User login
A tale of two sisters with liver disease
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
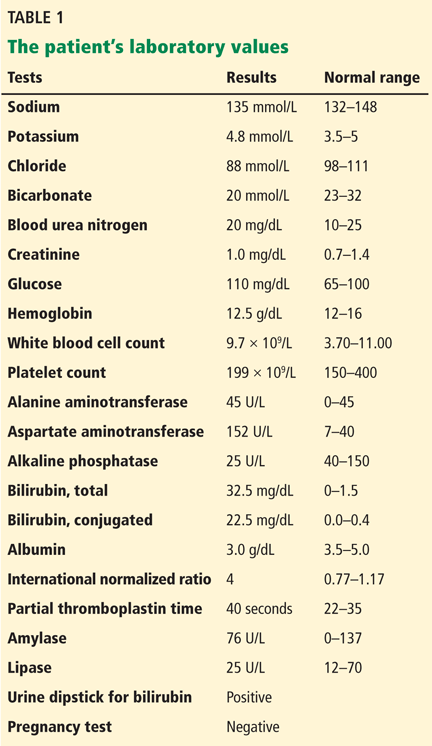
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
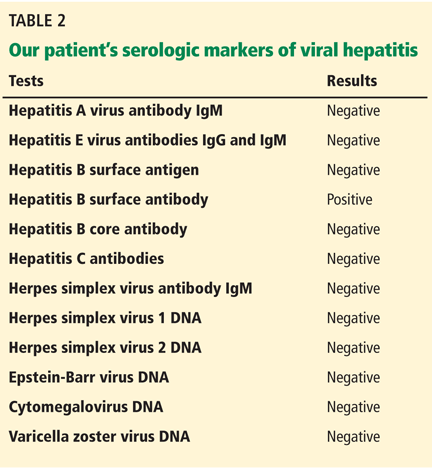
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
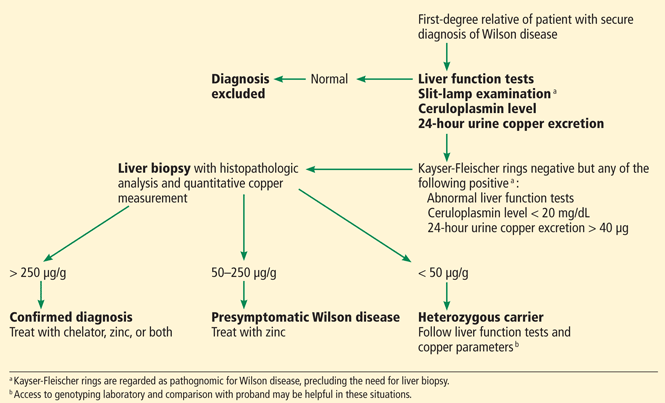
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
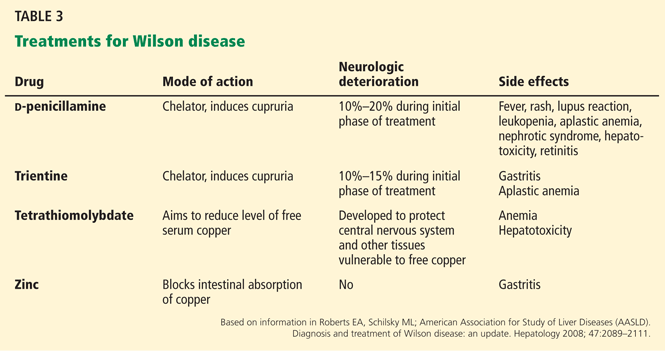
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
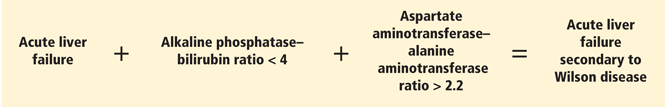
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.

Not all abdominal pain is gastrointestinal
A 31-year-old woman presents to the office with a chief complaint of right mid-abdominal pain that began 1 day ago. She says she did not seek medical attention earlier because she had to be at work that morning and she thought the pain would resolve on its own.
She reports no fever, headache, anorexia, nausea, vomiting, malaise, loss of weight, melena, or changes in bowel habits. She describes the pain as sharp, localized to the right side, and radiating to the vulva upon sitting up. She denies any association of pain with current dietary habits or bowel function. She has no recollection of precipitating or alleviating factors, including the use of analgesics to reduce the pain.
On further discussion, she mentions that 1 year ago she began experiencing chronic abdominal pain, which she says is sometimes exacerbated by coughing, by standing for extended periods of time, and during menses, and is alleviated upon lying down.
She has regular menstrual periods, and her last one ended 7 days ago.
Her surgical history includes two uncomplicated cesarean deliveries. She does not use tobacco, alcohol, or illicit substances. She is not aware of any allergies to drugs or foods.
She appears to be in no acute distress and has been sitting quietly thus far. She seems to have positioned her hand on her abdomen over the corresponding area of pain.
On physical examination, vital signs are within normal limits, and she is alert and oriented to person, place, and time. Her sclerae are anicteric, and the pupils are equal, round, and reactive to light.
Cardiovascular and pulmonary examinations are also within normal limits. Examination of the abdomen elicits tenderness and guarding along the lateral border of the rectus abdominis muscle on the right side at the level of umbilicus, with no rebound tenderness or rigidity. The liver and spleen are not enlarged, and no abdominal mass is detected. No skin rash, joint swelling, or peripheral edema is noted. A neurologic examination is normal.
1. With the information provided, which of the following is least likely to be causing her symptoms?
- Chronic mesenteric ischemia
- Peptic ulcer
- Acute cholecystitis
- Slipping rib syndrome
CHRONIC MESENTERIC ISCHEMIA
Chronic mesenteric ischemia is the least likely diagnosis because the patient lacks risk factors for atherosclerosis and because she does not have postprandial pain, which is pathognomonic for chronic mesenteric ischemia. It is thought to be caused by a decrease in blood flow through the splanchnic vessels.1 Symptoms tend to arise after eating because of a postprandial increase in metabolic demands.1 These patients also often have atherosclerotic risk factors such as hypertension, hyperlipidemia, and smoking causing coronary artery disease, or a history of stroke.
The primary symptom is abdominal pain, most often described as achy, crampy, or spastic episodes of pain, usually occurring within 2 hours of eating.2 Weight loss is common, as patients can develop a fear of eating. Postprandial pain may also be associated with nausea, vomiting, and bloating.
Findings on clinical examination are usually less severe than the actual symptoms. Visceral duplex or multidetector computed tomography (CT) is an excellent tool to detect blood flow in potential stenotic vessels.2
PEPTIC ULCER DISEASE
Peptic ulcer disease is not a likely diagnosis in this patient because she has no history of taking nonsteroidal anti-inflammatory drugs (NSAIDs).
A study of US patients between 1997 and 2007 reported an annual incidence of peptic ulcer disease of 0.05% to 0.19% depending on the method of diagnosis.3 Peptic ulcer is thought to result from increased gastric acid secretion with a resultant inflammatory response, leading to erosion and ulceration.
The most common possible catalysts include Helicobacter pylori infection, NSAIDs, smoking, alcohol use, and hypersecretory states such as Zollinger-Ellison syndrome.4–6 Complications include internal bleeding, perforation causing peritonitis, and penetration to adjacent organs.
Pathophysiology
Peptic ulcer is the result of an increase in the normal level of gastric acid and a decrease in the protective ability of the gastric mucosa.7 Cytoprotection may be lost through a decrease in the products of arachidonic acid metabolism (eg, prostaglandins, which have a protective effect) or an increase in leukotriene B4 (LTB4), which has a damaging effect. Prostaglandins are thought not only to protect the normal gastric mucosa, but also to provide an antisecretory effect.
On the other hand, leukotrienes—specifically LTB4 and LTC4—are proinflammatory agents and can damage the gastric mucosa. NSAIDs enhance the production of leukotrienes through the 5-lipoxygenase pathway. The ability of LTB4 to cause degranulation and release of lysosomal enzymes may play a vital role in the inflammatory response to NSAIDs.8–10 LTC4 may promote gastric mucosal damage through a reduction of tissue perfusion resulting from the promotion of vascular stasis.8,11,12
Symptoms help differentiate ulcer type
The classic symptom is burning epigastric pain after meals. Pain that occurs immediately after meals is a classic symptom of gastric ulcer. Pain that occurs 2 to 3 hours after meals and that is relieved by food or antacids is a strong indicator of duodenal ulcer.13 Other symptoms include dyspepsia, bloating, distention, heartburn, and chest discomfort.13
Accurate diagnosis is vital in selecting the proper treatment. Diagnostic tests may include H pylori testing, upper-gastrointestinal endoscopy, and radiography with barium swallow.
CHOLECYSTITIS
In cholecystitis, the primary complaint is pain, usually in the right upper quadrant of the abdomen. Patients describe sudden, sharp, and intense pain that radiates to the back or shoulder. Patients may report pain after heavy meals, and some report nausea and vomiting. Cholecystitis is in the differential diagnosis of this patient because of the anatomic location of her pain.
The diagnosis is confirmed by imaging. Abdominal ultrasonography, technetium-99m hepatic iminodiacetic acid scanning, and CT are the most commonly used studies.14
Cholecystitis can be acute or chronic. Acute cholecystitis is categorized as calculous or acalculous. Calculous cholecystitis is multifactorial, but the primary cause is blockage of the cystic duct by gallstones.15 Other factors include irritants such as lysolecithin (released during bile stasis), which can trigger gallbladder inflammation,15–17 and infection.18
When the cystic duct is blocked, bile builds up inside the gallbladder, causing irritation and inflammation of the walls of the gallbladder.14
Acalculous cholecystitis, which resembles calculous cholecystitis but without the gallstones,19 accounts for 2% to 15% of all cases of acute cholecystitis.19,20 It has been observed in hospitalized critically ill patients, but it can also present in an outpatient setting, most often in elderly men with vascular disease.21 Causes include infection, trauma, and tumor obstruction, resulting in endothelial injury, gallbladder stasis, ischemia, and eventually necrosis.14,20,22,23
SLIPPING RIB SYNDROME
Slipping rib syndrome, also known as Tietze syndrome, is believed to be caused by hypermobile costal cartilage. The affected rib slips behind the rib above on contraction of the abdominal wall. This displacement increases the probability of costal nerve impingement and tissue inflammation producing unilateral, sharp, subcostal and upper-abdominal pain.
In this patient, slipping rib syndrome is a possible diagnosis because of the location of the pain and because the pain described by the patient is highly suggestive of neuropathic pain.
Slipping rib syndrome is diagnosed clinically by a “hooking” maneuver: the clinician hooks his or her fingers at the patient’s subcostal area, reproducing the pain by movement of the ribs anteriorly.24 When this test is performed in our patient the result is negative, ruling out slipping rib syndrome.
THE WORKUP CONTINUES
A complete blood cell count and comprehensive metabolic panel are within normal limits. Abdominal duplex ultrasonography reveals no celiac or mesenteric occlusions, thus ruling out chronic mesenteric ischemia.
Noncontrast CT shows no renal or ureteric stones and no evidence of bleeding in the urinary tract. CT with contrast shows no bowel distention, no evidence of hernia, and a normal appendix and ovaries.
2. After exclusion of the previous choices, which of the following is the most likely cause of her symptoms?
- Anterior cutaneous nerve entrapment syndrome (ACNES)
- Ovarian cyst
- Renal stones
- Appendicitis
- Ventral hernia
- Median arcuate ligament syndrome
ANTERIOR CUTANEOUS NERVE ENTRAPMENT SYNDROME
ACNES is the most likely diagnosis. A study published in 2013 indicated that many cases of functional abdominal pain may actually be undiagnosed cases of chronic abdominal wall pain such as ACNES.25 The condition, first described in 1972,26 is thought to be caused by thoracic cutaneous intercostal nerve entrapment between the abdominal muscles, causing pain at the point of entrapment.
The patient may present with pain that is either acute or chronic. Acute pain is localized more on the right side close to an old scar, or at the outer edge of the rectus abdominis muscle. The pain may vary from dull to burning to sharp; it can radiate horizontally in the upper half of the abdomen or obliquely in the lower half of the abdomen with movements such as twisting and sitting up.27
Despite the acute pain, patients are able to carry on daily functions. The pain may be alleviated by lying down.
The pain may be misdiagnosed as gynecologic or renal. In younger men, the pain may raise concern about hernia, and in older patients, cancer.27 Patients may complain of chronic intermittent pain, usually unilateral, and to a lesser extent bilateral.27
The anatomic location of the pain usually reflects the intercostal nerve involved. The pain is not related to eating or to bowel movements.25 Some patients report exacerbation upon coughing or standing, during menses, and with use of oral contraceptives.28,29 When inquiring about surgical history, it is common to find that the patient has had multiple abdominal surgical procedures.
On examination, the patient has nondistressing pain, with a hand often placed over the painful area.27 On firm palpation, a tender spot of less than 2 cm can be detected.
The diagnosis can be confirmed with a positive Carnett test. The patient lies supine on the examination table with the arms crossed over the chest, then elevates the head or the feet to tense the abdominal muscles.26,27 If doing so reproduces the pain (ie, a positive test), this increases the suspicion of ACNES; if the pain decreases or is not reproducible, an intra-abdominal cause is more likely.
If the pain is difficult to localize, the “pinch test” can be done by using the thumb and index finger to pinch and lift the skin of the abdomen, including the subcutaneous layer of fat, first on one side and then on the other. This helps determine the side with greater pain.27
OVARIAN CYSTS
Ovarian cysts are fluid-filled sacs on the surface of or within the ovary. They are often benign and require no intervention. However, 5% to 10% of US women with a suspicious ovarian mass undergo a surgical procedure, and 13% to 21% of these are found to have a malignancy.30,31
Ovarian cysts are usually painless unless complicated by rupture or bleeding. Patients who present with pain describe it as dull and aching and in the abdomen or pelvis. In rare cases, ovarian cysts can be large enough to cause pain from torsion. Other symptoms may include delayed menses and bleeding outside of the menstrual period.32–34
Ovarian cysts are thought to be caused by hormonal changes during the menstrual cycle. They can be detected during pelvic examination or during pelvic ultrasonography. Cysts that are primarily fluid-filled are generally benign and require no intervention. On the other hand, cysts composed of solid material require intervention.
Treatment depends on several factors, including size and type of cyst, the patient’s age, and whether torsion is present. Treatment can range from observation to medical or surgical management. Laparoscopic surgery is commonly used when surgical treatment is warranted.
RENAL STONES
From 10% to 15% of US adults develop a kidney stone at some time during their life.35 There is no single cause, but one factor that promotes stone formation is a greater amount of crystal-forming substances in the urine, such as calcium, oxalate, and uric acid.36 Most renal stones are calcium oxalate, uric acid, struvite, or cysteine.
Symptoms arise when the stone moves within the urinary tract. Patients present to the emergency room in severe distress, usually with flank pain that radiates to the lower abdomen or groin. The pain is episodic, fluctuates in intensity, and may present with dysuria, frequency, or urgency. It is also associated with nausea and vomiting.37
Renal stones are diagnosed through a series of laboratory and imaging studies. Imaging studies include plain radiography (which can miss small stones), renal sonography, and computed tomography without contrast.
APPENDICITIS
In the United States, the lifetime risk of developing appendicitis is 8.6% in men and 6.7% in women.38 Appendicitis is one of the most common reasons for emergency surgery.
Appendicitis is thought to result from obstruction by fecal matter blocking the opening of the appendix or from a viral infection (eg, with an adenovirus).39,40 The resulting bacterial growth can cause the appendix to become inflamed and purulent.
Patients typically present with umbilical or epigastric pain radiating to the right lower quadrant of the abdomen. Over time, the pain becomes sharper. Certain movements can exacerbate the pain, and lying down may alleviate it. Other symptoms are nausea, vomiting, loss of appetite, and low-grade fever.
Findings on the abdominal examination that help to confirm the diagnosis include rigidity and tenderness, classically localized to a point two-thirds of the way from the umbilicus to the anterior superior iliac spine. Rebound tenderness is usually present. Up to 25% of cases in some series presented atypically, with variable location and findings on physical examination (eg, bowel irregularities, indigestion, flatulence, generalized malaise). In addition to the physical examination, laboratory testing and imaging (ultrasonography, CT) may aid in confirming the diagnosis of appendicitis or any other cause of the pain.38
VENTRAL HERNIA
Ventral hernia is a bulging of abdominal organs or other tissues through a defect of the musculature of the abdominal wall. Ventral hernia is categorized by its location as epigastric, abdominal, or incisional. An open abdominal procedure is the cause in nearly 10% of cases41; the herniation occurs with weakening of the surgical scar.
Ventral hernia is usually detected on physical examination, and patients may present after noting a bulge in the abdominal wall. Symptoms vary. Some patients have no symptoms, while others have mild abdominal discomfort or severe abdominal pain as well as nausea and vomiting. Imaging with CT, ultrasonography, or magnetic resonance imaging helps confirm the diagnosis. Complications of ventral hernia include incarceration and bowel strangulation.
MEDIAN ARCUATE LIGAMENT SYNDROME
Median arcuate ligament syndrome is a challenging diagnosis and a very rare cause of abdominal pain. It is thought to be caused by celiac artery compression by fibroligamentous bands. Pain fluctuates with respiration and is greater during expiration.
Patients may present with recurrent episodes of crampy postprandial pain that cause them to avoid eating, resulting in weight loss. The pain may be associated with nausea, vomiting, and bloating.
The diagnosis is confirmed by duplex ultrasonography, angiography, or magnetic resonance angiography. Treatment is surgical division of the fibroligamentous band and crus, and this is often done laparascopically. In patients with severe persistent celiac artery stenosis, angioplasty and stenting may be considered.2
CASE CONTINUED
Before the physical examination, our patient identifies the location of her pain. A Carnett test is performed, as for ACNES: the patient is placed in the supine position and is instructed to cross both arms over her chest. In an effort to promote muscle tension, she is asked to elevate her head off the examination table, as if performing a mini sit-up, and as she does this, pressure is applied to the identified tender area. The pain is easily reproduced, further confirming involvement of the abdominal wall rather than the viscera. After this, electromyography shows abnormal findings. The patient is then referred to the pain management clinic for a diagnostic nerve block.
3. Which of the following is the first-line treatment of ACNES?
- Local injection of anesthetic
- Surgical neurectomy
LOCAL INJECTION OF ANESTHETIC
Local injection of anesthetic is the first-line treatment of ACNES.
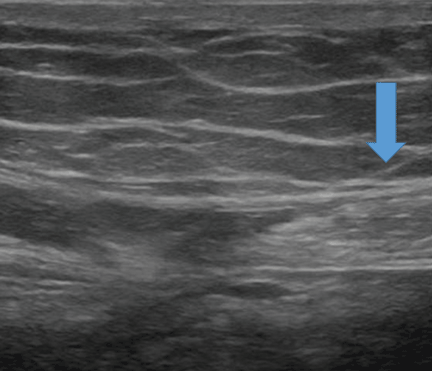
Since ACNES is underdiagnosed, the patient may be less likely to be familiar with it. He or she should receive a detailed explanation of the condition and its management; this will help achieve a successful outcome.
Local anesthetic injection is used for both diagnosis and treatment; 2% lidocaine (or an equivalent) or dehydrated (absolute) alcohol or both can eliminate the pain caused by ACNES. The injection is commonly done under ultrasonographic guidance (Figure 1).42
Complete pain relief may be achieved with a single injection, but some patients require up to five injections.
The adjuvant use of corticosteroids in ACNES to reduce inflammation is controversial.
If anesthetic injections bring only minimal pain relief or if the patient has nerve entrapment in a scar, then surgical neurectomy is an option.43 The procedure is performed under local anesthesia, as the patient’s response aids in identifying the specific nerve or nerves involved.
RETURNING TO THE PATIENT
After a long discussion with our patient about ACNES and the treatment options, she agrees to undergo nerve block in the hope of relieving her pain. She receives a 0.5-mL injection of 2% lidocaine subcutaneously, and within minutes she reports relief of pain. She cannot believe that with a simple injection her pain was relieved. We advise her to return if her pain recurs or if new symptoms arise.
KEEP ACNES IN MIND
ACNES is one of the most commonly misdiagnosed conditions of patients presenting to the outpatient clinic with acute or chronic abdominal pain. This is because the focus is directed to intra-abdominal causes. But if ACNES is kept in consideration from the beginning of the patient encounter, extensive testing, time, and patient anxiety may be reduced significantly. A simple physical examination and the Carnett test aid in raising suspicion of ACNES. If ACNES is confirmed, ultrasonographically guided local anesthetic injection is both diagnostic and therapeutic.
- American Gastroenterological Association Medical Position Statement: Guidelines On Intestinal Ischemia. Gastroenterology 2000; 118:951–953.
- Bobadilla JL. Mesenteric ischemia. Surg Clin North Am 2013; 93:925–940.
- Sung JJ, Kuipers EJ, El-Serag HB. Systematic review: the global incidence and prevalence of peptic ulcer disease. Aliment Pharmacol Ther 2009; 29:938–946.
- Najm WI. Peptic ulcer disease. Prim Care 2011; 38:383–394.
- Malfertheiner P, Chan FK, McColl KE. Peptic ulcer disease. Lancet 2009; 374:1449–1461.
- Chan FK, Leung WK. Peptic-ulcer disease. Lancet 2002; 360:933–941.
- Bright-Asare P, Habte T, Yirgou B, Benjamin J. Prostaglandins, H2-receptor antagonists and peptic ulcer disease. Drugs 1988; 35(suppl 3):1–9.
- Hudson N, Balsitis M, Everitt S, Hawkey CJ. Enhanced gastric mucosal leukotriene B4 synthesis in patients taking non-steroidal anti-inflammatory drugs. Gut 1993; 34:742–747.
- Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature 1980; 286:264–265.
- Bokoch GM, Reed PW. Effect of various lipoxygenase metabolites of arachidonic acid on degranulation of polymorphonuclear leukocytes. J Biol Chem 1981; 256:5317–5320.
- Whittle BJ, Oren-Wolman N, Guth PH. Gastric vasoconstrictor actions of leukotriene C4, PGF2 alpha, and thromboxane mimetic U-46619 on rat submucosal microcirculation in vivo. Am J Physiol 1985; 248:G580–G586.
- Pihan G, Rogers C, Szabo S. Vascular injury in acute gastric mucosal damage. Mediatory role of leukotrienes. Dig Dis Sci 1988; 33:625–632.
- Ramakrishnan K, Salinas RC. Peptic ulcer disease. Am Fam Physician 2007; 76:1005–1012.
- Parmet S, Lynm C, Glass RM. JAMA patient page. Acute cholecystitis. JAMA 2003; 289:124.
- Roslyn JJ, DenBesten L, Thompson JE Jr, Silverman BF. Roles of lithogenic bile and cystic duct occlusion in the pathogenesis of acute cholecystitis. Am J Surg 1980; 140:126–130.
- Kaminski DL. Arachidonic acid metabolites in hepatobiliary physiology and disease. Gastroenterology 1989; 97:781–792.
- Jivegård L, Thornell E, Svanvik J. Pathophysiology of acute obstructive cholecystitis: implications for non-operative management. Br J Surg 1987; 74:1084–1086.
- Csendes A, Burdiles P, Maluenda F, Diaz JC, Csendes P, Mitru N. Simultaneous bacteriologic assessment of bile from gallbladder and common bile duct in control subjects and patients with gallstones and common duct stones. Arch Surg 1996; 131:389–394.
- Barie PS, Fischer E. Acute acalculous cholecystitis. J Am Coll Surg 1995; 180:232–244.
- Shapiro MJ, Luchtefeld WB, Kurzweil S, Kaminski DL, Durham RM, Mazuski JE. Acute acalculous cholecystitis in the critically ill. Am Surg 1994; 60:335–339.
- Savoca PE, Longo WE, Zucker KA, McMillen MM, Modlin IM. The increasing prevalence of acalculous cholecystitis in outpatients. Results of a 7-year study. Ann Surg 1990; 211:433–437.
- Gofrit O, Eid A, Pikarsky A, Lebensart PD, Pizov G, Rivkind A. Cholesterol embolisation causing chronic acalculous cholecystitis. Eur J Surg 1996; 162:243–245.
- McChesney JA, Northup PG, Bickston SJ. Acute acalculous cholecystitis associated with systemic sepsis and visceral arterial hypoperfusion: a case series and review of pathophysiology. Dig Dis Sci 2003; 48:1960–1967.
- Aeschlimann A, Kahn MF. Tietze’s syndrome: a critical review. Clin Exp Rheumatol 1990; 8:407–412.
- van Assen T, de Jager-Kievit JW, Scheltinga MR, Roumen RM. Chronic abdominal wall pain misdiagnosed as functional abdominal pain. J Am Board Fam Med 2013; 26:738–744.
- Akhnikh S, de Korte N, de Winter P. Anterior cutaneous nerve entrapment syndrome (ACNES): the forgotten diagnosis. Eur J Pediatr 2014; 173:445–449.
- Applegate WV. Abdominal cutaneous nerve entrapment syndrome (ACNES): a commonly overlooked cause of abdominal pain. Perm J 2002; 6:20–27.
- Grover M. UNC Center for Functional GI & Motility Disorders. Chronic abdominal wall pain: a missed diagnosis. www.med.unc.edu/ibs/files/educational-gi-handouts/Chronic%20Abdominal%20Pain.pdf. Accessed September 9, 2015.
- Greenbaum D, Dawson F, Watson R. Chronic abdominal wall pain (CAWP): a common but frequently overlooked disorder. Poster presented at the World Congress of Gastroenterology, Sydney, Australia, August 26–31, 1990.
- National Institutes of Health Consensus Development Conference Statement. Ovarian cancer: screening, treatment, and follow-up. Gynecol Oncol 1994; 55:S4–S14.
- Koonings PP, Campbell K, Mishell DR Jr, Grimes DA. Relative frequency of primary ovarian neoplasms: a 10-year review. Obstet Gynecol 1989; 74:921–926.
- Givens V, Mitchell GE, Harraway-Smith C, Reddy A, Maness DL. Diagnosis and management of adnexal masses. Am Fam Physician 2009; 80:815–820.
- Goff BA, Mandel L, Muntz HG, Melancon CH. Ovarian carcinoma diagnosis. Cancer 2000; 89:2068–2075.
- Friedman GD, Skilling JS, Udaltsova NV, Smith LH. Early symptoms of ovarian cancer: a case-control study without recall bias. Fam Pract 2005; 22:548–553.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976-1994. Kidney Int 2003; 63:1817–1823.
- Worcester EM, Coe FL. Clinical practice. Calcium kidney stones. N Engl J Med 2010; 363:954–963.
- Miller NL, Lingeman JE. Management of kidney stones. BMJ 2007; 334:468–472.
- Lewis SR, Mahony PJ, Simpson J. Appendicitis. BMJ 2011; 343:d5976.
- Lamps LW. Infectious causes of appendicitis. Infect Dis Clin North Am 2010; 24:995–1018.
- Reif RM. Viral appendicitis. Hum Pathol 1981; 12:193–196.
- Akkary E, Panait L, Roberts K, Duffy A, Bell R. Sutureless laparoscopic ventral hernia repair in obese patients. JSLS 2011; 15:154–159.
- Boelens OB, Scheltinga MR, Houterman S, Roumen RM. Randomized clinical trial of trigger point infiltration with lidocaine to diagnose anterior cutaneous nerve entrapment syndrome. Br J Surg 2013; 100:217–221.
- Boelens OB, Scheltinga MR, Houterman S, Roumen RM. Management of anterior cutaneous nerve entrapment syndrome in a cohort of 139 patients. Ann Surg 2011; 254:1054–1058.
A 31-year-old woman presents to the office with a chief complaint of right mid-abdominal pain that began 1 day ago. She says she did not seek medical attention earlier because she had to be at work that morning and she thought the pain would resolve on its own.
She reports no fever, headache, anorexia, nausea, vomiting, malaise, loss of weight, melena, or changes in bowel habits. She describes the pain as sharp, localized to the right side, and radiating to the vulva upon sitting up. She denies any association of pain with current dietary habits or bowel function. She has no recollection of precipitating or alleviating factors, including the use of analgesics to reduce the pain.
On further discussion, she mentions that 1 year ago she began experiencing chronic abdominal pain, which she says is sometimes exacerbated by coughing, by standing for extended periods of time, and during menses, and is alleviated upon lying down.
She has regular menstrual periods, and her last one ended 7 days ago.
Her surgical history includes two uncomplicated cesarean deliveries. She does not use tobacco, alcohol, or illicit substances. She is not aware of any allergies to drugs or foods.
She appears to be in no acute distress and has been sitting quietly thus far. She seems to have positioned her hand on her abdomen over the corresponding area of pain.
On physical examination, vital signs are within normal limits, and she is alert and oriented to person, place, and time. Her sclerae are anicteric, and the pupils are equal, round, and reactive to light.
Cardiovascular and pulmonary examinations are also within normal limits. Examination of the abdomen elicits tenderness and guarding along the lateral border of the rectus abdominis muscle on the right side at the level of umbilicus, with no rebound tenderness or rigidity. The liver and spleen are not enlarged, and no abdominal mass is detected. No skin rash, joint swelling, or peripheral edema is noted. A neurologic examination is normal.
1. With the information provided, which of the following is least likely to be causing her symptoms?
- Chronic mesenteric ischemia
- Peptic ulcer
- Acute cholecystitis
- Slipping rib syndrome
CHRONIC MESENTERIC ISCHEMIA
Chronic mesenteric ischemia is the least likely diagnosis because the patient lacks risk factors for atherosclerosis and because she does not have postprandial pain, which is pathognomonic for chronic mesenteric ischemia. It is thought to be caused by a decrease in blood flow through the splanchnic vessels.1 Symptoms tend to arise after eating because of a postprandial increase in metabolic demands.1 These patients also often have atherosclerotic risk factors such as hypertension, hyperlipidemia, and smoking causing coronary artery disease, or a history of stroke.
The primary symptom is abdominal pain, most often described as achy, crampy, or spastic episodes of pain, usually occurring within 2 hours of eating.2 Weight loss is common, as patients can develop a fear of eating. Postprandial pain may also be associated with nausea, vomiting, and bloating.
Findings on clinical examination are usually less severe than the actual symptoms. Visceral duplex or multidetector computed tomography (CT) is an excellent tool to detect blood flow in potential stenotic vessels.2
PEPTIC ULCER DISEASE
Peptic ulcer disease is not a likely diagnosis in this patient because she has no history of taking nonsteroidal anti-inflammatory drugs (NSAIDs).
A study of US patients between 1997 and 2007 reported an annual incidence of peptic ulcer disease of 0.05% to 0.19% depending on the method of diagnosis.3 Peptic ulcer is thought to result from increased gastric acid secretion with a resultant inflammatory response, leading to erosion and ulceration.
The most common possible catalysts include Helicobacter pylori infection, NSAIDs, smoking, alcohol use, and hypersecretory states such as Zollinger-Ellison syndrome.4–6 Complications include internal bleeding, perforation causing peritonitis, and penetration to adjacent organs.
Pathophysiology
Peptic ulcer is the result of an increase in the normal level of gastric acid and a decrease in the protective ability of the gastric mucosa.7 Cytoprotection may be lost through a decrease in the products of arachidonic acid metabolism (eg, prostaglandins, which have a protective effect) or an increase in leukotriene B4 (LTB4), which has a damaging effect. Prostaglandins are thought not only to protect the normal gastric mucosa, but also to provide an antisecretory effect.
On the other hand, leukotrienes—specifically LTB4 and LTC4—are proinflammatory agents and can damage the gastric mucosa. NSAIDs enhance the production of leukotrienes through the 5-lipoxygenase pathway. The ability of LTB4 to cause degranulation and release of lysosomal enzymes may play a vital role in the inflammatory response to NSAIDs.8–10 LTC4 may promote gastric mucosal damage through a reduction of tissue perfusion resulting from the promotion of vascular stasis.8,11,12
Symptoms help differentiate ulcer type
The classic symptom is burning epigastric pain after meals. Pain that occurs immediately after meals is a classic symptom of gastric ulcer. Pain that occurs 2 to 3 hours after meals and that is relieved by food or antacids is a strong indicator of duodenal ulcer.13 Other symptoms include dyspepsia, bloating, distention, heartburn, and chest discomfort.13
Accurate diagnosis is vital in selecting the proper treatment. Diagnostic tests may include H pylori testing, upper-gastrointestinal endoscopy, and radiography with barium swallow.
CHOLECYSTITIS
In cholecystitis, the primary complaint is pain, usually in the right upper quadrant of the abdomen. Patients describe sudden, sharp, and intense pain that radiates to the back or shoulder. Patients may report pain after heavy meals, and some report nausea and vomiting. Cholecystitis is in the differential diagnosis of this patient because of the anatomic location of her pain.
The diagnosis is confirmed by imaging. Abdominal ultrasonography, technetium-99m hepatic iminodiacetic acid scanning, and CT are the most commonly used studies.14
Cholecystitis can be acute or chronic. Acute cholecystitis is categorized as calculous or acalculous. Calculous cholecystitis is multifactorial, but the primary cause is blockage of the cystic duct by gallstones.15 Other factors include irritants such as lysolecithin (released during bile stasis), which can trigger gallbladder inflammation,15–17 and infection.18
When the cystic duct is blocked, bile builds up inside the gallbladder, causing irritation and inflammation of the walls of the gallbladder.14
Acalculous cholecystitis, which resembles calculous cholecystitis but without the gallstones,19 accounts for 2% to 15% of all cases of acute cholecystitis.19,20 It has been observed in hospitalized critically ill patients, but it can also present in an outpatient setting, most often in elderly men with vascular disease.21 Causes include infection, trauma, and tumor obstruction, resulting in endothelial injury, gallbladder stasis, ischemia, and eventually necrosis.14,20,22,23
SLIPPING RIB SYNDROME
Slipping rib syndrome, also known as Tietze syndrome, is believed to be caused by hypermobile costal cartilage. The affected rib slips behind the rib above on contraction of the abdominal wall. This displacement increases the probability of costal nerve impingement and tissue inflammation producing unilateral, sharp, subcostal and upper-abdominal pain.
In this patient, slipping rib syndrome is a possible diagnosis because of the location of the pain and because the pain described by the patient is highly suggestive of neuropathic pain.
Slipping rib syndrome is diagnosed clinically by a “hooking” maneuver: the clinician hooks his or her fingers at the patient’s subcostal area, reproducing the pain by movement of the ribs anteriorly.24 When this test is performed in our patient the result is negative, ruling out slipping rib syndrome.
THE WORKUP CONTINUES
A complete blood cell count and comprehensive metabolic panel are within normal limits. Abdominal duplex ultrasonography reveals no celiac or mesenteric occlusions, thus ruling out chronic mesenteric ischemia.
Noncontrast CT shows no renal or ureteric stones and no evidence of bleeding in the urinary tract. CT with contrast shows no bowel distention, no evidence of hernia, and a normal appendix and ovaries.
2. After exclusion of the previous choices, which of the following is the most likely cause of her symptoms?
- Anterior cutaneous nerve entrapment syndrome (ACNES)
- Ovarian cyst
- Renal stones
- Appendicitis
- Ventral hernia
- Median arcuate ligament syndrome
ANTERIOR CUTANEOUS NERVE ENTRAPMENT SYNDROME
ACNES is the most likely diagnosis. A study published in 2013 indicated that many cases of functional abdominal pain may actually be undiagnosed cases of chronic abdominal wall pain such as ACNES.25 The condition, first described in 1972,26 is thought to be caused by thoracic cutaneous intercostal nerve entrapment between the abdominal muscles, causing pain at the point of entrapment.
The patient may present with pain that is either acute or chronic. Acute pain is localized more on the right side close to an old scar, or at the outer edge of the rectus abdominis muscle. The pain may vary from dull to burning to sharp; it can radiate horizontally in the upper half of the abdomen or obliquely in the lower half of the abdomen with movements such as twisting and sitting up.27
Despite the acute pain, patients are able to carry on daily functions. The pain may be alleviated by lying down.
The pain may be misdiagnosed as gynecologic or renal. In younger men, the pain may raise concern about hernia, and in older patients, cancer.27 Patients may complain of chronic intermittent pain, usually unilateral, and to a lesser extent bilateral.27
The anatomic location of the pain usually reflects the intercostal nerve involved. The pain is not related to eating or to bowel movements.25 Some patients report exacerbation upon coughing or standing, during menses, and with use of oral contraceptives.28,29 When inquiring about surgical history, it is common to find that the patient has had multiple abdominal surgical procedures.
On examination, the patient has nondistressing pain, with a hand often placed over the painful area.27 On firm palpation, a tender spot of less than 2 cm can be detected.
The diagnosis can be confirmed with a positive Carnett test. The patient lies supine on the examination table with the arms crossed over the chest, then elevates the head or the feet to tense the abdominal muscles.26,27 If doing so reproduces the pain (ie, a positive test), this increases the suspicion of ACNES; if the pain decreases or is not reproducible, an intra-abdominal cause is more likely.
If the pain is difficult to localize, the “pinch test” can be done by using the thumb and index finger to pinch and lift the skin of the abdomen, including the subcutaneous layer of fat, first on one side and then on the other. This helps determine the side with greater pain.27
OVARIAN CYSTS
Ovarian cysts are fluid-filled sacs on the surface of or within the ovary. They are often benign and require no intervention. However, 5% to 10% of US women with a suspicious ovarian mass undergo a surgical procedure, and 13% to 21% of these are found to have a malignancy.30,31
Ovarian cysts are usually painless unless complicated by rupture or bleeding. Patients who present with pain describe it as dull and aching and in the abdomen or pelvis. In rare cases, ovarian cysts can be large enough to cause pain from torsion. Other symptoms may include delayed menses and bleeding outside of the menstrual period.32–34
Ovarian cysts are thought to be caused by hormonal changes during the menstrual cycle. They can be detected during pelvic examination or during pelvic ultrasonography. Cysts that are primarily fluid-filled are generally benign and require no intervention. On the other hand, cysts composed of solid material require intervention.
Treatment depends on several factors, including size and type of cyst, the patient’s age, and whether torsion is present. Treatment can range from observation to medical or surgical management. Laparoscopic surgery is commonly used when surgical treatment is warranted.
RENAL STONES
From 10% to 15% of US adults develop a kidney stone at some time during their life.35 There is no single cause, but one factor that promotes stone formation is a greater amount of crystal-forming substances in the urine, such as calcium, oxalate, and uric acid.36 Most renal stones are calcium oxalate, uric acid, struvite, or cysteine.
Symptoms arise when the stone moves within the urinary tract. Patients present to the emergency room in severe distress, usually with flank pain that radiates to the lower abdomen or groin. The pain is episodic, fluctuates in intensity, and may present with dysuria, frequency, or urgency. It is also associated with nausea and vomiting.37
Renal stones are diagnosed through a series of laboratory and imaging studies. Imaging studies include plain radiography (which can miss small stones), renal sonography, and computed tomography without contrast.
APPENDICITIS
In the United States, the lifetime risk of developing appendicitis is 8.6% in men and 6.7% in women.38 Appendicitis is one of the most common reasons for emergency surgery.
Appendicitis is thought to result from obstruction by fecal matter blocking the opening of the appendix or from a viral infection (eg, with an adenovirus).39,40 The resulting bacterial growth can cause the appendix to become inflamed and purulent.
Patients typically present with umbilical or epigastric pain radiating to the right lower quadrant of the abdomen. Over time, the pain becomes sharper. Certain movements can exacerbate the pain, and lying down may alleviate it. Other symptoms are nausea, vomiting, loss of appetite, and low-grade fever.
Findings on the abdominal examination that help to confirm the diagnosis include rigidity and tenderness, classically localized to a point two-thirds of the way from the umbilicus to the anterior superior iliac spine. Rebound tenderness is usually present. Up to 25% of cases in some series presented atypically, with variable location and findings on physical examination (eg, bowel irregularities, indigestion, flatulence, generalized malaise). In addition to the physical examination, laboratory testing and imaging (ultrasonography, CT) may aid in confirming the diagnosis of appendicitis or any other cause of the pain.38
VENTRAL HERNIA
Ventral hernia is a bulging of abdominal organs or other tissues through a defect of the musculature of the abdominal wall. Ventral hernia is categorized by its location as epigastric, abdominal, or incisional. An open abdominal procedure is the cause in nearly 10% of cases41; the herniation occurs with weakening of the surgical scar.
Ventral hernia is usually detected on physical examination, and patients may present after noting a bulge in the abdominal wall. Symptoms vary. Some patients have no symptoms, while others have mild abdominal discomfort or severe abdominal pain as well as nausea and vomiting. Imaging with CT, ultrasonography, or magnetic resonance imaging helps confirm the diagnosis. Complications of ventral hernia include incarceration and bowel strangulation.
MEDIAN ARCUATE LIGAMENT SYNDROME
Median arcuate ligament syndrome is a challenging diagnosis and a very rare cause of abdominal pain. It is thought to be caused by celiac artery compression by fibroligamentous bands. Pain fluctuates with respiration and is greater during expiration.
Patients may present with recurrent episodes of crampy postprandial pain that cause them to avoid eating, resulting in weight loss. The pain may be associated with nausea, vomiting, and bloating.
The diagnosis is confirmed by duplex ultrasonography, angiography, or magnetic resonance angiography. Treatment is surgical division of the fibroligamentous band and crus, and this is often done laparascopically. In patients with severe persistent celiac artery stenosis, angioplasty and stenting may be considered.2
CASE CONTINUED
Before the physical examination, our patient identifies the location of her pain. A Carnett test is performed, as for ACNES: the patient is placed in the supine position and is instructed to cross both arms over her chest. In an effort to promote muscle tension, she is asked to elevate her head off the examination table, as if performing a mini sit-up, and as she does this, pressure is applied to the identified tender area. The pain is easily reproduced, further confirming involvement of the abdominal wall rather than the viscera. After this, electromyography shows abnormal findings. The patient is then referred to the pain management clinic for a diagnostic nerve block.
3. Which of the following is the first-line treatment of ACNES?
- Local injection of anesthetic
- Surgical neurectomy
LOCAL INJECTION OF ANESTHETIC
Local injection of anesthetic is the first-line treatment of ACNES.
Since ACNES is underdiagnosed, the patient may be less likely to be familiar with it. He or she should receive a detailed explanation of the condition and its management; this will help achieve a successful outcome.
Local anesthetic injection is used for both diagnosis and treatment; 2% lidocaine (or an equivalent) or dehydrated (absolute) alcohol or both can eliminate the pain caused by ACNES. The injection is commonly done under ultrasonographic guidance (Figure 1).42
Complete pain relief may be achieved with a single injection, but some patients require up to five injections.
The adjuvant use of corticosteroids in ACNES to reduce inflammation is controversial.
If anesthetic injections bring only minimal pain relief or if the patient has nerve entrapment in a scar, then surgical neurectomy is an option.43 The procedure is performed under local anesthesia, as the patient’s response aids in identifying the specific nerve or nerves involved.
RETURNING TO THE PATIENT
After a long discussion with our patient about ACNES and the treatment options, she agrees to undergo nerve block in the hope of relieving her pain. She receives a 0.5-mL injection of 2% lidocaine subcutaneously, and within minutes she reports relief of pain. She cannot believe that with a simple injection her pain was relieved. We advise her to return if her pain recurs or if new symptoms arise.
KEEP ACNES IN MIND
ACNES is one of the most commonly misdiagnosed conditions of patients presenting to the outpatient clinic with acute or chronic abdominal pain. This is because the focus is directed to intra-abdominal causes. But if ACNES is kept in consideration from the beginning of the patient encounter, extensive testing, time, and patient anxiety may be reduced significantly. A simple physical examination and the Carnett test aid in raising suspicion of ACNES. If ACNES is confirmed, ultrasonographically guided local anesthetic injection is both diagnostic and therapeutic.
A 31-year-old woman presents to the office with a chief complaint of right mid-abdominal pain that began 1 day ago. She says she did not seek medical attention earlier because she had to be at work that morning and she thought the pain would resolve on its own.
She reports no fever, headache, anorexia, nausea, vomiting, malaise, loss of weight, melena, or changes in bowel habits. She describes the pain as sharp, localized to the right side, and radiating to the vulva upon sitting up. She denies any association of pain with current dietary habits or bowel function. She has no recollection of precipitating or alleviating factors, including the use of analgesics to reduce the pain.
On further discussion, she mentions that 1 year ago she began experiencing chronic abdominal pain, which she says is sometimes exacerbated by coughing, by standing for extended periods of time, and during menses, and is alleviated upon lying down.
She has regular menstrual periods, and her last one ended 7 days ago.
Her surgical history includes two uncomplicated cesarean deliveries. She does not use tobacco, alcohol, or illicit substances. She is not aware of any allergies to drugs or foods.
She appears to be in no acute distress and has been sitting quietly thus far. She seems to have positioned her hand on her abdomen over the corresponding area of pain.
On physical examination, vital signs are within normal limits, and she is alert and oriented to person, place, and time. Her sclerae are anicteric, and the pupils are equal, round, and reactive to light.
Cardiovascular and pulmonary examinations are also within normal limits. Examination of the abdomen elicits tenderness and guarding along the lateral border of the rectus abdominis muscle on the right side at the level of umbilicus, with no rebound tenderness or rigidity. The liver and spleen are not enlarged, and no abdominal mass is detected. No skin rash, joint swelling, or peripheral edema is noted. A neurologic examination is normal.
1. With the information provided, which of the following is least likely to be causing her symptoms?
- Chronic mesenteric ischemia
- Peptic ulcer
- Acute cholecystitis
- Slipping rib syndrome
CHRONIC MESENTERIC ISCHEMIA
Chronic mesenteric ischemia is the least likely diagnosis because the patient lacks risk factors for atherosclerosis and because she does not have postprandial pain, which is pathognomonic for chronic mesenteric ischemia. It is thought to be caused by a decrease in blood flow through the splanchnic vessels.1 Symptoms tend to arise after eating because of a postprandial increase in metabolic demands.1 These patients also often have atherosclerotic risk factors such as hypertension, hyperlipidemia, and smoking causing coronary artery disease, or a history of stroke.
The primary symptom is abdominal pain, most often described as achy, crampy, or spastic episodes of pain, usually occurring within 2 hours of eating.2 Weight loss is common, as patients can develop a fear of eating. Postprandial pain may also be associated with nausea, vomiting, and bloating.
Findings on clinical examination are usually less severe than the actual symptoms. Visceral duplex or multidetector computed tomography (CT) is an excellent tool to detect blood flow in potential stenotic vessels.2
PEPTIC ULCER DISEASE
Peptic ulcer disease is not a likely diagnosis in this patient because she has no history of taking nonsteroidal anti-inflammatory drugs (NSAIDs).
A study of US patients between 1997 and 2007 reported an annual incidence of peptic ulcer disease of 0.05% to 0.19% depending on the method of diagnosis.3 Peptic ulcer is thought to result from increased gastric acid secretion with a resultant inflammatory response, leading to erosion and ulceration.
The most common possible catalysts include Helicobacter pylori infection, NSAIDs, smoking, alcohol use, and hypersecretory states such as Zollinger-Ellison syndrome.4–6 Complications include internal bleeding, perforation causing peritonitis, and penetration to adjacent organs.
Pathophysiology
Peptic ulcer is the result of an increase in the normal level of gastric acid and a decrease in the protective ability of the gastric mucosa.7 Cytoprotection may be lost through a decrease in the products of arachidonic acid metabolism (eg, prostaglandins, which have a protective effect) or an increase in leukotriene B4 (LTB4), which has a damaging effect. Prostaglandins are thought not only to protect the normal gastric mucosa, but also to provide an antisecretory effect.
On the other hand, leukotrienes—specifically LTB4 and LTC4—are proinflammatory agents and can damage the gastric mucosa. NSAIDs enhance the production of leukotrienes through the 5-lipoxygenase pathway. The ability of LTB4 to cause degranulation and release of lysosomal enzymes may play a vital role in the inflammatory response to NSAIDs.8–10 LTC4 may promote gastric mucosal damage through a reduction of tissue perfusion resulting from the promotion of vascular stasis.8,11,12
Symptoms help differentiate ulcer type
The classic symptom is burning epigastric pain after meals. Pain that occurs immediately after meals is a classic symptom of gastric ulcer. Pain that occurs 2 to 3 hours after meals and that is relieved by food or antacids is a strong indicator of duodenal ulcer.13 Other symptoms include dyspepsia, bloating, distention, heartburn, and chest discomfort.13
Accurate diagnosis is vital in selecting the proper treatment. Diagnostic tests may include H pylori testing, upper-gastrointestinal endoscopy, and radiography with barium swallow.
CHOLECYSTITIS
In cholecystitis, the primary complaint is pain, usually in the right upper quadrant of the abdomen. Patients describe sudden, sharp, and intense pain that radiates to the back or shoulder. Patients may report pain after heavy meals, and some report nausea and vomiting. Cholecystitis is in the differential diagnosis of this patient because of the anatomic location of her pain.
The diagnosis is confirmed by imaging. Abdominal ultrasonography, technetium-99m hepatic iminodiacetic acid scanning, and CT are the most commonly used studies.14
Cholecystitis can be acute or chronic. Acute cholecystitis is categorized as calculous or acalculous. Calculous cholecystitis is multifactorial, but the primary cause is blockage of the cystic duct by gallstones.15 Other factors include irritants such as lysolecithin (released during bile stasis), which can trigger gallbladder inflammation,15–17 and infection.18
When the cystic duct is blocked, bile builds up inside the gallbladder, causing irritation and inflammation of the walls of the gallbladder.14
Acalculous cholecystitis, which resembles calculous cholecystitis but without the gallstones,19 accounts for 2% to 15% of all cases of acute cholecystitis.19,20 It has been observed in hospitalized critically ill patients, but it can also present in an outpatient setting, most often in elderly men with vascular disease.21 Causes include infection, trauma, and tumor obstruction, resulting in endothelial injury, gallbladder stasis, ischemia, and eventually necrosis.14,20,22,23
SLIPPING RIB SYNDROME
Slipping rib syndrome, also known as Tietze syndrome, is believed to be caused by hypermobile costal cartilage. The affected rib slips behind the rib above on contraction of the abdominal wall. This displacement increases the probability of costal nerve impingement and tissue inflammation producing unilateral, sharp, subcostal and upper-abdominal pain.
In this patient, slipping rib syndrome is a possible diagnosis because of the location of the pain and because the pain described by the patient is highly suggestive of neuropathic pain.
Slipping rib syndrome is diagnosed clinically by a “hooking” maneuver: the clinician hooks his or her fingers at the patient’s subcostal area, reproducing the pain by movement of the ribs anteriorly.24 When this test is performed in our patient the result is negative, ruling out slipping rib syndrome.
THE WORKUP CONTINUES
A complete blood cell count and comprehensive metabolic panel are within normal limits. Abdominal duplex ultrasonography reveals no celiac or mesenteric occlusions, thus ruling out chronic mesenteric ischemia.
Noncontrast CT shows no renal or ureteric stones and no evidence of bleeding in the urinary tract. CT with contrast shows no bowel distention, no evidence of hernia, and a normal appendix and ovaries.
2. After exclusion of the previous choices, which of the following is the most likely cause of her symptoms?
- Anterior cutaneous nerve entrapment syndrome (ACNES)
- Ovarian cyst
- Renal stones
- Appendicitis
- Ventral hernia
- Median arcuate ligament syndrome
ANTERIOR CUTANEOUS NERVE ENTRAPMENT SYNDROME
ACNES is the most likely diagnosis. A study published in 2013 indicated that many cases of functional abdominal pain may actually be undiagnosed cases of chronic abdominal wall pain such as ACNES.25 The condition, first described in 1972,26 is thought to be caused by thoracic cutaneous intercostal nerve entrapment between the abdominal muscles, causing pain at the point of entrapment.
The patient may present with pain that is either acute or chronic. Acute pain is localized more on the right side close to an old scar, or at the outer edge of the rectus abdominis muscle. The pain may vary from dull to burning to sharp; it can radiate horizontally in the upper half of the abdomen or obliquely in the lower half of the abdomen with movements such as twisting and sitting up.27
Despite the acute pain, patients are able to carry on daily functions. The pain may be alleviated by lying down.
The pain may be misdiagnosed as gynecologic or renal. In younger men, the pain may raise concern about hernia, and in older patients, cancer.27 Patients may complain of chronic intermittent pain, usually unilateral, and to a lesser extent bilateral.27
The anatomic location of the pain usually reflects the intercostal nerve involved. The pain is not related to eating or to bowel movements.25 Some patients report exacerbation upon coughing or standing, during menses, and with use of oral contraceptives.28,29 When inquiring about surgical history, it is common to find that the patient has had multiple abdominal surgical procedures.
On examination, the patient has nondistressing pain, with a hand often placed over the painful area.27 On firm palpation, a tender spot of less than 2 cm can be detected.
