophaga infections. Int J Antimicrob Agents 2007; 29:367–373.
- Paul K, Patel SS. Eikenella corrodens infections in children and adolescents: case reports and review of the literature. Clin Infect Dis 2001; 33:54–61.
- Cunha BA, Hamid NS, Krol V, Eisenstein L. Safety of meropenem in patients reporting penicillin allergy: lack of allergic cross reactions. J Chemother 2008; 20:233–237.
- Verghese A, Hamati F, Berk S, Franzus B, Berk S, Smith JK. Susceptibility of dysgonic fermenter 2 to antimicrobial agents in vitro. Antimicrob Agents Chemother 1988; 32:78–80.
- Atkins RM, Duckworth T, Kanis JA. Algodystrophy following Colles’ fracture. J Hand Surg Br 1989; 14:161–164.
- Rommel O, Malin JP, Zenz M, Jänig W. Quantitative sensory testing, neurophysiological and psychological examination in patients with complex regional pain syndrome and hemisensory deficits. Pain 2001; 93:279–293.
- Sethna NF, Meier PM, Zurakowski D, Berde CB. Cutaneous sensory abnormalities in children and adolescents with complex regional pain syndromes. Pain 2007; 131:153–161.
- Harden RN, Bruehl S, Stanton-Hicks M, Wilson PR. Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med 2007; 8:326–331.
- Chelimsky TC, Low PA, Naessens JM, Wilson PR, Amadio PC, O’Brien PC. Value of autonomic testing in reflex sympathetic dystrophy. Mayo Clin Proc 1995; 70:1029–1040.
- Stanton-Hicks MD, Burton AW, Bruehl SP, et al. An updated interdisciplinary clinical pathway for CRPS: report of an expert panel. Pain Pract 2002; 2:1–16.
- Brummett CM, Cohen SP, eds. Managing pain: essentials of diagnosis and treatment. New York; Oxford University Press; 2013.
- Dirckx M, Stronks DL, Groeneweg G, Huygen FJ. Effect of immunomodulating medications in complex regional pain syndrome: a systematic review. Clin J Pain 2012; 28:355–363.
- Schwartzman RJ, Gurusinghe C, Gracely E. Prevalence of complex regional pain syndrome in a cohort of multiple sclerosis patients. Pain Physician 2008; 11:133–136.
- Sandroni P, Benrud-Larson LM, McClelland RL, Low PA. Complex regional pain syndrome type I: incidence and prevalence in Olmsted county, a population-based study. Pain 2003; 103:199–207.
- Kurvers HA, Jacobs MJ, Beuk RJ, et al. Reflex sympathetic dystrophy: evolution of microcirculatory disturbances in time. Pain 1995; 60:333–340.
A 48-year-old man with gout, multiple sclerosis, and previously treated methicillin-resistant Staphylococcus aureus (MRSA) infection presented to the emergency room with pain and significant swelling at the site of a dog bite on his left forearm. He had been bitten 2 weeks earlier by a friend’s dog, and the bite had punctured the skin. He also had red streaking on the skin of the left arm from the wrist to the elbow, and he reported feeling “feverish” and having night sweats.
At first, the bite had seemed to improve, then swelling and pain had developed and increased. He reported this to his primary care physician, along with the information that he had previously had an anaphylactic reaction to penicillin and a cephalosporin. His physician, considering a penicillin allergy, started him on ciprofloxacin (Cipro) plus clindamycin (Cleocin). The patient took this for 5 days, but without improvement. The appearance of the red streaking on his left forearm prompted his presentation to our emergency room.
ORGANISMS IN DOG BITES
1. Which is the most common cause of infected dog bite?
- Pasteurella canis
- Streptococci and S aureus
- Erysipelothrix rhusiopathiae
- Capnocytophaga canimorsus
- Eikenella corrodens
Streptococci (50%) and S aureus (20% to 40%) are the organisms most commonly responsible for infected dog bites, as they are for other skin and soft-tissue infections.1P canis is unique to dog bite infections but accounts for only 18%.2E rhusiopathiae is an unusual isolate from cat and dog bites and is more commonly isolated from the mouths of fish and aquatic mammals. C canimorsus is a normal inhabitant of the oral cavity of dogs and cats but an unusual cause of wound infection from a dog bite. It is notable for sepsis and central nervous system infections uniquely associated with veterinarians, dog owners, kennel workers, and mail carriers.3E corrodens infection is more common with human bites.4
THE EVALUATION BEGINS
On examination, the patient had marked edema of the left forearm and pain in the joints of the left hand. His temperature was 100.2°F (37.9°C). Because of the duration and severity of symptoms, the examining physician was concerned about septic arthritis of the wrist, and the patient was admitted to the hospital.
In the hospital, our patient was thermodynamically stable without documented fever or chills. There was no open wound to culture, and blood cultures were negative. Marked edema and joint involvement raised suspicion of erysipeloid. This “cousin” of erysipelas often involves the underlying joint, is associated with edema, and produces systemic manifestations of fever and arthralgia.
Radiography of the left forearm and hand demonstrated multiple foci of demineralization within the carpal bones and proximal radius, attributed to disuse. Magnetic resonance imaging (MRI) the next day showed multiple bone infarcts in the carpal bones and the distal radius, with synovitis and fluid in the carpal joints and without adjacent osteomyelitis. Fluid was also seen in the soft tissues in the ulnar aspect of the left wrist, and tenosynovitis involving the flexor carpi radialis tendon was noted.
Arthrocentesis of his left radiocarpal joint produced synovial fluid negative for crystals and negative on Gram stain; the fluid was also sent for culture. The patient’s tetanus immunization was current, and the dog was known to have been immunized against rabies.
ANTIBIOTICS FOR INFECTED DOG BITES
2. Which antibiotic regimen would you choose for this patient?
- Oral amoxicillin and clavulanate
- Meropenem
- Vancomycin, clindamycin, aztreonam
- Clindamycin and levofloxacin
- Clindamycin and trimethoprim-sulfamethoxazole
Oral amoxicillin and clavulanate (Augmentin) is a judicious choice for prophylactic treatment of deep bites in the early stages of infection. However, our patient’s wound was no longer in the early stages of infection, and he had a history of an adverse reaction to penicillin.
Meropenem (Merrem IV) cross-reacts minimally with penicillin allergy and is reported to be safe in patients with a history of anaphylactic reactions to penicillin,5 but overuse of carbapenems has led to the development of carbapenem-resistant strains of Klebsiella, Stenotrophomonas, and Acinetobacter organisms.
Given the rise of MRSA infections and the common involvement of staphylococci, streptococci, and anaerobic bacteria in complicated dog bites, the combination of vancomycin and clindamycin is a good choice, and aztreonam (Azactam) would add empiric coverage of gram-negative enteric organisms.
Levofloxacin (Levaquin) also covers gramnegative enteric organisms, but Fusobacterium canifelinum, a common anaerobe in the oral flora of dogs and cats, is intrinsically resistant to fluoroquinolones.
Clindamycin and levofloxacin would be a good step-down oral regimen. Pasteurella multocida has variable sensitivity to the commonly used agents dicloxacillin (Dynapen), cephalexin (Keflex), macrolides, and clindamycin, but it is a less likely pathogen at this late stage and could be covered with levofloxacin alone.
C canimorsus is resistant to trimethoprim-sulfamethoxazole (Bactrim) and cephalexin, but is well covered by clindamycin.6
CASE CONTINUED
Our patient was started on intravenous vancomycin, clindamycin, and aztreonam for coverage of dog-mouth flora. Blood cultures and cultures of synovial fluid of the left wrist were negative. Vancomycin was discontinued after 48 hours when blood cultures did not grow staphylococcal organisms, but clindamycin and aztreonam were continued for a total of 8 days to treat possible infection with anaerobic and gram-negative enteric pathogens.
To test for autonomic dysfunction, a plastic pen case drawn lightly across each forearm revealed a loss of tactile adherence (ie, areas where moist, sweaty skin impeded the movement of the pen case) on the affected forearm, a sign of underlying nerve injury. The affected forearm was sensitive to light touch, with pain out of proportion to the stimulus.
ARRIVING AT THE DIAGNOSIS
Based on the wide distribution of inflammation, autonomic dysfunction (shown by differences in temperature and sweating), radiographic evidence of demineralization, hyperesthesia, and lack of improvement in pain and swelling after two courses of antibiotics, the patient’s clinical course was determined to be consistent with complex regional pain syndrome type 1, previously referred to as reflex sympathetic dystrophy.
Symptoms of complex regional pain syndrome traditionally include pain, regional edema, joint stiffness, muscular atrophy, vasomotor disturbances (causing temperature variability and erythema), regional diaphoresis, and localized skeletal demineralization on radiography.
Complex regional pain syndrome type 1 occurs as regional pain and inflammation as an excessive sympathetic reaction to an often minor insult, without nerve injury. When the syndrome occurs in a patient with obvious partial nerve injury, it is categorized as type 2 (formerly known as causalgia). The two types are clinically indistinguishable and are not uncommon. About 10% of all patients with complex regional pain syndrome have obvious nerve injury (complex regional pain syndrome type 2). In a study of 109 patients with Colles fracture, 25% developed symptoms of complex regional pain syndrome.7
Complex regional pain syndrome is difficult to diagnose, as it resembles many other ailments, such as gout, infection, bone tumor, stress fracture, and arthritis. Its pathophysiology is poorly understood, but it is believed to result from a “short circuit” in the reflex arc between somatic afferent sensory fibers and autonomic sympathetic efferent fibers, and this is thought to explain the increased sympathetic stimulation.
Although the pathophysiology is likely the same in type 1 and type 2, electromyography with a nerve conduction study is a reliable way to detect nerve damage and thus distinguish between the two types of complex regional pain syndrome.8
Our understanding of this syndrome is evolving. A recent study using sensory testing showed that 33% of patients with type 1 had combinations of increased and decreased thresholds for the detection of thermal, vibratory, and mechanical stimuli in the distribution of discrete peripheral nerves, suggesting that the patients actually had type 2.9
CONFIRMING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
3. Which of the following is the best way to confirm complex regional pain syndrome type 1?
- Erythrocyte sedimentation rate, C-reactive protein, and complete blood cell count
- Plain radiography of the hand and forearm
- Three-phase technetium bone scan
- The Budapest diagnostic criteria
- MRI
- Autonomic testing
Complex regional pain syndrome type 1 is a clinical diagnosis. Diagnostic studies lack sensitivity and specificity but may confirm complex regional pain syndrome type 1 or rule out other diagnoses. The Budapest diagnostic criteria10 (Table 1) may be the best way to confirm this diagnosis. The criteria are as follows: continuing pain disproportionate to an inciting event, coupled with three of four symptoms plus at least one sign from sensory, vasomotor, sudomotor, and motor-trophic categories.
Laboratory tests are not helpful because acute-phase reactants and blood counts remain normal in these patients.
Plain radiography is not sensitive in early diagnosis, but at 2 weeks it may show patchy areas of osteopenia in adjacent bones throughout the region, as well as subsequent diffuse demineralization.
Three-phase bone scanning is more sensitive than plain radiography, with 75% of patients showing regional disparities in blood flow in early sequences and increased bone uptake in the later sequences.
MRI is a sensitive early test, as it better defines focal areas of bone loss and increased T2 bone signal in adjacent bone, as well as early soft-tissue changes. Computed tomography does not show early specific changes in muscle, tendon, or bone and so is not recommended.
THE EVALUATION CONTINUES
The admitting diagnosis was septic arthritis, and our patient underwent computed tomography, which showed focal demineralization that could have represented bone infarcts or infection, confounding the diagnosis of complex regional pain syndrome.
Autonomic nerve testing can help distinguish complex regional pain syndrome from other disorders. Complex regional pain syndrome is characterized by increased sympathetic activity and results in increased sweat output. Autonomic testing includes resting sweat output, resting skin temperature, and quantitative sudomotor axon reflex testing. In one study, an increase in resting sweat output used in conjunction with quantitative sudomotor axon reflex testing predicted the diagnosis of complex regional pain syndrome with a specificity of 98%.11 However, autonomic testing is limited to academic centers and is not readily available.
TREATING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
4. Which is the best first-line therapy for complex regional pain syndrome type 1?
- Stellate ganglion nerve block
- Occupational therapy to splint the wrist and forearm
- Oral corticosteroids
- Physical therapy to prevent loss of joint motion
- Tricyclic antidepressant drugs (eg, amitriptyline), pregabalin, and bisphosphonates
Physical therapy should be started early in all patients, with range-of-motion exercises to prevent contracture and enhance mobility.
Stellate ganglion nerve block has been used to counter severe sympathetic hyperactivity, but it also may aggravate symptoms of complex regional pain syndrome and so remains a controversial treatment.
Immobilization and splinting should be avoided, as this will augment edema, pain, and contracture of joints.
Corticosteroids do not shorten the course or assuage symptoms and may increase edema.
Amitriptyline (Elavil) and pregabalin (Lyrica) have been used successfully to counter extended courses of allodynia and hyperalgesia. Bisphosphonates may decrease bone loss and pain and may be needed should the course be complicated by myositis ossificans, a form of dystrophic bone formation in juxtaposed tendon and muscle related to neuroactivation of fibroblasts and osteoblasts.
THE COURSE OF COMPLEX REGIONAL PAIN SYNDROME
Traditionally, type 1 was divided into three stages—an early inflammatory stage, a dystrophic stage, and a late atrophic stage.12 Although there is no evidence to support a consistent three-stage evolution, the severity of symptoms may help determine the best approach to management.13
Patients initially exhibit burning or throbbing pain, diffuse aching, sensitivity to touch or cold (allodynia), localized edema, and vasomotor disturbances of variable intensity that may produce altered color and temperature. Topical capsaicin cream; a tricyclic antidepressant; an anticonvulsant such as gabapentin (Neurontin), pregabalin, or lamotrigine (Lamictal); or a nonsteroidal anti-inflammatory drug should be tried first. Some of these treatments are poorly tolerated in elderly patients. If pain persists, nasal calcitonin may be added. Trigger-point injections with an anesthetic or glucocorticoid may be tried.
The management of early complex regional pain syndrome is sometimes supplemented with systemic corticosteroids, but reviews of randomized controlled trials have failed to show efficacy.14
Later in the course, patients may suffer persistent soft-tissue edema, accompanied by thickening of the skin and periarticular soft tissues, muscle wasting, and the skin changes of brawny edema. Regional blockade of sympathetic ganglions, epidural administration of clonidine, implantable peripheral nerve stimulators, and spinal cord stimulators have all been applied by experts in pain management and may provide benefit. Progression of the syndrome may include cyanosis, mottling, increased sweating, abnormal hair growth, and diffuse swelling in nonarticular tissue.
It is always acceptable to refer to an experienced pain management specialist, and a multidisciplinary approach is recommended at the outset.12
OUR PATIENT’S CARE CONTINUED
Our patient’s forearm and wrist were placed in a sling to keep his left arm elevated when active. This helped control sympathetic vascular edema and throbbing pain. Physical therapy with range-of-motion exercises prevented contracture.
He was discharged home on limited oxycodone as needed, with close follow-up by his primary care physician to monitor his pain symptoms. The pain and swelling slowly improved over the next 2 months, but he suffered a fall, twisting his left wrist. This minor injury was followed by more intense pain and swelling of the forearm, hand, and wrist.
COMORBIDITIES
5. Which of the following statements about conditions associated with complex regional pain syndrome most likely applies to our patient?
- Gout is likely following minor trauma
- Minor trauma or surgical bone biopsy may reactivate complex regional pain syndrome
- Septic hip arthritis due to MRSA may have reemerged and seeded the wrist
- Patients with multiple sclerosis have a propensity for complex regional pain syndrome
- Complex regional pain syndrome type 1 begets type 2
Gout does follow minor injury, but our patient’s uric acid was well controlled on allopurinol (Zyloprim), and gout is unlikely to be causing polyarticular swelling of the hand, wrist, and forearm.
Minor trauma, sometimes inconsequential enough to have been completely forgotten, may either initiate complex regional pain syndrome or, as seen here, reactivate it. Bone changes seen on MRI sometimes trigger surgical bone biopsy, only to reactivate the dysesthesia and sympathetic vascular reaction. Surgery should be avoided. Trauma and surgery are causative rather than associative comorbidities.
Sepsis due to MRSA after total hip arthroplasty may be reactivated, especially in the setting of immunosuppressive treatment. But the diffuse bone changes seen in multiple carpal, radial, and ulnar bones suggest generalized vascular and sympathetic disarray, most consistent with complex regional pain syndrome type 1.
AN ASSOCIATION WITH MULTIPLE SCLEROSIS?
Multiple sclerosis and other central neuropathic conditions such as stroke are associated with complex regional pain syndrome type 1.15,16
A hypothetical cause for the higher prevalence of complex regional pain syndrome in patients with multiple sclerosis may be demyelination resulting in aberrant signaling and overreaction to distal pain receptors. Demyelination of neurons within the autonomic or spinothalamic tracts potentially increases susceptibility to development of the pain syndrome.
Our patient had an apparent stimulus for the development of the syndrome, ie, the initial dog bite, and the wrist injury later may have caused peripheral nerve injury. Such injury may lead to release of vasodilatory neuropeptides including substance P from stimulated cutaneous nerves with cell bodies in the dorsal root ganglia. Excessive vasodilation and increased vascular permeability result in the affected limb becoming edematous and causing cutaneous nerves to be further activated. Stimulated cutaneous neurons normally have an inhibitory influence on sympathetic activity at the level of entry of the dorsal root ganglia in the cord. In complex regional pain syndrome, this inhibition is lost, resulting in a hyperactive somatosympathetic reflex.17 Underlying multiple sclerosis may have contributed to the loss of inhibition by the cutaneous nerves on the sympathetic system.
CASE CONCLUDED
We continued to closely follow this patient, who was on a self-directed program of physical therapy. One year after the original dog bite, the complex regional pain syndrome had completely resolved.
A 48-year-old man with gout, multiple sclerosis, and previously treated methicillin-resistant Staphylococcus aureus (MRSA) infection presented to the emergency room with pain and significant swelling at the site of a dog bite on his left forearm. He had been bitten 2 weeks earlier by a friend’s dog, and the bite had punctured the skin. He also had red streaking on the skin of the left arm from the wrist to the elbow, and he reported feeling “feverish” and having night sweats.
At first, the bite had seemed to improve, then swelling and pain had developed and increased. He reported this to his primary care physician, along with the information that he had previously had an anaphylactic reaction to penicillin and a cephalosporin. His physician, considering a penicillin allergy, started him on ciprofloxacin (Cipro) plus clindamycin (Cleocin). The patient took this for 5 days, but without improvement. The appearance of the red streaking on his left forearm prompted his presentation to our emergency room.
ORGANISMS IN DOG BITES
1. Which is the most common cause of infected dog bite?
- Pasteurella canis
- Streptococci and S aureus
- Erysipelothrix rhusiopathiae
- Capnocytophaga canimorsus
- Eikenella corrodens
Streptococci (50%) and S aureus (20% to 40%) are the organisms most commonly responsible for infected dog bites, as they are for other skin and soft-tissue infections.1P canis is unique to dog bite infections but accounts for only 18%.2E rhusiopathiae is an unusual isolate from cat and dog bites and is more commonly isolated from the mouths of fish and aquatic mammals. C canimorsus is a normal inhabitant of the oral cavity of dogs and cats but an unusual cause of wound infection from a dog bite. It is notable for sepsis and central nervous system infections uniquely associated with veterinarians, dog owners, kennel workers, and mail carriers.3E corrodens infection is more common with human bites.4
THE EVALUATION BEGINS
On examination, the patient had marked edema of the left forearm and pain in the joints of the left hand. His temperature was 100.2°F (37.9°C). Because of the duration and severity of symptoms, the examining physician was concerned about septic arthritis of the wrist, and the patient was admitted to the hospital.
In the hospital, our patient was thermodynamically stable without documented fever or chills. There was no open wound to culture, and blood cultures were negative. Marked edema and joint involvement raised suspicion of erysipeloid. This “cousin” of erysipelas often involves the underlying joint, is associated with edema, and produces systemic manifestations of fever and arthralgia.
Radiography of the left forearm and hand demonstrated multiple foci of demineralization within the carpal bones and proximal radius, attributed to disuse. Magnetic resonance imaging (MRI) the next day showed multiple bone infarcts in the carpal bones and the distal radius, with synovitis and fluid in the carpal joints and without adjacent osteomyelitis. Fluid was also seen in the soft tissues in the ulnar aspect of the left wrist, and tenosynovitis involving the flexor carpi radialis tendon was noted.
Arthrocentesis of his left radiocarpal joint produced synovial fluid negative for crystals and negative on Gram stain; the fluid was also sent for culture. The patient’s tetanus immunization was current, and the dog was known to have been immunized against rabies.
ANTIBIOTICS FOR INFECTED DOG BITES
2. Which antibiotic regimen would you choose for this patient?
- Oral amoxicillin and clavulanate
- Meropenem
- Vancomycin, clindamycin, aztreonam
- Clindamycin and levofloxacin
- Clindamycin and trimethoprim-sulfamethoxazole
Oral amoxicillin and clavulanate (Augmentin) is a judicious choice for prophylactic treatment of deep bites in the early stages of infection. However, our patient’s wound was no longer in the early stages of infection, and he had a history of an adverse reaction to penicillin.
Meropenem (Merrem IV) cross-reacts minimally with penicillin allergy and is reported to be safe in patients with a history of anaphylactic reactions to penicillin,5 but overuse of carbapenems has led to the development of carbapenem-resistant strains of Klebsiella, Stenotrophomonas, and Acinetobacter organisms.
Given the rise of MRSA infections and the common involvement of staphylococci, streptococci, and anaerobic bacteria in complicated dog bites, the combination of vancomycin and clindamycin is a good choice, and aztreonam (Azactam) would add empiric coverage of gram-negative enteric organisms.
Levofloxacin (Levaquin) also covers gramnegative enteric organisms, but Fusobacterium canifelinum, a common anaerobe in the oral flora of dogs and cats, is intrinsically resistant to fluoroquinolones.
Clindamycin and levofloxacin would be a good step-down oral regimen. Pasteurella multocida has variable sensitivity to the commonly used agents dicloxacillin (Dynapen), cephalexin (Keflex), macrolides, and clindamycin, but it is a less likely pathogen at this late stage and could be covered with levofloxacin alone.
C canimorsus is resistant to trimethoprim-sulfamethoxazole (Bactrim) and cephalexin, but is well covered by clindamycin.6
CASE CONTINUED
Our patient was started on intravenous vancomycin, clindamycin, and aztreonam for coverage of dog-mouth flora. Blood cultures and cultures of synovial fluid of the left wrist were negative. Vancomycin was discontinued after 48 hours when blood cultures did not grow staphylococcal organisms, but clindamycin and aztreonam were continued for a total of 8 days to treat possible infection with anaerobic and gram-negative enteric pathogens.
To test for autonomic dysfunction, a plastic pen case drawn lightly across each forearm revealed a loss of tactile adherence (ie, areas where moist, sweaty skin impeded the movement of the pen case) on the affected forearm, a sign of underlying nerve injury. The affected forearm was sensitive to light touch, with pain out of proportion to the stimulus.
ARRIVING AT THE DIAGNOSIS
Based on the wide distribution of inflammation, autonomic dysfunction (shown by differences in temperature and sweating), radiographic evidence of demineralization, hyperesthesia, and lack of improvement in pain and swelling after two courses of antibiotics, the patient’s clinical course was determined to be consistent with complex regional pain syndrome type 1, previously referred to as reflex sympathetic dystrophy.
Symptoms of complex regional pain syndrome traditionally include pain, regional edema, joint stiffness, muscular atrophy, vasomotor disturbances (causing temperature variability and erythema), regional diaphoresis, and localized skeletal demineralization on radiography.
Complex regional pain syndrome type 1 occurs as regional pain and inflammation as an excessive sympathetic reaction to an often minor insult, without nerve injury. When the syndrome occurs in a patient with obvious partial nerve injury, it is categorized as type 2 (formerly known as causalgia). The two types are clinically indistinguishable and are not uncommon. About 10% of all patients with complex regional pain syndrome have obvious nerve injury (complex regional pain syndrome type 2). In a study of 109 patients with Colles fracture, 25% developed symptoms of complex regional pain syndrome.7
Complex regional pain syndrome is difficult to diagnose, as it resembles many other ailments, such as gout, infection, bone tumor, stress fracture, and arthritis. Its pathophysiology is poorly understood, but it is believed to result from a “short circuit” in the reflex arc between somatic afferent sensory fibers and autonomic sympathetic efferent fibers, and this is thought to explain the increased sympathetic stimulation.
Although the pathophysiology is likely the same in type 1 and type 2, electromyography with a nerve conduction study is a reliable way to detect nerve damage and thus distinguish between the two types of complex regional pain syndrome.8
Our understanding of this syndrome is evolving. A recent study using sensory testing showed that 33% of patients with type 1 had combinations of increased and decreased thresholds for the detection of thermal, vibratory, and mechanical stimuli in the distribution of discrete peripheral nerves, suggesting that the patients actually had type 2.9
CONFIRMING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
3. Which of the following is the best way to confirm complex regional pain syndrome type 1?
- Erythrocyte sedimentation rate, C-reactive protein, and complete blood cell count
- Plain radiography of the hand and forearm
- Three-phase technetium bone scan
- The Budapest diagnostic criteria
- MRI
- Autonomic testing
Complex regional pain syndrome type 1 is a clinical diagnosis. Diagnostic studies lack sensitivity and specificity but may confirm complex regional pain syndrome type 1 or rule out other diagnoses. The Budapest diagnostic criteria10 (Table 1) may be the best way to confirm this diagnosis. The criteria are as follows: continuing pain disproportionate to an inciting event, coupled with three of four symptoms plus at least one sign from sensory, vasomotor, sudomotor, and motor-trophic categories.
Laboratory tests are not helpful because acute-phase reactants and blood counts remain normal in these patients.
Plain radiography is not sensitive in early diagnosis, but at 2 weeks it may show patchy areas of osteopenia in adjacent bones throughout the region, as well as subsequent diffuse demineralization.
Three-phase bone scanning is more sensitive than plain radiography, with 75% of patients showing regional disparities in blood flow in early sequences and increased bone uptake in the later sequences.
MRI is a sensitive early test, as it better defines focal areas of bone loss and increased T2 bone signal in adjacent bone, as well as early soft-tissue changes. Computed tomography does not show early specific changes in muscle, tendon, or bone and so is not recommended.
THE EVALUATION CONTINUES
The admitting diagnosis was septic arthritis, and our patient underwent computed tomography, which showed focal demineralization that could have represented bone infarcts or infection, confounding the diagnosis of complex regional pain syndrome.
Autonomic nerve testing can help distinguish complex regional pain syndrome from other disorders. Complex regional pain syndrome is characterized by increased sympathetic activity and results in increased sweat output. Autonomic testing includes resting sweat output, resting skin temperature, and quantitative sudomotor axon reflex testing. In one study, an increase in resting sweat output used in conjunction with quantitative sudomotor axon reflex testing predicted the diagnosis of complex regional pain syndrome with a specificity of 98%.11 However, autonomic testing is limited to academic centers and is not readily available.
TREATING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
4. Which is the best first-line therapy for complex regional pain syndrome type 1?
- Stellate ganglion nerve block
- Occupational therapy to splint the wrist and forearm
- Oral corticosteroids
- Physical therapy to prevent loss of joint motion
- Tricyclic antidepressant drugs (eg, amitriptyline), pregabalin, and bisphosphonates
Physical therapy should be started early in all patients, with range-of-motion exercises to prevent contracture and enhance mobility.
Stellate ganglion nerve block has been used to counter severe sympathetic hyperactivity, but it also may aggravate symptoms of complex regional pain syndrome and so remains a controversial treatment.
Immobilization and splinting should be avoided, as this will augment edema, pain, and contracture of joints.
Corticosteroids do not shorten the course or assuage symptoms and may increase edema.
Amitriptyline (Elavil) and pregabalin (Lyrica) have been used successfully to counter extended courses of allodynia and hyperalgesia. Bisphosphonates may decrease bone loss and pain and may be needed should the course be complicated by myositis ossificans, a form of dystrophic bone formation in juxtaposed tendon and muscle related to neuroactivation of fibroblasts and osteoblasts.
THE COURSE OF COMPLEX REGIONAL PAIN SYNDROME
Traditionally, type 1 was divided into three stages—an early inflammatory stage, a dystrophic stage, and a late atrophic stage.12 Although there is no evidence to support a consistent three-stage evolution, the severity of symptoms may help determine the best approach to management.13
Patients initially exhibit burning or throbbing pain, diffuse aching, sensitivity to touch or cold (allodynia), localized edema, and vasomotor disturbances of variable intensity that may produce altered color and temperature. Topical capsaicin cream; a tricyclic antidepressant; an anticonvulsant such as gabapentin (Neurontin), pregabalin, or lamotrigine (Lamictal); or a nonsteroidal anti-inflammatory drug should be tried first. Some of these treatments are poorly tolerated in elderly patients. If pain persists, nasal calcitonin may be added. Trigger-point injections with an anesthetic or glucocorticoid may be tried.
The management of early complex regional pain syndrome is sometimes supplemented with systemic corticosteroids, but reviews of randomized controlled trials have failed to show efficacy.14
Later in the course, patients may suffer persistent soft-tissue edema, accompanied by thickening of the skin and periarticular soft tissues, muscle wasting, and the skin changes of brawny edema. Regional blockade of sympathetic ganglions, epidural administration of clonidine, implantable peripheral nerve stimulators, and spinal cord stimulators have all been applied by experts in pain management and may provide benefit. Progression of the syndrome may include cyanosis, mottling, increased sweating, abnormal hair growth, and diffuse swelling in nonarticular tissue.
It is always acceptable to refer to an experienced pain management specialist, and a multidisciplinary approach is recommended at the outset.12
OUR PATIENT’S CARE CONTINUED
Our patient’s forearm and wrist were placed in a sling to keep his left arm elevated when active. This helped control sympathetic vascular edema and throbbing pain. Physical therapy with range-of-motion exercises prevented contracture.
He was discharged home on limited oxycodone as needed, with close follow-up by his primary care physician to monitor his pain symptoms. The pain and swelling slowly improved over the next 2 months, but he suffered a fall, twisting his left wrist. This minor injury was followed by more intense pain and swelling of the forearm, hand, and wrist.
COMORBIDITIES
5. Which of the following statements about conditions associated with complex regional pain syndrome most likely applies to our patient?
- Gout is likely following minor trauma
- Minor trauma or surgical bone biopsy may reactivate complex regional pain syndrome
- Septic hip arthritis due to MRSA may have reemerged and seeded the wrist
- Patients with multiple sclerosis have a propensity for complex regional pain syndrome
- Complex regional pain syndrome type 1 begets type 2
Gout does follow minor injury, but our patient’s uric acid was well controlled on allopurinol (Zyloprim), and gout is unlikely to be causing polyarticular swelling of the hand, wrist, and forearm.
Minor trauma, sometimes inconsequential enough to have been completely forgotten, may either initiate complex regional pain syndrome or, as seen here, reactivate it. Bone changes seen on MRI sometimes trigger surgical bone biopsy, only to reactivate the dysesthesia and sympathetic vascular reaction. Surgery should be avoided. Trauma and surgery are causative rather than associative comorbidities.
Sepsis due to MRSA after total hip arthroplasty may be reactivated, especially in the setting of immunosuppressive treatment. But the diffuse bone changes seen in multiple carpal, radial, and ulnar bones suggest generalized vascular and sympathetic disarray, most consistent with complex regional pain syndrome type 1.
AN ASSOCIATION WITH MULTIPLE SCLEROSIS?
Multiple sclerosis and other central neuropathic conditions such as stroke are associated with complex regional pain syndrome type 1.15,16
A hypothetical cause for the higher prevalence of complex regional pain syndrome in patients with multiple sclerosis may be demyelination resulting in aberrant signaling and overreaction to distal pain receptors. Demyelination of neurons within the autonomic or spinothalamic tracts potentially increases susceptibility to development of the pain syndrome.
Our patient had an apparent stimulus for the development of the syndrome, ie, the initial dog bite, and the wrist injury later may have caused peripheral nerve injury. Such injury may lead to release of vasodilatory neuropeptides including substance P from stimulated cutaneous nerves with cell bodies in the dorsal root ganglia. Excessive vasodilation and increased vascular permeability result in the affected limb becoming edematous and causing cutaneous nerves to be further activated. Stimulated cutaneous neurons normally have an inhibitory influence on sympathetic activity at the level of entry of the dorsal root ganglia in the cord. In complex regional pain syndrome, this inhibition is lost, resulting in a hyperactive somatosympathetic reflex.17 Underlying multiple sclerosis may have contributed to the loss of inhibition by the cutaneous nerves on the sympathetic system.
CASE CONCLUDED
We continued to closely follow this patient, who was on a self-directed program of physical therapy. One year after the original dog bite, the complex regional pain syndrome had completely resolved.
- Talan DA, Citron DM, Abrahamian FM, Moran GJ, Goldstein EJ. Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group. N Engl J Med 1999; 340:85–92.
- Holst E, Rollof J, Larsson L, Nielsen JP. Characterization and distribution of Pasteurella species recovered from infected humans. J Clin Microbiol 1992; 30:2984–2987.
- Jolivet-Gougeon A, Sixou JL, Tamanai-Shacoori Z, Bonnaure-Mallet M. Antimicrobial treatment of Capnocytophaga infections. Int J Antimicrob Agents 2007; 29:367–373.
- Paul K, Patel SS. Eikenella corrodens infections in children and adolescents: case reports and review of the literature. Clin Infect Dis 2001; 33:54–61.
- Cunha BA, Hamid NS, Krol V, Eisenstein L. Safety of meropenem in patients reporting penicillin allergy: lack of allergic cross reactions. J Chemother 2008; 20:233–237.
- Verghese A, Hamati F, Berk S, Franzus B, Berk S, Smith JK. Susceptibility of dysgonic fermenter 2 to antimicrobial agents in vitro. Antimicrob Agents Chemother 1988; 32:78–80.
- Atkins RM, Duckworth T, Kanis JA. Algodystrophy following Colles’ fracture. J Hand Surg Br 1989; 14:161–164.
- Rommel O, Malin JP, Zenz M, Jänig W. Quantitative sensory testing, neurophysiological and psychological examination in patients with complex regional pain syndrome and hemisensory deficits. Pain 2001; 93:279–293.
- Sethna NF, Meier PM, Zurakowski D, Berde CB. Cutaneous sensory abnormalities in children and adolescents with complex regional pain syndromes. Pain 2007; 131:153–161.
- Harden RN, Bruehl S, Stanton-Hicks M, Wilson PR. Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med 2007; 8:326–331.
- Chelimsky TC, Low PA, Naessens JM, Wilson PR, Amadio PC, O’Brien PC. Value of autonomic testing in reflex sympathetic dystrophy. Mayo Clin Proc 1995; 70:1029–1040.
- Stanton-Hicks MD, Burton AW, Bruehl SP, et al. An updated interdisciplinary clinical pathway for CRPS: report of an expert panel. Pain Pract 2002; 2:1–16.
- Brummett CM, Cohen SP, eds. Managing pain: essentials of diagnosis and treatment. New York; Oxford University Press; 2013.
- Dirckx M, Stronks DL, Groeneweg G, Huygen FJ. Effect of immunomodulating medications in complex regional pain syndrome: a systematic review. Clin J Pain 2012; 28:355–363.
- Schwartzman RJ, Gurusinghe C, Gracely E. Prevalence of complex regional pain syndrome in a cohort of multiple sclerosis patients. Pain Physician 2008; 11:133–136.
- Sandroni P, Benrud-Larson LM, McClelland RL, Low PA. Complex regional pain syndrome type I: incidence and prevalence in Olmsted county, a population-based study. Pain 2003; 103:199–207.
- Kurvers HA, Jacobs MJ, Beuk RJ, et al. Reflex sympathetic dystrophy: evolution of microcirculatory disturbances in time. Pain 1995; 60:333–340.
- Talan DA, Citron DM, Abrahamian FM, Moran GJ, Goldstein EJ. Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group. N Engl J Med 1999; 340:85–92.
- Holst E, Rollof J, Larsson L, Nielsen JP. Characterization and distribution of Pasteurella species recovered from infected humans. J Clin Microbiol 1992; 30:2984–2987.
- Jolivet-Gougeon A, Sixou JL, Tamanai-Shacoori Z, Bonnaure-Mallet M. Antimicrobial treatment of Capnocytophaga infections. Int J Antimicrob Agents 2007; 29:367–373.
- Paul K, Patel SS. Eikenella corrodens infections in children and adolescents: case reports and review of the literature. Clin Infect Dis 2001; 33:54–61.
- Cunha BA, Hamid NS, Krol V, Eisenstein L. Safety of meropenem in patients reporting penicillin allergy: lack of allergic cross reactions. J Chemother 2008; 20:233–237.
- Verghese A, Hamati F, Berk S, Franzus B, Berk S, Smith JK. Susceptibility of dysgonic fermenter 2 to antimicrobial agents in vitro. Antimicrob Agents Chemother 1988; 32:78–80.
- Atkins RM, Duckworth T, Kanis JA. Algodystrophy following Colles’ fracture. J Hand Surg Br 1989; 14:161–164.
- Rommel O, Malin JP, Zenz M, Jänig W. Quantitative sensory testing, neurophysiological and psychological examination in patients with complex regional pain syndrome and hemisensory deficits. Pain 2001; 93:279–293.
- Sethna NF, Meier PM, Zurakowski D, Berde CB. Cutaneous sensory abnormalities in children and adolescents with complex regional pain syndromes. Pain 2007; 131:153–161.
- Harden RN, Bruehl S, Stanton-Hicks M, Wilson PR. Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med 2007; 8:326–331.
- Chelimsky TC, Low PA, Naessens JM, Wilson PR, Amadio PC, O’Brien PC. Value of autonomic testing in reflex sympathetic dystrophy. Mayo Clin Proc 1995; 70:1029–1040.
- Stanton-Hicks MD, Burton AW, Bruehl SP, et al. An updated interdisciplinary clinical pathway for CRPS: report of an expert panel. Pain Pract 2002; 2:1–16.
- Brummett CM, Cohen SP, eds. Managing pain: essentials of diagnosis and treatment. New York; Oxford University Press; 2013.
- Dirckx M, Stronks DL, Groeneweg G, Huygen FJ. Effect of immunomodulating medications in complex regional pain syndrome: a systematic review. Clin J Pain 2012; 28:355–363.
- Schwartzman RJ, Gurusinghe C, Gracely E. Prevalence of complex regional pain syndrome in a cohort of multiple sclerosis patients. Pain Physician 2008; 11:133–136.
- Sandroni P, Benrud-Larson LM, McClelland RL, Low PA. Complex regional pain syndrome type I: incidence and prevalence in Olmsted county, a population-based study. Pain 2003; 103:199–207.
- Kurvers HA, Jacobs MJ, Beuk RJ, et al. Reflex sympathetic dystrophy: evolution of microcirculatory disturbances in time. Pain 1995; 60:333–340.
A 20-year-old woman with fatigue and palpitations
A 20-year-old woman presents to the emergency department with fatigue and the sudden onset of palpitations. She reports no history of significant illness or surgery. She says she is not currently taking prescription or over-the-counter medications. She does not smoke, drink alcohol, or use illicit drugs.
Her weight is 52 kg (115 lb), her height is 170 cm (67 in), and her body mass index (BMI) is 18 kg/m2. Vital signs: temperature 35.7°C (96.4°F), blood pressure 92/48 mm Hg, heart rate 73 bpm, respiratory rate 5 breaths per minute, and oxygen saturation 98% on room air.
She appears tired but is alert, conversant, and cooperative. Her skin is normal, and dentition is fair. Her pulse is regular, and respirations are slow. The abdomen is soft, non-tender, and flat. Strength is 4 on a scale of 5 in all extremities. Deep-tendon reflexes are 2+ and symmetric.
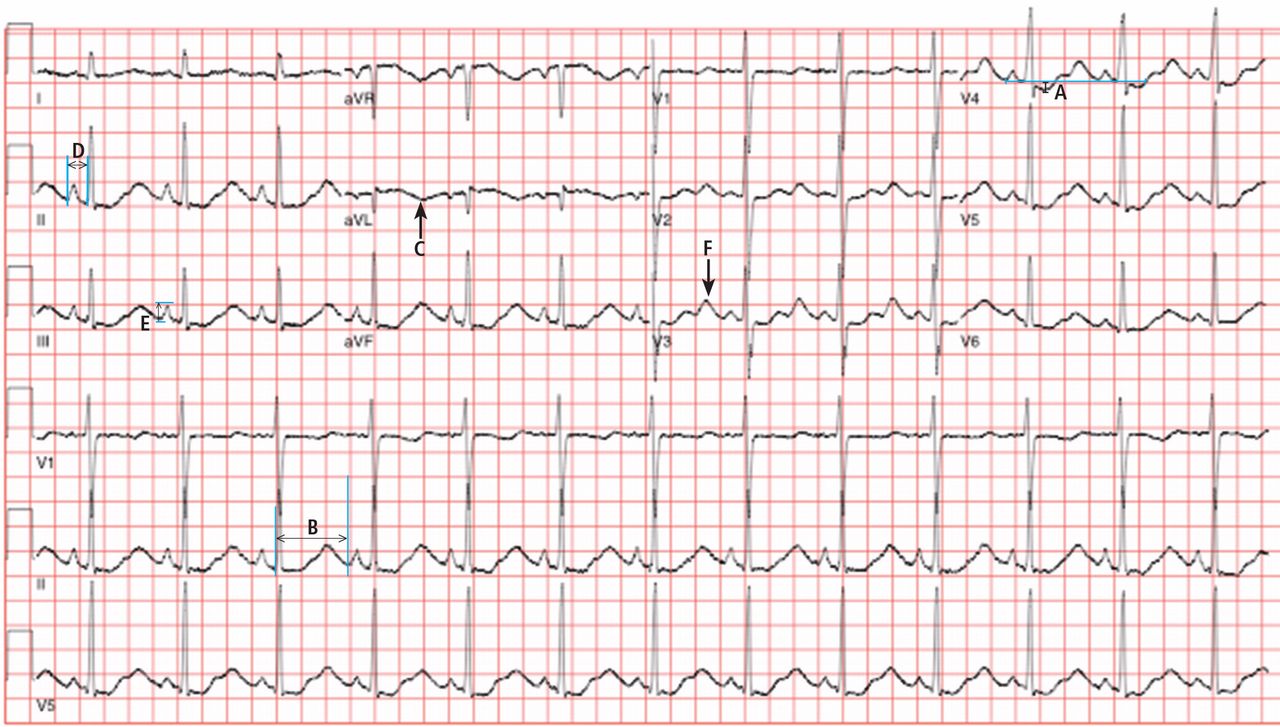
Electrocardiography (Figure 1) in the emergency department shows ST-segment depression, a prolonged corrected QT interval of 665 msec, T-wave inversion, PR prolongation, increased P-wave amplitude, and U waves.
1. Which electrolyte abnormality is associated with this electrocardiographic picture?
- Hypercalcemia
- Hyperkalemia
- Hypocalcemia
- Hypokalemia
Hypokalemia is the likely cause of these findings. The finding of U waves is considered significant when they are inverted, merged with the T wave, or have an amplitude greater than the T wave.1 U waves are best seen in the precordial leads. When severe, hypokalemia can lead to potentially fatal arrhythmias such as high-grade atrioventricular block, ventricular tachycardia, and ventricular fibrillation.2
Hyperkalemia is associated with peaked T waves, a prolonged PR interval, decreased P wave amplitude, and a widened QRS complex.2 When acute and severe, hyperkalemia is associated with ventricular arrhythmia.
Hypocalcemia is associated with a prolonged QT interval and ventricular dysrhythmia, but not U waves.2
Hypercalcemia is associated with bradydysrhythmia, as well as with a shortened QT interval.2
LABORATORY TESTING
Laboratory testing shows the following:
- Sodium 126 mmol/L (reference range 135–145)
- Potassium 1.5 mmol/L (3.5–5.1)
- Chloride 58 mmol/L (100–110)
- Bicarbonate 62 mmol/L (20–30)
- Blood urea nitrogen 16 mg/dL (7–18)
- Creatinine 0.8 mg/dL (0.5–1.0)
- Glucose 106 mg/dL (70–110)
- Ionized calcium 4.4 mg/dL (4.5–5.3)
- Magnesium 1.8 mg/dL (1.7–2.3)
- Phosphorus 4.1 mg/dL (2.5–4.5)
- Venous blood gases pH 7.56 (7.35–7.45), Pco2 69 mm Hg (35–45).
POTASSIUM HOMEOSTASIS
Ninety-eight percent of potassium is intracellular and only 2% is extracellular.3 The main cellular stores are myocytes and hepatocytes. Patients with decreased muscle mass may be at a higher risk of hypokalemia as a result of decreased skeletal muscle stores.4
The acute development of hypokalemia occurs from transcellular shifts. Alkalosis, insulin secretion, and beta-adrenergic stimulation promote the intracellular uptake of potassium. The major hormonal regulator of potassium excretion is aldosterone, which is stimulated by renal hypoperfusion and promotes potassium-ion secretion in the distal convoluted tubule.
Chronic hypokalemia develops in patients with ongoing renal or gastrointestinal potassium loss. If the cause of potassium loss is not elucidated by the history, the physical, and a review of medications, then one of two things is possible: either the patient has renal tubular disease affecting acid-base and potassium regulation, causing excessive mineralocorticoid secretion, which is associated with an abnormal response to aldosterone; or the patient is not being forthcoming in the history.
2. Which is the most likely cause of hypokalemia in this patient?
- Vomiting
- Liddle syndrome
- Bartter syndrome
- Gitelman syndrome
- Diuretic use
Her laboratory tests reveal hypokalemia and hyponatremic-hypochloremic metabolic alkalosis with compensatory respiratory acidosis. In metabolic alkalosis, the expected respiratory compensation is an increase of 0.7 mm Hg in Pco2 for each 1-mEq/L increase in bicarbonate. Therefore, the expected Pco2 is 67, close to the patient’s actual value of 69.
Protracted vomiting with loss of gastric acid juices could be a cause of the metabolic disturbances in this young woman, although she did not mention vomiting during the history.
Liddle syndrome, or pseudoaldosteronism, is a rare autosomal dominant disorder characterized by altered renal epithelial sodium channels, excessive sodium retention, and resultant hypertension. Hypokalemia and alkalosis are seen in Liddle syndrome, but the absence of hypertension in our patient makes Liddle syndrome unlikely.
Bartter syndrome is an inherited autosomal recessive disorder of the sodium-potassium-chloride cotransporter in the thick ascending loop of Henle, resulting in impaired reabsorption of chloride and sodium. Bartter syndrome mimics chronic loop-diuretic use and is associated with hypercalciuria. Bartter syndrome is possible in this patient; however, patients with Bartter syndrome are usually diagnosed in infancy or childhood and have evidence of growth impairment.
Gitelman syndrome is an autosomal recessive disorder of the thiazide-sensitive sodium-chloride cotransporter. Although Gitelman syndrome is more common than Bartter syndrome and presents at older ages, it is not usually associated with such profound metabolic alkalosis. Gitelman syndrome mimics chronic use of thiazide diuretics and is associated with hypocalciuria.
Diuretic use could also cause the metabolic disturbances described; however, the patient denied taking diuretics.
The most common cause of hypokalemia in clinical practice is diuretic use.4 In this young woman with unexplained hypokalemia, the most likely cause is either undisclosed self-induced vomiting or diuretic abuse. The degree of metabolic alkalosis suggests vomiting, since metabolic alkalosis this severe is usually seen only with protracted vomiting. Bartter and Gitelman syndromes are included in the differential diagnosis, but they are much less common than hypokalemia associated with diuretics or self-induced vomiting.5
3. Which test could help elucidate the cause of hypokalemia in this patient?
- Ratio of plasma aldosterone to rennin
- Urine chloride
- Ratio of urinary potassium to creatinine
- Urinary anion gap and urinary pH
APPROACH TO HYPOKALEMIA
Determining the cause of hypokalemia starts with a thorough history and physical examination. The history should focus on drugs such as diuretics and laxatives. Women should be asked about their menstrual history since irregular periods may suggest an eating disorder. The physical examination should focus on signs that suggest self-induced vomiting, such as dry skin, dental erosions, enlarged parotid glands, and calluses or scars on the knuckles.
Patients with an unclear cause of hypokalemia after a thorough history and physical examination can be categorized into one of three groups based on blood pressure and acid-base status:
- Hypokalemia, hypertension, metabolic alkalosis
- Hypokalemia, normal blood pressure, metabolic acidosis
- Hypokalemia, normal blood pressure, metabolic alkalosis.
Hypokalemia, hypertension, metabolic alkalosis
The blood pressure provides an important clue in the evaluation of hypokalemia. The combination of hypertension, hypokalemia, and alkalosis should raise concern for hyperaldosteronism or pseudoaldosteronism. Primary hyperaldosteronism from an adrenal adenoma (Conn syndrome) is characterized by a plasma aldosterone-renin ratio of greater than 20.6,7 In contrast, patients with secondary hyperaldosteronism due to renovascular disease have a plasma aldosterone-renin ratio of less than 10. Patients with pseudoaldosteronism have low aldosterone and renin levels and hypertension. Since our patient has a normal blood pressure, testing the plasma aldosterone and renin levels would not help determine the cause of her hypokalemia.
Hypokalemia, normal blood pressure, metabolic acidosis
Patients with normal blood pressure, hypokalemia, and normal plasma anion gap acidosis either have renal tubular acidosis or have lost potassium because of diarrhea or laxative abuse. In a patient who denies taking laxatives or denies a history of diarrhea, checking the urinary anion gap and urinary pH may help differentiate the cause of acidosis and hypokalemia.
The urinary anion gap, calculated by the equation sodium + potassium − chloride, is an indirect estimate of hydrogen excretion in the form of ammonium ion8; the normal value is 0 to 10 mEq/L. A negative value represents increased hydrogen excretion in response to systemic acidosis from gastrointestinal or renal loss of bicarbonate (proximal renal tubular acidosis). A urinary pH greater than 5.5 in the setting of systemic acidosis suggests impaired ability of the kidneys to acidify urine and raises the possibility of renal tubular acidosis.
This patient has metabolic alkalosis, so calculation of the urinary anion gap would not be helpful.
Hypokalemia, normal blood pressure, metabolic alkalosis
Patients such as ours, with normal blood pressure, hypokalemia, and alkalosis, have been vomiting, have used diuretics, or have an inherited renal tubulopathy such as Bartter or Gitelman syndrome. Usually, differentiating Bartter and Gitelman syndromes from chronic vomiting or diuretic use is done with the history and physical examination. However, in patients with a questionable history and a lack of findings on physical examination, checking the urinary chloride, potassium, calcium, and creatinine may be helpful.
A urinary potassium-creatinine ratio greater than 15 suggests renal loss, whereas a ratio less than 15 suggests extrarenal loss.9

Patients who are taking a diuretic or who have Bartter or Gitelman syndrome have a high urinary chloride concentration, ie, greater than 20 mmol/L, whereas patients with hypokalemia and alkalosis from chronic vomiting tend to have a concentration less than 10 mmol/L.10
Table 1 summarizes an approach to the evaluation of unexplained hypokalemia based on blood pressure and acid-base status.
A HIDDEN HISTORY
On further questioning, the patient admits to an 8-year history of daily self-induced vomiting in an attempt to lose weight, in addition to multiple hospitalizations for hypokalemia and a previous diagnosis of an eating disorder.
INITIAL MANAGEMENT OF HYPOKALEMIA
The initial management of hypokalemia should focus on life-threatening emergencies. While patients with potassium levels greater than 3 mmol/L are usually asymptomatic, those with levels below 3 mmol/L present with muscle weakness and rhabdomyolysis.4 An acute drop in serum potassium to less than 2 mmol/L is associated with respiratory muscle weakness and ventricular arrhythmias.4 If the patient has cardiac symptoms or hypoventilation due to respiratory muscle weakness, continuous monitoring in the intensive care unit and aggressive therapy are warranted.
4. Which potassium formulation is most appropriate for the treatment of hypokalemia in this patient?
- Potassium chloride
- Potassium phosphate
- Potassium acetate
Oral potassium is preferable in patients with a serum potassium above 2.5 mmol/L.4,11 Potassium phosphate should be used when supplementation with both potassium and phosphorus is needed. Potassium acetate should be reserved for patients with acidosis and hypokalemia. Otherwise, potassium chloride is typically preferred.4,12 It comes in liquid and tablet forms. Liquid forms have an unpleasant taste, whereas tablets are usually well tolerated. No more than 20 to 40 mEq of potassium chloride tablets should be given at a time, since higher doses are associated with gastrointestinal mucosal injury.12
Potassium chloride is particularly preferred in patients with metabolic alkalosis, since increased chloride intake and delivery to the distal tubule increases the expression of pendrin, a luminal chloride and bicarbonate exchanger in the cortical collecting duct.13 With metabolic alkalosis, increased excretion of bicarbonate occurs through up-regulation of pendrin. Potassium depletion down-regulates pendrin.13 Additionally, correction of metabolic alkalosis increases serum potassium by movement of potassium from the intracellular to the extracellular space.
Intravenous potassium should be reserved for patients with severe hypokalemia (< 2.5 mmol/L) or significant arrhythmias.11 Oral and intravenous potassium can safely be given simultaneously.11 The intravenous rate should not exceed more than 10 to 20 mEq of potassium chloride per hour unless the patient has a life-threatening arrhythmia, respiratory failure, or severe hypokalemia.14,15 In life-threatening situations, a femoral line should be placed, and potassium should be given as rapidly as 20 mEq over 15 to 20 minutes.14 Cannulation of the subclavian and internal jugular veins should be avoided in severe hypokalemia since mechanical irritation from guidewire placement can provoke ventricular arrhythmias.14
During intravenous administration of potassium, laboratory monitoring after every 20 mEq of potassium chloride is advised because of the possibility of rebound hyperkalemia. In patients with severe hypokalemia, avoidance of factors that can worsen intracellular shift of potassium is also important. Avoid dextrose-containing fluids to prevent insulin-induced shifting of potassium into cells. Restore intravascular volume to blunt hypovolemia-induced renin and aldosterone secretion. If a patient presents with severe hypokalemia and acidosis, correct the hypokalemia before the acidosis to avoid intracellular shift of potassium.
OUR PATIENT’S MANAGEMENT AND FOLLOW-UP PLAN
Given the severity of our patient’s hypokalemia and her complaint of palpitations, she was admitted to the hospital for monitoring. She required 180 mEq of intravenous potassium chloride and 140 mEq of oral potassium chloride during the first 24 hours in order to achieve a serum potassium level above 3 mmol/L. Electrocardiographic U waves resolved once the level was above 2 mmol/L, and ST depressions resolved once it was above 3 mmol/L. The QT interval normalized after 24 hours of hospitalization.
On discharge, she was prescribed oral potassium chloride 40 mEq daily and magnesium sulfate 400 mg twice daily, with plans for a followup visit with her outpatient therapy team, which includes a psychiatrist, a social worker, and her primary care provider. She declined a referral for inpatient therapy but agreed to a goal of decreasing the frequency of induced vomiting and outpatient visits. She was also educated on how and when to access emergency medical care.16
- Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT Interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
- Diercks DB, Shumaik GM, Harrigan RA, Brady WJ, Chan TC. Electrocardiographic manifestations: electrolyte abnormalities. J Emerg Med 2004; 27:153–160.
- Unwin RJ, Luft FC, Shirley DG. Pathophysiology and management of hypokalemia: a clinical perspective. Nat Rev Nephrol 2011; 7:75–84.
- Gennari FJ. Hypokalemia. N Engl J Med 1998; 339:451–458.
- Mehler PS. Clinical practice. Bulimia nervosa. N Engl J Med 2003; 349:875–881.
- Tzanela M, Effraimidis G, Vassiliadi D, et al. The aldosterone to renin ratio in the evaluation of patients with incidentally detected adrenal masses. Endocrine 2007; 32:136–142.
- Diederich S, Mai K, Bähr V, Helffrich S, Pfeiffer A, Perschel FH. The simultaneous measurement of plasma-aldosterone- and -renin-concentration allows rapid classification of all disorders of the renin-aldosterone system. Exp Clin Endocrinol Diabetes 2007; 115:433–438.
- Goldstein MB, Bear R, Richardson RM, Marsden PA, Halperin ML. The urine anion gap: a clinically useful index of ammonium excretion. Am J Med Sci 1986; 292:198–202.
- Groeneveld JH, Sijpkens YW, Lin SH, Davids MR, Halperin ML. An approach to the patient with severe hypokalaemia: the potassium quiz. QJM 2005; 98:305–316.
- Galla JH. Metabolic alkalosis. J Am Soc Nephrol 2000; 11:369–375.
- Asmar A, Mohandas R, Wingo CS. A physiologic-based approach to the treatment of a patient with hypokalemia. Am J Kidney Dis 2012; 60:492–497.
- Cohn JN, Kowey PR, Whelton PK, Prisant LM. New guidelines for potassium replacement in clinical practice: a contemporary review by the National Council on Potassium in Clinical Practice. Arch Intern Med 2000; 160:2429–2436.
- Luke RG, Galla JH. It is chloride depletion alkalosis, not contraction alkalosis. J Am Soc Nephrol 2012; 23:204–207.
- Kruse JA, Carlson RW. Rapid correction of hypokalemia using concentrated intravenous potassium chloride infusions. Arch Intern Med 1990; 150:613–617.
- Weiner ID, Wingo CS. Hypokalemia—consequences, causes, and correction. J Am Soc Nephrol 1997; 8:1179–1188.
- AED Medical Care Standards Task Force. Eating disorders: Critical Points for Early Recognition and Medical Risk Management in the Care of Individuals with Eating Disorders. AED Report 2012. www.aedweb.org/web/downloads/Guide-English.pdf. Accessed April 4, 2014.
A 20-year-old woman presents to the emergency department with fatigue and the sudden onset of palpitations. She reports no history of significant illness or surgery. She says she is not currently taking prescription or over-the-counter medications. She does not smoke, drink alcohol, or use illicit drugs.
Her weight is 52 kg (115 lb), her height is 170 cm (67 in), and her body mass index (BMI) is 18 kg/m2. Vital signs: temperature 35.7°C (96.4°F), blood pressure 92/48 mm Hg, heart rate 73 bpm, respiratory rate 5 breaths per minute, and oxygen saturation 98% on room air.
She appears tired but is alert, conversant, and cooperative. Her skin is normal, and dentition is fair. Her pulse is regular, and respirations are slow. The abdomen is soft, non-tender, and flat. Strength is 4 on a scale of 5 in all extremities. Deep-tendon reflexes are 2+ and symmetric.
Electrocardiography (Figure 1) in the emergency department shows ST-segment depression, a prolonged corrected QT interval of 665 msec, T-wave inversion, PR prolongation, increased P-wave amplitude, and U waves.
1. Which electrolyte abnormality is associated with this electrocardiographic picture?
- Hypercalcemia
- Hyperkalemia
- Hypocalcemia
- Hypokalemia
Hypokalemia is the likely cause of these findings. The finding of U waves is considered significant when they are inverted, merged with the T wave, or have an amplitude greater than the T wave.1 U waves are best seen in the precordial leads. When severe, hypokalemia can lead to potentially fatal arrhythmias such as high-grade atrioventricular block, ventricular tachycardia, and ventricular fibrillation.2
Hyperkalemia is associated with peaked T waves, a prolonged PR interval, decreased P wave amplitude, and a widened QRS complex.2 When acute and severe, hyperkalemia is associated with ventricular arrhythmia.
Hypocalcemia is associated with a prolonged QT interval and ventricular dysrhythmia, but not U waves.2
Hypercalcemia is associated with bradydysrhythmia, as well as with a shortened QT interval.2
LABORATORY TESTING
Laboratory testing shows the following:
- Sodium 126 mmol/L (reference range 135–145)
- Potassium 1.5 mmol/L (3.5–5.1)
- Chloride 58 mmol/L (100–110)
- Bicarbonate 62 mmol/L (20–30)
- Blood urea nitrogen 16 mg/dL (7–18)
- Creatinine 0.8 mg/dL (0.5–1.0)
- Glucose 106 mg/dL (70–110)
- Ionized calcium 4.4 mg/dL (4.5–5.3)
- Magnesium 1.8 mg/dL (1.7–2.3)
- Phosphorus 4.1 mg/dL (2.5–4.5)
- Venous blood gases pH 7.56 (7.35–7.45), Pco2 69 mm Hg (35–45).
POTASSIUM HOMEOSTASIS
Ninety-eight percent of potassium is intracellular and only 2% is extracellular.3 The main cellular stores are myocytes and hepatocytes. Patients with decreased muscle mass may be at a higher risk of hypokalemia as a result of decreased skeletal muscle stores.4
The acute development of hypokalemia occurs from transcellular shifts. Alkalosis, insulin secretion, and beta-adrenergic stimulation promote the intracellular uptake of potassium. The major hormonal regulator of potassium excretion is aldosterone, which is stimulated by renal hypoperfusion and promotes potassium-ion secretion in the distal convoluted tubule.
Chronic hypokalemia develops in patients with ongoing renal or gastrointestinal potassium loss. If the cause of potassium loss is not elucidated by the history, the physical, and a review of medications, then one of two things is possible: either the patient has renal tubular disease affecting acid-base and potassium regulation, causing excessive mineralocorticoid secretion, which is associated with an abnormal response to aldosterone; or the patient is not being forthcoming in the history.
2. Which is the most likely cause of hypokalemia in this patient?
- Vomiting
- Liddle syndrome
- Bartter syndrome
- Gitelman syndrome
- Diuretic use
Her laboratory tests reveal hypokalemia and hyponatremic-hypochloremic metabolic alkalosis with compensatory respiratory acidosis. In metabolic alkalosis, the expected respiratory compensation is an increase of 0.7 mm Hg in Pco2 for each 1-mEq/L increase in bicarbonate. Therefore, the expected Pco2 is 67, close to the patient’s actual value of 69.
Protracted vomiting with loss of gastric acid juices could be a cause of the metabolic disturbances in this young woman, although she did not mention vomiting during the history.
Liddle syndrome, or pseudoaldosteronism, is a rare autosomal dominant disorder characterized by altered renal epithelial sodium channels, excessive sodium retention, and resultant hypertension. Hypokalemia and alkalosis are seen in Liddle syndrome, but the absence of hypertension in our patient makes Liddle syndrome unlikely.
Bartter syndrome is an inherited autosomal recessive disorder of the sodium-potassium-chloride cotransporter in the thick ascending loop of Henle, resulting in impaired reabsorption of chloride and sodium. Bartter syndrome mimics chronic loop-diuretic use and is associated with hypercalciuria. Bartter syndrome is possible in this patient; however, patients with Bartter syndrome are usually diagnosed in infancy or childhood and have evidence of growth impairment.
Gitelman syndrome is an autosomal recessive disorder of the thiazide-sensitive sodium-chloride cotransporter. Although Gitelman syndrome is more common than Bartter syndrome and presents at older ages, it is not usually associated with such profound metabolic alkalosis. Gitelman syndrome mimics chronic use of thiazide diuretics and is associated with hypocalciuria.
Diuretic use could also cause the metabolic disturbances described; however, the patient denied taking diuretics.
The most common cause of hypokalemia in clinical practice is diuretic use.4 In this young woman with unexplained hypokalemia, the most likely cause is either undisclosed self-induced vomiting or diuretic abuse. The degree of metabolic alkalosis suggests vomiting, since metabolic alkalosis this severe is usually seen only with protracted vomiting. Bartter and Gitelman syndromes are included in the differential diagnosis, but they are much less common than hypokalemia associated with diuretics or self-induced vomiting.5
3. Which test could help elucidate the cause of hypokalemia in this patient?
- Ratio of plasma aldosterone to rennin
- Urine chloride
- Ratio of urinary potassium to creatinine
- Urinary anion gap and urinary pH
APPROACH TO HYPOKALEMIA
Determining the cause of hypokalemia starts with a thorough history and physical examination. The history should focus on drugs such as diuretics and laxatives. Women should be asked about their menstrual history since irregular periods may suggest an eating disorder. The physical examination should focus on signs that suggest self-induced vomiting, such as dry skin, dental erosions, enlarged parotid glands, and calluses or scars on the knuckles.
