User login
Severe pain in the frontotemporal area
Migraine is a neurologic disease characterized by episodes of throbbing, often unilateral, headache. These attacks are associated with visual or other sensory symptoms (classic aura) related to the central nervous system, nausea, and vomiting, and are often set off or exacerbated by physical activity. The age-adjusted prevalence is estimated at 15.9% across all adults, but migraine is much more common in women, with a prevalence of 21% in women and 10.7% in men.
On the basis of the patient's history, clinical suspicion for chronic migraine should be high. Hemiplegic migraine usually presents with temporary unilateral hemiparesis, sometimes with speech disturbance. Attacks of chronic paroxysmal hemicrania are also unilateral but are characterized by their highly intense but short duration. Clinical suspicion for a space-occupying lesion should be raised in cases where patients with a history of headache present with new symptoms or abnormal signs.
The diagnosis of chronic migraine is a clinical one. The American Headache Society defines chronic migraine as at least five attacks of migraine-like or tension type–like headache that must fulfill specific criteria. If the migraine occurs with aura, it must occur 8 days or more per month for more than 3 months and be relieved by a triptan or ergot derivative. If the migraine occurs without aura, the same criteria apply, but it is important that the aforementioned signs and symptoms cannot be accounted for by another diagnosis.
For moderate or severe attacks, migraine-specific agents are recommended: triptans, dihydroergotamine (DHE), small-molecule CGRP receptor antagonists (gepants), and selective serotonin (5-HT1F) receptor agonists (ditans). However, it is accepted that migraine treatment must be individualized, and that a trial-and-error period should be expected. Recent data suggest that about 30% of migraine patients who are prescribed a triptan have an insufficient response to this approach. Some research has shown that such patients have a better response after being switched to a second drug in the triptan class, while other studies have shown no difference. The patient in the current case might also be a candidate for preventive treatment, which should generally be considered when, in spite of acute treatment, migraine interferes with the patient's day-to-day routine or when attacks become frequent. The four CGRP monoclonal antibodies approved in the United States are eptinezumab, erenumab, fremanezumab, and galcanezumab.
Angeliki Vgontzas, MD, Instructor, Department of Neurology, Harvard Medical School; Associate Neurologist, Department of Neurology, Brigham and Women's Hospital/Brigham and Women's Faulkner Hospital, Boston, Massachusetts.
Angeliki Vgontzas, MD, has disclosed no relevant financial relationships.
Migraine is a neurologic disease characterized by episodes of throbbing, often unilateral, headache. These attacks are associated with visual or other sensory symptoms (classic aura) related to the central nervous system, nausea, and vomiting, and are often set off or exacerbated by physical activity. The age-adjusted prevalence is estimated at 15.9% across all adults, but migraine is much more common in women, with a prevalence of 21% in women and 10.7% in men.
On the basis of the patient's history, clinical suspicion for chronic migraine should be high. Hemiplegic migraine usually presents with temporary unilateral hemiparesis, sometimes with speech disturbance. Attacks of chronic paroxysmal hemicrania are also unilateral but are characterized by their highly intense but short duration. Clinical suspicion for a space-occupying lesion should be raised in cases where patients with a history of headache present with new symptoms or abnormal signs.
The diagnosis of chronic migraine is a clinical one. The American Headache Society defines chronic migraine as at least five attacks of migraine-like or tension type–like headache that must fulfill specific criteria. If the migraine occurs with aura, it must occur 8 days or more per month for more than 3 months and be relieved by a triptan or ergot derivative. If the migraine occurs without aura, the same criteria apply, but it is important that the aforementioned signs and symptoms cannot be accounted for by another diagnosis.
For moderate or severe attacks, migraine-specific agents are recommended: triptans, dihydroergotamine (DHE), small-molecule CGRP receptor antagonists (gepants), and selective serotonin (5-HT1F) receptor agonists (ditans). However, it is accepted that migraine treatment must be individualized, and that a trial-and-error period should be expected. Recent data suggest that about 30% of migraine patients who are prescribed a triptan have an insufficient response to this approach. Some research has shown that such patients have a better response after being switched to a second drug in the triptan class, while other studies have shown no difference. The patient in the current case might also be a candidate for preventive treatment, which should generally be considered when, in spite of acute treatment, migraine interferes with the patient's day-to-day routine or when attacks become frequent. The four CGRP monoclonal antibodies approved in the United States are eptinezumab, erenumab, fremanezumab, and galcanezumab.
Angeliki Vgontzas, MD, Instructor, Department of Neurology, Harvard Medical School; Associate Neurologist, Department of Neurology, Brigham and Women's Hospital/Brigham and Women's Faulkner Hospital, Boston, Massachusetts.
Angeliki Vgontzas, MD, has disclosed no relevant financial relationships.
Migraine is a neurologic disease characterized by episodes of throbbing, often unilateral, headache. These attacks are associated with visual or other sensory symptoms (classic aura) related to the central nervous system, nausea, and vomiting, and are often set off or exacerbated by physical activity. The age-adjusted prevalence is estimated at 15.9% across all adults, but migraine is much more common in women, with a prevalence of 21% in women and 10.7% in men.
On the basis of the patient's history, clinical suspicion for chronic migraine should be high. Hemiplegic migraine usually presents with temporary unilateral hemiparesis, sometimes with speech disturbance. Attacks of chronic paroxysmal hemicrania are also unilateral but are characterized by their highly intense but short duration. Clinical suspicion for a space-occupying lesion should be raised in cases where patients with a history of headache present with new symptoms or abnormal signs.
The diagnosis of chronic migraine is a clinical one. The American Headache Society defines chronic migraine as at least five attacks of migraine-like or tension type–like headache that must fulfill specific criteria. If the migraine occurs with aura, it must occur 8 days or more per month for more than 3 months and be relieved by a triptan or ergot derivative. If the migraine occurs without aura, the same criteria apply, but it is important that the aforementioned signs and symptoms cannot be accounted for by another diagnosis.
For moderate or severe attacks, migraine-specific agents are recommended: triptans, dihydroergotamine (DHE), small-molecule CGRP receptor antagonists (gepants), and selective serotonin (5-HT1F) receptor agonists (ditans). However, it is accepted that migraine treatment must be individualized, and that a trial-and-error period should be expected. Recent data suggest that about 30% of migraine patients who are prescribed a triptan have an insufficient response to this approach. Some research has shown that such patients have a better response after being switched to a second drug in the triptan class, while other studies have shown no difference. The patient in the current case might also be a candidate for preventive treatment, which should generally be considered when, in spite of acute treatment, migraine interferes with the patient's day-to-day routine or when attacks become frequent. The four CGRP monoclonal antibodies approved in the United States are eptinezumab, erenumab, fremanezumab, and galcanezumab.
Angeliki Vgontzas, MD, Instructor, Department of Neurology, Harvard Medical School; Associate Neurologist, Department of Neurology, Brigham and Women's Hospital/Brigham and Women's Faulkner Hospital, Boston, Massachusetts.
Angeliki Vgontzas, MD, has disclosed no relevant financial relationships.
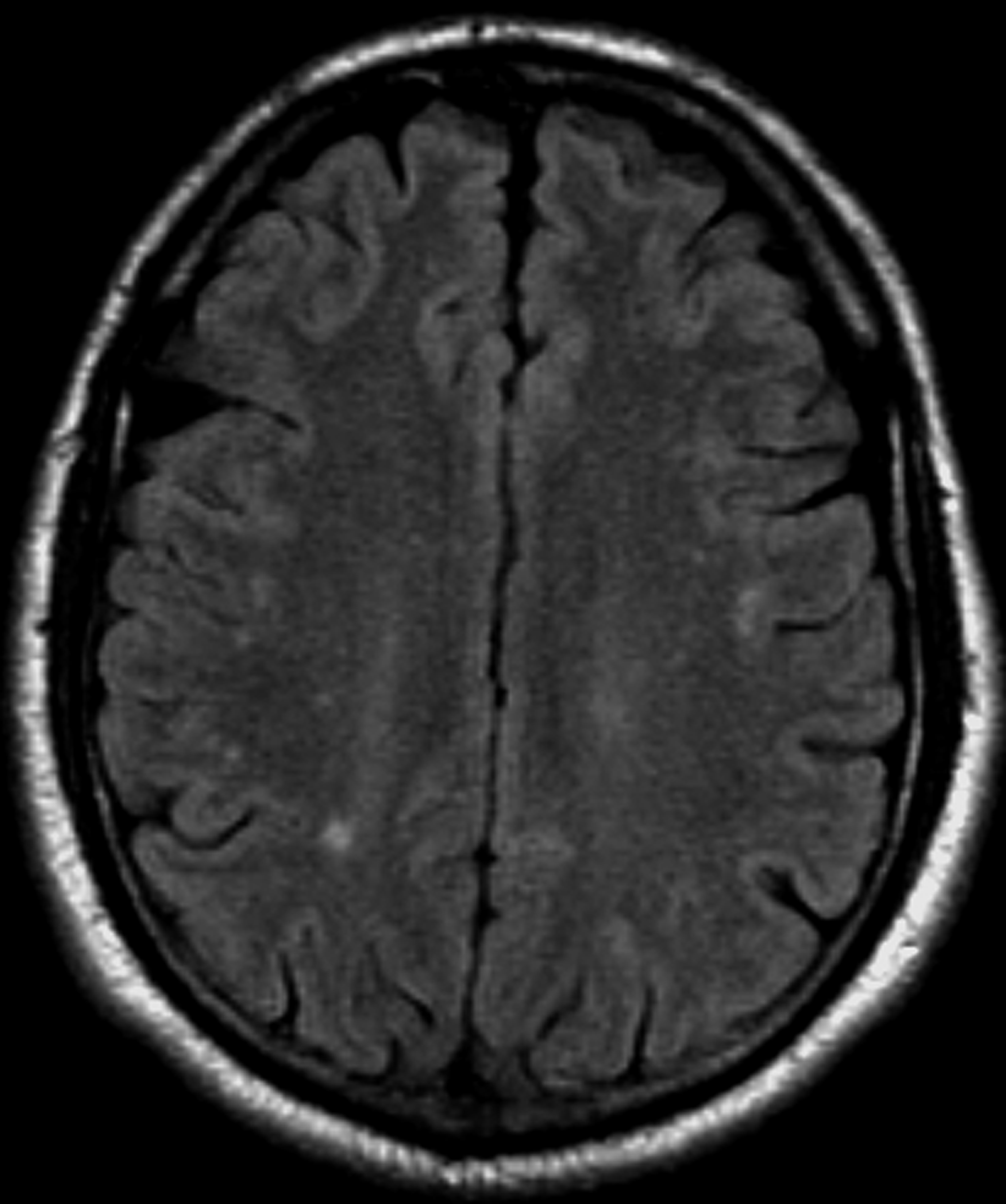
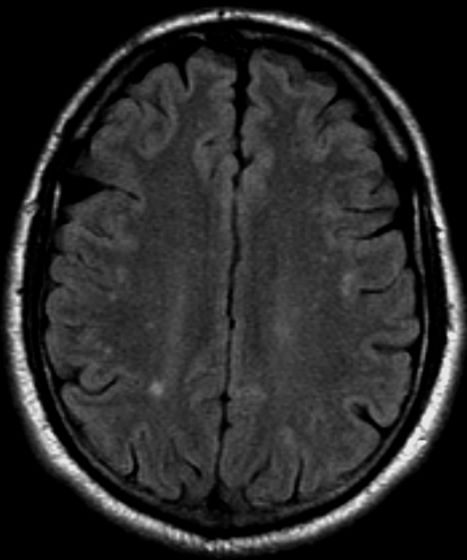
A 36-year-old man presents with severe pain in the frontotemporal area. He reports a history of severe headaches, sometimes with nausea. In the past year, these symptoms have begun to "knock him out" for nearly 2 weeks out of a month. The patient historically has been able to curtail his symptoms with a triptan. Physical examination is remarkable for Adie tonic pupil. A 1.5 T MRI of the brain is performed. Axial FLAIR sequence reveals scattered white matter lesions consistent with foci of demyelination.
Patient with tachycardia
On the basis of the patient's personal and family history together with his presentation, the likely diagnosis is latent autoimmune diabetes in adults (LADA). LADA is characterized by beta-cell loss and insulin resistance. This slowly evolving form of autoimmune diabetes comprises 2%-12% of all patients with adult-onset diabetes. Patients with LADA present with evidence of autoimmunity and varying C-peptide levels, which decrease more slowly in this subgroup than in patients with type 1 diabetes (T1D). They also have immunogenic markers associated with T1D, primarily anti-glutamic acid decarboxylase (GAD) antibodies.
Patients with LADA are often misdiagnosed as having T2D. The clinical picture of LADA overlaps with that of T2D, with patients being insulin resistant and often overweight. In addition, presenting symptoms of LADA — excessive thirst, blurred vision, and high blood glucose — are also seen in T2D. Although LADA is technically classified as T1D, some groups posit that the condition exists on a spectrum between T1D and T2D. Compared with patients with T2D, those with LADA are generally younger at diagnosis (often in their 30s), have lower BMI, and report a personal or family history of autoimmune diseases, such as the patient in this quiz. Throughout the disease course, individuals with LADA show a reduced frequency of metabolic syndrome compared with those with T2D.