The diagnosis can be confirmed with a positive Carnett test. The patient lies supine on the examination table with the arms crossed over the chest, then elevates the head or the feet to tense the abdominal muscles.26,27 If doing so reproduces the pain (ie, a positive test), this increases the suspicion of ACNES; if the pain decreases or is not reproducible, an intra-abdominal cause is more likely.
If the pain is difficult to localize, the “pinch test” can be done by using the thumb and index finger to pinch and lift the skin of the abdomen, including the subcutaneous layer of fat, first on one side and then on the other. This helps determine the side with greater pain.27
OVARIAN CYSTS
Ovarian cysts are fluid-filled sacs on the surface of or within the ovary. They are often benign and require no intervention. However, 5% to 10% of US women with a suspicious ovarian mass undergo a surgical procedure, and 13% to 21% of these are found to have a malignancy.30,31
Ovarian cysts are usually painless unless complicated by rupture or bleeding. Patients who present with pain describe it as dull and aching and in the abdomen or pelvis. In rare cases, ovarian cysts can be large enough to cause pain from torsion. Other symptoms may include delayed menses and bleeding outside of the menstrual period.32–34
Ovarian cysts are thought to be caused by hormonal changes during the menstrual cycle. They can be detected during pelvic examination or during pelvic ultrasonography. Cysts that are primarily fluid-filled are generally benign and require no intervention. On the other hand, cysts composed of solid material require intervention.
Treatment depends on several factors, including size and type of cyst, the patient’s age, and whether torsion is present. Treatment can range from observation to medical or surgical management. Laparoscopic surgery is commonly used when surgical treatment is warranted.
RENAL STONES
From 10% to 15% of US adults develop a kidney stone at some time during their life.35 There is no single cause, but one factor that promotes stone formation is a greater amount of crystal-forming substances in the urine, such as calcium, oxalate, and uric acid.36 Most renal stones are calcium oxalate, uric acid, struvite, or cysteine.
Symptoms arise when the stone moves within the urinary tract. Patients present to the emergency room in severe distress, usually with flank pain that radiates to the lower abdomen or groin. The pain is episodic, fluctuates in intensity, and may present with dysuria, frequency, or urgency. It is also associated with nausea and vomiting.37
Renal stones are diagnosed through a series of laboratory and imaging studies. Imaging studies include plain radiography (which can miss small stones), renal sonography, and computed tomography without contrast.
APPENDICITIS
In the United States, the lifetime risk of developing appendicitis is 8.6% in men and 6.7% in women.38 Appendicitis is one of the most common reasons for emergency surgery.
Appendicitis is thought to result from obstruction by fecal matter blocking the opening of the appendix or from a viral infection (eg, with an adenovirus).39,40 The resulting bacterial growth can cause the appendix to become inflamed and purulent.
Patients typically present with umbilical or epigastric pain radiating to the right lower quadrant of the abdomen. Over time, the pain becomes sharper. Certain movements can exacerbate the pain, and lying down may alleviate it. Other symptoms are nausea, vomiting, loss of appetite, and low-grade fever.
Findings on the abdominal examination that help to confirm the diagnosis include rigidity and tenderness, classically localized to a point two-thirds of the way from the umbilicus to the anterior superior iliac spine. Rebound tenderness is usually present. Up to 25% of cases in some series presented atypically, with variable location and findings on physical examination (eg, bowel irregularities, indigestion, flatulence, generalized malaise). In addition to the physical examination, laboratory testing and imaging (ultrasonography, CT) may aid in confirming the diagnosis of appendicitis or any other cause of the pain.38
VENTRAL HERNIA
Ventral hernia is a bulging of abdominal organs or other tissues through a defect of the musculature of the abdominal wall. Ventral hernia is categorized by its location as epigastric, abdominal, or incisional. An open abdominal procedure is the cause in nearly 10% of cases41; the herniation occurs with weakening of the surgical scar.
Ventral hernia is usually detected on physical examination, and patients may present after noting a bulge in the abdominal wall. Symptoms vary. Some patients have no symptoms, while others have mild abdominal discomfort or severe abdominal pain as well as nausea and vomiting. Imaging with CT, ultrasonography, or magnetic resonance imaging helps confirm the diagnosis. Complications of ventral hernia include incarceration and bowel strangulation.
MEDIAN ARCUATE LIGAMENT SYNDROME
Median arcuate ligament syndrome is a challenging diagnosis and a very rare cause of abdominal pain. It is thought to be caused by celiac artery compression by fibroligamentous bands. Pain fluctuates with respiration and is greater during expiration.
Patients may present with recurrent episodes of crampy postprandial pain that cause them to avoid eating, resulting in weight loss. The pain may be associated with nausea, vomiting, and bloating.
The diagnosis is confirmed by duplex ultrasonography, angiography, or magnetic resonance angiography. Treatment is surgical division of the fibroligamentous band and crus, and this is often done laparascopically. In patients with severe persistent celiac artery stenosis, angioplasty and stenting may be considered.2
CASE CONTINUED
Before the physical examination, our patient identifies the location of her pain. A Carnett test is performed, as for ACNES: the patient is placed in the supine position and is instructed to cross both arms over her chest. In an effort to promote muscle tension, she is asked to elevate her head off the examination table, as if performing a mini sit-up, and as she does this, pressure is applied to the identified tender area. The pain is easily reproduced, further confirming involvement of the abdominal wall rather than the viscera. After this, electromyography shows abnormal findings. The patient is then referred to the pain management clinic for a diagnostic nerve block.
3. Which of the following is the first-line treatment of ACNES?
- Local injection of anesthetic
- Surgical neurectomy
LOCAL INJECTION OF ANESTHETIC
Local injection of anesthetic is the first-line treatment of ACNES.
Since ACNES is underdiagnosed, the patient may be less likely to be familiar with it. He or she should receive a detailed explanation of the condition and its management; this will help achieve a successful outcome.
Local anesthetic injection is used for both diagnosis and treatment; 2% lidocaine (or an equivalent) or dehydrated (absolute) alcohol or both can eliminate the pain caused by ACNES. The injection is commonly done under ultrasonographic guidance (Figure 1).42
Complete pain relief may be achieved with a single injection, but some patients require up to five injections.
The adjuvant use of corticosteroids in ACNES to reduce inflammation is controversial.
If anesthetic injections bring only minimal pain relief or if the patient has nerve entrapment in a scar, then surgical neurectomy is an option.43 The procedure is performed under local anesthesia, as the patient’s response aids in identifying the specific nerve or nerves involved.
RETURNING TO THE PATIENT
After a long discussion with our patient about ACNES and the treatment options, she agrees to undergo nerve block in the hope of relieving her pain. She receives a 0.5-mL injection of 2% lidocaine subcutaneously, and within minutes she reports relief of pain. She cannot believe that with a simple injection her pain was relieved. We advise her to return if her pain recurs or if new symptoms arise.
KEEP ACNES IN MIND
ACNES is one of the most commonly misdiagnosed conditions of patients presenting to the outpatient clinic with acute or chronic abdominal pain. This is because the focus is directed to intra-abdominal causes. But if ACNES is kept in consideration from the beginning of the patient encounter, extensive testing, time, and patient anxiety may be reduced significantly. A simple physical examination and the Carnett test aid in raising suspicion of ACNES. If ACNES is confirmed, ultrasonographically guided local anesthetic injection is both diagnostic and therapeutic.
- American Gastroenterological Association Medical Position Statement: Guidelines On Intestinal Ischemia. Gastroenterology 2000; 118:951–953.
- Bobadilla JL. Mesenteric ischemia. Surg Clin North Am 2013; 93:925–940.
- Sung JJ, Kuipers EJ, El-Serag HB. Systematic review: the global incidence and prevalence of peptic ulcer disease. Aliment Pharmacol Ther 2009; 29:938–946.
- Najm WI. Peptic ulcer disease. Prim Care 2011; 38:383–394.
- Malfertheiner P, Chan FK, McColl KE. Peptic ulcer disease. Lancet 2009; 374:1449–1461.
- Chan FK, Leung WK. Peptic-ulcer disease. Lancet 2002; 360:933–941.
- Bright-Asare P, Habte T, Yirgou B, Benjamin J. Prostaglandins, H2-receptor antagonists and peptic ulcer disease. Drugs 1988; 35(suppl 3):1–9.
- Hudson N, Balsitis M, Everitt S, Hawkey CJ. Enhanced gastric mucosal leukotriene B4 synthesis in patients taking non-steroidal anti-inflammatory drugs. Gut 1993; 34:742–747.
- Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature 1980; 286:264–265.
- Bokoch GM, Reed PW. Effect of various lipoxygenase metabolites of arachidonic acid on degranulation of polymorphonuclear leukocytes. J Biol Chem 1981; 256:5317–5320.
- Whittle BJ, Oren-Wolman N, Guth PH. Gastric vasoconstrictor actions of leukotriene C4, PGF2 alpha, and thromboxane mimetic U-46619 on rat submucosal microcirculation in vivo. Am J Physiol 1985; 248:G580–G586.
- Pihan G, Rogers C, Szabo S. Vascular injury in acute gastric mucosal damage. Mediatory role of leukotrienes. Dig Dis Sci 1988; 33:625–632.
- Ramakrishnan K, Salinas RC. Peptic ulcer disease. Am Fam Physician 2007; 76:1005–1012.
- Parmet S, Lynm C, Glass RM. JAMA patient page. Acute cholecystitis. JAMA 2003; 289:124.
- Roslyn JJ, DenBesten L, Thompson JE Jr, Silverman BF. Roles of lithogenic bile and cystic duct occlusion in the pathogenesis of acute cholecystitis. Am J Surg 1980; 140:126–130.
- Kaminski DL. Arachidonic acid metabolites in hepatobiliary physiology and disease. Gastroenterology 1989; 97:781–792.
- Jivegård L, Thornell E, Svanvik J. Pathophysiology of acute obstructive cholecystitis: implications for non-operative management. Br J Surg 1987; 74:1084–1086.
- Csendes A, Burdiles P, Maluenda F, Diaz JC, Csendes P, Mitru N. Simultaneous bacteriologic assessment of bile from gallbladder and common bile duct in control subjects and patients with gallstones and common duct stones. Arch Surg 1996; 131:389–394.
- Barie PS, Fischer E. Acute acalculous cholecystitis. J Am Coll Surg 1995; 180:232–244.
- Shapiro MJ, Luchtefeld WB, Kurzweil S, Kaminski DL, Durham RM, Mazuski JE. Acute acalculous cholecystitis in the critically ill. Am Surg 1994; 60:335–339.
- Savoca PE, Longo WE, Zucker KA, McMillen MM, Modlin IM. The increasing prevalence of acalculous cholecystitis in outpatients. Results of a 7-year study. Ann Surg 1990; 211:433–437.
- Gofrit O, Eid A, Pikarsky A, Lebensart PD, Pizov G, Rivkind A. Cholesterol embolisation causing chronic acalculous cholecystitis. Eur J Surg 1996; 162:243–245.
- McChesney JA, Northup PG, Bickston SJ. Acute acalculous cholecystitis associated with systemic sepsis and visceral arterial hypoperfusion: a case series and review of pathophysiology. Dig Dis Sci 2003; 48:1960–1967.
- Aeschlimann A, Kahn MF. Tietze’s syndrome: a critical review. Clin Exp Rheumatol 1990; 8:407–412.
- van Assen T, de Jager-Kievit JW, Scheltinga MR, Roumen RM. Chronic abdominal wall pain misdiagnosed as functional abdominal pain. J Am Board Fam Med 2013; 26:738–744.
- Akhnikh S, de Korte N, de Winter P. Anterior cutaneous nerve entrapment syndrome (ACNES): the forgotten diagnosis. Eur J Pediatr 2014; 173:445–449.
- Applegate WV. Abdominal cutaneous nerve entrapment syndrome (ACNES): a commonly overlooked cause of abdominal pain. Perm J 2002; 6:20–27.
- Grover M. UNC Center for Functional GI & Motility Disorders. Chronic abdominal wall pain: a missed diagnosis. www.med.unc.edu/ibs/files/educational-gi-handouts/Chronic%20Abdominal%20Pain.pdf. Accessed September 9, 2015.
- Greenbaum D, Dawson F, Watson R. Chronic abdominal wall pain (CAWP): a common but frequently overlooked disorder. Poster presented at the World Congress of Gastroenterology, Sydney, Australia, August 26–31, 1990.
- National Institutes of Health Consensus Development Conference Statement. Ovarian cancer: screening, treatment, and follow-up. Gynecol Oncol 1994; 55:S4–S14.
- Koonings PP, Campbell K, Mishell DR Jr, Grimes DA. Relative frequency of primary ovarian neoplasms: a 10-year review. Obstet Gynecol 1989; 74:921–926.
- Givens V, Mitchell GE, Harraway-Smith C, Reddy A, Maness DL. Diagnosis and management of adnexal masses. Am Fam Physician 2009; 80:815–820.
- Goff BA, Mandel L, Muntz HG, Melancon CH. Ovarian carcinoma diagnosis. Cancer 2000; 89:2068–2075.
- Friedman GD, Skilling JS, Udaltsova NV, Smith LH. Early symptoms of ovarian cancer: a case-control study without recall bias. Fam Pract 2005; 22:548–553.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976-1994. Kidney Int 2003; 63:1817–1823.
- Worcester EM, Coe FL. Clinical practice. Calcium kidney stones. N Engl J Med 2010; 363:954–963.
- Miller NL, Lingeman JE. Management of kidney stones. BMJ 2007; 334:468–472.
- Lewis SR, Mahony PJ, Simpson J. Appendicitis. BMJ 2011; 343:d5976.
- Lamps LW. Infectious causes of appendicitis. Infect Dis Clin North Am 2010; 24:995–1018.
- Reif RM. Viral appendicitis. Hum Pathol 1981; 12:193–196.
- Akkary E, Panait L, Roberts K, Duffy A, Bell R. Sutureless laparoscopic ventral hernia repair in obese patients. JSLS 2011; 15:154–159.
- Boelens OB, Scheltinga MR, Houterman S, Roumen RM. Randomized clinical trial of trigger point infiltration with lidocaine to diagnose anterior cutaneous nerve entrapment syndrome. Br J Surg 2013; 100:217–221.
- Boelens OB, Scheltinga MR, Houterman S, Roumen RM. Management of anterior cutaneous nerve entrapment syndrome in a cohort of 139 patients. Ann Surg 2011; 254:1054–1058.
- American Gastroenterological Association Medical Position Statement: Guidelines On Intestinal Ischemia. Gastroenterology 2000; 118:951–953.
- Bobadilla JL. Mesenteric ischemia. Surg Clin North Am 2013; 93:925–940.
- Sung JJ, Kuipers EJ, El-Serag HB. Systematic review: the global incidence and prevalence of peptic ulcer disease. Aliment Pharmacol Ther 2009; 29:938–946.
- Najm WI. Peptic ulcer disease. Prim Care 2011; 38:383–394.
- Malfertheiner P, Chan FK, McColl KE. Peptic ulcer disease. Lancet 2009; 374:1449–1461.
- Chan FK, Leung WK. Peptic-ulcer disease. Lancet 2002; 360:933–941.
- Bright-Asare P, Habte T, Yirgou B, Benjamin J. Prostaglandins, H2-receptor antagonists and peptic ulcer disease. Drugs 1988; 35(suppl 3):1–9.
- Hudson N, Balsitis M, Everitt S, Hawkey CJ. Enhanced gastric mucosal leukotriene B4 synthesis in patients taking non-steroidal anti-inflammatory drugs. Gut 1993; 34:742–747.
- Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature 1980; 286:264–265.
- Bokoch GM, Reed PW. Effect of various lipoxygenase metabolites of arachidonic acid on degranulation of polymorphonuclear leukocytes. J Biol Chem 1981; 256:5317–5320.
- Whittle BJ, Oren-Wolman N, Guth PH. Gastric vasoconstrictor actions of leukotriene C4, PGF2 alpha, and thromboxane mimetic U-46619 on rat submucosal microcirculation in vivo. Am J Physiol 1985; 248:G580–G586.
- Pihan G, Rogers C, Szabo S. Vascular injury in acute gastric mucosal damage. Mediatory role of leukotrienes. Dig Dis Sci 1988; 33:625–632.
- Ramakrishnan K, Salinas RC. Peptic ulcer disease. Am Fam Physician 2007; 76:1005–1012.
- Parmet S, Lynm C, Glass RM. JAMA patient page. Acute cholecystitis. JAMA 2003; 289:124.
- Roslyn JJ, DenBesten L, Thompson JE Jr, Silverman BF. Roles of lithogenic bile and cystic duct occlusion in the pathogenesis of acute cholecystitis. Am J Surg 1980; 140:126–130.
- Kaminski DL. Arachidonic acid metabolites in hepatobiliary physiology and disease. Gastroenterology 1989; 97:781–792.
- Jivegård L, Thornell E, Svanvik J. Pathophysiology of acute obstructive cholecystitis: implications for non-operative management. Br J Surg 1987; 74:1084–1086.
- Csendes A, Burdiles P, Maluenda F, Diaz JC, Csendes P, Mitru N. Simultaneous bacteriologic assessment of bile from gallbladder and common bile duct in control subjects and patients with gallstones and common duct stones. Arch Surg 1996; 131:389–394.
- Barie PS, Fischer E. Acute acalculous cholecystitis. J Am Coll Surg 1995; 180:232–244.
- Shapiro MJ, Luchtefeld WB, Kurzweil S, Kaminski DL, Durham RM, Mazuski JE. Acute acalculous cholecystitis in the critically ill. Am Surg 1994; 60:335–339.
- Savoca PE, Longo WE, Zucker KA, McMillen MM, Modlin IM. The increasing prevalence of acalculous cholecystitis in outpatients. Results of a 7-year study. Ann Surg 1990; 211:433–437.
- Gofrit O, Eid A, Pikarsky A, Lebensart PD, Pizov G, Rivkind A. Cholesterol embolisation causing chronic acalculous cholecystitis. Eur J Surg 1996; 162:243–245.
- McChesney JA, Northup PG, Bickston SJ. Acute acalculous cholecystitis associated with systemic sepsis and visceral arterial hypoperfusion: a case series and review of pathophysiology. Dig Dis Sci 2003; 48:1960–1967.
- Aeschlimann A, Kahn MF. Tietze’s syndrome: a critical review. Clin Exp Rheumatol 1990; 8:407–412.
- van Assen T, de Jager-Kievit JW, Scheltinga MR, Roumen RM. Chronic abdominal wall pain misdiagnosed as functional abdominal pain. J Am Board Fam Med 2013; 26:738–744.
- Akhnikh S, de Korte N, de Winter P. Anterior cutaneous nerve entrapment syndrome (ACNES): the forgotten diagnosis. Eur J Pediatr 2014; 173:445–449.
- Applegate WV. Abdominal cutaneous nerve entrapment syndrome (ACNES): a commonly overlooked cause of abdominal pain. Perm J 2002; 6:20–27.
- Grover M. UNC Center for Functional GI & Motility Disorders. Chronic abdominal wall pain: a missed diagnosis. www.med.unc.edu/ibs/files/educational-gi-handouts/Chronic%20Abdominal%20Pain.pdf. Accessed September 9, 2015.
- Greenbaum D, Dawson F, Watson R. Chronic abdominal wall pain (CAWP): a common but frequently overlooked disorder. Poster presented at the World Congress of Gastroenterology, Sydney, Australia, August 26–31, 1990.
- National Institutes of Health Consensus Development Conference Statement. Ovarian cancer: screening, treatment, and follow-up. Gynecol Oncol 1994; 55:S4–S14.
- Koonings PP, Campbell K, Mishell DR Jr, Grimes DA. Relative frequency of primary ovarian neoplasms: a 10-year review. Obstet Gynecol 1989; 74:921–926.
- Givens V, Mitchell GE, Harraway-Smith C, Reddy A, Maness DL. Diagnosis and management of adnexal masses. Am Fam Physician 2009; 80:815–820.
- Goff BA, Mandel L, Muntz HG, Melancon CH. Ovarian carcinoma diagnosis. Cancer 2000; 89:2068–2075.
- Friedman GD, Skilling JS, Udaltsova NV, Smith LH. Early symptoms of ovarian cancer: a case-control study without recall bias. Fam Pract 2005; 22:548–553.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976-1994. Kidney Int 2003; 63:1817–1823.
- Worcester EM, Coe FL. Clinical practice. Calcium kidney stones. N Engl J Med 2010; 363:954–963.
- Miller NL, Lingeman JE. Management of kidney stones. BMJ 2007; 334:468–472.
- Lewis SR, Mahony PJ, Simpson J. Appendicitis. BMJ 2011; 343:d5976.
- Lamps LW. Infectious causes of appendicitis. Infect Dis Clin North Am 2010; 24:995–1018.
- Reif RM. Viral appendicitis. Hum Pathol 1981; 12:193–196.
- Akkary E, Panait L, Roberts K, Duffy A, Bell R. Sutureless laparoscopic ventral hernia repair in obese patients. JSLS 2011; 15:154–159.
- Boelens OB, Scheltinga MR, Houterman S, Roumen RM. Randomized clinical trial of trigger point infiltration with lidocaine to diagnose anterior cutaneous nerve entrapment syndrome. Br J Surg 2013; 100:217–221.
- Boelens OB, Scheltinga MR, Houterman S, Roumen RM. Management of anterior cutaneous nerve entrapment syndrome in a cohort of 139 patients. Ann Surg 2011; 254:1054–1058.
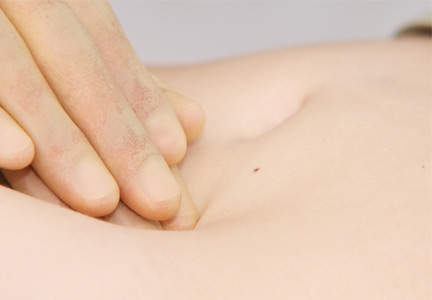
A young man with an unusual cause of palpitations
A 23-year-old man presents to the emergency department with the sudden onset of palpitations, lightheadedness, and dyspnea, accompanied by weakness and nausea, which started earlier in the evening. He estimates that he has experienced 15 similar episodes, lasting minutes to hours, since the age of 16, with the last one 3 years ago. These episodes typically end by themselves or with self-induced vomiting and lying supine. The current episode did not resolve with these maneuvers.
He has never received medical attention for these symptoms. He has no chest pain, orthopnea, paroxysmal nocturnal dyspnea, lower extremity edema, or syncope. He has had no recent illness, contacts with sick people, or travel.
The patient’s history includes a “childhood heart murmur,” which resolved, and also mild asthma. He is otherwise healthy but has not received regular medical care. He used to play competitive soccer but quit because playing made his symptoms of dyspnea on exertion and palpitations much worse.
He uses marijuana frequently and alcohol occasionally. He does not smoke tobacco or use other recreational drugs. Other than infrequent use of albuterol, he does not take any prescription or over-the-counter medications. He has no allergies. He knows of no family history of arrhythmia or sudden cardiac death.
Physical examination. On initial examination, his temperature is 36.4°C (97.5°F), heart rate 230 bpm, systolic blood pressure 60 mm Hg, respiratory rate 30 breaths per minute, oxygen saturation 100% while breathing room air, and body mass index 25 kg/m2.

He is awake, anxious, and appears ill. He speaks only in short sentences. A focused cardiac examination reveals a regular tachycardia with no appreciable murmur or extra heart sounds; the apical impulse is not displaced. His lungs are clear. His abdomen is soft and nontender. He has 2+ pulses on a scale of 0 to 4+, with no peripheral edema.
His initial electrocardiogram (ECG) (Figure 1) shows a heart rate of 260 bpm and a regular wide complex tachycardia, defined as a rate greater than 100 bpm and a QRS complex wider than 0.12 seconds.
FOCUS ON REGULAR WIDE COMPLEX TACHYCARDIA
1. Which of the following is not in the differential diagnosis of regular wide complex tachycardia?
- Monomorphic ventricular tachycardia
- Orthodromic atrioventricular reentrant tachycardia
- Antidromic atrioventricular reentrant tachycardia
- Sinus tachycardia with bundle branch block
Orthodromic atrioventricular reentrant tachycardia is not in the differential diagnosis.
Wide complex tachycardia can occur when the impulse originates outside the normal conduction system or when there is abnormal ventricular activation through the atrioventricular (AV) node and His-Purkinje system.
The main distinction to make when diagnosing the cause of a wide complex tachycardia is between the following:
Monomorphic ventricular tachycardia, which originates from a single ventricular focus that depolarizes the adjacent myocardium in a stepwise fashion, causing a wide QRS complex that does not begin in the native conduction system, and
Sinus tachycardia with bundle branch block, ie, supraventricular tachycardia with aberrant conduction within the normal conduction system.

Three different conduction patterns are seen with atrioventricular reentrant tachycardia (Figure 2):
Sinus depolarization (Figure 2), in which the atrial impulse travels down the AV node, and the accessory pathway can be hidden and not contribute to the surface ECG.
Orthodromic atrioventricular reentrant tachycardia (Figure2), in which the depolarizing impulse travels antegrade down the AV node, then propagates from the ventricle back to the atria via the accessory pathway, resulting in a narrow QRS.
Antidromic atrioventricular reentrant tachycardia (Figure 2), in which the depolarization travels antegrade down the accessory pathway then propagates from the ventricle back to the atria via the AV node, resulting in a wide complex QRS with a delta wave.
Important features of the patient’s electrocardiogram (Figure 1) are consistent with antidromic atrioventricular reentrant tachycardia:
- “Buried” retrograde P waves, which are best seen in the continuous strip of lead II as a positive deflection notching in the negative nadir of the wave
- The PR segment is short, suggesting retrograde atrial depolarization
- The P wave is followed by a slow slurred upstroke (delta wave), best seen in lead I.
Treatment depends on diagnosis
Distinguishing supraventricular tachycardia from ventricular tachycardia is important, as the treatments differ. Supraventricular tachycardia is treated with adenosine, calcium channel blockers, and beta-blockers, which are not only ineffective for ventricular tachycardia, but rarely may precipitate hemodynamic deterioration.
Also important is distinguishing pre-excitation atrial fibrillation from other types of supraventricular tachycardia with aberrancy, because the nodal blockade used to treat other causes of the condition may worsen the tachycardia via the accessory pathway. If pre-excitation atrial fibrillation is suspected on the basis of an irregular wide complex tachycardia with delta waves on ECG, then procainamide—a sodium channel blocker that affects the cardiac action potential and prolongs the refractory period of the accessory pathway—can be used to help control the arrhythmia.1
Brugada criteria aid diagnosis
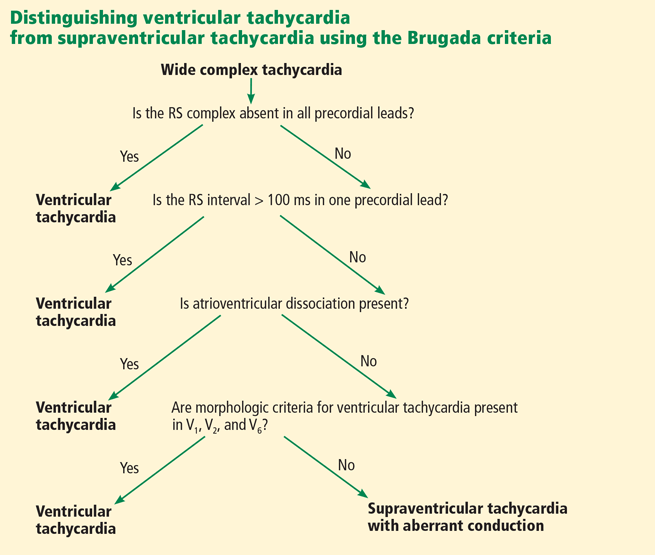
In 1991, Brugada et al2 devised an algorithm to differentiate ventricular tachycardia from supraventricular tachycardia with aberrancy in the setting of regular wide complex tachycardia (Figure 3). It has a sensitivity of 98.7% and a specificity of 96.5% for diagnosing ventricular tachycardia and 96.5% sensitivity and 98.7% specificity for diagnosing supraventricular tachycardia with aberrant conduction. Using the algorithm, only 11 (ie, 2%) of the 544 tachycardias in their study were misclassified.2–4
The Brugada algorithm consists of four criteria, with the presence of any leading to a diagnosis of ventricular tachycardia:
- Absence of an RS complex in all precordial leads (the QRS complexes in precordial leads have all negative or all positive deflections).
- An RS interval in at least one precordial lead of at least 100 ms (the interval is measured from the onset of R to the nadir of the S wave).
- AV dissociation, as determined by the existence of P waves marching out independent of the QRS complexes, capture beats (narrow QRS complexes resulting from the rare occasion when an intrinsic P wave conducts down the native pathway), or fusion beats (combined capture beat and ventricular beat, resulting in a different morphology than most of the wide QRS complexes present).
- Leads V1, V2, and V6 satisfying the classic morphologic criteria for ventricular tachycardia.
If none of these criteria are met, supraventricular tachycardia is diagnosed.
In our patient, we can further confirm the diagnosis of antidromic atrioventricular reentrant tachycardia by using Brugada criteria to exclude ventricular tachycardia (Figure 3): the ECG (Figure 1) shows an RS complex in multiple precordial leads, the maximum RS interval is less than 100 ms in the precordial leads, there is no evidence of AV dissociation (lead II in the continuous strip shows buried P waves associated with QRS), and morphologic criteria are not met for ventricular tachycardia in leads V1, V2, and V6.
IS CARDIOVERSION NEEDED?
According to the American Heart Association guidelines for advanced cardiopulmonary life support, patients with tachyarrhythmias who are hemodynamically unstable should undergo cardioversion immediately.5
Our patient, who has a heart rate faster than 200 bpm and a systolic blood pressure of only 60 mm Hg, undergoes synchronized cardioversion in the emergency department. Immediately afterward, his ECG (Figure 4) demonstrates sinus rhythm with pre-excitation consistent with type B Wolff-Parkinson-White syndrome.

Once he is hemodynamically stable, a more thorough physical examination is performed. Examination of the head, ears, eyes, nose, and throat is unremarkable. He has no jugular venous distention or carotid bruits. His lungs are clear to auscultation bilaterally, without wheezes. His cardiac examination shows a regular rate and rhythm, normal first and second heart sounds, and no murmurs, rubs, or gallops.
WHICH DIAGNOSTIC STUDIES ARE NEEDED?
Laboratory tests
In an otherwise healthy young patient presenting with an arrhythmia, the initial laboratory workup should focus on a precipitating illness or a disease state that may incite an arrhythmia.
Our patient is evaluated for infection or septic shock (white blood cell count with differential), anemia (hemoglobin), thyrotoxicosis (thyroid-stimulating hormone and free thyroxine levels), drug abuse (urine toxicology screen), and cardiac syndromes including structural heart disease and myocardial injury (cardiac enzymes and B-type natriuretic peptide).6
His initial laboratory tests show normal electrolyte levels and renal function, leukocytosis with a white blood cell count of 15.6 × 109/L (normal 4.0–10.0), mildly elevated thyroid-stimulating hormone, and a negative urine toxicology screen.
Transthoracic echocardiography
For a young patient presenting with pre-excitation on ECG and hemodynamic instability, transthoracic echocardiography to evaluate chamber size and look for structural abnormalities is a reasonable option.

Our patient undergoes transthoracic echocardiography, which demonstrates normal left ventricular size and function with a left ventricular ejection fraction of 69%, moderate right atrial enlargement, and mild right ventricular enlargement (Figure 5). The septal leaflet of the tricuspid valve is apically displaced, and there is mild regurgitation.
DIAGNOSIS: EBSTEIN ANOMALY
These findings are consistent with Ebstein anomaly. It can be recognized on transthoracic echocardiography as adherence of the septal and posterior tricuspid valve leaflets to the myocardium due to failure of the tissue to detach during embryogenesis, apical displacement of the annulus, right atrial enlargement, and right ventricular enlargement.7–10 Apical displacement of the tricuspid valve is a hallmark finding and must be more than 20 mm or 8 mm/m2 of body surface area to make the diagnosis.11–13 ECG often demonstrates right atrial enlargement, first-degree atrial ventricular block, and right bundle branch block.
Ebstein anomaly is a rare embryonic developmental abnormality of the tricuspid valve. It occurs in 1 to 5 of 200,000 live births, accounting for approximately 0.5% of all congenital heart disease.14,15 Most cases are sporadic and result from failure of the ventricle to delaminate during embryogenesis of the tricuspid valve, resulting in apical displacement of either the septal, posterior, or, very rarely, anterior leaflet of the tricuspid valve.7,8 The prevalence is higher in infants whose mothers took lithium during early pregnancy.16
2. Which of the following is not a common finding associated with Ebstein anomaly?
- Apical displacement of the septal leaflet of the tricuspid valve
- Wolff-Parkinson-White syndrome
- Accessory bypass tract
- Tachyarrhythmias
- Increased risk of sudden death
- Left-sided heart failure
The answer is left-sided heart failure. Ebstein anomaly is associated with increased risk of tachyarrhythmias, right-sided heart failure, and sudden death.7,8,17,18 In Ebstein anomaly, the tricuspid valve forms closer to the apex, so the part of the right ventricle that is superior to the displaced tricuspid valve functions as the right atrium, thus the term “atrialized” right ventricle. These abnormalities create an environment for accessory pathways, most commonly type B Wolff-Parkinson-White syndrome.19 Biventricular dysfunction can occur in rare severe cases.7,8,18
Our patient is found to have an accessory tract-mediated antidromic atrioventricular reentrant tachycardia in the setting of Wolff-Parkinson-White syndrome and Ebstein anomaly. This is further confirmed with an electrophysiology study demonstrating a right posterior accessory pathway.
TREATMENT FOR EBSTEIN ANOMALY
3. Which treatment is advised for Ebstein anomaly?
- Observation alone
- Standard heart failure medications
- Radiofrequency catheter ablation
- Tricuspid valve repair or replacement
- Biventricular reconstruction
- Heart transplant
The answer is all of the above. Observation alone is advised for patients with mild symptoms, no evidence of right-to-left shunting, and only mild cardiomegaly. Medical management includes an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker, a beta-blocker, and diuretics. Radiofrequency catheter ablation is the first-line therapy for patients with symptomatic Wolff-Parkinson-White syndrome.20 A patient who develops worsening right-sided heart failure, cyanosis, paradoxical emboli, or frequent tachyarrhythmias should be considered for corrective surgery, which may include tricuspid valve repair or replacement, or biventricular reconstruction.7,8,21 Cardiac transplant is reserved for severe cases.8
On hospital day 4, our patient undergoes successful radiofrequency catheter ablation without complications. At follow-up 3 months later, he continues to do well, with resolution of his symptoms and no further evidence of pre-excitation. His postprocedure ECG no longer shows delta waves.
TAKE-HOME POINTS
- For a patient with regular wide complex tachycardia, the first step is to assess hemodynamic stability. If the patient is hemodynamically unstable, emergent cardioversion is indicated.
- The differential diagnosis for regular wide complex tachycardia includes supraventricular tachycardia with aberrancy (orthodromic atrioventricular reentrant tachycardia, antidromic atrioventricular reentrant tachycardia, atrial tachycardia), and ventricular tachycardia.
- When pre-excited atrial fibrillation is suspected, AV nodal blocking agents should be avoided, as they may worsen tachyarrhythmia. Sodium channel blockers such as procainamide can help slow down the conduction of the accessory pathway.
- Ebstein anomaly is diagnosed on transthoracic echocardiography as apical displacement of the tricuspid valve resulting in atrialization of the right ventricle.
- Patients with Ebstein anomaly have a higher risk of death from right-sided heart failure and tachyarrhythmias, most commonly type B Wolff-Parkinson-White syndrome.
- Ebstein anomaly is medically managed with standard heart failure medications, including neurohormonal blockade therapies.
- Patients with Ebstein anomaly and cyanosis require surgical intervention with either valve repair or replacement.
Acknowledgment: We thank Dr. William Collins for his contribution in reviewing the manuscript and his technical expertise in developing some of the figures.
- January CT, Wann L, Alpert JS, et al; ACC/AHA Task Force Members. 2014 AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation 2014; 130:2071–2104.
- Brugada P, Brugada J, Mont L, Smeets J, Andries EW. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation 1991; 83:1649–1659.
- Alzand BS, Crijns HJ. Diagnostic criteria of broad QRS complex tachycardia: decades of evolution. Europace 2011; 13:465–472.
- Wellens HJ, Bar FW, Lie KI. The value of the electrocardiogram in the differential diagnosis of a tachycardia with a widened QRS complex. Am J Med 1978; 64:27–33.
- Field JM, Hazinski MF, Sayre MR, et al. Part 1: executive summary: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010; 122(suppl 3):S640–S656.
- Walkey AJ, Wiener RS, Ghobrial JM, Curtis LH, Benjamin EJ. Incident stroke and mortality associated with new-onset atrial fibrillation in patients hospitalized with severe sepsis. JAMA 2011; 306:2248–2254.
- Attenhofer Jost CH, Connolly HM, Edwards WD, Hayes D, Warnes CA, Danielson GK. Ebstein’s anomaly - review of a multifaceted congenital cardiac condition. Swiss Med Wkly 2005; 135:269–281.
- Attenhofer Jost CH, Connolly HM, Dearani JA, Edwards WD, Danielson GK. Ebstein’s anomaly. Circulation 2007; 115:277–285.
- Oechslin E, Buchholz S, Jenni R. Ebstein’s anomaly in adults: Doppler-echocardiographic evaluation. Thorac Cardiovasc Surg 2000; 48:209–213.
- Ali SK, Nimeri NA. Clinical and echocardiographic features of Ebstein’s malformation in Sudanese patients. Cardiol Young 2006; 16:147–151.
- Edwards WD. Embryology and pathologic features of Ebstein’s anomaly. Prog Pediatr Cardiol 1993; 2:5–15.
- Shiina A, Seward JB, Edwards WD, Hagler DJ, Tajik AJ. Two dimensional echocardiographic spectrum of Ebstein’s anomaly: detailed anatomic assessment. J Am Coll Cardiol 1984; 3:356–370.
- Gussenhoven EJ, Stewart PA, Becker AE, Essed CE, Ligtvoet KM, De Villeneuve VH. “Offsetting” of the septal tricuspid leaflet in normal hearts and in hearts with Ebstein’s anomaly. Anatomic and echographic correlation. Am J Cardiol 1984; 54:172–176.
- Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. Second of two parts. N Engl J Med 2000; 342:334–342.
- Report of the New England Regional Infant Cardiac Program. Pediatrics 1980; 65:375–461.
- Cohen LS, Friedman JM, Jefferson JW, Johnson EM, Weiner ML. A reevaluation of risk of in utero exposure to lithium. JAMA 1994; 271:146–150.
- Paranon S, Acar P. Ebstein’s anomaly of the tricuspid valve: from fetus to adult: congenital heart disease. Heart 2008; 94:237–243.
- Watson H. Natural history of Ebstein’s anomaly of tricuspid valve in childhood and adolescence. An international co-operative study of 505 cases. Br Heart J 1974; 36:417–427.
- Delhaas T, Sarvaas GJ, Rijlaarsdam ME, et al. A multicenter, long-term study on arrhythmias in children with Ebstein anomaly. Pediatr Cardiol 2010; 31:229–223.
- Tischenko A, Fox DJ, Yee R, et al. When should we recommend catheter ablation for patients with the Wolff-Parkinson-White syndrome? Curr Opin Cardiol 2008; 23:32–37.
- Misaki T, Watanabe G, Iwa T, et al. Surgical treatment of patients with Wolff-Parkinson-White syndrome and associated Ebstein’s anomaly. J Thorac Cardiovasc Surg 1995; 110:1702–1707.
A 23-year-old man presents to the emergency department with the sudden onset of palpitations, lightheadedness, and dyspnea, accompanied by weakness and nausea, which started earlier in the evening. He estimates that he has experienced 15 similar episodes, lasting minutes to hours, since the age of 16, with the last one 3 years ago. These episodes typically end by themselves or with self-induced vomiting and lying supine. The current episode did not resolve with these maneuvers.
He has never received medical attention for these symptoms. He has no chest pain, orthopnea, paroxysmal nocturnal dyspnea, lower extremity edema, or syncope. He has had no recent illness, contacts with sick people, or travel.
The patient’s history includes a “childhood heart murmur,” which resolved, and also mild asthma. He is otherwise healthy but has not received regular medical care. He used to play competitive soccer but quit because playing made his symptoms of dyspnea on exertion and palpitations much worse.
He uses marijuana frequently and alcohol occasionally. He does not smoke tobacco or use other recreational drugs. Other than infrequent use of albuterol, he does not take any prescription or over-the-counter medications. He has no allergies. He knows of no family history of arrhythmia or sudden cardiac death.
Physical examination. On initial examination, his temperature is 36.4°C (97.5°F), heart rate 230 bpm, systolic blood pressure 60 mm Hg, respiratory rate 30 breaths per minute, oxygen saturation 100% while breathing room air, and body mass index 25 kg/m2.
He is awake, anxious, and appears ill. He speaks only in short sentences. A focused cardiac examination reveals a regular tachycardia with no appreciable murmur or extra heart sounds; the apical impulse is not displaced. His lungs are clear. His abdomen is soft and nontender. He has 2+ pulses on a scale of 0 to 4+, with no peripheral edema.
His initial electrocardiogram (ECG) (Figure 1) shows a heart rate of 260 bpm and a regular wide complex tachycardia, defined as a rate greater than 100 bpm and a QRS complex wider than 0.12 seconds.
FOCUS ON REGULAR WIDE COMPLEX TACHYCARDIA
1. Which of the following is not in the differential diagnosis of regular wide complex tachycardia?
- Monomorphic ventricular tachycardia
- Orthodromic atrioventricular reentrant tachycardia
- Antidromic atrioventricular reentrant tachycardia
- Sinus tachycardia with bundle branch block
Orthodromic atrioventricular reentrant tachycardia is not in the differential diagnosis.
Wide complex tachycardia can occur when the impulse originates outside the normal conduction system or when there is abnormal ventricular activation through the atrioventricular (AV) node and His-Purkinje system.
The main distinction to make when diagnosing the cause of a wide complex tachycardia is between the following:
Monomorphic ventricular tachycardia, which originates from a single ventricular focus that depolarizes the adjacent myocardium in a stepwise fashion, causing a wide QRS complex that does not begin in the native conduction system, and
Sinus tachycardia with bundle branch block, ie, supraventricular tachycardia with aberrant conduction within the normal conduction system.
Three different conduction patterns are seen with atrioventricular reentrant tachycardia (Figure 2):
Sinus depolarization (Figure 2), in which the atrial impulse travels down the AV node, and the accessory pathway can be hidden and not contribute to the surface ECG.
Orthodromic atrioventricular reentrant tachycardia (Figure2), in which the depolarizing impulse travels antegrade down the AV node, then propagates from the ventricle back to the atria via the accessory pathway, resulting in a narrow QRS.
Antidromic atrioventricular reentrant tachycardia (Figure 2), in which the depolarization travels antegrade down the accessory pathway then propagates from the ventricle back to the atria via the AV node, resulting in a wide complex QRS with a delta wave.
Important features of the patient’s electrocardiogram (Figure 1) are consistent with antidromic atrioventricular reentrant tachycardia:
- “Buried” retrograde P waves, which are best seen in the continuous strip of lead II as a positive deflection notching in the negative nadir of the wave
- The PR segment is short, suggesting retrograde atrial depolarization
- The P wave is followed by a slow slurred upstroke (delta wave), best seen in lead I.
Treatment depends on diagnosis
Distinguishing supraventricular tachycardia from ventricular tachycardia is important, as the treatments differ. Supraventricular tachycardia is treated with adenosine, calcium channel blockers, and beta-blockers, which are not only ineffective for ventricular tachycardia, but rarely may precipitate hemodynamic deterioration.
Also important is distinguishing pre-excitation atrial fibrillation from other types of supraventricular tachycardia with aberrancy, because the nodal blockade used to treat other causes of the condition may worsen the tachycardia via the accessory pathway. If pre-excitation atrial fibrillation is suspected on the basis of an irregular wide complex tachycardia with delta waves on ECG, then procainamide—a sodium channel blocker that affects the cardiac action potential and prolongs the refractory period of the accessory pathway—can be used to help control the arrhythmia.1
Brugada criteria aid diagnosis
In 1991, Brugada et al2 devised an algorithm to differentiate ventricular tachycardia from supraventricular tachycardia with aberrancy in the setting of regular wide complex tachycardia (Figure 3). It has a sensitivity of 98.7% and a specificity of 96.5% for diagnosing ventricular tachycardia and 96.5% sensitivity and 98.7% specificity for diagnosing supraventricular tachycardia with aberrant conduction. Using the algorithm, only 11 (ie, 2%) of the 544 tachycardias in their study were misclassified.2–4
The Brugada algorithm consists of four criteria, with the presence of any leading to a diagnosis of ventricular tachycardia:
- Absence of an RS complex in all precordial leads (the QRS complexes in precordial leads have all negative or all positive deflections).
- An RS interval in at least one precordial lead of at least 100 ms (the interval is measured from the onset of R to the nadir of the S wave).
- AV dissociation, as determined by the existence of P waves marching out independent of the QRS complexes, capture beats (narrow QRS complexes resulting from the rare occasion when an intrinsic P wave conducts down the native pathway), or fusion beats (combined capture beat and ventricular beat, resulting in a different morphology than most of the wide QRS complexes present).
- Leads V1, V2, and V6 satisfying the classic morphologic criteria for ventricular tachycardia.
If none of these criteria are met, supraventricular tachycardia is diagnosed.
In our patient, we can further confirm the diagnosis of antidromic atrioventricular reentrant tachycardia by using Brugada criteria to exclude ventricular tachycardia (Figure 3): the ECG (Figure 1) shows an RS complex in multiple precordial leads, the maximum RS interval is less than 100 ms in the precordial leads, there is no evidence of AV dissociation (lead II in the continuous strip shows buried P waves associated with QRS), and morphologic criteria are not met for ventricular tachycardia in leads V1, V2, and V6.
IS CARDIOVERSION NEEDED?
According to the American Heart Association guidelines for advanced cardiopulmonary life support, patients with tachyarrhythmias who are hemodynamically unstable should undergo cardioversion immediately.5
Our patient, who has a heart rate faster than 200 bpm and a systolic blood pressure of only 60 mm Hg, undergoes synchronized cardioversion in the emergency department. Immediately afterward, his ECG (Figure 4) demonstrates sinus rhythm with pre-excitation consistent with type B Wolff-Parkinson-White syndrome.
Once he is hemodynamically stable, a more thorough physical examination is performed. Examination of the head, ears, eyes, nose, and throat is unremarkable. He has no jugular venous distention or carotid bruits. His lungs are clear to auscultation bilaterally, without wheezes. His cardiac examination shows a regular rate and rhythm, normal first and second heart sounds, and no murmurs, rubs, or gallops.
WHICH DIAGNOSTIC STUDIES ARE NEEDED?
Laboratory tests
In an otherwise healthy young patient presenting with an arrhythmia, the initial laboratory workup should focus on a precipitating illness or a disease state that may incite an arrhythmia.
Our patient is evaluated for infection or septic shock (white blood cell count with differential), anemia (hemoglobin), thyrotoxicosis (thyroid-stimulating hormone and free thyroxine levels), drug abuse (urine toxicology screen), and cardiac syndromes including structural heart disease and myocardial injury (cardiac enzymes and B-type natriuretic peptide).6
His initial laboratory tests show normal electrolyte levels and renal function, leukocytosis with a white blood cell count of 15.6 × 109/L (normal 4.0–10.0), mildly elevated thyroid-stimulating hormone, and a negative urine toxicology screen.
Transthoracic echocardiography
For a young patient presenting with pre-excitation on ECG and hemodynamic instability, transthoracic echocardiography to evaluate chamber size and look for structural abnormalities is a reasonable option.
Our patient undergoes transthoracic echocardiography, which demonstrates normal left ventricular size and function with a left ventricular ejection fraction of 69%, moderate right atrial enlargement, and mild right ventricular enlargement (Figure 5). The septal leaflet of the tricuspid valve is apically displaced, and there is mild regurgitation.
DIAGNOSIS: EBSTEIN ANOMALY
These findings are consistent with Ebstein anomaly. It can be recognized on transthoracic echocardiography as adherence of the septal and posterior tricuspid valve leaflets to the myocardium due to failure of the tissue to detach during embryogenesis, apical displacement of the annulus, right atrial enlargement, and right ventricular enlargement.7–10 Apical displacement of the tricuspid valve is a hallmark finding and must be more than 20 mm or 8 mm/m2 of body surface area to make the diagnosis.11–13 ECG often demonstrates right atrial enlargement, first-degree atrial ventricular block, and right bundle branch block.
Ebstein anomaly is a rare embryonic developmental abnormality of the tricuspid valve. It occurs in 1 to 5 of 200,000 live births, accounting for approximately 0.5% of all congenital heart disease.14,15 Most cases are sporadic and result from failure of the ventricle to delaminate during embryogenesis of the tricuspid valve, resulting in apical displacement of either the septal, posterior, or, very rarely, anterior leaflet of the tricuspid valve.7,8 The prevalence is higher in infants whose mothers took lithium during early pregnancy.16
2. Which of the following is not a common finding associated with Ebstein anomaly?
- Apical displacement of the septal leaflet of the tricuspid valve
- Wolff-Parkinson-White syndrome
- Accessory bypass tract
- Tachyarrhythmias
- Increased risk of sudden death
- Left-sided heart failure
The answer is left-sided heart failure. Ebstein anomaly is associated with increased risk of tachyarrhythmias, right-sided heart failure, and sudden death.7,8,17,18 In Ebstein anomaly, the tricuspid valve forms closer to the apex, so the part of the right ventricle that is superior to the displaced tricuspid valve functions as the right atrium, thus the term “atrialized” right ventricle. These abnormalities create an environment for accessory pathways, most commonly type B Wolff-Parkinson-White syndrome.19 Biventricular dysfunction can occur in rare severe cases.7,8,18
Our patient is found to have an accessory tract-mediated antidromic atrioventricular reentrant tachycardia in the setting of Wolff-Parkinson-White syndrome and Ebstein anomaly. This is further confirmed with an electrophysiology study demonstrating a right posterior accessory pathway.
TREATMENT FOR EBSTEIN ANOMALY
3. Which treatment is advised for Ebstein anomaly?
- Observation alone
- Standard heart failure medications
- Radiofrequency catheter ablation
- Tricuspid valve repair or replacement
- Biventricular reconstruction
- Heart transplant
The answer is all of the above. Observation alone is advised for patients with mild symptoms, no evidence of right-to-left shunting, and only mild cardiomegaly. Medical management includes an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker, a beta-blocker, and diuretics. Radiofrequency catheter ablation is the first-line therapy for patients with symptomatic Wolff-Parkinson-White syndrome.20 A patient who develops worsening right-sided heart failure, cyanosis, paradoxical emboli, or frequent tachyarrhythmias should be considered for corrective surgery, which may include tricuspid valve repair or replacement, or biventricular reconstruction.7,8,21 Cardiac transplant is reserved for severe cases.8
On hospital day 4, our patient undergoes successful radiofrequency catheter ablation without complications. At follow-up 3 months later, he continues to do well, with resolution of his symptoms and no further evidence of pre-excitation. His postprocedure ECG no longer shows delta waves.
TAKE-HOME POINTS
- For a patient with regular wide complex tachycardia, the first step is to assess hemodynamic stability. If the patient is hemodynamically unstable, emergent cardioversion is indicated.
- The differential diagnosis for regular wide complex tachycardia includes supraventricular tachycardia with aberrancy (orthodromic atrioventricular reentrant tachycardia, antidromic atrioventricular reentrant tachycardia, atrial tachycardia), and ventricular tachycardia.
- When pre-excited atrial fibrillation is suspected, AV nodal blocking agents should be avoided, as they may worsen tachyarrhythmia. Sodium channel blockers such as procainamide can help slow down the conduction of the accessory pathway.
- Ebstein anomaly is diagnosed on transthoracic echocardiography as apical displacement of the tricuspid valve resulting in atrialization of the right ventricle.
- Patients with Ebstein anomaly have a higher risk of death from right-sided heart failure and tachyarrhythmias, most commonly type B Wolff-Parkinson-White syndrome.
- Ebstein anomaly is medically managed with standard heart failure medications, including neurohormonal blockade therapies.
- Patients with Ebstein anomaly and cyanosis require surgical intervention with either valve repair or replacement.
Acknowledgment: We thank Dr. William Collins for his contribution in reviewing the manuscript and his technical expertise in developing some of the figures.
A 23-year-old man presents to the emergency department with the sudden onset of palpitations, lightheadedness, and dyspnea, accompanied by weakness and nausea, which started earlier in the evening. He estimates that he has experienced 15 similar episodes, lasting minutes to hours, since the age of 16, with the last one 3 years ago. These episodes typically end by themselves or with self-induced vomiting and lying supine. The current episode did not resolve with these maneuvers.
