User login
Lithium-associated hypercalcemia: Monitoring and management
Hypercalcemia is a well-known but underrecognized adverse effect of lithium. Most patients with lithium-associated hypercalcemia (LAH) have either nonspecific symptoms (eg, persistent tiredness, constipation, polyuria, polydipsia) or no symptoms. Clinically, LAH differs from primary hyperparathyroidism, though the management protocol of these 2 conditions is almost the same. In this article, we discuss how lithium can affect calcium and parathyroid hormone (PTH) levels and how LAH and lithium-associated hyperparathyroidism (LAHP) differs from primary hyperparathyroidism. We also outline a suggested approach to monitoring and management.
An insidious problem
Due to the varying definitions and methods used to assess hypercalcemia, the reported prevalence of LAH varies from 4.3% to 80%.1 McKnight et al2 conducted a systematic review and meta-analysis of studies of the relationship between lithium and parathyroid function that included 14 case-control studies, 36 case reports, and 6 cross-sectional studies without a control group. They found that the levels of calcium and PTH were 10% higher in lithium-treated patients than in controls.2
Pathophysiology. Lithium is known to increase both calcium and PTH levels. PTH is responsible for calcium homeostasis. It is secreted in response to low calcium levels, which it increases by its action on bones, intestines, and kidneys. Vitamin D also plays a crucial role in calcium homeostasis. A deficiency of vitamin D triggers a compensatory increase in PTH to maintain calcium levels.3
Calcium and PTH levels increase soon after administration of lithium, but the rise is usually mild and insidious. In a small proportion of patients who receive long-term lithium treatment, calcium levels can exceed the normal range. Patients who develop LAH typically have serum calcium levels slightly above the normal range and PTH levels ranging from the higher side of the normal range to several times the upper limit of the normal range. Patients might also experience elevated PTH levels without any increase in calcium levels. Lithium can affect calcium and PTH levels in multiple ways. For instance, it increases the reabsorption of calcium in the kidney as well as the reset point of calcium-sensing receptors. Therefore, only higher levels of calcium can inhibit the release of PTH. Hence, in cases where the PTH level is within the normal range, it is generally higher than would be expected for a given serum calcium level. Lithium can also directly affect the parathyroid glands and can lead to either single-nodule or multimodule hyperplasia.4
Long-term lithium use can cause chronic kidney disease (CKD), which in turn leads to vitamin D deficiency and hyperparathyroidism. However, secondary hyperparathyroidism with CKD is usually seen in the more advanced stages of CKD, and is associated with low-to-normal calcium levels (as opposed to the high levels seen in LAH).3-5
Lithium-associated hyperparathyroidism
Primary hyperparathyroidism is the most common cause of hypercalcemia. Its prevalence ranges from 1 to 7 cases per 1,000 adults. The incidence of LAH/LAHP is 4- to 6-fold higher compared to the general population.6 Similar to LAH/LAHP, primary hyperparathyroidism is more common in older adults (age >60) and females. Hence, some researchers have suggested that lithium probably unmasks hyperparathyroidism in patients who are susceptible to primary hyperparathyroidism.3
Look for these clinical manifestations
Symptoms of primary hyperparathyroidism are related to high calcium and PTH levels. They are commonly described as “painful bones, renal stones, abdominal groans (due to hypercalcemia-induced ileus), and psychic moans (lethargy, poor concentration, depression).” Common adverse outcomes associated with primary hyperparathyroidism are renal stones, high risk of fracture, constipation, peptic ulcer, and pancreatitis.3,7
Continue: In contrast...
In contrast, LAHP is characterized by mild, intermittent, and/or persistent hypercalcemia and mildly increased PTH (Table 1).1,3,4 In some patients, it could improve without active intervention. Because lithium increases the absorption of urinary calcium, it is associated with hypocalciuria and a lower risk of renal stones. Additionally, lithium has osteoprotective effects and has not been associated with an increased risk of fracture. Some researchers have suggested that the presentation of LAHP is more like familial hypocalciuric hypercalcemia (FHC), which is also associated with hypocalciuria. FHC is a benign condition and does not require active intervention.3,4 Similar to those with FHC, many patients with LAHP may live with chronic asymptomatic hypercalcemia without any significant adverse outcome.

A suggested approach to monitoring
In most cases, LAH is an insidious adverse effect that is usually detected on blood tests after many years of lithium therapy.8 For patients starting lithium therapy, International Society of Bipolar Disorder guidelines recommend testing calcium levels at baseline, 6 months, and annually thereafter, or as clinically indicated, to detect and monitor hypercalcemia and hyperparathyroidism. However, these guidelines do not provide any recommendations regarding how to manage abnormal findings.9
Clinical laboratories report both total and adjusted calcium values. The adjusted calcium value takes into account albumin levels. This is a way to compensate for an abnormal concentration of albumin (establishing what a patient’s total calcium concentration would be if the albumin concentration was normal). Table 25 shows the categorization of adjusted calcium values.For patients receiving lithium, some researchers have suggested monitoring PTH as well as calcium.1

The Figure outlines our proposed approach to monitoring for LAH in patients receiving lithium. An isolated high value of calcium could be due to prolonged venous stasis if a tourniquet is used for phlebotomy. In such instances, the calcium level should be tested again without a tourniquet.10 If the repeat blood test shows elevated calcium levels, then both PTH and serum calcium should be tested.