Patients with an unclear cause of hypokalemia after a thorough history and physical examination can be categorized into one of three groups based on blood pressure and acid-base status:
- Hypokalemia, hypertension, metabolic alkalosis
- Hypokalemia, normal blood pressure, metabolic acidosis
- Hypokalemia, normal blood pressure, metabolic alkalosis.
Hypokalemia, hypertension, metabolic alkalosis
The blood pressure provides an important clue in the evaluation of hypokalemia. The combination of hypertension, hypokalemia, and alkalosis should raise concern for hyperaldosteronism or pseudoaldosteronism. Primary hyperaldosteronism from an adrenal adenoma (Conn syndrome) is characterized by a plasma aldosterone-renin ratio of greater than 20.6,7 In contrast, patients with secondary hyperaldosteronism due to renovascular disease have a plasma aldosterone-renin ratio of less than 10. Patients with pseudoaldosteronism have low aldosterone and renin levels and hypertension. Since our patient has a normal blood pressure, testing the plasma aldosterone and renin levels would not help determine the cause of her hypokalemia.
Hypokalemia, normal blood pressure, metabolic acidosis
Patients with normal blood pressure, hypokalemia, and normal plasma anion gap acidosis either have renal tubular acidosis or have lost potassium because of diarrhea or laxative abuse. In a patient who denies taking laxatives or denies a history of diarrhea, checking the urinary anion gap and urinary pH may help differentiate the cause of acidosis and hypokalemia.
The urinary anion gap, calculated by the equation sodium + potassium − chloride, is an indirect estimate of hydrogen excretion in the form of ammonium ion8; the normal value is 0 to 10 mEq/L. A negative value represents increased hydrogen excretion in response to systemic acidosis from gastrointestinal or renal loss of bicarbonate (proximal renal tubular acidosis). A urinary pH greater than 5.5 in the setting of systemic acidosis suggests impaired ability of the kidneys to acidify urine and raises the possibility of renal tubular acidosis.
This patient has metabolic alkalosis, so calculation of the urinary anion gap would not be helpful.
Hypokalemia, normal blood pressure, metabolic alkalosis
Patients such as ours, with normal blood pressure, hypokalemia, and alkalosis, have been vomiting, have used diuretics, or have an inherited renal tubulopathy such as Bartter or Gitelman syndrome. Usually, differentiating Bartter and Gitelman syndromes from chronic vomiting or diuretic use is done with the history and physical examination. However, in patients with a questionable history and a lack of findings on physical examination, checking the urinary chloride, potassium, calcium, and creatinine may be helpful.
A urinary potassium-creatinine ratio greater than 15 suggests renal loss, whereas a ratio less than 15 suggests extrarenal loss.9
Patients who are taking a diuretic or who have Bartter or Gitelman syndrome have a high urinary chloride concentration, ie, greater than 20 mmol/L, whereas patients with hypokalemia and alkalosis from chronic vomiting tend to have a concentration less than 10 mmol/L.10
Table 1 summarizes an approach to the evaluation of unexplained hypokalemia based on blood pressure and acid-base status.
A HIDDEN HISTORY
On further questioning, the patient admits to an 8-year history of daily self-induced vomiting in an attempt to lose weight, in addition to multiple hospitalizations for hypokalemia and a previous diagnosis of an eating disorder.
INITIAL MANAGEMENT OF HYPOKALEMIA
The initial management of hypokalemia should focus on life-threatening emergencies. While patients with potassium levels greater than 3 mmol/L are usually asymptomatic, those with levels below 3 mmol/L present with muscle weakness and rhabdomyolysis.4 An acute drop in serum potassium to less than 2 mmol/L is associated with respiratory muscle weakness and ventricular arrhythmias.4 If the patient has cardiac symptoms or hypoventilation due to respiratory muscle weakness, continuous monitoring in the intensive care unit and aggressive therapy are warranted.
4. Which potassium formulation is most appropriate for the treatment of hypokalemia in this patient?
- Potassium chloride
- Potassium phosphate
- Potassium acetate
Oral potassium is preferable in patients with a serum potassium above 2.5 mmol/L.4,11 Potassium phosphate should be used when supplementation with both potassium and phosphorus is needed. Potassium acetate should be reserved for patients with acidosis and hypokalemia. Otherwise, potassium chloride is typically preferred.4,12 It comes in liquid and tablet forms. Liquid forms have an unpleasant taste, whereas tablets are usually well tolerated. No more than 20 to 40 mEq of potassium chloride tablets should be given at a time, since higher doses are associated with gastrointestinal mucosal injury.12
Potassium chloride is particularly preferred in patients with metabolic alkalosis, since increased chloride intake and delivery to the distal tubule increases the expression of pendrin, a luminal chloride and bicarbonate exchanger in the cortical collecting duct.13 With metabolic alkalosis, increased excretion of bicarbonate occurs through up-regulation of pendrin. Potassium depletion down-regulates pendrin.13 Additionally, correction of metabolic alkalosis increases serum potassium by movement of potassium from the intracellular to the extracellular space.
Intravenous potassium should be reserved for patients with severe hypokalemia (< 2.5 mmol/L) or significant arrhythmias.11 Oral and intravenous potassium can safely be given simultaneously.11 The intravenous rate should not exceed more than 10 to 20 mEq of potassium chloride per hour unless the patient has a life-threatening arrhythmia, respiratory failure, or severe hypokalemia.14,15 In life-threatening situations, a femoral line should be placed, and potassium should be given as rapidly as 20 mEq over 15 to 20 minutes.14 Cannulation of the subclavian and internal jugular veins should be avoided in severe hypokalemia since mechanical irritation from guidewire placement can provoke ventricular arrhythmias.14
During intravenous administration of potassium, laboratory monitoring after every 20 mEq of potassium chloride is advised because of the possibility of rebound hyperkalemia. In patients with severe hypokalemia, avoidance of factors that can worsen intracellular shift of potassium is also important. Avoid dextrose-containing fluids to prevent insulin-induced shifting of potassium into cells. Restore intravascular volume to blunt hypovolemia-induced renin and aldosterone secretion. If a patient presents with severe hypokalemia and acidosis, correct the hypokalemia before the acidosis to avoid intracellular shift of potassium.
OUR PATIENT’S MANAGEMENT AND FOLLOW-UP PLAN
Given the severity of our patient’s hypokalemia and her complaint of palpitations, she was admitted to the hospital for monitoring. She required 180 mEq of intravenous potassium chloride and 140 mEq of oral potassium chloride during the first 24 hours in order to achieve a serum potassium level above 3 mmol/L. Electrocardiographic U waves resolved once the level was above 2 mmol/L, and ST depressions resolved once it was above 3 mmol/L. The QT interval normalized after 24 hours of hospitalization.
On discharge, she was prescribed oral potassium chloride 40 mEq daily and magnesium sulfate 400 mg twice daily, with plans for a followup visit with her outpatient therapy team, which includes a psychiatrist, a social worker, and her primary care provider. She declined a referral for inpatient therapy but agreed to a goal of decreasing the frequency of induced vomiting and outpatient visits. She was also educated on how and when to access emergency medical care.16
A 20-year-old woman presents to the emergency department with fatigue and the sudden onset of palpitations. She reports no history of significant illness or surgery. She says she is not currently taking prescription or over-the-counter medications. She does not smoke, drink alcohol, or use illicit drugs.
Her weight is 52 kg (115 lb), her height is 170 cm (67 in), and her body mass index (BMI) is 18 kg/m2. Vital signs: temperature 35.7°C (96.4°F), blood pressure 92/48 mm Hg, heart rate 73 bpm, respiratory rate 5 breaths per minute, and oxygen saturation 98% on room air.
She appears tired but is alert, conversant, and cooperative. Her skin is normal, and dentition is fair. Her pulse is regular, and respirations are slow. The abdomen is soft, non-tender, and flat. Strength is 4 on a scale of 5 in all extremities. Deep-tendon reflexes are 2+ and symmetric.
Electrocardiography (Figure 1) in the emergency department shows ST-segment depression, a prolonged corrected QT interval of 665 msec, T-wave inversion, PR prolongation, increased P-wave amplitude, and U waves.
1. Which electrolyte abnormality is associated with this electrocardiographic picture?
- Hypercalcemia
- Hyperkalemia
- Hypocalcemia
- Hypokalemia
Hypokalemia is the likely cause of these findings. The finding of U waves is considered significant when they are inverted, merged with the T wave, or have an amplitude greater than the T wave.1 U waves are best seen in the precordial leads. When severe, hypokalemia can lead to potentially fatal arrhythmias such as high-grade atrioventricular block, ventricular tachycardia, and ventricular fibrillation.2
Hyperkalemia is associated with peaked T waves, a prolonged PR interval, decreased P wave amplitude, and a widened QRS complex.2 When acute and severe, hyperkalemia is associated with ventricular arrhythmia.
Hypocalcemia is associated with a prolonged QT interval and ventricular dysrhythmia, but not U waves.2
Hypercalcemia is associated with bradydysrhythmia, as well as with a shortened QT interval.2
LABORATORY TESTING
Laboratory testing shows the following:
- Sodium 126 mmol/L (reference range 135–145)
- Potassium 1.5 mmol/L (3.5–5.1)
- Chloride 58 mmol/L (100–110)
- Bicarbonate 62 mmol/L (20–30)
- Blood urea nitrogen 16 mg/dL (7–18)
- Creatinine 0.8 mg/dL (0.5–1.0)
- Glucose 106 mg/dL (70–110)
- Ionized calcium 4.4 mg/dL (4.5–5.3)
- Magnesium 1.8 mg/dL (1.7–2.3)
- Phosphorus 4.1 mg/dL (2.5–4.5)
- Venous blood gases pH 7.56 (7.35–7.45), Pco2 69 mm Hg (35–45).
POTASSIUM HOMEOSTASIS
Ninety-eight percent of potassium is intracellular and only 2% is extracellular.3 The main cellular stores are myocytes and hepatocytes. Patients with decreased muscle mass may be at a higher risk of hypokalemia as a result of decreased skeletal muscle stores.4
The acute development of hypokalemia occurs from transcellular shifts. Alkalosis, insulin secretion, and beta-adrenergic stimulation promote the intracellular uptake of potassium. The major hormonal regulator of potassium excretion is aldosterone, which is stimulated by renal hypoperfusion and promotes potassium-ion secretion in the distal convoluted tubule.
Chronic hypokalemia develops in patients with ongoing renal or gastrointestinal potassium loss. If the cause of potassium loss is not elucidated by the history, the physical, and a review of medications, then one of two things is possible: either the patient has renal tubular disease affecting acid-base and potassium regulation, causing excessive mineralocorticoid secretion, which is associated with an abnormal response to aldosterone; or the patient is not being forthcoming in the history.
2. Which is the most likely cause of hypokalemia in this patient?
- Vomiting
- Liddle syndrome
- Bartter syndrome
- Gitelman syndrome
- Diuretic use
Her laboratory tests reveal hypokalemia and hyponatremic-hypochloremic metabolic alkalosis with compensatory respiratory acidosis. In metabolic alkalosis, the expected respiratory compensation is an increase of 0.7 mm Hg in Pco2 for each 1-mEq/L increase in bicarbonate. Therefore, the expected Pco2 is 67, close to the patient’s actual value of 69.
Protracted vomiting with loss of gastric acid juices could be a cause of the metabolic disturbances in this young woman, although she did not mention vomiting during the history.
Liddle syndrome, or pseudoaldosteronism, is a rare autosomal dominant disorder characterized by altered renal epithelial sodium channels, excessive sodium retention, and resultant hypertension. Hypokalemia and alkalosis are seen in Liddle syndrome, but the absence of hypertension in our patient makes Liddle syndrome unlikely.
Bartter syndrome is an inherited autosomal recessive disorder of the sodium-potassium-chloride cotransporter in the thick ascending loop of Henle, resulting in impaired reabsorption of chloride and sodium. Bartter syndrome mimics chronic loop-diuretic use and is associated with hypercalciuria. Bartter syndrome is possible in this patient; however, patients with Bartter syndrome are usually diagnosed in infancy or childhood and have evidence of growth impairment.
Gitelman syndrome is an autosomal recessive disorder of the thiazide-sensitive sodium-chloride cotransporter. Although Gitelman syndrome is more common than Bartter syndrome and presents at older ages, it is not usually associated with such profound metabolic alkalosis. Gitelman syndrome mimics chronic use of thiazide diuretics and is associated with hypocalciuria.
Diuretic use could also cause the metabolic disturbances described; however, the patient denied taking diuretics.
The most common cause of hypokalemia in clinical practice is diuretic use.4 In this young woman with unexplained hypokalemia, the most likely cause is either undisclosed self-induced vomiting or diuretic abuse. The degree of metabolic alkalosis suggests vomiting, since metabolic alkalosis this severe is usually seen only with protracted vomiting. Bartter and Gitelman syndromes are included in the differential diagnosis, but they are much less common than hypokalemia associated with diuretics or self-induced vomiting.5
3. Which test could help elucidate the cause of hypokalemia in this patient?
- Ratio of plasma aldosterone to rennin
- Urine chloride
- Ratio of urinary potassium to creatinine
- Urinary anion gap and urinary pH
APPROACH TO HYPOKALEMIA
Determining the cause of hypokalemia starts with a thorough history and physical examination. The history should focus on drugs such as diuretics and laxatives. Women should be asked about their menstrual history since irregular periods may suggest an eating disorder. The physical examination should focus on signs that suggest self-induced vomiting, such as dry skin, dental erosions, enlarged parotid glands, and calluses or scars on the knuckles.
Patients with an unclear cause of hypokalemia after a thorough history and physical examination can be categorized into one of three groups based on blood pressure and acid-base status:
- Hypokalemia, hypertension, metabolic alkalosis
- Hypokalemia, normal blood pressure, metabolic acidosis
- Hypokalemia, normal blood pressure, metabolic alkalosis.
Hypokalemia, hypertension, metabolic alkalosis
The blood pressure provides an important clue in the evaluation of hypokalemia. The combination of hypertension, hypokalemia, and alkalosis should raise concern for hyperaldosteronism or pseudoaldosteronism. Primary hyperaldosteronism from an adrenal adenoma (Conn syndrome) is characterized by a plasma aldosterone-renin ratio of greater than 20.6,7 In contrast, patients with secondary hyperaldosteronism due to renovascular disease have a plasma aldosterone-renin ratio of less than 10. Patients with pseudoaldosteronism have low aldosterone and renin levels and hypertension. Since our patient has a normal blood pressure, testing the plasma aldosterone and renin levels would not help determine the cause of her hypokalemia.
Hypokalemia, normal blood pressure, metabolic acidosis
Patients with normal blood pressure, hypokalemia, and normal plasma anion gap acidosis either have renal tubular acidosis or have lost potassium because of diarrhea or laxative abuse. In a patient who denies taking laxatives or denies a history of diarrhea, checking the urinary anion gap and urinary pH may help differentiate the cause of acidosis and hypokalemia.
The urinary anion gap, calculated by the equation sodium + potassium − chloride, is an indirect estimate of hydrogen excretion in the form of ammonium ion8; the normal value is 0 to 10 mEq/L. A negative value represents increased hydrogen excretion in response to systemic acidosis from gastrointestinal or renal loss of bicarbonate (proximal renal tubular acidosis). A urinary pH greater than 5.5 in the setting of systemic acidosis suggests impaired ability of the kidneys to acidify urine and raises the possibility of renal tubular acidosis.
This patient has metabolic alkalosis, so calculation of the urinary anion gap would not be helpful.
Hypokalemia, normal blood pressure, metabolic alkalosis
Patients such as ours, with normal blood pressure, hypokalemia, and alkalosis, have been vomiting, have used diuretics, or have an inherited renal tubulopathy such as Bartter or Gitelman syndrome. Usually, differentiating Bartter and Gitelman syndromes from chronic vomiting or diuretic use is done with the history and physical examination. However, in patients with a questionable history and a lack of findings on physical examination, checking the urinary chloride, potassium, calcium, and creatinine may be helpful.
A urinary potassium-creatinine ratio greater than 15 suggests renal loss, whereas a ratio less than 15 suggests extrarenal loss.9
Patients who are taking a diuretic or who have Bartter or Gitelman syndrome have a high urinary chloride concentration, ie, greater than 20 mmol/L, whereas patients with hypokalemia and alkalosis from chronic vomiting tend to have a concentration less than 10 mmol/L.10
Table 1 summarizes an approach to the evaluation of unexplained hypokalemia based on blood pressure and acid-base status.
A HIDDEN HISTORY
On further questioning, the patient admits to an 8-year history of daily self-induced vomiting in an attempt to lose weight, in addition to multiple hospitalizations for hypokalemia and a previous diagnosis of an eating disorder.
INITIAL MANAGEMENT OF HYPOKALEMIA
The initial management of hypokalemia should focus on life-threatening emergencies. While patients with potassium levels greater than 3 mmol/L are usually asymptomatic, those with levels below 3 mmol/L present with muscle weakness and rhabdomyolysis.4 An acute drop in serum potassium to less than 2 mmol/L is associated with respiratory muscle weakness and ventricular arrhythmias.4 If the patient has cardiac symptoms or hypoventilation due to respiratory muscle weakness, continuous monitoring in the intensive care unit and aggressive therapy are warranted.
4. Which potassium formulation is most appropriate for the treatment of hypokalemia in this patient?
- Potassium chloride
- Potassium phosphate
- Potassium acetate
Oral potassium is preferable in patients with a serum potassium above 2.5 mmol/L.4,11 Potassium phosphate should be used when supplementation with both potassium and phosphorus is needed. Potassium acetate should be reserved for patients with acidosis and hypokalemia. Otherwise, potassium chloride is typically preferred.4,12 It comes in liquid and tablet forms. Liquid forms have an unpleasant taste, whereas tablets are usually well tolerated. No more than 20 to 40 mEq of potassium chloride tablets should be given at a time, since higher doses are associated with gastrointestinal mucosal injury.12
Potassium chloride is particularly preferred in patients with metabolic alkalosis, since increased chloride intake and delivery to the distal tubule increases the expression of pendrin, a luminal chloride and bicarbonate exchanger in the cortical collecting duct.13 With metabolic alkalosis, increased excretion of bicarbonate occurs through up-regulation of pendrin. Potassium depletion down-regulates pendrin.13 Additionally, correction of metabolic alkalosis increases serum potassium by movement of potassium from the intracellular to the extracellular space.
Intravenous potassium should be reserved for patients with severe hypokalemia (< 2.5 mmol/L) or significant arrhythmias.11 Oral and intravenous potassium can safely be given simultaneously.11 The intravenous rate should not exceed more than 10 to 20 mEq of potassium chloride per hour unless the patient has a life-threatening arrhythmia, respiratory failure, or severe hypokalemia.14,15 In life-threatening situations, a femoral line should be placed, and potassium should be given as rapidly as 20 mEq over 15 to 20 minutes.14 Cannulation of the subclavian and internal jugular veins should be avoided in severe hypokalemia since mechanical irritation from guidewire placement can provoke ventricular arrhythmias.14
During intravenous administration of potassium, laboratory monitoring after every 20 mEq of potassium chloride is advised because of the possibility of rebound hyperkalemia. In patients with severe hypokalemia, avoidance of factors that can worsen intracellular shift of potassium is also important. Avoid dextrose-containing fluids to prevent insulin-induced shifting of potassium into cells. Restore intravascular volume to blunt hypovolemia-induced renin and aldosterone secretion. If a patient presents with severe hypokalemia and acidosis, correct the hypokalemia before the acidosis to avoid intracellular shift of potassium.
OUR PATIENT’S MANAGEMENT AND FOLLOW-UP PLAN
Given the severity of our patient’s hypokalemia and her complaint of palpitations, she was admitted to the hospital for monitoring. She required 180 mEq of intravenous potassium chloride and 140 mEq of oral potassium chloride during the first 24 hours in order to achieve a serum potassium level above 3 mmol/L. Electrocardiographic U waves resolved once the level was above 2 mmol/L, and ST depressions resolved once it was above 3 mmol/L. The QT interval normalized after 24 hours of hospitalization.
On discharge, she was prescribed oral potassium chloride 40 mEq daily and magnesium sulfate 400 mg twice daily, with plans for a followup visit with her outpatient therapy team, which includes a psychiatrist, a social worker, and her primary care provider. She declined a referral for inpatient therapy but agreed to a goal of decreasing the frequency of induced vomiting and outpatient visits. She was also educated on how and when to access emergency medical care.16
- Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT Interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
- Diercks DB, Shumaik GM, Harrigan RA, Brady WJ, Chan TC. Electrocardiographic manifestations: electrolyte abnormalities. J Emerg Med 2004; 27:153–160.
- Unwin RJ, Luft FC, Shirley DG. Pathophysiology and management of hypokalemia: a clinical perspective. Nat Rev Nephrol 2011; 7:75–84.
- Gennari FJ. Hypokalemia. N Engl J Med 1998; 339:451–458.
- Mehler PS. Clinical practice. Bulimia nervosa. N Engl J Med 2003; 349:875–881.
- Tzanela M, Effraimidis G, Vassiliadi D, et al. The aldosterone to renin ratio in the evaluation of patients with incidentally detected adrenal masses. Endocrine 2007; 32:136–142.
- Diederich S, Mai K, Bähr V, Helffrich S, Pfeiffer A, Perschel FH. The simultaneous measurement of plasma-aldosterone- and -renin-concentration allows rapid classification of all disorders of the renin-aldosterone system. Exp Clin Endocrinol Diabetes 2007; 115:433–438.
- Goldstein MB, Bear R, Richardson RM, Marsden PA, Halperin ML. The urine anion gap: a clinically useful index of ammonium excretion. Am J Med Sci 1986; 292:198–202.
- Groeneveld JH, Sijpkens YW, Lin SH, Davids MR, Halperin ML. An approach to the patient with severe hypokalaemia: the potassium quiz. QJM 2005; 98:305–316.
- Galla JH. Metabolic alkalosis. J Am Soc Nephrol 2000; 11:369–375.
- Asmar A, Mohandas R, Wingo CS. A physiologic-based approach to the treatment of a patient with hypokalemia. Am J Kidney Dis 2012; 60:492–497.
- Cohn JN, Kowey PR, Whelton PK, Prisant LM. New guidelines for potassium replacement in clinical practice: a contemporary review by the National Council on Potassium in Clinical Practice. Arch Intern Med 2000; 160:2429–2436.
- Luke RG, Galla JH. It is chloride depletion alkalosis, not contraction alkalosis. J Am Soc Nephrol 2012; 23:204–207.
- Kruse JA, Carlson RW. Rapid correction of hypokalemia using concentrated intravenous potassium chloride infusions. Arch Intern Med 1990; 150:613–617.
- Weiner ID, Wingo CS. Hypokalemia—consequences, causes, and correction. J Am Soc Nephrol 1997; 8:1179–1188.
- AED Medical Care Standards Task Force. Eating disorders: Critical Points for Early Recognition and Medical Risk Management in the Care of Individuals with Eating Disorders. AED Report 2012. www.aedweb.org/web/downloads/Guide-English.pdf. Accessed April 4, 2014.
- Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT Interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
- Diercks DB, Shumaik GM, Harrigan RA, Brady WJ, Chan TC. Electrocardiographic manifestations: electrolyte abnormalities. J Emerg Med 2004; 27:153–160.
- Unwin RJ, Luft FC, Shirley DG. Pathophysiology and management of hypokalemia: a clinical perspective. Nat Rev Nephrol 2011; 7:75–84.
- Gennari FJ. Hypokalemia. N Engl J Med 1998; 339:451–458.
- Mehler PS. Clinical practice. Bulimia nervosa. N Engl J Med 2003; 349:875–881.
- Tzanela M, Effraimidis G, Vassiliadi D, et al. The aldosterone to renin ratio in the evaluation of patients with incidentally detected adrenal masses. Endocrine 2007; 32:136–142.
- Diederich S, Mai K, Bähr V, Helffrich S, Pfeiffer A, Perschel FH. The simultaneous measurement of plasma-aldosterone- and -renin-concentration allows rapid classification of all disorders of the renin-aldosterone system. Exp Clin Endocrinol Diabetes 2007; 115:433–438.
- Goldstein MB, Bear R, Richardson RM, Marsden PA, Halperin ML. The urine anion gap: a clinically useful index of ammonium excretion. Am J Med Sci 1986; 292:198–202.
- Groeneveld JH, Sijpkens YW, Lin SH, Davids MR, Halperin ML. An approach to the patient with severe hypokalaemia: the potassium quiz. QJM 2005; 98:305–316.
- Galla JH. Metabolic alkalosis. J Am Soc Nephrol 2000; 11:369–375.
- Asmar A, Mohandas R, Wingo CS. A physiologic-based approach to the treatment of a patient with hypokalemia. Am J Kidney Dis 2012; 60:492–497.
- Cohn JN, Kowey PR, Whelton PK, Prisant LM. New guidelines for potassium replacement in clinical practice: a contemporary review by the National Council on Potassium in Clinical Practice. Arch Intern Med 2000; 160:2429–2436.
- Luke RG, Galla JH. It is chloride depletion alkalosis, not contraction alkalosis. J Am Soc Nephrol 2012; 23:204–207.
- Kruse JA, Carlson RW. Rapid correction of hypokalemia using concentrated intravenous potassium chloride infusions. Arch Intern Med 1990; 150:613–617.
- Weiner ID, Wingo CS. Hypokalemia—consequences, causes, and correction. J Am Soc Nephrol 1997; 8:1179–1188.
- AED Medical Care Standards Task Force. Eating disorders: Critical Points for Early Recognition and Medical Risk Management in the Care of Individuals with Eating Disorders. AED Report 2012. www.aedweb.org/web/downloads/Guide-English.pdf. Accessed April 4, 2014.
An 85-year-old with muscle pain
An 85-year-old man with hypertension, hyperlipidemia, and coronary artery disease presented to our clinic with diffuse muscle pain. The pain had been present for about 3 months, but it had become noticeably worse over the past few weeks.
He was not aware of any trauma. He described the muscle pain as dull and particularly severe in his lower extremities (his thighs and calves). The pain did not limit his daily activities, nor did physical exertion or the time of day have any effect on the level of the pain.
His medications at that time included metoprolol, aspirin, hydrochlorothiazide, simvastatin, and a daily multivitamin.
He was not in acute distress. On neurologic and musculoskeletal examinations, all deep-tendon reflexes were intact, with no tenderness to palpation of the upper and lower extremities. No abnormalities were noted on the joint examination. He had full range of motion, with 5/5 muscle strength in the upper and lower extremities bilaterally and normal muscle tone. He was able to walk with ease. Results of initial laboratory testing, including creatine kinase and erythrocyte sedimentation rate, were normal.
1. What should be the next best step in the evaluation of this patient’s muscle pain?
- Order tests for cyclic citrullinated peptide (CCP) antibody and rheumatoid factor
- Advise him to refrain from physical activity until his symptoms resolve
- Take a more detailed history, including a review of medications and supplements
- Recommend a trial of a nonsteroidal anti-inflammatory drug (NSAID)
- Send him for radiographic imaging
Since his muscle pain has persisted for several months without improvement, a more detailed history should be taken, including a review of current medications and supplements.
Testing CCP antibody and rheumatoid factor would be useful if rheumatoid arthritis were suspected, but in the absence of demonstrable arthritis on examination, these tests would have low specificity even if the results were positive.
An NSAID may temporarily alleviate his pain, but it will not help establish a diagnosis. And in elderly patients, NSAIDs are not without complications and so should be prescribed only in appropriate situations.
Imaging would be appropriate at this point only if there was clinical suspicion of a specific disease. However, our patient has no focal deficits, and the suspicion of fracture or malignancy is low.
The medical history should include asking about current drug regimens, recent medication changes, and the use of herbal supplements, since polypharmacy is common in elderly patients with multiple comorbidities.
On further questioning, our patient said that his dose of simvastatin had been increased from 40 mg daily to 80 mg daily about 1 month before his symptoms appeared. He was taking a daily multivitamin but was not using herbal supplements or other over-the-counter products. He did not recall any constitutional symptoms before the onset of his current symptoms, and he had never had similar muscle pain in the past.
2. Based on the additional information from the history, what is the most likely cause of his muscle pain?
- Limited myositis secondary to recent viral infection
- Rhabdomyolysis
- Hypothyroidism
- Drug-drug interaction
- Statin-induced myalgia
Our patient’s history provided nothing to suggest viral myositis. Hypothyroidism should always be considered in patients with myalgia, but this is not likely in our patient, as he does not display other characteristics, such as diminished reflexes, hypotonia, cold intolerance, and mood instability. Even though calcium channel blockers have been known to cause myalgia in patients on statins, a drug-drug reaction is not likely, as he had not started taking a calcium channel blocker before his symptoms began. This patient did not show signs or symptoms of rhabdomyolysis, a type of myopathy in which necrosis of the muscle tissue occurs, generally causing profound weakness and pain.1
Therefore, statin-induced myopathy is the most likely cause of his diffuse muscle pain, particularly since his simvastatin had been increased 1 month before the onset of symptoms.
3. What should be the next step in his management?
- Decrease the dose of simvastatin to the last known dose he was able to tolerate
- Continue simvastatin at the same dose and then monitor
- Switch to another statin
- Add coenzyme Q10
- Stop simvastatin
Decreasing the statin dosage to the last well-tolerated dose would not be appropriate in a patient with myopathy, as the symptoms would probably not improve.2–4 Also, one should not switch to a different statin while a patient is experiencing symptoms. Rather, the statin should be stopped for at least 6 weeks or until the symptoms have fully resolved.1
Adding coenzyme Q10 is another option, especially in a patient with previously diagnosed coronary artery disease,5 when continued statin therapy is thought necessary to reduce the likelihood of repeat coronary events.
We discontinued his simvastatin. Followup 3 weeks later in the outpatient clinic showed that his symptoms were slowly improving. The symptoms had resolved completely 4 months later.
4. How should we manage our patient’s hyperlipidemia once his symptoms have resolved?
- Restart simvastatin at the 80-mg dose
- Restart simvastatin at the 40-mg dose
- Start a hydrophilic statin at full dose
- Use a drug from another class of lipid-lowering drugs
- Wait another 3 months before prescribing any lipid-lowering drug
His treatment for hyperlipidemia should be continued, considering his comorbidities. However, restarting the same statin, even at a lower dose, will likely cause his symptoms to recur. Thus, a different statin should be tried once his muscle pain has resolved.
Other classes of lipid-lowering drugs are usually less efficacious than statins, particularly when trying to control low-density lipoprotein (LDL) cholesterol, so a drug from another class should not be used until other statin options have been attempted.2,6,7
Simvastatin is lipophilic. Trying a statin with hydrophilic properties (eg, pravastatin, rosuvastatin, fluvastatin) has been shown to convey similar cardioprotective effects with a lower propensity for myalgia, as lipophilic statins have a higher propensity to penetrate muscle tissue than do hydrophilic statins.3,4,8
Once his symptoms resolved, our patient was started on a hydrophilic statin, fluvastatin 20 mg daily. Unfortunately, his pain recurred 3 weeks later. The statin was stopped, and his symptoms again resolved.
5. Since our patient was unable to tolerate a second statin, what should be the next step in his management?
- Restart simvastatin
- Use a drug from another class to control the hyperlipidemia
- Wait at least 6 months after symptoms resolve before trying any lipid-lowering drug
- Initiate therapy with coenzyme Q10 and fish oil
- Wait for symptoms to resolve, then restart a hydrophilic statin at a lower dose and lower frequency
Restarting simvastatin will likely cause a recurrence of the myalgia. Other lipid-lowering drugs such as nicotinic acid, bile acid resins, and fibrates are not as efficacious as statins. Coenzyme Q10 and fish oil can reduce lipid levels, but they are not as efficacious as statins.
In view of our patient’s lipid profile—LDL cholesterol elevated at 167 mg/dL, high-density lipoprotein cholesterol 31 mg/dL, triglycerides 47 mg/dL—it is important to treat his hyperlipidemia. Therefore, another attempt at statin therapy should be made once his symptoms have resolved.
Studies have shown that restarting a statin at a low dose and low frequency is effective in patients who have experienced intolerance to a statin.3,4 Our patient was treated with low-dose pravastatin (20 mg), resulting in a moderate improvement in his LDL cholesterol to 123 mg/dL.
STATIN-INDUCED MYOPATHY: ADDRESSING THE DILEMMA
Treating hyperlipidemia is important to prevent vascular events in patients with or without coronary artery disease. Statins are the most effective agents available for controlling hypercholesterolemia, specifically LDL levels, as well as for preventing myocardial infarction.
Unfortunately, significant side effects have been reported, and myopathy is the most prevalent. Statin-induced myopathy includes a combination of muscle tenderness, myalgia, and weakness.2–11 In randomized controlled trials, the risk of myopathy was estimated to be between 1.5% and 5%.6 In unselected clinic patients on high-dose statins, the rate of muscle complaints may be as high as 20%.12
The cause of statin-induced myopathy is not known, although studies have linked it to genetic defects.7 Risk factors have been identified and include personal and family history of myalgia, Asian ethnicity, hypothyroidism, and type 1 diabetes. The incidence of statin-induced myalgia is two to three times higher in patients on corticosteroid therapy. Other risk factors include female sex, liver disease, and renal dysfunction.7,8
A less common etiology is anti-HMG coenzyme A reductase antibodies. Studies have shown that these antibody levels correlate well with the amount of myositis as measured by creatine kinase levels. However, there is no consensus yet on screening for these antibodies.13
Statin therapy poses a dilemma, as there is a thin line between the benefits and the risks of side effects, especially statin-induced myopathy.3,4 Current recommendations include discontinuing the statin until symptoms fully resolve. Creatine kinase levels may be useful in assessing for potential muscle breakdown, especially in patients with reduced renal function, as this predisposes them to statin-induced myopathy, yet normal values do not preclude the diagnosis of statin-induced myopathy.3,4,7,8
Once symptoms resolve and laboratory test results normalize, a trial of a different statin is recommended. If patients become symptomatic, a trial of a low-dose hydrophilic statin at a once- or twice-weekly interval has been recommended. Several studies have assessed the efficacy of a low-dose statin with decreased frequency of administration and have consistently shown significant improvement in lipid levels.3,4 For instance, once-weekly rosuvastatin at a dose between 5 mg and 20 mg resulted in a 29% reduction in LDL cholesterol levels, and 80% of patients did not experience a recurrence of myalgia.3 Furthermore, a study of patients treated with 5 mg to 10 mg of rosuvastatin twice a week resulted in a 26% decrease in LDL cholesterol levels.4 This study also showed that when an additional non-statin lipid-lowering drug was prescribed (eg, ezetimibe, bile acid resin, nicotinic acid), more than half of the patients reached their goal lipid level.4
The addition of coenzyme Q10 and fish oil has also been suggested. Although, the evidence to support this is inconclusive, the potential benefit outweighs the risk, since the side effects are minimal.1 However, no study yet has evaluated the risks vs the benefits in patients with elevated creatine kinase.
Statin-induced myopathy is a commonly encountered adverse effect. Currently, there are no guidelines on restarting statin therapy after statin-induced myopathy; however, data suggest that statin therapy should be restarted once symptoms resolve, and that variations in dose and frequency may be necessary.1–8,14
- Fernandez G, Spatz ES, Jablecki C, Phillips PS. Statin myopathy: a common dilemma not reflected in clinical trials. Cleve Clin J Med 2011; 78:393–403.
- Foley KA, Simpson RJ, Crouse JR, Weiss TW, Markson LE, Alexander CM. Effectiveness of statin titration on low-density lipoprotein cholesterol goal attainment in patients at high risk of atherogenic events. Am J Cardiol 2003; 92:79–81.
- Backes JM, Moriarty PM, Ruisinger JF, Gibson CA. Effects of once weekly rosuvastatin among patients with a prior statin intolerance. Am J Cardiol 2007; 100:554–555.
- Gadarla M, Kearns AK, Thompson PD. Efficacy of rosuvastatin (5 mg and 10 mg) twice a week in patients intolerant to daily statins. Am J Cardiol 2008; 101:1747–1748.
- Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme q10 on myopathic symptoms in patients treated with statins. Am J Cardiol 2007; 99:1409–1412.
- Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Tomaszewski M, Stepien KM, Tomaszewska J, Czuczwar SJ. Statin-induced myopathies. Pharmacol Rep 2011; 63:859–866.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Heart Protection Study Collaborative Group. MRC/BHF heart protection study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Guyton JR. Benefit versus risk in statin treatment. Am J Cardiol 2006; 97:95C–97C.
- Buettner C, Davis RB, Leveille SG, Mittleman MA, Mukamal KJ. Prevalence of musculoskeletal pain and statin use. J Gen Intern Med 2008; 23:1182–1186.
- Werner JL, Christopher-Stine L, Ghazarian SR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum 2012; 64:4087–4093.
- The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 1998; 339:1349–1357.
An 85-year-old man with hypertension, hyperlipidemia, and coronary artery disease presented to our clinic with diffuse muscle pain. The pain had been present for about 3 months, but it had become noticeably worse over the past few weeks.
He was not aware of any trauma. He described the muscle pain as dull and particularly severe in his lower extremities (his thighs and calves). The pain did not limit his daily activities, nor did physical exertion or the time of day have any effect on the level of the pain.
His medications at that time included metoprolol, aspirin, hydrochlorothiazide, simvastatin, and a daily multivitamin.
He was not in acute distress. On neurologic and musculoskeletal examinations, all deep-tendon reflexes were intact, with no tenderness to palpation of the upper and lower extremities. No abnormalities were noted on the joint examination. He had full range of motion, with 5/5 muscle strength in the upper and lower extremities bilaterally and normal muscle tone. He was able to walk with ease. Results of initial laboratory testing, including creatine kinase and erythrocyte sedimentation rate, were normal.
1. What should be the next best step in the evaluation of this patient’s muscle pain?
- Order tests for cyclic citrullinated peptide (CCP) antibody and rheumatoid factor
- Advise him to refrain from physical activity until his symptoms resolve
- Take a more detailed history, including a review of medications and supplements
- Recommend a trial of a nonsteroidal anti-inflammatory drug (NSAID)
- Send him for radiographic imaging
Since his muscle pain has persisted for several months without improvement, a more detailed history should be taken, including a review of current medications and supplements.
Testing CCP antibody and rheumatoid factor would be useful if rheumatoid arthritis were suspected, but in the absence of demonstrable arthritis on examination, these tests would have low specificity even if the results were positive.
An NSAID may temporarily alleviate his pain, but it will not help establish a diagnosis. And in elderly patients, NSAIDs are not without complications and so should be prescribed only in appropriate situations.
Imaging would be appropriate at this point only if there was clinical suspicion of a specific disease. However, our patient has no focal deficits, and the suspicion of fracture or malignancy is low.
The medical history should include asking about current drug regimens, recent medication changes, and the use of herbal supplements, since polypharmacy is common in elderly patients with multiple comorbidities.
On further questioning, our patient said that his dose of simvastatin had been increased from 40 mg daily to 80 mg daily about 1 month before his symptoms appeared. He was taking a daily multivitamin but was not using herbal supplements or other over-the-counter products. He did not recall any constitutional symptoms before the onset of his current symptoms, and he had never had similar muscle pain in the past.
2. Based on the additional information from the history, what is the most likely cause of his muscle pain?
- Limited myositis secondary to recent viral infection
- Rhabdomyolysis
- Hypothyroidism
- Drug-drug interaction
- Statin-induced myalgia
Our patient’s history provided nothing to suggest viral myositis. Hypothyroidism should always be considered in patients with myalgia, but this is not likely in our patient, as he does not display other characteristics, such as diminished reflexes, hypotonia, cold intolerance, and mood instability. Even though calcium channel blockers have been known to cause myalgia in patients on statins, a drug-drug reaction is not likely, as he had not started taking a calcium channel blocker before his symptoms began. This patient did not show signs or symptoms of rhabdomyolysis, a type of myopathy in which necrosis of the muscle tissue occurs, generally causing profound weakness and pain.1
Therefore, statin-induced myopathy is the most likely cause of his diffuse muscle pain, particularly since his simvastatin had been increased 1 month before the onset of symptoms.
3. What should be the next step in his management?
- Decrease the dose of simvastatin to the last known dose he was able to tolerate
- Continue simvastatin at the same dose and then monitor
- Switch to another statin
- Add coenzyme Q10
- Stop simvastatin
Decreasing the statin dosage to the last well-tolerated dose would not be appropriate in a patient with myopathy, as the symptoms would probably not improve.2–4 Also, one should not switch to a different statin while a patient is experiencing symptoms. Rather, the statin should be stopped for at least 6 weeks or until the symptoms have fully resolved.1
Adding coenzyme Q10 is another option, especially in a patient with previously diagnosed coronary artery disease,5 when continued statin therapy is thought necessary to reduce the likelihood of repeat coronary events.
We discontinued his simvastatin. Followup 3 weeks later in the outpatient clinic showed that his symptoms were slowly improving. The symptoms had resolved completely 4 months later.
4. How should we manage our patient’s hyperlipidemia once his symptoms have resolved?
- Restart simvastatin at the 80-mg dose
- Restart simvastatin at the 40-mg dose
- Start a hydrophilic statin at full dose
- Use a drug from another class of lipid-lowering drugs
- Wait another 3 months before prescribing any lipid-lowering drug
His treatment for hyperlipidemia should be continued, considering his comorbidities. However, restarting the same statin, even at a lower dose, will likely cause his symptoms to recur. Thus, a different statin should be tried once his muscle pain has resolved.
Other classes of lipid-lowering drugs are usually less efficacious than statins, particularly when trying to control low-density lipoprotein (LDL) cholesterol, so a drug from another class should not be used until other statin options have been attempted.2,6,7
Simvastatin is lipophilic. Trying a statin with hydrophilic properties (eg, pravastatin, rosuvastatin, fluvastatin) has been shown to convey similar cardioprotective effects with a lower propensity for myalgia, as lipophilic statins have a higher propensity to penetrate muscle tissue than do hydrophilic statins.3,4,8
Once his symptoms resolved, our patient was started on a hydrophilic statin, fluvastatin 20 mg daily. Unfortunately, his pain recurred 3 weeks later. The statin was stopped, and his symptoms again resolved.
5. Since our patient was unable to tolerate a second statin, what should be the next step in his management?
- Restart simvastatin
- Use a drug from another class to control the hyperlipidemia
- Wait at least 6 months after symptoms resolve before trying any lipid-lowering drug
- Initiate therapy with coenzyme Q10 and fish oil
- Wait for symptoms to resolve, then restart a hydrophilic statin at a lower dose and lower frequency
Restarting simvastatin will likely cause a recurrence of the myalgia. Other lipid-lowering drugs such as nicotinic acid, bile acid resins, and fibrates are not as efficacious as statins. Coenzyme Q10 and fish oil can reduce lipid levels, but they are not as efficacious as statins.
In view of our patient’s lipid profile—LDL cholesterol elevated at 167 mg/dL, high-density lipoprotein cholesterol 31 mg/dL, triglycerides 47 mg/dL—it is important to treat his hyperlipidemia. Therefore, another attempt at statin therapy should be made once his symptoms have resolved.
Studies have shown that restarting a statin at a low dose and low frequency is effective in patients who have experienced intolerance to a statin.3,4 Our patient was treated with low-dose pravastatin (20 mg), resulting in a moderate improvement in his LDL cholesterol to 123 mg/dL.
STATIN-INDUCED MYOPATHY: ADDRESSING THE DILEMMA
Treating hyperlipidemia is important to prevent vascular events in patients with or without coronary artery disease. Statins are the most effective agents available for controlling hypercholesterolemia, specifically LDL levels, as well as for preventing myocardial infarction.
Unfortunately, significant side effects have been reported, and myopathy is the most prevalent. Statin-induced myopathy includes a combination of muscle tenderness, myalgia, and weakness.2–11 In randomized controlled trials, the risk of myopathy was estimated to be between 1.5% and 5%.6 In unselected clinic patients on high-dose statins, the rate of muscle complaints may be as high as 20%.12
The cause of statin-induced myopathy is not known, although studies have linked it to genetic defects.7 Risk factors have been identified and include personal and family history of myalgia, Asian ethnicity, hypothyroidism, and type 1 diabetes. The incidence of statin-induced myalgia is two to three times higher in patients on corticosteroid therapy. Other risk factors include female sex, liver disease, and renal dysfunction.7,8
A less common etiology is anti-HMG coenzyme A reductase antibodies. Studies have shown that these antibody levels correlate well with the amount of myositis as measured by creatine kinase levels. However, there is no consensus yet on screening for these antibodies.13
Statin therapy poses a dilemma, as there is a thin line between the benefits and the risks of side effects, especially statin-induced myopathy.3,4 Current recommendations include discontinuing the statin until symptoms fully resolve. Creatine kinase levels may be useful in assessing for potential muscle breakdown, especially in patients with reduced renal function, as this predisposes them to statin-induced myopathy, yet normal values do not preclude the diagnosis of statin-induced myopathy.3,4,7,8
Once symptoms resolve and laboratory test results normalize, a trial of a different statin is recommended. If patients become symptomatic, a trial of a low-dose hydrophilic statin at a once- or twice-weekly interval has been recommended. Several studies have assessed the efficacy of a low-dose statin with decreased frequency of administration and have consistently shown significant improvement in lipid levels.3,4 For instance, once-weekly rosuvastatin at a dose between 5 mg and 20 mg resulted in a 29% reduction in LDL cholesterol levels, and 80% of patients did not experience a recurrence of myalgia.3 Furthermore, a study of patients treated with 5 mg to 10 mg of rosuvastatin twice a week resulted in a 26% decrease in LDL cholesterol levels.4 This study also showed that when an additional non-statin lipid-lowering drug was prescribed (eg, ezetimibe, bile acid resin, nicotinic acid), more than half of the patients reached their goal lipid level.4
The addition of coenzyme Q10 and fish oil has also been suggested. Although, the evidence to support this is inconclusive, the potential benefit outweighs the risk, since the side effects are minimal.1 However, no study yet has evaluated the risks vs the benefits in patients with elevated creatine kinase.
Statin-induced myopathy is a commonly encountered adverse effect. Currently, there are no guidelines on restarting statin therapy after statin-induced myopathy; however, data suggest that statin therapy should be restarted once symptoms resolve, and that variations in dose and frequency may be necessary.1–8,14
An 85-year-old man with hypertension, hyperlipidemia, and coronary artery disease presented to our clinic with diffuse muscle pain. The pain had been present for about 3 months, but it had become noticeably worse over the past few weeks.
He was not aware of any trauma. He described the muscle pain as dull and particularly severe in his lower extremities (his thighs and calves). The pain did not limit his daily activities, nor did physical exertion or the time of day have any effect on the level of the pain.
His medications at that time included metoprolol, aspirin, hydrochlorothiazide, simvastatin, and a daily multivitamin.
He was not in acute distress. On neurologic and musculoskeletal examinations, all deep-tendon reflexes were intact, with no tenderness to palpation of the upper and lower extremities. No abnormalities were noted on the joint examination. He had full range of motion, with 5/5 muscle strength in the upper and lower extremities bilaterally and normal muscle tone. He was able to walk with ease. Results of initial laboratory testing, including creatine kinase and erythrocyte sedimentation rate, were normal.
1. What should be the next best step in the evaluation of this patient’s muscle pain?
- Order tests for cyclic citrullinated peptide (CCP) antibody and rheumatoid factor
- Advise him to refrain from physical activity until his symptoms resolve
- Take a more detailed history, including a review of medications and supplements
- Recommend a trial of a nonsteroidal anti-inflammatory drug (NSAID)
- Send him for radiographic imaging
Since his muscle pain has persisted for several months without improvement, a more detailed history should be taken, including a review of current medications and supplements.
Testing CCP antibody and rheumatoid factor would be useful if rheumatoid arthritis were suspected, but in the absence of demonstrable arthritis on examination, these tests would have low specificity even if the results were positive.
An NSAID may temporarily alleviate his pain, but it will not help establish a diagnosis. And in elderly patients, NSAIDs are not without complications and so should be prescribed only in appropriate situations.
Imaging would be appropriate at this point only if there was clinical suspicion of a specific disease. However, our patient has no focal deficits, and the suspicion of fracture or malignancy is low.
The medical history should include asking about current drug regimens, recent medication changes, and the use of herbal supplements, since polypharmacy is common in elderly patients with multiple comorbidities.
On further questioning, our patient said that his dose of simvastatin had been increased from 40 mg daily to 80 mg daily about 1 month before his symptoms appeared. He was taking a daily multivitamin but was not using herbal supplements or other over-the-counter products. He did not recall any constitutional symptoms before the onset of his current symptoms, and he had never had similar muscle pain in the past.
2. Based on the additional information from the history, what is the most likely cause of his muscle pain?
- Limited myositis secondary to recent viral infection
- Rhabdomyolysis
- Hypothyroidism
- Drug-drug interaction
- Statin-induced myalgia
Our patient’s history provided nothing to suggest viral myositis. Hypothyroidism should always be considered in patients with myalgia, but this is not likely in our patient, as he does not display other characteristics, such as diminished reflexes, hypotonia, cold intolerance, and mood instability. Even though calcium channel blockers have been known to cause myalgia in patients on statins, a drug-drug reaction is not likely, as he had not started taking a calcium channel blocker before his symptoms began. This patient did not show signs or symptoms of rhabdomyolysis, a type of myopathy in which necrosis of the muscle tissue occurs, generally causing profound weakness and pain.1
Therefore, statin-induced myopathy is the most likely cause of his diffuse muscle pain, particularly since his simvastatin had been increased 1 month before the onset of symptoms.
3. What should be the next step in his management?
- Decrease the dose of simvastatin to the last known dose he was able to tolerate
- Continue simvastatin at the same dose and then monitor
- Switch to another statin
- Add coenzyme Q10
- Stop simvastatin
Decreasing the statin dosage to the last well-tolerated dose would not be appropriate in a patient with myopathy, as the symptoms would probably not improve.2–4 Also, one should not switch to a different statin while a patient is experiencing symptoms. Rather, the statin should be stopped for at least 6 weeks or until the symptoms have fully resolved.1
Adding coenzyme Q10 is another option, especially in a patient with previously diagnosed coronary artery disease,5 when continued statin therapy is thought necessary to reduce the likelihood of repeat coronary events.
We discontinued his simvastatin. Followup 3 weeks later in the outpatient clinic showed that his symptoms were slowly improving. The symptoms had resolved completely 4 months later.
4. How should we manage our patient’s hyperlipidemia once his symptoms have resolved?
- Restart simvastatin at the 80-mg dose
- Restart simvastatin at the 40-mg dose
- Start a hydrophilic statin at full dose
- Use a drug from another class of lipid-lowering drugs
- Wait another 3 months before prescribing any lipid-lowering drug
His treatment for hyperlipidemia should be continued, considering his comorbidities. However, restarting the same statin, even at a lower dose, will likely cause his symptoms to recur. Thus, a different statin should be tried once his muscle pain has resolved.
Other classes of lipid-lowering drugs are usually less efficacious than statins, particularly when trying to control low-density lipoprotein (LDL) cholesterol, so a drug from another class should not be used until other statin options have been attempted.2,6,7
Simvastatin is lipophilic. Trying a statin with hydrophilic properties (eg, pravastatin, rosuvastatin, fluvastatin) has been shown to convey similar cardioprotective effects with a lower propensity for myalgia, as lipophilic statins have a higher propensity to penetrate muscle tissue than do hydrophilic statins.3,4,8
Once his symptoms resolved, our patient was started on a hydrophilic statin, fluvastatin 20 mg daily. Unfortunately, his pain recurred 3 weeks later. The statin was stopped, and his symptoms again resolved.
5. Since our patient was unable to tolerate a second statin, what should be the next step in his management?
- Restart simvastatin
- Use a drug from another class to control the hyperlipidemia
- Wait at least 6 months after symptoms resolve before trying any lipid-lowering drug
- Initiate therapy with coenzyme Q10 and fish oil
- Wait for symptoms to resolve, then restart a hydrophilic statin at a lower dose and lower frequency
Restarting simvastatin will likely cause a recurrence of the myalgia. Other lipid-lowering drugs such as nicotinic acid, bile acid resins, and fibrates are not as efficacious as statins. Coenzyme Q10 and fish oil can reduce lipid levels, but they are not as efficacious as statins.
In view of our patient’s lipid profile—LDL cholesterol elevated at 167 mg/dL, high-density lipoprotein cholesterol 31 mg/dL, triglycerides 47 mg/dL—it is important to treat his hyperlipidemia. Therefore, another attempt at statin therapy should be made once his symptoms have resolved.
Studies have shown that restarting a statin at a low dose and low frequency is effective in patients who have experienced intolerance to a statin.3,4 Our patient was treated with low-dose pravastatin (20 mg), resulting in a moderate improvement in his LDL cholesterol to 123 mg/dL.
STATIN-INDUCED MYOPATHY: ADDRESSING THE DILEMMA
Treating hyperlipidemia is important to prevent vascular events in patients with or without coronary artery disease. Statins are the most effective agents available for controlling hypercholesterolemia, specifically LDL levels, as well as for preventing myocardial infarction.
Unfortunately, significant side effects have been reported, and myopathy is the most prevalent. Statin-induced myopathy includes a combination of muscle tenderness, myalgia, and weakness.2–11 In randomized controlled trials, the risk of myopathy was estimated to be between 1.5% and 5%.6 In unselected clinic patients on high-dose statins, the rate of muscle complaints may be as high as 20%.12
The cause of statin-induced myopathy is not known, although studies have linked it to genetic defects.7 Risk factors have been identified and include personal and family history of myalgia, Asian ethnicity, hypothyroidism, and type 1 diabetes. The incidence of statin-induced myalgia is two to three times higher in patients on corticosteroid therapy. Other risk factors include female sex, liver disease, and renal dysfunction.7,8
A less common etiology is anti-HMG coenzyme A reductase antibodies. Studies have shown that these antibody levels correlate well with the amount of myositis as measured by creatine kinase levels. However, there is no consensus yet on screening for these antibodies.13
Statin therapy poses a dilemma, as there is a thin line between the benefits and the risks of side effects, especially statin-induced myopathy.3,4 Current recommendations include discontinuing the statin until symptoms fully resolve. Creatine kinase levels may be useful in assessing for potential muscle breakdown, especially in patients with reduced renal function, as this predisposes them to statin-induced myopathy, yet normal values do not preclude the diagnosis of statin-induced myopathy.3,4,7,8
Once symptoms resolve and laboratory test results normalize, a trial of a different statin is recommended. If patients become symptomatic, a trial of a low-dose hydrophilic statin at a once- or twice-weekly interval has been recommended. Several studies have assessed the efficacy of a low-dose statin with decreased frequency of administration and have consistently shown significant improvement in lipid levels.3,4 For instance, once-weekly rosuvastatin at a dose between 5 mg and 20 mg resulted in a 29% reduction in LDL cholesterol levels, and 80% of patients did not experience a recurrence of myalgia.3 Furthermore, a study of patients treated with 5 mg to 10 mg of rosuvastatin twice a week resulted in a 26% decrease in LDL cholesterol levels.4 This study also showed that when an additional non-statin lipid-lowering drug was prescribed (eg, ezetimibe, bile acid resin, nicotinic acid), more than half of the patients reached their goal lipid level.4
The addition of coenzyme Q10 and fish oil has also been suggested. Although, the evidence to support this is inconclusive, the potential benefit outweighs the risk, since the side effects are minimal.1 However, no study yet has evaluated the risks vs the benefits in patients with elevated creatine kinase.
Statin-induced myopathy is a commonly encountered adverse effect. Currently, there are no guidelines on restarting statin therapy after statin-induced myopathy; however, data suggest that statin therapy should be restarted once symptoms resolve, and that variations in dose and frequency may be necessary.1–8,14
- Fernandez G, Spatz ES, Jablecki C, Phillips PS. Statin myopathy: a common dilemma not reflected in clinical trials. Cleve Clin J Med 2011; 78:393–403.
- Foley KA, Simpson RJ, Crouse JR, Weiss TW, Markson LE, Alexander CM. Effectiveness of statin titration on low-density lipoprotein cholesterol goal attainment in patients at high risk of atherogenic events. Am J Cardiol 2003; 92:79–81.
- Backes JM, Moriarty PM, Ruisinger JF, Gibson CA. Effects of once weekly rosuvastatin among patients with a prior statin intolerance. Am J Cardiol 2007; 100:554–555.
- Gadarla M, Kearns AK, Thompson PD. Efficacy of rosuvastatin (5 mg and 10 mg) twice a week in patients intolerant to daily statins. Am J Cardiol 2008; 101:1747–1748.
- Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme q10 on myopathic symptoms in patients treated with statins. Am J Cardiol 2007; 99:1409–1412.
- Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Tomaszewski M, Stepien KM, Tomaszewska J, Czuczwar SJ. Statin-induced myopathies. Pharmacol Rep 2011; 63:859–866.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Heart Protection Study Collaborative Group. MRC/BHF heart protection study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Guyton JR. Benefit versus risk in statin treatment. Am J Cardiol 2006; 97:95C–97C.
- Buettner C, Davis RB, Leveille SG, Mittleman MA, Mukamal KJ. Prevalence of musculoskeletal pain and statin use. J Gen Intern Med 2008; 23:1182–1186.
- Werner JL, Christopher-Stine L, Ghazarian SR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum 2012; 64:4087–4093.
- The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 1998; 339:1349–1357.
- Fernandez G, Spatz ES, Jablecki C, Phillips PS. Statin myopathy: a common dilemma not reflected in clinical trials. Cleve Clin J Med 2011; 78:393–403.
- Foley KA, Simpson RJ, Crouse JR, Weiss TW, Markson LE, Alexander CM. Effectiveness of statin titration on low-density lipoprotein cholesterol goal attainment in patients at high risk of atherogenic events. Am J Cardiol 2003; 92:79–81.
- Backes JM, Moriarty PM, Ruisinger JF, Gibson CA. Effects of once weekly rosuvastatin among patients with a prior statin intolerance. Am J Cardiol 2007; 100:554–555.
- Gadarla M, Kearns AK, Thompson PD. Efficacy of rosuvastatin (5 mg and 10 mg) twice a week in patients intolerant to daily statins. Am J Cardiol 2008; 101:1747–1748.
- Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme q10 on myopathic symptoms in patients treated with statins. Am J Cardiol 2007; 99:1409–1412.
- Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Tomaszewski M, Stepien KM, Tomaszewska J, Czuczwar SJ. Statin-induced myopathies. Pharmacol Rep 2011; 63:859–866.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Heart Protection Study Collaborative Group. MRC/BHF heart protection study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Guyton JR. Benefit versus risk in statin treatment. Am J Cardiol 2006; 97:95C–97C.
- Buettner C, Davis RB, Leveille SG, Mittleman MA, Mukamal KJ. Prevalence of musculoskeletal pain and statin use. J Gen Intern Med 2008; 23:1182–1186.
- Werner JL, Christopher-Stine L, Ghazarian SR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum 2012; 64:4087–4093.
- The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 1998; 339:1349–1357.
Ascites in a 42-year-old woman
A 42-year-old woman is admitted to the hospital with worsening shortness of breath on exertion, poor exercise tolerance, leg edema, and swelling of the abdomen. Her symptoms have been getting worse over the last 4 months. She reports no history of fever, chills, night sweats, bleeding disorder, joint pain, weight loss, or loss of appetite.
She has type 2 diabetes mellitus and hypothyroidism. She had rheumatoid arthritis but said it was “inactive,” not requiring treatment for the last 18 years. Three months ago, she underwent a total hysterectomy and salpingo-oophorectomy for a complex adnexal mass, biopsy of which revealed a benign mucinous ovarian cyst.
Her current medications include furosemide, levothyroxine, and metformin. She is an ex-smoker with a 7 pack-year history. She drinks a glass of wine on social occasions only. Her family history is unremarkable.
On examination, she is not in distress and she has no fever. She has jugular venous distention of 5 cm, tense ascites, and marked edema of the legs, as well as hyperpigmented patches and erythematous plaques over both shins. Neck palpation reveals no lymphadenopathy or thyromegaly.
Her liver and the tip of the spleen are palpable following paracentesis, once ascitic fluid is removed.
The cardiovascular examination is normal. Chest auscultation reveals decreased breath sounds at the right lung base with bibasilar crackles. No focal neurologic deficit is noted on clinical examination.
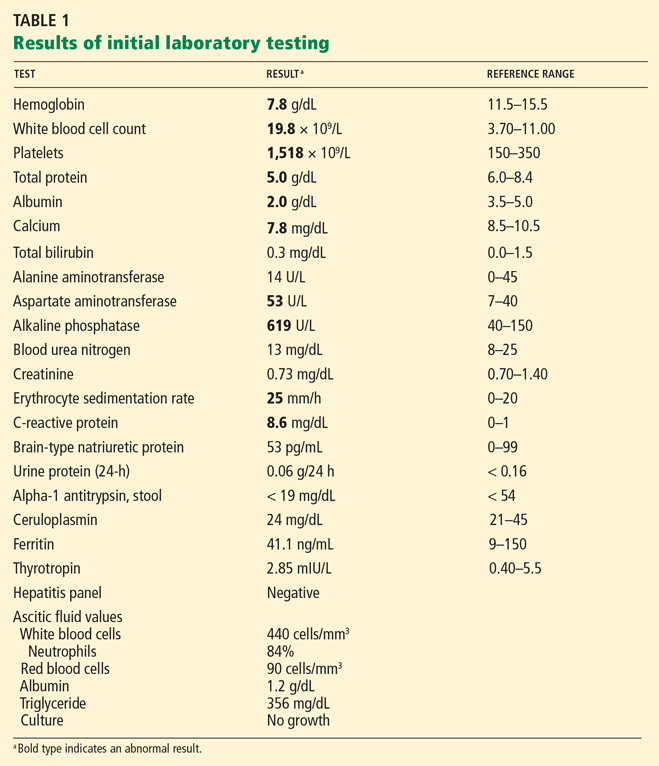
Laboratory testing at the time of hospital admission (Table 1) includes a hepatitis panel (negative for exposure to hepatitis A, B, and C) and ascitic fluid studies. Chest radiography shows a right pleural effusion. Echocardiography demonstrates moderate pericardial effusion without tamponade; left and right ventricular function is normal. Cardiac magnetic resonance imaging finds no evidence of pericardial constriction or restrictive cardiomyopathy. Pressures are normal on pulmonary artery catheterization.
FINDING THE CAUSE OF ASCITES
1. What is the most likely cause of ascites in this patient?
- Cirrhosis
- Recent abdominal surgery
- Congestive heart failure
- Abdominal malignancy
- Nephrotic syndrome
The serum-ascites albumin gradient—ie, the serum albumin concentration minus the ascitic fluid albumin concentration—helps determine whether ascites is related to portal hypertension.1 A high gradient (ie, above 1.1 g/dL) is seen in cirrhosis, alcoholic hepatitis, congestive heart failure, vascular occlusion syndromes (eg, Budd-Chiari syndrome), and metastatic liver disease.
From the values in Table 1, our patient’s gradient is 0.8 g/dL, which is considered low. However, we cannot completely rule out cirrhosis as the cause of her ascites because she was taking a diuretic, and diuretics can falsely decrease the gradient. Heart failure is unlikely, based on the results of echocardiography and catheterization. In addition, the 24-hour urinary protein concentration is normal, as is alpha-1 antitrypsin secretion in the stool, ruling out protein-losing nephropathy or enteropathy as the cause of her low albumin and ascites.