He has never received medical attention for these symptoms. He has no chest pain, orthopnea, paroxysmal nocturnal dyspnea, lower extremity edema, or syncope. He has had no recent illness, contacts with sick people, or travel.
The patient’s history includes a “childhood heart murmur,” which resolved, and also mild asthma. He is otherwise healthy but has not received regular medical care. He used to play competitive soccer but quit because playing made his symptoms of dyspnea on exertion and palpitations much worse.
He uses marijuana frequently and alcohol occasionally. He does not smoke tobacco or use other recreational drugs. Other than infrequent use of albuterol, he does not take any prescription or over-the-counter medications. He has no allergies. He knows of no family history of arrhythmia or sudden cardiac death.
Physical examination. On initial examination, his temperature is 36.4°C (97.5°F), heart rate 230 bpm, systolic blood pressure 60 mm Hg, respiratory rate 30 breaths per minute, oxygen saturation 100% while breathing room air, and body mass index 25 kg/m2.
He is awake, anxious, and appears ill. He speaks only in short sentences. A focused cardiac examination reveals a regular tachycardia with no appreciable murmur or extra heart sounds; the apical impulse is not displaced. His lungs are clear. His abdomen is soft and nontender. He has 2+ pulses on a scale of 0 to 4+, with no peripheral edema.
His initial electrocardiogram (ECG) (Figure 1) shows a heart rate of 260 bpm and a regular wide complex tachycardia, defined as a rate greater than 100 bpm and a QRS complex wider than 0.12 seconds.
FOCUS ON REGULAR WIDE COMPLEX TACHYCARDIA
1. Which of the following is not in the differential diagnosis of regular wide complex tachycardia?
- Monomorphic ventricular tachycardia
- Orthodromic atrioventricular reentrant tachycardia
- Antidromic atrioventricular reentrant tachycardia
- Sinus tachycardia with bundle branch block
Orthodromic atrioventricular reentrant tachycardia is not in the differential diagnosis.
Wide complex tachycardia can occur when the impulse originates outside the normal conduction system or when there is abnormal ventricular activation through the atrioventricular (AV) node and His-Purkinje system.
The main distinction to make when diagnosing the cause of a wide complex tachycardia is between the following:
Monomorphic ventricular tachycardia, which originates from a single ventricular focus that depolarizes the adjacent myocardium in a stepwise fashion, causing a wide QRS complex that does not begin in the native conduction system, and
Sinus tachycardia with bundle branch block, ie, supraventricular tachycardia with aberrant conduction within the normal conduction system.
Three different conduction patterns are seen with atrioventricular reentrant tachycardia (Figure 2):
Sinus depolarization (Figure 2), in which the atrial impulse travels down the AV node, and the accessory pathway can be hidden and not contribute to the surface ECG.
Orthodromic atrioventricular reentrant tachycardia (Figure2), in which the depolarizing impulse travels antegrade down the AV node, then propagates from the ventricle back to the atria via the accessory pathway, resulting in a narrow QRS.
Antidromic atrioventricular reentrant tachycardia (Figure 2), in which the depolarization travels antegrade down the accessory pathway then propagates from the ventricle back to the atria via the AV node, resulting in a wide complex QRS with a delta wave.
Important features of the patient’s electrocardiogram (Figure 1) are consistent with antidromic atrioventricular reentrant tachycardia:
- “Buried” retrograde P waves, which are best seen in the continuous strip of lead II as a positive deflection notching in the negative nadir of the wave
- The PR segment is short, suggesting retrograde atrial depolarization
- The P wave is followed by a slow slurred upstroke (delta wave), best seen in lead I.
Treatment depends on diagnosis
Distinguishing supraventricular tachycardia from ventricular tachycardia is important, as the treatments differ. Supraventricular tachycardia is treated with adenosine, calcium channel blockers, and beta-blockers, which are not only ineffective for ventricular tachycardia, but rarely may precipitate hemodynamic deterioration.
Also important is distinguishing pre-excitation atrial fibrillation from other types of supraventricular tachycardia with aberrancy, because the nodal blockade used to treat other causes of the condition may worsen the tachycardia via the accessory pathway. If pre-excitation atrial fibrillation is suspected on the basis of an irregular wide complex tachycardia with delta waves on ECG, then procainamide—a sodium channel blocker that affects the cardiac action potential and prolongs the refractory period of the accessory pathway—can be used to help control the arrhythmia.1
Brugada criteria aid diagnosis
In 1991, Brugada et al2 devised an algorithm to differentiate ventricular tachycardia from supraventricular tachycardia with aberrancy in the setting of regular wide complex tachycardia (Figure 3). It has a sensitivity of 98.7% and a specificity of 96.5% for diagnosing ventricular tachycardia and 96.5% sensitivity and 98.7% specificity for diagnosing supraventricular tachycardia with aberrant conduction. Using the algorithm, only 11 (ie, 2%) of the 544 tachycardias in their study were misclassified.2–4
The Brugada algorithm consists of four criteria, with the presence of any leading to a diagnosis of ventricular tachycardia:
- Absence of an RS complex in all precordial leads (the QRS complexes in precordial leads have all negative or all positive deflections).
- An RS interval in at least one precordial lead of at least 100 ms (the interval is measured from the onset of R to the nadir of the S wave).
- AV dissociation, as determined by the existence of P waves marching out independent of the QRS complexes, capture beats (narrow QRS complexes resulting from the rare occasion when an intrinsic P wave conducts down the native pathway), or fusion beats (combined capture beat and ventricular beat, resulting in a different morphology than most of the wide QRS complexes present).
- Leads V1, V2, and V6 satisfying the classic morphologic criteria for ventricular tachycardia.
If none of these criteria are met, supraventricular tachycardia is diagnosed.
In our patient, we can further confirm the diagnosis of antidromic atrioventricular reentrant tachycardia by using Brugada criteria to exclude ventricular tachycardia (Figure 3): the ECG (Figure 1) shows an RS complex in multiple precordial leads, the maximum RS interval is less than 100 ms in the precordial leads, there is no evidence of AV dissociation (lead II in the continuous strip shows buried P waves associated with QRS), and morphologic criteria are not met for ventricular tachycardia in leads V1, V2, and V6.
IS CARDIOVERSION NEEDED?
According to the American Heart Association guidelines for advanced cardiopulmonary life support, patients with tachyarrhythmias who are hemodynamically unstable should undergo cardioversion immediately.5
Our patient, who has a heart rate faster than 200 bpm and a systolic blood pressure of only 60 mm Hg, undergoes synchronized cardioversion in the emergency department. Immediately afterward, his ECG (Figure 4) demonstrates sinus rhythm with pre-excitation consistent with type B Wolff-Parkinson-White syndrome.
Once he is hemodynamically stable, a more thorough physical examination is performed. Examination of the head, ears, eyes, nose, and throat is unremarkable. He has no jugular venous distention or carotid bruits. His lungs are clear to auscultation bilaterally, without wheezes. His cardiac examination shows a regular rate and rhythm, normal first and second heart sounds, and no murmurs, rubs, or gallops.
WHICH DIAGNOSTIC STUDIES ARE NEEDED?
Laboratory tests
In an otherwise healthy young patient presenting with an arrhythmia, the initial laboratory workup should focus on a precipitating illness or a disease state that may incite an arrhythmia.
Our patient is evaluated for infection or septic shock (white blood cell count with differential), anemia (hemoglobin), thyrotoxicosis (thyroid-stimulating hormone and free thyroxine levels), drug abuse (urine toxicology screen), and cardiac syndromes including structural heart disease and myocardial injury (cardiac enzymes and B-type natriuretic peptide).6
His initial laboratory tests show normal electrolyte levels and renal function, leukocytosis with a white blood cell count of 15.6 × 109/L (normal 4.0–10.0), mildly elevated thyroid-stimulating hormone, and a negative urine toxicology screen.
Transthoracic echocardiography
For a young patient presenting with pre-excitation on ECG and hemodynamic instability, transthoracic echocardiography to evaluate chamber size and look for structural abnormalities is a reasonable option.
Our patient undergoes transthoracic echocardiography, which demonstrates normal left ventricular size and function with a left ventricular ejection fraction of 69%, moderate right atrial enlargement, and mild right ventricular enlargement (Figure 5). The septal leaflet of the tricuspid valve is apically displaced, and there is mild regurgitation.
DIAGNOSIS: EBSTEIN ANOMALY
These findings are consistent with Ebstein anomaly. It can be recognized on transthoracic echocardiography as adherence of the septal and posterior tricuspid valve leaflets to the myocardium due to failure of the tissue to detach during embryogenesis, apical displacement of the annulus, right atrial enlargement, and right ventricular enlargement.7–10 Apical displacement of the tricuspid valve is a hallmark finding and must be more than 20 mm or 8 mm/m2 of body surface area to make the diagnosis.11–13 ECG often demonstrates right atrial enlargement, first-degree atrial ventricular block, and right bundle branch block.
Ebstein anomaly is a rare embryonic developmental abnormality of the tricuspid valve. It occurs in 1 to 5 of 200,000 live births, accounting for approximately 0.5% of all congenital heart disease.14,15 Most cases are sporadic and result from failure of the ventricle to delaminate during embryogenesis of the tricuspid valve, resulting in apical displacement of either the septal, posterior, or, very rarely, anterior leaflet of the tricuspid valve.7,8 The prevalence is higher in infants whose mothers took lithium during early pregnancy.16
2. Which of the following is not a common finding associated with Ebstein anomaly?
- Apical displacement of the septal leaflet of the tricuspid valve
- Wolff-Parkinson-White syndrome
- Accessory bypass tract
- Tachyarrhythmias
- Increased risk of sudden death
- Left-sided heart failure
The answer is left-sided heart failure. Ebstein anomaly is associated with increased risk of tachyarrhythmias, right-sided heart failure, and sudden death.7,8,17,18 In Ebstein anomaly, the tricuspid valve forms closer to the apex, so the part of the right ventricle that is superior to the displaced tricuspid valve functions as the right atrium, thus the term “atrialized” right ventricle. These abnormalities create an environment for accessory pathways, most commonly type B Wolff-Parkinson-White syndrome.19 Biventricular dysfunction can occur in rare severe cases.7,8,18
Our patient is found to have an accessory tract-mediated antidromic atrioventricular reentrant tachycardia in the setting of Wolff-Parkinson-White syndrome and Ebstein anomaly. This is further confirmed with an electrophysiology study demonstrating a right posterior accessory pathway.
TREATMENT FOR EBSTEIN ANOMALY
3. Which treatment is advised for Ebstein anomaly?
- Observation alone
- Standard heart failure medications
- Radiofrequency catheter ablation
- Tricuspid valve repair or replacement
- Biventricular reconstruction
- Heart transplant
The answer is all of the above. Observation alone is advised for patients with mild symptoms, no evidence of right-to-left shunting, and only mild cardiomegaly. Medical management includes an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker, a beta-blocker, and diuretics. Radiofrequency catheter ablation is the first-line therapy for patients with symptomatic Wolff-Parkinson-White syndrome.20 A patient who develops worsening right-sided heart failure, cyanosis, paradoxical emboli, or frequent tachyarrhythmias should be considered for corrective surgery, which may include tricuspid valve repair or replacement, or biventricular reconstruction.7,8,21 Cardiac transplant is reserved for severe cases.8
On hospital day 4, our patient undergoes successful radiofrequency catheter ablation without complications. At follow-up 3 months later, he continues to do well, with resolution of his symptoms and no further evidence of pre-excitation. His postprocedure ECG no longer shows delta waves.
TAKE-HOME POINTS
- For a patient with regular wide complex tachycardia, the first step is to assess hemodynamic stability. If the patient is hemodynamically unstable, emergent cardioversion is indicated.
- The differential diagnosis for regular wide complex tachycardia includes supraventricular tachycardia with aberrancy (orthodromic atrioventricular reentrant tachycardia, antidromic atrioventricular reentrant tachycardia, atrial tachycardia), and ventricular tachycardia.
- When pre-excited atrial fibrillation is suspected, AV nodal blocking agents should be avoided, as they may worsen tachyarrhythmia. Sodium channel blockers such as procainamide can help slow down the conduction of the accessory pathway.
- Ebstein anomaly is diagnosed on transthoracic echocardiography as apical displacement of the tricuspid valve resulting in atrialization of the right ventricle.
- Patients with Ebstein anomaly have a higher risk of death from right-sided heart failure and tachyarrhythmias, most commonly type B Wolff-Parkinson-White syndrome.
- Ebstein anomaly is medically managed with standard heart failure medications, including neurohormonal blockade therapies.
- Patients with Ebstein anomaly and cyanosis require surgical intervention with either valve repair or replacement.
Acknowledgment: We thank Dr. William Collins for his contribution in reviewing the manuscript and his technical expertise in developing some of the figures.
- January CT, Wann L, Alpert JS, et al; ACC/AHA Task Force Members. 2014 AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation 2014; 130:2071–2104.
- Brugada P, Brugada J, Mont L, Smeets J, Andries EW. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation 1991; 83:1649–1659.
- Alzand BS, Crijns HJ. Diagnostic criteria of broad QRS complex tachycardia: decades of evolution. Europace 2011; 13:465–472.
- Wellens HJ, Bar FW, Lie KI. The value of the electrocardiogram in the differential diagnosis of a tachycardia with a widened QRS complex. Am J Med 1978; 64:27–33.
- Field JM, Hazinski MF, Sayre MR, et al. Part 1: executive summary: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010; 122(suppl 3):S640–S656.
- Walkey AJ, Wiener RS, Ghobrial JM, Curtis LH, Benjamin EJ. Incident stroke and mortality associated with new-onset atrial fibrillation in patients hospitalized with severe sepsis. JAMA 2011; 306:2248–2254.
- Attenhofer Jost CH, Connolly HM, Edwards WD, Hayes D, Warnes CA, Danielson GK. Ebstein’s anomaly - review of a multifaceted congenital cardiac condition. Swiss Med Wkly 2005; 135:269–281.
- Attenhofer Jost CH, Connolly HM, Dearani JA, Edwards WD, Danielson GK. Ebstein’s anomaly. Circulation 2007; 115:277–285.
- Oechslin E, Buchholz S, Jenni R. Ebstein’s anomaly in adults: Doppler-echocardiographic evaluation. Thorac Cardiovasc Surg 2000; 48:209–213.
- Ali SK, Nimeri NA. Clinical and echocardiographic features of Ebstein’s malformation in Sudanese patients. Cardiol Young 2006; 16:147–151.
- Edwards WD. Embryology and pathologic features of Ebstein’s anomaly. Prog Pediatr Cardiol 1993; 2:5–15.
- Shiina A, Seward JB, Edwards WD, Hagler DJ, Tajik AJ. Two dimensional echocardiographic spectrum of Ebstein’s anomaly: detailed anatomic assessment. J Am Coll Cardiol 1984; 3:356–370.
- Gussenhoven EJ, Stewart PA, Becker AE, Essed CE, Ligtvoet KM, De Villeneuve VH. “Offsetting” of the septal tricuspid leaflet in normal hearts and in hearts with Ebstein’s anomaly. Anatomic and echographic correlation. Am J Cardiol 1984; 54:172–176.
- Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. Second of two parts. N Engl J Med 2000; 342:334–342.
- Report of the New England Regional Infant Cardiac Program. Pediatrics 1980; 65:375–461.
- Cohen LS, Friedman JM, Jefferson JW, Johnson EM, Weiner ML. A reevaluation of risk of in utero exposure to lithium. JAMA 1994; 271:146–150.
- Paranon S, Acar P. Ebstein’s anomaly of the tricuspid valve: from fetus to adult: congenital heart disease. Heart 2008; 94:237–243.
- Watson H. Natural history of Ebstein’s anomaly of tricuspid valve in childhood and adolescence. An international co-operative study of 505 cases. Br Heart J 1974; 36:417–427.
- Delhaas T, Sarvaas GJ, Rijlaarsdam ME, et al. A multicenter, long-term study on arrhythmias in children with Ebstein anomaly. Pediatr Cardiol 2010; 31:229–223.
- Tischenko A, Fox DJ, Yee R, et al. When should we recommend catheter ablation for patients with the Wolff-Parkinson-White syndrome? Curr Opin Cardiol 2008; 23:32–37.
- Misaki T, Watanabe G, Iwa T, et al. Surgical treatment of patients with Wolff-Parkinson-White syndrome and associated Ebstein’s anomaly. J Thorac Cardiovasc Surg 1995; 110:1702–1707.
- January CT, Wann L, Alpert JS, et al; ACC/AHA Task Force Members. 2014 AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation 2014; 130:2071–2104.
- Brugada P, Brugada J, Mont L, Smeets J, Andries EW. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation 1991; 83:1649–1659.
- Alzand BS, Crijns HJ. Diagnostic criteria of broad QRS complex tachycardia: decades of evolution. Europace 2011; 13:465–472.
- Wellens HJ, Bar FW, Lie KI. The value of the electrocardiogram in the differential diagnosis of a tachycardia with a widened QRS complex. Am J Med 1978; 64:27–33.
- Field JM, Hazinski MF, Sayre MR, et al. Part 1: executive summary: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010; 122(suppl 3):S640–S656.
- Walkey AJ, Wiener RS, Ghobrial JM, Curtis LH, Benjamin EJ. Incident stroke and mortality associated with new-onset atrial fibrillation in patients hospitalized with severe sepsis. JAMA 2011; 306:2248–2254.
- Attenhofer Jost CH, Connolly HM, Edwards WD, Hayes D, Warnes CA, Danielson GK. Ebstein’s anomaly - review of a multifaceted congenital cardiac condition. Swiss Med Wkly 2005; 135:269–281.
- Attenhofer Jost CH, Connolly HM, Dearani JA, Edwards WD, Danielson GK. Ebstein’s anomaly. Circulation 2007; 115:277–285.
- Oechslin E, Buchholz S, Jenni R. Ebstein’s anomaly in adults: Doppler-echocardiographic evaluation. Thorac Cardiovasc Surg 2000; 48:209–213.
- Ali SK, Nimeri NA. Clinical and echocardiographic features of Ebstein’s malformation in Sudanese patients. Cardiol Young 2006; 16:147–151.
- Edwards WD. Embryology and pathologic features of Ebstein’s anomaly. Prog Pediatr Cardiol 1993; 2:5–15.
- Shiina A, Seward JB, Edwards WD, Hagler DJ, Tajik AJ. Two dimensional echocardiographic spectrum of Ebstein’s anomaly: detailed anatomic assessment. J Am Coll Cardiol 1984; 3:356–370.
- Gussenhoven EJ, Stewart PA, Becker AE, Essed CE, Ligtvoet KM, De Villeneuve VH. “Offsetting” of the septal tricuspid leaflet in normal hearts and in hearts with Ebstein’s anomaly. Anatomic and echographic correlation. Am J Cardiol 1984; 54:172–176.
- Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. Second of two parts. N Engl J Med 2000; 342:334–342.
- Report of the New England Regional Infant Cardiac Program. Pediatrics 1980; 65:375–461.
- Cohen LS, Friedman JM, Jefferson JW, Johnson EM, Weiner ML. A reevaluation of risk of in utero exposure to lithium. JAMA 1994; 271:146–150.
- Paranon S, Acar P. Ebstein’s anomaly of the tricuspid valve: from fetus to adult: congenital heart disease. Heart 2008; 94:237–243.
- Watson H. Natural history of Ebstein’s anomaly of tricuspid valve in childhood and adolescence. An international co-operative study of 505 cases. Br Heart J 1974; 36:417–427.
- Delhaas T, Sarvaas GJ, Rijlaarsdam ME, et al. A multicenter, long-term study on arrhythmias in children with Ebstein anomaly. Pediatr Cardiol 2010; 31:229–223.
- Tischenko A, Fox DJ, Yee R, et al. When should we recommend catheter ablation for patients with the Wolff-Parkinson-White syndrome? Curr Opin Cardiol 2008; 23:32–37.
- Misaki T, Watanabe G, Iwa T, et al. Surgical treatment of patients with Wolff-Parkinson-White syndrome and associated Ebstein’s anomaly. J Thorac Cardiovasc Surg 1995; 110:1702–1707.
A 56-year-old with diarrhea and weakness
A 56-year-old man presents to the emergency department with nausea, weakness, and exertional dyspnea, which have been going on for 1 week. He is sent by his primary care physician after being noted to be hypotensive with a weak, thready pulse.
He has had diarrhea with intermittent abdominal pain over the past year, with 10 stools daily, including 3 or 4 at night. The stools are described as large, nonbloody, sticky, greasy, and occasionally watery. Stools are fewer when he curtails his food intake. The diarrhea is associated with occasional diffuse abdominal pain he describes as a burning sensation. He has no incontinence or tenesmus. He reports that he has unintentionally lost 137 lb (62 kg) over the past year. He has not taken over-the-counter antidiarrheal agents.
CHRONIC DIARRHEA
1. Chronic diarrhea is defined as lasting for at least how long?
- 1 week
- 2 weeks
- 3 weeks
- 4 weeks
Chronic diarrhea is defined as looser stools for more than 4 weeks,1 a period that allows most cases of acute, self-limited, infectious diarrhea to resolve.
Because individuals perceive diarrhea differently, reported prevalence rates of chronic diarrhea vary.2 Based on the definition of having excessive stool frequency, the prevalence in the United States is about 5%.1
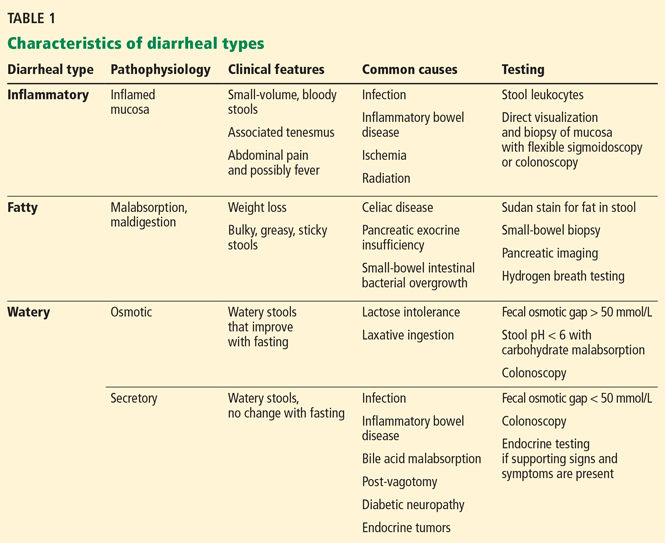
In developing countries, the most common cause of chronic diarrhea is infection. In developed nations, irritable bowel syndrome, inflammatory bowel disease, malabsorption syndrome, and chronic infection predominate.1
Once chronicity is established, diarrhea should be characterized as inflammatory, fatty, or watery (Table 1).3
CASE CONTINUED: HISTORY OF HYPERTENSION, DIABETES
Our patient reports that he has never traveled outside the United States. He has a history of hypertension and type 2 diabetes mellitus that is controlled on oral agents. He has had surgery for a radial fracture and for reconstruction of his knees. He uses no tobacco, alcohol, or illicit drugs and works as a train engineer. He has no pets. He knows of no family history of inflammatory bowel disease or chronic diarrhea.
Comment. Patients with diabetes are at increased risk of gastrointestinal problems, with severity increasing with poorer control.4 Although our patient’s diabetes puts him at risk of diabetic autonomic neuropathy, his blood glucose control has been consistently good since his diagnosis, and his last measured hemoglobin A1c was 7.3% (reference range 4%–7%). His description of greasy stools in conjunction with his marked weight loss puts fatty diarrhea higher on the differential diagnosis.
DRUG-INDUCED DIARRHEA
His medications include glimepiride 1 mg twice daily, lisinopril 10 mg daily, metformin 500 mg twice daily, omeprazole 40 mg daily, and naproxen 220 mg daily. He has been taking metformin for at least 2 years. He is allergic to pentobarbital.
2. Which of his medications is least likely to be associated with his diarrhea?
- Lisinopril
- Metformin
- Glimepiride
- Naproxen
More than 700 drugs are known to cause diarrhea, often through the interplay of simultaneous mechanisms.5 The diagnosis of drug-induced diarrhea requires taking a careful medication history and establishing a temporal relationship between the drug and the diarrheal symptoms. Treatment consists of withdrawing the offending agent.
Nonsteroidal anti-inflammatory drugs (eg, naproxen) are associated with collagenous colitis that occurs mostly after long-term use (> 6 months). Metformin-induced diarrhea is related to fat malabsorption. Olmesartan, an angiotensin II receptor antagonist, has been associated with severe sprue-like enteropathy. On the other hand, the incidence of diarrhea with lisinopril is similar to that with placebo.7
CASE CONTINUED: EXAMINATION AND LABORATORY VALUES
The patient’s primary care physician had recently referred him to a gastroenterologist, and 4 days before presenting to the emergency department he had undergone abdominal and pelvic computed tomography (CT) with iodinated contrast, which had showed hepatic steatosis and pancreatic atrophy.
On examination now, the patient’s temperature is 97.5°F (36.4°C), heart rate 90 beats per minute, respirations 18 breaths per minute, oxygen saturation 99% on room air, and blood pressure 85/55 mm Hg. His body mass index is 32.5 kg/m2. His oral mucosa is dry. The rest of the examination is normal. No rash or ulcers are noted.
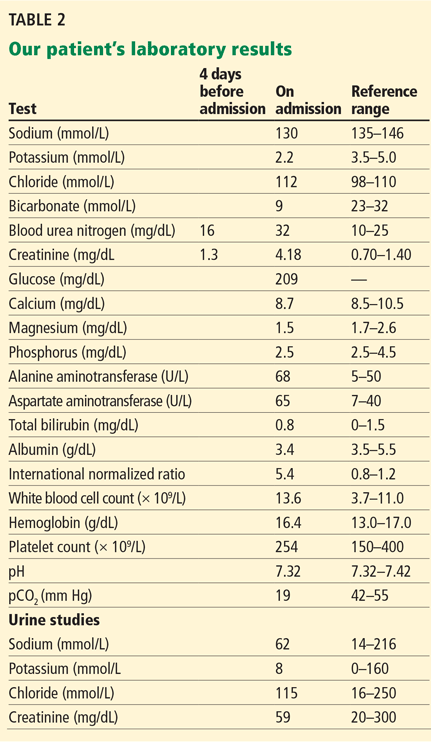
His laboratory values (Table 2) are notable for sodium 130 mmol/L, potassium 2.2 mmol/L, bicarbonate 9 mmol/L, blood urea nitrogen 32 mg/dL, creatinine 4.18 mg/dL, and international normalized ratio 5.4. Arterial blood gases drawn on admission reveal pH 7.32 and pCO2 19 mm Hg.
ACID-BASE DISTURBANCES
3. The patient’s acidosis is most likely related to which of the following?
- Sepsis
- Diarrhea
- Metformin
- Acute kidney injury
It is most likely related to diarrhea. The patient has a non-anion-gap metabolic acidosis. (The anion gap can be calculated by subtracting the sum of the serum bicarbonate and chloride values from the sodium—here, 130 – [112 + 9] = 9—and most textbooks list the reference range as 10–12 mmol/L.) Non-anion-gap metabolic acidosis results from excessive loss of bicarbonate or impaired ability of the kidney to excrete acid. Bicarbonate losses can occur in diarrhea or in ureteral diversion to the colon. Impairment in urinary acidification can occur in renal tubular acidosis.

To determine the cause of non-anion-gap acidosis, calculating the urine anion gap can be useful (Table 3), as it reflects the ability of the kidneys to excrete acid and is an indirect measure of ammonium excretion. Our patient’s urine anion gap is –45 mmol/L ([62 + 8] – 115), which supports diarrhea as the cause of his non-anion-gap acidosis. Sepsis, metformin use, or acute kidney injury would result in an anion-gap acidosis.
To manage acid-base disturbances, it is important to first determine whether there is a single primary disturbance with compensation or a mixed disorder. In the case of metabolic acidosis, for every 1-mmol/L decrease in bicarbonate, there should be a corresponding 1.3-mm Hg decrease in pCO2. Our patient’s laboratory data show that he had a pure non-anion-gap metabolic acidosis.8 His sensation of dyspnea was likely related to respiratory compensation as evidenced by an appropriately low pCO2.
CASE CONTINUED: HIS LABORATORY VALUES IMPROVE
The patient is admitted to the hospital for fluid resuscitation with normal saline and potassium and magnesium replacement.
Renal ultrasonography reveals normal-appearing kidneys without obstruction. The calculated fractional excretion of sodium is 3.4%. Urine microscopy reveals two to five hyaline casts per low-power field. His urine output remains adequate, and 3 days after hospitalization, his renal function starts to improve, as reflected in falling serum creatinine and blood urea nitrogen levels: his creatinine level has declined to 1.91 mg/dL and his blood urea nitrogen level has declined to 24 mg/dL. His acute kidney injury is attributed to intravenous contrast given for computed tomography, as well as to volume depletion and hypotension.
Stool studies for ova, parasites, and Clostridium difficile are negative. Fecal calprotectin and lactoferrin are useful noninvasive markers of intestinal inflammation but were not checked in this case.
Loperamide, taken as needed, is started for his diarrhea, along with empiric pancreatic enzyme replacement. After 3 days of treatment with oral vitamin K 10 mg, his international normalized ratio comes down to 1.4, from his admission value of 5.4. Given the clinical concern for fat malabsorption, vitamin D levels are also checked: his 25-hydroxyvitamin D level is less than 10 ng/mL (lower limit of normal 20). His fecal neutral fats are reported as normal, but split fats are increased.
STOOL FAT STUDIES
4. What does increased fecal split fats but normal fecal neutral fats imply?
- Pancreatic insufficiency
- Intestinal malabsorption
- Does not differentiate between the two
The finding does not differentiate between pancreatic insufficiency and intestinal malabsorption. The two-step Sudan stain has been used to differentiate maldigestion (eg, caused by pancreatic insufficiency) from malabsorption. Although patients with impaired digestion were once thought to excrete excessive amounts of intact triglyceride whereas those with malabsorption excrete more of the lipolytic or “split” product, the Sudan stain does not differentiate between the two.10 Stool fecal-elastase 1 testing correlates well with pancreatic exocrine function but was not performed in our patient.11
CASE CONTINUED: CELIAC DISEASE IS DIAGNOSED

Given the description of his stools, unintentional weight loss, and improvement of stool frequency with fasting, serologic testing for celiac disease is performed (Table 4). The patient undergoes esophagogastroduodenoscopy, which shows mild duodenitis. Small-bowel biopsy reveals blunted villous architecture and increased mixed inflammatory cells of the epithelium and lamina propria, suggestive of celiac disease.
The patient is diagnosed with celiac disease and is counseled to follow a gluten-free diet. He is discharged home and scheduled to follow up with a gastroenterologist and nephrologist. His liver function test abnormalities are attributed to a combination of nonalcoholic steatohepatitis and celiac disease.
CELIAC DISEASE AND MALABSORPTION
Celiac disease is an immune-mediated disorder that causes mucosal injury to the small intestine, leading to malabsorption. It is triggered by gluten intake in genetically susceptible individuals. The HLA-DQ2 haplotype is expressed in nearly 90% of patients with the disease.
The worldwide prevalence of celiac disease is about 0.6% to 1%. Those with an affected first-degree relative, type 1 diabetes, Hashimoto thyroiditis, an autoimmune disease, Down syndrome, Turner syndrome, or IgA deficiency have an increased risk.
Celiac disease presents with chronic diarrhea, weight loss, and abdominal distention and pain. Sequelae of nutrient malabsorption such as iron-deficiency anemia, short stature, and osteopenia may be evident. Liver function may also be impaired. Dermatitis herpetiformis and gluten ataxia are rarer presentations of celiac disease.12
In the absence of immunoglobulin (Ig) A deficiency, measurement of serum IgA anti-tissue transglutaminase antibodies is recommended for initial testing. IgG antitissue transglutaminase antibodies can be measured in those with IgA deficiency.12
Duodenal biopsies to confirm the diagnosis are recommended in adults unless they have previously had biopsy-proven dermatitis herpetiformis.
Gluten-free diet
The treatment for celiac disease is avoidance of gluten. Patients who consult with a nutritionist and participate in an advocacy group are more likely to adhere to a gluten-free diet, and the physician should strongly encourage and facilitate these activities.13
Untreated disease can lead to osteoporosis, impaired splenic function with increased risk of infection with encapsulated organisms, infertility or recurrent abortion, ulcerative jejunoileitis, and lymphoma.12 Patients should be monitored annually for adherence to the gluten-free diet and for the development of any associated condition. Despite adherence to a gluten-free diet, calcium absorption and bone mineral density are lower in patients with celiac disease than in controls.14 Careful monitoring of fracture risk and adequate calcium and vitamin D replacement are also important.
Our patient undergoes dual-emission x-ray absorptiometry after discharge, with results consistent with osteopenia. His T scores range from –0.2 at the right hip to –1.1 in the left femoral neck.
Recurrence or persistently abnormal levels of IgA anti-tissue transglutaminase antibodies usually indicates poor dietary compliance.12
5. In patients whose symptoms do not improve on gluten restriction, there should be concern for which of the following?
- Lymphoma
- Nonadherence to gluten restriction
- Microscopic colitis
- All of the above
The answer is all of the above. Up to 30% of patients have persistent symptoms on a gluten-free diet. Persistent exposure to gluten is the most common reason for lack of clinical improvement. In addition, bacterial overgrowth of the small bowel, lactose intolerance, pancreatic insufficiency, and microscopic colitis may coexist with celiac disease and may contribute to ongoing symptoms. In a small subset of patients with persistent villous atrophy and symptoms despite strict adherence to a gluten-free diet for 12 months, the disease is deemed “refractory.” Refractory celiac disease can be a precursor to enteropathy-associated T-cell lymphoma.13
CASE CONCLUDED
On telephone follow-up 3 weeks after discharge, the patient reports complete resolution of diarrhea and stabilization of his weight. He reports strict adherence to a gluten-free diet and feels he is coping well.
Diagnoses
- Presenting weakness secondary to dehydration and hypokalemia
- Dyspnea secondary to respiratory compensation for metabolic acidosis
- Non-anion-gap metabolic acidosis secondary to diarrhea
- Acute kidney injury secondary to iodinated contrast, volume depletion, hypotension
- Chronic diarrhea secondary to celiac disease
- Coagulopathy secondary to fat malabsorption secondary to celiac disease.
- Fine KD, Schiller LR. AGA technical review on the evaluation and management of chronic diarrhea. Gastroenterology 1999; 116:1464–1486.
- Talley NJ, Weaver AL, Zinsmeister AR, Melton LJ 3rd. Self-reported diarrhea: what does it mean? Am J Gastroenterol 1994; 89:1160–1164.
- Sweetser S. Evaluating the patient with diarrhea: a case-based approach. Mayo Clin Proc 2012; 87:596–602.
- Bytzer P, Talley NJ, Leemon M, Young LJ, Jones MP, Horowitz M. Prevalence of gastrointestinal symptoms associated with diabetes mellitus: a population-based survey of 15,000 adults. Arch Intern Med 2001; 161:1989–1996.
- Chassany O, Michaux A, Bergmann JF. Drug-induced diarrhoea. Drug Saf 2000; 22:53–72.
- Rubio-Tapia A, Herman ML, Ludvigsson JF, et al. Severe spruelike enteropathy associated with olmesartan. Mayo Clin Proc 2012; 87:732–738.
- Zestril (lisinopril) tablets. www.accessdata.fda.gov/drugsatfda_docs/label/2012/019777s062lbl.pdf. Accessed September 8, 2015.
- Whittier WL, Rutecki GW. Primer on clinical acid-base problem solving. Dis Mon 2004; 50:122–162.
- Langhorst J, Elsenbruch S, Koelzer J, Rueffer A, Michalsen A, Dobos GJ. Noninvasive markers in the assessment of intestinal inflammation in inflammatory bowel diseases: performance of fecal lactoferrin, calprotectin, and PMN-elastase, CRP, and clinical indices. Am J Gastroenterol 2008; 103:162–169.
- Khouri MR, Ng SN, Huang G, Shiau YF. Fecal triglyceride excretion is not excessive in pancreatic insufficiency. Gastroenterology 1989; 96:848–852.
- Dominici R, Franzini C. Fecal elastase-1 as a test for pancreatic function: a review. Clin Chem Lab Med 2002; 40:325–332.
- Fasano A, Catassi C. Celiac disease. New Engl J Med 2012; 367:2419–2426.
- Mooney PD, Hadjivassiliou M, Sanders DS. Coeliac disease. BMJ 2014; 348:g1561–g1561.
- Pazianas M, Butcher GP, Subhani JM, et al. Calcium absorption and bone mineral density in celiacs after long term treatment with gluten-free diet and adequate calcium intake. Osteoporos Int 2005; 16:56–63.
A 56-year-old man presents to the emergency department with nausea, weakness, and exertional dyspnea, which have been going on for 1 week. He is sent by his primary care physician after being noted to be hypotensive with a weak, thready pulse.
He has had diarrhea with intermittent abdominal pain over the past year, with 10 stools daily, including 3 or 4 at night. The stools are described as large, nonbloody, sticky, greasy, and occasionally watery. Stools are fewer when he curtails his food intake. The diarrhea is associated with occasional diffuse abdominal pain he describes as a burning sensation. He has no incontinence or tenesmus. He reports that he has unintentionally lost 137 lb (62 kg) over the past year. He has not taken over-the-counter antidiarrheal agents.
CHRONIC DIARRHEA
1. Chronic diarrhea is defined as lasting for at least how long?
- 1 week
- 2 weeks
- 3 weeks
- 4 weeks
Chronic diarrhea is defined as looser stools for more than 4 weeks,1 a period that allows most cases of acute, self-limited, infectious diarrhea to resolve.
Because individuals perceive diarrhea differently, reported prevalence rates of chronic diarrhea vary.2 Based on the definition of having excessive stool frequency, the prevalence in the United States is about 5%.1
In developing countries, the most common cause of chronic diarrhea is infection. In developed nations, irritable bowel syndrome, inflammatory bowel disease, malabsorption syndrome, and chronic infection predominate.1
Once chronicity is established, diarrhea should be characterized as inflammatory, fatty, or watery (Table 1).3
CASE CONTINUED: HISTORY OF HYPERTENSION, DIABETES
Our patient reports that he has never traveled outside the United States. He has a history of hypertension and type 2 diabetes mellitus that is controlled on oral agents. He has had surgery for a radial fracture and for reconstruction of his knees. He uses no tobacco, alcohol, or illicit drugs and works as a train engineer. He has no pets. He knows of no family history of inflammatory bowel disease or chronic diarrhea.
Comment. Patients with diabetes are at increased risk of gastrointestinal problems, with severity increasing with poorer control.4 Although our patient’s diabetes puts him at risk of diabetic autonomic neuropathy, his blood glucose control has been consistently good since his diagnosis, and his last measured hemoglobin A1c was 7.3% (reference range 4%–7%). His description of greasy stools in conjunction with his marked weight loss puts fatty diarrhea higher on the differential diagnosis.
DRUG-INDUCED DIARRHEA
His medications include glimepiride 1 mg twice daily, lisinopril 10 mg daily, metformin 500 mg twice daily, omeprazole 40 mg daily, and naproxen 220 mg daily. He has been taking metformin for at least 2 years. He is allergic to pentobarbital.
2. Which of his medications is least likely to be associated with his diarrhea?
- Lisinopril
- Metformin
- Glimepiride
- Naproxen
More than 700 drugs are known to cause diarrhea, often through the interplay of simultaneous mechanisms.5 The diagnosis of drug-induced diarrhea requires taking a careful medication history and establishing a temporal relationship between the drug and the diarrheal symptoms. Treatment consists of withdrawing the offending agent.
Nonsteroidal anti-inflammatory drugs (eg, naproxen) are associated with collagenous colitis that occurs mostly after long-term use (> 6 months). Metformin-induced diarrhea is related to fat malabsorption. Olmesartan, an angiotensin II receptor antagonist, has been associated with severe sprue-like enteropathy. On the other hand, the incidence of diarrhea with lisinopril is similar to that with placebo.7
CASE CONTINUED: EXAMINATION AND LABORATORY VALUES
The patient’s primary care physician had recently referred him to a gastroenterologist, and 4 days before presenting to the emergency department he had undergone abdominal and pelvic computed tomography (CT) with iodinated contrast, which had showed hepatic steatosis and pancreatic atrophy.
On examination now, the patient’s temperature is 97.5°F (36.4°C), heart rate 90 beats per minute, respirations 18 breaths per minute, oxygen saturation 99% on room air, and blood pressure 85/55 mm Hg. His body mass index is 32.5 kg/m2. His oral mucosa is dry. The rest of the examination is normal. No rash or ulcers are noted.
His laboratory values (Table 2) are notable for sodium 130 mmol/L, potassium 2.2 mmol/L, bicarbonate 9 mmol/L, blood urea nitrogen 32 mg/dL, creatinine 4.18 mg/dL, and international normalized ratio 5.4. Arterial blood gases drawn on admission reveal pH 7.32 and pCO2 19 mm Hg.
ACID-BASE DISTURBANCES
3. The patient’s acidosis is most likely related to which of the following?
- Sepsis
- Diarrhea
- Metformin
- Acute kidney injury
It is most likely related to diarrhea. The patient has a non-anion-gap metabolic acidosis. (The anion gap can be calculated by subtracting the sum of the serum bicarbonate and chloride values from the sodium—here, 130 – [112 + 9] = 9—and most textbooks list the reference range as 10–12 mmol/L.) Non-anion-gap metabolic acidosis results from excessive loss of bicarbonate or impaired ability of the kidney to excrete acid. Bicarbonate losses can occur in diarrhea or in ureteral diversion to the colon. Impairment in urinary acidification can occur in renal tubular acidosis.
To determine the cause of non-anion-gap acidosis, calculating the urine anion gap can be useful (Table 3), as it reflects the ability of the kidneys to excrete acid and is an indirect measure of ammonium excretion. Our patient’s urine anion gap is –45 mmol/L ([62 + 8] – 115), which supports diarrhea as the cause of his non-anion-gap acidosis. Sepsis, metformin use, or acute kidney injury would result in an anion-gap acidosis.
To manage acid-base disturbances, it is important to first determine whether there is a single primary disturbance with compensation or a mixed disorder. In the case of metabolic acidosis, for every 1-mmol/L decrease in bicarbonate, there should be a corresponding 1.3-mm Hg decrease in pCO2. Our patient’s laboratory data show that he had a pure non-anion-gap metabolic acidosis.8 His sensation of dyspnea was likely related to respiratory compensation as evidenced by an appropriately low pCO2.
CASE CONTINUED: HIS LABORATORY VALUES IMPROVE
The patient is admitted to the hospital for fluid resuscitation with normal saline and potassium and magnesium replacement.
Renal ultrasonography reveals normal-appearing kidneys without obstruction. The calculated fractional excretion of sodium is 3.4%. Urine microscopy reveals two to five hyaline casts per low-power field. His urine output remains adequate, and 3 days after hospitalization, his renal function starts to improve, as reflected in falling serum creatinine and blood urea nitrogen levels: his creatinine level has declined to 1.91 mg/dL and his blood urea nitrogen level has declined to 24 mg/dL. His acute kidney injury is attributed to intravenous contrast given for computed tomography, as well as to volume depletion and hypotension.
Stool studies for ova, parasites, and Clostridium difficile are negative. Fecal calprotectin and lactoferrin are useful noninvasive markers of intestinal inflammation but were not checked in this case.
Loperamide, taken as needed, is started for his diarrhea, along with empiric pancreatic enzyme replacement. After 3 days of treatment with oral vitamin K 10 mg, his international normalized ratio comes down to 1.4, from his admission value of 5.4. Given the clinical concern for fat malabsorption, vitamin D levels are also checked: his 25-hydroxyvitamin D level is less than 10 ng/mL (lower limit of normal 20). His fecal neutral fats are reported as normal, but split fats are increased.
STOOL FAT STUDIES
4. What does increased fecal split fats but normal fecal neutral fats imply?
- Pancreatic insufficiency
- Intestinal malabsorption
- Does not differentiate between the two
The finding does not differentiate between pancreatic insufficiency and intestinal malabsorption. The two-step Sudan stain has been used to differentiate maldigestion (eg, caused by pancreatic insufficiency) from malabsorption. Although patients with impaired digestion were once thought to excrete excessive amounts of intact triglyceride whereas those with malabsorption excrete more of the lipolytic or “split” product, the Sudan stain does not differentiate between the two.10 Stool fecal-elastase 1 testing correlates well with pancreatic exocrine function but was not performed in our patient.11
CASE CONTINUED: CELIAC DISEASE IS DIAGNOSED
Given the description of his stools, unintentional weight loss, and improvement of stool frequency with fasting, serologic testing for celiac disease is performed (Table 4). The patient undergoes esophagogastroduodenoscopy, which shows mild duodenitis. Small-bowel biopsy reveals blunted villous architecture and increased mixed inflammatory cells of the epithelium and lamina propria, suggestive of celiac disease.
The patient is diagnosed with celiac disease and is counseled to follow a gluten-free diet. He is discharged home and scheduled to follow up with a gastroenterologist and nephrologist. His liver function test abnormalities are attributed to a combination of nonalcoholic steatohepatitis and celiac disease.
CELIAC DISEASE AND MALABSORPTION
Celiac disease is an immune-mediated disorder that causes mucosal injury to the small intestine, leading to malabsorption. It is triggered by gluten intake in genetically susceptible individuals. The HLA-DQ2 haplotype is expressed in nearly 90% of patients with the disease.
The worldwide prevalence of celiac disease is about 0.6% to 1%. Those with an affected first-degree relative, type 1 diabetes, Hashimoto thyroiditis, an autoimmune disease, Down syndrome, Turner syndrome, or IgA deficiency have an increased risk.
Celiac disease presents with chronic diarrhea, weight loss, and abdominal distention and pain. Sequelae of nutrient malabsorption such as iron-deficiency anemia, short stature, and osteopenia may be evident. Liver function may also be impaired. Dermatitis herpetiformis and gluten ataxia are rarer presentations of celiac disease.12
In the absence of immunoglobulin (Ig) A deficiency, measurement of serum IgA anti-tissue transglutaminase antibodies is recommended for initial testing. IgG antitissue transglutaminase antibodies can be measured in those with IgA deficiency.12
Duodenal biopsies to confirm the diagnosis are recommended in adults unless they have previously had biopsy-proven dermatitis herpetiformis.
Gluten-free diet
The treatment for celiac disease is avoidance of gluten. Patients who consult with a nutritionist and participate in an advocacy group are more likely to adhere to a gluten-free diet, and the physician should strongly encourage and facilitate these activities.13
Untreated disease can lead to osteoporosis, impaired splenic function with increased risk of infection with encapsulated organisms, infertility or recurrent abortion, ulcerative jejunoileitis, and lymphoma.12 Patients should be monitored annually for adherence to the gluten-free diet and for the development of any associated condition. Despite adherence to a gluten-free diet, calcium absorption and bone mineral density are lower in patients with celiac disease than in controls.14 Careful monitoring of fracture risk and adequate calcium and vitamin D replacement are also important.
Our patient undergoes dual-emission x-ray absorptiometry after discharge, with results consistent with osteopenia. His T scores range from –0.2 at the right hip to –1.1 in the left femoral neck.
Recurrence or persistently abnormal levels of IgA anti-tissue transglutaminase antibodies usually indicates poor dietary compliance.12
5. In patients whose symptoms do not improve on gluten restriction, there should be concern for which of the following?
- Lymphoma
- Nonadherence to gluten restriction
- Microscopic colitis
- All of the above
The answer is all of the above. Up to 30% of patients have persistent symptoms on a gluten-free diet. Persistent exposure to gluten is the most common reason for lack of clinical improvement. In addition, bacterial overgrowth of the small bowel, lactose intolerance, pancreatic insufficiency, and microscopic colitis may coexist with celiac disease and may contribute to ongoing symptoms. In a small subset of patients with persistent villous atrophy and symptoms despite strict adherence to a gluten-free diet for 12 months, the disease is deemed “refractory.” Refractory celiac disease can be a precursor to enteropathy-associated T-cell lymphoma.13
CASE CONCLUDED
On telephone follow-up 3 weeks after discharge, the patient reports complete resolution of diarrhea and stabilization of his weight. He reports strict adherence to a gluten-free diet and feels he is coping well.
Diagnoses
- Presenting weakness secondary to dehydration and hypokalemia
- Dyspnea secondary to respiratory compensation for metabolic acidosis
- Non-anion-gap metabolic acidosis secondary to diarrhea
- Acute kidney injury secondary to iodinated contrast, volume depletion, hypotension
- Chronic diarrhea secondary to celiac disease
- Coagulopathy secondary to fat malabsorption secondary to celiac disease.
A 56-year-old man presents to the emergency department with nausea, weakness, and exertional dyspnea, which have been going on for 1 week. He is sent by his primary care physician after being noted to be hypotensive with a weak, thready pulse.
He has had diarrhea with intermittent abdominal pain over the past year, with 10 stools daily, including 3 or 4 at night. The stools are described as large, nonbloody, sticky, greasy, and occasionally watery. Stools are fewer when he curtails his food intake. The diarrhea is associated with occasional diffuse abdominal pain he describes as a burning sensation. He has no incontinence or tenesmus. He reports that he has unintentionally lost 137 lb (62 kg) over the past year. He has not taken over-the-counter antidiarrheal agents.
CHRONIC DIARRHEA
1. Chronic diarrhea is defined as lasting for at least how long?
- 1 week
- 2 weeks
- 3 weeks
- 4 weeks
Chronic diarrhea is defined as looser stools for more than 4 weeks,1 a period that allows most cases of acute, self-limited, infectious diarrhea to resolve.
Because individuals perceive diarrhea differently, reported prevalence rates of chronic diarrhea vary.2 Based on the definition of having excessive stool frequency, the prevalence in the United States is about 5%.1
In developing countries, the most common cause of chronic diarrhea is infection. In developed nations, irritable bowel syndrome, inflammatory bowel disease, malabsorption syndrome, and chronic infection predominate.1
Once chronicity is established, diarrhea should be characterized as inflammatory, fatty, or watery (Table 1).3
CASE CONTINUED: HISTORY OF HYPERTENSION, DIABETES
Our patient reports that he has never traveled outside the United States. He has a history of hypertension and type 2 diabetes mellitus that is controlled on oral agents. He has had surgery for a radial fracture and for reconstruction of his knees. He uses no tobacco, alcohol, or illicit drugs and works as a train engineer. He has no pets. He knows of no family history of inflammatory bowel disease or chronic diarrhea.
Comment. Patients with diabetes are at increased risk of gastrointestinal problems, with severity increasing with poorer control.4 Although our patient’s diabetes puts him at risk of diabetic autonomic neuropathy, his blood glucose control has been consistently good since his diagnosis, and his last measured hemoglobin A1c was 7.3% (reference range 4%–7%). His description of greasy stools in conjunction with his marked weight loss puts fatty diarrhea higher on the differential diagnosis.
DRUG-INDUCED DIARRHEA
His medications include glimepiride 1 mg twice daily, lisinopril 10 mg daily, metformin 500 mg twice daily, omeprazole 40 mg daily, and naproxen 220 mg daily. He has been taking metformin for at least 2 years. He is allergic to pentobarbital.
2. Which of his medications is least likely to be associated with his diarrhea?
- Lisinopril
- Metformin
- Glimepiride
- Naproxen
More than 700 drugs are known to cause diarrhea, often through the interplay of simultaneous mechanisms.5 The diagnosis of drug-induced diarrhea requires taking a careful medication history and establishing a temporal relationship between the drug and the diarrheal symptoms. Treatment consists of withdrawing the offending agent.
Nonsteroidal anti-inflammatory drugs (eg, naproxen) are associated with collagenous colitis that occurs mostly after long-term use (> 6 months). Metformin-induced diarrhea is related to fat malabsorption. Olmesartan, an angiotensin II receptor antagonist, has been associated with severe sprue-like enteropathy. On the other hand, the incidence of diarrhea with lisinopril is similar to that with placebo.7
CASE CONTINUED: EXAMINATION AND LABORATORY VALUES
The patient’s primary care physician had recently referred him to a gastroenterologist, and 4 days before presenting to the emergency department he had undergone abdominal and pelvic computed tomography (CT) with iodinated contrast, which had showed hepatic steatosis and pancreatic atrophy.
On examination now, the patient’s temperature is 97.5°F (36.4°C), heart rate 90 beats per minute, respirations 18 breaths per minute, oxygen saturation 99% on room air, and blood pressure 85/55 mm Hg. His body mass index is 32.5 kg/m2. His oral mucosa is dry. The rest of the examination is normal. No rash or ulcers are noted.
His laboratory values (Table 2) are notable for sodium 130 mmol/L, potassium 2.2 mmol/L, bicarbonate 9 mmol/L, blood urea nitrogen 32 mg/dL, creatinine 4.18 mg/dL, and international normalized ratio 5.4. Arterial blood gases drawn on admission reveal pH 7.32 and pCO2 19 mm Hg.
