User login
A 79-year-old with acute portal vein thrombosis
A 79-year-old man presented with chills and fever. He had a history of polymyalgia rheumatica and had been tapered off corticosteroids 1 month before admission. One week before he presented, he had developed generalized myalgia, chills, and fatigue. A cortisol stimulation test at that time was normal, prednisone was restarted, and his symptoms had improved. But 1 day before he presented, the chills had returned, this time with fever. Laboratory testing at an outpatient clinic had revealed abnormal liver enzyme levels.
On the day he presented, he felt worse, with persistent chills, fever, and vague lower abdominal pain, but he denied nausea, vomiting, changes in bowel habits, melena, hematochezia, and hematemesis. He was admitted for additional evaluation.
His medical history also included coronary artery disease (for which he had undergone coronary artery bypass grafting), hypertension, stable liver cysts, and gout. He had no known inflammatory bowel disease and no recent abdominal surgery. His medications included prednisone, atorvastatin, atenolol, aspirin, niacin, and cholecalciferol. He had no history of smoking, significant drinking, or use of illicit drugs. He had no respiratory or cardiac symptoms or neurologic symptoms consistent with a transient ischemic attack or stroke. He denied any rashes.
On admission, he was febrile, with temperatures reaching 102˚F (38.9˚C). His blood pressure was 137/63 mm Hg, pulse 54 beats per minute, respiration rate 18 breaths per minute, and oxygen saturation 97% on room air. A harsh systolic murmur was noted on physical examination. His abdomen was nondistended, nontender, and without bruits.
Laboratory testing (Table 1) revealed leukocytosis, anemia, mildly abnormal aminotransferase levels, elevated alkaline phosphatase, and markedly elevated C-reactive protein.
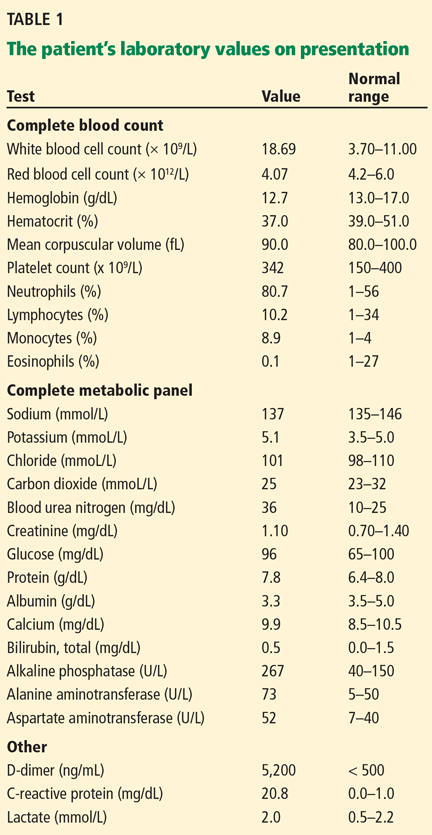
A full workup for fever was performed, including blood and urine cultures; chest radiography; contrast-enhanced computed tomography (CT) of the chest, abdomen, and pelvis; magnetic resonance imaging (MRI) of the abdomen; and colonoscopy. No source of infection—bacterial, viral, or fungal—was found. However, CT revealed new extensive thrombosis of the right portal vein and its branches (Figure 1).
CLINICAL PRESENTATION
1. Which of the following is least consistent with the clinical presentation of acute portal vein thrombosis?
- Abdominal pain
- Fever and chills
- Hematemesis
- Leukocytosis
- Absence of symptoms
Of these signs and symptoms, hematemesis is the least likely to be associated with acute portal vein thrombosis, although it can be associated with chronic cases.
Symptoms of portal vein thrombosis
Portal vein thrombosis causes extrahepatic obstruction of the portal venous system, which provides two-thirds of the total hepatic blood flow.
Acute. Often, thrombotic occlusion of the portal vein produces no acute symptoms because of immediate, compensatory vasodilation of the hepatic arterial system.1 Additionally, in the ensuing days, the thrombus becomes an organized collagenous plug, and collateral veins develop to bypass the blocked vein and maintain portal perfusion in a process called cavernous transformation.1,2 Thus, many patients have no symptoms.
If symptoms occur, portal vein thrombosis can initially present as transient abdominal pain with fever, as seen in this patient.3 Many patients with acute portal vein thrombosis experience abdominal pain due to intra-abdominal sepsis, also referred to as pylephlebitis.2,4 High, spiking fevers and chills also occur, caused by infected thrombi associated with intra-abdominal infections such as appendicitis, diverticulitis, and pancreatitis.5,6
Chronic. In contrast, symptomatic chronic portal vein thrombosis commonly presents with sequelae of portal hypertension, most notably gastrointestinal bleeding. Hematemesis from ruptured esophageal varices is the most frequent reason for seeking medical attention, though varices also develop in the stomach, duodenum, jejunum, gallbladder, and bile ducts.2,7 Abdominal pain is less common in chronic portal vein thrombosis unless the thrombus extends into the mesenteric veins and causes bowel ischemia or infarction. Long-standing portal vein thrombosis may also lead to dilated venous collaterals that compress large bile ducts, resulting in portal cholangiopathy.1,8
Portal vein thrombosis may present as acute intestinal ischemia and bowel infarction, though this is uncommon. This is generally seen with extensive occlusive portal vein thrombosis and concomitant mesenteric venous thrombosis.1,2
Other symptoms that are common but nonspecific are nausea, vomiting, diarrhea, weight loss, and anorexia.2
Signs of portal vein thrombosis
On examination, patients with acute portal vein thrombosis have minimal physical signs unless they have other contributing conditions. For example, acute portal vein thrombosis can result in abdominal distention secondary to ileus, or guarding and ascites secondary to intestinal infarction.3,9
Some patients with chronic portal vein thrombosis also have normal physical findings, but many have signs. Splenomegaly is seen in 75% to 100% of patients.2,7 Hepatomegaly, abdominal tenderness, and low-grade fever are common as well.2,10 Ascites is usually not present without underlying cirrhosis; however, mild and transient ascites can develop immediately after the thrombotic event before the patient develops collateral circulation.2
Laboratory testing for portal vein thrombosis
Laboratory test results are typically unremarkable. Liver function tests show preserved hepatic function but may reveal mild increases in aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase, and bilirubin.2,10
In acute cases, elevations of acute-phase reactant levels can occur.9 Leukocytosis and blood cultures growing Bacteroides species are seen in septic cases or pylephlebitis.11,12 There may be mild anemia, particularly after a recent bleeding episode, or mild leukopenia and thrombocytopenia due to hypersplenism. Suspicion of an underlying myeloproliferative disorder is high if thrombocytosis is present.2
DIAGNOSIS
2. All of the following would be appropriate initial diagnostic studies for portal vein thrombosis except which one?
- Doppler ultrasonography
- Contrast-enhanced CT
- Contrast-enhanced MRI
- Angiography
Portal vein thrombosis is most often diagnosed with noninvasive techniques, namely Doppler ultrasonography, CT, and MRI—not angiography.
Ultrasonography can reveal an echogenic thrombus in the vessel lumen with distention of the portal vein proximal to the occlusion and extensive collateral vessels. Plain ultrasonography fails to reveal the thrombus in up to one-third of patients. However, duplex ultrasonography with color flow Doppler imaging can confirm partial or complete absence of flow in the vein with 89% sensitivity and 92% specificity.13,14
On contrast-enhanced CT, the thrombus appears as a filling defect within the portal venous segment. Complete occlusion of the vein may produce a “train track” appearance due to contrast around the vessel.10 Without contrast, the clot will appear as hyperattenuating material in the portal vein, but contrast-enhanced imaging may be necessary to differentiate the thrombus from the vessel wall.15 Gas within the portal venous system is specific for pylephlebitis.4 Evidence of cavernous transformation is seen in chronic portal vein thrombosis.
Contrast-enhanced magnetic resonance angiography can also be used to evaluate patency and flow direction. In addition, it provides detailed anatomic information about the entire portal venous system, including the intrahepatic portal vessels, which is limited in CT imaging.2,10 CT and MRI can also help to identify predisposing conditions (eg, intra-abdominal infection, hepatocellular carcinoma) and complications (eg, intestinal infarction) associated with portal vein thrombosis.
Angiography can be considered if noninvasive techniques are inconclusive but is generally not necessary, given the increased use of CT and MRI.
In our patient, abdominal CT revealed occlusive thrombosis of the right portal vein and its branches (Figure 1). The left and main portal veins were patent. There was no evidence of intra-abdominal infection or infarction.
FINDING THE CAUSE
3. Which of the following is not a common cause
of portal vein thrombosis?
- A hypercoagulable state
- Immune deficiency
- Intra-abdominal infection
- Malignancy
- Portal hypertension

Once portal vein thrombosis has been diagnosed, the cause should be identified (Table 2). The differential diagnosis is broad, including both local factors (eg, injury to the portal vein, local inflammation, infection) and general factors (eg, inherited and acquired hypercoagulable conditions). Thrombophilias are identified in 60% of patients with portal vein thrombosis and local factors in 40%.7 Moreover, the etiology is often multifactorial. However, immune deficiency is not a common cause.
Hypercoagulability
Prothrombotic disorders can be either inherited or acquired.
Inherited deficiencies in the natural anticoagulants antithrombin, protein C, and protein S are associated with a high risk of thrombosis but have a low prevalence in the general population. In the setting of liver abnormalities, familial testing may be helpful to distinguish inherited causes of portal vein thrombosis from defective liver function as a consequence of portal vein thrombosis. The factor V Leiden mutation (G1691A) and the G20210A mutation in the prothrombin gene are more prevalent (> 2%) but generally confer a lower thrombosis risk.16 The prothrombin gene mutation G20210A is the most common risk factor for portal vein thrombosis, with prevalence of 2% to 22% in adults with nonmalignant, noncirrhotic portal vein thrombosis.3
Hyperhomocysteinemia due to a methylene tetrahydrofolate reductase (MTHFR) mutation (C677T) is another inherited associated risk factor for portal vein thrombosis, but hyperhomocysteinemia can also arise as a complication of portal vein thrombosis-related liver disease.3
Acquired prothrombotic disorders, particularly myeloproliferative diseases, are found in 22% to 48% of cases of portal vein thrombosis. Many young patients with myeloproliferative disorders present with portal vein thrombosis as the first symptom, and testing for the G1849T point mutation in JAK2 can make the diagnosis.17 Splenectomy with underlying myeloproliferative disorder confers a particularly high risk for portal vein thrombosis.18
Other thrombophilic disorders including antiphospholipid antibody syndrome, paroxysmal nocturnal hemoglobinuria, and malignancy can contribute to portal vein thrombosis.3 Pregnancy and oral contraceptive use have also been associated with hypercoagulability, and cessation of oral estrogen is recommended in such cases. The risk may be further increased in patients on oral contraceptives who have a previously unrecognized hypercoagulable state.3
Inflammation and infection
Inflammation and infection are local risk factors for portal vein thrombosis. Acute portal vein thrombosis has been associated with intra-abdominal infections (eg, appendicitis, cholecystitis) and with inflammatory conditions such as inflammatory bowel disease and pancreatitis.16,19 From 3% to 5% of all portal vein thrombosis cases result from pancreatitis, either from a single acute episode or from repeat inflammation of chronic pancreatitis.10 Portal vein thrombosis in the setting of inflammatory bowel disease can occur even when the disease is in remission, particularly in ulcerative colitis.20,21
Injury to the portal venous system
Abdominal surgery, particularly splenectomy, portosystemic shunting, colectomy, and blunt abdominal trauma can cause injury to the portal venous system, resulting in portal vein thrombosis. This is usually seen only in patients with portal hypertension, an underlying prothrombotic condition such as myeloproliferative disease, or inflammatory bowel disease.10,19,22
Impaired portal vein flow
Cirrhosis and malignancy are major risk factors for portal vein thrombosis. In case series, cirrhosis was found in 24% to 32% of patients with portal vein thrombosis.2,23 However, the overall prevalence of portal vein thrombosis in cirrhotic patients varies widely, from 0.6% to 28%, depending on the degree of cirrhosis.10
The pathogenesis of portal vein thrombosis in cirrhosis is unclear but may be multifactorial. Decreased portal blood flow (with subsequent stasis) and periportal lymphangitis and fibrosis are thought to stimulate thrombus formation.3,10 Additionally, patients with advanced cirrhosis are prothrombotic because of reduced hepatic synthesis of antithrombin, protein C, protein S, and coagulation factors.
Malignancy is associated with 21% to 24% of cases of portal vein thrombosis in adults, with pancreatic cancer and hepatocellular carcinoma being the most common.2,3 Others include cholangiocarcinoma and carcinomas of the stomach, lung, prostate, uterus, and kidney. Cancer causes portal vein thrombosis through a combination of tumor invasion into the portal vein, extrinsic compression by the tumor, periportal fibrosis following surgery or radiation, and hypercoagulability secondary to malignancy.9,16,24
Idiopathic portal vein thrombosis
Portal vein thrombosis is usually caused by one or more of the underlying factors mentioned above but is idiopathic in 8% to 15% of cases.10
Back to our patient
The cause of this patient’s portal vein thrombosis is unclear. He did not have a history of cirrhosis, inflammatory bowel disease, trauma, or abdominal surgery. His febrile illness could have precipitated the formation of a thrombus, but no definitive source of infection or inflammation was discovered. His workup was negative for pancreatitis, appendicitis, cholecystitis, diverticulitis, and prostatitis. No occult malignancy was found. It is also possible that his fever was the result of the thrombosis.
A full hypercoagulability panel revealed no striking abnormalities. He did have elevated fibrinogen and factor VIII levels that were consistent with an acute-phase reaction, along with an elevated erythrocyte sedimentation rate (> 90 mm/hr) and C-reactive protein level. Aside from the portal vein thrombosis, no potential source of inflammation could be identified.
Mildly reduced levels of antithrombin III activity were attributed to enoxaparin therapy and ultimately normalized on repeated testing. The patient had very minimally elevated titers of anticardiolipin immunoglobulin G (1:10 GPL) and anti-beta-2 glycoprotein immunoglobulin M (21 SMU), which were not thought to be significant. Tests for lupus anticoagulant, prothrombin gene mutation, activated protein C resistance, and JAK2 mutation were negative.
TREATMENT
4. Treatment of symptomatic portal vein thrombosis generally includes which two of the following?
- Anticoagulation
- Intravenous gamma globulin
- Broad-spectrum antibiotics
Anticoagulant therapy
Treatment of acute, symptomatic portal vein thrombosis involves anticoagulant therapy to prevent extension of the thrombus and, ultimately, to allow for recanalization of the obstructed veins. Anticoagulant therapy is initially intravenous unfractionated heparin or subcutaneous low-molecular-weight heparin, eventually bridged to an oral agent such as warfarin.3,9 Currently, there are inadequate data on the use of oral or parenteral factor Xa inhibitors or direct thrombin inhibitors in the treatment of this disease.
When started immediately, anticoagulation therapy is associated with complete recanalization in 38.3% and partial recanalization in 14% of patients presenting with complete thrombosis. Without anticoagulation, spontaneous recanalization is unusual.25
Although the optimal duration of anticoagulant therapy is unclear, a minimum of 3 months is generally recommended.9,26 If a hypercoagulable state is present or if the portal vein thrombosis is unprovoked (eg, by surgery, trauma, or an intra-abdominal infection), long-term treatment should be considered.26
Experience with thrombolytic therapy or mechanical recanalization has been limited, but the use of catheter-based techniques for pharmacomechanical thrombolysis has been reported.27–29 Transjugular intrahepatic portosystemic shunting is also an alternative to anticoagulation, but its role in treating portal vein thrombosis is complicated by technical difficulties of the procedure, postoperative complications, and recurrent occlusion of the shunt.25
Currently, there are no data comparing the risk-benefit ratio of early anticoagulation and that of invasive procedures. These more aggressive treatments are generally considered only when there is extensive thrombosis or ascites (which are both predictive factors of poor response to anticoagulation alone) and in patients for whom anticoagulation has failed.3 Surgical thrombectomy is rarely indicated, typically only in instances in which laparotomy is being performed for suspected bowel infarction.3
Antibiotics
In addition to anticoagulation, broad-spectrum antibiotics covering gram-negative and anaerobic bacteria are indicated for those cases of portal vein thrombosis associated with underlying infection.9
For chronic cases, the goals of management are to prevent and treat gastroesophageal variceal bleeding and to prevent recurrent thrombosis.9 Nonselective beta-blockers (eg, propranolol) and endoscopic band ligation have shown evidence of reducing the incidence of recurrent bleeding and prolonging survival in retrospective studies.9,30,31 Long-term anticoagulation is generally indicated to prevent further thrombosis and to increase the likelihood of recanalization only for patients with a permanent prothrombotic condition.9 In patients with clinically significant portal hypertension, the benefit of continued anticoagulation therapy must be weighed against the risk of esophageal and gastric variceal bleeding.
There is controversy regarding how to manage portal vein thrombosis that is incidentally identified and asymptomatic (eg, if it is discovered on an imaging study for another indication). Current guidelines recommend against anticoagulation in patients with incidentally discovered and asymptomatic splanchnic vein thrombosis, including portal vein thrombosis.26
Intravenous gamma globulin is not part of the treatment.
CASE CONTINUED
The patient’s presenting symptoms of fever, chills, and abdominal pain completely resolved after a course of antibiotic therapy. The erythrocyte sedimentation rate subsequently normalized and factor VIII activity improved. We believed that an underlying infectious or inflammatory process had contributed to the development of portal vein thrombosis, though the specific cause could not be identified. The patient was treated with enoxaparin 1 mg/kg twice a day and transitioned to warfarin.
Magnetic resonance venography done 3 months after diagnosis showed persistent right portal vein thrombosis that was largely unchanged. Anticoagulation was continued for 1 year with no change in his portal vein thrombosis on sequential imaging and was subsequently discontinued. To date, no malignancy or infectious process has been found, and the patient continues to do well 2 years later.
- Ponziani FR, Zocco MA, Campanale C, et al. Portal vein thrombosis: insight into physiopathology, diagnosis, and treatment. World J Gastroenterol 2010; 16:143–155.
- Cohen J, Edelman RR, Chopra S. Portal vein thrombosis: a review. Am J Med 1992; 92:173–182.
- Primignani M. Portal vein thrombosis, revisited. Dig Liver Dis 2010; 42:163–170.
- Condat B, Valla D. Nonmalignant portal vein thrombosis in adults. Nat Clin Pract Gastroenterol Hepatol 2006; 3:505–515.
- Condat B, Pessione F, Helene Denninger M, Hillaire S, Valla D. Recent portal or mesenteric venous thrombosis: increased recognition and frequent recanalization on anticoagulant therapy. Hepatology 2000; 32:466–470.
- Sheen CL, Lamparelli H, Milne A, Green I, Ramage JK. Clinical features, diagnosis and outcome of acute portal vein thrombosis. QJM 2000; 93:531–534.
- Sogaard KK, Astrup LB, Vilstrup H, Gronbaek H. Portal vein thrombosis; risk factors, clinical presentation and treatment. BMC Gastroenterol 2007; 7:34.
- Llop E, de Juan C, Seijo S, et al. Portal cholangiopathy: radiological classification and natural history. Gut 2011; 60:853–860.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Sobhonslidsuk A, Reddy KR. Portal vein thrombosis: a concise review. Am J Gastroenterol 2002; 97:535–541.
- Ni YH, Wang NC, Peng MY, Chou YY, Chang FY. Bacteroides fragilis bacteremia associated with portal vein and superior mesentery vein thrombosis secondary to antithrombin III and protein C deficiency: a case report. J Microbiol Immunol Infect 2002; 35:255–258.
- Trum J, Valla D, Cohen G, et al. Bacteroides bacteraemia of undetermined origin: strong association with portal vein thrombosis and cryptogenic pylephlebitis. Eur J Gastroenterol Hepatol 1993; 5:655–659.
- Ueno N, Sasaki A, Tomiyama T, Tano S, Kimura K. Color Doppler ultrasonography in the diagnosis of cavernous transformation of the portal vein. J Clin Ultrasound 1997; 25:227–233.
- Tessler FN, Gehring BJ, Gomes AS, et al. Diagnosis of portal vein thrombosis: value of color Doppler imaging. AJR Am J Roentgenol 1991; 157:293–296.
- Hidajat N, Stobbe H, Griesshaber V, Felix R, Schroder RJ. Imaging and radiological interventions of portal vein thrombosis. Acta Radiol 2005; 46:336–343.
- Valla DC, Condat B. Portal vein thrombosis in adults: pathophysiology, pathogenesis and management. J Hepatol 2000; 32:865–871.
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352:1779–1790.
- Krauth MT, Lechner K, Neugebauer EA, Pabinger I. The postoperative splenic/portal vein thrombosis after splenectomy and its prevention—an unresolved issue. Haematologica 2008; 93:1227–1232.
- Sinagra E, Aragona E, Romano C, et al. The role of portal vein thrombosis in the clinical course of inflammatory bowel diseases: report on three cases and review of the literature. Gastroenterol Res Pract 2012; 2012:916428.
- Maconi G, Bolzacchini E, Dell’Era A, Russo U, Ardizzone S, de Franchis R. Portal vein thrombosis in inflammatory bowel diseases: a single-center case series. J Crohns Colitis 2012; 6:362–367.
- Jackson LM, O’Gorman PJ, O’Connell J, Cronin CC, Cotter KP, Shanahan F. Thrombosis in inflammatory bowel disease: clinical setting, procoagulant profile and factor V Leiden. QJM 1997; 90:183–188.
- Eguchi A, Hashizume M, Kitano S, Tanoue K, Wada H, Sugimachi K. High rate of portal thrombosis after splenectomy in patients with esophageal varices and idiopathic portal hypertension. Arch Surg 1991; 126:752–755.
- Ogren M, Bergqvist D, Björck M, Acosta S, Eriksson H, Sternby NH. Portal vein thrombosis: prevalence, patient characteristics and lifetime risk: a population study based on 23,796 consecutive autopsies. World J Gastroenterol 2006; 12:2115–2119.
- Falanga A, Marchetti M, Vignoli A. Coagulation and cancer: biological and clinical aspects. J Thromb Haemost 2013; 11:223–233.
- Congly SE, Lee SS. Portal vein thrombosis: should anticoagulation be used? Curr Gastroenterol Rep 2013; 15:306.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Uflacker R. Applications of percutaneous mechanical thrombectomy in transjugular intrahepatic portosystemic shunt and portal vein thrombosis. Tech Vasc Interv Radiol 2003; 6:59–69.
- Takahashi N, Kuroki K, Yanaga K. Percutaneous transhepatic mechanical thrombectomy for acute mesenteric venous thrombosis. J Endovasc Ther 2005; 12:508–511.
- Lopera JE, Correa G, Brazzini A, et al. Percutaneous transhepatic treatment of symptomatic mesenteric venous thrombosis. J Vasc Surg 2002; 36:1058–1061.
- Orr DW, Harrison PM, Devlin J, et al. Chronic mesenteric venous thrombosis: evaluation and determinants of survival during long-term follow-up. Clin Gastroenterol Hepatol 2007; 5:80–86.
- Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant therapy. Gastroenterology 2001; 120:490–497.
A 79-year-old man presented with chills and fever. He had a history of polymyalgia rheumatica and had been tapered off corticosteroids 1 month before admission. One week before he presented, he had developed generalized myalgia, chills, and fatigue. A cortisol stimulation test at that time was normal, prednisone was restarted, and his symptoms had improved. But 1 day before he presented, the chills had returned, this time with fever. Laboratory testing at an outpatient clinic had revealed abnormal liver enzyme levels.
On the day he presented, he felt worse, with persistent chills, fever, and vague lower abdominal pain, but he denied nausea, vomiting, changes in bowel habits, melena, hematochezia, and hematemesis. He was admitted for additional evaluation.
His medical history also included coronary artery disease (for which he had undergone coronary artery bypass grafting), hypertension, stable liver cysts, and gout. He had no known inflammatory bowel disease and no recent abdominal surgery. His medications included prednisone, atorvastatin, atenolol, aspirin, niacin, and cholecalciferol. He had no history of smoking, significant drinking, or use of illicit drugs. He had no respiratory or cardiac symptoms or neurologic symptoms consistent with a transient ischemic attack or stroke. He denied any rashes.
On admission, he was febrile, with temperatures reaching 102˚F (38.9˚C). His blood pressure was 137/63 mm Hg, pulse 54 beats per minute, respiration rate 18 breaths per minute, and oxygen saturation 97% on room air. A harsh systolic murmur was noted on physical examination. His abdomen was nondistended, nontender, and without bruits.
Laboratory testing (Table 1) revealed leukocytosis, anemia, mildly abnormal aminotransferase levels, elevated alkaline phosphatase, and markedly elevated C-reactive protein.
A full workup for fever was performed, including blood and urine cultures; chest radiography; contrast-enhanced computed tomography (CT) of the chest, abdomen, and pelvis; magnetic resonance imaging (MRI) of the abdomen; and colonoscopy. No source of infection—bacterial, viral, or fungal—was found. However, CT revealed new extensive thrombosis of the right portal vein and its branches (Figure 1).
CLINICAL PRESENTATION
1. Which of the following is least consistent with the clinical presentation of acute portal vein thrombosis?
- Abdominal pain
- Fever and chills
- Hematemesis
- Leukocytosis
- Absence of symptoms
Of these signs and symptoms, hematemesis is the least likely to be associated with acute portal vein thrombosis, although it can be associated with chronic cases.
Symptoms of portal vein thrombosis
Portal vein thrombosis causes extrahepatic obstruction of the portal venous system, which provides two-thirds of the total hepatic blood flow.
Acute. Often, thrombotic occlusion of the portal vein produces no acute symptoms because of immediate, compensatory vasodilation of the hepatic arterial system.1 Additionally, in the ensuing days, the thrombus becomes an organized collagenous plug, and collateral veins develop to bypass the blocked vein and maintain portal perfusion in a process called cavernous transformation.1,2 Thus, many patients have no symptoms.
If symptoms occur, portal vein thrombosis can initially present as transient abdominal pain with fever, as seen in this patient.3 Many patients with acute portal vein thrombosis experience abdominal pain due to intra-abdominal sepsis, also referred to as pylephlebitis.2,4 High, spiking fevers and chills also occur, caused by infected thrombi associated with intra-abdominal infections such as appendicitis, diverticulitis, and pancreatitis.5,6
Chronic. In contrast, symptomatic chronic portal vein thrombosis commonly presents with sequelae of portal hypertension, most notably gastrointestinal bleeding. Hematemesis from ruptured esophageal varices is the most frequent reason for seeking medical attention, though varices also develop in the stomach, duodenum, jejunum, gallbladder, and bile ducts.2,7 Abdominal pain is less common in chronic portal vein thrombosis unless the thrombus extends into the mesenteric veins and causes bowel ischemia or infarction. Long-standing portal vein thrombosis may also lead to dilated venous collaterals that compress large bile ducts, resulting in portal cholangiopathy.1,8
Portal vein thrombosis may present as acute intestinal ischemia and bowel infarction, though this is uncommon. This is generally seen with extensive occlusive portal vein thrombosis and concomitant mesenteric venous thrombosis.1,2
Other symptoms that are common but nonspecific are nausea, vomiting, diarrhea, weight loss, and anorexia.2
Signs of portal vein thrombosis
On examination, patients with acute portal vein thrombosis have minimal physical signs unless they have other contributing conditions. For example, acute portal vein thrombosis can result in abdominal distention secondary to ileus, or guarding and ascites secondary to intestinal infarction.3,9
Some patients with chronic portal vein thrombosis also have normal physical findings, but many have signs. Splenomegaly is seen in 75% to 100% of patients.2,7 Hepatomegaly, abdominal tenderness, and low-grade fever are common as well.2,10 Ascites is usually not present without underlying cirrhosis; however, mild and transient ascites can develop immediately after the thrombotic event before the patient develops collateral circulation.2
Laboratory testing for portal vein thrombosis
Laboratory test results are typically unremarkable. Liver function tests show preserved hepatic function but may reveal mild increases in aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase, and bilirubin.2,10
In acute cases, elevations of acute-phase reactant levels can occur.9 Leukocytosis and blood cultures growing Bacteroides species are seen in septic cases or pylephlebitis.11,12 There may be mild anemia, particularly after a recent bleeding episode, or mild leukopenia and thrombocytopenia due to hypersplenism. Suspicion of an underlying myeloproliferative disorder is high if thrombocytosis is present.2
DIAGNOSIS
2. All of the following would be appropriate initial diagnostic studies for portal vein thrombosis except which one?
- Doppler ultrasonography
- Contrast-enhanced CT
- Contrast-enhanced MRI
- Angiography
Portal vein thrombosis is most often diagnosed with noninvasive techniques, namely Doppler ultrasonography, CT, and MRI—not angiography.
Ultrasonography can reveal an echogenic thrombus in the vessel lumen with distention of the portal vein proximal to the occlusion and extensive collateral vessels. Plain ultrasonography fails to reveal the thrombus in up to one-third of patients. However, duplex ultrasonography with color flow Doppler imaging can confirm partial or complete absence of flow in the vein with 89% sensitivity and 92% specificity.13,14
On contrast-enhanced CT, the thrombus appears as a filling defect within the portal venous segment. Complete occlusion of the vein may produce a “train track” appearance due to contrast around the vessel.10 Without contrast, the clot will appear as hyperattenuating material in the portal vein, but contrast-enhanced imaging may be necessary to differentiate the thrombus from the vessel wall.15 Gas within the portal venous system is specific for pylephlebitis.4 Evidence of cavernous transformation is seen in chronic portal vein thrombosis.
Contrast-enhanced magnetic resonance angiography can also be used to evaluate patency and flow direction. In addition, it provides detailed anatomic information about the entire portal venous system, including the intrahepatic portal vessels, which is limited in CT imaging.2,10 CT and MRI can also help to identify predisposing conditions (eg, intra-abdominal infection, hepatocellular carcinoma) and complications (eg, intestinal infarction) associated with portal vein thrombosis.
Angiography can be considered if noninvasive techniques are inconclusive but is generally not necessary, given the increased use of CT and MRI.
In our patient, abdominal CT revealed occlusive thrombosis of the right portal vein and its branches (Figure 1). The left and main portal veins were patent. There was no evidence of intra-abdominal infection or infarction.
FINDING THE CAUSE
3. Which of the following is not a common cause
of portal vein thrombosis?
- A hypercoagulable state
- Immune deficiency
- Intra-abdominal infection
- Malignancy
- Portal hypertension
Once portal vein thrombosis has been diagnosed, the cause should be identified (Table 2). The differential diagnosis is broad, including both local factors (eg, injury to the portal vein, local inflammation, infection) and general factors (eg, inherited and acquired hypercoagulable conditions). Thrombophilias are identified in 60% of patients with portal vein thrombosis and local factors in 40%.7 Moreover, the etiology is often multifactorial. However, immune deficiency is not a common cause.
Hypercoagulability
Prothrombotic disorders can be either inherited or acquired.
Inherited deficiencies in the natural anticoagulants antithrombin, protein C, and protein S are associated with a high risk of thrombosis but have a low prevalence in the general population. In the setting of liver abnormalities, familial testing may be helpful to distinguish inherited causes of portal vein thrombosis from defective liver function as a consequence of portal vein thrombosis. The factor V Leiden mutation (G1691A) and the G20210A mutation in the prothrombin gene are more prevalent (> 2%) but generally confer a lower thrombosis risk.16 The prothrombin gene mutation G20210A is the most common risk factor for portal vein thrombosis, with prevalence of 2% to 22% in adults with nonmalignant, noncirrhotic portal vein thrombosis.3
Hyperhomocysteinemia due to a methylene tetrahydrofolate reductase (MTHFR) mutation (C677T) is another inherited associated risk factor for portal vein thrombosis, but hyperhomocysteinemia can also arise as a complication of portal vein thrombosis-related liver disease.3
Acquired prothrombotic disorders, particularly myeloproliferative diseases, are found in 22% to 48% of cases of portal vein thrombosis. Many young patients with myeloproliferative disorders present with portal vein thrombosis as the first symptom, and testing for the G1849T point mutation in JAK2 can make the diagnosis.17 Splenectomy with underlying myeloproliferative disorder confers a particularly high risk for portal vein thrombosis.18
Other thrombophilic disorders including antiphospholipid antibody syndrome, paroxysmal nocturnal hemoglobinuria, and malignancy can contribute to portal vein thrombosis.3 Pregnancy and oral contraceptive use have also been associated with hypercoagulability, and cessation of oral estrogen is recommended in such cases. The risk may be further increased in patients on oral contraceptives who have a previously unrecognized hypercoagulable state.3
Inflammation and infection
Inflammation and infection are local risk factors for portal vein thrombosis. Acute portal vein thrombosis has been associated with intra-abdominal infections (eg, appendicitis, cholecystitis) and with inflammatory conditions such as inflammatory bowel disease and pancreatitis.16,19 From 3% to 5% of all portal vein thrombosis cases result from pancreatitis, either from a single acute episode or from repeat inflammation of chronic pancreatitis.10 Portal vein thrombosis in the setting of inflammatory bowel disease can occur even when the disease is in remission, particularly in ulcerative colitis.20,21
Injury to the portal venous system
Abdominal surgery, particularly splenectomy, portosystemic shunting, colectomy, and blunt abdominal trauma can cause injury to the portal venous system, resulting in portal vein thrombosis. This is usually seen only in patients with portal hypertension, an underlying prothrombotic condition such as myeloproliferative disease, or inflammatory bowel disease.10,19,22
Impaired portal vein flow
Cirrhosis and malignancy are major risk factors for portal vein thrombosis. In case series, cirrhosis was found in 24% to 32% of patients with portal vein thrombosis.2,23 However, the overall prevalence of portal vein thrombosis in cirrhotic patients varies widely, from 0.6% to 28%, depending on the degree of cirrhosis.10
The pathogenesis of portal vein thrombosis in cirrhosis is unclear but may be multifactorial. Decreased portal blood flow (with subsequent stasis) and periportal lymphangitis and fibrosis are thought to stimulate thrombus formation.3,10 Additionally, patients with advanced cirrhosis are prothrombotic because of reduced hepatic synthesis of antithrombin, protein C, protein S, and coagulation factors.
Malignancy is associated with 21% to 24% of cases of portal vein thrombosis in adults, with pancreatic cancer and hepatocellular carcinoma being the most common.2,3 Others include cholangiocarcinoma and carcinomas of the stomach, lung, prostate, uterus, and kidney. Cancer causes portal vein thrombosis through a combination of tumor invasion into the portal vein, extrinsic compression by the tumor, periportal fibrosis following surgery or radiation, and hypercoagulability secondary to malignancy.9,16,24
Idiopathic portal vein thrombosis
Portal vein thrombosis is usually caused by one or more of the underlying factors mentioned above but is idiopathic in 8% to 15% of cases.10
Back to our patient
The cause of this patient’s portal vein thrombosis is unclear. He did not have a history of cirrhosis, inflammatory bowel disease, trauma, or abdominal surgery. His febrile illness could have precipitated the formation of a thrombus, but no definitive source of infection or inflammation was discovered. His workup was negative for pancreatitis, appendicitis, cholecystitis, diverticulitis, and prostatitis. No occult malignancy was found. It is also possible that his fever was the result of the thrombosis.
A full hypercoagulability panel revealed no striking abnormalities. He did have elevated fibrinogen and factor VIII levels that were consistent with an acute-phase reaction, along with an elevated erythrocyte sedimentation rate (> 90 mm/hr) and C-reactive protein level. Aside from the portal vein thrombosis, no potential source of inflammation could be identified.
Mildly reduced levels of antithrombin III activity were attributed to enoxaparin therapy and ultimately normalized on repeated testing. The patient had very minimally elevated titers of anticardiolipin immunoglobulin G (1:10 GPL) and anti-beta-2 glycoprotein immunoglobulin M (21 SMU), which were not thought to be significant. Tests for lupus anticoagulant, prothrombin gene mutation, activated protein C resistance, and JAK2 mutation were negative.
TREATMENT
4. Treatment of symptomatic portal vein thrombosis generally includes which two of the following?
- Anticoagulation
- Intravenous gamma globulin
- Broad-spectrum antibiotics
Anticoagulant therapy
Treatment of acute, symptomatic portal vein thrombosis involves anticoagulant therapy to prevent extension of the thrombus and, ultimately, to allow for recanalization of the obstructed veins. Anticoagulant therapy is initially intravenous unfractionated heparin or subcutaneous low-molecular-weight heparin, eventually bridged to an oral agent such as warfarin.3,9 Currently, there are inadequate data on the use of oral or parenteral factor Xa inhibitors or direct thrombin inhibitors in the treatment of this disease.
When started immediately, anticoagulation therapy is associated with complete recanalization in 38.3% and partial recanalization in 14% of patients presenting with complete thrombosis. Without anticoagulation, spontaneous recanalization is unusual.25
Although the optimal duration of anticoagulant therapy is unclear, a minimum of 3 months is generally recommended.9,26 If a hypercoagulable state is present or if the portal vein thrombosis is unprovoked (eg, by surgery, trauma, or an intra-abdominal infection), long-term treatment should be considered.26
Experience with thrombolytic therapy or mechanical recanalization has been limited, but the use of catheter-based techniques for pharmacomechanical thrombolysis has been reported.27–29 Transjugular intrahepatic portosystemic shunting is also an alternative to anticoagulation, but its role in treating portal vein thrombosis is complicated by technical difficulties of the procedure, postoperative complications, and recurrent occlusion of the shunt.25
Currently, there are no data comparing the risk-benefit ratio of early anticoagulation and that of invasive procedures. These more aggressive treatments are generally considered only when there is extensive thrombosis or ascites (which are both predictive factors of poor response to anticoagulation alone) and in patients for whom anticoagulation has failed.3 Surgical thrombectomy is rarely indicated, typically only in instances in which laparotomy is being performed for suspected bowel infarction.3
Antibiotics
In addition to anticoagulation, broad-spectrum antibiotics covering gram-negative and anaerobic bacteria are indicated for those cases of portal vein thrombosis associated with underlying infection.9
For chronic cases, the goals of management are to prevent and treat gastroesophageal variceal bleeding and to prevent recurrent thrombosis.9 Nonselective beta-blockers (eg, propranolol) and endoscopic band ligation have shown evidence of reducing the incidence of recurrent bleeding and prolonging survival in retrospective studies.9,30,31 Long-term anticoagulation is generally indicated to prevent further thrombosis and to increase the likelihood of recanalization only for patients with a permanent prothrombotic condition.9 In patients with clinically significant portal hypertension, the benefit of continued anticoagulation therapy must be weighed against the risk of esophageal and gastric variceal bleeding.
There is controversy regarding how to manage portal vein thrombosis that is incidentally identified and asymptomatic (eg, if it is discovered on an imaging study for another indication). Current guidelines recommend against anticoagulation in patients with incidentally discovered and asymptomatic splanchnic vein thrombosis, including portal vein thrombosis.26
Intravenous gamma globulin is not part of the treatment.
CASE CONTINUED
The patient’s presenting symptoms of fever, chills, and abdominal pain completely resolved after a course of antibiotic therapy. The erythrocyte sedimentation rate subsequently normalized and factor VIII activity improved. We believed that an underlying infectious or inflammatory process had contributed to the development of portal vein thrombosis, though the specific cause could not be identified. The patient was treated with enoxaparin 1 mg/kg twice a day and transitioned to warfarin.
Magnetic resonance venography done 3 months after diagnosis showed persistent right portal vein thrombosis that was largely unchanged. Anticoagulation was continued for 1 year with no change in his portal vein thrombosis on sequential imaging and was subsequently discontinued. To date, no malignancy or infectious process has been found, and the patient continues to do well 2 years later.
A 79-year-old man presented with chills and fever. He had a history of polymyalgia rheumatica and had been tapered off corticosteroids 1 month before admission. One week before he presented, he had developed generalized myalgia, chills, and fatigue. A cortisol stimulation test at that time was normal, prednisone was restarted, and his symptoms had improved. But 1 day before he presented, the chills had returned, this time with fever. Laboratory testing at an outpatient clinic had revealed abnormal liver enzyme levels.
On the day he presented, he felt worse, with persistent chills, fever, and vague lower abdominal pain, but he denied nausea, vomiting, changes in bowel habits, melena, hematochezia, and hematemesis. He was admitted for additional evaluation.
His medical history also included coronary artery disease (for which he had undergone coronary artery bypass grafting), hypertension, stable liver cysts, and gout. He had no known inflammatory bowel disease and no recent abdominal surgery. His medications included prednisone, atorvastatin, atenolol, aspirin, niacin, and cholecalciferol. He had no history of smoking, significant drinking, or use of illicit drugs. He had no respiratory or cardiac symptoms or neurologic symptoms consistent with a transient ischemic attack or stroke. He denied any rashes.
On admission, he was febrile, with temperatures reaching 102˚F (38.9˚C). His blood pressure was 137/63 mm Hg, pulse 54 beats per minute, respiration rate 18 breaths per minute, and oxygen saturation 97% on room air. A harsh systolic murmur was noted on physical examination. His abdomen was nondistended, nontender, and without bruits.
Laboratory testing (Table 1) revealed leukocytosis, anemia, mildly abnormal aminotransferase levels, elevated alkaline phosphatase, and markedly elevated C-reactive protein.
A full workup for fever was performed, including blood and urine cultures; chest radiography; contrast-enhanced computed tomography (CT) of the chest, abdomen, and pelvis; magnetic resonance imaging (MRI) of the abdomen; and colonoscopy. No source of infection—bacterial, viral, or fungal—was found. However, CT revealed new extensive thrombosis of the right portal vein and its branches (Figure 1).
CLINICAL PRESENTATION
1. Which of the following is least consistent with the clinical presentation of acute portal vein thrombosis?
- Abdominal pain
- Fever and chills
- Hematemesis
- Leukocytosis
- Absence of symptoms
Of these signs and symptoms, hematemesis is the least likely to be associated with acute portal vein thrombosis, although it can be associated with chronic cases.