A high triglyceride content in her ascitic fluid (> 150 mg/dL) is consistent with chylous ascites, which is seen in patients with previous abdominal surgery or with lymphatic obstruction due to malignancy. A high neutrophil count in the ascitic fluid and a negative culture are also consistent with chylous ascites. However, in this patient, recent surgery as the cause of chylous ascites does not explain the systemic features of hepatosplenomegaly, anemia, thrombocytosis, and low albumin. Moreover, her high C-reactive protein value suggests an ongoing inflammatory process, although her erythrocyte sedimentation rate is not significantly elevated.
Therefore, the most likely cause of ascites in this patient is abdominal malignancy.
WHAT SHOULD BE DONE NEXT?
2. Which of the following studies is reasonable in this patient at this point?
- Serum protein electrophoresis
- Computed tomography (CT) of the chest, abdomen, and pelvis
- Liver biopsy
- Cytologic study of the ascitic fluid
All of these studies would be reasonable and in fact were done in this patient.
Serum protein electrophoresis (Table 2) identified a monoclonal protein band in the immunoglobulin G (IgG) kappa region.
Cytologic study of the ascitic fluid was negative for malignant cells.
Chest CT revealed bilateral pleural effusions, pericardial effusion, and bilateral axillary lymphadenopathy. CT of the abdomen and pelvis was normal, except for ascites, and no pelvic tumor was noted.
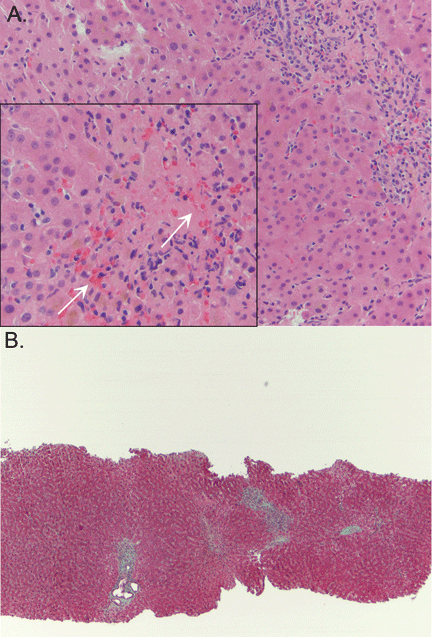
Liver biopsy was done to look for the source of her unexplained ascites with elevated alkaline phosphatase, as all other investigations so far were normal. It revealed mild centrilobular scarring, but the rest of the parenchymal architecture was normal, with no evidence of bridging fibrosis or nodular regenerative hyperplasia (Figure 1).
Transjugular measurement of the hepatic vein pressure revealed a hepatic vein pressure gradient of 9 mm Hg, indicating mild portal hypertension. Venography showed widely patent hepatic and portal veins. Her high inflammatory marker levels could have been caused by smoldering rheumatoid arthritis; however, since the patient has had no joint symptoms for 18 years, this is very unlikely. It is more likely to be caused by a plasma cell disorder, as suggested by a monoclonal protein on electrophoresis.
WHAT IS THE DIAGNOSIS?
3. What is the most likely diagnosis in our patient?
- Rheumatoid arthritis
- Cryoglobulinemia
- Capillary leak syndrome
- Hematologic malignancy
- Syndrome of polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes (POEMS syndrome)
Rheumatoid arthritis can present with hepatosplenomegaly, lymphadenopathy, ascites, and skin rash, particularly if antinuclear antibody and rheumatoid factor are elevated. Ascites is known to occur in association with rheumatoid arthritis in the setting of Felty syndrome or nodular regenerative hyperplasia of the liver.2 However, our patient did not have leukopenia or evidence of regenerative hyperplasia on liver biopsy. Moreover, her rheumatoid arthritis had remained clinically inactive for a long time.
Cryoglobulinemia was possible, given her ascites, neuropathy, and splenomegaly, but her serum hepatic antibody and C4 complement values were normal.3 Also, the appearance of her rash was not typical of cryoglobulinemia.
Capillary leak syndrome was ruled out by the absence of hypotensive episodes, edema of the face or upper extremities, or renal failure.4
Lymphoma was excluded by flow cytometry.
A monoclonal protein on serum electrophoresis may suggest multiple myeloma, but this patient had multisystem involvement including organomegaly, endocrinopathy, and skin abnormalities. Thus, POEMS syndrome is the most likely diagnosis.
4. Which test should be done at this time to confirm the diagnosis of POEMS syndrome?
- Bone marrow biopsy
- Vascular endothelial growth factor testing
- Nerve conduction study
- Complete x-ray bone survey
A test for vascular endothelial growth factor should be done. This growth factor is almost always elevated in POEMS, and a positive test helps confirm the diagnosis of POEMS. Our patient’s level was elevated at 1,664 pg/mL (reference range 31–86).
POEMS is thought to be a variant of plasma cell dyscrasia, and all patients with POEMS have a monoclonal protein on electrophoresis. On this background, multiple myeloma is an important consideration.
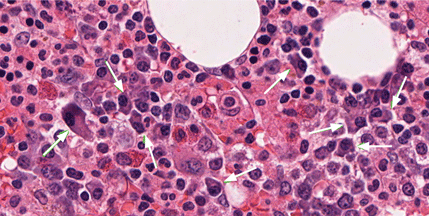
Our patient underwent bone marrow biopsy, which revealed mild plasmacytosis (< 10%) (Figure 2). A complete bone survey showed generalized osteopenia without blastic or lytic lesions. To complete the workup for POEMS syndrome, a nerve conduction study was done to look for neuropathy; it showed bilateral sensory motor neuropathy with features of both a demyelinating process and axonal loss.
POEMS SYNDROME
POEMS syndrome is a constellation of features such as organomegaly and endocrine and skin abnormalities in association with neuropathy and a monoclonal protein on electrophoresis.5 In 2003, Dispenzieri et al6 described the major and minor diagnostic criteria based on a retrospective analysis of 99 patients with POEMS syndrome.6 Later, elevated vascular endothelial growth factor was added as a confirmatory diagnostic criterion.7 This growth factor is also an indicator of prognosis in POEMS syndrome, and its level can be used to monitor the response to treatment.7
Our patient met both major criteria for POEMS syndrome, ie, polyneuropathy (based on nerve conduction studies) and a monoclonal protein. Polyneuropathy in POEMS syndrome usually occurs as sensorimotor peripheral neuropathy of insidious onset and is seldom painful. Nerve biopsy study reveals demyelination with features of axonal loss. Interestingly, although our patient had neuropathy as diagnosed by electromyography, she remained clinically asymptomatic.
The monoclonal protein in POEMS syndrome is commonly IgA or IgG. Light chains are always present and are mainly the lambda type; kappa light chains are also reported in rare cases. Our patient had IgG kappa light chains.
Our patient met a number of the minor criteria for POEMS syndrome: ie, organomegaly (hepatosplenomegaly, lymphadenopathy), endocrinopathy (hypothyroidism, diabetes), skin changes (hyperpigmentation and plaques of the lower extremities), edema, pleural effusion, and ascites.
Endocrine disorders in POEMS syndrome
The endocrine abnormalities most often described in POEMS syndrome are hypogonadism, hypothyroidism, and diabetes mellitus. But because hypothyroidism and diabetes are common in the general population, it is debatable whether either of these could constitute the endocrine component of POEMS syndrome. Nevertheless, in three large series,6,7 occurrences of these two disorders were common, although less specific than adrenal or pituitary involvement.
In the analysis by Dispenzieri et al,6 67% of patients had at least one endocrine abnormality. Our patient had no evidence of an adrenal disorder.
Skin, skeletal, and other changes
The skin changes in POEMS syndrome are often nonspecific and include hyperpigmentation, sclerodema-like thickening, and plaques.
Skeletal changes are noted in up to 97% of patients. A skeletal survey in our patient revealed generalized osteopenia as opposed to osteosclerotic lesions, which are common in POEMS syndrome.
Anemia and thrombocytosis (as in our patient) are usually seen in POEMS syndrome and are induced by cytokines.6 POEMS syndrome also leads to increased thrombotic complications from the release of inflammatory cytokines.
Hypoalbuminemia and anasarca including ascites are often seen in POEMS syndrome (prevalence 29% to 89%) and are attributed to cytokine-induced increased vascular permeability. In POEMS syndrome, the serum-ascites albumin gradient is usually less than 1.1 g/dL, as in our patient.
Stepani et al8 reported one case of culture-negative neutrocytic ascites with portal hypertension in POEMS syndrome.8 (Culture-negative neutrocytic ascites is defined as an ascitic fluid polymorphonuclear count greater than 250/mm3 and a negative ascitic fluid culture in the absence of previous antibiotic therapy.) Chylous ascites has not yet been described in POEMS syndrome. However, chylous ascites is predominantly lymphocytic, whereas our patient had neutrocytic ascites.
We concluded that the cause of our patient’s ascites was multifactorial and included previous surgery and POEMS syndrome.
Nonclassic presentation
In addition to its classic presentation, POEMS syndrome is often reported in association with other “unusual features” such as cardiomyopathy, pulmonary hypertension, and cryoglobulinemia.6
So far, very few cases of portal hypertension in POEMS syndrome have been reported. Stepani et al8 described a patient who had POEMS syndrome and portal hypertension with extensive portal fibrosis without cirrhosis on liver biopsy. Inoue et al9 reported a liver biopsy feature consistent with idiopathic portal hypertension, also noting a case with mild fibrosis and few lymphocytic infiltrates in the portal tract.9
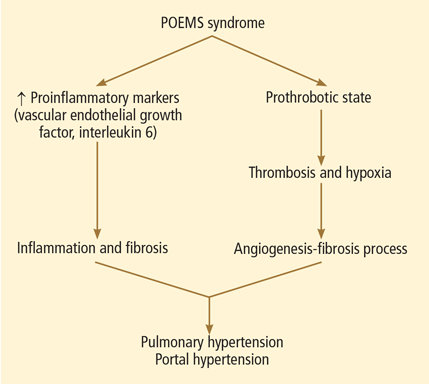
The etiopathogenesis of POEMS syndrome is attributed to proangiogenic vascular endothelial growth factor, and other inflammatory cytokines (interleukin 6, interleukin 1 beta, tumor necrosis factor alpha) also play a key role in pulmonary hypertension.10,11 A similar pathogenesis could also contribute to the development of portal hypertension (Figure 3).
CASE CONCLUDED
We started our patient on oral prednisone 60 mg daily for a month, tapered to a maintenance dose of 15 mg to suppress clonal proliferation of plasma cells. Her symptoms improved. Her vascular endothelial growth factor level decreased from 1,664 to 624 pg/mL. She was enrolled in a National Institutes of Health study to evaluate the effect of a potential new immunomodulator treatment for POEMS syndrome.
In conclusion, POEMS syndrome is rare and can present with many atypical features. A high index of suspicion is needed to detect it in a patient who has noncirrhotic portal hypertension with ascites and multisystem involvement.
- Runyon BA, Montano AA, Akriviadis EA, Antillon MR, Irving MA, McHutchison JG. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med 1992; 117:215–220.
- Harris M, Rash RM, Dymock IW. Nodular, non-cirrhotic liver associated with portal hypertension in a patient with rheumatoid arthritis. J Clin Pathol 1974; 27:963–966.
- Ramos-Casals M, Stone JH, Cid MC, Bosch X. The cryoglobulinaemias. Lancet 2012; 379:348–360.
- Druey KM, Greipp PR. Narrative review: the systemic capillary leak syndrome. Ann Intern Med 2010; 153:90–98.
- Bardwick PA, Zvaifler NJ, Gill GN, Newman D, Greenway GD, Resnick DL. Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome. Report on two cases and a review of the literature. Medicine (Baltimore) 1980; 59:311–322.
- Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood 2003; 101:2496–2506.
- Dispenzieri A. POEMS syndrome. Blood Rev 2007; 21:285–299.
- Stepani P, Courouble Y, Postel P, et al. Portal hypertension and neutrocytic ascites in POEMS syndrome. Gastroenterol Clin Biol 1998; 22:1095–1097. Article in French.
- Inoue R, Nakazawa A, Tsukada N, et al. POEMS syndrome with idiopathic portal hypertension: autopsy case and review of the literature. Pathol Int 2010; 60:316–320.
- Gherardi RK, Bélec L, Soubrier M, et al. Overproduction of proinflammatory cytokines imbalanced by their antagonists in POEMS syndrome. Blood 1996; 87:1458–1465.
- Mukerjee D, Kingdon E, Vanderpump M, Coghlan JG. Pathophysiological insights from a case of reversible pulmonary arterial hypertension. J R Soc Med 2003; 96:403–404.
A 42-year-old woman is admitted to the hospital with worsening shortness of breath on exertion, poor exercise tolerance, leg edema, and swelling of the abdomen. Her symptoms have been getting worse over the last 4 months. She reports no history of fever, chills, night sweats, bleeding disorder, joint pain, weight loss, or loss of appetite.
She has type 2 diabetes mellitus and hypothyroidism. She had rheumatoid arthritis but said it was “inactive,” not requiring treatment for the last 18 years. Three months ago, she underwent a total hysterectomy and salpingo-oophorectomy for a complex adnexal mass, biopsy of which revealed a benign mucinous ovarian cyst.
Her current medications include furosemide, levothyroxine, and metformin. She is an ex-smoker with a 7 pack-year history. She drinks a glass of wine on social occasions only. Her family history is unremarkable.
On examination, she is not in distress and she has no fever. She has jugular venous distention of 5 cm, tense ascites, and marked edema of the legs, as well as hyperpigmented patches and erythematous plaques over both shins. Neck palpation reveals no lymphadenopathy or thyromegaly.
Her liver and the tip of the spleen are palpable following paracentesis, once ascitic fluid is removed.
The cardiovascular examination is normal. Chest auscultation reveals decreased breath sounds at the right lung base with bibasilar crackles. No focal neurologic deficit is noted on clinical examination.
Laboratory testing at the time of hospital admission (Table 1) includes a hepatitis panel (negative for exposure to hepatitis A, B, and C) and ascitic fluid studies. Chest radiography shows a right pleural effusion. Echocardiography demonstrates moderate pericardial effusion without tamponade; left and right ventricular function is normal. Cardiac magnetic resonance imaging finds no evidence of pericardial constriction or restrictive cardiomyopathy. Pressures are normal on pulmonary artery catheterization.
FINDING THE CAUSE OF ASCITES
1. What is the most likely cause of ascites in this patient?
- Cirrhosis
- Recent abdominal surgery
- Congestive heart failure
- Abdominal malignancy
- Nephrotic syndrome
The serum-ascites albumin gradient—ie, the serum albumin concentration minus the ascitic fluid albumin concentration—helps determine whether ascites is related to portal hypertension.1 A high gradient (ie, above 1.1 g/dL) is seen in cirrhosis, alcoholic hepatitis, congestive heart failure, vascular occlusion syndromes (eg, Budd-Chiari syndrome), and metastatic liver disease.
From the values in Table 1, our patient’s gradient is 0.8 g/dL, which is considered low. However, we cannot completely rule out cirrhosis as the cause of her ascites because she was taking a diuretic, and diuretics can falsely decrease the gradient. Heart failure is unlikely, based on the results of echocardiography and catheterization. In addition, the 24-hour urinary protein concentration is normal, as is alpha-1 antitrypsin secretion in the stool, ruling out protein-losing nephropathy or enteropathy as the cause of her low albumin and ascites.
A high triglyceride content in her ascitic fluid (> 150 mg/dL) is consistent with chylous ascites, which is seen in patients with previous abdominal surgery or with lymphatic obstruction due to malignancy. A high neutrophil count in the ascitic fluid and a negative culture are also consistent with chylous ascites. However, in this patient, recent surgery as the cause of chylous ascites does not explain the systemic features of hepatosplenomegaly, anemia, thrombocytosis, and low albumin. Moreover, her high C-reactive protein value suggests an ongoing inflammatory process, although her erythrocyte sedimentation rate is not significantly elevated.
Therefore, the most likely cause of ascites in this patient is abdominal malignancy.
WHAT SHOULD BE DONE NEXT?
2. Which of the following studies is reasonable in this patient at this point?
- Serum protein electrophoresis
- Computed tomography (CT) of the chest, abdomen, and pelvis
- Liver biopsy
- Cytologic study of the ascitic fluid
All of these studies would be reasonable and in fact were done in this patient.
Serum protein electrophoresis (Table 2) identified a monoclonal protein band in the immunoglobulin G (IgG) kappa region.
Cytologic study of the ascitic fluid was negative for malignant cells.
Chest CT revealed bilateral pleural effusions, pericardial effusion, and bilateral axillary lymphadenopathy. CT of the abdomen and pelvis was normal, except for ascites, and no pelvic tumor was noted.
Liver biopsy was done to look for the source of her unexplained ascites with elevated alkaline phosphatase, as all other investigations so far were normal. It revealed mild centrilobular scarring, but the rest of the parenchymal architecture was normal, with no evidence of bridging fibrosis or nodular regenerative hyperplasia (Figure 1).
Transjugular measurement of the hepatic vein pressure revealed a hepatic vein pressure gradient of 9 mm Hg, indicating mild portal hypertension. Venography showed widely patent hepatic and portal veins. Her high inflammatory marker levels could have been caused by smoldering rheumatoid arthritis; however, since the patient has had no joint symptoms for 18 years, this is very unlikely. It is more likely to be caused by a plasma cell disorder, as suggested by a monoclonal protein on electrophoresis.
WHAT IS THE DIAGNOSIS?
3. What is the most likely diagnosis in our patient?
- Rheumatoid arthritis
- Cryoglobulinemia
- Capillary leak syndrome
- Hematologic malignancy
- Syndrome of polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes (POEMS syndrome)
Rheumatoid arthritis can present with hepatosplenomegaly, lymphadenopathy, ascites, and skin rash, particularly if antinuclear antibody and rheumatoid factor are elevated. Ascites is known to occur in association with rheumatoid arthritis in the setting of Felty syndrome or nodular regenerative hyperplasia of the liver.2 However, our patient did not have leukopenia or evidence of regenerative hyperplasia on liver biopsy. Moreover, her rheumatoid arthritis had remained clinically inactive for a long time.
Cryoglobulinemia was possible, given her ascites, neuropathy, and splenomegaly, but her serum hepatic antibody and C4 complement values were normal.3 Also, the appearance of her rash was not typical of cryoglobulinemia.
Capillary leak syndrome was ruled out by the absence of hypotensive episodes, edema of the face or upper extremities, or renal failure.4
Lymphoma was excluded by flow cytometry.
A monoclonal protein on serum electrophoresis may suggest multiple myeloma, but this patient had multisystem involvement including organomegaly, endocrinopathy, and skin abnormalities. Thus, POEMS syndrome is the most likely diagnosis.
4. Which test should be done at this time to confirm the diagnosis of POEMS syndrome?
- Bone marrow biopsy
- Vascular endothelial growth factor testing
- Nerve conduction study
- Complete x-ray bone survey
A test for vascular endothelial growth factor should be done. This growth factor is almost always elevated in POEMS, and a positive test helps confirm the diagnosis of POEMS. Our patient’s level was elevated at 1,664 pg/mL (reference range 31–86).
POEMS is thought to be a variant of plasma cell dyscrasia, and all patients with POEMS have a monoclonal protein on electrophoresis. On this background, multiple myeloma is an important consideration.
Our patient underwent bone marrow biopsy, which revealed mild plasmacytosis (< 10%) (Figure 2). A complete bone survey showed generalized osteopenia without blastic or lytic lesions. To complete the workup for POEMS syndrome, a nerve conduction study was done to look for neuropathy; it showed bilateral sensory motor neuropathy with features of both a demyelinating process and axonal loss.
POEMS SYNDROME
POEMS syndrome is a constellation of features such as organomegaly and endocrine and skin abnormalities in association with neuropathy and a monoclonal protein on electrophoresis.5 In 2003, Dispenzieri et al6 described the major and minor diagnostic criteria based on a retrospective analysis of 99 patients with POEMS syndrome.6 Later, elevated vascular endothelial growth factor was added as a confirmatory diagnostic criterion.7 This growth factor is also an indicator of prognosis in POEMS syndrome, and its level can be used to monitor the response to treatment.7
Our patient met both major criteria for POEMS syndrome, ie, polyneuropathy (based on nerve conduction studies) and a monoclonal protein. Polyneuropathy in POEMS syndrome usually occurs as sensorimotor peripheral neuropathy of insidious onset and is seldom painful. Nerve biopsy study reveals demyelination with features of axonal loss. Interestingly, although our patient had neuropathy as diagnosed by electromyography, she remained clinically asymptomatic.
The monoclonal protein in POEMS syndrome is commonly IgA or IgG. Light chains are always present and are mainly the lambda type; kappa light chains are also reported in rare cases. Our patient had IgG kappa light chains.
Our patient met a number of the minor criteria for POEMS syndrome: ie, organomegaly (hepatosplenomegaly, lymphadenopathy), endocrinopathy (hypothyroidism, diabetes), skin changes (hyperpigmentation and plaques of the lower extremities), edema, pleural effusion, and ascites.
Endocrine disorders in POEMS syndrome
The endocrine abnormalities most often described in POEMS syndrome are hypogonadism, hypothyroidism, and diabetes mellitus. But because hypothyroidism and diabetes are common in the general population, it is debatable whether either of these could constitute the endocrine component of POEMS syndrome. Nevertheless, in three large series,6,7 occurrences of these two disorders were common, although less specific than adrenal or pituitary involvement.
In the analysis by Dispenzieri et al,6 67% of patients had at least one endocrine abnormality. Our patient had no evidence of an adrenal disorder.
Skin, skeletal, and other changes
The skin changes in POEMS syndrome are often nonspecific and include hyperpigmentation, sclerodema-like thickening, and plaques.
Skeletal changes are noted in up to 97% of patients. A skeletal survey in our patient revealed generalized osteopenia as opposed to osteosclerotic lesions, which are common in POEMS syndrome.
Anemia and thrombocytosis (as in our patient) are usually seen in POEMS syndrome and are induced by cytokines.6 POEMS syndrome also leads to increased thrombotic complications from the release of inflammatory cytokines.
Hypoalbuminemia and anasarca including ascites are often seen in POEMS syndrome (prevalence 29% to 89%) and are attributed to cytokine-induced increased vascular permeability. In POEMS syndrome, the serum-ascites albumin gradient is usually less than 1.1 g/dL, as in our patient.
Stepani et al8 reported one case of culture-negative neutrocytic ascites with portal hypertension in POEMS syndrome.8 (Culture-negative neutrocytic ascites is defined as an ascitic fluid polymorphonuclear count greater than 250/mm3 and a negative ascitic fluid culture in the absence of previous antibiotic therapy.) Chylous ascites has not yet been described in POEMS syndrome. However, chylous ascites is predominantly lymphocytic, whereas our patient had neutrocytic ascites.
We concluded that the cause of our patient’s ascites was multifactorial and included previous surgery and POEMS syndrome.
Nonclassic presentation
In addition to its classic presentation, POEMS syndrome is often reported in association with other “unusual features” such as cardiomyopathy, pulmonary hypertension, and cryoglobulinemia.6
So far, very few cases of portal hypertension in POEMS syndrome have been reported. Stepani et al8 described a patient who had POEMS syndrome and portal hypertension with extensive portal fibrosis without cirrhosis on liver biopsy. Inoue et al9 reported a liver biopsy feature consistent with idiopathic portal hypertension, also noting a case with mild fibrosis and few lymphocytic infiltrates in the portal tract.9
The etiopathogenesis of POEMS syndrome is attributed to proangiogenic vascular endothelial growth factor, and other inflammatory cytokines (interleukin 6, interleukin 1 beta, tumor necrosis factor alpha) also play a key role in pulmonary hypertension.10,11 A similar pathogenesis could also contribute to the development of portal hypertension (Figure 3).
CASE CONCLUDED
We started our patient on oral prednisone 60 mg daily for a month, tapered to a maintenance dose of 15 mg to suppress clonal proliferation of plasma cells. Her symptoms improved. Her vascular endothelial growth factor level decreased from 1,664 to 624 pg/mL. She was enrolled in a National Institutes of Health study to evaluate the effect of a potential new immunomodulator treatment for POEMS syndrome.
In conclusion, POEMS syndrome is rare and can present with many atypical features. A high index of suspicion is needed to detect it in a patient who has noncirrhotic portal hypertension with ascites and multisystem involvement.
A 42-year-old woman is admitted to the hospital with worsening shortness of breath on exertion, poor exercise tolerance, leg edema, and swelling of the abdomen. Her symptoms have been getting worse over the last 4 months. She reports no history of fever, chills, night sweats, bleeding disorder, joint pain, weight loss, or loss of appetite.
She has type 2 diabetes mellitus and hypothyroidism. She had rheumatoid arthritis but said it was “inactive,” not requiring treatment for the last 18 years. Three months ago, she underwent a total hysterectomy and salpingo-oophorectomy for a complex adnexal mass, biopsy of which revealed a benign mucinous ovarian cyst.
Her current medications include furosemide, levothyroxine, and metformin. She is an ex-smoker with a 7 pack-year history. She drinks a glass of wine on social occasions only. Her family history is unremarkable.
On examination, she is not in distress and she has no fever. She has jugular venous distention of 5 cm, tense ascites, and marked edema of the legs, as well as hyperpigmented patches and erythematous plaques over both shins. Neck palpation reveals no lymphadenopathy or thyromegaly.
Her liver and the tip of the spleen are palpable following paracentesis, once ascitic fluid is removed.
The cardiovascular examination is normal. Chest auscultation reveals decreased breath sounds at the right lung base with bibasilar crackles. No focal neurologic deficit is noted on clinical examination.
Laboratory testing at the time of hospital admission (Table 1) includes a hepatitis panel (negative for exposure to hepatitis A, B, and C) and ascitic fluid studies. Chest radiography shows a right pleural effusion. Echocardiography demonstrates moderate pericardial effusion without tamponade; left and right ventricular function is normal. Cardiac magnetic resonance imaging finds no evidence of pericardial constriction or restrictive cardiomyopathy. Pressures are normal on pulmonary artery catheterization.
FINDING THE CAUSE OF ASCITES
1. What is the most likely cause of ascites in this patient?
- Cirrhosis
- Recent abdominal surgery
- Congestive heart failure
- Abdominal malignancy
- Nephrotic syndrome
The serum-ascites albumin gradient—ie, the serum albumin concentration minus the ascitic fluid albumin concentration—helps determine whether ascites is related to portal hypertension.1 A high gradient (ie, above 1.1 g/dL) is seen in cirrhosis, alcoholic hepatitis, congestive heart failure, vascular occlusion syndromes (eg, Budd-Chiari syndrome), and metastatic liver disease.
From the values in Table 1, our patient’s gradient is 0.8 g/dL, which is considered low. However, we cannot completely rule out cirrhosis as the cause of her ascites because she was taking a diuretic, and diuretics can falsely decrease the gradient. Heart failure is unlikely, based on the results of echocardiography and catheterization. In addition, the 24-hour urinary protein concentration is normal, as is alpha-1 antitrypsin secretion in the stool, ruling out protein-losing nephropathy or enteropathy as the cause of her low albumin and ascites.
A high triglyceride content in her ascitic fluid (> 150 mg/dL) is consistent with chylous ascites, which is seen in patients with previous abdominal surgery or with lymphatic obstruction due to malignancy. A high neutrophil count in the ascitic fluid and a negative culture are also consistent with chylous ascites. However, in this patient, recent surgery as the cause of chylous ascites does not explain the systemic features of hepatosplenomegaly, anemia, thrombocytosis, and low albumin. Moreover, her high C-reactive protein value suggests an ongoing inflammatory process, although her erythrocyte sedimentation rate is not significantly elevated.
Therefore, the most likely cause of ascites in this patient is abdominal malignancy.
WHAT SHOULD BE DONE NEXT?
2. Which of the following studies is reasonable in this patient at this point?
- Serum protein electrophoresis
- Computed tomography (CT) of the chest, abdomen, and pelvis
- Liver biopsy
- Cytologic study of the ascitic fluid
All of these studies would be reasonable and in fact were done in this patient.
Serum protein electrophoresis (Table 2) identified a monoclonal protein band in the immunoglobulin G (IgG) kappa region.
Cytologic study of the ascitic fluid was negative for malignant cells.
Chest CT revealed bilateral pleural effusions, pericardial effusion, and bilateral axillary lymphadenopathy. CT of the abdomen and pelvis was normal, except for ascites, and no pelvic tumor was noted.
Liver biopsy was done to look for the source of her unexplained ascites with elevated alkaline phosphatase, as all other investigations so far were normal. It revealed mild centrilobular scarring, but the rest of the parenchymal architecture was normal, with no evidence of bridging fibrosis or nodular regenerative hyperplasia (Figure 1).
Transjugular measurement of the hepatic vein pressure revealed a hepatic vein pressure gradient of 9 mm Hg, indicating mild portal hypertension. Venography showed widely patent hepatic and portal veins. Her high inflammatory marker levels could have been caused by smoldering rheumatoid arthritis; however, since the patient has had no joint symptoms for 18 years, this is very unlikely. It is more likely to be caused by a plasma cell disorder, as suggested by a monoclonal protein on electrophoresis.
WHAT IS THE DIAGNOSIS?
3. What is the most likely diagnosis in our patient?
- Rheumatoid arthritis
- Cryoglobulinemia
- Capillary leak syndrome
- Hematologic malignancy
- Syndrome of polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes (POEMS syndrome)
Rheumatoid arthritis can present with hepatosplenomegaly, lymphadenopathy, ascites, and skin rash, particularly if antinuclear antibody and rheumatoid factor are elevated. Ascites is known to occur in association with rheumatoid arthritis in the setting of Felty syndrome or nodular regenerative hyperplasia of the liver.2 However, our patient did not have leukopenia or evidence of regenerative hyperplasia on liver biopsy. Moreover, her rheumatoid arthritis had remained clinically inactive for a long time.
Cryoglobulinemia was possible, given her ascites, neuropathy, and splenomegaly, but her serum hepatic antibody and C4 complement values were normal.3 Also, the appearance of her rash was not typical of cryoglobulinemia.
Capillary leak syndrome was ruled out by the absence of hypotensive episodes, edema of the face or upper extremities, or renal failure.4
Lymphoma was excluded by flow cytometry.
A monoclonal protein on serum electrophoresis may suggest multiple myeloma, but this patient had multisystem involvement including organomegaly, endocrinopathy, and skin abnormalities. Thus, POEMS syndrome is the most likely diagnosis.
4. Which test should be done at this time to confirm the diagnosis of POEMS syndrome?
- Bone marrow biopsy
- Vascular endothelial growth factor testing
- Nerve conduction study
- Complete x-ray bone survey
A test for vascular endothelial growth factor should be done. This growth factor is almost always elevated in POEMS, and a positive test helps confirm the diagnosis of POEMS. Our patient’s level was elevated at 1,664 pg/mL (reference range 31–86).
POEMS is thought to be a variant of plasma cell dyscrasia, and all patients with POEMS have a monoclonal protein on electrophoresis. On this background, multiple myeloma is an important consideration.
Our patient underwent bone marrow biopsy, which revealed mild plasmacytosis (< 10%) (Figure 2). A complete bone survey showed generalized osteopenia without blastic or lytic lesions. To complete the workup for POEMS syndrome, a nerve conduction study was done to look for neuropathy; it showed bilateral sensory motor neuropathy with features of both a demyelinating process and axonal loss.
POEMS SYNDROME
POEMS syndrome is a constellation of features such as organomegaly and endocrine and skin abnormalities in association with neuropathy and a monoclonal protein on electrophoresis.5 In 2003, Dispenzieri et al6 described the major and minor diagnostic criteria based on a retrospective analysis of 99 patients with POEMS syndrome.6 Later, elevated vascular endothelial growth factor was added as a confirmatory diagnostic criterion.7 This growth factor is also an indicator of prognosis in POEMS syndrome, and its level can be used to monitor the response to treatment.7
Our patient met both major criteria for POEMS syndrome, ie, polyneuropathy (based on nerve conduction studies) and a monoclonal protein. Polyneuropathy in POEMS syndrome usually occurs as sensorimotor peripheral neuropathy of insidious onset and is seldom painful. Nerve biopsy study reveals demyelination with features of axonal loss. Interestingly, although our patient had neuropathy as diagnosed by electromyography, she remained clinically asymptomatic.
The monoclonal protein in POEMS syndrome is commonly IgA or IgG. Light chains are always present and are mainly the lambda type; kappa light chains are also reported in rare cases. Our patient had IgG kappa light chains.
Our patient met a number of the minor criteria for POEMS syndrome: ie, organomegaly (hepatosplenomegaly, lymphadenopathy), endocrinopathy (hypothyroidism, diabetes), skin changes (hyperpigmentation and plaques of the lower extremities), edema, pleural effusion, and ascites.
Endocrine disorders in POEMS syndrome
The endocrine abnormalities most often described in POEMS syndrome are hypogonadism, hypothyroidism, and diabetes mellitus. But because hypothyroidism and diabetes are common in the general population, it is debatable whether either of these could constitute the endocrine component of POEMS syndrome. Nevertheless, in three large series,6,7 occurrences of these two disorders were common, although less specific than adrenal or pituitary involvement.
In the analysis by Dispenzieri et al,6 67% of patients had at least one endocrine abnormality. Our patient had no evidence of an adrenal disorder.
Skin, skeletal, and other changes
The skin changes in POEMS syndrome are often nonspecific and include hyperpigmentation, sclerodema-like thickening, and plaques.
Skeletal changes are noted in up to 97% of patients. A skeletal survey in our patient revealed generalized osteopenia as opposed to osteosclerotic lesions, which are common in POEMS syndrome.
Anemia and thrombocytosis (as in our patient) are usually seen in POEMS syndrome and are induced by cytokines.6 POEMS syndrome also leads to increased thrombotic complications from the release of inflammatory cytokines.
Hypoalbuminemia and anasarca including ascites are often seen in POEMS syndrome (prevalence 29% to 89%) and are attributed to cytokine-induced increased vascular permeability. In POEMS syndrome, the serum-ascites albumin gradient is usually less than 1.1 g/dL, as in our patient.
Stepani et al8 reported one case of culture-negative neutrocytic ascites with portal hypertension in POEMS syndrome.8 (Culture-negative neutrocytic ascites is defined as an ascitic fluid polymorphonuclear count greater than 250/mm3 and a negative ascitic fluid culture in the absence of previous antibiotic therapy.) Chylous ascites has not yet been described in POEMS syndrome. However, chylous ascites is predominantly lymphocytic, whereas our patient had neutrocytic ascites.
We concluded that the cause of our patient’s ascites was multifactorial and included previous surgery and POEMS syndrome.
Nonclassic presentation
In addition to its classic presentation, POEMS syndrome is often reported in association with other “unusual features” such as cardiomyopathy, pulmonary hypertension, and cryoglobulinemia.6
So far, very few cases of portal hypertension in POEMS syndrome have been reported. Stepani et al8 described a patient who had POEMS syndrome and portal hypertension with extensive portal fibrosis without cirrhosis on liver biopsy. Inoue et al9 reported a liver biopsy feature consistent with idiopathic portal hypertension, also noting a case with mild fibrosis and few lymphocytic infiltrates in the portal tract.9
The etiopathogenesis of POEMS syndrome is attributed to proangiogenic vascular endothelial growth factor, and other inflammatory cytokines (interleukin 6, interleukin 1 beta, tumor necrosis factor alpha) also play a key role in pulmonary hypertension.10,11 A similar pathogenesis could also contribute to the development of portal hypertension (Figure 3).
CASE CONCLUDED
We started our patient on oral prednisone 60 mg daily for a month, tapered to a maintenance dose of 15 mg to suppress clonal proliferation of plasma cells. Her symptoms improved. Her vascular endothelial growth factor level decreased from 1,664 to 624 pg/mL. She was enrolled in a National Institutes of Health study to evaluate the effect of a potential new immunomodulator treatment for POEMS syndrome.
In conclusion, POEMS syndrome is rare and can present with many atypical features. A high index of suspicion is needed to detect it in a patient who has noncirrhotic portal hypertension with ascites and multisystem involvement.
- Runyon BA, Montano AA, Akriviadis EA, Antillon MR, Irving MA, McHutchison JG. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med 1992; 117:215–220.
- Harris M, Rash RM, Dymock IW. Nodular, non-cirrhotic liver associated with portal hypertension in a patient with rheumatoid arthritis. J Clin Pathol 1974; 27:963–966.
- Ramos-Casals M, Stone JH, Cid MC, Bosch X. The cryoglobulinaemias. Lancet 2012; 379:348–360.
- Druey KM, Greipp PR. Narrative review: the systemic capillary leak syndrome. Ann Intern Med 2010; 153:90–98.
- Bardwick PA, Zvaifler NJ, Gill GN, Newman D, Greenway GD, Resnick DL. Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome. Report on two cases and a review of the literature. Medicine (Baltimore) 1980; 59:311–322.
- Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood 2003; 101:2496–2506.
- Dispenzieri A. POEMS syndrome. Blood Rev 2007; 21:285–299.
- Stepani P, Courouble Y, Postel P, et al. Portal hypertension and neutrocytic ascites in POEMS syndrome. Gastroenterol Clin Biol 1998; 22:1095–1097. Article in French.
- Inoue R, Nakazawa A, Tsukada N, et al. POEMS syndrome with idiopathic portal hypertension: autopsy case and review of the literature. Pathol Int 2010; 60:316–320.
- Gherardi RK, Bélec L, Soubrier M, et al. Overproduction of proinflammatory cytokines imbalanced by their antagonists in POEMS syndrome. Blood 1996; 87:1458–1465.
- Mukerjee D, Kingdon E, Vanderpump M, Coghlan JG. Pathophysiological insights from a case of reversible pulmonary arterial hypertension. J R Soc Med 2003; 96:403–404.
- Runyon BA, Montano AA, Akriviadis EA, Antillon MR, Irving MA, McHutchison JG. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med 1992; 117:215–220.
- Harris M, Rash RM, Dymock IW. Nodular, non-cirrhotic liver associated with portal hypertension in a patient with rheumatoid arthritis. J Clin Pathol 1974; 27:963–966.
- Ramos-Casals M, Stone JH, Cid MC, Bosch X. The cryoglobulinaemias. Lancet 2012; 379:348–360.
- Druey KM, Greipp PR. Narrative review: the systemic capillary leak syndrome. Ann Intern Med 2010; 153:90–98.
- Bardwick PA, Zvaifler NJ, Gill GN, Newman D, Greenway GD, Resnick DL. Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome. Report on two cases and a review of the literature. Medicine (Baltimore) 1980; 59:311–322.
- Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood 2003; 101:2496–2506.
- Dispenzieri A. POEMS syndrome. Blood Rev 2007; 21:285–299.
- Stepani P, Courouble Y, Postel P, et al. Portal hypertension and neutrocytic ascites in POEMS syndrome. Gastroenterol Clin Biol 1998; 22:1095–1097. Article in French.
- Inoue R, Nakazawa A, Tsukada N, et al. POEMS syndrome with idiopathic portal hypertension: autopsy case and review of the literature. Pathol Int 2010; 60:316–320.
- Gherardi RK, Bélec L, Soubrier M, et al. Overproduction of proinflammatory cytokines imbalanced by their antagonists in POEMS syndrome. Blood 1996; 87:1458–1465.
- Mukerjee D, Kingdon E, Vanderpump M, Coghlan JG. Pathophysiological insights from a case of reversible pulmonary arterial hypertension. J R Soc Med 2003; 96:403–404.
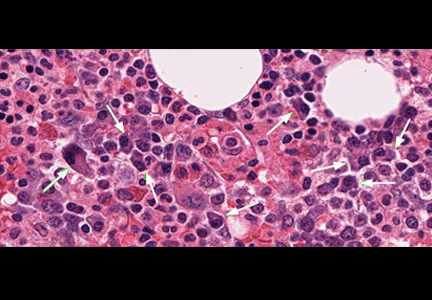
A 51-year-old woman with dyspnea
A 51-year-old woman presents to the emergency department with dyspnea, which began 4 days ago. She reports no chest pain, palpitations, hemoptysis, fevers, chills, weight loss, recent travel, immobility, or surgery. One week ago she noticed cramping in her right calf, but that has since resolved.
Her history includes hypertension, hypothyroidism, and immune-mediated glomerulonephritis with proteinuria. She is premenopausal. She takes losartan and levothyroxine; she is not taking oral contraceptives or herbal supplements. She is up to date with her cancer screening and has had negative findings on colonoscopy and mammography within the past year.
She has never smoked and she does not drink alcohol or use illicit drugs. Her mother has a history of provoked deep vein thrombosis and colon cancer.
Her temperature is 36.2°C (97.2°F), heart rate 163 beats per minute, blood pressure 158/102 mm Hg, respiratory rate 40 breaths per minute, and oxygen saturation by pulse oximetry 80% while breathing room air, corrected to 94% with oxygen 6 L/min via nasal cannula.
On physical examination, she is sitting upright on a stretcher and appears uncomfortable and anxious. She is awake and able to communicate clearly. Examination of the head, ears, eyes, nose, and throat is unremarkable, with moist mucous membranes. Her lungs are clear to auscultation. Her heart beat is very rapid, with a regular rhythm and an accentuated P2 heart sound. A right parasternal heave can be palpated in addition to a rightwardly displaced point of maximal impulse. The abdomen is normal, with no tenderness or organomegaly. She has no pain, edema, or erythema in the legs or feet, and she has strong, symmetric pulses (2+) in all extremities. The neurologic examination is nonfocal.
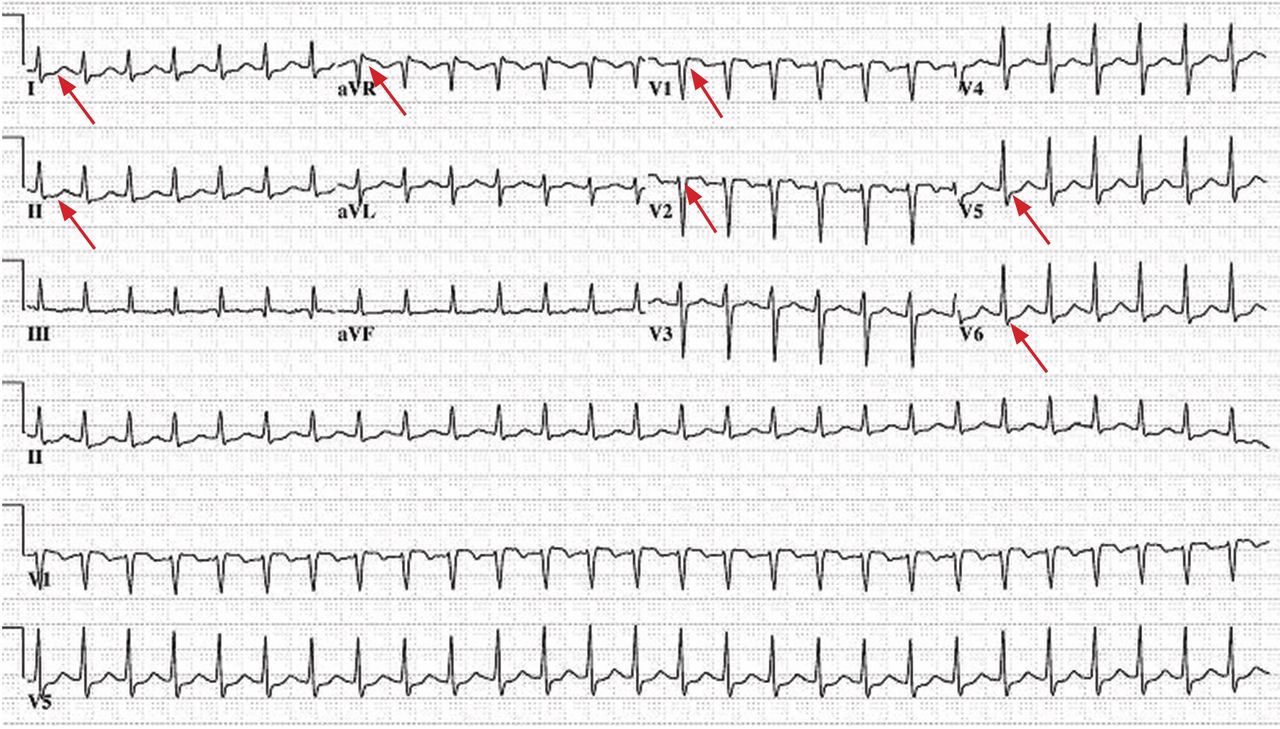
Electrocardiography (ECG) done on arrival (Figure 1) reveals supraventricular tachycardia, a normal axis, and nonspecific ST-segment and T-wave abnormalities, findings commonly seen in pulmonary embolism.1,2 On the other hand, her ECG does not show some of the other signs of right ventricular strain due to pulmonary embolism such as atrial arrhythmias, complete right bundle branch block, or inferior Q-waves.3,4
In view of her ECG findings and her symptoms of dyspnea, calf pain, tachypnea, tachycardia, and a pronounced P2 heart sound, her physician concludes that she very likely has a pulmonary embolism1 and orders an intravenous infusion of unfractionated heparin to be started immediately.
TESTING FOR PULMONARY EMBOLISM
1. Which of the following would be the best initial diagnostic imaging study to perform in this patient, who has a high pretest probability of pulmonary embolism?
- Multidetector computed tomographic (CT) pulmonary angiography
- Transthoracic echocardiography
- Magnetic resonance imaging
- Lower-extremity duplex ultrasonography
- Pulmonary angiography
- Ventilation-perfusion scintigraphy
Multidetector CT angiography is rapid, noninvasive, and highly sensitive (83%–90%) and specific (96%) for pulmonary embolism.5,6 In patients such as ours who have a high pretest probability of having the disease, its positive predictive value is 96%.5 Therefore, it would be the initial diagnostic study to perform in our patient.
Although transthoracic echocardiography is noninvasive and can detect right ventricular strain in the setting of pulmonary embolism, it may miss half of all pulmonary emboli detected by angiography.7,8
When technically adequate images are obtained, the combination of magnetic resonance angiography and magnetic resonance venography is very sensitive (92%) and specific (96%) for pulmonary embolism.9 However, one-fourth of patients undergoing these studies may have technically inadequate results, so this is not the best choice for diagnosis.9
As our patient complained of recent cramping in the right calf, lower-extremity duplex ultrasonography would be a reasonable test to screen for acute deep vein thrombosis as the source of pulmonary embolism. However, given her worrisome vital signs and impending hemodynamic collapse, CT pulmonary angiography would be a better initial test as it may guide more aggressive therapy. Furthermore, even if ultrasonography showed no evidence of deep vein thrombosis, clinical suspicion for pulmonary embolism would remain high enough that therapeutic anticoagulation would be continued until further testing ruled out this diagnosis.
Pulmonary angiography is the gold-standard test for pulmonary embolism. However, it is time-consuming, expensive, and invasive and so is not usually done unless the diagnosis cannot be made with other imaging studies.
Ventilation-perfusion scintigraphy is an established and safe diagnostic test for pulmonary embolism. It is particularly helpful in patients who have renal dysfunction or contrast allergy. The sensitivity of a high-probability scan is 78%, while the specificity of a very-low-probability scan is 97%.10 However, this study is often nondiagnostic (in 26.5% of cases),10 and further imaging may be required.
RESULTS OF CT ANGIOGRAPHY
Our patient undergoes CT angiography, which reveals multiple bilateral pulmonary emboli and right ventricular enlargement (Figure 2). Transthoracic echocardiography shows dilation of the right ventricle, with severely reduced systolic function, an underfilled and hyperdynamic left ventricle (ejection fraction 75%), and moderate tricuspid valve regurgitation. Her right ventricular systolic pressure is estimated to be 47 mm Hg.
Doppler ultrasonography of the legs reveals an occlusive thrombus within the right small saphenous vein that bulges and extends into the right popliteal vein. Also noted is a nonocclusive thrombus in the upper right popliteal vein that likely originated from the thrombus in the small saphenous vein.
Initial laboratory testing (Table 1) shows elevations of the cardiac enzymes troponin T and N-terminal pro-B-type natriuretic peptide (NT-pro-BNP).
ESTIMATING PROGNOSIS IN PULMONARY EMBOLISM
2. Which of the following laboratory results at presentation is independently associated with a worse outcome in patients with pulmonary embolism?
- Elevated NT-pro-BNP
- Hypercalcemia
- Thrombocytosis
- Hypernatremia
- Elevated procalcitonin
The Pulmonary Embolism Severity Index11 and the Simplified Pulmonary Embolism Severity Index12 (Table 2) are clinical calculators that help predict 30-day risk of death in patients with pulmonary embolism. Our patient’s Pulmonary Embolism Severity Index score is 60, indicating a very low risk, but her simplified severity index score is 2, indicating a high risk.
A shock index score (the heart rate divided by the systolic blood pressure) greater than 1 is also a sensitive measure of risk.13 (Our patient’s shock index score is 1.03.) Although the simplified version is more accurate,14 the shock index is also helpful when deciding whether patients with suspected pulmonary embolism should receive early fibrinolysis.15
In a large registry of patients with confirmed pulmonary embolism, risk factors for death were age greater than 70, cancer, clinical congestive heart failure, chronic obstructive pulmonary disease, systolic blood pressure lower than 90 mm Hg, respiratory rate less than 20 per minute, and right ventricular hypokinesis.16 Right ventricular dysfunction progressing to right ventricular failure and cardiogenic shock is the most common cause of death in patients with pulmonary embolism.16–18
Post hoc analysis has also shown that elevations of the biomarkers BNP, NT-pro-BNP, and cardiac troponins I and T are associated with a prolonged hospital course and a higher risk of death within 30 days.19 Interestingly, a recent retrospective analysis found hyponatremia to be an independent risk factor for death in the short term.20
Thrombocytopenia, not thrombocytosis, is associated with worse outcomes in patients with pulmonary embolism.16 Procalcitonin is elevated in bacterial pneumonia but is normal in pulmonary embolism and so may be helpful in differentiating between the two.21,22 Hypernatremia, hypercalcemia, and elevated procalcitonin have not been shown to be independently associated with worse outcomes in acute pulmonary embolism.
Thus, of the answer choices shown above, elevated NT-pro-BNP is the correct answer.
Classified as massive, submassive, or low-risk
Pulmonary embolism is often stratified as massive, submassive, or low-risk, reflecting the severity and the degree of cardiovascular collapse. The treatment depends on the classification.
Pulmonary embolism is classified as massive if the patient has a cardiac arrest or a systolic blood pressure lower than 90 mm Hg for more than 15 minutes.23 Nearly half of patients in this category die.24
Pulmonary embolism is submassive if the patient has systolic pressure greater than 90 mm Hg but has right ventricular dysfunction, as evidenced by physical examination, elevated cardiac biomarkers, electrocardiography, transthoracic echocardiography, or computed tomography. The death rate is as high as 15%.24
Pulmonary embolism in a normotensive patient with no right ventricular dysfunction is defined as low-risk.
Our patient so far
Our patient has bilateral pulmonary emboli, most likely originating from a deep vein thrombosis in her right lower leg. Her pulmonary embolism would be classified as submassive, as her systolic pressure is greater than 90 mm Hg and right ventricular dysfunction—significant right ventricular strain—was noted on both transthoracic echocardiography and computed tomography. Also, cardiac biomarkers are elevated, and the physical examination revealed a prominent P2 sound and right parasternal heave, also suggestive of right ventricular dysfunction.
Now, 6 hours have passed, and even though she has been receiving intravenous heparin during this time, her shock index remains greater than 1, indicating hemodynamic instability. Her pulse rate is still markedly high—over 160 bpm—and she still appears quite anxious and uncomfortable.
HOW SHOULD THIS PATIENT BE TREATED?
3. Which of the following is the most appropriate treatment for this patient?
- Start warfarin immediately while bridging with unfractionated heparin, low-molecular-weight heparin, or fondaparinux
- Start fibrinolysis with alteplase
- Give metoprolol intravenously to control her heart rate
- Start dabigatran immediately while bridging with unfractionated heparin
- Place an inferior vena cava filter
- Consult cardiothoracic surgery for emergency pulmonary embolectomy
All patients with confirmed pulmonary embolism and no contraindications to anticoagulation should begin treatment with low-molecular-weight heparin, unfractionated heparin, or fondaparinux.23 In addition, this therapy should be started empirically while the patient is still undergoing diagnostic testing if the pretest probability of pulmonary embolism is intermediate or high.23
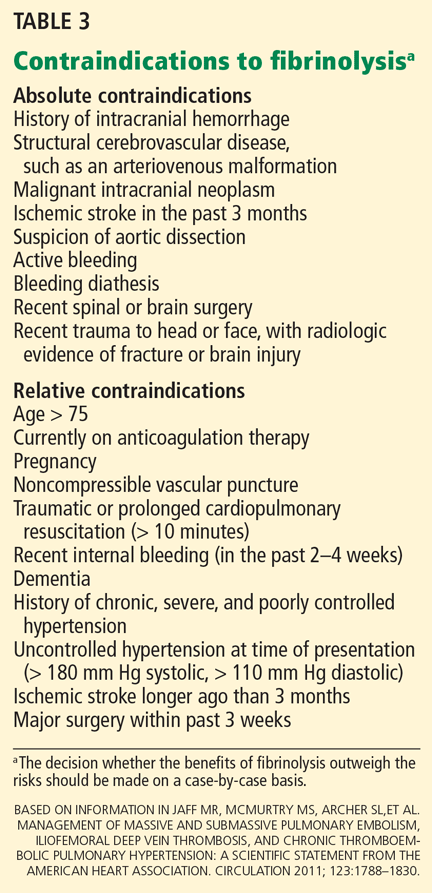
Warfarin is indicated for all patients with pulmonary embolism who do not have contraindications to it (Table 3). If unfractionated heparin, low-molecular-weight heparin, or fondaparinux has not already been started, it should be started at the same time as warfarin and should be continued until the international normalized ratio (INR) is within the therapeutic range.
Fibrinolysis. Treatment with a fibrinolytic agent in addition to heparin results in faster improvement of right ventricular function and pulmonary perfusion than with heparin alone.25 It may also decrease the incidence of pulmonary hypertension secondary to chronic thromboembolic disease.26 It should be considered in patients with massive pulmonary embolism.23
Whether fibrinolysis is appropriate for all patients with submassive pulmonary embolism remains controversial. Currently, it is not recommended for minor right ventricular dysfunction or myocardial necrosis if the patient has no signs of overt clinical decline.23 The Pulmonary Embolism Thrombolysis trial27 is an ongoing prospective randomized comparison of tenecteplase in a single bolus plus heparin vs heparin alone in normotensive patients with submassive pulmonary embolism, such as our patient. This trial may elucidate the benefit of fibrinolytic therapy in patients with submassive pulmonary embolism.
Patients at low risk are generally treated with heparin and warfarin anticoagulation alone. Fibrinolysis is not recommended in these patients, as the risk of bleeding outweighs the potential benefits.23
Metoprolol may not be advisable for our patient, as her tachycardia is likely compensatory, and beta-blocker therapy could blunt this compensatory response, leading to inadequate systemic perfusion.
Dabigatran is an oral direct thrombin inhibitor that does not require laboratory monitoring. It is currently approved for the prevention of stroke in patients with atrial fibrillation. It has been shown to be as effective as warfarin in the treatment of acute venous thromboembolism28 and may be a viable option in the future, but as of this writing it has not yet been approved in the United States for this indication. Furthermore, dabigatran inhibits thrombin immediately, so continued heparin bridging would not be necessary.
An inferior vena cava filter may prevent recurrent pulmonary embolism for patients who have absolute contraindications to anticoagulation, most significantly in the short term, ie, in the first few weeks after placement. However, these devices have not yet been shown to improve long-term mortality rates.
Embolectomy, percutaneous or surgical, is also an option. For patients in whom thrombolytic therapy is not effective, “rescue” surgical embolectomy has been associated with better outcomes compared with secondary thrombolysis and so should be considered.29
Back to our patient
An intravenous infusion of alteplase is started, and the patient’s tachycardia improves. Her oxygen requirements normalize, and she is transferred to the general medical floor the next day. She receives subcutaneous dalteparin as a bridge therapy, and warfarin is titrated to a goal INR of 2.0 to 3.0. Because of the acute deep vein thrombosis in her right lower leg, she is instructed to wear knee-high fitted compression hose for primary prevention of postphlebitic syndrome.
HOW LONG TO TREAT? IS GENETIC TESTING INDICATED?
Patients with a first episode of unprovoked venous thromboembolism should receive oral anticoagulants for 6 months, while those with recurrent unprovoked venous thromboembolism require lifelong oral anticoagulation.23
Whether to test for inherited thrombophilia after a first episode of venous thromboembolism to guide the duration of anticoagulation is controversial.30 Indiscriminate testing has not been recommended in these patients,31 but the American College of Medical Genetics recommends genetic screening for factor V Leiden in patients who have an unprovoked incident of venous thromboembolism before age 50.32
No randomized controlled trial has assessed whether thrombophilia testing decreases the recurrence rate of venous thromboembolism.33 One uncontrolled study suggested that testing for inherited thrombophilias in patients with a first episode does not affect the risk of recurrence.34 Testing is costly and may cause psychological distress for patients and family members.
Our patient is discharged home on warfarin for 6 months with subsequent follow-up evaluation in the thrombophilia clinic.
WHEN SHOULD WARFARIN BE RESTARTED?
4. If our patient were to discontinue oral anticoagulation in 6 months, which of the following, if present 1 month afterwards, would be a reason to restart oral anticoagulation?
- Elevated serum cotinine
- Positive pregnancy test
- Elevated follicle-stimulating hormone and luteinizing hormone and low estradiol levels
- Elevated D-dimer
Cotinine is a nicotine metabolite, and serum levels are elevated in smokers. Smoking and pregnancy both increase the risk of venous thromboembolism. However, smoking or pregnancy alone would not be a reason to increase the duration of anticoagulation.
Warfarin is contraindicated in pregnancy because of its teratogenic effects, particularly in the first trimester. Follicle-stimulating hormone and luteinizing hormone levels increase in response to decreased estradiol at menopause. Postmenopausal women are not at increased risk of venous thromboembolism unless they are taking oral estrogen hormone replacement therapy.
An elevated D-dimer level 1 month after stopping of oral anticoagulation has been associated with a higher rate of recurrence of venous thromboembolism, which is reduced by a resumption of anticoagulation.35 Therefore, consideration should be given to resuming anticoagulation in patients who have an elevated D-dimer level.
TAKE-HOME POINTS
It is important to distinguish between massive, submassive, and low-risk pulmonary embolism, since each type has a different treatment and prognosis.
Fibrinolytic therapy is indicated in patients with massive pulmonary embolism when no contraindication is present, whereas it is not indicated in those with low-risk pulmonary embolism.
Management of submassive pulmonary embolism continues to be an area of considerable debate. Current American Heart Association guidelines recommend consideration of fibrinolysis initially in patients with submassive acute pulmonary embolism if there is concurrent clinical evidence of adverse prognosis—eg, new hemodynamic instability, worsening respiratory insufficiency, severe right ventricular dysfunction, or major myocardial necrosis.23 On the other hand, the American College of Chest Physicians recommends against initial systemic fibrinolysis in submassive acute pulmonary embolism based on biomarkers or findings on ECG, transthoracic echocardiography, or CT, recommending it only in therapeutically anticoagulated patients deemed to be at high risk of hypotension.36
Since the optimal treatment of submassive pulmonary embolism is still not known, it is important that clinicians remain up to date on the evidence and guidelines.
- Stein PD, Beemath A, Matta F, et al. Clinical characteristics of patients with acute pulmonary embolism: data from PIOPED II. Am J Med 2007; 120:871–879.
- Stein PD, Saltzman HA, Weg JG. Clinical characteristics of patients with acute pulmonary embolism. Am J Cardiol 1991; 68:1723–1724.
- Ferrari E, Imbert A, Chevalier T, Mihoubi A, Morand P, Baudouy M. The ECG in pulmonary embolism. Predictive value of negative T waves in precordial leads—80 case reports. Chest 1997; 111:537–543.
- Geibel A, Zehender M, Kasper W, Olschewski M, Klima C, Konstantinides SV. Prognostic value of the ECG on admission in patients with acute major pulmonary embolism. Eur Respir J 2005; 25:843–848.
- Perrier A, Roy PM, Sanchez O, et al. Multidetector-row computed tomography in suspected pulmonary embolism. N Engl J Med 2005; 352:1760–1768.
- Qanadli SD, Hajjam ME, Mesurolle B, et al. Pulmonary embolism detection: prospective evaluation of dual-section helical CT versus selective pulmonary arteriography in 157 patients. Radiology 2000; 217:447–455.
- Miniati M, Monti S, Pratali L, et al. Value of transthoracic echocardiography in the diagnosis of pulmonary embolism: results of a prospective study in unselected patients. Am J Med 2001; 110:528–535.
- Bova C, Greco F, Misuraca G, et al. Diagnostic utility of echocardiography in patients with suspected pulmonary embolism. Am J Emerg Med 2003; 21:180–183.
- Stein PD, Chenevert TL, Fowler SE, et al; PIOPED III (Prospective Investigation of Pulmonary Embolism Diagnosis III) Investigators. Gadolinium-enhanced magnetic resonance angiography for pulmonary embolism: a multicenter prospective study (PIOPED III). Ann Intern Med 2010; 152:434–443,W142–W143.
- Sostman HD, Stein PD, Gottschalk A, Matta F, Hull R, Goodman L. Acute pulmonary embolism: sensitivity and specificity of ventilation-perfusion scintigraphy in PIOPED II study. Radiology 2008; 246:941–946.
- Aujesky D, Obrosky DS, Stone RA, et al. Derivation and validation of a prognostic model for pulmonary embolism. Am J Respir Crit Care Med 2005; 172:1041–1046.
- Jiménez D, Aujesky D, Moores L, et al; RIETE Investigators. Simplification of the pulmonary embolism severity index for prognostication in patients with acute symptomatic pulmonary embolism. Arch Intern Med 2010; 170:1383–1389.
- Otero R, Trujillo-Santos J, Cayuela A, et al; Registro Informatizado de la Enfermedad Tromboembólica (RIETE) Investigators. Haemodynamically unstable pulmonary embolism in the RIETE Registry: systolic blood pressure or shock index? Eur Respir J 2007; 30:1111–1116.
- Sam A, Sánchez D, Gómez V, et al. The shock index and the simplified PESI for identification of low-risk patients with acute pulmonary embolism. Eur Respir J 2011; 37:762–766.
- Kucher N, Luder CM, Dörnhöfer T, Windecker S, Meier B, Hess OM. Novel management strategy for patients with suspected pulmonary embolism. Eur Heart J 2003; 24:366–376.
- Goldhaber SZ, Visani L, De Rosa M. Acute pulmonary embolism: clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER). Lancet 1999; 353:1386–1389.
- ten Wolde M, Söhne M, Quak E, Mac Gillavry MR, Büller HR. Prognostic value of echocardiographically assessed right ventricular dysfunction in patients with pulmonary embolism. Arch Intern Med 2004; 164:1685–1689.
- Sanchez O, Trinquart L, Colombet I, et al. Prognostic value of right ventricular dysfunction in patients with haemodynamically stable pulmonary embolism: a systematic review. Eur Heart J 2008; 29:1569–1577.
- Kucher N, Goldhaber SZ. Cardiac biomarkers for risk stratification of patients with acute pulmonary embolism. Circulation 2003; 108:2191–2194.
- Scherz N, Labarère J, Méan M, Ibrahim SA, Fine MJ, Aujesky D. Prognostic importance of hyponatremia in patients with acute pulmonary embolism. Am J Respir Crit Care Med 2010; 182:1178–1183.
- Delèvaux I, André M, Aumaître O, Bègue RJ, Colombier M, Piette JC. Procalcitonin measurement for differential diagnosis between pulmonary embolism and pneumonia. Crit Care Med 2003; 31:661.