ACID-BASE DISTURBANCES
3. The patient’s acidosis is most likely related to which of the following?
- Sepsis
- Diarrhea
- Metformin
- Acute kidney injury
It is most likely related to diarrhea. The patient has a non-anion-gap metabolic acidosis. (The anion gap can be calculated by subtracting the sum of the serum bicarbonate and chloride values from the sodium—here, 130 – [112 + 9] = 9—and most textbooks list the reference range as 10–12 mmol/L.) Non-anion-gap metabolic acidosis results from excessive loss of bicarbonate or impaired ability of the kidney to excrete acid. Bicarbonate losses can occur in diarrhea or in ureteral diversion to the colon. Impairment in urinary acidification can occur in renal tubular acidosis.
To determine the cause of non-anion-gap acidosis, calculating the urine anion gap can be useful (Table 3), as it reflects the ability of the kidneys to excrete acid and is an indirect measure of ammonium excretion. Our patient’s urine anion gap is –45 mmol/L ([62 + 8] – 115), which supports diarrhea as the cause of his non-anion-gap acidosis. Sepsis, metformin use, or acute kidney injury would result in an anion-gap acidosis.
To manage acid-base disturbances, it is important to first determine whether there is a single primary disturbance with compensation or a mixed disorder. In the case of metabolic acidosis, for every 1-mmol/L decrease in bicarbonate, there should be a corresponding 1.3-mm Hg decrease in pCO2. Our patient’s laboratory data show that he had a pure non-anion-gap metabolic acidosis.8 His sensation of dyspnea was likely related to respiratory compensation as evidenced by an appropriately low pCO2.
CASE CONTINUED: HIS LABORATORY VALUES IMPROVE
The patient is admitted to the hospital for fluid resuscitation with normal saline and potassium and magnesium replacement.
Renal ultrasonography reveals normal-appearing kidneys without obstruction. The calculated fractional excretion of sodium is 3.4%. Urine microscopy reveals two to five hyaline casts per low-power field. His urine output remains adequate, and 3 days after hospitalization, his renal function starts to improve, as reflected in falling serum creatinine and blood urea nitrogen levels: his creatinine level has declined to 1.91 mg/dL and his blood urea nitrogen level has declined to 24 mg/dL. His acute kidney injury is attributed to intravenous contrast given for computed tomography, as well as to volume depletion and hypotension.
Stool studies for ova, parasites, and Clostridium difficile are negative. Fecal calprotectin and lactoferrin are useful noninvasive markers of intestinal inflammation but were not checked in this case.
Loperamide, taken as needed, is started for his diarrhea, along with empiric pancreatic enzyme replacement. After 3 days of treatment with oral vitamin K 10 mg, his international normalized ratio comes down to 1.4, from his admission value of 5.4. Given the clinical concern for fat malabsorption, vitamin D levels are also checked: his 25-hydroxyvitamin D level is less than 10 ng/mL (lower limit of normal 20). His fecal neutral fats are reported as normal, but split fats are increased.
STOOL FAT STUDIES
4. What does increased fecal split fats but normal fecal neutral fats imply?
- Pancreatic insufficiency
- Intestinal malabsorption
- Does not differentiate between the two
The finding does not differentiate between pancreatic insufficiency and intestinal malabsorption. The two-step Sudan stain has been used to differentiate maldigestion (eg, caused by pancreatic insufficiency) from malabsorption. Although patients with impaired digestion were once thought to excrete excessive amounts of intact triglyceride whereas those with malabsorption excrete more of the lipolytic or “split” product, the Sudan stain does not differentiate between the two.10 Stool fecal-elastase 1 testing correlates well with pancreatic exocrine function but was not performed in our patient.11
CASE CONTINUED: CELIAC DISEASE IS DIAGNOSED
Given the description of his stools, unintentional weight loss, and improvement of stool frequency with fasting, serologic testing for celiac disease is performed (Table 4). The patient undergoes esophagogastroduodenoscopy, which shows mild duodenitis. Small-bowel biopsy reveals blunted villous architecture and increased mixed inflammatory cells of the epithelium and lamina propria, suggestive of celiac disease.
The patient is diagnosed with celiac disease and is counseled to follow a gluten-free diet. He is discharged home and scheduled to follow up with a gastroenterologist and nephrologist. His liver function test abnormalities are attributed to a combination of nonalcoholic steatohepatitis and celiac disease.
CELIAC DISEASE AND MALABSORPTION
Celiac disease is an immune-mediated disorder that causes mucosal injury to the small intestine, leading to malabsorption. It is triggered by gluten intake in genetically susceptible individuals. The HLA-DQ2 haplotype is expressed in nearly 90% of patients with the disease.
The worldwide prevalence of celiac disease is about 0.6% to 1%. Those with an affected first-degree relative, type 1 diabetes, Hashimoto thyroiditis, an autoimmune disease, Down syndrome, Turner syndrome, or IgA deficiency have an increased risk.
Celiac disease presents with chronic diarrhea, weight loss, and abdominal distention and pain. Sequelae of nutrient malabsorption such as iron-deficiency anemia, short stature, and osteopenia may be evident. Liver function may also be impaired. Dermatitis herpetiformis and gluten ataxia are rarer presentations of celiac disease.12
In the absence of immunoglobulin (Ig) A deficiency, measurement of serum IgA anti-tissue transglutaminase antibodies is recommended for initial testing. IgG antitissue transglutaminase antibodies can be measured in those with IgA deficiency.12
Duodenal biopsies to confirm the diagnosis are recommended in adults unless they have previously had biopsy-proven dermatitis herpetiformis.
Gluten-free diet
The treatment for celiac disease is avoidance of gluten. Patients who consult with a nutritionist and participate in an advocacy group are more likely to adhere to a gluten-free diet, and the physician should strongly encourage and facilitate these activities.13
Untreated disease can lead to osteoporosis, impaired splenic function with increased risk of infection with encapsulated organisms, infertility or recurrent abortion, ulcerative jejunoileitis, and lymphoma.12 Patients should be monitored annually for adherence to the gluten-free diet and for the development of any associated condition. Despite adherence to a gluten-free diet, calcium absorption and bone mineral density are lower in patients with celiac disease than in controls.14 Careful monitoring of fracture risk and adequate calcium and vitamin D replacement are also important.
Our patient undergoes dual-emission x-ray absorptiometry after discharge, with results consistent with osteopenia. His T scores range from –0.2 at the right hip to –1.1 in the left femoral neck.
Recurrence or persistently abnormal levels of IgA anti-tissue transglutaminase antibodies usually indicates poor dietary compliance.12
5. In patients whose symptoms do not improve on gluten restriction, there should be concern for which of the following?
- Lymphoma
- Nonadherence to gluten restriction
- Microscopic colitis
- All of the above
The answer is all of the above. Up to 30% of patients have persistent symptoms on a gluten-free diet. Persistent exposure to gluten is the most common reason for lack of clinical improvement. In addition, bacterial overgrowth of the small bowel, lactose intolerance, pancreatic insufficiency, and microscopic colitis may coexist with celiac disease and may contribute to ongoing symptoms. In a small subset of patients with persistent villous atrophy and symptoms despite strict adherence to a gluten-free diet for 12 months, the disease is deemed “refractory.” Refractory celiac disease can be a precursor to enteropathy-associated T-cell lymphoma.13
CASE CONCLUDED
On telephone follow-up 3 weeks after discharge, the patient reports complete resolution of diarrhea and stabilization of his weight. He reports strict adherence to a gluten-free diet and feels he is coping well.
Diagnoses
- Presenting weakness secondary to dehydration and hypokalemia
- Dyspnea secondary to respiratory compensation for metabolic acidosis
- Non-anion-gap metabolic acidosis secondary to diarrhea
- Acute kidney injury secondary to iodinated contrast, volume depletion, hypotension
- Chronic diarrhea secondary to celiac disease
- Coagulopathy secondary to fat malabsorption secondary to celiac disease.
- Fine KD, Schiller LR. AGA technical review on the evaluation and management of chronic diarrhea. Gastroenterology 1999; 116:1464–1486.
- Talley NJ, Weaver AL, Zinsmeister AR, Melton LJ 3rd. Self-reported diarrhea: what does it mean? Am J Gastroenterol 1994; 89:1160–1164.
- Sweetser S. Evaluating the patient with diarrhea: a case-based approach. Mayo Clin Proc 2012; 87:596–602.
- Bytzer P, Talley NJ, Leemon M, Young LJ, Jones MP, Horowitz M. Prevalence of gastrointestinal symptoms associated with diabetes mellitus: a population-based survey of 15,000 adults. Arch Intern Med 2001; 161:1989–1996.
- Chassany O, Michaux A, Bergmann JF. Drug-induced diarrhoea. Drug Saf 2000; 22:53–72.
- Rubio-Tapia A, Herman ML, Ludvigsson JF, et al. Severe spruelike enteropathy associated with olmesartan. Mayo Clin Proc 2012; 87:732–738.
- Zestril (lisinopril) tablets. www.accessdata.fda.gov/drugsatfda_docs/label/2012/019777s062lbl.pdf. Accessed September 8, 2015.
- Whittier WL, Rutecki GW. Primer on clinical acid-base problem solving. Dis Mon 2004; 50:122–162.
- Langhorst J, Elsenbruch S, Koelzer J, Rueffer A, Michalsen A, Dobos GJ. Noninvasive markers in the assessment of intestinal inflammation in inflammatory bowel diseases: performance of fecal lactoferrin, calprotectin, and PMN-elastase, CRP, and clinical indices. Am J Gastroenterol 2008; 103:162–169.
- Khouri MR, Ng SN, Huang G, Shiau YF. Fecal triglyceride excretion is not excessive in pancreatic insufficiency. Gastroenterology 1989; 96:848–852.
- Dominici R, Franzini C. Fecal elastase-1 as a test for pancreatic function: a review. Clin Chem Lab Med 2002; 40:325–332.
- Fasano A, Catassi C. Celiac disease. New Engl J Med 2012; 367:2419–2426.
- Mooney PD, Hadjivassiliou M, Sanders DS. Coeliac disease. BMJ 2014; 348:g1561–g1561.
- Pazianas M, Butcher GP, Subhani JM, et al. Calcium absorption and bone mineral density in celiacs after long term treatment with gluten-free diet and adequate calcium intake. Osteoporos Int 2005; 16:56–63.
- Fine KD, Schiller LR. AGA technical review on the evaluation and management of chronic diarrhea. Gastroenterology 1999; 116:1464–1486.
- Talley NJ, Weaver AL, Zinsmeister AR, Melton LJ 3rd. Self-reported diarrhea: what does it mean? Am J Gastroenterol 1994; 89:1160–1164.
- Sweetser S. Evaluating the patient with diarrhea: a case-based approach. Mayo Clin Proc 2012; 87:596–602.
- Bytzer P, Talley NJ, Leemon M, Young LJ, Jones MP, Horowitz M. Prevalence of gastrointestinal symptoms associated with diabetes mellitus: a population-based survey of 15,000 adults. Arch Intern Med 2001; 161:1989–1996.
- Chassany O, Michaux A, Bergmann JF. Drug-induced diarrhoea. Drug Saf 2000; 22:53–72.
- Rubio-Tapia A, Herman ML, Ludvigsson JF, et al. Severe spruelike enteropathy associated with olmesartan. Mayo Clin Proc 2012; 87:732–738.
- Zestril (lisinopril) tablets. www.accessdata.fda.gov/drugsatfda_docs/label/2012/019777s062lbl.pdf. Accessed September 8, 2015.
- Whittier WL, Rutecki GW. Primer on clinical acid-base problem solving. Dis Mon 2004; 50:122–162.
- Langhorst J, Elsenbruch S, Koelzer J, Rueffer A, Michalsen A, Dobos GJ. Noninvasive markers in the assessment of intestinal inflammation in inflammatory bowel diseases: performance of fecal lactoferrin, calprotectin, and PMN-elastase, CRP, and clinical indices. Am J Gastroenterol 2008; 103:162–169.
- Khouri MR, Ng SN, Huang G, Shiau YF. Fecal triglyceride excretion is not excessive in pancreatic insufficiency. Gastroenterology 1989; 96:848–852.
- Dominici R, Franzini C. Fecal elastase-1 as a test for pancreatic function: a review. Clin Chem Lab Med 2002; 40:325–332.
- Fasano A, Catassi C. Celiac disease. New Engl J Med 2012; 367:2419–2426.
- Mooney PD, Hadjivassiliou M, Sanders DS. Coeliac disease. BMJ 2014; 348:g1561–g1561.
- Pazianas M, Butcher GP, Subhani JM, et al. Calcium absorption and bone mineral density in celiacs after long term treatment with gluten-free diet and adequate calcium intake. Osteoporos Int 2005; 16:56–63.
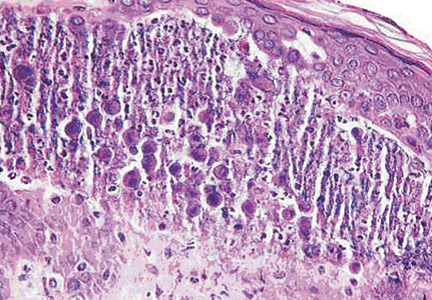
Ankle pain in a young woman with Gaucher disease
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
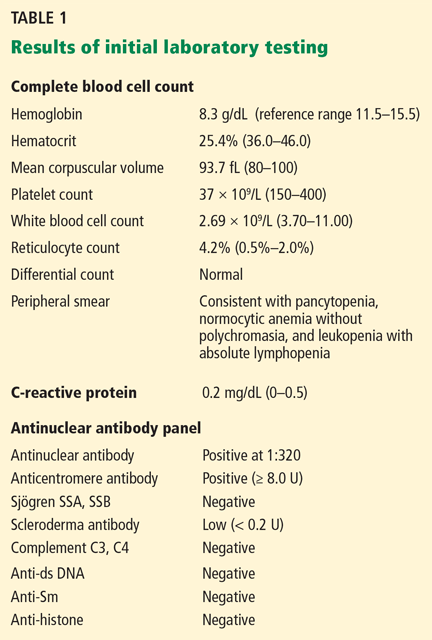
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
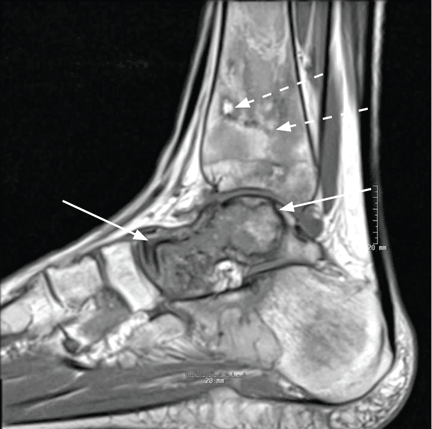
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.
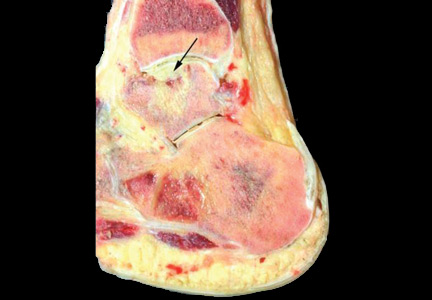
A 79-year-old with acute portal vein thrombosis
A 79-year-old man presented with chills and fever. He had a history of polymyalgia rheumatica and had been tapered off corticosteroids 1 month before admission. One week before he presented, he had developed generalized myalgia, chills, and fatigue. A cortisol stimulation test at that time was normal, prednisone was restarted, and his symptoms had improved. But 1 day before he presented, the chills had returned, this time with fever. Laboratory testing at an outpatient clinic had revealed abnormal liver enzyme levels.
On the day he presented, he felt worse, with persistent chills, fever, and vague lower abdominal pain, but he denied nausea, vomiting, changes in bowel habits, melena, hematochezia, and hematemesis. He was admitted for additional evaluation.
His medical history also included coronary artery disease (for which he had undergone coronary artery bypass grafting), hypertension, stable liver cysts, and gout. He had no known inflammatory bowel disease and no recent abdominal surgery. His medications included prednisone, atorvastatin, atenolol, aspirin, niacin, and cholecalciferol. He had no history of smoking, significant drinking, or use of illicit drugs. He had no respiratory or cardiac symptoms or neurologic symptoms consistent with a transient ischemic attack or stroke. He denied any rashes.
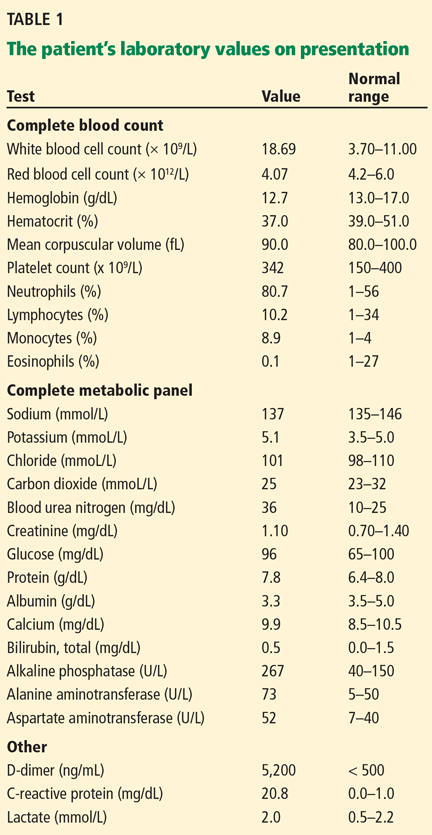
On admission, he was febrile, with temperatures reaching 102˚F (38.9˚C). His blood pressure was 137/63 mm Hg, pulse 54 beats per minute, respiration rate 18 breaths per minute, and oxygen saturation 97% on room air. A harsh systolic murmur was noted on physical examination. His abdomen was nondistended, nontender, and without bruits.
Laboratory testing (Table 1) revealed leukocytosis, anemia, mildly abnormal aminotransferase levels, elevated alkaline phosphatase, and markedly elevated C-reactive protein.
A full workup for fever was performed, including blood and urine cultures; chest radiography; contrast-enhanced computed tomography (CT) of the chest, abdomen, and pelvis; magnetic resonance imaging (MRI) of the abdomen; and colonoscopy. No source of infection—bacterial, viral, or fungal—was found. However, CT revealed new extensive thrombosis of the right portal vein and its branches (Figure 1).
CLINICAL PRESENTATION
1. Which of the following is least consistent with the clinical presentation of acute portal vein thrombosis?
- Abdominal pain
- Fever and chills
- Hematemesis
- Leukocytosis
- Absence of symptoms
Of these signs and symptoms, hematemesis is the least likely to be associated with acute portal vein thrombosis, although it can be associated with chronic cases.
Symptoms of portal vein thrombosis
Portal vein thrombosis causes extrahepatic obstruction of the portal venous system, which provides two-thirds of the total hepatic blood flow.
Acute. Often, thrombotic occlusion of the portal vein produces no acute symptoms because of immediate, compensatory vasodilation of the hepatic arterial system.1 Additionally, in the ensuing days, the thrombus becomes an organized collagenous plug, and collateral veins develop to bypass the blocked vein and maintain portal perfusion in a process called cavernous transformation.1,2 Thus, many patients have no symptoms.
If symptoms occur, portal vein thrombosis can initially present as transient abdominal pain with fever, as seen in this patient.3 Many patients with acute portal vein thrombosis experience abdominal pain due to intra-abdominal sepsis, also referred to as pylephlebitis.2,4 High, spiking fevers and chills also occur, caused by infected thrombi associated with intra-abdominal infections such as appendicitis, diverticulitis, and pancreatitis.5,6
Chronic. In contrast, symptomatic chronic portal vein thrombosis commonly presents with sequelae of portal hypertension, most notably gastrointestinal bleeding. Hematemesis from ruptured esophageal varices is the most frequent reason for seeking medical attention, though varices also develop in the stomach, duodenum, jejunum, gallbladder, and bile ducts.2,7 Abdominal pain is less common in chronic portal vein thrombosis unless the thrombus extends into the mesenteric veins and causes bowel ischemia or infarction. Long-standing portal vein thrombosis may also lead to dilated venous collaterals that compress large bile ducts, resulting in portal cholangiopathy.1,8
Portal vein thrombosis may present as acute intestinal ischemia and bowel infarction, though this is uncommon. This is generally seen with extensive occlusive portal vein thrombosis and concomitant mesenteric venous thrombosis.1,2
Other symptoms that are common but nonspecific are nausea, vomiting, diarrhea, weight loss, and anorexia.2
Signs of portal vein thrombosis
On examination, patients with acute portal vein thrombosis have minimal physical signs unless they have other contributing conditions. For example, acute portal vein thrombosis can result in abdominal distention secondary to ileus, or guarding and ascites secondary to intestinal infarction.3,9
Some patients with chronic portal vein thrombosis also have normal physical findings, but many have signs. Splenomegaly is seen in 75% to 100% of patients.2,7 Hepatomegaly, abdominal tenderness, and low-grade fever are common as well.2,10 Ascites is usually not present without underlying cirrhosis; however, mild and transient ascites can develop immediately after the thrombotic event before the patient develops collateral circulation.2
Laboratory testing for portal vein thrombosis
Laboratory test results are typically unremarkable. Liver function tests show preserved hepatic function but may reveal mild increases in aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase, and bilirubin.2,10
In acute cases, elevations of acute-phase reactant levels can occur.9 Leukocytosis and blood cultures growing Bacteroides species are seen in septic cases or pylephlebitis.11,12 There may be mild anemia, particularly after a recent bleeding episode, or mild leukopenia and thrombocytopenia due to hypersplenism. Suspicion of an underlying myeloproliferative disorder is high if thrombocytosis is present.2
DIAGNOSIS
2. All of the following would be appropriate initial diagnostic studies for portal vein thrombosis except which one?
- Doppler ultrasonography
- Contrast-enhanced CT
- Contrast-enhanced MRI
- Angiography
Portal vein thrombosis is most often diagnosed with noninvasive techniques, namely Doppler ultrasonography, CT, and MRI—not angiography.
Ultrasonography can reveal an echogenic thrombus in the vessel lumen with distention of the portal vein proximal to the occlusion and extensive collateral vessels. Plain ultrasonography fails to reveal the thrombus in up to one-third of patients. However, duplex ultrasonography with color flow Doppler imaging can confirm partial or complete absence of flow in the vein with 89% sensitivity and 92% specificity.13,14
On contrast-enhanced CT, the thrombus appears as a filling defect within the portal venous segment. Complete occlusion of the vein may produce a “train track” appearance due to contrast around the vessel.10 Without contrast, the clot will appear as hyperattenuating material in the portal vein, but contrast-enhanced imaging may be necessary to differentiate the thrombus from the vessel wall.15 Gas within the portal venous system is specific for pylephlebitis.4 Evidence of cavernous transformation is seen in chronic portal vein thrombosis.
Contrast-enhanced magnetic resonance angiography can also be used to evaluate patency and flow direction. In addition, it provides detailed anatomic information about the entire portal venous system, including the intrahepatic portal vessels, which is limited in CT imaging.2,10 CT and MRI can also help to identify predisposing conditions (eg, intra-abdominal infection, hepatocellular carcinoma) and complications (eg, intestinal infarction) associated with portal vein thrombosis.
Angiography can be considered if noninvasive techniques are inconclusive but is generally not necessary, given the increased use of CT and MRI.
In our patient, abdominal CT revealed occlusive thrombosis of the right portal vein and its branches (Figure 1). The left and main portal veins were patent. There was no evidence of intra-abdominal infection or infarction.
FINDING THE CAUSE
3. Which of the following is not a common cause
of portal vein thrombosis?
- A hypercoagulable state
- Immune deficiency
- Intra-abdominal infection
- Malignancy
- Portal hypertension

Once portal vein thrombosis has been diagnosed, the cause should be identified (Table 2). The differential diagnosis is broad, including both local factors (eg, injury to the portal vein, local inflammation, infection) and general factors (eg, inherited and acquired hypercoagulable conditions). Thrombophilias are identified in 60% of patients with portal vein thrombosis and local factors in 40%.7 Moreover, the etiology is often multifactorial. However, immune deficiency is not a common cause.
Hypercoagulability
Prothrombotic disorders can be either inherited or acquired.
Inherited deficiencies in the natural anticoagulants antithrombin, protein C, and protein S are associated with a high risk of thrombosis but have a low prevalence in the general population. In the setting of liver abnormalities, familial testing may be helpful to distinguish inherited causes of portal vein thrombosis from defective liver function as a consequence of portal vein thrombosis. The factor V Leiden mutation (G1691A) and the G20210A mutation in the prothrombin gene are more prevalent (> 2%) but generally confer a lower thrombosis risk.16 The prothrombin gene mutation G20210A is the most common risk factor for portal vein thrombosis, with prevalence of 2% to 22% in adults with nonmalignant, noncirrhotic portal vein thrombosis.3
Hyperhomocysteinemia due to a methylene tetrahydrofolate reductase (MTHFR) mutation (C677T) is another inherited associated risk factor for portal vein thrombosis, but hyperhomocysteinemia can also arise as a complication of portal vein thrombosis-related liver disease.3
Acquired prothrombotic disorders, particularly myeloproliferative diseases, are found in 22% to 48% of cases of portal vein thrombosis. Many young patients with myeloproliferative disorders present with portal vein thrombosis as the first symptom, and testing for the G1849T point mutation in JAK2 can make the diagnosis.17 Splenectomy with underlying myeloproliferative disorder confers a particularly high risk for portal vein thrombosis.18
Other thrombophilic disorders including antiphospholipid antibody syndrome, paroxysmal nocturnal hemoglobinuria, and malignancy can contribute to portal vein thrombosis.3 Pregnancy and oral contraceptive use have also been associated with hypercoagulability, and cessation of oral estrogen is recommended in such cases. The risk may be further increased in patients on oral contraceptives who have a previously unrecognized hypercoagulable state.3
Inflammation and infection
Inflammation and infection are local risk factors for portal vein thrombosis. Acute portal vein thrombosis has been associated with intra-abdominal infections (eg, appendicitis, cholecystitis) and with inflammatory conditions such as inflammatory bowel disease and pancreatitis.16,19 From 3% to 5% of all portal vein thrombosis cases result from pancreatitis, either from a single acute episode or from repeat inflammation of chronic pancreatitis.10 Portal vein thrombosis in the setting of inflammatory bowel disease can occur even when the disease is in remission, particularly in ulcerative colitis.20,21
Injury to the portal venous system
Abdominal surgery, particularly splenectomy, portosystemic shunting, colectomy, and blunt abdominal trauma can cause injury to the portal venous system, resulting in portal vein thrombosis. This is usually seen only in patients with portal hypertension, an underlying prothrombotic condition such as myeloproliferative disease, or inflammatory bowel disease.10,19,22
Impaired portal vein flow
Cirrhosis and malignancy are major risk factors for portal vein thrombosis. In case series, cirrhosis was found in 24% to 32% of patients with portal vein thrombosis.2,23 However, the overall prevalence of portal vein thrombosis in cirrhotic patients varies widely, from 0.6% to 28%, depending on the degree of cirrhosis.10
The pathogenesis of portal vein thrombosis in cirrhosis is unclear but may be multifactorial. Decreased portal blood flow (with subsequent stasis) and periportal lymphangitis and fibrosis are thought to stimulate thrombus formation.3,10 Additionally, patients with advanced cirrhosis are prothrombotic because of reduced hepatic synthesis of antithrombin, protein C, protein S, and coagulation factors.
Malignancy is associated with 21% to 24% of cases of portal vein thrombosis in adults, with pancreatic cancer and hepatocellular carcinoma being the most common.2,3 Others include cholangiocarcinoma and carcinomas of the stomach, lung, prostate, uterus, and kidney. Cancer causes portal vein thrombosis through a combination of tumor invasion into the portal vein, extrinsic compression by the tumor, periportal fibrosis following surgery or radiation, and hypercoagulability secondary to malignancy.9,16,24
Idiopathic portal vein thrombosis
Portal vein thrombosis is usually caused by one or more of the underlying factors mentioned above but is idiopathic in 8% to 15% of cases.10
Back to our patient
The cause of this patient’s portal vein thrombosis is unclear. He did not have a history of cirrhosis, inflammatory bowel disease, trauma, or abdominal surgery. His febrile illness could have precipitated the formation of a thrombus, but no definitive source of infection or inflammation was discovered. His workup was negative for pancreatitis, appendicitis, cholecystitis, diverticulitis, and prostatitis. No occult malignancy was found. It is also possible that his fever was the result of the thrombosis.
A full hypercoagulability panel revealed no striking abnormalities. He did have elevated fibrinogen and factor VIII levels that were consistent with an acute-phase reaction, along with an elevated erythrocyte sedimentation rate (> 90 mm/hr) and C-reactive protein level. Aside from the portal vein thrombosis, no potential source of inflammation could be identified.
Mildly reduced levels of antithrombin III activity were attributed to enoxaparin therapy and ultimately normalized on repeated testing. The patient had very minimally elevated titers of anticardiolipin immunoglobulin G (1:10 GPL) and anti-beta-2 glycoprotein immunoglobulin M (21 SMU), which were not thought to be significant. Tests for lupus anticoagulant, prothrombin gene mutation, activated protein C resistance, and JAK2 mutation were negative.
TREATMENT
4. Treatment of symptomatic portal vein thrombosis generally includes which two of the following?
- Anticoagulation
- Intravenous gamma globulin
- Broad-spectrum antibiotics
Anticoagulant therapy
Treatment of acute, symptomatic portal vein thrombosis involves anticoagulant therapy to prevent extension of the thrombus and, ultimately, to allow for recanalization of the obstructed veins. Anticoagulant therapy is initially intravenous unfractionated heparin or subcutaneous low-molecular-weight heparin, eventually bridged to an oral agent such as warfarin.3,9 Currently, there are inadequate data on the use of oral or parenteral factor Xa inhibitors or direct thrombin inhibitors in the treatment of this disease.
When started immediately, anticoagulation therapy is associated with complete recanalization in 38.3% and partial recanalization in 14% of patients presenting with complete thrombosis. Without anticoagulation, spontaneous recanalization is unusual.25
Although the optimal duration of anticoagulant therapy is unclear, a minimum of 3 months is generally recommended.9,26 If a hypercoagulable state is present or if the portal vein thrombosis is unprovoked (eg, by surgery, trauma, or an intra-abdominal infection), long-term treatment should be considered.26
Experience with thrombolytic therapy or mechanical recanalization has been limited, but the use of catheter-based techniques for pharmacomechanical thrombolysis has been reported.27–29 Transjugular intrahepatic portosystemic shunting is also an alternative to anticoagulation, but its role in treating portal vein thrombosis is complicated by technical difficulties of the procedure, postoperative complications, and recurrent occlusion of the shunt.25
Currently, there are no data comparing the risk-benefit ratio of early anticoagulation and that of invasive procedures. These more aggressive treatments are generally considered only when there is extensive thrombosis or ascites (which are both predictive factors of poor response to anticoagulation alone) and in patients for whom anticoagulation has failed.3 Surgical thrombectomy is rarely indicated, typically only in instances in which laparotomy is being performed for suspected bowel infarction.3
Antibiotics
In addition to anticoagulation, broad-spectrum antibiotics covering gram-negative and anaerobic bacteria are indicated for those cases of portal vein thrombosis associated with underlying infection.9
For chronic cases, the goals of management are to prevent and treat gastroesophageal variceal bleeding and to prevent recurrent thrombosis.9 Nonselective beta-blockers (eg, propranolol) and endoscopic band ligation have shown evidence of reducing the incidence of recurrent bleeding and prolonging survival in retrospective studies.9,30,31 Long-term anticoagulation is generally indicated to prevent further thrombosis and to increase the likelihood of recanalization only for patients with a permanent prothrombotic condition.9 In patients with clinically significant portal hypertension, the benefit of continued anticoagulation therapy must be weighed against the risk of esophageal and gastric variceal bleeding.
There is controversy regarding how to manage portal vein thrombosis that is incidentally identified and asymptomatic (eg, if it is discovered on an imaging study for another indication). Current guidelines recommend against anticoagulation in patients with incidentally discovered and asymptomatic splanchnic vein thrombosis, including portal vein thrombosis.26
Intravenous gamma globulin is not part of the treatment.
CASE CONTINUED
The patient’s presenting symptoms of fever, chills, and abdominal pain completely resolved after a course of antibiotic therapy. The erythrocyte sedimentation rate subsequently normalized and factor VIII activity improved. We believed that an underlying infectious or inflammatory process had contributed to the development of portal vein thrombosis, though the specific cause could not be identified. The patient was treated with enoxaparin 1 mg/kg twice a day and transitioned to warfarin.
Magnetic resonance venography done 3 months after diagnosis showed persistent right portal vein thrombosis that was largely unchanged. Anticoagulation was continued for 1 year with no change in his portal vein thrombosis on sequential imaging and was subsequently discontinued. To date, no malignancy or infectious process has been found, and the patient continues to do well 2 years later.
- Ponziani FR, Zocco MA, Campanale C, et al. Portal vein thrombosis: insight into physiopathology, diagnosis, and treatment. World J Gastroenterol 2010; 16:143–155.
- Cohen J, Edelman RR, Chopra S. Portal vein thrombosis: a review. Am J Med 1992; 92:173–182.
- Primignani M. Portal vein thrombosis, revisited. Dig Liver Dis 2010; 42:163–170.
- Condat B, Valla D. Nonmalignant portal vein thrombosis in adults. Nat Clin Pract Gastroenterol Hepatol 2006; 3:505–515.
- Condat B, Pessione F, Helene Denninger M, Hillaire S, Valla D. Recent portal or mesenteric venous thrombosis: increased recognition and frequent recanalization on anticoagulant therapy. Hepatology 2000; 32:466–470.
- Sheen CL, Lamparelli H, Milne A, Green I, Ramage JK. Clinical features, diagnosis and outcome of acute portal vein thrombosis. QJM 2000; 93:531–534.
- Sogaard KK, Astrup LB, Vilstrup H, Gronbaek H. Portal vein thrombosis; risk factors, clinical presentation and treatment. BMC Gastroenterol 2007; 7:34.
- Llop E, de Juan C, Seijo S, et al. Portal cholangiopathy: radiological classification and natural history. Gut 2011; 60:853–860.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Sobhonslidsuk A, Reddy KR. Portal vein thrombosis: a concise review. Am J Gastroenterol 2002; 97:535–541.
- Ni YH, Wang NC, Peng MY, Chou YY, Chang FY. Bacteroides fragilis bacteremia associated with portal vein and superior mesentery vein thrombosis secondary to antithrombin III and protein C deficiency: a case report. J Microbiol Immunol Infect 2002; 35:255–258.
- Trum J, Valla D, Cohen G, et al. Bacteroides bacteraemia of undetermined origin: strong association with portal vein thrombosis and cryptogenic pylephlebitis. Eur J Gastroenterol Hepatol 1993; 5:655–659.
- Ueno N, Sasaki A, Tomiyama T, Tano S, Kimura K. Color Doppler ultrasonography in the diagnosis of cavernous transformation of the portal vein. J Clin Ultrasound 1997; 25:227–233.
- Tessler FN, Gehring BJ, Gomes AS, et al. Diagnosis of portal vein thrombosis: value of color Doppler imaging. AJR Am J Roentgenol 1991; 157:293–296.
- Hidajat N, Stobbe H, Griesshaber V, Felix R, Schroder RJ. Imaging and radiological interventions of portal vein thrombosis. Acta Radiol 2005; 46:336–343.
- Valla DC, Condat B. Portal vein thrombosis in adults: pathophysiology, pathogenesis and management. J Hepatol 2000; 32:865–871.
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352:1779–1790.
- Krauth MT, Lechner K, Neugebauer EA, Pabinger I. The postoperative splenic/portal vein thrombosis after splenectomy and its prevention—an unresolved issue. Haematologica 2008; 93:1227–1232.
- Sinagra E, Aragona E, Romano C, et al. The role of portal vein thrombosis in the clinical course of inflammatory bowel diseases: report on three cases and review of the literature. Gastroenterol Res Pract 2012; 2012:916428.
- Maconi G, Bolzacchini E, Dell’Era A, Russo U, Ardizzone S, de Franchis R. Portal vein thrombosis in inflammatory bowel diseases: a single-center case series. J Crohns Colitis 2012; 6:362–367.
- Jackson LM, O’Gorman PJ, O’Connell J, Cronin CC, Cotter KP, Shanahan F. Thrombosis in inflammatory bowel disease: clinical setting, procoagulant profile and factor V Leiden. QJM 1997; 90:183–188.
- Eguchi A, Hashizume M, Kitano S, Tanoue K, Wada H, Sugimachi K. High rate of portal thrombosis after splenectomy in patients with esophageal varices and idiopathic portal hypertension. Arch Surg 1991; 126:752–755.
- Ogren M, Bergqvist D, Björck M, Acosta S, Eriksson H, Sternby NH. Portal vein thrombosis: prevalence, patient characteristics and lifetime risk: a population study based on 23,796 consecutive autopsies. World J Gastroenterol 2006; 12:2115–2119.
- Falanga A, Marchetti M, Vignoli A. Coagulation and cancer: biological and clinical aspects. J Thromb Haemost 2013; 11:223–233.
- Congly SE, Lee SS. Portal vein thrombosis: should anticoagulation be used? Curr Gastroenterol Rep 2013; 15:306.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Uflacker R. Applications of percutaneous mechanical thrombectomy in transjugular intrahepatic portosystemic shunt and portal vein thrombosis. Tech Vasc Interv Radiol 2003; 6:59–69.
- Takahashi N, Kuroki K, Yanaga K. Percutaneous transhepatic mechanical thrombectomy for acute mesenteric venous thrombosis. J Endovasc Ther 2005; 12:508–511.
- Lopera JE, Correa G, Brazzini A, et al. Percutaneous transhepatic treatment of symptomatic mesenteric venous thrombosis. J Vasc Surg 2002; 36:1058–1061.
- Orr DW, Harrison PM, Devlin J, et al. Chronic mesenteric venous thrombosis: evaluation and determinants of survival during long-term follow-up. Clin Gastroenterol Hepatol 2007; 5:80–86.
- Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant therapy. Gastroenterology 2001; 120:490–497.
A 79-year-old man presented with chills and fever. He had a history of polymyalgia rheumatica and had been tapered off corticosteroids 1 month before admission. One week before he presented, he had developed generalized myalgia, chills, and fatigue. A cortisol stimulation test at that time was normal, prednisone was restarted, and his symptoms had improved. But 1 day before he presented, the chills had returned, this time with fever. Laboratory testing at an outpatient clinic had revealed abnormal liver enzyme levels.
On the day he presented, he felt worse, with persistent chills, fever, and vague lower abdominal pain, but he denied nausea, vomiting, changes in bowel habits, melena, hematochezia, and hematemesis. He was admitted for additional evaluation.
His medical history also included coronary artery disease (for which he had undergone coronary artery bypass grafting), hypertension, stable liver cysts, and gout. He had no known inflammatory bowel disease and no recent abdominal surgery. His medications included prednisone, atorvastatin, atenolol, aspirin, niacin, and cholecalciferol. He had no history of smoking, significant drinking, or use of illicit drugs. He had no respiratory or cardiac symptoms or neurologic symptoms consistent with a transient ischemic attack or stroke. He denied any rashes.
On admission, he was febrile, with temperatures reaching 102˚F (38.9˚C). His blood pressure was 137/63 mm Hg, pulse 54 beats per minute, respiration rate 18 breaths per minute, and oxygen saturation 97% on room air. A harsh systolic murmur was noted on physical examination. His abdomen was nondistended, nontender, and without bruits.
Laboratory testing (Table 1) revealed leukocytosis, anemia, mildly abnormal aminotransferase levels, elevated alkaline phosphatase, and markedly elevated C-reactive protein.
A full workup for fever was performed, including blood and urine cultures; chest radiography; contrast-enhanced computed tomography (CT) of the chest, abdomen, and pelvis; magnetic resonance imaging (MRI) of the abdomen; and colonoscopy. No source of infection—bacterial, viral, or fungal—was found. However, CT revealed new extensive thrombosis of the right portal vein and its branches (Figure 1).
CLINICAL PRESENTATION
1. Which of the following is least consistent with the clinical presentation of acute portal vein thrombosis?
- Abdominal pain
- Fever and chills
- Hematemesis
- Leukocytosis
- Absence of symptoms
Of these signs and symptoms, hematemesis is the least likely to be associated with acute portal vein thrombosis, although it can be associated with chronic cases.
Symptoms of portal vein thrombosis
Portal vein thrombosis causes extrahepatic obstruction of the portal venous system, which provides two-thirds of the total hepatic blood flow.
Acute. Often, thrombotic occlusion of the portal vein produces no acute symptoms because of immediate, compensatory vasodilation of the hepatic arterial system.1 Additionally, in the ensuing days, the thrombus becomes an organized collagenous plug, and collateral veins develop to bypass the blocked vein and maintain portal perfusion in a process called cavernous transformation.1,2 Thus, many patients have no symptoms.
If symptoms occur, portal vein thrombosis can initially present as transient abdominal pain with fever, as seen in this patient.3 Many patients with acute portal vein thrombosis experience abdominal pain due to intra-abdominal sepsis, also referred to as pylephlebitis.2,4 High, spiking fevers and chills also occur, caused by infected thrombi associated with intra-abdominal infections such as appendicitis, diverticulitis, and pancreatitis.5,6
Chronic. In contrast, symptomatic chronic portal vein thrombosis commonly presents with sequelae of portal hypertension, most notably gastrointestinal bleeding. Hematemesis from ruptured esophageal varices is the most frequent reason for seeking medical attention, though varices also develop in the stomach, duodenum, jejunum, gallbladder, and bile ducts.2,7 Abdominal pain is less common in chronic portal vein thrombosis unless the thrombus extends into the mesenteric veins and causes bowel ischemia or infarction. Long-standing portal vein thrombosis may also lead to dilated venous collaterals that compress large bile ducts, resulting in portal cholangiopathy.1,8
Portal vein thrombosis may present as acute intestinal ischemia and bowel infarction, though this is uncommon. This is generally seen with extensive occlusive portal vein thrombosis and concomitant mesenteric venous thrombosis.1,2
Other symptoms that are common but nonspecific are nausea, vomiting, diarrhea, weight loss, and anorexia.2
Signs of portal vein thrombosis
On examination, patients with acute portal vein thrombosis have minimal physical signs unless they have other contributing conditions. For example, acute portal vein thrombosis can result in abdominal distention secondary to ileus, or guarding and ascites secondary to intestinal infarction.3,9
Some patients with chronic portal vein thrombosis also have normal physical findings, but many have signs. Splenomegaly is seen in 75% to 100% of patients.2,7 Hepatomegaly, abdominal tenderness, and low-grade fever are common as well.2,10 Ascites is usually not present without underlying cirrhosis; however, mild and transient ascites can develop immediately after the thrombotic event before the patient develops collateral circulation.2
Laboratory testing for portal vein thrombosis
Laboratory test results are typically unremarkable. Liver function tests show preserved hepatic function but may reveal mild increases in aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase, and bilirubin.2,10
In acute cases, elevations of acute-phase reactant levels can occur.9 Leukocytosis and blood cultures growing Bacteroides species are seen in septic cases or pylephlebitis.11,12 There may be mild anemia, particularly after a recent bleeding episode, or mild leukopenia and thrombocytopenia due to hypersplenism. Suspicion of an underlying myeloproliferative disorder is high if thrombocytosis is present.2
DIAGNOSIS
2. All of the following would be appropriate initial diagnostic studies for portal vein thrombosis except which one?
- Doppler ultrasonography
- Contrast-enhanced CT
- Contrast-enhanced MRI
- Angiography
Portal vein thrombosis is most often diagnosed with noninvasive techniques, namely Doppler ultrasonography, CT, and MRI—not angiography.
Ultrasonography can reveal an echogenic thrombus in the vessel lumen with distention of the portal vein proximal to the occlusion and extensive collateral vessels. Plain ultrasonography fails to reveal the thrombus in up to one-third of patients. However, duplex ultrasonography with color flow Doppler imaging can confirm partial or complete absence of flow in the vein with 89% sensitivity and 92% specificity.13,14
On contrast-enhanced CT, the thrombus appears as a filling defect within the portal venous segment. Complete occlusion of the vein may produce a “train track” appearance due to contrast around the vessel.10 Without contrast, the clot will appear as hyperattenuating material in the portal vein, but contrast-enhanced imaging may be necessary to differentiate the thrombus from the vessel wall.15 Gas within the portal venous system is specific for pylephlebitis.4 Evidence of cavernous transformation is seen in chronic portal vein thrombosis.
Contrast-enhanced magnetic resonance angiography can also be used to evaluate patency and flow direction. In addition, it provides detailed anatomic information about the entire portal venous system, including the intrahepatic portal vessels, which is limited in CT imaging.2,10 CT and MRI can also help to identify predisposing conditions (eg, intra-abdominal infection, hepatocellular carcinoma) and complications (eg, intestinal infarction) associated with portal vein thrombosis.
Angiography can be considered if noninvasive techniques are inconclusive but is generally not necessary, given the increased use of CT and MRI.
In our patient, abdominal CT revealed occlusive thrombosis of the right portal vein and its branches (Figure 1). The left and main portal veins were patent. There was no evidence of intra-abdominal infection or infarction.
FINDING THE CAUSE
3. Which of the following is not a common cause
of portal vein thrombosis?
- A hypercoagulable state
- Immune deficiency
- Intra-abdominal infection
- Malignancy
- Portal hypertension
Once portal vein thrombosis has been diagnosed, the cause should be identified (Table 2). The differential diagnosis is broad, including both local factors (eg, injury to the portal vein, local inflammation, infection) and general factors (eg, inherited and acquired hypercoagulable conditions). Thrombophilias are identified in 60% of patients with portal vein thrombosis and local factors in 40%.7 Moreover, the etiology is often multifactorial. However, immune deficiency is not a common cause.
Hypercoagulability
Prothrombotic disorders can be either inherited or acquired.
Inherited deficiencies in the natural anticoagulants antithrombin, protein C, and protein S are associated with a high risk of thrombosis but have a low prevalence in the general population. In the setting of liver abnormalities, familial testing may be helpful to distinguish inherited causes of portal vein thrombosis from defective liver function as a consequence of portal vein thrombosis. The factor V Leiden mutation (G1691A) and the G20210A mutation in the prothrombin gene are more prevalent (> 2%) but generally confer a lower thrombosis risk.16 The prothrombin gene mutation G20210A is the most common risk factor for portal vein thrombosis, with prevalence of 2% to 22% in adults with nonmalignant, noncirrhotic portal vein thrombosis.3
Hyperhomocysteinemia due to a methylene tetrahydrofolate reductase (MTHFR) mutation (C677T) is another inherited associated risk factor for portal vein thrombosis, but hyperhomocysteinemia can also arise as a complication of portal vein thrombosis-related liver disease.3
Acquired prothrombotic disorders, particularly myeloproliferative diseases, are found in 22% to 48% of cases of portal vein thrombosis. Many young patients with myeloproliferative disorders present with portal vein thrombosis as the first symptom, and testing for the G1849T point mutation in JAK2 can make the diagnosis.17 Splenectomy with underlying myeloproliferative disorder confers a particularly high risk for portal vein thrombosis.18
Other thrombophilic disorders including antiphospholipid antibody syndrome, paroxysmal nocturnal hemoglobinuria, and malignancy can contribute to portal vein thrombosis.3 Pregnancy and oral contraceptive use have also been associated with hypercoagulability, and cessation of oral estrogen is recommended in such cases. The risk may be further increased in patients on oral contraceptives who have a previously unrecognized hypercoagulable state.3
Inflammation and infection
Inflammation and infection are local risk factors for portal vein thrombosis. Acute portal vein thrombosis has been associated with intra-abdominal infections (eg, appendicitis, cholecystitis) and with inflammatory conditions such as inflammatory bowel disease and pancreatitis.16,19 From 3% to 5% of all portal vein thrombosis cases result from pancreatitis, either from a single acute episode or from repeat inflammation of chronic pancreatitis.10 Portal vein thrombosis in the setting of inflammatory bowel disease can occur even when the disease is in remission, particularly in ulcerative colitis.20,21
Injury to the portal venous system
Abdominal surgery, particularly splenectomy, portosystemic shunting, colectomy, and blunt abdominal trauma can cause injury to the portal venous system, resulting in portal vein thrombosis. This is usually seen only in patients with portal hypertension, an underlying prothrombotic condition such as myeloproliferative disease, or inflammatory bowel disease.10,19,22
Impaired portal vein flow
Cirrhosis and malignancy are major risk factors for portal vein thrombosis. In case series, cirrhosis was found in 24% to 32% of patients with portal vein thrombosis.2,23 However, the overall prevalence of portal vein thrombosis in cirrhotic patients varies widely, from 0.6% to 28%, depending on the degree of cirrhosis.10
The pathogenesis of portal vein thrombosis in cirrhosis is unclear but may be multifactorial. Decreased portal blood flow (with subsequent stasis) and periportal lymphangitis and fibrosis are thought to stimulate thrombus formation.3,10 Additionally, patients with advanced cirrhosis are prothrombotic because of reduced hepatic synthesis of antithrombin, protein C, protein S, and coagulation factors.
Malignancy is associated with 21% to 24% of cases of portal vein thrombosis in adults, with pancreatic cancer and hepatocellular carcinoma being the most common.2,3 Others include cholangiocarcinoma and carcinomas of the stomach, lung, prostate, uterus, and kidney. Cancer causes portal vein thrombosis through a combination of tumor invasion into the portal vein, extrinsic compression by the tumor, periportal fibrosis following surgery or radiation, and hypercoagulability secondary to malignancy.9,16,24
Idiopathic portal vein thrombosis
Portal vein thrombosis is usually caused by one or more of the underlying factors mentioned above but is idiopathic in 8% to 15% of cases.10
Back to our patient
The cause of this patient’s portal vein thrombosis is unclear. He did not have a history of cirrhosis, inflammatory bowel disease, trauma, or abdominal surgery. His febrile illness could have precipitated the formation of a thrombus, but no definitive source of infection or inflammation was discovered. His workup was negative for pancreatitis, appendicitis, cholecystitis, diverticulitis, and prostatitis. No occult malignancy was found. It is also possible that his fever was the result of the thrombosis.
A full hypercoagulability panel revealed no striking abnormalities. He did have elevated fibrinogen and factor VIII levels that were consistent with an acute-phase reaction, along with an elevated erythrocyte sedimentation rate (> 90 mm/hr) and C-reactive protein level. Aside from the portal vein thrombosis, no potential source of inflammation could be identified.
Mildly reduced levels of antithrombin III activity were attributed to enoxaparin therapy and ultimately normalized on repeated testing. The patient had very minimally elevated titers of anticardiolipin immunoglobulin G (1:10 GPL) and anti-beta-2 glycoprotein immunoglobulin M (21 SMU), which were not thought to be significant. Tests for lupus anticoagulant, prothrombin gene mutation, activated protein C resistance, and JAK2 mutation were negative.
TREATMENT
4. Treatment of symptomatic portal vein thrombosis generally includes which two of the following?
- Anticoagulation
- Intravenous gamma globulin
- Broad-spectrum antibiotics
Anticoagulant therapy
Treatment of acute, symptomatic portal vein thrombosis involves anticoagulant therapy to prevent extension of the thrombus and, ultimately, to allow for recanalization of the obstructed veins. Anticoagulant therapy is initially intravenous unfractionated heparin or subcutaneous low-molecular-weight heparin, eventually bridged to an oral agent such as warfarin.3,9 Currently, there are inadequate data on the use of oral or parenteral factor Xa inhibitors or direct thrombin inhibitors in the treatment of this disease.
When started immediately, anticoagulation therapy is associated with complete recanalization in 38.3% and partial recanalization in 14% of patients presenting with complete thrombosis. Without anticoagulation, spontaneous recanalization is unusual.25
Although the optimal duration of anticoagulant therapy is unclear, a minimum of 3 months is generally recommended.9,26 If a hypercoagulable state is present or if the portal vein thrombosis is unprovoked (eg, by surgery, trauma, or an intra-abdominal infection), long-term treatment should be considered.26
Experience with thrombolytic therapy or mechanical recanalization has been limited, but the use of catheter-based techniques for pharmacomechanical thrombolysis has been reported.27–29 Transjugular intrahepatic portosystemic shunting is also an alternative to anticoagulation, but its role in treating portal vein thrombosis is complicated by technical difficulties of the procedure, postoperative complications, and recurrent occlusion of the shunt.25
Currently, there are no data comparing the risk-benefit ratio of early anticoagulation and that of invasive procedures. These more aggressive treatments are generally considered only when there is extensive thrombosis or ascites (which are both predictive factors of poor response to anticoagulation alone) and in patients for whom anticoagulation has failed.3 Surgical thrombectomy is rarely indicated, typically only in instances in which laparotomy is being performed for suspected bowel infarction.3
Antibiotics
In addition to anticoagulation, broad-spectrum antibiotics covering gram-negative and anaerobic bacteria are indicated for those cases of portal vein thrombosis associated with underlying infection.9
For chronic cases, the goals of management are to prevent and treat gastroesophageal variceal bleeding and to prevent recurrent thrombosis.9 Nonselective beta-blockers (eg, propranolol) and endoscopic band ligation have shown evidence of reducing the incidence of recurrent bleeding and prolonging survival in retrospective studies.9,30,31 Long-term anticoagulation is generally indicated to prevent further thrombosis and to increase the likelihood of recanalization only for patients with a permanent prothrombotic condition.9 In patients with clinically significant portal hypertension, the benefit of continued anticoagulation therapy must be weighed against the risk of esophageal and gastric variceal bleeding.