Key to diagnosis is the absence of insulin requirement for at least 6 months. Anti-GAD antibodies are the most sensitive marker for LADA; other autoantibodies that occur less frequently include ICA, IA-2A, ZnT8A, and tetraspanin 7 autoantibodies. With a paucity of large-scale clinical trials in LADA, current treatment strategies are not based on consensus guidelines, though an expert panel has published management recommendations. Category 1 patients (defined as a C-peptide level < 0.3 nmol/L) are treated with intensive insulin therapy. The recommendation for category 2 patients (defined as C-peptide values ≥ 0.3 and ≤ 0.7 nmol/L) is a modified American Diabetes Association/European Association for the Study of Diabetes algorithm for T2D. However, patients with category 2 LADA may need to initiate insulin therapy earlier to combat beta-cell failure (ostensibly because LADA is an autoimmune disease beta-cell function declines much faster than in T2D). For category 3 patients (defined as C-peptide values > 0.7 nmol/L), treatment decisions are made in response to changing C-peptide levels.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia.
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
On the basis of the patient's personal and family history together with his presentation, the likely diagnosis is latent autoimmune diabetes in adults (LADA). LADA is characterized by beta-cell loss and insulin resistance. This slowly evolving form of autoimmune diabetes comprises 2%-12% of all patients with adult-onset diabetes. Patients with LADA present with evidence of autoimmunity and varying C-peptide levels, which decrease more slowly in this subgroup than in patients with type 1 diabetes (T1D). They also have immunogenic markers associated with T1D, primarily anti-glutamic acid decarboxylase (GAD) antibodies.
Patients with LADA are often misdiagnosed as having T2D. The clinical picture of LADA overlaps with that of T2D, with patients being insulin resistant and often overweight. In addition, presenting symptoms of LADA — excessive thirst, blurred vision, and high blood glucose — are also seen in T2D. Although LADA is technically classified as T1D, some groups posit that the condition exists on a spectrum between T1D and T2D. Compared with patients with T2D, those with LADA are generally younger at diagnosis (often in their 30s), have lower BMI, and report a personal or family history of autoimmune diseases, such as the patient in this quiz. Throughout the disease course, individuals with LADA show a reduced frequency of metabolic syndrome compared with those with T2D.
Key to diagnosis is the absence of insulin requirement for at least 6 months. Anti-GAD antibodies are the most sensitive marker for LADA; other autoantibodies that occur less frequently include ICA, IA-2A, ZnT8A, and tetraspanin 7 autoantibodies. With a paucity of large-scale clinical trials in LADA, current treatment strategies are not based on consensus guidelines, though an expert panel has published management recommendations. Category 1 patients (defined as a C-peptide level < 0.3 nmol/L) are treated with intensive insulin therapy. The recommendation for category 2 patients (defined as C-peptide values ≥ 0.3 and ≤ 0.7 nmol/L) is a modified American Diabetes Association/European Association for the Study of Diabetes algorithm for T2D. However, patients with category 2 LADA may need to initiate insulin therapy earlier to combat beta-cell failure (ostensibly because LADA is an autoimmune disease beta-cell function declines much faster than in T2D). For category 3 patients (defined as C-peptide values > 0.7 nmol/L), treatment decisions are made in response to changing C-peptide levels.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia.
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
On the basis of the patient's personal and family history together with his presentation, the likely diagnosis is latent autoimmune diabetes in adults (LADA). LADA is characterized by beta-cell loss and insulin resistance. This slowly evolving form of autoimmune diabetes comprises 2%-12% of all patients with adult-onset diabetes. Patients with LADA present with evidence of autoimmunity and varying C-peptide levels, which decrease more slowly in this subgroup than in patients with type 1 diabetes (T1D). They also have immunogenic markers associated with T1D, primarily anti-glutamic acid decarboxylase (GAD) antibodies.
Patients with LADA are often misdiagnosed as having T2D. The clinical picture of LADA overlaps with that of T2D, with patients being insulin resistant and often overweight. In addition, presenting symptoms of LADA — excessive thirst, blurred vision, and high blood glucose — are also seen in T2D. Although LADA is technically classified as T1D, some groups posit that the condition exists on a spectrum between T1D and T2D. Compared with patients with T2D, those with LADA are generally younger at diagnosis (often in their 30s), have lower BMI, and report a personal or family history of autoimmune diseases, such as the patient in this quiz. Throughout the disease course, individuals with LADA show a reduced frequency of metabolic syndrome compared with those with T2D.
Key to diagnosis is the absence of insulin requirement for at least 6 months. Anti-GAD antibodies are the most sensitive marker for LADA; other autoantibodies that occur less frequently include ICA, IA-2A, ZnT8A, and tetraspanin 7 autoantibodies. With a paucity of large-scale clinical trials in LADA, current treatment strategies are not based on consensus guidelines, though an expert panel has published management recommendations. Category 1 patients (defined as a C-peptide level < 0.3 nmol/L) are treated with intensive insulin therapy. The recommendation for category 2 patients (defined as C-peptide values ≥ 0.3 and ≤ 0.7 nmol/L) is a modified American Diabetes Association/European Association for the Study of Diabetes algorithm for T2D. However, patients with category 2 LADA may need to initiate insulin therapy earlier to combat beta-cell failure (ostensibly because LADA is an autoimmune disease beta-cell function declines much faster than in T2D). For category 3 patients (defined as C-peptide values > 0.7 nmol/L), treatment decisions are made in response to changing C-peptide levels.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia.
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
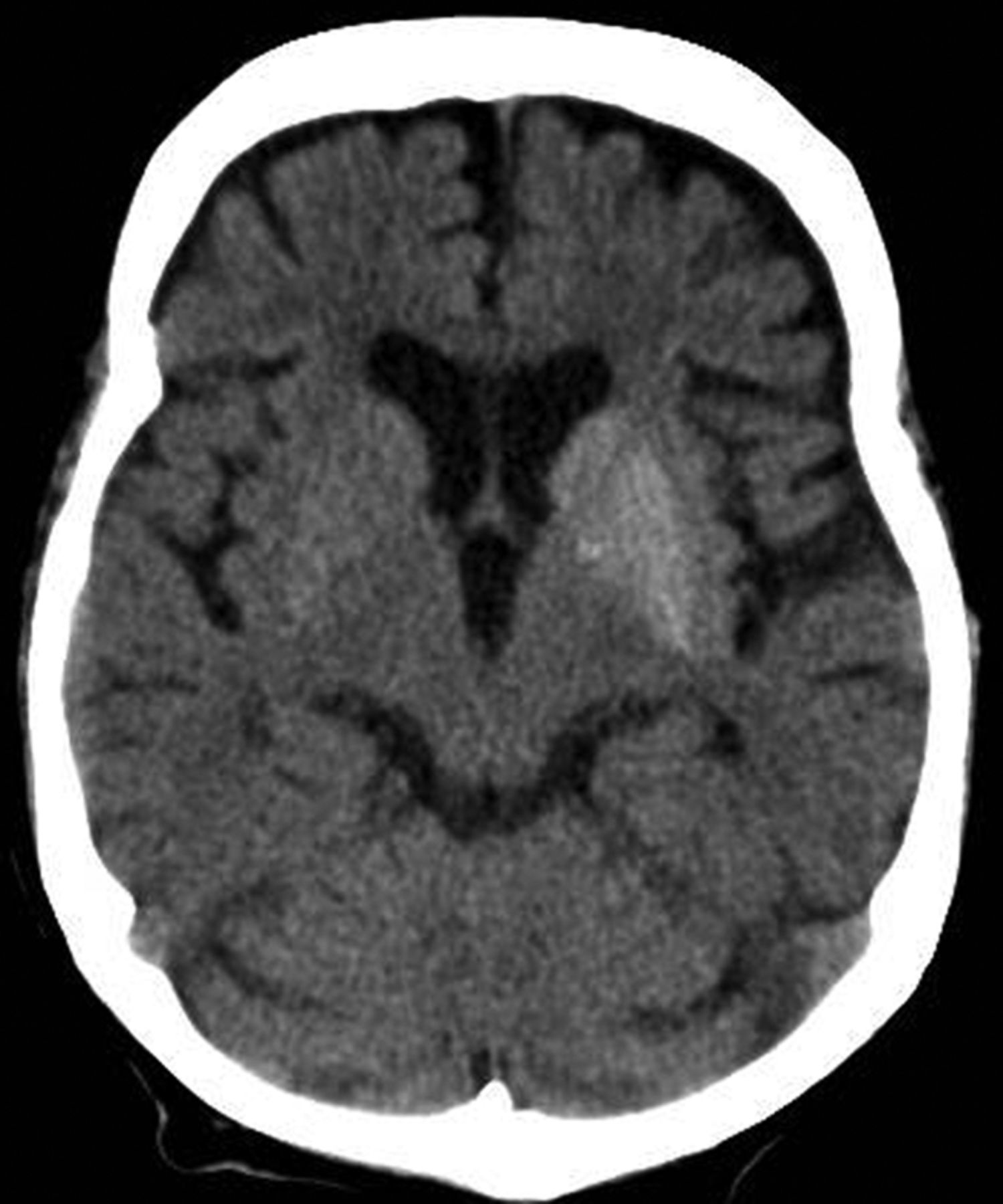
A 33-year-old man presents with blurred vision and tachycardia. Physical examination is remarkable for a BMI of 27 kg/m2. The patient explains that he feels he has lost weight. However, he attributes this change to a new exercise regimen he undertook when he was diagnosed with type 2 diabetes (T2D) about 8 months ago. The patient also notes polydipsia over a series of weeks. He reports that his first cousin may have lupus, though her diagnosis is still uncertain. Axial noncontrast CT demonstrates hyperattenuation that is more pronounced on the left side and involves the lentiform and caudate nuclei bilaterally.
History of AD with progressing flare
The patient is empirically diagnosed with AD complicated by bacterial infection. A skin swab culture is positive for Staphylococcus aureus and Streptococcus pyogenes.
AD is a common chronic inflammatory skin disease characterized by pruritus, eczematous lesions, xerosis, and lichenification. Individuals of all ages may be affected by AD, although it normally begins in infancy. Studies suggest that as many as 17.1% of adults and 22.6% of children are affected by AD. The disease is associated with diminished quality of life, sleep disturbance, depression, and anxiety. To further complicate matters, patients with AD have a significantly increased risk for recurrent skin infections, including bacterial, viral, and fungal infections.
The underlying mechanisms of bacterial infection in AD are multifactorial and involve both host and bacterial factors. Factors implicated in the increased risk for infection in patients with AD include skin barrier defects, suppression of cutaneous innate immunity by type 2 inflammation, S aureus colonization, and cutaneous dysbiosis. Up to 90% of patients with AD are colonized with S aureus. It has been theorized that the host skin microbiota may play a role in protecting against S aureus colonization and infection in patients with AD. Additionally, bacterial virulence factors, such as the superantigens, proteases, and cytolytic phenol‐soluble modulins secreted by S aureus, trigger skin inflammation and may also contribute to bacterial persistence and/or epithelial penetration and infection.
Overt bacterial infection in patients with AD can be recognized by the presence of weeping lesions, honey‐colored crusts, and pustules. However, cutaneous erythema and warmth, oozing associated with edema, and regional lymphadenopathy are seen in both AD exacerbations and in patients with infection, making clinical diagnosis challenging. In addition, anatomical site‐ and skin type-specific features may disguise signs of infection, and the high frequency of S aureus colonization in AD makes positive skin swab culture of suspected infection an unreliable diagnostic tool.
S pyogenes is the second most common cause of skin and soft tissue infections in AD (S aureus is the leading cause, although data suggest that pediatric patients are not likely to be affected by superinfections caused by methicillin-resistant S aureus [MRSA]). S pyogenes may cause infections in patients with AD alone or in combination with S aureus. Patients with these skin infections usually present with pustules or impetigo. The lesions may appear as punched-out erosions with scalloped borders that mimic eczema herpeticum or eczema coxsackium. According to guidelines from the American Academy of Dermatology, the presence of purulent exudate and pustules on skin examination may suggest a diagnosis of secondary bacterial infection over inflammation from dermatitis.
The use of systemic antibiotics in the treatment of noninfected AD is not recommended; however, systemic antibiotics can be recommended for patients with clinical evidence of bacterial infection, in addition to standard treatment for AD, including the concurrent application of topical steroids. For patients with AD who have signs and symptoms of systemic illness, hospitalization and empirical intravenous antibiotics are recommended. The antibiotic regimen should provide coverage against S aureus because this is the most frequently identified bacterial pathogen in AD.
When treating critically ill patients, treatment that provides coverage for both MRSA and methicillin-susceptible S aureus (MSSA) with vancomycin and an antistaphylococcal beta-lactam is appropriate. In patients with severe but non–life-threatening infections, vancomycin may be used alone as empirical therapy, pending culture results. Clindamycin can also be considered, particularly if there is no concern for an endovascular infection and the local incidence of clindamycin resistance is less than 15%.