- Köktürk N, Kanbay A, Bukan N, Ekim N. The value of serum procalcitonin in differential diagnosis of pulmonary embolism and community-acquired pneumonia. Clin Appl Thromb Hemost 2011; 17:519–525.
- Jaff MR, McMurtry MS, Archer SL, et al; American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation. Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation 2011; 123:1788–1830.
- Kucher N, Rossi E, De Rosa M, Goldhaber SZ. Massive pulmonary embolism. Circulation 2006; 113:577–582.
- Goldhaber SZ, Haire WD, Feldstein ML, et al. Alteplase versus heparin in acute pulmonary embolism: randomised trial assessing right-ventricular function and pulmonary perfusion. Lancet 1993; 341:507–511.
- Kline JA, Steuerwald MT, Marchick MR, Hernandez-Nino J, Rose GA. Prospective evaluation of right ventricular function and functional status 6 months after acute submassive pulmonary embolism: frequency of persistent or subsequent elevation in estimated pulmonary artery pressure. Chest 2009; 136:1202–1210.
- Steering Committee of PEITHO Investigators. Single-bolus tenecteplase plus heparin compared with heparin alone for normotensive patients with acute pulmonary embolism who have evidence of right ventricular dysfunction and myocardial injury: rationale and design of the Pulmonary Embolism Thrombolysis (PEITHO) trial. Am Heart J 2012; 163:33–38.e1.
- Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- Meneveau N, Séronde MF, Blonde MC, et al. Management of unsuccessful thrombolysis in acute massive pulmonary embolism. Chest 2006; 129:1043–1050.
- Ho WK, Hankey GJ, Eikelboom JW. Should adult patients be routinely tested for heritable thrombophilia after an episode of venous thromboembolism? Med J Aust 2011; 195:139–142.
- Baglin T, Gray E, Greaves M, et al; British Committee for Standards in Haematology. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010; 149:209–220.
- Grody WW, Griffin JH, Taylor AK, Korf BR, Heit JA; ACMG Factor V Leiden Working Group. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med 2001; 3:139–148.
- Cohn D, Vansenne F, de Borgie C, Middeldorp S. Thrombophilia testing for prevention of recurrent venous thromboembolism. Cochrane Database Syst Rev 2009; ( 1):CD007069.
- Coppens M, Reijnders JH, Middeldorp S, Doggen CJ, Rosendaal FR. Testing for inherited thrombophilia does not reduce the recurrence of venous thrombosis. J Thromb Haemost 2008; 6:1474–1477.
- Palareti G, Cosmi B, Legnani C, et al; PROLONG Investigators. Ddimer testing to determine the duration of anticoagulation therapy. N Engl J Med 2006; 355:1780–1789.
- Guyatt GH, Akl EA, Crowther M, Gutterman DD, Schuünemann HJ; American College of Chest Physicians Antithrombotic Therapy and Prevention of Thrombosis Panel. Executive summary: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl):7S–47S.
A 51-year-old woman presents to the emergency department with dyspnea, which began 4 days ago. She reports no chest pain, palpitations, hemoptysis, fevers, chills, weight loss, recent travel, immobility, or surgery. One week ago she noticed cramping in her right calf, but that has since resolved.
Her history includes hypertension, hypothyroidism, and immune-mediated glomerulonephritis with proteinuria. She is premenopausal. She takes losartan and levothyroxine; she is not taking oral contraceptives or herbal supplements. She is up to date with her cancer screening and has had negative findings on colonoscopy and mammography within the past year.
She has never smoked and she does not drink alcohol or use illicit drugs. Her mother has a history of provoked deep vein thrombosis and colon cancer.
Her temperature is 36.2°C (97.2°F), heart rate 163 beats per minute, blood pressure 158/102 mm Hg, respiratory rate 40 breaths per minute, and oxygen saturation by pulse oximetry 80% while breathing room air, corrected to 94% with oxygen 6 L/min via nasal cannula.
On physical examination, she is sitting upright on a stretcher and appears uncomfortable and anxious. She is awake and able to communicate clearly. Examination of the head, ears, eyes, nose, and throat is unremarkable, with moist mucous membranes. Her lungs are clear to auscultation. Her heart beat is very rapid, with a regular rhythm and an accentuated P2 heart sound. A right parasternal heave can be palpated in addition to a rightwardly displaced point of maximal impulse. The abdomen is normal, with no tenderness or organomegaly. She has no pain, edema, or erythema in the legs or feet, and she has strong, symmetric pulses (2+) in all extremities. The neurologic examination is nonfocal.
Electrocardiography (ECG) done on arrival (Figure 1) reveals supraventricular tachycardia, a normal axis, and nonspecific ST-segment and T-wave abnormalities, findings commonly seen in pulmonary embolism.1,2 On the other hand, her ECG does not show some of the other signs of right ventricular strain due to pulmonary embolism such as atrial arrhythmias, complete right bundle branch block, or inferior Q-waves.3,4
In view of her ECG findings and her symptoms of dyspnea, calf pain, tachypnea, tachycardia, and a pronounced P2 heart sound, her physician concludes that she very likely has a pulmonary embolism1 and orders an intravenous infusion of unfractionated heparin to be started immediately.
TESTING FOR PULMONARY EMBOLISM
1. Which of the following would be the best initial diagnostic imaging study to perform in this patient, who has a high pretest probability of pulmonary embolism?
- Multidetector computed tomographic (CT) pulmonary angiography
- Transthoracic echocardiography
- Magnetic resonance imaging
- Lower-extremity duplex ultrasonography
- Pulmonary angiography
- Ventilation-perfusion scintigraphy
Multidetector CT angiography is rapid, noninvasive, and highly sensitive (83%–90%) and specific (96%) for pulmonary embolism.5,6 In patients such as ours who have a high pretest probability of having the disease, its positive predictive value is 96%.5 Therefore, it would be the initial diagnostic study to perform in our patient.
Although transthoracic echocardiography is noninvasive and can detect right ventricular strain in the setting of pulmonary embolism, it may miss half of all pulmonary emboli detected by angiography.7,8
When technically adequate images are obtained, the combination of magnetic resonance angiography and magnetic resonance venography is very sensitive (92%) and specific (96%) for pulmonary embolism.9 However, one-fourth of patients undergoing these studies may have technically inadequate results, so this is not the best choice for diagnosis.9
As our patient complained of recent cramping in the right calf, lower-extremity duplex ultrasonography would be a reasonable test to screen for acute deep vein thrombosis as the source of pulmonary embolism. However, given her worrisome vital signs and impending hemodynamic collapse, CT pulmonary angiography would be a better initial test as it may guide more aggressive therapy. Furthermore, even if ultrasonography showed no evidence of deep vein thrombosis, clinical suspicion for pulmonary embolism would remain high enough that therapeutic anticoagulation would be continued until further testing ruled out this diagnosis.
Pulmonary angiography is the gold-standard test for pulmonary embolism. However, it is time-consuming, expensive, and invasive and so is not usually done unless the diagnosis cannot be made with other imaging studies.
Ventilation-perfusion scintigraphy is an established and safe diagnostic test for pulmonary embolism. It is particularly helpful in patients who have renal dysfunction or contrast allergy. The sensitivity of a high-probability scan is 78%, while the specificity of a very-low-probability scan is 97%.10 However, this study is often nondiagnostic (in 26.5% of cases),10 and further imaging may be required.
RESULTS OF CT ANGIOGRAPHY
Our patient undergoes CT angiography, which reveals multiple bilateral pulmonary emboli and right ventricular enlargement (Figure 2). Transthoracic echocardiography shows dilation of the right ventricle, with severely reduced systolic function, an underfilled and hyperdynamic left ventricle (ejection fraction 75%), and moderate tricuspid valve regurgitation. Her right ventricular systolic pressure is estimated to be 47 mm Hg.
Doppler ultrasonography of the legs reveals an occlusive thrombus within the right small saphenous vein that bulges and extends into the right popliteal vein. Also noted is a nonocclusive thrombus in the upper right popliteal vein that likely originated from the thrombus in the small saphenous vein.
Initial laboratory testing (Table 1) shows elevations of the cardiac enzymes troponin T and N-terminal pro-B-type natriuretic peptide (NT-pro-BNP).
ESTIMATING PROGNOSIS IN PULMONARY EMBOLISM
2. Which of the following laboratory results at presentation is independently associated with a worse outcome in patients with pulmonary embolism?
- Elevated NT-pro-BNP
- Hypercalcemia
- Thrombocytosis
- Hypernatremia
- Elevated procalcitonin
The Pulmonary Embolism Severity Index11 and the Simplified Pulmonary Embolism Severity Index12 (Table 2) are clinical calculators that help predict 30-day risk of death in patients with pulmonary embolism. Our patient’s Pulmonary Embolism Severity Index score is 60, indicating a very low risk, but her simplified severity index score is 2, indicating a high risk.
A shock index score (the heart rate divided by the systolic blood pressure) greater than 1 is also a sensitive measure of risk.13 (Our patient’s shock index score is 1.03.) Although the simplified version is more accurate,14 the shock index is also helpful when deciding whether patients with suspected pulmonary embolism should receive early fibrinolysis.15
In a large registry of patients with confirmed pulmonary embolism, risk factors for death were age greater than 70, cancer, clinical congestive heart failure, chronic obstructive pulmonary disease, systolic blood pressure lower than 90 mm Hg, respiratory rate less than 20 per minute, and right ventricular hypokinesis.16 Right ventricular dysfunction progressing to right ventricular failure and cardiogenic shock is the most common cause of death in patients with pulmonary embolism.16–18
Post hoc analysis has also shown that elevations of the biomarkers BNP, NT-pro-BNP, and cardiac troponins I and T are associated with a prolonged hospital course and a higher risk of death within 30 days.19 Interestingly, a recent retrospective analysis found hyponatremia to be an independent risk factor for death in the short term.20
Thrombocytopenia, not thrombocytosis, is associated with worse outcomes in patients with pulmonary embolism.16 Procalcitonin is elevated in bacterial pneumonia but is normal in pulmonary embolism and so may be helpful in differentiating between the two.21,22 Hypernatremia, hypercalcemia, and elevated procalcitonin have not been shown to be independently associated with worse outcomes in acute pulmonary embolism.
Thus, of the answer choices shown above, elevated NT-pro-BNP is the correct answer.
Classified as massive, submassive, or low-risk
Pulmonary embolism is often stratified as massive, submassive, or low-risk, reflecting the severity and the degree of cardiovascular collapse. The treatment depends on the classification.
Pulmonary embolism is classified as massive if the patient has a cardiac arrest or a systolic blood pressure lower than 90 mm Hg for more than 15 minutes.23 Nearly half of patients in this category die.24
Pulmonary embolism is submassive if the patient has systolic pressure greater than 90 mm Hg but has right ventricular dysfunction, as evidenced by physical examination, elevated cardiac biomarkers, electrocardiography, transthoracic echocardiography, or computed tomography. The death rate is as high as 15%.24
Pulmonary embolism in a normotensive patient with no right ventricular dysfunction is defined as low-risk.
Our patient so far
Our patient has bilateral pulmonary emboli, most likely originating from a deep vein thrombosis in her right lower leg. Her pulmonary embolism would be classified as submassive, as her systolic pressure is greater than 90 mm Hg and right ventricular dysfunction—significant right ventricular strain—was noted on both transthoracic echocardiography and computed tomography. Also, cardiac biomarkers are elevated, and the physical examination revealed a prominent P2 sound and right parasternal heave, also suggestive of right ventricular dysfunction.
Now, 6 hours have passed, and even though she has been receiving intravenous heparin during this time, her shock index remains greater than 1, indicating hemodynamic instability. Her pulse rate is still markedly high—over 160 bpm—and she still appears quite anxious and uncomfortable.
HOW SHOULD THIS PATIENT BE TREATED?
3. Which of the following is the most appropriate treatment for this patient?
- Start warfarin immediately while bridging with unfractionated heparin, low-molecular-weight heparin, or fondaparinux
- Start fibrinolysis with alteplase
- Give metoprolol intravenously to control her heart rate
- Start dabigatran immediately while bridging with unfractionated heparin
- Place an inferior vena cava filter
- Consult cardiothoracic surgery for emergency pulmonary embolectomy
All patients with confirmed pulmonary embolism and no contraindications to anticoagulation should begin treatment with low-molecular-weight heparin, unfractionated heparin, or fondaparinux.23 In addition, this therapy should be started empirically while the patient is still undergoing diagnostic testing if the pretest probability of pulmonary embolism is intermediate or high.23
Warfarin is indicated for all patients with pulmonary embolism who do not have contraindications to it (Table 3). If unfractionated heparin, low-molecular-weight heparin, or fondaparinux has not already been started, it should be started at the same time as warfarin and should be continued until the international normalized ratio (INR) is within the therapeutic range.
Fibrinolysis. Treatment with a fibrinolytic agent in addition to heparin results in faster improvement of right ventricular function and pulmonary perfusion than with heparin alone.25 It may also decrease the incidence of pulmonary hypertension secondary to chronic thromboembolic disease.26 It should be considered in patients with massive pulmonary embolism.23
Whether fibrinolysis is appropriate for all patients with submassive pulmonary embolism remains controversial. Currently, it is not recommended for minor right ventricular dysfunction or myocardial necrosis if the patient has no signs of overt clinical decline.23 The Pulmonary Embolism Thrombolysis trial27 is an ongoing prospective randomized comparison of tenecteplase in a single bolus plus heparin vs heparin alone in normotensive patients with submassive pulmonary embolism, such as our patient. This trial may elucidate the benefit of fibrinolytic therapy in patients with submassive pulmonary embolism.
Patients at low risk are generally treated with heparin and warfarin anticoagulation alone. Fibrinolysis is not recommended in these patients, as the risk of bleeding outweighs the potential benefits.23
Metoprolol may not be advisable for our patient, as her tachycardia is likely compensatory, and beta-blocker therapy could blunt this compensatory response, leading to inadequate systemic perfusion.
Dabigatran is an oral direct thrombin inhibitor that does not require laboratory monitoring. It is currently approved for the prevention of stroke in patients with atrial fibrillation. It has been shown to be as effective as warfarin in the treatment of acute venous thromboembolism28 and may be a viable option in the future, but as of this writing it has not yet been approved in the United States for this indication. Furthermore, dabigatran inhibits thrombin immediately, so continued heparin bridging would not be necessary.
An inferior vena cava filter may prevent recurrent pulmonary embolism for patients who have absolute contraindications to anticoagulation, most significantly in the short term, ie, in the first few weeks after placement. However, these devices have not yet been shown to improve long-term mortality rates.
Embolectomy, percutaneous or surgical, is also an option. For patients in whom thrombolytic therapy is not effective, “rescue” surgical embolectomy has been associated with better outcomes compared with secondary thrombolysis and so should be considered.29
Back to our patient
An intravenous infusion of alteplase is started, and the patient’s tachycardia improves. Her oxygen requirements normalize, and she is transferred to the general medical floor the next day. She receives subcutaneous dalteparin as a bridge therapy, and warfarin is titrated to a goal INR of 2.0 to 3.0. Because of the acute deep vein thrombosis in her right lower leg, she is instructed to wear knee-high fitted compression hose for primary prevention of postphlebitic syndrome.
HOW LONG TO TREAT? IS GENETIC TESTING INDICATED?
Patients with a first episode of unprovoked venous thromboembolism should receive oral anticoagulants for 6 months, while those with recurrent unprovoked venous thromboembolism require lifelong oral anticoagulation.23
Whether to test for inherited thrombophilia after a first episode of venous thromboembolism to guide the duration of anticoagulation is controversial.30 Indiscriminate testing has not been recommended in these patients,31 but the American College of Medical Genetics recommends genetic screening for factor V Leiden in patients who have an unprovoked incident of venous thromboembolism before age 50.32
No randomized controlled trial has assessed whether thrombophilia testing decreases the recurrence rate of venous thromboembolism.33 One uncontrolled study suggested that testing for inherited thrombophilias in patients with a first episode does not affect the risk of recurrence.34 Testing is costly and may cause psychological distress for patients and family members.
Our patient is discharged home on warfarin for 6 months with subsequent follow-up evaluation in the thrombophilia clinic.
WHEN SHOULD WARFARIN BE RESTARTED?
4. If our patient were to discontinue oral anticoagulation in 6 months, which of the following, if present 1 month afterwards, would be a reason to restart oral anticoagulation?
- Elevated serum cotinine
- Positive pregnancy test
- Elevated follicle-stimulating hormone and luteinizing hormone and low estradiol levels
- Elevated D-dimer
Cotinine is a nicotine metabolite, and serum levels are elevated in smokers. Smoking and pregnancy both increase the risk of venous thromboembolism. However, smoking or pregnancy alone would not be a reason to increase the duration of anticoagulation.
Warfarin is contraindicated in pregnancy because of its teratogenic effects, particularly in the first trimester. Follicle-stimulating hormone and luteinizing hormone levels increase in response to decreased estradiol at menopause. Postmenopausal women are not at increased risk of venous thromboembolism unless they are taking oral estrogen hormone replacement therapy.
An elevated D-dimer level 1 month after stopping of oral anticoagulation has been associated with a higher rate of recurrence of venous thromboembolism, which is reduced by a resumption of anticoagulation.35 Therefore, consideration should be given to resuming anticoagulation in patients who have an elevated D-dimer level.
TAKE-HOME POINTS
It is important to distinguish between massive, submassive, and low-risk pulmonary embolism, since each type has a different treatment and prognosis.
Fibrinolytic therapy is indicated in patients with massive pulmonary embolism when no contraindication is present, whereas it is not indicated in those with low-risk pulmonary embolism.
Management of submassive pulmonary embolism continues to be an area of considerable debate. Current American Heart Association guidelines recommend consideration of fibrinolysis initially in patients with submassive acute pulmonary embolism if there is concurrent clinical evidence of adverse prognosis—eg, new hemodynamic instability, worsening respiratory insufficiency, severe right ventricular dysfunction, or major myocardial necrosis.23 On the other hand, the American College of Chest Physicians recommends against initial systemic fibrinolysis in submassive acute pulmonary embolism based on biomarkers or findings on ECG, transthoracic echocardiography, or CT, recommending it only in therapeutically anticoagulated patients deemed to be at high risk of hypotension.36
Since the optimal treatment of submassive pulmonary embolism is still not known, it is important that clinicians remain up to date on the evidence and guidelines.
A 51-year-old woman presents to the emergency department with dyspnea, which began 4 days ago. She reports no chest pain, palpitations, hemoptysis, fevers, chills, weight loss, recent travel, immobility, or surgery. One week ago she noticed cramping in her right calf, but that has since resolved.
Her history includes hypertension, hypothyroidism, and immune-mediated glomerulonephritis with proteinuria. She is premenopausal. She takes losartan and levothyroxine; she is not taking oral contraceptives or herbal supplements. She is up to date with her cancer screening and has had negative findings on colonoscopy and mammography within the past year.
She has never smoked and she does not drink alcohol or use illicit drugs. Her mother has a history of provoked deep vein thrombosis and colon cancer.
Her temperature is 36.2°C (97.2°F), heart rate 163 beats per minute, blood pressure 158/102 mm Hg, respiratory rate 40 breaths per minute, and oxygen saturation by pulse oximetry 80% while breathing room air, corrected to 94% with oxygen 6 L/min via nasal cannula.
On physical examination, she is sitting upright on a stretcher and appears uncomfortable and anxious. She is awake and able to communicate clearly. Examination of the head, ears, eyes, nose, and throat is unremarkable, with moist mucous membranes. Her lungs are clear to auscultation. Her heart beat is very rapid, with a regular rhythm and an accentuated P2 heart sound. A right parasternal heave can be palpated in addition to a rightwardly displaced point of maximal impulse. The abdomen is normal, with no tenderness or organomegaly. She has no pain, edema, or erythema in the legs or feet, and she has strong, symmetric pulses (2+) in all extremities. The neurologic examination is nonfocal.
Electrocardiography (ECG) done on arrival (Figure 1) reveals supraventricular tachycardia, a normal axis, and nonspecific ST-segment and T-wave abnormalities, findings commonly seen in pulmonary embolism.1,2 On the other hand, her ECG does not show some of the other signs of right ventricular strain due to pulmonary embolism such as atrial arrhythmias, complete right bundle branch block, or inferior Q-waves.3,4
In view of her ECG findings and her symptoms of dyspnea, calf pain, tachypnea, tachycardia, and a pronounced P2 heart sound, her physician concludes that she very likely has a pulmonary embolism1 and orders an intravenous infusion of unfractionated heparin to be started immediately.
TESTING FOR PULMONARY EMBOLISM
1. Which of the following would be the best initial diagnostic imaging study to perform in this patient, who has a high pretest probability of pulmonary embolism?
- Multidetector computed tomographic (CT) pulmonary angiography
- Transthoracic echocardiography
- Magnetic resonance imaging
- Lower-extremity duplex ultrasonography
- Pulmonary angiography
- Ventilation-perfusion scintigraphy
Multidetector CT angiography is rapid, noninvasive, and highly sensitive (83%–90%) and specific (96%) for pulmonary embolism.5,6 In patients such as ours who have a high pretest probability of having the disease, its positive predictive value is 96%.5 Therefore, it would be the initial diagnostic study to perform in our patient.
Although transthoracic echocardiography is noninvasive and can detect right ventricular strain in the setting of pulmonary embolism, it may miss half of all pulmonary emboli detected by angiography.7,8
When technically adequate images are obtained, the combination of magnetic resonance angiography and magnetic resonance venography is very sensitive (92%) and specific (96%) for pulmonary embolism.9 However, one-fourth of patients undergoing these studies may have technically inadequate results, so this is not the best choice for diagnosis.9
As our patient complained of recent cramping in the right calf, lower-extremity duplex ultrasonography would be a reasonable test to screen for acute deep vein thrombosis as the source of pulmonary embolism. However, given her worrisome vital signs and impending hemodynamic collapse, CT pulmonary angiography would be a better initial test as it may guide more aggressive therapy. Furthermore, even if ultrasonography showed no evidence of deep vein thrombosis, clinical suspicion for pulmonary embolism would remain high enough that therapeutic anticoagulation would be continued until further testing ruled out this diagnosis.
Pulmonary angiography is the gold-standard test for pulmonary embolism. However, it is time-consuming, expensive, and invasive and so is not usually done unless the diagnosis cannot be made with other imaging studies.
Ventilation-perfusion scintigraphy is an established and safe diagnostic test for pulmonary embolism. It is particularly helpful in patients who have renal dysfunction or contrast allergy. The sensitivity of a high-probability scan is 78%, while the specificity of a very-low-probability scan is 97%.10 However, this study is often nondiagnostic (in 26.5% of cases),10 and further imaging may be required.
RESULTS OF CT ANGIOGRAPHY
Our patient undergoes CT angiography, which reveals multiple bilateral pulmonary emboli and right ventricular enlargement (Figure 2). Transthoracic echocardiography shows dilation of the right ventricle, with severely reduced systolic function, an underfilled and hyperdynamic left ventricle (ejection fraction 75%), and moderate tricuspid valve regurgitation. Her right ventricular systolic pressure is estimated to be 47 mm Hg.
Doppler ultrasonography of the legs reveals an occlusive thrombus within the right small saphenous vein that bulges and extends into the right popliteal vein. Also noted is a nonocclusive thrombus in the upper right popliteal vein that likely originated from the thrombus in the small saphenous vein.
Initial laboratory testing (Table 1) shows elevations of the cardiac enzymes troponin T and N-terminal pro-B-type natriuretic peptide (NT-pro-BNP).
ESTIMATING PROGNOSIS IN PULMONARY EMBOLISM
2. Which of the following laboratory results at presentation is independently associated with a worse outcome in patients with pulmonary embolism?
- Elevated NT-pro-BNP
- Hypercalcemia
- Thrombocytosis
- Hypernatremia
- Elevated procalcitonin
The Pulmonary Embolism Severity Index11 and the Simplified Pulmonary Embolism Severity Index12 (Table 2) are clinical calculators that help predict 30-day risk of death in patients with pulmonary embolism. Our patient’s Pulmonary Embolism Severity Index score is 60, indicating a very low risk, but her simplified severity index score is 2, indicating a high risk.
A shock index score (the heart rate divided by the systolic blood pressure) greater than 1 is also a sensitive measure of risk.13 (Our patient’s shock index score is 1.03.) Although the simplified version is more accurate,14 the shock index is also helpful when deciding whether patients with suspected pulmonary embolism should receive early fibrinolysis.15
In a large registry of patients with confirmed pulmonary embolism, risk factors for death were age greater than 70, cancer, clinical congestive heart failure, chronic obstructive pulmonary disease, systolic blood pressure lower than 90 mm Hg, respiratory rate less than 20 per minute, and right ventricular hypokinesis.16 Right ventricular dysfunction progressing to right ventricular failure and cardiogenic shock is the most common cause of death in patients with pulmonary embolism.16–18
Post hoc analysis has also shown that elevations of the biomarkers BNP, NT-pro-BNP, and cardiac troponins I and T are associated with a prolonged hospital course and a higher risk of death within 30 days.19 Interestingly, a recent retrospective analysis found hyponatremia to be an independent risk factor for death in the short term.20
Thrombocytopenia, not thrombocytosis, is associated with worse outcomes in patients with pulmonary embolism.16 Procalcitonin is elevated in bacterial pneumonia but is normal in pulmonary embolism and so may be helpful in differentiating between the two.21,22 Hypernatremia, hypercalcemia, and elevated procalcitonin have not been shown to be independently associated with worse outcomes in acute pulmonary embolism.
Thus, of the answer choices shown above, elevated NT-pro-BNP is the correct answer.
Classified as massive, submassive, or low-risk
Pulmonary embolism is often stratified as massive, submassive, or low-risk, reflecting the severity and the degree of cardiovascular collapse. The treatment depends on the classification.
Pulmonary embolism is classified as massive if the patient has a cardiac arrest or a systolic blood pressure lower than 90 mm Hg for more than 15 minutes.23 Nearly half of patients in this category die.24
Pulmonary embolism is submassive if the patient has systolic pressure greater than 90 mm Hg but has right ventricular dysfunction, as evidenced by physical examination, elevated cardiac biomarkers, electrocardiography, transthoracic echocardiography, or computed tomography. The death rate is as high as 15%.24
Pulmonary embolism in a normotensive patient with no right ventricular dysfunction is defined as low-risk.
Our patient so far
Our patient has bilateral pulmonary emboli, most likely originating from a deep vein thrombosis in her right lower leg. Her pulmonary embolism would be classified as submassive, as her systolic pressure is greater than 90 mm Hg and right ventricular dysfunction—significant right ventricular strain—was noted on both transthoracic echocardiography and computed tomography. Also, cardiac biomarkers are elevated, and the physical examination revealed a prominent P2 sound and right parasternal heave, also suggestive of right ventricular dysfunction.
Now, 6 hours have passed, and even though she has been receiving intravenous heparin during this time, her shock index remains greater than 1, indicating hemodynamic instability. Her pulse rate is still markedly high—over 160 bpm—and she still appears quite anxious and uncomfortable.
HOW SHOULD THIS PATIENT BE TREATED?
3. Which of the following is the most appropriate treatment for this patient?
- Start warfarin immediately while bridging with unfractionated heparin, low-molecular-weight heparin, or fondaparinux
- Start fibrinolysis with alteplase
- Give metoprolol intravenously to control her heart rate
- Start dabigatran immediately while bridging with unfractionated heparin
- Place an inferior vena cava filter
- Consult cardiothoracic surgery for emergency pulmonary embolectomy
All patients with confirmed pulmonary embolism and no contraindications to anticoagulation should begin treatment with low-molecular-weight heparin, unfractionated heparin, or fondaparinux.23 In addition, this therapy should be started empirically while the patient is still undergoing diagnostic testing if the pretest probability of pulmonary embolism is intermediate or high.23
Warfarin is indicated for all patients with pulmonary embolism who do not have contraindications to it (Table 3). If unfractionated heparin, low-molecular-weight heparin, or fondaparinux has not already been started, it should be started at the same time as warfarin and should be continued until the international normalized ratio (INR) is within the therapeutic range.
Fibrinolysis. Treatment with a fibrinolytic agent in addition to heparin results in faster improvement of right ventricular function and pulmonary perfusion than with heparin alone.25 It may also decrease the incidence of pulmonary hypertension secondary to chronic thromboembolic disease.26 It should be considered in patients with massive pulmonary embolism.23
Whether fibrinolysis is appropriate for all patients with submassive pulmonary embolism remains controversial. Currently, it is not recommended for minor right ventricular dysfunction or myocardial necrosis if the patient has no signs of overt clinical decline.23 The Pulmonary Embolism Thrombolysis trial27 is an ongoing prospective randomized comparison of tenecteplase in a single bolus plus heparin vs heparin alone in normotensive patients with submassive pulmonary embolism, such as our patient. This trial may elucidate the benefit of fibrinolytic therapy in patients with submassive pulmonary embolism.
Patients at low risk are generally treated with heparin and warfarin anticoagulation alone. Fibrinolysis is not recommended in these patients, as the risk of bleeding outweighs the potential benefits.23
Metoprolol may not be advisable for our patient, as her tachycardia is likely compensatory, and beta-blocker therapy could blunt this compensatory response, leading to inadequate systemic perfusion.
Dabigatran is an oral direct thrombin inhibitor that does not require laboratory monitoring. It is currently approved for the prevention of stroke in patients with atrial fibrillation. It has been shown to be as effective as warfarin in the treatment of acute venous thromboembolism28 and may be a viable option in the future, but as of this writing it has not yet been approved in the United States for this indication. Furthermore, dabigatran inhibits thrombin immediately, so continued heparin bridging would not be necessary.
An inferior vena cava filter may prevent recurrent pulmonary embolism for patients who have absolute contraindications to anticoagulation, most significantly in the short term, ie, in the first few weeks after placement. However, these devices have not yet been shown to improve long-term mortality rates.
Embolectomy, percutaneous or surgical, is also an option. For patients in whom thrombolytic therapy is not effective, “rescue” surgical embolectomy has been associated with better outcomes compared with secondary thrombolysis and so should be considered.29
Back to our patient
An intravenous infusion of alteplase is started, and the patient’s tachycardia improves. Her oxygen requirements normalize, and she is transferred to the general medical floor the next day. She receives subcutaneous dalteparin as a bridge therapy, and warfarin is titrated to a goal INR of 2.0 to 3.0. Because of the acute deep vein thrombosis in her right lower leg, she is instructed to wear knee-high fitted compression hose for primary prevention of postphlebitic syndrome.
HOW LONG TO TREAT? IS GENETIC TESTING INDICATED?
Patients with a first episode of unprovoked venous thromboembolism should receive oral anticoagulants for 6 months, while those with recurrent unprovoked venous thromboembolism require lifelong oral anticoagulation.23
Whether to test for inherited thrombophilia after a first episode of venous thromboembolism to guide the duration of anticoagulation is controversial.30 Indiscriminate testing has not been recommended in these patients,31 but the American College of Medical Genetics recommends genetic screening for factor V Leiden in patients who have an unprovoked incident of venous thromboembolism before age 50.32
No randomized controlled trial has assessed whether thrombophilia testing decreases the recurrence rate of venous thromboembolism.33 One uncontrolled study suggested that testing for inherited thrombophilias in patients with a first episode does not affect the risk of recurrence.34 Testing is costly and may cause psychological distress for patients and family members.
Our patient is discharged home on warfarin for 6 months with subsequent follow-up evaluation in the thrombophilia clinic.
WHEN SHOULD WARFARIN BE RESTARTED?
4. If our patient were to discontinue oral anticoagulation in 6 months, which of the following, if present 1 month afterwards, would be a reason to restart oral anticoagulation?
- Elevated serum cotinine
- Positive pregnancy test
- Elevated follicle-stimulating hormone and luteinizing hormone and low estradiol levels
- Elevated D-dimer
Cotinine is a nicotine metabolite, and serum levels are elevated in smokers. Smoking and pregnancy both increase the risk of venous thromboembolism. However, smoking or pregnancy alone would not be a reason to increase the duration of anticoagulation.
Warfarin is contraindicated in pregnancy because of its teratogenic effects, particularly in the first trimester. Follicle-stimulating hormone and luteinizing hormone levels increase in response to decreased estradiol at menopause. Postmenopausal women are not at increased risk of venous thromboembolism unless they are taking oral estrogen hormone replacement therapy.
An elevated D-dimer level 1 month after stopping of oral anticoagulation has been associated with a higher rate of recurrence of venous thromboembolism, which is reduced by a resumption of anticoagulation.35 Therefore, consideration should be given to resuming anticoagulation in patients who have an elevated D-dimer level.
TAKE-HOME POINTS
It is important to distinguish between massive, submassive, and low-risk pulmonary embolism, since each type has a different treatment and prognosis.
Fibrinolytic therapy is indicated in patients with massive pulmonary embolism when no contraindication is present, whereas it is not indicated in those with low-risk pulmonary embolism.
Management of submassive pulmonary embolism continues to be an area of considerable debate. Current American Heart Association guidelines recommend consideration of fibrinolysis initially in patients with submassive acute pulmonary embolism if there is concurrent clinical evidence of adverse prognosis—eg, new hemodynamic instability, worsening respiratory insufficiency, severe right ventricular dysfunction, or major myocardial necrosis.23 On the other hand, the American College of Chest Physicians recommends against initial systemic fibrinolysis in submassive acute pulmonary embolism based on biomarkers or findings on ECG, transthoracic echocardiography, or CT, recommending it only in therapeutically anticoagulated patients deemed to be at high risk of hypotension.36
Since the optimal treatment of submassive pulmonary embolism is still not known, it is important that clinicians remain up to date on the evidence and guidelines.
- Stein PD, Beemath A, Matta F, et al. Clinical characteristics of patients with acute pulmonary embolism: data from PIOPED II. Am J Med 2007; 120:871–879.
- Stein PD, Saltzman HA, Weg JG. Clinical characteristics of patients with acute pulmonary embolism. Am J Cardiol 1991; 68:1723–1724.
- Ferrari E, Imbert A, Chevalier T, Mihoubi A, Morand P, Baudouy M. The ECG in pulmonary embolism. Predictive value of negative T waves in precordial leads—80 case reports. Chest 1997; 111:537–543.
- Geibel A, Zehender M, Kasper W, Olschewski M, Klima C, Konstantinides SV. Prognostic value of the ECG on admission in patients with acute major pulmonary embolism. Eur Respir J 2005; 25:843–848.
- Perrier A, Roy PM, Sanchez O, et al. Multidetector-row computed tomography in suspected pulmonary embolism. N Engl J Med 2005; 352:1760–1768.
- Qanadli SD, Hajjam ME, Mesurolle B, et al. Pulmonary embolism detection: prospective evaluation of dual-section helical CT versus selective pulmonary arteriography in 157 patients. Radiology 2000; 217:447–455.
- Miniati M, Monti S, Pratali L, et al. Value of transthoracic echocardiography in the diagnosis of pulmonary embolism: results of a prospective study in unselected patients. Am J Med 2001; 110:528–535.
- Bova C, Greco F, Misuraca G, et al. Diagnostic utility of echocardiography in patients with suspected pulmonary embolism. Am J Emerg Med 2003; 21:180–183.
- Stein PD, Chenevert TL, Fowler SE, et al; PIOPED III (Prospective Investigation of Pulmonary Embolism Diagnosis III) Investigators. Gadolinium-enhanced magnetic resonance angiography for pulmonary embolism: a multicenter prospective study (PIOPED III). Ann Intern Med 2010; 152:434–443,W142–W143.
- Sostman HD, Stein PD, Gottschalk A, Matta F, Hull R, Goodman L. Acute pulmonary embolism: sensitivity and specificity of ventilation-perfusion scintigraphy in PIOPED II study. Radiology 2008; 246:941–946.
- Aujesky D, Obrosky DS, Stone RA, et al. Derivation and validation of a prognostic model for pulmonary embolism. Am J Respir Crit Care Med 2005; 172:1041–1046.
- Jiménez D, Aujesky D, Moores L, et al; RIETE Investigators. Simplification of the pulmonary embolism severity index for prognostication in patients with acute symptomatic pulmonary embolism. Arch Intern Med 2010; 170:1383–1389.
- Otero R, Trujillo-Santos J, Cayuela A, et al; Registro Informatizado de la Enfermedad Tromboembólica (RIETE) Investigators. Haemodynamically unstable pulmonary embolism in the RIETE Registry: systolic blood pressure or shock index? Eur Respir J 2007; 30:1111–1116.
- Sam A, Sánchez D, Gómez V, et al. The shock index and the simplified PESI for identification of low-risk patients with acute pulmonary embolism. Eur Respir J 2011; 37:762–766.
- Kucher N, Luder CM, Dörnhöfer T, Windecker S, Meier B, Hess OM. Novel management strategy for patients with suspected pulmonary embolism. Eur Heart J 2003; 24:366–376.
- Goldhaber SZ, Visani L, De Rosa M. Acute pulmonary embolism: clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER). Lancet 1999; 353:1386–1389.
- ten Wolde M, Söhne M, Quak E, Mac Gillavry MR, Büller HR. Prognostic value of echocardiographically assessed right ventricular dysfunction in patients with pulmonary embolism. Arch Intern Med 2004; 164:1685–1689.
- Sanchez O, Trinquart L, Colombet I, et al. Prognostic value of right ventricular dysfunction in patients with haemodynamically stable pulmonary embolism: a systematic review. Eur Heart J 2008; 29:1569–1577.
- Kucher N, Goldhaber SZ. Cardiac biomarkers for risk stratification of patients with acute pulmonary embolism. Circulation 2003; 108:2191–2194.
- Scherz N, Labarère J, Méan M, Ibrahim SA, Fine MJ, Aujesky D. Prognostic importance of hyponatremia in patients with acute pulmonary embolism. Am J Respir Crit Care Med 2010; 182:1178–1183.
- Delèvaux I, André M, Aumaître O, Bègue RJ, Colombier M, Piette JC. Procalcitonin measurement for differential diagnosis between pulmonary embolism and pneumonia. Crit Care Med 2003; 31:661.
- Köktürk N, Kanbay A, Bukan N, Ekim N. The value of serum procalcitonin in differential diagnosis of pulmonary embolism and community-acquired pneumonia. Clin Appl Thromb Hemost 2011; 17:519–525.
- Jaff MR, McMurtry MS, Archer SL, et al; American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation. Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation 2011; 123:1788–1830.
- Kucher N, Rossi E, De Rosa M, Goldhaber SZ. Massive pulmonary embolism. Circulation 2006; 113:577–582.
- Goldhaber SZ, Haire WD, Feldstein ML, et al. Alteplase versus heparin in acute pulmonary embolism: randomised trial assessing right-ventricular function and pulmonary perfusion. Lancet 1993; 341:507–511.
- Kline JA, Steuerwald MT, Marchick MR, Hernandez-Nino J, Rose GA. Prospective evaluation of right ventricular function and functional status 6 months after acute submassive pulmonary embolism: frequency of persistent or subsequent elevation in estimated pulmonary artery pressure. Chest 2009; 136:1202–1210.
- Steering Committee of PEITHO Investigators. Single-bolus tenecteplase plus heparin compared with heparin alone for normotensive patients with acute pulmonary embolism who have evidence of right ventricular dysfunction and myocardial injury: rationale and design of the Pulmonary Embolism Thrombolysis (PEITHO) trial. Am Heart J 2012; 163:33–38.e1.
- Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- Meneveau N, Séronde MF, Blonde MC, et al. Management of unsuccessful thrombolysis in acute massive pulmonary embolism. Chest 2006; 129:1043–1050.
- Ho WK, Hankey GJ, Eikelboom JW. Should adult patients be routinely tested for heritable thrombophilia after an episode of venous thromboembolism? Med J Aust 2011; 195:139–142.
- Baglin T, Gray E, Greaves M, et al; British Committee for Standards in Haematology. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010; 149:209–220.
- Grody WW, Griffin JH, Taylor AK, Korf BR, Heit JA; ACMG Factor V Leiden Working Group. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med 2001; 3:139–148.
- Cohn D, Vansenne F, de Borgie C, Middeldorp S. Thrombophilia testing for prevention of recurrent venous thromboembolism. Cochrane Database Syst Rev 2009; ( 1):CD007069.
- Coppens M, Reijnders JH, Middeldorp S, Doggen CJ, Rosendaal FR. Testing for inherited thrombophilia does not reduce the recurrence of venous thrombosis. J Thromb Haemost 2008; 6:1474–1477.
- Palareti G, Cosmi B, Legnani C, et al; PROLONG Investigators. Ddimer testing to determine the duration of anticoagulation therapy. N Engl J Med 2006; 355:1780–1789.
- Guyatt GH, Akl EA, Crowther M, Gutterman DD, Schuünemann HJ; American College of Chest Physicians Antithrombotic Therapy and Prevention of Thrombosis Panel. Executive summary: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl):7S–47S.
- Stein PD, Beemath A, Matta F, et al. Clinical characteristics of patients with acute pulmonary embolism: data from PIOPED II. Am J Med 2007; 120:871–879.
- Stein PD, Saltzman HA, Weg JG. Clinical characteristics of patients with acute pulmonary embolism. Am J Cardiol 1991; 68:1723–1724.
- Ferrari E, Imbert A, Chevalier T, Mihoubi A, Morand P, Baudouy M. The ECG in pulmonary embolism. Predictive value of negative T waves in precordial leads—80 case reports. Chest 1997; 111:537–543.
- Geibel A, Zehender M, Kasper W, Olschewski M, Klima C, Konstantinides SV. Prognostic value of the ECG on admission in patients with acute major pulmonary embolism. Eur Respir J 2005; 25:843–848.
- Perrier A, Roy PM, Sanchez O, et al. Multidetector-row computed tomography in suspected pulmonary embolism. N Engl J Med 2005; 352:1760–1768.
- Qanadli SD, Hajjam ME, Mesurolle B, et al. Pulmonary embolism detection: prospective evaluation of dual-section helical CT versus selective pulmonary arteriography in 157 patients. Radiology 2000; 217:447–455.
- Miniati M, Monti S, Pratali L, et al. Value of transthoracic echocardiography in the diagnosis of pulmonary embolism: results of a prospective study in unselected patients. Am J Med 2001; 110:528–535.
- Bova C, Greco F, Misuraca G, et al. Diagnostic utility of echocardiography in patients with suspected pulmonary embolism. Am J Emerg Med 2003; 21:180–183.
- Stein PD, Chenevert TL, Fowler SE, et al; PIOPED III (Prospective Investigation of Pulmonary Embolism Diagnosis III) Investigators. Gadolinium-enhanced magnetic resonance angiography for pulmonary embolism: a multicenter prospective study (PIOPED III). Ann Intern Med 2010; 152:434–443,W142–W143.
- Sostman HD, Stein PD, Gottschalk A, Matta F, Hull R, Goodman L. Acute pulmonary embolism: sensitivity and specificity of ventilation-perfusion scintigraphy in PIOPED II study. Radiology 2008; 246:941–946.
- Aujesky D, Obrosky DS, Stone RA, et al. Derivation and validation of a prognostic model for pulmonary embolism. Am J Respir Crit Care Med 2005; 172:1041–1046.
- Jiménez D, Aujesky D, Moores L, et al; RIETE Investigators. Simplification of the pulmonary embolism severity index for prognostication in patients with acute symptomatic pulmonary embolism. Arch Intern Med 2010; 170:1383–1389.
- Otero R, Trujillo-Santos J, Cayuela A, et al; Registro Informatizado de la Enfermedad Tromboembólica (RIETE) Investigators. Haemodynamically unstable pulmonary embolism in the RIETE Registry: systolic blood pressure or shock index? Eur Respir J 2007; 30:1111–1116.
- Sam A, Sánchez D, Gómez V, et al. The shock index and the simplified PESI for identification of low-risk patients with acute pulmonary embolism. Eur Respir J 2011; 37:762–766.
- Kucher N, Luder CM, Dörnhöfer T, Windecker S, Meier B, Hess OM. Novel management strategy for patients with suspected pulmonary embolism. Eur Heart J 2003; 24:366–376.
- Goldhaber SZ, Visani L, De Rosa M. Acute pulmonary embolism: clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER). Lancet 1999; 353:1386–1389.
- ten Wolde M, Söhne M, Quak E, Mac Gillavry MR, Büller HR. Prognostic value of echocardiographically assessed right ventricular dysfunction in patients with pulmonary embolism. Arch Intern Med 2004; 164:1685–1689.
- Sanchez O, Trinquart L, Colombet I, et al. Prognostic value of right ventricular dysfunction in patients with haemodynamically stable pulmonary embolism: a systematic review. Eur Heart J 2008; 29:1569–1577.
- Kucher N, Goldhaber SZ. Cardiac biomarkers for risk stratification of patients with acute pulmonary embolism. Circulation 2003; 108:2191–2194.
- Scherz N, Labarère J, Méan M, Ibrahim SA, Fine MJ, Aujesky D. Prognostic importance of hyponatremia in patients with acute pulmonary embolism. Am J Respir Crit Care Med 2010; 182:1178–1183.
- Delèvaux I, André M, Aumaître O, Bègue RJ, Colombier M, Piette JC. Procalcitonin measurement for differential diagnosis between pulmonary embolism and pneumonia. Crit Care Med 2003; 31:661.
- Köktürk N, Kanbay A, Bukan N, Ekim N. The value of serum procalcitonin in differential diagnosis of pulmonary embolism and community-acquired pneumonia. Clin Appl Thromb Hemost 2011; 17:519–525.
- Jaff MR, McMurtry MS, Archer SL, et al; American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation. Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation 2011; 123:1788–1830.
- Kucher N, Rossi E, De Rosa M, Goldhaber SZ. Massive pulmonary embolism. Circulation 2006; 113:577–582.
- Goldhaber SZ, Haire WD, Feldstein ML, et al. Alteplase versus heparin in acute pulmonary embolism: randomised trial assessing right-ventricular function and pulmonary perfusion. Lancet 1993; 341:507–511.
- Kline JA, Steuerwald MT, Marchick MR, Hernandez-Nino J, Rose GA. Prospective evaluation of right ventricular function and functional status 6 months after acute submassive pulmonary embolism: frequency of persistent or subsequent elevation in estimated pulmonary artery pressure. Chest 2009; 136:1202–1210.
- Steering Committee of PEITHO Investigators. Single-bolus tenecteplase plus heparin compared with heparin alone for normotensive patients with acute pulmonary embolism who have evidence of right ventricular dysfunction and myocardial injury: rationale and design of the Pulmonary Embolism Thrombolysis (PEITHO) trial. Am Heart J 2012; 163:33–38.e1.
- Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- Meneveau N, Séronde MF, Blonde MC, et al. Management of unsuccessful thrombolysis in acute massive pulmonary embolism. Chest 2006; 129:1043–1050.
- Ho WK, Hankey GJ, Eikelboom JW. Should adult patients be routinely tested for heritable thrombophilia after an episode of venous thromboembolism? Med J Aust 2011; 195:139–142.
- Baglin T, Gray E, Greaves M, et al; British Committee for Standards in Haematology. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010; 149:209–220.
- Grody WW, Griffin JH, Taylor AK, Korf BR, Heit JA; ACMG Factor V Leiden Working Group. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med 2001; 3:139–148.
- Cohn D, Vansenne F, de Borgie C, Middeldorp S. Thrombophilia testing for prevention of recurrent venous thromboembolism. Cochrane Database Syst Rev 2009; ( 1):CD007069.
- Coppens M, Reijnders JH, Middeldorp S, Doggen CJ, Rosendaal FR. Testing for inherited thrombophilia does not reduce the recurrence of venous thrombosis. J Thromb Haemost 2008; 6:1474–1477.
- Palareti G, Cosmi B, Legnani C, et al; PROLONG Investigators. Ddimer testing to determine the duration of anticoagulation therapy. N Engl J Med 2006; 355:1780–1789.
- Guyatt GH, Akl EA, Crowther M, Gutterman DD, Schuünemann HJ; American College of Chest Physicians Antithrombotic Therapy and Prevention of Thrombosis Panel. Executive summary: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl):7S–47S.
Recurrent abdominal pain and vomiting
A 32-year-old man presents to the emergency department with excruciating abdominal pain associated with multiple episodes of vomiting for the past 2 days. He reports no fevers, headaches, diarrhea, constipation, hematochezia, melena, musculoskeletal symptoms, or weight loss. His abdominal pain is generalized and crampy. It does not radiate and has no precipitating factors. The pain is relieved only with intravenous narcotics.
He does not smoke, drink alcohol, or use illicit drugs. He has no known drug or food allergies. He says that his current condition causes him emotional stress that affects his performance at work.
About a year ago, after a complicated surgical procedure, he needed chronic high-dose narcotics. A few months later, he developed multiple bouts of abdominal pain and vomiting that required hospital visits. He now takes oral oxycodone 10–15 mg every 4–6 hours.
On admission, his vital signs are stable, but he is in excruciating pain. He is alert and oriented to person, place, and time. His sclera are anicteric, and the pupils are equal, round, and reactive to light. Lung and heart examinations are normal. The abdomen is soft and nondistended but tender in all four quadrants without guarding; the liver and spleen are not palpable, and no abdominal masses are detected. He has no skin rash, joint swelling or tenderness, or peripheral edema. The neurologic examination is normal. Computed tomography (CT) of the abdomen with contrast shows no signs of bowel obstruction, pancreatic calcifications or edema, cholecystitis, or hepatobiliary disease. Results of initial laboratory testing are shown in Table 1.
1. Based on the information available, which is the least likely cause of his symptoms?
- Acute pancreatitis
- Cyclic vomiting syndrome
- Acute intermittent porphyria
- Gastroparesis
Acute pancreatitis
Acute pancreatitis is the least likely cause of his symptoms. It is commonly caused by gallstones, alcohol, hypertriglyceridemia, and certain drugs.1 The associated abdominal pain is usually epigastric, radiates to the back, and is accompanied by nausea or vomiting, or both. The onset of pain is sudden and rapidly increases in severity within 30 minutes. CT shows enlargement of the pancreas with diffuse edema, heterogeneity of pancreatic parenchyma, peripancreatic stranding, and peripancreatic fluid collections.1 The diagnosis is based on two of the following three criteria: abdominal pain characteristic of acute pancreatitis; a serum amylase or lipase concentration three or more times the upper limit of normal; and characteristic findings of acute pancreatitis on CT.1
Cyclic vomiting syndrome
Cyclic vomiting syndrome is thought to be caused by episodic dysautonomia, mitochondrial DNA mutations, and hypothalamic emetic response oversensitivity,2–4 but the exact pathogenesis is unknown. The syndrome has been strongly linked to migraine and to the chronic excessive use of cannabinoids.5–9 The Rome III diagnostic criteria10 are the following: the vomiting episodes are stereotypical, ie, they are acute and last for less than 1 week; the patient has had three or more episodes in the previous year; and the patient has no nausea or vomiting between episodes. The patient must meet all three criteria. A history of migraine or a family history of migraine further supports the diagnosis.
Acute intermittent porphyria
Acute intermittent porphyria is characterized by neurovisceral symptoms such as convulsions, paresis, autonomic dysfunction, constipation, and diarrhea that result from the overproduction of porphyrin precursors and deficiency of porphobilinogen deaminase.11
Most patients have poorly localized, severe, steady abdominal pain that develops over hours to days and that may persist for days to weeks.11 Since the pain is neuropathic, abdominal tenderness is usually minimal during an acute attack. Other clues include signs of ileus, such as constipation, nausea, abdominal distention, or decreased bowel sounds; bladder dysfunction, eg, urinary retention, incontinence, or dysuria; reddish-brown urine; and sensory neuropathy of the chest, back, and extremities.11 Blistering skin lesions are usually not seen. The presence of porphobilinogen in the urine confirms the diagnosis.11
Gastroparesis
Gastroparesis is a result of discoordination between the sympathetic and parasympathetic nervous systems, neurons, and smooth muscles within the stomach, causing a decrease in gastric motility. Common causes are diabetes,12 scleroderma,13 and neurologic disorders.14 It can also be iatrogenic,15 resulting from visceral nerve injury and drug treatment with narcotics, calcium channel blockers, muscarinic cholinergic antagonists, or certain antidepressants. Symptoms are related to gastric stasis, ie, abdominal pain from gastric distention, bloating, vomiting, and early satiety.15 Abdominal pain may worsen after eating, and vomitus usually consists of recently ingested food. These patients may have abdominal distension or tenderness and succussion splash. After excluding possible mechanical obstruction, a gastric-emptying study may be necessary to make the diagnosis.15
CASE CONTINUED
A serum and urine drug screen in our patient is positive only for opioids. Urine measures of delta-aminolevulinic acid and porphobilinogen are normal. CT angiography of the abdomen shows no signs of mesenteric vascular occlusion. Esophagogastroduodenoscopy shows antral gastritis, but the esophagus and duodenum appear normal, and colonoscopy is normal as well. Histologic study of biopsy specimens obtained during endoscopy is unrevealing. A gastric-emptying study shows delayed emptying. The patient’s abdominal pain and vomiting persist with the initial dose of intravenous narcotic but resolve with escalating doses. When asked, the patient denies an excessive need for hot baths.
2. Which is the most likely diagnosis at this point?
- Narcotic bowel syndrome
- Opioid withdrawal
- Crohn disease
- Chronic pancreatitis
- Chronic mesenteric ischemia
- Cannabinoid hyperemesis
Narcotic bowel syndrome
Narcotic bowel syndrome is the most likely diagnosis. Grunkemeier et al16 described it as chronic or frequently recurring abdominal pain that is treated with narcotics, either chronically or acutely with high doses, and that includes all the following features16:
- The pain worsens or resolves incompletely with continued or increasing doses of narcotics
- The pain markedly worsens when the narcotic dose is decreased, and decreases when the drug is reinstituted (the “soarand-crash” effect)
- The frequency, duration, and intensity of the pain episodes gradually increase
- The nature of the pain and its intensity are not explained by a current or previous gastrointestinal diagnosis.16
This syndrome is common in patients who receive high doses of narcotics for postoperative pain or for other, nonmalignant causes of pain. Patients eventually become dependent on the drugs but are not aware that chronic use activates and facilitates areas in the brain that enhance the perception of pain.16 A study of a rat model of narcotic bowel syndrome17 showed that morphine-induced hyperalgesia depends on central sensitization involving the activation of spinal microglia. This eventually results in concomitant peripheral sensitization involving the colonic mucosal neuroimmune system, and also in central or peripheral activation of opioid kappa-receptors by dynorphin release.17
Patients tend to present with chronic or intermittent colicky abdominal pain that requires escalating doses of narcotics. Eventually, they develop tachyphylaxis and shortened pain-free periods and will require even higher doses of narcotics. This ultimately enhances the perception of pain and worsens opioid bowel symptoms, causing a vicious circle of pain and more narcotic use.16
Laboratory tests are usually normal, and imaging may show only ileus. Gastric emptying may be delayed in patients who have either narcotic bowel syndrome or gastroparesis, but since abdominal pain from narcotic bowel syndrome is a result of central and visceral hypersensitivity, these patients perceive more severe abdominal pain than patients with gastroparesis alone.
Opioid withdrawal
Symptoms of opioid withdrawal may appear as soon as 6 to 24 hours after cessation of the opioid in patients known to be dependent on opioids. These patients present with crampy abdominal pain with nausea.18 Other symptoms include agitation, rhinorrhea, lacrimation, excessive yawning, arthralgias, papillary dilation, and piloerection.18
Our patient did not have the typical signs of opioid withdrawal.
Crohn disease
Crohn disease is a multisystem disorder with specific clinical and pathologic features. It is characterized by focal, asymmetric, transmural, and occasionally granulomatous inflammation primarily affecting the gastrointestinal tract.19 Characteristic symptoms include abdominal pain, chronic diarrhea with or without rectal bleeding, and weight loss. Extraintestinal signs may include anemia and inflammatory changes in the eyes, skin, and joints. The diagnosis is based on endoscopic, radiographic, and pathologic findings.19
Our patient did not have diarrhea or signs of Crohn disease on CT, endoscopy, or histology.
Chronic pancreatitis
Chronic pancreatitis involves progressive inflammatory changes resulting in permanent structural damage to the pancreas and subsequent exocrine and endocrine dysfunction.20 Patients have epigastric abdominal pain that often radiates to the back20; it is associated with eating and is partly relieved with leaning forward. Symptoms of pancreatic insufficiency such as fat malabsorption (resulting in steatorrhea and fat-soluble vitamin deficiency) and diabetes are common. Calcifications within the pancreas on CT suggest chronic pancreatitis.20 Serum lipase and amylase levels may be normal or slightly elevated.20
Our patient’s abdominal pain was not typical of pancreatitis. He had no signs or symptoms of pancreatic insufficiency and no calcifications within the pancreas.
Chronic mesenteric ischemia
Chronic mesenteric ischemia (“intestinal angina”) is caused by a reduction in intestinal blood flow as a result of occlusion, vasospasm, or hypoperfusion of the mesenteric vasculature.21 It is commonly seen in patients who smoke or who have atherosclerotic vascular disease. These patients have chronic dull or crampy abdominal pain that usually occurs within 1 hour after eating.21 To avoid pain, patients avoid eating, resulting in weight loss.21 CT angiography with multi-detector CT is as effective as angiography (the gold standard) in depicting splanchnic arterial anatomy.22
Our patient is young and has no known risk factors for atherosclerosis such as smoking. His abdominal pain is more intermittent than chronic and is not associated with eating.
Cannabinoid hyperemesis
Cannabinoid hyperemesis should be considered in patients with long-term cannabis use presenting with cyclic vomiting, abdominal pain, compulsive use of hot showers, and improvement of symptoms with cannabis cessation.23 Although cannabinoids have been recognized for their antiemetic effects, long-term use may eventually cause autonomic instability and disturbances in the hypothalamic-pituitary-adrenal axis, resulting in cyclic vomiting and thermoregulatory impairment.23
Although our patient presented with multiple episodes of vomiting and abdominal pain, he denied using marijuana, he tested negative for tetrahydrocannabinol, and he did not associate any relief of his symptoms with hot baths.
CASE CONTINUED
Our patient receives intravenous hydration, antiemetics, and a narcotic in tapering intravenous doses, and his symptoms gradually improve. He is discharged from the hospital. However, a few weeks later he is readmitted with the same symptoms of abdominal pain and nausea.
3. What is the cornerstone of treatment for narcotic bowel syndrome?
- Establishing a therapeutic relationship
- Detoxification
- Supportive management with intravenous fluids, antiemetics, and stool-softeners
- Medical management with a short-acting narcotic, clonidine, lorazepam, and desipramine
MANAGEMENT OF NARCOTIC BOWEL SYNDROME
An effective therapeutic relationship with the patient is the cornerstone of treatment and should be established before starting detoxification.17 The physician must first learn to accept that the patient’s condition is real and must show genuine empathy as well as provide information about the pathophysiologic basis of the condition, the rationale for withholding narcotics, and the detrimental role narcotics play in the vicious circle of pain.
Detoxification involves gradually withdrawing the narcotic and substituting a nonnarcotic such as an antidepressant for pain control, as well as prescribing a drug such as a benzodiazepine or clonidine to prevent withdrawal symptoms and a laxative to prevent constipation.17,24 The physician must reassure the patient that he or she will not be abandoned in pain and that all medications will be continuously adjusted as needed to keep him or her comfortable throughout the detoxification process.17,24 The physician must continuously gauge the patient’s willingness to continue with treatment and must also be readily available to address the patient’s concerns in a timely manner.17,24 Involving family members and friends may provide additional support to the patient. Referral to a functional gastrointestinal motility program, a pain specialist, and a psychologist may also be considered.17,24 Follow-up care is essential, even after the withdrawal program has ended.17,24
BACK TO THE PATIENT
After successfully establishing a therapeutic relationship and discussing the treatment plan with our patient, we started him on the same dosage of narcotic that he had been receiving, calculated in intravenous morphine equivalents to achieve maximal comfort, and then decreased the dosage by 10% to 33% daily until he was completely off narcotics. An antidepressant and a benzodiazepine were given simultaneously with narcotic tapering. Oral clonidine (0.1–0.4 mg/day) was given after the narcotic dosage was reduced to about half, and polyethylene glycol was given as needed for constipation. The total duration of detoxification was 7 days.
The patient was referred to a psychologist for cognitive-behavioral and relaxation therapy, as well as for encouragement and support. At 6 months, he had had no recurrence of symptoms.
TAKE-HOME MESSAGE
In the United States, the number of patients taking a narcotic for nonmalignant pain is increasing, 25 and physicians should be more aware of complications such as narcotic bowel syndrome.
Narcotic bowel syndrome should be suspected in any patient with prolonged narcotic use presenting with multiple recurrent episodes of abdominal pain after other causes are ruled out.
Establishing a good therapeutic relationship with the patient is the cornerstone of successful treatment. Patients who understand their condition and are willing to be treated tend to have better outcomes.
Supportive treatment, symptom relief, and emotional support during detoxification increase compliance.
- Banks PA, Freeman ML; Practice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol 2006; 101:2379–2400.
- Boles RG, Adams K, Ito M, Li BU. Maternal inheritance in cyclic vomiting syndrome with neuromuscular disease. Am J Med Genet A 2003; 120A:474–482.
- Wang Q, Ito M, Adams K, et al. Mitochondrial DNA control region sequence variation in migraine headache and cyclic vomiting syndrome. Am J Med Genet A 2004; 131:50–58.
- Taché Y. Cyclic vomiting syndrome: the corticotropinreleasing-factor hypothesis. Dig Dis Sci 1999; 44(suppl 8):79S–86S.
- Withers GD, Silburn SR, Forbes DA. Precipitants and aetiology of cyclic vomiting syndrome. Acta Paediatr 1998; 87:272–277.
- Whitney HB. Cyclic vomiting. A brief review of this affection as illustrated by a typical case. Arch Pediatr 1898; 15:839–845.
- Stickler GB. Relationship between cyclic vomiting syndrome and migraine. Clin Pediatr (Phila) 2005; 44:505–508.
- Li BU, Murray RD, Heitlinger LA, Robbins JL, Hayes JR. Is cyclic vomiting syndrome related to migraine? J Pediatr 1999; 134:567–572.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Rome Foundation. Rome III disorders and diagnostic criteria. http://www.romecriteria.org/criteria/. Accessed February 27, 2013.
- Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med 2005; 142:439–450.
- Camilleri M. Clinical practice. Diabetic gastroparesis. N Engl J Med 2007; 356:820–829.
- Maddern GJ, Horowitz M, Jamieson GG, Chatterton BE, Collins PJ, Roberts-Thomson P. Abnormalities of esophageal and gastric emptying in progressive systemic sclerosis. Gastroenterology 1984; 87:922–926.
- Jost WH. Gastrointestinal dysfunction in Parkinson’s disease. J Neurol Sci 2010; 289:69–73.
- Parkman HP, Hasler WL, Fisher RS; American Gastroenterological Association. American Gastroenterological Association technical review on the diagnosis and treatment of gastroparesis. Gastroenterology 2004; 127:1592–1622.
- Grunkemeier DM, Cassara JE, Dalton CB, Drossman DA. The narcotic bowel syndrome: clinical features, pathophysiology, and management. Clin Gastroenterol Hepatol 2007; 5:1126–1139.
- Agostini S, Eutamene H, Cartier C, et al. Evidence of central and peripheral sensitization in a rat model of narcotic bowel-like syndrome. Gastroenterology 2010; 139:553–563,563.e1–e5.
- Nicholls L, Bragaw L, Ruetsch C. Opioid dependence treatment and guidelines. J Manag Care Pharm 2010; 16(1 suppl B):S14–S21.
- Lichtenstein GR, Hanauer SB, Sandborn WJ; Practice Parameters Committee of American College of Gastroenterology. Management of Crohn’s disease in adults. Am J Gastroenterol 2009; 104:465–483.