There is controversy regarding how to manage portal vein thrombosis that is incidentally identified and asymptomatic (eg, if it is discovered on an imaging study for another indication). Current guidelines recommend against anticoagulation in patients with incidentally discovered and asymptomatic splanchnic vein thrombosis, including portal vein thrombosis.26
Intravenous gamma globulin is not part of the treatment.
CASE CONTINUED
The patient’s presenting symptoms of fever, chills, and abdominal pain completely resolved after a course of antibiotic therapy. The erythrocyte sedimentation rate subsequently normalized and factor VIII activity improved. We believed that an underlying infectious or inflammatory process had contributed to the development of portal vein thrombosis, though the specific cause could not be identified. The patient was treated with enoxaparin 1 mg/kg twice a day and transitioned to warfarin.
Magnetic resonance venography done 3 months after diagnosis showed persistent right portal vein thrombosis that was largely unchanged. Anticoagulation was continued for 1 year with no change in his portal vein thrombosis on sequential imaging and was subsequently discontinued. To date, no malignancy or infectious process has been found, and the patient continues to do well 2 years later.
A 79-year-old man presented with chills and fever. He had a history of polymyalgia rheumatica and had been tapered off corticosteroids 1 month before admission. One week before he presented, he had developed generalized myalgia, chills, and fatigue. A cortisol stimulation test at that time was normal, prednisone was restarted, and his symptoms had improved. But 1 day before he presented, the chills had returned, this time with fever. Laboratory testing at an outpatient clinic had revealed abnormal liver enzyme levels.
On the day he presented, he felt worse, with persistent chills, fever, and vague lower abdominal pain, but he denied nausea, vomiting, changes in bowel habits, melena, hematochezia, and hematemesis. He was admitted for additional evaluation.
His medical history also included coronary artery disease (for which he had undergone coronary artery bypass grafting), hypertension, stable liver cysts, and gout. He had no known inflammatory bowel disease and no recent abdominal surgery. His medications included prednisone, atorvastatin, atenolol, aspirin, niacin, and cholecalciferol. He had no history of smoking, significant drinking, or use of illicit drugs. He had no respiratory or cardiac symptoms or neurologic symptoms consistent with a transient ischemic attack or stroke. He denied any rashes.
On admission, he was febrile, with temperatures reaching 102˚F (38.9˚C). His blood pressure was 137/63 mm Hg, pulse 54 beats per minute, respiration rate 18 breaths per minute, and oxygen saturation 97% on room air. A harsh systolic murmur was noted on physical examination. His abdomen was nondistended, nontender, and without bruits.
Laboratory testing (Table 1) revealed leukocytosis, anemia, mildly abnormal aminotransferase levels, elevated alkaline phosphatase, and markedly elevated C-reactive protein.
A full workup for fever was performed, including blood and urine cultures; chest radiography; contrast-enhanced computed tomography (CT) of the chest, abdomen, and pelvis; magnetic resonance imaging (MRI) of the abdomen; and colonoscopy. No source of infection—bacterial, viral, or fungal—was found. However, CT revealed new extensive thrombosis of the right portal vein and its branches (Figure 1).
CLINICAL PRESENTATION
1. Which of the following is least consistent with the clinical presentation of acute portal vein thrombosis?
- Abdominal pain
- Fever and chills
- Hematemesis
- Leukocytosis
- Absence of symptoms
Of these signs and symptoms, hematemesis is the least likely to be associated with acute portal vein thrombosis, although it can be associated with chronic cases.
Symptoms of portal vein thrombosis
Portal vein thrombosis causes extrahepatic obstruction of the portal venous system, which provides two-thirds of the total hepatic blood flow.
Acute. Often, thrombotic occlusion of the portal vein produces no acute symptoms because of immediate, compensatory vasodilation of the hepatic arterial system.1 Additionally, in the ensuing days, the thrombus becomes an organized collagenous plug, and collateral veins develop to bypass the blocked vein and maintain portal perfusion in a process called cavernous transformation.1,2 Thus, many patients have no symptoms.
If symptoms occur, portal vein thrombosis can initially present as transient abdominal pain with fever, as seen in this patient.3 Many patients with acute portal vein thrombosis experience abdominal pain due to intra-abdominal sepsis, also referred to as pylephlebitis.2,4 High, spiking fevers and chills also occur, caused by infected thrombi associated with intra-abdominal infections such as appendicitis, diverticulitis, and pancreatitis.5,6
Chronic. In contrast, symptomatic chronic portal vein thrombosis commonly presents with sequelae of portal hypertension, most notably gastrointestinal bleeding. Hematemesis from ruptured esophageal varices is the most frequent reason for seeking medical attention, though varices also develop in the stomach, duodenum, jejunum, gallbladder, and bile ducts.2,7 Abdominal pain is less common in chronic portal vein thrombosis unless the thrombus extends into the mesenteric veins and causes bowel ischemia or infarction. Long-standing portal vein thrombosis may also lead to dilated venous collaterals that compress large bile ducts, resulting in portal cholangiopathy.1,8
Portal vein thrombosis may present as acute intestinal ischemia and bowel infarction, though this is uncommon. This is generally seen with extensive occlusive portal vein thrombosis and concomitant mesenteric venous thrombosis.1,2
Other symptoms that are common but nonspecific are nausea, vomiting, diarrhea, weight loss, and anorexia.2
Signs of portal vein thrombosis
On examination, patients with acute portal vein thrombosis have minimal physical signs unless they have other contributing conditions. For example, acute portal vein thrombosis can result in abdominal distention secondary to ileus, or guarding and ascites secondary to intestinal infarction.3,9
Some patients with chronic portal vein thrombosis also have normal physical findings, but many have signs. Splenomegaly is seen in 75% to 100% of patients.2,7 Hepatomegaly, abdominal tenderness, and low-grade fever are common as well.2,10 Ascites is usually not present without underlying cirrhosis; however, mild and transient ascites can develop immediately after the thrombotic event before the patient develops collateral circulation.2
Laboratory testing for portal vein thrombosis
Laboratory test results are typically unremarkable. Liver function tests show preserved hepatic function but may reveal mild increases in aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase, and bilirubin.2,10
In acute cases, elevations of acute-phase reactant levels can occur.9 Leukocytosis and blood cultures growing Bacteroides species are seen in septic cases or pylephlebitis.11,12 There may be mild anemia, particularly after a recent bleeding episode, or mild leukopenia and thrombocytopenia due to hypersplenism. Suspicion of an underlying myeloproliferative disorder is high if thrombocytosis is present.2
DIAGNOSIS
2. All of the following would be appropriate initial diagnostic studies for portal vein thrombosis except which one?
- Doppler ultrasonography
- Contrast-enhanced CT
- Contrast-enhanced MRI
- Angiography
Portal vein thrombosis is most often diagnosed with noninvasive techniques, namely Doppler ultrasonography, CT, and MRI—not angiography.
Ultrasonography can reveal an echogenic thrombus in the vessel lumen with distention of the portal vein proximal to the occlusion and extensive collateral vessels. Plain ultrasonography fails to reveal the thrombus in up to one-third of patients. However, duplex ultrasonography with color flow Doppler imaging can confirm partial or complete absence of flow in the vein with 89% sensitivity and 92% specificity.13,14
On contrast-enhanced CT, the thrombus appears as a filling defect within the portal venous segment. Complete occlusion of the vein may produce a “train track” appearance due to contrast around the vessel.10 Without contrast, the clot will appear as hyperattenuating material in the portal vein, but contrast-enhanced imaging may be necessary to differentiate the thrombus from the vessel wall.15 Gas within the portal venous system is specific for pylephlebitis.4 Evidence of cavernous transformation is seen in chronic portal vein thrombosis.
Contrast-enhanced magnetic resonance angiography can also be used to evaluate patency and flow direction. In addition, it provides detailed anatomic information about the entire portal venous system, including the intrahepatic portal vessels, which is limited in CT imaging.2,10 CT and MRI can also help to identify predisposing conditions (eg, intra-abdominal infection, hepatocellular carcinoma) and complications (eg, intestinal infarction) associated with portal vein thrombosis.
Angiography can be considered if noninvasive techniques are inconclusive but is generally not necessary, given the increased use of CT and MRI.
In our patient, abdominal CT revealed occlusive thrombosis of the right portal vein and its branches (Figure 1). The left and main portal veins were patent. There was no evidence of intra-abdominal infection or infarction.
FINDING THE CAUSE
3. Which of the following is not a common cause
of portal vein thrombosis?
- A hypercoagulable state
- Immune deficiency
- Intra-abdominal infection
- Malignancy
- Portal hypertension
Once portal vein thrombosis has been diagnosed, the cause should be identified (Table 2). The differential diagnosis is broad, including both local factors (eg, injury to the portal vein, local inflammation, infection) and general factors (eg, inherited and acquired hypercoagulable conditions). Thrombophilias are identified in 60% of patients with portal vein thrombosis and local factors in 40%.7 Moreover, the etiology is often multifactorial. However, immune deficiency is not a common cause.
Hypercoagulability
Prothrombotic disorders can be either inherited or acquired.
Inherited deficiencies in the natural anticoagulants antithrombin, protein C, and protein S are associated with a high risk of thrombosis but have a low prevalence in the general population. In the setting of liver abnormalities, familial testing may be helpful to distinguish inherited causes of portal vein thrombosis from defective liver function as a consequence of portal vein thrombosis. The factor V Leiden mutation (G1691A) and the G20210A mutation in the prothrombin gene are more prevalent (> 2%) but generally confer a lower thrombosis risk.16 The prothrombin gene mutation G20210A is the most common risk factor for portal vein thrombosis, with prevalence of 2% to 22% in adults with nonmalignant, noncirrhotic portal vein thrombosis.3
Hyperhomocysteinemia due to a methylene tetrahydrofolate reductase (MTHFR) mutation (C677T) is another inherited associated risk factor for portal vein thrombosis, but hyperhomocysteinemia can also arise as a complication of portal vein thrombosis-related liver disease.3
Acquired prothrombotic disorders, particularly myeloproliferative diseases, are found in 22% to 48% of cases of portal vein thrombosis. Many young patients with myeloproliferative disorders present with portal vein thrombosis as the first symptom, and testing for the G1849T point mutation in JAK2 can make the diagnosis.17 Splenectomy with underlying myeloproliferative disorder confers a particularly high risk for portal vein thrombosis.18
Other thrombophilic disorders including antiphospholipid antibody syndrome, paroxysmal nocturnal hemoglobinuria, and malignancy can contribute to portal vein thrombosis.3 Pregnancy and oral contraceptive use have also been associated with hypercoagulability, and cessation of oral estrogen is recommended in such cases. The risk may be further increased in patients on oral contraceptives who have a previously unrecognized hypercoagulable state.3
Inflammation and infection
Inflammation and infection are local risk factors for portal vein thrombosis. Acute portal vein thrombosis has been associated with intra-abdominal infections (eg, appendicitis, cholecystitis) and with inflammatory conditions such as inflammatory bowel disease and pancreatitis.16,19 From 3% to 5% of all portal vein thrombosis cases result from pancreatitis, either from a single acute episode or from repeat inflammation of chronic pancreatitis.10 Portal vein thrombosis in the setting of inflammatory bowel disease can occur even when the disease is in remission, particularly in ulcerative colitis.20,21
Injury to the portal venous system
Abdominal surgery, particularly splenectomy, portosystemic shunting, colectomy, and blunt abdominal trauma can cause injury to the portal venous system, resulting in portal vein thrombosis. This is usually seen only in patients with portal hypertension, an underlying prothrombotic condition such as myeloproliferative disease, or inflammatory bowel disease.10,19,22
Impaired portal vein flow
Cirrhosis and malignancy are major risk factors for portal vein thrombosis. In case series, cirrhosis was found in 24% to 32% of patients with portal vein thrombosis.2,23 However, the overall prevalence of portal vein thrombosis in cirrhotic patients varies widely, from 0.6% to 28%, depending on the degree of cirrhosis.10
The pathogenesis of portal vein thrombosis in cirrhosis is unclear but may be multifactorial. Decreased portal blood flow (with subsequent stasis) and periportal lymphangitis and fibrosis are thought to stimulate thrombus formation.3,10 Additionally, patients with advanced cirrhosis are prothrombotic because of reduced hepatic synthesis of antithrombin, protein C, protein S, and coagulation factors.
Malignancy is associated with 21% to 24% of cases of portal vein thrombosis in adults, with pancreatic cancer and hepatocellular carcinoma being the most common.2,3 Others include cholangiocarcinoma and carcinomas of the stomach, lung, prostate, uterus, and kidney. Cancer causes portal vein thrombosis through a combination of tumor invasion into the portal vein, extrinsic compression by the tumor, periportal fibrosis following surgery or radiation, and hypercoagulability secondary to malignancy.9,16,24
Idiopathic portal vein thrombosis
Portal vein thrombosis is usually caused by one or more of the underlying factors mentioned above but is idiopathic in 8% to 15% of cases.10
Back to our patient
The cause of this patient’s portal vein thrombosis is unclear. He did not have a history of cirrhosis, inflammatory bowel disease, trauma, or abdominal surgery. His febrile illness could have precipitated the formation of a thrombus, but no definitive source of infection or inflammation was discovered. His workup was negative for pancreatitis, appendicitis, cholecystitis, diverticulitis, and prostatitis. No occult malignancy was found. It is also possible that his fever was the result of the thrombosis.
A full hypercoagulability panel revealed no striking abnormalities. He did have elevated fibrinogen and factor VIII levels that were consistent with an acute-phase reaction, along with an elevated erythrocyte sedimentation rate (> 90 mm/hr) and C-reactive protein level. Aside from the portal vein thrombosis, no potential source of inflammation could be identified.
Mildly reduced levels of antithrombin III activity were attributed to enoxaparin therapy and ultimately normalized on repeated testing. The patient had very minimally elevated titers of anticardiolipin immunoglobulin G (1:10 GPL) and anti-beta-2 glycoprotein immunoglobulin M (21 SMU), which were not thought to be significant. Tests for lupus anticoagulant, prothrombin gene mutation, activated protein C resistance, and JAK2 mutation were negative.
TREATMENT
4. Treatment of symptomatic portal vein thrombosis generally includes which two of the following?
- Anticoagulation
- Intravenous gamma globulin
- Broad-spectrum antibiotics
Anticoagulant therapy
Treatment of acute, symptomatic portal vein thrombosis involves anticoagulant therapy to prevent extension of the thrombus and, ultimately, to allow for recanalization of the obstructed veins. Anticoagulant therapy is initially intravenous unfractionated heparin or subcutaneous low-molecular-weight heparin, eventually bridged to an oral agent such as warfarin.3,9 Currently, there are inadequate data on the use of oral or parenteral factor Xa inhibitors or direct thrombin inhibitors in the treatment of this disease.
When started immediately, anticoagulation therapy is associated with complete recanalization in 38.3% and partial recanalization in 14% of patients presenting with complete thrombosis. Without anticoagulation, spontaneous recanalization is unusual.25
Although the optimal duration of anticoagulant therapy is unclear, a minimum of 3 months is generally recommended.9,26 If a hypercoagulable state is present or if the portal vein thrombosis is unprovoked (eg, by surgery, trauma, or an intra-abdominal infection), long-term treatment should be considered.26
Experience with thrombolytic therapy or mechanical recanalization has been limited, but the use of catheter-based techniques for pharmacomechanical thrombolysis has been reported.27–29 Transjugular intrahepatic portosystemic shunting is also an alternative to anticoagulation, but its role in treating portal vein thrombosis is complicated by technical difficulties of the procedure, postoperative complications, and recurrent occlusion of the shunt.25
Currently, there are no data comparing the risk-benefit ratio of early anticoagulation and that of invasive procedures. These more aggressive treatments are generally considered only when there is extensive thrombosis or ascites (which are both predictive factors of poor response to anticoagulation alone) and in patients for whom anticoagulation has failed.3 Surgical thrombectomy is rarely indicated, typically only in instances in which laparotomy is being performed for suspected bowel infarction.3
Antibiotics
In addition to anticoagulation, broad-spectrum antibiotics covering gram-negative and anaerobic bacteria are indicated for those cases of portal vein thrombosis associated with underlying infection.9
For chronic cases, the goals of management are to prevent and treat gastroesophageal variceal bleeding and to prevent recurrent thrombosis.9 Nonselective beta-blockers (eg, propranolol) and endoscopic band ligation have shown evidence of reducing the incidence of recurrent bleeding and prolonging survival in retrospective studies.9,30,31 Long-term anticoagulation is generally indicated to prevent further thrombosis and to increase the likelihood of recanalization only for patients with a permanent prothrombotic condition.9 In patients with clinically significant portal hypertension, the benefit of continued anticoagulation therapy must be weighed against the risk of esophageal and gastric variceal bleeding.
There is controversy regarding how to manage portal vein thrombosis that is incidentally identified and asymptomatic (eg, if it is discovered on an imaging study for another indication). Current guidelines recommend against anticoagulation in patients with incidentally discovered and asymptomatic splanchnic vein thrombosis, including portal vein thrombosis.26
Intravenous gamma globulin is not part of the treatment.
CASE CONTINUED
The patient’s presenting symptoms of fever, chills, and abdominal pain completely resolved after a course of antibiotic therapy. The erythrocyte sedimentation rate subsequently normalized and factor VIII activity improved. We believed that an underlying infectious or inflammatory process had contributed to the development of portal vein thrombosis, though the specific cause could not be identified. The patient was treated with enoxaparin 1 mg/kg twice a day and transitioned to warfarin.
Magnetic resonance venography done 3 months after diagnosis showed persistent right portal vein thrombosis that was largely unchanged. Anticoagulation was continued for 1 year with no change in his portal vein thrombosis on sequential imaging and was subsequently discontinued. To date, no malignancy or infectious process has been found, and the patient continues to do well 2 years later.
- Ponziani FR, Zocco MA, Campanale C, et al. Portal vein thrombosis: insight into physiopathology, diagnosis, and treatment. World J Gastroenterol 2010; 16:143–155.
- Cohen J, Edelman RR, Chopra S. Portal vein thrombosis: a review. Am J Med 1992; 92:173–182.
- Primignani M. Portal vein thrombosis, revisited. Dig Liver Dis 2010; 42:163–170.
- Condat B, Valla D. Nonmalignant portal vein thrombosis in adults. Nat Clin Pract Gastroenterol Hepatol 2006; 3:505–515.
- Condat B, Pessione F, Helene Denninger M, Hillaire S, Valla D. Recent portal or mesenteric venous thrombosis: increased recognition and frequent recanalization on anticoagulant therapy. Hepatology 2000; 32:466–470.
- Sheen CL, Lamparelli H, Milne A, Green I, Ramage JK. Clinical features, diagnosis and outcome of acute portal vein thrombosis. QJM 2000; 93:531–534.
- Sogaard KK, Astrup LB, Vilstrup H, Gronbaek H. Portal vein thrombosis; risk factors, clinical presentation and treatment. BMC Gastroenterol 2007; 7:34.
- Llop E, de Juan C, Seijo S, et al. Portal cholangiopathy: radiological classification and natural history. Gut 2011; 60:853–860.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Sobhonslidsuk A, Reddy KR. Portal vein thrombosis: a concise review. Am J Gastroenterol 2002; 97:535–541.
- Ni YH, Wang NC, Peng MY, Chou YY, Chang FY. Bacteroides fragilis bacteremia associated with portal vein and superior mesentery vein thrombosis secondary to antithrombin III and protein C deficiency: a case report. J Microbiol Immunol Infect 2002; 35:255–258.
- Trum J, Valla D, Cohen G, et al. Bacteroides bacteraemia of undetermined origin: strong association with portal vein thrombosis and cryptogenic pylephlebitis. Eur J Gastroenterol Hepatol 1993; 5:655–659.
- Ueno N, Sasaki A, Tomiyama T, Tano S, Kimura K. Color Doppler ultrasonography in the diagnosis of cavernous transformation of the portal vein. J Clin Ultrasound 1997; 25:227–233.
- Tessler FN, Gehring BJ, Gomes AS, et al. Diagnosis of portal vein thrombosis: value of color Doppler imaging. AJR Am J Roentgenol 1991; 157:293–296.
- Hidajat N, Stobbe H, Griesshaber V, Felix R, Schroder RJ. Imaging and radiological interventions of portal vein thrombosis. Acta Radiol 2005; 46:336–343.
- Valla DC, Condat B. Portal vein thrombosis in adults: pathophysiology, pathogenesis and management. J Hepatol 2000; 32:865–871.
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352:1779–1790.
- Krauth MT, Lechner K, Neugebauer EA, Pabinger I. The postoperative splenic/portal vein thrombosis after splenectomy and its prevention—an unresolved issue. Haematologica 2008; 93:1227–1232.
- Sinagra E, Aragona E, Romano C, et al. The role of portal vein thrombosis in the clinical course of inflammatory bowel diseases: report on three cases and review of the literature. Gastroenterol Res Pract 2012; 2012:916428.
- Maconi G, Bolzacchini E, Dell’Era A, Russo U, Ardizzone S, de Franchis R. Portal vein thrombosis in inflammatory bowel diseases: a single-center case series. J Crohns Colitis 2012; 6:362–367.
- Jackson LM, O’Gorman PJ, O’Connell J, Cronin CC, Cotter KP, Shanahan F. Thrombosis in inflammatory bowel disease: clinical setting, procoagulant profile and factor V Leiden. QJM 1997; 90:183–188.
- Eguchi A, Hashizume M, Kitano S, Tanoue K, Wada H, Sugimachi K. High rate of portal thrombosis after splenectomy in patients with esophageal varices and idiopathic portal hypertension. Arch Surg 1991; 126:752–755.
- Ogren M, Bergqvist D, Björck M, Acosta S, Eriksson H, Sternby NH. Portal vein thrombosis: prevalence, patient characteristics and lifetime risk: a population study based on 23,796 consecutive autopsies. World J Gastroenterol 2006; 12:2115–2119.
- Falanga A, Marchetti M, Vignoli A. Coagulation and cancer: biological and clinical aspects. J Thromb Haemost 2013; 11:223–233.
- Congly SE, Lee SS. Portal vein thrombosis: should anticoagulation be used? Curr Gastroenterol Rep 2013; 15:306.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Uflacker R. Applications of percutaneous mechanical thrombectomy in transjugular intrahepatic portosystemic shunt and portal vein thrombosis. Tech Vasc Interv Radiol 2003; 6:59–69.
- Takahashi N, Kuroki K, Yanaga K. Percutaneous transhepatic mechanical thrombectomy for acute mesenteric venous thrombosis. J Endovasc Ther 2005; 12:508–511.
- Lopera JE, Correa G, Brazzini A, et al. Percutaneous transhepatic treatment of symptomatic mesenteric venous thrombosis. J Vasc Surg 2002; 36:1058–1061.
- Orr DW, Harrison PM, Devlin J, et al. Chronic mesenteric venous thrombosis: evaluation and determinants of survival during long-term follow-up. Clin Gastroenterol Hepatol 2007; 5:80–86.
- Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant therapy. Gastroenterology 2001; 120:490–497.
- Ponziani FR, Zocco MA, Campanale C, et al. Portal vein thrombosis: insight into physiopathology, diagnosis, and treatment. World J Gastroenterol 2010; 16:143–155.
- Cohen J, Edelman RR, Chopra S. Portal vein thrombosis: a review. Am J Med 1992; 92:173–182.
- Primignani M. Portal vein thrombosis, revisited. Dig Liver Dis 2010; 42:163–170.
- Condat B, Valla D. Nonmalignant portal vein thrombosis in adults. Nat Clin Pract Gastroenterol Hepatol 2006; 3:505–515.
- Condat B, Pessione F, Helene Denninger M, Hillaire S, Valla D. Recent portal or mesenteric venous thrombosis: increased recognition and frequent recanalization on anticoagulant therapy. Hepatology 2000; 32:466–470.
- Sheen CL, Lamparelli H, Milne A, Green I, Ramage JK. Clinical features, diagnosis and outcome of acute portal vein thrombosis. QJM 2000; 93:531–534.
- Sogaard KK, Astrup LB, Vilstrup H, Gronbaek H. Portal vein thrombosis; risk factors, clinical presentation and treatment. BMC Gastroenterol 2007; 7:34.
- Llop E, de Juan C, Seijo S, et al. Portal cholangiopathy: radiological classification and natural history. Gut 2011; 60:853–860.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Sobhonslidsuk A, Reddy KR. Portal vein thrombosis: a concise review. Am J Gastroenterol 2002; 97:535–541.
- Ni YH, Wang NC, Peng MY, Chou YY, Chang FY. Bacteroides fragilis bacteremia associated with portal vein and superior mesentery vein thrombosis secondary to antithrombin III and protein C deficiency: a case report. J Microbiol Immunol Infect 2002; 35:255–258.
- Trum J, Valla D, Cohen G, et al. Bacteroides bacteraemia of undetermined origin: strong association with portal vein thrombosis and cryptogenic pylephlebitis. Eur J Gastroenterol Hepatol 1993; 5:655–659.
- Ueno N, Sasaki A, Tomiyama T, Tano S, Kimura K. Color Doppler ultrasonography in the diagnosis of cavernous transformation of the portal vein. J Clin Ultrasound 1997; 25:227–233.
- Tessler FN, Gehring BJ, Gomes AS, et al. Diagnosis of portal vein thrombosis: value of color Doppler imaging. AJR Am J Roentgenol 1991; 157:293–296.
- Hidajat N, Stobbe H, Griesshaber V, Felix R, Schroder RJ. Imaging and radiological interventions of portal vein thrombosis. Acta Radiol 2005; 46:336–343.
- Valla DC, Condat B. Portal vein thrombosis in adults: pathophysiology, pathogenesis and management. J Hepatol 2000; 32:865–871.
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352:1779–1790.
- Krauth MT, Lechner K, Neugebauer EA, Pabinger I. The postoperative splenic/portal vein thrombosis after splenectomy and its prevention—an unresolved issue. Haematologica 2008; 93:1227–1232.
- Sinagra E, Aragona E, Romano C, et al. The role of portal vein thrombosis in the clinical course of inflammatory bowel diseases: report on three cases and review of the literature. Gastroenterol Res Pract 2012; 2012:916428.
- Maconi G, Bolzacchini E, Dell’Era A, Russo U, Ardizzone S, de Franchis R. Portal vein thrombosis in inflammatory bowel diseases: a single-center case series. J Crohns Colitis 2012; 6:362–367.
- Jackson LM, O’Gorman PJ, O’Connell J, Cronin CC, Cotter KP, Shanahan F. Thrombosis in inflammatory bowel disease: clinical setting, procoagulant profile and factor V Leiden. QJM 1997; 90:183–188.
- Eguchi A, Hashizume M, Kitano S, Tanoue K, Wada H, Sugimachi K. High rate of portal thrombosis after splenectomy in patients with esophageal varices and idiopathic portal hypertension. Arch Surg 1991; 126:752–755.
- Ogren M, Bergqvist D, Björck M, Acosta S, Eriksson H, Sternby NH. Portal vein thrombosis: prevalence, patient characteristics and lifetime risk: a population study based on 23,796 consecutive autopsies. World J Gastroenterol 2006; 12:2115–2119.
- Falanga A, Marchetti M, Vignoli A. Coagulation and cancer: biological and clinical aspects. J Thromb Haemost 2013; 11:223–233.
- Congly SE, Lee SS. Portal vein thrombosis: should anticoagulation be used? Curr Gastroenterol Rep 2013; 15:306.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Uflacker R. Applications of percutaneous mechanical thrombectomy in transjugular intrahepatic portosystemic shunt and portal vein thrombosis. Tech Vasc Interv Radiol 2003; 6:59–69.
- Takahashi N, Kuroki K, Yanaga K. Percutaneous transhepatic mechanical thrombectomy for acute mesenteric venous thrombosis. J Endovasc Ther 2005; 12:508–511.
- Lopera JE, Correa G, Brazzini A, et al. Percutaneous transhepatic treatment of symptomatic mesenteric venous thrombosis. J Vasc Surg 2002; 36:1058–1061.
- Orr DW, Harrison PM, Devlin J, et al. Chronic mesenteric venous thrombosis: evaluation and determinants of survival during long-term follow-up. Clin Gastroenterol Hepatol 2007; 5:80–86.
- Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant therapy. Gastroenterology 2001; 120:490–497.
A 57-year-old woman with abdominal pain
A 57-year-old woman presented to the emergency department with left lower quadrant pain, which had started 1 week earlier and was constant, dull, aching, and nonradiating. There were no aggravating or alleviating factors. The pain was associated with low-grade fever and nausea. She reported no vomiting, no change in bowel habits, and no hematemesis, hematochezia, or melena. She did not have urinary urgency, frequency, or dysuria. She had no cardiac, respiratory, or neurologic symptoms.
Her medical history included hypothyroidism, type 2 diabetes mellitus, diverticulosis, and chronic obstructive pulmonary disease. Her medications included metformin, insulin, levothyroxine, and inhaled tiotropium. She had no allergies. She had never undergone surgery, including cesarean delivery. She was postmenopausal. She had two children, both of whom had been born vaginally at full term. She denied using alcohol, tobacco, and illicit drugs. Her family history was noncontributory.
On examination, she was not in acute distress. Her temperature was 36.7°C (98.1°F), blood pressure 130/90 mm Hg, heart rate 86 beats per minute and regular, respiratory rate 16 breaths per minute, and oxygen saturation 98% on ambient air. Examination of her head and neck was unremarkable. Cardiopulmonary examination was normal. Abdominal examination revealed normal bowel sounds, mild tenderness in the left lower quadrant with localized guarding, and rebound tenderness. Neurologic examination was unremarkable.
Initial laboratory data showed mild leukocytosis. Computed tomography with contrast of the abdomen and pelvis suggested acute diverticulitis.
ATRIAL FIBRILLATION, AND THEN A TURN FOR THE WORSE
The patient was admitted with an initial diagnosis of acute diverticulitis. She was started on antibiotics, hydration, and pain medications, and her abdominal pain gradually improved.
On the third hospital day, she suddenly experienced shortness of breath and palpitations. At the time of admission her electrocardiogram had been normal, but it now showed atrial fibrillation with a rapid ventricular response. She also developed elevated troponin levels, which were thought to represent type 2 non-ST-elevation myocardial infarction.
She was started on aspirin, clopidogrel, and anticoagulation with heparin bridged with warfarin for the new-onset atrial fibrillation. Her heart rate was controlled with metoprolol, and her shortness of breath improved. An echocardiogram was normal.
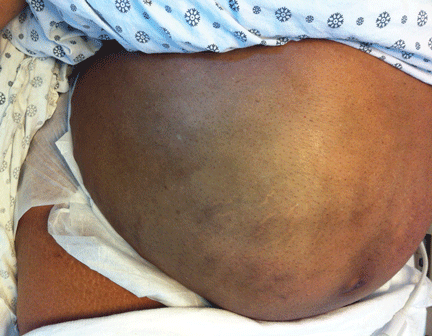
On the seventh hospital day, she developed severe right-sided lower abdominal pain and bruising. Her blood pressure was 90/60 mm Hg, heart rate 110 beats per minute and irregularly irregular, respiratory rate 22 breaths per minute, and oxygen saturation 97% on room air. Her abdomen was diffusely tender with a palpable mass in the right lower quadrant and hypoactive bowel sounds. Ecchymosis was noted (Figure 1).
DIFFERENTIAL DIAGNOSIS
1. What is the likely cause of her decompensation?
- Acute mesenteric ischemia
- Perforation of the gastrointestinal tract
- Rectus sheath hematoma
- Abdominal compartment syndrome due to acute pancreatitis
Acute mesenteric ischemia
Signs and symptoms of acute mesenteric ischemia can be vague. Moreover, when it leads to bowel necrosis the mortality rate is high, ranging from 30% to 65%.1 Hence, one should suspect it and try to diagnose it early.
Most patients with this condition have comorbidities; risk factors include atherosclerotic disease, cardiac conditions (congestive heart failure, recent myocardial infarction, and atrial fibrillation), systemic illness, and inherited or acquired hypercoagulable states.2
The four major causes are:
- Acute thromboembolic occlusion of the superior mesenteric artery (the most common site of occlusion because of the acute angle of origin from the aorta)
- Acute thrombosis of the mesenteric vein
- Acute thrombosis of the mesenteric artery
- Nonocclusive disease affecting the mesenteric vessels2
Nonocclusive disease is seen in conditions in which the mesenteric vessels are already compromised due to background stenosis owing to atherosclerosis. Also, conditions such as septic and cardiogenic shock can compromise these arteries, leading to ischemia, which, if it persists, can lead to bowel infarction. Ischemic colitis falls under this category. It commonly involves the descending and sigmoid colon.3
The initial symptom of ischemia may be abdominal pain that is brought on by eating large meals (“postprandial intestinal angina.”2 When the ischemia worsens to infarction, patients may have a diffusely tender abdomen and constant pain that does not vary with palpation. Surprisingly, patients do not exhibit peritoneal signs early on. This gives rise to the description of “pain out of proportion to the physical findings” traditionally associated with acute mesenteric ischemia.2
Diagnosis. Supportive laboratory data include marked leukocytosis, elevated hematocrit due to hemoconcentration, metabolic acidosis, and elevated lactate.4 Newer markers such as serum alpha-glutathione S-transferase (alpha-GST) and intestinal fatty acid-binding protein (I-FABP) may be used to support the diagnosis.
Elevated alpha-GST has 72% sensitivity and 77% specificity in the diagnosis of acute mesenteric ischemia.5 The caveat is that it cannot reliably differentiate ischemia from infarction. Its sensitivity can be improved to 97% to 100% by using the white blood cell count and lactate levels in combination.5
An I-FABP level higher than 100 ng/mL has 100% sensitivity for diagnosing mesenteric infarction but only 25% sensitivity for bowel strangulation.6
Early use of abdominal computed tomography with contrast can aid in recognizing this diagnosis.7 Thus, it should be ordered in suspected cases, even in patients who have elevated creatinine levels (which would normally preclude the use of contrast), since early diagnosis followed by endovascular therapy is associated with survival benefit, and the risk of contrast-induced nephropathy appears to be small.8 Computed tomography helps to determine the state of mesenteric vessels and bowel perfusion before ischemia progresses to infarction. It also helps to rule out other common diagnoses. Findings that suggest acute mesenteric ischemia include segmental bowel wall thickening, intestinal pneumatosis with gas in the portal vein, bowel dilation, mesenteric stranding, portomesenteric thrombosis, and solid-organ infarction.9
Treatment. If superior mesenteric artery occlusion is diagnosed on computed tomography, the next step is to determine if there is peritonitis.10 In patients who have evidence of peritonitis, exploratory laparotomy is performed. For emboli in such patients, open embolectomy followed by on-table angiography is carried out in combination with damage-control surgery. For patients with peritonitis and acute thrombosis, stenting along with damage-control surgery is preferred.10
On the other hand, if there is no peritonitis, the thrombosis may be amenable to endovascular intervention. For patients with acute embolic occlusion with no contraindications to thrombolysis, aspiration embolectomy in combination with local catheter-directed thrombolysis with recombinant tissue plasminogen activator can be performed. This can be combined with endovascular mechanical embolectomy for more complete management.10 Patients with contraindications to thrombolysis can be treated either with aspiration and mechanical embolectomy or with open embolectomy with angiography.10
During laparotomy, the surgeon carefully inspects the bowel for signs of necrosis. Signs that bowel is still viable include pink color, bleeding from cut surfaces, good peristalsis, and visible pulsations in the arterial arcade of the mesentery.
Acute mesenteric artery thrombosis arising from chronic atherosclerotic disease can be treated with stenting of the stenotic lesion.10 Patients with this condition would also benefit from aggressive management of atherosclerotic disease with statins along with antiplatelet agents.10
Mesenteric vein thrombosis requires prompt institution of anticoagulation. However, in advanced cases leading to bowel infarction, exploratory laparotomy with resection of the necrotic bowel may be required. Anticoagulation should be continued for at least 6 months, and further therapy should be determined by the underlying precipitating condition.10
Critically ill patients who develop mesenteric ischemia secondary to persistent hypotension usually respond to adequate volume resuscitation, cessation of vasopressors, and overall improvement in their hemodynamic status. These patients must be closely monitored for development of gangrene of the bowel because they may be intubated and not able to complain. Any sudden deterioration in their condition should prompt physicians to consider bowel necrosis developing in these patients. Elevation of lactate levels out of proportion to the degree of hypotension may be corroborative evidence.4
Our patient had risk factors for acute mesenteric ischemia that included atrial fibrillation and recent non-ST-elevation myocardial infarction. She could have had arterial emboli due to atrial fibrillation, in situ superior mesenteric arterial thrombosis, or splanchnic arterial vasoconstriction due to the myocardial infarction associated with transient hypotension.
Arguing against this diagnosis, although she had a grossly distended abdomen, abdominal bruising usually is not seen. Also, a palpable mass in the right lower quadrant is uncommon except when acute mesenteric ischemia occurs due to segmental intestinal strangulation, as with strangulated hernia or volvulus. She also had therapeutic international normalized ratio (INR) levels constantly while on anticoagulation. Nevertheless, acute mesenteric ischemia should be strongly considered in the initial differential diagnosis of this patient’s acute decompensation.
Perforation of the gastrointestinal tract
Diverticulitis is the acute inflammation of one or more diverticula, which are small pouches created by herniation of the mucosa into the wall of the colon. The condition is caused by microscopic or macroscopic perforation of the diverticula. Microscopic perforation is usually self-limited (uncomplicated diverticulitis) and responds to conservative treatment, whereas macroscopic perforation can be associated with fecal or purulent peritonitis, abscess, enteric fistula, bowel obstruction, and stricture (complicated diverticulitis), in which case surgery may be necessary.
Patients with peritonitis due to free perforation present with generalized tenderness with rebound tenderness and guarding on abdominal examination. The abdomen may be distended and tympanic to percussion, with diminished or absent bowel sounds. Patients may have hemodynamic compromise.
Plain upright abdominal radiographs may show free air under the diaphragm. Computed tomography may show oral contrast outside the lumen and detect even small amounts of free intraperitoneal air (more clearly seen on a lung window setting).
Our patient initially presented with acute diverticulitis. She later developed diffuse abdominal tenderness with hypoactive bowel sounds. Bowel perforation is certainly a possibility at this stage, though it is usually not associated with abdominal bruising. She would need additional imaging to rule out this complication.
Other differential diagnoses to be considered in this patient with right lower-quadrant pain include acute appendicitis, incarcerated inguinal hernia, volvulus (particularly cecal volvulus), small-bowel obstruction, pyelonephritis, and gynecologic causes such as adnexal torsion, ruptured ovarian cyst, and tubo-ovarian abscess. Computed tomography helps to differentiate most of these causes.
Rectus sheath hematoma
Rectus sheath hematoma is relatively uncommon and often not considered in the initial differential diagnosis of an acute abdomen. This gives it the rightful term “a great masquerader.” It usually results from bleeding into the rectus sheath from damage to the superior (more common) or inferior epigastric arteries and occasionally from a direct tear of the rectus abdominis muscle. Predisposing factors include anticoagulant therapy (most common), advanced age, hypertension, previous abdominal surgery, trauma, paroxysmal coughing, medication injections, pregnancy, blood dyscrasias, severe vomiting, violent physical activity, and leukemia.11
Clinical manifestations include acute abdominal pain, often associated with fever, nausea, and vomiting. Physical examination may reveal signs of hypovolemic shock, a palpable nonpulsatile abdominal mass, and signs of local peritoneal irritation. The Carnett sign11 (tenderness within the abdominal wall that persists and does not improve with raising the head) and the Fothergill sign11 (a tender abdominal mass that does not cross the midline and remains palpable with tensing of the rectus sheath) may be elicited.
Computed tomography is more sensitive than abdominal ultrasonography in differentiating rectus sheath hematoma from an intra-abdominal pathology.11 In addition, computed tomography also helps to determine if the bleeding is active or not, which has therapeutic implications.
In our patient, rectus sheath hematoma is a possibility because of her ongoing anticoagulation, findings of localized abdominal bruising, and palpable right lower-quadrant mass, and it is high on the list of differential diagnoses. Rectus sheath hematoma should be considered in the differential diagnosis of lower abdominal pain particularly in elderly women who are on anticoagulation and in whom the onset of pain coincides with a paroxysm of cough.12 Women are twice as likely as men to develop rectus sheath hematoma, owing to their different muscle mass.13 In addition, anterior abdominal wall muscles are stretched during pregnancy.13
Abdominal compartment syndrome
Abdominal compartment syndrome has been classically associated with surgical patients. However, it is being increasingly recognized in critically ill medical patients, in whom detecting and treating it early may result in significant reduction in rates of morbidity and death.14
Abdominal compartment syndrome is of three types: primary, secondary, and recurrent. Primary abdominal compartment syndrome refers to the classic surgical patients with evidence of direct injury to the abdominal or pelvic organs through major trauma or extensive abdominal surgeries. Secondary abdominal compartment syndrome refers to its development in critically ill intensive care patients in whom the pathology does not directly involve the abdominal or pelvic organs.
Various medical conditions can culminate in abdominal compartment syndrome and result in multiorgan failure. Recurrent abdominal compartment syndrome refers to its development after management of either primary or secondary intra-abdominal hypertension or abdominal compartment syndrome.15 Clinicians thus must be aware of secondary and recurrent abdominal compartment syndrome occurring in critically ill patients.
The normal intra-abdominal pressure is around 5 to 7 mm Hg, even in most critically ill patients. Persistent elevation, ie, higher than 12 mm Hg, is referred to as intra-abdominal hypertension.16–18 Intra-abdominal hypertension is subdivided into four grades:
- Grade I: 12–15 mm Hg
- Grade II: 16–20 mm Hg
- Grade III: 21–25 mm Hg
- Grade IV: > 25 mm Hg.
The World Society of the Abdominal Compartment Syndrome (WSACS) defines abdominal compartment syndrome as pressure higher than 20 mm Hg along with organ damage.18 It may or may not be associated with an abdominal perfusion pressure less than 60 mm Hg.18
Risk factors associated with abdominal compartment syndrome include conditions causing decreased gut motility (gastroparesis, ileus, and colonic pseudo-obstruction), intra-abdominal or retroperitoneal masses or abscesses, ascites, hemoperitoneum, acute pancreatitis, third-spacing due to massive fluid resuscitation with transfusions, peritoneal dialysis, and shock.18,19
Physical examination has a sensitivity of only 40% to 60% in detecting intra-abdominal hypertension.20 The gold-standard method of measuring the intra-abdominal pressure is the modified Kron technique,18 using a Foley catheter in the bladder connected to a pressure transducer. With the patient in the supine position, the transducer is zeroed at the mid-axillary line at the level of the iliac crest, and 25 mL of normal saline is instilled into the bladder and maintained for 30 to 60 seconds to let the detrusor muscle relax.15 Pressure tracings are then recorded at the end of expiration. Factors that are known to affect the transbladder pressure include patient position, respiratory movement, and body mass index, and should be taken into account when reading the pressure recordings.15,21 Other techniques that can be used include intragastric, intra-inferior vena cava, and intrarectal approaches.15
The WSACS recommends that any patient admitted to a critical care unit or in whom new organ failure develops should be screened for risk factors for intra-abdominal hypertension and abdominal compartment syndrome. If a patient has at least two of the risk factors suggested by WSACS, a baseline intra-abdominal pressure measurement should be obtained. Patients at risk for intra-abdominal hypertension should have the intra-abdominal pressure measured every 4 to 6 hours. However, in the face of hemodynamic instability and worsening multiorgan failure, the pressure may need to be measured hourly.18
Clinicians managing patients in the intensive care unit should think of intra-abdominal pressure alongside blood pressure, urine output, and mental status when evaluating hemodynamic status. Clinical manifestations of abdominal compartment syndrome reflect the underlying organ dysfunction and include hypotension, refractory shock, decreased urine output, intracranial hypertension, progressive hypoxemia and hypercarbia, elevated pulmonary peak pressures, and worsening of metabolic acidosis.22
Treatment. The standard treatment for primary abdominal compartment syndrome is surgical decompression. According to WSACS guidelines, insertion of a percutaneous drainage catheter should be advocated in patients with gross ascites and in whom decompressive surgery is not feasible. A damage-control resuscitation strategy used for patients undergoing damage-control laparotomy has been found to increase the 30-day survival rate.23 A damage-control resuscitation strategy consists of increasing the use of plasma and platelet transfusions over packed red cell transfusions, limiting the use of crystalloid solutions in early fluid resuscitation, and allowing for permissive hypotension.
Secondary abdominal compartment syndrome is treated conservatively in most cases, since patients with this condition are very poor surgical candidates owing to their comorbidities.18 However, in patients with progressive organ dysfunction in whom medical management has failed, surgical decompression should be considered.18 Medical management of secondary abdominal compartment syndrome depends on the underlying etiology. Strategies include nasogastric or colonic decompression, use of prokinetic agents, paracentesis in cases with gross ascites, and maintaining a cumulative negative fluid balance. The WSACS does not recommend routine use of diuretics, albumin infusion, or renal replacement strategies. Pain should be adequately controlled to improve abdominal wall compliance.18,24 Neuromuscular blockade agents may be used to aid this process. Neostigmine may be used to treat colonic pseudo-obstruction when other conservative methods fail. Use of enteral nutrition should be minimized.18
Our patient might have abdominal compartment syndrome, but a definitive diagnosis cannot be made at this point without measuring the intra-abdominal pressure.
WHICH IMAGING TEST WOULD BE BEST?
2. Which imaging test would be best for establishing the diagnosis in this patient?
- Plain abdominal radiography
- Abdominal ultrasonography
- Computed tomography of the abdomen and pelvis with contrast
- Magnetic resonance imaging of the abdomen and pelvis
Plain abdominal radiography
Plain abdominal radiography can help to determine if there is free gas under the diaphragm (due to bowel perforation), obstructed bowel, sentinel loop, volvulus, or fecoliths causing the abdominal pain. It cannot diagnose rectus sheath hematoma or acute mesenteric ischemia.
Abdominal ultrasonography
Abdominal ultrasonography can be used as the first diagnostic test, as it is widely available, safe, effective, and tolerable. It does not expose the patient to radiation or intravenous contrast agents. It helps to diagnose rectus sheath hematoma and helps to follow its maturation and resolution once a diagnosis is made. It can provide a rapid assessment of the size, location, extent, and physical characteristics of the mass.
Rectus sheath hematoma appears spindle-shaped on sagittal sections and ovoid on coronal sections. It often appears sonolucent in the early stages and sonodense in the late stage, but the appearance may be heterogeneous depending on the combined presence of clot and fresh blood. These findings are sufficient to make the diagnosis.
Abdominal ultrasonography has 85% to 96% sensitivity in diagnosing rectus sheath hematoma.25 It can help diagnose other causes of the abdominal pain, such as renal stones and cholecystitis. It is the preferred imaging test in pediatric patients, pregnant patients, and those with renal insufficiency.
Abdominal computed tomography
Abdominal computed tomography has a sensitivity and specificity of 100% for diagnosing acute rectus sheath hematoma with a duration of less than 5 days.25 It not only helps to determine the precise location and extent, but also helps to determine if there is active extravasation. Even in patients with renal insufficiency, noncontrast computed tomography will help to confirm the diagnosis, although it may not show extravasation or it may miss certain abdominal pathologies because of the lack of contrast.
Acute rectus sheath hematoma appears as a hyperdense mass posterior to the rectus abdominis muscle with ipsilateral anterolateral muscular enlargement. Chronic rectus sheath hematoma appears isodense or hypodense relative to the surrounding muscle. Above the arcuate line, rectus sheath hematoma has a spindle shape; below the arcuate line, it is typically spherical.
In 1996, Berná et al26 classified rectus sheath hematoma into three grades based on findings of computed tomography:
- Grade I is intramuscular and unilateral
- Grade II may involve bilateral rectus muscles without extension into the prevesicular space
- Grade III extends into the peritoneum and prevesicular space
Magnetic resonance imaging
Magnetic resonance imaging is useful to differentiate chronic rectus sheath hematoma (greater than 5-day duration) from an anterior abdominal wall mass. Chronic rectus sheath hematoma will have high signal intensity on both T1- and T2-weighted images up to 10 months after the onset of the hematoma.27
Back to our patient
Since our patient’s symptoms are acute and of less than 5 days’ duration, computed tomography of the abdomen and pelvis would be the best diagnostic test, with therapeutic implications if there is ongoing extravasation.
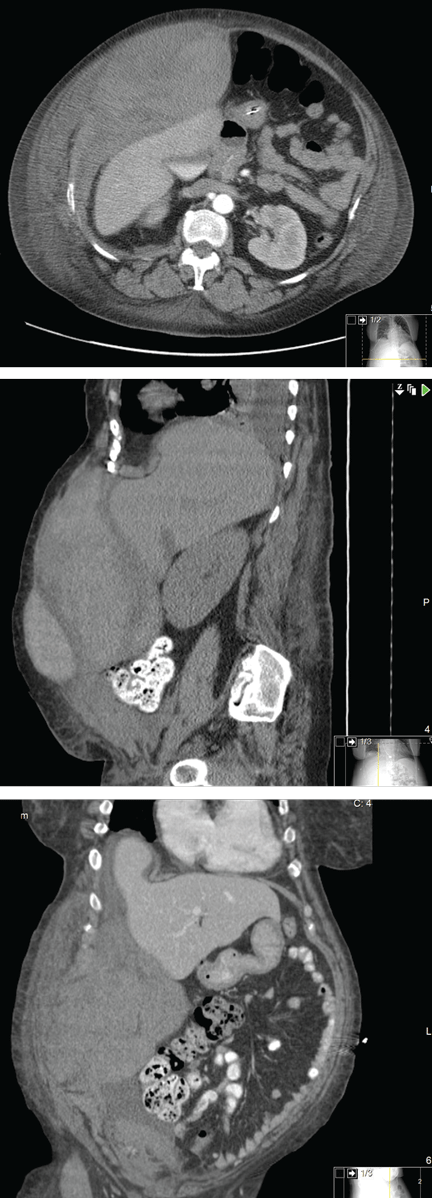
Computed tomography of the abdomen with contrast showed a new hematoma, measuring 25 by 14 by 13.5 cm, in the right rectus sheath (Figure 2), with no other findings. The hematoma was grade I, since it was intramuscular and unilateral without extension elsewhere.
Laboratory workup showed that the patient’s hematocrit was falling, from 34% to 24%, and her INR was elevated at 2.5. She was resuscitated with fluids, blood transfusion, and fresh-frozen plasma. Anticoagulation was withheld. In spite of resuscitation, her hematocrit kept falling, though she remained hemodynamically stable.
THE WAY FORWARD
3. At this point, what would be the best approach to management in this patient?
- Serial clinical examinations and frequent monitoring of the complete blood cell count
- Urgent surgical consult for exploratory laparotomy with evaluation of the hematoma and ligation of the bleeding vessel
- Repeat computed tomographic angiography to identify a possible bleeding vessel; consideration of radiographically guided arterial embolization
- Measuring the intra-abdominal pressure using the intrabladder pressure for abdominal compartment syndrome and consideration of surgical drainage
The key clinical concern in a patient with a rectus sheath hematoma who is hemodynamically stable is whether the hematoma is expanding. This patient responded to initial resuscitation, but her falling hematocrit was evidence of ongoing bleeding leading to an expanding rectus sheath hematoma. Thus, serial clinical examinations and frequent monitoring of the complete blood cell count would not be enough, as it could miss fatal ongoing bleeding.
Radiographically guided embolization with Gelfoam, thrombin, or coils should be attempted first, as this is less invasive than exploratory laparotomy.28 It can achieve hemostasis, decrease the size of the hematoma, limit the need for blood products, and prevent rupture into the abdomen. If this is unsuccessful, the next step is ligation of the bleeding vessel.29
Surgical treatment includes evacuation of the hematoma, repair of the rectus sheath, ligation of bleeding vessels, and abdominal wall closure. Surgical evacuation or guided drainage of a rectus sheath hematoma on its own is not normally indicated and may indeed cause persistent bleeding by diminishing a potential tamponade effect. However, it may become necessary if the hematoma is very large or infected, if it causes marked respiratory impairment, or if abdominal compartment syndrome is suspected.