Bacteremia triggered by S aureus initially requires the use of a bactericidal intravenous agent. For MRSA, vancomycin is the first-line agent. Cefazolin and nafcillin are both acceptable first-line agents for MSSA, although nafcillin can cause venous irritation and phlebitis when administered peripherally. Among children with S aureus bacteremia, an oral agent to which the isolate is susceptible is appropriate, as long as there are no concerns for ongoing bacteremia or endovascular complications. Duration of therapy should be determined by the clinical response; 7-14 days is usually recommended.
For patients with AD with uncomplicated, nonpurulent skin infection, a beta-lactam antibiotic that covers both S aureus and beta-hemolytic streptococci (eg, cefazolin or cephalexin) may be appropriate pending clinical response or culture and considering local epidemiology and resistance patterns. In patients who present with a skin abscess, history of MRSA colonization, close contacts with a history of skin infections, or recent hospitalization, consideration of coverage for MRSA is recommended. Acceptable oral options for MRSA skin infections in both children and adults include clindamycin, doxycycline, trimethoprim-sulfamethoxazole, and linezolid, assuming that the isolate is susceptible in vitro. Finally, topical mupirocin ointment for 5-10 days is an appropriate treatment for patients with AD with minor, localized skin infections such as impetigo.
William D. James, MD, Professor, Department of Dermatology, University of Pennsylvania, Philadelphia
Disclosure: William D. James, MD, has disclosed the following relevant financial relationships:
Received income in an amount equal to or greater than $250 from: Elsevier
The patient is empirically diagnosed with AD complicated by bacterial infection. A skin swab culture is positive for Staphylococcus aureus and Streptococcus pyogenes.
AD is a common chronic inflammatory skin disease characterized by pruritus, eczematous lesions, xerosis, and lichenification. Individuals of all ages may be affected by AD, although it normally begins in infancy. Studies suggest that as many as 17.1% of adults and 22.6% of children are affected by AD. The disease is associated with diminished quality of life, sleep disturbance, depression, and anxiety. To further complicate matters, patients with AD have a significantly increased risk for recurrent skin infections, including bacterial, viral, and fungal infections.
The underlying mechanisms of bacterial infection in AD are multifactorial and involve both host and bacterial factors. Factors implicated in the increased risk for infection in patients with AD include skin barrier defects, suppression of cutaneous innate immunity by type 2 inflammation, S aureus colonization, and cutaneous dysbiosis. Up to 90% of patients with AD are colonized with S aureus. It has been theorized that the host skin microbiota may play a role in protecting against S aureus colonization and infection in patients with AD. Additionally, bacterial virulence factors, such as the superantigens, proteases, and cytolytic phenol‐soluble modulins secreted by S aureus, trigger skin inflammation and may also contribute to bacterial persistence and/or epithelial penetration and infection.
Overt bacterial infection in patients with AD can be recognized by the presence of weeping lesions, honey‐colored crusts, and pustules. However, cutaneous erythema and warmth, oozing associated with edema, and regional lymphadenopathy are seen in both AD exacerbations and in patients with infection, making clinical diagnosis challenging. In addition, anatomical site‐ and skin type-specific features may disguise signs of infection, and the high frequency of S aureus colonization in AD makes positive skin swab culture of suspected infection an unreliable diagnostic tool.
S pyogenes is the second most common cause of skin and soft tissue infections in AD (S aureus is the leading cause, although data suggest that pediatric patients are not likely to be affected by superinfections caused by methicillin-resistant S aureus [MRSA]). S pyogenes may cause infections in patients with AD alone or in combination with S aureus. Patients with these skin infections usually present with pustules or impetigo. The lesions may appear as punched-out erosions with scalloped borders that mimic eczema herpeticum or eczema coxsackium. According to guidelines from the American Academy of Dermatology, the presence of purulent exudate and pustules on skin examination may suggest a diagnosis of secondary bacterial infection over inflammation from dermatitis.
The use of systemic antibiotics in the treatment of noninfected AD is not recommended; however, systemic antibiotics can be recommended for patients with clinical evidence of bacterial infection, in addition to standard treatment for AD, including the concurrent application of topical steroids. For patients with AD who have signs and symptoms of systemic illness, hospitalization and empirical intravenous antibiotics are recommended. The antibiotic regimen should provide coverage against S aureus because this is the most frequently identified bacterial pathogen in AD.
When treating critically ill patients, treatment that provides coverage for both MRSA and methicillin-susceptible S aureus (MSSA) with vancomycin and an antistaphylococcal beta-lactam is appropriate. In patients with severe but non–life-threatening infections, vancomycin may be used alone as empirical therapy, pending culture results. Clindamycin can also be considered, particularly if there is no concern for an endovascular infection and the local incidence of clindamycin resistance is less than 15%.
Bacteremia triggered by S aureus initially requires the use of a bactericidal intravenous agent. For MRSA, vancomycin is the first-line agent. Cefazolin and nafcillin are both acceptable first-line agents for MSSA, although nafcillin can cause venous irritation and phlebitis when administered peripherally. Among children with S aureus bacteremia, an oral agent to which the isolate is susceptible is appropriate, as long as there are no concerns for ongoing bacteremia or endovascular complications. Duration of therapy should be determined by the clinical response; 7-14 days is usually recommended.
For patients with AD with uncomplicated, nonpurulent skin infection, a beta-lactam antibiotic that covers both S aureus and beta-hemolytic streptococci (eg, cefazolin or cephalexin) may be appropriate pending clinical response or culture and considering local epidemiology and resistance patterns. In patients who present with a skin abscess, history of MRSA colonization, close contacts with a history of skin infections, or recent hospitalization, consideration of coverage for MRSA is recommended. Acceptable oral options for MRSA skin infections in both children and adults include clindamycin, doxycycline, trimethoprim-sulfamethoxazole, and linezolid, assuming that the isolate is susceptible in vitro. Finally, topical mupirocin ointment for 5-10 days is an appropriate treatment for patients with AD with minor, localized skin infections such as impetigo.
William D. James, MD, Professor, Department of Dermatology, University of Pennsylvania, Philadelphia
Disclosure: William D. James, MD, has disclosed the following relevant financial relationships:
Received income in an amount equal to or greater than $250 from: Elsevier
The patient is empirically diagnosed with AD complicated by bacterial infection. A skin swab culture is positive for Staphylococcus aureus and Streptococcus pyogenes.
AD is a common chronic inflammatory skin disease characterized by pruritus, eczematous lesions, xerosis, and lichenification. Individuals of all ages may be affected by AD, although it normally begins in infancy. Studies suggest that as many as 17.1% of adults and 22.6% of children are affected by AD. The disease is associated with diminished quality of life, sleep disturbance, depression, and anxiety. To further complicate matters, patients with AD have a significantly increased risk for recurrent skin infections, including bacterial, viral, and fungal infections.
The underlying mechanisms of bacterial infection in AD are multifactorial and involve both host and bacterial factors. Factors implicated in the increased risk for infection in patients with AD include skin barrier defects, suppression of cutaneous innate immunity by type 2 inflammation, S aureus colonization, and cutaneous dysbiosis. Up to 90% of patients with AD are colonized with S aureus. It has been theorized that the host skin microbiota may play a role in protecting against S aureus colonization and infection in patients with AD. Additionally, bacterial virulence factors, such as the superantigens, proteases, and cytolytic phenol‐soluble modulins secreted by S aureus, trigger skin inflammation and may also contribute to bacterial persistence and/or epithelial penetration and infection.
Overt bacterial infection in patients with AD can be recognized by the presence of weeping lesions, honey‐colored crusts, and pustules. However, cutaneous erythema and warmth, oozing associated with edema, and regional lymphadenopathy are seen in both AD exacerbations and in patients with infection, making clinical diagnosis challenging. In addition, anatomical site‐ and skin type-specific features may disguise signs of infection, and the high frequency of S aureus colonization in AD makes positive skin swab culture of suspected infection an unreliable diagnostic tool.
S pyogenes is the second most common cause of skin and soft tissue infections in AD (S aureus is the leading cause, although data suggest that pediatric patients are not likely to be affected by superinfections caused by methicillin-resistant S aureus [MRSA]). S pyogenes may cause infections in patients with AD alone or in combination with S aureus. Patients with these skin infections usually present with pustules or impetigo. The lesions may appear as punched-out erosions with scalloped borders that mimic eczema herpeticum or eczema coxsackium. According to guidelines from the American Academy of Dermatology, the presence of purulent exudate and pustules on skin examination may suggest a diagnosis of secondary bacterial infection over inflammation from dermatitis.
The use of systemic antibiotics in the treatment of noninfected AD is not recommended; however, systemic antibiotics can be recommended for patients with clinical evidence of bacterial infection, in addition to standard treatment for AD, including the concurrent application of topical steroids. For patients with AD who have signs and symptoms of systemic illness, hospitalization and empirical intravenous antibiotics are recommended. The antibiotic regimen should provide coverage against S aureus because this is the most frequently identified bacterial pathogen in AD.
When treating critically ill patients, treatment that provides coverage for both MRSA and methicillin-susceptible S aureus (MSSA) with vancomycin and an antistaphylococcal beta-lactam is appropriate. In patients with severe but non–life-threatening infections, vancomycin may be used alone as empirical therapy, pending culture results. Clindamycin can also be considered, particularly if there is no concern for an endovascular infection and the local incidence of clindamycin resistance is less than 15%.
Bacteremia triggered by S aureus initially requires the use of a bactericidal intravenous agent. For MRSA, vancomycin is the first-line agent. Cefazolin and nafcillin are both acceptable first-line agents for MSSA, although nafcillin can cause venous irritation and phlebitis when administered peripherally. Among children with S aureus bacteremia, an oral agent to which the isolate is susceptible is appropriate, as long as there are no concerns for ongoing bacteremia or endovascular complications. Duration of therapy should be determined by the clinical response; 7-14 days is usually recommended.
For patients with AD with uncomplicated, nonpurulent skin infection, a beta-lactam antibiotic that covers both S aureus and beta-hemolytic streptococci (eg, cefazolin or cephalexin) may be appropriate pending clinical response or culture and considering local epidemiology and resistance patterns. In patients who present with a skin abscess, history of MRSA colonization, close contacts with a history of skin infections, or recent hospitalization, consideration of coverage for MRSA is recommended. Acceptable oral options for MRSA skin infections in both children and adults include clindamycin, doxycycline, trimethoprim-sulfamethoxazole, and linezolid, assuming that the isolate is susceptible in vitro. Finally, topical mupirocin ointment for 5-10 days is an appropriate treatment for patients with AD with minor, localized skin infections such as impetigo.
William D. James, MD, Professor, Department of Dermatology, University of Pennsylvania, Philadelphia
Disclosure: William D. James, MD, has disclosed the following relevant financial relationships:
Received income in an amount equal to or greater than $250 from: Elsevier
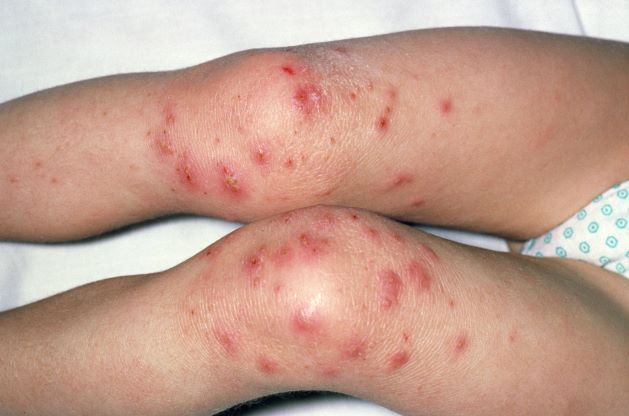
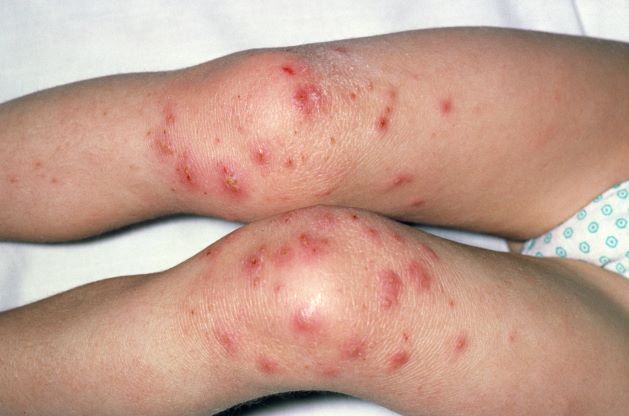
A 9-year-old girl with a history of moderate atopic dermatitis (AD) presents with a rapidly progressing AD flare. The patient had been stable over the past 6 months with the use of daily emollients. Over the past 36-48 hours, the patient developed pruritic lesions and pustules on her knees and elbows, and erythema and scaling around the eyes. Physical examination reveals a temperature of 101.5°F (38.6°C), a heart rate of 112 beats/min, a respiratory rate of 32 breaths/min, and a blood pressure of 100/95 mm Hg. Physical findings include cutaneous erythema and warmth surrounding the affected areas, pustules with yellow fluid, and regional lymphadenopathy.