Symptoms of portal vein thrombosis
Portal vein thrombosis causes extrahepatic obstruction of the portal venous system, which provides two-thirds of the total hepatic blood flow.
Acute. Often, thrombotic occlusion of the portal vein produces no acute symptoms because of immediate, compensatory vasodilation of the hepatic arterial system.1 Additionally, in the ensuing days, the thrombus becomes an organized collagenous plug, and collateral veins develop to bypass the blocked vein and maintain portal perfusion in a process called cavernous transformation.1,2 Thus, many patients have no symptoms.
If symptoms occur, portal vein thrombosis can initially present as transient abdominal pain with fever, as seen in this patient.3 Many patients with acute portal vein thrombosis experience abdominal pain due to intra-abdominal sepsis, also referred to as pylephlebitis.2,4 High, spiking fevers and chills also occur, caused by infected thrombi associated with intra-abdominal infections such as appendicitis, diverticulitis, and pancreatitis.5,6
Chronic. In contrast, symptomatic chronic portal vein thrombosis commonly presents with sequelae of portal hypertension, most notably gastrointestinal bleeding. Hematemesis from ruptured esophageal varices is the most frequent reason for seeking medical attention, though varices also develop in the stomach, duodenum, jejunum, gallbladder, and bile ducts.2,7 Abdominal pain is less common in chronic portal vein thrombosis unless the thrombus extends into the mesenteric veins and causes bowel ischemia or infarction. Long-standing portal vein thrombosis may also lead to dilated venous collaterals that compress large bile ducts, resulting in portal cholangiopathy.1,8
Portal vein thrombosis may present as acute intestinal ischemia and bowel infarction, though this is uncommon. This is generally seen with extensive occlusive portal vein thrombosis and concomitant mesenteric venous thrombosis.1,2
Other symptoms that are common but nonspecific are nausea, vomiting, diarrhea, weight loss, and anorexia.2
Signs of portal vein thrombosis
On examination, patients with acute portal vein thrombosis have minimal physical signs unless they have other contributing conditions. For example, acute portal vein thrombosis can result in abdominal distention secondary to ileus, or guarding and ascites secondary to intestinal infarction.3,9
Some patients with chronic portal vein thrombosis also have normal physical findings, but many have signs. Splenomegaly is seen in 75% to 100% of patients.2,7 Hepatomegaly, abdominal tenderness, and low-grade fever are common as well.2,10 Ascites is usually not present without underlying cirrhosis; however, mild and transient ascites can develop immediately after the thrombotic event before the patient develops collateral circulation.2
Laboratory testing for portal vein thrombosis
Laboratory test results are typically unremarkable. Liver function tests show preserved hepatic function but may reveal mild increases in aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase, and bilirubin.2,10
In acute cases, elevations of acute-phase reactant levels can occur.9 Leukocytosis and blood cultures growing Bacteroides species are seen in septic cases or pylephlebitis.11,12 There may be mild anemia, particularly after a recent bleeding episode, or mild leukopenia and thrombocytopenia due to hypersplenism. Suspicion of an underlying myeloproliferative disorder is high if thrombocytosis is present.2
DIAGNOSIS
2. All of the following would be appropriate initial diagnostic studies for portal vein thrombosis except which one?
- Doppler ultrasonography
- Contrast-enhanced CT
- Contrast-enhanced MRI
- Angiography
Portal vein thrombosis is most often diagnosed with noninvasive techniques, namely Doppler ultrasonography, CT, and MRI—not angiography.
Ultrasonography can reveal an echogenic thrombus in the vessel lumen with distention of the portal vein proximal to the occlusion and extensive collateral vessels. Plain ultrasonography fails to reveal the thrombus in up to one-third of patients. However, duplex ultrasonography with color flow Doppler imaging can confirm partial or complete absence of flow in the vein with 89% sensitivity and 92% specificity.13,14
On contrast-enhanced CT, the thrombus appears as a filling defect within the portal venous segment. Complete occlusion of the vein may produce a “train track” appearance due to contrast around the vessel.10 Without contrast, the clot will appear as hyperattenuating material in the portal vein, but contrast-enhanced imaging may be necessary to differentiate the thrombus from the vessel wall.15 Gas within the portal venous system is specific for pylephlebitis.4 Evidence of cavernous transformation is seen in chronic portal vein thrombosis.
Contrast-enhanced magnetic resonance angiography can also be used to evaluate patency and flow direction. In addition, it provides detailed anatomic information about the entire portal venous system, including the intrahepatic portal vessels, which is limited in CT imaging.2,10 CT and MRI can also help to identify predisposing conditions (eg, intra-abdominal infection, hepatocellular carcinoma) and complications (eg, intestinal infarction) associated with portal vein thrombosis.
Angiography can be considered if noninvasive techniques are inconclusive but is generally not necessary, given the increased use of CT and MRI.
In our patient, abdominal CT revealed occlusive thrombosis of the right portal vein and its branches (Figure 1). The left and main portal veins were patent. There was no evidence of intra-abdominal infection or infarction.
FINDING THE CAUSE
3. Which of the following is not a common cause
of portal vein thrombosis?
- A hypercoagulable state
- Immune deficiency
- Intra-abdominal infection
- Malignancy
- Portal hypertension
Once portal vein thrombosis has been diagnosed, the cause should be identified (Table 2). The differential diagnosis is broad, including both local factors (eg, injury to the portal vein, local inflammation, infection) and general factors (eg, inherited and acquired hypercoagulable conditions). Thrombophilias are identified in 60% of patients with portal vein thrombosis and local factors in 40%.7 Moreover, the etiology is often multifactorial. However, immune deficiency is not a common cause.
Hypercoagulability
Prothrombotic disorders can be either inherited or acquired.
Inherited deficiencies in the natural anticoagulants antithrombin, protein C, and protein S are associated with a high risk of thrombosis but have a low prevalence in the general population. In the setting of liver abnormalities, familial testing may be helpful to distinguish inherited causes of portal vein thrombosis from defective liver function as a consequence of portal vein thrombosis. The factor V Leiden mutation (G1691A) and the G20210A mutation in the prothrombin gene are more prevalent (> 2%) but generally confer a lower thrombosis risk.16 The prothrombin gene mutation G20210A is the most common risk factor for portal vein thrombosis, with prevalence of 2% to 22% in adults with nonmalignant, noncirrhotic portal vein thrombosis.3
Hyperhomocysteinemia due to a methylene tetrahydrofolate reductase (MTHFR) mutation (C677T) is another inherited associated risk factor for portal vein thrombosis, but hyperhomocysteinemia can also arise as a complication of portal vein thrombosis-related liver disease.3
Acquired prothrombotic disorders, particularly myeloproliferative diseases, are found in 22% to 48% of cases of portal vein thrombosis. Many young patients with myeloproliferative disorders present with portal vein thrombosis as the first symptom, and testing for the G1849T point mutation in JAK2 can make the diagnosis.17 Splenectomy with underlying myeloproliferative disorder confers a particularly high risk for portal vein thrombosis.18
Other thrombophilic disorders including antiphospholipid antibody syndrome, paroxysmal nocturnal hemoglobinuria, and malignancy can contribute to portal vein thrombosis.3 Pregnancy and oral contraceptive use have also been associated with hypercoagulability, and cessation of oral estrogen is recommended in such cases. The risk may be further increased in patients on oral contraceptives who have a previously unrecognized hypercoagulable state.3
Inflammation and infection
Inflammation and infection are local risk factors for portal vein thrombosis. Acute portal vein thrombosis has been associated with intra-abdominal infections (eg, appendicitis, cholecystitis) and with inflammatory conditions such as inflammatory bowel disease and pancreatitis.16,19 From 3% to 5% of all portal vein thrombosis cases result from pancreatitis, either from a single acute episode or from repeat inflammation of chronic pancreatitis.10 Portal vein thrombosis in the setting of inflammatory bowel disease can occur even when the disease is in remission, particularly in ulcerative colitis.20,21
Injury to the portal venous system
Abdominal surgery, particularly splenectomy, portosystemic shunting, colectomy, and blunt abdominal trauma can cause injury to the portal venous system, resulting in portal vein thrombosis. This is usually seen only in patients with portal hypertension, an underlying prothrombotic condition such as myeloproliferative disease, or inflammatory bowel disease.10,19,22
Impaired portal vein flow
Cirrhosis and malignancy are major risk factors for portal vein thrombosis. In case series, cirrhosis was found in 24% to 32% of patients with portal vein thrombosis.2,23 However, the overall prevalence of portal vein thrombosis in cirrhotic patients varies widely, from 0.6% to 28%, depending on the degree of cirrhosis.10
The pathogenesis of portal vein thrombosis in cirrhosis is unclear but may be multifactorial. Decreased portal blood flow (with subsequent stasis) and periportal lymphangitis and fibrosis are thought to stimulate thrombus formation.3,10 Additionally, patients with advanced cirrhosis are prothrombotic because of reduced hepatic synthesis of antithrombin, protein C, protein S, and coagulation factors.
Malignancy is associated with 21% to 24% of cases of portal vein thrombosis in adults, with pancreatic cancer and hepatocellular carcinoma being the most common.2,3 Others include cholangiocarcinoma and carcinomas of the stomach, lung, prostate, uterus, and kidney. Cancer causes portal vein thrombosis through a combination of tumor invasion into the portal vein, extrinsic compression by the tumor, periportal fibrosis following surgery or radiation, and hypercoagulability secondary to malignancy.9,16,24
Idiopathic portal vein thrombosis
Portal vein thrombosis is usually caused by one or more of the underlying factors mentioned above but is idiopathic in 8% to 15% of cases.10
Back to our patient
The cause of this patient’s portal vein thrombosis is unclear. He did not have a history of cirrhosis, inflammatory bowel disease, trauma, or abdominal surgery. His febrile illness could have precipitated the formation of a thrombus, but no definitive source of infection or inflammation was discovered. His workup was negative for pancreatitis, appendicitis, cholecystitis, diverticulitis, and prostatitis. No occult malignancy was found. It is also possible that his fever was the result of the thrombosis.
A full hypercoagulability panel revealed no striking abnormalities. He did have elevated fibrinogen and factor VIII levels that were consistent with an acute-phase reaction, along with an elevated erythrocyte sedimentation rate (> 90 mm/hr) and C-reactive protein level. Aside from the portal vein thrombosis, no potential source of inflammation could be identified.
Mildly reduced levels of antithrombin III activity were attributed to enoxaparin therapy and ultimately normalized on repeated testing. The patient had very minimally elevated titers of anticardiolipin immunoglobulin G (1:10 GPL) and anti-beta-2 glycoprotein immunoglobulin M (21 SMU), which were not thought to be significant. Tests for lupus anticoagulant, prothrombin gene mutation, activated protein C resistance, and JAK2 mutation were negative.
TREATMENT
4. Treatment of symptomatic portal vein thrombosis generally includes which two of the following?
- Anticoagulation
- Intravenous gamma globulin
- Broad-spectrum antibiotics
Anticoagulant therapy
Treatment of acute, symptomatic portal vein thrombosis involves anticoagulant therapy to prevent extension of the thrombus and, ultimately, to allow for recanalization of the obstructed veins. Anticoagulant therapy is initially intravenous unfractionated heparin or subcutaneous low-molecular-weight heparin, eventually bridged to an oral agent such as warfarin.3,9 Currently, there are inadequate data on the use of oral or parenteral factor Xa inhibitors or direct thrombin inhibitors in the treatment of this disease.
When started immediately, anticoagulation therapy is associated with complete recanalization in 38.3% and partial recanalization in 14% of patients presenting with complete thrombosis. Without anticoagulation, spontaneous recanalization is unusual.25
Although the optimal duration of anticoagulant therapy is unclear, a minimum of 3 months is generally recommended.9,26 If a hypercoagulable state is present or if the portal vein thrombosis is unprovoked (eg, by surgery, trauma, or an intra-abdominal infection), long-term treatment should be considered.26
Experience with thrombolytic therapy or mechanical recanalization has been limited, but the use of catheter-based techniques for pharmacomechanical thrombolysis has been reported.27–29 Transjugular intrahepatic portosystemic shunting is also an alternative to anticoagulation, but its role in treating portal vein thrombosis is complicated by technical difficulties of the procedure, postoperative complications, and recurrent occlusion of the shunt.25
Currently, there are no data comparing the risk-benefit ratio of early anticoagulation and that of invasive procedures. These more aggressive treatments are generally considered only when there is extensive thrombosis or ascites (which are both predictive factors of poor response to anticoagulation alone) and in patients for whom anticoagulation has failed.3 Surgical thrombectomy is rarely indicated, typically only in instances in which laparotomy is being performed for suspected bowel infarction.3
Antibiotics
In addition to anticoagulation, broad-spectrum antibiotics covering gram-negative and anaerobic bacteria are indicated for those cases of portal vein thrombosis associated with underlying infection.9
For chronic cases, the goals of management are to prevent and treat gastroesophageal variceal bleeding and to prevent recurrent thrombosis.9 Nonselective beta-blockers (eg, propranolol) and endoscopic band ligation have shown evidence of reducing the incidence of recurrent bleeding and prolonging survival in retrospective studies.9,30,31 Long-term anticoagulation is generally indicated to prevent further thrombosis and to increase the likelihood of recanalization only for patients with a permanent prothrombotic condition.9 In patients with clinically significant portal hypertension, the benefit of continued anticoagulation therapy must be weighed against the risk of esophageal and gastric variceal bleeding.
There is controversy regarding how to manage portal vein thrombosis that is incidentally identified and asymptomatic (eg, if it is discovered on an imaging study for another indication). Current guidelines recommend against anticoagulation in patients with incidentally discovered and asymptomatic splanchnic vein thrombosis, including portal vein thrombosis.26
Intravenous gamma globulin is not part of the treatment.
CASE CONTINUED
The patient’s presenting symptoms of fever, chills, and abdominal pain completely resolved after a course of antibiotic therapy. The erythrocyte sedimentation rate subsequently normalized and factor VIII activity improved. We believed that an underlying infectious or inflammatory process had contributed to the development of portal vein thrombosis, though the specific cause could not be identified. The patient was treated with enoxaparin 1 mg/kg twice a day and transitioned to warfarin.
Magnetic resonance venography done 3 months after diagnosis showed persistent right portal vein thrombosis that was largely unchanged. Anticoagulation was continued for 1 year with no change in his portal vein thrombosis on sequential imaging and was subsequently discontinued. To date, no malignancy or infectious process has been found, and the patient continues to do well 2 years later.
- Ponziani FR, Zocco MA, Campanale C, et al. Portal vein thrombosis: insight into physiopathology, diagnosis, and treatment. World J Gastroenterol 2010; 16:143–155.
- Cohen J, Edelman RR, Chopra S. Portal vein thrombosis: a review. Am J Med 1992; 92:173–182.
- Primignani M. Portal vein thrombosis, revisited. Dig Liver Dis 2010; 42:163–170.
- Condat B, Valla D. Nonmalignant portal vein thrombosis in adults. Nat Clin Pract Gastroenterol Hepatol 2006; 3:505–515.
- Condat B, Pessione F, Helene Denninger M, Hillaire S, Valla D. Recent portal or mesenteric venous thrombosis: increased recognition and frequent recanalization on anticoagulant therapy. Hepatology 2000; 32:466–470.
- Sheen CL, Lamparelli H, Milne A, Green I, Ramage JK. Clinical features, diagnosis and outcome of acute portal vein thrombosis. QJM 2000; 93:531–534.
- Sogaard KK, Astrup LB, Vilstrup H, Gronbaek H. Portal vein thrombosis; risk factors, clinical presentation and treatment. BMC Gastroenterol 2007; 7:34.
- Llop E, de Juan C, Seijo S, et al. Portal cholangiopathy: radiological classification and natural history. Gut 2011; 60:853–860.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Sobhonslidsuk A, Reddy KR. Portal vein thrombosis: a concise review. Am J Gastroenterol 2002; 97:535–541.
- Ni YH, Wang NC, Peng MY, Chou YY, Chang FY. Bacteroides fragilis bacteremia associated with portal vein and superior mesentery vein thrombosis secondary to antithrombin III and protein C deficiency: a case report. J Microbiol Immunol Infect 2002; 35:255–258.
- Trum J, Valla D, Cohen G, et al. Bacteroides bacteraemia of undetermined origin: strong association with portal vein thrombosis and cryptogenic pylephlebitis. Eur J Gastroenterol Hepatol 1993; 5:655–659.
- Ueno N, Sasaki A, Tomiyama T, Tano S, Kimura K. Color Doppler ultrasonography in the diagnosis of cavernous transformation of the portal vein. J Clin Ultrasound 1997; 25:227–233.
- Tessler FN, Gehring BJ, Gomes AS, et al. Diagnosis of portal vein thrombosis: value of color Doppler imaging. AJR Am J Roentgenol 1991; 157:293–296.
- Hidajat N, Stobbe H, Griesshaber V, Felix R, Schroder RJ. Imaging and radiological interventions of portal vein thrombosis. Acta Radiol 2005; 46:336–343.
- Valla DC, Condat B. Portal vein thrombosis in adults: pathophysiology, pathogenesis and management. J Hepatol 2000; 32:865–871.
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352:1779–1790.
- Krauth MT, Lechner K, Neugebauer EA, Pabinger I. The postoperative splenic/portal vein thrombosis after splenectomy and its prevention—an unresolved issue. Haematologica 2008; 93:1227–1232.
- Sinagra E, Aragona E, Romano C, et al. The role of portal vein thrombosis in the clinical course of inflammatory bowel diseases: report on three cases and review of the literature. Gastroenterol Res Pract 2012; 2012:916428.
- Maconi G, Bolzacchini E, Dell’Era A, Russo U, Ardizzone S, de Franchis R. Portal vein thrombosis in inflammatory bowel diseases: a single-center case series. J Crohns Colitis 2012; 6:362–367.
- Jackson LM, O’Gorman PJ, O’Connell J, Cronin CC, Cotter KP, Shanahan F. Thrombosis in inflammatory bowel disease: clinical setting, procoagulant profile and factor V Leiden. QJM 1997; 90:183–188.
- Eguchi A, Hashizume M, Kitano S, Tanoue K, Wada H, Sugimachi K. High rate of portal thrombosis after splenectomy in patients with esophageal varices and idiopathic portal hypertension. Arch Surg 1991; 126:752–755.
- Ogren M, Bergqvist D, Björck M, Acosta S, Eriksson H, Sternby NH. Portal vein thrombosis: prevalence, patient characteristics and lifetime risk: a population study based on 23,796 consecutive autopsies. World J Gastroenterol 2006; 12:2115–2119.
- Falanga A, Marchetti M, Vignoli A. Coagulation and cancer: biological and clinical aspects. J Thromb Haemost 2013; 11:223–233.
- Congly SE, Lee SS. Portal vein thrombosis: should anticoagulation be used? Curr Gastroenterol Rep 2013; 15:306.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Uflacker R. Applications of percutaneous mechanical thrombectomy in transjugular intrahepatic portosystemic shunt and portal vein thrombosis. Tech Vasc Interv Radiol 2003; 6:59–69.
- Takahashi N, Kuroki K, Yanaga K. Percutaneous transhepatic mechanical thrombectomy for acute mesenteric venous thrombosis. J Endovasc Ther 2005; 12:508–511.
- Lopera JE, Correa G, Brazzini A, et al. Percutaneous transhepatic treatment of symptomatic mesenteric venous thrombosis. J Vasc Surg 2002; 36:1058–1061.
- Orr DW, Harrison PM, Devlin J, et al. Chronic mesenteric venous thrombosis: evaluation and determinants of survival during long-term follow-up. Clin Gastroenterol Hepatol 2007; 5:80–86.
- Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant therapy. Gastroenterology 2001; 120:490–497.
- Ponziani FR, Zocco MA, Campanale C, et al. Portal vein thrombosis: insight into physiopathology, diagnosis, and treatment. World J Gastroenterol 2010; 16:143–155.
- Cohen J, Edelman RR, Chopra S. Portal vein thrombosis: a review. Am J Med 1992; 92:173–182.
- Primignani M. Portal vein thrombosis, revisited. Dig Liver Dis 2010; 42:163–170.
- Condat B, Valla D. Nonmalignant portal vein thrombosis in adults. Nat Clin Pract Gastroenterol Hepatol 2006; 3:505–515.
- Condat B, Pessione F, Helene Denninger M, Hillaire S, Valla D. Recent portal or mesenteric venous thrombosis: increased recognition and frequent recanalization on anticoagulant therapy. Hepatology 2000; 32:466–470.
- Sheen CL, Lamparelli H, Milne A, Green I, Ramage JK. Clinical features, diagnosis and outcome of acute portal vein thrombosis. QJM 2000; 93:531–534.
- Sogaard KK, Astrup LB, Vilstrup H, Gronbaek H. Portal vein thrombosis; risk factors, clinical presentation and treatment. BMC Gastroenterol 2007; 7:34.
- Llop E, de Juan C, Seijo S, et al. Portal cholangiopathy: radiological classification and natural history. Gut 2011; 60:853–860.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Sobhonslidsuk A, Reddy KR. Portal vein thrombosis: a concise review. Am J Gastroenterol 2002; 97:535–541.
- Ni YH, Wang NC, Peng MY, Chou YY, Chang FY. Bacteroides fragilis bacteremia associated with portal vein and superior mesentery vein thrombosis secondary to antithrombin III and protein C deficiency: a case report. J Microbiol Immunol Infect 2002; 35:255–258.
- Trum J, Valla D, Cohen G, et al. Bacteroides bacteraemia of undetermined origin: strong association with portal vein thrombosis and cryptogenic pylephlebitis. Eur J Gastroenterol Hepatol 1993; 5:655–659.
- Ueno N, Sasaki A, Tomiyama T, Tano S, Kimura K. Color Doppler ultrasonography in the diagnosis of cavernous transformation of the portal vein. J Clin Ultrasound 1997; 25:227–233.
- Tessler FN, Gehring BJ, Gomes AS, et al. Diagnosis of portal vein thrombosis: value of color Doppler imaging. AJR Am J Roentgenol 1991; 157:293–296.
- Hidajat N, Stobbe H, Griesshaber V, Felix R, Schroder RJ. Imaging and radiological interventions of portal vein thrombosis. Acta Radiol 2005; 46:336–343.
- Valla DC, Condat B. Portal vein thrombosis in adults: pathophysiology, pathogenesis and management. J Hepatol 2000; 32:865–871.
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352:1779–1790.
- Krauth MT, Lechner K, Neugebauer EA, Pabinger I. The postoperative splenic/portal vein thrombosis after splenectomy and its prevention—an unresolved issue. Haematologica 2008; 93:1227–1232.
- Sinagra E, Aragona E, Romano C, et al. The role of portal vein thrombosis in the clinical course of inflammatory bowel diseases: report on three cases and review of the literature. Gastroenterol Res Pract 2012; 2012:916428.
- Maconi G, Bolzacchini E, Dell’Era A, Russo U, Ardizzone S, de Franchis R. Portal vein thrombosis in inflammatory bowel diseases: a single-center case series. J Crohns Colitis 2012; 6:362–367.
- Jackson LM, O’Gorman PJ, O’Connell J, Cronin CC, Cotter KP, Shanahan F. Thrombosis in inflammatory bowel disease: clinical setting, procoagulant profile and factor V Leiden. QJM 1997; 90:183–188.
- Eguchi A, Hashizume M, Kitano S, Tanoue K, Wada H, Sugimachi K. High rate of portal thrombosis after splenectomy in patients with esophageal varices and idiopathic portal hypertension. Arch Surg 1991; 126:752–755.
- Ogren M, Bergqvist D, Björck M, Acosta S, Eriksson H, Sternby NH. Portal vein thrombosis: prevalence, patient characteristics and lifetime risk: a population study based on 23,796 consecutive autopsies. World J Gastroenterol 2006; 12:2115–2119.
- Falanga A, Marchetti M, Vignoli A. Coagulation and cancer: biological and clinical aspects. J Thromb Haemost 2013; 11:223–233.
- Congly SE, Lee SS. Portal vein thrombosis: should anticoagulation be used? Curr Gastroenterol Rep 2013; 15:306.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Uflacker R. Applications of percutaneous mechanical thrombectomy in transjugular intrahepatic portosystemic shunt and portal vein thrombosis. Tech Vasc Interv Radiol 2003; 6:59–69.
- Takahashi N, Kuroki K, Yanaga K. Percutaneous transhepatic mechanical thrombectomy for acute mesenteric venous thrombosis. J Endovasc Ther 2005; 12:508–511.
- Lopera JE, Correa G, Brazzini A, et al. Percutaneous transhepatic treatment of symptomatic mesenteric venous thrombosis. J Vasc Surg 2002; 36:1058–1061.
- Orr DW, Harrison PM, Devlin J, et al. Chronic mesenteric venous thrombosis: evaluation and determinants of survival during long-term follow-up. Clin Gastroenterol Hepatol 2007; 5:80–86.
- Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant therapy. Gastroenterology 2001; 120:490–497.
Overcoming health care disparities via better cross-cultural communication and health literacy
An english-speaking middle-aged woman from an ethnic minority group presents to her internist for follow-up of her chronic medical problems, which include diabetes, high blood pressure, asthma, and high cholesterol. Although she sees her physician regularly, her medical conditions are not optimally controlled.
At one of the visits, her physician gives her a list of her medications and, while reviewing it, explains—not for the first time—the importance of taking all of them as prescribed. The patient looks at the paper for a while, and then cautiously tells the physician, “But I can’t read.”
This patient presented to our practice several years ago. The scenario may be familiar to many primary physicians, except for the ending— ie, the patient telling her physician that she cannot read.
Her case raises several questions:
- Why did the physician not realize at the first encounter that she could not read the names of her prescribed medications?
- Why did the patient wait to tell her physician that important fact?
- And to what extent did her inability to read contribute to the poor control of her chronic medical problems?
Patients like this one are the human faces behind the statistics about health disparities—the worse outcomes noted in minority populations. Here, we discuss the issues of cross-cultural communication and health literacy as they relate to health care disparities.
DISPARITY IS NOT ONLY DUE TO LACK OF ACCESS
Health care disparity has been an important topic of discussion in medicine in the past decade.
In a 2003 publication,1 the Institute of Medicine identified lower quality of health care in minority populations as a serious problem. Further, it disputed the long-held belief that the differences in health care between minority and nonminority populations could be explained by lack of access to medical services in minority groups. Instead, it cited factors at the level of the health care system, the level of the patient, and the “care-process level” (ie, the physician-patient encounter) as contributing in distinct ways to the problem.1
A CALL FOR CULTURAL COMPETENCE
In a policy paper published in 2010, the American College of Physicians2 reviewed the progress made in addressing health care disparities. In addition, noting that an individual’s environment, income, level of education, and other factors all affect health, it called for a concerted effort to improve insurance coverage, health literacy, and the health care delivery system; to address stressors both within and outside the health care system; and to recruit more minority health care workers.
None of these things seems like anything a busy practicing clinician could do much about. However, we can try to improve our cultural competence in our interactions with patients on an individual level.
The report recommends that physicians and other health care professionals be sensitive to cultural diversity among patients. It also says we should recognize our preconceived perceptions of minority patients that may affect their treatment and contribute to disparities in health care in minorities. To those ends, it calls for cultural competence training in medical school to improve cultural awareness and sensitivity.2
The Office of Minority Health broadly defines cultural and linguistic competence in health as “a set of congruent behaviors, attitudes, and policies that come together in a system, agency, or among professionals that enables effective work in cross-cultural situations.”3 Cultural competence training should focus on being aware of one’s personal bias, as well as on education about culture-specific norms or knowledge of possible causes of mistrust in minority groups.
For example, many African Americans may mistrust the medical system, given the awareness of previous inequities such as the notorious Tuskegee syphilis study (in which informed consent was not used and treatment that was needed was withheld). Further, beliefs about health in minority populations may be discordant with the Western medical model.4
RECOGNIZING OUR OWN BIASES
Preconceived perceptions on the part of the physician may be shaped by previous experiences with patients from a specific minority group or by personal bias. Unfortunately, even a well-meaning physician who has tried to learn about cultural norms of specific minority groups can be at risk of stereotyping by assuming that all members of that group hold the same beliefs. From the patient’s viewpoint, they can also be molded by previous experiences of health care inequities or unfavorable interactions with physicians.
For example, in the case we described above, perhaps the physician had assumed that the patient was noncompliant and therefore did not look for reasons for the poor control of her medical problems, or maybe the patient did not trust the physician enough to explain the reason for her difficulty with understanding how to take her medications.
Being aware of our own unconscious stereotyping of minority groups is an important step in effectively communicating with patients from different cultural backgrounds or with low health literacy. We also need to reflect about our own health belief system and try to incorporate the patient’s viewpoint into decision-making.
If, on reflection, we recognize that we do harbor biases, we ought to think about ways to better accommodate patients from different backgrounds and literacy levels, including trying to learn more about their culture or mastering techniques to effectively explain treatment plans to low-literacy patients.
ALL ENCOUNTERS WITH PATIENTS ARE ‘CROSS-CULTURAL’
In health care, “cross-cultural communication” does not refer only to interactions between persons from different ethnic backgrounds or with different beliefs about health. Health care has a culture of its own, creating a cross-cultural encounter the moment a person enters your office or clinic in the role of “patient.”
Carillo et al5 categorized issues that may pose difficulties in a cross-cultural encounter as those of authority, physical contact, communication styles, gender, sexuality, and family.
Physician-patient communication is a complicated issue. Many patients will not question a physician if their own cultural norms view it as disrespectful—even if they have very specific fears about the diagnosis or treatment plan. They may also defer any important decision to a family member who has the authority to make decisions for the family.
Frequently, miscommunication is unintentional. In a recent study of hospitalized patients,6 77% of the physicians believed that their patients understood their diagnoses, while only 57% of patients could correctly state this information.
WHAT DOES THE PATIENT THINK?
A key issue in cross-cultural communication, and one that is often neglected, is to address a patient’s fears about his or her illness. In the study mentioned above, more than half of the patients who reported having anxieties or fears in the hospital stated that their physicians did not discuss their fears.6 But if we fail to do so, patients may be less satisfied with the treatment plan and may not accept our recommendations.
A patient’s understanding of his or her illness may be very different from the biomedical explanation. For example, we once saw an elderly man who was admitted to the hospital with back pain due to metastatic prostate cancer, but who was convinced that his symptoms were caused by a voodoo “hex” placed on him by his ex-wife.

For example, for the man who thought that his ex-wife put a hex on him, asking him “What do you think has caused your problem?” during the initial history-taking would allow him to express his concern about the hex and give the physician an opportunity to learn of this fear and then to offer the biomedical explanation for the problem and for the recommended treatment.
What happens more often in practice is that the specific fear is not addressed at the start of the encounter. Consequently, the patient is less likely to follow through with the treatment plan, as he or she does not feel the prescribed treatment is fixing the real problem. This process of exploring the explanatory model of illness may be viewed on a practical level as a way of managing expectations in the clinical care of culturally diverse populations.
HEALTH LITERACY: MORE THAN THE ABILITY TO READ
The better you know how to read, the healthier you probably are. In fact, a study found that a person’s literacy level correlated more strongly with health than did race or formal education level.9 (Apparently, attending school does not necessarily mean that people know how to read, and not attending school doesn’t mean that they don’t.)
Even more important than literacy may be health literacy, defined by Ratzan and Parker as “the degree to which individuals have the capacity to obtain, process, and understand basic health information and services needed to make appropriate health decisions.”8 It includes basic math and critical-thinking skills that allow patients to use medications properly and participate in treatment decisions. Thus, health literacy is much more than the ability to read.
Even people who read and write very well may have trouble when confronted with the complexities of navigating our health care system, such as appointment scheduling, specialty referrals, and follow-up testing and procedures: their health literacy may be lower than their general literacy. We had a patient, a highly trained professional, who was confused by instructions for preparing for colonoscopy on a patient handout. Another similar patient could not understand the dosing of eye drops after cataract surgery because the instructions on the discharge paperwork were unclear.
However, limited health literacy disproportionately affects minority groups and is linked to poorer health care outcomes. Thus, addressing limited health literacy is important in addressing health care disparities. Effective physician-patient communication about treatment plans is fundamental to providing equitable care to patients from minority groups, some of whom may be at high risk for low health literacy.
Below, we will review some of the data on health literacy and offer suggestions for screening and interventions for those whose health literacy is limited.
36% have basic or below-basic reading skills
Every 10 years, the US Department of Education completes its National Assessment of Adult Literacy. Its 2003 survey—the most recent—included 19,000 adults in the community and in prison, interviewed at their place of residence.10 Each participant completed a set of tasks to measure his or her ability to read, understand, and interpret text and to use and interpret numbers.
Participants were divided into four categories based on the results: proficient (12%), intermediate (53%), basic (22%), and below basic (14%). Additionally, 5% of potential participants could not be tested because they had insufficient skills to participate in the survey.
Low literacy puts patients at risk
Although literacy is not the same as health literacy, functionally, those who have basic or below-basic literacy skills (36% of the US population) are at high risk for encountering problems in the US health care system. For example, they would have difficulty with most patient education handouts and health insurance forms.
Limited health literacy exacts both personal and financial costs. Patients with low health literacy are less likely to understand how to take their medications, what prescription warning labels mean, how to schedule follow-up appointments, and how to fill out health insurance forms.11–14
Medicare managed-care enrollees are more likely to be hospitalized if they have limited health literacy,15 and diabetic Medicaid patients who have limited health literacy are less likely to have good glycemic control.16 One study showed annual health care costs of $10,688 for Medicaid enrollees with limited health literacy compared with $2,891 for all enrollees.17 The total cost of limited health literacy to the US health care system is estimated to be between $50 and $73 billion per year.18
Screening for limited health literacy: You can’t tell just by looking
Given the high costs of low health literacy, identifying patients who have it is of paramount importance.
Groups who are more likely to have limited health literacy include the elderly, the poor, the unemployed, high school dropouts, members of minority groups, recent immigrants, and people for whom English is a second language.
However, these demographic factors are not sufficient as a screen for low health literacy—you can't tell just by looking. Red flags for low health literacy include difficulty filling out forms in the office, missed appointments, nonadherence to medication regimens, failure to follow up with scheduled testing, and difficulty reading written materials, often masked with a statement such as “I forgot my glasses and will read this at home.”
A number of screening tests have been developed, including the Rapid Estimate of Adult Literacy in Medicine (REALM)19 and the Test for Functional Health Literacy in Adults (TOFHLA).20 These tests are long, making them difficult to incorporate into a patient visit in a busy primary care practice, but they are useful for research. A newer screening test asks the patient to review a nutrition label and answer six questions.21
The most useful screening test for clinical use may consist of a single question. Questions that have been validated:
- “How often do you need to have someone help you when you read instructions, pamphlets, or other written material from your doctor or pharmacy?” Positive answers are “sometimes,” “often,” or “always.”
- “How confident are you filling out medical forms by yourself?” Positive answers are “somewhat,” “a little bit,” or “not at all.”22–24
These questions can be included either in the initial screening by a nurse or medical assistant or as part of the social history portion of the interview with the physician.
A “brown bag review” can also be helpful. Patients are asked to bring in their medications (often in a brown bag—hence the name). Asking the patient to identify each medication by name and the indication for it can uncover knowledge gaps that indicate low health literacy.
The point to remember is that patients with low health literacy will probably not tell you that they do not understand. However, they would appreciate being asked in a nonthreatening manner.
Make your office a shame-free environment
Many experts advocate a “universal precautions approach,” in which interventions to address low health literacy are incorporated into routine office practice for all patients. Practice sites should adopt a culture of a “shame-free environment,” in which support staff encourage patients to ask questions and are trained to offer assistance to those having difficulty reading or filling out forms.
On a broader level, medical offices and hospitals can partner with adult-learning specialists to help patients gain skills to navigate the health care system. All signage should be clear and should use plain language as opposed to medical terms. Medical forms and questionnaires should be designed to collect only essential information and should be written at a sixth-grade reading level or below. Patient instructions and educational materials should also be clear and free of jargon.
The ‘teach-back’ technique
The “teach-back” technique is a simple method to confirm patient understanding at the end of the visit. This involves asking patients in a nonthreatening way to explain or demonstrate what they have been told. Examples:
- “I want to make sure I have explained things correctly. Can you tell me how you plan to take your medication when you go home?”
- “I want to make sure I have done a good job explaining things to you. When you go home and tell your spouse about your visit today, what will you say?”
These questions should be asked in a nonthreatening way. Put the burden of explanation on yourself as the first step, and let the patient know you are willing to explain again more thoroughly any instructions that may have not been clearly understood.
Other measures
Pictures and computer-based education may be useful for some patients who have difficulty reading.
Weiss25 advocates six steps to improve communication with patients in all encounters: slow down; use plain, nonmedical language; show or draw pictures; limit the amount of information provided; use the teach-back technique; and create a shame-free environment, encouraging questions.
Improving health literacy, as it relates to cross-cultural communication of treatment plans, must encompass understanding of health beliefs often based on cultural norms, in order to come to agreement on a mutually acceptable plan of care. Physicians should be aware of preferences for nontraditional or complementary treatments that may reflect specific cultural beliefs.
IF THE PATIENT DOES NOT SPEAK ENGLISH
Verbal communication across language barriers poses another layer of challenge. A trained interpreter should be used whenever possible when treating a patient who speaks a different language than that of the practitioner. When family members are used as interpreters, there are risks that the patient may not fully disclose facts about the history of illness or specific symptoms, and also that family members may place their own “twist” on the story when translating.
The physician should speak directly to the patient in a normal tone of voice. In this setting, also remember that nonverbal communication can be misinterpreted. Gestures should be avoided. Finally, be aware that personal space is viewed differently depending on cultural background, as is eye contact.
It is helpful to have a pre-interview meeting with the interpreter to explain the format of the interview, as well as a post-interview meeting to ensure all parties felt they effectively communicated during the encounter.
TOWARD EQUITABLE CARE
Health care disparities are the result of multiple determinants. In December 2008, a National Institutes of Health summit conference cited not only barriers to access, but also the interaction of biological, behavioral, social, environmental, economic, cultural, and political factors, and noted that the causes and effects of health disparities transcend health care.26
Clearly, an individual physician’s efforts will not be all that is needed to eliminate health disparities. A team-based approach is essential, using skills of nonphysician members of the health care team such as nurses, medical assistants, social workers, and case managers. Continued opportunity for professional training and development in provider-patient communication skills should be offered.
However, the impact of effective cross-cultural communication and managing low health literacy populations on the physician-patient level should not be understated. As practitioners treating patients from diverse backgrounds, improving self-awareness, eliciting the patient’s explanatory model, and assuring understanding of treatment plans for patients with low health literacy or with language barriers, we can do our part in working toward equitable care for all patients.
- Institute of Medicine of the National Academies. Unequal Treatment: Confronting Racial and Ethnic Disparities in Healthcare; 2003. http://www.nap.edu/openbook.php?record_id=12875&page=R1. Accessed January 5, 2012.
- American College of Physicians. Racial and Ethnic Disparities in Health Care, Updated 2010. Philadelphia: American College of Physicians; 2010: Policy Paper.
- US Department of Health and Human Services. The Office of Minority Health. What Is Cultural Competency? http://minorityhealth.hhs.gov/templates/browse.aspx?lvl=2&lvlid=11. Accessed January 5, 2012.
- Eiser AR, Ellis G. Viewpoint: cultural competence and the African American experience with health care: the case for specific content in cross-cultural education. Acad Med 2007; 82:176–183.
- Carrillo JE, Green AR, Betancourt JR. Cross-cultural primary care: a patient-based approach. Ann Intern Med 1999; 130:829–834.
- Olson DP, Windish DM. Communication discrepancies between physicians and hospitalized patients. Arch Intern Med 2010; 170:1302–1307.
- Kleinman A, Eisenberg L, Good B. Culture, illness, and care: clinical lessons from anthropologic and cross-cultural research. Ann Intern Med 1978; 88:251–258.
- National Library of Medicine. Current bibliographies in medicine 2000–1. Health Literacy. www.nlm.nih.gov/archive//20061214/pubs/cbm/hliteracy.html. Accessed January 5, 2012.
- Sentell TL, Halpin HA. Importance of adult literacy in understanding health disparities. J Gen Intern Med 2006; 21:862–866.
- Kutner M, Greenberg E, Jin Y, Paulsen C. The Health Literacy of America’s Adults: Results From the 2003 National Assessment of Adult Literacy (NCES 2006–483). US Department of Education. Washington, DC: National Center for Education Statistics; 2006. http://nces.ed.gov/pubs2006/2006483.pdf. Accessed January 5, 2012.
- Williams MV, Parker RM, Baker DW, et al. Inadequate functional health literacy among patients at two public hospitals. JAMA 1995; 274:1677–1682.
- Baker DW, Parker RM, Williams MV, et al. The health care experience of patients with low literacy. Arch Fam Med 1996; 5:329–334.
- Fact Sheet: health literacy and understanding medical information. Lawrenceville, NJ: Center for Health Care Strategies; 2002.
- Wolf MS, Davis TC, Tilson HH, Bass PF, Parker RM. Misunderstanding of prescription drug warning labels among patients with low literacy. Am J Health Syst Pharm 2006; 63:1048–1055.
- Baker DW, Gazmararian JA, Williams MV, et al. Functional health literacy and the risk of hospital admission among Medicare managed care enrollees. Am J Public Health 2002; 92:1278–1283.
- Schillinger D, Barton LR, Karter AJ, Wang F, Adler N. Does literacy mediate the relationship between education and health outcomes? A study of a low-income population with diabetes. Public Health Rep 2006; 121:245–254.
- Weiss BD, Palmer R. Relationship between health care costs and very low literacy skills in a medically needy and indigent Medicaid population. J Am Board Fam Pract 2004; 17:44–47.