Abdominal compartment syndrome is very rare but is associated with a 50% mortality rate.30 It should be suspected in patients with oliguria, low cardiac output, changes in minute ventilation, and altered splanchnic blood flow. The diagnosis is confirmed with indwelling catheter manometry of the bladder to measure intra-abdominal pressure. Intra-abominal pressure above 25 mm Hg should be treated with decompressive laparotomy.30 However, the clinical suspicion of abdominal compartment syndrome was low in this patient.
The best choice at this point would be urgent computed tomographic angiography to identify a bleeding vessel, along with consideration of radiographically guided arterial embolization.
TREATMENT IS USUALLY CONSERVATIVE
Treatment of rectus sheath hematoma is conservative in most hemodynamically stable patients, with embolization or surgical intervention reserved for unstable patients or those in whom the hematoma is expanding.
Knowledge of the grading system of Berná et al26 helps to assess the patient’s risk and to anticipate potential complications. Grade I hematomas are mild and do not necessitate admission. Patients with grade II hematoma can be admitted to the floor for 24 to 48 hours for observation. Grade III usually occurs in patients receiving anticoagulant therapy and frequently requires blood products. These patients have a prolonged hospital stay and more complications, including hypovolemic shock, myonecrosis, acute coronary syndrome, arrhythmias, acute renal failure, small-bowel infarction, and abdominal compartment syndrome—all of which increases the risk of morbidity and death. Thus, patients who are on anticoagulation who develop grade III rectus sheath hematoma should be admitted to the hospital, preferably to the intensive care unit, to ensure that the hematoma is not expanding and to plan reinstitution of anticoagulation as appropriate.
In most cases, rectus sheath hematomas resolve within 1 to 3 months. Resolution of large hematomas may be hastened with the use of pulsed ultrasound.31 However, this treatment should be used only after the acute phase is over, when there is evidence of an organized thrombus and coagulation measures have returned to the target range. This helps to reduce the risk of bleeding and to prevent symptoms from worsening.31
OUR PATIENT’S COURSE
Our patient underwent urgent computed tomographic angiography, which showed a modest increase in the size of the rectus sheath hematoma. However, no definitive blush of contrast was seen to suggest active arterial bleeding. Her hematocrit stabilized, and she remained hemodynamically stable without requiring additional intervention. Most likely her bleeding was self-contained. She had normal intra-abdominal pressure on serial monitoring. She was later transferred to acute inpatient rehabilitation in view of deconditioning and is currently doing well. The hematoma persisted, decreasing only slightly in size over the next 3 weeks.
- Kougias P, Lau D, El Sayed HF, Zhou W, Huynh TT, Lin PH. Determinants of mortality and treatment outcome following surgical interventions for acute mesenteric ischemia. J Vasc Surg 2007; 46:467–474.
- Sise MJ. Acute mesenteric ischemia. Surg Clin North Am 2014; 94:165–181.
- Scharff JR, Longo WE, Vartanian SM, Jacobs DL, Bahadursingh AN, Kaminski DL. Ischemic colitis: spectrum of disease and outcome. Surgery 2003; 134:624–629.
- Lange H, Jäckel R. Usefulness of plasma lactate concentration in the diagnosis of acute abdominal disease. Eur J Surg 1994; 160:381–384.
- Gearhart SL, Delaney CP, Senagore AJ, et al. Prospective assessment of the predictive value of alpha-glutathione S-transferase for intestinal ischemia. Am Surg 2003; 69:324–329.
- Kanda T, Fujii H, Tani T, et al. Intestinal fatty acid-binding protein is a useful diagnostic marker for mesenteric infarction in humans. Gastroenterology 1996; 110:339–343.
- Menke J. Diagnostic accuracy of multidetector CT in acute mesenteric ischemia: systematic review and meta-analysis. Radiology 2010; 256:93–101.
- Acosta S, Björnsson S, Ekberg O, Resch T. CT angiography followed by endovascular intervention for acute superior mesenteric artery occlusion does not increase risk of contrast-induced renal failure. Eur J Vasc Endovasc Surg 2010; 39:726–730.
- Clark RA. Computed tomography of bowel infarction. J Comput Assist Tomogr 1987; 11:757–762.
- Acosta S, Björck M. Modern treatment of acute mesenteric ischaemia. Br J Surg 2014; 101:e100–e108.
- Smithson A, Ruiz J, Perello R, Valverde M, Ramos J, Garzo L. Diagnostic and management of spontaneous rectus sheath hematoma. Eur J Intern Med 2013; 24:579–582.
- Moreno Gallego A, Aguayo JL, Flores B, et al. Ultrasonography and computed tomography reduce unnecessary surgery in abdominal rectus sheath haematoma. Br J Surg 1997; 84:1295–1297.
- Dubinsky IL. Hematoma of the rectus abdominis muscle: case report and review of the literature. J Emerg Med 1997; 15:165–167.
- Yi M, Yao G, Bai Y. The monitoring of intra-abdominal pressure in critically ill patients. (In Chinese.) Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2014; 26:175–178.
- Hunt L, Frost SA, Hillman K, Newton PJ, Davidson PM. Management of intra-abdominal hypertension and abdominal compartment syndrome: a review. J Trauma Manag Outcomes 2014; 8:2.
- Malbrain ML, Cheatham ML, Kirkpatrick A, et al. Results from the International Conference of Experts on Intra-abdominal Hypertension and Abdominal Compartment Syndrome. I. Definitions. Intensive Care Med 2006; 32:1722–1732.
- Malbrain ML, Chiumello D, Cesana BM, et al; WAKE-Up! Investigators. A systematic review and individual patient data meta-analysis on intra-abdominal hypertension in critically ill patients: the wake-up project. World initiative on Abdominal Hypertension Epidemiology, a Unifying Project (WAKE-Up!). Minerva Anestesiol 2014; 80:293–306.
- Kirkpatrick AW, Roberts DJ, De Waele J, et al; Pediatric Guidelines Sub-Committee for the World Society of the Abdominal Compartment Syndrome. Intra-abdominal hypertension and the abdominal compartment syndrome: updated consensus definitions and clinical practice guidelines from the World Society of the Abdominal Compartment Syndrome. Intensive Care Med 2013; 39:1190–1206.
- Holodinsky JK, Roberts DJ, Ball CG, et al. Risk factors for intra-abdominal hypertension and abdominal compartment syndrome among adult intensive care unit patients: a systematic review and meta-analysis. Crit Care 2013; 17:R249.
- Sugrue M, Bauman A, Jones F, et al. Clinical examination is an inaccurate predictor of intraabdominal pressure. World J Surg 2002; 26:1428–1431.
- Cheatham ML, De Waele JJ, De Laet I, et al; World Society of the Abdominal Compartment Syndrome (WSACS) Clinical Trials Working Group. The impact of body position on intra-abdominal pressure measurement: a multicenter analysis. Crit Care Med 2009; 37:2187–2190.
- Ortiz-Diaz E, Lan CK. Intra-abdominal hypertension in medical critically ill patients: a narrative review. Shock 2014; 41:175–180.
- Cotton BA, Reddy N, Hatch QM, et al. Damage control resuscitation is associated with a reduction in resuscitation volumes and improvement in survival in 390 damage control laparotomy patients. Ann Surg 2011; 254:598–605.
- An G, West MA. Abdominal compartment syndrome: a concise clinical review. Crit Care Med 2008; 36:1304–1310.
- Tolcher MC, Nitsche JF, Arendt KW, Rose CH. Spontaneous rectus sheath hematoma pregnancy: case report and review of the literature. Obstet Gynecol Surv 2010; 65:517–522.
- Berná JD, Garcia-Medina V, Guirao J, Garcia-Medina J. Rectus sheath hematoma: diagnostic classification by CT. Abdom Imaging 1996; 21:62–64.
- Unger EC, Glazer HS, Lee JK, Ling D. MRI of extracranial hematomas: preliminary observations. AJR Am J Roentgenol 1986; 146:403–407.
- Rimola J, Perendreu J, Falcó J, Fortuño JR, Massuet A, Branera J. Percutaneous arterial embolization in the management of rectus sheath hematoma. AJR Am J Roentgenol 2007; 188:W497–W502.
- Titone C, Lipsius M, Krakauer JS. “Spontaneous” hematoma of the rectus abdominis muscle: critical review of 50 cases with emphasis on early diagnosis and treatment. Surgery 1972; 72:568–572.
- Osinbowale O, Bartholomew JR. Rectus sheath hematoma. Vasc Med 2008; 13:275–279.
- Berná-Serna JD, Sánchez-Garre J, Madrigal M, Zuazu I, Berná-Mestre JD. Ultrasound therapy in rectus sheath hematoma. Phys Ther 2005; 85:352–357.
A 57-year-old woman presented to the emergency department with left lower quadrant pain, which had started 1 week earlier and was constant, dull, aching, and nonradiating. There were no aggravating or alleviating factors. The pain was associated with low-grade fever and nausea. She reported no vomiting, no change in bowel habits, and no hematemesis, hematochezia, or melena. She did not have urinary urgency, frequency, or dysuria. She had no cardiac, respiratory, or neurologic symptoms.
Her medical history included hypothyroidism, type 2 diabetes mellitus, diverticulosis, and chronic obstructive pulmonary disease. Her medications included metformin, insulin, levothyroxine, and inhaled tiotropium. She had no allergies. She had never undergone surgery, including cesarean delivery. She was postmenopausal. She had two children, both of whom had been born vaginally at full term. She denied using alcohol, tobacco, and illicit drugs. Her family history was noncontributory.
On examination, she was not in acute distress. Her temperature was 36.7°C (98.1°F), blood pressure 130/90 mm Hg, heart rate 86 beats per minute and regular, respiratory rate 16 breaths per minute, and oxygen saturation 98% on ambient air. Examination of her head and neck was unremarkable. Cardiopulmonary examination was normal. Abdominal examination revealed normal bowel sounds, mild tenderness in the left lower quadrant with localized guarding, and rebound tenderness. Neurologic examination was unremarkable.
Initial laboratory data showed mild leukocytosis. Computed tomography with contrast of the abdomen and pelvis suggested acute diverticulitis.
ATRIAL FIBRILLATION, AND THEN A TURN FOR THE WORSE
The patient was admitted with an initial diagnosis of acute diverticulitis. She was started on antibiotics, hydration, and pain medications, and her abdominal pain gradually improved.
On the third hospital day, she suddenly experienced shortness of breath and palpitations. At the time of admission her electrocardiogram had been normal, but it now showed atrial fibrillation with a rapid ventricular response. She also developed elevated troponin levels, which were thought to represent type 2 non-ST-elevation myocardial infarction.
She was started on aspirin, clopidogrel, and anticoagulation with heparin bridged with warfarin for the new-onset atrial fibrillation. Her heart rate was controlled with metoprolol, and her shortness of breath improved. An echocardiogram was normal.
On the seventh hospital day, she developed severe right-sided lower abdominal pain and bruising. Her blood pressure was 90/60 mm Hg, heart rate 110 beats per minute and irregularly irregular, respiratory rate 22 breaths per minute, and oxygen saturation 97% on room air. Her abdomen was diffusely tender with a palpable mass in the right lower quadrant and hypoactive bowel sounds. Ecchymosis was noted (Figure 1).
DIFFERENTIAL DIAGNOSIS
1. What is the likely cause of her decompensation?
- Acute mesenteric ischemia
- Perforation of the gastrointestinal tract
- Rectus sheath hematoma
- Abdominal compartment syndrome due to acute pancreatitis
Acute mesenteric ischemia
Signs and symptoms of acute mesenteric ischemia can be vague. Moreover, when it leads to bowel necrosis the mortality rate is high, ranging from 30% to 65%.1 Hence, one should suspect it and try to diagnose it early.
Most patients with this condition have comorbidities; risk factors include atherosclerotic disease, cardiac conditions (congestive heart failure, recent myocardial infarction, and atrial fibrillation), systemic illness, and inherited or acquired hypercoagulable states.2
The four major causes are:
- Acute thromboembolic occlusion of the superior mesenteric artery (the most common site of occlusion because of the acute angle of origin from the aorta)
- Acute thrombosis of the mesenteric vein
- Acute thrombosis of the mesenteric artery
- Nonocclusive disease affecting the mesenteric vessels2
Nonocclusive disease is seen in conditions in which the mesenteric vessels are already compromised due to background stenosis owing to atherosclerosis. Also, conditions such as septic and cardiogenic shock can compromise these arteries, leading to ischemia, which, if it persists, can lead to bowel infarction. Ischemic colitis falls under this category. It commonly involves the descending and sigmoid colon.3
The initial symptom of ischemia may be abdominal pain that is brought on by eating large meals (“postprandial intestinal angina.”2 When the ischemia worsens to infarction, patients may have a diffusely tender abdomen and constant pain that does not vary with palpation. Surprisingly, patients do not exhibit peritoneal signs early on. This gives rise to the description of “pain out of proportion to the physical findings” traditionally associated with acute mesenteric ischemia.2
Diagnosis. Supportive laboratory data include marked leukocytosis, elevated hematocrit due to hemoconcentration, metabolic acidosis, and elevated lactate.4 Newer markers such as serum alpha-glutathione S-transferase (alpha-GST) and intestinal fatty acid-binding protein (I-FABP) may be used to support the diagnosis.
Elevated alpha-GST has 72% sensitivity and 77% specificity in the diagnosis of acute mesenteric ischemia.5 The caveat is that it cannot reliably differentiate ischemia from infarction. Its sensitivity can be improved to 97% to 100% by using the white blood cell count and lactate levels in combination.5
An I-FABP level higher than 100 ng/mL has 100% sensitivity for diagnosing mesenteric infarction but only 25% sensitivity for bowel strangulation.6
Early use of abdominal computed tomography with contrast can aid in recognizing this diagnosis.7 Thus, it should be ordered in suspected cases, even in patients who have elevated creatinine levels (which would normally preclude the use of contrast), since early diagnosis followed by endovascular therapy is associated with survival benefit, and the risk of contrast-induced nephropathy appears to be small.8 Computed tomography helps to determine the state of mesenteric vessels and bowel perfusion before ischemia progresses to infarction. It also helps to rule out other common diagnoses. Findings that suggest acute mesenteric ischemia include segmental bowel wall thickening, intestinal pneumatosis with gas in the portal vein, bowel dilation, mesenteric stranding, portomesenteric thrombosis, and solid-organ infarction.9
Treatment. If superior mesenteric artery occlusion is diagnosed on computed tomography, the next step is to determine if there is peritonitis.10 In patients who have evidence of peritonitis, exploratory laparotomy is performed. For emboli in such patients, open embolectomy followed by on-table angiography is carried out in combination with damage-control surgery. For patients with peritonitis and acute thrombosis, stenting along with damage-control surgery is preferred.10
On the other hand, if there is no peritonitis, the thrombosis may be amenable to endovascular intervention. For patients with acute embolic occlusion with no contraindications to thrombolysis, aspiration embolectomy in combination with local catheter-directed thrombolysis with recombinant tissue plasminogen activator can be performed. This can be combined with endovascular mechanical embolectomy for more complete management.10 Patients with contraindications to thrombolysis can be treated either with aspiration and mechanical embolectomy or with open embolectomy with angiography.10
During laparotomy, the surgeon carefully inspects the bowel for signs of necrosis. Signs that bowel is still viable include pink color, bleeding from cut surfaces, good peristalsis, and visible pulsations in the arterial arcade of the mesentery.
Acute mesenteric artery thrombosis arising from chronic atherosclerotic disease can be treated with stenting of the stenotic lesion.10 Patients with this condition would also benefit from aggressive management of atherosclerotic disease with statins along with antiplatelet agents.10
Mesenteric vein thrombosis requires prompt institution of anticoagulation. However, in advanced cases leading to bowel infarction, exploratory laparotomy with resection of the necrotic bowel may be required. Anticoagulation should be continued for at least 6 months, and further therapy should be determined by the underlying precipitating condition.10
Critically ill patients who develop mesenteric ischemia secondary to persistent hypotension usually respond to adequate volume resuscitation, cessation of vasopressors, and overall improvement in their hemodynamic status. These patients must be closely monitored for development of gangrene of the bowel because they may be intubated and not able to complain. Any sudden deterioration in their condition should prompt physicians to consider bowel necrosis developing in these patients. Elevation of lactate levels out of proportion to the degree of hypotension may be corroborative evidence.4
Our patient had risk factors for acute mesenteric ischemia that included atrial fibrillation and recent non-ST-elevation myocardial infarction. She could have had arterial emboli due to atrial fibrillation, in situ superior mesenteric arterial thrombosis, or splanchnic arterial vasoconstriction due to the myocardial infarction associated with transient hypotension.
Arguing against this diagnosis, although she had a grossly distended abdomen, abdominal bruising usually is not seen. Also, a palpable mass in the right lower quadrant is uncommon except when acute mesenteric ischemia occurs due to segmental intestinal strangulation, as with strangulated hernia or volvulus. She also had therapeutic international normalized ratio (INR) levels constantly while on anticoagulation. Nevertheless, acute mesenteric ischemia should be strongly considered in the initial differential diagnosis of this patient’s acute decompensation.
Perforation of the gastrointestinal tract
Diverticulitis is the acute inflammation of one or more diverticula, which are small pouches created by herniation of the mucosa into the wall of the colon. The condition is caused by microscopic or macroscopic perforation of the diverticula. Microscopic perforation is usually self-limited (uncomplicated diverticulitis) and responds to conservative treatment, whereas macroscopic perforation can be associated with fecal or purulent peritonitis, abscess, enteric fistula, bowel obstruction, and stricture (complicated diverticulitis), in which case surgery may be necessary.
Patients with peritonitis due to free perforation present with generalized tenderness with rebound tenderness and guarding on abdominal examination. The abdomen may be distended and tympanic to percussion, with diminished or absent bowel sounds. Patients may have hemodynamic compromise.
Plain upright abdominal radiographs may show free air under the diaphragm. Computed tomography may show oral contrast outside the lumen and detect even small amounts of free intraperitoneal air (more clearly seen on a lung window setting).
Our patient initially presented with acute diverticulitis. She later developed diffuse abdominal tenderness with hypoactive bowel sounds. Bowel perforation is certainly a possibility at this stage, though it is usually not associated with abdominal bruising. She would need additional imaging to rule out this complication.
Other differential diagnoses to be considered in this patient with right lower-quadrant pain include acute appendicitis, incarcerated inguinal hernia, volvulus (particularly cecal volvulus), small-bowel obstruction, pyelonephritis, and gynecologic causes such as adnexal torsion, ruptured ovarian cyst, and tubo-ovarian abscess. Computed tomography helps to differentiate most of these causes.
Rectus sheath hematoma
Rectus sheath hematoma is relatively uncommon and often not considered in the initial differential diagnosis of an acute abdomen. This gives it the rightful term “a great masquerader.” It usually results from bleeding into the rectus sheath from damage to the superior (more common) or inferior epigastric arteries and occasionally from a direct tear of the rectus abdominis muscle. Predisposing factors include anticoagulant therapy (most common), advanced age, hypertension, previous abdominal surgery, trauma, paroxysmal coughing, medication injections, pregnancy, blood dyscrasias, severe vomiting, violent physical activity, and leukemia.11
Clinical manifestations include acute abdominal pain, often associated with fever, nausea, and vomiting. Physical examination may reveal signs of hypovolemic shock, a palpable nonpulsatile abdominal mass, and signs of local peritoneal irritation. The Carnett sign11 (tenderness within the abdominal wall that persists and does not improve with raising the head) and the Fothergill sign11 (a tender abdominal mass that does not cross the midline and remains palpable with tensing of the rectus sheath) may be elicited.
Computed tomography is more sensitive than abdominal ultrasonography in differentiating rectus sheath hematoma from an intra-abdominal pathology.11 In addition, computed tomography also helps to determine if the bleeding is active or not, which has therapeutic implications.
In our patient, rectus sheath hematoma is a possibility because of her ongoing anticoagulation, findings of localized abdominal bruising, and palpable right lower-quadrant mass, and it is high on the list of differential diagnoses. Rectus sheath hematoma should be considered in the differential diagnosis of lower abdominal pain particularly in elderly women who are on anticoagulation and in whom the onset of pain coincides with a paroxysm of cough.12 Women are twice as likely as men to develop rectus sheath hematoma, owing to their different muscle mass.13 In addition, anterior abdominal wall muscles are stretched during pregnancy.13
Abdominal compartment syndrome
Abdominal compartment syndrome has been classically associated with surgical patients. However, it is being increasingly recognized in critically ill medical patients, in whom detecting and treating it early may result in significant reduction in rates of morbidity and death.14
Abdominal compartment syndrome is of three types: primary, secondary, and recurrent. Primary abdominal compartment syndrome refers to the classic surgical patients with evidence of direct injury to the abdominal or pelvic organs through major trauma or extensive abdominal surgeries. Secondary abdominal compartment syndrome refers to its development in critically ill intensive care patients in whom the pathology does not directly involve the abdominal or pelvic organs.
Various medical conditions can culminate in abdominal compartment syndrome and result in multiorgan failure. Recurrent abdominal compartment syndrome refers to its development after management of either primary or secondary intra-abdominal hypertension or abdominal compartment syndrome.15 Clinicians thus must be aware of secondary and recurrent abdominal compartment syndrome occurring in critically ill patients.
The normal intra-abdominal pressure is around 5 to 7 mm Hg, even in most critically ill patients. Persistent elevation, ie, higher than 12 mm Hg, is referred to as intra-abdominal hypertension.16–18 Intra-abdominal hypertension is subdivided into four grades:
- Grade I: 12–15 mm Hg
- Grade II: 16–20 mm Hg
- Grade III: 21–25 mm Hg
- Grade IV: > 25 mm Hg.
The World Society of the Abdominal Compartment Syndrome (WSACS) defines abdominal compartment syndrome as pressure higher than 20 mm Hg along with organ damage.18 It may or may not be associated with an abdominal perfusion pressure less than 60 mm Hg.18
Risk factors associated with abdominal compartment syndrome include conditions causing decreased gut motility (gastroparesis, ileus, and colonic pseudo-obstruction), intra-abdominal or retroperitoneal masses or abscesses, ascites, hemoperitoneum, acute pancreatitis, third-spacing due to massive fluid resuscitation with transfusions, peritoneal dialysis, and shock.18,19
Physical examination has a sensitivity of only 40% to 60% in detecting intra-abdominal hypertension.20 The gold-standard method of measuring the intra-abdominal pressure is the modified Kron technique,18 using a Foley catheter in the bladder connected to a pressure transducer. With the patient in the supine position, the transducer is zeroed at the mid-axillary line at the level of the iliac crest, and 25 mL of normal saline is instilled into the bladder and maintained for 30 to 60 seconds to let the detrusor muscle relax.15 Pressure tracings are then recorded at the end of expiration. Factors that are known to affect the transbladder pressure include patient position, respiratory movement, and body mass index, and should be taken into account when reading the pressure recordings.15,21 Other techniques that can be used include intragastric, intra-inferior vena cava, and intrarectal approaches.15
The WSACS recommends that any patient admitted to a critical care unit or in whom new organ failure develops should be screened for risk factors for intra-abdominal hypertension and abdominal compartment syndrome. If a patient has at least two of the risk factors suggested by WSACS, a baseline intra-abdominal pressure measurement should be obtained. Patients at risk for intra-abdominal hypertension should have the intra-abdominal pressure measured every 4 to 6 hours. However, in the face of hemodynamic instability and worsening multiorgan failure, the pressure may need to be measured hourly.18
Clinicians managing patients in the intensive care unit should think of intra-abdominal pressure alongside blood pressure, urine output, and mental status when evaluating hemodynamic status. Clinical manifestations of abdominal compartment syndrome reflect the underlying organ dysfunction and include hypotension, refractory shock, decreased urine output, intracranial hypertension, progressive hypoxemia and hypercarbia, elevated pulmonary peak pressures, and worsening of metabolic acidosis.22
Treatment. The standard treatment for primary abdominal compartment syndrome is surgical decompression. According to WSACS guidelines, insertion of a percutaneous drainage catheter should be advocated in patients with gross ascites and in whom decompressive surgery is not feasible. A damage-control resuscitation strategy used for patients undergoing damage-control laparotomy has been found to increase the 30-day survival rate.23 A damage-control resuscitation strategy consists of increasing the use of plasma and platelet transfusions over packed red cell transfusions, limiting the use of crystalloid solutions in early fluid resuscitation, and allowing for permissive hypotension.
Secondary abdominal compartment syndrome is treated conservatively in most cases, since patients with this condition are very poor surgical candidates owing to their comorbidities.18 However, in patients with progressive organ dysfunction in whom medical management has failed, surgical decompression should be considered.18 Medical management of secondary abdominal compartment syndrome depends on the underlying etiology. Strategies include nasogastric or colonic decompression, use of prokinetic agents, paracentesis in cases with gross ascites, and maintaining a cumulative negative fluid balance. The WSACS does not recommend routine use of diuretics, albumin infusion, or renal replacement strategies. Pain should be adequately controlled to improve abdominal wall compliance.18,24 Neuromuscular blockade agents may be used to aid this process. Neostigmine may be used to treat colonic pseudo-obstruction when other conservative methods fail. Use of enteral nutrition should be minimized.18
Our patient might have abdominal compartment syndrome, but a definitive diagnosis cannot be made at this point without measuring the intra-abdominal pressure.
WHICH IMAGING TEST WOULD BE BEST?
2. Which imaging test would be best for establishing the diagnosis in this patient?
- Plain abdominal radiography
- Abdominal ultrasonography
- Computed tomography of the abdomen and pelvis with contrast
- Magnetic resonance imaging of the abdomen and pelvis
Plain abdominal radiography
Plain abdominal radiography can help to determine if there is free gas under the diaphragm (due to bowel perforation), obstructed bowel, sentinel loop, volvulus, or fecoliths causing the abdominal pain. It cannot diagnose rectus sheath hematoma or acute mesenteric ischemia.
Abdominal ultrasonography
Abdominal ultrasonography can be used as the first diagnostic test, as it is widely available, safe, effective, and tolerable. It does not expose the patient to radiation or intravenous contrast agents. It helps to diagnose rectus sheath hematoma and helps to follow its maturation and resolution once a diagnosis is made. It can provide a rapid assessment of the size, location, extent, and physical characteristics of the mass.
Rectus sheath hematoma appears spindle-shaped on sagittal sections and ovoid on coronal sections. It often appears sonolucent in the early stages and sonodense in the late stage, but the appearance may be heterogeneous depending on the combined presence of clot and fresh blood. These findings are sufficient to make the diagnosis.
Abdominal ultrasonography has 85% to 96% sensitivity in diagnosing rectus sheath hematoma.25 It can help diagnose other causes of the abdominal pain, such as renal stones and cholecystitis. It is the preferred imaging test in pediatric patients, pregnant patients, and those with renal insufficiency.
Abdominal computed tomography
Abdominal computed tomography has a sensitivity and specificity of 100% for diagnosing acute rectus sheath hematoma with a duration of less than 5 days.25 It not only helps to determine the precise location and extent, but also helps to determine if there is active extravasation. Even in patients with renal insufficiency, noncontrast computed tomography will help to confirm the diagnosis, although it may not show extravasation or it may miss certain abdominal pathologies because of the lack of contrast.
Acute rectus sheath hematoma appears as a hyperdense mass posterior to the rectus abdominis muscle with ipsilateral anterolateral muscular enlargement. Chronic rectus sheath hematoma appears isodense or hypodense relative to the surrounding muscle. Above the arcuate line, rectus sheath hematoma has a spindle shape; below the arcuate line, it is typically spherical.
In 1996, Berná et al26 classified rectus sheath hematoma into three grades based on findings of computed tomography:
- Grade I is intramuscular and unilateral
- Grade II may involve bilateral rectus muscles without extension into the prevesicular space
- Grade III extends into the peritoneum and prevesicular space
Magnetic resonance imaging
Magnetic resonance imaging is useful to differentiate chronic rectus sheath hematoma (greater than 5-day duration) from an anterior abdominal wall mass. Chronic rectus sheath hematoma will have high signal intensity on both T1- and T2-weighted images up to 10 months after the onset of the hematoma.27
Back to our patient
Since our patient’s symptoms are acute and of less than 5 days’ duration, computed tomography of the abdomen and pelvis would be the best diagnostic test, with therapeutic implications if there is ongoing extravasation.
Computed tomography of the abdomen with contrast showed a new hematoma, measuring 25 by 14 by 13.5 cm, in the right rectus sheath (Figure 2), with no other findings. The hematoma was grade I, since it was intramuscular and unilateral without extension elsewhere.
Laboratory workup showed that the patient’s hematocrit was falling, from 34% to 24%, and her INR was elevated at 2.5. She was resuscitated with fluids, blood transfusion, and fresh-frozen plasma. Anticoagulation was withheld. In spite of resuscitation, her hematocrit kept falling, though she remained hemodynamically stable.
THE WAY FORWARD
3. At this point, what would be the best approach to management in this patient?
- Serial clinical examinations and frequent monitoring of the complete blood cell count
- Urgent surgical consult for exploratory laparotomy with evaluation of the hematoma and ligation of the bleeding vessel
- Repeat computed tomographic angiography to identify a possible bleeding vessel; consideration of radiographically guided arterial embolization
- Measuring the intra-abdominal pressure using the intrabladder pressure for abdominal compartment syndrome and consideration of surgical drainage
The key clinical concern in a patient with a rectus sheath hematoma who is hemodynamically stable is whether the hematoma is expanding. This patient responded to initial resuscitation, but her falling hematocrit was evidence of ongoing bleeding leading to an expanding rectus sheath hematoma. Thus, serial clinical examinations and frequent monitoring of the complete blood cell count would not be enough, as it could miss fatal ongoing bleeding.
Radiographically guided embolization with Gelfoam, thrombin, or coils should be attempted first, as this is less invasive than exploratory laparotomy.28 It can achieve hemostasis, decrease the size of the hematoma, limit the need for blood products, and prevent rupture into the abdomen. If this is unsuccessful, the next step is ligation of the bleeding vessel.29
Surgical treatment includes evacuation of the hematoma, repair of the rectus sheath, ligation of bleeding vessels, and abdominal wall closure. Surgical evacuation or guided drainage of a rectus sheath hematoma on its own is not normally indicated and may indeed cause persistent bleeding by diminishing a potential tamponade effect. However, it may become necessary if the hematoma is very large or infected, if it causes marked respiratory impairment, or if abdominal compartment syndrome is suspected.
Abdominal compartment syndrome is very rare but is associated with a 50% mortality rate.30 It should be suspected in patients with oliguria, low cardiac output, changes in minute ventilation, and altered splanchnic blood flow. The diagnosis is confirmed with indwelling catheter manometry of the bladder to measure intra-abdominal pressure. Intra-abominal pressure above 25 mm Hg should be treated with decompressive laparotomy.30 However, the clinical suspicion of abdominal compartment syndrome was low in this patient.
The best choice at this point would be urgent computed tomographic angiography to identify a bleeding vessel, along with consideration of radiographically guided arterial embolization.
TREATMENT IS USUALLY CONSERVATIVE
Treatment of rectus sheath hematoma is conservative in most hemodynamically stable patients, with embolization or surgical intervention reserved for unstable patients or those in whom the hematoma is expanding.
Knowledge of the grading system of Berná et al26 helps to assess the patient’s risk and to anticipate potential complications. Grade I hematomas are mild and do not necessitate admission. Patients with grade II hematoma can be admitted to the floor for 24 to 48 hours for observation. Grade III usually occurs in patients receiving anticoagulant therapy and frequently requires blood products. These patients have a prolonged hospital stay and more complications, including hypovolemic shock, myonecrosis, acute coronary syndrome, arrhythmias, acute renal failure, small-bowel infarction, and abdominal compartment syndrome—all of which increases the risk of morbidity and death. Thus, patients who are on anticoagulation who develop grade III rectus sheath hematoma should be admitted to the hospital, preferably to the intensive care unit, to ensure that the hematoma is not expanding and to plan reinstitution of anticoagulation as appropriate.
In most cases, rectus sheath hematomas resolve within 1 to 3 months. Resolution of large hematomas may be hastened with the use of pulsed ultrasound.31 However, this treatment should be used only after the acute phase is over, when there is evidence of an organized thrombus and coagulation measures have returned to the target range. This helps to reduce the risk of bleeding and to prevent symptoms from worsening.31
OUR PATIENT’S COURSE
Our patient underwent urgent computed tomographic angiography, which showed a modest increase in the size of the rectus sheath hematoma. However, no definitive blush of contrast was seen to suggest active arterial bleeding. Her hematocrit stabilized, and she remained hemodynamically stable without requiring additional intervention. Most likely her bleeding was self-contained. She had normal intra-abdominal pressure on serial monitoring. She was later transferred to acute inpatient rehabilitation in view of deconditioning and is currently doing well. The hematoma persisted, decreasing only slightly in size over the next 3 weeks.
A 57-year-old woman presented to the emergency department with left lower quadrant pain, which had started 1 week earlier and was constant, dull, aching, and nonradiating. There were no aggravating or alleviating factors. The pain was associated with low-grade fever and nausea. She reported no vomiting, no change in bowel habits, and no hematemesis, hematochezia, or melena. She did not have urinary urgency, frequency, or dysuria. She had no cardiac, respiratory, or neurologic symptoms.
Her medical history included hypothyroidism, type 2 diabetes mellitus, diverticulosis, and chronic obstructive pulmonary disease. Her medications included metformin, insulin, levothyroxine, and inhaled tiotropium. She had no allergies. She had never undergone surgery, including cesarean delivery. She was postmenopausal. She had two children, both of whom had been born vaginally at full term. She denied using alcohol, tobacco, and illicit drugs. Her family history was noncontributory.
On examination, she was not in acute distress. Her temperature was 36.7°C (98.1°F), blood pressure 130/90 mm Hg, heart rate 86 beats per minute and regular, respiratory rate 16 breaths per minute, and oxygen saturation 98% on ambient air. Examination of her head and neck was unremarkable. Cardiopulmonary examination was normal. Abdominal examination revealed normal bowel sounds, mild tenderness in the left lower quadrant with localized guarding, and rebound tenderness. Neurologic examination was unremarkable.
Initial laboratory data showed mild leukocytosis. Computed tomography with contrast of the abdomen and pelvis suggested acute diverticulitis.
ATRIAL FIBRILLATION, AND THEN A TURN FOR THE WORSE
The patient was admitted with an initial diagnosis of acute diverticulitis. She was started on antibiotics, hydration, and pain medications, and her abdominal pain gradually improved.
On the third hospital day, she suddenly experienced shortness of breath and palpitations. At the time of admission her electrocardiogram had been normal, but it now showed atrial fibrillation with a rapid ventricular response. She also developed elevated troponin levels, which were thought to represent type 2 non-ST-elevation myocardial infarction.
She was started on aspirin, clopidogrel, and anticoagulation with heparin bridged with warfarin for the new-onset atrial fibrillation. Her heart rate was controlled with metoprolol, and her shortness of breath improved. An echocardiogram was normal.
On the seventh hospital day, she developed severe right-sided lower abdominal pain and bruising. Her blood pressure was 90/60 mm Hg, heart rate 110 beats per minute and irregularly irregular, respiratory rate 22 breaths per minute, and oxygen saturation 97% on room air. Her abdomen was diffusely tender with a palpable mass in the right lower quadrant and hypoactive bowel sounds. Ecchymosis was noted (Figure 1).
DIFFERENTIAL DIAGNOSIS
1. What is the likely cause of her decompensation?
- Acute mesenteric ischemia
- Perforation of the gastrointestinal tract
- Rectus sheath hematoma
- Abdominal compartment syndrome due to acute pancreatitis
Acute mesenteric ischemia
Signs and symptoms of acute mesenteric ischemia can be vague. Moreover, when it leads to bowel necrosis the mortality rate is high, ranging from 30% to 65%.1 Hence, one should suspect it and try to diagnose it early.
Most patients with this condition have comorbidities; risk factors include atherosclerotic disease, cardiac conditions (congestive heart failure, recent myocardial infarction, and atrial fibrillation), systemic illness, and inherited or acquired hypercoagulable states.2
The four major causes are:
- Acute thromboembolic occlusion of the superior mesenteric artery (the most common site of occlusion because of the acute angle of origin from the aorta)
- Acute thrombosis of the mesenteric vein
- Acute thrombosis of the mesenteric artery
- Nonocclusive disease affecting the mesenteric vessels2
Nonocclusive disease is seen in conditions in which the mesenteric vessels are already compromised due to background stenosis owing to atherosclerosis. Also, conditions such as septic and cardiogenic shock can compromise these arteries, leading to ischemia, which, if it persists, can lead to bowel infarction. Ischemic colitis falls under this category. It commonly involves the descending and sigmoid colon.3
The initial symptom of ischemia may be abdominal pain that is brought on by eating large meals (“postprandial intestinal angina.”2 When the ischemia worsens to infarction, patients may have a diffusely tender abdomen and constant pain that does not vary with palpation. Surprisingly, patients do not exhibit peritoneal signs early on. This gives rise to the description of “pain out of proportion to the physical findings” traditionally associated with acute mesenteric ischemia.2
Diagnosis. Supportive laboratory data include marked leukocytosis, elevated hematocrit due to hemoconcentration, metabolic acidosis, and elevated lactate.4 Newer markers such as serum alpha-glutathione S-transferase (alpha-GST) and intestinal fatty acid-binding protein (I-FABP) may be used to support the diagnosis.
Elevated alpha-GST has 72% sensitivity and 77% specificity in the diagnosis of acute mesenteric ischemia.5 The caveat is that it cannot reliably differentiate ischemia from infarction. Its sensitivity can be improved to 97% to 100% by using the white blood cell count and lactate levels in combination.5
An I-FABP level higher than 100 ng/mL has 100% sensitivity for diagnosing mesenteric infarction but only 25% sensitivity for bowel strangulation.6
Early use of abdominal computed tomography with contrast can aid in recognizing this diagnosis.7 Thus, it should be ordered in suspected cases, even in patients who have elevated creatinine levels (which would normally preclude the use of contrast), since early diagnosis followed by endovascular therapy is associated with survival benefit, and the risk of contrast-induced nephropathy appears to be small.8 Computed tomography helps to determine the state of mesenteric vessels and bowel perfusion before ischemia progresses to infarction. It also helps to rule out other common diagnoses. Findings that suggest acute mesenteric ischemia include segmental bowel wall thickening, intestinal pneumatosis with gas in the portal vein, bowel dilation, mesenteric stranding, portomesenteric thrombosis, and solid-organ infarction.9
Treatment. If superior mesenteric artery occlusion is diagnosed on computed tomography, the next step is to determine if there is peritonitis.10 In patients who have evidence of peritonitis, exploratory laparotomy is performed. For emboli in such patients, open embolectomy followed by on-table angiography is carried out in combination with damage-control surgery. For patients with peritonitis and acute thrombosis, stenting along with damage-control surgery is preferred.10
On the other hand, if there is no peritonitis, the thrombosis may be amenable to endovascular intervention. For patients with acute embolic occlusion with no contraindications to thrombolysis, aspiration embolectomy in combination with local catheter-directed thrombolysis with recombinant tissue plasminogen activator can be performed. This can be combined with endovascular mechanical embolectomy for more complete management.10 Patients with contraindications to thrombolysis can be treated either with aspiration and mechanical embolectomy or with open embolectomy with angiography.10
During laparotomy, the surgeon carefully inspects the bowel for signs of necrosis. Signs that bowel is still viable include pink color, bleeding from cut surfaces, good peristalsis, and visible pulsations in the arterial arcade of the mesentery.
Acute mesenteric artery thrombosis arising from chronic atherosclerotic disease can be treated with stenting of the stenotic lesion.10 Patients with this condition would also benefit from aggressive management of atherosclerotic disease with statins along with antiplatelet agents.10
Mesenteric vein thrombosis requires prompt institution of anticoagulation. However, in advanced cases leading to bowel infarction, exploratory laparotomy with resection of the necrotic bowel may be required. Anticoagulation should be continued for at least 6 months, and further therapy should be determined by the underlying precipitating condition.10
Critically ill patients who develop mesenteric ischemia secondary to persistent hypotension usually respond to adequate volume resuscitation, cessation of vasopressors, and overall improvement in their hemodynamic status. These patients must be closely monitored for development of gangrene of the bowel because they may be intubated and not able to complain. Any sudden deterioration in their condition should prompt physicians to consider bowel necrosis developing in these patients. Elevation of lactate levels out of proportion to the degree of hypotension may be corroborative evidence.4
Our patient had risk factors for acute mesenteric ischemia that included atrial fibrillation and recent non-ST-elevation myocardial infarction. She could have had arterial emboli due to atrial fibrillation, in situ superior mesenteric arterial thrombosis, or splanchnic arterial vasoconstriction due to the myocardial infarction associated with transient hypotension.
Arguing against this diagnosis, although she had a grossly distended abdomen, abdominal bruising usually is not seen. Also, a palpable mass in the right lower quadrant is uncommon except when acute mesenteric ischemia occurs due to segmental intestinal strangulation, as with strangulated hernia or volvulus. She also had therapeutic international normalized ratio (INR) levels constantly while on anticoagulation. Nevertheless, acute mesenteric ischemia should be strongly considered in the initial differential diagnosis of this patient’s acute decompensation.
Perforation of the gastrointestinal tract
Diverticulitis is the acute inflammation of one or more diverticula, which are small pouches created by herniation of the mucosa into the wall of the colon. The condition is caused by microscopic or macroscopic perforation of the diverticula. Microscopic perforation is usually self-limited (uncomplicated diverticulitis) and responds to conservative treatment, whereas macroscopic perforation can be associated with fecal or purulent peritonitis, abscess, enteric fistula, bowel obstruction, and stricture (complicated diverticulitis), in which case surgery may be necessary.
Patients with peritonitis due to free perforation present with generalized tenderness with rebound tenderness and guarding on abdominal examination. The abdomen may be distended and tympanic to percussion, with diminished or absent bowel sounds. Patients may have hemodynamic compromise.
Plain upright abdominal radiographs may show free air under the diaphragm. Computed tomography may show oral contrast outside the lumen and detect even small amounts of free intraperitoneal air (more clearly seen on a lung window setting).
Our patient initially presented with acute diverticulitis. She later developed diffuse abdominal tenderness with hypoactive bowel sounds. Bowel perforation is certainly a possibility at this stage, though it is usually not associated with abdominal bruising. She would need additional imaging to rule out this complication.
Other differential diagnoses to be considered in this patient with right lower-quadrant pain include acute appendicitis, incarcerated inguinal hernia, volvulus (particularly cecal volvulus), small-bowel obstruction, pyelonephritis, and gynecologic causes such as adnexal torsion, ruptured ovarian cyst, and tubo-ovarian abscess. Computed tomography helps to differentiate most of these causes.
Rectus sheath hematoma
Rectus sheath hematoma is relatively uncommon and often not considered in the initial differential diagnosis of an acute abdomen. This gives it the rightful term “a great masquerader.” It usually results from bleeding into the rectus sheath from damage to the superior (more common) or inferior epigastric arteries and occasionally from a direct tear of the rectus abdominis muscle. Predisposing factors include anticoagulant therapy (most common), advanced age, hypertension, previous abdominal surgery, trauma, paroxysmal coughing, medication injections, pregnancy, blood dyscrasias, severe vomiting, violent physical activity, and leukemia.11
Clinical manifestations include acute abdominal pain, often associated with fever, nausea, and vomiting. Physical examination may reveal signs of hypovolemic shock, a palpable nonpulsatile abdominal mass, and signs of local peritoneal irritation. The Carnett sign11 (tenderness within the abdominal wall that persists and does not improve with raising the head) and the Fothergill sign11 (a tender abdominal mass that does not cross the midline and remains palpable with tensing of the rectus sheath) may be elicited.
Computed tomography is more sensitive than abdominal ultrasonography in differentiating rectus sheath hematoma from an intra-abdominal pathology.11 In addition, computed tomography also helps to determine if the bleeding is active or not, which has therapeutic implications.
In our patient, rectus sheath hematoma is a possibility because of her ongoing anticoagulation, findings of localized abdominal bruising, and palpable right lower-quadrant mass, and it is high on the list of differential diagnoses. Rectus sheath hematoma should be considered in the differential diagnosis of lower abdominal pain particularly in elderly women who are on anticoagulation and in whom the onset of pain coincides with a paroxysm of cough.12 Women are twice as likely as men to develop rectus sheath hematoma, owing to their different muscle mass.13 In addition, anterior abdominal wall muscles are stretched during pregnancy.13
Abdominal compartment syndrome
Abdominal compartment syndrome has been classically associated with surgical patients. However, it is being increasingly recognized in critically ill medical patients, in whom detecting and treating it early may result in significant reduction in rates of morbidity and death.14
Abdominal compartment syndrome is of three types: primary, secondary, and recurrent. Primary abdominal compartment syndrome refers to the classic surgical patients with evidence of direct injury to the abdominal or pelvic organs through major trauma or extensive abdominal surgeries. Secondary abdominal compartment syndrome refers to its development in critically ill intensive care patients in whom the pathology does not directly involve the abdominal or pelvic organs.
Various medical conditions can culminate in abdominal compartment syndrome and result in multiorgan failure. Recurrent abdominal compartment syndrome refers to its development after management of either primary or secondary intra-abdominal hypertension or abdominal compartment syndrome.15 Clinicians thus must be aware of secondary and recurrent abdominal compartment syndrome occurring in critically ill patients.
The normal intra-abdominal pressure is around 5 to 7 mm Hg, even in most critically ill patients. Persistent elevation, ie, higher than 12 mm Hg, is referred to as intra-abdominal hypertension.16–18 Intra-abdominal hypertension is subdivided into four grades:
- Grade I: 12–15 mm Hg
- Grade II: 16–20 mm Hg
- Grade III: 21–25 mm Hg
- Grade IV: > 25 mm Hg.
The World Society of the Abdominal Compartment Syndrome (WSACS) defines abdominal compartment syndrome as pressure higher than 20 mm Hg along with organ damage.18 It may or may not be associated with an abdominal perfusion pressure less than 60 mm Hg.18
Risk factors associated with abdominal compartment syndrome include conditions causing decreased gut motility (gastroparesis, ileus, and colonic pseudo-obstruction), intra-abdominal or retroperitoneal masses or abscesses, ascites, hemoperitoneum, acute pancreatitis, third-spacing due to massive fluid resuscitation with transfusions, peritoneal dialysis, and shock.18,19
Physical examination has a sensitivity of only 40% to 60% in detecting intra-abdominal hypertension.20 The gold-standard method of measuring the intra-abdominal pressure is the modified Kron technique,18 using a Foley catheter in the bladder connected to a pressure transducer. With the patient in the supine position, the transducer is zeroed at the mid-axillary line at the level of the iliac crest, and 25 mL of normal saline is instilled into the bladder and maintained for 30 to 60 seconds to let the detrusor muscle relax.15 Pressure tracings are then recorded at the end of expiration. Factors that are known to affect the transbladder pressure include patient position, respiratory movement, and body mass index, and should be taken into account when reading the pressure recordings.15,21 Other techniques that can be used include intragastric, intra-inferior vena cava, and intrarectal approaches.15
The WSACS recommends that any patient admitted to a critical care unit or in whom new organ failure develops should be screened for risk factors for intra-abdominal hypertension and abdominal compartment syndrome. If a patient has at least two of the risk factors suggested by WSACS, a baseline intra-abdominal pressure measurement should be obtained. Patients at risk for intra-abdominal hypertension should have the intra-abdominal pressure measured every 4 to 6 hours. However, in the face of hemodynamic instability and worsening multiorgan failure, the pressure may need to be measured hourly.18
Clinicians managing patients in the intensive care unit should think of intra-abdominal pressure alongside blood pressure, urine output, and mental status when evaluating hemodynamic status. Clinical manifestations of abdominal compartment syndrome reflect the underlying organ dysfunction and include hypotension, refractory shock, decreased urine output, intracranial hypertension, progressive hypoxemia and hypercarbia, elevated pulmonary peak pressures, and worsening of metabolic acidosis.22
Treatment. The standard treatment for primary abdominal compartment syndrome is surgical decompression. According to WSACS guidelines, insertion of a percutaneous drainage catheter should be advocated in patients with gross ascites and in whom decompressive surgery is not feasible. A damage-control resuscitation strategy used for patients undergoing damage-control laparotomy has been found to increase the 30-day survival rate.23 A damage-control resuscitation strategy consists of increasing the use of plasma and platelet transfusions over packed red cell transfusions, limiting the use of crystalloid solutions in early fluid resuscitation, and allowing for permissive hypotension.
Secondary abdominal compartment syndrome is treated conservatively in most cases, since patients with this condition are very poor surgical candidates owing to their comorbidities.18 However, in patients with progressive organ dysfunction in whom medical management has failed, surgical decompression should be considered.18 Medical management of secondary abdominal compartment syndrome depends on the underlying etiology. Strategies include nasogastric or colonic decompression, use of prokinetic agents, paracentesis in cases with gross ascites, and maintaining a cumulative negative fluid balance. The WSACS does not recommend routine use of diuretics, albumin infusion, or renal replacement strategies. Pain should be adequately controlled to improve abdominal wall compliance.18,24 Neuromuscular blockade agents may be used to aid this process. Neostigmine may be used to treat colonic pseudo-obstruction when other conservative methods fail. Use of enteral nutrition should be minimized.18
Our patient might have abdominal compartment syndrome, but a definitive diagnosis cannot be made at this point without measuring the intra-abdominal pressure.
WHICH IMAGING TEST WOULD BE BEST?
2. Which imaging test would be best for establishing the diagnosis in this patient?
- Plain abdominal radiography
- Abdominal ultrasonography
- Computed tomography of the abdomen and pelvis with contrast
- Magnetic resonance imaging of the abdomen and pelvis
Plain abdominal radiography
Plain abdominal radiography can help to determine if there is free gas under the diaphragm (due to bowel perforation), obstructed bowel, sentinel loop, volvulus, or fecoliths causing the abdominal pain. It cannot diagnose rectus sheath hematoma or acute mesenteric ischemia.
Abdominal ultrasonography
Abdominal ultrasonography can be used as the first diagnostic test, as it is widely available, safe, effective, and tolerable. It does not expose the patient to radiation or intravenous contrast agents. It helps to diagnose rectus sheath hematoma and helps to follow its maturation and resolution once a diagnosis is made. It can provide a rapid assessment of the size, location, extent, and physical characteristics of the mass.
Rectus sheath hematoma appears spindle-shaped on sagittal sections and ovoid on coronal sections. It often appears sonolucent in the early stages and sonodense in the late stage, but the appearance may be heterogeneous depending on the combined presence of clot and fresh blood. These findings are sufficient to make the diagnosis.
Abdominal ultrasonography has 85% to 96% sensitivity in diagnosing rectus sheath hematoma.25 It can help diagnose other causes of the abdominal pain, such as renal stones and cholecystitis. It is the preferred imaging test in pediatric patients, pregnant patients, and those with renal insufficiency.
Abdominal computed tomography
Abdominal computed tomography has a sensitivity and specificity of 100% for diagnosing acute rectus sheath hematoma with a duration of less than 5 days.25 It not only helps to determine the precise location and extent, but also helps to determine if there is active extravasation. Even in patients with renal insufficiency, noncontrast computed tomography will help to confirm the diagnosis, although it may not show extravasation or it may miss certain abdominal pathologies because of the lack of contrast.
Acute rectus sheath hematoma appears as a hyperdense mass posterior to the rectus abdominis muscle with ipsilateral anterolateral muscular enlargement. Chronic rectus sheath hematoma appears isodense or hypodense relative to the surrounding muscle. Above the arcuate line, rectus sheath hematoma has a spindle shape; below the arcuate line, it is typically spherical.
In 1996, Berná et al26 classified rectus sheath hematoma into three grades based on findings of computed tomography:
- Grade I is intramuscular and unilateral
- Grade II may involve bilateral rectus muscles without extension into the prevesicular space
- Grade III extends into the peritoneum and prevesicular space
Magnetic resonance imaging
Magnetic resonance imaging is useful to differentiate chronic rectus sheath hematoma (greater than 5-day duration) from an anterior abdominal wall mass. Chronic rectus sheath hematoma will have high signal intensity on both T1- and T2-weighted images up to 10 months after the onset of the hematoma.27
Back to our patient
Since our patient’s symptoms are acute and of less than 5 days’ duration, computed tomography of the abdomen and pelvis would be the best diagnostic test, with therapeutic implications if there is ongoing extravasation.
Computed tomography of the abdomen with contrast showed a new hematoma, measuring 25 by 14 by 13.5 cm, in the right rectus sheath (Figure 2), with no other findings. The hematoma was grade I, since it was intramuscular and unilateral without extension elsewhere.
Laboratory workup showed that the patient’s hematocrit was falling, from 34% to 24%, and her INR was elevated at 2.5. She was resuscitated with fluids, blood transfusion, and fresh-frozen plasma. Anticoagulation was withheld. In spite of resuscitation, her hematocrit kept falling, though she remained hemodynamically stable.