- Steer ML, Waxman I, Freedman S. Chronic pancreatitis. N Engl J Med 1995; 332:1482–1490.
- American Gastroenterological Association Medical Position Statement: guidelines on intestinal ischemia. Gastroenterology 2000; 118:951–953.
- Savastano S, Teso S, Corrà S, Fantozzi O, Miotto D. Multislice CT angiography of the celiac and superior mesenteric arteries: comparison with arteriographic findings. Radiol Med 2002; 103:456–463.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Drossman DA, Morris CB, Edwards H, et al. Diagnosis, characterization, and 3-month outcome after detoxification of 39 patients with narcotic bowel syndrome. Am J Gastroenterol 2012; 107:1426–1440.
- Trescot AM, Boswell MV, Atluri SL, et al. Opioid guidelines in the management of chronic non-cancer pain. Pain Physician 2006; 9:1–39.
A 32-year-old man presents to the emergency department with excruciating abdominal pain associated with multiple episodes of vomiting for the past 2 days. He reports no fevers, headaches, diarrhea, constipation, hematochezia, melena, musculoskeletal symptoms, or weight loss. His abdominal pain is generalized and crampy. It does not radiate and has no precipitating factors. The pain is relieved only with intravenous narcotics.
He does not smoke, drink alcohol, or use illicit drugs. He has no known drug or food allergies. He says that his current condition causes him emotional stress that affects his performance at work.
About a year ago, after a complicated surgical procedure, he needed chronic high-dose narcotics. A few months later, he developed multiple bouts of abdominal pain and vomiting that required hospital visits. He now takes oral oxycodone 10–15 mg every 4–6 hours.
On admission, his vital signs are stable, but he is in excruciating pain. He is alert and oriented to person, place, and time. His sclera are anicteric, and the pupils are equal, round, and reactive to light. Lung and heart examinations are normal. The abdomen is soft and nondistended but tender in all four quadrants without guarding; the liver and spleen are not palpable, and no abdominal masses are detected. He has no skin rash, joint swelling or tenderness, or peripheral edema. The neurologic examination is normal. Computed tomography (CT) of the abdomen with contrast shows no signs of bowel obstruction, pancreatic calcifications or edema, cholecystitis, or hepatobiliary disease. Results of initial laboratory testing are shown in Table 1.
1. Based on the information available, which is the least likely cause of his symptoms?
- Acute pancreatitis
- Cyclic vomiting syndrome
- Acute intermittent porphyria
- Gastroparesis
Acute pancreatitis
Acute pancreatitis is the least likely cause of his symptoms. It is commonly caused by gallstones, alcohol, hypertriglyceridemia, and certain drugs.1 The associated abdominal pain is usually epigastric, radiates to the back, and is accompanied by nausea or vomiting, or both. The onset of pain is sudden and rapidly increases in severity within 30 minutes. CT shows enlargement of the pancreas with diffuse edema, heterogeneity of pancreatic parenchyma, peripancreatic stranding, and peripancreatic fluid collections.1 The diagnosis is based on two of the following three criteria: abdominal pain characteristic of acute pancreatitis; a serum amylase or lipase concentration three or more times the upper limit of normal; and characteristic findings of acute pancreatitis on CT.1
Cyclic vomiting syndrome
Cyclic vomiting syndrome is thought to be caused by episodic dysautonomia, mitochondrial DNA mutations, and hypothalamic emetic response oversensitivity,2–4 but the exact pathogenesis is unknown. The syndrome has been strongly linked to migraine and to the chronic excessive use of cannabinoids.5–9 The Rome III diagnostic criteria10 are the following: the vomiting episodes are stereotypical, ie, they are acute and last for less than 1 week; the patient has had three or more episodes in the previous year; and the patient has no nausea or vomiting between episodes. The patient must meet all three criteria. A history of migraine or a family history of migraine further supports the diagnosis.
Acute intermittent porphyria
Acute intermittent porphyria is characterized by neurovisceral symptoms such as convulsions, paresis, autonomic dysfunction, constipation, and diarrhea that result from the overproduction of porphyrin precursors and deficiency of porphobilinogen deaminase.11
Most patients have poorly localized, severe, steady abdominal pain that develops over hours to days and that may persist for days to weeks.11 Since the pain is neuropathic, abdominal tenderness is usually minimal during an acute attack. Other clues include signs of ileus, such as constipation, nausea, abdominal distention, or decreased bowel sounds; bladder dysfunction, eg, urinary retention, incontinence, or dysuria; reddish-brown urine; and sensory neuropathy of the chest, back, and extremities.11 Blistering skin lesions are usually not seen. The presence of porphobilinogen in the urine confirms the diagnosis.11
Gastroparesis
Gastroparesis is a result of discoordination between the sympathetic and parasympathetic nervous systems, neurons, and smooth muscles within the stomach, causing a decrease in gastric motility. Common causes are diabetes,12 scleroderma,13 and neurologic disorders.14 It can also be iatrogenic,15 resulting from visceral nerve injury and drug treatment with narcotics, calcium channel blockers, muscarinic cholinergic antagonists, or certain antidepressants. Symptoms are related to gastric stasis, ie, abdominal pain from gastric distention, bloating, vomiting, and early satiety.15 Abdominal pain may worsen after eating, and vomitus usually consists of recently ingested food. These patients may have abdominal distension or tenderness and succussion splash. After excluding possible mechanical obstruction, a gastric-emptying study may be necessary to make the diagnosis.15
CASE CONTINUED
A serum and urine drug screen in our patient is positive only for opioids. Urine measures of delta-aminolevulinic acid and porphobilinogen are normal. CT angiography of the abdomen shows no signs of mesenteric vascular occlusion. Esophagogastroduodenoscopy shows antral gastritis, but the esophagus and duodenum appear normal, and colonoscopy is normal as well. Histologic study of biopsy specimens obtained during endoscopy is unrevealing. A gastric-emptying study shows delayed emptying. The patient’s abdominal pain and vomiting persist with the initial dose of intravenous narcotic but resolve with escalating doses. When asked, the patient denies an excessive need for hot baths.
2. Which is the most likely diagnosis at this point?
- Narcotic bowel syndrome
- Opioid withdrawal
- Crohn disease
- Chronic pancreatitis
- Chronic mesenteric ischemia
- Cannabinoid hyperemesis
Narcotic bowel syndrome
Narcotic bowel syndrome is the most likely diagnosis. Grunkemeier et al16 described it as chronic or frequently recurring abdominal pain that is treated with narcotics, either chronically or acutely with high doses, and that includes all the following features16:
- The pain worsens or resolves incompletely with continued or increasing doses of narcotics
- The pain markedly worsens when the narcotic dose is decreased, and decreases when the drug is reinstituted (the “soarand-crash” effect)
- The frequency, duration, and intensity of the pain episodes gradually increase
- The nature of the pain and its intensity are not explained by a current or previous gastrointestinal diagnosis.16
This syndrome is common in patients who receive high doses of narcotics for postoperative pain or for other, nonmalignant causes of pain. Patients eventually become dependent on the drugs but are not aware that chronic use activates and facilitates areas in the brain that enhance the perception of pain.16 A study of a rat model of narcotic bowel syndrome17 showed that morphine-induced hyperalgesia depends on central sensitization involving the activation of spinal microglia. This eventually results in concomitant peripheral sensitization involving the colonic mucosal neuroimmune system, and also in central or peripheral activation of opioid kappa-receptors by dynorphin release.17
Patients tend to present with chronic or intermittent colicky abdominal pain that requires escalating doses of narcotics. Eventually, they develop tachyphylaxis and shortened pain-free periods and will require even higher doses of narcotics. This ultimately enhances the perception of pain and worsens opioid bowel symptoms, causing a vicious circle of pain and more narcotic use.16
Laboratory tests are usually normal, and imaging may show only ileus. Gastric emptying may be delayed in patients who have either narcotic bowel syndrome or gastroparesis, but since abdominal pain from narcotic bowel syndrome is a result of central and visceral hypersensitivity, these patients perceive more severe abdominal pain than patients with gastroparesis alone.
Opioid withdrawal
Symptoms of opioid withdrawal may appear as soon as 6 to 24 hours after cessation of the opioid in patients known to be dependent on opioids. These patients present with crampy abdominal pain with nausea.18 Other symptoms include agitation, rhinorrhea, lacrimation, excessive yawning, arthralgias, papillary dilation, and piloerection.18
Our patient did not have the typical signs of opioid withdrawal.
Crohn disease
Crohn disease is a multisystem disorder with specific clinical and pathologic features. It is characterized by focal, asymmetric, transmural, and occasionally granulomatous inflammation primarily affecting the gastrointestinal tract.19 Characteristic symptoms include abdominal pain, chronic diarrhea with or without rectal bleeding, and weight loss. Extraintestinal signs may include anemia and inflammatory changes in the eyes, skin, and joints. The diagnosis is based on endoscopic, radiographic, and pathologic findings.19
Our patient did not have diarrhea or signs of Crohn disease on CT, endoscopy, or histology.
Chronic pancreatitis
Chronic pancreatitis involves progressive inflammatory changes resulting in permanent structural damage to the pancreas and subsequent exocrine and endocrine dysfunction.20 Patients have epigastric abdominal pain that often radiates to the back20; it is associated with eating and is partly relieved with leaning forward. Symptoms of pancreatic insufficiency such as fat malabsorption (resulting in steatorrhea and fat-soluble vitamin deficiency) and diabetes are common. Calcifications within the pancreas on CT suggest chronic pancreatitis.20 Serum lipase and amylase levels may be normal or slightly elevated.20
Our patient’s abdominal pain was not typical of pancreatitis. He had no signs or symptoms of pancreatic insufficiency and no calcifications within the pancreas.
Chronic mesenteric ischemia
Chronic mesenteric ischemia (“intestinal angina”) is caused by a reduction in intestinal blood flow as a result of occlusion, vasospasm, or hypoperfusion of the mesenteric vasculature.21 It is commonly seen in patients who smoke or who have atherosclerotic vascular disease. These patients have chronic dull or crampy abdominal pain that usually occurs within 1 hour after eating.21 To avoid pain, patients avoid eating, resulting in weight loss.21 CT angiography with multi-detector CT is as effective as angiography (the gold standard) in depicting splanchnic arterial anatomy.22
Our patient is young and has no known risk factors for atherosclerosis such as smoking. His abdominal pain is more intermittent than chronic and is not associated with eating.
Cannabinoid hyperemesis
Cannabinoid hyperemesis should be considered in patients with long-term cannabis use presenting with cyclic vomiting, abdominal pain, compulsive use of hot showers, and improvement of symptoms with cannabis cessation.23 Although cannabinoids have been recognized for their antiemetic effects, long-term use may eventually cause autonomic instability and disturbances in the hypothalamic-pituitary-adrenal axis, resulting in cyclic vomiting and thermoregulatory impairment.23
Although our patient presented with multiple episodes of vomiting and abdominal pain, he denied using marijuana, he tested negative for tetrahydrocannabinol, and he did not associate any relief of his symptoms with hot baths.
CASE CONTINUED
Our patient receives intravenous hydration, antiemetics, and a narcotic in tapering intravenous doses, and his symptoms gradually improve. He is discharged from the hospital. However, a few weeks later he is readmitted with the same symptoms of abdominal pain and nausea.
3. What is the cornerstone of treatment for narcotic bowel syndrome?
- Establishing a therapeutic relationship
- Detoxification
- Supportive management with intravenous fluids, antiemetics, and stool-softeners
- Medical management with a short-acting narcotic, clonidine, lorazepam, and desipramine
MANAGEMENT OF NARCOTIC BOWEL SYNDROME
An effective therapeutic relationship with the patient is the cornerstone of treatment and should be established before starting detoxification.17 The physician must first learn to accept that the patient’s condition is real and must show genuine empathy as well as provide information about the pathophysiologic basis of the condition, the rationale for withholding narcotics, and the detrimental role narcotics play in the vicious circle of pain.
Detoxification involves gradually withdrawing the narcotic and substituting a nonnarcotic such as an antidepressant for pain control, as well as prescribing a drug such as a benzodiazepine or clonidine to prevent withdrawal symptoms and a laxative to prevent constipation.17,24 The physician must reassure the patient that he or she will not be abandoned in pain and that all medications will be continuously adjusted as needed to keep him or her comfortable throughout the detoxification process.17,24 The physician must continuously gauge the patient’s willingness to continue with treatment and must also be readily available to address the patient’s concerns in a timely manner.17,24 Involving family members and friends may provide additional support to the patient. Referral to a functional gastrointestinal motility program, a pain specialist, and a psychologist may also be considered.17,24 Follow-up care is essential, even after the withdrawal program has ended.17,24
BACK TO THE PATIENT
After successfully establishing a therapeutic relationship and discussing the treatment plan with our patient, we started him on the same dosage of narcotic that he had been receiving, calculated in intravenous morphine equivalents to achieve maximal comfort, and then decreased the dosage by 10% to 33% daily until he was completely off narcotics. An antidepressant and a benzodiazepine were given simultaneously with narcotic tapering. Oral clonidine (0.1–0.4 mg/day) was given after the narcotic dosage was reduced to about half, and polyethylene glycol was given as needed for constipation. The total duration of detoxification was 7 days.
The patient was referred to a psychologist for cognitive-behavioral and relaxation therapy, as well as for encouragement and support. At 6 months, he had had no recurrence of symptoms.
TAKE-HOME MESSAGE
In the United States, the number of patients taking a narcotic for nonmalignant pain is increasing, 25 and physicians should be more aware of complications such as narcotic bowel syndrome.
Narcotic bowel syndrome should be suspected in any patient with prolonged narcotic use presenting with multiple recurrent episodes of abdominal pain after other causes are ruled out.
Establishing a good therapeutic relationship with the patient is the cornerstone of successful treatment. Patients who understand their condition and are willing to be treated tend to have better outcomes.
Supportive treatment, symptom relief, and emotional support during detoxification increase compliance.
A 32-year-old man presents to the emergency department with excruciating abdominal pain associated with multiple episodes of vomiting for the past 2 days. He reports no fevers, headaches, diarrhea, constipation, hematochezia, melena, musculoskeletal symptoms, or weight loss. His abdominal pain is generalized and crampy. It does not radiate and has no precipitating factors. The pain is relieved only with intravenous narcotics.
He does not smoke, drink alcohol, or use illicit drugs. He has no known drug or food allergies. He says that his current condition causes him emotional stress that affects his performance at work.
About a year ago, after a complicated surgical procedure, he needed chronic high-dose narcotics. A few months later, he developed multiple bouts of abdominal pain and vomiting that required hospital visits. He now takes oral oxycodone 10–15 mg every 4–6 hours.
On admission, his vital signs are stable, but he is in excruciating pain. He is alert and oriented to person, place, and time. His sclera are anicteric, and the pupils are equal, round, and reactive to light. Lung and heart examinations are normal. The abdomen is soft and nondistended but tender in all four quadrants without guarding; the liver and spleen are not palpable, and no abdominal masses are detected. He has no skin rash, joint swelling or tenderness, or peripheral edema. The neurologic examination is normal. Computed tomography (CT) of the abdomen with contrast shows no signs of bowel obstruction, pancreatic calcifications or edema, cholecystitis, or hepatobiliary disease. Results of initial laboratory testing are shown in Table 1.
1. Based on the information available, which is the least likely cause of his symptoms?
- Acute pancreatitis
- Cyclic vomiting syndrome
- Acute intermittent porphyria
- Gastroparesis
Acute pancreatitis
Acute pancreatitis is the least likely cause of his symptoms. It is commonly caused by gallstones, alcohol, hypertriglyceridemia, and certain drugs.1 The associated abdominal pain is usually epigastric, radiates to the back, and is accompanied by nausea or vomiting, or both. The onset of pain is sudden and rapidly increases in severity within 30 minutes. CT shows enlargement of the pancreas with diffuse edema, heterogeneity of pancreatic parenchyma, peripancreatic stranding, and peripancreatic fluid collections.1 The diagnosis is based on two of the following three criteria: abdominal pain characteristic of acute pancreatitis; a serum amylase or lipase concentration three or more times the upper limit of normal; and characteristic findings of acute pancreatitis on CT.1
Cyclic vomiting syndrome
Cyclic vomiting syndrome is thought to be caused by episodic dysautonomia, mitochondrial DNA mutations, and hypothalamic emetic response oversensitivity,2–4 but the exact pathogenesis is unknown. The syndrome has been strongly linked to migraine and to the chronic excessive use of cannabinoids.5–9 The Rome III diagnostic criteria10 are the following: the vomiting episodes are stereotypical, ie, they are acute and last for less than 1 week; the patient has had three or more episodes in the previous year; and the patient has no nausea or vomiting between episodes. The patient must meet all three criteria. A history of migraine or a family history of migraine further supports the diagnosis.
Acute intermittent porphyria
Acute intermittent porphyria is characterized by neurovisceral symptoms such as convulsions, paresis, autonomic dysfunction, constipation, and diarrhea that result from the overproduction of porphyrin precursors and deficiency of porphobilinogen deaminase.11
Most patients have poorly localized, severe, steady abdominal pain that develops over hours to days and that may persist for days to weeks.11 Since the pain is neuropathic, abdominal tenderness is usually minimal during an acute attack. Other clues include signs of ileus, such as constipation, nausea, abdominal distention, or decreased bowel sounds; bladder dysfunction, eg, urinary retention, incontinence, or dysuria; reddish-brown urine; and sensory neuropathy of the chest, back, and extremities.11 Blistering skin lesions are usually not seen. The presence of porphobilinogen in the urine confirms the diagnosis.11
Gastroparesis
Gastroparesis is a result of discoordination between the sympathetic and parasympathetic nervous systems, neurons, and smooth muscles within the stomach, causing a decrease in gastric motility. Common causes are diabetes,12 scleroderma,13 and neurologic disorders.14 It can also be iatrogenic,15 resulting from visceral nerve injury and drug treatment with narcotics, calcium channel blockers, muscarinic cholinergic antagonists, or certain antidepressants. Symptoms are related to gastric stasis, ie, abdominal pain from gastric distention, bloating, vomiting, and early satiety.15 Abdominal pain may worsen after eating, and vomitus usually consists of recently ingested food. These patients may have abdominal distension or tenderness and succussion splash. After excluding possible mechanical obstruction, a gastric-emptying study may be necessary to make the diagnosis.15
CASE CONTINUED
A serum and urine drug screen in our patient is positive only for opioids. Urine measures of delta-aminolevulinic acid and porphobilinogen are normal. CT angiography of the abdomen shows no signs of mesenteric vascular occlusion. Esophagogastroduodenoscopy shows antral gastritis, but the esophagus and duodenum appear normal, and colonoscopy is normal as well. Histologic study of biopsy specimens obtained during endoscopy is unrevealing. A gastric-emptying study shows delayed emptying. The patient’s abdominal pain and vomiting persist with the initial dose of intravenous narcotic but resolve with escalating doses. When asked, the patient denies an excessive need for hot baths.
2. Which is the most likely diagnosis at this point?
- Narcotic bowel syndrome
- Opioid withdrawal
- Crohn disease
- Chronic pancreatitis
- Chronic mesenteric ischemia
- Cannabinoid hyperemesis
Narcotic bowel syndrome
Narcotic bowel syndrome is the most likely diagnosis. Grunkemeier et al16 described it as chronic or frequently recurring abdominal pain that is treated with narcotics, either chronically or acutely with high doses, and that includes all the following features16:
- The pain worsens or resolves incompletely with continued or increasing doses of narcotics
- The pain markedly worsens when the narcotic dose is decreased, and decreases when the drug is reinstituted (the “soarand-crash” effect)
- The frequency, duration, and intensity of the pain episodes gradually increase
- The nature of the pain and its intensity are not explained by a current or previous gastrointestinal diagnosis.16
This syndrome is common in patients who receive high doses of narcotics for postoperative pain or for other, nonmalignant causes of pain. Patients eventually become dependent on the drugs but are not aware that chronic use activates and facilitates areas in the brain that enhance the perception of pain.16 A study of a rat model of narcotic bowel syndrome17 showed that morphine-induced hyperalgesia depends on central sensitization involving the activation of spinal microglia. This eventually results in concomitant peripheral sensitization involving the colonic mucosal neuroimmune system, and also in central or peripheral activation of opioid kappa-receptors by dynorphin release.17
Patients tend to present with chronic or intermittent colicky abdominal pain that requires escalating doses of narcotics. Eventually, they develop tachyphylaxis and shortened pain-free periods and will require even higher doses of narcotics. This ultimately enhances the perception of pain and worsens opioid bowel symptoms, causing a vicious circle of pain and more narcotic use.16
Laboratory tests are usually normal, and imaging may show only ileus. Gastric emptying may be delayed in patients who have either narcotic bowel syndrome or gastroparesis, but since abdominal pain from narcotic bowel syndrome is a result of central and visceral hypersensitivity, these patients perceive more severe abdominal pain than patients with gastroparesis alone.
Opioid withdrawal
Symptoms of opioid withdrawal may appear as soon as 6 to 24 hours after cessation of the opioid in patients known to be dependent on opioids. These patients present with crampy abdominal pain with nausea.18 Other symptoms include agitation, rhinorrhea, lacrimation, excessive yawning, arthralgias, papillary dilation, and piloerection.18
Our patient did not have the typical signs of opioid withdrawal.
Crohn disease
Crohn disease is a multisystem disorder with specific clinical and pathologic features. It is characterized by focal, asymmetric, transmural, and occasionally granulomatous inflammation primarily affecting the gastrointestinal tract.19 Characteristic symptoms include abdominal pain, chronic diarrhea with or without rectal bleeding, and weight loss. Extraintestinal signs may include anemia and inflammatory changes in the eyes, skin, and joints. The diagnosis is based on endoscopic, radiographic, and pathologic findings.19
Our patient did not have diarrhea or signs of Crohn disease on CT, endoscopy, or histology.
Chronic pancreatitis
Chronic pancreatitis involves progressive inflammatory changes resulting in permanent structural damage to the pancreas and subsequent exocrine and endocrine dysfunction.20 Patients have epigastric abdominal pain that often radiates to the back20; it is associated with eating and is partly relieved with leaning forward. Symptoms of pancreatic insufficiency such as fat malabsorption (resulting in steatorrhea and fat-soluble vitamin deficiency) and diabetes are common. Calcifications within the pancreas on CT suggest chronic pancreatitis.20 Serum lipase and amylase levels may be normal or slightly elevated.20
Our patient’s abdominal pain was not typical of pancreatitis. He had no signs or symptoms of pancreatic insufficiency and no calcifications within the pancreas.
Chronic mesenteric ischemia
Chronic mesenteric ischemia (“intestinal angina”) is caused by a reduction in intestinal blood flow as a result of occlusion, vasospasm, or hypoperfusion of the mesenteric vasculature.21 It is commonly seen in patients who smoke or who have atherosclerotic vascular disease. These patients have chronic dull or crampy abdominal pain that usually occurs within 1 hour after eating.21 To avoid pain, patients avoid eating, resulting in weight loss.21 CT angiography with multi-detector CT is as effective as angiography (the gold standard) in depicting splanchnic arterial anatomy.22
Our patient is young and has no known risk factors for atherosclerosis such as smoking. His abdominal pain is more intermittent than chronic and is not associated with eating.
Cannabinoid hyperemesis
Cannabinoid hyperemesis should be considered in patients with long-term cannabis use presenting with cyclic vomiting, abdominal pain, compulsive use of hot showers, and improvement of symptoms with cannabis cessation.23 Although cannabinoids have been recognized for their antiemetic effects, long-term use may eventually cause autonomic instability and disturbances in the hypothalamic-pituitary-adrenal axis, resulting in cyclic vomiting and thermoregulatory impairment.23
Although our patient presented with multiple episodes of vomiting and abdominal pain, he denied using marijuana, he tested negative for tetrahydrocannabinol, and he did not associate any relief of his symptoms with hot baths.
CASE CONTINUED
Our patient receives intravenous hydration, antiemetics, and a narcotic in tapering intravenous doses, and his symptoms gradually improve. He is discharged from the hospital. However, a few weeks later he is readmitted with the same symptoms of abdominal pain and nausea.
3. What is the cornerstone of treatment for narcotic bowel syndrome?
- Establishing a therapeutic relationship
- Detoxification
- Supportive management with intravenous fluids, antiemetics, and stool-softeners
- Medical management with a short-acting narcotic, clonidine, lorazepam, and desipramine
MANAGEMENT OF NARCOTIC BOWEL SYNDROME
An effective therapeutic relationship with the patient is the cornerstone of treatment and should be established before starting detoxification.17 The physician must first learn to accept that the patient’s condition is real and must show genuine empathy as well as provide information about the pathophysiologic basis of the condition, the rationale for withholding narcotics, and the detrimental role narcotics play in the vicious circle of pain.
Detoxification involves gradually withdrawing the narcotic and substituting a nonnarcotic such as an antidepressant for pain control, as well as prescribing a drug such as a benzodiazepine or clonidine to prevent withdrawal symptoms and a laxative to prevent constipation.17,24 The physician must reassure the patient that he or she will not be abandoned in pain and that all medications will be continuously adjusted as needed to keep him or her comfortable throughout the detoxification process.17,24 The physician must continuously gauge the patient’s willingness to continue with treatment and must also be readily available to address the patient’s concerns in a timely manner.17,24 Involving family members and friends may provide additional support to the patient. Referral to a functional gastrointestinal motility program, a pain specialist, and a psychologist may also be considered.17,24 Follow-up care is essential, even after the withdrawal program has ended.17,24
BACK TO THE PATIENT
After successfully establishing a therapeutic relationship and discussing the treatment plan with our patient, we started him on the same dosage of narcotic that he had been receiving, calculated in intravenous morphine equivalents to achieve maximal comfort, and then decreased the dosage by 10% to 33% daily until he was completely off narcotics. An antidepressant and a benzodiazepine were given simultaneously with narcotic tapering. Oral clonidine (0.1–0.4 mg/day) was given after the narcotic dosage was reduced to about half, and polyethylene glycol was given as needed for constipation. The total duration of detoxification was 7 days.
The patient was referred to a psychologist for cognitive-behavioral and relaxation therapy, as well as for encouragement and support. At 6 months, he had had no recurrence of symptoms.
TAKE-HOME MESSAGE
In the United States, the number of patients taking a narcotic for nonmalignant pain is increasing, 25 and physicians should be more aware of complications such as narcotic bowel syndrome.
Narcotic bowel syndrome should be suspected in any patient with prolonged narcotic use presenting with multiple recurrent episodes of abdominal pain after other causes are ruled out.
Establishing a good therapeutic relationship with the patient is the cornerstone of successful treatment. Patients who understand their condition and are willing to be treated tend to have better outcomes.
Supportive treatment, symptom relief, and emotional support during detoxification increase compliance.
- Banks PA, Freeman ML; Practice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol 2006; 101:2379–2400.
- Boles RG, Adams K, Ito M, Li BU. Maternal inheritance in cyclic vomiting syndrome with neuromuscular disease. Am J Med Genet A 2003; 120A:474–482.
- Wang Q, Ito M, Adams K, et al. Mitochondrial DNA control region sequence variation in migraine headache and cyclic vomiting syndrome. Am J Med Genet A 2004; 131:50–58.
- Taché Y. Cyclic vomiting syndrome: the corticotropinreleasing-factor hypothesis. Dig Dis Sci 1999; 44(suppl 8):79S–86S.
- Withers GD, Silburn SR, Forbes DA. Precipitants and aetiology of cyclic vomiting syndrome. Acta Paediatr 1998; 87:272–277.
- Whitney HB. Cyclic vomiting. A brief review of this affection as illustrated by a typical case. Arch Pediatr 1898; 15:839–845.
- Stickler GB. Relationship between cyclic vomiting syndrome and migraine. Clin Pediatr (Phila) 2005; 44:505–508.
- Li BU, Murray RD, Heitlinger LA, Robbins JL, Hayes JR. Is cyclic vomiting syndrome related to migraine? J Pediatr 1999; 134:567–572.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Rome Foundation. Rome III disorders and diagnostic criteria. http://www.romecriteria.org/criteria/. Accessed February 27, 2013.
- Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med 2005; 142:439–450.
- Camilleri M. Clinical practice. Diabetic gastroparesis. N Engl J Med 2007; 356:820–829.
- Maddern GJ, Horowitz M, Jamieson GG, Chatterton BE, Collins PJ, Roberts-Thomson P. Abnormalities of esophageal and gastric emptying in progressive systemic sclerosis. Gastroenterology 1984; 87:922–926.
- Jost WH. Gastrointestinal dysfunction in Parkinson’s disease. J Neurol Sci 2010; 289:69–73.
- Parkman HP, Hasler WL, Fisher RS; American Gastroenterological Association. American Gastroenterological Association technical review on the diagnosis and treatment of gastroparesis. Gastroenterology 2004; 127:1592–1622.
- Grunkemeier DM, Cassara JE, Dalton CB, Drossman DA. The narcotic bowel syndrome: clinical features, pathophysiology, and management. Clin Gastroenterol Hepatol 2007; 5:1126–1139.
- Agostini S, Eutamene H, Cartier C, et al. Evidence of central and peripheral sensitization in a rat model of narcotic bowel-like syndrome. Gastroenterology 2010; 139:553–563,563.e1–e5.
- Nicholls L, Bragaw L, Ruetsch C. Opioid dependence treatment and guidelines. J Manag Care Pharm 2010; 16(1 suppl B):S14–S21.
- Lichtenstein GR, Hanauer SB, Sandborn WJ; Practice Parameters Committee of American College of Gastroenterology. Management of Crohn’s disease in adults. Am J Gastroenterol 2009; 104:465–483.
- Steer ML, Waxman I, Freedman S. Chronic pancreatitis. N Engl J Med 1995; 332:1482–1490.
- American Gastroenterological Association Medical Position Statement: guidelines on intestinal ischemia. Gastroenterology 2000; 118:951–953.
- Savastano S, Teso S, Corrà S, Fantozzi O, Miotto D. Multislice CT angiography of the celiac and superior mesenteric arteries: comparison with arteriographic findings. Radiol Med 2002; 103:456–463.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Drossman DA, Morris CB, Edwards H, et al. Diagnosis, characterization, and 3-month outcome after detoxification of 39 patients with narcotic bowel syndrome. Am J Gastroenterol 2012; 107:1426–1440.
- Trescot AM, Boswell MV, Atluri SL, et al. Opioid guidelines in the management of chronic non-cancer pain. Pain Physician 2006; 9:1–39.
- Banks PA, Freeman ML; Practice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol 2006; 101:2379–2400.
- Boles RG, Adams K, Ito M, Li BU. Maternal inheritance in cyclic vomiting syndrome with neuromuscular disease. Am J Med Genet A 2003; 120A:474–482.
- Wang Q, Ito M, Adams K, et al. Mitochondrial DNA control region sequence variation in migraine headache and cyclic vomiting syndrome. Am J Med Genet A 2004; 131:50–58.
- Taché Y. Cyclic vomiting syndrome: the corticotropinreleasing-factor hypothesis. Dig Dis Sci 1999; 44(suppl 8):79S–86S.
- Withers GD, Silburn SR, Forbes DA. Precipitants and aetiology of cyclic vomiting syndrome. Acta Paediatr 1998; 87:272–277.
- Whitney HB. Cyclic vomiting. A brief review of this affection as illustrated by a typical case. Arch Pediatr 1898; 15:839–845.
- Stickler GB. Relationship between cyclic vomiting syndrome and migraine. Clin Pediatr (Phila) 2005; 44:505–508.
- Li BU, Murray RD, Heitlinger LA, Robbins JL, Hayes JR. Is cyclic vomiting syndrome related to migraine? J Pediatr 1999; 134:567–572.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Rome Foundation. Rome III disorders and diagnostic criteria. http://www.romecriteria.org/criteria/. Accessed February 27, 2013.
- Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med 2005; 142:439–450.
- Camilleri M. Clinical practice. Diabetic gastroparesis. N Engl J Med 2007; 356:820–829.
- Maddern GJ, Horowitz M, Jamieson GG, Chatterton BE, Collins PJ, Roberts-Thomson P. Abnormalities of esophageal and gastric emptying in progressive systemic sclerosis. Gastroenterology 1984; 87:922–926.
- Jost WH. Gastrointestinal dysfunction in Parkinson’s disease. J Neurol Sci 2010; 289:69–73.
- Parkman HP, Hasler WL, Fisher RS; American Gastroenterological Association. American Gastroenterological Association technical review on the diagnosis and treatment of gastroparesis. Gastroenterology 2004; 127:1592–1622.
- Grunkemeier DM, Cassara JE, Dalton CB, Drossman DA. The narcotic bowel syndrome: clinical features, pathophysiology, and management. Clin Gastroenterol Hepatol 2007; 5:1126–1139.
- Agostini S, Eutamene H, Cartier C, et al. Evidence of central and peripheral sensitization in a rat model of narcotic bowel-like syndrome. Gastroenterology 2010; 139:553–563,563.e1–e5.
- Nicholls L, Bragaw L, Ruetsch C. Opioid dependence treatment and guidelines. J Manag Care Pharm 2010; 16(1 suppl B):S14–S21.
- Lichtenstein GR, Hanauer SB, Sandborn WJ; Practice Parameters Committee of American College of Gastroenterology. Management of Crohn’s disease in adults. Am J Gastroenterol 2009; 104:465–483.
- Steer ML, Waxman I, Freedman S. Chronic pancreatitis. N Engl J Med 1995; 332:1482–1490.
- American Gastroenterological Association Medical Position Statement: guidelines on intestinal ischemia. Gastroenterology 2000; 118:951–953.
- Savastano S, Teso S, Corrà S, Fantozzi O, Miotto D. Multislice CT angiography of the celiac and superior mesenteric arteries: comparison with arteriographic findings. Radiol Med 2002; 103:456–463.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Drossman DA, Morris CB, Edwards H, et al. Diagnosis, characterization, and 3-month outcome after detoxification of 39 patients with narcotic bowel syndrome. Am J Gastroenterol 2012; 107:1426–1440.
- Trescot AM, Boswell MV, Atluri SL, et al. Opioid guidelines in the management of chronic non-cancer pain. Pain Physician 2006; 9:1–39.
A 74-year-old man with abdominal pain
A 74-year-old man presented to the emergency department in December 2011 with a 1-week history of worsening abdominal pain, nausea with emesis, and decreased appetite. The pain was dull, diffuse, and not related to oral intake or bowel movements. He denied any bloody stools, melena, or hematemesis, but he had not had a bowel movement in the past week.
He was already known to have stage IV colon cancer with metastases to the lungs and liver. He had undergone a partial colectomy in 2009 and was receiving chemotherapy at the time of admission.
He also had an infrarenal abdominal aortic aneurysm that had been repaired in 2003 with endovascular placement of a Gore Excluder stent graft. This was complicated by a type II endoleak, treated with coil embolization. The same endoleak later recurred and was treated with injection of Onyx liquid embolic agent.
His medical history also included hypertension, type 2 diabetes mellitus, and hyperlipidemia. He had undergone a laparoscopic cholecystectomy in 2007.
He denied any fevers, chills, headache, lightheadedness, or change in vision. He had no respiratory, cardiac, or urinary symptoms. He had been constipated for the past few weeks and had recently been started on a bowel regimen, with mild relief. There had been no other changes to his medications.
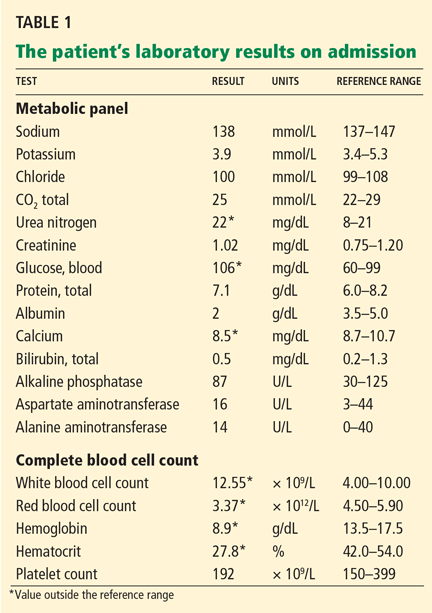
His temperature on presentation was 97.5°F (36.4°C), blood pressure 120/64 mm Hg, pulse 96, respiratory rate 22, and oxygen saturation 95% on room air. He was awake, alert, oriented, and in no acute distress. His mucous membranes were dry. His lungs were clear to auscultation, and his heart sounds were normal. His bowel sounds were hyperactive and his abdomen was slightly tender diffusely, but there was no abdominal distention, rebound tenderness, guarding, or palpable masses. His joints, muscle strength, and muscle tone were normal. Table 1 shows his initial laboratory values.
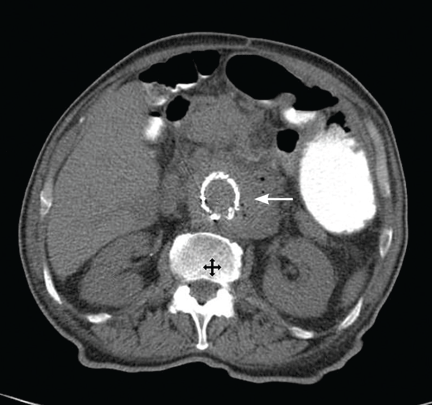
Given the patient’s history of colon cancer, the emergency department physician ordered computed tomography (CT) of the abdomen to assess the state of his disease and to evaluate for bowel obstruction. The scan revealed a large abdominal aortic aneurysm with foci of gas within the aneurysmal sac. Metastases in the liver, lung, and retroperitoneum appeared stable; abundant colonic stool suggested constipation (Figure 1).
CAUSES OF PERIAORTIC GAS AFTER ANEURYSM REPAIR
1. What is the most common cause of periaortic ectopic gas in a patient with a repaired abdominal aortic aneurysm?
- Endoleak
- Stent graft infection
- Retroperitoneal fibrosis
- Aortoenteric fistula
Endoleak
Endoleak, a complication of endovascular abdominal aortic aneurysm repair, is defined as blood flow within the aneurysm sac but outside the endoluminal graft.1 It occurs in up to 15% of patients after endograft placement in the first month alone, and in up to 47% of patients eventually.2 It can lead to aneurysm enlargement and rupture. Endoleaks are classified into five types, each with different causes and management options.3,4 Contrast-enhanced CT is the most commonly used diagnostic tool.5
Endoleak cannot be ruled out in our patient, since CT was done without contrast. However, gas within the aneurysm is not consistent with this diagnosis.
Stent graft infection
Infection has been reported in 1% to 6% of patients receiving a stent graft for aortic aneurysm.6 They occur most commonly in the first year after placement; one study showed that 42% of patients diagnosed with graft infection presented within 3 months of endovascular repair.7
The leading cause of graft infection is contamination during the original procedure, but secondary infection from hematologic seeding and contamination from adjacent bowel are also possible.8 In our patient, who underwent graft placement followed by endovascular repairs of endoleaks, bacterial seeding of his aortic aneurysm from the procedures should be considered.9
The most common organisms are staphylococcal species, with Staphylococcus aureus more common in early infection and coagulase-negative staphylococci more common in late infection.10 Methicillin-resistant S aureus has been reported in as many as 25% of cases of graft infection. Diphtheroids and gram-negative enteric organisms should also be considered.11
CT is the most effective imaging test for graft infection. Perigraft soft tissue, fluid, and gas are the major CT findings.12
Given that our patient presented with abdominal pain, leukocytosis, and the CT finding of perigraft gas, graft infection should be high on our list differential diagnoses.
Retroperitoneal fibrosis
Retroperitoneal fibrosis is most often idiopathic, although many believe it is due to an exaggerated local inflammatory reaction to aortic atherosclerosis or is a manifestation of a systemic autoimmune disease.13 Secondary retroperitoneal fibrosis may be due to drugs, infection, or malignancy.
Pathologic findings include sclerotic plaques, typically around the abdominal vessels and ureters. Clinical presentations are often nonspecific, with early symptoms that include back or abdominal pain, malaise, anorexia, edema, and hematuria.14,15 Progressive ureteral obstruction can occur in later stages. CT with contrast is the imaging test of choice to visualize the extent of disease, with the fibrosis exhibiting attenuation similar to that of muscle.16
Initial treatment of idiopathic retroperitoneal fibrosis is with a glucocorticoid or other immunosuppressive agent, whereas treatment of secondary retroperitoneal fibrosis is aimed at the underlying cause.17 Late stages complicated by ureteral obstruction typically require surgery.18
Our patient did have some nonspecific complaints that could be due to retroperitoneal fibrosis. He also had an intra-abdominal malignancy, which could lead to secondary retroperitoneal fibrosis. However, his CT findings of periaortic gas are not consistent with this diagnosis.19
Aortoenteric fistula
Aortoenteric fistulas can be either primary or secondary.
Primary aortoenteric fistulas occur de novo in patients who have never undergone any surgery or procedure in the aorta. This type of fistula usually results from pressure erosion of an atherosclerotic abdominal aortic aneurysm into the gastrointestinal tract. They are rare, with an annual incidence of 0.04% to 0.07% in the general population.20,21
Secondary aortoenteric fistulas are complications of aortic reconstructive therapy. After open repair, a perianastomotic or pseudoaneurysmal fistula can develop into the gastrointestinal tract.4 Endovascular repair leaves the aortic wall intact with no exposed suture lines, but an aortoenteric fistula can still develop22 and in fact occur in 0.4% to 3.1% of recipients of stent grafts for abdominal aortic aneurysm repair.23 In such cases, it is commonly thought that graft infection can lead to formation of an aortoenteric fistula, but a penetrating gastrointestinal ulcer, tumor invasion, radiation therapy, and trauma have also been implicated.19,24–26 An aortoenteric fistula can present several months to several years after either open or endovascular abdominal aortic aneurysm repair.4,23
One of the main CT signs of an aortoenteric fistula is periaortic ectopic gas at least 3 to 4 weeks after surgery or endovascular repair.19 Gas around the stent graft is most commonly caused by infection, but an aortoenteric fistula must also be considered in our patient, as roughly one-third of graft infections present as aortoenteric fistula.27 Our patient denied having any gastrointestinal bleeding, but his hemoglobin concentration at presentation was 8.9 g/dL.
Highlight point. Perigraft gas after abdominal aortic aneurysm repair can be seen in graft infection and aortoenteric fistula.
SIGNS AND SYMPTOMS OF AORTOENTERIC FISTULA
2. What is the most common clinical sign or symptom of an aortoenteric fistula?
- Gastrointestinal bleeding
- Sepsis
- Abdominal pain
- Back pain
Gastrointestinal bleeding occurs in 80% of patients who have an aortoenteric fistula, sepsis in 40%, abdominal pain in 30%, and back pain in 15%.19 The classic triad of symptoms is gastrointestinal bleeding, abdominal pain, and a pulsatile abdominal mass. However, symptoms can vary widely, and the classic triad is present in fewer than 25% of cases.28 Sepsis may be the predominant clinical manifestation, particularly in the early stages of fistula formation. Unexplained fever is an underrecognized early manifestation.24
Highlight point. The classic triad of symptoms of an aortoenteric fistula (gastrointestinal bleeding, abdominal pain, and a pulsatile abdominal mass) is seen in fewer than 25% of cases.
Case continued: The patient develops frank bleeding
The vascular surgery service was consulted because of concern for an aortic graft infection, since surgical removal of the infected material is recommended.10 The patient was deemed to be a poor surgical candidate, given his stage IV colon cancer, so he was treated conservatively with broad-spectrum antibiotics.
Over the next 2 days, he had two episodes of dark, bloody bowel movements, but he remained hemodynamically stable. He subsequently developed frank bleeding per rectum with symptoms of lightheadedness, and his hemoglobin concentration fell to 6.9 g/dL. He was given a total of 3 units of packed red blood cells, which raised his hemoglobin level, but only to 8.3 g/dL. The gastroenterology service was consulted to evaluate for the source of the bleeding.
Comment. In a situation like this, an aortoenteric fistula is high on our list of differential diagnoses as the cause of bleeding, but other causes of frank bleeding per rectum such as diverticulosis, arteriovenous malformation, hemorrhoids, or a rapid upper-gastrointestinal bleed cannot be ruled out.
Upper-gastrointestinal endoscopy is the most commonly used diagnostic test for aortoenteric fistulas. It can also find other possible sources of gastrointestinal bleeding. CT with contrast is another option. It can depict the fistula itself or reveal signs of infection, such as gas or liquid surrounding the graft. In an emergency, when there is not enough time for diagnostic testing and an aortoenteric fistula is strongly suspected on clinical grounds, emergency surgical exploration is warranted.4,24
In our patient, the gastrointestinal service elected to first perform endoscopy to look for an aortoenteric fistula.
WHERE DO AORTOENTERIC FISTULAS OCCUR?
3. In which part of the gastrointestinal tract is an aortoenteric fistula most commonly located?
- Esophagus
- Stomach
- Duodenum
- Jejunum
Aortoenteric fistulas can occur at any of these locations, but 80% of cases of secondary aortoenteric fistula are in the duodenum, most often in the third or fourth (horizontal or ascending) part.19 Endoscopic visualization of a pulsatile bleeding mass in this area is diagnostic. However, even if no fistula is seen, upper endoscopy cannot rule out an aortoenteric fistula because the lesion can be located more distal than the scope can reach, which is typically no farther than the first or second parts.4,24
Case continued: What endoscopy showed
The esophagus was normal. There was old clotted blood in the stomach, but no lesions or ulcers. The duodenal bulb and second portion of the duodenum were normal. Three ulcers were noted in the third and fourth portions of the duodenum. The largest and deepest ulcer had an adherent blood clot, and the bowel wall was pulsatile in this region (Figure 2). These findings revealed the source of the gastrointestinal bleeding and were consistent with an aortoenteric fistula.
The patient’s initial bloody bowel movements were herald bleeds, ie, transient and self-limited episodes resulting from necrosis and mucosal ulceration. Herald bleeds can precede a massive gastrointestinal hemorrhage resulting from a true aortoenteric communication.19
Highlight point. Herald bleeds are self-limited and precede hemorrhage that results from a true aortoenteric communication.
TREATMENT OF AORTOENTERIC FISTULA
4. How are aortoenteric fistulas treated?
- Surgery
- Antibiotics
- Endoscopic intervention
Surgery is the definitive treatment. The traditional procedure is open surgical resection of the affected portion of the aorta followed by extra-anatomic (axillobifemoral) bypass or in situ aortic reconstruction using an antibiotic-impregnated prosthetic graft, autogenous femoral vein graft, or cryopreserved allograft.9,29 There have been cases of successful endovascular repair of aortoenteric fistulas, but this approach is generally used as a palliative bridge to definitive surgery.30
Antibiotics should be used if graft infection is suspected, ie, in most cases. However, surgery is still needed to repair the fistula and remove the source of infection. Cultures taken during surgical repair can help guide the choice of antibiotic after surgery.
Endoscopy can aid in diagnosing an aortoenteric fistula, as in the case of our patient. However, vascular surgery is necessary to close the communication between the aorta and the gastrointestinal tract.
Case continued: The patient declines treatment
In view of the patient’s enteroscopic findings, the vascular surgery service was again consulted for surgical correction of the aortoenteric fistula. Treatment was discussed with the patient and his family, but they declined any intervention in view of the high risk of morbidity and death that surgery would entail. Nearing the end of life with advanced cancer and a newly diagnosed aortoenteric fistula, the patient preferred comfort measures with hospice care.
Take-home points
Abdominal pain is the reason for 5% to 10% of emergency department visits, and between 35% to 41% of patients admitted to the hospital because of abdominal pain do not have a definitive diagnosis.31 It is crucial to think about an aortoenteric fistula in such patients who have a history of abdominal aortic aneurysm repair and gastrointestinal bleeding. Timely diagnosis and intervention are necessary to manage this otherwise-fatal condition.
- Hong C, Heiken JP, Sicard GA, Pilgram TK, Bae KT. Clinical significance of endoleak detected on follow-up CT after endovascular repair of abdominal aortic aneurysm. AJR Am J Roentgenol 2008; 191:808–813.
- Veith FJ, Baum RA, Ohki T, et al. Nature and significance of endoleaks and endotension: summary of opinions expressed at an international conference. J Vasc Surg 2002; 35:1029–1035.
- Corriere MA, Feurer ID, Becker SY, et al. Endoleak following endovascular abdominal aortic aneurysm repair: implications for duration of screening. Ann Surg 2004; 239:800–805.
- Saratzis N, Saratzis A, Melas N, Ktenidis K, Kiskinis D. Aortoduodenal fistulas after endovascular stent-graft repair of abdominal aortic aneurysms: single-center experience and review of the literature. J Endovasc Ther 2008; 15:441–448.
- Demko TM, Diamond JR, Groff J. Obstructive nephropathy as a result of retroperitoneal fibrosis: a review of its pathogenesis and associations. J Am Soc Nephrol 1997; 8:684–688.
- Zetrenne E, McIntosh BC, McRae MH, Gusberg R, Evans GR, Narayan D. Prosthetic vascular graft infection: a multi-center review of surgical management. Yale J Biol Med 2007; 80:113–121.
- Vogel TR, Symons R, Flum DR. The incidence and factors associated with graft infection after aortic aneurysm repair. J Vasc Surg 2008; 47:264–269.
- Swain TW, Calligaro KD, Dougherty MD. Management of infected aortic prosthetic grafts. Vasc Endovascular Surg 2004; 38:75–82.
- Cernohorsky P, Reijnen MM, Tielliu IF, van Sterkenburg SM, van den Dungen JJ, Zeebregts CJ. The relevance of aortic endograft prosthetic infection. J Vasc Surg 2011; 54:327–333.
- FitzGerald SF, Kelly C, Humphreys H. Diagnosis and treatment of prosthetic aortic graft infections: confusion and inconsistency in the absence of evidence or consensus. J Antimicrob Chemother 2005; 56:996–999.
- Orton DF, LeVeen RF, Saigh JA, et al. Aortic prosthetic graft infections: radiologic manifestations and implications for management. Radiographics 2000; 20:977–993.
- Pacanowski JP, Dieter RS, Stevens SL, Freeman MB, Goldman MH. Endoleak: the achilles heel of endovascular abdominal aortic aneurysm exclusion—a case report. WMJ 2002; 101:57–58,63.
- van Bommel EF. Retroperitoneal fibrosis. Neth J Med 2002; 60:231–242.
- Utz DC, Henry JD. Retroperitoneal fibrosis. Med Clin North Am 1966; 50:1091–1099.
- Dalla-Palma L, Rocca-Rossetti S, Pozzi-Mucelli RS, Rizzatto G. Computed tomography in the diagnosis of retroperitoneal fibrosis. Urol Radiol 1981; 3:77–83.
- Harreby M, Bilde T, Helin P, Meyhoff HH, Vinterberg H, Nielsen VA. Retroperitoneal fibrosis treated with methylprednisolon pulse and disease-modifying antirheumatic drugs. Scand J Urol Nephrol 1994; 28:237–242.
- Jois RN, Gaffney K, Marshall T, Scott DG. Chronic periaortitis. Rheumatology (Oxford) 2004; 43:1441–1446.
- Saers SJ, Scheltinga MR. Primary aortoenteric fistula. Br J Surg 2005; 92:143–152.
- Baril DT, Carroccio A, Ellozy SH, et al. Evolving strategies for the treatment of aortoenteric fistulas. J Vasc Surg 2006; 44:250–257.
- Vu QD, Menias CO, Bhalla S, Peterson C, Wang LL, Balfe DM. Aortoenteric fistulas: CT features and potential mimics. Radiographics 2009; 29:197–209.
- Jayarajan S, Napolitano LM, Rectenwald JE, Upchurch GR. Primary aortoenteric fistula and endovascular repair. Vasc Endovascular Surg 2009; 43:592–596.
- Ruby BJ, Cogbill TH. Aortoduodenal fistula 5 years after endovascular abdominal aortic aneurysm repair with the Ancure stent graft. J Vasc Surg 2007; 45:834–836.
- Senadhi V, Brown JC, Arora D, Shaffer R, Shetty D, Mackrell P. A mysterious cause of gastrointestinal bleeding disguising itself as diverticulosis and peptic ulcer disease: a review of diagnostic modalities for aortoenteric fistula. Case Rep Gastroenterol 2010; 4:510–517.
- Simon T, Feller E. Diverse presentation of secondary aortoenteric fistulae. Case Report Med 2011; 2011:406730.
- Schwab CW, McMahon DJ, Phillips G, Pentecost MJ. Aortic balloon control of a traumatic aortoenteric fistula after damage control laparotomy: a case report. J Trauma 1996; 40:1021–1023.
- Napoli PJ, Meade PC, Adams CW. Primary aortoenteric fistula from a posttraumatic pseudoaneurysm. J Trauma 1996; 41:149–152.
- Laser A, Baker N, Rectenwald J, Eliason JL, Criado-Pallares E, Upchurch GR. Graft infection after endovascular abdominal aortic aneurysm repair. J Vasc Surg 2011; 54:58–63.
- Luo CY, Lai CH, Wen JS, Lin BW. Secondary aortocolic fistula: case report and review of the literature. Ann Vasc Surg 2010; 24:256.e5–256.e12.
- Kim JY, Kim YW, Kim CJ, Lim HI, Kim DI, Huh S. Successful surgical treatment of aortoenteric fistula. J Korean Med Sci 2007; 22:846–850.
- Verhey P, Best A, Lakin P, Nachiondo J, Petersen B. Successful endovascular treatment of aortoenteric fistula secondary to eroding duodenal stent. J Vasc Interv Radiol 2006; 17:1345–1348.
- Kendall JL, Moreira ME. Evaluation of the adult with abdominal pain in the emergency department. In:Hockberger RS, editor: UpToDate. Waltham, MA: UpToDate, 2012.
A 74-year-old man presented to the emergency department in December 2011 with a 1-week history of worsening abdominal pain, nausea with emesis, and decreased appetite. The pain was dull, diffuse, and not related to oral intake or bowel movements. He denied any bloody stools, melena, or hematemesis, but he had not had a bowel movement in the past week.
He was already known to have stage IV colon cancer with metastases to the lungs and liver. He had undergone a partial colectomy in 2009 and was receiving chemotherapy at the time of admission.
He also had an infrarenal abdominal aortic aneurysm that had been repaired in 2003 with endovascular placement of a Gore Excluder stent graft. This was complicated by a type II endoleak, treated with coil embolization. The same endoleak later recurred and was treated with injection of Onyx liquid embolic agent.
His medical history also included hypertension, type 2 diabetes mellitus, and hyperlipidemia. He had undergone a laparoscopic cholecystectomy in 2007.
He denied any fevers, chills, headache, lightheadedness, or change in vision. He had no respiratory, cardiac, or urinary symptoms. He had been constipated for the past few weeks and had recently been started on a bowel regimen, with mild relief. There had been no other changes to his medications.
His temperature on presentation was 97.5°F (36.4°C), blood pressure 120/64 mm Hg, pulse 96, respiratory rate 22, and oxygen saturation 95% on room air. He was awake, alert, oriented, and in no acute distress. His mucous membranes were dry. His lungs were clear to auscultation, and his heart sounds were normal. His bowel sounds were hyperactive and his abdomen was slightly tender diffusely, but there was no abdominal distention, rebound tenderness, guarding, or palpable masses. His joints, muscle strength, and muscle tone were normal. Table 1 shows his initial laboratory values.
Given the patient’s history of colon cancer, the emergency department physician ordered computed tomography (CT) of the abdomen to assess the state of his disease and to evaluate for bowel obstruction. The scan revealed a large abdominal aortic aneurysm with foci of gas within the aneurysmal sac. Metastases in the liver, lung, and retroperitoneum appeared stable; abundant colonic stool suggested constipation (Figure 1).
CAUSES OF PERIAORTIC GAS AFTER ANEURYSM REPAIR
1. What is the most common cause of periaortic ectopic gas in a patient with a repaired abdominal aortic aneurysm?
- Endoleak
- Stent graft infection
- Retroperitoneal fibrosis
- Aortoenteric fistula
Endoleak
Endoleak, a complication of endovascular abdominal aortic aneurysm repair, is defined as blood flow within the aneurysm sac but outside the endoluminal graft.1 It occurs in up to 15% of patients after endograft placement in the first month alone, and in up to 47% of patients eventually.2 It can lead to aneurysm enlargement and rupture. Endoleaks are classified into five types, each with different causes and management options.3,4 Contrast-enhanced CT is the most commonly used diagnostic tool.5
Endoleak cannot be ruled out in our patient, since CT was done without contrast. However, gas within the aneurysm is not consistent with this diagnosis.
Stent graft infection
Infection has been reported in 1% to 6% of patients receiving a stent graft for aortic aneurysm.6 They occur most commonly in the first year after placement; one study showed that 42% of patients diagnosed with graft infection presented within 3 months of endovascular repair.7
The leading cause of graft infection is contamination during the original procedure, but secondary infection from hematologic seeding and contamination from adjacent bowel are also possible.8 In our patient, who underwent graft placement followed by endovascular repairs of endoleaks, bacterial seeding of his aortic aneurysm from the procedures should be considered.9
The most common organisms are staphylococcal species, with Staphylococcus aureus more common in early infection and coagulase-negative staphylococci more common in late infection.10 Methicillin-resistant S aureus has been reported in as many as 25% of cases of graft infection. Diphtheroids and gram-negative enteric organisms should also be considered.11
CT is the most effective imaging test for graft infection. Perigraft soft tissue, fluid, and gas are the major CT findings.12
Given that our patient presented with abdominal pain, leukocytosis, and the CT finding of perigraft gas, graft infection should be high on our list differential diagnoses.
Retroperitoneal fibrosis
Retroperitoneal fibrosis is most often idiopathic, although many believe it is due to an exaggerated local inflammatory reaction to aortic atherosclerosis or is a manifestation of a systemic autoimmune disease.13 Secondary retroperitoneal fibrosis may be due to drugs, infection, or malignancy.
Pathologic findings include sclerotic plaques, typically around the abdominal vessels and ureters. Clinical presentations are often nonspecific, with early symptoms that include back or abdominal pain, malaise, anorexia, edema, and hematuria.14,15 Progressive ureteral obstruction can occur in later stages. CT with contrast is the imaging test of choice to visualize the extent of disease, with the fibrosis exhibiting attenuation similar to that of muscle.16
Initial treatment of idiopathic retroperitoneal fibrosis is with a glucocorticoid or other immunosuppressive agent, whereas treatment of secondary retroperitoneal fibrosis is aimed at the underlying cause.17 Late stages complicated by ureteral obstruction typically require surgery.18
Our patient did have some nonspecific complaints that could be due to retroperitoneal fibrosis. He also had an intra-abdominal malignancy, which could lead to secondary retroperitoneal fibrosis. However, his CT findings of periaortic gas are not consistent with this diagnosis.19
Aortoenteric fistula
Aortoenteric fistulas can be either primary or secondary.
Primary aortoenteric fistulas occur de novo in patients who have never undergone any surgery or procedure in the aorta. This type of fistula usually results from pressure erosion of an atherosclerotic abdominal aortic aneurysm into the gastrointestinal tract. They are rare, with an annual incidence of 0.04% to 0.07% in the general population.20,21
Secondary aortoenteric fistulas are complications of aortic reconstructive therapy. After open repair, a perianastomotic or pseudoaneurysmal fistula can develop into the gastrointestinal tract.4 Endovascular repair leaves the aortic wall intact with no exposed suture lines, but an aortoenteric fistula can still develop22 and in fact occur in 0.4% to 3.1% of recipients of stent grafts for abdominal aortic aneurysm repair.23 In such cases, it is commonly thought that graft infection can lead to formation of an aortoenteric fistula, but a penetrating gastrointestinal ulcer, tumor invasion, radiation therapy, and trauma have also been implicated.19,24–26 An aortoenteric fistula can present several months to several years after either open or endovascular abdominal aortic aneurysm repair.4,23
One of the main CT signs of an aortoenteric fistula is periaortic ectopic gas at least 3 to 4 weeks after surgery or endovascular repair.19 Gas around the stent graft is most commonly caused by infection, but an aortoenteric fistula must also be considered in our patient, as roughly one-third of graft infections present as aortoenteric fistula.27 Our patient denied having any gastrointestinal bleeding, but his hemoglobin concentration at presentation was 8.9 g/dL.
Highlight point. Perigraft gas after abdominal aortic aneurysm repair can be seen in graft infection and aortoenteric fistula.
SIGNS AND SYMPTOMS OF AORTOENTERIC FISTULA
2. What is the most common clinical sign or symptom of an aortoenteric fistula?
- Gastrointestinal bleeding
- Sepsis
- Abdominal pain
- Back pain
Gastrointestinal bleeding occurs in 80% of patients who have an aortoenteric fistula, sepsis in 40%, abdominal pain in 30%, and back pain in 15%.19 The classic triad of symptoms is gastrointestinal bleeding, abdominal pain, and a pulsatile abdominal mass. However, symptoms can vary widely, and the classic triad is present in fewer than 25% of cases.28 Sepsis may be the predominant clinical manifestation, particularly in the early stages of fistula formation. Unexplained fever is an underrecognized early manifestation.24
Highlight point. The classic triad of symptoms of an aortoenteric fistula (gastrointestinal bleeding, abdominal pain, and a pulsatile abdominal mass) is seen in fewer than 25% of cases.
Case continued: The patient develops frank bleeding
The vascular surgery service was consulted because of concern for an aortic graft infection, since surgical removal of the infected material is recommended.10 The patient was deemed to be a poor surgical candidate, given his stage IV colon cancer, so he was treated conservatively with broad-spectrum antibiotics.
Over the next 2 days, he had two episodes of dark, bloody bowel movements, but he remained hemodynamically stable. He subsequently developed frank bleeding per rectum with symptoms of lightheadedness, and his hemoglobin concentration fell to 6.9 g/dL. He was given a total of 3 units of packed red blood cells, which raised his hemoglobin level, but only to 8.3 g/dL. The gastroenterology service was consulted to evaluate for the source of the bleeding.
Comment. In a situation like this, an aortoenteric fistula is high on our list of differential diagnoses as the cause of bleeding, but other causes of frank bleeding per rectum such as diverticulosis, arteriovenous malformation, hemorrhoids, or a rapid upper-gastrointestinal bleed cannot be ruled out.
Upper-gastrointestinal endoscopy is the most commonly used diagnostic test for aortoenteric fistulas. It can also find other possible sources of gastrointestinal bleeding. CT with contrast is another option. It can depict the fistula itself or reveal signs of infection, such as gas or liquid surrounding the graft. In an emergency, when there is not enough time for diagnostic testing and an aortoenteric fistula is strongly suspected on clinical grounds, emergency surgical exploration is warranted.4,24
In our patient, the gastrointestinal service elected to first perform endoscopy to look for an aortoenteric fistula.
WHERE DO AORTOENTERIC FISTULAS OCCUR?
3. In which part of the gastrointestinal tract is an aortoenteric fistula most commonly located?
- Esophagus
- Stomach
- Duodenum
- Jejunum
Aortoenteric fistulas can occur at any of these locations, but 80% of cases of secondary aortoenteric fistula are in the duodenum, most often in the third or fourth (horizontal or ascending) part.19 Endoscopic visualization of a pulsatile bleeding mass in this area is diagnostic. However, even if no fistula is seen, upper endoscopy cannot rule out an aortoenteric fistula because the lesion can be located more distal than the scope can reach, which is typically no farther than the first or second parts.4,24
Case continued: What endoscopy showed
The esophagus was normal. There was old clotted blood in the stomach, but no lesions or ulcers. The duodenal bulb and second portion of the duodenum were normal. Three ulcers were noted in the third and fourth portions of the duodenum. The largest and deepest ulcer had an adherent blood clot, and the bowel wall was pulsatile in this region (Figure 2). These findings revealed the source of the gastrointestinal bleeding and were consistent with an aortoenteric fistula.