Complaints of incomplete bladder emptying
Many patients with nonmetastatic prostate cancer are asymptomatic at the time of diagnosis due to widespread routine screening. When localized symptoms do occur, they may include urinary frequency, decreased urine stream, urinary urgency, and hematuria. An increasing proportion of patients with localized disease are asymptomatic, however; such signs and symptoms may well be related to age-associated prostate enlargement or other conditions. Nevertheless, men over the age of 50 years who present with urinary symptoms should be screened for prostate cancer using DRE and PSA. Benign prostatic hyperplasia, for example, can manifest in urinary symptoms and even elevate PSA. Acute prostatitis, on the other hand, presents as a urinary tract infection.
Because this patient showed elevated PSA levels, albeit with normal DRE findings, needle biopsy of the prostate is indicated for tissue diagnosis, usually performed with transrectal ultrasound. A pathologic evaluation of the biopsy specimen will determine the patient's Gleason score. PSA density (amount of PSA per gram of prostate tissue) and PSA doubling time should be collected as well. As seen in the present case, MRI can be used to assess lesions concerning for prostate cancer prior to biopsy. Lesions are then assigned Prostate Imaging–Reporting and Data System (PI-RADS) scores depending on their location within the prostatic zones. Imaging is also useful in staging and active surveillance. Staging is based on the tumor, node, and metastasis (TNM), with clinically localized prostate cancers including any T, N0, M0, NX, or MX cases. The clinician should pursue genetic testing to determine the presence of high-risk germline mutations.
The NCCN Guidelines recommend that for clinically localized prostate cancer, approaches include watchful waiting, active surveillance, radical prostatectomy, and radiation therapy. For asymptomatic patients who are older and/or have other serious comorbidities, active surveillance is often suggested. Radical prostatectomy is typically reserved for patients with a life expectancy of 10 years or more. Pelvic lymph node dissection may be performed on the basis of probability of nodal metastasis. Radiotherapy is also potentially curative in localized prostate cancer and may be delivered via brachytherapy, proton radiation, or external beam radiation therapy (EBRT). EBRT techniques include intensity-modulated radiation therapy (IMRT) and hypofractionated, image-guided stereotactic body radiation therapy (SBRT).
Chad R. Tracy, MD, Professor; Director, Minimally Invasive Surgery, Department of Urology, University of Iowa Hospitals and Clinics, Iowa City, Iowa
Chad R. Tracy, MD, has disclosed the following relevant financial relationships:
Serve(d) as a consultant for: Cvico Medical Solutions
Many patients with nonmetastatic prostate cancer are asymptomatic at the time of diagnosis due to widespread routine screening. When localized symptoms do occur, they may include urinary frequency, decreased urine stream, urinary urgency, and hematuria. An increasing proportion of patients with localized disease are asymptomatic, however; such signs and symptoms may well be related to age-associated prostate enlargement or other conditions. Nevertheless, men over the age of 50 years who present with urinary symptoms should be screened for prostate cancer using DRE and PSA. Benign prostatic hyperplasia, for example, can manifest in urinary symptoms and even elevate PSA. Acute prostatitis, on the other hand, presents as a urinary tract infection.
Because this patient showed elevated PSA levels, albeit with normal DRE findings, needle biopsy of the prostate is indicated for tissue diagnosis, usually performed with transrectal ultrasound. A pathologic evaluation of the biopsy specimen will determine the patient's Gleason score. PSA density (amount of PSA per gram of prostate tissue) and PSA doubling time should be collected as well. As seen in the present case, MRI can be used to assess lesions concerning for prostate cancer prior to biopsy. Lesions are then assigned Prostate Imaging–Reporting and Data System (PI-RADS) scores depending on their location within the prostatic zones. Imaging is also useful in staging and active surveillance. Staging is based on the tumor, node, and metastasis (TNM), with clinically localized prostate cancers including any T, N0, M0, NX, or MX cases. The clinician should pursue genetic testing to determine the presence of high-risk germline mutations.
The NCCN Guidelines recommend that for clinically localized prostate cancer, approaches include watchful waiting, active surveillance, radical prostatectomy, and radiation therapy. For asymptomatic patients who are older and/or have other serious comorbidities, active surveillance is often suggested. Radical prostatectomy is typically reserved for patients with a life expectancy of 10 years or more. Pelvic lymph node dissection may be performed on the basis of probability of nodal metastasis. Radiotherapy is also potentially curative in localized prostate cancer and may be delivered via brachytherapy, proton radiation, or external beam radiation therapy (EBRT). EBRT techniques include intensity-modulated radiation therapy (IMRT) and hypofractionated, image-guided stereotactic body radiation therapy (SBRT).
Chad R. Tracy, MD, Professor; Director, Minimally Invasive Surgery, Department of Urology, University of Iowa Hospitals and Clinics, Iowa City, Iowa
Chad R. Tracy, MD, has disclosed the following relevant financial relationships:
Serve(d) as a consultant for: Cvico Medical Solutions
Many patients with nonmetastatic prostate cancer are asymptomatic at the time of diagnosis due to widespread routine screening. When localized symptoms do occur, they may include urinary frequency, decreased urine stream, urinary urgency, and hematuria. An increasing proportion of patients with localized disease are asymptomatic, however; such signs and symptoms may well be related to age-associated prostate enlargement or other conditions. Nevertheless, men over the age of 50 years who present with urinary symptoms should be screened for prostate cancer using DRE and PSA. Benign prostatic hyperplasia, for example, can manifest in urinary symptoms and even elevate PSA. Acute prostatitis, on the other hand, presents as a urinary tract infection.
Because this patient showed elevated PSA levels, albeit with normal DRE findings, needle biopsy of the prostate is indicated for tissue diagnosis, usually performed with transrectal ultrasound. A pathologic evaluation of the biopsy specimen will determine the patient's Gleason score. PSA density (amount of PSA per gram of prostate tissue) and PSA doubling time should be collected as well. As seen in the present case, MRI can be used to assess lesions concerning for prostate cancer prior to biopsy. Lesions are then assigned Prostate Imaging–Reporting and Data System (PI-RADS) scores depending on their location within the prostatic zones. Imaging is also useful in staging and active surveillance. Staging is based on the tumor, node, and metastasis (TNM), with clinically localized prostate cancers including any T, N0, M0, NX, or MX cases. The clinician should pursue genetic testing to determine the presence of high-risk germline mutations.
The NCCN Guidelines recommend that for clinically localized prostate cancer, approaches include watchful waiting, active surveillance, radical prostatectomy, and radiation therapy. For asymptomatic patients who are older and/or have other serious comorbidities, active surveillance is often suggested. Radical prostatectomy is typically reserved for patients with a life expectancy of 10 years or more. Pelvic lymph node dissection may be performed on the basis of probability of nodal metastasis. Radiotherapy is also potentially curative in localized prostate cancer and may be delivered via brachytherapy, proton radiation, or external beam radiation therapy (EBRT). EBRT techniques include intensity-modulated radiation therapy (IMRT) and hypofractionated, image-guided stereotactic body radiation therapy (SBRT).
Chad R. Tracy, MD, Professor; Director, Minimally Invasive Surgery, Department of Urology, University of Iowa Hospitals and Clinics, Iowa City, Iowa
Chad R. Tracy, MD, has disclosed the following relevant financial relationships:
Serve(d) as a consultant for: Cvico Medical Solutions
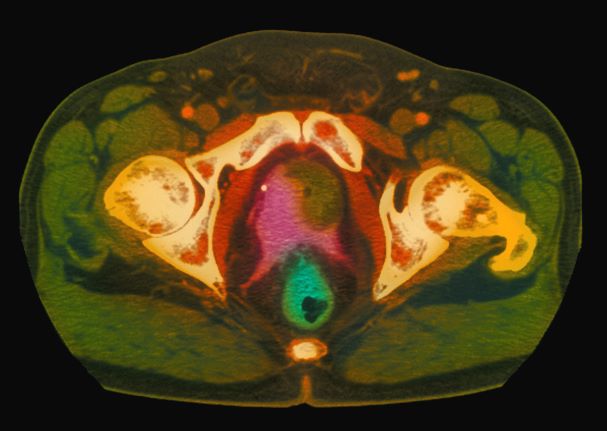
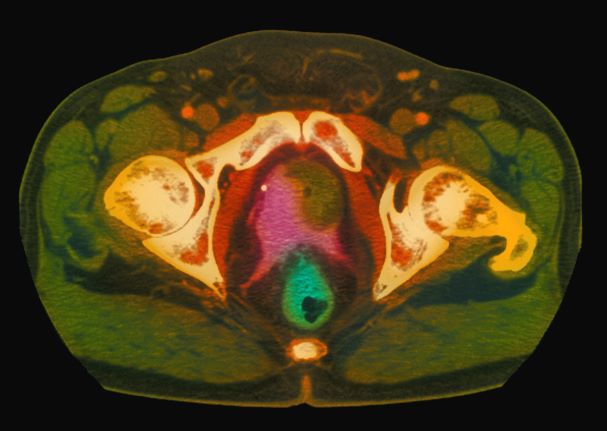
A 61-year-old man presents with complaints of frequent urination and incomplete bladder emptying. He also has been feeling fatigued but cannot tell if this is because his symptoms are worse at night and he has not been sleeping well. Despite a family history of atrial fibrillation, he reports no significant medical history beyond appendicitis many years ago. The patient underwent a prostate cancer screening about 18 months ago, which was normal. During a recent office visit, digital rectal examination (DRE) was normal, but prostate-specific antigen (PSA) levels were elevated at 10.2 ng/mL. An MRI is performed as part of the workup.
Boy presents with abdominal cramping
Ulcerative colitis (UC) is an autoimmune-related inflammatory bowel disease (IBD). It typically develops in the rectum and extends to involve the large intestine. Pediatric UC can have a more severe phenotype than adult disease and may affect a child's pubertal development, bone mineral density, nutrition levels, and social life. It is currently theorized that the age at diagnosis and sex of the patient do not predict disease activity.
UC disease can announce itself as mild, moderate, or severe, and the Pediatric Ulcerative Colitis Activity Index (PUCAI) endoscopic grading is used as a clinical scoring system. The most common presenting symptoms are rectal bleeding, diarrhea, and abdominal pain; among children, the presentation can vary.
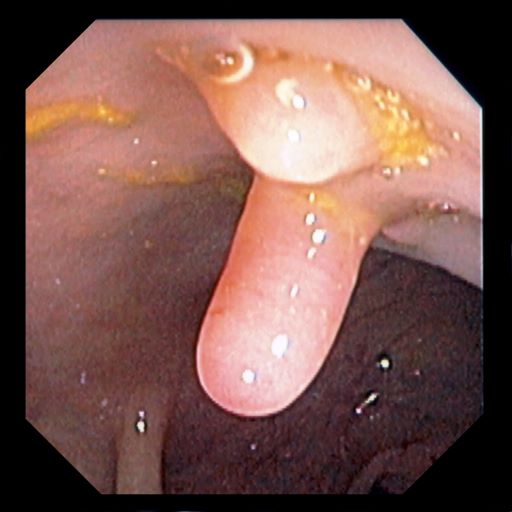
Crohns disease, another IBD, must be carefully ruled out of the differential. Colonoscopy represents the first-line approach in the diagnosis of IBD. The findings that would suggest Crohns disease are sparing of the rectal mucosa, aphthous ulceration, and noncontiguous (or skip) lesions. Micronutrient and vitamin levels are usually low in Crohns disease. And although weight loss, perineal disease, fistulae, and obstruction are commonly seen in the context of Crohns disease, they are uncommon or rare in UC. Bleeding is observed much more frequently in UC.
During UC workup, elevated erythrocyte sedimentation rate and C-reactive protein level often serve as markers of disease activity. Antineutrophil cytoplasmic antibody (ANCA) test is frequently used with suspected UC (though this measure may not correlate with disease activity). In addition, a broad metabolic panel should be performed, along with stool cultures, to rule out infection.
The goals of pediatric UC management are to maintain control of the disease, extend periods of remission, and reduce long-term damage caused by inflammation, all while potentially allowing the patient to function as normally as possible. Anti-inflammatory therapy with 5-aminosalicylic acid agents, such as sulfasalazine and mesalamine, is foundational to treatment. Acute flares of UC in the pediatric population are usually responsive to corticosteroids, but these regimens should be short-term only. Immunomodulatory agents, tumor necrosis factor inhibitors, and newer therapies such as monoclonal antibodies are also used during flares, but only a minority of patients will require these therapies. These are also considered treatment alternatives for patients who are steroid-dependent or steroid-refractory.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships
Ulcerative colitis (UC) is an autoimmune-related inflammatory bowel disease (IBD). It typically develops in the rectum and extends to involve the large intestine. Pediatric UC can have a more severe phenotype than adult disease and may affect a child's pubertal development, bone mineral density, nutrition levels, and social life. It is currently theorized that the age at diagnosis and sex of the patient do not predict disease activity.