- Friedland RB. Understanding health literacy: new estimates of the costs of inadequate health literacy. Washington, DC: National Academy on an Aging Society; 1998.
- Davis TC, Long SW, Jackson RH, et al. Rapid estimate of adult literacy in medicine: a shortened screening instrument. Fam Med 1993; 25:391–395.
- Baker DW, Williams MV, Parker RM, Gazmararian JA, Nurss J. Development of a brief test to measure functional health literacy. Patient Educ Couns 1999; 38:33–42.
- Weiss BD, Mays MZ, Martz W, et al. Quick assessment of literacy in primary care: the newest vital sign. Ann Fam Med 2005; 3:514–522.
- Chew LD, Bradley KA, Boyko EJ. Brief questions to identify patients with inadequate health literacy. Fam Med 2004; 36:588–594.
- Morris NS, MacLean CD, Chew LD, Littenberg B. The Single Item Literacy Screener: evaluation of a brief instrument to identify limited reading ability. BMC Fam Pract 2006; 7:21.
- Wallace LS, Rogers ES, Roskos SE, Holiday DB, Weiss BD. Brief report: screening items to identify patients with limited health literacy skills. J Gen Intern Med 2006; 21:874–877.
- Weiss BD. Health Literacy and Patient Safety: Help Patients Understand. 2nd ed. American Medical Association Foundation and American Medical Association. www.ama-assn.org/ama1/pub/upload/mm/367/healthlitclinicians.pdf. Accessed January 5, 2012.
- Dankwa-Mullan I, Rhee KB, Williams K, et al. The science of eliminating health disparities: summary and analysis of the NIH summit recommendations. Am J Public Health 2010; 100(suppl 1):S12–S18.
An english-speaking middle-aged woman from an ethnic minority group presents to her internist for follow-up of her chronic medical problems, which include diabetes, high blood pressure, asthma, and high cholesterol. Although she sees her physician regularly, her medical conditions are not optimally controlled.
At one of the visits, her physician gives her a list of her medications and, while reviewing it, explains—not for the first time—the importance of taking all of them as prescribed. The patient looks at the paper for a while, and then cautiously tells the physician, “But I can’t read.”
This patient presented to our practice several years ago. The scenario may be familiar to many primary physicians, except for the ending— ie, the patient telling her physician that she cannot read.
Her case raises several questions:
- Why did the physician not realize at the first encounter that she could not read the names of her prescribed medications?
- Why did the patient wait to tell her physician that important fact?
- And to what extent did her inability to read contribute to the poor control of her chronic medical problems?
Patients like this one are the human faces behind the statistics about health disparities—the worse outcomes noted in minority populations. Here, we discuss the issues of cross-cultural communication and health literacy as they relate to health care disparities.
DISPARITY IS NOT ONLY DUE TO LACK OF ACCESS
Health care disparity has been an important topic of discussion in medicine in the past decade.
In a 2003 publication,1 the Institute of Medicine identified lower quality of health care in minority populations as a serious problem. Further, it disputed the long-held belief that the differences in health care between minority and nonminority populations could be explained by lack of access to medical services in minority groups. Instead, it cited factors at the level of the health care system, the level of the patient, and the “care-process level” (ie, the physician-patient encounter) as contributing in distinct ways to the problem.1
A CALL FOR CULTURAL COMPETENCE
In a policy paper published in 2010, the American College of Physicians2 reviewed the progress made in addressing health care disparities. In addition, noting that an individual’s environment, income, level of education, and other factors all affect health, it called for a concerted effort to improve insurance coverage, health literacy, and the health care delivery system; to address stressors both within and outside the health care system; and to recruit more minority health care workers.
None of these things seems like anything a busy practicing clinician could do much about. However, we can try to improve our cultural competence in our interactions with patients on an individual level.
The report recommends that physicians and other health care professionals be sensitive to cultural diversity among patients. It also says we should recognize our preconceived perceptions of minority patients that may affect their treatment and contribute to disparities in health care in minorities. To those ends, it calls for cultural competence training in medical school to improve cultural awareness and sensitivity.2
The Office of Minority Health broadly defines cultural and linguistic competence in health as “a set of congruent behaviors, attitudes, and policies that come together in a system, agency, or among professionals that enables effective work in cross-cultural situations.”3 Cultural competence training should focus on being aware of one’s personal bias, as well as on education about culture-specific norms or knowledge of possible causes of mistrust in minority groups.
For example, many African Americans may mistrust the medical system, given the awareness of previous inequities such as the notorious Tuskegee syphilis study (in which informed consent was not used and treatment that was needed was withheld). Further, beliefs about health in minority populations may be discordant with the Western medical model.4
RECOGNIZING OUR OWN BIASES
Preconceived perceptions on the part of the physician may be shaped by previous experiences with patients from a specific minority group or by personal bias. Unfortunately, even a well-meaning physician who has tried to learn about cultural norms of specific minority groups can be at risk of stereotyping by assuming that all members of that group hold the same beliefs. From the patient’s viewpoint, they can also be molded by previous experiences of health care inequities or unfavorable interactions with physicians.
For example, in the case we described above, perhaps the physician had assumed that the patient was noncompliant and therefore did not look for reasons for the poor control of her medical problems, or maybe the patient did not trust the physician enough to explain the reason for her difficulty with understanding how to take her medications.
Being aware of our own unconscious stereotyping of minority groups is an important step in effectively communicating with patients from different cultural backgrounds or with low health literacy. We also need to reflect about our own health belief system and try to incorporate the patient’s viewpoint into decision-making.
If, on reflection, we recognize that we do harbor biases, we ought to think about ways to better accommodate patients from different backgrounds and literacy levels, including trying to learn more about their culture or mastering techniques to effectively explain treatment plans to low-literacy patients.
ALL ENCOUNTERS WITH PATIENTS ARE ‘CROSS-CULTURAL’
In health care, “cross-cultural communication” does not refer only to interactions between persons from different ethnic backgrounds or with different beliefs about health. Health care has a culture of its own, creating a cross-cultural encounter the moment a person enters your office or clinic in the role of “patient.”
Carillo et al5 categorized issues that may pose difficulties in a cross-cultural encounter as those of authority, physical contact, communication styles, gender, sexuality, and family.
Physician-patient communication is a complicated issue. Many patients will not question a physician if their own cultural norms view it as disrespectful—even if they have very specific fears about the diagnosis or treatment plan. They may also defer any important decision to a family member who has the authority to make decisions for the family.
Frequently, miscommunication is unintentional. In a recent study of hospitalized patients,6 77% of the physicians believed that their patients understood their diagnoses, while only 57% of patients could correctly state this information.
WHAT DOES THE PATIENT THINK?
A key issue in cross-cultural communication, and one that is often neglected, is to address a patient’s fears about his or her illness. In the study mentioned above, more than half of the patients who reported having anxieties or fears in the hospital stated that their physicians did not discuss their fears.6 But if we fail to do so, patients may be less satisfied with the treatment plan and may not accept our recommendations.
A patient’s understanding of his or her illness may be very different from the biomedical explanation. For example, we once saw an elderly man who was admitted to the hospital with back pain due to metastatic prostate cancer, but who was convinced that his symptoms were caused by a voodoo “hex” placed on him by his ex-wife.
For example, for the man who thought that his ex-wife put a hex on him, asking him “What do you think has caused your problem?” during the initial history-taking would allow him to express his concern about the hex and give the physician an opportunity to learn of this fear and then to offer the biomedical explanation for the problem and for the recommended treatment.
What happens more often in practice is that the specific fear is not addressed at the start of the encounter. Consequently, the patient is less likely to follow through with the treatment plan, as he or she does not feel the prescribed treatment is fixing the real problem. This process of exploring the explanatory model of illness may be viewed on a practical level as a way of managing expectations in the clinical care of culturally diverse populations.
HEALTH LITERACY: MORE THAN THE ABILITY TO READ
The better you know how to read, the healthier you probably are. In fact, a study found that a person’s literacy level correlated more strongly with health than did race or formal education level.9 (Apparently, attending school does not necessarily mean that people know how to read, and not attending school doesn’t mean that they don’t.)
Even more important than literacy may be health literacy, defined by Ratzan and Parker as “the degree to which individuals have the capacity to obtain, process, and understand basic health information and services needed to make appropriate health decisions.”8 It includes basic math and critical-thinking skills that allow patients to use medications properly and participate in treatment decisions. Thus, health literacy is much more than the ability to read.
Even people who read and write very well may have trouble when confronted with the complexities of navigating our health care system, such as appointment scheduling, specialty referrals, and follow-up testing and procedures: their health literacy may be lower than their general literacy. We had a patient, a highly trained professional, who was confused by instructions for preparing for colonoscopy on a patient handout. Another similar patient could not understand the dosing of eye drops after cataract surgery because the instructions on the discharge paperwork were unclear.
However, limited health literacy disproportionately affects minority groups and is linked to poorer health care outcomes. Thus, addressing limited health literacy is important in addressing health care disparities. Effective physician-patient communication about treatment plans is fundamental to providing equitable care to patients from minority groups, some of whom may be at high risk for low health literacy.
Below, we will review some of the data on health literacy and offer suggestions for screening and interventions for those whose health literacy is limited.
36% have basic or below-basic reading skills
Every 10 years, the US Department of Education completes its National Assessment of Adult Literacy. Its 2003 survey—the most recent—included 19,000 adults in the community and in prison, interviewed at their place of residence.10 Each participant completed a set of tasks to measure his or her ability to read, understand, and interpret text and to use and interpret numbers.
Participants were divided into four categories based on the results: proficient (12%), intermediate (53%), basic (22%), and below basic (14%). Additionally, 5% of potential participants could not be tested because they had insufficient skills to participate in the survey.
Low literacy puts patients at risk
Although literacy is not the same as health literacy, functionally, those who have basic or below-basic literacy skills (36% of the US population) are at high risk for encountering problems in the US health care system. For example, they would have difficulty with most patient education handouts and health insurance forms.
Limited health literacy exacts both personal and financial costs. Patients with low health literacy are less likely to understand how to take their medications, what prescription warning labels mean, how to schedule follow-up appointments, and how to fill out health insurance forms.11–14
Medicare managed-care enrollees are more likely to be hospitalized if they have limited health literacy,15 and diabetic Medicaid patients who have limited health literacy are less likely to have good glycemic control.16 One study showed annual health care costs of $10,688 for Medicaid enrollees with limited health literacy compared with $2,891 for all enrollees.17 The total cost of limited health literacy to the US health care system is estimated to be between $50 and $73 billion per year.18
Screening for limited health literacy: You can’t tell just by looking
Given the high costs of low health literacy, identifying patients who have it is of paramount importance.
Groups who are more likely to have limited health literacy include the elderly, the poor, the unemployed, high school dropouts, members of minority groups, recent immigrants, and people for whom English is a second language.
However, these demographic factors are not sufficient as a screen for low health literacy—you can't tell just by looking. Red flags for low health literacy include difficulty filling out forms in the office, missed appointments, nonadherence to medication regimens, failure to follow up with scheduled testing, and difficulty reading written materials, often masked with a statement such as “I forgot my glasses and will read this at home.”
A number of screening tests have been developed, including the Rapid Estimate of Adult Literacy in Medicine (REALM)19 and the Test for Functional Health Literacy in Adults (TOFHLA).20 These tests are long, making them difficult to incorporate into a patient visit in a busy primary care practice, but they are useful for research. A newer screening test asks the patient to review a nutrition label and answer six questions.21
The most useful screening test for clinical use may consist of a single question. Questions that have been validated:
- “How often do you need to have someone help you when you read instructions, pamphlets, or other written material from your doctor or pharmacy?” Positive answers are “sometimes,” “often,” or “always.”
- “How confident are you filling out medical forms by yourself?” Positive answers are “somewhat,” “a little bit,” or “not at all.”22–24
These questions can be included either in the initial screening by a nurse or medical assistant or as part of the social history portion of the interview with the physician.
A “brown bag review” can also be helpful. Patients are asked to bring in their medications (often in a brown bag—hence the name). Asking the patient to identify each medication by name and the indication for it can uncover knowledge gaps that indicate low health literacy.
The point to remember is that patients with low health literacy will probably not tell you that they do not understand. However, they would appreciate being asked in a nonthreatening manner.
Make your office a shame-free environment
Many experts advocate a “universal precautions approach,” in which interventions to address low health literacy are incorporated into routine office practice for all patients. Practice sites should adopt a culture of a “shame-free environment,” in which support staff encourage patients to ask questions and are trained to offer assistance to those having difficulty reading or filling out forms.
On a broader level, medical offices and hospitals can partner with adult-learning specialists to help patients gain skills to navigate the health care system. All signage should be clear and should use plain language as opposed to medical terms. Medical forms and questionnaires should be designed to collect only essential information and should be written at a sixth-grade reading level or below. Patient instructions and educational materials should also be clear and free of jargon.
The ‘teach-back’ technique
The “teach-back” technique is a simple method to confirm patient understanding at the end of the visit. This involves asking patients in a nonthreatening way to explain or demonstrate what they have been told. Examples:
- “I want to make sure I have explained things correctly. Can you tell me how you plan to take your medication when you go home?”
- “I want to make sure I have done a good job explaining things to you. When you go home and tell your spouse about your visit today, what will you say?”
These questions should be asked in a nonthreatening way. Put the burden of explanation on yourself as the first step, and let the patient know you are willing to explain again more thoroughly any instructions that may have not been clearly understood.
Other measures
Pictures and computer-based education may be useful for some patients who have difficulty reading.
Weiss25 advocates six steps to improve communication with patients in all encounters: slow down; use plain, nonmedical language; show or draw pictures; limit the amount of information provided; use the teach-back technique; and create a shame-free environment, encouraging questions.
Improving health literacy, as it relates to cross-cultural communication of treatment plans, must encompass understanding of health beliefs often based on cultural norms, in order to come to agreement on a mutually acceptable plan of care. Physicians should be aware of preferences for nontraditional or complementary treatments that may reflect specific cultural beliefs.
IF THE PATIENT DOES NOT SPEAK ENGLISH
Verbal communication across language barriers poses another layer of challenge. A trained interpreter should be used whenever possible when treating a patient who speaks a different language than that of the practitioner. When family members are used as interpreters, there are risks that the patient may not fully disclose facts about the history of illness or specific symptoms, and also that family members may place their own “twist” on the story when translating.
The physician should speak directly to the patient in a normal tone of voice. In this setting, also remember that nonverbal communication can be misinterpreted. Gestures should be avoided. Finally, be aware that personal space is viewed differently depending on cultural background, as is eye contact.
It is helpful to have a pre-interview meeting with the interpreter to explain the format of the interview, as well as a post-interview meeting to ensure all parties felt they effectively communicated during the encounter.
TOWARD EQUITABLE CARE
Health care disparities are the result of multiple determinants. In December 2008, a National Institutes of Health summit conference cited not only barriers to access, but also the interaction of biological, behavioral, social, environmental, economic, cultural, and political factors, and noted that the causes and effects of health disparities transcend health care.26
Clearly, an individual physician’s efforts will not be all that is needed to eliminate health disparities. A team-based approach is essential, using skills of nonphysician members of the health care team such as nurses, medical assistants, social workers, and case managers. Continued opportunity for professional training and development in provider-patient communication skills should be offered.
However, the impact of effective cross-cultural communication and managing low health literacy populations on the physician-patient level should not be understated. As practitioners treating patients from diverse backgrounds, improving self-awareness, eliciting the patient’s explanatory model, and assuring understanding of treatment plans for patients with low health literacy or with language barriers, we can do our part in working toward equitable care for all patients.
An english-speaking middle-aged woman from an ethnic minority group presents to her internist for follow-up of her chronic medical problems, which include diabetes, high blood pressure, asthma, and high cholesterol. Although she sees her physician regularly, her medical conditions are not optimally controlled.
At one of the visits, her physician gives her a list of her medications and, while reviewing it, explains—not for the first time—the importance of taking all of them as prescribed. The patient looks at the paper for a while, and then cautiously tells the physician, “But I can’t read.”
This patient presented to our practice several years ago. The scenario may be familiar to many primary physicians, except for the ending— ie, the patient telling her physician that she cannot read.
Her case raises several questions:
- Why did the physician not realize at the first encounter that she could not read the names of her prescribed medications?
- Why did the patient wait to tell her physician that important fact?
- And to what extent did her inability to read contribute to the poor control of her chronic medical problems?
Patients like this one are the human faces behind the statistics about health disparities—the worse outcomes noted in minority populations. Here, we discuss the issues of cross-cultural communication and health literacy as they relate to health care disparities.
DISPARITY IS NOT ONLY DUE TO LACK OF ACCESS
Health care disparity has been an important topic of discussion in medicine in the past decade.
In a 2003 publication,1 the Institute of Medicine identified lower quality of health care in minority populations as a serious problem. Further, it disputed the long-held belief that the differences in health care between minority and nonminority populations could be explained by lack of access to medical services in minority groups. Instead, it cited factors at the level of the health care system, the level of the patient, and the “care-process level” (ie, the physician-patient encounter) as contributing in distinct ways to the problem.1
A CALL FOR CULTURAL COMPETENCE
In a policy paper published in 2010, the American College of Physicians2 reviewed the progress made in addressing health care disparities. In addition, noting that an individual’s environment, income, level of education, and other factors all affect health, it called for a concerted effort to improve insurance coverage, health literacy, and the health care delivery system; to address stressors both within and outside the health care system; and to recruit more minority health care workers.
None of these things seems like anything a busy practicing clinician could do much about. However, we can try to improve our cultural competence in our interactions with patients on an individual level.
The report recommends that physicians and other health care professionals be sensitive to cultural diversity among patients. It also says we should recognize our preconceived perceptions of minority patients that may affect their treatment and contribute to disparities in health care in minorities. To those ends, it calls for cultural competence training in medical school to improve cultural awareness and sensitivity.2
The Office of Minority Health broadly defines cultural and linguistic competence in health as “a set of congruent behaviors, attitudes, and policies that come together in a system, agency, or among professionals that enables effective work in cross-cultural situations.”3 Cultural competence training should focus on being aware of one’s personal bias, as well as on education about culture-specific norms or knowledge of possible causes of mistrust in minority groups.
For example, many African Americans may mistrust the medical system, given the awareness of previous inequities such as the notorious Tuskegee syphilis study (in which informed consent was not used and treatment that was needed was withheld). Further, beliefs about health in minority populations may be discordant with the Western medical model.4
RECOGNIZING OUR OWN BIASES
Preconceived perceptions on the part of the physician may be shaped by previous experiences with patients from a specific minority group or by personal bias. Unfortunately, even a well-meaning physician who has tried to learn about cultural norms of specific minority groups can be at risk of stereotyping by assuming that all members of that group hold the same beliefs. From the patient’s viewpoint, they can also be molded by previous experiences of health care inequities or unfavorable interactions with physicians.
For example, in the case we described above, perhaps the physician had assumed that the patient was noncompliant and therefore did not look for reasons for the poor control of her medical problems, or maybe the patient did not trust the physician enough to explain the reason for her difficulty with understanding how to take her medications.
Being aware of our own unconscious stereotyping of minority groups is an important step in effectively communicating with patients from different cultural backgrounds or with low health literacy. We also need to reflect about our own health belief system and try to incorporate the patient’s viewpoint into decision-making.
If, on reflection, we recognize that we do harbor biases, we ought to think about ways to better accommodate patients from different backgrounds and literacy levels, including trying to learn more about their culture or mastering techniques to effectively explain treatment plans to low-literacy patients.
ALL ENCOUNTERS WITH PATIENTS ARE ‘CROSS-CULTURAL’
In health care, “cross-cultural communication” does not refer only to interactions between persons from different ethnic backgrounds or with different beliefs about health. Health care has a culture of its own, creating a cross-cultural encounter the moment a person enters your office or clinic in the role of “patient.”
Carillo et al5 categorized issues that may pose difficulties in a cross-cultural encounter as those of authority, physical contact, communication styles, gender, sexuality, and family.
Physician-patient communication is a complicated issue. Many patients will not question a physician if their own cultural norms view it as disrespectful—even if they have very specific fears about the diagnosis or treatment plan. They may also defer any important decision to a family member who has the authority to make decisions for the family.
Frequently, miscommunication is unintentional. In a recent study of hospitalized patients,6 77% of the physicians believed that their patients understood their diagnoses, while only 57% of patients could correctly state this information.
WHAT DOES THE PATIENT THINK?
A key issue in cross-cultural communication, and one that is often neglected, is to address a patient’s fears about his or her illness. In the study mentioned above, more than half of the patients who reported having anxieties or fears in the hospital stated that their physicians did not discuss their fears.6 But if we fail to do so, patients may be less satisfied with the treatment plan and may not accept our recommendations.
A patient’s understanding of his or her illness may be very different from the biomedical explanation. For example, we once saw an elderly man who was admitted to the hospital with back pain due to metastatic prostate cancer, but who was convinced that his symptoms were caused by a voodoo “hex” placed on him by his ex-wife.
For example, for the man who thought that his ex-wife put a hex on him, asking him “What do you think has caused your problem?” during the initial history-taking would allow him to express his concern about the hex and give the physician an opportunity to learn of this fear and then to offer the biomedical explanation for the problem and for the recommended treatment.
What happens more often in practice is that the specific fear is not addressed at the start of the encounter. Consequently, the patient is less likely to follow through with the treatment plan, as he or she does not feel the prescribed treatment is fixing the real problem. This process of exploring the explanatory model of illness may be viewed on a practical level as a way of managing expectations in the clinical care of culturally diverse populations.
HEALTH LITERACY: MORE THAN THE ABILITY TO READ
The better you know how to read, the healthier you probably are. In fact, a study found that a person’s literacy level correlated more strongly with health than did race or formal education level.9 (Apparently, attending school does not necessarily mean that people know how to read, and not attending school doesn’t mean that they don’t.)
Even more important than literacy may be health literacy, defined by Ratzan and Parker as “the degree to which individuals have the capacity to obtain, process, and understand basic health information and services needed to make appropriate health decisions.”8 It includes basic math and critical-thinking skills that allow patients to use medications properly and participate in treatment decisions. Thus, health literacy is much more than the ability to read.
Even people who read and write very well may have trouble when confronted with the complexities of navigating our health care system, such as appointment scheduling, specialty referrals, and follow-up testing and procedures: their health literacy may be lower than their general literacy. We had a patient, a highly trained professional, who was confused by instructions for preparing for colonoscopy on a patient handout. Another similar patient could not understand the dosing of eye drops after cataract surgery because the instructions on the discharge paperwork were unclear.
However, limited health literacy disproportionately affects minority groups and is linked to poorer health care outcomes. Thus, addressing limited health literacy is important in addressing health care disparities. Effective physician-patient communication about treatment plans is fundamental to providing equitable care to patients from minority groups, some of whom may be at high risk for low health literacy.
Below, we will review some of the data on health literacy and offer suggestions for screening and interventions for those whose health literacy is limited.
36% have basic or below-basic reading skills
Every 10 years, the US Department of Education completes its National Assessment of Adult Literacy. Its 2003 survey—the most recent—included 19,000 adults in the community and in prison, interviewed at their place of residence.10 Each participant completed a set of tasks to measure his or her ability to read, understand, and interpret text and to use and interpret numbers.
Participants were divided into four categories based on the results: proficient (12%), intermediate (53%), basic (22%), and below basic (14%). Additionally, 5% of potential participants could not be tested because they had insufficient skills to participate in the survey.
Low literacy puts patients at risk
Although literacy is not the same as health literacy, functionally, those who have basic or below-basic literacy skills (36% of the US population) are at high risk for encountering problems in the US health care system. For example, they would have difficulty with most patient education handouts and health insurance forms.
Limited health literacy exacts both personal and financial costs. Patients with low health literacy are less likely to understand how to take their medications, what prescription warning labels mean, how to schedule follow-up appointments, and how to fill out health insurance forms.11–14
Medicare managed-care enrollees are more likely to be hospitalized if they have limited health literacy,15 and diabetic Medicaid patients who have limited health literacy are less likely to have good glycemic control.16 One study showed annual health care costs of $10,688 for Medicaid enrollees with limited health literacy compared with $2,891 for all enrollees.17 The total cost of limited health literacy to the US health care system is estimated to be between $50 and $73 billion per year.18
Screening for limited health literacy: You can’t tell just by looking
Given the high costs of low health literacy, identifying patients who have it is of paramount importance.
Groups who are more likely to have limited health literacy include the elderly, the poor, the unemployed, high school dropouts, members of minority groups, recent immigrants, and people for whom English is a second language.
However, these demographic factors are not sufficient as a screen for low health literacy—you can't tell just by looking. Red flags for low health literacy include difficulty filling out forms in the office, missed appointments, nonadherence to medication regimens, failure to follow up with scheduled testing, and difficulty reading written materials, often masked with a statement such as “I forgot my glasses and will read this at home.”
A number of screening tests have been developed, including the Rapid Estimate of Adult Literacy in Medicine (REALM)19 and the Test for Functional Health Literacy in Adults (TOFHLA).20 These tests are long, making them difficult to incorporate into a patient visit in a busy primary care practice, but they are useful for research. A newer screening test asks the patient to review a nutrition label and answer six questions.21
The most useful screening test for clinical use may consist of a single question. Questions that have been validated:
- “How often do you need to have someone help you when you read instructions, pamphlets, or other written material from your doctor or pharmacy?” Positive answers are “sometimes,” “often,” or “always.”
- “How confident are you filling out medical forms by yourself?” Positive answers are “somewhat,” “a little bit,” or “not at all.”22–24
These questions can be included either in the initial screening by a nurse or medical assistant or as part of the social history portion of the interview with the physician.
A “brown bag review” can also be helpful. Patients are asked to bring in their medications (often in a brown bag—hence the name). Asking the patient to identify each medication by name and the indication for it can uncover knowledge gaps that indicate low health literacy.
The point to remember is that patients with low health literacy will probably not tell you that they do not understand. However, they would appreciate being asked in a nonthreatening manner.
Make your office a shame-free environment
Many experts advocate a “universal precautions approach,” in which interventions to address low health literacy are incorporated into routine office practice for all patients. Practice sites should adopt a culture of a “shame-free environment,” in which support staff encourage patients to ask questions and are trained to offer assistance to those having difficulty reading or filling out forms.
On a broader level, medical offices and hospitals can partner with adult-learning specialists to help patients gain skills to navigate the health care system. All signage should be clear and should use plain language as opposed to medical terms. Medical forms and questionnaires should be designed to collect only essential information and should be written at a sixth-grade reading level or below. Patient instructions and educational materials should also be clear and free of jargon.
The ‘teach-back’ technique
The “teach-back” technique is a simple method to confirm patient understanding at the end of the visit. This involves asking patients in a nonthreatening way to explain or demonstrate what they have been told. Examples:
- “I want to make sure I have explained things correctly. Can you tell me how you plan to take your medication when you go home?”
- “I want to make sure I have done a good job explaining things to you. When you go home and tell your spouse about your visit today, what will you say?”
These questions should be asked in a nonthreatening way. Put the burden of explanation on yourself as the first step, and let the patient know you are willing to explain again more thoroughly any instructions that may have not been clearly understood.
Other measures
Pictures and computer-based education may be useful for some patients who have difficulty reading.
Weiss25 advocates six steps to improve communication with patients in all encounters: slow down; use plain, nonmedical language; show or draw pictures; limit the amount of information provided; use the teach-back technique; and create a shame-free environment, encouraging questions.
Improving health literacy, as it relates to cross-cultural communication of treatment plans, must encompass understanding of health beliefs often based on cultural norms, in order to come to agreement on a mutually acceptable plan of care. Physicians should be aware of preferences for nontraditional or complementary treatments that may reflect specific cultural beliefs.
IF THE PATIENT DOES NOT SPEAK ENGLISH
Verbal communication across language barriers poses another layer of challenge. A trained interpreter should be used whenever possible when treating a patient who speaks a different language than that of the practitioner. When family members are used as interpreters, there are risks that the patient may not fully disclose facts about the history of illness or specific symptoms, and also that family members may place their own “twist” on the story when translating.
The physician should speak directly to the patient in a normal tone of voice. In this setting, also remember that nonverbal communication can be misinterpreted. Gestures should be avoided. Finally, be aware that personal space is viewed differently depending on cultural background, as is eye contact.
It is helpful to have a pre-interview meeting with the interpreter to explain the format of the interview, as well as a post-interview meeting to ensure all parties felt they effectively communicated during the encounter.
TOWARD EQUITABLE CARE
Health care disparities are the result of multiple determinants. In December 2008, a National Institutes of Health summit conference cited not only barriers to access, but also the interaction of biological, behavioral, social, environmental, economic, cultural, and political factors, and noted that the causes and effects of health disparities transcend health care.26
Clearly, an individual physician’s efforts will not be all that is needed to eliminate health disparities. A team-based approach is essential, using skills of nonphysician members of the health care team such as nurses, medical assistants, social workers, and case managers. Continued opportunity for professional training and development in provider-patient communication skills should be offered.
However, the impact of effective cross-cultural communication and managing low health literacy populations on the physician-patient level should not be understated. As practitioners treating patients from diverse backgrounds, improving self-awareness, eliciting the patient’s explanatory model, and assuring understanding of treatment plans for patients with low health literacy or with language barriers, we can do our part in working toward equitable care for all patients.
- Institute of Medicine of the National Academies. Unequal Treatment: Confronting Racial and Ethnic Disparities in Healthcare; 2003. http://www.nap.edu/openbook.php?record_id=12875&page=R1. Accessed January 5, 2012.
- American College of Physicians. Racial and Ethnic Disparities in Health Care, Updated 2010. Philadelphia: American College of Physicians; 2010: Policy Paper.
- US Department of Health and Human Services. The Office of Minority Health. What Is Cultural Competency? http://minorityhealth.hhs.gov/templates/browse.aspx?lvl=2&lvlid=11. Accessed January 5, 2012.
- Eiser AR, Ellis G. Viewpoint: cultural competence and the African American experience with health care: the case for specific content in cross-cultural education. Acad Med 2007; 82:176–183.
- Carrillo JE, Green AR, Betancourt JR. Cross-cultural primary care: a patient-based approach. Ann Intern Med 1999; 130:829–834.
- Olson DP, Windish DM. Communication discrepancies between physicians and hospitalized patients. Arch Intern Med 2010; 170:1302–1307.
- Kleinman A, Eisenberg L, Good B. Culture, illness, and care: clinical lessons from anthropologic and cross-cultural research. Ann Intern Med 1978; 88:251–258.
- National Library of Medicine. Current bibliographies in medicine 2000–1. Health Literacy. www.nlm.nih.gov/archive//20061214/pubs/cbm/hliteracy.html. Accessed January 5, 2012.
- Sentell TL, Halpin HA. Importance of adult literacy in understanding health disparities. J Gen Intern Med 2006; 21:862–866.
- Kutner M, Greenberg E, Jin Y, Paulsen C. The Health Literacy of America’s Adults: Results From the 2003 National Assessment of Adult Literacy (NCES 2006–483). US Department of Education. Washington, DC: National Center for Education Statistics; 2006. http://nces.ed.gov/pubs2006/2006483.pdf. Accessed January 5, 2012.
- Williams MV, Parker RM, Baker DW, et al. Inadequate functional health literacy among patients at two public hospitals. JAMA 1995; 274:1677–1682.
- Baker DW, Parker RM, Williams MV, et al. The health care experience of patients with low literacy. Arch Fam Med 1996; 5:329–334.
- Fact Sheet: health literacy and understanding medical information. Lawrenceville, NJ: Center for Health Care Strategies; 2002.
- Wolf MS, Davis TC, Tilson HH, Bass PF, Parker RM. Misunderstanding of prescription drug warning labels among patients with low literacy. Am J Health Syst Pharm 2006; 63:1048–1055.
- Baker DW, Gazmararian JA, Williams MV, et al. Functional health literacy and the risk of hospital admission among Medicare managed care enrollees. Am J Public Health 2002; 92:1278–1283.
- Schillinger D, Barton LR, Karter AJ, Wang F, Adler N. Does literacy mediate the relationship between education and health outcomes? A study of a low-income population with diabetes. Public Health Rep 2006; 121:245–254.
- Weiss BD, Palmer R. Relationship between health care costs and very low literacy skills in a medically needy and indigent Medicaid population. J Am Board Fam Pract 2004; 17:44–47.
- Friedland RB. Understanding health literacy: new estimates of the costs of inadequate health literacy. Washington, DC: National Academy on an Aging Society; 1998.
- Davis TC, Long SW, Jackson RH, et al. Rapid estimate of adult literacy in medicine: a shortened screening instrument. Fam Med 1993; 25:391–395.
- Baker DW, Williams MV, Parker RM, Gazmararian JA, Nurss J. Development of a brief test to measure functional health literacy. Patient Educ Couns 1999; 38:33–42.
- Weiss BD, Mays MZ, Martz W, et al. Quick assessment of literacy in primary care: the newest vital sign. Ann Fam Med 2005; 3:514–522.
- Chew LD, Bradley KA, Boyko EJ. Brief questions to identify patients with inadequate health literacy. Fam Med 2004; 36:588–594.
- Morris NS, MacLean CD, Chew LD, Littenberg B. The Single Item Literacy Screener: evaluation of a brief instrument to identify limited reading ability. BMC Fam Pract 2006; 7:21.
- Wallace LS, Rogers ES, Roskos SE, Holiday DB, Weiss BD. Brief report: screening items to identify patients with limited health literacy skills. J Gen Intern Med 2006; 21:874–877.
- Weiss BD. Health Literacy and Patient Safety: Help Patients Understand. 2nd ed. American Medical Association Foundation and American Medical Association. www.ama-assn.org/ama1/pub/upload/mm/367/healthlitclinicians.pdf. Accessed January 5, 2012.
- Dankwa-Mullan I, Rhee KB, Williams K, et al. The science of eliminating health disparities: summary and analysis of the NIH summit recommendations. Am J Public Health 2010; 100(suppl 1):S12–S18.
- Institute of Medicine of the National Academies. Unequal Treatment: Confronting Racial and Ethnic Disparities in Healthcare; 2003. http://www.nap.edu/openbook.php?record_id=12875&page=R1. Accessed January 5, 2012.
- American College of Physicians. Racial and Ethnic Disparities in Health Care, Updated 2010. Philadelphia: American College of Physicians; 2010: Policy Paper.
- US Department of Health and Human Services. The Office of Minority Health. What Is Cultural Competency? http://minorityhealth.hhs.gov/templates/browse.aspx?lvl=2&lvlid=11. Accessed January 5, 2012.
- Eiser AR, Ellis G. Viewpoint: cultural competence and the African American experience with health care: the case for specific content in cross-cultural education. Acad Med 2007; 82:176–183.
- Carrillo JE, Green AR, Betancourt JR. Cross-cultural primary care: a patient-based approach. Ann Intern Med 1999; 130:829–834.
- Olson DP, Windish DM. Communication discrepancies between physicians and hospitalized patients. Arch Intern Med 2010; 170:1302–1307.
- Kleinman A, Eisenberg L, Good B. Culture, illness, and care: clinical lessons from anthropologic and cross-cultural research. Ann Intern Med 1978; 88:251–258.
- National Library of Medicine. Current bibliographies in medicine 2000–1. Health Literacy. www.nlm.nih.gov/archive//20061214/pubs/cbm/hliteracy.html. Accessed January 5, 2012.
- Sentell TL, Halpin HA. Importance of adult literacy in understanding health disparities. J Gen Intern Med 2006; 21:862–866.
- Kutner M, Greenberg E, Jin Y, Paulsen C. The Health Literacy of America’s Adults: Results From the 2003 National Assessment of Adult Literacy (NCES 2006–483). US Department of Education. Washington, DC: National Center for Education Statistics; 2006. http://nces.ed.gov/pubs2006/2006483.pdf. Accessed January 5, 2012.
- Williams MV, Parker RM, Baker DW, et al. Inadequate functional health literacy among patients at two public hospitals. JAMA 1995; 274:1677–1682.
- Baker DW, Parker RM, Williams MV, et al. The health care experience of patients with low literacy. Arch Fam Med 1996; 5:329–334.
- Fact Sheet: health literacy and understanding medical information. Lawrenceville, NJ: Center for Health Care Strategies; 2002.
- Wolf MS, Davis TC, Tilson HH, Bass PF, Parker RM. Misunderstanding of prescription drug warning labels among patients with low literacy. Am J Health Syst Pharm 2006; 63:1048–1055.
- Baker DW, Gazmararian JA, Williams MV, et al. Functional health literacy and the risk of hospital admission among Medicare managed care enrollees. Am J Public Health 2002; 92:1278–1283.
- Schillinger D, Barton LR, Karter AJ, Wang F, Adler N. Does literacy mediate the relationship between education and health outcomes? A study of a low-income population with diabetes. Public Health Rep 2006; 121:245–254.
- Weiss BD, Palmer R. Relationship between health care costs and very low literacy skills in a medically needy and indigent Medicaid population. J Am Board Fam Pract 2004; 17:44–47.
- Friedland RB. Understanding health literacy: new estimates of the costs of inadequate health literacy. Washington, DC: National Academy on an Aging Society; 1998.
- Davis TC, Long SW, Jackson RH, et al. Rapid estimate of adult literacy in medicine: a shortened screening instrument. Fam Med 1993; 25:391–395.
- Baker DW, Williams MV, Parker RM, Gazmararian JA, Nurss J. Development of a brief test to measure functional health literacy. Patient Educ Couns 1999; 38:33–42.
- Weiss BD, Mays MZ, Martz W, et al. Quick assessment of literacy in primary care: the newest vital sign. Ann Fam Med 2005; 3:514–522.
- Chew LD, Bradley KA, Boyko EJ. Brief questions to identify patients with inadequate health literacy. Fam Med 2004; 36:588–594.
- Morris NS, MacLean CD, Chew LD, Littenberg B. The Single Item Literacy Screener: evaluation of a brief instrument to identify limited reading ability. BMC Fam Pract 2006; 7:21.
- Wallace LS, Rogers ES, Roskos SE, Holiday DB, Weiss BD. Brief report: screening items to identify patients with limited health literacy skills. J Gen Intern Med 2006; 21:874–877.
- Weiss BD. Health Literacy and Patient Safety: Help Patients Understand. 2nd ed. American Medical Association Foundation and American Medical Association. www.ama-assn.org/ama1/pub/upload/mm/367/healthlitclinicians.pdf. Accessed January 5, 2012.
- Dankwa-Mullan I, Rhee KB, Williams K, et al. The science of eliminating health disparities: summary and analysis of the NIH summit recommendations. Am J Public Health 2010; 100(suppl 1):S12–S18.
KEY POINTS
- To provide optimal care, physicians and staff need to think about ways to accommodate patients of other cultures and backgrounds, in particular by learning more about the patient’s culture and by examining themselves for possible bias.
- Even people who read and write very well may have limited health literacy. We should not assume that patients understand what we are talking about.
- Weiss (2011) advocates six steps to improve communication with patients in all encounters: slow down; use plain, nonmedical language; show or draw pictures; limit the amount of information provided; use the “teach-back” technique; and create a shame-free environment, encouraging questions.
- The “teach-back” technique is a simple way to confirm a patient’s understanding at the end of the visit. This involves asking the patient in a nonthreatening way to explain or show what he or she has been told.
A 49-year-old woman with a persistent cough
A 49-year-old woman presents with a cough that has persisted for 3 weeks.
Two weeks ago, she was seen in the outpatient clinic for a nonproductive cough, rhinorrhea, sneezing, and a sore throat. At that time, she described coughing spells that were occasionally accompanied by posttussive chest pain and vomiting. The cough was worse at night and was occasionally associated with wheezing. She reported no fevers, chills, rigors, night sweats, or dyspnea. She said she has tried over-the-counter cough suppressants, antihistamines, and decongestants, but they provided no relief. Since she had a history of well-controlled asthma, she was diagnosed with an asthma exacerbation and was given prednisone 20 mg to take orally every day for 5 days, to be followed by an inhaled corticosteroid until her symptoms resolved.
Now, she has returned because her symptoms have persisted despite treatment, and she is seeking a second medical opinion. Her paroxysmal cough has become more frequent and more severe.
In addition to asthma, she has a history of allergic rhinitis. Her current medications include the over-the-counter histamine H1 antagonist cetirizine (Zyrtec), a fluticasone-salmeterol inhaler (Advair), and an albuterol inhaler (Proventil HFA). She reports having had mild asthma exacerbations in the past during the winter, which were managed well with her albuterol inhaler.
She has never smoked; she drinks alcohol socially. She has not traveled outside the United States during the past several months. She is married and has two children, ages 25 and 23. She lives at home with only her husband, and he has not been sick. However, she works at a greeting card store, and two of her coworkers have similar upper respiratory symptoms, although they have only a mild cough.
Her immunizations are not up-to-date. She last received the tetanus-diphtheria toxoid (Td) vaccine 12 years ago, and she never received the pediatric tetanus, diphtheria, and acellular pertussis (Tdap) vaccine. She generally receives the influenza vaccine annually, and she received it about 6 weeks before this presentation.
She is not in distress, but she has paroxysms of severe coughing throughout her examination. Her pulse is 100 beats per minute, respiratory rate 18, and blood pressure 130/86 mm Hg. Her oropharynx is clear. The pulmonary examination reveals poor inspiratory effort due to coughing but is otherwise normal. The rest of the examination is normal, as is her chest radiograph.
WHAT DOES SHE HAVE?
1. Which of the following would best explain her symptoms?
- Asthma
- Postviral cough
- Pertussis
- Chronic bronchitis
- Pneumonia
- Gastroesophageal reflux disease
Asthma is a reasonable consideration, given her medical history, her occasional wheezing, and her nonproductive cough that is worse at night. However, asthma typically responds well to corticosteroid therapy. She has already received a course of prednisone, but her symptoms have not improved.
Postviral cough could also be considered in this patient. However, postviral cough does not typically occur in paroxysms, nor does it lead to posttussive vomiting. It is also generally regarded as a diagnosis of exclusion.