The patient’s initial bloody bowel movements were herald bleeds, ie, transient and self-limited episodes resulting from necrosis and mucosal ulceration. Herald bleeds can precede a massive gastrointestinal hemorrhage resulting from a true aortoenteric communication.19
Highlight point. Herald bleeds are self-limited and precede hemorrhage that results from a true aortoenteric communication.
TREATMENT OF AORTOENTERIC FISTULA
4. How are aortoenteric fistulas treated?
- Surgery
- Antibiotics
- Endoscopic intervention
Surgery is the definitive treatment. The traditional procedure is open surgical resection of the affected portion of the aorta followed by extra-anatomic (axillobifemoral) bypass or in situ aortic reconstruction using an antibiotic-impregnated prosthetic graft, autogenous femoral vein graft, or cryopreserved allograft.9,29 There have been cases of successful endovascular repair of aortoenteric fistulas, but this approach is generally used as a palliative bridge to definitive surgery.30
Antibiotics should be used if graft infection is suspected, ie, in most cases. However, surgery is still needed to repair the fistula and remove the source of infection. Cultures taken during surgical repair can help guide the choice of antibiotic after surgery.
Endoscopy can aid in diagnosing an aortoenteric fistula, as in the case of our patient. However, vascular surgery is necessary to close the communication between the aorta and the gastrointestinal tract.
Case continued: The patient declines treatment
In view of the patient’s enteroscopic findings, the vascular surgery service was again consulted for surgical correction of the aortoenteric fistula. Treatment was discussed with the patient and his family, but they declined any intervention in view of the high risk of morbidity and death that surgery would entail. Nearing the end of life with advanced cancer and a newly diagnosed aortoenteric fistula, the patient preferred comfort measures with hospice care.
Take-home points
Abdominal pain is the reason for 5% to 10% of emergency department visits, and between 35% to 41% of patients admitted to the hospital because of abdominal pain do not have a definitive diagnosis.31 It is crucial to think about an aortoenteric fistula in such patients who have a history of abdominal aortic aneurysm repair and gastrointestinal bleeding. Timely diagnosis and intervention are necessary to manage this otherwise-fatal condition.
A 74-year-old man presented to the emergency department in December 2011 with a 1-week history of worsening abdominal pain, nausea with emesis, and decreased appetite. The pain was dull, diffuse, and not related to oral intake or bowel movements. He denied any bloody stools, melena, or hematemesis, but he had not had a bowel movement in the past week.
He was already known to have stage IV colon cancer with metastases to the lungs and liver. He had undergone a partial colectomy in 2009 and was receiving chemotherapy at the time of admission.
He also had an infrarenal abdominal aortic aneurysm that had been repaired in 2003 with endovascular placement of a Gore Excluder stent graft. This was complicated by a type II endoleak, treated with coil embolization. The same endoleak later recurred and was treated with injection of Onyx liquid embolic agent.
His medical history also included hypertension, type 2 diabetes mellitus, and hyperlipidemia. He had undergone a laparoscopic cholecystectomy in 2007.
He denied any fevers, chills, headache, lightheadedness, or change in vision. He had no respiratory, cardiac, or urinary symptoms. He had been constipated for the past few weeks and had recently been started on a bowel regimen, with mild relief. There had been no other changes to his medications.
His temperature on presentation was 97.5°F (36.4°C), blood pressure 120/64 mm Hg, pulse 96, respiratory rate 22, and oxygen saturation 95% on room air. He was awake, alert, oriented, and in no acute distress. His mucous membranes were dry. His lungs were clear to auscultation, and his heart sounds were normal. His bowel sounds were hyperactive and his abdomen was slightly tender diffusely, but there was no abdominal distention, rebound tenderness, guarding, or palpable masses. His joints, muscle strength, and muscle tone were normal. Table 1 shows his initial laboratory values.
Given the patient’s history of colon cancer, the emergency department physician ordered computed tomography (CT) of the abdomen to assess the state of his disease and to evaluate for bowel obstruction. The scan revealed a large abdominal aortic aneurysm with foci of gas within the aneurysmal sac. Metastases in the liver, lung, and retroperitoneum appeared stable; abundant colonic stool suggested constipation (Figure 1).
CAUSES OF PERIAORTIC GAS AFTER ANEURYSM REPAIR
1. What is the most common cause of periaortic ectopic gas in a patient with a repaired abdominal aortic aneurysm?
- Endoleak
- Stent graft infection
- Retroperitoneal fibrosis
- Aortoenteric fistula
Endoleak
Endoleak, a complication of endovascular abdominal aortic aneurysm repair, is defined as blood flow within the aneurysm sac but outside the endoluminal graft.1 It occurs in up to 15% of patients after endograft placement in the first month alone, and in up to 47% of patients eventually.2 It can lead to aneurysm enlargement and rupture. Endoleaks are classified into five types, each with different causes and management options.3,4 Contrast-enhanced CT is the most commonly used diagnostic tool.5
Endoleak cannot be ruled out in our patient, since CT was done without contrast. However, gas within the aneurysm is not consistent with this diagnosis.
Stent graft infection
Infection has been reported in 1% to 6% of patients receiving a stent graft for aortic aneurysm.6 They occur most commonly in the first year after placement; one study showed that 42% of patients diagnosed with graft infection presented within 3 months of endovascular repair.7
The leading cause of graft infection is contamination during the original procedure, but secondary infection from hematologic seeding and contamination from adjacent bowel are also possible.8 In our patient, who underwent graft placement followed by endovascular repairs of endoleaks, bacterial seeding of his aortic aneurysm from the procedures should be considered.9
The most common organisms are staphylococcal species, with Staphylococcus aureus more common in early infection and coagulase-negative staphylococci more common in late infection.10 Methicillin-resistant S aureus has been reported in as many as 25% of cases of graft infection. Diphtheroids and gram-negative enteric organisms should also be considered.11
CT is the most effective imaging test for graft infection. Perigraft soft tissue, fluid, and gas are the major CT findings.12
Given that our patient presented with abdominal pain, leukocytosis, and the CT finding of perigraft gas, graft infection should be high on our list differential diagnoses.
Retroperitoneal fibrosis
Retroperitoneal fibrosis is most often idiopathic, although many believe it is due to an exaggerated local inflammatory reaction to aortic atherosclerosis or is a manifestation of a systemic autoimmune disease.13 Secondary retroperitoneal fibrosis may be due to drugs, infection, or malignancy.
Pathologic findings include sclerotic plaques, typically around the abdominal vessels and ureters. Clinical presentations are often nonspecific, with early symptoms that include back or abdominal pain, malaise, anorexia, edema, and hematuria.14,15 Progressive ureteral obstruction can occur in later stages. CT with contrast is the imaging test of choice to visualize the extent of disease, with the fibrosis exhibiting attenuation similar to that of muscle.16
Initial treatment of idiopathic retroperitoneal fibrosis is with a glucocorticoid or other immunosuppressive agent, whereas treatment of secondary retroperitoneal fibrosis is aimed at the underlying cause.17 Late stages complicated by ureteral obstruction typically require surgery.18
Our patient did have some nonspecific complaints that could be due to retroperitoneal fibrosis. He also had an intra-abdominal malignancy, which could lead to secondary retroperitoneal fibrosis. However, his CT findings of periaortic gas are not consistent with this diagnosis.19
Aortoenteric fistula
Aortoenteric fistulas can be either primary or secondary.
Primary aortoenteric fistulas occur de novo in patients who have never undergone any surgery or procedure in the aorta. This type of fistula usually results from pressure erosion of an atherosclerotic abdominal aortic aneurysm into the gastrointestinal tract. They are rare, with an annual incidence of 0.04% to 0.07% in the general population.20,21
Secondary aortoenteric fistulas are complications of aortic reconstructive therapy. After open repair, a perianastomotic or pseudoaneurysmal fistula can develop into the gastrointestinal tract.4 Endovascular repair leaves the aortic wall intact with no exposed suture lines, but an aortoenteric fistula can still develop22 and in fact occur in 0.4% to 3.1% of recipients of stent grafts for abdominal aortic aneurysm repair.23 In such cases, it is commonly thought that graft infection can lead to formation of an aortoenteric fistula, but a penetrating gastrointestinal ulcer, tumor invasion, radiation therapy, and trauma have also been implicated.19,24–26 An aortoenteric fistula can present several months to several years after either open or endovascular abdominal aortic aneurysm repair.4,23
One of the main CT signs of an aortoenteric fistula is periaortic ectopic gas at least 3 to 4 weeks after surgery or endovascular repair.19 Gas around the stent graft is most commonly caused by infection, but an aortoenteric fistula must also be considered in our patient, as roughly one-third of graft infections present as aortoenteric fistula.27 Our patient denied having any gastrointestinal bleeding, but his hemoglobin concentration at presentation was 8.9 g/dL.
Highlight point. Perigraft gas after abdominal aortic aneurysm repair can be seen in graft infection and aortoenteric fistula.
SIGNS AND SYMPTOMS OF AORTOENTERIC FISTULA
2. What is the most common clinical sign or symptom of an aortoenteric fistula?
- Gastrointestinal bleeding
- Sepsis
- Abdominal pain
- Back pain
Gastrointestinal bleeding occurs in 80% of patients who have an aortoenteric fistula, sepsis in 40%, abdominal pain in 30%, and back pain in 15%.19 The classic triad of symptoms is gastrointestinal bleeding, abdominal pain, and a pulsatile abdominal mass. However, symptoms can vary widely, and the classic triad is present in fewer than 25% of cases.28 Sepsis may be the predominant clinical manifestation, particularly in the early stages of fistula formation. Unexplained fever is an underrecognized early manifestation.24
Highlight point. The classic triad of symptoms of an aortoenteric fistula (gastrointestinal bleeding, abdominal pain, and a pulsatile abdominal mass) is seen in fewer than 25% of cases.
Case continued: The patient develops frank bleeding
The vascular surgery service was consulted because of concern for an aortic graft infection, since surgical removal of the infected material is recommended.10 The patient was deemed to be a poor surgical candidate, given his stage IV colon cancer, so he was treated conservatively with broad-spectrum antibiotics.
Over the next 2 days, he had two episodes of dark, bloody bowel movements, but he remained hemodynamically stable. He subsequently developed frank bleeding per rectum with symptoms of lightheadedness, and his hemoglobin concentration fell to 6.9 g/dL. He was given a total of 3 units of packed red blood cells, which raised his hemoglobin level, but only to 8.3 g/dL. The gastroenterology service was consulted to evaluate for the source of the bleeding.
Comment. In a situation like this, an aortoenteric fistula is high on our list of differential diagnoses as the cause of bleeding, but other causes of frank bleeding per rectum such as diverticulosis, arteriovenous malformation, hemorrhoids, or a rapid upper-gastrointestinal bleed cannot be ruled out.
Upper-gastrointestinal endoscopy is the most commonly used diagnostic test for aortoenteric fistulas. It can also find other possible sources of gastrointestinal bleeding. CT with contrast is another option. It can depict the fistula itself or reveal signs of infection, such as gas or liquid surrounding the graft. In an emergency, when there is not enough time for diagnostic testing and an aortoenteric fistula is strongly suspected on clinical grounds, emergency surgical exploration is warranted.4,24
In our patient, the gastrointestinal service elected to first perform endoscopy to look for an aortoenteric fistula.
WHERE DO AORTOENTERIC FISTULAS OCCUR?
3. In which part of the gastrointestinal tract is an aortoenteric fistula most commonly located?
- Esophagus
- Stomach
- Duodenum
- Jejunum
Aortoenteric fistulas can occur at any of these locations, but 80% of cases of secondary aortoenteric fistula are in the duodenum, most often in the third or fourth (horizontal or ascending) part.19 Endoscopic visualization of a pulsatile bleeding mass in this area is diagnostic. However, even if no fistula is seen, upper endoscopy cannot rule out an aortoenteric fistula because the lesion can be located more distal than the scope can reach, which is typically no farther than the first or second parts.4,24
Case continued: What endoscopy showed
The esophagus was normal. There was old clotted blood in the stomach, but no lesions or ulcers. The duodenal bulb and second portion of the duodenum were normal. Three ulcers were noted in the third and fourth portions of the duodenum. The largest and deepest ulcer had an adherent blood clot, and the bowel wall was pulsatile in this region (Figure 2). These findings revealed the source of the gastrointestinal bleeding and were consistent with an aortoenteric fistula.
The patient’s initial bloody bowel movements were herald bleeds, ie, transient and self-limited episodes resulting from necrosis and mucosal ulceration. Herald bleeds can precede a massive gastrointestinal hemorrhage resulting from a true aortoenteric communication.19
Highlight point. Herald bleeds are self-limited and precede hemorrhage that results from a true aortoenteric communication.
TREATMENT OF AORTOENTERIC FISTULA
4. How are aortoenteric fistulas treated?
- Surgery
- Antibiotics
- Endoscopic intervention
Surgery is the definitive treatment. The traditional procedure is open surgical resection of the affected portion of the aorta followed by extra-anatomic (axillobifemoral) bypass or in situ aortic reconstruction using an antibiotic-impregnated prosthetic graft, autogenous femoral vein graft, or cryopreserved allograft.9,29 There have been cases of successful endovascular repair of aortoenteric fistulas, but this approach is generally used as a palliative bridge to definitive surgery.30
Antibiotics should be used if graft infection is suspected, ie, in most cases. However, surgery is still needed to repair the fistula and remove the source of infection. Cultures taken during surgical repair can help guide the choice of antibiotic after surgery.
Endoscopy can aid in diagnosing an aortoenteric fistula, as in the case of our patient. However, vascular surgery is necessary to close the communication between the aorta and the gastrointestinal tract.
Case continued: The patient declines treatment
In view of the patient’s enteroscopic findings, the vascular surgery service was again consulted for surgical correction of the aortoenteric fistula. Treatment was discussed with the patient and his family, but they declined any intervention in view of the high risk of morbidity and death that surgery would entail. Nearing the end of life with advanced cancer and a newly diagnosed aortoenteric fistula, the patient preferred comfort measures with hospice care.
Take-home points
Abdominal pain is the reason for 5% to 10% of emergency department visits, and between 35% to 41% of patients admitted to the hospital because of abdominal pain do not have a definitive diagnosis.31 It is crucial to think about an aortoenteric fistula in such patients who have a history of abdominal aortic aneurysm repair and gastrointestinal bleeding. Timely diagnosis and intervention are necessary to manage this otherwise-fatal condition.
- Hong C, Heiken JP, Sicard GA, Pilgram TK, Bae KT. Clinical significance of endoleak detected on follow-up CT after endovascular repair of abdominal aortic aneurysm. AJR Am J Roentgenol 2008; 191:808–813.
- Veith FJ, Baum RA, Ohki T, et al. Nature and significance of endoleaks and endotension: summary of opinions expressed at an international conference. J Vasc Surg 2002; 35:1029–1035.
- Corriere MA, Feurer ID, Becker SY, et al. Endoleak following endovascular abdominal aortic aneurysm repair: implications for duration of screening. Ann Surg 2004; 239:800–805.
- Saratzis N, Saratzis A, Melas N, Ktenidis K, Kiskinis D. Aortoduodenal fistulas after endovascular stent-graft repair of abdominal aortic aneurysms: single-center experience and review of the literature. J Endovasc Ther 2008; 15:441–448.
- Demko TM, Diamond JR, Groff J. Obstructive nephropathy as a result of retroperitoneal fibrosis: a review of its pathogenesis and associations. J Am Soc Nephrol 1997; 8:684–688.
- Zetrenne E, McIntosh BC, McRae MH, Gusberg R, Evans GR, Narayan D. Prosthetic vascular graft infection: a multi-center review of surgical management. Yale J Biol Med 2007; 80:113–121.
- Vogel TR, Symons R, Flum DR. The incidence and factors associated with graft infection after aortic aneurysm repair. J Vasc Surg 2008; 47:264–269.
- Swain TW, Calligaro KD, Dougherty MD. Management of infected aortic prosthetic grafts. Vasc Endovascular Surg 2004; 38:75–82.
- Cernohorsky P, Reijnen MM, Tielliu IF, van Sterkenburg SM, van den Dungen JJ, Zeebregts CJ. The relevance of aortic endograft prosthetic infection. J Vasc Surg 2011; 54:327–333.
- FitzGerald SF, Kelly C, Humphreys H. Diagnosis and treatment of prosthetic aortic graft infections: confusion and inconsistency in the absence of evidence or consensus. J Antimicrob Chemother 2005; 56:996–999.
- Orton DF, LeVeen RF, Saigh JA, et al. Aortic prosthetic graft infections: radiologic manifestations and implications for management. Radiographics 2000; 20:977–993.
- Pacanowski JP, Dieter RS, Stevens SL, Freeman MB, Goldman MH. Endoleak: the achilles heel of endovascular abdominal aortic aneurysm exclusion—a case report. WMJ 2002; 101:57–58,63.
- van Bommel EF. Retroperitoneal fibrosis. Neth J Med 2002; 60:231–242.
- Utz DC, Henry JD. Retroperitoneal fibrosis. Med Clin North Am 1966; 50:1091–1099.
- Dalla-Palma L, Rocca-Rossetti S, Pozzi-Mucelli RS, Rizzatto G. Computed tomography in the diagnosis of retroperitoneal fibrosis. Urol Radiol 1981; 3:77–83.
- Harreby M, Bilde T, Helin P, Meyhoff HH, Vinterberg H, Nielsen VA. Retroperitoneal fibrosis treated with methylprednisolon pulse and disease-modifying antirheumatic drugs. Scand J Urol Nephrol 1994; 28:237–242.
- Jois RN, Gaffney K, Marshall T, Scott DG. Chronic periaortitis. Rheumatology (Oxford) 2004; 43:1441–1446.
- Saers SJ, Scheltinga MR. Primary aortoenteric fistula. Br J Surg 2005; 92:143–152.
- Baril DT, Carroccio A, Ellozy SH, et al. Evolving strategies for the treatment of aortoenteric fistulas. J Vasc Surg 2006; 44:250–257.
- Vu QD, Menias CO, Bhalla S, Peterson C, Wang LL, Balfe DM. Aortoenteric fistulas: CT features and potential mimics. Radiographics 2009; 29:197–209.
- Jayarajan S, Napolitano LM, Rectenwald JE, Upchurch GR. Primary aortoenteric fistula and endovascular repair. Vasc Endovascular Surg 2009; 43:592–596.
- Ruby BJ, Cogbill TH. Aortoduodenal fistula 5 years after endovascular abdominal aortic aneurysm repair with the Ancure stent graft. J Vasc Surg 2007; 45:834–836.
- Senadhi V, Brown JC, Arora D, Shaffer R, Shetty D, Mackrell P. A mysterious cause of gastrointestinal bleeding disguising itself as diverticulosis and peptic ulcer disease: a review of diagnostic modalities for aortoenteric fistula. Case Rep Gastroenterol 2010; 4:510–517.
- Simon T, Feller E. Diverse presentation of secondary aortoenteric fistulae. Case Report Med 2011; 2011:406730.
- Schwab CW, McMahon DJ, Phillips G, Pentecost MJ. Aortic balloon control of a traumatic aortoenteric fistula after damage control laparotomy: a case report. J Trauma 1996; 40:1021–1023.
- Napoli PJ, Meade PC, Adams CW. Primary aortoenteric fistula from a posttraumatic pseudoaneurysm. J Trauma 1996; 41:149–152.
- Laser A, Baker N, Rectenwald J, Eliason JL, Criado-Pallares E, Upchurch GR. Graft infection after endovascular abdominal aortic aneurysm repair. J Vasc Surg 2011; 54:58–63.
- Luo CY, Lai CH, Wen JS, Lin BW. Secondary aortocolic fistula: case report and review of the literature. Ann Vasc Surg 2010; 24:256.e5–256.e12.
- Kim JY, Kim YW, Kim CJ, Lim HI, Kim DI, Huh S. Successful surgical treatment of aortoenteric fistula. J Korean Med Sci 2007; 22:846–850.
- Verhey P, Best A, Lakin P, Nachiondo J, Petersen B. Successful endovascular treatment of aortoenteric fistula secondary to eroding duodenal stent. J Vasc Interv Radiol 2006; 17:1345–1348.
- Kendall JL, Moreira ME. Evaluation of the adult with abdominal pain in the emergency department. In:Hockberger RS, editor: UpToDate. Waltham, MA: UpToDate, 2012.
- Hong C, Heiken JP, Sicard GA, Pilgram TK, Bae KT. Clinical significance of endoleak detected on follow-up CT after endovascular repair of abdominal aortic aneurysm. AJR Am J Roentgenol 2008; 191:808–813.
- Veith FJ, Baum RA, Ohki T, et al. Nature and significance of endoleaks and endotension: summary of opinions expressed at an international conference. J Vasc Surg 2002; 35:1029–1035.
- Corriere MA, Feurer ID, Becker SY, et al. Endoleak following endovascular abdominal aortic aneurysm repair: implications for duration of screening. Ann Surg 2004; 239:800–805.
- Saratzis N, Saratzis A, Melas N, Ktenidis K, Kiskinis D. Aortoduodenal fistulas after endovascular stent-graft repair of abdominal aortic aneurysms: single-center experience and review of the literature. J Endovasc Ther 2008; 15:441–448.
- Demko TM, Diamond JR, Groff J. Obstructive nephropathy as a result of retroperitoneal fibrosis: a review of its pathogenesis and associations. J Am Soc Nephrol 1997; 8:684–688.
- Zetrenne E, McIntosh BC, McRae MH, Gusberg R, Evans GR, Narayan D. Prosthetic vascular graft infection: a multi-center review of surgical management. Yale J Biol Med 2007; 80:113–121.
- Vogel TR, Symons R, Flum DR. The incidence and factors associated with graft infection after aortic aneurysm repair. J Vasc Surg 2008; 47:264–269.
- Swain TW, Calligaro KD, Dougherty MD. Management of infected aortic prosthetic grafts. Vasc Endovascular Surg 2004; 38:75–82.
- Cernohorsky P, Reijnen MM, Tielliu IF, van Sterkenburg SM, van den Dungen JJ, Zeebregts CJ. The relevance of aortic endograft prosthetic infection. J Vasc Surg 2011; 54:327–333.
- FitzGerald SF, Kelly C, Humphreys H. Diagnosis and treatment of prosthetic aortic graft infections: confusion and inconsistency in the absence of evidence or consensus. J Antimicrob Chemother 2005; 56:996–999.
- Orton DF, LeVeen RF, Saigh JA, et al. Aortic prosthetic graft infections: radiologic manifestations and implications for management. Radiographics 2000; 20:977–993.
- Pacanowski JP, Dieter RS, Stevens SL, Freeman MB, Goldman MH. Endoleak: the achilles heel of endovascular abdominal aortic aneurysm exclusion—a case report. WMJ 2002; 101:57–58,63.
- van Bommel EF. Retroperitoneal fibrosis. Neth J Med 2002; 60:231–242.
- Utz DC, Henry JD. Retroperitoneal fibrosis. Med Clin North Am 1966; 50:1091–1099.
- Dalla-Palma L, Rocca-Rossetti S, Pozzi-Mucelli RS, Rizzatto G. Computed tomography in the diagnosis of retroperitoneal fibrosis. Urol Radiol 1981; 3:77–83.
- Harreby M, Bilde T, Helin P, Meyhoff HH, Vinterberg H, Nielsen VA. Retroperitoneal fibrosis treated with methylprednisolon pulse and disease-modifying antirheumatic drugs. Scand J Urol Nephrol 1994; 28:237–242.
- Jois RN, Gaffney K, Marshall T, Scott DG. Chronic periaortitis. Rheumatology (Oxford) 2004; 43:1441–1446.
- Saers SJ, Scheltinga MR. Primary aortoenteric fistula. Br J Surg 2005; 92:143–152.
- Baril DT, Carroccio A, Ellozy SH, et al. Evolving strategies for the treatment of aortoenteric fistulas. J Vasc Surg 2006; 44:250–257.
- Vu QD, Menias CO, Bhalla S, Peterson C, Wang LL, Balfe DM. Aortoenteric fistulas: CT features and potential mimics. Radiographics 2009; 29:197–209.
- Jayarajan S, Napolitano LM, Rectenwald JE, Upchurch GR. Primary aortoenteric fistula and endovascular repair. Vasc Endovascular Surg 2009; 43:592–596.
- Ruby BJ, Cogbill TH. Aortoduodenal fistula 5 years after endovascular abdominal aortic aneurysm repair with the Ancure stent graft. J Vasc Surg 2007; 45:834–836.
- Senadhi V, Brown JC, Arora D, Shaffer R, Shetty D, Mackrell P. A mysterious cause of gastrointestinal bleeding disguising itself as diverticulosis and peptic ulcer disease: a review of diagnostic modalities for aortoenteric fistula. Case Rep Gastroenterol 2010; 4:510–517.
- Simon T, Feller E. Diverse presentation of secondary aortoenteric fistulae. Case Report Med 2011; 2011:406730.
- Schwab CW, McMahon DJ, Phillips G, Pentecost MJ. Aortic balloon control of a traumatic aortoenteric fistula after damage control laparotomy: a case report. J Trauma 1996; 40:1021–1023.
- Napoli PJ, Meade PC, Adams CW. Primary aortoenteric fistula from a posttraumatic pseudoaneurysm. J Trauma 1996; 41:149–152.
- Laser A, Baker N, Rectenwald J, Eliason JL, Criado-Pallares E, Upchurch GR. Graft infection after endovascular abdominal aortic aneurysm repair. J Vasc Surg 2011; 54:58–63.
- Luo CY, Lai CH, Wen JS, Lin BW. Secondary aortocolic fistula: case report and review of the literature. Ann Vasc Surg 2010; 24:256.e5–256.e12.
- Kim JY, Kim YW, Kim CJ, Lim HI, Kim DI, Huh S. Successful surgical treatment of aortoenteric fistula. J Korean Med Sci 2007; 22:846–850.
- Verhey P, Best A, Lakin P, Nachiondo J, Petersen B. Successful endovascular treatment of aortoenteric fistula secondary to eroding duodenal stent. J Vasc Interv Radiol 2006; 17:1345–1348.
- Kendall JL, Moreira ME. Evaluation of the adult with abdominal pain in the emergency department. In:Hockberger RS, editor: UpToDate. Waltham, MA: UpToDate, 2012.
A young woman with enlarged lymph nodes
A previously healthy 25-year-old woman presents to her primary care physician with a “lump in the neck”—a painless, swollen area under the lower part of her left jaw that she noticed several weeks ago and that continues to enlarge. She has also noted a recent increase in fatigue, as well as the onset of generalized headaches and mild sinus congestion. Presumed by another physician to have sinusitis, she had already received a 2-week course of an antibiotic (she could not recall which antibiotic), with no improvement in her symptoms. She has been trying to lose weight and has lost 5 pounds in the last 4 months. She reports no fevers, chills, or night sweats.
She works as a special-education teacher and lives in a rural area. She has not travelled during the past year, inside or outside the United States. When she was an adolescent, she underwent tonsillectomy and had her wisdom teeth extracted. Her family has no history of hematologic dyscrasia or malignancy. She has two dogs, which are indoor pets, and she utilizes a city water supply.
1. Which of the following causes of a lump in the neck is most important to exclude?
- Viral or bacterial infection
- Lymphoma
- Oral cavity abscess
- Infectious mononucleosis
- Congenital anomaly
A lump in the neck can be broadly categorized as congenital, inflammatory, or malignant. Congenital causes include branchial cleft cyst (anterior to the sternocleidomastoid muscle) and thyroglossal duct cyst (usually in the midline between the hyoid bone and the isthmus of the thyroid gland). Other possibilities include lipoma and, less frequently, a salivary gland disorder such as sialadenitis.
A complaint of a neck lump very often correlates with the physical finding of lymphadenopathy, and a standard approach in evaluation should be undertaken, based on the mnemonic “PAAA”—ie, palpation, age, area, and associated symptoms.
Palpation. In palpation of the lymph node group, one should note the size and tactile quality of the lymph nodes and assess for abnormal temperature, tenderness, fluctuance, and mobility. In general, lymph nodes larger than 1.5 cm by 1.5 cm are more likely to be of granulomatous or neoplastic origin.1 Nodes that are tender, warm, or fluctuant are likely reactive to a local infectious process; nodes that are firm, matted, and fixed are most characteristic of malignancy; and rubbery, mobile nodes may represent either granulomatous disease or lymphoma.2
Age helps stratify the risk of malignancy as an underlying cause, which is increased in people over age 50 presenting with lymphadenopathy.1
Area. An assessment of the extent of the lymphadenopathy can guide the search either for a cause of generalized lymphadenopathy or for pathology in the anatomic area drained by the particular lymph node group, including the scalp (occipital or preauricular); external ear (posterior auricular); oral cavity (submandibular, submental); soft tissues of the face and neck (superficial cervical); upper respiratory tract and thyroid (deep cervical); and thoracic cavity and abdominal cavity (supraclavicular).

Asking the patient about occupational, environmental, and behavioral risk factors and associated signs and symptoms such as fever, rash, diaphoresis, unintentional weight loss, and splenomegaly helps to narrow the differential diagnosis. Common diagnoses to consider in the evaluation of peripheral lymphadenopathy are listed in Table 1.
A viral or bacterial upper respiratory infection is one of the most common causes of cervical lymphadenopathy, although this usually does not persist for many weeks. Mononucleosis more commonly involves the posterior cervical chain and is often accompanied by splenomegaly. Because of the prolonged presence of the lump, malignancy, including lymphoma, is the most important of the answer choices to consider and rule out in a timely fashion.
INITIAL PHYSICAL EXAMINATION
The woman appears to be well and is in no acute distress. Her oral temperature is 98.1°F (36.7°C), blood pressure 119/72 mm Hg, heart rate 86 beats per minute, and respiratory rate 18 breaths per minute.
The head and neck appear normal. The nares are patent with normal mucosa and no visible drainage. There is no tenderness during palpation of the facial sinuses. The ear canals, tympanic membranes, oropharynx, and tongue appear normal. Several firm, mobile, nontender lymph nodes about 1 cm in diameter are palpable in the left submandibular and right supraclavicular area. No other occipital, submental, axillary, or inguinal lymphadenopathy is noted. There is no overlying erythema or warmth. The cardiac examination is normal, and the lungs are clear on auscultation. The abdomen is soft, nontender, and nondistended, with no organomegaly. The skin appears normal, and the neurologic examination is normal.
INITIAL LABORATORY TESTS
Results of initial laboratory tests are as follows:
- White blood cell count 5.74 × 109/L (reference range 3.70–11.00)
- Red blood cell count 4.49 × 109/L (3.90–5.20)
- Hemoglobin 13.0 g/dL (11.5–15.5)
- Hematocrit 38.4% (36.0–46.0)
- Platelet count 210 × 109/L (150–400)
- Mean corpuscular volume 85 fL (80–100)
- Absolute neutrophil count 3.26 × 109/L (1.45–7.50)
- Blood urea nitrogen 8 mg/dL (8–25)
- Creatinine 0.65 mg/dL (0.70–1.40)
- Lactate dehydrogenase 146 U/L (100–220)
- Uric acid 4.0 mg/dL (2.0–7.0)
- Thyrotropin (thyroid-stimulating hormone) 1.86 μIU/mL (0.4–5.5).
A recent tuberculin skin test obtained as part of her employment screening was negative, and so was a test for antibody to human immunodeficiency virus (HIV), obtained recently before donating plasma. A urine pregnancy test done in the office was also negative. A peripheral blood smear showed slight toxic granulation with rare reactive lymphocytes.
2. Which test would provide the greatest diagnostic yield at this point?
- Needle aspiration biopsy of lymph node
- Excisional lymph node biopsy
- Polymerase chain reaction (PCR) testing for HIV
- Antistreptolysin-O (ASO) titer
Because of the persistent enlargement of the patient’s lymph nodes despite several weeks of antibiotic treatment, and because submandibular and supraclavicular nodes were involved, excisional lymph node biopsy would be the best of these choices to evaluate for malignancy. Compared with needle aspiration biopsy, it is the gold standard, preserving the nodal architecture and providing ample tissue for immunostaining and additional studies.
Needle aspiration biopsy is safe, inexpensive, and easy to do and can be useful in situations of limited resources, but it does not reliably distinguish between a reactive and a neoplastic process.1 Its collection and interpretation are highly variable and personnel-dependent, and its sensitivity for detecting lymphoma is reported to be as low as 7.1% (95% confidence interval 0.9% to 23.5%).2
An acute retroviral syndrome can cause adenopathy, especially before seroconversion is evident, but it is usually associated with an influenza-like illness and monocytosis. Although this patient had no apparent risk factors for HIV, ordering PCR testing for HIV is also an important step when the clinical situation is suggestive. In the absence of an abnormal-appearing oropharynx, tonsillar exudate, or high fever, the pretest probability of streptococcal pharyngitis is low, and an ASO titer is unlikely to be diagnostic in this case.
CASE CONTINUED: BIOPSY PERFORMED
An incisional lymph node biopsy was obtained (Figure 1).
3. Which can confirm the suspected diagnosis?
- Tissue culture
- Test for immunoglobulin (Ig) G and IgM antibodies to Toxoplasma gondii
- Serum PCR testing for T gondii
- T gondii IgG avidity testing
DIAGNOSIS OF TOXOPLASMOSIS
In this patient, acute toxoplasmosis was suspected based on recognition of the morphologic triad seen in Toxoplasma lymphadenitis— ie, follicular hyperplasia, abundance of monocytoid cells, and clusters of epithelioid lymphocytes.3,4
Detection and measurement of IgM antibodies against T gondii is the most widely used serologic test for acute toxoplasmosis and is often considered the reference standard among the most common commercially available agglutination screening assays. It has a sensitivity between 93.7% and 100% and a specificity of 97.1% to 99.2%.5 Confirmation is generally done with enzyme-linked immunoassay or chemiluminescent-based tests, which can detect lower levels of IgG and IgM.5
A positive serum IgG confirms seroconversion but by itself cannot distinguish between acute and chronic infection, although it is commonly obtained in conjunction with IgM levels.6 Since both IgG and IgM can be elevated months after initial infection, serum IgA and IgE levels can more accurately suggest the timing of infection if clarification is needed.6,7 In addition, IgG avidity testing can distinguish acute infection from chronic infection: a high avidity index suggests the acute infection occurred at least 3 to 5 months ago, whereas the avidity index may be low or zero if acute infection occurred within the past 4 weeks.7 The sensitivity of avidity testing is 91.3% to 94.4%, and the specificity 87.8% to 98.5%.7
Serum PCR testing for T gondii is useful when toxoplasmosis is suspected in patients whose immune system may not be able to mount an adequate antibody response or in patients in the hyperacute phase of infection, even before a detectable antibody response can be formed.8,9 However, because of limitations of equipment, expertise, and overall cost, this method is not universally available. Additionally, blood cultures and PCR testing or tissue culture of pathologic specimens cannot routinely be relied on for diagnosis, as often the burden of microorganisms present in these specimens is low. When positive, culture specimens may yield bradyzoites or tachyzoites, but only after considerable latency of many days to weeks.10
How people acquire acute toxoplasmosis
T gondii is an obligate intracellular protozoan parasite. Sexual replication of the organism takes place within the intestines of cats (the definitive host), with subsequent excretion of infective oocysts in feces.11 These hardy oocysts can contaminate soil or water supplies and can survive for months, depending on ambient temperature and humidity. Ingestion of oocysts can lead to infection of a variety of mammals, including sheep, pigs, chicken, and cattle.
Infection in humans can occur with consumption of raw or undercooked foods contaminated with oocysts, and inadequate hand-washing and poor kitchen hygiene substantially increase the risk of infection.12 Activities such as gardening can expose humans to oocysts in contaminated water and soil. In addition, direct contact with cat feces, such as when cleaning the litterbox, is a known exposure risk. Vertical transmission can manifest as congenital toxoplasmosis in a fetus when transmitted from an infected pregnant mother.
Eating raw or undercooked food is considered to be the greatest risk factor for acquired toxoplasmosis and is believed to be responsible for about 50% of all cases.12 However, in pooled data from 14 case-control studies, no clear risk factor for Toxoplasma infection could be identified in up to 60% of affected people, leading many experts to believe contaminated water may play a larger role in acquisition than previously surmised.12
Toxoplasma cysts have a predilection for muscle and neural tissue, resulting in myositis, myocarditis, encephalitis, and chorioretinitis. Severe systemic manifestations are seen in people with impaired T-cell immunity, such as those with HIV infection and acquired immunodeficiency syndrome; or hematologic malignancy; in recipients of solid-organ transplants; or in people taking corticosteroids or cytotoxic drugs. Congenital infection can result in stillbirth, microcephaly, developmental delay, or deafness in the developing fetus and is an important cause of infant morbidity and death worldwide.13
Infection in immunocompetent people is usually asymptomatic.14 However, up to 20% of immunocompetent patients develop symptoms that tend to be nonspecific and include muscle aches and lymphadenopathy, and these are often mistaken for an influenza-like illness.14,15 Other symptoms include malaise, fevers, night sweats, pharyngitis, abdominal pain, hepatosplenomegaly, maculopapular rash, and atypical lymphocytosis (less than 10% of peripheral blood).11 The most common physical manifestation of acute toxoplasmosis is isolated cervical lymphadenopathy, although any lymph node group can be affected.14 Lymph nodes are not fixed or matted and generally are neither tender nor suppurative.16
4. What is the correct treatment strategy for acute toxoplasmosis in this case?
- Symptomatic treatment only
- Trimethoprim 160 mg and sulfamethoxazole 800 mg daily
- Combination of atovaquone and clindamycin
- Combination pyrimethamine, sulfadiazine, and folinic acid
TREATMENT AND PROGNOSIS OF ACUTE TOXOPLASMOSIS
No antimicrobial treatment is required for most immunocompetent patients. Symptoms are self-limited and resolve within 1 to 2 months in 60% of patients.14 A substantial proportion of patients—25%—will have lingering symptoms at 2 to 4 months, and some (10%) can have mild symptoms for 6 months or longer.16
Symptomatic treatment with analgesics such as nonsteroidal anti-inflammatory drugs (NSAIDs) is appropriate.
Immunocompromised and critically ill patients and those with ocular manifestations require combination therapy with pyrimethamine, sulfadiazine, and folinic acid.17 Trimethoprim-sulfamethoxazole is effective as prophylaxis against T gondii infection in immunocompromised patients at a dosage of 160 mg trimethoprim/800 mg sulfamethoxazole daily, but it is also an alternative for treatment at higher dosages (5 mg/kg trimethoprim and 25 mg/kg sulfamethoxazole twice daily).
Atovaquone and clindamycin can be used in sulfa-sensitive patients17 and also in those with latent toxoplasmosis for better penetration of tissue cysts. Corticosteroids are used as adjuncts in those with ocular involvement.
Spiramycin is the treatment of choice in pregnant women and can be given throughout the pregnancy.17,18 A recent comparative study by Hotop et al18 reported a reduction in the rate of fetal transmission (1.6% vs 4.8%) when spiramycin was given from the time of diagnosis through the 16th week of pregnancy, followed by a minimum of 4 weeks of combination therapy with pyrimethamine, sulfadiazine, and folinic acid.18
CASE CONCLUDED
Serologic testing was positive for IgM and IgG antibodies to T gondii, which suggested subacute infection. The patient received no antimicrobial therapy and her lymphadenopathy eventually resolved. Her generalized fatigue gradually resolved over the next year without antimicrobial treatment.
A thorough re-review of potential exposures was done at subsequent office visits to help elucidate how she may have acquired the infection. She recalled no recent exposure to cats or rodents, nor consumption of raw meat. We could only suppose that there may have been inadvertent exposure to oocyst-containing soil or water or to undercooked meat products. Thus, the diagnosis of acute toxoplasmosis should be kept in mind in the evaluation of lymphadenopathy, even in the absence of a clear history of exposure.
- Habermann TM, Steensma DP. Lymphadenopathy. Mayo Clin Proc 2000; 75:723–732.
- Khillan R, Sidhu G, Axiotis C, Braverman AS. Fine needle aspiration (FNA) cytology for diagnosis of cervical lymphadenopathy. Int J Hematol 2012; 95:282–284.
- Dorfman RF, Remington JS. Value of lymph-node biopsy in the diagnosis of acute acquired toxoplasmosis. N Engl J Med 1973; 289:878–881.
- Eapen M, Mathew CF, Aravindan KP. Evidence based criteria for the histopathological diagnosis of toxoplasmic lymphadenopathy. J Clin Pathol 2005; 58:1143–1146.
- Villard O, Cimon B, Franck J, et al; Network from the French National Reference Center for Toxoplasmosis. Evaluation of the usefulness of six commercial agglutination assays for serologic diagnosis of toxoplasmosis. Diagn Microbiol Infect Dis 2012; 73:231–235.
- Suzuki LA, Rocha RJ, Rossi CL. Evaluation of serological markers for the immunodiagnosis of acute acquired toxoplasmosis. J Med Microbiol 2001; 50:62–70.
- Lachaud L, Calas O, Picot MC, Albaba S, Bourgeois N, Pratlong F. Value of 2 IgG avidity commercial tests used alone or in association to date toxoplasmosis contamination. Diagn Microbiol Infect Dis 2009; 64:267–274.
- Rahumatullah A, Khoo BY, Noordin R. Triplex PCR using new primers for the detection of Toxoplasma gondii. Exp Parasitol 2012; 131:231–238.
- Contini C, Giuliodori M, Cultrera R, Seraceni S. Detection of clinical-stage specific molecular Toxoplasma gondii gene patterns in patients with toxoplasmic lymphadenitis. J Med Microbiol 2006; 55:771–774.
- Silveira C, Vallochi AL, Rodrigues da Silva U, et al. Toxoplasma gondii in the peripheral blood of patients with acute and chronic toxoplasmosis. Br J Ophthalmol 2011; 95:396–400.
- Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet 2004; 363:1965–1976.
- Petersen E, Vesco G, Villari S, Buffolano W. What do we know about risk factors for infection in humans with Toxoplasma gondii and how can we prevent infections? Zoonoses Public Health 2010; 57:8–17.
- Feldman DM, Timms D, Borgida AF. Toxoplasmosis, parvovirus, and cytomegalovirus in pregnancy. Clin Lab Med 2010; 30:709–720.
- Weiss LM, Dubey JP. Toxoplasmosis: a history of clinical observations. Int J Parasitol 2009; 39:895–901.
- Remington JS. Toxoplasmosis in the adult. Bull NY Acad Med 1974; 50:211–227.
- McCabe RE, Brooks RG, Dorfman RF, Remington JS. Clinical spectrum in 107 cases of toxoplasmic lymphadenopathy. Rev Infect Dis 1987; 9:754–774.
- Toxoplasmosis. The Medical Letter, Drugs for Parasitic Infections. New Rochelle, NY: The Medical Letter Inc, June 1, 2010:57–58.
- Hotop A, Hlobil H, Gross U. Efficacy of rapid treatment initiation following primary Toxoplasma gondii infection during pregnancy. Clin Infect Dis 2012; 54:1545–1552.
A previously healthy 25-year-old woman presents to her primary care physician with a “lump in the neck”—a painless, swollen area under the lower part of her left jaw that she noticed several weeks ago and that continues to enlarge. She has also noted a recent increase in fatigue, as well as the onset of generalized headaches and mild sinus congestion. Presumed by another physician to have sinusitis, she had already received a 2-week course of an antibiotic (she could not recall which antibiotic), with no improvement in her symptoms. She has been trying to lose weight and has lost 5 pounds in the last 4 months. She reports no fevers, chills, or night sweats.
She works as a special-education teacher and lives in a rural area. She has not travelled during the past year, inside or outside the United States. When she was an adolescent, she underwent tonsillectomy and had her wisdom teeth extracted. Her family has no history of hematologic dyscrasia or malignancy. She has two dogs, which are indoor pets, and she utilizes a city water supply.
1. Which of the following causes of a lump in the neck is most important to exclude?
- Viral or bacterial infection
- Lymphoma
- Oral cavity abscess
- Infectious mononucleosis
- Congenital anomaly
A lump in the neck can be broadly categorized as congenital, inflammatory, or malignant. Congenital causes include branchial cleft cyst (anterior to the sternocleidomastoid muscle) and thyroglossal duct cyst (usually in the midline between the hyoid bone and the isthmus of the thyroid gland). Other possibilities include lipoma and, less frequently, a salivary gland disorder such as sialadenitis.
A complaint of a neck lump very often correlates with the physical finding of lymphadenopathy, and a standard approach in evaluation should be undertaken, based on the mnemonic “PAAA”—ie, palpation, age, area, and associated symptoms.
Palpation. In palpation of the lymph node group, one should note the size and tactile quality of the lymph nodes and assess for abnormal temperature, tenderness, fluctuance, and mobility. In general, lymph nodes larger than 1.5 cm by 1.5 cm are more likely to be of granulomatous or neoplastic origin.1 Nodes that are tender, warm, or fluctuant are likely reactive to a local infectious process; nodes that are firm, matted, and fixed are most characteristic of malignancy; and rubbery, mobile nodes may represent either granulomatous disease or lymphoma.2
Age helps stratify the risk of malignancy as an underlying cause, which is increased in people over age 50 presenting with lymphadenopathy.1
Area. An assessment of the extent of the lymphadenopathy can guide the search either for a cause of generalized lymphadenopathy or for pathology in the anatomic area drained by the particular lymph node group, including the scalp (occipital or preauricular); external ear (posterior auricular); oral cavity (submandibular, submental); soft tissues of the face and neck (superficial cervical); upper respiratory tract and thyroid (deep cervical); and thoracic cavity and abdominal cavity (supraclavicular).
Asking the patient about occupational, environmental, and behavioral risk factors and associated signs and symptoms such as fever, rash, diaphoresis, unintentional weight loss, and splenomegaly helps to narrow the differential diagnosis. Common diagnoses to consider in the evaluation of peripheral lymphadenopathy are listed in Table 1.
A viral or bacterial upper respiratory infection is one of the most common causes of cervical lymphadenopathy, although this usually does not persist for many weeks. Mononucleosis more commonly involves the posterior cervical chain and is often accompanied by splenomegaly. Because of the prolonged presence of the lump, malignancy, including lymphoma, is the most important of the answer choices to consider and rule out in a timely fashion.
INITIAL PHYSICAL EXAMINATION
The woman appears to be well and is in no acute distress. Her oral temperature is 98.1°F (36.7°C), blood pressure 119/72 mm Hg, heart rate 86 beats per minute, and respiratory rate 18 breaths per minute.
The head and neck appear normal. The nares are patent with normal mucosa and no visible drainage. There is no tenderness during palpation of the facial sinuses. The ear canals, tympanic membranes, oropharynx, and tongue appear normal. Several firm, mobile, nontender lymph nodes about 1 cm in diameter are palpable in the left submandibular and right supraclavicular area. No other occipital, submental, axillary, or inguinal lymphadenopathy is noted. There is no overlying erythema or warmth. The cardiac examination is normal, and the lungs are clear on auscultation. The abdomen is soft, nontender, and nondistended, with no organomegaly. The skin appears normal, and the neurologic examination is normal.
INITIAL LABORATORY TESTS
Results of initial laboratory tests are as follows:
- White blood cell count 5.74 × 109/L (reference range 3.70–11.00)
- Red blood cell count 4.49 × 109/L (3.90–5.20)
- Hemoglobin 13.0 g/dL (11.5–15.5)
- Hematocrit 38.4% (36.0–46.0)
- Platelet count 210 × 109/L (150–400)
- Mean corpuscular volume 85 fL (80–100)
- Absolute neutrophil count 3.26 × 109/L (1.45–7.50)
- Blood urea nitrogen 8 mg/dL (8–25)
- Creatinine 0.65 mg/dL (0.70–1.40)
- Lactate dehydrogenase 146 U/L (100–220)
- Uric acid 4.0 mg/dL (2.0–7.0)
- Thyrotropin (thyroid-stimulating hormone) 1.86 μIU/mL (0.4–5.5).
A recent tuberculin skin test obtained as part of her employment screening was negative, and so was a test for antibody to human immunodeficiency virus (HIV), obtained recently before donating plasma. A urine pregnancy test done in the office was also negative. A peripheral blood smear showed slight toxic granulation with rare reactive lymphocytes.
2. Which test would provide the greatest diagnostic yield at this point?
- Needle aspiration biopsy of lymph node
- Excisional lymph node biopsy
- Polymerase chain reaction (PCR) testing for HIV
- Antistreptolysin-O (ASO) titer
Because of the persistent enlargement of the patient’s lymph nodes despite several weeks of antibiotic treatment, and because submandibular and supraclavicular nodes were involved, excisional lymph node biopsy would be the best of these choices to evaluate for malignancy. Compared with needle aspiration biopsy, it is the gold standard, preserving the nodal architecture and providing ample tissue for immunostaining and additional studies.
Needle aspiration biopsy is safe, inexpensive, and easy to do and can be useful in situations of limited resources, but it does not reliably distinguish between a reactive and a neoplastic process.1 Its collection and interpretation are highly variable and personnel-dependent, and its sensitivity for detecting lymphoma is reported to be as low as 7.1% (95% confidence interval 0.9% to 23.5%).2
An acute retroviral syndrome can cause adenopathy, especially before seroconversion is evident, but it is usually associated with an influenza-like illness and monocytosis. Although this patient had no apparent risk factors for HIV, ordering PCR testing for HIV is also an important step when the clinical situation is suggestive. In the absence of an abnormal-appearing oropharynx, tonsillar exudate, or high fever, the pretest probability of streptococcal pharyngitis is low, and an ASO titer is unlikely to be diagnostic in this case.
CASE CONTINUED: BIOPSY PERFORMED
An incisional lymph node biopsy was obtained (Figure 1).
3. Which can confirm the suspected diagnosis?
- Tissue culture
- Test for immunoglobulin (Ig) G and IgM antibodies to Toxoplasma gondii
- Serum PCR testing for T gondii
- T gondii IgG avidity testing
DIAGNOSIS OF TOXOPLASMOSIS
In this patient, acute toxoplasmosis was suspected based on recognition of the morphologic triad seen in Toxoplasma lymphadenitis— ie, follicular hyperplasia, abundance of monocytoid cells, and clusters of epithelioid lymphocytes.3,4
Detection and measurement of IgM antibodies against T gondii is the most widely used serologic test for acute toxoplasmosis and is often considered the reference standard among the most common commercially available agglutination screening assays. It has a sensitivity between 93.7% and 100% and a specificity of 97.1% to 99.2%.5 Confirmation is generally done with enzyme-linked immunoassay or chemiluminescent-based tests, which can detect lower levels of IgG and IgM.5
A positive serum IgG confirms seroconversion but by itself cannot distinguish between acute and chronic infection, although it is commonly obtained in conjunction with IgM levels.6 Since both IgG and IgM can be elevated months after initial infection, serum IgA and IgE levels can more accurately suggest the timing of infection if clarification is needed.6,7 In addition, IgG avidity testing can distinguish acute infection from chronic infection: a high avidity index suggests the acute infection occurred at least 3 to 5 months ago, whereas the avidity index may be low or zero if acute infection occurred within the past 4 weeks.7 The sensitivity of avidity testing is 91.3% to 94.4%, and the specificity 87.8% to 98.5%.7
Serum PCR testing for T gondii is useful when toxoplasmosis is suspected in patients whose immune system may not be able to mount an adequate antibody response or in patients in the hyperacute phase of infection, even before a detectable antibody response can be formed.8,9 However, because of limitations of equipment, expertise, and overall cost, this method is not universally available. Additionally, blood cultures and PCR testing or tissue culture of pathologic specimens cannot routinely be relied on for diagnosis, as often the burden of microorganisms present in these specimens is low. When positive, culture specimens may yield bradyzoites or tachyzoites, but only after considerable latency of many days to weeks.10
How people acquire acute toxoplasmosis
T gondii is an obligate intracellular protozoan parasite. Sexual replication of the organism takes place within the intestines of cats (the definitive host), with subsequent excretion of infective oocysts in feces.11 These hardy oocysts can contaminate soil or water supplies and can survive for months, depending on ambient temperature and humidity. Ingestion of oocysts can lead to infection of a variety of mammals, including sheep, pigs, chicken, and cattle.
Infection in humans can occur with consumption of raw or undercooked foods contaminated with oocysts, and inadequate hand-washing and poor kitchen hygiene substantially increase the risk of infection.12 Activities such as gardening can expose humans to oocysts in contaminated water and soil. In addition, direct contact with cat feces, such as when cleaning the litterbox, is a known exposure risk. Vertical transmission can manifest as congenital toxoplasmosis in a fetus when transmitted from an infected pregnant mother.
Eating raw or undercooked food is considered to be the greatest risk factor for acquired toxoplasmosis and is believed to be responsible for about 50% of all cases.12 However, in pooled data from 14 case-control studies, no clear risk factor for Toxoplasma infection could be identified in up to 60% of affected people, leading many experts to believe contaminated water may play a larger role in acquisition than previously surmised.12
Toxoplasma cysts have a predilection for muscle and neural tissue, resulting in myositis, myocarditis, encephalitis, and chorioretinitis. Severe systemic manifestations are seen in people with impaired T-cell immunity, such as those with HIV infection and acquired immunodeficiency syndrome; or hematologic malignancy; in recipients of solid-organ transplants; or in people taking corticosteroids or cytotoxic drugs. Congenital infection can result in stillbirth, microcephaly, developmental delay, or deafness in the developing fetus and is an important cause of infant morbidity and death worldwide.13
Infection in immunocompetent people is usually asymptomatic.14 However, up to 20% of immunocompetent patients develop symptoms that tend to be nonspecific and include muscle aches and lymphadenopathy, and these are often mistaken for an influenza-like illness.14,15 Other symptoms include malaise, fevers, night sweats, pharyngitis, abdominal pain, hepatosplenomegaly, maculopapular rash, and atypical lymphocytosis (less than 10% of peripheral blood).11 The most common physical manifestation of acute toxoplasmosis is isolated cervical lymphadenopathy, although any lymph node group can be affected.14 Lymph nodes are not fixed or matted and generally are neither tender nor suppurative.16
4. What is the correct treatment strategy for acute toxoplasmosis in this case?
- Symptomatic treatment only
- Trimethoprim 160 mg and sulfamethoxazole 800 mg daily
- Combination of atovaquone and clindamycin
- Combination pyrimethamine, sulfadiazine, and folinic acid
TREATMENT AND PROGNOSIS OF ACUTE TOXOPLASMOSIS
No antimicrobial treatment is required for most immunocompetent patients. Symptoms are self-limited and resolve within 1 to 2 months in 60% of patients.14 A substantial proportion of patients—25%—will have lingering symptoms at 2 to 4 months, and some (10%) can have mild symptoms for 6 months or longer.16
Symptomatic treatment with analgesics such as nonsteroidal anti-inflammatory drugs (NSAIDs) is appropriate.
Immunocompromised and critically ill patients and those with ocular manifestations require combination therapy with pyrimethamine, sulfadiazine, and folinic acid.17 Trimethoprim-sulfamethoxazole is effective as prophylaxis against T gondii infection in immunocompromised patients at a dosage of 160 mg trimethoprim/800 mg sulfamethoxazole daily, but it is also an alternative for treatment at higher dosages (5 mg/kg trimethoprim and 25 mg/kg sulfamethoxazole twice daily).
Atovaquone and clindamycin can be used in sulfa-sensitive patients17 and also in those with latent toxoplasmosis for better penetration of tissue cysts. Corticosteroids are used as adjuncts in those with ocular involvement.
Spiramycin is the treatment of choice in pregnant women and can be given throughout the pregnancy.17,18 A recent comparative study by Hotop et al18 reported a reduction in the rate of fetal transmission (1.6% vs 4.8%) when spiramycin was given from the time of diagnosis through the 16th week of pregnancy, followed by a minimum of 4 weeks of combination therapy with pyrimethamine, sulfadiazine, and folinic acid.18
CASE CONCLUDED
Serologic testing was positive for IgM and IgG antibodies to T gondii, which suggested subacute infection. The patient received no antimicrobial therapy and her lymphadenopathy eventually resolved. Her generalized fatigue gradually resolved over the next year without antimicrobial treatment.
A thorough re-review of potential exposures was done at subsequent office visits to help elucidate how she may have acquired the infection. She recalled no recent exposure to cats or rodents, nor consumption of raw meat. We could only suppose that there may have been inadvertent exposure to oocyst-containing soil or water or to undercooked meat products. Thus, the diagnosis of acute toxoplasmosis should be kept in mind in the evaluation of lymphadenopathy, even in the absence of a clear history of exposure.
A previously healthy 25-year-old woman presents to her primary care physician with a “lump in the neck”—a painless, swollen area under the lower part of her left jaw that she noticed several weeks ago and that continues to enlarge. She has also noted a recent increase in fatigue, as well as the onset of generalized headaches and mild sinus congestion. Presumed by another physician to have sinusitis, she had already received a 2-week course of an antibiotic (she could not recall which antibiotic), with no improvement in her symptoms. She has been trying to lose weight and has lost 5 pounds in the last 4 months. She reports no fevers, chills, or night sweats.
She works as a special-education teacher and lives in a rural area. She has not travelled during the past year, inside or outside the United States. When she was an adolescent, she underwent tonsillectomy and had her wisdom teeth extracted. Her family has no history of hematologic dyscrasia or malignancy. She has two dogs, which are indoor pets, and she utilizes a city water supply.
1. Which of the following causes of a lump in the neck is most important to exclude?
- Viral or bacterial infection
- Lymphoma
- Oral cavity abscess
- Infectious mononucleosis
- Congenital anomaly
A lump in the neck can be broadly categorized as congenital, inflammatory, or malignant. Congenital causes include branchial cleft cyst (anterior to the sternocleidomastoid muscle) and thyroglossal duct cyst (usually in the midline between the hyoid bone and the isthmus of the thyroid gland). Other possibilities include lipoma and, less frequently, a salivary gland disorder such as sialadenitis.
A complaint of a neck lump very often correlates with the physical finding of lymphadenopathy, and a standard approach in evaluation should be undertaken, based on the mnemonic “PAAA”—ie, palpation, age, area, and associated symptoms.
Palpation. In palpation of the lymph node group, one should note the size and tactile quality of the lymph nodes and assess for abnormal temperature, tenderness, fluctuance, and mobility. In general, lymph nodes larger than 1.5 cm by 1.5 cm are more likely to be of granulomatous or neoplastic origin.1 Nodes that are tender, warm, or fluctuant are likely reactive to a local infectious process; nodes that are firm, matted, and fixed are most characteristic of malignancy; and rubbery, mobile nodes may represent either granulomatous disease or lymphoma.2
Age helps stratify the risk of malignancy as an underlying cause, which is increased in people over age 50 presenting with lymphadenopathy.1
Area. An assessment of the extent of the lymphadenopathy can guide the search either for a cause of generalized lymphadenopathy or for pathology in the anatomic area drained by the particular lymph node group, including the scalp (occipital or preauricular); external ear (posterior auricular); oral cavity (submandibular, submental); soft tissues of the face and neck (superficial cervical); upper respiratory tract and thyroid (deep cervical); and thoracic cavity and abdominal cavity (supraclavicular).
Asking the patient about occupational, environmental, and behavioral risk factors and associated signs and symptoms such as fever, rash, diaphoresis, unintentional weight loss, and splenomegaly helps to narrow the differential diagnosis. Common diagnoses to consider in the evaluation of peripheral lymphadenopathy are listed in Table 1.
A viral or bacterial upper respiratory infection is one of the most common causes of cervical lymphadenopathy, although this usually does not persist for many weeks. Mononucleosis more commonly involves the posterior cervical chain and is often accompanied by splenomegaly. Because of the prolonged presence of the lump, malignancy, including lymphoma, is the most important of the answer choices to consider and rule out in a timely fashion.
INITIAL PHYSICAL EXAMINATION
The woman appears to be well and is in no acute distress. Her oral temperature is 98.1°F (36.7°C), blood pressure 119/72 mm Hg, heart rate 86 beats per minute, and respiratory rate 18 breaths per minute.
The head and neck appear normal. The nares are patent with normal mucosa and no visible drainage. There is no tenderness during palpation of the facial sinuses. The ear canals, tympanic membranes, oropharynx, and tongue appear normal. Several firm, mobile, nontender lymph nodes about 1 cm in diameter are palpable in the left submandibular and right supraclavicular area. No other occipital, submental, axillary, or inguinal lymphadenopathy is noted. There is no overlying erythema or warmth. The cardiac examination is normal, and the lungs are clear on auscultation. The abdomen is soft, nontender, and nondistended, with no organomegaly. The skin appears normal, and the neurologic examination is normal.
INITIAL LABORATORY TESTS
Results of initial laboratory tests are as follows:
- White blood cell count 5.74 × 109/L (reference range 3.70–11.00)
- Red blood cell count 4.49 × 109/L (3.90–5.20)
- Hemoglobin 13.0 g/dL (11.5–15.5)
- Hematocrit 38.4% (36.0–46.0)
- Platelet count 210 × 109/L (150–400)
- Mean corpuscular volume 85 fL (80–100)
- Absolute neutrophil count 3.26 × 109/L (1.45–7.50)
- Blood urea nitrogen 8 mg/dL (8–25)
- Creatinine 0.65 mg/dL (0.70–1.40)
- Lactate dehydrogenase 146 U/L (100–220)
- Uric acid 4.0 mg/dL (2.0–7.0)
- Thyrotropin (thyroid-stimulating hormone) 1.86 μIU/mL (0.4–5.5).
A recent tuberculin skin test obtained as part of her employment screening was negative, and so was a test for antibody to human immunodeficiency virus (HIV), obtained recently before donating plasma. A urine pregnancy test done in the office was also negative. A peripheral blood smear showed slight toxic granulation with rare reactive lymphocytes.
2. Which test would provide the greatest diagnostic yield at this point?
- Needle aspiration biopsy of lymph node
- Excisional lymph node biopsy
- Polymerase chain reaction (PCR) testing for HIV
- Antistreptolysin-O (ASO) titer
Because of the persistent enlargement of the patient’s lymph nodes despite several weeks of antibiotic treatment, and because submandibular and supraclavicular nodes were involved, excisional lymph node biopsy would be the best of these choices to evaluate for malignancy. Compared with needle aspiration biopsy, it is the gold standard, preserving the nodal architecture and providing ample tissue for immunostaining and additional studies.
Needle aspiration biopsy is safe, inexpensive, and easy to do and can be useful in situations of limited resources, but it does not reliably distinguish between a reactive and a neoplastic process.1 Its collection and interpretation are highly variable and personnel-dependent, and its sensitivity for detecting lymphoma is reported to be as low as 7.1% (95% confidence interval 0.9% to 23.5%).2
An acute retroviral syndrome can cause adenopathy, especially before seroconversion is evident, but it is usually associated with an influenza-like illness and monocytosis. Although this patient had no apparent risk factors for HIV, ordering PCR testing for HIV is also an important step when the clinical situation is suggestive. In the absence of an abnormal-appearing oropharynx, tonsillar exudate, or high fever, the pretest probability of streptococcal pharyngitis is low, and an ASO titer is unlikely to be diagnostic in this case.
CASE CONTINUED: BIOPSY PERFORMED
An incisional lymph node biopsy was obtained (Figure 1).