UC disease can announce itself as mild, moderate, or severe, and the Pediatric Ulcerative Colitis Activity Index (PUCAI) endoscopic grading is used as a clinical scoring system. The most common presenting symptoms are rectal bleeding, diarrhea, and abdominal pain; among children, the presentation can vary.
Crohns disease, another IBD, must be carefully ruled out of the differential. Colonoscopy represents the first-line approach in the diagnosis of IBD. The findings that would suggest Crohns disease are sparing of the rectal mucosa, aphthous ulceration, and noncontiguous (or skip) lesions. Micronutrient and vitamin levels are usually low in Crohns disease. And although weight loss, perineal disease, fistulae, and obstruction are commonly seen in the context of Crohns disease, they are uncommon or rare in UC. Bleeding is observed much more frequently in UC.
During UC workup, elevated erythrocyte sedimentation rate and C-reactive protein level often serve as markers of disease activity. Antineutrophil cytoplasmic antibody (ANCA) test is frequently used with suspected UC (though this measure may not correlate with disease activity). In addition, a broad metabolic panel should be performed, along with stool cultures, to rule out infection.
The goals of pediatric UC management are to maintain control of the disease, extend periods of remission, and reduce long-term damage caused by inflammation, all while potentially allowing the patient to function as normally as possible. Anti-inflammatory therapy with 5-aminosalicylic acid agents, such as sulfasalazine and mesalamine, is foundational to treatment. Acute flares of UC in the pediatric population are usually responsive to corticosteroids, but these regimens should be short-term only. Immunomodulatory agents, tumor necrosis factor inhibitors, and newer therapies such as monoclonal antibodies are also used during flares, but only a minority of patients will require these therapies. These are also considered treatment alternatives for patients who are steroid-dependent or steroid-refractory.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships
Ulcerative colitis (UC) is an autoimmune-related inflammatory bowel disease (IBD). It typically develops in the rectum and extends to involve the large intestine. Pediatric UC can have a more severe phenotype than adult disease and may affect a child's pubertal development, bone mineral density, nutrition levels, and social life. It is currently theorized that the age at diagnosis and sex of the patient do not predict disease activity.
UC disease can announce itself as mild, moderate, or severe, and the Pediatric Ulcerative Colitis Activity Index (PUCAI) endoscopic grading is used as a clinical scoring system. The most common presenting symptoms are rectal bleeding, diarrhea, and abdominal pain; among children, the presentation can vary.
Crohns disease, another IBD, must be carefully ruled out of the differential. Colonoscopy represents the first-line approach in the diagnosis of IBD. The findings that would suggest Crohns disease are sparing of the rectal mucosa, aphthous ulceration, and noncontiguous (or skip) lesions. Micronutrient and vitamin levels are usually low in Crohns disease. And although weight loss, perineal disease, fistulae, and obstruction are commonly seen in the context of Crohns disease, they are uncommon or rare in UC. Bleeding is observed much more frequently in UC.
During UC workup, elevated erythrocyte sedimentation rate and C-reactive protein level often serve as markers of disease activity. Antineutrophil cytoplasmic antibody (ANCA) test is frequently used with suspected UC (though this measure may not correlate with disease activity). In addition, a broad metabolic panel should be performed, along with stool cultures, to rule out infection.
The goals of pediatric UC management are to maintain control of the disease, extend periods of remission, and reduce long-term damage caused by inflammation, all while potentially allowing the patient to function as normally as possible. Anti-inflammatory therapy with 5-aminosalicylic acid agents, such as sulfasalazine and mesalamine, is foundational to treatment. Acute flares of UC in the pediatric population are usually responsive to corticosteroids, but these regimens should be short-term only. Immunomodulatory agents, tumor necrosis factor inhibitors, and newer therapies such as monoclonal antibodies are also used during flares, but only a minority of patients will require these therapies. These are also considered treatment alternatives for patients who are steroid-dependent or steroid-refractory.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships
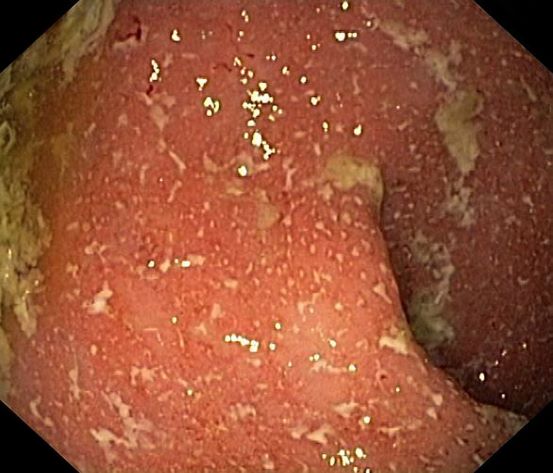
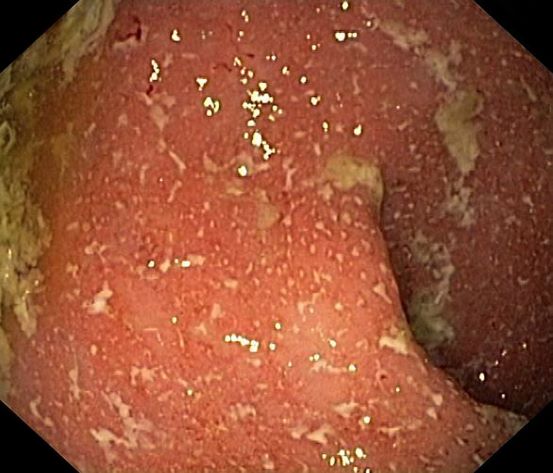
A 5-year-old boy presents with abdominal cramping and bloody stools over the course of 2 days. His mother explains that the onset of diarrhea was insidious. Because the patient has a sensitive stomach, she tries to keep his diet relatively bland, but she worries about what he eats at school. He is slightly underweight for his age group. The family has not traveled recently. The patient does not have a fever, but skin turgor is decreased. There is no evidence of fistulae or abscesses. His complete blood cell count is 10.6 g/dL.
Decreased visual acuity and paresthesia
All of the above conditions can have ophthalmic manifestations, but the majority of optic neuritis cases seen in clinical practice are either sporadic or MS related. Optic neuritis is the first demyelinating event in approximately 20% of patients with MS. It develops in approximately 40% of MS patients during the course of their disease.
Optic neuritis is characterized by loss of vision (or loss of color vision) in the affected eye and pain on movement of the eye (painful ophthalmoplegia). Less often, patients with optic neuritis may describe phosphenes (transient flashes of light or black squares) lasting from hours to months. Phosphenes may occur before or during an optic neuritis event or even several months after recovery.
The diagnosis of optic neuritis is usually made clinically, with direct imaging of the optic nerves showing evidence of optic disc swelling with blurred margins. The real contribution of imaging in the setting of optic neuritis, however, is made by imaging of the brain, not of the optic nerves themselves. MRI of the brain provides information that can change the management of optic neuritis and yields prognostic information regarding the patient's future risk of developing MS. The most valuable predictor of the development of subsequent MS is the presence of white matter abnormalities. Between 27% and 70% of patients (in various studies) with isolated optic neuritis showed abnormal MRI brain findings, as defined by the presence of two or more white matter lesions on T2-weighted images. Patients with two or more lesions may have up to an 80% chance of meeting criteria for MS within the next 5 years.
A gradual recovery of visual acuity with time is characteristic of optic neuritis, although permanent residual deficits in color vision and contrast and brightness sensitivity are common. The symptoms of optic neuritis will usually resolve without medical treatment, although continuing to take regular MS disease-modulating medication is usually helpful. An intravenous steroid or oral prednisone is sometimes recommended to speed recovery. A 3- to 5-day course of high-dose (1 g) IV methylprednisolone, followed by a rapid oral taper of prednisone, has been shown to provide rapid recovery of symptoms in the acute phase. However, IV steroids do little to affect the ultimate visual acuity in patients with optic neuritis.
Typically, patients begin to recover 2-4 weeks after the onset of the vision loss. The optic nerve may take up to 6-12 months to heal completely, but most patients recover as much vision as they are going to within the first few months.
For patients with optic neuritis whose brain lesions on MRI indicate a high risk of developing clinically definite MS, treatment with immunomodulators may be considered. IV immunoglobulin treatment of acute optic neuritis has been shown to have no beneficial effect. In severe cases, plasma exchange may be considered.
Krupa Pandey, MD, Director, Multiple Sclerosis Center, Department of Neurology & Neuroscience Institute, Hackensack University Medical Center; Neurologist, Department of Neurology, Hackensack Meridian Health, Hackensack, NJ.
Krupa Pandey, MD, has serve(d) as a speaker or a member of a speakers bureau for: Bristol-Myers Squibb; Biogen; Alexion; Genentech; Sanofi-Genzyme.
All of the above conditions can have ophthalmic manifestations, but the majority of optic neuritis cases seen in clinical practice are either sporadic or MS related. Optic neuritis is the first demyelinating event in approximately 20% of patients with MS. It develops in approximately 40% of MS patients during the course of their disease.
Optic neuritis is characterized by loss of vision (or loss of color vision) in the affected eye and pain on movement of the eye (painful ophthalmoplegia). Less often, patients with optic neuritis may describe phosphenes (transient flashes of light or black squares) lasting from hours to months. Phosphenes may occur before or during an optic neuritis event or even several months after recovery.
The diagnosis of optic neuritis is usually made clinically, with direct imaging of the optic nerves showing evidence of optic disc swelling with blurred margins. The real contribution of imaging in the setting of optic neuritis, however, is made by imaging of the brain, not of the optic nerves themselves. MRI of the brain provides information that can change the management of optic neuritis and yields prognostic information regarding the patient's future risk of developing MS. The most valuable predictor of the development of subsequent MS is the presence of white matter abnormalities. Between 27% and 70% of patients (in various studies) with isolated optic neuritis showed abnormal MRI brain findings, as defined by the presence of two or more white matter lesions on T2-weighted images. Patients with two or more lesions may have up to an 80% chance of meeting criteria for MS within the next 5 years.
A gradual recovery of visual acuity with time is characteristic of optic neuritis, although permanent residual deficits in color vision and contrast and brightness sensitivity are common. The symptoms of optic neuritis will usually resolve without medical treatment, although continuing to take regular MS disease-modulating medication is usually helpful. An intravenous steroid or oral prednisone is sometimes recommended to speed recovery. A 3- to 5-day course of high-dose (1 g) IV methylprednisolone, followed by a rapid oral taper of prednisone, has been shown to provide rapid recovery of symptoms in the acute phase. However, IV steroids do little to affect the ultimate visual acuity in patients with optic neuritis.
Typically, patients begin to recover 2-4 weeks after the onset of the vision loss. The optic nerve may take up to 6-12 months to heal completely, but most patients recover as much vision as they are going to within the first few months.
For patients with optic neuritis whose brain lesions on MRI indicate a high risk of developing clinically definite MS, treatment with immunomodulators may be considered. IV immunoglobulin treatment of acute optic neuritis has been shown to have no beneficial effect. In severe cases, plasma exchange may be considered.
Krupa Pandey, MD, Director, Multiple Sclerosis Center, Department of Neurology & Neuroscience Institute, Hackensack University Medical Center; Neurologist, Department of Neurology, Hackensack Meridian Health, Hackensack, NJ.
Krupa Pandey, MD, has serve(d) as a speaker or a member of a speakers bureau for: Bristol-Myers Squibb; Biogen; Alexion; Genentech; Sanofi-Genzyme.
All of the above conditions can have ophthalmic manifestations, but the majority of optic neuritis cases seen in clinical practice are either sporadic or MS related. Optic neuritis is the first demyelinating event in approximately 20% of patients with MS. It develops in approximately 40% of MS patients during the course of their disease.
Optic neuritis is characterized by loss of vision (or loss of color vision) in the affected eye and pain on movement of the eye (painful ophthalmoplegia). Less often, patients with optic neuritis may describe phosphenes (transient flashes of light or black squares) lasting from hours to months. Phosphenes may occur before or during an optic neuritis event or even several months after recovery.
The diagnosis of optic neuritis is usually made clinically, with direct imaging of the optic nerves showing evidence of optic disc swelling with blurred margins. The real contribution of imaging in the setting of optic neuritis, however, is made by imaging of the brain, not of the optic nerves themselves. MRI of the brain provides information that can change the management of optic neuritis and yields prognostic information regarding the patient's future risk of developing MS. The most valuable predictor of the development of subsequent MS is the presence of white matter abnormalities. Between 27% and 70% of patients (in various studies) with isolated optic neuritis showed abnormal MRI brain findings, as defined by the presence of two or more white matter lesions on T2-weighted images. Patients with two or more lesions may have up to an 80% chance of meeting criteria for MS within the next 5 years.
A gradual recovery of visual acuity with time is characteristic of optic neuritis, although permanent residual deficits in color vision and contrast and brightness sensitivity are common. The symptoms of optic neuritis will usually resolve without medical treatment, although continuing to take regular MS disease-modulating medication is usually helpful. An intravenous steroid or oral prednisone is sometimes recommended to speed recovery. A 3- to 5-day course of high-dose (1 g) IV methylprednisolone, followed by a rapid oral taper of prednisone, has been shown to provide rapid recovery of symptoms in the acute phase. However, IV steroids do little to affect the ultimate visual acuity in patients with optic neuritis.