Pertussis (whooping cough) should be suspected in this patient, given the time course of her symptoms, the paroxysmal cough, and the posttussive vomiting. In addition, at her job she interacts with hundreds of people a day, increasing her risk of exposure to respiratory tract pathogens, including Bordetella pertussis.
Chronic bronchitis is defined by cough (typically productive) lasting at least 3 months per year for at least 2 consecutive years, which does not fit the time course for this patient. It is vastly more common in smokers.
Pneumonia typically presents with a cough that can be productive or nonproductive, but also with fever, chills, and radiologic evidence of a pulmonary infiltrate or consolidation. This woman has none of these.
Gastroesophageal reflux disease is one of the most common causes of chronic cough, with symptoms typically worse at night. However, it is generally associated with symptoms such as heartburn, a sour taste in the mouth, or regurgitation, which our patient did not report.
Thus, pertussis is the most likely diagnosis.
PERTUSSIS IS ON THE RISE
Pertussis is an acute and highly contagious disease caused by infection of the respiratory tract by B pertussis, a small, aerobic, gramnegative, pleomorphic coccobacillus that produces a number of antigenic and biologically active products, including pertussis toxin, filamentous hemagglutinin, agglutinogens, and tracheal cytotoxin. Transmitted by aerosolized droplets, it attaches to the ciliated epithelial cells of the lower respiratory tract, paralyzes the cilia via toxins, and causes inflammation, thus interfering with the clearing of respiratory secretions.
The incidence of pertussis is on the rise. In 2005, 25,827 cases were reported in the United States, the highest number since 1959.1 Pertussis is now epidemic in California. At the time of this writing, the number of confirmed, probable, and suspected cases in California was 9,477 (including 10 infant deaths) for the year 2010—the most cases reported in the past 65 years.2,3
In 2010, outbreaks were also reported in Michigan, Texas, Ohio, upstate New York, and Arizona.4 The overall incidence of pertussis is likely even higher than what is reported, since many cases go unrecognized or unreported.
Highly contagious
Pertussis is transmitted person-to-person, primarily through aerosolized droplets from coughing or sneezing or by direct contact with secretions from the respiratory tract of infected persons. It is highly contagious, with secondary attack rates of up to 80% in susceptible people.
A three-stage clinical course
The clinical definition of pertussis used by the US Centers for Disease Control and Prevention (CDC) and the Council of State and Territorial Epidemiologists is an acute cough illness lasting at least 2 weeks, with paroxysms of coughing, an inspiratory “whoop,” or posttussive vomiting without another apparent cause.5
The clinical course of the illness is traditionally divided into three stages:
The catarrhal phase typically lasts 1 to 2 weeks and is clinically indistinguishable from a viral upper respiratory infection. It is characterized by the insidious onset of malaise, coryza, sneezing, low-grade fever, and a mild cough that gradually becomes severe.6
The paroxysmal phase normally lasts 1 to 6 weeks but may persist for up to 10 weeks. The diagnosis of pertussis is usually suspected during this phase. The classic features of this phase are bursts or paroxysms of numerous, rapid coughs. These are followed by a long inspiratory effort usually accompanied by a characteristic high-pitched whoop, most notably observed in infants and children. Infants and children may appear very ill and distressed during this time and may become cyanotic, but cyanosis is uncommon in adults and adolescents. The paroxysms may also be followed by exhaustion and posttussive vomiting. In some cases, the cough is not paroxysmal, but rather simply persistent. The coughing attacks tend to occur more often at night, with an average of 15 attacks per 24 hours. During the first 1 to 2 weeks of this stage, the attacks generally increase in frequency, remain at the same intensity level for 2 to 3 weeks, and then gradually decrease over 1 to 2 weeks.1,7
The convalescent phase can have a variable course, ranging from weeks to months, with an average duration of 2 to 3 weeks. During this stage, the paroxysms of coughing become less frequent and gradually resolve. Paroxysms often recur with subsequent respiratory infections.
In infants and young children, pertussis tends to follow these stages in a predictable sequence. Adolescents and adults, however, tend to go through the stages without being as ill and typically do not exhibit the characteristic whoop.
TESTING FOR PERTUSSIS
2. Which would be the test of choice to confirm pertussis in this patient?
- Bacterial culture of nasopharyngeal secretions
- Polymerase chain reaction (PCR) testing of nasopharyngeal secretions
- Direct fluorescent antibody testing of nasopharyngeal secretions
- Enzyme-linked immunosorbent assay (ELISA) serologic testing
Establishing the diagnosis of pertussis is often rather challenging.
Bacterial culture: Very specific, but slow and not so sensitive
Bacterial culture is still the gold standard for diagnosing pertussis, as a positive culture for B pertussis is 100% specific.5
However, this test has drawbacks. Its sensitivity has a wide range (15% to 80%) and depends very much on the time from the onset of symptoms to the time the culture specimen is collected. The yield drops off significantly after 1 week, and after 3 weeks the test has a sensitivity of only 1% to 3%.8 Therefore, for our patient, who has had symptoms for 3 weeks already, bacterial culture would not be the best test. In addition, the results are usually not known for 7 to 14 days, which is too slow to be useful in managing acute cases.
For swabbing, a Dacron swab is inserted through the nostril to the posterior pharynx and is left in place for 10 seconds to maximize the yield of the specimen. Recovery rates for B pertussis are low if the throat or the anterior nasal passage is swabbed instead of the posterior pharynx.9
Nasopharyngeal aspiration is a more complicated procedure, requiring a suction device to trap the mucus, but it may provide higher yields than swabbing.10 In this method, the specimen is obtained by inserting a small tube (eg, an infant feeding tube) connected to a mucus trap into the nostril back to the posterior pharynx.
Often, direct inoculation of medium for B pertussis is not possible. In such cases, clinical specimens are placed in Regan Lowe transport medium (half-strength charcoal agar supplemented with horse blood and cephalexin).11,12
Polymerase chain reaction testing: Faster, more sensitive, but less specific
PCR testing of nasopharyngeal specimens is now being used instead of bacterial culture to diagnose pertussis in many situations. Alternatively, nasopharyngeal aspirate (or secretions collected with two Dacron swabs) can be obtained and divided at the time of collection and the specimens sent for both culture and PCR testing. Because bacterial culture is time-consuming and has poor sensitivity, the CDC states that a positive PCR test, along with the clinical symptoms and epidemiologic information, is sufficient for diagnosis.5
PCR testing can detect B pertussis with greater sensitivity and more rapidly than bacterial culture.12–14 Its sensitivity ranges from 61% to 99%, its specificity ranges from 88% to 98%,12,15,16 and its results can be available in 2 to 24 hours.12
PCR testing’s advantage in terms of sensitivity is especially pronounced in the later stages of the disease (as in our patient), when clinical suspicion of pertussis typically arises. It can be used effectively for up to 4 weeks from the onset of cough.14 Our patient, who presented nearly 3 weeks after the onset of symptoms, underwent nasopharyngeal sampling for PCR testing.
However, PCR testing is not as specific for B pertussis as is bacterial culture, since other Bordetella species can cause positive results on PCR testing. Also, as with culture, a negative test does not reliably rule out the disease, especially if the sample is collected late in the course.
Therefore, basing the diagnosis on PCR testing alone without the proper clinical context is not advised: pertussis outbreaks have been mistakenly declared on the basis of false-positive PCR test results. Three so-called “pertussis outbreaks” in three different states from 2004 to 200617 were largely the result of overdiagnosis based on equivocal or false-positive PCR test results without the appropriate clinical circumstances. Retrospective review of these pseudo-outbreaks revealed that few cases actually met the CDC’s diagnostic criteria.17 Many patients were not tested (by any method) for pertussis and were treated as having probable cases of pertussis on the basis of their symptoms. Patients who were tested and who had a positive PCR test did not meet the clinical definition of pertussis according to the Council of State and Territorial Epidemiologists.17
Since PCR testing varies in sensitivity and specificity, obtaining culture confirmation of pertussis for at least one suspicious case is recommended any time an outbreak is suspected. This is necessary for monitoring for continued presence of the agent among cases of disease, recruitment of isolates for epidemiologic studies, and surveillance for antibiotic resistance.
Direct fluorescence antibody testing
The CDC does not recommend direct fluorescence antibody testing to diagnose pertussis. This test is commercially available and is sometimes used to screen patients for B pertussis infection, but it lacks sensitivity and specificity for this organism. Cross-reaction with normal nasopharyngeal flora can lead to a false-positive result.18 In addition, the interpretation of the test is subjective, so the sensitivity and specificity are quite variable: the sensitivity is reported as 52% to 65%, while the specificity can vary from 15% to 99%.
Enzyme-linked immunosorbent assay
ELISA testing has been used in epidemiologic studies to measure serum antibodies to B pertussis. Many serologic tests exist, but none is commercially available. Many of these tests are used by the CDC and state health departments to help confirm the diagnosis, especially during outbreaks. Generally, serologic tests are more useful for diagnosis in later phases of the disease. Currently used ELISA tests use both paired and single serology techniques measuring elevated immunoglobulin G serum antibody concentrations against an array of antigens, including pertussis toxin, filamentous hemagglutinin, pertactin, and fimbrae. As a result, a range of sensitivities (33%–95%) and specificities (72%–100%) has been reported.12,14,19
TREATING PERTUSSIS
Our patient’s PCR test result comes back positive. In view of her symptoms and this result, we decide to treat her empirically for pertussis, even though she has had no known contact with anyone with the disease and there is currently no outbreak of it in the community.
3. According to the most recent evidence, which of the following would be the treatment of choice for pertussis in this patient?
- Azithromycin (Zithromax)
- Amoxicillin (Moxatag)
- Levofloxacin (Levaquin)
- Sulfamethoxazole-trimethoprim (Bactrim)
- Supportive measures (hydration, humidifier, antitussives, antihistamines, decongestants)
Azithromycin and the other macrolide antibiotics erythromycin and clarithromycin are first-line therapies for pertussis in adolescents and adults. If given during the catarrhal phase, they can reduce the duration and severity of symptoms and lessen the period of communicability.20,21 After the catarrhal phase, however, it is uncertain whether antibiotics change the clinical course of pertussis, as the data are conflicting.20–22
Factors to consider when selecting a macrolide antibiotic are tolerability, the potential for adverse events and drug interactions, ease of compliance, and cost. All three macrolides are equally effective against pertussis, but azithromycin and clarithromycin are generally better tolerated and are associated with milder and less frequent side effects than erythromycin, including lower rates of gastrointestinal side effects.
Erythromycin and clarithromycin inhibit the cytochrome P450 enzyme system, specifically CYP3A4, and can interact with a great many commonly prescribed drugs metabolized by this enzyme. Therefore, azithromycin may be a better choice for patients already taking other medications, like our patient.
Azithromycin and clarithromycin have longer half-lives and achieve higher tissue concentrations than erythromycin, allowing for less-frequent dosing (daily for azithromycin and twice daily for clarithromycin) and shorter treatment duration (5 days for azithromycin and 7 days for clarithromycin).
An advantage of erythromycin, though, is its lower cost. The cost of a recommended course of erythromycin treatment for pertussis (ie, 500 mg every 6 hours for 14 days) is roughly $20, compared with $75 for azithromycin.
Amoxicillin is not effective in clearing B pertussis from the nasopharynx and thus is not a reasonable option for the treatment of pertussis.23
Levofloxacin is also not recommended for the treatment of pertussis.
Sulfamethoxazole-trimethoprim is a second-line agent for pertussis. It is effective in eradicating B pertussis from the nasopharynx20 and is generally used as an alternative to the macrolide agents in patients who cannot tolerate or have contraindications to macrolides. Sulfamethoxazole-trimethoprim can also be an option for patients infected with rare macrolide-resistant strains of B pertussis.
Supportive measures by themselves are reasonable for patients with pertussis beyond the catarrhal phase, since antibiotics are typically not effective at that stage of the disease.
From 80% to 90% of patients with untreated pertussis spontaneously clear the bacteria from the nasopharynx within 3 to 4 weeks from the onset of cough symptoms.20 However, supportive measures, including antitussives (both over-the-counter and prescription), tend to have very little effect on the severity or duration of the illness, especially when used past the early stage of the illness.
POSTEXPOSURE CHEMOPROPHYLAXIS FOR CLOSE CONTACTS
Postexposure chemoprophylaxis should be given to close contacts of patients who have pertussis to help prevent secondary cases.22 The CDC defines a close contact as someone who has had face-to-face exposure within 3 feet of a symptomatic patient within 21 days after the onset of symptoms in the patient. Close contacts should be treated with antibiotic regimens similar to those used in confirmed cases of pertussis.
In our patient’s case, the diagnosis of pertussis was reported to the Ohio Department of Health. Shortly afterward, the department contacted the patient and obtained information about her close contacts. These people were then contacted and encouraged to complete a course of antibiotics for postexposure chemoprophylaxis, given the high secondary attack rates.
PERTUSSIS VACCINES
4. Which of the following vaccines could have reduced our patient’s chance of contracting the disease or reduced the severity or time course of the illness?
- DTaP
- Tdap
- Whole-cell pertussis vaccine
- No vaccine would have reduced her risk
It is important to prevent pertussis, given its associated morbidities and its generally poor response to drug therapy. Continued vigilance is imperative to maintain high levels of vaccine coverage, including the timely completion of the pertussis vaccination schedule.
The two vaccines in current use in the United States to produce immunity to pertussis—DTaP and Tdap—also confer immunity to diphtheria and tetanus. DTaP is used for children under 7 years of age, and Tdap is for ages 10 to 64. Thus, our patient should have received a series of DTaP injections as an infant and small child, and a Tdap booster at age 11 or 12 years and every 10 years after that.
The upper case “D,” “T,” and “P” in the abbreviations signifies full-strength doses and the lower case “d,” “t,” and “p” indicate that the doses of those components have been reduced. The “a” in both vaccines stands for “acellular”: ie, the pertussis component does not contain cellular elements.
DTaP for initial pertussis vaccination
The current recommendation for initial pertussis vaccination consists of a primary series of DTaP. DTaP vaccination is recommended for infants at 2 months of age, then again at 4 months of age, and again at 6 months of age. A fourth dose is given between the ages of 15 and 18 months, and a fifth dose is given between the ages of 4 to 6 years. If the fourth dose was given after age 4, then no fifth dose is needed.20
Tdap as a booster
The booster vaccine for adolescents and adults is Tdap. In 2005, two Tdap vaccines were licensed in the United States: Adacel for people ages 11 to 64 years, and Boostrix for people ages 10 to 18 years.
The CDC’s Advisory Committee on Immunization Practices (ACIP) recommends a booster dose of Tdap at age 11 or 12 years. Every 10 years thereafter, a booster of tetanus and diphtheria toxoid (Td) vaccine is recommended, except that one of the Td doses can be replaced by Tdap if the patient hasn’t received Tdap yet.
For adults ages 19 to 64, the ACIP currently recommends routine use of a single booster dose of Tdap to replace a single dose of Td if they received the last dose of toxoid vaccine 10 or more years earlier. If the previous dose of Td was given within the past 10 years, a single dose of Tdap is appropriate to protect patients against pertussis. This is especially true for patients at increased risk of pertussis or its complications, as well as for health care professionals and adults who have close contact with infants, such as new parents, grandparents, and child-care providers. The minimum interval since the last Td vaccination is ideally 2 years, although shorter intervals can be used for control of pertussis outbreaks and for those who have close contact with infants.24
In 2010, the ACIP decided that, for those ages 65 and older, a single dose of Tdap vaccine may be given in place of Td if the patient has not previously received Tdap, regardless of how much time has elapsed since the last vaccination with a Td-containing vaccine.25 Data from the Vaccine Adverse Event Reporting System suggest that Tdap vaccine in this age group is as safe as the Td vaccine.25
Subsequent tetanus vaccine doses, in the form of Td, should be given at 10-year intervals throughout adulthood. Administration of Tdap at 10-year intervals appears to be highly immunogenic and well tolerated,25 suggesting that it is possible that Tdap will become part of routine booster dosing instead of Td, pending further study.
Tdap is not contraindicated in pregnant women. Ideally, women should be vaccinated with Tdap before becoming pregnant if they have not previously received it. If the pregnant woman is not at risk of acquiring or transmitting pertussis during pregnancy, the ACIP recommends deferring Tdap vaccination until the immediate postpartum period.
Adults who require a vaccine containing tetanus toxoid for wound management should receive Tdap instead of Td if they have never received Tdap. Adults who have never received vaccine containing tetanus and diphtheria toxoid should receive a series of three vaccinations. The preferred schedule is a dose of Tdap, followed by a dose of Td more than 4 weeks later, and a second dose of Td 6 to 12 months later, though Tdap can be substituted for Td for any one of the three doses in the series. Adults with a history of pertussis generally should receive Tdap according to routine recommendations.
Tdap is contraindicated in people with a history of serious allergic reaction to any component of the Tdap vaccine or with a history of encephalopathy not attributable to an identifiable cause within 7 days of receiving a pertussis vaccine. Tdap is relatively contraindicated and should be deferred in people with current moderate to severe acute illness, current unstable neurologic condition, or a history of Arthus hypersensitivity reaction to a tetanus-toxoid-containing vaccine within the past 10 years, and in people who have developed Guillain-Barré syndrome, within 6 weeks of receiving a tetanus-toxoid–containing vaccine.
Tdap is generally well tolerated. Adverse effects are typically mild and may include localized pain, redness, and swelling; low-grade fever; headache; fatigue; and, less commonly, gastrointestinal upset, myalgia, arthralgia, rash, and swollen glands.
Whole-cell pertussis vaccine is no longer available in the United States
Whole-cell pertussis vaccine provides good protection against pertussis, with 70% to 90% efficacy after three doses. It is less expensive-than acellular formulations and therefore is used in many parts of the world where cost is an issue. It is no longer available in the United States, however, due to high rates of local reactions such as redness, swelling, and pain at the injection site.
The importance of staying up-to-date with booster shots
Booster vaccination for pertussis in adolescents and adults is critical, since the largest recent outbreaks have occurred in these groups.21 The high rate of outbreaks is presumably the result of waning immunity from childhood immunizations and of high interpersonal contact rates. Vaccination has been shown to reduce the chance of contracting the disease and to reduce the severity and time course of the illness.21
Adolescents and adults are an important reservoir for potentially serious infections in infants who are either unvaccinated or whose vaccination schedule has not been completed. These infants are at risk of severe illness, including pneumonia, seizures, encephalopathy, and apnea, or even death. Adults and teens can also suffer complications from pertussis, although these tend to be less serious, especially in those who have been vaccinated. Complications in teens and adults are often caused by malaise and the cough itself, including weight loss (33%), urinary stress incontinence (28%), syncope (6%), rib fractures from severe coughing (4%), and pneumonia (2%).26 Thus, it is important that adolescents and adults stay up-to-date with pertussis vaccination.
CASE CONTINUED
Our patient was treated with a short (5-day) course of azithromycin 500 mg daily. It did not improve her symptoms very much, but this was not unexpected, given her late presentation and duration of symptoms. Her cough persisted for about 2 months afterwards, but it improved with time and with supportive care at home.
CONTINUED CHALLENGES
Pertussis is a reemerging disease with an increased incidence over the past 30 years, and even more so over the past 10 years. Unfortunately, treatments are not very effective, especially since the disease is often diagnosed late in the course.
We are fortunate to have a vaccine that can prevent pertussis, yet pertussis persists, in large part because of waning immunity from childhood vaccination. The duration of immunity from childhood vaccination is not yet clear. Many adolescents and adults do not follow up on these booster vaccines, thus increasing their susceptibility to pertussis. Consequently, they can transmit the disease to children who are not fully immunized. Prevention by maintaining active immunity is the key to controlling this terrible disease.
- Centers for Disease Control and Prevention. Pertussis. National Immunization Program, 2005. http://www.cdc.gov/vaccines/pubs/pinkbook/downloads/pert.pdf. Accessed July 6, 2011.
- California Department of Public Health. Pertussis report. www.cdph.ca.gov/programs/immunize/Documents/PertussisReport2011-01-07.pdf. Accessed July 6, 2011.
- Centers for Disease Control and Prevention. Pertussis (whooping cough). www.cdc.gov/pertussis/outbreaks.html. Accessed July 3, 2011.
- Centers for Disease Control and Prevention. Notifiable diseases and mortality tables. MMWR Morb Mortal Wkly Rep 2010; 59:847–861. http://www.cdc.gov/mmwr/PDF/wk/mm5927.pdf. Accessed July 1, 2011.
- Centers for Disease Control and Prevention. Pertussis. Vaccines and preventable diseases: pertussis (whooping cough) vaccination, 2010. http://www.cdc.gov/vaccines/vpd-vac/pertussis/default.htm. Accessed July 6, 2011.
- Hewlett EL, Edwards KM. Clinical practice. Pertussis—not just for kids. N Engl J Med 2005; 352:1215–1222.
- Hewlett E. Bordetella species. In: Mandell GL, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. 5th ed, Philadelphia, PA: Churchill Livingstone; 2000:2701.
- Viljanen MK, Ruuskanen O, Granberg C, Salmi TT. Serological diagnosis of pertussis: IgM, IgA and IgG antibodies against Bordetella pertussis measured by enzyme-linked immunosorbent assay (ELISA). Scand J Infect Dis 1982; 14:117–122.
- Bejuk D, Begovac J, Bace A, Kuzmanovic-Sterk N, Aleraj B. Culture of Bordetella pertussis from three upper respiratory tract specimens. Pediatr Infect Dis J 1995; 14:64–65.
- Hallander HO, Reizenstein E, Renemar B, Rasmuson G, Mardin L, Olin P. Comparison of nasopharyngeal aspirates with swabs for culture of Bordetella pertussis. J Clin Microbiol 1993; 31:50–52.
- Regan J, Lowe F. Enrichment medium for the isolation of Bordetella. J Clin Microbiol 1977; 6:303–309.
- World Health Organization. Laboratory manual for the diagnosis of whooping cough caused by Bordetella pertussis/Bordetella para-pertussis. Department of Immunization, Vaccines and Biologicals. Printed 2004. Revised 2007. www.who.int/vaccines-documents/. Accessed July 6, 2011.
- Meade BD, Bollen A. Recommendations for use of the polymerase chain reaction in the diagnosis of Bordetella pertussis infections. J Med Microbiol 1994; 41:51–55.
- Wendelboe AM, Van Rie A. Diagnosis of pertussis: a historical review and recent developments. Expert Rev Mol Diagn 2006; 6:857–864.
- Knorr L, Fox JD, Tilley PA, Ahmed-Bentley J. Evaluation of real-time PCR for diagnosis of Bordetella pertussis infection. BMC Infect Dis 2006; 6:62.
- Sotir MJ, Cappozzo DL, Warshauer DM, et al. Evaluation of polymerase chain reaction and culture for diagnosis of pertussis in the control of a county-wide outbreak focused among adolescents and adults. Clin Infect Dis 2007; 44:1216–1219.
- Centers for Disease Control and Prevention (CDC). Outbreaks of respiratory illness mistakenly attributed to pertussis—New Hampshire, Massachusetts, and Tennessee, 2004–2006. MMWR Morb Mortal Wkly Rep 2007; 56:837–842.
- Ewanowich CA, Chui LW, Paranchych MG, Peppler MS, Marusyk RG, Albritton WL. Major outbreak of pertussis in northern Alberta, Canada: analysis of discrepant direct fluorescent-antibody and culture results by using polymerase chain reaction methodology. J Clin Microbiol 1993; 31:1715–1725.
- Müller FM, Hoppe JE, Wirsing von König CH. Laboratory diagnosis of pertussis: state of the art in 1997. J Clin Microbiol 1997; 35:2435–2443.
- Tiwari T, Murphy TV, Moran J; National Immunization Program, CDC. Recommended antimicrobial agents for the treatment and postexposure prophylaxis of pertussis: 2005 CDC Guidelines. MMWR Recomm Rep 2005; 54:1–16.
- Wirsing von König CH, Postels-Multani S, Bock HL, Schmitt HJ. Pertussis in adults: frequency of transmission after household exposure. Lancet 1995; 346:1326–1329.
- von König CH. Use of antibiotics in the prevention and treatment of pertussis. Pediatr Infect Dis J 2005; 24(suppl 5):S66–S68.
- Trollfors B. Effect of erythromycin and amoxycillin on Bordetella pertussis in the nasopharynx. Infection 1978; 6:228–230.
- Broder KR, Cortese MM, Iskander JK, et al; Advisory Committee on Immunization Practices (ACIP). Preventing tetanus, diphtheria, and pertussis among adolescents: use of tetanus toxoid, reduced diphtheria toxoid and acellular pertussis vaccines recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2006; 55:1–34.
- Centers for Disease Control and Prevention. Recommendations and Guidelines. ACIP presentation slides: October 2010 meeting. http://www.cdc.gov/vaccines/recs/acip/slides-oct10.htm. Accessed July 6, 2011.
- Cortese MM, Bisgard KM. Pertussis. In:Wallace RB, Kohatsu N, Last JM, editors. Wallace/Maxcy-Rosenau-Last Public Health & Preventive Medicine. 15th ed. New York, NY: McGraw-Hill Medical, 2008:111–114.
A 49-year-old woman presents with a cough that has persisted for 3 weeks.
Two weeks ago, she was seen in the outpatient clinic for a nonproductive cough, rhinorrhea, sneezing, and a sore throat. At that time, she described coughing spells that were occasionally accompanied by posttussive chest pain and vomiting. The cough was worse at night and was occasionally associated with wheezing. She reported no fevers, chills, rigors, night sweats, or dyspnea. She said she has tried over-the-counter cough suppressants, antihistamines, and decongestants, but they provided no relief. Since she had a history of well-controlled asthma, she was diagnosed with an asthma exacerbation and was given prednisone 20 mg to take orally every day for 5 days, to be followed by an inhaled corticosteroid until her symptoms resolved.
Now, she has returned because her symptoms have persisted despite treatment, and she is seeking a second medical opinion. Her paroxysmal cough has become more frequent and more severe.
In addition to asthma, she has a history of allergic rhinitis. Her current medications include the over-the-counter histamine H1 antagonist cetirizine (Zyrtec), a fluticasone-salmeterol inhaler (Advair), and an albuterol inhaler (Proventil HFA). She reports having had mild asthma exacerbations in the past during the winter, which were managed well with her albuterol inhaler.
She has never smoked; she drinks alcohol socially. She has not traveled outside the United States during the past several months. She is married and has two children, ages 25 and 23. She lives at home with only her husband, and he has not been sick. However, she works at a greeting card store, and two of her coworkers have similar upper respiratory symptoms, although they have only a mild cough.
Her immunizations are not up-to-date. She last received the tetanus-diphtheria toxoid (Td) vaccine 12 years ago, and she never received the pediatric tetanus, diphtheria, and acellular pertussis (Tdap) vaccine. She generally receives the influenza vaccine annually, and she received it about 6 weeks before this presentation.
She is not in distress, but she has paroxysms of severe coughing throughout her examination. Her pulse is 100 beats per minute, respiratory rate 18, and blood pressure 130/86 mm Hg. Her oropharynx is clear. The pulmonary examination reveals poor inspiratory effort due to coughing but is otherwise normal. The rest of the examination is normal, as is her chest radiograph.
WHAT DOES SHE HAVE?
1. Which of the following would best explain her symptoms?
- Asthma
- Postviral cough
- Pertussis
- Chronic bronchitis
- Pneumonia
- Gastroesophageal reflux disease
Asthma is a reasonable consideration, given her medical history, her occasional wheezing, and her nonproductive cough that is worse at night. However, asthma typically responds well to corticosteroid therapy. She has already received a course of prednisone, but her symptoms have not improved.
Postviral cough could also be considered in this patient. However, postviral cough does not typically occur in paroxysms, nor does it lead to posttussive vomiting. It is also generally regarded as a diagnosis of exclusion.
Pertussis (whooping cough) should be suspected in this patient, given the time course of her symptoms, the paroxysmal cough, and the posttussive vomiting. In addition, at her job she interacts with hundreds of people a day, increasing her risk of exposure to respiratory tract pathogens, including Bordetella pertussis.
Chronic bronchitis is defined by cough (typically productive) lasting at least 3 months per year for at least 2 consecutive years, which does not fit the time course for this patient. It is vastly more common in smokers.
Pneumonia typically presents with a cough that can be productive or nonproductive, but also with fever, chills, and radiologic evidence of a pulmonary infiltrate or consolidation. This woman has none of these.
Gastroesophageal reflux disease is one of the most common causes of chronic cough, with symptoms typically worse at night. However, it is generally associated with symptoms such as heartburn, a sour taste in the mouth, or regurgitation, which our patient did not report.
Thus, pertussis is the most likely diagnosis.
PERTUSSIS IS ON THE RISE
Pertussis is an acute and highly contagious disease caused by infection of the respiratory tract by B pertussis, a small, aerobic, gramnegative, pleomorphic coccobacillus that produces a number of antigenic and biologically active products, including pertussis toxin, filamentous hemagglutinin, agglutinogens, and tracheal cytotoxin. Transmitted by aerosolized droplets, it attaches to the ciliated epithelial cells of the lower respiratory tract, paralyzes the cilia via toxins, and causes inflammation, thus interfering with the clearing of respiratory secretions.
The incidence of pertussis is on the rise. In 2005, 25,827 cases were reported in the United States, the highest number since 1959.1 Pertussis is now epidemic in California. At the time of this writing, the number of confirmed, probable, and suspected cases in California was 9,477 (including 10 infant deaths) for the year 2010—the most cases reported in the past 65 years.2,3
In 2010, outbreaks were also reported in Michigan, Texas, Ohio, upstate New York, and Arizona.4 The overall incidence of pertussis is likely even higher than what is reported, since many cases go unrecognized or unreported.
Highly contagious
Pertussis is transmitted person-to-person, primarily through aerosolized droplets from coughing or sneezing or by direct contact with secretions from the respiratory tract of infected persons. It is highly contagious, with secondary attack rates of up to 80% in susceptible people.
A three-stage clinical course
The clinical definition of pertussis used by the US Centers for Disease Control and Prevention (CDC) and the Council of State and Territorial Epidemiologists is an acute cough illness lasting at least 2 weeks, with paroxysms of coughing, an inspiratory “whoop,” or posttussive vomiting without another apparent cause.5
The clinical course of the illness is traditionally divided into three stages:
The catarrhal phase typically lasts 1 to 2 weeks and is clinically indistinguishable from a viral upper respiratory infection. It is characterized by the insidious onset of malaise, coryza, sneezing, low-grade fever, and a mild cough that gradually becomes severe.6
The paroxysmal phase normally lasts 1 to 6 weeks but may persist for up to 10 weeks. The diagnosis of pertussis is usually suspected during this phase. The classic features of this phase are bursts or paroxysms of numerous, rapid coughs. These are followed by a long inspiratory effort usually accompanied by a characteristic high-pitched whoop, most notably observed in infants and children. Infants and children may appear very ill and distressed during this time and may become cyanotic, but cyanosis is uncommon in adults and adolescents. The paroxysms may also be followed by exhaustion and posttussive vomiting. In some cases, the cough is not paroxysmal, but rather simply persistent. The coughing attacks tend to occur more often at night, with an average of 15 attacks per 24 hours. During the first 1 to 2 weeks of this stage, the attacks generally increase in frequency, remain at the same intensity level for 2 to 3 weeks, and then gradually decrease over 1 to 2 weeks.1,7
The convalescent phase can have a variable course, ranging from weeks to months, with an average duration of 2 to 3 weeks. During this stage, the paroxysms of coughing become less frequent and gradually resolve. Paroxysms often recur with subsequent respiratory infections.
In infants and young children, pertussis tends to follow these stages in a predictable sequence. Adolescents and adults, however, tend to go through the stages without being as ill and typically do not exhibit the characteristic whoop.
TESTING FOR PERTUSSIS
2. Which would be the test of choice to confirm pertussis in this patient?
- Bacterial culture of nasopharyngeal secretions
- Polymerase chain reaction (PCR) testing of nasopharyngeal secretions
- Direct fluorescent antibody testing of nasopharyngeal secretions
- Enzyme-linked immunosorbent assay (ELISA) serologic testing
Establishing the diagnosis of pertussis is often rather challenging.
Bacterial culture: Very specific, but slow and not so sensitive
Bacterial culture is still the gold standard for diagnosing pertussis, as a positive culture for B pertussis is 100% specific.5
However, this test has drawbacks. Its sensitivity has a wide range (15% to 80%) and depends very much on the time from the onset of symptoms to the time the culture specimen is collected. The yield drops off significantly after 1 week, and after 3 weeks the test has a sensitivity of only 1% to 3%.8 Therefore, for our patient, who has had symptoms for 3 weeks already, bacterial culture would not be the best test. In addition, the results are usually not known for 7 to 14 days, which is too slow to be useful in managing acute cases.
For swabbing, a Dacron swab is inserted through the nostril to the posterior pharynx and is left in place for 10 seconds to maximize the yield of the specimen. Recovery rates for B pertussis are low if the throat or the anterior nasal passage is swabbed instead of the posterior pharynx.9
Nasopharyngeal aspiration is a more complicated procedure, requiring a suction device to trap the mucus, but it may provide higher yields than swabbing.10 In this method, the specimen is obtained by inserting a small tube (eg, an infant feeding tube) connected to a mucus trap into the nostril back to the posterior pharynx.
Often, direct inoculation of medium for B pertussis is not possible. In such cases, clinical specimens are placed in Regan Lowe transport medium (half-strength charcoal agar supplemented with horse blood and cephalexin).11,12
Polymerase chain reaction testing: Faster, more sensitive, but less specific
PCR testing of nasopharyngeal specimens is now being used instead of bacterial culture to diagnose pertussis in many situations. Alternatively, nasopharyngeal aspirate (or secretions collected with two Dacron swabs) can be obtained and divided at the time of collection and the specimens sent for both culture and PCR testing. Because bacterial culture is time-consuming and has poor sensitivity, the CDC states that a positive PCR test, along with the clinical symptoms and epidemiologic information, is sufficient for diagnosis.5
PCR testing can detect B pertussis with greater sensitivity and more rapidly than bacterial culture.12–14 Its sensitivity ranges from 61% to 99%, its specificity ranges from 88% to 98%,12,15,16 and its results can be available in 2 to 24 hours.12
PCR testing’s advantage in terms of sensitivity is especially pronounced in the later stages of the disease (as in our patient), when clinical suspicion of pertussis typically arises. It can be used effectively for up to 4 weeks from the onset of cough.14 Our patient, who presented nearly 3 weeks after the onset of symptoms, underwent nasopharyngeal sampling for PCR testing.
However, PCR testing is not as specific for B pertussis as is bacterial culture, since other Bordetella species can cause positive results on PCR testing. Also, as with culture, a negative test does not reliably rule out the disease, especially if the sample is collected late in the course.
Therefore, basing the diagnosis on PCR testing alone without the proper clinical context is not advised: pertussis outbreaks have been mistakenly declared on the basis of false-positive PCR test results. Three so-called “pertussis outbreaks” in three different states from 2004 to 200617 were largely the result of overdiagnosis based on equivocal or false-positive PCR test results without the appropriate clinical circumstances. Retrospective review of these pseudo-outbreaks revealed that few cases actually met the CDC’s diagnostic criteria.17 Many patients were not tested (by any method) for pertussis and were treated as having probable cases of pertussis on the basis of their symptoms. Patients who were tested and who had a positive PCR test did not meet the clinical definition of pertussis according to the Council of State and Territorial Epidemiologists.17
Since PCR testing varies in sensitivity and specificity, obtaining culture confirmation of pertussis for at least one suspicious case is recommended any time an outbreak is suspected. This is necessary for monitoring for continued presence of the agent among cases of disease, recruitment of isolates for epidemiologic studies, and surveillance for antibiotic resistance.
Direct fluorescence antibody testing
The CDC does not recommend direct fluorescence antibody testing to diagnose pertussis. This test is commercially available and is sometimes used to screen patients for B pertussis infection, but it lacks sensitivity and specificity for this organism. Cross-reaction with normal nasopharyngeal flora can lead to a false-positive result.18 In addition, the interpretation of the test is subjective, so the sensitivity and specificity are quite variable: the sensitivity is reported as 52% to 65%, while the specificity can vary from 15% to 99%.
Enzyme-linked immunosorbent assay
ELISA testing has been used in epidemiologic studies to measure serum antibodies to B pertussis. Many serologic tests exist, but none is commercially available. Many of these tests are used by the CDC and state health departments to help confirm the diagnosis, especially during outbreaks. Generally, serologic tests are more useful for diagnosis in later phases of the disease. Currently used ELISA tests use both paired and single serology techniques measuring elevated immunoglobulin G serum antibody concentrations against an array of antigens, including pertussis toxin, filamentous hemagglutinin, pertactin, and fimbrae. As a result, a range of sensitivities (33%–95%) and specificities (72%–100%) has been reported.12,14,19
TREATING PERTUSSIS
Our patient’s PCR test result comes back positive. In view of her symptoms and this result, we decide to treat her empirically for pertussis, even though she has had no known contact with anyone with the disease and there is currently no outbreak of it in the community.
3. According to the most recent evidence, which of the following would be the treatment of choice for pertussis in this patient?
- Azithromycin (Zithromax)
- Amoxicillin (Moxatag)
- Levofloxacin (Levaquin)
- Sulfamethoxazole-trimethoprim (Bactrim)
- Supportive measures (hydration, humidifier, antitussives, antihistamines, decongestants)
Azithromycin and the other macrolide antibiotics erythromycin and clarithromycin are first-line therapies for pertussis in adolescents and adults. If given during the catarrhal phase, they can reduce the duration and severity of symptoms and lessen the period of communicability.20,21 After the catarrhal phase, however, it is uncertain whether antibiotics change the clinical course of pertussis, as the data are conflicting.20–22
Factors to consider when selecting a macrolide antibiotic are tolerability, the potential for adverse events and drug interactions, ease of compliance, and cost. All three macrolides are equally effective against pertussis, but azithromycin and clarithromycin are generally better tolerated and are associated with milder and less frequent side effects than erythromycin, including lower rates of gastrointestinal side effects.
Erythromycin and clarithromycin inhibit the cytochrome P450 enzyme system, specifically CYP3A4, and can interact with a great many commonly prescribed drugs metabolized by this enzyme. Therefore, azithromycin may be a better choice for patients already taking other medications, like our patient.
Azithromycin and clarithromycin have longer half-lives and achieve higher tissue concentrations than erythromycin, allowing for less-frequent dosing (daily for azithromycin and twice daily for clarithromycin) and shorter treatment duration (5 days for azithromycin and 7 days for clarithromycin).
An advantage of erythromycin, though, is its lower cost. The cost of a recommended course of erythromycin treatment for pertussis (ie, 500 mg every 6 hours for 14 days) is roughly $20, compared with $75 for azithromycin.
Amoxicillin is not effective in clearing B pertussis from the nasopharynx and thus is not a reasonable option for the treatment of pertussis.23
Levofloxacin is also not recommended for the treatment of pertussis.
Sulfamethoxazole-trimethoprim is a second-line agent for pertussis. It is effective in eradicating B pertussis from the nasopharynx20 and is generally used as an alternative to the macrolide agents in patients who cannot tolerate or have contraindications to macrolides. Sulfamethoxazole-trimethoprim can also be an option for patients infected with rare macrolide-resistant strains of B pertussis.
Supportive measures by themselves are reasonable for patients with pertussis beyond the catarrhal phase, since antibiotics are typically not effective at that stage of the disease.
From 80% to 90% of patients with untreated pertussis spontaneously clear the bacteria from the nasopharynx within 3 to 4 weeks from the onset of cough symptoms.20 However, supportive measures, including antitussives (both over-the-counter and prescription), tend to have very little effect on the severity or duration of the illness, especially when used past the early stage of the illness.
POSTEXPOSURE CHEMOPROPHYLAXIS FOR CLOSE CONTACTS
Postexposure chemoprophylaxis should be given to close contacts of patients who have pertussis to help prevent secondary cases.22 The CDC defines a close contact as someone who has had face-to-face exposure within 3 feet of a symptomatic patient within 21 days after the onset of symptoms in the patient. Close contacts should be treated with antibiotic regimens similar to those used in confirmed cases of pertussis.
In our patient’s case, the diagnosis of pertussis was reported to the Ohio Department of Health. Shortly afterward, the department contacted the patient and obtained information about her close contacts. These people were then contacted and encouraged to complete a course of antibiotics for postexposure chemoprophylaxis, given the high secondary attack rates.
PERTUSSIS VACCINES
4. Which of the following vaccines could have reduced our patient’s chance of contracting the disease or reduced the severity or time course of the illness?
- DTaP
- Tdap
- Whole-cell pertussis vaccine
- No vaccine would have reduced her risk
It is important to prevent pertussis, given its associated morbidities and its generally poor response to drug therapy. Continued vigilance is imperative to maintain high levels of vaccine coverage, including the timely completion of the pertussis vaccination schedule.
The two vaccines in current use in the United States to produce immunity to pertussis—DTaP and Tdap—also confer immunity to diphtheria and tetanus. DTaP is used for children under 7 years of age, and Tdap is for ages 10 to 64. Thus, our patient should have received a series of DTaP injections as an infant and small child, and a Tdap booster at age 11 or 12 years and every 10 years after that.
The upper case “D,” “T,” and “P” in the abbreviations signifies full-strength doses and the lower case “d,” “t,” and “p” indicate that the doses of those components have been reduced. The “a” in both vaccines stands for “acellular”: ie, the pertussis component does not contain cellular elements.
DTaP for initial pertussis vaccination
The current recommendation for initial pertussis vaccination consists of a primary series of DTaP. DTaP vaccination is recommended for infants at 2 months of age, then again at 4 months of age, and again at 6 months of age. A fourth dose is given between the ages of 15 and 18 months, and a fifth dose is given between the ages of 4 to 6 years. If the fourth dose was given after age 4, then no fifth dose is needed.20
Tdap as a booster
The booster vaccine for adolescents and adults is Tdap. In 2005, two Tdap vaccines were licensed in the United States: Adacel for people ages 11 to 64 years, and Boostrix for people ages 10 to 18 years.