THE WAY FORWARD
3. At this point, what would be the best approach to management in this patient?
- Serial clinical examinations and frequent monitoring of the complete blood cell count
- Urgent surgical consult for exploratory laparotomy with evaluation of the hematoma and ligation of the bleeding vessel
- Repeat computed tomographic angiography to identify a possible bleeding vessel; consideration of radiographically guided arterial embolization
- Measuring the intra-abdominal pressure using the intrabladder pressure for abdominal compartment syndrome and consideration of surgical drainage
The key clinical concern in a patient with a rectus sheath hematoma who is hemodynamically stable is whether the hematoma is expanding. This patient responded to initial resuscitation, but her falling hematocrit was evidence of ongoing bleeding leading to an expanding rectus sheath hematoma. Thus, serial clinical examinations and frequent monitoring of the complete blood cell count would not be enough, as it could miss fatal ongoing bleeding.
Radiographically guided embolization with Gelfoam, thrombin, or coils should be attempted first, as this is less invasive than exploratory laparotomy.28 It can achieve hemostasis, decrease the size of the hematoma, limit the need for blood products, and prevent rupture into the abdomen. If this is unsuccessful, the next step is ligation of the bleeding vessel.29
Surgical treatment includes evacuation of the hematoma, repair of the rectus sheath, ligation of bleeding vessels, and abdominal wall closure. Surgical evacuation or guided drainage of a rectus sheath hematoma on its own is not normally indicated and may indeed cause persistent bleeding by diminishing a potential tamponade effect. However, it may become necessary if the hematoma is very large or infected, if it causes marked respiratory impairment, or if abdominal compartment syndrome is suspected.
Abdominal compartment syndrome is very rare but is associated with a 50% mortality rate.30 It should be suspected in patients with oliguria, low cardiac output, changes in minute ventilation, and altered splanchnic blood flow. The diagnosis is confirmed with indwelling catheter manometry of the bladder to measure intra-abdominal pressure. Intra-abominal pressure above 25 mm Hg should be treated with decompressive laparotomy.30 However, the clinical suspicion of abdominal compartment syndrome was low in this patient.
The best choice at this point would be urgent computed tomographic angiography to identify a bleeding vessel, along with consideration of radiographically guided arterial embolization.
TREATMENT IS USUALLY CONSERVATIVE
Treatment of rectus sheath hematoma is conservative in most hemodynamically stable patients, with embolization or surgical intervention reserved for unstable patients or those in whom the hematoma is expanding.
Knowledge of the grading system of Berná et al26 helps to assess the patient’s risk and to anticipate potential complications. Grade I hematomas are mild and do not necessitate admission. Patients with grade II hematoma can be admitted to the floor for 24 to 48 hours for observation. Grade III usually occurs in patients receiving anticoagulant therapy and frequently requires blood products. These patients have a prolonged hospital stay and more complications, including hypovolemic shock, myonecrosis, acute coronary syndrome, arrhythmias, acute renal failure, small-bowel infarction, and abdominal compartment syndrome—all of which increases the risk of morbidity and death. Thus, patients who are on anticoagulation who develop grade III rectus sheath hematoma should be admitted to the hospital, preferably to the intensive care unit, to ensure that the hematoma is not expanding and to plan reinstitution of anticoagulation as appropriate.
In most cases, rectus sheath hematomas resolve within 1 to 3 months. Resolution of large hematomas may be hastened with the use of pulsed ultrasound.31 However, this treatment should be used only after the acute phase is over, when there is evidence of an organized thrombus and coagulation measures have returned to the target range. This helps to reduce the risk of bleeding and to prevent symptoms from worsening.31
OUR PATIENT’S COURSE
Our patient underwent urgent computed tomographic angiography, which showed a modest increase in the size of the rectus sheath hematoma. However, no definitive blush of contrast was seen to suggest active arterial bleeding. Her hematocrit stabilized, and she remained hemodynamically stable without requiring additional intervention. Most likely her bleeding was self-contained. She had normal intra-abdominal pressure on serial monitoring. She was later transferred to acute inpatient rehabilitation in view of deconditioning and is currently doing well. The hematoma persisted, decreasing only slightly in size over the next 3 weeks.
- Kougias P, Lau D, El Sayed HF, Zhou W, Huynh TT, Lin PH. Determinants of mortality and treatment outcome following surgical interventions for acute mesenteric ischemia. J Vasc Surg 2007; 46:467–474.
- Sise MJ. Acute mesenteric ischemia. Surg Clin North Am 2014; 94:165–181.
- Scharff JR, Longo WE, Vartanian SM, Jacobs DL, Bahadursingh AN, Kaminski DL. Ischemic colitis: spectrum of disease and outcome. Surgery 2003; 134:624–629.
- Lange H, Jäckel R. Usefulness of plasma lactate concentration in the diagnosis of acute abdominal disease. Eur J Surg 1994; 160:381–384.
- Gearhart SL, Delaney CP, Senagore AJ, et al. Prospective assessment of the predictive value of alpha-glutathione S-transferase for intestinal ischemia. Am Surg 2003; 69:324–329.
- Kanda T, Fujii H, Tani T, et al. Intestinal fatty acid-binding protein is a useful diagnostic marker for mesenteric infarction in humans. Gastroenterology 1996; 110:339–343.
- Menke J. Diagnostic accuracy of multidetector CT in acute mesenteric ischemia: systematic review and meta-analysis. Radiology 2010; 256:93–101.
- Acosta S, Björnsson S, Ekberg O, Resch T. CT angiography followed by endovascular intervention for acute superior mesenteric artery occlusion does not increase risk of contrast-induced renal failure. Eur J Vasc Endovasc Surg 2010; 39:726–730.
- Clark RA. Computed tomography of bowel infarction. J Comput Assist Tomogr 1987; 11:757–762.
- Acosta S, Björck M. Modern treatment of acute mesenteric ischaemia. Br J Surg 2014; 101:e100–e108.
- Smithson A, Ruiz J, Perello R, Valverde M, Ramos J, Garzo L. Diagnostic and management of spontaneous rectus sheath hematoma. Eur J Intern Med 2013; 24:579–582.
- Moreno Gallego A, Aguayo JL, Flores B, et al. Ultrasonography and computed tomography reduce unnecessary surgery in abdominal rectus sheath haematoma. Br J Surg 1997; 84:1295–1297.
- Dubinsky IL. Hematoma of the rectus abdominis muscle: case report and review of the literature. J Emerg Med 1997; 15:165–167.
- Yi M, Yao G, Bai Y. The monitoring of intra-abdominal pressure in critically ill patients. (In Chinese.) Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2014; 26:175–178.
- Hunt L, Frost SA, Hillman K, Newton PJ, Davidson PM. Management of intra-abdominal hypertension and abdominal compartment syndrome: a review. J Trauma Manag Outcomes 2014; 8:2.
- Malbrain ML, Cheatham ML, Kirkpatrick A, et al. Results from the International Conference of Experts on Intra-abdominal Hypertension and Abdominal Compartment Syndrome. I. Definitions. Intensive Care Med 2006; 32:1722–1732.
- Malbrain ML, Chiumello D, Cesana BM, et al; WAKE-Up! Investigators. A systematic review and individual patient data meta-analysis on intra-abdominal hypertension in critically ill patients: the wake-up project. World initiative on Abdominal Hypertension Epidemiology, a Unifying Project (WAKE-Up!). Minerva Anestesiol 2014; 80:293–306.
- Kirkpatrick AW, Roberts DJ, De Waele J, et al; Pediatric Guidelines Sub-Committee for the World Society of the Abdominal Compartment Syndrome. Intra-abdominal hypertension and the abdominal compartment syndrome: updated consensus definitions and clinical practice guidelines from the World Society of the Abdominal Compartment Syndrome. Intensive Care Med 2013; 39:1190–1206.
- Holodinsky JK, Roberts DJ, Ball CG, et al. Risk factors for intra-abdominal hypertension and abdominal compartment syndrome among adult intensive care unit patients: a systematic review and meta-analysis. Crit Care 2013; 17:R249.
- Sugrue M, Bauman A, Jones F, et al. Clinical examination is an inaccurate predictor of intraabdominal pressure. World J Surg 2002; 26:1428–1431.
- Cheatham ML, De Waele JJ, De Laet I, et al; World Society of the Abdominal Compartment Syndrome (WSACS) Clinical Trials Working Group. The impact of body position on intra-abdominal pressure measurement: a multicenter analysis. Crit Care Med 2009; 37:2187–2190.
- Ortiz-Diaz E, Lan CK. Intra-abdominal hypertension in medical critically ill patients: a narrative review. Shock 2014; 41:175–180.
- Cotton BA, Reddy N, Hatch QM, et al. Damage control resuscitation is associated with a reduction in resuscitation volumes and improvement in survival in 390 damage control laparotomy patients. Ann Surg 2011; 254:598–605.
- An G, West MA. Abdominal compartment syndrome: a concise clinical review. Crit Care Med 2008; 36:1304–1310.
- Tolcher MC, Nitsche JF, Arendt KW, Rose CH. Spontaneous rectus sheath hematoma pregnancy: case report and review of the literature. Obstet Gynecol Surv 2010; 65:517–522.
- Berná JD, Garcia-Medina V, Guirao J, Garcia-Medina J. Rectus sheath hematoma: diagnostic classification by CT. Abdom Imaging 1996; 21:62–64.
- Unger EC, Glazer HS, Lee JK, Ling D. MRI of extracranial hematomas: preliminary observations. AJR Am J Roentgenol 1986; 146:403–407.
- Rimola J, Perendreu J, Falcó J, Fortuño JR, Massuet A, Branera J. Percutaneous arterial embolization in the management of rectus sheath hematoma. AJR Am J Roentgenol 2007; 188:W497–W502.
- Titone C, Lipsius M, Krakauer JS. “Spontaneous” hematoma of the rectus abdominis muscle: critical review of 50 cases with emphasis on early diagnosis and treatment. Surgery 1972; 72:568–572.
- Osinbowale O, Bartholomew JR. Rectus sheath hematoma. Vasc Med 2008; 13:275–279.
- Berná-Serna JD, Sánchez-Garre J, Madrigal M, Zuazu I, Berná-Mestre JD. Ultrasound therapy in rectus sheath hematoma. Phys Ther 2005; 85:352–357.
- Kougias P, Lau D, El Sayed HF, Zhou W, Huynh TT, Lin PH. Determinants of mortality and treatment outcome following surgical interventions for acute mesenteric ischemia. J Vasc Surg 2007; 46:467–474.
- Sise MJ. Acute mesenteric ischemia. Surg Clin North Am 2014; 94:165–181.
- Scharff JR, Longo WE, Vartanian SM, Jacobs DL, Bahadursingh AN, Kaminski DL. Ischemic colitis: spectrum of disease and outcome. Surgery 2003; 134:624–629.
- Lange H, Jäckel R. Usefulness of plasma lactate concentration in the diagnosis of acute abdominal disease. Eur J Surg 1994; 160:381–384.
- Gearhart SL, Delaney CP, Senagore AJ, et al. Prospective assessment of the predictive value of alpha-glutathione S-transferase for intestinal ischemia. Am Surg 2003; 69:324–329.
- Kanda T, Fujii H, Tani T, et al. Intestinal fatty acid-binding protein is a useful diagnostic marker for mesenteric infarction in humans. Gastroenterology 1996; 110:339–343.
- Menke J. Diagnostic accuracy of multidetector CT in acute mesenteric ischemia: systematic review and meta-analysis. Radiology 2010; 256:93–101.
- Acosta S, Björnsson S, Ekberg O, Resch T. CT angiography followed by endovascular intervention for acute superior mesenteric artery occlusion does not increase risk of contrast-induced renal failure. Eur J Vasc Endovasc Surg 2010; 39:726–730.
- Clark RA. Computed tomography of bowel infarction. J Comput Assist Tomogr 1987; 11:757–762.
- Acosta S, Björck M. Modern treatment of acute mesenteric ischaemia. Br J Surg 2014; 101:e100–e108.
- Smithson A, Ruiz J, Perello R, Valverde M, Ramos J, Garzo L. Diagnostic and management of spontaneous rectus sheath hematoma. Eur J Intern Med 2013; 24:579–582.
- Moreno Gallego A, Aguayo JL, Flores B, et al. Ultrasonography and computed tomography reduce unnecessary surgery in abdominal rectus sheath haematoma. Br J Surg 1997; 84:1295–1297.
- Dubinsky IL. Hematoma of the rectus abdominis muscle: case report and review of the literature. J Emerg Med 1997; 15:165–167.
- Yi M, Yao G, Bai Y. The monitoring of intra-abdominal pressure in critically ill patients. (In Chinese.) Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2014; 26:175–178.
- Hunt L, Frost SA, Hillman K, Newton PJ, Davidson PM. Management of intra-abdominal hypertension and abdominal compartment syndrome: a review. J Trauma Manag Outcomes 2014; 8:2.
- Malbrain ML, Cheatham ML, Kirkpatrick A, et al. Results from the International Conference of Experts on Intra-abdominal Hypertension and Abdominal Compartment Syndrome. I. Definitions. Intensive Care Med 2006; 32:1722–1732.
- Malbrain ML, Chiumello D, Cesana BM, et al; WAKE-Up! Investigators. A systematic review and individual patient data meta-analysis on intra-abdominal hypertension in critically ill patients: the wake-up project. World initiative on Abdominal Hypertension Epidemiology, a Unifying Project (WAKE-Up!). Minerva Anestesiol 2014; 80:293–306.
- Kirkpatrick AW, Roberts DJ, De Waele J, et al; Pediatric Guidelines Sub-Committee for the World Society of the Abdominal Compartment Syndrome. Intra-abdominal hypertension and the abdominal compartment syndrome: updated consensus definitions and clinical practice guidelines from the World Society of the Abdominal Compartment Syndrome. Intensive Care Med 2013; 39:1190–1206.
- Holodinsky JK, Roberts DJ, Ball CG, et al. Risk factors for intra-abdominal hypertension and abdominal compartment syndrome among adult intensive care unit patients: a systematic review and meta-analysis. Crit Care 2013; 17:R249.
- Sugrue M, Bauman A, Jones F, et al. Clinical examination is an inaccurate predictor of intraabdominal pressure. World J Surg 2002; 26:1428–1431.
- Cheatham ML, De Waele JJ, De Laet I, et al; World Society of the Abdominal Compartment Syndrome (WSACS) Clinical Trials Working Group. The impact of body position on intra-abdominal pressure measurement: a multicenter analysis. Crit Care Med 2009; 37:2187–2190.
- Ortiz-Diaz E, Lan CK. Intra-abdominal hypertension in medical critically ill patients: a narrative review. Shock 2014; 41:175–180.
- Cotton BA, Reddy N, Hatch QM, et al. Damage control resuscitation is associated with a reduction in resuscitation volumes and improvement in survival in 390 damage control laparotomy patients. Ann Surg 2011; 254:598–605.
- An G, West MA. Abdominal compartment syndrome: a concise clinical review. Crit Care Med 2008; 36:1304–1310.
- Tolcher MC, Nitsche JF, Arendt KW, Rose CH. Spontaneous rectus sheath hematoma pregnancy: case report and review of the literature. Obstet Gynecol Surv 2010; 65:517–522.
- Berná JD, Garcia-Medina V, Guirao J, Garcia-Medina J. Rectus sheath hematoma: diagnostic classification by CT. Abdom Imaging 1996; 21:62–64.
- Unger EC, Glazer HS, Lee JK, Ling D. MRI of extracranial hematomas: preliminary observations. AJR Am J Roentgenol 1986; 146:403–407.
- Rimola J, Perendreu J, Falcó J, Fortuño JR, Massuet A, Branera J. Percutaneous arterial embolization in the management of rectus sheath hematoma. AJR Am J Roentgenol 2007; 188:W497–W502.
- Titone C, Lipsius M, Krakauer JS. “Spontaneous” hematoma of the rectus abdominis muscle: critical review of 50 cases with emphasis on early diagnosis and treatment. Surgery 1972; 72:568–572.
- Osinbowale O, Bartholomew JR. Rectus sheath hematoma. Vasc Med 2008; 13:275–279.
- Berná-Serna JD, Sánchez-Garre J, Madrigal M, Zuazu I, Berná-Mestre JD. Ultrasound therapy in rectus sheath hematoma. Phys Ther 2005; 85:352–357.
When the dissociation curve shifts to the left
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.

Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.
Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.
Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.
A 41-year-old man with abdominal pain
A 41-year-old man presented with pain in the left upper quadrant for 4 days. The pain was constant, was worse on inspiration, and did not radiate. He denied fevers, night sweats, nausea, vomiting, diarrhea, and urinary symptoms. He had been diagnosed with multiple sclerosis a few years earlier, and he had undergone aortofemoral bypass surgery on the left side 2 years ago. He denied smoking or using illicit drugs and described himself as a social drinker.
In the emergency room, he appeared comfortable. He was afebrile, blood pressure 136/69 mm Hg, pulse rate 98 per minute, and respiratory rate 16. All pulses were palpable and equal, the jugular venous pressure was not elevated, and no cardiac murmurs were heard. The abdomen was tender in the left upper quadrant, with no guarding or rigidity. Examination of the nervous, musculoskeletal, and respiratory systems was unremarkable. Skin examination revealed only scars from previous surgery.
LABORATORY AND IMAGING RESULTS
- White blood cell count 7.2 × 109/L (reference range 4.0–10.0) with a normal differential
- Hemoglobin 134 g/dL (140–180)
- Platelet count 167 × 109/L (150–400)
- Renal and liver panels were normal
- Erythrocyte sedimentation rate 30 mm/hour
- C-reactive protein level 14.3 mg/L
- D-dimer level 2,670 ng/mL (< 500)
- International normalized ratio (INR) 1.0 (0.9–1.3)
- Activated partial thromboplastin time (aPTT) 44 seconds (25–38)
- Fibrinogen level 3.0 g/L (1.8–3.5)
- Urinalysis negative for leukocytes and casts.
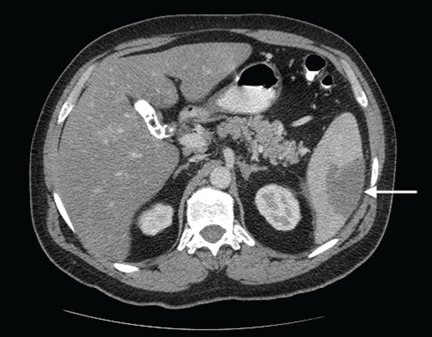
Computed tomography of the abdomen showed a wedge-shaped area of hypodensity along the inferolateral aspect of the spleen measuring 6 × 3.8 cm, consistent with a recent infarct (Figure 1). There was also evidence of a previous infarct in the posterolateral aspect of the spleen. Splenic, celiac, superior mesenteric, and inferior mesenteric arteries were patent.
1. Given these findings, which of the following diagnoses should be considered?
- Subacute infective endocarditis
- Inherited thrombophilia
- Antiphospholipid syndrome
All three diagnoses should be considered in this case.
Endocarditis
Embolism from a source in the heart caused by subacute bacterial endocarditis is more common than the other two conditions listed here and must be excluded.
Our patient lacks key features of this condition: he has no predisposing factors (artificial valve, cyanotic congenital heart disease, previous endocarditis, intravenous drug abuse); no constitutional symptoms of fever, night sweats, and weight loss; no findings on examination of skin and cardiovascular systems; and a normal white blood cell count. Nevertheless, even though the absence of these features makes bacterial endocarditis unlikely, it does not exclude it. Blood cultures and transesophageal echocardiography are indicated to rule out bacterial endocarditis.
We obtained serial blood cultures, which were negative, and transesophageal echocardiography showed normal valves and no evidence of thrombus or vegetation, thus excluding a cardiac source of emboli.
Thrombophilia
Our patient has a history of recurrent thromboembolic episodes, and this warrants testing to rule out an inherited thrombophilia. A family history of thromboembolic disease should also be sought.1
In our patient, tests for prothrombotic activity including protein C chromogen, activated protein C ratio, free protein S, functional protein S, antithrombin factor V Leiden, and the prothrombin 20210G>A mutation were either negative or within the reference range. A negative family history of thromboembolic disease and the negative laboratory tests make inherited thrombophilia unlikely in our patient.
Sickle cell disease, polycythemia vera, and essential thrombocythemia may also cause splenic infarction but can be ruled out in this patient on the basis of history and initial blood tests.
Antiphospholipid syndrome
A history of vascular disease (aortofemoral bypass surgery), a recent splenic infarct, and an elevated aPTT makes antiphospholipid syndrome the likeliest diagnosis in this patient.
Appropriate tests are for lupus anticoagulant, immunoglobulin G (IgG) or IgM cardiolipin antibody, and beta-2 glycoprotein 1 (beta-2 GP1) antibody, as well as the dilute Russell viper venom time (dRVVT) and the dRVVT ratio. The IgG and IgM cardiolipin antibody and beta-2 GP1 antibody tests have the same diagnostic value, and only medium to high titers should be considered positive.
Our patient’s IgG cardiolipin antibody level was in the normal range at 15 IgG phospholipid units (reference range 0–22); his IgM cardiolipin antibody level was high at 41 IgM phospholipid units (0–10). The dRVVT was 57 seconds (24–42), and the dRVVT ratio was 2.0 (0.0–1.3).
2. What further investigations are indicated before starting treatment?
- No further investigations required
- Repeat testing for phospholipid antibodies in 12 weeks
- Test for antinuclear antibodies
Antiphospholipid antibodies may appear transiently in certain infections, such as syphilis, Lyme disease, Epstein-Barr virus, cytomegalovirus, hepatitis C, and human immunodeficiency virus. Therefore, the presence of antiphospholipid antibodies must be confirmed over time, with two positive results at least 12 weeks apart.2
When repeated 12 weeks later, our patient’s IgG anticardiolipin antibody level was 14 GPL units, and the IgM anticardiolipin antibody level was 30 MPL units; the dRVVT was 55 seconds, and the dRVVT ratio was 1.8. These results, along with a history of recurrent arterial thrombosis, confirmed antiphospholipid syndrome.
The 2009 update of the International Society of Thrombosis and Haemostasis guidelines recommend two tests, the dRVVT and the aPTT, since no single test is 100% sensitive for lupus anticoagulant.3 The dRVVT has a high specificity for lupus anticoagulant in patients at high risk of thrombosis.
A SYNDROME WITH A WIDE RANGE OF EFFECTS AND COMPLICATIONS
Antiphospholipid syndrome is a systemic autoimmune disease that manifests as arterial and venous thrombosis and as obstetric complications. Thrombosis tends to be recurrent and may involve any site. For example, it can cause blurred vision in one or both eyes; amaurosis fugax; visual field defects; central or branch retinal artery or vein occlusion; deep vein thrombosis; pulmonary embolism; myocardial infarction; transient ischemic attack and stroke; cerebral vein thrombosis; and portal, renal, and mesenteric infarction involving veins or arteries.4 Pulmonary capillaritis may cause diffuse alveolar hemorrhage. Livedo reticularis, digital gangrene, cutaneous necrosis, splinter hemorrhages, chorea, and transverse myelopathy may also occur.
Obstetric complications of antiphospholipid syndrome include recurrent miscarriage and pregnancy loss at or after 10 weeks of gestation, eclampsia, preeclampsia, and placental insufficiency.5 The syndrome also has a potentially lethal variant characterized by multiorgan thrombosis affecting mainly small vessels.
The diagnosis of antiphospholipid syndrome requires relevant clinical features and symptoms and the presence of at least one of the antiphospholipid antibodies. Because the rate of false-positive tests for antiphospholipid antibodies ranges from 3% to 20% in the general population, asymptomatic patients should not be tested.6
Antiphospholipid syndrome may occur in the setting of other autoimmune diseases, most commonly systemic lupus erythematosus, when it is termed “secondary” antiphospholipid syndrome. Although only 40% of patients with lupus have antiphospholipid antibodies and less than 40% will have a thrombotic event, thrombotic antiphospholipid syndrome is a major adverse prognostic factor in these patients.7,8 Therefore, it is prudent to consider systemic lupus erythematosus and to do appropriate tests if the patient has other features suggestive of lupus, such as renal, skin, or musculoskeletal lesions.
In our patient, antinuclear antibody testing was positive, with a titer of 1:320, and showed a finely speckled staining pattern. Tests for antibodies to Sjögren syndrome A and B antigens were negative. The complement C3 level was 1.28 g/L (reference range 0.74–1.85) and the C4 level was 0.24 g/L (0.16–0.44). Although the speckled staining pattern can be seen in lupus, it is more common in Sjögren syndrome, mixed connective tissue disease, scleroderma, and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia).9 Moreover, normal levels of complement C3 and C4, in the absence of clinical features, make lupus unlikely. Similarly, our patient had no clinical features of other connective tissue disorders. Therefore, he had primary antiphospholipid syndrome.
3. How should this patient be managed?
- Antiplatelet therapy
- Warfarin to maintain an INR between 2.0 and 3.0
- Warfarin to maintain an INR above 3.0
The risk of recurrent thrombosis is high in patients who test positive for lupus anticoagulant, and the risk is highest in patients who are also positive for anticardiolipin and anti-beta-2 GP1 antibodies: the incidence of thrombosis is 12.2% at 1 year, 26.1% at 5 years, 44.2% at 10 years.10
Since our patient is positive for lupus anticoagulant (prolonged aPTT and elevated dRVVT, both indicating lupus anticoagulant positivity) and for anticardiolipin antibodies (anti-beta-2 GP1 not tested), his risk of recurrent thrombosis is high, and he requires lifelong anticoagulation therapy.
The intensity of anticoagulation in different subgroups of patients is controversial. Based on retrospective trials, indefinite anticoagulation at an INR of 2.0 to 3.0 has been suggested for patients with antiphospholipid syndrome presenting with venous thrombosis, and more intense anticoagulation with an INR above 3.0 in patients with recurrent or arterial thrombosis.11 The combination of warfarin with an INR between 2.0 and 3.0 and aspirin 100 mg daily has also been proposed for patients with arterial thrombosis.12
Modifiable risk factors such as smoking, obesity, and use of estrogens should be addressed in all patients with antiphospholipid syndrome.
In pregnant women with complications such as preeclampsia, low-dose aspirin can be used, and in women with a history of miscarriage, the combination of low-dose aspirin and heparin is recommended throughout the prenatal period.4
In patients who have recurrent thrombosis despite adequate anticoagulation, an expert committee12 has proposed that alternative regimens could include long-term low-molecular-weight heparin instead of warfarin, the combination of warfarin and aspirin, or warfarin and hydroxychloroquine. Adding a statin can also be considered.
Treatment of catastrophic antiphospholipid syndrome is based on expert opinion. A combination of anticoagulation, corticosteroids, plasma exchange, intravenous immunoglobulins, and rituximab has been tried, but the mortality rate remains high.13
OUR PATIENT'S COURSE
Our patient was started on warfarin, with a target INR above 3.0, and was doing well at 6 months of follow-up.
- De Stefano V, Rossi E. Testing for inherited thrombophilia and consequences for antithrombotic prophylaxis in patients with venous thromboembolism and their relatives. A review of the Guidelines from Scientific Societies and Working Groups. Thromb Haemost 2013; 110:697–705.
- Galli M. Interpretation and recommended testing for antiphospholipid antibodies. Semin Thromb Hemost 2012; 38:348–352.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Keeling D, Mackie I, Moore GW, Greer IA, Greaves M; British Committee for Standards in Haematology. Guidelines on the investigation and management of antiphospholipid syndrome. Br J Haematol 2012; 157:47–58.
- Misita CP, Moll S. Antiphospholipid antibodies. Circulation 2005; 112:e39–e44.
- Rand JH, Wolgast LR. Do’s and don’t’s in diagnosing antiphospholipid syndrome. Hematology Am Soc Hematol Educ Program 2012; 2012:455–459.
- Mok CC, Tang SS, To CH, Petri M. Incidence and risk factors of thromboembolism in systemic lupus erythematosus: a comparison of three ethnic groups. Arthritis Rheum 2005; 52:2774–2782.
- Ruiz-Irastorza G, Egurbide MV, Ugalde J, Aguirre C. High impact of antiphospholipid syndrome on irreversible organ damage and survival of patients with systemic lupus erythematosus. Arch Intern Med 2004; 164:77–82.
- Locht H, Pelck R, Manthorpe R. Clinical manifestations correlated to the prevalence of autoantibodies in a large (n = 321) cohort of patients with primary Sjögren’s syndrome: a comparison of patients initially diagnosed according to the Copenhagen classification criteria with the American-European consensus criteria. Autoimmun Rev 2005; 4:276–281.
- Pengo V, Ruffatti A, Legnani C, et al. Clinical course of high-risk patients diagnosed with antiphospholipid syndrome. J Thromb Haemost 2010; 8:237–242.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza I, et al. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: report of a task force at the 13th International Congress on antiphospholipid antibodies. Lupus 2011; 20:206–218.
- Cervera R. Update on the diagnosis, treatment, and prognosis of the catastrophic antiphospholipid syndrome. Curr Rheumatol Rep 2010; 12:70–76.
A 41-year-old man presented with pain in the left upper quadrant for 4 days. The pain was constant, was worse on inspiration, and did not radiate. He denied fevers, night sweats, nausea, vomiting, diarrhea, and urinary symptoms. He had been diagnosed with multiple sclerosis a few years earlier, and he had undergone aortofemoral bypass surgery on the left side 2 years ago. He denied smoking or using illicit drugs and described himself as a social drinker.
In the emergency room, he appeared comfortable. He was afebrile, blood pressure 136/69 mm Hg, pulse rate 98 per minute, and respiratory rate 16. All pulses were palpable and equal, the jugular venous pressure was not elevated, and no cardiac murmurs were heard. The abdomen was tender in the left upper quadrant, with no guarding or rigidity. Examination of the nervous, musculoskeletal, and respiratory systems was unremarkable. Skin examination revealed only scars from previous surgery.
LABORATORY AND IMAGING RESULTS
- White blood cell count 7.2 × 109/L (reference range 4.0–10.0) with a normal differential
- Hemoglobin 134 g/dL (140–180)
- Platelet count 167 × 109/L (150–400)
- Renal and liver panels were normal
- Erythrocyte sedimentation rate 30 mm/hour
- C-reactive protein level 14.3 mg/L
- D-dimer level 2,670 ng/mL (< 500)
- International normalized ratio (INR) 1.0 (0.9–1.3)
- Activated partial thromboplastin time (aPTT) 44 seconds (25–38)
- Fibrinogen level 3.0 g/L (1.8–3.5)
- Urinalysis negative for leukocytes and casts.
Computed tomography of the abdomen showed a wedge-shaped area of hypodensity along the inferolateral aspect of the spleen measuring 6 × 3.8 cm, consistent with a recent infarct (Figure 1). There was also evidence of a previous infarct in the posterolateral aspect of the spleen. Splenic, celiac, superior mesenteric, and inferior mesenteric arteries were patent.
1. Given these findings, which of the following diagnoses should be considered?
- Subacute infective endocarditis
- Inherited thrombophilia
- Antiphospholipid syndrome
All three diagnoses should be considered in this case.
Endocarditis
Embolism from a source in the heart caused by subacute bacterial endocarditis is more common than the other two conditions listed here and must be excluded.
Our patient lacks key features of this condition: he has no predisposing factors (artificial valve, cyanotic congenital heart disease, previous endocarditis, intravenous drug abuse); no constitutional symptoms of fever, night sweats, and weight loss; no findings on examination of skin and cardiovascular systems; and a normal white blood cell count. Nevertheless, even though the absence of these features makes bacterial endocarditis unlikely, it does not exclude it. Blood cultures and transesophageal echocardiography are indicated to rule out bacterial endocarditis.
We obtained serial blood cultures, which were negative, and transesophageal echocardiography showed normal valves and no evidence of thrombus or vegetation, thus excluding a cardiac source of emboli.
Thrombophilia
Our patient has a history of recurrent thromboembolic episodes, and this warrants testing to rule out an inherited thrombophilia. A family history of thromboembolic disease should also be sought.1
In our patient, tests for prothrombotic activity including protein C chromogen, activated protein C ratio, free protein S, functional protein S, antithrombin factor V Leiden, and the prothrombin 20210G>A mutation were either negative or within the reference range. A negative family history of thromboembolic disease and the negative laboratory tests make inherited thrombophilia unlikely in our patient.
Sickle cell disease, polycythemia vera, and essential thrombocythemia may also cause splenic infarction but can be ruled out in this patient on the basis of history and initial blood tests.
Antiphospholipid syndrome
A history of vascular disease (aortofemoral bypass surgery), a recent splenic infarct, and an elevated aPTT makes antiphospholipid syndrome the likeliest diagnosis in this patient.
Appropriate tests are for lupus anticoagulant, immunoglobulin G (IgG) or IgM cardiolipin antibody, and beta-2 glycoprotein 1 (beta-2 GP1) antibody, as well as the dilute Russell viper venom time (dRVVT) and the dRVVT ratio. The IgG and IgM cardiolipin antibody and beta-2 GP1 antibody tests have the same diagnostic value, and only medium to high titers should be considered positive.
Our patient’s IgG cardiolipin antibody level was in the normal range at 15 IgG phospholipid units (reference range 0–22); his IgM cardiolipin antibody level was high at 41 IgM phospholipid units (0–10). The dRVVT was 57 seconds (24–42), and the dRVVT ratio was 2.0 (0.0–1.3).
2. What further investigations are indicated before starting treatment?
- No further investigations required
- Repeat testing for phospholipid antibodies in 12 weeks
- Test for antinuclear antibodies
Antiphospholipid antibodies may appear transiently in certain infections, such as syphilis, Lyme disease, Epstein-Barr virus, cytomegalovirus, hepatitis C, and human immunodeficiency virus. Therefore, the presence of antiphospholipid antibodies must be confirmed over time, with two positive results at least 12 weeks apart.2
When repeated 12 weeks later, our patient’s IgG anticardiolipin antibody level was 14 GPL units, and the IgM anticardiolipin antibody level was 30 MPL units; the dRVVT was 55 seconds, and the dRVVT ratio was 1.8. These results, along with a history of recurrent arterial thrombosis, confirmed antiphospholipid syndrome.
The 2009 update of the International Society of Thrombosis and Haemostasis guidelines recommend two tests, the dRVVT and the aPTT, since no single test is 100% sensitive for lupus anticoagulant.3 The dRVVT has a high specificity for lupus anticoagulant in patients at high risk of thrombosis.
A SYNDROME WITH A WIDE RANGE OF EFFECTS AND COMPLICATIONS
Antiphospholipid syndrome is a systemic autoimmune disease that manifests as arterial and venous thrombosis and as obstetric complications. Thrombosis tends to be recurrent and may involve any site. For example, it can cause blurred vision in one or both eyes; amaurosis fugax; visual field defects; central or branch retinal artery or vein occlusion; deep vein thrombosis; pulmonary embolism; myocardial infarction; transient ischemic attack and stroke; cerebral vein thrombosis; and portal, renal, and mesenteric infarction involving veins or arteries.4 Pulmonary capillaritis may cause diffuse alveolar hemorrhage. Livedo reticularis, digital gangrene, cutaneous necrosis, splinter hemorrhages, chorea, and transverse myelopathy may also occur.
Obstetric complications of antiphospholipid syndrome include recurrent miscarriage and pregnancy loss at or after 10 weeks of gestation, eclampsia, preeclampsia, and placental insufficiency.5 The syndrome also has a potentially lethal variant characterized by multiorgan thrombosis affecting mainly small vessels.
The diagnosis of antiphospholipid syndrome requires relevant clinical features and symptoms and the presence of at least one of the antiphospholipid antibodies. Because the rate of false-positive tests for antiphospholipid antibodies ranges from 3% to 20% in the general population, asymptomatic patients should not be tested.6
Antiphospholipid syndrome may occur in the setting of other autoimmune diseases, most commonly systemic lupus erythematosus, when it is termed “secondary” antiphospholipid syndrome. Although only 40% of patients with lupus have antiphospholipid antibodies and less than 40% will have a thrombotic event, thrombotic antiphospholipid syndrome is a major adverse prognostic factor in these patients.7,8 Therefore, it is prudent to consider systemic lupus erythematosus and to do appropriate tests if the patient has other features suggestive of lupus, such as renal, skin, or musculoskeletal lesions.
In our patient, antinuclear antibody testing was positive, with a titer of 1:320, and showed a finely speckled staining pattern. Tests for antibodies to Sjögren syndrome A and B antigens were negative. The complement C3 level was 1.28 g/L (reference range 0.74–1.85) and the C4 level was 0.24 g/L (0.16–0.44). Although the speckled staining pattern can be seen in lupus, it is more common in Sjögren syndrome, mixed connective tissue disease, scleroderma, and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia).9 Moreover, normal levels of complement C3 and C4, in the absence of clinical features, make lupus unlikely. Similarly, our patient had no clinical features of other connective tissue disorders. Therefore, he had primary antiphospholipid syndrome.
3. How should this patient be managed?
- Antiplatelet therapy
- Warfarin to maintain an INR between 2.0 and 3.0
- Warfarin to maintain an INR above 3.0
The risk of recurrent thrombosis is high in patients who test positive for lupus anticoagulant, and the risk is highest in patients who are also positive for anticardiolipin and anti-beta-2 GP1 antibodies: the incidence of thrombosis is 12.2% at 1 year, 26.1% at 5 years, 44.2% at 10 years.10
Since our patient is positive for lupus anticoagulant (prolonged aPTT and elevated dRVVT, both indicating lupus anticoagulant positivity) and for anticardiolipin antibodies (anti-beta-2 GP1 not tested), his risk of recurrent thrombosis is high, and he requires lifelong anticoagulation therapy.
The intensity of anticoagulation in different subgroups of patients is controversial. Based on retrospective trials, indefinite anticoagulation at an INR of 2.0 to 3.0 has been suggested for patients with antiphospholipid syndrome presenting with venous thrombosis, and more intense anticoagulation with an INR above 3.0 in patients with recurrent or arterial thrombosis.11 The combination of warfarin with an INR between 2.0 and 3.0 and aspirin 100 mg daily has also been proposed for patients with arterial thrombosis.12
Modifiable risk factors such as smoking, obesity, and use of estrogens should be addressed in all patients with antiphospholipid syndrome.
In pregnant women with complications such as preeclampsia, low-dose aspirin can be used, and in women with a history of miscarriage, the combination of low-dose aspirin and heparin is recommended throughout the prenatal period.4
In patients who have recurrent thrombosis despite adequate anticoagulation, an expert committee12 has proposed that alternative regimens could include long-term low-molecular-weight heparin instead of warfarin, the combination of warfarin and aspirin, or warfarin and hydroxychloroquine. Adding a statin can also be considered.
Treatment of catastrophic antiphospholipid syndrome is based on expert opinion. A combination of anticoagulation, corticosteroids, plasma exchange, intravenous immunoglobulins, and rituximab has been tried, but the mortality rate remains high.13
OUR PATIENT'S COURSE
Our patient was started on warfarin, with a target INR above 3.0, and was doing well at 6 months of follow-up.
A 41-year-old man presented with pain in the left upper quadrant for 4 days. The pain was constant, was worse on inspiration, and did not radiate. He denied fevers, night sweats, nausea, vomiting, diarrhea, and urinary symptoms. He had been diagnosed with multiple sclerosis a few years earlier, and he had undergone aortofemoral bypass surgery on the left side 2 years ago. He denied smoking or using illicit drugs and described himself as a social drinker.
In the emergency room, he appeared comfortable. He was afebrile, blood pressure 136/69 mm Hg, pulse rate 98 per minute, and respiratory rate 16. All pulses were palpable and equal, the jugular venous pressure was not elevated, and no cardiac murmurs were heard. The abdomen was tender in the left upper quadrant, with no guarding or rigidity. Examination of the nervous, musculoskeletal, and respiratory systems was unremarkable. Skin examination revealed only scars from previous surgery.
LABORATORY AND IMAGING RESULTS
- White blood cell count 7.2 × 109/L (reference range 4.0–10.0) with a normal differential
- Hemoglobin 134 g/dL (140–180)
- Platelet count 167 × 109/L (150–400)
- Renal and liver panels were normal
- Erythrocyte sedimentation rate 30 mm/hour
- C-reactive protein level 14.3 mg/L
- D-dimer level 2,670 ng/mL (< 500)
- International normalized ratio (INR) 1.0 (0.9–1.3)
- Activated partial thromboplastin time (aPTT) 44 seconds (25–38)
- Fibrinogen level 3.0 g/L (1.8–3.5)
- Urinalysis negative for leukocytes and casts.
Computed tomography of the abdomen showed a wedge-shaped area of hypodensity along the inferolateral aspect of the spleen measuring 6 × 3.8 cm, consistent with a recent infarct (Figure 1). There was also evidence of a previous infarct in the posterolateral aspect of the spleen. Splenic, celiac, superior mesenteric, and inferior mesenteric arteries were patent.
1. Given these findings, which of the following diagnoses should be considered?
- Subacute infective endocarditis
- Inherited thrombophilia
- Antiphospholipid syndrome
All three diagnoses should be considered in this case.
Endocarditis
Embolism from a source in the heart caused by subacute bacterial endocarditis is more common than the other two conditions listed here and must be excluded.
Our patient lacks key features of this condition: he has no predisposing factors (artificial valve, cyanotic congenital heart disease, previous endocarditis, intravenous drug abuse); no constitutional symptoms of fever, night sweats, and weight loss; no findings on examination of skin and cardiovascular systems; and a normal white blood cell count. Nevertheless, even though the absence of these features makes bacterial endocarditis unlikely, it does not exclude it. Blood cultures and transesophageal echocardiography are indicated to rule out bacterial endocarditis.
We obtained serial blood cultures, which were negative, and transesophageal echocardiography showed normal valves and no evidence of thrombus or vegetation, thus excluding a cardiac source of emboli.
Thrombophilia
Our patient has a history of recurrent thromboembolic episodes, and this warrants testing to rule out an inherited thrombophilia. A family history of thromboembolic disease should also be sought.1
In our patient, tests for prothrombotic activity including protein C chromogen, activated protein C ratio, free protein S, functional protein S, antithrombin factor V Leiden, and the prothrombin 20210G>A mutation were either negative or within the reference range. A negative family history of thromboembolic disease and the negative laboratory tests make inherited thrombophilia unlikely in our patient.
Sickle cell disease, polycythemia vera, and essential thrombocythemia may also cause splenic infarction but can be ruled out in this patient on the basis of history and initial blood tests.
Antiphospholipid syndrome
A history of vascular disease (aortofemoral bypass surgery), a recent splenic infarct, and an elevated aPTT makes antiphospholipid syndrome the likeliest diagnosis in this patient.
Appropriate tests are for lupus anticoagulant, immunoglobulin G (IgG) or IgM cardiolipin antibody, and beta-2 glycoprotein 1 (beta-2 GP1) antibody, as well as the dilute Russell viper venom time (dRVVT) and the dRVVT ratio. The IgG and IgM cardiolipin antibody and beta-2 GP1 antibody tests have the same diagnostic value, and only medium to high titers should be considered positive.
Our patient’s IgG cardiolipin antibody level was in the normal range at 15 IgG phospholipid units (reference range 0–22); his IgM cardiolipin antibody level was high at 41 IgM phospholipid units (0–10). The dRVVT was 57 seconds (24–42), and the dRVVT ratio was 2.0 (0.0–1.3).
2. What further investigations are indicated before starting treatment?
- No further investigations required
- Repeat testing for phospholipid antibodies in 12 weeks
- Test for antinuclear antibodies
Antiphospholipid antibodies may appear transiently in certain infections, such as syphilis, Lyme disease, Epstein-Barr virus, cytomegalovirus, hepatitis C, and human immunodeficiency virus. Therefore, the presence of antiphospholipid antibodies must be confirmed over time, with two positive results at least 12 weeks apart.2
When repeated 12 weeks later, our patient’s IgG anticardiolipin antibody level was 14 GPL units, and the IgM anticardiolipin antibody level was 30 MPL units; the dRVVT was 55 seconds, and the dRVVT ratio was 1.8. These results, along with a history of recurrent arterial thrombosis, confirmed antiphospholipid syndrome.
The 2009 update of the International Society of Thrombosis and Haemostasis guidelines recommend two tests, the dRVVT and the aPTT, since no single test is 100% sensitive for lupus anticoagulant.3 The dRVVT has a high specificity for lupus anticoagulant in patients at high risk of thrombosis.
A SYNDROME WITH A WIDE RANGE OF EFFECTS AND COMPLICATIONS
Antiphospholipid syndrome is a systemic autoimmune disease that manifests as arterial and venous thrombosis and as obstetric complications. Thrombosis tends to be recurrent and may involve any site. For example, it can cause blurred vision in one or both eyes; amaurosis fugax; visual field defects; central or branch retinal artery or vein occlusion; deep vein thrombosis; pulmonary embolism; myocardial infarction; transient ischemic attack and stroke; cerebral vein thrombosis; and portal, renal, and mesenteric infarction involving veins or arteries.4 Pulmonary capillaritis may cause diffuse alveolar hemorrhage. Livedo reticularis, digital gangrene, cutaneous necrosis, splinter hemorrhages, chorea, and transverse myelopathy may also occur.
Obstetric complications of antiphospholipid syndrome include recurrent miscarriage and pregnancy loss at or after 10 weeks of gestation, eclampsia, preeclampsia, and placental insufficiency.5 The syndrome also has a potentially lethal variant characterized by multiorgan thrombosis affecting mainly small vessels.
The diagnosis of antiphospholipid syndrome requires relevant clinical features and symptoms and the presence of at least one of the antiphospholipid antibodies. Because the rate of false-positive tests for antiphospholipid antibodies ranges from 3% to 20% in the general population, asymptomatic patients should not be tested.6
Antiphospholipid syndrome may occur in the setting of other autoimmune diseases, most commonly systemic lupus erythematosus, when it is termed “secondary” antiphospholipid syndrome. Although only 40% of patients with lupus have antiphospholipid antibodies and less than 40% will have a thrombotic event, thrombotic antiphospholipid syndrome is a major adverse prognostic factor in these patients.7,8 Therefore, it is prudent to consider systemic lupus erythematosus and to do appropriate tests if the patient has other features suggestive of lupus, such as renal, skin, or musculoskeletal lesions.
In our patient, antinuclear antibody testing was positive, with a titer of 1:320, and showed a finely speckled staining pattern. Tests for antibodies to Sjögren syndrome A and B antigens were negative. The complement C3 level was 1.28 g/L (reference range 0.74–1.85) and the C4 level was 0.24 g/L (0.16–0.44). Although the speckled staining pattern can be seen in lupus, it is more common in Sjögren syndrome, mixed connective tissue disease, scleroderma, and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia).9 Moreover, normal levels of complement C3 and C4, in the absence of clinical features, make lupus unlikely. Similarly, our patient had no clinical features of other connective tissue disorders. Therefore, he had primary antiphospholipid syndrome.
3. How should this patient be managed?
- Antiplatelet therapy
- Warfarin to maintain an INR between 2.0 and 3.0
- Warfarin to maintain an INR above 3.0
The risk of recurrent thrombosis is high in patients who test positive for lupus anticoagulant, and the risk is highest in patients who are also positive for anticardiolipin and anti-beta-2 GP1 antibodies: the incidence of thrombosis is 12.2% at 1 year, 26.1% at 5 years, 44.2% at 10 years.10
Since our patient is positive for lupus anticoagulant (prolonged aPTT and elevated dRVVT, both indicating lupus anticoagulant positivity) and for anticardiolipin antibodies (anti-beta-2 GP1 not tested), his risk of recurrent thrombosis is high, and he requires lifelong anticoagulation therapy.
The intensity of anticoagulation in different subgroups of patients is controversial. Based on retrospective trials, indefinite anticoagulation at an INR of 2.0 to 3.0 has been suggested for patients with antiphospholipid syndrome presenting with venous thrombosis, and more intense anticoagulation with an INR above 3.0 in patients with recurrent or arterial thrombosis.11 The combination of warfarin with an INR between 2.0 and 3.0 and aspirin 100 mg daily has also been proposed for patients with arterial thrombosis.12
Modifiable risk factors such as smoking, obesity, and use of estrogens should be addressed in all patients with antiphospholipid syndrome.
In pregnant women with complications such as preeclampsia, low-dose aspirin can be used, and in women with a history of miscarriage, the combination of low-dose aspirin and heparin is recommended throughout the prenatal period.4
In patients who have recurrent thrombosis despite adequate anticoagulation, an expert committee12 has proposed that alternative regimens could include long-term low-molecular-weight heparin instead of warfarin, the combination of warfarin and aspirin, or warfarin and hydroxychloroquine. Adding a statin can also be considered.
Treatment of catastrophic antiphospholipid syndrome is based on expert opinion. A combination of anticoagulation, corticosteroids, plasma exchange, intravenous immunoglobulins, and rituximab has been tried, but the mortality rate remains high.13
OUR PATIENT'S COURSE
Our patient was started on warfarin, with a target INR above 3.0, and was doing well at 6 months of follow-up.
- De Stefano V, Rossi E. Testing for inherited thrombophilia and consequences for antithrombotic prophylaxis in patients with venous thromboembolism and their relatives. A review of the Guidelines from Scientific Societies and Working Groups. Thromb Haemost 2013; 110:697–705.
- Galli M. Interpretation and recommended testing for antiphospholipid antibodies. Semin Thromb Hemost 2012; 38:348–352.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Keeling D, Mackie I, Moore GW, Greer IA, Greaves M; British Committee for Standards in Haematology. Guidelines on the investigation and management of antiphospholipid syndrome. Br J Haematol 2012; 157:47–58.
- Misita CP, Moll S. Antiphospholipid antibodies. Circulation 2005; 112:e39–e44.
- Rand JH, Wolgast LR. Do’s and don’t’s in diagnosing antiphospholipid syndrome. Hematology Am Soc Hematol Educ Program 2012; 2012:455–459.
- Mok CC, Tang SS, To CH, Petri M. Incidence and risk factors of thromboembolism in systemic lupus erythematosus: a comparison of three ethnic groups. Arthritis Rheum 2005; 52:2774–2782.
- Ruiz-Irastorza G, Egurbide MV, Ugalde J, Aguirre C. High impact of antiphospholipid syndrome on irreversible organ damage and survival of patients with systemic lupus erythematosus. Arch Intern Med 2004; 164:77–82.
- Locht H, Pelck R, Manthorpe R. Clinical manifestations correlated to the prevalence of autoantibodies in a large (n = 321) cohort of patients with primary Sjögren’s syndrome: a comparison of patients initially diagnosed according to the Copenhagen classification criteria with the American-European consensus criteria. Autoimmun Rev 2005; 4:276–281.
- Pengo V, Ruffatti A, Legnani C, et al. Clinical course of high-risk patients diagnosed with antiphospholipid syndrome. J Thromb Haemost 2010; 8:237–242.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza I, et al. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: report of a task force at the 13th International Congress on antiphospholipid antibodies. Lupus 2011; 20:206–218.
- Cervera R. Update on the diagnosis, treatment, and prognosis of the catastrophic antiphospholipid syndrome. Curr Rheumatol Rep 2010; 12:70–76.
- De Stefano V, Rossi E. Testing for inherited thrombophilia and consequences for antithrombotic prophylaxis in patients with venous thromboembolism and their relatives. A review of the Guidelines from Scientific Societies and Working Groups. Thromb Haemost 2013; 110:697–705.
- Galli M. Interpretation and recommended testing for antiphospholipid antibodies. Semin Thromb Hemost 2012; 38:348–352.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Keeling D, Mackie I, Moore GW, Greer IA, Greaves M; British Committee for Standards in Haematology. Guidelines on the investigation and management of antiphospholipid syndrome. Br J Haematol 2012; 157:47–58.
- Misita CP, Moll S. Antiphospholipid antibodies. Circulation 2005; 112:e39–e44.
- Rand JH, Wolgast LR. Do’s and don’t’s in diagnosing antiphospholipid syndrome. Hematology Am Soc Hematol Educ Program 2012; 2012:455–459.
- Mok CC, Tang SS, To CH, Petri M. Incidence and risk factors of thromboembolism in systemic lupus erythematosus: a comparison of three ethnic groups. Arthritis Rheum 2005; 52:2774–2782.
- Ruiz-Irastorza G, Egurbide MV, Ugalde J, Aguirre C. High impact of antiphospholipid syndrome on irreversible organ damage and survival of patients with systemic lupus erythematosus. Arch Intern Med 2004; 164:77–82.
- Locht H, Pelck R, Manthorpe R. Clinical manifestations correlated to the prevalence of autoantibodies in a large (n = 321) cohort of patients with primary Sjögren’s syndrome: a comparison of patients initially diagnosed according to the Copenhagen classification criteria with the American-European consensus criteria. Autoimmun Rev 2005; 4:276–281.