Typically, patients begin to recover 2-4 weeks after the onset of the vision loss. The optic nerve may take up to 6-12 months to heal completely, but most patients recover as much vision as they are going to within the first few months.
For patients with optic neuritis whose brain lesions on MRI indicate a high risk of developing clinically definite MS, treatment with immunomodulators may be considered. IV immunoglobulin treatment of acute optic neuritis has been shown to have no beneficial effect. In severe cases, plasma exchange may be considered.
Krupa Pandey, MD, Director, Multiple Sclerosis Center, Department of Neurology & Neuroscience Institute, Hackensack University Medical Center; Neurologist, Department of Neurology, Hackensack Meridian Health, Hackensack, NJ.
Krupa Pandey, MD, has serve(d) as a speaker or a member of a speakers bureau for: Bristol-Myers Squibb; Biogen; Alexion; Genentech; Sanofi-Genzyme.
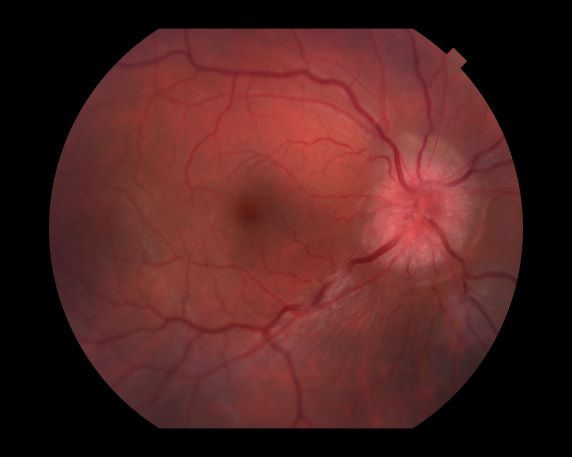
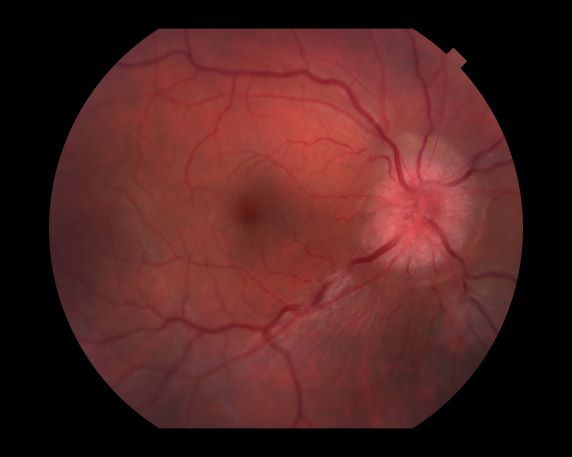
A 44-year-old woman presents with decreased visual acuity, painful ophthalmoplegia, photophobia, and paresthesia of the left hand. The patient's ocular history was unremarkable. Her medical history was significant only for recurrent urinary tract infections. She did not have a history of neurologic problems and reported that she did not have dizziness, tingling, tremors, sensory changes, speech changes, or focal weaknesses. Besides current use of naproxen, she said she was not taking any other medications. Her family ocular history was significant for glaucoma in her father and paternal grandfather. Her maternal grandfather died at age 58 of multiple sclerosis (MS).
Woman with dyspnea and persistent cough
On the basis of the patient's presentation and imaging results, the likely diagnosis is non–small cell cancer (NSCLC) of an adenocarcinoma subtype. NSCLC makes up about 80% of all lung cancer cases. Adenocarcinoma in particular is the most common type of lung cancer in the United States, accounting for about 40% of cases, and it is the most common histology among nonsmokers. Women are more likely to develop this subtype of NSCLC and are generally younger when they present with symptoms. This type of cancer arises from the bronchial mucosal glands and usually develops in a peripheral location within the lung.
In the course of workup, immunohistochemical (IHC) analyses are used to identify tumor type and lineage (adenocarcinoma, squamous cell carcinoma, metastatic malignancy, or primary pleural mesothelioma). Separate IHC analyses are then used to guide treatment decisions, identifying whether ALK inhibitor therapy or programmed cell death protein ligand 1 (PD-L1) inhibitor therapy would be appropriate.
Tissue should also be conserved for molecular testing. NCCN guidelines advise that all patients with adenocarcinoma should be tested for EGFR mutations, and DNA mutational analysis is the preferred method for assessment. Patients should also undergo routine biomarker testing, with an eye toward ALK, RET, and ROS1 rearrangements, BRAF mutations, c-MET and exon 14 skipping mutations, and PD-L1 expression levels. For patients with metastatic NSCLC, PD-L1 IHC testing is recommended.
Most cases of lung cancer are diagnosed at a late stage, when symptoms have already begun to manifest. Of note, however, women with adenocarcinoma are more likely to present with localized disease. Treatment is largely influenced by the presence of targetable mutations. Among adenocarcinoma cases, the most common mutations are in the EGFR and KRAS genes.
For patients who are EGFR mutation positive (exon 10 deletion or L858R), osimertinib is the recommended first-line therapy. For patients who are positive for the EGFR exon 20 insertion mutation, initial systemic therapy options for adenocarcinoma are appropriate; the preferred regimen being pembrolizumab-carboplatin-pemetrexed if there are no contraindications to programmed cell death protein 1 (PD-1) or PD-L1 inhibitors.
KRAS mutations, unlike EGFR mutations, are associated with smoking. Because overlapping targetable alterations are uncommon, identification of KRAS mutations suggests that these patients will not benefit from additional molecular testing. Again, initial systemic therapy options for adenocarcinoma are appropriate, but the presence of KRAS mutation predicts a poor response to EGFR tyrosine kinase inhibitors. The FDA approved a KRAS inhibitor in June 2021 and immune checkpoint inhibitors appear to be beneficial in this population.
Maurie Markman, MD, President, Department of Medical Oncology, Cancer Treatment Centers of America.
Maurie Markman, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Merck
Serve(d) as a speaker or a member of a speakers bureau for: AstraZeneca; Novis; Glaxo Smith Kline
Received research grant from: AstraZeneca; Novis; GSK; Merck
On the basis of the patient's presentation and imaging results, the likely diagnosis is non–small cell cancer (NSCLC) of an adenocarcinoma subtype. NSCLC makes up about 80% of all lung cancer cases. Adenocarcinoma in particular is the most common type of lung cancer in the United States, accounting for about 40% of cases, and it is the most common histology among nonsmokers. Women are more likely to develop this subtype of NSCLC and are generally younger when they present with symptoms. This type of cancer arises from the bronchial mucosal glands and usually develops in a peripheral location within the lung.
In the course of workup, immunohistochemical (IHC) analyses are used to identify tumor type and lineage (adenocarcinoma, squamous cell carcinoma, metastatic malignancy, or primary pleural mesothelioma). Separate IHC analyses are then used to guide treatment decisions, identifying whether ALK inhibitor therapy or programmed cell death protein ligand 1 (PD-L1) inhibitor therapy would be appropriate.
Tissue should also be conserved for molecular testing. NCCN guidelines advise that all patients with adenocarcinoma should be tested for EGFR mutations, and DNA mutational analysis is the preferred method for assessment. Patients should also undergo routine biomarker testing, with an eye toward ALK, RET, and ROS1 rearrangements, BRAF mutations, c-MET and exon 14 skipping mutations, and PD-L1 expression levels. For patients with metastatic NSCLC, PD-L1 IHC testing is recommended.
Most cases of lung cancer are diagnosed at a late stage, when symptoms have already begun to manifest. Of note, however, women with adenocarcinoma are more likely to present with localized disease. Treatment is largely influenced by the presence of targetable mutations. Among adenocarcinoma cases, the most common mutations are in the EGFR and KRAS genes.
For patients who are EGFR mutation positive (exon 10 deletion or L858R), osimertinib is the recommended first-line therapy. For patients who are positive for the EGFR exon 20 insertion mutation, initial systemic therapy options for adenocarcinoma are appropriate; the preferred regimen being pembrolizumab-carboplatin-pemetrexed if there are no contraindications to programmed cell death protein 1 (PD-1) or PD-L1 inhibitors.
KRAS mutations, unlike EGFR mutations, are associated with smoking. Because overlapping targetable alterations are uncommon, identification of KRAS mutations suggests that these patients will not benefit from additional molecular testing. Again, initial systemic therapy options for adenocarcinoma are appropriate, but the presence of KRAS mutation predicts a poor response to EGFR tyrosine kinase inhibitors. The FDA approved a KRAS inhibitor in June 2021 and immune checkpoint inhibitors appear to be beneficial in this population.
Maurie Markman, MD, President, Department of Medical Oncology, Cancer Treatment Centers of America.
Maurie Markman, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Merck
Serve(d) as a speaker or a member of a speakers bureau for: AstraZeneca; Novis; Glaxo Smith Kline
Received research grant from: AstraZeneca; Novis; GSK; Merck
On the basis of the patient's presentation and imaging results, the likely diagnosis is non–small cell cancer (NSCLC) of an adenocarcinoma subtype. NSCLC makes up about 80% of all lung cancer cases. Adenocarcinoma in particular is the most common type of lung cancer in the United States, accounting for about 40% of cases, and it is the most common histology among nonsmokers. Women are more likely to develop this subtype of NSCLC and are generally younger when they present with symptoms. This type of cancer arises from the bronchial mucosal glands and usually develops in a peripheral location within the lung.
In the course of workup, immunohistochemical (IHC) analyses are used to identify tumor type and lineage (adenocarcinoma, squamous cell carcinoma, metastatic malignancy, or primary pleural mesothelioma). Separate IHC analyses are then used to guide treatment decisions, identifying whether ALK inhibitor therapy or programmed cell death protein ligand 1 (PD-L1) inhibitor therapy would be appropriate.
Tissue should also be conserved for molecular testing. NCCN guidelines advise that all patients with adenocarcinoma should be tested for EGFR mutations, and DNA mutational analysis is the preferred method for assessment. Patients should also undergo routine biomarker testing, with an eye toward ALK, RET, and ROS1 rearrangements, BRAF mutations, c-MET and exon 14 skipping mutations, and PD-L1 expression levels. For patients with metastatic NSCLC, PD-L1 IHC testing is recommended.
Most cases of lung cancer are diagnosed at a late stage, when symptoms have already begun to manifest. Of note, however, women with adenocarcinoma are more likely to present with localized disease. Treatment is largely influenced by the presence of targetable mutations. Among adenocarcinoma cases, the most common mutations are in the EGFR and KRAS genes.
For patients who are EGFR mutation positive (exon 10 deletion or L858R), osimertinib is the recommended first-line therapy. For patients who are positive for the EGFR exon 20 insertion mutation, initial systemic therapy options for adenocarcinoma are appropriate; the preferred regimen being pembrolizumab-carboplatin-pemetrexed if there are no contraindications to programmed cell death protein 1 (PD-1) or PD-L1 inhibitors.
KRAS mutations, unlike EGFR mutations, are associated with smoking. Because overlapping targetable alterations are uncommon, identification of KRAS mutations suggests that these patients will not benefit from additional molecular testing. Again, initial systemic therapy options for adenocarcinoma are appropriate, but the presence of KRAS mutation predicts a poor response to EGFR tyrosine kinase inhibitors. The FDA approved a KRAS inhibitor in June 2021 and immune checkpoint inhibitors appear to be beneficial in this population.
Maurie Markman, MD, President, Department of Medical Oncology, Cancer Treatment Centers of America.
Maurie Markman, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Merck
Serve(d) as a speaker or a member of a speakers bureau for: AstraZeneca; Novis; Glaxo Smith Kline
Received research grant from: AstraZeneca; Novis; GSK; Merck
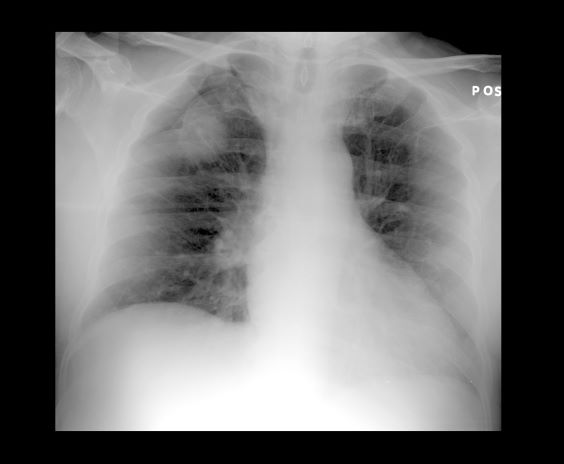
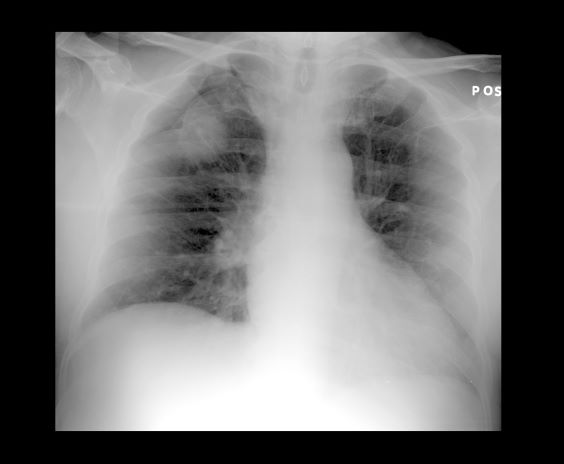
A 52-year-old woman presents with dyspnea and a persistent cough. She is 5 ft 5 in and weighs 155 lb, with no recent significant weight loss. She has been experiencing symptoms for a few months, which she originally thought might be related to her history of GERD. She reports that she was a light smoker before she had children but has not smoked regularly in about 20 years. Because of the patient's respiratory symptoms, chest radiography is ordered.