3. Which can confirm the suspected diagnosis?
- Tissue culture
- Test for immunoglobulin (Ig) G and IgM antibodies to Toxoplasma gondii
- Serum PCR testing for T gondii
- T gondii IgG avidity testing
DIAGNOSIS OF TOXOPLASMOSIS
In this patient, acute toxoplasmosis was suspected based on recognition of the morphologic triad seen in Toxoplasma lymphadenitis— ie, follicular hyperplasia, abundance of monocytoid cells, and clusters of epithelioid lymphocytes.3,4
Detection and measurement of IgM antibodies against T gondii is the most widely used serologic test for acute toxoplasmosis and is often considered the reference standard among the most common commercially available agglutination screening assays. It has a sensitivity between 93.7% and 100% and a specificity of 97.1% to 99.2%.5 Confirmation is generally done with enzyme-linked immunoassay or chemiluminescent-based tests, which can detect lower levels of IgG and IgM.5
A positive serum IgG confirms seroconversion but by itself cannot distinguish between acute and chronic infection, although it is commonly obtained in conjunction with IgM levels.6 Since both IgG and IgM can be elevated months after initial infection, serum IgA and IgE levels can more accurately suggest the timing of infection if clarification is needed.6,7 In addition, IgG avidity testing can distinguish acute infection from chronic infection: a high avidity index suggests the acute infection occurred at least 3 to 5 months ago, whereas the avidity index may be low or zero if acute infection occurred within the past 4 weeks.7 The sensitivity of avidity testing is 91.3% to 94.4%, and the specificity 87.8% to 98.5%.7
Serum PCR testing for T gondii is useful when toxoplasmosis is suspected in patients whose immune system may not be able to mount an adequate antibody response or in patients in the hyperacute phase of infection, even before a detectable antibody response can be formed.8,9 However, because of limitations of equipment, expertise, and overall cost, this method is not universally available. Additionally, blood cultures and PCR testing or tissue culture of pathologic specimens cannot routinely be relied on for diagnosis, as often the burden of microorganisms present in these specimens is low. When positive, culture specimens may yield bradyzoites or tachyzoites, but only after considerable latency of many days to weeks.10
How people acquire acute toxoplasmosis
T gondii is an obligate intracellular protozoan parasite. Sexual replication of the organism takes place within the intestines of cats (the definitive host), with subsequent excretion of infective oocysts in feces.11 These hardy oocysts can contaminate soil or water supplies and can survive for months, depending on ambient temperature and humidity. Ingestion of oocysts can lead to infection of a variety of mammals, including sheep, pigs, chicken, and cattle.
Infection in humans can occur with consumption of raw or undercooked foods contaminated with oocysts, and inadequate hand-washing and poor kitchen hygiene substantially increase the risk of infection.12 Activities such as gardening can expose humans to oocysts in contaminated water and soil. In addition, direct contact with cat feces, such as when cleaning the litterbox, is a known exposure risk. Vertical transmission can manifest as congenital toxoplasmosis in a fetus when transmitted from an infected pregnant mother.
Eating raw or undercooked food is considered to be the greatest risk factor for acquired toxoplasmosis and is believed to be responsible for about 50% of all cases.12 However, in pooled data from 14 case-control studies, no clear risk factor for Toxoplasma infection could be identified in up to 60% of affected people, leading many experts to believe contaminated water may play a larger role in acquisition than previously surmised.12
Toxoplasma cysts have a predilection for muscle and neural tissue, resulting in myositis, myocarditis, encephalitis, and chorioretinitis. Severe systemic manifestations are seen in people with impaired T-cell immunity, such as those with HIV infection and acquired immunodeficiency syndrome; or hematologic malignancy; in recipients of solid-organ transplants; or in people taking corticosteroids or cytotoxic drugs. Congenital infection can result in stillbirth, microcephaly, developmental delay, or deafness in the developing fetus and is an important cause of infant morbidity and death worldwide.13
Infection in immunocompetent people is usually asymptomatic.14 However, up to 20% of immunocompetent patients develop symptoms that tend to be nonspecific and include muscle aches and lymphadenopathy, and these are often mistaken for an influenza-like illness.14,15 Other symptoms include malaise, fevers, night sweats, pharyngitis, abdominal pain, hepatosplenomegaly, maculopapular rash, and atypical lymphocytosis (less than 10% of peripheral blood).11 The most common physical manifestation of acute toxoplasmosis is isolated cervical lymphadenopathy, although any lymph node group can be affected.14 Lymph nodes are not fixed or matted and generally are neither tender nor suppurative.16
4. What is the correct treatment strategy for acute toxoplasmosis in this case?
- Symptomatic treatment only
- Trimethoprim 160 mg and sulfamethoxazole 800 mg daily
- Combination of atovaquone and clindamycin
- Combination pyrimethamine, sulfadiazine, and folinic acid
TREATMENT AND PROGNOSIS OF ACUTE TOXOPLASMOSIS
No antimicrobial treatment is required for most immunocompetent patients. Symptoms are self-limited and resolve within 1 to 2 months in 60% of patients.14 A substantial proportion of patients—25%—will have lingering symptoms at 2 to 4 months, and some (10%) can have mild symptoms for 6 months or longer.16
Symptomatic treatment with analgesics such as nonsteroidal anti-inflammatory drugs (NSAIDs) is appropriate.
Immunocompromised and critically ill patients and those with ocular manifestations require combination therapy with pyrimethamine, sulfadiazine, and folinic acid.17 Trimethoprim-sulfamethoxazole is effective as prophylaxis against T gondii infection in immunocompromised patients at a dosage of 160 mg trimethoprim/800 mg sulfamethoxazole daily, but it is also an alternative for treatment at higher dosages (5 mg/kg trimethoprim and 25 mg/kg sulfamethoxazole twice daily).
Atovaquone and clindamycin can be used in sulfa-sensitive patients17 and also in those with latent toxoplasmosis for better penetration of tissue cysts. Corticosteroids are used as adjuncts in those with ocular involvement.
Spiramycin is the treatment of choice in pregnant women and can be given throughout the pregnancy.17,18 A recent comparative study by Hotop et al18 reported a reduction in the rate of fetal transmission (1.6% vs 4.8%) when spiramycin was given from the time of diagnosis through the 16th week of pregnancy, followed by a minimum of 4 weeks of combination therapy with pyrimethamine, sulfadiazine, and folinic acid.18
CASE CONCLUDED
Serologic testing was positive for IgM and IgG antibodies to T gondii, which suggested subacute infection. The patient received no antimicrobial therapy and her lymphadenopathy eventually resolved. Her generalized fatigue gradually resolved over the next year without antimicrobial treatment.
A thorough re-review of potential exposures was done at subsequent office visits to help elucidate how she may have acquired the infection. She recalled no recent exposure to cats or rodents, nor consumption of raw meat. We could only suppose that there may have been inadvertent exposure to oocyst-containing soil or water or to undercooked meat products. Thus, the diagnosis of acute toxoplasmosis should be kept in mind in the evaluation of lymphadenopathy, even in the absence of a clear history of exposure.
- Habermann TM, Steensma DP. Lymphadenopathy. Mayo Clin Proc 2000; 75:723–732.
- Khillan R, Sidhu G, Axiotis C, Braverman AS. Fine needle aspiration (FNA) cytology for diagnosis of cervical lymphadenopathy. Int J Hematol 2012; 95:282–284.
- Dorfman RF, Remington JS. Value of lymph-node biopsy in the diagnosis of acute acquired toxoplasmosis. N Engl J Med 1973; 289:878–881.
- Eapen M, Mathew CF, Aravindan KP. Evidence based criteria for the histopathological diagnosis of toxoplasmic lymphadenopathy. J Clin Pathol 2005; 58:1143–1146.
- Villard O, Cimon B, Franck J, et al; Network from the French National Reference Center for Toxoplasmosis. Evaluation of the usefulness of six commercial agglutination assays for serologic diagnosis of toxoplasmosis. Diagn Microbiol Infect Dis 2012; 73:231–235.
- Suzuki LA, Rocha RJ, Rossi CL. Evaluation of serological markers for the immunodiagnosis of acute acquired toxoplasmosis. J Med Microbiol 2001; 50:62–70.
- Lachaud L, Calas O, Picot MC, Albaba S, Bourgeois N, Pratlong F. Value of 2 IgG avidity commercial tests used alone or in association to date toxoplasmosis contamination. Diagn Microbiol Infect Dis 2009; 64:267–274.
- Rahumatullah A, Khoo BY, Noordin R. Triplex PCR using new primers for the detection of Toxoplasma gondii. Exp Parasitol 2012; 131:231–238.
- Contini C, Giuliodori M, Cultrera R, Seraceni S. Detection of clinical-stage specific molecular Toxoplasma gondii gene patterns in patients with toxoplasmic lymphadenitis. J Med Microbiol 2006; 55:771–774.
- Silveira C, Vallochi AL, Rodrigues da Silva U, et al. Toxoplasma gondii in the peripheral blood of patients with acute and chronic toxoplasmosis. Br J Ophthalmol 2011; 95:396–400.
- Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet 2004; 363:1965–1976.
- Petersen E, Vesco G, Villari S, Buffolano W. What do we know about risk factors for infection in humans with Toxoplasma gondii and how can we prevent infections? Zoonoses Public Health 2010; 57:8–17.
- Feldman DM, Timms D, Borgida AF. Toxoplasmosis, parvovirus, and cytomegalovirus in pregnancy. Clin Lab Med 2010; 30:709–720.
- Weiss LM, Dubey JP. Toxoplasmosis: a history of clinical observations. Int J Parasitol 2009; 39:895–901.
- Remington JS. Toxoplasmosis in the adult. Bull NY Acad Med 1974; 50:211–227.
- McCabe RE, Brooks RG, Dorfman RF, Remington JS. Clinical spectrum in 107 cases of toxoplasmic lymphadenopathy. Rev Infect Dis 1987; 9:754–774.
- Toxoplasmosis. The Medical Letter, Drugs for Parasitic Infections. New Rochelle, NY: The Medical Letter Inc, June 1, 2010:57–58.
- Hotop A, Hlobil H, Gross U. Efficacy of rapid treatment initiation following primary Toxoplasma gondii infection during pregnancy. Clin Infect Dis 2012; 54:1545–1552.
- Habermann TM, Steensma DP. Lymphadenopathy. Mayo Clin Proc 2000; 75:723–732.
- Khillan R, Sidhu G, Axiotis C, Braverman AS. Fine needle aspiration (FNA) cytology for diagnosis of cervical lymphadenopathy. Int J Hematol 2012; 95:282–284.
- Dorfman RF, Remington JS. Value of lymph-node biopsy in the diagnosis of acute acquired toxoplasmosis. N Engl J Med 1973; 289:878–881.
- Eapen M, Mathew CF, Aravindan KP. Evidence based criteria for the histopathological diagnosis of toxoplasmic lymphadenopathy. J Clin Pathol 2005; 58:1143–1146.
- Villard O, Cimon B, Franck J, et al; Network from the French National Reference Center for Toxoplasmosis. Evaluation of the usefulness of six commercial agglutination assays for serologic diagnosis of toxoplasmosis. Diagn Microbiol Infect Dis 2012; 73:231–235.
- Suzuki LA, Rocha RJ, Rossi CL. Evaluation of serological markers for the immunodiagnosis of acute acquired toxoplasmosis. J Med Microbiol 2001; 50:62–70.
- Lachaud L, Calas O, Picot MC, Albaba S, Bourgeois N, Pratlong F. Value of 2 IgG avidity commercial tests used alone or in association to date toxoplasmosis contamination. Diagn Microbiol Infect Dis 2009; 64:267–274.
- Rahumatullah A, Khoo BY, Noordin R. Triplex PCR using new primers for the detection of Toxoplasma gondii. Exp Parasitol 2012; 131:231–238.
- Contini C, Giuliodori M, Cultrera R, Seraceni S. Detection of clinical-stage specific molecular Toxoplasma gondii gene patterns in patients with toxoplasmic lymphadenitis. J Med Microbiol 2006; 55:771–774.
- Silveira C, Vallochi AL, Rodrigues da Silva U, et al. Toxoplasma gondii in the peripheral blood of patients with acute and chronic toxoplasmosis. Br J Ophthalmol 2011; 95:396–400.
- Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet 2004; 363:1965–1976.
- Petersen E, Vesco G, Villari S, Buffolano W. What do we know about risk factors for infection in humans with Toxoplasma gondii and how can we prevent infections? Zoonoses Public Health 2010; 57:8–17.
- Feldman DM, Timms D, Borgida AF. Toxoplasmosis, parvovirus, and cytomegalovirus in pregnancy. Clin Lab Med 2010; 30:709–720.
- Weiss LM, Dubey JP. Toxoplasmosis: a history of clinical observations. Int J Parasitol 2009; 39:895–901.
- Remington JS. Toxoplasmosis in the adult. Bull NY Acad Med 1974; 50:211–227.
- McCabe RE, Brooks RG, Dorfman RF, Remington JS. Clinical spectrum in 107 cases of toxoplasmic lymphadenopathy. Rev Infect Dis 1987; 9:754–774.
- Toxoplasmosis. The Medical Letter, Drugs for Parasitic Infections. New Rochelle, NY: The Medical Letter Inc, June 1, 2010:57–58.
- Hotop A, Hlobil H, Gross U. Efficacy of rapid treatment initiation following primary Toxoplasma gondii infection during pregnancy. Clin Infect Dis 2012; 54:1545–1552.
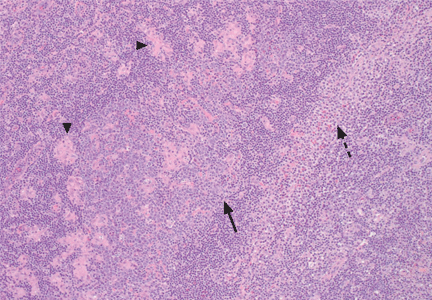
Chronic lymphocytic leukemia and apparent hyperkalemia
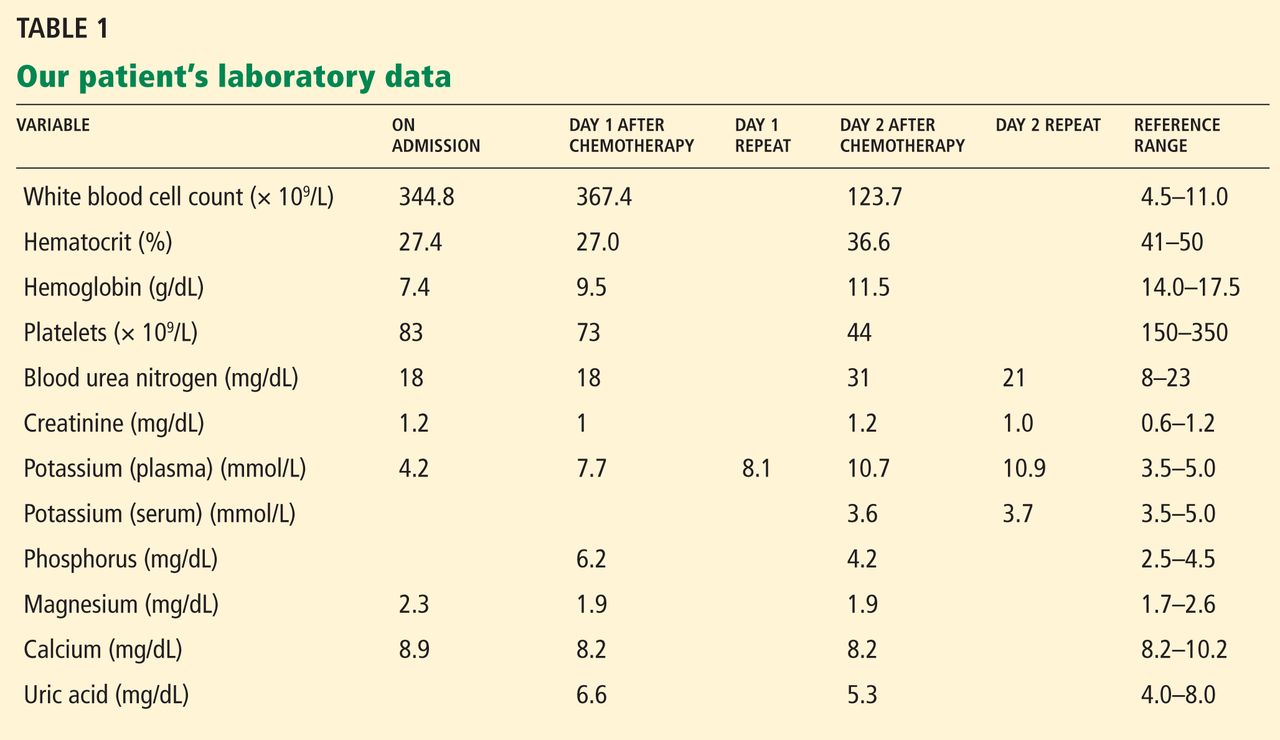
A 64-year-old man with chronic lymphocytic leukemia (CLL), Rai stage IV, was admitted to the hospital to undergo his first cycle of chemotherapy with fludarabine, cyclophosphamide, and rituximab. The physical examination at this time was normal except for splenomegaly and painless bilateral inguinal lymphadenopathy. His laboratory results on admission are shown in Table 1.
On the first day after chemotherapy, laboratory testing revealed an elevated plasma potassium level of 7.7 mmol/L. The specimen was drawn into a BD Vacutainer plasma separator tube with lithium-heparin additive (Becton, Dickinson, and Company, Franklin Lake, NJ) and analyzed on a Unicel DXC 800 chemistry analyzer (Beckman Coulter, Inc, Brea, CA).
1. Which electrocardiographic feature is not associated with hyperkalemia?
- Peaked P waves
- Prolonged PR interval
- Shortened QT interval
- Widened QRS
- Asystole
EVALUATING CARDIAC TOXICITY FROM HYPERKALEMIA
Hyperkalemia is not associated with peaked P waves, but rather with a reduction in the size of the P waves.
Hyperkalemia, defined as a plasma potassium concentration above 5.5 mmol/L, occurs as a result either of a release or a shift of intracellular potassium into the intravascular space or of decreased renal excretion. The earliest changes noted on electrocardiography are peaking and narrowing of T waves, followed by shortening of the QT interval. As hyperkalemia progresses, electrocardiography may show bradycardia, absent P waves, and PR interval prolongation, including second- or third-degree atrioventricular block.
At a plasma potassium concentration greater than 7 mmol/L, there may be a junctional escape rhythm, a sine wave pattern with widening of the QRS complex merging with T waves, ventricular fibrillation, or asystole. However, even with high potassium levels, electrocardiographic changes may be absent.1
The patient denied fatigue, muscle weakness, or palpitations. Electrocardiography did not show peaked T waves, shortened QT intervals, decreased P waves, prolonged PR interval, or widening of the QRS interval.
2. Which is an appropriate intervention for hyperkalemia with cardiac toxicity?
- Membrane stabilization with calcium gluconate
- Shifting potassium into the cells with a beta-adrenergic agonist given by nebulization
- Shifting potassium into the cells with insulin and dextrose
- Removal of potassium with sodium polystyrene sulfonate (Kayexalate)
- Removal of potassium with dialysis
In the setting of cardiac toxicity, the management of hyperkalemia involves stabilizing the cardiac muscle membrane, shifting potassium into the cells, and removing potassium from the body. It is important to do all three interventions when cardiac toxicity is present to provide sustained therapeutic benefit.2
MANAGING HYPERKALEMIA
General principles
When potassium levels are above 6 mmol/L, electrocardiography should always be done. Immediately stop potassium supplementation and any drugs that can cause hyperkalemia, such as potassium-sparing diuretics, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, nonsteroidal anti-inflammatory drugs, and trimethoprim-sulfamethoxazole (Bactrim). If the potassium level is greater than 6.5 mmol/L, the patient should be placed on telemetric monitoring, and the potassium level should be measured often.
Hyperkalemia with cardiac toxicity
Membrane stabilization involves intravenous infusion of 10 mL of 10% calcium gluconate. The onset of action is 1 to 3 minutes, and the duration of action is 30 to 60 minutes.
Shifting of potassium is done either with insulin or with a beta-adrenergic agonist nebulizer. With insulin, 10 U of regular insulin is given intravenously along with 50 mL of 50% dextrose; the onset of action is 20 minutes, and the duration is 4 to 6 hours. The dose of beta-adrenergic agonist depends on the type used; the onset of action is 20 minutes, with a duration of 2 to 4 hours.
Removal of potassium is achieved either by drug therapy or by dialysis. Sodium or calcium polystyrene sulfonate is given by mouth, 15 g every 6 hours, or 30 to 60 g by retention enema; the onset of action is 1 to 2 hours, with a duration of 4 to 6 hours. If dialysis is used, 2 to 3 hours is recommended; the onset of action is immediate and lasts for the duration of the dialysis session.
CASE CONTINUED
The patient was moved to a telemetry unit. A repeat plasma potassium measurement after 30 minutes confirmed hyperkalemia (Table 1). The specimen was transported to the laboratory via a pneumatic tube system, centrifuged at 3,300 rpm for 10 minutes, and analyzed on a Beckman Unicel DXC 800 chemistry analyzer. The time from specimen collection to analysis was approximately 60 minutes.
He was treated with oral sodium polystyrene sulfonate because no electrocardiographic changes were observed. Subsequent plasma potassium levels drawn, collected, and analyzed with the same technique described above were persistently high. Repeated electrocardiography continued to show no changes related to hyperkalemia.
DIAGNOSING THE CAUSE OF THE APPARENT HYPERKALEMIA
3. Which is the most likely cause of hyperkalemia in this patient?
- Acute renal failure
- Tumor lysis syndrome
- Hemolysis
- Reverse pseudohyperkalemia
- Pseudohyperkalemia
DIFFERENTIAL DIAGNOSIS
The patient had normal levels of blood urea nitrogen and creatinine and adequate urine output, thus ruling out acute renal failure. Hyperuricemia, hyperphosphatemia, and hypocalcemia were not found, thus ruling out tumor lysis syndrome.
In vitro hemolysis is assessed by visual inspection showing a pink or red hue to serum or plasma, or by hemolysis index calculation using spectrophotometric measurements. Factors associated with in vitro hemolysis include vein fragility, the phlebotomist’s skill and technique, and transportation of the specimen, including duration, mode, and temperature. The plasma potassium level was repeatedly measured from a lithium-heparin tube, thus minimizing the possibility of laboratory error. No evidence of hemolysis was observed during the phlebotomy, transportation, or specimen analysis.
Serum and plasma potassium levels were simultaneously measured to test for pseudohyperkalemia, a falsely elevated serum potassium concentration caused by the release of platelet potassium during clot formation or after venipuncture. Contributing factors include the prolonged use of a tourniquet, hemolysis, and marked leukocytosis or thrombocytosis. A serum-to-plasma potassium gradient greater than 0.4 mmol/L is diagnostic of pseudohyperkalemia.
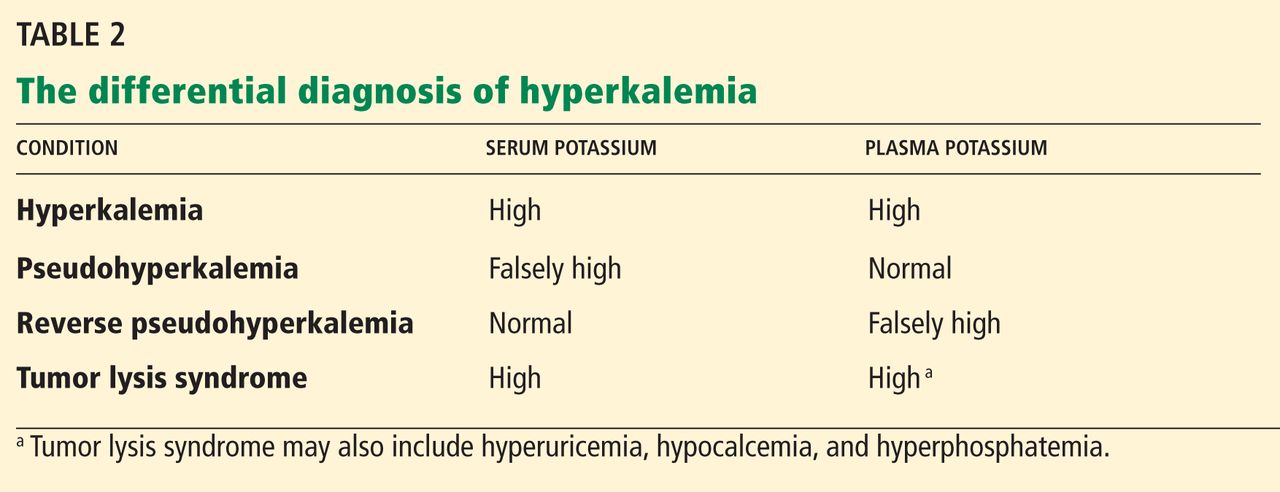
Reverse pseudohyperkalemia, a falsely high potassium level in plasma samples, is defined as a serum-to-plasma potassium gradient less than 0.4 mmol/L (Table 2). This was the most likely cause of the hyperkalemia in our patient.
On the second day after chemotherapy, two blood samples were collected simultaneously—one into a lithium heparin BD Vacutainer plasma separator tube, and the other into a plain, red-top BD Vacutainer serum collection tube without heparin. The specimens were transported to the laboratory by pneumatic tube and were centrifuged at 3,300 rpm for 10 minutes. The specimens were analyzed simultaneously 20 minutes after collection on a Unicel DXC 800 chemistry analyzer. The analysis revealed a serum-to-plasma potassium gradient of −7.1 mmol/L (serum potassium 3.6 mmol/L and plasma potassium 10.7 mmol/L).3 Repeated potassium measurements drawn in similar fashion after 1 hour continued to show a markedly elevated potassium concentration compared with the serum concentration (Table 1).
Serum and plasma samples were again measured simultaneously 24 hours after detecting hyperkalemia to evaluate for pseudohyperkalemia in this patient. In another published report, serum and plasma measurements were obtained 1 week after observing hyperkalemia.4 Blood gas analysis was done the same day in two other reported cases.4,5
Of note, further review of our patient’s medical history noted hyperkalemia at the time he was diagnosed with CLL. At that time, his plasma potassium level was 7.5 mmol/L, a repeated plasma potassium level was 6.9 mmol/L, and a subsequent blood gas analysis—done 30 minutes after the repeated plasma potassium measurement using a Rapidlab analyzer (Siemens Healthcare Diagnostics, Washington, DC)—showed a potassium level of 3.4 mmol/L. The phenomenon of reverse pseudohyperkalemia was not recognized at that time.
The true mechanism of reverse pseudohyperkalemia has not yet been established. Even minor leakage of intracellular potassium from leukemic cells can have a major effect on the extracellular potassium level. Mechanical stressors in the form of pneumatic tube transport and specimen sampling into vacuum tubes have been implicated as causes of this artifact.5,6 Another possible mechanism is heparin-induced lysis of leukocytes in the setting of hematologic malignancy during laboratory processing.4,7,8
LESSONS LEARNED
In patients with hematologic proliferative disorders who develop hyperkalemia in the absence of electrocardiographic changes and an obvious cause of increased potassium levels (eg, acute renal failure, tumor lysis syndrome), we should entertain the possibility of hemolysis, laboratory error, pseudohyperkalemia, and reverse pseudohyperkalemia. The potassium level should be remeasured to rule out laboratory error and hemolysis. In patients with marked leukocytosis or thrombocytosis, simultaneous measurement of serum and plasma potassium levels helps diagnose pseudohyperkalemia and reverse pseudohyperkalemia. Also, prompt blood gas analysis can help identify spurious hyperkalemia.
- Mirvis DM, Goldberger AL. Electrocardiography. In:Bonow RO, Mann DL, Zipes DP, Libby P, editors. Braunwald's Heart Disease—A Textbook of Cardiovascular Medicine. 9th ed. Boston, MA: Elsevier Saunders; 2011:126–167.
- Rastergar A, Soleimani M. Hypokalaemia and hyperkalaemia. Postgrad Med J 2001; 77:759–764.
- Sevastos N, Theodossiades G, Archimandritis AJ. Pseudohyperkalemia in serum: a new insight into an old phenomenon. Clin Med Res 2008; 6:30–32.
- Meng QH, Krahn J. Reverse pseudohyperkalemia in heparin plasma samples from a patient with chronic lymphocytic leukemia. Clin Biochem 2011; 44:728–730.
- Garwicz D, Karlman M, Øra I. Reverse pseudohyperkalemia in heparin plasma samples from a child with T cell acute lymphoblastic leukemia with hyperleukocytosis [Letter]. Clin Chim Acta 2011; 412:396–397.
- Kellerman PS, Thornbery JM. Pseudohyperkalemia due to pneumatic tube transport in a leukemic patient. Am J Kidney Dis 2005; 46:746–748.
- Abraham B, Fakhar I, Tikaria A, et al. Reverse pseudohyperkalemia in a leukemic patient. Clin Chem 2008; 54:449–551.
- Singh PJ, Zawada ET, Santella RN. A case of ‘reverse’ pseudohyperkalemia. Miner Electrolyte Metab 1997; 23:58–61.
A 64-year-old man with chronic lymphocytic leukemia (CLL), Rai stage IV, was admitted to the hospital to undergo his first cycle of chemotherapy with fludarabine, cyclophosphamide, and rituximab. The physical examination at this time was normal except for splenomegaly and painless bilateral inguinal lymphadenopathy. His laboratory results on admission are shown in Table 1.
On the first day after chemotherapy, laboratory testing revealed an elevated plasma potassium level of 7.7 mmol/L. The specimen was drawn into a BD Vacutainer plasma separator tube with lithium-heparin additive (Becton, Dickinson, and Company, Franklin Lake, NJ) and analyzed on a Unicel DXC 800 chemistry analyzer (Beckman Coulter, Inc, Brea, CA).
1. Which electrocardiographic feature is not associated with hyperkalemia?
- Peaked P waves
- Prolonged PR interval
- Shortened QT interval
- Widened QRS
- Asystole
EVALUATING CARDIAC TOXICITY FROM HYPERKALEMIA
Hyperkalemia is not associated with peaked P waves, but rather with a reduction in the size of the P waves.
Hyperkalemia, defined as a plasma potassium concentration above 5.5 mmol/L, occurs as a result either of a release or a shift of intracellular potassium into the intravascular space or of decreased renal excretion. The earliest changes noted on electrocardiography are peaking and narrowing of T waves, followed by shortening of the QT interval. As hyperkalemia progresses, electrocardiography may show bradycardia, absent P waves, and PR interval prolongation, including second- or third-degree atrioventricular block.
At a plasma potassium concentration greater than 7 mmol/L, there may be a junctional escape rhythm, a sine wave pattern with widening of the QRS complex merging with T waves, ventricular fibrillation, or asystole. However, even with high potassium levels, electrocardiographic changes may be absent.1
The patient denied fatigue, muscle weakness, or palpitations. Electrocardiography did not show peaked T waves, shortened QT intervals, decreased P waves, prolonged PR interval, or widening of the QRS interval.
2. Which is an appropriate intervention for hyperkalemia with cardiac toxicity?
- Membrane stabilization with calcium gluconate
- Shifting potassium into the cells with a beta-adrenergic agonist given by nebulization
- Shifting potassium into the cells with insulin and dextrose
- Removal of potassium with sodium polystyrene sulfonate (Kayexalate)
- Removal of potassium with dialysis
In the setting of cardiac toxicity, the management of hyperkalemia involves stabilizing the cardiac muscle membrane, shifting potassium into the cells, and removing potassium from the body. It is important to do all three interventions when cardiac toxicity is present to provide sustained therapeutic benefit.2
MANAGING HYPERKALEMIA
General principles
When potassium levels are above 6 mmol/L, electrocardiography should always be done. Immediately stop potassium supplementation and any drugs that can cause hyperkalemia, such as potassium-sparing diuretics, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, nonsteroidal anti-inflammatory drugs, and trimethoprim-sulfamethoxazole (Bactrim). If the potassium level is greater than 6.5 mmol/L, the patient should be placed on telemetric monitoring, and the potassium level should be measured often.
Hyperkalemia with cardiac toxicity
Membrane stabilization involves intravenous infusion of 10 mL of 10% calcium gluconate. The onset of action is 1 to 3 minutes, and the duration of action is 30 to 60 minutes.
Shifting of potassium is done either with insulin or with a beta-adrenergic agonist nebulizer. With insulin, 10 U of regular insulin is given intravenously along with 50 mL of 50% dextrose; the onset of action is 20 minutes, and the duration is 4 to 6 hours. The dose of beta-adrenergic agonist depends on the type used; the onset of action is 20 minutes, with a duration of 2 to 4 hours.
Removal of potassium is achieved either by drug therapy or by dialysis. Sodium or calcium polystyrene sulfonate is given by mouth, 15 g every 6 hours, or 30 to 60 g by retention enema; the onset of action is 1 to 2 hours, with a duration of 4 to 6 hours. If dialysis is used, 2 to 3 hours is recommended; the onset of action is immediate and lasts for the duration of the dialysis session.
CASE CONTINUED
The patient was moved to a telemetry unit. A repeat plasma potassium measurement after 30 minutes confirmed hyperkalemia (Table 1). The specimen was transported to the laboratory via a pneumatic tube system, centrifuged at 3,300 rpm for 10 minutes, and analyzed on a Beckman Unicel DXC 800 chemistry analyzer. The time from specimen collection to analysis was approximately 60 minutes.
He was treated with oral sodium polystyrene sulfonate because no electrocardiographic changes were observed. Subsequent plasma potassium levels drawn, collected, and analyzed with the same technique described above were persistently high. Repeated electrocardiography continued to show no changes related to hyperkalemia.
DIAGNOSING THE CAUSE OF THE APPARENT HYPERKALEMIA
3. Which is the most likely cause of hyperkalemia in this patient?
- Acute renal failure
- Tumor lysis syndrome
- Hemolysis
- Reverse pseudohyperkalemia
- Pseudohyperkalemia
DIFFERENTIAL DIAGNOSIS
The patient had normal levels of blood urea nitrogen and creatinine and adequate urine output, thus ruling out acute renal failure. Hyperuricemia, hyperphosphatemia, and hypocalcemia were not found, thus ruling out tumor lysis syndrome.
In vitro hemolysis is assessed by visual inspection showing a pink or red hue to serum or plasma, or by hemolysis index calculation using spectrophotometric measurements. Factors associated with in vitro hemolysis include vein fragility, the phlebotomist’s skill and technique, and transportation of the specimen, including duration, mode, and temperature. The plasma potassium level was repeatedly measured from a lithium-heparin tube, thus minimizing the possibility of laboratory error. No evidence of hemolysis was observed during the phlebotomy, transportation, or specimen analysis.
Serum and plasma potassium levels were simultaneously measured to test for pseudohyperkalemia, a falsely elevated serum potassium concentration caused by the release of platelet potassium during clot formation or after venipuncture. Contributing factors include the prolonged use of a tourniquet, hemolysis, and marked leukocytosis or thrombocytosis. A serum-to-plasma potassium gradient greater than 0.4 mmol/L is diagnostic of pseudohyperkalemia.
Reverse pseudohyperkalemia, a falsely high potassium level in plasma samples, is defined as a serum-to-plasma potassium gradient less than 0.4 mmol/L (Table 2). This was the most likely cause of the hyperkalemia in our patient.
On the second day after chemotherapy, two blood samples were collected simultaneously—one into a lithium heparin BD Vacutainer plasma separator tube, and the other into a plain, red-top BD Vacutainer serum collection tube without heparin. The specimens were transported to the laboratory by pneumatic tube and were centrifuged at 3,300 rpm for 10 minutes. The specimens were analyzed simultaneously 20 minutes after collection on a Unicel DXC 800 chemistry analyzer. The analysis revealed a serum-to-plasma potassium gradient of −7.1 mmol/L (serum potassium 3.6 mmol/L and plasma potassium 10.7 mmol/L).3 Repeated potassium measurements drawn in similar fashion after 1 hour continued to show a markedly elevated potassium concentration compared with the serum concentration (Table 1).
Serum and plasma samples were again measured simultaneously 24 hours after detecting hyperkalemia to evaluate for pseudohyperkalemia in this patient. In another published report, serum and plasma measurements were obtained 1 week after observing hyperkalemia.4 Blood gas analysis was done the same day in two other reported cases.4,5
Of note, further review of our patient’s medical history noted hyperkalemia at the time he was diagnosed with CLL. At that time, his plasma potassium level was 7.5 mmol/L, a repeated plasma potassium level was 6.9 mmol/L, and a subsequent blood gas analysis—done 30 minutes after the repeated plasma potassium measurement using a Rapidlab analyzer (Siemens Healthcare Diagnostics, Washington, DC)—showed a potassium level of 3.4 mmol/L. The phenomenon of reverse pseudohyperkalemia was not recognized at that time.
The true mechanism of reverse pseudohyperkalemia has not yet been established. Even minor leakage of intracellular potassium from leukemic cells can have a major effect on the extracellular potassium level. Mechanical stressors in the form of pneumatic tube transport and specimen sampling into vacuum tubes have been implicated as causes of this artifact.5,6 Another possible mechanism is heparin-induced lysis of leukocytes in the setting of hematologic malignancy during laboratory processing.4,7,8
LESSONS LEARNED
In patients with hematologic proliferative disorders who develop hyperkalemia in the absence of electrocardiographic changes and an obvious cause of increased potassium levels (eg, acute renal failure, tumor lysis syndrome), we should entertain the possibility of hemolysis, laboratory error, pseudohyperkalemia, and reverse pseudohyperkalemia. The potassium level should be remeasured to rule out laboratory error and hemolysis. In patients with marked leukocytosis or thrombocytosis, simultaneous measurement of serum and plasma potassium levels helps diagnose pseudohyperkalemia and reverse pseudohyperkalemia. Also, prompt blood gas analysis can help identify spurious hyperkalemia.
A 64-year-old man with chronic lymphocytic leukemia (CLL), Rai stage IV, was admitted to the hospital to undergo his first cycle of chemotherapy with fludarabine, cyclophosphamide, and rituximab. The physical examination at this time was normal except for splenomegaly and painless bilateral inguinal lymphadenopathy. His laboratory results on admission are shown in Table 1.
On the first day after chemotherapy, laboratory testing revealed an elevated plasma potassium level of 7.7 mmol/L. The specimen was drawn into a BD Vacutainer plasma separator tube with lithium-heparin additive (Becton, Dickinson, and Company, Franklin Lake, NJ) and analyzed on a Unicel DXC 800 chemistry analyzer (Beckman Coulter, Inc, Brea, CA).
1. Which electrocardiographic feature is not associated with hyperkalemia?
- Peaked P waves
- Prolonged PR interval
- Shortened QT interval
- Widened QRS
- Asystole
EVALUATING CARDIAC TOXICITY FROM HYPERKALEMIA
Hyperkalemia is not associated with peaked P waves, but rather with a reduction in the size of the P waves.
Hyperkalemia, defined as a plasma potassium concentration above 5.5 mmol/L, occurs as a result either of a release or a shift of intracellular potassium into the intravascular space or of decreased renal excretion. The earliest changes noted on electrocardiography are peaking and narrowing of T waves, followed by shortening of the QT interval. As hyperkalemia progresses, electrocardiography may show bradycardia, absent P waves, and PR interval prolongation, including second- or third-degree atrioventricular block.
At a plasma potassium concentration greater than 7 mmol/L, there may be a junctional escape rhythm, a sine wave pattern with widening of the QRS complex merging with T waves, ventricular fibrillation, or asystole. However, even with high potassium levels, electrocardiographic changes may be absent.1
The patient denied fatigue, muscle weakness, or palpitations. Electrocardiography did not show peaked T waves, shortened QT intervals, decreased P waves, prolonged PR interval, or widening of the QRS interval.
2. Which is an appropriate intervention for hyperkalemia with cardiac toxicity?
- Membrane stabilization with calcium gluconate
- Shifting potassium into the cells with a beta-adrenergic agonist given by nebulization
- Shifting potassium into the cells with insulin and dextrose
- Removal of potassium with sodium polystyrene sulfonate (Kayexalate)
- Removal of potassium with dialysis
In the setting of cardiac toxicity, the management of hyperkalemia involves stabilizing the cardiac muscle membrane, shifting potassium into the cells, and removing potassium from the body. It is important to do all three interventions when cardiac toxicity is present to provide sustained therapeutic benefit.2
MANAGING HYPERKALEMIA
General principles
When potassium levels are above 6 mmol/L, electrocardiography should always be done. Immediately stop potassium supplementation and any drugs that can cause hyperkalemia, such as potassium-sparing diuretics, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, nonsteroidal anti-inflammatory drugs, and trimethoprim-sulfamethoxazole (Bactrim). If the potassium level is greater than 6.5 mmol/L, the patient should be placed on telemetric monitoring, and the potassium level should be measured often.
Hyperkalemia with cardiac toxicity
Membrane stabilization involves intravenous infusion of 10 mL of 10% calcium gluconate. The onset of action is 1 to 3 minutes, and the duration of action is 30 to 60 minutes.
Shifting of potassium is done either with insulin or with a beta-adrenergic agonist nebulizer. With insulin, 10 U of regular insulin is given intravenously along with 50 mL of 50% dextrose; the onset of action is 20 minutes, and the duration is 4 to 6 hours. The dose of beta-adrenergic agonist depends on the type used; the onset of action is 20 minutes, with a duration of 2 to 4 hours.
Removal of potassium is achieved either by drug therapy or by dialysis. Sodium or calcium polystyrene sulfonate is given by mouth, 15 g every 6 hours, or 30 to 60 g by retention enema; the onset of action is 1 to 2 hours, with a duration of 4 to 6 hours. If dialysis is used, 2 to 3 hours is recommended; the onset of action is immediate and lasts for the duration of the dialysis session.
CASE CONTINUED
The patient was moved to a telemetry unit. A repeat plasma potassium measurement after 30 minutes confirmed hyperkalemia (Table 1). The specimen was transported to the laboratory via a pneumatic tube system, centrifuged at 3,300 rpm for 10 minutes, and analyzed on a Beckman Unicel DXC 800 chemistry analyzer. The time from specimen collection to analysis was approximately 60 minutes.
He was treated with oral sodium polystyrene sulfonate because no electrocardiographic changes were observed. Subsequent plasma potassium levels drawn, collected, and analyzed with the same technique described above were persistently high. Repeated electrocardiography continued to show no changes related to hyperkalemia.
DIAGNOSING THE CAUSE OF THE APPARENT HYPERKALEMIA
3. Which is the most likely cause of hyperkalemia in this patient?
- Acute renal failure
- Tumor lysis syndrome
- Hemolysis
- Reverse pseudohyperkalemia
- Pseudohyperkalemia
DIFFERENTIAL DIAGNOSIS
The patient had normal levels of blood urea nitrogen and creatinine and adequate urine output, thus ruling out acute renal failure. Hyperuricemia, hyperphosphatemia, and hypocalcemia were not found, thus ruling out tumor lysis syndrome.
In vitro hemolysis is assessed by visual inspection showing a pink or red hue to serum or plasma, or by hemolysis index calculation using spectrophotometric measurements. Factors associated with in vitro hemolysis include vein fragility, the phlebotomist’s skill and technique, and transportation of the specimen, including duration, mode, and temperature. The plasma potassium level was repeatedly measured from a lithium-heparin tube, thus minimizing the possibility of laboratory error. No evidence of hemolysis was observed during the phlebotomy, transportation, or specimen analysis.
Serum and plasma potassium levels were simultaneously measured to test for pseudohyperkalemia, a falsely elevated serum potassium concentration caused by the release of platelet potassium during clot formation or after venipuncture. Contributing factors include the prolonged use of a tourniquet, hemolysis, and marked leukocytosis or thrombocytosis. A serum-to-plasma potassium gradient greater than 0.4 mmol/L is diagnostic of pseudohyperkalemia.
Reverse pseudohyperkalemia, a falsely high potassium level in plasma samples, is defined as a serum-to-plasma potassium gradient less than 0.4 mmol/L (Table 2). This was the most likely cause of the hyperkalemia in our patient.
On the second day after chemotherapy, two blood samples were collected simultaneously—one into a lithium heparin BD Vacutainer plasma separator tube, and the other into a plain, red-top BD Vacutainer serum collection tube without heparin. The specimens were transported to the laboratory by pneumatic tube and were centrifuged at 3,300 rpm for 10 minutes. The specimens were analyzed simultaneously 20 minutes after collection on a Unicel DXC 800 chemistry analyzer. The analysis revealed a serum-to-plasma potassium gradient of −7.1 mmol/L (serum potassium 3.6 mmol/L and plasma potassium 10.7 mmol/L).3 Repeated potassium measurements drawn in similar fashion after 1 hour continued to show a markedly elevated potassium concentration compared with the serum concentration (Table 1).
Serum and plasma samples were again measured simultaneously 24 hours after detecting hyperkalemia to evaluate for pseudohyperkalemia in this patient. In another published report, serum and plasma measurements were obtained 1 week after observing hyperkalemia.4 Blood gas analysis was done the same day in two other reported cases.4,5
Of note, further review of our patient’s medical history noted hyperkalemia at the time he was diagnosed with CLL. At that time, his plasma potassium level was 7.5 mmol/L, a repeated plasma potassium level was 6.9 mmol/L, and a subsequent blood gas analysis—done 30 minutes after the repeated plasma potassium measurement using a Rapidlab analyzer (Siemens Healthcare Diagnostics, Washington, DC)—showed a potassium level of 3.4 mmol/L. The phenomenon of reverse pseudohyperkalemia was not recognized at that time.
The true mechanism of reverse pseudohyperkalemia has not yet been established. Even minor leakage of intracellular potassium from leukemic cells can have a major effect on the extracellular potassium level. Mechanical stressors in the form of pneumatic tube transport and specimen sampling into vacuum tubes have been implicated as causes of this artifact.5,6 Another possible mechanism is heparin-induced lysis of leukocytes in the setting of hematologic malignancy during laboratory processing.4,7,8
LESSONS LEARNED
In patients with hematologic proliferative disorders who develop hyperkalemia in the absence of electrocardiographic changes and an obvious cause of increased potassium levels (eg, acute renal failure, tumor lysis syndrome), we should entertain the possibility of hemolysis, laboratory error, pseudohyperkalemia, and reverse pseudohyperkalemia. The potassium level should be remeasured to rule out laboratory error and hemolysis. In patients with marked leukocytosis or thrombocytosis, simultaneous measurement of serum and plasma potassium levels helps diagnose pseudohyperkalemia and reverse pseudohyperkalemia. Also, prompt blood gas analysis can help identify spurious hyperkalemia.
- Mirvis DM, Goldberger AL. Electrocardiography. In:Bonow RO, Mann DL, Zipes DP, Libby P, editors. Braunwald's Heart Disease—A Textbook of Cardiovascular Medicine. 9th ed. Boston, MA: Elsevier Saunders; 2011:126–167.
- Rastergar A, Soleimani M. Hypokalaemia and hyperkalaemia. Postgrad Med J 2001; 77:759–764.
- Sevastos N, Theodossiades G, Archimandritis AJ. Pseudohyperkalemia in serum: a new insight into an old phenomenon. Clin Med Res 2008; 6:30–32.
- Meng QH, Krahn J. Reverse pseudohyperkalemia in heparin plasma samples from a patient with chronic lymphocytic leukemia. Clin Biochem 2011; 44:728–730.
- Garwicz D, Karlman M, Øra I. Reverse pseudohyperkalemia in heparin plasma samples from a child with T cell acute lymphoblastic leukemia with hyperleukocytosis [Letter]. Clin Chim Acta 2011; 412:396–397.
- Kellerman PS, Thornbery JM. Pseudohyperkalemia due to pneumatic tube transport in a leukemic patient. Am J Kidney Dis 2005; 46:746–748.
- Abraham B, Fakhar I, Tikaria A, et al. Reverse pseudohyperkalemia in a leukemic patient. Clin Chem 2008; 54:449–551.
- Singh PJ, Zawada ET, Santella RN. A case of ‘reverse’ pseudohyperkalemia. Miner Electrolyte Metab 1997; 23:58–61.
- Mirvis DM, Goldberger AL. Electrocardiography. In:Bonow RO, Mann DL, Zipes DP, Libby P, editors. Braunwald's Heart Disease—A Textbook of Cardiovascular Medicine. 9th ed. Boston, MA: Elsevier Saunders; 2011:126–167.
- Rastergar A, Soleimani M. Hypokalaemia and hyperkalaemia. Postgrad Med J 2001; 77:759–764.
- Sevastos N, Theodossiades G, Archimandritis AJ. Pseudohyperkalemia in serum: a new insight into an old phenomenon. Clin Med Res 2008; 6:30–32.
- Meng QH, Krahn J. Reverse pseudohyperkalemia in heparin plasma samples from a patient with chronic lymphocytic leukemia. Clin Biochem 2011; 44:728–730.
- Garwicz D, Karlman M, Øra I. Reverse pseudohyperkalemia in heparin plasma samples from a child with T cell acute lymphoblastic leukemia with hyperleukocytosis [Letter]. Clin Chim Acta 2011; 412:396–397.
- Kellerman PS, Thornbery JM. Pseudohyperkalemia due to pneumatic tube transport in a leukemic patient. Am J Kidney Dis 2005; 46:746–748.
- Abraham B, Fakhar I, Tikaria A, et al. Reverse pseudohyperkalemia in a leukemic patient. Clin Chem 2008; 54:449–551.
- Singh PJ, Zawada ET, Santella RN. A case of ‘reverse’ pseudohyperkalemia. Miner Electrolyte Metab 1997; 23:58–61.
Fever, dyspnea, and hepatitis in an Iraq veteran
A healthy 42-year-old US Army reservist returned home to Oregon in early April after a 12-month deployment in Iraq. About 6 weeks later, he developed a mild nonproductive cough; then, over the next 2 weeks, his symptoms progressed to myalgia, mild headache, fever, chills, drenching night sweats, and dyspnea on exertion.
About 2 weeks after the onset of his symptoms, he saw his primary care provider. The results of laboratory tests at that time were normal except for the following:
- Platelet count 110 × 109/L (reference range 150–400)
- Alkaline phosphatase 354 IU/L (40–100)
- Alanine aminotransferase 99 IU/L (5–36)
- Aspartate aminotransferase 220 IU/L (7–33).
Chest radiography was negative. He was told he had a viral infection and was sent home with no treatment.
1. Which of the following is the most likely diagnosis in this patient?
- Influenza
- Ehrlichiosis
- Q fever
- Visceral leishmaniasis
- Malaria
Military operations in Iraq and Afghanistan have involved large numbers of US Army Reserve and National Guard personnel: by 2007, more than 500,000 Reserve and National Guard personnel had served in these combat operations.1 Although these personnel are generally healthy and receive mandatory travel screenings, prophylactic drug treatment, and vaccinations, their close, long-term exposure to local populations and environments puts them at risk of many infections.2
Often, these veterans develop symptoms after returning home, and they seek medical care from providers outside the military medical system.3,4 Civilian health care providers are thus increasingly called on to recognize clinical syndromes associated with military operations.
FEVER IN RETURNED SOLDIERS
The presentation of this 42-year-old veteran has an extensive differential diagnosis. His symptoms arose more than a month after his return from Iraq, meaning he could have acquired an infection in Iraq, on his trip home, or even after arriving home.
A number of common viral and atypical respiratory pathogens could be involved, and although circulating influenza was not common at the time of year he happened to return (spring), it remains a possibility. However, the duration of his illness, with symptoms that gradually worsened over 12 days, argues against influenza and community-acquired respiratory and other viral illnesses.
Aronson et al5 have reviewed the infectious risks in deployed military personnel.5 Infectious syndromes that have manifested in military personnel a month or more after returning from Iraq or Afghanistan include malaria, Q fever, brucellosis, typhoid fever, and leishmaniasis.5
Malaria
Malaria should be considered in all travelers from endemic areas presenting with fever, especially if they have thrombocytopenia and anemia. Plasmodium vivax is present in Iraq, but transmission is rare and isolated. Defense Medical Surveillance System data show that most of the recent malaria cases in US military personnel were acquired in Afghanistan or Korea. Many of these cases were caused by P vivax and manifested weeks to months after exposure, and diagnosis was significantly delayed because the provider did not consider malaria in the differential diagnosis.4,6,7
Testing for malaria with serial thick and thin blood smears and the BinaxNOW (Iverness Medical, Princeton, NJ) rapid test, when available, should be done in all those who have served in malaria-endemic regions and who present with unexplained fever or consistent symptoms. Testing should be done even if prophylaxis was taken or the potential exposure was weeks to months before presentation.
Brucellosis
Brucellosis, a zoonosis typically acquired by ingesting unpasteurized dairy products, has a high prevalence in Eurasia. A nonspecific, multisystem illness with fever, hepatitis, and arthritis (classically sacroiliitis) is commonly described.
Brucellosis is less likely in our patient, given that he denied consumption of local dairy products while deployed. Also, he had prominent respiratory symptoms, which would not be typical of brucellosis.
Leishmaniasis
Leishmaniasis, a parasitic disease transmitted by sand flies, manifests in one of three ways, ie, as a cutaneous, a mucosal, or a visceral disease. Most infections recently reported in US military personnel have been cutaneous and were acquired in Iraq, where Leishmania major is the primary species.8 Visceral disease mimics lymphoma (fever, hepatosplenomegaly, and cytopenia), but only a handful of cases have been reported from Iraq and Afghanistan.9 The incubation period of visceral leishmaniasis is prolonged, and civilian providers should consider it even if the patient’s period of deployment was relatively long ago.
Q fever in military personnel
Q fever is caused by the intracellular bacterium Coxiella burnetii.
Q fever has been reported in more than 150 US military personnel deployed to Iraq and Afghanistan.10–12 However, it may be more common than that. In one report, 10% of patients admitted to a combat support hospital in Iraq with International Classification of Diseases, Ninth Revision codes potentially consistent with Q fever tested positive for it.13 And in several cases that manifested after deployment, Q fever was not considered initially by the health care provider.11,14 In response, the US Centers for Disease Control and Prevention (CDC) released a health advisory in May 2010 alerting providers about Q fever in travelers returning from Iraq and the Netherlands.15
Q fever is a zoonosis associated with a wide range of animal reservoirs, primarily agricultural livestock such as cattle, goats, and sheep, but also a variety of other animals. There are multiple routes of transmission, including direct animal contact, ingestion of unpasteurized dairy products, and, most commonly, inhalation of aerosolized particles contaminated by animal droppings or secretions.16 Tick-borne and sexual transmission have been reported in rare instances.17,18 Importantly, in many cases from Iraq and from an outbreak in the Netherlands there was no obvious exposure.19
Q fever is a potential agent of bioterrorism; therefore, a large-scale, single-point outbreak should raise concern about a possible intentional release of the organism.20
Q fever has myriad presentations
About 60% of cases of Q fever infection are asymptomatic.21 In the United States, the estimated seroprevalence is 3%. Such a high seroprevalence, despite the relatively small number of reported cases, suggests that this infection is often subclinical.22

After 2 to 3 weeks of incubation, Q fever infection can produce a wide range of presentations involving almost any organ system (Table 1).16 An influenza-like illness with fever, pneumonia, and hepatitis is classic. Often, headache is severe enough to warrant lumbar puncture. Atypical and often severe presentations include gastrointestinal or neurologic manifestations.23–25 Rates of hospitalization and in-hospital death are low in acute disease: hospitalization occurs in roughly 2% of cases, and death in about 1% of those hospitalized.26,27
The presentation may mimic that of conditions caused by common community pathogens such as Legionella, Rickettsia, cytomegalovirus, Ebola virus, influenza, Mycoplasma, and human immunodeficiency virus (primary infection). Heightened suspicion is needed to prevent delays in diagnosis and treatment.
This patient’s symptoms and his recent deployment made Q fever very likely.
CASE CONTINUED
The patient continued to feel sick and reported having three to four loose bowel movements per day and mild abdominal pain. His cough and dyspnea persisted.
He had not had contact with anyone who was ill, denied being exposed to animals or insects, and had not consumed unpasteurized dairy products; he recalled having cleaned his military-issue uniforms and equipment 2 to 3 weeks before the symptoms began. He called a military physician to get advice on what else could be causing his symptoms. This physician recommended tests based on potential exposures in Iraq. Tests for brucellosis, visceral leishmaniasis, and Q fever were ordered.
Over the next several days, he began to feel better, and at 3 to 4 weeks after the onset of his symptoms, he felt that he had returned to normal. All tests were negative.
TESTING FOR ATYPICAL PATHOGENS
2. Which of the following would be the most readily available method to confirm the diagnosis?
- Culture
- Polymerase chain reaction (PCR) testing
- Histopathologic testing
- Serologic testing
Testing for atypical pathogens was reasonable in this patient. In addition to an evaluation for parasitic causes of persistent and chronic diarrhea, an evaluation for Q fever, brucellosis, and visceral leishmaniasis via serologic testing was warranted. A variety of tests exist for all of these infections, but serologic tests are the most readily available.
Leishmaniasis testing
Visceral leishmaniasis was traditionally diagnosed by visualizing organisms in splenic or bone marrow aspirates,28 but now serologic tests are available through commercial and public health laboratories such as the CDC. Immunochromatographic tests using recombinant k39 antigen are highly sensitive and specific and have been used in military cases.29,30
Brucellosis testing
Brucella can be cultured from blood or tissue samples. The laboratory must be alerted, as special media can be used to increase the yield and precautions must be taken to prevent laboratory-acquired infection. Serologic testing is the method most commonly used for diagnosis.31
Q fever testing
Q fever can be diagnosed with serologic testing during its acute and convalescent phases.
PCR testing of blood is useful for diagnosing acute disease and is positive before serologic conversion, thus allowing rapid diagnosis and treatment.32 The Joint Biological Agent Identification and Diagnostic System (JBAIDS) PCR platform was studied in a Combat Support Hospital in Iraq for making the rapid diagnosis of Q fever and has since been approved by the US Food and Drug Administration for military use.33
Culture is beyond the scope of most clinical laboratories and requires specialized cell culture or egg yolk media. In tissue, usually liver tissue obtained in an effort to evaluate hepatitis, the histologic finding of “doughnut” granulomas, or fibrin-encased granulomas, can be suggestive of C burnetii but may be nonspecific and seen with other infections.23,34
Serologic testing with an immunofluorescence assay (IFA) remains the most common method of diagnosis. It is based on the detection of immunoglobulin G (IgG) and IgM responses against phase I and phase II antigens of C burnetii. After initial infection, the organism displays phase I antigens and is highly infectious. When grown in culture, the organism undergoes phase shifting to a less infectious form with predominantly phase II antigens. Paradoxically, after initial infection in humans, antibody response against phase II antigens is seen first, whereas in chronic infection, a phase I antibody response dominates.26,35 Phase II antibodies appear around week 2, and 90% of samples from infected people are positive by week 3. A fourfold rise in titer between the acute-phase and convalescent-phase samples confirms the diagnosis.35
A number of serologic assays are available worldwide, but they have different methods and cutoff values, so questions have arisen about the equivalence of the results.14,36 Serologic cutoffs have been defined in Europe, where most cases of Q fever have been reported.37
CHRONIC Q FEVER
3. Which of the following would be the most likely chronic manifestation of Q fever?
- Pneumonia
- Hepatitis
- Endocarditis
- Chronic fatigue
- Osteomyelitis
Chronic syndromes can develop years to months after untreated or inadequately treated infection and can be serious. Chronic infection can also result after a clinically silent initial infection.26,38 Culture-negative endocarditis, which occurs in fewer than 1% of patients diagnosed with acute infection, is the most common chronic manifestation (Table 1). Patients with underlying valvular disease, malignancy, or immunosuppression are at greater risk.38–40
Challenges and controversies
The diagnosis of chronic Q fever remains challenging. Traditionally, elevated phase I IgG titers were considered highly predictive of chronic disease. A cutoff of 1:800 was set, based on retrospective data from chronic cases in Europe, but its generalizability to different assays and patient populations has been unclear. 14,36,37 Recent reviews and prospective analysis with serial serologic studies in the Dutch outbreak and other sources suggest the positive predictive value (PPV) of phase I IgG titers greater than 1:800 to be lower than previously estimated, largely due to widespread testing and resultant increased seroprevalence assessments. It has been suggested the cutoff be raised to 1:1,600, which still only carries a 59% positive predictive value.41,42
Chronic fatigue due to Q fever remains a controversial topic and has only been described in Europe, Asia, and Australia. A direct link has yet to be established.43 Additional research is needed, but small studies of prolonged antibiotic treatment have not shown benefit in these cases.44
CASE CONTINUED

This patient did well. During a routine physical while enrolled at the Army War College in Carlisle, PA, 6 months after the original presentation, he mentioned his illness to the physician, who then repeated testing for Q fever; the test was positive (Table 2). Subsequently, serum samples from before and after his deployment were tested along with another convalescent-phase sample, and the results demonstrated Q fever seroconversion. He was well and had no physical complaints. He had no heart murmur, and a complete blood count and tests of liver enzymes and inflammatory markers were normal.
TREATMENT AND PREVENTION OF Q FEVER
4. Which of the following treatments would be appropriate, given his diagnosis of Q fever?
- Doxycycline (Vibramycin) 100 mg twice daily for 14 days
- Levofloxacin (Levaquin) 500 mg daily for 5 days
- Doxycycline 100 mg twice daily for 14 days and hydroxychloroquine (Plaquenil) 200 mg three times per day for 18 months
- No treatment
The treatment goals in Q fever are to hasten the resolution of symptoms and to prevent chronic disease. Generally, if there are no clinical findings or symptoms, treatment is not indicated. If the patient has symptoms, early treatment is preferred, but a response may be seen even when there is a delay in diagnosis.
Doxycycline 100 mg twice a day for 14 days is the treatment of choice. In addition, quinolones have in vitro activity,45 and a recent study suggests moxifloxacin (Avelox) may be the preferred antibiotic for those who cannot tolerate doxycycline.46 In pregnant women and in children, macrolides and trimethoprim-sulfamethoxazole (Bactrim) are preferred.47,48
Treatment of chronic Q fever, in particular endocarditis, warrants more intensive therapy. A retrospective review of treated cases of endocarditis suggested that monotherapy with doxycycline often failed, and combination therapy with hydroxychloroquine has been advocated based on in vivo and in vitro experience.49
PREVENTING LONG-TERM SEQUELAE OF CHRONIC Q FEVER
5. At this point, what is the next step in the management of this patient?
- No further follow-up is indicated
- Transthoracic echocardiography (TTE)
- Repeat Q fever serologic testing in 3 to 6 months
- Whole-blood PCR testing and transesophageal echocardiography (TEE)
Long-term follow-up of patients with Q fever has been advocated to monitor for the development of chronic Q fever, but recent studies question the previously devised algorithms.50
The data the algorithms were based on suggested that preexisting valvular heart disease could be associated with up to a 39% risk of endocarditis, and a two-step approach was devised to prevent and identify early chronic infection.51,52 Patients with Q fever would undergo TTE at baseline, and if the findings were abnormal (including mild regurgitation), then 12 months of prophylactic treatment with hydroxychloroquine and doxycycline was recommended. If TTE was normal, serial serologic testing every 3 months was recommended. If the anti-phase I IgG titer was greater than 1:800 at any point, TEE and a whole-blood PCR assay were recommended to evaluate for endocarditis.51
These recommendations were based on data from the French National Reference Center and had not been prospectively evaluated. The 2007–2008 Dutch outbreak provided a large cohort of Q fever cases. After initial screening with TTE and serologic follow-up, 59% of patients were noted to have mild valvular abnormalities, and many had phase I IgG levels greater than 1:800 during follow-up despite being clinically free of disease. The Dutch subsequently stopped screening with TTE as part of routine follow-up and elected to follow patients clinically.
Similar findings have been noted from case follow-up in France and Taiwan, also supporting using serologic cutoffs alone in determining the need for evaluation (with TEE) or treatment of chronic disease.53,54 The usefulness of serologic testing every 3 months has also been questioned, and some have advocated extending the interval, especially since less emphasis is being placed on the results in favor of more practical clinical follow-up.39
One such clinical approach at follow-up is presented in Figure 1. TTE should be reserved for patients with known valvular disease or a clear murmur. Those with underlying valvular disease and acute Q fever should be managed on an individual basis by a specialist in infectious disease, and antibiotic prophylaxis should be considered. Patients without underlying disease should have regular follow-up examinations and serologic testing every 6 months, and clinical symptoms should guide further testing (eg, with TEE and PCR testing) for chronic disease.