The CDC’s Advisory Committee on Immunization Practices (ACIP) recommends a booster dose of Tdap at age 11 or 12 years. Every 10 years thereafter, a booster of tetanus and diphtheria toxoid (Td) vaccine is recommended, except that one of the Td doses can be replaced by Tdap if the patient hasn’t received Tdap yet.
For adults ages 19 to 64, the ACIP currently recommends routine use of a single booster dose of Tdap to replace a single dose of Td if they received the last dose of toxoid vaccine 10 or more years earlier. If the previous dose of Td was given within the past 10 years, a single dose of Tdap is appropriate to protect patients against pertussis. This is especially true for patients at increased risk of pertussis or its complications, as well as for health care professionals and adults who have close contact with infants, such as new parents, grandparents, and child-care providers. The minimum interval since the last Td vaccination is ideally 2 years, although shorter intervals can be used for control of pertussis outbreaks and for those who have close contact with infants.24
In 2010, the ACIP decided that, for those ages 65 and older, a single dose of Tdap vaccine may be given in place of Td if the patient has not previously received Tdap, regardless of how much time has elapsed since the last vaccination with a Td-containing vaccine.25 Data from the Vaccine Adverse Event Reporting System suggest that Tdap vaccine in this age group is as safe as the Td vaccine.25
Subsequent tetanus vaccine doses, in the form of Td, should be given at 10-year intervals throughout adulthood. Administration of Tdap at 10-year intervals appears to be highly immunogenic and well tolerated,25 suggesting that it is possible that Tdap will become part of routine booster dosing instead of Td, pending further study.
Tdap is not contraindicated in pregnant women. Ideally, women should be vaccinated with Tdap before becoming pregnant if they have not previously received it. If the pregnant woman is not at risk of acquiring or transmitting pertussis during pregnancy, the ACIP recommends deferring Tdap vaccination until the immediate postpartum period.
Adults who require a vaccine containing tetanus toxoid for wound management should receive Tdap instead of Td if they have never received Tdap. Adults who have never received vaccine containing tetanus and diphtheria toxoid should receive a series of three vaccinations. The preferred schedule is a dose of Tdap, followed by a dose of Td more than 4 weeks later, and a second dose of Td 6 to 12 months later, though Tdap can be substituted for Td for any one of the three doses in the series. Adults with a history of pertussis generally should receive Tdap according to routine recommendations.
Tdap is contraindicated in people with a history of serious allergic reaction to any component of the Tdap vaccine or with a history of encephalopathy not attributable to an identifiable cause within 7 days of receiving a pertussis vaccine. Tdap is relatively contraindicated and should be deferred in people with current moderate to severe acute illness, current unstable neurologic condition, or a history of Arthus hypersensitivity reaction to a tetanus-toxoid-containing vaccine within the past 10 years, and in people who have developed Guillain-Barré syndrome, within 6 weeks of receiving a tetanus-toxoid–containing vaccine.
Tdap is generally well tolerated. Adverse effects are typically mild and may include localized pain, redness, and swelling; low-grade fever; headache; fatigue; and, less commonly, gastrointestinal upset, myalgia, arthralgia, rash, and swollen glands.
Whole-cell pertussis vaccine is no longer available in the United States
Whole-cell pertussis vaccine provides good protection against pertussis, with 70% to 90% efficacy after three doses. It is less expensive-than acellular formulations and therefore is used in many parts of the world where cost is an issue. It is no longer available in the United States, however, due to high rates of local reactions such as redness, swelling, and pain at the injection site.
The importance of staying up-to-date with booster shots
Booster vaccination for pertussis in adolescents and adults is critical, since the largest recent outbreaks have occurred in these groups.21 The high rate of outbreaks is presumably the result of waning immunity from childhood immunizations and of high interpersonal contact rates. Vaccination has been shown to reduce the chance of contracting the disease and to reduce the severity and time course of the illness.21
Adolescents and adults are an important reservoir for potentially serious infections in infants who are either unvaccinated or whose vaccination schedule has not been completed. These infants are at risk of severe illness, including pneumonia, seizures, encephalopathy, and apnea, or even death. Adults and teens can also suffer complications from pertussis, although these tend to be less serious, especially in those who have been vaccinated. Complications in teens and adults are often caused by malaise and the cough itself, including weight loss (33%), urinary stress incontinence (28%), syncope (6%), rib fractures from severe coughing (4%), and pneumonia (2%).26 Thus, it is important that adolescents and adults stay up-to-date with pertussis vaccination.
CASE CONTINUED
Our patient was treated with a short (5-day) course of azithromycin 500 mg daily. It did not improve her symptoms very much, but this was not unexpected, given her late presentation and duration of symptoms. Her cough persisted for about 2 months afterwards, but it improved with time and with supportive care at home.
CONTINUED CHALLENGES
Pertussis is a reemerging disease with an increased incidence over the past 30 years, and even more so over the past 10 years. Unfortunately, treatments are not very effective, especially since the disease is often diagnosed late in the course.
We are fortunate to have a vaccine that can prevent pertussis, yet pertussis persists, in large part because of waning immunity from childhood vaccination. The duration of immunity from childhood vaccination is not yet clear. Many adolescents and adults do not follow up on these booster vaccines, thus increasing their susceptibility to pertussis. Consequently, they can transmit the disease to children who are not fully immunized. Prevention by maintaining active immunity is the key to controlling this terrible disease.
A 49-year-old woman presents with a cough that has persisted for 3 weeks.
Two weeks ago, she was seen in the outpatient clinic for a nonproductive cough, rhinorrhea, sneezing, and a sore throat. At that time, she described coughing spells that were occasionally accompanied by posttussive chest pain and vomiting. The cough was worse at night and was occasionally associated with wheezing. She reported no fevers, chills, rigors, night sweats, or dyspnea. She said she has tried over-the-counter cough suppressants, antihistamines, and decongestants, but they provided no relief. Since she had a history of well-controlled asthma, she was diagnosed with an asthma exacerbation and was given prednisone 20 mg to take orally every day for 5 days, to be followed by an inhaled corticosteroid until her symptoms resolved.
Now, she has returned because her symptoms have persisted despite treatment, and she is seeking a second medical opinion. Her paroxysmal cough has become more frequent and more severe.
In addition to asthma, she has a history of allergic rhinitis. Her current medications include the over-the-counter histamine H1 antagonist cetirizine (Zyrtec), a fluticasone-salmeterol inhaler (Advair), and an albuterol inhaler (Proventil HFA). She reports having had mild asthma exacerbations in the past during the winter, which were managed well with her albuterol inhaler.
She has never smoked; she drinks alcohol socially. She has not traveled outside the United States during the past several months. She is married and has two children, ages 25 and 23. She lives at home with only her husband, and he has not been sick. However, she works at a greeting card store, and two of her coworkers have similar upper respiratory symptoms, although they have only a mild cough.
Her immunizations are not up-to-date. She last received the tetanus-diphtheria toxoid (Td) vaccine 12 years ago, and she never received the pediatric tetanus, diphtheria, and acellular pertussis (Tdap) vaccine. She generally receives the influenza vaccine annually, and she received it about 6 weeks before this presentation.
She is not in distress, but she has paroxysms of severe coughing throughout her examination. Her pulse is 100 beats per minute, respiratory rate 18, and blood pressure 130/86 mm Hg. Her oropharynx is clear. The pulmonary examination reveals poor inspiratory effort due to coughing but is otherwise normal. The rest of the examination is normal, as is her chest radiograph.
WHAT DOES SHE HAVE?
1. Which of the following would best explain her symptoms?
- Asthma
- Postviral cough
- Pertussis
- Chronic bronchitis
- Pneumonia
- Gastroesophageal reflux disease
Asthma is a reasonable consideration, given her medical history, her occasional wheezing, and her nonproductive cough that is worse at night. However, asthma typically responds well to corticosteroid therapy. She has already received a course of prednisone, but her symptoms have not improved.
Postviral cough could also be considered in this patient. However, postviral cough does not typically occur in paroxysms, nor does it lead to posttussive vomiting. It is also generally regarded as a diagnosis of exclusion.
Pertussis (whooping cough) should be suspected in this patient, given the time course of her symptoms, the paroxysmal cough, and the posttussive vomiting. In addition, at her job she interacts with hundreds of people a day, increasing her risk of exposure to respiratory tract pathogens, including Bordetella pertussis.
Chronic bronchitis is defined by cough (typically productive) lasting at least 3 months per year for at least 2 consecutive years, which does not fit the time course for this patient. It is vastly more common in smokers.
Pneumonia typically presents with a cough that can be productive or nonproductive, but also with fever, chills, and radiologic evidence of a pulmonary infiltrate or consolidation. This woman has none of these.
Gastroesophageal reflux disease is one of the most common causes of chronic cough, with symptoms typically worse at night. However, it is generally associated with symptoms such as heartburn, a sour taste in the mouth, or regurgitation, which our patient did not report.
Thus, pertussis is the most likely diagnosis.
PERTUSSIS IS ON THE RISE
Pertussis is an acute and highly contagious disease caused by infection of the respiratory tract by B pertussis, a small, aerobic, gramnegative, pleomorphic coccobacillus that produces a number of antigenic and biologically active products, including pertussis toxin, filamentous hemagglutinin, agglutinogens, and tracheal cytotoxin. Transmitted by aerosolized droplets, it attaches to the ciliated epithelial cells of the lower respiratory tract, paralyzes the cilia via toxins, and causes inflammation, thus interfering with the clearing of respiratory secretions.
The incidence of pertussis is on the rise. In 2005, 25,827 cases were reported in the United States, the highest number since 1959.1 Pertussis is now epidemic in California. At the time of this writing, the number of confirmed, probable, and suspected cases in California was 9,477 (including 10 infant deaths) for the year 2010—the most cases reported in the past 65 years.2,3
In 2010, outbreaks were also reported in Michigan, Texas, Ohio, upstate New York, and Arizona.4 The overall incidence of pertussis is likely even higher than what is reported, since many cases go unrecognized or unreported.
Highly contagious
Pertussis is transmitted person-to-person, primarily through aerosolized droplets from coughing or sneezing or by direct contact with secretions from the respiratory tract of infected persons. It is highly contagious, with secondary attack rates of up to 80% in susceptible people.
A three-stage clinical course
The clinical definition of pertussis used by the US Centers for Disease Control and Prevention (CDC) and the Council of State and Territorial Epidemiologists is an acute cough illness lasting at least 2 weeks, with paroxysms of coughing, an inspiratory “whoop,” or posttussive vomiting without another apparent cause.5
The clinical course of the illness is traditionally divided into three stages:
The catarrhal phase typically lasts 1 to 2 weeks and is clinically indistinguishable from a viral upper respiratory infection. It is characterized by the insidious onset of malaise, coryza, sneezing, low-grade fever, and a mild cough that gradually becomes severe.6
The paroxysmal phase normally lasts 1 to 6 weeks but may persist for up to 10 weeks. The diagnosis of pertussis is usually suspected during this phase. The classic features of this phase are bursts or paroxysms of numerous, rapid coughs. These are followed by a long inspiratory effort usually accompanied by a characteristic high-pitched whoop, most notably observed in infants and children. Infants and children may appear very ill and distressed during this time and may become cyanotic, but cyanosis is uncommon in adults and adolescents. The paroxysms may also be followed by exhaustion and posttussive vomiting. In some cases, the cough is not paroxysmal, but rather simply persistent. The coughing attacks tend to occur more often at night, with an average of 15 attacks per 24 hours. During the first 1 to 2 weeks of this stage, the attacks generally increase in frequency, remain at the same intensity level for 2 to 3 weeks, and then gradually decrease over 1 to 2 weeks.1,7
The convalescent phase can have a variable course, ranging from weeks to months, with an average duration of 2 to 3 weeks. During this stage, the paroxysms of coughing become less frequent and gradually resolve. Paroxysms often recur with subsequent respiratory infections.
In infants and young children, pertussis tends to follow these stages in a predictable sequence. Adolescents and adults, however, tend to go through the stages without being as ill and typically do not exhibit the characteristic whoop.
TESTING FOR PERTUSSIS
2. Which would be the test of choice to confirm pertussis in this patient?
- Bacterial culture of nasopharyngeal secretions
- Polymerase chain reaction (PCR) testing of nasopharyngeal secretions
- Direct fluorescent antibody testing of nasopharyngeal secretions
- Enzyme-linked immunosorbent assay (ELISA) serologic testing
Establishing the diagnosis of pertussis is often rather challenging.
Bacterial culture: Very specific, but slow and not so sensitive
Bacterial culture is still the gold standard for diagnosing pertussis, as a positive culture for B pertussis is 100% specific.5
However, this test has drawbacks. Its sensitivity has a wide range (15% to 80%) and depends very much on the time from the onset of symptoms to the time the culture specimen is collected. The yield drops off significantly after 1 week, and after 3 weeks the test has a sensitivity of only 1% to 3%.8 Therefore, for our patient, who has had symptoms for 3 weeks already, bacterial culture would not be the best test. In addition, the results are usually not known for 7 to 14 days, which is too slow to be useful in managing acute cases.
For swabbing, a Dacron swab is inserted through the nostril to the posterior pharynx and is left in place for 10 seconds to maximize the yield of the specimen. Recovery rates for B pertussis are low if the throat or the anterior nasal passage is swabbed instead of the posterior pharynx.9
Nasopharyngeal aspiration is a more complicated procedure, requiring a suction device to trap the mucus, but it may provide higher yields than swabbing.10 In this method, the specimen is obtained by inserting a small tube (eg, an infant feeding tube) connected to a mucus trap into the nostril back to the posterior pharynx.
Often, direct inoculation of medium for B pertussis is not possible. In such cases, clinical specimens are placed in Regan Lowe transport medium (half-strength charcoal agar supplemented with horse blood and cephalexin).11,12
Polymerase chain reaction testing: Faster, more sensitive, but less specific
PCR testing of nasopharyngeal specimens is now being used instead of bacterial culture to diagnose pertussis in many situations. Alternatively, nasopharyngeal aspirate (or secretions collected with two Dacron swabs) can be obtained and divided at the time of collection and the specimens sent for both culture and PCR testing. Because bacterial culture is time-consuming and has poor sensitivity, the CDC states that a positive PCR test, along with the clinical symptoms and epidemiologic information, is sufficient for diagnosis.5
PCR testing can detect B pertussis with greater sensitivity and more rapidly than bacterial culture.12–14 Its sensitivity ranges from 61% to 99%, its specificity ranges from 88% to 98%,12,15,16 and its results can be available in 2 to 24 hours.12
PCR testing’s advantage in terms of sensitivity is especially pronounced in the later stages of the disease (as in our patient), when clinical suspicion of pertussis typically arises. It can be used effectively for up to 4 weeks from the onset of cough.14 Our patient, who presented nearly 3 weeks after the onset of symptoms, underwent nasopharyngeal sampling for PCR testing.
However, PCR testing is not as specific for B pertussis as is bacterial culture, since other Bordetella species can cause positive results on PCR testing. Also, as with culture, a negative test does not reliably rule out the disease, especially if the sample is collected late in the course.
Therefore, basing the diagnosis on PCR testing alone without the proper clinical context is not advised: pertussis outbreaks have been mistakenly declared on the basis of false-positive PCR test results. Three so-called “pertussis outbreaks” in three different states from 2004 to 200617 were largely the result of overdiagnosis based on equivocal or false-positive PCR test results without the appropriate clinical circumstances. Retrospective review of these pseudo-outbreaks revealed that few cases actually met the CDC’s diagnostic criteria.17 Many patients were not tested (by any method) for pertussis and were treated as having probable cases of pertussis on the basis of their symptoms. Patients who were tested and who had a positive PCR test did not meet the clinical definition of pertussis according to the Council of State and Territorial Epidemiologists.17
Since PCR testing varies in sensitivity and specificity, obtaining culture confirmation of pertussis for at least one suspicious case is recommended any time an outbreak is suspected. This is necessary for monitoring for continued presence of the agent among cases of disease, recruitment of isolates for epidemiologic studies, and surveillance for antibiotic resistance.
Direct fluorescence antibody testing
The CDC does not recommend direct fluorescence antibody testing to diagnose pertussis. This test is commercially available and is sometimes used to screen patients for B pertussis infection, but it lacks sensitivity and specificity for this organism. Cross-reaction with normal nasopharyngeal flora can lead to a false-positive result.18 In addition, the interpretation of the test is subjective, so the sensitivity and specificity are quite variable: the sensitivity is reported as 52% to 65%, while the specificity can vary from 15% to 99%.
Enzyme-linked immunosorbent assay
ELISA testing has been used in epidemiologic studies to measure serum antibodies to B pertussis. Many serologic tests exist, but none is commercially available. Many of these tests are used by the CDC and state health departments to help confirm the diagnosis, especially during outbreaks. Generally, serologic tests are more useful for diagnosis in later phases of the disease. Currently used ELISA tests use both paired and single serology techniques measuring elevated immunoglobulin G serum antibody concentrations against an array of antigens, including pertussis toxin, filamentous hemagglutinin, pertactin, and fimbrae. As a result, a range of sensitivities (33%–95%) and specificities (72%–100%) has been reported.12,14,19
TREATING PERTUSSIS
Our patient’s PCR test result comes back positive. In view of her symptoms and this result, we decide to treat her empirically for pertussis, even though she has had no known contact with anyone with the disease and there is currently no outbreak of it in the community.
3. According to the most recent evidence, which of the following would be the treatment of choice for pertussis in this patient?
- Azithromycin (Zithromax)
- Amoxicillin (Moxatag)
- Levofloxacin (Levaquin)
- Sulfamethoxazole-trimethoprim (Bactrim)
- Supportive measures (hydration, humidifier, antitussives, antihistamines, decongestants)
Azithromycin and the other macrolide antibiotics erythromycin and clarithromycin are first-line therapies for pertussis in adolescents and adults. If given during the catarrhal phase, they can reduce the duration and severity of symptoms and lessen the period of communicability.20,21 After the catarrhal phase, however, it is uncertain whether antibiotics change the clinical course of pertussis, as the data are conflicting.20–22
Factors to consider when selecting a macrolide antibiotic are tolerability, the potential for adverse events and drug interactions, ease of compliance, and cost. All three macrolides are equally effective against pertussis, but azithromycin and clarithromycin are generally better tolerated and are associated with milder and less frequent side effects than erythromycin, including lower rates of gastrointestinal side effects.
Erythromycin and clarithromycin inhibit the cytochrome P450 enzyme system, specifically CYP3A4, and can interact with a great many commonly prescribed drugs metabolized by this enzyme. Therefore, azithromycin may be a better choice for patients already taking other medications, like our patient.
Azithromycin and clarithromycin have longer half-lives and achieve higher tissue concentrations than erythromycin, allowing for less-frequent dosing (daily for azithromycin and twice daily for clarithromycin) and shorter treatment duration (5 days for azithromycin and 7 days for clarithromycin).
An advantage of erythromycin, though, is its lower cost. The cost of a recommended course of erythromycin treatment for pertussis (ie, 500 mg every 6 hours for 14 days) is roughly $20, compared with $75 for azithromycin.
Amoxicillin is not effective in clearing B pertussis from the nasopharynx and thus is not a reasonable option for the treatment of pertussis.23
Levofloxacin is also not recommended for the treatment of pertussis.
Sulfamethoxazole-trimethoprim is a second-line agent for pertussis. It is effective in eradicating B pertussis from the nasopharynx20 and is generally used as an alternative to the macrolide agents in patients who cannot tolerate or have contraindications to macrolides. Sulfamethoxazole-trimethoprim can also be an option for patients infected with rare macrolide-resistant strains of B pertussis.
Supportive measures by themselves are reasonable for patients with pertussis beyond the catarrhal phase, since antibiotics are typically not effective at that stage of the disease.
From 80% to 90% of patients with untreated pertussis spontaneously clear the bacteria from the nasopharynx within 3 to 4 weeks from the onset of cough symptoms.20 However, supportive measures, including antitussives (both over-the-counter and prescription), tend to have very little effect on the severity or duration of the illness, especially when used past the early stage of the illness.
POSTEXPOSURE CHEMOPROPHYLAXIS FOR CLOSE CONTACTS
Postexposure chemoprophylaxis should be given to close contacts of patients who have pertussis to help prevent secondary cases.22 The CDC defines a close contact as someone who has had face-to-face exposure within 3 feet of a symptomatic patient within 21 days after the onset of symptoms in the patient. Close contacts should be treated with antibiotic regimens similar to those used in confirmed cases of pertussis.
In our patient’s case, the diagnosis of pertussis was reported to the Ohio Department of Health. Shortly afterward, the department contacted the patient and obtained information about her close contacts. These people were then contacted and encouraged to complete a course of antibiotics for postexposure chemoprophylaxis, given the high secondary attack rates.
PERTUSSIS VACCINES
4. Which of the following vaccines could have reduced our patient’s chance of contracting the disease or reduced the severity or time course of the illness?
- DTaP
- Tdap
- Whole-cell pertussis vaccine
- No vaccine would have reduced her risk
It is important to prevent pertussis, given its associated morbidities and its generally poor response to drug therapy. Continued vigilance is imperative to maintain high levels of vaccine coverage, including the timely completion of the pertussis vaccination schedule.
The two vaccines in current use in the United States to produce immunity to pertussis—DTaP and Tdap—also confer immunity to diphtheria and tetanus. DTaP is used for children under 7 years of age, and Tdap is for ages 10 to 64. Thus, our patient should have received a series of DTaP injections as an infant and small child, and a Tdap booster at age 11 or 12 years and every 10 years after that.
The upper case “D,” “T,” and “P” in the abbreviations signifies full-strength doses and the lower case “d,” “t,” and “p” indicate that the doses of those components have been reduced. The “a” in both vaccines stands for “acellular”: ie, the pertussis component does not contain cellular elements.
DTaP for initial pertussis vaccination
The current recommendation for initial pertussis vaccination consists of a primary series of DTaP. DTaP vaccination is recommended for infants at 2 months of age, then again at 4 months of age, and again at 6 months of age. A fourth dose is given between the ages of 15 and 18 months, and a fifth dose is given between the ages of 4 to 6 years. If the fourth dose was given after age 4, then no fifth dose is needed.20
Tdap as a booster
The booster vaccine for adolescents and adults is Tdap. In 2005, two Tdap vaccines were licensed in the United States: Adacel for people ages 11 to 64 years, and Boostrix for people ages 10 to 18 years.
The CDC’s Advisory Committee on Immunization Practices (ACIP) recommends a booster dose of Tdap at age 11 or 12 years. Every 10 years thereafter, a booster of tetanus and diphtheria toxoid (Td) vaccine is recommended, except that one of the Td doses can be replaced by Tdap if the patient hasn’t received Tdap yet.
For adults ages 19 to 64, the ACIP currently recommends routine use of a single booster dose of Tdap to replace a single dose of Td if they received the last dose of toxoid vaccine 10 or more years earlier. If the previous dose of Td was given within the past 10 years, a single dose of Tdap is appropriate to protect patients against pertussis. This is especially true for patients at increased risk of pertussis or its complications, as well as for health care professionals and adults who have close contact with infants, such as new parents, grandparents, and child-care providers. The minimum interval since the last Td vaccination is ideally 2 years, although shorter intervals can be used for control of pertussis outbreaks and for those who have close contact with infants.24
In 2010, the ACIP decided that, for those ages 65 and older, a single dose of Tdap vaccine may be given in place of Td if the patient has not previously received Tdap, regardless of how much time has elapsed since the last vaccination with a Td-containing vaccine.25 Data from the Vaccine Adverse Event Reporting System suggest that Tdap vaccine in this age group is as safe as the Td vaccine.25
Subsequent tetanus vaccine doses, in the form of Td, should be given at 10-year intervals throughout adulthood. Administration of Tdap at 10-year intervals appears to be highly immunogenic and well tolerated,25 suggesting that it is possible that Tdap will become part of routine booster dosing instead of Td, pending further study.
Tdap is not contraindicated in pregnant women. Ideally, women should be vaccinated with Tdap before becoming pregnant if they have not previously received it. If the pregnant woman is not at risk of acquiring or transmitting pertussis during pregnancy, the ACIP recommends deferring Tdap vaccination until the immediate postpartum period.
Adults who require a vaccine containing tetanus toxoid for wound management should receive Tdap instead of Td if they have never received Tdap. Adults who have never received vaccine containing tetanus and diphtheria toxoid should receive a series of three vaccinations. The preferred schedule is a dose of Tdap, followed by a dose of Td more than 4 weeks later, and a second dose of Td 6 to 12 months later, though Tdap can be substituted for Td for any one of the three doses in the series. Adults with a history of pertussis generally should receive Tdap according to routine recommendations.
Tdap is contraindicated in people with a history of serious allergic reaction to any component of the Tdap vaccine or with a history of encephalopathy not attributable to an identifiable cause within 7 days of receiving a pertussis vaccine. Tdap is relatively contraindicated and should be deferred in people with current moderate to severe acute illness, current unstable neurologic condition, or a history of Arthus hypersensitivity reaction to a tetanus-toxoid-containing vaccine within the past 10 years, and in people who have developed Guillain-Barré syndrome, within 6 weeks of receiving a tetanus-toxoid–containing vaccine.
Tdap is generally well tolerated. Adverse effects are typically mild and may include localized pain, redness, and swelling; low-grade fever; headache; fatigue; and, less commonly, gastrointestinal upset, myalgia, arthralgia, rash, and swollen glands.
Whole-cell pertussis vaccine is no longer available in the United States
Whole-cell pertussis vaccine provides good protection against pertussis, with 70% to 90% efficacy after three doses. It is less expensive-than acellular formulations and therefore is used in many parts of the world where cost is an issue. It is no longer available in the United States, however, due to high rates of local reactions such as redness, swelling, and pain at the injection site.
The importance of staying up-to-date with booster shots
Booster vaccination for pertussis in adolescents and adults is critical, since the largest recent outbreaks have occurred in these groups.21 The high rate of outbreaks is presumably the result of waning immunity from childhood immunizations and of high interpersonal contact rates. Vaccination has been shown to reduce the chance of contracting the disease and to reduce the severity and time course of the illness.21
Adolescents and adults are an important reservoir for potentially serious infections in infants who are either unvaccinated or whose vaccination schedule has not been completed. These infants are at risk of severe illness, including pneumonia, seizures, encephalopathy, and apnea, or even death. Adults and teens can also suffer complications from pertussis, although these tend to be less serious, especially in those who have been vaccinated. Complications in teens and adults are often caused by malaise and the cough itself, including weight loss (33%), urinary stress incontinence (28%), syncope (6%), rib fractures from severe coughing (4%), and pneumonia (2%).26 Thus, it is important that adolescents and adults stay up-to-date with pertussis vaccination.
CASE CONTINUED
Our patient was treated with a short (5-day) course of azithromycin 500 mg daily. It did not improve her symptoms very much, but this was not unexpected, given her late presentation and duration of symptoms. Her cough persisted for about 2 months afterwards, but it improved with time and with supportive care at home.
CONTINUED CHALLENGES
Pertussis is a reemerging disease with an increased incidence over the past 30 years, and even more so over the past 10 years. Unfortunately, treatments are not very effective, especially since the disease is often diagnosed late in the course.
We are fortunate to have a vaccine that can prevent pertussis, yet pertussis persists, in large part because of waning immunity from childhood vaccination. The duration of immunity from childhood vaccination is not yet clear. Many adolescents and adults do not follow up on these booster vaccines, thus increasing their susceptibility to pertussis. Consequently, they can transmit the disease to children who are not fully immunized. Prevention by maintaining active immunity is the key to controlling this terrible disease.
- Centers for Disease Control and Prevention. Pertussis. National Immunization Program, 2005. http://www.cdc.gov/vaccines/pubs/pinkbook/downloads/pert.pdf. Accessed July 6, 2011.
- California Department of Public Health. Pertussis report. www.cdph.ca.gov/programs/immunize/Documents/PertussisReport2011-01-07.pdf. Accessed July 6, 2011.
- Centers for Disease Control and Prevention. Pertussis (whooping cough). www.cdc.gov/pertussis/outbreaks.html. Accessed July 3, 2011.
- Centers for Disease Control and Prevention. Notifiable diseases and mortality tables. MMWR Morb Mortal Wkly Rep 2010; 59:847–861. http://www.cdc.gov/mmwr/PDF/wk/mm5927.pdf. Accessed July 1, 2011.
- Centers for Disease Control and Prevention. Pertussis. Vaccines and preventable diseases: pertussis (whooping cough) vaccination, 2010. http://www.cdc.gov/vaccines/vpd-vac/pertussis/default.htm. Accessed July 6, 2011.
- Hewlett EL, Edwards KM. Clinical practice. Pertussis—not just for kids. N Engl J Med 2005; 352:1215–1222.
- Hewlett E. Bordetella species. In: Mandell GL, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. 5th ed, Philadelphia, PA: Churchill Livingstone; 2000:2701.
- Viljanen MK, Ruuskanen O, Granberg C, Salmi TT. Serological diagnosis of pertussis: IgM, IgA and IgG antibodies against Bordetella pertussis measured by enzyme-linked immunosorbent assay (ELISA). Scand J Infect Dis 1982; 14:117–122.
- Bejuk D, Begovac J, Bace A, Kuzmanovic-Sterk N, Aleraj B. Culture of Bordetella pertussis from three upper respiratory tract specimens. Pediatr Infect Dis J 1995; 14:64–65.
- Hallander HO, Reizenstein E, Renemar B, Rasmuson G, Mardin L, Olin P. Comparison of nasopharyngeal aspirates with swabs for culture of Bordetella pertussis. J Clin Microbiol 1993; 31:50–52.
- Regan J, Lowe F. Enrichment medium for the isolation of Bordetella. J Clin Microbiol 1977; 6:303–309.
- World Health Organization. Laboratory manual for the diagnosis of whooping cough caused by Bordetella pertussis/Bordetella para-pertussis. Department of Immunization, Vaccines and Biologicals. Printed 2004. Revised 2007. www.who.int/vaccines-documents/. Accessed July 6, 2011.
- Meade BD, Bollen A. Recommendations for use of the polymerase chain reaction in the diagnosis of Bordetella pertussis infections. J Med Microbiol 1994; 41:51–55.
- Wendelboe AM, Van Rie A. Diagnosis of pertussis: a historical review and recent developments. Expert Rev Mol Diagn 2006; 6:857–864.
- Knorr L, Fox JD, Tilley PA, Ahmed-Bentley J. Evaluation of real-time PCR for diagnosis of Bordetella pertussis infection. BMC Infect Dis 2006; 6:62.
- Sotir MJ, Cappozzo DL, Warshauer DM, et al. Evaluation of polymerase chain reaction and culture for diagnosis of pertussis in the control of a county-wide outbreak focused among adolescents and adults. Clin Infect Dis 2007; 44:1216–1219.
- Centers for Disease Control and Prevention (CDC). Outbreaks of respiratory illness mistakenly attributed to pertussis—New Hampshire, Massachusetts, and Tennessee, 2004–2006. MMWR Morb Mortal Wkly Rep 2007; 56:837–842.
- Ewanowich CA, Chui LW, Paranchych MG, Peppler MS, Marusyk RG, Albritton WL. Major outbreak of pertussis in northern Alberta, Canada: analysis of discrepant direct fluorescent-antibody and culture results by using polymerase chain reaction methodology. J Clin Microbiol 1993; 31:1715–1725.
- Müller FM, Hoppe JE, Wirsing von König CH. Laboratory diagnosis of pertussis: state of the art in 1997. J Clin Microbiol 1997; 35:2435–2443.
- Tiwari T, Murphy TV, Moran J; National Immunization Program, CDC. Recommended antimicrobial agents for the treatment and postexposure prophylaxis of pertussis: 2005 CDC Guidelines. MMWR Recomm Rep 2005; 54:1–16.
- Wirsing von König CH, Postels-Multani S, Bock HL, Schmitt HJ. Pertussis in adults: frequency of transmission after household exposure. Lancet 1995; 346:1326–1329.
- von König CH. Use of antibiotics in the prevention and treatment of pertussis. Pediatr Infect Dis J 2005; 24(suppl 5):S66–S68.
- Trollfors B. Effect of erythromycin and amoxycillin on Bordetella pertussis in the nasopharynx. Infection 1978; 6:228–230.
- Broder KR, Cortese MM, Iskander JK, et al; Advisory Committee on Immunization Practices (ACIP). Preventing tetanus, diphtheria, and pertussis among adolescents: use of tetanus toxoid, reduced diphtheria toxoid and acellular pertussis vaccines recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2006; 55:1–34.
- Centers for Disease Control and Prevention. Recommendations and Guidelines. ACIP presentation slides: October 2010 meeting. http://www.cdc.gov/vaccines/recs/acip/slides-oct10.htm. Accessed July 6, 2011.
- Cortese MM, Bisgard KM. Pertussis. In:Wallace RB, Kohatsu N, Last JM, editors. Wallace/Maxcy-Rosenau-Last Public Health & Preventive Medicine. 15th ed. New York, NY: McGraw-Hill Medical, 2008:111–114.
- Centers for Disease Control and Prevention. Pertussis. National Immunization Program, 2005. http://www.cdc.gov/vaccines/pubs/pinkbook/downloads/pert.pdf. Accessed July 6, 2011.
- California Department of Public Health. Pertussis report. www.cdph.ca.gov/programs/immunize/Documents/PertussisReport2011-01-07.pdf. Accessed July 6, 2011.
- Centers for Disease Control and Prevention. Pertussis (whooping cough). www.cdc.gov/pertussis/outbreaks.html. Accessed July 3, 2011.
- Centers for Disease Control and Prevention. Notifiable diseases and mortality tables. MMWR Morb Mortal Wkly Rep 2010; 59:847–861. http://www.cdc.gov/mmwr/PDF/wk/mm5927.pdf. Accessed July 1, 2011.
- Centers for Disease Control and Prevention. Pertussis. Vaccines and preventable diseases: pertussis (whooping cough) vaccination, 2010. http://www.cdc.gov/vaccines/vpd-vac/pertussis/default.htm. Accessed July 6, 2011.
- Hewlett EL, Edwards KM. Clinical practice. Pertussis—not just for kids. N Engl J Med 2005; 352:1215–1222.
- Hewlett E. Bordetella species. In: Mandell GL, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. 5th ed, Philadelphia, PA: Churchill Livingstone; 2000:2701.
- Viljanen MK, Ruuskanen O, Granberg C, Salmi TT. Serological diagnosis of pertussis: IgM, IgA and IgG antibodies against Bordetella pertussis measured by enzyme-linked immunosorbent assay (ELISA). Scand J Infect Dis 1982; 14:117–122.
- Bejuk D, Begovac J, Bace A, Kuzmanovic-Sterk N, Aleraj B. Culture of Bordetella pertussis from three upper respiratory tract specimens. Pediatr Infect Dis J 1995; 14:64–65.
- Hallander HO, Reizenstein E, Renemar B, Rasmuson G, Mardin L, Olin P. Comparison of nasopharyngeal aspirates with swabs for culture of Bordetella pertussis. J Clin Microbiol 1993; 31:50–52.
- Regan J, Lowe F. Enrichment medium for the isolation of Bordetella. J Clin Microbiol 1977; 6:303–309.
- World Health Organization. Laboratory manual for the diagnosis of whooping cough caused by Bordetella pertussis/Bordetella para-pertussis. Department of Immunization, Vaccines and Biologicals. Printed 2004. Revised 2007. www.who.int/vaccines-documents/. Accessed July 6, 2011.
- Meade BD, Bollen A. Recommendations for use of the polymerase chain reaction in the diagnosis of Bordetella pertussis infections. J Med Microbiol 1994; 41:51–55.
- Wendelboe AM, Van Rie A. Diagnosis of pertussis: a historical review and recent developments. Expert Rev Mol Diagn 2006; 6:857–864.
- Knorr L, Fox JD, Tilley PA, Ahmed-Bentley J. Evaluation of real-time PCR for diagnosis of Bordetella pertussis infection. BMC Infect Dis 2006; 6:62.
- Sotir MJ, Cappozzo DL, Warshauer DM, et al. Evaluation of polymerase chain reaction and culture for diagnosis of pertussis in the control of a county-wide outbreak focused among adolescents and adults. Clin Infect Dis 2007; 44:1216–1219.
- Centers for Disease Control and Prevention (CDC). Outbreaks of respiratory illness mistakenly attributed to pertussis—New Hampshire, Massachusetts, and Tennessee, 2004–2006. MMWR Morb Mortal Wkly Rep 2007; 56:837–842.
- Ewanowich CA, Chui LW, Paranchych MG, Peppler MS, Marusyk RG, Albritton WL. Major outbreak of pertussis in northern Alberta, Canada: analysis of discrepant direct fluorescent-antibody and culture results by using polymerase chain reaction methodology. J Clin Microbiol 1993; 31:1715–1725.
- Müller FM, Hoppe JE, Wirsing von König CH. Laboratory diagnosis of pertussis: state of the art in 1997. J Clin Microbiol 1997; 35:2435–2443.
- Tiwari T, Murphy TV, Moran J; National Immunization Program, CDC. Recommended antimicrobial agents for the treatment and postexposure prophylaxis of pertussis: 2005 CDC Guidelines. MMWR Recomm Rep 2005; 54:1–16.
- Wirsing von König CH, Postels-Multani S, Bock HL, Schmitt HJ. Pertussis in adults: frequency of transmission after household exposure. Lancet 1995; 346:1326–1329.
- von König CH. Use of antibiotics in the prevention and treatment of pertussis. Pediatr Infect Dis J 2005; 24(suppl 5):S66–S68.
- Trollfors B. Effect of erythromycin and amoxycillin on Bordetella pertussis in the nasopharynx. Infection 1978; 6:228–230.
- Broder KR, Cortese MM, Iskander JK, et al; Advisory Committee on Immunization Practices (ACIP). Preventing tetanus, diphtheria, and pertussis among adolescents: use of tetanus toxoid, reduced diphtheria toxoid and acellular pertussis vaccines recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2006; 55:1–34.
- Centers for Disease Control and Prevention. Recommendations and Guidelines. ACIP presentation slides: October 2010 meeting. http://www.cdc.gov/vaccines/recs/acip/slides-oct10.htm. Accessed July 6, 2011.
- Cortese MM, Bisgard KM. Pertussis. In:Wallace RB, Kohatsu N, Last JM, editors. Wallace/Maxcy-Rosenau-Last Public Health & Preventive Medicine. 15th ed. New York, NY: McGraw-Hill Medical, 2008:111–114.
A 31-year-old man with abdominal pain and a rectal nodule
A 31-year-old man presents to the emergency department with abdominal pain and diarrhea, which began 4 days ago. The pain is in both of the lower quadrants, is crampy and persistent, and is relieved with bowel movements. He has been having watery stools five to six times per day, without frank blood.
He reports no fevers, chills, nausea, or vomiting, and he has never travelled outside the country. He underwent laparotomy 6 months ago for a gunshot wound. He takes no prescription drugs. He smokes and he drinks alcohol, and he says he has used heroin and oxycodone recreationally.
His blood pressure is 134/74 mm Hg, and he is afebrile. An abdominal examination reveals no mass or tenderness.
Results of a complete blood count, serum chemistry panel, and serum amylase level are normal. His lipase level is slightly elevated at 80 U/L (reference range 12–70). His stool is negative for Clostridium difficile toxin on enzyme immunoassay.
Computed tomography of the abdomen reveals diffuse pericolonic hyperemia and possible thickening of the rectosigmoid colon, raising the concern that he might have infectious or inflammatory colitis. The patient is admitted for further evaluation.
Colonoscopy to evaluate the abnormalities on computed tomography finds only a 5-mm submucosal nodule in the rectum (Figure 1). Biopsy of the nodule shows it to be a well-differentiated neuroendocrine neoplasm (carcinoid tumor). Random colon biopsy samples are normal.
The patient’s symptoms resolve over the next 24 hours without any treatment.
WHAT EXPLAINS THE PATIENT’S SYMPTOMS?
1. Which of the following best explains the patient’s clinical presentation?
- Narcotic withdrawal
- Carcinoid syndrome
- Viral gastroenteritis
- Acute pancreatitis
Viral gastroenteritis is common and affects people of all ages. The very young and the elderly are at higher risk of adverse outcomes, but few people die of it in the United States.
Our patient’s symptoms were consistent with viral gastroenteritis that resolved spontaneously while he received only supportive care.
Narcotic withdrawal can also cause watery stools and abdominal pain. However, this patient lacked other signs and symptoms of withdrawal, and his symptoms improved without any detoxification or maintenance treatment.
Pancreatitis. Although the patient had a mildly elevated lipase level, his lack of nausea and vomiting and the location of the pain were not consistent with acute pancreatitis.
Carcinoid syndrome. Carcinoid tumors are rare, typically indolent neuroendocrine neoplasms. The carcinoid syndrome consists of cutaneous flushing, gut hypermotility with diarrhea, and bronchospasm.1–5 Our patient did not have the full range of these symptoms. However, the presentation of carcinoid tumors varies broadly depending on the location, morphology, or biology of the tumor.6 Although our patient had diarrhea, his symptoms improved without any specific treatment. Rectal carcinoid tumors rarely cause diarrhea, and therefore the tumor noted on colonoscopy was almost certainly an incidental finding unrelated to his clinical presentation.