- Pengo V, Ruffatti A, Legnani C, et al. Clinical course of high-risk patients diagnosed with antiphospholipid syndrome. J Thromb Haemost 2010; 8:237–242.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza I, et al. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: report of a task force at the 13th International Congress on antiphospholipid antibodies. Lupus 2011; 20:206–218.
- Cervera R. Update on the diagnosis, treatment, and prognosis of the catastrophic antiphospholipid syndrome. Curr Rheumatol Rep 2010; 12:70–76.
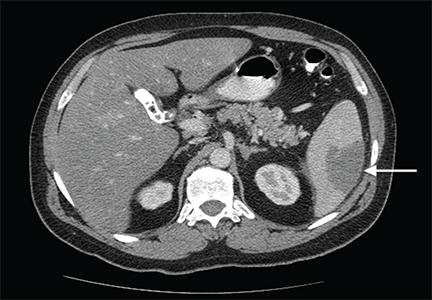
A 61-year-old man with fluctuating hypertension
A 61-year-old man with type 2 diabetes mellitus on glimepiride therapy presented with somnolence and slurred speech. His capillary glucose level was 17 mg/dL and his serum glucose level was 28 mg/dL. He was treated with intravenous dextrose, and his glucose level promptly returned to normal.
He had been adherent to his medication regimen and denied overmedicating or accidental overdosing. Over the past 7 months, he had noted redness on his palms, a rash on his legs, intermittent moderate to severe headaches, weight loss, and decreased appetite. In addition, his blood pressure had been labile, which his physicians had attributed to autonomic instability. He had continued on the same dose of glimepiride despite losing weight.
His history included multivessel coronary artery disease treated with angioplasty and placement of multiple coronary stents; ischemic cardiomyopathy with a left ventricular ejection fraction of 28%; implantation of a cardioverter-defibrillator for secondary prevention of ventricular arrhythmia; an ischemic stroke; and multiple sclerosis complicated by bilateral blindness, with optic nerve involvement and autonomic instability, present for over a year and manifested by labile blood pressure. He was a long-time tobacco user. His daily medications included ticagrelor 90 mg, aspirin 81 mg, metoprolol 50 mg, ramipril 10 mg, simvastatin 20 mg, glimepiride 2 mg, and esomeprazole 40 mg. He needed help taking his medications.
At the time of hospital admission, his heart rate was 69 beats per minute with a regular rhythm, blood pressure 115/73 mm Hg, respiratory rate 11 breaths per minute with an oxygen saturation of 99% on room air, and oral temperature 34.7°C (94.5°F). He appeared to be in no distress.
Cardiovascular examination revealed no murmurs or gallops; there was mild nonpitting edema of the lower extremities. Pulmonary, abdominal, and neurologic examinations were unrevealing except for bilateral blindness. Vascular examination revealed no bruits. Results of a complete blood cell count and metabolic panel were normal except for a hemoglobin level of 9.9 g/dL (reference range 13.5–17.5) and a platelet count of 477 × 109/L (150–450).
Although he continued to receive the same medications he had been taking at home, his blood pressure fluctuated. On the second hospital day, it reached 186/135 mm Hg, at which time he also had palpitations, dyspnea, and crackles in the lower lobes of both lungs. Volume resuscitation on admission was suspected to have played a role, and he received furosemide, which improved his symptoms. But several hours later, his blood pressure rose again, and he became diaphoretic. Despite aggressive treatment with different antihypertensive agents, his blood pressure remained high and his symptoms persisted. Chest radiography showed no evidence of pulmonary edema. Because of his progressive dyspnea, the diagnosis of pulmonary embolism was entertained.
CAUSES OF RESISTANT HYPERTENSION
1. What could explain this patient’s high blood pressure?
- A drug effect
- Renovascular disease
- Excess circulating catecholamines
- Obstructive sleep apnea
- Primary aldosteronism
Sympathomimetic drugs such as epinephrine, norepinephrine, dopamine, and vasopressin, which are used when hemodynamic support is required, can raise both systolic and diastolic blood pressure. Nonsteroidal anti-inflammatory drugs and nasal decongestants are common culprits in the community. However, our patient was using none of these drugs.
Renovascular disease is one of many causes of resistant hypertension, accounting for 8% of all cases.1,2 Despite fluctuations, the blood pressure often remains chronically elevated, its changes are less paroxysmal than in our patient, and a precipitating factor such as a dietary indiscretion is sometimes identified.1
Excess circulating catecholamines can be a result of stress, exogenous administration, or endogenous oversecretion. Our patient’s clinical presentation is highly suspicious for a high-catecholamine state, and this should be further evaluated.
Obstructive sleep apnea is common in patients with resistant hypertension, with an estimated prevalence as high as 60% in this group.3,4
Primary aldosteronism has an estimated prevalence of about 20% in patients evaluated for resistant hypertension.5
AN ADRENAL MASS IS INCIDENTALLY DISCOVERED
Computed tomographic angiography of the chest revealed no evidence of pulmonary emboli. There was mild dilation of the central pulmonary arteries and an incidental, incompletely imaged 4.7-by-3.4-cm mass of mixed attenuation in the right adrenal gland, with macroscopic fat within the lesion.
Computed tomography (CT) of the abdomen with dedicated cuts through the adrenal glands revealed a 4.7-cm heterogeneous right adrenal mass with a density of 34 Hounsfield units (HU). The left adrenal gland appeared diffusely enlarged without a discretely seen mass, consistent with hyperplasticity (Figure 1).
2. Based on the patient’s clinical presentation and findings on CT, what would be the most likely diagnosis for this incidentally found adrenal mass?
- Adrenocortical adenoma
- Adrenocortical carcinoma
- Metastatic mass
- Pheochromocytoma
Adrenocortical adenoma can present as a small homogeneous mass of variable size, with smooth margins, and rarely containing hemorrhagic tissue or calcifications. The typical density on nonenhanced CT is less than 10 HU. On enhanced CT, it is nonvascular. T2-weighted magnetic resonance imaging (MRI) shows a lesion of the same intensity as liver tissue.6
Adrenocortical adenoma is not classically associated with autologous activity and thus is less likely to explain our patient’s symptoms.
Adrenocortical carcinoma can present as a large heterogeneous mass, usually greater than 4 cm in diameter, with irregular margins and areas of necrosis, hemorrhage, or calcification. The typical density on nonenhanced CT is greater than 10 HU. On enhanced CT, the mass is usually vascular, and T2-weighted MRI will show a lesion more intense than liver tissue.6
Adrenocortical carcinoma is also not classically associated with autologous activity, and so is not likely to explain our patient’s symptoms.6
Metastatic disease can present with masses of variable size, often bilaterally, and occasionally with cysts or areas of hemorrhage. The typical density of metastatic lesions on nonenhanced CT is greater than 10 HU. On enhanced CT, they are usually vascular, and on T2-weighted MRI they are hyperintense.6 The characteristics of the mass and the absence of a primary malignancy on CT of the chest and abdomen do not support the diagnosis of metastatic disease.
Pheochromocytoma is a neuroendocrine tumor of the adrenal medulla that can present as a large heterogeneous mass, greater than 3 cm in diameter, with clear margins and cysts or areas of hemorrhage. Extra-adrenal neuroendocrine tumors are typically called paragangliomas and have features similar to those of pheochromocytoma. The typical density of pheochromocytoma on nonenhanced CT is greater than 10 HU. On enhanced CT, it is usually vascular, and T2-weighted MRI shows a hyperintense lesion. Pheochromocytoma can be biochemically active and thus can cause signs and symptoms that will lead to the diagnosis.6
Other imaging tests may play a role in the evaluation of adrenal masses but are not required for the diagnosis of pheochromocytoma. Functional positron emission tomography using metaiodobenzylguanidine labeled with iodine 123 or-iodine 131 or using the glucose analogue F-18 fluorodeoxyglucose has been used in the initial assessment of pheochromocytoma, with good sensitivity and specificity.7,8
Our patient’s pacemaker-defibrillator precluded him from undergoing MRI.
DIAGNOSIS: PHEOCHROMOCYTOMA
Pheochromocytoma was highly suspected on the basis of the patient’s clinical presentation, and metoprolol was immediately discontinued. He was started on the calcium channel blocker verapamil and the alpha-blocker phenoxybenzamine.
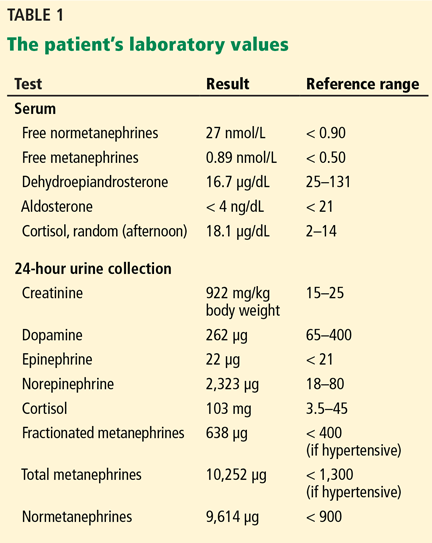
Serum samples were obtained to measure metanephrines, dehydroepiandrosterone, aldosterone, and cortisol, and a 24-hour urine collection was obtained to measure creatinine, dopamine, epinephrine, norepinephrine, cortisol, and metanephrines. Based on the results (Table 1) and on the findings on imaging, the patient was diagnosed with pheochromocytoma. A surgical consultation was obtained, and surgery was recommended.
WHEN DOES PHEOCHROMOCYTOMA CALL FOR SURGERY?
3. Which criterion is most important when determining the need for surgery for pheochromocytoma?
- Findings on fine-needle aspiration biopsy
- Biochemical activity
- Size of the mass
- Bilateral masses
Fine-needle aspiration biopsy can be done when a mass is found incidentally and no evidence of biochemical activity is detected, although it is not an essential part of the diagnostic workup.9 In most cases, the sampling from fine-needle aspiration is not sufficient to achieve a diagnosis.
Biochemical activity is the most important factor when determining the need for prompt surgical intervention. The excess circulating catecholamines have been associated with increased risk of cardiovascular morbidity and death independent of the morbidity associated with hypertension alone.10 Biochemical activity can be independent of the size of the mass, but larger masses typically present with symptoms.
Bilateral masses have been associated with metastatic disease.11 In retrospect, the patient’s history of hypertension and cerebrovascular accident could be associated with the development of a catecholamine-releasing tumor.
A GOOD OUTCOME FROM SURGERY
The patient was continued on phenoxybenzamine for 7 days and responded well to this therapy.
After this preoperative preparation, he underwent laparoscopic right adrenalectomy with excision of a retroperitoneal adrenal mass. His postoperative course was complicated by transient hypotension requiring low-dose vasopressin support for less than 24 hours. He was then restarted on his previous dosage of metoprolol and was discharged home on postoperative day 5 with stable blood pressure. Follow-up 24-hour urine collection 1 month after he was discharged showed normalization of metanephrine, normetanephrine, epinephrine, and norepinephrine levels.
Despite low suspicion for an underlying genetic syndrome, he was referred for genetic testing and was scheduled to have a repeat 24-hour urine collection and imaging in 6 months to follow his enlarged left adrenal gland, which did not appear to be metabolically hyperactive.
4. What is the most common perioperative complication of pheochromocytoma excision?
- Hypoglycemia
- Hypotension
- Hypocortisolism
- Hypertension
- Tachycardia
Hypoglycemia has been observed after removal of pheochromocytoma, as levels of catecholamines (which normally inhibit pancreatic beta cells) decrease and insulin secretion consequently increases.12 Our patient developed hypoglycemia before surgery, not after, and it was likely due to the combination of his antidiabetic therapy, weight loss, and decreased oral intake.
Hypotension is the most common complication in the perioperative period. It is associated with excessive loss of catecholamine secretion. It is usually short-lived but may require aggressive administration of intravenous fluids and use of sympathomimetic agents.
Hypocortisolism is unlikely in patients with pheochromocytoma, but it is likely after removal of adrenocortical adenoma.
Hypertension and tachycardia affect up to 40% of pheochromocytoma patients in some case series.12
PHEOCHROMOCYTOMA: A CATECHOLAMINE-SECRETING TUMOR
The pathophysiology of pheochromocytoma is complex. It is characterized by accelerated growth of cells producing catecholamines, which may produce symptoms when secreted into the bloodstream. The classic triad of symptoms is headache, hypertension, and hyperglycemia, although our patient had very low blood sugar levels. Other common symptoms are nausea, orthostasis, and tremor, although not all symptoms are invariably seen.
Genetic testing recommended
Genetic associations have been described and are thought to be responsible for 20% to 30% of cases of pheochromocytoma. All associated germline mutations are autosomal dominant, some with variable penetrance. These include:
- Succinate dehydrogenase subunit B, C, and D mutations
- von Hippel-Lindau syndrome
- Multiple endocrine neoplasia type 1 and type 2 syndromes
- Neurofibromatosis type 1.13,14
The succinate dehydrogenase subunit mutations have been associated with, but not limited to, extra-adrenal adenomas (paragangliomas) and carry a worse prognosis.
Some experts recommend genetic testing in all cases of pheochromocytoma, sporadic or familial, and this testing should be followed by counseling if a mutation is found.15 Others recommend genetic testing based on the patient’s age (under age 50), history, imaging, and biochemical features of the tumor (metanephrines predominate in multiple endocrine neoplasia syndromes, and normetanephrines in von Hippel-Lindau syndrome).13
Serious consequences
A thorough evaluation is recommended, since pheochromocytoma has been associated with increased cardiovascular morbidity. In a retrospective series, Stolk et al10 reported that patients with pheochromocytoma had a higher incidence of myocardial infarction, angina, and stroke in the years preceding the diagnosis than did patients with essential hypertension (13.8% vs 1.1%, P < .001).10
Catecholamine cardiomyopathy has been described and shares clinical features with Takotsubo or stress cardiomyopathy, with global left ventricular systolic and diastolic dysfunction that improve or resolve after the adrenergic insult is removed.16
Conditions that warrant further evaluation or that may suggest pheochromocytoma are malignant hypertension, hypertensive encephalopathy, ischemic stroke, subarachnoid hemorrhage, acute pulmonary edema, angina pectoris, myocardial infarction, aortic dissection, and kidney injury.
When to suspect pheochromocytoma
Pheochromocytoma should be suspected in a patient with resistant hypertension, family history, or imaging findings that suggest an adrenal mass with a heterogeneous appearance. The diagnostic algorithm follows the same pathway as for the evaluation of an incidentally found adrenal mass, with determination of its dimension and characteristics by CT or MRI, and with biochemical testing of urine catecholamines, plasma free metanephrines, renin, aldosterone, and cortisol.
The diagnosis of pheochromocytoma is established by obtaining fractionated metanephrines and catecholamines in a 24-hour urine collection (sensitivity 90%, specificity 98%). Analysis of plasma metanephrines has a higher sensitivity (97%) but lower specificity (85%).17 The combination of typical signs, symptoms, and laboratory findings makes the diagnosis likely, especially in combination with a unilateral adrenal mass.
Laparoscopic surgery after medical preparation for active tumors
If the mass appears benign and not biochemically hyperactive, then follow-up at 1 year is recommended, with repeat testing. Surgical evaluation and intervention is recommended for lesions that appear malignant or that are biochemically active and clinically symptomatic.9
Preoperative hemodynamic control is essential in the management of pheochromocytoma to prevent or minimize hemodynamic changes that can be driven by increased catecholamines. Control is typically achieved with initial alpha-blockade and then beta-blockade to avoid worsening hypertension and to prevent an acute hypertensive crisis during surgical intervention. Phenoxybenzamine, the mainstay of therapy, is a nonselective alpha-blocker with a long duration of action that requires titration over several days up to 3 weeks.
A selective alpha-1-blocker such as doxazosin can be used to control postoperative hypotension, as it has a shorter half-life than phenoxybenzamine. Alternative strategies include calcium channel blockers, centrally acting sympathetic blockers, and magnesium.18
Laparoscopic adrenalectomy by an experienced surgeon after excellent medical preparation is often considered the treatment of choice, but for larger or malignant masses, an open procedure is recommended. The risk of perioperative morbidity and death can be reduced by adequate medical management. With successful surgical resection, the long-term prognosis is favorable.
- Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2008; 51:1403–1419.
- Kumar N, Calhoun DA, Dudenbostel T. Management of patients with resistant hypertension: current treatment options. Integr Blood Press Control 2013; 6:139–151.
- Pedrosa RP, Drager LF, Gonzaga CC, et al. Obstructive sleep apnea: the most common secondary cause of hypertension associated with resistant hypertension. Hypertension 2011; 58:811–817.
- Marcus JA, Pothineni A, Marcus CZ, Bisognano JD. The role of obesity and obstructive sleep apnea in the pathogenesis and treatment of resistant hypertension. Curr Hypertens Rep 2014; 16:411.
- Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB, Weissmann P. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension 2002; 40:892–896.
- Young WF Clinical practice. The incidentally discovered adrenal mass. N Engl J Med 2007; 356:601–610.
- Lin M, Wong V, Yap J, Jin R, Leong P, Campbell P. FDG PET in the evaluation of phaeochromocytoma: a correlative study with MIBG scintigraphy and Ki-67 proliferative index. Clin Imaging 2013; 37:1084–1088.
- Raja A, Leung K, Stamm M, Girgis S, Low G. Multimodality imaging findings of pheochromocytoma with associated clinical and biochemical features in 53 patients with histologically confirmed tumors. AJR Am J Roentgenol 2013; 201:825–833.
- Nieman LK. Approach to the patient with an adrenal incidentaloma. J Clin Endocrinol Metab 2010; 95:4106–4113.
- Stolk RF, Bakx C, Mulder J, Timmers HJ, Lenders JW. Is the excess cardiovascular morbidity in pheochromocytoma related to blood pressure or to catecholamines? J Clin Endocrinol Metab 2013; 98:1100–1106.
- Grumbach MM, Biller BM, Braunstein GD, et al. Management of the clinically inapparent adrenal mass (‘incidentaloma’). Ann Intern Med 2003; 138:424–429.
- Lentschener C, Gaujoux S, Tesniere A, Dousset B. Point of controversy: perioperative care of patients undergoing pheochromocytoma removal—time for a reappraisal? Eur J Endocrinol 2011; 165:365–373.
- Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst 2003; 95:1196–1204.
- Lee P, Leonard J. Textbook on endocrinology. BMJ 1994; 308:1512.
- Fishbein L, Merrill S, Fraker DL, Cohen DL, Nathanson KL. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol 2013; 20:1444–1450.
- Agarwal G, Sadacharan D, Kapoor A, et al. Cardiovascular dysfunction and catecholamine cardiomyopathy in pheochromocytoma patients and their reversal following surgical cure: results of a prospective case-control study. Surgery 2011; 150:1202–1211.
- Sawka AM, Jaeschke R, Singh RJ, Young WF A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 2003; 88:553–558.
- Domi R, Laho H. Management of pheochromocytoma: old ideas and new drugs. Niger J Clin Pract 2012; 15:253–257.
A 61-year-old man with type 2 diabetes mellitus on glimepiride therapy presented with somnolence and slurred speech. His capillary glucose level was 17 mg/dL and his serum glucose level was 28 mg/dL. He was treated with intravenous dextrose, and his glucose level promptly returned to normal.
He had been adherent to his medication regimen and denied overmedicating or accidental overdosing. Over the past 7 months, he had noted redness on his palms, a rash on his legs, intermittent moderate to severe headaches, weight loss, and decreased appetite. In addition, his blood pressure had been labile, which his physicians had attributed to autonomic instability. He had continued on the same dose of glimepiride despite losing weight.
His history included multivessel coronary artery disease treated with angioplasty and placement of multiple coronary stents; ischemic cardiomyopathy with a left ventricular ejection fraction of 28%; implantation of a cardioverter-defibrillator for secondary prevention of ventricular arrhythmia; an ischemic stroke; and multiple sclerosis complicated by bilateral blindness, with optic nerve involvement and autonomic instability, present for over a year and manifested by labile blood pressure. He was a long-time tobacco user. His daily medications included ticagrelor 90 mg, aspirin 81 mg, metoprolol 50 mg, ramipril 10 mg, simvastatin 20 mg, glimepiride 2 mg, and esomeprazole 40 mg. He needed help taking his medications.
At the time of hospital admission, his heart rate was 69 beats per minute with a regular rhythm, blood pressure 115/73 mm Hg, respiratory rate 11 breaths per minute with an oxygen saturation of 99% on room air, and oral temperature 34.7°C (94.5°F). He appeared to be in no distress.
Cardiovascular examination revealed no murmurs or gallops; there was mild nonpitting edema of the lower extremities. Pulmonary, abdominal, and neurologic examinations were unrevealing except for bilateral blindness. Vascular examination revealed no bruits. Results of a complete blood cell count and metabolic panel were normal except for a hemoglobin level of 9.9 g/dL (reference range 13.5–17.5) and a platelet count of 477 × 109/L (150–450).
Although he continued to receive the same medications he had been taking at home, his blood pressure fluctuated. On the second hospital day, it reached 186/135 mm Hg, at which time he also had palpitations, dyspnea, and crackles in the lower lobes of both lungs. Volume resuscitation on admission was suspected to have played a role, and he received furosemide, which improved his symptoms. But several hours later, his blood pressure rose again, and he became diaphoretic. Despite aggressive treatment with different antihypertensive agents, his blood pressure remained high and his symptoms persisted. Chest radiography showed no evidence of pulmonary edema. Because of his progressive dyspnea, the diagnosis of pulmonary embolism was entertained.
CAUSES OF RESISTANT HYPERTENSION
1. What could explain this patient’s high blood pressure?
- A drug effect
- Renovascular disease
- Excess circulating catecholamines
- Obstructive sleep apnea
- Primary aldosteronism
Sympathomimetic drugs such as epinephrine, norepinephrine, dopamine, and vasopressin, which are used when hemodynamic support is required, can raise both systolic and diastolic blood pressure. Nonsteroidal anti-inflammatory drugs and nasal decongestants are common culprits in the community. However, our patient was using none of these drugs.
Renovascular disease is one of many causes of resistant hypertension, accounting for 8% of all cases.1,2 Despite fluctuations, the blood pressure often remains chronically elevated, its changes are less paroxysmal than in our patient, and a precipitating factor such as a dietary indiscretion is sometimes identified.1
Excess circulating catecholamines can be a result of stress, exogenous administration, or endogenous oversecretion. Our patient’s clinical presentation is highly suspicious for a high-catecholamine state, and this should be further evaluated.
Obstructive sleep apnea is common in patients with resistant hypertension, with an estimated prevalence as high as 60% in this group.3,4
Primary aldosteronism has an estimated prevalence of about 20% in patients evaluated for resistant hypertension.5
AN ADRENAL MASS IS INCIDENTALLY DISCOVERED
Computed tomographic angiography of the chest revealed no evidence of pulmonary emboli. There was mild dilation of the central pulmonary arteries and an incidental, incompletely imaged 4.7-by-3.4-cm mass of mixed attenuation in the right adrenal gland, with macroscopic fat within the lesion.
Computed tomography (CT) of the abdomen with dedicated cuts through the adrenal glands revealed a 4.7-cm heterogeneous right adrenal mass with a density of 34 Hounsfield units (HU). The left adrenal gland appeared diffusely enlarged without a discretely seen mass, consistent with hyperplasticity (Figure 1).
2. Based on the patient’s clinical presentation and findings on CT, what would be the most likely diagnosis for this incidentally found adrenal mass?
- Adrenocortical adenoma
- Adrenocortical carcinoma
- Metastatic mass
- Pheochromocytoma
Adrenocortical adenoma can present as a small homogeneous mass of variable size, with smooth margins, and rarely containing hemorrhagic tissue or calcifications. The typical density on nonenhanced CT is less than 10 HU. On enhanced CT, it is nonvascular. T2-weighted magnetic resonance imaging (MRI) shows a lesion of the same intensity as liver tissue.6
Adrenocortical adenoma is not classically associated with autologous activity and thus is less likely to explain our patient’s symptoms.
Adrenocortical carcinoma can present as a large heterogeneous mass, usually greater than 4 cm in diameter, with irregular margins and areas of necrosis, hemorrhage, or calcification. The typical density on nonenhanced CT is greater than 10 HU. On enhanced CT, the mass is usually vascular, and T2-weighted MRI will show a lesion more intense than liver tissue.6
Adrenocortical carcinoma is also not classically associated with autologous activity, and so is not likely to explain our patient’s symptoms.6
Metastatic disease can present with masses of variable size, often bilaterally, and occasionally with cysts or areas of hemorrhage. The typical density of metastatic lesions on nonenhanced CT is greater than 10 HU. On enhanced CT, they are usually vascular, and on T2-weighted MRI they are hyperintense.6 The characteristics of the mass and the absence of a primary malignancy on CT of the chest and abdomen do not support the diagnosis of metastatic disease.
Pheochromocytoma is a neuroendocrine tumor of the adrenal medulla that can present as a large heterogeneous mass, greater than 3 cm in diameter, with clear margins and cysts or areas of hemorrhage. Extra-adrenal neuroendocrine tumors are typically called paragangliomas and have features similar to those of pheochromocytoma. The typical density of pheochromocytoma on nonenhanced CT is greater than 10 HU. On enhanced CT, it is usually vascular, and T2-weighted MRI shows a hyperintense lesion. Pheochromocytoma can be biochemically active and thus can cause signs and symptoms that will lead to the diagnosis.6
Other imaging tests may play a role in the evaluation of adrenal masses but are not required for the diagnosis of pheochromocytoma. Functional positron emission tomography using metaiodobenzylguanidine labeled with iodine 123 or-iodine 131 or using the glucose analogue F-18 fluorodeoxyglucose has been used in the initial assessment of pheochromocytoma, with good sensitivity and specificity.7,8
Our patient’s pacemaker-defibrillator precluded him from undergoing MRI.
DIAGNOSIS: PHEOCHROMOCYTOMA
Pheochromocytoma was highly suspected on the basis of the patient’s clinical presentation, and metoprolol was immediately discontinued. He was started on the calcium channel blocker verapamil and the alpha-blocker phenoxybenzamine.
Serum samples were obtained to measure metanephrines, dehydroepiandrosterone, aldosterone, and cortisol, and a 24-hour urine collection was obtained to measure creatinine, dopamine, epinephrine, norepinephrine, cortisol, and metanephrines. Based on the results (Table 1) and on the findings on imaging, the patient was diagnosed with pheochromocytoma. A surgical consultation was obtained, and surgery was recommended.
WHEN DOES PHEOCHROMOCYTOMA CALL FOR SURGERY?
3. Which criterion is most important when determining the need for surgery for pheochromocytoma?
- Findings on fine-needle aspiration biopsy
- Biochemical activity
- Size of the mass
- Bilateral masses
Fine-needle aspiration biopsy can be done when a mass is found incidentally and no evidence of biochemical activity is detected, although it is not an essential part of the diagnostic workup.9 In most cases, the sampling from fine-needle aspiration is not sufficient to achieve a diagnosis.
Biochemical activity is the most important factor when determining the need for prompt surgical intervention. The excess circulating catecholamines have been associated with increased risk of cardiovascular morbidity and death independent of the morbidity associated with hypertension alone.10 Biochemical activity can be independent of the size of the mass, but larger masses typically present with symptoms.
Bilateral masses have been associated with metastatic disease.11 In retrospect, the patient’s history of hypertension and cerebrovascular accident could be associated with the development of a catecholamine-releasing tumor.
A GOOD OUTCOME FROM SURGERY
The patient was continued on phenoxybenzamine for 7 days and responded well to this therapy.
After this preoperative preparation, he underwent laparoscopic right adrenalectomy with excision of a retroperitoneal adrenal mass. His postoperative course was complicated by transient hypotension requiring low-dose vasopressin support for less than 24 hours. He was then restarted on his previous dosage of metoprolol and was discharged home on postoperative day 5 with stable blood pressure. Follow-up 24-hour urine collection 1 month after he was discharged showed normalization of metanephrine, normetanephrine, epinephrine, and norepinephrine levels.
Despite low suspicion for an underlying genetic syndrome, he was referred for genetic testing and was scheduled to have a repeat 24-hour urine collection and imaging in 6 months to follow his enlarged left adrenal gland, which did not appear to be metabolically hyperactive.
4. What is the most common perioperative complication of pheochromocytoma excision?
- Hypoglycemia
- Hypotension
- Hypocortisolism
- Hypertension
- Tachycardia
Hypoglycemia has been observed after removal of pheochromocytoma, as levels of catecholamines (which normally inhibit pancreatic beta cells) decrease and insulin secretion consequently increases.12 Our patient developed hypoglycemia before surgery, not after, and it was likely due to the combination of his antidiabetic therapy, weight loss, and decreased oral intake.
Hypotension is the most common complication in the perioperative period. It is associated with excessive loss of catecholamine secretion. It is usually short-lived but may require aggressive administration of intravenous fluids and use of sympathomimetic agents.
Hypocortisolism is unlikely in patients with pheochromocytoma, but it is likely after removal of adrenocortical adenoma.
Hypertension and tachycardia affect up to 40% of pheochromocytoma patients in some case series.12
PHEOCHROMOCYTOMA: A CATECHOLAMINE-SECRETING TUMOR
The pathophysiology of pheochromocytoma is complex. It is characterized by accelerated growth of cells producing catecholamines, which may produce symptoms when secreted into the bloodstream. The classic triad of symptoms is headache, hypertension, and hyperglycemia, although our patient had very low blood sugar levels. Other common symptoms are nausea, orthostasis, and tremor, although not all symptoms are invariably seen.
Genetic testing recommended
Genetic associations have been described and are thought to be responsible for 20% to 30% of cases of pheochromocytoma. All associated germline mutations are autosomal dominant, some with variable penetrance. These include:
- Succinate dehydrogenase subunit B, C, and D mutations
- von Hippel-Lindau syndrome
- Multiple endocrine neoplasia type 1 and type 2 syndromes
- Neurofibromatosis type 1.13,14
The succinate dehydrogenase subunit mutations have been associated with, but not limited to, extra-adrenal adenomas (paragangliomas) and carry a worse prognosis.
Some experts recommend genetic testing in all cases of pheochromocytoma, sporadic or familial, and this testing should be followed by counseling if a mutation is found.15 Others recommend genetic testing based on the patient’s age (under age 50), history, imaging, and biochemical features of the tumor (metanephrines predominate in multiple endocrine neoplasia syndromes, and normetanephrines in von Hippel-Lindau syndrome).13
Serious consequences
A thorough evaluation is recommended, since pheochromocytoma has been associated with increased cardiovascular morbidity. In a retrospective series, Stolk et al10 reported that patients with pheochromocytoma had a higher incidence of myocardial infarction, angina, and stroke in the years preceding the diagnosis than did patients with essential hypertension (13.8% vs 1.1%, P < .001).10
Catecholamine cardiomyopathy has been described and shares clinical features with Takotsubo or stress cardiomyopathy, with global left ventricular systolic and diastolic dysfunction that improve or resolve after the adrenergic insult is removed.16
Conditions that warrant further evaluation or that may suggest pheochromocytoma are malignant hypertension, hypertensive encephalopathy, ischemic stroke, subarachnoid hemorrhage, acute pulmonary edema, angina pectoris, myocardial infarction, aortic dissection, and kidney injury.
When to suspect pheochromocytoma
Pheochromocytoma should be suspected in a patient with resistant hypertension, family history, or imaging findings that suggest an adrenal mass with a heterogeneous appearance. The diagnostic algorithm follows the same pathway as for the evaluation of an incidentally found adrenal mass, with determination of its dimension and characteristics by CT or MRI, and with biochemical testing of urine catecholamines, plasma free metanephrines, renin, aldosterone, and cortisol.
The diagnosis of pheochromocytoma is established by obtaining fractionated metanephrines and catecholamines in a 24-hour urine collection (sensitivity 90%, specificity 98%). Analysis of plasma metanephrines has a higher sensitivity (97%) but lower specificity (85%).17 The combination of typical signs, symptoms, and laboratory findings makes the diagnosis likely, especially in combination with a unilateral adrenal mass.
Laparoscopic surgery after medical preparation for active tumors
If the mass appears benign and not biochemically hyperactive, then follow-up at 1 year is recommended, with repeat testing. Surgical evaluation and intervention is recommended for lesions that appear malignant or that are biochemically active and clinically symptomatic.9
Preoperative hemodynamic control is essential in the management of pheochromocytoma to prevent or minimize hemodynamic changes that can be driven by increased catecholamines. Control is typically achieved with initial alpha-blockade and then beta-blockade to avoid worsening hypertension and to prevent an acute hypertensive crisis during surgical intervention. Phenoxybenzamine, the mainstay of therapy, is a nonselective alpha-blocker with a long duration of action that requires titration over several days up to 3 weeks.
A selective alpha-1-blocker such as doxazosin can be used to control postoperative hypotension, as it has a shorter half-life than phenoxybenzamine. Alternative strategies include calcium channel blockers, centrally acting sympathetic blockers, and magnesium.18
Laparoscopic adrenalectomy by an experienced surgeon after excellent medical preparation is often considered the treatment of choice, but for larger or malignant masses, an open procedure is recommended. The risk of perioperative morbidity and death can be reduced by adequate medical management. With successful surgical resection, the long-term prognosis is favorable.
A 61-year-old man with type 2 diabetes mellitus on glimepiride therapy presented with somnolence and slurred speech. His capillary glucose level was 17 mg/dL and his serum glucose level was 28 mg/dL. He was treated with intravenous dextrose, and his glucose level promptly returned to normal.
He had been adherent to his medication regimen and denied overmedicating or accidental overdosing. Over the past 7 months, he had noted redness on his palms, a rash on his legs, intermittent moderate to severe headaches, weight loss, and decreased appetite. In addition, his blood pressure had been labile, which his physicians had attributed to autonomic instability. He had continued on the same dose of glimepiride despite losing weight.
His history included multivessel coronary artery disease treated with angioplasty and placement of multiple coronary stents; ischemic cardiomyopathy with a left ventricular ejection fraction of 28%; implantation of a cardioverter-defibrillator for secondary prevention of ventricular arrhythmia; an ischemic stroke; and multiple sclerosis complicated by bilateral blindness, with optic nerve involvement and autonomic instability, present for over a year and manifested by labile blood pressure. He was a long-time tobacco user. His daily medications included ticagrelor 90 mg, aspirin 81 mg, metoprolol 50 mg, ramipril 10 mg, simvastatin 20 mg, glimepiride 2 mg, and esomeprazole 40 mg. He needed help taking his medications.
At the time of hospital admission, his heart rate was 69 beats per minute with a regular rhythm, blood pressure 115/73 mm Hg, respiratory rate 11 breaths per minute with an oxygen saturation of 99% on room air, and oral temperature 34.7°C (94.5°F). He appeared to be in no distress.
Cardiovascular examination revealed no murmurs or gallops; there was mild nonpitting edema of the lower extremities. Pulmonary, abdominal, and neurologic examinations were unrevealing except for bilateral blindness. Vascular examination revealed no bruits. Results of a complete blood cell count and metabolic panel were normal except for a hemoglobin level of 9.9 g/dL (reference range 13.5–17.5) and a platelet count of 477 × 109/L (150–450).
Although he continued to receive the same medications he had been taking at home, his blood pressure fluctuated. On the second hospital day, it reached 186/135 mm Hg, at which time he also had palpitations, dyspnea, and crackles in the lower lobes of both lungs. Volume resuscitation on admission was suspected to have played a role, and he received furosemide, which improved his symptoms. But several hours later, his blood pressure rose again, and he became diaphoretic. Despite aggressive treatment with different antihypertensive agents, his blood pressure remained high and his symptoms persisted. Chest radiography showed no evidence of pulmonary edema. Because of his progressive dyspnea, the diagnosis of pulmonary embolism was entertained.
CAUSES OF RESISTANT HYPERTENSION
1. What could explain this patient’s high blood pressure?
- A drug effect
- Renovascular disease
- Excess circulating catecholamines
- Obstructive sleep apnea
- Primary aldosteronism
Sympathomimetic drugs such as epinephrine, norepinephrine, dopamine, and vasopressin, which are used when hemodynamic support is required, can raise both systolic and diastolic blood pressure. Nonsteroidal anti-inflammatory drugs and nasal decongestants are common culprits in the community. However, our patient was using none of these drugs.
Renovascular disease is one of many causes of resistant hypertension, accounting for 8% of all cases.1,2 Despite fluctuations, the blood pressure often remains chronically elevated, its changes are less paroxysmal than in our patient, and a precipitating factor such as a dietary indiscretion is sometimes identified.1
Excess circulating catecholamines can be a result of stress, exogenous administration, or endogenous oversecretion. Our patient’s clinical presentation is highly suspicious for a high-catecholamine state, and this should be further evaluated.
Obstructive sleep apnea is common in patients with resistant hypertension, with an estimated prevalence as high as 60% in this group.3,4
Primary aldosteronism has an estimated prevalence of about 20% in patients evaluated for resistant hypertension.5
AN ADRENAL MASS IS INCIDENTALLY DISCOVERED
Computed tomographic angiography of the chest revealed no evidence of pulmonary emboli. There was mild dilation of the central pulmonary arteries and an incidental, incompletely imaged 4.7-by-3.4-cm mass of mixed attenuation in the right adrenal gland, with macroscopic fat within the lesion.
Computed tomography (CT) of the abdomen with dedicated cuts through the adrenal glands revealed a 4.7-cm heterogeneous right adrenal mass with a density of 34 Hounsfield units (HU). The left adrenal gland appeared diffusely enlarged without a discretely seen mass, consistent with hyperplasticity (Figure 1).
2. Based on the patient’s clinical presentation and findings on CT, what would be the most likely diagnosis for this incidentally found adrenal mass?
- Adrenocortical adenoma
- Adrenocortical carcinoma
- Metastatic mass
- Pheochromocytoma
Adrenocortical adenoma can present as a small homogeneous mass of variable size, with smooth margins, and rarely containing hemorrhagic tissue or calcifications. The typical density on nonenhanced CT is less than 10 HU. On enhanced CT, it is nonvascular. T2-weighted magnetic resonance imaging (MRI) shows a lesion of the same intensity as liver tissue.6
Adrenocortical adenoma is not classically associated with autologous activity and thus is less likely to explain our patient’s symptoms.
Adrenocortical carcinoma can present as a large heterogeneous mass, usually greater than 4 cm in diameter, with irregular margins and areas of necrosis, hemorrhage, or calcification. The typical density on nonenhanced CT is greater than 10 HU. On enhanced CT, the mass is usually vascular, and T2-weighted MRI will show a lesion more intense than liver tissue.6
Adrenocortical carcinoma is also not classically associated with autologous activity, and so is not likely to explain our patient’s symptoms.6
Metastatic disease can present with masses of variable size, often bilaterally, and occasionally with cysts or areas of hemorrhage. The typical density of metastatic lesions on nonenhanced CT is greater than 10 HU. On enhanced CT, they are usually vascular, and on T2-weighted MRI they are hyperintense.6 The characteristics of the mass and the absence of a primary malignancy on CT of the chest and abdomen do not support the diagnosis of metastatic disease.
Pheochromocytoma is a neuroendocrine tumor of the adrenal medulla that can present as a large heterogeneous mass, greater than 3 cm in diameter, with clear margins and cysts or areas of hemorrhage. Extra-adrenal neuroendocrine tumors are typically called paragangliomas and have features similar to those of pheochromocytoma. The typical density of pheochromocytoma on nonenhanced CT is greater than 10 HU. On enhanced CT, it is usually vascular, and T2-weighted MRI shows a hyperintense lesion. Pheochromocytoma can be biochemically active and thus can cause signs and symptoms that will lead to the diagnosis.6
Other imaging tests may play a role in the evaluation of adrenal masses but are not required for the diagnosis of pheochromocytoma. Functional positron emission tomography using metaiodobenzylguanidine labeled with iodine 123 or-iodine 131 or using the glucose analogue F-18 fluorodeoxyglucose has been used in the initial assessment of pheochromocytoma, with good sensitivity and specificity.7,8
Our patient’s pacemaker-defibrillator precluded him from undergoing MRI.
DIAGNOSIS: PHEOCHROMOCYTOMA
Pheochromocytoma was highly suspected on the basis of the patient’s clinical presentation, and metoprolol was immediately discontinued. He was started on the calcium channel blocker verapamil and the alpha-blocker phenoxybenzamine.
Serum samples were obtained to measure metanephrines, dehydroepiandrosterone, aldosterone, and cortisol, and a 24-hour urine collection was obtained to measure creatinine, dopamine, epinephrine, norepinephrine, cortisol, and metanephrines. Based on the results (Table 1) and on the findings on imaging, the patient was diagnosed with pheochromocytoma. A surgical consultation was obtained, and surgery was recommended.
WHEN DOES PHEOCHROMOCYTOMA CALL FOR SURGERY?
3. Which criterion is most important when determining the need for surgery for pheochromocytoma?
- Findings on fine-needle aspiration biopsy
- Biochemical activity
- Size of the mass
- Bilateral masses
Fine-needle aspiration biopsy can be done when a mass is found incidentally and no evidence of biochemical activity is detected, although it is not an essential part of the diagnostic workup.9 In most cases, the sampling from fine-needle aspiration is not sufficient to achieve a diagnosis.
Biochemical activity is the most important factor when determining the need for prompt surgical intervention. The excess circulating catecholamines have been associated with increased risk of cardiovascular morbidity and death independent of the morbidity associated with hypertension alone.10 Biochemical activity can be independent of the size of the mass, but larger masses typically present with symptoms.
Bilateral masses have been associated with metastatic disease.11 In retrospect, the patient’s history of hypertension and cerebrovascular accident could be associated with the development of a catecholamine-releasing tumor.
A GOOD OUTCOME FROM SURGERY
The patient was continued on phenoxybenzamine for 7 days and responded well to this therapy.
After this preoperative preparation, he underwent laparoscopic right adrenalectomy with excision of a retroperitoneal adrenal mass. His postoperative course was complicated by transient hypotension requiring low-dose vasopressin support for less than 24 hours. He was then restarted on his previous dosage of metoprolol and was discharged home on postoperative day 5 with stable blood pressure. Follow-up 24-hour urine collection 1 month after he was discharged showed normalization of metanephrine, normetanephrine, epinephrine, and norepinephrine levels.
Despite low suspicion for an underlying genetic syndrome, he was referred for genetic testing and was scheduled to have a repeat 24-hour urine collection and imaging in 6 months to follow his enlarged left adrenal gland, which did not appear to be metabolically hyperactive.
4. What is the most common perioperative complication of pheochromocytoma excision?
- Hypoglycemia
- Hypotension
- Hypocortisolism
- Hypertension
- Tachycardia
Hypoglycemia has been observed after removal of pheochromocytoma, as levels of catecholamines (which normally inhibit pancreatic beta cells) decrease and insulin secretion consequently increases.12 Our patient developed hypoglycemia before surgery, not after, and it was likely due to the combination of his antidiabetic therapy, weight loss, and decreased oral intake.
Hypotension is the most common complication in the perioperative period. It is associated with excessive loss of catecholamine secretion. It is usually short-lived but may require aggressive administration of intravenous fluids and use of sympathomimetic agents.
Hypocortisolism is unlikely in patients with pheochromocytoma, but it is likely after removal of adrenocortical adenoma.
Hypertension and tachycardia affect up to 40% of pheochromocytoma patients in some case series.12
PHEOCHROMOCYTOMA: A CATECHOLAMINE-SECRETING TUMOR
The pathophysiology of pheochromocytoma is complex. It is characterized by accelerated growth of cells producing catecholamines, which may produce symptoms when secreted into the bloodstream. The classic triad of symptoms is headache, hypertension, and hyperglycemia, although our patient had very low blood sugar levels. Other common symptoms are nausea, orthostasis, and tremor, although not all symptoms are invariably seen.
Genetic testing recommended
Genetic associations have been described and are thought to be responsible for 20% to 30% of cases of pheochromocytoma. All associated germline mutations are autosomal dominant, some with variable penetrance. These include:
- Succinate dehydrogenase subunit B, C, and D mutations
- von Hippel-Lindau syndrome
- Multiple endocrine neoplasia type 1 and type 2 syndromes
- Neurofibromatosis type 1.13,14
The succinate dehydrogenase subunit mutations have been associated with, but not limited to, extra-adrenal adenomas (paragangliomas) and carry a worse prognosis.
Some experts recommend genetic testing in all cases of pheochromocytoma, sporadic or familial, and this testing should be followed by counseling if a mutation is found.15 Others recommend genetic testing based on the patient’s age (under age 50), history, imaging, and biochemical features of the tumor (metanephrines predominate in multiple endocrine neoplasia syndromes, and normetanephrines in von Hippel-Lindau syndrome).13
Serious consequences
A thorough evaluation is recommended, since pheochromocytoma has been associated with increased cardiovascular morbidity. In a retrospective series, Stolk et al10 reported that patients with pheochromocytoma had a higher incidence of myocardial infarction, angina, and stroke in the years preceding the diagnosis than did patients with essential hypertension (13.8% vs 1.1%, P < .001).10
Catecholamine cardiomyopathy has been described and shares clinical features with Takotsubo or stress cardiomyopathy, with global left ventricular systolic and diastolic dysfunction that improve or resolve after the adrenergic insult is removed.16
Conditions that warrant further evaluation or that may suggest pheochromocytoma are malignant hypertension, hypertensive encephalopathy, ischemic stroke, subarachnoid hemorrhage, acute pulmonary edema, angina pectoris, myocardial infarction, aortic dissection, and kidney injury.
When to suspect pheochromocytoma
Pheochromocytoma should be suspected in a patient with resistant hypertension, family history, or imaging findings that suggest an adrenal mass with a heterogeneous appearance. The diagnostic algorithm follows the same pathway as for the evaluation of an incidentally found adrenal mass, with determination of its dimension and characteristics by CT or MRI, and with biochemical testing of urine catecholamines, plasma free metanephrines, renin, aldosterone, and cortisol.
The diagnosis of pheochromocytoma is established by obtaining fractionated metanephrines and catecholamines in a 24-hour urine collection (sensitivity 90%, specificity 98%). Analysis of plasma metanephrines has a higher sensitivity (97%) but lower specificity (85%).17 The combination of typical signs, symptoms, and laboratory findings makes the diagnosis likely, especially in combination with a unilateral adrenal mass.
Laparoscopic surgery after medical preparation for active tumors
If the mass appears benign and not biochemically hyperactive, then follow-up at 1 year is recommended, with repeat testing. Surgical evaluation and intervention is recommended for lesions that appear malignant or that are biochemically active and clinically symptomatic.9
Preoperative hemodynamic control is essential in the management of pheochromocytoma to prevent or minimize hemodynamic changes that can be driven by increased catecholamines. Control is typically achieved with initial alpha-blockade and then beta-blockade to avoid worsening hypertension and to prevent an acute hypertensive crisis during surgical intervention. Phenoxybenzamine, the mainstay of therapy, is a nonselective alpha-blocker with a long duration of action that requires titration over several days up to 3 weeks.
A selective alpha-1-blocker such as doxazosin can be used to control postoperative hypotension, as it has a shorter half-life than phenoxybenzamine. Alternative strategies include calcium channel blockers, centrally acting sympathetic blockers, and magnesium.18
Laparoscopic adrenalectomy by an experienced surgeon after excellent medical preparation is often considered the treatment of choice, but for larger or malignant masses, an open procedure is recommended. The risk of perioperative morbidity and death can be reduced by adequate medical management. With successful surgical resection, the long-term prognosis is favorable.
- Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2008; 51:1403–1419.
- Kumar N, Calhoun DA, Dudenbostel T. Management of patients with resistant hypertension: current treatment options. Integr Blood Press Control 2013; 6:139–151.
- Pedrosa RP, Drager LF, Gonzaga CC, et al. Obstructive sleep apnea: the most common secondary cause of hypertension associated with resistant hypertension. Hypertension 2011; 58:811–817.
- Marcus JA, Pothineni A, Marcus CZ, Bisognano JD. The role of obesity and obstructive sleep apnea in the pathogenesis and treatment of resistant hypertension. Curr Hypertens Rep 2014; 16:411.
- Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB, Weissmann P. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension 2002; 40:892–896.
- Young WF Clinical practice. The incidentally discovered adrenal mass. N Engl J Med 2007; 356:601–610.
- Lin M, Wong V, Yap J, Jin R, Leong P, Campbell P. FDG PET in the evaluation of phaeochromocytoma: a correlative study with MIBG scintigraphy and Ki-67 proliferative index. Clin Imaging 2013; 37:1084–1088.
- Raja A, Leung K, Stamm M, Girgis S, Low G. Multimodality imaging findings of pheochromocytoma with associated clinical and biochemical features in 53 patients with histologically confirmed tumors. AJR Am J Roentgenol 2013; 201:825–833.
- Nieman LK. Approach to the patient with an adrenal incidentaloma. J Clin Endocrinol Metab 2010; 95:4106–4113.
- Stolk RF, Bakx C, Mulder J, Timmers HJ, Lenders JW. Is the excess cardiovascular morbidity in pheochromocytoma related to blood pressure or to catecholamines? J Clin Endocrinol Metab 2013; 98:1100–1106.
- Grumbach MM, Biller BM, Braunstein GD, et al. Management of the clinically inapparent adrenal mass (‘incidentaloma’). Ann Intern Med 2003; 138:424–429.
- Lentschener C, Gaujoux S, Tesniere A, Dousset B. Point of controversy: perioperative care of patients undergoing pheochromocytoma removal—time for a reappraisal? Eur J Endocrinol 2011; 165:365–373.
- Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst 2003; 95:1196–1204.
- Lee P, Leonard J. Textbook on endocrinology. BMJ 1994; 308:1512.
- Fishbein L, Merrill S, Fraker DL, Cohen DL, Nathanson KL. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol 2013; 20:1444–1450.
- Agarwal G, Sadacharan D, Kapoor A, et al. Cardiovascular dysfunction and catecholamine cardiomyopathy in pheochromocytoma patients and their reversal following surgical cure: results of a prospective case-control study. Surgery 2011; 150:1202–1211.
- Sawka AM, Jaeschke R, Singh RJ, Young WF A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 2003; 88:553–558.
- Domi R, Laho H. Management of pheochromocytoma: old ideas and new drugs. Niger J Clin Pract 2012; 15:253–257.
- Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2008; 51:1403–1419.
- Kumar N, Calhoun DA, Dudenbostel T. Management of patients with resistant hypertension: current treatment options. Integr Blood Press Control 2013; 6:139–151.
- Pedrosa RP, Drager LF, Gonzaga CC, et al. Obstructive sleep apnea: the most common secondary cause of hypertension associated with resistant hypertension. Hypertension 2011; 58:811–817.
- Marcus JA, Pothineni A, Marcus CZ, Bisognano JD. The role of obesity and obstructive sleep apnea in the pathogenesis and treatment of resistant hypertension. Curr Hypertens Rep 2014; 16:411.
- Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB, Weissmann P. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension 2002; 40:892–896.
- Young WF Clinical practice. The incidentally discovered adrenal mass. N Engl J Med 2007; 356:601–610.
- Lin M, Wong V, Yap J, Jin R, Leong P, Campbell P. FDG PET in the evaluation of phaeochromocytoma: a correlative study with MIBG scintigraphy and Ki-67 proliferative index. Clin Imaging 2013; 37:1084–1088.
- Raja A, Leung K, Stamm M, Girgis S, Low G. Multimodality imaging findings of pheochromocytoma with associated clinical and biochemical features in 53 patients with histologically confirmed tumors. AJR Am J Roentgenol 2013; 201:825–833.
- Nieman LK. Approach to the patient with an adrenal incidentaloma. J Clin Endocrinol Metab 2010; 95:4106–4113.
- Stolk RF, Bakx C, Mulder J, Timmers HJ, Lenders JW. Is the excess cardiovascular morbidity in pheochromocytoma related to blood pressure or to catecholamines? J Clin Endocrinol Metab 2013; 98:1100–1106.
- Grumbach MM, Biller BM, Braunstein GD, et al. Management of the clinically inapparent adrenal mass (‘incidentaloma’). Ann Intern Med 2003; 138:424–429.
- Lentschener C, Gaujoux S, Tesniere A, Dousset B. Point of controversy: perioperative care of patients undergoing pheochromocytoma removal—time for a reappraisal? Eur J Endocrinol 2011; 165:365–373.
- Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst 2003; 95:1196–1204.
- Lee P, Leonard J. Textbook on endocrinology. BMJ 1994; 308:1512.
- Fishbein L, Merrill S, Fraker DL, Cohen DL, Nathanson KL. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol 2013; 20:1444–1450.
- Agarwal G, Sadacharan D, Kapoor A, et al. Cardiovascular dysfunction and catecholamine cardiomyopathy in pheochromocytoma patients and their reversal following surgical cure: results of a prospective case-control study. Surgery 2011; 150:1202–1211.
- Sawka AM, Jaeschke R, Singh RJ, Young WF A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 2003; 88:553–558.
- Domi R, Laho H. Management of pheochromocytoma: old ideas and new drugs. Niger J Clin Pract 2012; 15:253–257.