This frontal projection chest radiography clearly demonstrates a mass in the upper lobe of the right lung that represents the appearance of lung cancer (malignancy).
3-year-history of difficulty walking
The patient has probably transitioned to the secondary progressive form of multiple sclerosis (MS). Four phenotypes have been identified in MS, with relapsing-remitting MS (RRMS) representing the most common and secondary progressive MS (SPMS) the second most common. RRMS is thought to begin as an inflammatory disease that over time becomes primarily neurodegenerative. The course of RRMS is marked by episodes of neurologic deficit followed by periods of remission which may be asymptomatic. When symptoms do not resolve — becoming fixed without remission — this is a sign of progression to SPMS. One in two RRMS patients will develop SPMS within 15 years of their diagnosis, leading to a progressive decrease of neurologic function and limitation of daily activities. Risk factors for developing SPMS include older age at onset of RRMS, longer duration of RRMS, and more cortical inflammatory lesions at baseline.
RRMS is diagnosed through clinical findings and laboratory results, the main approaches being MRI of the brain and spinal cord, and examination of cerebrospinal fluid. Neurologic symptoms must be consistent with those typically seen in MS, with deficit lasting for days to weeks. MRI is useful in monitoring disease progression (ie, new lesions that develop during relapses in RRMS). There are no universally accepted diagnostic criteria for SPMS, however. A patient usually can be diagnosed upon meeting these criteria: The patient was previously diagnosed with RRMS; the patient's symptoms are gradually worsening; this worsening is not tied to a relapse; and this worsening has been observed for 6 months or longer. Of note, SPMS' symptom-worsening characteristics can be subtle and difficult for patients to detect, and delays in diagnosis of up to several years are common.
Recognizing the onset of transition to SPMS is critical, as early initiation of therapy is thought to slow disease progression, the primary goal of treatment. In patients with SPMS, adhering to a holistic health program and managing comorbidities, especially vascular risk factors, can help preserve the health and functions of both the central nervous system and brain. Patients with SPMS who experience relapses or demonstrate new lesion formation as captured on MRI are thought to have active SPMS (aSPMS) and generally benefit from disease-modifying therapy (DMT). There is generally a transition period of about 5 years during which SPMS patients will still have a relapsing form of the disease, meaning that DMTs have proven to be effective in managing progressive MS should theoretically be beneficial for SPMS during this period. There are FDA-approved treatments for aSPMS, but off-label use is acceptable of those medications indicated for relapsing MS in those patients with evidence of relapses or new MRI activity.
Krupa Pandey, MD, Director, Multiple Sclerosis Center, Department of Neurology & Neuroscience Institute, Hackensack University Medical Center; Neurologist, Department of Neurology, Hackensack Meridian Health, Hackensack, NJ
Krupa Pandey, MD, has serve(d) as a speaker or a member of a speakers bureau for: Bristol-Myers Squibb; Biogen; Alexion; Genentech; Sanofi-Genzyme
The patient has probably transitioned to the secondary progressive form of multiple sclerosis (MS). Four phenotypes have been identified in MS, with relapsing-remitting MS (RRMS) representing the most common and secondary progressive MS (SPMS) the second most common. RRMS is thought to begin as an inflammatory disease that over time becomes primarily neurodegenerative. The course of RRMS is marked by episodes of neurologic deficit followed by periods of remission which may be asymptomatic. When symptoms do not resolve — becoming fixed without remission — this is a sign of progression to SPMS. One in two RRMS patients will develop SPMS within 15 years of their diagnosis, leading to a progressive decrease of neurologic function and limitation of daily activities. Risk factors for developing SPMS include older age at onset of RRMS, longer duration of RRMS, and more cortical inflammatory lesions at baseline.
RRMS is diagnosed through clinical findings and laboratory results, the main approaches being MRI of the brain and spinal cord, and examination of cerebrospinal fluid. Neurologic symptoms must be consistent with those typically seen in MS, with deficit lasting for days to weeks. MRI is useful in monitoring disease progression (ie, new lesions that develop during relapses in RRMS). There are no universally accepted diagnostic criteria for SPMS, however. A patient usually can be diagnosed upon meeting these criteria: The patient was previously diagnosed with RRMS; the patient's symptoms are gradually worsening; this worsening is not tied to a relapse; and this worsening has been observed for 6 months or longer. Of note, SPMS' symptom-worsening characteristics can be subtle and difficult for patients to detect, and delays in diagnosis of up to several years are common.
Recognizing the onset of transition to SPMS is critical, as early initiation of therapy is thought to slow disease progression, the primary goal of treatment. In patients with SPMS, adhering to a holistic health program and managing comorbidities, especially vascular risk factors, can help preserve the health and functions of both the central nervous system and brain. Patients with SPMS who experience relapses or demonstrate new lesion formation as captured on MRI are thought to have active SPMS (aSPMS) and generally benefit from disease-modifying therapy (DMT). There is generally a transition period of about 5 years during which SPMS patients will still have a relapsing form of the disease, meaning that DMTs have proven to be effective in managing progressive MS should theoretically be beneficial for SPMS during this period. There are FDA-approved treatments for aSPMS, but off-label use is acceptable of those medications indicated for relapsing MS in those patients with evidence of relapses or new MRI activity.
Krupa Pandey, MD, Director, Multiple Sclerosis Center, Department of Neurology & Neuroscience Institute, Hackensack University Medical Center; Neurologist, Department of Neurology, Hackensack Meridian Health, Hackensack, NJ
Krupa Pandey, MD, has serve(d) as a speaker or a member of a speakers bureau for: Bristol-Myers Squibb; Biogen; Alexion; Genentech; Sanofi-Genzyme
The patient has probably transitioned to the secondary progressive form of multiple sclerosis (MS). Four phenotypes have been identified in MS, with relapsing-remitting MS (RRMS) representing the most common and secondary progressive MS (SPMS) the second most common. RRMS is thought to begin as an inflammatory disease that over time becomes primarily neurodegenerative. The course of RRMS is marked by episodes of neurologic deficit followed by periods of remission which may be asymptomatic. When symptoms do not resolve — becoming fixed without remission — this is a sign of progression to SPMS. One in two RRMS patients will develop SPMS within 15 years of their diagnosis, leading to a progressive decrease of neurologic function and limitation of daily activities. Risk factors for developing SPMS include older age at onset of RRMS, longer duration of RRMS, and more cortical inflammatory lesions at baseline.
RRMS is diagnosed through clinical findings and laboratory results, the main approaches being MRI of the brain and spinal cord, and examination of cerebrospinal fluid. Neurologic symptoms must be consistent with those typically seen in MS, with deficit lasting for days to weeks. MRI is useful in monitoring disease progression (ie, new lesions that develop during relapses in RRMS). There are no universally accepted diagnostic criteria for SPMS, however. A patient usually can be diagnosed upon meeting these criteria: The patient was previously diagnosed with RRMS; the patient's symptoms are gradually worsening; this worsening is not tied to a relapse; and this worsening has been observed for 6 months or longer. Of note, SPMS' symptom-worsening characteristics can be subtle and difficult for patients to detect, and delays in diagnosis of up to several years are common.
Recognizing the onset of transition to SPMS is critical, as early initiation of therapy is thought to slow disease progression, the primary goal of treatment. In patients with SPMS, adhering to a holistic health program and managing comorbidities, especially vascular risk factors, can help preserve the health and functions of both the central nervous system and brain. Patients with SPMS who experience relapses or demonstrate new lesion formation as captured on MRI are thought to have active SPMS (aSPMS) and generally benefit from disease-modifying therapy (DMT). There is generally a transition period of about 5 years during which SPMS patients will still have a relapsing form of the disease, meaning that DMTs have proven to be effective in managing progressive MS should theoretically be beneficial for SPMS during this period. There are FDA-approved treatments for aSPMS, but off-label use is acceptable of those medications indicated for relapsing MS in those patients with evidence of relapses or new MRI activity.
Krupa Pandey, MD, Director, Multiple Sclerosis Center, Department of Neurology & Neuroscience Institute, Hackensack University Medical Center; Neurologist, Department of Neurology, Hackensack Meridian Health, Hackensack, NJ
Krupa Pandey, MD, has serve(d) as a speaker or a member of a speakers bureau for: Bristol-Myers Squibb; Biogen; Alexion; Genentech; Sanofi-Genzyme
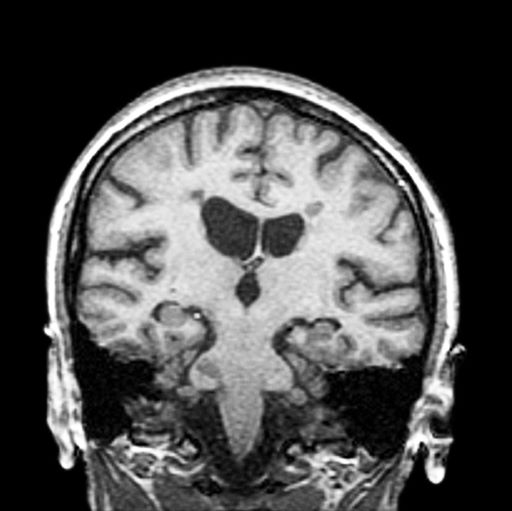
A 51-year-old woman presents with a 3-year history of difficulty walking. She says that it is difficult to pinpoint when her walking problems began but reports that it has been gradual. She recalls about 10 years back a history of numbness and tingling in her hands that improved over the course of a few weeks without any further workup. She also recalls blurry vision and loss of color perception in her left eye 5 years ago while traveling for work. Because the symptoms resolved on their own over 6-8 weeks, she never sought care. MRI shows plaques of demyelination.
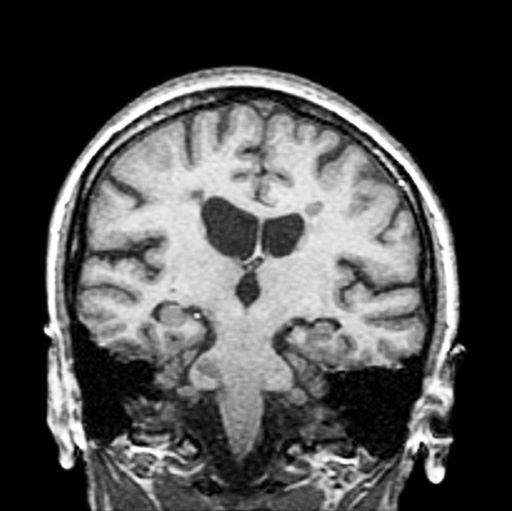
History of dysphagia and abdominal pain
The diagnosis is squamous cell carcinoma. A central or hilar mass is most likely to be a squamous cell carcinoma or a small cell tumor and less commonly an adenocarcinoma. Histologically, when there is lack of cohesion among the epithelial cells due to malignant changes, the cells get arranged in a concentric manner. The fate of a squamous cell is to form keratin, so these cells lay down keratin in a concentric manner and then appear as keratin pearls.
This patient's tumor is found to have programmed cell death–ligand 1 ≥ 1% and has no actionable molecular markers. The patient has a performance status score of 1. In a patient with advanced or metastatic squamous cell carcinoma with a performance status score of 1, the National Comprehensive Cancer Network recommends pembrolizumab/carboplatin/paclitaxel or pembrolizumab/carboplatin/albumin-bound paclitaxel as preferred regimens. The pembrolizumab component is based on the results of the KEYNOTE-407 trial. In patients with previously untreated metastatic, squamous non-small cell lung cancer, the addition of pembrolizumab to chemotherapy with carboplatin plus paclitaxel or nab-paclitaxel resulted in significantly longer overall survival and progression-free survival than chemotherapy alone.
Maurie Markman, MD, President, Department of Medical Oncology, Cancer Treatment Centers of America.
Maurie Markman, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Merck
Serve(d) as a speaker or a member of a speakers bureau for: AstraZeneca; Novis; Glaxo Smith Kline
Received research grant from: AstraZeneca; Novis; GSK; Merck
The diagnosis is squamous cell carcinoma. A central or hilar mass is most likely to be a squamous cell carcinoma or a small cell tumor and less commonly an adenocarcinoma. Histologically, when there is lack of cohesion among the epithelial cells due to malignant changes, the cells get arranged in a concentric manner. The fate of a squamous cell is to form keratin, so these cells lay down keratin in a concentric manner and then appear as keratin pearls.