In this patient, phase I and II antibody titers were notably elevated (in TABLE 2, phase I titers > 1:800 and 1:1600 cutoffs). Such high titers have been common in military cases from Iraq and Afghanistan, and to date no cases of endocarditis have been diagnosed despite close follow-up. Most cases in military personnel are in relatively young patients who lack risk factors for endocarditis. Based on emerging data from large overseas outbreaks and the potential toxicity of intensive preemptive dual-antimicrobial therapy, an approach of close follow-up was taken.
PRIMARY PREVENTION OF Q FEVER
Prevention of Q fever remains a challenge, as the organism is highly persistent in the environment. An effective licensed vaccine exists in Australia under the brand name Q-Vax, but no approved vaccine is currently available in the United States.55
THE PATIENT’S COURSE
The patient returned for follow-up about 1 year after his first presentation. He noted some ongoing fatigue but attributed this to his course work, and he said he otherwise felt well. He exercises regularly, with no shortness of breath, fevers, chills, or weight loss. He continued to have elevated Q fever titers. Because he had no symptoms, no heart murmur, and normal inflammatory markers, he had no further workup and continued to be followed with serial serologic testing and examinations.
- Defense Science Board Task Force on Deployment of Members of the National Guard and Reserve in the Global War on Terrorism. Washington, DC. September 2007.
- Sanders JW, Putnam SD, Frankart C, et al. Impact of illness and non-combat injury during Operations Iraqi Freedom and Enduring Freedom (Afghanistan). Am J Trop Med Hyg 2005; 73:713–719.
- Gleeson TD, Decker CF, Johnson MD, Hartzell JD, Mascola JR. Q fever in US military returning from Iraq. Am J Med 2007; 120:e11–e12.
- Hagan JE, Marcos LA, Steinberg TH. Fever in a soldier returned from Afghanistan. J Travel Med 2010; 17:351–352.
- Aronson NE, Sanders JW, Moran KA. In harm’s way: infections in deployed American military forces. Clin Infect Dis 2006; 43:1045–1051.
- Klein TA, Pacha LA, Lee HC, et al. Plasmodium vivax malaria among U.S. forces Korea in the Republic of Korea, 1993–2007. Mil Med 2009; 174:412–418.
- Ciminera P, Brundage J. Malaria in U.S. military forces: a description of deployment exposures from 2003 through 2005. Am J Trop Med Hyg 2007; 76:275–279.
- Lesho EP, Wortmann G, Neafie R, Aronson N. Nonhealing skin lesions in a sailor and a journalist returning from Iraq. Cleve Clin J Med 2005; 72:93–96,
- Myles O, Wortmann GW, Cummings JF, et al. Visceral leishmaniasis: clinical observations in 4 US army soldiers deployed to Afghanistan or Iraq, 2002–2004. Arch Intern Med 2007; 167:1899–1901.
- Faix DJ, Harrison DJ, Riddle MS, et al. Outbreak of Q fever among US military in western Iraq, June–July 2005. Clin Infect Dis 2008; 46:e65–e68.
- Leung-Shea C, Danaher PJ. Q fever in members of the United States armed forces returning from Iraq. Clin Infect Dis 2006; 43:e77–e82.
- Anderson AD, Smoak B, Shuping E, Ockenhouse C, Petruccelli B. Q fever and the US military. Emerg Infect Dis 2005; 11:1320–1322.
- Anderson AD, Baker TR, Littrell AC, Mott RL, Niebuhr DW, Smoak BL. Seroepidemiologic survey for Coxiella burnetii among hospitalized US troops deployed to Iraq. Zoonoses Public Health 2011; 58:276–283.
- Ake JA, Massung RF, Whitman TJ, Gleeson TD. Difficulties in the diagnosis and management of a US servicemember presenting with possible chronic Q fever. J Infect 2010; 60:175–177.
- Centers for Disease Control and Prevention (CDC). Potential for Q fever infection among travelers returning from Iraq and the Netherlands http://www.bt.cdc.gov/HAN/han00313.asp. Accessed July 5, 2012.
- Parker NR, Barralet JH, Bell AM. Q fever. Lancet 2006; 367:679–688.
- Miceli MH, Veryser AK, Anderson AD, Hofinger D, Lee SA, Tancik C. A case of person-to-person transmission of Q fever from an active duty serviceman to his spouse. Vector Borne Zoonotic Dis 2010; 10:539–541.
- Milazzo A, Hall R, Storm PA, Harris RJ, Winslow W, Marmion BP. Sexually transmitted Q fever. Clin Infect Dis 2001; 33:399–402.
- Hartzell JD, Peng SW, Wood-Morris RN, et al. Atypical Q fever in US soldiers. Emerg Infect Dis 2007; 13:1247–1249.
- Bossi P, Tegnell A, Baka A, et al; Task Force on Biological and Chemical Agent Threats, Public Health Directorate, European Commission, Luxembourg. Bichat guidelines for the clinical management of Q fever and bioterrorism-related Q fever. Euro Surveill 2004; 9:E19–E20.
- Roest HI, Tilburg JJ, van der Hoek W, et al. The Q fever epidemic in The Netherlands: history, onset, response and reflection. Epidemiol Infect 2011; 139:1–12.
- Anderson AD, Kruszon-Moran D, Loftis AD, et al. Seroprevalence of Q fever in the United States, 2003–2004. Am J Trop Med Hyg 2009; 81:691–694.
- Hatchette TF, Marrie TJ. Atypical manifestations of chronic Q fever. Clin Infect Dis 2001; 33:1347–1351.
- Bernit E, Pouget J, Janbon F, et al. Neurological involvement in acute Q fever: a report of 29 cases and review of the literature. Arch Intern Med 2002; 162:693–700.
- Kofteridis DP, Mazokopakis EE, Tselentis Y, Gikas A. Neurological complications of acute Q fever infection. Eur J Epidemiol 2004; 19:1051–1054.
- Raoult D, Marrie T, Mege J. Natural history and pathophysiology of Q fever. Lancet Infect Dis 2005; 5:219–226.
- Kampschreur LM, Wegdam-Blans MC, Thijsen SF, et al. Acute Q fever related in-hospital mortality in the Netherlands. Neth J Med 2010; 68:408–413.
- Srivastava P, Dayama A, Mehrotra S, Sundar S. Diagnosis of visceral leishmaniasis. Trans R Soc Trop Med Hyg 2011; 105:1–6.
- Chappuis F, Rijal S, Soto A, Menten J, Boelaert M. A meta-analysis of the diagnostic performance of the direct agglutination test and rK39 dipstick for visceral leishmaniasis. BMJ 2006; 333:723.
- Hartzell JD, Aronson NE, Weina PJ, Howard RS, Yadava A, Wortmann GW. Positive rK39 serologic assay results in US servicemen with cutaneous leishmaniasis. Am J Trop Med Hyg 2008; 79:843–846.
- Pappas G, Akritidis N, Bosilkovski M, Tsianos E. Brucellosis. N Engl J Med 2005; 352:2325–2336.
- Schneeberger PM, Hermans MH, van Hannen EJ, Schellekens JJ, Leenders AC, Wever PC. Real-time PCR with serum samples is indispensable for early diagnosis of acute Q fever. Clin Vaccine Immunol 2010; 17:286–290.
- Hamilton LR, George DL, Scoville SL, Hospenthal DR, Griffith ME. PCR for rapid diagnosis of acute Q fever at a combat support hospital in Iraq. Mil Med 2011; 176:103–105.
- Bonilla MF, Kaul DR, Saint S, Isada CM, Brotman DJ. Clinical problem-solving. Ring around the diagnosis. N Engl J Med 2006; 354:1937–1942.
- Fournier PE, Marrie TJ, Raoult D. Diagnosis of Q fever. J Clin Microbiol 1998; 36:1823–1834.
- Healy B, van Woerden H, Raoult D, et al. Chronic Q fever: different serological results in three countries—results of a follow-up study 6 years after a point source outbreak. Clin Infect Dis 2011; 52:1013–1019.
- Dupont HT, Thirion X, Raoult D. Q fever serology: cutoff determination for microimmunofluorescence. Clin Diagn Lab Immunol 1994; 1:189–196.
- Karakousis PC, Trucksis M, Dumler JS. Chronic Q fever in the United States. J Clin Microbiol 2006; 44:2283–2287.
- van der Hoek W, Versteeg B, Meekelenkamp JC, et al. Follow-up of 686 patients with acute Q fever and detection of chronic infection. Clin Infect Dis 2011; 52:1431–1436.
- Fenollar F, Fournier PE, Carrieri MP, Habib G, Messana T, Raoult D. Risks factors and prevention of Q fever endocarditis. Clin Infect Dis 2001; 33:312–316.
- Frankel D, Richet H, Renvoisé A, Raoult D. Q fever in France, 1985–2009. Emerg Infect Dis 2011; 17:350–356.
- Baddour LM, Wilson WR, Bayer AS, et al; Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease; Council on Cardiovascular Disease in the Young; Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia; American Heart Association; Infectious Diseases Society of America. Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications: a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation 2005; 111:e394–e434.
- Wildman MJ, Smith EG, Groves J, Beattie JM, Caul EO, Ayres JG. Chronic fatigue following infection by Coxiella burnetii (Q fever): ten-year follow-up of the 1989 UK outbreak cohort. QJM 2002; 95:527–538.
- Iwakami E, Arashima Y, Kato K, et al. Treatment of chronic fatigue syndrome with antibiotics: pilot study assessing the involvement of Coxiella burnetii infection. Intern Med 2005; 44:1258–1263.
- Rolain JM, Maurin M, Raoult D. Bacteriostatic and bactericidal activities of moxifloxacin against Coxiella burnetii. Antimicrob Agents Chemother 2001; 45:301–302.
- Dijkstra F, Riphagen-Dalhuisen J, Wijers N, et al. Antibiotic therapy for acute Q fever in The Netherlands in 2007 and 2008 and its relation to hospitalization. Epidemiol Infect 2011; 139:1332–1341.
- Raoult D. Use of macrolides for Q fever. Antimicrob Agents Chemother 2003; 47:446.
- Carcopino X, Raoult D, Bretelle F, Boubli L, Stein A. Managing Q fever during pregnancy: the benefits of long-term cotrimoxazole therapy. Clin Infect Dis 2007; 45:548–555.
- Raoult D, Houpikian P, Tissot Dupont H, Riss JM, Arditi-Djiane J, Brouqui P. Treatment of Q fever endocarditis: comparison of 2 regimens containing doxycycline and ofloxacin or hydroxychloroquine. Arch Intern Med 1999; 159:167–173.
- Healy B, Llewelyn M, Westmoreland D, Lloyd G, Brown N. The value of follow-up after acute Q fever infection. J Infect 2006; 52:e109–e112.
- Landais C, Fenollar F, Thuny F, Raoult D. From acute Q fever to endocarditis: serological follow-up strategy. Clin Infect Dis 2007; 44:1337–1340.
- Hartzell JD, Wood-Morris RN, Martinez LJ, Trotta RF. Q fever: epidemiology, diagnosis, and treatment. Mayo Clin Proc 2008; 83:574–579.
- Hung MN, Lin LJ, Hou MY, et al. Serologic assessment of the risk of developing chronic Q fever in cohorts of acutely infected individuals. J Infect 2011; 62:39–44.
- Sunder S, Gras G, Bastides F, De Gialluly C, Choutet P, Bernard L. Chronic Q fever: relevance of serology. Clin Infect Dis 2011; 53:749–750.
- Gefenaite G, Munster JM, van Houdt R, Hak E. Effectiveness of the Q fever vaccine: a meta-analysis. Vaccine 2011; 29:395–398.
A healthy 42-year-old US Army reservist returned home to Oregon in early April after a 12-month deployment in Iraq. About 6 weeks later, he developed a mild nonproductive cough; then, over the next 2 weeks, his symptoms progressed to myalgia, mild headache, fever, chills, drenching night sweats, and dyspnea on exertion.
About 2 weeks after the onset of his symptoms, he saw his primary care provider. The results of laboratory tests at that time were normal except for the following:
- Platelet count 110 × 109/L (reference range 150–400)
- Alkaline phosphatase 354 IU/L (40–100)
- Alanine aminotransferase 99 IU/L (5–36)
- Aspartate aminotransferase 220 IU/L (7–33).
Chest radiography was negative. He was told he had a viral infection and was sent home with no treatment.
1. Which of the following is the most likely diagnosis in this patient?
- Influenza
- Ehrlichiosis
- Q fever
- Visceral leishmaniasis
- Malaria
Military operations in Iraq and Afghanistan have involved large numbers of US Army Reserve and National Guard personnel: by 2007, more than 500,000 Reserve and National Guard personnel had served in these combat operations.1 Although these personnel are generally healthy and receive mandatory travel screenings, prophylactic drug treatment, and vaccinations, their close, long-term exposure to local populations and environments puts them at risk of many infections.2
Often, these veterans develop symptoms after returning home, and they seek medical care from providers outside the military medical system.3,4 Civilian health care providers are thus increasingly called on to recognize clinical syndromes associated with military operations.
FEVER IN RETURNED SOLDIERS
The presentation of this 42-year-old veteran has an extensive differential diagnosis. His symptoms arose more than a month after his return from Iraq, meaning he could have acquired an infection in Iraq, on his trip home, or even after arriving home.
A number of common viral and atypical respiratory pathogens could be involved, and although circulating influenza was not common at the time of year he happened to return (spring), it remains a possibility. However, the duration of his illness, with symptoms that gradually worsened over 12 days, argues against influenza and community-acquired respiratory and other viral illnesses.
Aronson et al5 have reviewed the infectious risks in deployed military personnel.5 Infectious syndromes that have manifested in military personnel a month or more after returning from Iraq or Afghanistan include malaria, Q fever, brucellosis, typhoid fever, and leishmaniasis.5
Malaria
Malaria should be considered in all travelers from endemic areas presenting with fever, especially if they have thrombocytopenia and anemia. Plasmodium vivax is present in Iraq, but transmission is rare and isolated. Defense Medical Surveillance System data show that most of the recent malaria cases in US military personnel were acquired in Afghanistan or Korea. Many of these cases were caused by P vivax and manifested weeks to months after exposure, and diagnosis was significantly delayed because the provider did not consider malaria in the differential diagnosis.4,6,7
Testing for malaria with serial thick and thin blood smears and the BinaxNOW (Iverness Medical, Princeton, NJ) rapid test, when available, should be done in all those who have served in malaria-endemic regions and who present with unexplained fever or consistent symptoms. Testing should be done even if prophylaxis was taken or the potential exposure was weeks to months before presentation.
Brucellosis
Brucellosis, a zoonosis typically acquired by ingesting unpasteurized dairy products, has a high prevalence in Eurasia. A nonspecific, multisystem illness with fever, hepatitis, and arthritis (classically sacroiliitis) is commonly described.
Brucellosis is less likely in our patient, given that he denied consumption of local dairy products while deployed. Also, he had prominent respiratory symptoms, which would not be typical of brucellosis.
Leishmaniasis
Leishmaniasis, a parasitic disease transmitted by sand flies, manifests in one of three ways, ie, as a cutaneous, a mucosal, or a visceral disease. Most infections recently reported in US military personnel have been cutaneous and were acquired in Iraq, where Leishmania major is the primary species.8 Visceral disease mimics lymphoma (fever, hepatosplenomegaly, and cytopenia), but only a handful of cases have been reported from Iraq and Afghanistan.9 The incubation period of visceral leishmaniasis is prolonged, and civilian providers should consider it even if the patient’s period of deployment was relatively long ago.
Q fever in military personnel
Q fever is caused by the intracellular bacterium Coxiella burnetii.
Q fever has been reported in more than 150 US military personnel deployed to Iraq and Afghanistan.10–12 However, it may be more common than that. In one report, 10% of patients admitted to a combat support hospital in Iraq with International Classification of Diseases, Ninth Revision codes potentially consistent with Q fever tested positive for it.13 And in several cases that manifested after deployment, Q fever was not considered initially by the health care provider.11,14 In response, the US Centers for Disease Control and Prevention (CDC) released a health advisory in May 2010 alerting providers about Q fever in travelers returning from Iraq and the Netherlands.15
Q fever is a zoonosis associated with a wide range of animal reservoirs, primarily agricultural livestock such as cattle, goats, and sheep, but also a variety of other animals. There are multiple routes of transmission, including direct animal contact, ingestion of unpasteurized dairy products, and, most commonly, inhalation of aerosolized particles contaminated by animal droppings or secretions.16 Tick-borne and sexual transmission have been reported in rare instances.17,18 Importantly, in many cases from Iraq and from an outbreak in the Netherlands there was no obvious exposure.19
Q fever is a potential agent of bioterrorism; therefore, a large-scale, single-point outbreak should raise concern about a possible intentional release of the organism.20
Q fever has myriad presentations
About 60% of cases of Q fever infection are asymptomatic.21 In the United States, the estimated seroprevalence is 3%. Such a high seroprevalence, despite the relatively small number of reported cases, suggests that this infection is often subclinical.22
After 2 to 3 weeks of incubation, Q fever infection can produce a wide range of presentations involving almost any organ system (Table 1).16 An influenza-like illness with fever, pneumonia, and hepatitis is classic. Often, headache is severe enough to warrant lumbar puncture. Atypical and often severe presentations include gastrointestinal or neurologic manifestations.23–25 Rates of hospitalization and in-hospital death are low in acute disease: hospitalization occurs in roughly 2% of cases, and death in about 1% of those hospitalized.26,27
The presentation may mimic that of conditions caused by common community pathogens such as Legionella, Rickettsia, cytomegalovirus, Ebola virus, influenza, Mycoplasma, and human immunodeficiency virus (primary infection). Heightened suspicion is needed to prevent delays in diagnosis and treatment.
This patient’s symptoms and his recent deployment made Q fever very likely.
CASE CONTINUED
The patient continued to feel sick and reported having three to four loose bowel movements per day and mild abdominal pain. His cough and dyspnea persisted.
He had not had contact with anyone who was ill, denied being exposed to animals or insects, and had not consumed unpasteurized dairy products; he recalled having cleaned his military-issue uniforms and equipment 2 to 3 weeks before the symptoms began. He called a military physician to get advice on what else could be causing his symptoms. This physician recommended tests based on potential exposures in Iraq. Tests for brucellosis, visceral leishmaniasis, and Q fever were ordered.
Over the next several days, he began to feel better, and at 3 to 4 weeks after the onset of his symptoms, he felt that he had returned to normal. All tests were negative.
TESTING FOR ATYPICAL PATHOGENS
2. Which of the following would be the most readily available method to confirm the diagnosis?
- Culture
- Polymerase chain reaction (PCR) testing
- Histopathologic testing
- Serologic testing
Testing for atypical pathogens was reasonable in this patient. In addition to an evaluation for parasitic causes of persistent and chronic diarrhea, an evaluation for Q fever, brucellosis, and visceral leishmaniasis via serologic testing was warranted. A variety of tests exist for all of these infections, but serologic tests are the most readily available.
Leishmaniasis testing
Visceral leishmaniasis was traditionally diagnosed by visualizing organisms in splenic or bone marrow aspirates,28 but now serologic tests are available through commercial and public health laboratories such as the CDC. Immunochromatographic tests using recombinant k39 antigen are highly sensitive and specific and have been used in military cases.29,30
Brucellosis testing
Brucella can be cultured from blood or tissue samples. The laboratory must be alerted, as special media can be used to increase the yield and precautions must be taken to prevent laboratory-acquired infection. Serologic testing is the method most commonly used for diagnosis.31
Q fever testing
Q fever can be diagnosed with serologic testing during its acute and convalescent phases.
PCR testing of blood is useful for diagnosing acute disease and is positive before serologic conversion, thus allowing rapid diagnosis and treatment.32 The Joint Biological Agent Identification and Diagnostic System (JBAIDS) PCR platform was studied in a Combat Support Hospital in Iraq for making the rapid diagnosis of Q fever and has since been approved by the US Food and Drug Administration for military use.33
Culture is beyond the scope of most clinical laboratories and requires specialized cell culture or egg yolk media. In tissue, usually liver tissue obtained in an effort to evaluate hepatitis, the histologic finding of “doughnut” granulomas, or fibrin-encased granulomas, can be suggestive of C burnetii but may be nonspecific and seen with other infections.23,34
Serologic testing with an immunofluorescence assay (IFA) remains the most common method of diagnosis. It is based on the detection of immunoglobulin G (IgG) and IgM responses against phase I and phase II antigens of C burnetii. After initial infection, the organism displays phase I antigens and is highly infectious. When grown in culture, the organism undergoes phase shifting to a less infectious form with predominantly phase II antigens. Paradoxically, after initial infection in humans, antibody response against phase II antigens is seen first, whereas in chronic infection, a phase I antibody response dominates.26,35 Phase II antibodies appear around week 2, and 90% of samples from infected people are positive by week 3. A fourfold rise in titer between the acute-phase and convalescent-phase samples confirms the diagnosis.35
A number of serologic assays are available worldwide, but they have different methods and cutoff values, so questions have arisen about the equivalence of the results.14,36 Serologic cutoffs have been defined in Europe, where most cases of Q fever have been reported.37
CHRONIC Q FEVER
3. Which of the following would be the most likely chronic manifestation of Q fever?
- Pneumonia
- Hepatitis
- Endocarditis
- Chronic fatigue
- Osteomyelitis
Chronic syndromes can develop years to months after untreated or inadequately treated infection and can be serious. Chronic infection can also result after a clinically silent initial infection.26,38 Culture-negative endocarditis, which occurs in fewer than 1% of patients diagnosed with acute infection, is the most common chronic manifestation (Table 1). Patients with underlying valvular disease, malignancy, or immunosuppression are at greater risk.38–40
Challenges and controversies
The diagnosis of chronic Q fever remains challenging. Traditionally, elevated phase I IgG titers were considered highly predictive of chronic disease. A cutoff of 1:800 was set, based on retrospective data from chronic cases in Europe, but its generalizability to different assays and patient populations has been unclear. 14,36,37 Recent reviews and prospective analysis with serial serologic studies in the Dutch outbreak and other sources suggest the positive predictive value (PPV) of phase I IgG titers greater than 1:800 to be lower than previously estimated, largely due to widespread testing and resultant increased seroprevalence assessments. It has been suggested the cutoff be raised to 1:1,600, which still only carries a 59% positive predictive value.41,42
Chronic fatigue due to Q fever remains a controversial topic and has only been described in Europe, Asia, and Australia. A direct link has yet to be established.43 Additional research is needed, but small studies of prolonged antibiotic treatment have not shown benefit in these cases.44
CASE CONTINUED
This patient did well. During a routine physical while enrolled at the Army War College in Carlisle, PA, 6 months after the original presentation, he mentioned his illness to the physician, who then repeated testing for Q fever; the test was positive (Table 2). Subsequently, serum samples from before and after his deployment were tested along with another convalescent-phase sample, and the results demonstrated Q fever seroconversion. He was well and had no physical complaints. He had no heart murmur, and a complete blood count and tests of liver enzymes and inflammatory markers were normal.
TREATMENT AND PREVENTION OF Q FEVER
4. Which of the following treatments would be appropriate, given his diagnosis of Q fever?
- Doxycycline (Vibramycin) 100 mg twice daily for 14 days
- Levofloxacin (Levaquin) 500 mg daily for 5 days
- Doxycycline 100 mg twice daily for 14 days and hydroxychloroquine (Plaquenil) 200 mg three times per day for 18 months
- No treatment
The treatment goals in Q fever are to hasten the resolution of symptoms and to prevent chronic disease. Generally, if there are no clinical findings or symptoms, treatment is not indicated. If the patient has symptoms, early treatment is preferred, but a response may be seen even when there is a delay in diagnosis.
Doxycycline 100 mg twice a day for 14 days is the treatment of choice. In addition, quinolones have in vitro activity,45 and a recent study suggests moxifloxacin (Avelox) may be the preferred antibiotic for those who cannot tolerate doxycycline.46 In pregnant women and in children, macrolides and trimethoprim-sulfamethoxazole (Bactrim) are preferred.47,48
Treatment of chronic Q fever, in particular endocarditis, warrants more intensive therapy. A retrospective review of treated cases of endocarditis suggested that monotherapy with doxycycline often failed, and combination therapy with hydroxychloroquine has been advocated based on in vivo and in vitro experience.49
PREVENTING LONG-TERM SEQUELAE OF CHRONIC Q FEVER
5. At this point, what is the next step in the management of this patient?
- No further follow-up is indicated
- Transthoracic echocardiography (TTE)
- Repeat Q fever serologic testing in 3 to 6 months
- Whole-blood PCR testing and transesophageal echocardiography (TEE)
Long-term follow-up of patients with Q fever has been advocated to monitor for the development of chronic Q fever, but recent studies question the previously devised algorithms.50
The data the algorithms were based on suggested that preexisting valvular heart disease could be associated with up to a 39% risk of endocarditis, and a two-step approach was devised to prevent and identify early chronic infection.51,52 Patients with Q fever would undergo TTE at baseline, and if the findings were abnormal (including mild regurgitation), then 12 months of prophylactic treatment with hydroxychloroquine and doxycycline was recommended. If TTE was normal, serial serologic testing every 3 months was recommended. If the anti-phase I IgG titer was greater than 1:800 at any point, TEE and a whole-blood PCR assay were recommended to evaluate for endocarditis.51
These recommendations were based on data from the French National Reference Center and had not been prospectively evaluated. The 2007–2008 Dutch outbreak provided a large cohort of Q fever cases. After initial screening with TTE and serologic follow-up, 59% of patients were noted to have mild valvular abnormalities, and many had phase I IgG levels greater than 1:800 during follow-up despite being clinically free of disease. The Dutch subsequently stopped screening with TTE as part of routine follow-up and elected to follow patients clinically.
Similar findings have been noted from case follow-up in France and Taiwan, also supporting using serologic cutoffs alone in determining the need for evaluation (with TEE) or treatment of chronic disease.53,54 The usefulness of serologic testing every 3 months has also been questioned, and some have advocated extending the interval, especially since less emphasis is being placed on the results in favor of more practical clinical follow-up.39
One such clinical approach at follow-up is presented in Figure 1. TTE should be reserved for patients with known valvular disease or a clear murmur. Those with underlying valvular disease and acute Q fever should be managed on an individual basis by a specialist in infectious disease, and antibiotic prophylaxis should be considered. Patients without underlying disease should have regular follow-up examinations and serologic testing every 6 months, and clinical symptoms should guide further testing (eg, with TEE and PCR testing) for chronic disease.
In this patient, phase I and II antibody titers were notably elevated (in TABLE 2, phase I titers > 1:800 and 1:1600 cutoffs). Such high titers have been common in military cases from Iraq and Afghanistan, and to date no cases of endocarditis have been diagnosed despite close follow-up. Most cases in military personnel are in relatively young patients who lack risk factors for endocarditis. Based on emerging data from large overseas outbreaks and the potential toxicity of intensive preemptive dual-antimicrobial therapy, an approach of close follow-up was taken.
PRIMARY PREVENTION OF Q FEVER
Prevention of Q fever remains a challenge, as the organism is highly persistent in the environment. An effective licensed vaccine exists in Australia under the brand name Q-Vax, but no approved vaccine is currently available in the United States.55
THE PATIENT’S COURSE
The patient returned for follow-up about 1 year after his first presentation. He noted some ongoing fatigue but attributed this to his course work, and he said he otherwise felt well. He exercises regularly, with no shortness of breath, fevers, chills, or weight loss. He continued to have elevated Q fever titers. Because he had no symptoms, no heart murmur, and normal inflammatory markers, he had no further workup and continued to be followed with serial serologic testing and examinations.
A healthy 42-year-old US Army reservist returned home to Oregon in early April after a 12-month deployment in Iraq. About 6 weeks later, he developed a mild nonproductive cough; then, over the next 2 weeks, his symptoms progressed to myalgia, mild headache, fever, chills, drenching night sweats, and dyspnea on exertion.
About 2 weeks after the onset of his symptoms, he saw his primary care provider. The results of laboratory tests at that time were normal except for the following:
- Platelet count 110 × 109/L (reference range 150–400)
- Alkaline phosphatase 354 IU/L (40–100)
- Alanine aminotransferase 99 IU/L (5–36)
- Aspartate aminotransferase 220 IU/L (7–33).
Chest radiography was negative. He was told he had a viral infection and was sent home with no treatment.
1. Which of the following is the most likely diagnosis in this patient?
- Influenza
- Ehrlichiosis
- Q fever
- Visceral leishmaniasis
- Malaria
Military operations in Iraq and Afghanistan have involved large numbers of US Army Reserve and National Guard personnel: by 2007, more than 500,000 Reserve and National Guard personnel had served in these combat operations.1 Although these personnel are generally healthy and receive mandatory travel screenings, prophylactic drug treatment, and vaccinations, their close, long-term exposure to local populations and environments puts them at risk of many infections.2
Often, these veterans develop symptoms after returning home, and they seek medical care from providers outside the military medical system.3,4 Civilian health care providers are thus increasingly called on to recognize clinical syndromes associated with military operations.
FEVER IN RETURNED SOLDIERS
The presentation of this 42-year-old veteran has an extensive differential diagnosis. His symptoms arose more than a month after his return from Iraq, meaning he could have acquired an infection in Iraq, on his trip home, or even after arriving home.
A number of common viral and atypical respiratory pathogens could be involved, and although circulating influenza was not common at the time of year he happened to return (spring), it remains a possibility. However, the duration of his illness, with symptoms that gradually worsened over 12 days, argues against influenza and community-acquired respiratory and other viral illnesses.
Aronson et al5 have reviewed the infectious risks in deployed military personnel.5 Infectious syndromes that have manifested in military personnel a month or more after returning from Iraq or Afghanistan include malaria, Q fever, brucellosis, typhoid fever, and leishmaniasis.5
Malaria
Malaria should be considered in all travelers from endemic areas presenting with fever, especially if they have thrombocytopenia and anemia. Plasmodium vivax is present in Iraq, but transmission is rare and isolated. Defense Medical Surveillance System data show that most of the recent malaria cases in US military personnel were acquired in Afghanistan or Korea. Many of these cases were caused by P vivax and manifested weeks to months after exposure, and diagnosis was significantly delayed because the provider did not consider malaria in the differential diagnosis.4,6,7
Testing for malaria with serial thick and thin blood smears and the BinaxNOW (Iverness Medical, Princeton, NJ) rapid test, when available, should be done in all those who have served in malaria-endemic regions and who present with unexplained fever or consistent symptoms. Testing should be done even if prophylaxis was taken or the potential exposure was weeks to months before presentation.
Brucellosis
Brucellosis, a zoonosis typically acquired by ingesting unpasteurized dairy products, has a high prevalence in Eurasia. A nonspecific, multisystem illness with fever, hepatitis, and arthritis (classically sacroiliitis) is commonly described.
Brucellosis is less likely in our patient, given that he denied consumption of local dairy products while deployed. Also, he had prominent respiratory symptoms, which would not be typical of brucellosis.
Leishmaniasis
Leishmaniasis, a parasitic disease transmitted by sand flies, manifests in one of three ways, ie, as a cutaneous, a mucosal, or a visceral disease. Most infections recently reported in US military personnel have been cutaneous and were acquired in Iraq, where Leishmania major is the primary species.8 Visceral disease mimics lymphoma (fever, hepatosplenomegaly, and cytopenia), but only a handful of cases have been reported from Iraq and Afghanistan.9 The incubation period of visceral leishmaniasis is prolonged, and civilian providers should consider it even if the patient’s period of deployment was relatively long ago.
Q fever in military personnel
Q fever is caused by the intracellular bacterium Coxiella burnetii.
Q fever has been reported in more than 150 US military personnel deployed to Iraq and Afghanistan.10–12 However, it may be more common than that. In one report, 10% of patients admitted to a combat support hospital in Iraq with International Classification of Diseases, Ninth Revision codes potentially consistent with Q fever tested positive for it.13 And in several cases that manifested after deployment, Q fever was not considered initially by the health care provider.11,14 In response, the US Centers for Disease Control and Prevention (CDC) released a health advisory in May 2010 alerting providers about Q fever in travelers returning from Iraq and the Netherlands.15
Q fever is a zoonosis associated with a wide range of animal reservoirs, primarily agricultural livestock such as cattle, goats, and sheep, but also a variety of other animals. There are multiple routes of transmission, including direct animal contact, ingestion of unpasteurized dairy products, and, most commonly, inhalation of aerosolized particles contaminated by animal droppings or secretions.16 Tick-borne and sexual transmission have been reported in rare instances.17,18 Importantly, in many cases from Iraq and from an outbreak in the Netherlands there was no obvious exposure.19
Q fever is a potential agent of bioterrorism; therefore, a large-scale, single-point outbreak should raise concern about a possible intentional release of the organism.20
Q fever has myriad presentations
About 60% of cases of Q fever infection are asymptomatic.21 In the United States, the estimated seroprevalence is 3%. Such a high seroprevalence, despite the relatively small number of reported cases, suggests that this infection is often subclinical.22
After 2 to 3 weeks of incubation, Q fever infection can produce a wide range of presentations involving almost any organ system (Table 1).16 An influenza-like illness with fever, pneumonia, and hepatitis is classic. Often, headache is severe enough to warrant lumbar puncture. Atypical and often severe presentations include gastrointestinal or neurologic manifestations.23–25 Rates of hospitalization and in-hospital death are low in acute disease: hospitalization occurs in roughly 2% of cases, and death in about 1% of those hospitalized.26,27
The presentation may mimic that of conditions caused by common community pathogens such as Legionella, Rickettsia, cytomegalovirus, Ebola virus, influenza, Mycoplasma, and human immunodeficiency virus (primary infection). Heightened suspicion is needed to prevent delays in diagnosis and treatment.
This patient’s symptoms and his recent deployment made Q fever very likely.
CASE CONTINUED
The patient continued to feel sick and reported having three to four loose bowel movements per day and mild abdominal pain. His cough and dyspnea persisted.
He had not had contact with anyone who was ill, denied being exposed to animals or insects, and had not consumed unpasteurized dairy products; he recalled having cleaned his military-issue uniforms and equipment 2 to 3 weeks before the symptoms began. He called a military physician to get advice on what else could be causing his symptoms. This physician recommended tests based on potential exposures in Iraq. Tests for brucellosis, visceral leishmaniasis, and Q fever were ordered.
Over the next several days, he began to feel better, and at 3 to 4 weeks after the onset of his symptoms, he felt that he had returned to normal. All tests were negative.
TESTING FOR ATYPICAL PATHOGENS
2. Which of the following would be the most readily available method to confirm the diagnosis?
- Culture
- Polymerase chain reaction (PCR) testing
- Histopathologic testing
- Serologic testing
Testing for atypical pathogens was reasonable in this patient. In addition to an evaluation for parasitic causes of persistent and chronic diarrhea, an evaluation for Q fever, brucellosis, and visceral leishmaniasis via serologic testing was warranted. A variety of tests exist for all of these infections, but serologic tests are the most readily available.
Leishmaniasis testing
Visceral leishmaniasis was traditionally diagnosed by visualizing organisms in splenic or bone marrow aspirates,28 but now serologic tests are available through commercial and public health laboratories such as the CDC. Immunochromatographic tests using recombinant k39 antigen are highly sensitive and specific and have been used in military cases.29,30
Brucellosis testing
Brucella can be cultured from blood or tissue samples. The laboratory must be alerted, as special media can be used to increase the yield and precautions must be taken to prevent laboratory-acquired infection. Serologic testing is the method most commonly used for diagnosis.31
Q fever testing
Q fever can be diagnosed with serologic testing during its acute and convalescent phases.
PCR testing of blood is useful for diagnosing acute disease and is positive before serologic conversion, thus allowing rapid diagnosis and treatment.32 The Joint Biological Agent Identification and Diagnostic System (JBAIDS) PCR platform was studied in a Combat Support Hospital in Iraq for making the rapid diagnosis of Q fever and has since been approved by the US Food and Drug Administration for military use.33
Culture is beyond the scope of most clinical laboratories and requires specialized cell culture or egg yolk media. In tissue, usually liver tissue obtained in an effort to evaluate hepatitis, the histologic finding of “doughnut” granulomas, or fibrin-encased granulomas, can be suggestive of C burnetii but may be nonspecific and seen with other infections.23,34
Serologic testing with an immunofluorescence assay (IFA) remains the most common method of diagnosis. It is based on the detection of immunoglobulin G (IgG) and IgM responses against phase I and phase II antigens of C burnetii. After initial infection, the organism displays phase I antigens and is highly infectious. When grown in culture, the organism undergoes phase shifting to a less infectious form with predominantly phase II antigens. Paradoxically, after initial infection in humans, antibody response against phase II antigens is seen first, whereas in chronic infection, a phase I antibody response dominates.26,35 Phase II antibodies appear around week 2, and 90% of samples from infected people are positive by week 3. A fourfold rise in titer between the acute-phase and convalescent-phase samples confirms the diagnosis.35
A number of serologic assays are available worldwide, but they have different methods and cutoff values, so questions have arisen about the equivalence of the results.14,36 Serologic cutoffs have been defined in Europe, where most cases of Q fever have been reported.37
CHRONIC Q FEVER
3. Which of the following would be the most likely chronic manifestation of Q fever?
- Pneumonia
- Hepatitis
- Endocarditis
- Chronic fatigue
- Osteomyelitis
Chronic syndromes can develop years to months after untreated or inadequately treated infection and can be serious. Chronic infection can also result after a clinically silent initial infection.26,38 Culture-negative endocarditis, which occurs in fewer than 1% of patients diagnosed with acute infection, is the most common chronic manifestation (Table 1). Patients with underlying valvular disease, malignancy, or immunosuppression are at greater risk.38–40
Challenges and controversies
The diagnosis of chronic Q fever remains challenging. Traditionally, elevated phase I IgG titers were considered highly predictive of chronic disease. A cutoff of 1:800 was set, based on retrospective data from chronic cases in Europe, but its generalizability to different assays and patient populations has been unclear. 14,36,37 Recent reviews and prospective analysis with serial serologic studies in the Dutch outbreak and other sources suggest the positive predictive value (PPV) of phase I IgG titers greater than 1:800 to be lower than previously estimated, largely due to widespread testing and resultant increased seroprevalence assessments. It has been suggested the cutoff be raised to 1:1,600, which still only carries a 59% positive predictive value.41,42
Chronic fatigue due to Q fever remains a controversial topic and has only been described in Europe, Asia, and Australia. A direct link has yet to be established.43 Additional research is needed, but small studies of prolonged antibiotic treatment have not shown benefit in these cases.44
CASE CONTINUED
This patient did well. During a routine physical while enrolled at the Army War College in Carlisle, PA, 6 months after the original presentation, he mentioned his illness to the physician, who then repeated testing for Q fever; the test was positive (Table 2). Subsequently, serum samples from before and after his deployment were tested along with another convalescent-phase sample, and the results demonstrated Q fever seroconversion. He was well and had no physical complaints. He had no heart murmur, and a complete blood count and tests of liver enzymes and inflammatory markers were normal.
TREATMENT AND PREVENTION OF Q FEVER
4. Which of the following treatments would be appropriate, given his diagnosis of Q fever?
- Doxycycline (Vibramycin) 100 mg twice daily for 14 days
- Levofloxacin (Levaquin) 500 mg daily for 5 days
- Doxycycline 100 mg twice daily for 14 days and hydroxychloroquine (Plaquenil) 200 mg three times per day for 18 months
- No treatment
The treatment goals in Q fever are to hasten the resolution of symptoms and to prevent chronic disease. Generally, if there are no clinical findings or symptoms, treatment is not indicated. If the patient has symptoms, early treatment is preferred, but a response may be seen even when there is a delay in diagnosis.
Doxycycline 100 mg twice a day for 14 days is the treatment of choice. In addition, quinolones have in vitro activity,45 and a recent study suggests moxifloxacin (Avelox) may be the preferred antibiotic for those who cannot tolerate doxycycline.46 In pregnant women and in children, macrolides and trimethoprim-sulfamethoxazole (Bactrim) are preferred.47,48
Treatment of chronic Q fever, in particular endocarditis, warrants more intensive therapy. A retrospective review of treated cases of endocarditis suggested that monotherapy with doxycycline often failed, and combination therapy with hydroxychloroquine has been advocated based on in vivo and in vitro experience.49
PREVENTING LONG-TERM SEQUELAE OF CHRONIC Q FEVER
5. At this point, what is the next step in the management of this patient?
- No further follow-up is indicated
- Transthoracic echocardiography (TTE)
- Repeat Q fever serologic testing in 3 to 6 months
- Whole-blood PCR testing and transesophageal echocardiography (TEE)
Long-term follow-up of patients with Q fever has been advocated to monitor for the development of chronic Q fever, but recent studies question the previously devised algorithms.50
The data the algorithms were based on suggested that preexisting valvular heart disease could be associated with up to a 39% risk of endocarditis, and a two-step approach was devised to prevent and identify early chronic infection.51,52 Patients with Q fever would undergo TTE at baseline, and if the findings were abnormal (including mild regurgitation), then 12 months of prophylactic treatment with hydroxychloroquine and doxycycline was recommended. If TTE was normal, serial serologic testing every 3 months was recommended. If the anti-phase I IgG titer was greater than 1:800 at any point, TEE and a whole-blood PCR assay were recommended to evaluate for endocarditis.51
These recommendations were based on data from the French National Reference Center and had not been prospectively evaluated. The 2007–2008 Dutch outbreak provided a large cohort of Q fever cases. After initial screening with TTE and serologic follow-up, 59% of patients were noted to have mild valvular abnormalities, and many had phase I IgG levels greater than 1:800 during follow-up despite being clinically free of disease. The Dutch subsequently stopped screening with TTE as part of routine follow-up and elected to follow patients clinically.
Similar findings have been noted from case follow-up in France and Taiwan, also supporting using serologic cutoffs alone in determining the need for evaluation (with TEE) or treatment of chronic disease.53,54 The usefulness of serologic testing every 3 months has also been questioned, and some have advocated extending the interval, especially since less emphasis is being placed on the results in favor of more practical clinical follow-up.39
One such clinical approach at follow-up is presented in Figure 1. TTE should be reserved for patients with known valvular disease or a clear murmur. Those with underlying valvular disease and acute Q fever should be managed on an individual basis by a specialist in infectious disease, and antibiotic prophylaxis should be considered. Patients without underlying disease should have regular follow-up examinations and serologic testing every 6 months, and clinical symptoms should guide further testing (eg, with TEE and PCR testing) for chronic disease.
In this patient, phase I and II antibody titers were notably elevated (in TABLE 2, phase I titers > 1:800 and 1:1600 cutoffs). Such high titers have been common in military cases from Iraq and Afghanistan, and to date no cases of endocarditis have been diagnosed despite close follow-up. Most cases in military personnel are in relatively young patients who lack risk factors for endocarditis. Based on emerging data from large overseas outbreaks and the potential toxicity of intensive preemptive dual-antimicrobial therapy, an approach of close follow-up was taken.
PRIMARY PREVENTION OF Q FEVER
Prevention of Q fever remains a challenge, as the organism is highly persistent in the environment. An effective licensed vaccine exists in Australia under the brand name Q-Vax, but no approved vaccine is currently available in the United States.55
THE PATIENT’S COURSE
The patient returned for follow-up about 1 year after his first presentation. He noted some ongoing fatigue but attributed this to his course work, and he said he otherwise felt well. He exercises regularly, with no shortness of breath, fevers, chills, or weight loss. He continued to have elevated Q fever titers. Because he had no symptoms, no heart murmur, and normal inflammatory markers, he had no further workup and continued to be followed with serial serologic testing and examinations.
- Defense Science Board Task Force on Deployment of Members of the National Guard and Reserve in the Global War on Terrorism. Washington, DC. September 2007.
- Sanders JW, Putnam SD, Frankart C, et al. Impact of illness and non-combat injury during Operations Iraqi Freedom and Enduring Freedom (Afghanistan). Am J Trop Med Hyg 2005; 73:713–719.
- Gleeson TD, Decker CF, Johnson MD, Hartzell JD, Mascola JR. Q fever in US military returning from Iraq. Am J Med 2007; 120:e11–e12.
- Hagan JE, Marcos LA, Steinberg TH. Fever in a soldier returned from Afghanistan. J Travel Med 2010; 17:351–352.
- Aronson NE, Sanders JW, Moran KA. In harm’s way: infections in deployed American military forces. Clin Infect Dis 2006; 43:1045–1051.
- Klein TA, Pacha LA, Lee HC, et al. Plasmodium vivax malaria among U.S. forces Korea in the Republic of Korea, 1993–2007. Mil Med 2009; 174:412–418.
- Ciminera P, Brundage J. Malaria in U.S. military forces: a description of deployment exposures from 2003 through 2005. Am J Trop Med Hyg 2007; 76:275–279.
- Lesho EP, Wortmann G, Neafie R, Aronson N. Nonhealing skin lesions in a sailor and a journalist returning from Iraq. Cleve Clin J Med 2005; 72:93–96,
- Myles O, Wortmann GW, Cummings JF, et al. Visceral leishmaniasis: clinical observations in 4 US army soldiers deployed to Afghanistan or Iraq, 2002–2004. Arch Intern Med 2007; 167:1899–1901.
- Faix DJ, Harrison DJ, Riddle MS, et al. Outbreak of Q fever among US military in western Iraq, June–July 2005. Clin Infect Dis 2008; 46:e65–e68.
- Leung-Shea C, Danaher PJ. Q fever in members of the United States armed forces returning from Iraq. Clin Infect Dis 2006; 43:e77–e82.
- Anderson AD, Smoak B, Shuping E, Ockenhouse C, Petruccelli B. Q fever and the US military. Emerg Infect Dis 2005; 11:1320–1322.
- Anderson AD, Baker TR, Littrell AC, Mott RL, Niebuhr DW, Smoak BL. Seroepidemiologic survey for Coxiella burnetii among hospitalized US troops deployed to Iraq. Zoonoses Public Health 2011; 58:276–283.
- Ake JA, Massung RF, Whitman TJ, Gleeson TD. Difficulties in the diagnosis and management of a US servicemember presenting with possible chronic Q fever. J Infect 2010; 60:175–177.
- Centers for Disease Control and Prevention (CDC). Potential for Q fever infection among travelers returning from Iraq and the Netherlands http://www.bt.cdc.gov/HAN/han00313.asp. Accessed July 5, 2012.
- Parker NR, Barralet JH, Bell AM. Q fever. Lancet 2006; 367:679–688.
- Miceli MH, Veryser AK, Anderson AD, Hofinger D, Lee SA, Tancik C. A case of person-to-person transmission of Q fever from an active duty serviceman to his spouse. Vector Borne Zoonotic Dis 2010; 10:539–541.
- Milazzo A, Hall R, Storm PA, Harris RJ, Winslow W, Marmion BP. Sexually transmitted Q fever. Clin Infect Dis 2001; 33:399–402.
- Hartzell JD, Peng SW, Wood-Morris RN, et al. Atypical Q fever in US soldiers. Emerg Infect Dis 2007; 13:1247–1249.
- Bossi P, Tegnell A, Baka A, et al; Task Force on Biological and Chemical Agent Threats, Public Health Directorate, European Commission, Luxembourg. Bichat guidelines for the clinical management of Q fever and bioterrorism-related Q fever. Euro Surveill 2004; 9:E19–E20.
- Roest HI, Tilburg JJ, van der Hoek W, et al. The Q fever epidemic in The Netherlands: history, onset, response and reflection. Epidemiol Infect 2011; 139:1–12.
- Anderson AD, Kruszon-Moran D, Loftis AD, et al. Seroprevalence of Q fever in the United States, 2003–2004. Am J Trop Med Hyg 2009; 81:691–694.
- Hatchette TF, Marrie TJ. Atypical manifestations of chronic Q fever. Clin Infect Dis 2001; 33:1347–1351.
- Bernit E, Pouget J, Janbon F, et al. Neurological involvement in acute Q fever: a report of 29 cases and review of the literature. Arch Intern Med 2002; 162:693–700.
- Kofteridis DP, Mazokopakis EE, Tselentis Y, Gikas A. Neurological complications of acute Q fever infection. Eur J Epidemiol 2004; 19:1051–1054.
- Raoult D, Marrie T, Mege J. Natural history and pathophysiology of Q fever. Lancet Infect Dis 2005; 5:219–226.
- Kampschreur LM, Wegdam-Blans MC, Thijsen SF, et al. Acute Q fever related in-hospital mortality in the Netherlands. Neth J Med 2010; 68:408–413.
- Srivastava P, Dayama A, Mehrotra S, Sundar S. Diagnosis of visceral leishmaniasis. Trans R Soc Trop Med Hyg 2011; 105:1–6.
- Chappuis F, Rijal S, Soto A, Menten J, Boelaert M. A meta-analysis of the diagnostic performance of the direct agglutination test and rK39 dipstick for visceral leishmaniasis. BMJ 2006; 333:723.
- Hartzell JD, Aronson NE, Weina PJ, Howard RS, Yadava A, Wortmann GW. Positive rK39 serologic assay results in US servicemen with cutaneous leishmaniasis. Am J Trop Med Hyg 2008; 79:843–846.
- Pappas G, Akritidis N, Bosilkovski M, Tsianos E. Brucellosis. N Engl J Med 2005; 352:2325–2336.
- Schneeberger PM, Hermans MH, van Hannen EJ, Schellekens JJ, Leenders AC, Wever PC. Real-time PCR with serum samples is indispensable for early diagnosis of acute Q fever. Clin Vaccine Immunol 2010; 17:286–290.
- Hamilton LR, George DL, Scoville SL, Hospenthal DR, Griffith ME. PCR for rapid diagnosis of acute Q fever at a combat support hospital in Iraq. Mil Med 2011; 176:103–105.
- Bonilla MF, Kaul DR, Saint S, Isada CM, Brotman DJ. Clinical problem-solving. Ring around the diagnosis. N Engl J Med 2006; 354:1937–1942.
- Fournier PE, Marrie TJ, Raoult D. Diagnosis of Q fever. J Clin Microbiol 1998; 36:1823–1834.
- Healy B, van Woerden H, Raoult D, et al. Chronic Q fever: different serological results in three countries—results of a follow-up study 6 years after a point source outbreak. Clin Infect Dis 2011; 52:1013–1019.
- Dupont HT, Thirion X, Raoult D. Q fever serology: cutoff determination for microimmunofluorescence. Clin Diagn Lab Immunol 1994; 1:189–196.
- Karakousis PC, Trucksis M, Dumler JS. Chronic Q fever in the United States. J Clin Microbiol 2006; 44:2283–2287.
- van der Hoek W, Versteeg B, Meekelenkamp JC, et al. Follow-up of 686 patients with acute Q fever and detection of chronic infection. Clin Infect Dis 2011; 52:1431–1436.
- Fenollar F, Fournier PE, Carrieri MP, Habib G, Messana T, Raoult D. Risks factors and prevention of Q fever endocarditis. Clin Infect Dis 2001; 33:312–316.
- Frankel D, Richet H, Renvoisé A, Raoult D. Q fever in France, 1985–2009. Emerg Infect Dis 2011; 17:350–356.
- Baddour LM, Wilson WR, Bayer AS, et al; Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease; Council on Cardiovascular Disease in the Young; Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia; American Heart Association; Infectious Diseases Society of America. Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications: a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation 2005; 111:e394–e434.
- Wildman MJ, Smith EG, Groves J, Beattie JM, Caul EO, Ayres JG. Chronic fatigue following infection by Coxiella burnetii (Q fever): ten-year follow-up of the 1989 UK outbreak cohort. QJM 2002; 95:527–538.
- Iwakami E, Arashima Y, Kato K, et al. Treatment of chronic fatigue syndrome with antibiotics: pilot study assessing the involvement of Coxiella burnetii infection. Intern Med 2005; 44:1258–1263.
- Rolain JM, Maurin M, Raoult D. Bacteriostatic and bactericidal activities of moxifloxacin against Coxiella burnetii. Antimicrob Agents Chemother 2001; 45:301–302.
- Dijkstra F, Riphagen-Dalhuisen J, Wijers N, et al. Antibiotic therapy for acute Q fever in The Netherlands in 2007 and 2008 and its relation to hospitalization. Epidemiol Infect 2011; 139:1332–1341.
- Raoult D. Use of macrolides for Q fever. Antimicrob Agents Chemother 2003; 47:446.
- Carcopino X, Raoult D, Bretelle F, Boubli L, Stein A. Managing Q fever during pregnancy: the benefits of long-term cotrimoxazole therapy. Clin Infect Dis 2007; 45:548–555.
- Raoult D, Houpikian P, Tissot Dupont H, Riss JM, Arditi-Djiane J, Brouqui P. Treatment of Q fever endocarditis: comparison of 2 regimens containing doxycycline and ofloxacin or hydroxychloroquine. Arch Intern Med 1999; 159:167–173.
- Healy B, Llewelyn M, Westmoreland D, Lloyd G, Brown N. The value of follow-up after acute Q fever infection. J Infect 2006; 52:e109–e112.
- Landais C, Fenollar F, Thuny F, Raoult D. From acute Q fever to endocarditis: serological follow-up strategy. Clin Infect Dis 2007; 44:1337–1340.
- Hartzell JD, Wood-Morris RN, Martinez LJ, Trotta RF. Q fever: epidemiology, diagnosis, and treatment. Mayo Clin Proc 2008; 83:574–579.
- Hung MN, Lin LJ, Hou MY, et al. Serologic assessment of the risk of developing chronic Q fever in cohorts of acutely infected individuals. J Infect 2011; 62:39–44.
- Sunder S, Gras G, Bastides F, De Gialluly C, Choutet P, Bernard L. Chronic Q fever: relevance of serology. Clin Infect Dis 2011; 53:749–750.
- Gefenaite G, Munster JM, van Houdt R, Hak E. Effectiveness of the Q fever vaccine: a meta-analysis. Vaccine 2011; 29:395–398.
- Defense Science Board Task Force on Deployment of Members of the National Guard and Reserve in the Global War on Terrorism. Washington, DC. September 2007.
- Sanders JW, Putnam SD, Frankart C, et al. Impact of illness and non-combat injury during Operations Iraqi Freedom and Enduring Freedom (Afghanistan). Am J Trop Med Hyg 2005; 73:713–719.
- Gleeson TD, Decker CF, Johnson MD, Hartzell JD, Mascola JR. Q fever in US military returning from Iraq. Am J Med 2007; 120:e11–e12.
- Hagan JE, Marcos LA, Steinberg TH. Fever in a soldier returned from Afghanistan. J Travel Med 2010; 17:351–352.
- Aronson NE, Sanders JW, Moran KA. In harm’s way: infections in deployed American military forces. Clin Infect Dis 2006; 43:1045–1051.
- Klein TA, Pacha LA, Lee HC, et al. Plasmodium vivax malaria among U.S. forces Korea in the Republic of Korea, 1993–2007. Mil Med 2009; 174:412–418.
- Ciminera P, Brundage J. Malaria in U.S. military forces: a description of deployment exposures from 2003 through 2005. Am J Trop Med Hyg 2007; 76:275–279.
- Lesho EP, Wortmann G, Neafie R, Aronson N. Nonhealing skin lesions in a sailor and a journalist returning from Iraq. Cleve Clin J Med 2005; 72:93–96,
- Myles O, Wortmann GW, Cummings JF, et al. Visceral leishmaniasis: clinical observations in 4 US army soldiers deployed to Afghanistan or Iraq, 2002–2004. Arch Intern Med 2007; 167:1899–1901.
- Faix DJ, Harrison DJ, Riddle MS, et al. Outbreak of Q fever among US military in western Iraq, June–July 2005. Clin Infect Dis 2008; 46:e65–e68.
- Leung-Shea C, Danaher PJ. Q fever in members of the United States armed forces returning from Iraq. Clin Infect Dis 2006; 43:e77–e82.
- Anderson AD, Smoak B, Shuping E, Ockenhouse C, Petruccelli B. Q fever and the US military. Emerg Infect Dis 2005; 11:1320–1322.
- Anderson AD, Baker TR, Littrell AC, Mott RL, Niebuhr DW, Smoak BL. Seroepidemiologic survey for Coxiella burnetii among hospitalized US troops deployed to Iraq. Zoonoses Public Health 2011; 58:276–283.
- Ake JA, Massung RF, Whitman TJ, Gleeson TD. Difficulties in the diagnosis and management of a US servicemember presenting with possible chronic Q fever. J Infect 2010; 60:175–177.
- Centers for Disease Control and Prevention (CDC). Potential for Q fever infection among travelers returning from Iraq and the Netherlands http://www.bt.cdc.gov/HAN/han00313.asp. Accessed July 5, 2012.
- Parker NR, Barralet JH, Bell AM. Q fever. Lancet 2006; 367:679–688.
- Miceli MH, Veryser AK, Anderson AD, Hofinger D, Lee SA, Tancik C. A case of person-to-person transmission of Q fever from an active duty serviceman to his spouse. Vector Borne Zoonotic Dis 2010; 10:539–541.
- Milazzo A, Hall R, Storm PA, Harris RJ, Winslow W, Marmion BP. Sexually transmitted Q fever. Clin Infect Dis 2001; 33:399–402.
- Hartzell JD, Peng SW, Wood-Morris RN, et al. Atypical Q fever in US soldiers. Emerg Infect Dis 2007; 13:1247–1249.
- Bossi P, Tegnell A, Baka A, et al; Task Force on Biological and Chemical Agent Threats, Public Health Directorate, European Commission, Luxembourg. Bichat guidelines for the clinical management of Q fever and bioterrorism-related Q fever. Euro Surveill 2004; 9:E19–E20.
- Roest HI, Tilburg JJ, van der Hoek W, et al. The Q fever epidemic in The Netherlands: history, onset, response and reflection. Epidemiol Infect 2011; 139:1–12.
- Anderson AD, Kruszon-Moran D, Loftis AD, et al. Seroprevalence of Q fever in the United States, 2003–2004. Am J Trop Med Hyg 2009; 81:691–694.
- Hatchette TF, Marrie TJ. Atypical manifestations of chronic Q fever. Clin Infect Dis 2001; 33:1347–1351.
- Bernit E, Pouget J, Janbon F, et al. Neurological involvement in acute Q fever: a report of 29 cases and review of the literature. Arch Intern Med 2002; 162:693–700.
- Kofteridis DP, Mazokopakis EE, Tselentis Y, Gikas A. Neurological complications of acute Q fever infection. Eur J Epidemiol 2004; 19:1051–1054.
- Raoult D, Marrie T, Mege J. Natural history and pathophysiology of Q fever. Lancet Infect Dis 2005; 5:219–226.
- Kampschreur LM, Wegdam-Blans MC, Thijsen SF, et al. Acute Q fever related in-hospital mortality in the Netherlands. Neth J Med 2010; 68:408–413.
- Srivastava P, Dayama A, Mehrotra S, Sundar S. Diagnosis of visceral leishmaniasis. Trans R Soc Trop Med Hyg 2011; 105:1–6.
- Chappuis F, Rijal S, Soto A, Menten J, Boelaert M. A meta-analysis of the diagnostic performance of the direct agglutination test and rK39 dipstick for visceral leishmaniasis. BMJ 2006; 333:723.
- Hartzell JD, Aronson NE, Weina PJ, Howard RS, Yadava A, Wortmann GW. Positive rK39 serologic assay results in US servicemen with cutaneous leishmaniasis. Am J Trop Med Hyg 2008; 79:843–846.
- Pappas G, Akritidis N, Bosilkovski M, Tsianos E. Brucellosis. N Engl J Med 2005; 352:2325–2336.
- Schneeberger PM, Hermans MH, van Hannen EJ, Schellekens JJ, Leenders AC, Wever PC. Real-time PCR with serum samples is indispensable for early diagnosis of acute Q fever. Clin Vaccine Immunol 2010; 17:286–290.
- Hamilton LR, George DL, Scoville SL, Hospenthal DR, Griffith ME. PCR for rapid diagnosis of acute Q fever at a combat support hospital in Iraq. Mil Med 2011; 176:103–105.
- Bonilla MF, Kaul DR, Saint S, Isada CM, Brotman DJ. Clinical problem-solving. Ring around the diagnosis. N Engl J Med 2006; 354:1937–1942.
- Fournier PE, Marrie TJ, Raoult D. Diagnosis of Q fever. J Clin Microbiol 1998; 36:1823–1834.
- Healy B, van Woerden H, Raoult D, et al. Chronic Q fever: different serological results in three countries—results of a follow-up study 6 years after a point source outbreak. Clin Infect Dis 2011; 52:1013–1019.
- Dupont HT, Thirion X, Raoult D. Q fever serology: cutoff determination for microimmunofluorescence. Clin Diagn Lab Immunol 1994; 1:189–196.
- Karakousis PC, Trucksis M, Dumler JS. Chronic Q fever in the United States. J Clin Microbiol 2006; 44:2283–2287.
- van der Hoek W, Versteeg B, Meekelenkamp JC, et al. Follow-up of 686 patients with acute Q fever and detection of chronic infection. Clin Infect Dis 2011; 52:1431–1436.
- Fenollar F, Fournier PE, Carrieri MP, Habib G, Messana T, Raoult D. Risks factors and prevention of Q fever endocarditis. Clin Infect Dis 2001; 33:312–316.
- Frankel D, Richet H, Renvoisé A, Raoult D. Q fever in France, 1985–2009. Emerg Infect Dis 2011; 17:350–356.
- Baddour LM, Wilson WR, Bayer AS, et al; Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease; Council on Cardiovascular Disease in the Young; Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia; American Heart Association; Infectious Diseases Society of America. Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications: a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation 2005; 111:e394–e434.
- Wildman MJ, Smith EG, Groves J, Beattie JM, Caul EO, Ayres JG. Chronic fatigue following infection by Coxiella burnetii (Q fever): ten-year follow-up of the 1989 UK outbreak cohort. QJM 2002; 95:527–538.
- Iwakami E, Arashima Y, Kato K, et al. Treatment of chronic fatigue syndrome with antibiotics: pilot study assessing the involvement of Coxiella burnetii infection. Intern Med 2005; 44:1258–1263.
- Rolain JM, Maurin M, Raoult D. Bacteriostatic and bactericidal activities of moxifloxacin against Coxiella burnetii. Antimicrob Agents Chemother 2001; 45:301–302.
- Dijkstra F, Riphagen-Dalhuisen J, Wijers N, et al. Antibiotic therapy for acute Q fever in The Netherlands in 2007 and 2008 and its relation to hospitalization. Epidemiol Infect 2011; 139:1332–1341.
- Raoult D. Use of macrolides for Q fever. Antimicrob Agents Chemother 2003; 47:446.
- Carcopino X, Raoult D, Bretelle F, Boubli L, Stein A. Managing Q fever during pregnancy: the benefits of long-term cotrimoxazole therapy. Clin Infect Dis 2007; 45:548–555.
- Raoult D, Houpikian P, Tissot Dupont H, Riss JM, Arditi-Djiane J, Brouqui P. Treatment of Q fever endocarditis: comparison of 2 regimens containing doxycycline and ofloxacin or hydroxychloroquine. Arch Intern Med 1999; 159:167–173.
- Healy B, Llewelyn M, Westmoreland D, Lloyd G, Brown N. The value of follow-up after acute Q fever infection. J Infect 2006; 52:e109–e112.
- Landais C, Fenollar F, Thuny F, Raoult D. From acute Q fever to endocarditis: serological follow-up strategy. Clin Infect Dis 2007; 44:1337–1340.
- Hartzell JD, Wood-Morris RN, Martinez LJ, Trotta RF. Q fever: epidemiology, diagnosis, and treatment. Mayo Clin Proc 2008; 83:574–579.
- Hung MN, Lin LJ, Hou MY, et al. Serologic assessment of the risk of developing chronic Q fever in cohorts of acutely infected individuals. J Infect 2011; 62:39–44.
- Sunder S, Gras G, Bastides F, De Gialluly C, Choutet P, Bernard L. Chronic Q fever: relevance of serology. Clin Infect Dis 2011; 53:749–750.
- Gefenaite G, Munster JM, van Houdt R, Hak E. Effectiveness of the Q fever vaccine: a meta-analysis. Vaccine 2011; 29:395–398.