The classic symptoms are caused by production of 5-hydroxyindoleacetic acid, typically by a carcinoid tumor of the small bowel. Rectal carcinoids do not produce the 5-hydroxyindoleacetic acid responsible for this “malignant” serotonin-driven syndrome and are typically asymptomatic. When rectal carcinoid tumors are symptomatic, patients may have symptoms of local irritation or obstruction, such as hematochezia, constipation, other changes in bowel habits, rectal pain, pruritis ani, or weight loss.2,7
Nearly 50% of rectal carcinoid tumors are discovered incidentally. The National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) registry database documented a 10-fold increase in the incidence of rectal carcinoids in the last 35 years, attributed in part to an increase in screening colonoscopy.8 Furthermore, although studies of large national or multicenter databases have found that 65% to 80% of all rectal carcinoid tumors are smaller than 1.0 cm, 93.3% to 100% of those discovered on screening endoscopy were 1.0 cm or smaller.8
Rectal carcinoid tumors have a characteristic feel on digital examination, with a hard, “buckshot” consistency, and are freely mobile.5 They have also been described as firm, nodular, rubbery, yellow, submucosal, and polypoid.8
WHERE DO CARCINOID TUMORS TEND TO ARISE?
2. Which of the following sites is the most commonly recognized site of a primary carcinoid tumor?
- Small bowel
- Lung
- Liver
- Pancreas
- Rectum
The small bowel is the most common site.
Carcinoid tumors derive from neoplastic proliferation of cells of the diffuse neuroendocrine system. Therefore, they can be found anywhere neuroendocrine cells are present, commonly in the gastrointestinal tract, urogenital tract, and the bronchial epithelium.
Traditionally, neuroendocrine tumors were classified by their embryologic origin: foregut (including the respiratory tract, thymus, stomach, and pancreas), midgut (including the small intestine, appendix, and right colon), and hindgut (including the transverse, descending, and sigmoid colon and rectum). Functionally, this was sensible, as each class of tumors presented similarly due to the similar hormonal secretory products.2,3,9
A 2004 population-based review of the SEER database10 classified incidence rates of carcinoid tumors and their distribution throughout the body. Most (54.5%) were discovered in the gastrointestinal tract, and of these, 44.7% were in the small intestine, 19.6% were in the rectum, 16.7% were in the appendix, 10.6% were in the colon, 7.2% were in the stomach, and the remaining 1.2% were at other gastrointestinal sites. Nongastrointestinal sites included the lungs and bronchi (30.1%), pancreas (2.3%), female reproductive tract and ovaries (1.2%), biliary system (1.1%), and head and neck (0.4%).10
The incidence rates have increased and the distribution of sites in the body has changed over time. For example, the appendix was once considered the site of highest incidence, with tumors often discovered incidentally during surgical resection. However, these data were based on anecdotal or single-institution reports and so may have been subject to reporting bias. According to the SEER data, the small intestine is now the leading site, perhaps because of increased awareness or improved diagnostic technology and imaging.10,11
The liver is a common site of metastasis, but it is an exceptionally rare location for a primary tumor.
HOW SHOULD THIS PATIENT BE MANAGED?
3. What is the appropriate management of rectal carcinoid in this patient?
- Since the nodule is 1.0 cm or smaller, watchful waiting is acceptable
- Since the nodule is 1.0 cm or smaller, local excision is appropriate, and no follow-up is required
- Because all carcinoid tumors are potentially malignant, radical resection (eg, abdominal perineal resection) is appropriate
- Because all carcinoid tumors are potentially malignant, radical resection with chemotherapy with 5-fluorouracil (Adrucil) and doxorubicin (Adriamycin) is required
Since the nodule is 1.0 cm or smaller, local excision is appropriate, and no follow-up will be required. Rectal carcinoid tumors generally have a favorable prognosis, with a 5-year survival rate of 87.5%.10
PROGNOSIS DEPENDS ON TUMOR SIZE, OTHER FACTORS
Many studies have examined risk factors contributing to poor prognosis, and this is an area of active study. Early research categorized rectal carcinoid risk in terms of tumor diameter, and this is still widely used to guide management. As early as 1959, Hanley et al5 recognized that tumors that were likely to metastasize were often larger than 1 cm, had infiltrated the muscularis, or were ulcerated. Today, it is understood that only 3% to 10% of rectal carcinoids smaller than 1 cm metastasize, whereas 17% to 42% of those 1 to 2 cm and 60% to 80% of those larger than 2 cm do.2,8,12,13
However, size is not the only consideration. Wang et al12 showed that muscular invasion is an independent risk factor for survival, and that tumor diameter is a significant predictor of invasion and metastasis. Similarly, a metaanalysis by Mani et al13 recognized tumor size and muscularis invasion as the most important predictors of malignancy in these neoplasms.
To aid in predicting prognosis, staging systems have been developed from institutional or national registries. Landry et al14 developed a TNM (tumor, node, metastasis) staging system for rectal carcinoids, in which the T value was based on tumor size and degree of invasion. A group at Memorial-Sloan Kettering Cancer Center15 developed a system for risk stratification of carcinoid of the rectum that is based on tumor size, muscularis invasion, lymphovascular invasion, and the mitotic rate.
TREATMENT IS BY EXCISION
Despite these new prognostic systems, there is no new guidance on therapeutic management. Surgical therapy is still largely guided by tumor size.
Lesions smaller than 1 cm are resected endoscopically or by another local transanal technique.2,3,15,16 Standard endoscopic mucosal resection is performed, and recent studies have suggested that endoscopic submucosal dissection is as effective17 or even preferred, because it resects to the deeper submucosa (as the name suggests).18 This en bloc technique may be appropriate for lesions with evidence of local invasion.18 Other situations may call for deeper resection, such as transanal resection for higher lesions and full-thickness mucosal-muscularis resection.
Tumors 1 to 2 cm are currently evaluated for other factors such as ulceration and umbilication, which influence the choice of local vs radical resection. Otherwise, there is little guidance for tumors of 1 to 2 cm.
Tumors larger than 2 cm have a high risk of muscularis invasion and metastasis, and hence they are resected with wide margins and imaging is then used to evaluate for metastasis.8,19 In cases of metastasis, local resection is often palliative, providing local symptom relief.19
AN INCIDENTALLY DISCOVERED CASE; PATIENT LOST TO FOLLOW-UP
Our patient’s case is typical of rectal carcinoid in that it was discovered incidentally during colonoscopy. His clinical presentation was likely unrelated to his carcinoid tumor, and he improved without specific treatment. His symptoms resolved within 24 hours with supportive treatment and he was discharged.
Pathologic confirmation of carcinoid tumor occurred after his discharge. Despite persistent attempts to contact the patient, he never returned for a follow-up appointment.
TAKE-HOME POINTS
- Carcinoid tumors are rare neoplasms of neuroendocrine origin.
- Rectal carcinoids are the third most common carcinoid of the gastrointestinal tract.
- Most rectal carcinoids are asymptomatic.
- Diagnosis is most often incidental and histologic.
- Treatment is by excision.
- Prognosis is favorable for smaller carcinoids and depends on size (and therefore, invasion).
- Thorson A, Biorck G, Bjorkman G, Waldenstrom J. Malignant carcinoid of the small intestine with metastases to the liver, valvular disease of the right side of the heart (pulmonary stenosis and tricuspid regurgitation without septal defects), peripheral vasomotor symptoms, bronchoconstriction, and an unusual type of cyanosis; a clinical and pathologic syndrome. Am Heart J 1954; 47:795–817.
- Wang AY, Ahmad NA. Rectal carcinoids. Curr Opin Gastroenterol 2006; 22:529–535.
- Modlin IM, Kidd M, Latich I, Zikusoka MN, Shapiro MD. Current status of gastrointestinal carcinoids. Gastroenterology 2005; 128:1717–1751.
- Aggarwal G, Obideen K, Wehbi M. Carcinoid tumors: what should increase our suspicion? Cleve Clin J Med 2008; 75:849–855.
- Hanley PH, Hines MO, Ray J, Armstrong R. Carcinoid tumors of the rectum. Experience with 26 cases. Proc R Soc Med 1959; 52(suppl):113–117.
- Pasieka JL. Carcinoid tumors. Surg Clin North Am 2009; 89:1123–1137.
- Jetmore AB, Ray JE, Gathright JB, McMullen KM, Hicks TC, Timmcke AE. Rectal carcinoids: the most frequent carcinoid tumor. Dis Colon Rectum 1992; 35:717–725.
- Scherübl H. Rectal carcinoids are on the rise: early detection by screening endoscopy. Endoscopy 2009; 41:162–165.
- Wilander E, Lundqvist M, Oberg K. Gastrointestinal carcinoid tumours. Histogenetic, histochemical, immunohistochemical, clinical and therapeutic aspects. Prog Histochem Cytochem 1989; 19:1–88.
- Maggard MA, O’Connell JB, Ko CY. Updated population-based review of carcinoid tumors. Ann Surg 2004; 240:117–122.
- Modlin IM, Sandor A. An analyisis of 8,305 cases of carcinoid tumors. Cancer 1997; 79:813–829.
- Wang M, Peng J, Yang W, Chen W, Mo S, Cai S. Prognostic analysis for carcinoid tumors of the rectum: a single institutional analysis of 106 cases. Colorectal Dis 2009; Epub ahead of print.
- Mani S, Modlin IM, Ballantyne G, Ahlman H, West B. Carcinoids of the rectum. J Am Coll Surg 1994; 179:231–248.
- Landry CS, Brock G, Scoggins CR, McMasters KM, Martin RC. A proposed staging system for rectal carcinoid tumors based on an analysis of 4701 patients. Surgery 2008; 144:460–466.
- Fahy BN, Tang LH, Klimstra D, et al. Carcinoid of the rectum risk stratification (CaRRs): a strategy for preoperative outcome assessment. Ann Surg Oncol 2007; 14:1735–1743.
- Shirouzu K, Isomoto H, Kakegawa T, Morimatsu M. Treatment of rectal carcinoid tumors. Am J Surg 1990; 160:262–265.
- Baek IH. Endoscopic submucosal dissection or conventional endoscopic mucosal resection is an effective and safe treatment for rectal carcinoid tumors: a retrospective study. J Laparoendosc Adv Surg Tech A 2010; 20:329–331.
- Yamaguchi N, Isomoto H, Nishiyama H, et al. Endoscopic submucosal dissection for rectal carcinoid tumors. Surg Endosc 2010; 24:504–508.
- Ramage JK, Goretzki PE, Manfredi R, et al; Frascati Consensus Conference participants. Consensus guidelines for the management of patients with digestive neuroendocrine tumours: well-differentiated colon and rectum tumour/carcinoma. Neuroendocrinology 2008; 87:31–39.
A 31-year-old man presents to the emergency department with abdominal pain and diarrhea, which began 4 days ago. The pain is in both of the lower quadrants, is crampy and persistent, and is relieved with bowel movements. He has been having watery stools five to six times per day, without frank blood.
He reports no fevers, chills, nausea, or vomiting, and he has never travelled outside the country. He underwent laparotomy 6 months ago for a gunshot wound. He takes no prescription drugs. He smokes and he drinks alcohol, and he says he has used heroin and oxycodone recreationally.
His blood pressure is 134/74 mm Hg, and he is afebrile. An abdominal examination reveals no mass or tenderness.
Results of a complete blood count, serum chemistry panel, and serum amylase level are normal. His lipase level is slightly elevated at 80 U/L (reference range 12–70). His stool is negative for Clostridium difficile toxin on enzyme immunoassay.
Computed tomography of the abdomen reveals diffuse pericolonic hyperemia and possible thickening of the rectosigmoid colon, raising the concern that he might have infectious or inflammatory colitis. The patient is admitted for further evaluation.
Colonoscopy to evaluate the abnormalities on computed tomography finds only a 5-mm submucosal nodule in the rectum (Figure 1). Biopsy of the nodule shows it to be a well-differentiated neuroendocrine neoplasm (carcinoid tumor). Random colon biopsy samples are normal.
The patient’s symptoms resolve over the next 24 hours without any treatment.
WHAT EXPLAINS THE PATIENT’S SYMPTOMS?
1. Which of the following best explains the patient’s clinical presentation?
- Narcotic withdrawal
- Carcinoid syndrome
- Viral gastroenteritis
- Acute pancreatitis
Viral gastroenteritis is common and affects people of all ages. The very young and the elderly are at higher risk of adverse outcomes, but few people die of it in the United States.
Our patient’s symptoms were consistent with viral gastroenteritis that resolved spontaneously while he received only supportive care.
Narcotic withdrawal can also cause watery stools and abdominal pain. However, this patient lacked other signs and symptoms of withdrawal, and his symptoms improved without any detoxification or maintenance treatment.
Pancreatitis. Although the patient had a mildly elevated lipase level, his lack of nausea and vomiting and the location of the pain were not consistent with acute pancreatitis.
Carcinoid syndrome. Carcinoid tumors are rare, typically indolent neuroendocrine neoplasms. The carcinoid syndrome consists of cutaneous flushing, gut hypermotility with diarrhea, and bronchospasm.1–5 Our patient did not have the full range of these symptoms. However, the presentation of carcinoid tumors varies broadly depending on the location, morphology, or biology of the tumor.6 Although our patient had diarrhea, his symptoms improved without any specific treatment. Rectal carcinoid tumors rarely cause diarrhea, and therefore the tumor noted on colonoscopy was almost certainly an incidental finding unrelated to his clinical presentation.
The classic symptoms are caused by production of 5-hydroxyindoleacetic acid, typically by a carcinoid tumor of the small bowel. Rectal carcinoids do not produce the 5-hydroxyindoleacetic acid responsible for this “malignant” serotonin-driven syndrome and are typically asymptomatic. When rectal carcinoid tumors are symptomatic, patients may have symptoms of local irritation or obstruction, such as hematochezia, constipation, other changes in bowel habits, rectal pain, pruritis ani, or weight loss.2,7
Nearly 50% of rectal carcinoid tumors are discovered incidentally. The National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) registry database documented a 10-fold increase in the incidence of rectal carcinoids in the last 35 years, attributed in part to an increase in screening colonoscopy.8 Furthermore, although studies of large national or multicenter databases have found that 65% to 80% of all rectal carcinoid tumors are smaller than 1.0 cm, 93.3% to 100% of those discovered on screening endoscopy were 1.0 cm or smaller.8
Rectal carcinoid tumors have a characteristic feel on digital examination, with a hard, “buckshot” consistency, and are freely mobile.5 They have also been described as firm, nodular, rubbery, yellow, submucosal, and polypoid.8
WHERE DO CARCINOID TUMORS TEND TO ARISE?
2. Which of the following sites is the most commonly recognized site of a primary carcinoid tumor?
- Small bowel
- Lung
- Liver
- Pancreas
- Rectum
The small bowel is the most common site.
Carcinoid tumors derive from neoplastic proliferation of cells of the diffuse neuroendocrine system. Therefore, they can be found anywhere neuroendocrine cells are present, commonly in the gastrointestinal tract, urogenital tract, and the bronchial epithelium.
Traditionally, neuroendocrine tumors were classified by their embryologic origin: foregut (including the respiratory tract, thymus, stomach, and pancreas), midgut (including the small intestine, appendix, and right colon), and hindgut (including the transverse, descending, and sigmoid colon and rectum). Functionally, this was sensible, as each class of tumors presented similarly due to the similar hormonal secretory products.2,3,9
A 2004 population-based review of the SEER database10 classified incidence rates of carcinoid tumors and their distribution throughout the body. Most (54.5%) were discovered in the gastrointestinal tract, and of these, 44.7% were in the small intestine, 19.6% were in the rectum, 16.7% were in the appendix, 10.6% were in the colon, 7.2% were in the stomach, and the remaining 1.2% were at other gastrointestinal sites. Nongastrointestinal sites included the lungs and bronchi (30.1%), pancreas (2.3%), female reproductive tract and ovaries (1.2%), biliary system (1.1%), and head and neck (0.4%).10
The incidence rates have increased and the distribution of sites in the body has changed over time. For example, the appendix was once considered the site of highest incidence, with tumors often discovered incidentally during surgical resection. However, these data were based on anecdotal or single-institution reports and so may have been subject to reporting bias. According to the SEER data, the small intestine is now the leading site, perhaps because of increased awareness or improved diagnostic technology and imaging.10,11
The liver is a common site of metastasis, but it is an exceptionally rare location for a primary tumor.
HOW SHOULD THIS PATIENT BE MANAGED?
3. What is the appropriate management of rectal carcinoid in this patient?
- Since the nodule is 1.0 cm or smaller, watchful waiting is acceptable
- Since the nodule is 1.0 cm or smaller, local excision is appropriate, and no follow-up is required
- Because all carcinoid tumors are potentially malignant, radical resection (eg, abdominal perineal resection) is appropriate
- Because all carcinoid tumors are potentially malignant, radical resection with chemotherapy with 5-fluorouracil (Adrucil) and doxorubicin (Adriamycin) is required
Since the nodule is 1.0 cm or smaller, local excision is appropriate, and no follow-up will be required. Rectal carcinoid tumors generally have a favorable prognosis, with a 5-year survival rate of 87.5%.10
PROGNOSIS DEPENDS ON TUMOR SIZE, OTHER FACTORS
Many studies have examined risk factors contributing to poor prognosis, and this is an area of active study. Early research categorized rectal carcinoid risk in terms of tumor diameter, and this is still widely used to guide management. As early as 1959, Hanley et al5 recognized that tumors that were likely to metastasize were often larger than 1 cm, had infiltrated the muscularis, or were ulcerated. Today, it is understood that only 3% to 10% of rectal carcinoids smaller than 1 cm metastasize, whereas 17% to 42% of those 1 to 2 cm and 60% to 80% of those larger than 2 cm do.2,8,12,13
However, size is not the only consideration. Wang et al12 showed that muscular invasion is an independent risk factor for survival, and that tumor diameter is a significant predictor of invasion and metastasis. Similarly, a metaanalysis by Mani et al13 recognized tumor size and muscularis invasion as the most important predictors of malignancy in these neoplasms.
To aid in predicting prognosis, staging systems have been developed from institutional or national registries. Landry et al14 developed a TNM (tumor, node, metastasis) staging system for rectal carcinoids, in which the T value was based on tumor size and degree of invasion. A group at Memorial-Sloan Kettering Cancer Center15 developed a system for risk stratification of carcinoid of the rectum that is based on tumor size, muscularis invasion, lymphovascular invasion, and the mitotic rate.
TREATMENT IS BY EXCISION
Despite these new prognostic systems, there is no new guidance on therapeutic management. Surgical therapy is still largely guided by tumor size.
Lesions smaller than 1 cm are resected endoscopically or by another local transanal technique.2,3,15,16 Standard endoscopic mucosal resection is performed, and recent studies have suggested that endoscopic submucosal dissection is as effective17 or even preferred, because it resects to the deeper submucosa (as the name suggests).18 This en bloc technique may be appropriate for lesions with evidence of local invasion.18 Other situations may call for deeper resection, such as transanal resection for higher lesions and full-thickness mucosal-muscularis resection.
Tumors 1 to 2 cm are currently evaluated for other factors such as ulceration and umbilication, which influence the choice of local vs radical resection. Otherwise, there is little guidance for tumors of 1 to 2 cm.
Tumors larger than 2 cm have a high risk of muscularis invasion and metastasis, and hence they are resected with wide margins and imaging is then used to evaluate for metastasis.8,19 In cases of metastasis, local resection is often palliative, providing local symptom relief.19
AN INCIDENTALLY DISCOVERED CASE; PATIENT LOST TO FOLLOW-UP
Our patient’s case is typical of rectal carcinoid in that it was discovered incidentally during colonoscopy. His clinical presentation was likely unrelated to his carcinoid tumor, and he improved without specific treatment. His symptoms resolved within 24 hours with supportive treatment and he was discharged.
Pathologic confirmation of carcinoid tumor occurred after his discharge. Despite persistent attempts to contact the patient, he never returned for a follow-up appointment.
TAKE-HOME POINTS
- Carcinoid tumors are rare neoplasms of neuroendocrine origin.
- Rectal carcinoids are the third most common carcinoid of the gastrointestinal tract.
- Most rectal carcinoids are asymptomatic.
- Diagnosis is most often incidental and histologic.
- Treatment is by excision.
- Prognosis is favorable for smaller carcinoids and depends on size (and therefore, invasion).
A 31-year-old man presents to the emergency department with abdominal pain and diarrhea, which began 4 days ago. The pain is in both of the lower quadrants, is crampy and persistent, and is relieved with bowel movements. He has been having watery stools five to six times per day, without frank blood.
He reports no fevers, chills, nausea, or vomiting, and he has never travelled outside the country. He underwent laparotomy 6 months ago for a gunshot wound. He takes no prescription drugs. He smokes and he drinks alcohol, and he says he has used heroin and oxycodone recreationally.
His blood pressure is 134/74 mm Hg, and he is afebrile. An abdominal examination reveals no mass or tenderness.
Results of a complete blood count, serum chemistry panel, and serum amylase level are normal. His lipase level is slightly elevated at 80 U/L (reference range 12–70). His stool is negative for Clostridium difficile toxin on enzyme immunoassay.
Computed tomography of the abdomen reveals diffuse pericolonic hyperemia and possible thickening of the rectosigmoid colon, raising the concern that he might have infectious or inflammatory colitis. The patient is admitted for further evaluation.
Colonoscopy to evaluate the abnormalities on computed tomography finds only a 5-mm submucosal nodule in the rectum (Figure 1). Biopsy of the nodule shows it to be a well-differentiated neuroendocrine neoplasm (carcinoid tumor). Random colon biopsy samples are normal.
The patient’s symptoms resolve over the next 24 hours without any treatment.
WHAT EXPLAINS THE PATIENT’S SYMPTOMS?
1. Which of the following best explains the patient’s clinical presentation?
- Narcotic withdrawal
- Carcinoid syndrome
- Viral gastroenteritis
- Acute pancreatitis
Viral gastroenteritis is common and affects people of all ages. The very young and the elderly are at higher risk of adverse outcomes, but few people die of it in the United States.
Our patient’s symptoms were consistent with viral gastroenteritis that resolved spontaneously while he received only supportive care.
Narcotic withdrawal can also cause watery stools and abdominal pain. However, this patient lacked other signs and symptoms of withdrawal, and his symptoms improved without any detoxification or maintenance treatment.
Pancreatitis. Although the patient had a mildly elevated lipase level, his lack of nausea and vomiting and the location of the pain were not consistent with acute pancreatitis.
Carcinoid syndrome. Carcinoid tumors are rare, typically indolent neuroendocrine neoplasms. The carcinoid syndrome consists of cutaneous flushing, gut hypermotility with diarrhea, and bronchospasm.1–5 Our patient did not have the full range of these symptoms. However, the presentation of carcinoid tumors varies broadly depending on the location, morphology, or biology of the tumor.6 Although our patient had diarrhea, his symptoms improved without any specific treatment. Rectal carcinoid tumors rarely cause diarrhea, and therefore the tumor noted on colonoscopy was almost certainly an incidental finding unrelated to his clinical presentation.
The classic symptoms are caused by production of 5-hydroxyindoleacetic acid, typically by a carcinoid tumor of the small bowel. Rectal carcinoids do not produce the 5-hydroxyindoleacetic acid responsible for this “malignant” serotonin-driven syndrome and are typically asymptomatic. When rectal carcinoid tumors are symptomatic, patients may have symptoms of local irritation or obstruction, such as hematochezia, constipation, other changes in bowel habits, rectal pain, pruritis ani, or weight loss.2,7
Nearly 50% of rectal carcinoid tumors are discovered incidentally. The National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) registry database documented a 10-fold increase in the incidence of rectal carcinoids in the last 35 years, attributed in part to an increase in screening colonoscopy.8 Furthermore, although studies of large national or multicenter databases have found that 65% to 80% of all rectal carcinoid tumors are smaller than 1.0 cm, 93.3% to 100% of those discovered on screening endoscopy were 1.0 cm or smaller.8
Rectal carcinoid tumors have a characteristic feel on digital examination, with a hard, “buckshot” consistency, and are freely mobile.5 They have also been described as firm, nodular, rubbery, yellow, submucosal, and polypoid.8
WHERE DO CARCINOID TUMORS TEND TO ARISE?
2. Which of the following sites is the most commonly recognized site of a primary carcinoid tumor?
- Small bowel
- Lung
- Liver
- Pancreas
- Rectum
The small bowel is the most common site.
Carcinoid tumors derive from neoplastic proliferation of cells of the diffuse neuroendocrine system. Therefore, they can be found anywhere neuroendocrine cells are present, commonly in the gastrointestinal tract, urogenital tract, and the bronchial epithelium.
Traditionally, neuroendocrine tumors were classified by their embryologic origin: foregut (including the respiratory tract, thymus, stomach, and pancreas), midgut (including the small intestine, appendix, and right colon), and hindgut (including the transverse, descending, and sigmoid colon and rectum). Functionally, this was sensible, as each class of tumors presented similarly due to the similar hormonal secretory products.2,3,9
A 2004 population-based review of the SEER database10 classified incidence rates of carcinoid tumors and their distribution throughout the body. Most (54.5%) were discovered in the gastrointestinal tract, and of these, 44.7% were in the small intestine, 19.6% were in the rectum, 16.7% were in the appendix, 10.6% were in the colon, 7.2% were in the stomach, and the remaining 1.2% were at other gastrointestinal sites. Nongastrointestinal sites included the lungs and bronchi (30.1%), pancreas (2.3%), female reproductive tract and ovaries (1.2%), biliary system (1.1%), and head and neck (0.4%).10
The incidence rates have increased and the distribution of sites in the body has changed over time. For example, the appendix was once considered the site of highest incidence, with tumors often discovered incidentally during surgical resection. However, these data were based on anecdotal or single-institution reports and so may have been subject to reporting bias. According to the SEER data, the small intestine is now the leading site, perhaps because of increased awareness or improved diagnostic technology and imaging.10,11
The liver is a common site of metastasis, but it is an exceptionally rare location for a primary tumor.
HOW SHOULD THIS PATIENT BE MANAGED?
3. What is the appropriate management of rectal carcinoid in this patient?
- Since the nodule is 1.0 cm or smaller, watchful waiting is acceptable
- Since the nodule is 1.0 cm or smaller, local excision is appropriate, and no follow-up is required
- Because all carcinoid tumors are potentially malignant, radical resection (eg, abdominal perineal resection) is appropriate
- Because all carcinoid tumors are potentially malignant, radical resection with chemotherapy with 5-fluorouracil (Adrucil) and doxorubicin (Adriamycin) is required
Since the nodule is 1.0 cm or smaller, local excision is appropriate, and no follow-up will be required. Rectal carcinoid tumors generally have a favorable prognosis, with a 5-year survival rate of 87.5%.10
PROGNOSIS DEPENDS ON TUMOR SIZE, OTHER FACTORS
Many studies have examined risk factors contributing to poor prognosis, and this is an area of active study. Early research categorized rectal carcinoid risk in terms of tumor diameter, and this is still widely used to guide management. As early as 1959, Hanley et al5 recognized that tumors that were likely to metastasize were often larger than 1 cm, had infiltrated the muscularis, or were ulcerated. Today, it is understood that only 3% to 10% of rectal carcinoids smaller than 1 cm metastasize, whereas 17% to 42% of those 1 to 2 cm and 60% to 80% of those larger than 2 cm do.2,8,12,13
However, size is not the only consideration. Wang et al12 showed that muscular invasion is an independent risk factor for survival, and that tumor diameter is a significant predictor of invasion and metastasis. Similarly, a metaanalysis by Mani et al13 recognized tumor size and muscularis invasion as the most important predictors of malignancy in these neoplasms.
To aid in predicting prognosis, staging systems have been developed from institutional or national registries. Landry et al14 developed a TNM (tumor, node, metastasis) staging system for rectal carcinoids, in which the T value was based on tumor size and degree of invasion. A group at Memorial-Sloan Kettering Cancer Center15 developed a system for risk stratification of carcinoid of the rectum that is based on tumor size, muscularis invasion, lymphovascular invasion, and the mitotic rate.
TREATMENT IS BY EXCISION
Despite these new prognostic systems, there is no new guidance on therapeutic management. Surgical therapy is still largely guided by tumor size.
Lesions smaller than 1 cm are resected endoscopically or by another local transanal technique.2,3,15,16 Standard endoscopic mucosal resection is performed, and recent studies have suggested that endoscopic submucosal dissection is as effective17 or even preferred, because it resects to the deeper submucosa (as the name suggests).18 This en bloc technique may be appropriate for lesions with evidence of local invasion.18 Other situations may call for deeper resection, such as transanal resection for higher lesions and full-thickness mucosal-muscularis resection.
Tumors 1 to 2 cm are currently evaluated for other factors such as ulceration and umbilication, which influence the choice of local vs radical resection. Otherwise, there is little guidance for tumors of 1 to 2 cm.
Tumors larger than 2 cm have a high risk of muscularis invasion and metastasis, and hence they are resected with wide margins and imaging is then used to evaluate for metastasis.8,19 In cases of metastasis, local resection is often palliative, providing local symptom relief.19
AN INCIDENTALLY DISCOVERED CASE; PATIENT LOST TO FOLLOW-UP
Our patient’s case is typical of rectal carcinoid in that it was discovered incidentally during colonoscopy. His clinical presentation was likely unrelated to his carcinoid tumor, and he improved without specific treatment. His symptoms resolved within 24 hours with supportive treatment and he was discharged.
Pathologic confirmation of carcinoid tumor occurred after his discharge. Despite persistent attempts to contact the patient, he never returned for a follow-up appointment.
TAKE-HOME POINTS
- Carcinoid tumors are rare neoplasms of neuroendocrine origin.
- Rectal carcinoids are the third most common carcinoid of the gastrointestinal tract.
- Most rectal carcinoids are asymptomatic.
- Diagnosis is most often incidental and histologic.
- Treatment is by excision.
- Prognosis is favorable for smaller carcinoids and depends on size (and therefore, invasion).
- Thorson A, Biorck G, Bjorkman G, Waldenstrom J. Malignant carcinoid of the small intestine with metastases to the liver, valvular disease of the right side of the heart (pulmonary stenosis and tricuspid regurgitation without septal defects), peripheral vasomotor symptoms, bronchoconstriction, and an unusual type of cyanosis; a clinical and pathologic syndrome. Am Heart J 1954; 47:795–817.
- Wang AY, Ahmad NA. Rectal carcinoids. Curr Opin Gastroenterol 2006; 22:529–535.
- Modlin IM, Kidd M, Latich I, Zikusoka MN, Shapiro MD. Current status of gastrointestinal carcinoids. Gastroenterology 2005; 128:1717–1751.
- Aggarwal G, Obideen K, Wehbi M. Carcinoid tumors: what should increase our suspicion? Cleve Clin J Med 2008; 75:849–855.
- Hanley PH, Hines MO, Ray J, Armstrong R. Carcinoid tumors of the rectum. Experience with 26 cases. Proc R Soc Med 1959; 52(suppl):113–117.
- Pasieka JL. Carcinoid tumors. Surg Clin North Am 2009; 89:1123–1137.
- Jetmore AB, Ray JE, Gathright JB, McMullen KM, Hicks TC, Timmcke AE. Rectal carcinoids: the most frequent carcinoid tumor. Dis Colon Rectum 1992; 35:717–725.
- Scherübl H. Rectal carcinoids are on the rise: early detection by screening endoscopy. Endoscopy 2009; 41:162–165.
- Wilander E, Lundqvist M, Oberg K. Gastrointestinal carcinoid tumours. Histogenetic, histochemical, immunohistochemical, clinical and therapeutic aspects. Prog Histochem Cytochem 1989; 19:1–88.
- Maggard MA, O’Connell JB, Ko CY. Updated population-based review of carcinoid tumors. Ann Surg 2004; 240:117–122.
- Modlin IM, Sandor A. An analyisis of 8,305 cases of carcinoid tumors. Cancer 1997; 79:813–829.
- Wang M, Peng J, Yang W, Chen W, Mo S, Cai S. Prognostic analysis for carcinoid tumors of the rectum: a single institutional analysis of 106 cases. Colorectal Dis 2009; Epub ahead of print.
- Mani S, Modlin IM, Ballantyne G, Ahlman H, West B. Carcinoids of the rectum. J Am Coll Surg 1994; 179:231–248.
- Landry CS, Brock G, Scoggins CR, McMasters KM, Martin RC. A proposed staging system for rectal carcinoid tumors based on an analysis of 4701 patients. Surgery 2008; 144:460–466.
- Fahy BN, Tang LH, Klimstra D, et al. Carcinoid of the rectum risk stratification (CaRRs): a strategy for preoperative outcome assessment. Ann Surg Oncol 2007; 14:1735–1743.
- Shirouzu K, Isomoto H, Kakegawa T, Morimatsu M. Treatment of rectal carcinoid tumors. Am J Surg 1990; 160:262–265.
- Baek IH. Endoscopic submucosal dissection or conventional endoscopic mucosal resection is an effective and safe treatment for rectal carcinoid tumors: a retrospective study. J Laparoendosc Adv Surg Tech A 2010; 20:329–331.
- Yamaguchi N, Isomoto H, Nishiyama H, et al. Endoscopic submucosal dissection for rectal carcinoid tumors. Surg Endosc 2010; 24:504–508.
- Ramage JK, Goretzki PE, Manfredi R, et al; Frascati Consensus Conference participants. Consensus guidelines for the management of patients with digestive neuroendocrine tumours: well-differentiated colon and rectum tumour/carcinoma. Neuroendocrinology 2008; 87:31–39.
- Thorson A, Biorck G, Bjorkman G, Waldenstrom J. Malignant carcinoid of the small intestine with metastases to the liver, valvular disease of the right side of the heart (pulmonary stenosis and tricuspid regurgitation without septal defects), peripheral vasomotor symptoms, bronchoconstriction, and an unusual type of cyanosis; a clinical and pathologic syndrome. Am Heart J 1954; 47:795–817.
- Wang AY, Ahmad NA. Rectal carcinoids. Curr Opin Gastroenterol 2006; 22:529–535.
- Modlin IM, Kidd M, Latich I, Zikusoka MN, Shapiro MD. Current status of gastrointestinal carcinoids. Gastroenterology 2005; 128:1717–1751.
- Aggarwal G, Obideen K, Wehbi M. Carcinoid tumors: what should increase our suspicion? Cleve Clin J Med 2008; 75:849–855.
- Hanley PH, Hines MO, Ray J, Armstrong R. Carcinoid tumors of the rectum. Experience with 26 cases. Proc R Soc Med 1959; 52(suppl):113–117.
- Pasieka JL. Carcinoid tumors. Surg Clin North Am 2009; 89:1123–1137.
- Jetmore AB, Ray JE, Gathright JB, McMullen KM, Hicks TC, Timmcke AE. Rectal carcinoids: the most frequent carcinoid tumor. Dis Colon Rectum 1992; 35:717–725.
- Scherübl H. Rectal carcinoids are on the rise: early detection by screening endoscopy. Endoscopy 2009; 41:162–165.
- Wilander E, Lundqvist M, Oberg K. Gastrointestinal carcinoid tumours. Histogenetic, histochemical, immunohistochemical, clinical and therapeutic aspects. Prog Histochem Cytochem 1989; 19:1–88.
- Maggard MA, O’Connell JB, Ko CY. Updated population-based review of carcinoid tumors. Ann Surg 2004; 240:117–122.
- Modlin IM, Sandor A. An analyisis of 8,305 cases of carcinoid tumors. Cancer 1997; 79:813–829.
- Wang M, Peng J, Yang W, Chen W, Mo S, Cai S. Prognostic analysis for carcinoid tumors of the rectum: a single institutional analysis of 106 cases. Colorectal Dis 2009; Epub ahead of print.
- Mani S, Modlin IM, Ballantyne G, Ahlman H, West B. Carcinoids of the rectum. J Am Coll Surg 1994; 179:231–248.
- Landry CS, Brock G, Scoggins CR, McMasters KM, Martin RC. A proposed staging system for rectal carcinoid tumors based on an analysis of 4701 patients. Surgery 2008; 144:460–466.
- Fahy BN, Tang LH, Klimstra D, et al. Carcinoid of the rectum risk stratification (CaRRs): a strategy for preoperative outcome assessment. Ann Surg Oncol 2007; 14:1735–1743.
- Shirouzu K, Isomoto H, Kakegawa T, Morimatsu M. Treatment of rectal carcinoid tumors. Am J Surg 1990; 160:262–265.
- Baek IH. Endoscopic submucosal dissection or conventional endoscopic mucosal resection is an effective and safe treatment for rectal carcinoid tumors: a retrospective study. J Laparoendosc Adv Surg Tech A 2010; 20:329–331.
- Yamaguchi N, Isomoto H, Nishiyama H, et al. Endoscopic submucosal dissection for rectal carcinoid tumors. Surg Endosc 2010; 24:504–508.
- Ramage JK, Goretzki PE, Manfredi R, et al; Frascati Consensus Conference participants. Consensus guidelines for the management of patients with digestive neuroendocrine tumours: well-differentiated colon and rectum tumour/carcinoma. Neuroendocrinology 2008; 87:31–39.
In reply: Diabetic ketoacidosis
In Reply: Dr. Jenkins brings up an important issue in his letter, and in fact we endorse his approach of ordering tests only if they will lead to a change in management. As we outlined in our case, clinical information alone strongly supported the diagnosis of type 2 diabetes mellitus.
In Reply: Dr. Jenkins brings up an important issue in his letter, and in fact we endorse his approach of ordering tests only if they will lead to a change in management. As we outlined in our case, clinical information alone strongly supported the diagnosis of type 2 diabetes mellitus.
In Reply: Dr. Jenkins brings up an important issue in his letter, and in fact we endorse his approach of ordering tests only if they will lead to a change in management. As we outlined in our case, clinical information alone strongly supported the diagnosis of type 2 diabetes mellitus.
A 48-year-old man with uncontrolled diabetes
A 48-year-old white man who has had diabetes mellitus for 6 years presents to the outpatient clinic because his blood sugar levels have been rising for the past week.
Both his parents had diabetes, and at the time of his diagnosis he weighed 278 pounds, all of which supported a diagnosis of type 2 diabetes mellitus. His disease was initially managed with diet, exercise, and metformin (Glucophage). Four months later, with weight loss and exercise, his blood sugar levels were consistently under 100 mg/dL, and metformin was discontinued.
He did well until 1 week ago, when he noted polyuria, polydipsia, and rising fingerstick glucose values, higher than 200 mg/dL. He has been eating well, with no nausea, vomiting, or symptoms of dehydration. He denies having any fever, chills, cough, nasal congestion, chest pain, abdominal pain, or dysuria.
In addition to his type 2 diabetes, he has hypertension, for which he takes losartan (Cozaar); hyperlipidemia, for which he takes atorvastatin (Lipitor); and gout, for which he takes allopurinol (Zyloprim).
His blood pressure is 148/70 mm Hg, pulse 100, and weight 273 pounds, and he is afebrile. On examination, his skin, head, eyes, ears, nose, throat, lungs, heart, and abdomen are normal. Urinalysis in the clinic shows large amounts of glucose and ketones.
WHAT IS THE LEAST LIKELY CAUSE OF HIS POOR CONTROL?
1. Which of the following is the least likely cause of his poorly controlled diabetes?
- Occult infection
- Poor adherence to diet and exercise
- Diabetic ketoacidosis
- Pancreatitis
Until 1 week ago, this patient’s diabetes had been well controlled for several years. Pancreatitis is the least likely cause of his uncontrolled diabetes, because he has no history of pancreatitis and has none of the symptoms of acute pancreatitis (fever, vomiting, or severe midepigastric pain radiating into the back).
Poor adherence to medication and lifestyle issues are very common in patients with poorly controlled diabetes and should always be included in the differential diagnosis.
Occult infection should also be considered in a patient with uncontrolled diabetes. Although this patient had no symptoms or signs of infection, urinalysis was done to look for an occult urinary tract infection and, surprisingly, it showed a large amount of ketones.
Case continued: He is treated for diabetic ketoacidosis
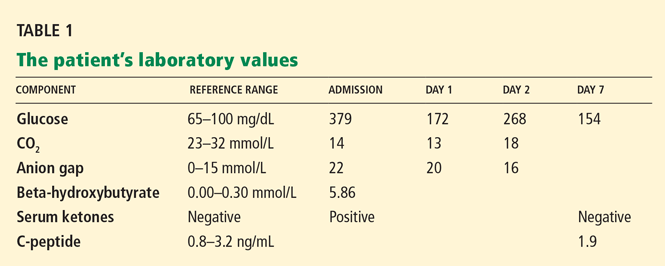
Diabetic ketoacidosis in ‘atypical diabetes’
Diabetic ketoacidosis is one of the most serious complications of diabetes. Many patients present with nausea, vomiting, and abdominal pain. Dehydration is often present because hyperglycemia leads to glucosuria and volume depletion. Interestingly, our patient showed none of these symptoms or signs.
Diabetic ketoacidosis is increasingly being recognized as a complication in patients with type 2 diabetes mellitus.1–4 Since the mid-1990s, clinicians have become increasingly aware of a condition variably termed “atypical diabetes,” “Flatbush diabetes,” “diabetes type 1B,” and “ketosis-prone type 2 diabetes mellitus,” in which patients, usually obese, present with diabetic ketoacidosis as their first manifestation, but are subsequently found to have type 2 diabetes mellitus. These patients typically are African American or of African, Hispanic, or Caribbean descent.
Ketoacidosis results from transient suppression of beta-cell function, the cause of which is unknown. A recent study comparing patients who have type 2 diabetes mellitus with and without diabetic ketoacidosis presenting with decompensated diabetes suggested insulinopenia was the predominant mechanism.5 For many of these patients, insulinopenia is transient: as the ketoacidosis resolves, betacell function improves and, with adequate insulin, lipolysis is reduced.
WHAT CAUSES DIABETIC KETOACIDOSIS?
2. Which of the following hormonal changes underlies the development of diabetic ketoacidosis?
- Insulin resistance
- Insulin deficiency
- Glucagon excess
- Glucagon deficiency
- Insulin deficiency and glucagon excess
- Insulin deficiency and glucagon deficiency
Diabetic ketoacidosis can occur when there is too much glucagon and not enough insulin. Insulin lowers the serum glucose level by promoting glucose uptake in peripheral tissues and by inhibiting gluconeogenesis and glycogenolysis in the liver. Insulin is also anabolic: it inhibits lipolysis in adipocytes and thus decreases the amount of substrate for ketogenesis.