This patient's tumor is found to have programmed cell death–ligand 1 ≥ 1% and has no actionable molecular markers. The patient has a performance status score of 1. In a patient with advanced or metastatic squamous cell carcinoma with a performance status score of 1, the National Comprehensive Cancer Network recommends pembrolizumab/carboplatin/paclitaxel or pembrolizumab/carboplatin/albumin-bound paclitaxel as preferred regimens. The pembrolizumab component is based on the results of the KEYNOTE-407 trial. In patients with previously untreated metastatic, squamous non-small cell lung cancer, the addition of pembrolizumab to chemotherapy with carboplatin plus paclitaxel or nab-paclitaxel resulted in significantly longer overall survival and progression-free survival than chemotherapy alone.
Maurie Markman, MD, President, Department of Medical Oncology, Cancer Treatment Centers of America.
Maurie Markman, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Merck
Serve(d) as a speaker or a member of a speakers bureau for: AstraZeneca; Novis; Glaxo Smith Kline
Received research grant from: AstraZeneca; Novis; GSK; Merck
The diagnosis is squamous cell carcinoma. A central or hilar mass is most likely to be a squamous cell carcinoma or a small cell tumor and less commonly an adenocarcinoma. Histologically, when there is lack of cohesion among the epithelial cells due to malignant changes, the cells get arranged in a concentric manner. The fate of a squamous cell is to form keratin, so these cells lay down keratin in a concentric manner and then appear as keratin pearls.
This patient's tumor is found to have programmed cell death–ligand 1 ≥ 1% and has no actionable molecular markers. The patient has a performance status score of 1. In a patient with advanced or metastatic squamous cell carcinoma with a performance status score of 1, the National Comprehensive Cancer Network recommends pembrolizumab/carboplatin/paclitaxel or pembrolizumab/carboplatin/albumin-bound paclitaxel as preferred regimens. The pembrolizumab component is based on the results of the KEYNOTE-407 trial. In patients with previously untreated metastatic, squamous non-small cell lung cancer, the addition of pembrolizumab to chemotherapy with carboplatin plus paclitaxel or nab-paclitaxel resulted in significantly longer overall survival and progression-free survival than chemotherapy alone.
Maurie Markman, MD, President, Department of Medical Oncology, Cancer Treatment Centers of America.
Maurie Markman, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Merck
Serve(d) as a speaker or a member of a speakers bureau for: AstraZeneca; Novis; Glaxo Smith Kline
Received research grant from: AstraZeneca; Novis; GSK; Merck
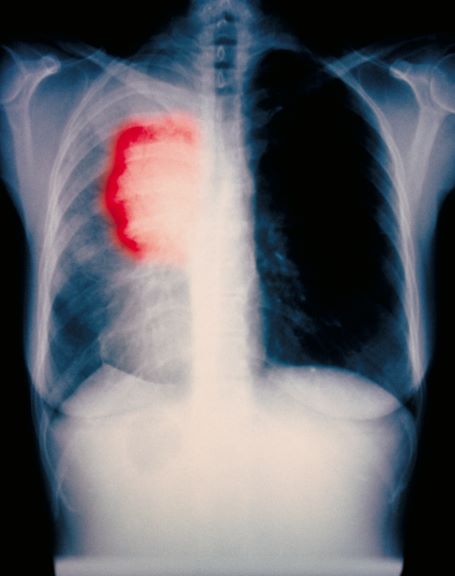
A 59-year-old woman presents with a 4-month history of dysphagia when eating solids in addition to nausea and abdominal pain. She also reports recent hemoptysis and the onset of hoarseness. She has had an unintentional 22-lb weight loss over the past 6 months. She has a history of emphysema. She takes no medication. She has a 26 pack-year history of cigarette smoking. She is 5 feet 4 in tall and weighs 105 lb, with a BMI of 18. Her vital signs are within normal limits. Chest auscultation reveals diminished breath sounds over the right lung fields. Chest radiography reveals a right-sided 6-cm hilar mass. Laboratory studies show a serum calcium level of 12 mg/dL (normal range, 8.5-10.5 mg/dL). A CT scan revealed a spiculated lesion and hepatic metastases. A biopsy was performed. Keratinization was found in the form of keratin pearls.
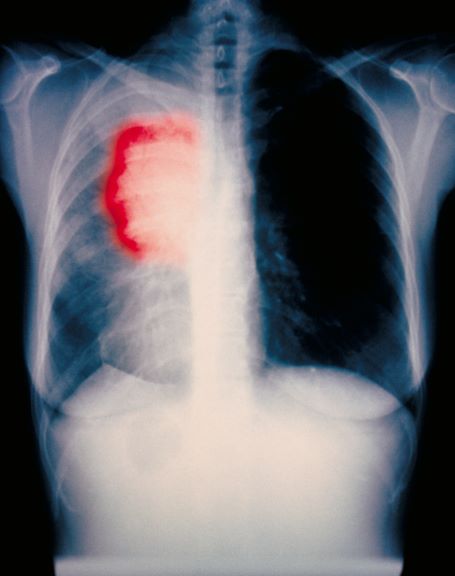
Woman presents with weight loss and nausea
It is likely that the polypoid appearance of the colonic lining is a result of chronic inflammation of longstanding Crohn disease with an ileocolonic manifestation. Crohn disease is an idiopathic, chronic inflammatory bowel disease characterized by cycles of relapse and remission. These asymptomatic periods can last for several months up to a few years, as reported by the patient in this case. Up to 50% of cases of Crohn disease are characterized by ileocolitis, or inflammation of the ileum and the colon. Although postinflammatory polyps are a cancer risk factor for inflammatory bowel disease and pseudopolyps are associated with severe disease, their appearance is not necessarily a poor prognostic factor.
When assessing ongoing disease activity in ileocolonic Crohn disease or ulcerative colitis, colonoscopy represents the first-line approach. Endoscopic visualization and biopsy are critical components of the diagnosis. Alternatively, cross-sectional imaging can be used to assess disease phenotype. In addition, plain radiography or a CT scan of the abdomen can identify bowel obstruction and scanning of the pelvis can detect any intra-abdominal abscesses. Ulcerative colitis looms large in the differential diagnosis. Although weight loss, perineal disease, fistulae, and obstruction are common in Crohn disease, they are uncommon or rare in ulcerative colitis, although bleeding is observed much more frequently in ulcerative colitis.
Treatment of Crohn disease is based on the severity, location, and subtype (inflammatory, stricturing, or penetrating). There is also now a focus on determining which patients are at risk for a more severe disease course and may require earlier and more aggressive therapies. Crohn disease is primarily managed through the introduction of early immunosuppressive or combination therapy with biologic agents in high-risk patients, as well as complementary diet modification. Although most patients will ultimately undergo surgery, there is no curative approach, unlike in ulcerative colitis.
In its clinical care pathway, the American Gastroenterological Association supports a top-down approach to therapy for adult patients with moderate to severe luminal Crohn disease (defining moderate to severe disease as having a Crohn Disease Activity Index score of 220 or higher, or having a high risk of complications). This approach supports the early use of biologic agents, with or without immunomodulators, over a stepwise strategy. The patient’s response to this new regimen should be determined in the 12-week period after the initiation of therapy. Endoscopy or transmural responses to therapy should be assessed after 6 months.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships.
It is likely that the polypoid appearance of the colonic lining is a result of chronic inflammation of longstanding Crohn disease with an ileocolonic manifestation. Crohn disease is an idiopathic, chronic inflammatory bowel disease characterized by cycles of relapse and remission. These asymptomatic periods can last for several months up to a few years, as reported by the patient in this case. Up to 50% of cases of Crohn disease are characterized by ileocolitis, or inflammation of the ileum and the colon. Although postinflammatory polyps are a cancer risk factor for inflammatory bowel disease and pseudopolyps are associated with severe disease, their appearance is not necessarily a poor prognostic factor.
When assessing ongoing disease activity in ileocolonic Crohn disease or ulcerative colitis, colonoscopy represents the first-line approach. Endoscopic visualization and biopsy are critical components of the diagnosis. Alternatively, cross-sectional imaging can be used to assess disease phenotype. In addition, plain radiography or a CT scan of the abdomen can identify bowel obstruction and scanning of the pelvis can detect any intra-abdominal abscesses. Ulcerative colitis looms large in the differential diagnosis. Although weight loss, perineal disease, fistulae, and obstruction are common in Crohn disease, they are uncommon or rare in ulcerative colitis, although bleeding is observed much more frequently in ulcerative colitis.
Treatment of Crohn disease is based on the severity, location, and subtype (inflammatory, stricturing, or penetrating). There is also now a focus on determining which patients are at risk for a more severe disease course and may require earlier and more aggressive therapies. Crohn disease is primarily managed through the introduction of early immunosuppressive or combination therapy with biologic agents in high-risk patients, as well as complementary diet modification. Although most patients will ultimately undergo surgery, there is no curative approach, unlike in ulcerative colitis.
In its clinical care pathway, the American Gastroenterological Association supports a top-down approach to therapy for adult patients with moderate to severe luminal Crohn disease (defining moderate to severe disease as having a Crohn Disease Activity Index score of 220 or higher, or having a high risk of complications). This approach supports the early use of biologic agents, with or without immunomodulators, over a stepwise strategy. The patient’s response to this new regimen should be determined in the 12-week period after the initiation of therapy. Endoscopy or transmural responses to therapy should be assessed after 6 months.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships.
It is likely that the polypoid appearance of the colonic lining is a result of chronic inflammation of longstanding Crohn disease with an ileocolonic manifestation. Crohn disease is an idiopathic, chronic inflammatory bowel disease characterized by cycles of relapse and remission. These asymptomatic periods can last for several months up to a few years, as reported by the patient in this case. Up to 50% of cases of Crohn disease are characterized by ileocolitis, or inflammation of the ileum and the colon. Although postinflammatory polyps are a cancer risk factor for inflammatory bowel disease and pseudopolyps are associated with severe disease, their appearance is not necessarily a poor prognostic factor.
When assessing ongoing disease activity in ileocolonic Crohn disease or ulcerative colitis, colonoscopy represents the first-line approach. Endoscopic visualization and biopsy are critical components of the diagnosis. Alternatively, cross-sectional imaging can be used to assess disease phenotype. In addition, plain radiography or a CT scan of the abdomen can identify bowel obstruction and scanning of the pelvis can detect any intra-abdominal abscesses. Ulcerative colitis looms large in the differential diagnosis. Although weight loss, perineal disease, fistulae, and obstruction are common in Crohn disease, they are uncommon or rare in ulcerative colitis, although bleeding is observed much more frequently in ulcerative colitis.
Treatment of Crohn disease is based on the severity, location, and subtype (inflammatory, stricturing, or penetrating). There is also now a focus on determining which patients are at risk for a more severe disease course and may require earlier and more aggressive therapies. Crohn disease is primarily managed through the introduction of early immunosuppressive or combination therapy with biologic agents in high-risk patients, as well as complementary diet modification. Although most patients will ultimately undergo surgery, there is no curative approach, unlike in ulcerative colitis.
In its clinical care pathway, the American Gastroenterological Association supports a top-down approach to therapy for adult patients with moderate to severe luminal Crohn disease (defining moderate to severe disease as having a Crohn Disease Activity Index score of 220 or higher, or having a high risk of complications). This approach supports the early use of biologic agents, with or without immunomodulators, over a stepwise strategy. The patient’s response to this new regimen should be determined in the 12-week period after the initiation of therapy. Endoscopy or transmural responses to therapy should be assessed after 6 months.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships.
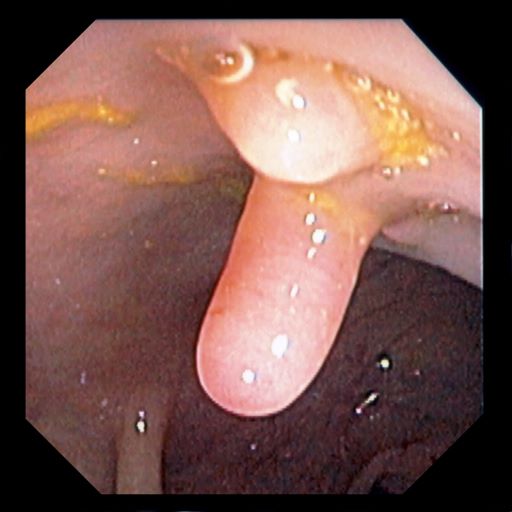
A 42-year-old woman presents with pain in her right abdomen, nausea, and diarrhea. She reports a weight loss of about 12 lb in the past several weeks because of a disinterest in food, which typically exacerbates her symptoms. She explains that she has been experiencing mounting stress at work and abdominal cramping and fatigue. Her family medical history is significant for pancreatic cancer and multiple sclerosis. She has not experienced any significant medical events in the past few years. Endoscopy shows polypoid appearance of the colonic lining.