Glucagon is the primary counterregulatory hormone responsible for ketogenesis.6 In the presence of glucagon excess, malonyl CoA production decreases, causing unblocking of carnitine acyltransferase I (CAT I) and allowing beta-oxidation to occur.6
Therefore, the sequence initiating ketogenesis begins with a shift in the ratio of glucagon to insulin, so that there is a relative or absolute excess of glucagon and a deficiency of insulin. A deficiency of insulin accelerates lipolysis, providing more substrate for ketogenesis, while excess glucagon turns on the oxidative sequence for fatty acids in the liver.
Three ketone bodies are produced in diabetic ketoacidosis: two ketoacids (beta-hydroxybutyric acid and acetoacetic acid), and one neutral ketone (acetone). The concentration of insulin required to suppress lipolysis is only one-tenth of that required to promote glucose utilization.7 Diabetic ketoacidosis is uncommon in patients with type 2 diabetes because they typically have enough insulin to inhibit lipolysis (and therefore ketoacid formation) but not enough to promote glucose utilization.
RISK FACTORS FOR DIABETIC KETOACIDOSIS
3. Which of the following is not a risk factor for diabetic ketoacidosis in type 2 diabetes mellitus?
- Acute illness
- Age > 65
- Inadequate insulin doses
- Antipsychotic drugs
- Ethnicity
Diabetic ketoacidosis is often precipitated by an acute illness such as an infection, cerebrovascular accident, myocardial infarction, or acute pancreatitis.8–12 These acute illnesses induce stress in the body and elevate counterregulatory hormones.
Inadequate insulin doses can also lead to diabetic ketoacidosis.
Drugs that affect carbohydrate metabolism are also risk factors. These include glucocorticoids, thiazide diuretics in high doses (> 50 mg daily), sympathomimetic agents, and second-generation antipsychotic agents (also called “atypical” antipsychotics) such as clozapine (Clozaril) and olanzapine (Zyprexa), although some are worse than others.13,14
Ketosis-prone type 2 diabetes mellitus is more prevalent in African Americans and Hispanics.8,15,16
Age is not a risk factor for developing diabetic ketoacidosis. In fact, diabetic ketoacidosis is the leading cause of morbidity and death in children with type 1 diabetes and can also occur in children with type 2 diabetes, particularly in obese African American adolescents.2
DISTINGUISHING TYPE 1 FROM TYPE 2
4. Which of the following is most specific in distinguishing type 1 from type 2 diabetes mellitus?
- C-peptide levels
- Islet cell antibodies
- Body mass index
- Family history
- Hemoglobin A1c level
Type 1 diabetes is characterized by destruction of pancreatic beta cells, leading to absolute insulin deficiency. The process is usually mediated by autoimmunity; therefore, testing for antibodies to islet cells, glutamic acid decarboxylase, insulin, and tyrosine phosphatase is the most specific way to distinguish type 1 from type 2 diabetes mellitus.
The hemoglobin A1c level correlates with the mean blood glucose level over the previous 8 to 12 weeks. The hemoglobin A1c is typically elevated in both type 1 and type 2 diabetes mellitus and therefore is not a useful distinguishing feature.
C-peptide is made when proinsulin is cleaved into insulin and C-peptide. It is released from endocytic vesicles with insulin in a one-to-one molar ratio. Thus, the level of C-peptide in the blood can show how much insulin is being made by the pancreas. C-peptide levels can help distinguish between type 1 and type 2 diabetes mellitus later in the course of the disease (levels are usually lower in a patient with type 1 diabetes), but they are not as useful early on because they can be normal early in the course of type 1 diabetes.17
A family history of diabetes is more common in type 2 diabetes, but patients with either type 1 or type 2 can have an affected close relative.
Patients with type 2 diabetes are generally overweight, with a body mass index greater than the 85th percentile for their age and sex. In contrast, patients with type 1 diabetes are usually not overweight and often have a recent history of weight loss. There are exceptions, however, and some patients with type 1 diabetes have an elevated body mass index, while some patients with type 2 diabetes are thin.
Although individually, C-peptide, family history, and body mass index are not very specific in distinguishing type 1 from type 2 diabetes mellitus, together they often give the clinician a good idea of the type of diabetes the patient has. In our case, although islet cell antibodies were not drawn, the normal C-peptide level, high body mass index, and family history all support a diagnosis of type 2 diabetes mellitus.
THE PATIENT CONTINUES TO DO WELL
The patient is discharged from the hospital on an insulin regimen. His blood sugar levels are closely monitored and remain near normal. Six months after the episode of diabetic ketoacidosis, his insulin is discontinued.
TAKE-HOME POINTS
Diabetic ketoacidosis is not unique to type 1 diabetes mellitus. It can occur in type 2, more commonly in patients who are nonwhite and who have precipitating factors such as acute illness, inadequate insulin treatment, or newly diagnosed diabetes. Clinicians should be aware of the possibility of diabetic ketoacidosis even in patients with type 2 diabetes who may not have these risk factors.
One approach to recognizing diabetic ketoacidosis better in patients with type 2 diabetes mellitus would include checking urine for ketones and serum electrolytes for high anion gap acidosis when patients with type 2 diabetes present with uncontrolled blood sugar levels. If ketonuria or acidosis is present, serum ketone and beta-hydroxybutyrate levels should be obtained to evaluate for diabetic ketoacidosis.
Patients should take insulin for an indeterminate period of time after initial treatment of diabetic ketoacidosis. As our case illustrates, in many cases, beta-cell function will return sufficiently to allow insulin to be discontinued. There are no clear guidelines for how long to continue insulin, but most practitioners continue it for weeks to months and discontinue it when glucose levels are stable and remain so with tapering doses. Sometimes oral agents need to be added as insulin is tapered.
Insulin therapy is tailored to the individual patient on the basis of blood glucose values. There are no data on which type of insulin is the most effective, and there are no data on whether these patients are at greater risk of hypoglycemia than other patients taking insulin. In general, there is no evidence that “prophylactic” insulin (ie, giving insulin to prevent diabetic ketoacidosis during times of illness or stress) is required. However, blood glucose monitoring is appropriate during infection or stress, and if hyperglycemia occurs in these situations, insulin use is prudent to reduce the risks of recurrent diabetic ketoacidosis.
- Umpierrez GE, Casals MM, Gebhart SP, Mixon PS, Clark WS, Phillips LS. Diabetic ketoacidosis in obese African-Americans. Diabetes 1995; 44:790–795.
- Valabhji J, Watson M, Cox J, Poulter C, Elwig C, Elkeles RS. Type 2 diabetes presenting as diabetic ketacidosis in adolescence. Diabet Med 2003; 20:416–417.
- Westphal SA. The occurrence of diabetic ketoacidosis in non-insulin-dependent diabetes and newly diagnosed diabetic adults. Am J Med 1996; 101:19–24.
- Welch B, Zib I. Case study: diabetic ketoacidosis in type 2 diabetes: “look under the sheets.” Clin Diabetes 2004; 22:198–200.
- Linfoot P, Bergstrom C, Ipp E. Pathophysiology of ketoacidosis in type 2 diabetes mellitus. Diabet Med 2005; 22:1414–1419.
- Foster DW, McGarry JD. The regulation of ketogenesis. Ciba Found Symp 1982; 87:120–131.
- Zierler KL, Rabinowitz D. Effect of very small concentrations of insulin on forearm metabolism: persistence of its action on potassium and free fatty acids without its effect on glucose. J Clin Invest 1964; 43:950–962.
- Newton CA, Raskin P. Diabetic ketoacidosis in type 1 and type 2 diabetes mellitus: clinical and biochemical differences. Arch Intern Med 2004; 164:1925–1931.
- Umpierrez GE, Kelly JP, Navarrete JE, Casals MM, Kitabchi AE. Hyperglycemic crises in urban blacks. Arch Intern Med 1997; 157:669–675.
- Jabbour SA, Miller JL. Uncontrolled diabetes mellitus. Clin Lab Med 2001; 21:99–110.
- Ennis ED, Kreisberg RA. Diabetic ketoacidosis and the hyperglycemic hyperosmolar syndrome. In: Leroith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus. Lippincott-Raven Publishers; Philadelphia, 1996:276–286.
- Case CC, Maldonado M. Diabetic ketoacidosis associated with Metabolife: a report of two cases. Diabetes Obes Metab 2002; 4:402–406.
- Kitabchi AE, Umpierrez GE, Murphy MB. Diabetic ketoacidosis and hyperglycemic hyperosmolar state. In:DeFronzo RA, Ferrannini E, Keen H, Zimmet P, editors. International Textbook of Diabetes Mellitus, 3rd ed. John Wiley and Sons, Ltd: Chichester, UK, 2004:1101–1119.
- Newcomer JW. Second generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs 2005; 19( suppl 1):1–93.
- Balasubramanyam A, Zern JW, Hyman DJ, Pavlik V. New profiles of diabetic ketoacidosis: type 1 vs. type 2 diabetes and the effect of ethnicity. Arch Intern Med 1999; 159:2317–2322.
- Davis SN, Umpierrez GE. Diabetic ketoacidosis in type 2 diabetes mellitus—pathophsyiology and clinical presentation. Nat Clin Pract Endocrinol Metab 2007; 3:730–731.
- Hoogwerf B, Rich S, Barbosa J. Meal-stimulated Cpeptide and insulin antibodies in type I diabetic subjects and their nondiabetic siblings characterized by HLA-DR antigens. Diabetes 1985; 34:440–445.
A 48-year-old white man who has had diabetes mellitus for 6 years presents to the outpatient clinic because his blood sugar levels have been rising for the past week.
Both his parents had diabetes, and at the time of his diagnosis he weighed 278 pounds, all of which supported a diagnosis of type 2 diabetes mellitus. His disease was initially managed with diet, exercise, and metformin (Glucophage). Four months later, with weight loss and exercise, his blood sugar levels were consistently under 100 mg/dL, and metformin was discontinued.
He did well until 1 week ago, when he noted polyuria, polydipsia, and rising fingerstick glucose values, higher than 200 mg/dL. He has been eating well, with no nausea, vomiting, or symptoms of dehydration. He denies having any fever, chills, cough, nasal congestion, chest pain, abdominal pain, or dysuria.
In addition to his type 2 diabetes, he has hypertension, for which he takes losartan (Cozaar); hyperlipidemia, for which he takes atorvastatin (Lipitor); and gout, for which he takes allopurinol (Zyloprim).
His blood pressure is 148/70 mm Hg, pulse 100, and weight 273 pounds, and he is afebrile. On examination, his skin, head, eyes, ears, nose, throat, lungs, heart, and abdomen are normal. Urinalysis in the clinic shows large amounts of glucose and ketones.
WHAT IS THE LEAST LIKELY CAUSE OF HIS POOR CONTROL?
1. Which of the following is the least likely cause of his poorly controlled diabetes?
- Occult infection
- Poor adherence to diet and exercise
- Diabetic ketoacidosis
- Pancreatitis
Until 1 week ago, this patient’s diabetes had been well controlled for several years. Pancreatitis is the least likely cause of his uncontrolled diabetes, because he has no history of pancreatitis and has none of the symptoms of acute pancreatitis (fever, vomiting, or severe midepigastric pain radiating into the back).
Poor adherence to medication and lifestyle issues are very common in patients with poorly controlled diabetes and should always be included in the differential diagnosis.
Occult infection should also be considered in a patient with uncontrolled diabetes. Although this patient had no symptoms or signs of infection, urinalysis was done to look for an occult urinary tract infection and, surprisingly, it showed a large amount of ketones.
Case continued: He is treated for diabetic ketoacidosis
Diabetic ketoacidosis in ‘atypical diabetes’
Diabetic ketoacidosis is one of the most serious complications of diabetes. Many patients present with nausea, vomiting, and abdominal pain. Dehydration is often present because hyperglycemia leads to glucosuria and volume depletion. Interestingly, our patient showed none of these symptoms or signs.
Diabetic ketoacidosis is increasingly being recognized as a complication in patients with type 2 diabetes mellitus.1–4 Since the mid-1990s, clinicians have become increasingly aware of a condition variably termed “atypical diabetes,” “Flatbush diabetes,” “diabetes type 1B,” and “ketosis-prone type 2 diabetes mellitus,” in which patients, usually obese, present with diabetic ketoacidosis as their first manifestation, but are subsequently found to have type 2 diabetes mellitus. These patients typically are African American or of African, Hispanic, or Caribbean descent.
Ketoacidosis results from transient suppression of beta-cell function, the cause of which is unknown. A recent study comparing patients who have type 2 diabetes mellitus with and without diabetic ketoacidosis presenting with decompensated diabetes suggested insulinopenia was the predominant mechanism.5 For many of these patients, insulinopenia is transient: as the ketoacidosis resolves, betacell function improves and, with adequate insulin, lipolysis is reduced.
WHAT CAUSES DIABETIC KETOACIDOSIS?
2. Which of the following hormonal changes underlies the development of diabetic ketoacidosis?
- Insulin resistance
- Insulin deficiency
- Glucagon excess
- Glucagon deficiency
- Insulin deficiency and glucagon excess
- Insulin deficiency and glucagon deficiency
Diabetic ketoacidosis can occur when there is too much glucagon and not enough insulin. Insulin lowers the serum glucose level by promoting glucose uptake in peripheral tissues and by inhibiting gluconeogenesis and glycogenolysis in the liver. Insulin is also anabolic: it inhibits lipolysis in adipocytes and thus decreases the amount of substrate for ketogenesis.
Glucagon is the primary counterregulatory hormone responsible for ketogenesis.6 In the presence of glucagon excess, malonyl CoA production decreases, causing unblocking of carnitine acyltransferase I (CAT I) and allowing beta-oxidation to occur.6
Therefore, the sequence initiating ketogenesis begins with a shift in the ratio of glucagon to insulin, so that there is a relative or absolute excess of glucagon and a deficiency of insulin. A deficiency of insulin accelerates lipolysis, providing more substrate for ketogenesis, while excess glucagon turns on the oxidative sequence for fatty acids in the liver.
Three ketone bodies are produced in diabetic ketoacidosis: two ketoacids (beta-hydroxybutyric acid and acetoacetic acid), and one neutral ketone (acetone). The concentration of insulin required to suppress lipolysis is only one-tenth of that required to promote glucose utilization.7 Diabetic ketoacidosis is uncommon in patients with type 2 diabetes because they typically have enough insulin to inhibit lipolysis (and therefore ketoacid formation) but not enough to promote glucose utilization.
RISK FACTORS FOR DIABETIC KETOACIDOSIS
3. Which of the following is not a risk factor for diabetic ketoacidosis in type 2 diabetes mellitus?
- Acute illness
- Age > 65
- Inadequate insulin doses
- Antipsychotic drugs
- Ethnicity
Diabetic ketoacidosis is often precipitated by an acute illness such as an infection, cerebrovascular accident, myocardial infarction, or acute pancreatitis.8–12 These acute illnesses induce stress in the body and elevate counterregulatory hormones.
Inadequate insulin doses can also lead to diabetic ketoacidosis.
Drugs that affect carbohydrate metabolism are also risk factors. These include glucocorticoids, thiazide diuretics in high doses (> 50 mg daily), sympathomimetic agents, and second-generation antipsychotic agents (also called “atypical” antipsychotics) such as clozapine (Clozaril) and olanzapine (Zyprexa), although some are worse than others.13,14
Ketosis-prone type 2 diabetes mellitus is more prevalent in African Americans and Hispanics.8,15,16
Age is not a risk factor for developing diabetic ketoacidosis. In fact, diabetic ketoacidosis is the leading cause of morbidity and death in children with type 1 diabetes and can also occur in children with type 2 diabetes, particularly in obese African American adolescents.2
DISTINGUISHING TYPE 1 FROM TYPE 2
4. Which of the following is most specific in distinguishing type 1 from type 2 diabetes mellitus?
- C-peptide levels
- Islet cell antibodies
- Body mass index
- Family history
- Hemoglobin A1c level
Type 1 diabetes is characterized by destruction of pancreatic beta cells, leading to absolute insulin deficiency. The process is usually mediated by autoimmunity; therefore, testing for antibodies to islet cells, glutamic acid decarboxylase, insulin, and tyrosine phosphatase is the most specific way to distinguish type 1 from type 2 diabetes mellitus.
The hemoglobin A1c level correlates with the mean blood glucose level over the previous 8 to 12 weeks. The hemoglobin A1c is typically elevated in both type 1 and type 2 diabetes mellitus and therefore is not a useful distinguishing feature.
C-peptide is made when proinsulin is cleaved into insulin and C-peptide. It is released from endocytic vesicles with insulin in a one-to-one molar ratio. Thus, the level of C-peptide in the blood can show how much insulin is being made by the pancreas. C-peptide levels can help distinguish between type 1 and type 2 diabetes mellitus later in the course of the disease (levels are usually lower in a patient with type 1 diabetes), but they are not as useful early on because they can be normal early in the course of type 1 diabetes.17
A family history of diabetes is more common in type 2 diabetes, but patients with either type 1 or type 2 can have an affected close relative.
Patients with type 2 diabetes are generally overweight, with a body mass index greater than the 85th percentile for their age and sex. In contrast, patients with type 1 diabetes are usually not overweight and often have a recent history of weight loss. There are exceptions, however, and some patients with type 1 diabetes have an elevated body mass index, while some patients with type 2 diabetes are thin.
Although individually, C-peptide, family history, and body mass index are not very specific in distinguishing type 1 from type 2 diabetes mellitus, together they often give the clinician a good idea of the type of diabetes the patient has. In our case, although islet cell antibodies were not drawn, the normal C-peptide level, high body mass index, and family history all support a diagnosis of type 2 diabetes mellitus.
THE PATIENT CONTINUES TO DO WELL
The patient is discharged from the hospital on an insulin regimen. His blood sugar levels are closely monitored and remain near normal. Six months after the episode of diabetic ketoacidosis, his insulin is discontinued.
TAKE-HOME POINTS
Diabetic ketoacidosis is not unique to type 1 diabetes mellitus. It can occur in type 2, more commonly in patients who are nonwhite and who have precipitating factors such as acute illness, inadequate insulin treatment, or newly diagnosed diabetes. Clinicians should be aware of the possibility of diabetic ketoacidosis even in patients with type 2 diabetes who may not have these risk factors.
One approach to recognizing diabetic ketoacidosis better in patients with type 2 diabetes mellitus would include checking urine for ketones and serum electrolytes for high anion gap acidosis when patients with type 2 diabetes present with uncontrolled blood sugar levels. If ketonuria or acidosis is present, serum ketone and beta-hydroxybutyrate levels should be obtained to evaluate for diabetic ketoacidosis.
Patients should take insulin for an indeterminate period of time after initial treatment of diabetic ketoacidosis. As our case illustrates, in many cases, beta-cell function will return sufficiently to allow insulin to be discontinued. There are no clear guidelines for how long to continue insulin, but most practitioners continue it for weeks to months and discontinue it when glucose levels are stable and remain so with tapering doses. Sometimes oral agents need to be added as insulin is tapered.
Insulin therapy is tailored to the individual patient on the basis of blood glucose values. There are no data on which type of insulin is the most effective, and there are no data on whether these patients are at greater risk of hypoglycemia than other patients taking insulin. In general, there is no evidence that “prophylactic” insulin (ie, giving insulin to prevent diabetic ketoacidosis during times of illness or stress) is required. However, blood glucose monitoring is appropriate during infection or stress, and if hyperglycemia occurs in these situations, insulin use is prudent to reduce the risks of recurrent diabetic ketoacidosis.
A 48-year-old white man who has had diabetes mellitus for 6 years presents to the outpatient clinic because his blood sugar levels have been rising for the past week.
Both his parents had diabetes, and at the time of his diagnosis he weighed 278 pounds, all of which supported a diagnosis of type 2 diabetes mellitus. His disease was initially managed with diet, exercise, and metformin (Glucophage). Four months later, with weight loss and exercise, his blood sugar levels were consistently under 100 mg/dL, and metformin was discontinued.
He did well until 1 week ago, when he noted polyuria, polydipsia, and rising fingerstick glucose values, higher than 200 mg/dL. He has been eating well, with no nausea, vomiting, or symptoms of dehydration. He denies having any fever, chills, cough, nasal congestion, chest pain, abdominal pain, or dysuria.
In addition to his type 2 diabetes, he has hypertension, for which he takes losartan (Cozaar); hyperlipidemia, for which he takes atorvastatin (Lipitor); and gout, for which he takes allopurinol (Zyloprim).
His blood pressure is 148/70 mm Hg, pulse 100, and weight 273 pounds, and he is afebrile. On examination, his skin, head, eyes, ears, nose, throat, lungs, heart, and abdomen are normal. Urinalysis in the clinic shows large amounts of glucose and ketones.
WHAT IS THE LEAST LIKELY CAUSE OF HIS POOR CONTROL?
1. Which of the following is the least likely cause of his poorly controlled diabetes?
- Occult infection
- Poor adherence to diet and exercise
- Diabetic ketoacidosis
- Pancreatitis
Until 1 week ago, this patient’s diabetes had been well controlled for several years. Pancreatitis is the least likely cause of his uncontrolled diabetes, because he has no history of pancreatitis and has none of the symptoms of acute pancreatitis (fever, vomiting, or severe midepigastric pain radiating into the back).
Poor adherence to medication and lifestyle issues are very common in patients with poorly controlled diabetes and should always be included in the differential diagnosis.
Occult infection should also be considered in a patient with uncontrolled diabetes. Although this patient had no symptoms or signs of infection, urinalysis was done to look for an occult urinary tract infection and, surprisingly, it showed a large amount of ketones.
Case continued: He is treated for diabetic ketoacidosis
Diabetic ketoacidosis in ‘atypical diabetes’
Diabetic ketoacidosis is one of the most serious complications of diabetes. Many patients present with nausea, vomiting, and abdominal pain. Dehydration is often present because hyperglycemia leads to glucosuria and volume depletion. Interestingly, our patient showed none of these symptoms or signs.
Diabetic ketoacidosis is increasingly being recognized as a complication in patients with type 2 diabetes mellitus.1–4 Since the mid-1990s, clinicians have become increasingly aware of a condition variably termed “atypical diabetes,” “Flatbush diabetes,” “diabetes type 1B,” and “ketosis-prone type 2 diabetes mellitus,” in which patients, usually obese, present with diabetic ketoacidosis as their first manifestation, but are subsequently found to have type 2 diabetes mellitus. These patients typically are African American or of African, Hispanic, or Caribbean descent.
Ketoacidosis results from transient suppression of beta-cell function, the cause of which is unknown. A recent study comparing patients who have type 2 diabetes mellitus with and without diabetic ketoacidosis presenting with decompensated diabetes suggested insulinopenia was the predominant mechanism.5 For many of these patients, insulinopenia is transient: as the ketoacidosis resolves, betacell function improves and, with adequate insulin, lipolysis is reduced.
WHAT CAUSES DIABETIC KETOACIDOSIS?
2. Which of the following hormonal changes underlies the development of diabetic ketoacidosis?
- Insulin resistance
- Insulin deficiency
- Glucagon excess
- Glucagon deficiency
- Insulin deficiency and glucagon excess
- Insulin deficiency and glucagon deficiency
Diabetic ketoacidosis can occur when there is too much glucagon and not enough insulin. Insulin lowers the serum glucose level by promoting glucose uptake in peripheral tissues and by inhibiting gluconeogenesis and glycogenolysis in the liver. Insulin is also anabolic: it inhibits lipolysis in adipocytes and thus decreases the amount of substrate for ketogenesis.
Glucagon is the primary counterregulatory hormone responsible for ketogenesis.6 In the presence of glucagon excess, malonyl CoA production decreases, causing unblocking of carnitine acyltransferase I (CAT I) and allowing beta-oxidation to occur.6
Therefore, the sequence initiating ketogenesis begins with a shift in the ratio of glucagon to insulin, so that there is a relative or absolute excess of glucagon and a deficiency of insulin. A deficiency of insulin accelerates lipolysis, providing more substrate for ketogenesis, while excess glucagon turns on the oxidative sequence for fatty acids in the liver.
Three ketone bodies are produced in diabetic ketoacidosis: two ketoacids (beta-hydroxybutyric acid and acetoacetic acid), and one neutral ketone (acetone). The concentration of insulin required to suppress lipolysis is only one-tenth of that required to promote glucose utilization.7 Diabetic ketoacidosis is uncommon in patients with type 2 diabetes because they typically have enough insulin to inhibit lipolysis (and therefore ketoacid formation) but not enough to promote glucose utilization.
RISK FACTORS FOR DIABETIC KETOACIDOSIS
3. Which of the following is not a risk factor for diabetic ketoacidosis in type 2 diabetes mellitus?
- Acute illness
- Age > 65
- Inadequate insulin doses
- Antipsychotic drugs
- Ethnicity
Diabetic ketoacidosis is often precipitated by an acute illness such as an infection, cerebrovascular accident, myocardial infarction, or acute pancreatitis.8–12 These acute illnesses induce stress in the body and elevate counterregulatory hormones.
Inadequate insulin doses can also lead to diabetic ketoacidosis.
Drugs that affect carbohydrate metabolism are also risk factors. These include glucocorticoids, thiazide diuretics in high doses (> 50 mg daily), sympathomimetic agents, and second-generation antipsychotic agents (also called “atypical” antipsychotics) such as clozapine (Clozaril) and olanzapine (Zyprexa), although some are worse than others.13,14
Ketosis-prone type 2 diabetes mellitus is more prevalent in African Americans and Hispanics.8,15,16
Age is not a risk factor for developing diabetic ketoacidosis. In fact, diabetic ketoacidosis is the leading cause of morbidity and death in children with type 1 diabetes and can also occur in children with type 2 diabetes, particularly in obese African American adolescents.2
DISTINGUISHING TYPE 1 FROM TYPE 2
4. Which of the following is most specific in distinguishing type 1 from type 2 diabetes mellitus?
- C-peptide levels
- Islet cell antibodies
- Body mass index
- Family history
- Hemoglobin A1c level
Type 1 diabetes is characterized by destruction of pancreatic beta cells, leading to absolute insulin deficiency. The process is usually mediated by autoimmunity; therefore, testing for antibodies to islet cells, glutamic acid decarboxylase, insulin, and tyrosine phosphatase is the most specific way to distinguish type 1 from type 2 diabetes mellitus.
The hemoglobin A1c level correlates with the mean blood glucose level over the previous 8 to 12 weeks. The hemoglobin A1c is typically elevated in both type 1 and type 2 diabetes mellitus and therefore is not a useful distinguishing feature.
C-peptide is made when proinsulin is cleaved into insulin and C-peptide. It is released from endocytic vesicles with insulin in a one-to-one molar ratio. Thus, the level of C-peptide in the blood can show how much insulin is being made by the pancreas. C-peptide levels can help distinguish between type 1 and type 2 diabetes mellitus later in the course of the disease (levels are usually lower in a patient with type 1 diabetes), but they are not as useful early on because they can be normal early in the course of type 1 diabetes.17
A family history of diabetes is more common in type 2 diabetes, but patients with either type 1 or type 2 can have an affected close relative.
Patients with type 2 diabetes are generally overweight, with a body mass index greater than the 85th percentile for their age and sex. In contrast, patients with type 1 diabetes are usually not overweight and often have a recent history of weight loss. There are exceptions, however, and some patients with type 1 diabetes have an elevated body mass index, while some patients with type 2 diabetes are thin.
Although individually, C-peptide, family history, and body mass index are not very specific in distinguishing type 1 from type 2 diabetes mellitus, together they often give the clinician a good idea of the type of diabetes the patient has. In our case, although islet cell antibodies were not drawn, the normal C-peptide level, high body mass index, and family history all support a diagnosis of type 2 diabetes mellitus.
THE PATIENT CONTINUES TO DO WELL
The patient is discharged from the hospital on an insulin regimen. His blood sugar levels are closely monitored and remain near normal. Six months after the episode of diabetic ketoacidosis, his insulin is discontinued.
TAKE-HOME POINTS
Diabetic ketoacidosis is not unique to type 1 diabetes mellitus. It can occur in type 2, more commonly in patients who are nonwhite and who have precipitating factors such as acute illness, inadequate insulin treatment, or newly diagnosed diabetes. Clinicians should be aware of the possibility of diabetic ketoacidosis even in patients with type 2 diabetes who may not have these risk factors.
One approach to recognizing diabetic ketoacidosis better in patients with type 2 diabetes mellitus would include checking urine for ketones and serum electrolytes for high anion gap acidosis when patients with type 2 diabetes present with uncontrolled blood sugar levels. If ketonuria or acidosis is present, serum ketone and beta-hydroxybutyrate levels should be obtained to evaluate for diabetic ketoacidosis.
Patients should take insulin for an indeterminate period of time after initial treatment of diabetic ketoacidosis. As our case illustrates, in many cases, beta-cell function will return sufficiently to allow insulin to be discontinued. There are no clear guidelines for how long to continue insulin, but most practitioners continue it for weeks to months and discontinue it when glucose levels are stable and remain so with tapering doses. Sometimes oral agents need to be added as insulin is tapered.
Insulin therapy is tailored to the individual patient on the basis of blood glucose values. There are no data on which type of insulin is the most effective, and there are no data on whether these patients are at greater risk of hypoglycemia than other patients taking insulin. In general, there is no evidence that “prophylactic” insulin (ie, giving insulin to prevent diabetic ketoacidosis during times of illness or stress) is required. However, blood glucose monitoring is appropriate during infection or stress, and if hyperglycemia occurs in these situations, insulin use is prudent to reduce the risks of recurrent diabetic ketoacidosis.
- Umpierrez GE, Casals MM, Gebhart SP, Mixon PS, Clark WS, Phillips LS. Diabetic ketoacidosis in obese African-Americans. Diabetes 1995; 44:790–795.
- Valabhji J, Watson M, Cox J, Poulter C, Elwig C, Elkeles RS. Type 2 diabetes presenting as diabetic ketacidosis in adolescence. Diabet Med 2003; 20:416–417.
- Westphal SA. The occurrence of diabetic ketoacidosis in non-insulin-dependent diabetes and newly diagnosed diabetic adults. Am J Med 1996; 101:19–24.
- Welch B, Zib I. Case study: diabetic ketoacidosis in type 2 diabetes: “look under the sheets.” Clin Diabetes 2004; 22:198–200.
- Linfoot P, Bergstrom C, Ipp E. Pathophysiology of ketoacidosis in type 2 diabetes mellitus. Diabet Med 2005; 22:1414–1419.
- Foster DW, McGarry JD. The regulation of ketogenesis. Ciba Found Symp 1982; 87:120–131.
- Zierler KL, Rabinowitz D. Effect of very small concentrations of insulin on forearm metabolism: persistence of its action on potassium and free fatty acids without its effect on glucose. J Clin Invest 1964; 43:950–962.
- Newton CA, Raskin P. Diabetic ketoacidosis in type 1 and type 2 diabetes mellitus: clinical and biochemical differences. Arch Intern Med 2004; 164:1925–1931.
- Umpierrez GE, Kelly JP, Navarrete JE, Casals MM, Kitabchi AE. Hyperglycemic crises in urban blacks. Arch Intern Med 1997; 157:669–675.
- Jabbour SA, Miller JL. Uncontrolled diabetes mellitus. Clin Lab Med 2001; 21:99–110.
- Ennis ED, Kreisberg RA. Diabetic ketoacidosis and the hyperglycemic hyperosmolar syndrome. In: Leroith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus. Lippincott-Raven Publishers; Philadelphia, 1996:276–286.
- Case CC, Maldonado M. Diabetic ketoacidosis associated with Metabolife: a report of two cases. Diabetes Obes Metab 2002; 4:402–406.
- Kitabchi AE, Umpierrez GE, Murphy MB. Diabetic ketoacidosis and hyperglycemic hyperosmolar state. In:DeFronzo RA, Ferrannini E, Keen H, Zimmet P, editors. International Textbook of Diabetes Mellitus, 3rd ed. John Wiley and Sons, Ltd: Chichester, UK, 2004:1101–1119.
- Newcomer JW. Second generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs 2005; 19( suppl 1):1–93.
- Balasubramanyam A, Zern JW, Hyman DJ, Pavlik V. New profiles of diabetic ketoacidosis: type 1 vs. type 2 diabetes and the effect of ethnicity. Arch Intern Med 1999; 159:2317–2322.
- Davis SN, Umpierrez GE. Diabetic ketoacidosis in type 2 diabetes mellitus—pathophsyiology and clinical presentation. Nat Clin Pract Endocrinol Metab 2007; 3:730–731.
- Hoogwerf B, Rich S, Barbosa J. Meal-stimulated Cpeptide and insulin antibodies in type I diabetic subjects and their nondiabetic siblings characterized by HLA-DR antigens. Diabetes 1985; 34:440–445.
- Umpierrez GE, Casals MM, Gebhart SP, Mixon PS, Clark WS, Phillips LS. Diabetic ketoacidosis in obese African-Americans. Diabetes 1995; 44:790–795.
- Valabhji J, Watson M, Cox J, Poulter C, Elwig C, Elkeles RS. Type 2 diabetes presenting as diabetic ketacidosis in adolescence. Diabet Med 2003; 20:416–417.
- Westphal SA. The occurrence of diabetic ketoacidosis in non-insulin-dependent diabetes and newly diagnosed diabetic adults. Am J Med 1996; 101:19–24.
- Welch B, Zib I. Case study: diabetic ketoacidosis in type 2 diabetes: “look under the sheets.” Clin Diabetes 2004; 22:198–200.
- Linfoot P, Bergstrom C, Ipp E. Pathophysiology of ketoacidosis in type 2 diabetes mellitus. Diabet Med 2005; 22:1414–1419.
- Foster DW, McGarry JD. The regulation of ketogenesis. Ciba Found Symp 1982; 87:120–131.
- Zierler KL, Rabinowitz D. Effect of very small concentrations of insulin on forearm metabolism: persistence of its action on potassium and free fatty acids without its effect on glucose. J Clin Invest 1964; 43:950–962.
- Newton CA, Raskin P. Diabetic ketoacidosis in type 1 and type 2 diabetes mellitus: clinical and biochemical differences. Arch Intern Med 2004; 164:1925–1931.
- Umpierrez GE, Kelly JP, Navarrete JE, Casals MM, Kitabchi AE. Hyperglycemic crises in urban blacks. Arch Intern Med 1997; 157:669–675.
- Jabbour SA, Miller JL. Uncontrolled diabetes mellitus. Clin Lab Med 2001; 21:99–110.
- Ennis ED, Kreisberg RA. Diabetic ketoacidosis and the hyperglycemic hyperosmolar syndrome. In: Leroith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus. Lippincott-Raven Publishers; Philadelphia, 1996:276–286.
- Case CC, Maldonado M. Diabetic ketoacidosis associated with Metabolife: a report of two cases. Diabetes Obes Metab 2002; 4:402–406.
- Kitabchi AE, Umpierrez GE, Murphy MB. Diabetic ketoacidosis and hyperglycemic hyperosmolar state. In:DeFronzo RA, Ferrannini E, Keen H, Zimmet P, editors. International Textbook of Diabetes Mellitus, 3rd ed. John Wiley and Sons, Ltd: Chichester, UK, 2004:1101–1119.
- Newcomer JW. Second generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs 2005; 19( suppl 1):1–93.
- Balasubramanyam A, Zern JW, Hyman DJ, Pavlik V. New profiles of diabetic ketoacidosis: type 1 vs. type 2 diabetes and the effect of ethnicity. Arch Intern Med 1999; 159:2317–2322.
- Davis SN, Umpierrez GE. Diabetic ketoacidosis in type 2 diabetes mellitus—pathophsyiology and clinical presentation. Nat Clin Pract Endocrinol Metab 2007; 3:730–731.
- Hoogwerf B, Rich S, Barbosa J. Meal-stimulated Cpeptide and insulin antibodies in type I diabetic subjects and their nondiabetic siblings characterized by HLA-DR antigens. Diabetes 1985; 34:440–445.
A case of refractory diarrhea
A 68-year-old white woman with irritable bowel syndrome has had worsening symptoms of right-sided abdominal pain, excessive bloating, and loose stools. Her bowel movements have increased from one a day to two or three a day. She has not noted any mucus or blood in the stool. She cannot identify any alleviating or aggravating factors, and the pain is not related to eating.
She consumes a normal diet, including meat and dairy. Over-the-counter antidiarrheal medications do not relieve the symptoms. She has had no fevers, chills, or night sweats, and she has not lost weight over the past year.
Her medical history includes breast cancer (in remission), alcohol abuse (in remission), and hypothyroidism, osteoporosis, and supraventricular tachycardia, all controlled with treatment as noted below. She has never undergone abdominal surgery.
A general review of systems is normal. Her current medications include oxybutynin (available as Ditropan, others), calcium polycarbophil (FiberCon, others), risedronate (Actonel), levothyroxine (Synthroid, others), simethicone (Maalox Anti-Gas, others), atenolol (Tenormin), trazodone (Desyrel), a calcium supplement, and aspirin. She began taking duloxetine (Cymbalta) 18 months ago, and the dose was increased from 60 mg to 90 mg 1 week before this visit.
She has never smoked, and she has abstained from alcohol for 10 years. She has no family history of colon cancer, celiac disease, or inflammatory bowel disease. She has not traveled outside the country in the past several years, and she notes no change in her source of drinking water.
On physical examination, she does not appear to be in acute distress. Her pulse is 64 and her blood pressure is 112/78 mm Hg. The cardiopulmonary examination is normal. Her abdomen is soft, symmetrical, nondistended, and nontender. Bowel sounds are normal. No abdominal masses, palpable organomegaly, or abdominal bruits are noted.
Results of basic laboratory tests, including thyroid-stimulating hormone (TSH), complete blood count, blood chemistries, renal function, and liver function, are normal. Colonoscopy shows normal mucosa as far as the cecum.
DIFFERENTIAL DIAGNOSIS
1. In addition to irritable bowel syndrome, which of these can explain her symptoms?
- Ulcerative colitis
- Celiac disease
- Microscopic colitis
- Hyperthyroidism
- Lactase deficiency
Ulcerative colitis typically presents with blood and mucus in the stool and gross abnormalities on colonoscopy, none of which is present in this patient.
Hyperthyroidism can be ruled out by the normal TSH level.
Lactase deficiency or lactose intolerance is unlikely because it is present in only 15% of people of northern European descent (compared with 80% of blacks and Hispanics and up to 100% of Native Americans and Asians).1 Furthermore, her pain is apparently not related to consuming dairy products.
The hydrogen breath test can aid in the diagnosis of lactase deficiency. This test relies on the breakdown of malabsorbed lactose by colonic flora. This is the most widely used test for this deficiency, but its high false-negative rate of 25% means that a negative result does not exclude the diagnosis and should not be relied on in working up a patient with chronic diarrhea.2 Simply noting whether symptoms develop after ingesting 50 g of lactose is clinically useful when lactase deficiency is suspected.
Based on the information so far, it is reasonable in this patient to evaluate for celiac disease and for microscopic colitis.
Celiac disease, also called gluten-sensitive enteropathy, has a varied presentation that includes nonspecific symptoms such as those in this patient. Classically, it causes diarrhea, but patients may present with a single nutrient deficiency and no diarrhea.
This patient lacks the elevated alkaline phosphatase or evidence of vitamin deficiencies characteristic of malabsorption in celiac disease (ie, vitamins A, B12, D, K, and folate)3. She also lacks evidence of malnutrition, such as iron deficiency anemia, weight loss, or low serum albumin. Finally, she does not have the dermatitis herpetiformis rash to suggest autoimmune gluten-sensitive enteropathy, nor does she have evidence of follicular hyperplasia or petechiae due to vitamin malabsorption.3
Because no single serologic test is ideal for diagnosing gluten-sensitive enteropathy, several tests are typically used: immunoglobulin A (IgA) antigliadin antibody, IgG antigliadin antibody, IgA antitransglutaminase antibody, and IgA antiendomysial antibody. IgA antitransglutaminase antibody is 92% to 98% sensitive and 91% to 100% specific for celiac disease. IgG antigliadin antibody is 92% to 97% sensitive and 99% specific. The positive predictive value of the IgA and IgG antigliadin antibody tests is less than 2% in the general population, whereas the positive predictive value for antiendomysial antibody and antitransglutaminase antibody are 15.7% and 21.8%, respectively.4 A positive serologic test for antiendomysial antibody is nearly 100% specific.
Our patient’s entire celiac antibody panel is negative, and thus celiac disease is unlikely.
Case continued: Features of microscopic colitis
In our patient, colonic biopsy reveals a mildly expanded lamina propria, intraepithelial lymphocytes, and a patchy but prominent thickening of the subepithelial collagen table. This set of features is consistent with collagenous colitis, a variant of microscopic colitis. Histologic signs on biopsy specimens are fairly specific for the disease.5
Chronic, intermittent, secretory diarrhea without bleeding is the hallmark of microscopic colitis. Associated symptoms may include abdominal pain, weight loss, and fatigue. If biopsies are not taken at the time of the initial evaluation, and the colonic pathology is overlooked, patients with collagenous colitis may be diagnosed with irritable bowel syndrome with diarrhea.6 The sedimentation rate is often elevated, and the antinuclear antibody test can be positive.7 Steatorrhea or protein-losing enteropathy can occur, and fecal leukocytes are present in more than 50% of patients.8
This patient fits well the demographics of the typical collagenous colitis patient: ie, a middle-aged woman in her 6th decade in otherwise good general health. The female-to-male ratio is 15:1 overall, although the relative frequency of collagenous colitis in women is greater than that of lymphocytic colitis.9 In a population-based study, the incidence of collagenous colitis was 5.1 per 100,000 per year, with a prevalence of 36 per 100,000; the incidence of lymphocytic colitis was 9.8 per 100,000 per year, with a prevalence of 64 per 100,000.10
Symptoms are typically vague and range from an annoyance to more than 20 non-bloody stools per day. The course of the disease also varies. Case series have reported a spontaneous remission rate of 15% to 20%,11 though flare-ups are common. Microscopic colitis is largely a benign disease. It does not increase a person’s risk of colon cancer.
CAUSES OF COLLAGENOUS COLITIS
2. What causes of collagenous colitis have been identified?
- Alcohol abuse
- Previous gastrointestinal surgery
- Drug-induced injury to colon
Neither alcohol use nor previous gastrointestinal surgery has been associated with the development of collagenous colitis.
Collagenous colitis has, however, been linked to several causes. Abnormal collagen metabolism has been demonstrated in patients as a result of increased expression of procollagen I and metalloproteinase inhibitor TIMP-1.12 Bacterial toxins and a bile-acid malabsorption defect in the terminal ileum and subsequent exposure of the colon to high concentrations of bile acids have also been linked to the development of collagenous colitis.
Many drugs have been linked to the development of collagenous colitis. Damage to the large intestine related to the use of non-steroidal anti-inflammatory drugs has been attributed to the blockage of prostaglandin synthesis.13 Simvastatin (Zocor), lansoprazole (Prilosec), and ticlopidine (Ticlid) have been linked to collagenous colitis; ticlopidine, flutamide (Eulexin), gold salts, lansoprazole, and sertraline (Zoloft) have been linked to the development of lymphocytic colitis.14 In one small series, patients developed colitis after switching from omeprazole (Prevacid) to lansoprazole. All patients had their symptoms and biopsy findings resolve within 1 week of stopping the drug.15
WHICH DRUG IS BEST?
3. Which drug is best for microscopic colitis, based on the current evidence?
- Bismuth (eg, Kaopectate, Pepto-Bismol)
- Sulfasalazine (Sulfazine)
- Budesonide (Entocort)
- Prednisolone
Studies have evaluated bismuth subsalicylate, Boswellia serrata extract, probiotics, prednisolone, budesonide, and other drugs for treating collagenous colitis.16
Bismuth trials have been small. In an open-label study of bismuth,17 symptoms improved in 11 of 12 patients.
Prednisolone recipients had a trend towards clinical response with treatment vs placebo, but it was not statistically significant, and there was incomplete remission of disease.18
Boswellia serrataextract19 and probiotics20 showed no clinical improvement.
Cholestyramine has been shown to be helpful when used in conjunction with an anti-inflammatory agent,21 and it may be helpful when used alone.
Aminosalicylate compounds have not been tested in prospective randomized trials, even though they are the cornerstone of treatment for ulcerative colitis. Retrospective trials have been equivocal.22
Budesonide currently has the best evidence of efficacy in collagenous colitis,23,24 and some evidence suggests it is also effective for other variants of microscopic colitis.
A total of 94 patients were enrolled in three placebo-controlled trials of budesonide at 9 mg daily or on a tapering schedule for 6 to 8 weeks. The pooled odds ratio for clinical response to treatment with budesonide was 12.32 (95% confidence interval 5.53–27.46), with a number needed to treat of 1.58. Significant histologic improvement with treatment was noted in all three trials.23
Quality of life has also been studied in patients with microscopic colitis who take budesonide. Symptoms, emotional functioning, and physical functioning are improved. Budesonide also improved stool consistency and significantly reduced the mean stool frequency compared with placebo.24
Compared with cortisol, budesonide has a 200 times greater affinity for the glucocorticoid receptor, and a 1,000 times greater topical anti-inflammatory potency. It is also well absorbed in the gastrointestinal tract but is substantially modified into very weak metabolites as a result of first-pass metabolism in the liver.25 This localized effect further supports the use of budesonide in patients with any form of microscopic colitis.
Although studies have shown budesonide to be effective, not every patient with a histologic diagnosis of microscopic colitis needs it. It is reasonable to try antidiarrheal agents, bismuth, or both as a first step because they are inexpensive and have few side effects. If budesonide is used, it should be given for 6 to 8 weeks, then stopped, and the patient should then be monitored for symptom recurrence. If a flare does occur, budesonide can be restarted and continued as maintenance therapy.
KEY CONSIDERATIONS
Microscopic colitis is diagnosed histologically, while irritable bowel syndrome is a clinical diagnosis. In population-based cohorts of histologically confirmed microscopic colitis, 50% to 70% met symptom-based Rome criteria for the diagnosis of irritable bowel syndrome. The clinical symptom-based criteria for irritable bowel syndrome are not specific enough to rule out the diagnosis of microscopic colitis. Therefore, patients with suspected diarrhea-predominant irritable bowel syndrome should undergo colonoscopy with biopsy to investigate microscopic colitis if symptoms are not well controlled by antidiarrheal therapy.26 The patient’s management may be very different depending on whether colonoscopy is done.
Management of microscopic colitis should include stopping any drugs associated with it. Simple antidiarrheal agents should be tried first to manage symptoms. If symptoms persist, patients can be treated with budesonide (Entocort EC) 9 mg by mouth daily for 8 weeks to induce remission, or 6 mg by mouth daily for 3 months as maintenance therapy.
OUR PATIENT’S COURSE
Our patient’s medication list includes duloxetine, a serotonin-norepinephrine reuptake inhibitor related to drugs that have been associated with the development of microscopic colitis. We tapered the duloxetine, and her symptoms improved by 50%. Her symptoms were eventually controlled after an 8-week course of oral budesonide 9 mg and ongoing intermittent use of loperamide (Imodium).
- Swagerty DL, Walling AD, Klein RM. Lactose intolerance. Am Fam Physician 2002; 65:1845–1856.
- Thomas PD, Forbes A, Green J, et al. Guidelines for the investigation of chronic diarrhea, 2nd edition. Gut 2003; 52(suppl 5):1–5.
- Nelsen DA. Gluten-sensitive enteropathy (celiac disease): more common than you think. Am Fam Physician 2002; 66:2259–2266.
- Bardella MT, Trovato C, Cesana BM, Pagliari C, Gebbia C, Peracchi M. Serological markers for coeliac disease: is it time to change? Dig Liver Dis 2001; 33:426–431.
- Barta Z, Mekkel G, Csipo I, et al. Micropscopic colitis: a retrospective study of clinical presentation in 53 patients. World J Gastroenterol 2005; 11:1351–1355.
- Tremaine WJ. Diagnosing collagenous colitis: does it make a difference? Eur J Gastroenterol Hepatol 1999; 11:477–479.
- Bohr J, Tysk C, Yang P, Danielsson D, Järnerot G. Autoantibodies and immunoglobulins in collagenous colitis. Gut 1996; 39:77–81.
- Zins BJ, Tremaine WJ, Carpenter HA. Collagenous colitis: mucosal biopsies and association with fecal leukocytes. Mayo Clin Proc 1995; 70:430–433.
- Olsen M, Eriksson S, Bohr J, Järnerot G, Tysk C. Lymphocytic colitis: a retrospective clinical study of 199 Swedish patients. Gut 2004; 53:536–541.
- Pardi DS. Microscopic colitis: an update. Inflamm Bowel Dis 2004; 10:860–870.
- Fernandez-Banares F, Salas A, Esteve M, Espinos J, Forne M, Viver JM. Collagenous and lymphocytic colitis: evaluation of clinical and histological features, response to treatment, and long-term follow-up. Am J Gastroenterol 2003; 98:340–347.
- Aignet T, Neureiter D, Müller S, Küspert G, Belke J, Kirchner T. Extracellular matrix composition and gene expression in collagenous colitis. Gastroenterology 1997; 113:136–143.
- Parfitt JR, Driman DK. Pathological effects of drugs on the gastrointestinal tract: a review. Hum Pathol 2007; 38:527–536.
- Fernández-Bañares F, Esteve M, Espinós JC, et al. Drug consumption and the risk of microscopic colitis. Am J Gastroenterol 2007; 102:324–330.
- Thomson RD, Lestine LS, Bensen SP, et al. Lansoprazole-associated microscopic colitis: a case series. Am J Gastroenterol 2002; 97:2908–2913.
- Chande N, McDonald JWD, MacDonald JK. Cochrane Inflammatory Bowel Disease and Functional Bowel Disorders Group. Interventions for treating collagenous colitis. Cochrane Database Syst Rev 2007 Jan 24;(1):CD006096.
- Fine K, Lee E. Efficacy of open-label bismuth subsalicylate for the treatment of microscopic colitis. Gastroenterology 1998; 114:29–36.
- Munck LK, Kjeldsen J, Philipsen E, Fscher Hansen B. Incomplete remission with short-term prednisolone treatment in collagenous colitis: a randomized study. Scand J Gastroenterol 2003; 38:606–610.
- Madisch A, Miehlke S, Eichele E, et al. Boswellia serrata extract for the treatment of collagenous colitis: a randomized, double-blind, placebo-controlled, multicenter trial. Int J Colorectal Dis 2007; 22:1445–1451.
- Wildt S, Munck LK, Vinter-Jensen L, et al. Probiotic treatment of collagenous colitis: a randomized, double-blind, placebo-controlled trial with Lactobacillus acidophilus and Bifidobacterium animalis subsp. lactis. Inflamm Bowel Dis 2006; 12:395–401.
- Calabrese C, Fabbri A, Areni A, Zahlane D, Scialpi C, Di Febo G. Mesalazine with or without cholestyramine in the treatment of microscopic colitis: randomized controlled trial. J Gastroenterol Hepatol 2007; 22:809–814.
- Wall GC, Schirmer LL, Page MJ. Pharmacotherapy for microscopic colitis. Pharmacotherapy 2007; 27:425–433.
- Feyen B, Wall GC, Finnerty EP, DeWitt JE, Reyes RS. Meta-analysis: budesonide treatment for collagenous colitis. Aliment Pharmacol Ther 2004; 20:745–749.
- Madisch A, Heymer P, Voss C, et al. Oral budesonide therapy improves quality of life in patients with collagenous colitis. Int J Colorectal Dis 2005; 20:312–316.
- Craig CR, editor. Modern Pharmacology With Clinical Application. 6th edition. Philadelphia: Lippincott Williams and Wilkins, 2003:481.
- Limsui D, Pardi DS, Camilleri M, et al. Symptomatic overlap between irritable bowel syndrome and microscopic colitis. Inflamm Bowel Dis 2007; 13:175–181.
A 68-year-old white woman with irritable bowel syndrome has had worsening symptoms of right-sided abdominal pain, excessive bloating, and loose stools. Her bowel movements have increased from one a day to two or three a day. She has not noted any mucus or blood in the stool. She cannot identify any alleviating or aggravating factors, and the pain is not related to eating.
She consumes a normal diet, including meat and dairy. Over-the-counter antidiarrheal medications do not relieve the symptoms. She has had no fevers, chills, or night sweats, and she has not lost weight over the past year.
Her medical history includes breast cancer (in remission), alcohol abuse (in remission), and hypothyroidism, osteoporosis, and supraventricular tachycardia, all controlled with treatment as noted below. She has never undergone abdominal surgery.
A general review of systems is normal. Her current medications include oxybutynin (available as Ditropan, others), calcium polycarbophil (FiberCon, others), risedronate (Actonel), levothyroxine (Synthroid, others), simethicone (Maalox Anti-Gas, others), atenolol (Tenormin), trazodone (Desyrel), a calcium supplement, and aspirin. She began taking duloxetine (Cymbalta) 18 months ago, and the dose was increased from 60 mg to 90 mg 1 week before this visit.
She has never smoked, and she has abstained from alcohol for 10 years. She has no family history of colon cancer, celiac disease, or inflammatory bowel disease. She has not traveled outside the country in the past several years, and she notes no change in her source of drinking water.
On physical examination, she does not appear to be in acute distress. Her pulse is 64 and her blood pressure is 112/78 mm Hg. The cardiopulmonary examination is normal. Her abdomen is soft, symmetrical, nondistended, and nontender. Bowel sounds are normal. No abdominal masses, palpable organomegaly, or abdominal bruits are noted.
Results of basic laboratory tests, including thyroid-stimulating hormone (TSH), complete blood count, blood chemistries, renal function, and liver function, are normal. Colonoscopy shows normal mucosa as far as the cecum.
DIFFERENTIAL DIAGNOSIS
1. In addition to irritable bowel syndrome, which of these can explain her symptoms?
- Ulcerative colitis
- Celiac disease
- Microscopic colitis
- Hyperthyroidism
- Lactase deficiency
Ulcerative colitis typically presents with blood and mucus in the stool and gross abnormalities on colonoscopy, none of which is present in this patient.
Hyperthyroidism can be ruled out by the normal TSH level.
Lactase deficiency or lactose intolerance is unlikely because it is present in only 15% of people of northern European descent (compared with 80% of blacks and Hispanics and up to 100% of Native Americans and Asians).1 Furthermore, her pain is apparently not related to consuming dairy products.
The hydrogen breath test can aid in the diagnosis of lactase deficiency. This test relies on the breakdown of malabsorbed lactose by colonic flora. This is the most widely used test for this deficiency, but its high false-negative rate of 25% means that a negative result does not exclude the diagnosis and should not be relied on in working up a patient with chronic diarrhea.2 Simply noting whether symptoms develop after ingesting 50 g of lactose is clinically useful when lactase deficiency is suspected.
Based on the information so far, it is reasonable in this patient to evaluate for celiac disease and for microscopic colitis.
Celiac disease, also called gluten-sensitive enteropathy, has a varied presentation that includes nonspecific symptoms such as those in this patient. Classically, it causes diarrhea, but patients may present with a single nutrient deficiency and no diarrhea.
This patient lacks the elevated alkaline phosphatase or evidence of vitamin deficiencies characteristic of malabsorption in celiac disease (ie, vitamins A, B12, D, K, and folate)3. She also lacks evidence of malnutrition, such as iron deficiency anemia, weight loss, or low serum albumin. Finally, she does not have the dermatitis herpetiformis rash to suggest autoimmune gluten-sensitive enteropathy, nor does she have evidence of follicular hyperplasia or petechiae due to vitamin malabsorption.3
Because no single serologic test is ideal for diagnosing gluten-sensitive enteropathy, several tests are typically used: immunoglobulin A (IgA) antigliadin antibody, IgG antigliadin antibody, IgA antitransglutaminase antibody, and IgA antiendomysial antibody. IgA antitransglutaminase antibody is 92% to 98% sensitive and 91% to 100% specific for celiac disease. IgG antigliadin antibody is 92% to 97% sensitive and 99% specific. The positive predictive value of the IgA and IgG antigliadin antibody tests is less than 2% in the general population, whereas the positive predictive value for antiendomysial antibody and antitransglutaminase antibody are 15.7% and 21.8%, respectively.4 A positive serologic test for antiendomysial antibody is nearly 100% specific.
Our patient’s entire celiac antibody panel is negative, and thus celiac disease is unlikely.
Case continued: Features of microscopic colitis
In our patient, colonic biopsy reveals a mildly expanded lamina propria, intraepithelial lymphocytes, and a patchy but prominent thickening of the subepithelial collagen table. This set of features is consistent with collagenous colitis, a variant of microscopic colitis. Histologic signs on biopsy specimens are fairly specific for the disease.5
Chronic, intermittent, secretory diarrhea without bleeding is the hallmark of microscopic colitis. Associated symptoms may include abdominal pain, weight loss, and fatigue. If biopsies are not taken at the time of the initial evaluation, and the colonic pathology is overlooked, patients with collagenous colitis may be diagnosed with irritable bowel syndrome with diarrhea.6 The sedimentation rate is often elevated, and the antinuclear antibody test can be positive.7 Steatorrhea or protein-losing enteropathy can occur, and fecal leukocytes are present in more than 50% of patients.8
This patient fits well the demographics of the typical collagenous colitis patient: ie, a middle-aged woman in her 6th decade in otherwise good general health. The female-to-male ratio is 15:1 overall, although the relative frequency of collagenous colitis in women is greater than that of lymphocytic colitis.9 In a population-based study, the incidence of collagenous colitis was 5.1 per 100,000 per year, with a prevalence of 36 per 100,000; the incidence of lymphocytic colitis was 9.8 per 100,000 per year, with a prevalence of 64 per 100,000.10
Symptoms are typically vague and range from an annoyance to more than 20 non-bloody stools per day. The course of the disease also varies. Case series have reported a spontaneous remission rate of 15% to 20%,11 though flare-ups are common. Microscopic colitis is largely a benign disease. It does not increase a person’s risk of colon cancer.
CAUSES OF COLLAGENOUS COLITIS
2. What causes of collagenous colitis have been identified?
- Alcohol abuse
- Previous gastrointestinal surgery
- Drug-induced injury to colon
Neither alcohol use nor previous gastrointestinal surgery has been associated with the development of collagenous colitis.
Collagenous colitis has, however, been linked to several causes. Abnormal collagen metabolism has been demonstrated in patients as a result of increased expression of procollagen I and metalloproteinase inhibitor TIMP-1.12 Bacterial toxins and a bile-acid malabsorption defect in the terminal ileum and subsequent exposure of the colon to high concentrations of bile acids have also been linked to the development of collagenous colitis.
Many drugs have been linked to the development of collagenous colitis. Damage to the large intestine related to the use of non-steroidal anti-inflammatory drugs has been attributed to the blockage of prostaglandin synthesis.13 Simvastatin (Zocor), lansoprazole (Prilosec), and ticlopidine (Ticlid) have been linked to collagenous colitis; ticlopidine, flutamide (Eulexin), gold salts, lansoprazole, and sertraline (Zoloft) have been linked to the development of lymphocytic colitis.14 In one small series, patients developed colitis after switching from omeprazole (Prevacid) to lansoprazole. All patients had their symptoms and biopsy findings resolve within 1 week of stopping the drug.15
WHICH DRUG IS BEST?
3. Which drug is best for microscopic colitis, based on the current evidence?
- Bismuth (eg, Kaopectate, Pepto-Bismol)
- Sulfasalazine (Sulfazine)
- Budesonide (Entocort)
- Prednisolone
Studies have evaluated bismuth subsalicylate, Boswellia serrata extract, probiotics, prednisolone, budesonide, and other drugs for treating collagenous colitis.16
Bismuth trials have been small. In an open-label study of bismuth,17 symptoms improved in 11 of 12 patients.
Prednisolone recipients had a trend towards clinical response with treatment vs placebo, but it was not statistically significant, and there was incomplete remission of disease.18
Boswellia serrataextract19 and probiotics20 showed no clinical improvement.
Cholestyramine has been shown to be helpful when used in conjunction with an anti-inflammatory agent,21 and it may be helpful when used alone.
Aminosalicylate compounds have not been tested in prospective randomized trials, even though they are the cornerstone of treatment for ulcerative colitis. Retrospective trials have been equivocal.22
Budesonide currently has the best evidence of efficacy in collagenous colitis,23,24 and some evidence suggests it is also effective for other variants of microscopic colitis.
A total of 94 patients were enrolled in three placebo-controlled trials of budesonide at 9 mg daily or on a tapering schedule for 6 to 8 weeks. The pooled odds ratio for clinical response to treatment with budesonide was 12.32 (95% confidence interval 5.53–27.46), with a number needed to treat of 1.58. Significant histologic improvement with treatment was noted in all three trials.23
Quality of life has also been studied in patients with microscopic colitis who take budesonide. Symptoms, emotional functioning, and physical functioning are improved. Budesonide also improved stool consistency and significantly reduced the mean stool frequency compared with placebo.24
Compared with cortisol, budesonide has a 200 times greater affinity for the glucocorticoid receptor, and a 1,000 times greater topical anti-inflammatory potency. It is also well absorbed in the gastrointestinal tract but is substantially modified into very weak metabolites as a result of first-pass metabolism in the liver.25 This localized effect further supports the use of budesonide in patients with any form of microscopic colitis.
Although studies have shown budesonide to be effective, not every patient with a histologic diagnosis of microscopic colitis needs it. It is reasonable to try antidiarrheal agents, bismuth, or both as a first step because they are inexpensive and have few side effects. If budesonide is used, it should be given for 6 to 8 weeks, then stopped, and the patient should then be monitored for symptom recurrence. If a flare does occur, budesonide can be restarted and continued as maintenance therapy.
KEY CONSIDERATIONS
Microscopic colitis is diagnosed histologically, while irritable bowel syndrome is a clinical diagnosis. In population-based cohorts of histologically confirmed microscopic colitis, 50% to 70% met symptom-based Rome criteria for the diagnosis of irritable bowel syndrome. The clinical symptom-based criteria for irritable bowel syndrome are not specific enough to rule out the diagnosis of microscopic colitis. Therefore, patients with suspected diarrhea-predominant irritable bowel syndrome should undergo colonoscopy with biopsy to investigate microscopic colitis if symptoms are not well controlled by antidiarrheal therapy.26 The patient’s management may be very different depending on whether colonoscopy is done.
Management of microscopic colitis should include stopping any drugs associated with it. Simple antidiarrheal agents should be tried first to manage symptoms. If symptoms persist, patients can be treated with budesonide (Entocort EC) 9 mg by mouth daily for 8 weeks to induce remission, or 6 mg by mouth daily for 3 months as maintenance therapy.
OUR PATIENT’S COURSE
Our patient’s medication list includes duloxetine, a serotonin-norepinephrine reuptake inhibitor related to drugs that have been associated with the development of microscopic colitis. We tapered the duloxetine, and her symptoms improved by 50%. Her symptoms were eventually controlled after an 8-week course of oral budesonide 9 mg and ongoing intermittent use of loperamide (Imodium).
A 68-year-old white woman with irritable bowel syndrome has had worsening symptoms of right-sided abdominal pain, excessive bloating, and loose stools. Her bowel movements have increased from one a day to two or three a day. She has not noted any mucus or blood in the stool. She cannot identify any alleviating or aggravating factors, and the pain is not related to eating.
She consumes a normal diet, including meat and dairy. Over-the-counter antidiarrheal medications do not relieve the symptoms. She has had no fevers, chills, or night sweats, and she has not lost weight over the past year.
Her medical history includes breast cancer (in remission), alcohol abuse (in remission), and hypothyroidism, osteoporosis, and supraventricular tachycardia, all controlled with treatment as noted below. She has never undergone abdominal surgery.
A general review of systems is normal. Her current medications include oxybutynin (available as Ditropan, others), calcium polycarbophil (FiberCon, others), risedronate (Actonel), levothyroxine (Synthroid, others), simethicone (Maalox Anti-Gas, others), atenolol (Tenormin), trazodone (Desyrel), a calcium supplement, and aspirin. She began taking duloxetine (Cymbalta) 18 months ago, and the dose was increased from 60 mg to 90 mg 1 week before this visit.
She has never smoked, and she has abstained from alcohol for 10 years. She has no family history of colon cancer, celiac disease, or inflammatory bowel disease. She has not traveled outside the country in the past several years, and she notes no change in her source of drinking water.
On physical examination, she does not appear to be in acute distress. Her pulse is 64 and her blood pressure is 112/78 mm Hg. The cardiopulmonary examination is normal. Her abdomen is soft, symmetrical, nondistended, and nontender. Bowel sounds are normal. No abdominal masses, palpable organomegaly, or abdominal bruits are noted.
Results of basic laboratory tests, including thyroid-stimulating hormone (TSH), complete blood count, blood chemistries, renal function, and liver function, are normal. Colonoscopy shows normal mucosa as far as the cecum.
DIFFERENTIAL DIAGNOSIS
1. In addition to irritable bowel syndrome, which of these can explain her symptoms?
- Ulcerative colitis
- Celiac disease
- Microscopic colitis
- Hyperthyroidism
- Lactase deficiency
Ulcerative colitis typically presents with blood and mucus in the stool and gross abnormalities on colonoscopy, none of which is present in this patient.
Hyperthyroidism can be ruled out by the normal TSH level.
Lactase deficiency or lactose intolerance is unlikely because it is present in only 15% of people of northern European descent (compared with 80% of blacks and Hispanics and up to 100% of Native Americans and Asians).1 Furthermore, her pain is apparently not related to consuming dairy products.
The hydrogen breath test can aid in the diagnosis of lactase deficiency. This test relies on the breakdown of malabsorbed lactose by colonic flora. This is the most widely used test for this deficiency, but its high false-negative rate of 25% means that a negative result does not exclude the diagnosis and should not be relied on in working up a patient with chronic diarrhea.2 Simply noting whether symptoms develop after ingesting 50 g of lactose is clinically useful when lactase deficiency is suspected.
Based on the information so far, it is reasonable in this patient to evaluate for celiac disease and for microscopic colitis.
Celiac disease, also called gluten-sensitive enteropathy, has a varied presentation that includes nonspecific symptoms such as those in this patient. Classically, it causes diarrhea, but patients may present with a single nutrient deficiency and no diarrhea.
This patient lacks the elevated alkaline phosphatase or evidence of vitamin deficiencies characteristic of malabsorption in celiac disease (ie, vitamins A, B12, D, K, and folate)3. She also lacks evidence of malnutrition, such as iron deficiency anemia, weight loss, or low serum albumin. Finally, she does not have the dermatitis herpetiformis rash to suggest autoimmune gluten-sensitive enteropathy, nor does she have evidence of follicular hyperplasia or petechiae due to vitamin malabsorption.3
Because no single serologic test is ideal for diagnosing gluten-sensitive enteropathy, several tests are typically used: immunoglobulin A (IgA) antigliadin antibody, IgG antigliadin antibody, IgA antitransglutaminase antibody, and IgA antiendomysial antibody. IgA antitransglutaminase antibody is 92% to 98% sensitive and 91% to 100% specific for celiac disease. IgG antigliadin antibody is 92% to 97% sensitive and 99% specific. The positive predictive value of the IgA and IgG antigliadin antibody tests is less than 2% in the general population, whereas the positive predictive value for antiendomysial antibody and antitransglutaminase antibody are 15.7% and 21.8%, respectively.4 A positive serologic test for antiendomysial antibody is nearly 100% specific.
Our patient’s entire celiac antibody panel is negative, and thus celiac disease is unlikely.
Case continued: Features of microscopic colitis
In our patient, colonic biopsy reveals a mildly expanded lamina propria, intraepithelial lymphocytes, and a patchy but prominent thickening of the subepithelial collagen table. This set of features is consistent with collagenous colitis, a variant of microscopic colitis. Histologic signs on biopsy specimens are fairly specific for the disease.5
Chronic, intermittent, secretory diarrhea without bleeding is the hallmark of microscopic colitis. Associated symptoms may include abdominal pain, weight loss, and fatigue. If biopsies are not taken at the time of the initial evaluation, and the colonic pathology is overlooked, patients with collagenous colitis may be diagnosed with irritable bowel syndrome with diarrhea.6 The sedimentation rate is often elevated, and the antinuclear antibody test can be positive.7 Steatorrhea or protein-losing enteropathy can occur, and fecal leukocytes are present in more than 50% of patients.8
This patient fits well the demographics of the typical collagenous colitis patient: ie, a middle-aged woman in her 6th decade in otherwise good general health. The female-to-male ratio is 15:1 overall, although the relative frequency of collagenous colitis in women is greater than that of lymphocytic colitis.9 In a population-based study, the incidence of collagenous colitis was 5.1 per 100,000 per year, with a prevalence of 36 per 100,000; the incidence of lymphocytic colitis was 9.8 per 100,000 per year, with a prevalence of 64 per 100,000.10
Symptoms are typically vague and range from an annoyance to more than 20 non-bloody stools per day. The course of the disease also varies. Case series have reported a spontaneous remission rate of 15% to 20%,11 though flare-ups are common. Microscopic colitis is largely a benign disease. It does not increase a person’s risk of colon cancer.
CAUSES OF COLLAGENOUS COLITIS
2. What causes of collagenous colitis have been identified?
- Alcohol abuse
- Previous gastrointestinal surgery
- Drug-induced injury to colon
Neither alcohol use nor previous gastrointestinal surgery has been associated with the development of collagenous colitis.
Collagenous colitis has, however, been linked to several causes. Abnormal collagen metabolism has been demonstrated in patients as a result of increased expression of procollagen I and metalloproteinase inhibitor TIMP-1.12 Bacterial toxins and a bile-acid malabsorption defect in the terminal ileum and subsequent exposure of the colon to high concentrations of bile acids have also been linked to the development of collagenous colitis.
Many drugs have been linked to the development of collagenous colitis. Damage to the large intestine related to the use of non-steroidal anti-inflammatory drugs has been attributed to the blockage of prostaglandin synthesis.13 Simvastatin (Zocor), lansoprazole (Prilosec), and ticlopidine (Ticlid) have been linked to collagenous colitis; ticlopidine, flutamide (Eulexin), gold salts, lansoprazole, and sertraline (Zoloft) have been linked to the development of lymphocytic colitis.14 In one small series, patients developed colitis after switching from omeprazole (Prevacid) to lansoprazole. All patients had their symptoms and biopsy findings resolve within 1 week of stopping the drug.15
WHICH DRUG IS BEST?
3. Which drug is best for microscopic colitis, based on the current evidence?
- Bismuth (eg, Kaopectate, Pepto-Bismol)
- Sulfasalazine (Sulfazine)
- Budesonide (Entocort)
- Prednisolone
Studies have evaluated bismuth subsalicylate, Boswellia serrata extract, probiotics, prednisolone, budesonide, and other drugs for treating collagenous colitis.16
Bismuth trials have been small. In an open-label study of bismuth,17 symptoms improved in 11 of 12 patients.
Prednisolone recipients had a trend towards clinical response with treatment vs placebo, but it was not statistically significant, and there was incomplete remission of disease.18
Boswellia serrataextract19 and probiotics20 showed no clinical improvement.
Cholestyramine has been shown to be helpful when used in conjunction with an anti-inflammatory agent,21 and it may be helpful when used alone.
Aminosalicylate compounds have not been tested in prospective randomized trials, even though they are the cornerstone of treatment for ulcerative colitis. Retrospective trials have been equivocal.22
Budesonide currently has the best evidence of efficacy in collagenous colitis,23,24 and some evidence suggests it is also effective for other variants of microscopic colitis.
A total of 94 patients were enrolled in three placebo-controlled trials of budesonide at 9 mg daily or on a tapering schedule for 6 to 8 weeks. The pooled odds ratio for clinical response to treatment with budesonide was 12.32 (95% confidence interval 5.53–27.46), with a number needed to treat of 1.58. Significant histologic improvement with treatment was noted in all three trials.23
Quality of life has also been studied in patients with microscopic colitis who take budesonide. Symptoms, emotional functioning, and physical functioning are improved. Budesonide also improved stool consistency and significantly reduced the mean stool frequency compared with placebo.24
Compared with cortisol, budesonide has a 200 times greater affinity for the glucocorticoid receptor, and a 1,000 times greater topical anti-inflammatory potency. It is also well absorbed in the gastrointestinal tract but is substantially modified into very weak metabolites as a result of first-pass metabolism in the liver.25 This localized effect further supports the use of budesonide in patients with any form of microscopic colitis.
Although studies have shown budesonide to be effective, not every patient with a histologic diagnosis of microscopic colitis needs it. It is reasonable to try antidiarrheal agents, bismuth, or both as a first step because they are inexpensive and have few side effects. If budesonide is used, it should be given for 6 to 8 weeks, then stopped, and the patient should then be monitored for symptom recurrence. If a flare does occur, budesonide can be restarted and continued as maintenance therapy.
KEY CONSIDERATIONS
Microscopic colitis is diagnosed histologically, while irritable bowel syndrome is a clinical diagnosis. In population-based cohorts of histologically confirmed microscopic colitis, 50% to 70% met symptom-based Rome criteria for the diagnosis of irritable bowel syndrome. The clinical symptom-based criteria for irritable bowel syndrome are not specific enough to rule out the diagnosis of microscopic colitis. Therefore, patients with suspected diarrhea-predominant irritable bowel syndrome should undergo colonoscopy with biopsy to investigate microscopic colitis if symptoms are not well controlled by antidiarrheal therapy.26 The patient’s management may be very different depending on whether colonoscopy is done.
Management of microscopic colitis should include stopping any drugs associated with it. Simple antidiarrheal agents should be tried first to manage symptoms. If symptoms persist, patients can be treated with budesonide (Entocort EC) 9 mg by mouth daily for 8 weeks to induce remission, or 6 mg by mouth daily for 3 months as maintenance therapy.
OUR PATIENT’S COURSE
Our patient’s medication list includes duloxetine, a serotonin-norepinephrine reuptake inhibitor related to drugs that have been associated with the development of microscopic colitis. We tapered the duloxetine, and her symptoms improved by 50%. Her symptoms were eventually controlled after an 8-week course of oral budesonide 9 mg and ongoing intermittent use of loperamide (Imodium).
- Swagerty DL, Walling AD, Klein RM. Lactose intolerance. Am Fam Physician 2002; 65:1845–1856.
- Thomas PD, Forbes A, Green J, et al. Guidelines for the investigation of chronic diarrhea, 2nd edition. Gut 2003; 52(suppl 5):1–5.
- Nelsen DA. Gluten-sensitive enteropathy (celiac disease): more common than you think. Am Fam Physician 2002; 66:2259–2266.
- Bardella MT, Trovato C, Cesana BM, Pagliari C, Gebbia C, Peracchi M. Serological markers for coeliac disease: is it time to change? Dig Liver Dis 2001; 33:426–431.
- Barta Z, Mekkel G, Csipo I, et al. Micropscopic colitis: a retrospective study of clinical presentation in 53 patients. World J Gastroenterol 2005; 11:1351–1355.
- Tremaine WJ. Diagnosing collagenous colitis: does it make a difference? Eur J Gastroenterol Hepatol 1999; 11:477–479.
- Bohr J, Tysk C, Yang P, Danielsson D, Järnerot G. Autoantibodies and immunoglobulins in collagenous colitis. Gut 1996; 39:77–81.
- Zins BJ, Tremaine WJ, Carpenter HA. Collagenous colitis: mucosal biopsies and association with fecal leukocytes. Mayo Clin Proc 1995; 70:430–433.
- Olsen M, Eriksson S, Bohr J, Järnerot G, Tysk C. Lymphocytic colitis: a retrospective clinical study of 199 Swedish patients. Gut 2004; 53:536–541.
- Pardi DS. Microscopic colitis: an update. Inflamm Bowel Dis 2004; 10:860–870.
- Fernandez-Banares F, Salas A, Esteve M, Espinos J, Forne M, Viver JM. Collagenous and lymphocytic colitis: evaluation of clinical and histological features, response to treatment, and long-term follow-up. Am J Gastroenterol 2003; 98:340–347.
- Aignet T, Neureiter D, Müller S, Küspert G, Belke J, Kirchner T. Extracellular matrix composition and gene expression in collagenous colitis. Gastroenterology 1997; 113:136–143.
- Parfitt JR, Driman DK. Pathological effects of drugs on the gastrointestinal tract: a review. Hum Pathol 2007; 38:527–536.
- Fernández-Bañares F, Esteve M, Espinós JC, et al. Drug consumption and the risk of microscopic colitis. Am J Gastroenterol 2007; 102:324–330.
- Thomson RD, Lestine LS, Bensen SP, et al. Lansoprazole-associated microscopic colitis: a case series. Am J Gastroenterol 2002; 97:2908–2913.
- Chande N, McDonald JWD, MacDonald JK. Cochrane Inflammatory Bowel Disease and Functional Bowel Disorders Group. Interventions for treating collagenous colitis. Cochrane Database Syst Rev 2007 Jan 24;(1):CD006096.
- Fine K, Lee E. Efficacy of open-label bismuth subsalicylate for the treatment of microscopic colitis. Gastroenterology 1998; 114:29–36.
- Munck LK, Kjeldsen J, Philipsen E, Fscher Hansen B. Incomplete remission with short-term prednisolone treatment in collagenous colitis: a randomized study. Scand J Gastroenterol 2003; 38:606–610.
- Madisch A, Miehlke S, Eichele E, et al. Boswellia serrata extract for the treatment of collagenous colitis: a randomized, double-blind, placebo-controlled, multicenter trial. Int J Colorectal Dis 2007; 22:1445–1451.
- Wildt S, Munck LK, Vinter-Jensen L, et al. Probiotic treatment of collagenous colitis: a randomized, double-blind, placebo-controlled trial with Lactobacillus acidophilus and Bifidobacterium animalis subsp. lactis. Inflamm Bowel Dis 2006; 12:395–401.
- Calabrese C, Fabbri A, Areni A, Zahlane D, Scialpi C, Di Febo G. Mesalazine with or without cholestyramine in the treatment of microscopic colitis: randomized controlled trial. J Gastroenterol Hepatol 2007; 22:809–814.
- Wall GC, Schirmer LL, Page MJ. Pharmacotherapy for microscopic colitis. Pharmacotherapy 2007; 27:425–433.
- Feyen B, Wall GC, Finnerty EP, DeWitt JE, Reyes RS. Meta-analysis: budesonide treatment for collagenous colitis. Aliment Pharmacol Ther 2004; 20:745–749.
- Madisch A, Heymer P, Voss C, et al. Oral budesonide therapy improves quality of life in patients with collagenous colitis. Int J Colorectal Dis 2005; 20:312–316.
- Craig CR, editor. Modern Pharmacology With Clinical Application. 6th edition. Philadelphia: Lippincott Williams and Wilkins, 2003:481.
- Limsui D, Pardi DS, Camilleri M, et al. Symptomatic overlap between irritable bowel syndrome and microscopic colitis. Inflamm Bowel Dis 2007; 13:175–181.
- Swagerty DL, Walling AD, Klein RM. Lactose intolerance. Am Fam Physician 2002; 65:1845–1856.
- Thomas PD, Forbes A, Green J, et al. Guidelines for the investigation of chronic diarrhea, 2nd edition. Gut 2003; 52(suppl 5):1–5.
- Nelsen DA. Gluten-sensitive enteropathy (celiac disease): more common than you think. Am Fam Physician 2002; 66:2259–2266.
- Bardella MT, Trovato C, Cesana BM, Pagliari C, Gebbia C, Peracchi M. Serological markers for coeliac disease: is it time to change? Dig Liver Dis 2001; 33:426–431.
- Barta Z, Mekkel G, Csipo I, et al. Micropscopic colitis: a retrospective study of clinical presentation in 53 patients. World J Gastroenterol 2005; 11:1351–1355.
- Tremaine WJ. Diagnosing collagenous colitis: does it make a difference? Eur J Gastroenterol Hepatol 1999; 11:477–479.
- Bohr J, Tysk C, Yang P, Danielsson D, Järnerot G. Autoantibodies and immunoglobulins in collagenous colitis. Gut 1996; 39:77–81.
- Zins BJ, Tremaine WJ, Carpenter HA. Collagenous colitis: mucosal biopsies and association with fecal leukocytes. Mayo Clin Proc 1995; 70:430–433.
- Olsen M, Eriksson S, Bohr J, Järnerot G, Tysk C. Lymphocytic colitis: a retrospective clinical study of 199 Swedish patients. Gut 2004; 53:536–541.
- Pardi DS. Microscopic colitis: an update. Inflamm Bowel Dis 2004; 10:860–870.
- Fernandez-Banares F, Salas A, Esteve M, Espinos J, Forne M, Viver JM. Collagenous and lymphocytic colitis: evaluation of clinical and histological features, response to treatment, and long-term follow-up. Am J Gastroenterol 2003; 98:340–347.
- Aignet T, Neureiter D, Müller S, Küspert G, Belke J, Kirchner T. Extracellular matrix composition and gene expression in collagenous colitis. Gastroenterology 1997; 113:136–143.
- Parfitt JR, Driman DK. Pathological effects of drugs on the gastrointestinal tract: a review. Hum Pathol 2007; 38:527–536.
- Fernández-Bañares F, Esteve M, Espinós JC, et al. Drug consumption and the risk of microscopic colitis. Am J Gastroenterol 2007; 102:324–330.
- Thomson RD, Lestine LS, Bensen SP, et al. Lansoprazole-associated microscopic colitis: a case series. Am J Gastroenterol 2002; 97:2908–2913.
- Chande N, McDonald JWD, MacDonald JK. Cochrane Inflammatory Bowel Disease and Functional Bowel Disorders Group. Interventions for treating collagenous colitis. Cochrane Database Syst Rev 2007 Jan 24;(1):CD006096.
- Fine K, Lee E. Efficacy of open-label bismuth subsalicylate for the treatment of microscopic colitis. Gastroenterology 1998; 114:29–36.
- Munck LK, Kjeldsen J, Philipsen E, Fscher Hansen B. Incomplete remission with short-term prednisolone treatment in collagenous colitis: a randomized study. Scand J Gastroenterol 2003; 38:606–610.
- Madisch A, Miehlke S, Eichele E, et al. Boswellia serrata extract for the treatment of collagenous colitis: a randomized, double-blind, placebo-controlled, multicenter trial. Int J Colorectal Dis 2007; 22:1445–1451.
- Wildt S, Munck LK, Vinter-Jensen L, et al. Probiotic treatment of collagenous colitis: a randomized, double-blind, placebo-controlled trial with Lactobacillus acidophilus and Bifidobacterium animalis subsp. lactis. Inflamm Bowel Dis 2006; 12:395–401.
- Calabrese C, Fabbri A, Areni A, Zahlane D, Scialpi C, Di Febo G. Mesalazine with or without cholestyramine in the treatment of microscopic colitis: randomized controlled trial. J Gastroenterol Hepatol 2007; 22:809–814.
- Wall GC, Schirmer LL, Page MJ. Pharmacotherapy for microscopic colitis. Pharmacotherapy 2007; 27:425–433.
- Feyen B, Wall GC, Finnerty EP, DeWitt JE, Reyes RS. Meta-analysis: budesonide treatment for collagenous colitis. Aliment Pharmacol Ther 2004; 20:745–749.
- Madisch A, Heymer P, Voss C, et al. Oral budesonide therapy improves quality of life in patients with collagenous colitis. Int J Colorectal Dis 2005; 20:312–316.
- Craig CR, editor. Modern Pharmacology With Clinical Application. 6th edition. Philadelphia: Lippincott Williams and Wilkins, 2003:481.
- Limsui D, Pardi DS, Camilleri M, et al. Symptomatic overlap between irritable bowel syndrome and microscopic colitis. Inflamm Bowel Dis 2007; 13:175–181.