If the PTH level is higher than the midpoint of the reference range, LAH should be suspected, though sometimes hypercalcemia can present without raised PTH. LAH has also been reported to cause a transient increase in calcium levels. If hypercalcemia frequently recurs, PTH levels should be monitored. If PTH is suppressed, then the raised calcium levels are probably secondary to something other than lithium; common reasons for this include the use of vitamin D supplements or thiazide diuretics, or malignancies such as multiple myeloma.3,5,8
Continue to: Treatment
Treatment: Continue lithium?
There are several options for treating LAH. Lithium may be continued or discontinued following close monitoring of calcium and PTH levels, with or without active interventions such as surgery or pharmacotherapy, and as deemed appropriate after consultation with an endocrinologist. The decision should be informed by evaluating the risks and benefits to the patient’s physical and mental health. LAH can be reversed by discontinuing lithium, but this might not be the case in patients receiving long-term lithium therapy, especially if their elevated calcium levels are associated with parathyroid adenomas or hyperplasia. Hence, close monitoring of calcium and PTH is required even after discontinuing lithium.3,8
Surgical treatment. The primary treatment of LAH and primary hyperparathyroidism is parathyroidectomy. The possibility of recovery after parathyroidectomy for primary hyperparathyroidism is 60% to 80%, though a small proportion of patients might experience recurrence. This figure might be higher for LAH, because it is more likely to affect multiple glands.1,11 Other potential complications of parathyroidectomy are recurrent laryngeal nerve injury causing paralysis of vocal cords leading to hoarseness of voice, stridor, or aspiration, and local hematoma and hypocalcemia (requiring vitamin D and/or calcium supplements).12
Pharmacotherapy. Cinacalcet is a calcimimetic drug that decreases the reset point of the calcium-sensing receptor. It can be used if a patient is not suitable for or apprehensive about surgical intervention.1,8
Bottom Line
Calcium levels should be regularly monitored in patients receiving lithium. If calcium levels are persistently high, parathyroid hormone levels should also be measured. Management of lithium-associated hypercalcemia includes watchful waiting, discontinuing lithium, parathyroidectomy, and pharmacotherapy with cinacalcet.
Related Resources
- Laski M, Foreman R, Hancock H, et al. Lithium: an underutilized element. Current Psychiatry. 2021;20(12):27-30,34. doi:10.12788/cp.0193
- Pelekanos M, Foo K. A resident’s guide to lithium. Current Psychiatry. 2021;20(4):e3-e7. doi:10.12788/cp.0113
Drug Brand Names
Cinacalcet • Sensipar
1. Meehan AD, Udumyan R, Kardell M, et al. Lithium-associated hypercalcemia: pathophysiology, prevalence, management. World J Surg. 2018;42(2):415-424.
2. McKnight RF, Adida M, Budge K, et al. Lithium toxicity profile: a systematic review and meta-analysis. Lancet. 2012;379(9817):721-728.
3. Shapiro HI, Davis KA. Hypercalcemia and “primary” hyperparathyroidism during lithium therapy. Am J Psychiatry. 2015;172(1):12-15.
4. Lerena VS, León NS, Sosa S, et al. Lithium and endocrine dysfunction. Medicina (B Aires). 2022;82(1):130-137.
5. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966.
6. Yeh MW, Ituarte PH, Zhou HC, et al. Incidence and prevalence of primary hyperparathyroidism in a racially mixed population. J Clin Endocrinol Metab. 2013;98(3):1122-1129.
7. Dandurand K, Ali DS, Khan AA. Primary hyperparathyroidism: a narrative review of diagnosis and medical management. J Clin Med. 2021;10(8):1604.
8. Mifsud S, Cilia K, Mifsud EL, et al. Lithium-associated hyperparathyroidism. Br J Hosp Med (Lond). 2020;81(11):1-9.
9. Yatham LN, Kennedy SH, Parikh SV, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) 2018 guidelines for the management of patients with bipolar disorder. Bipolar Disord. 2018;20(2):97-170.
10. Mieebi WM, Solomon AE, Wabote AP. The effect of tourniquet application on serum calcium and inorganic phosphorus determination. Journal of Health, Medicine and Nursing. 2019;65:51-54.
11. Awad SS, Miskulin J, Thompson N. Parathyroid adenomas versus four-gland hyperplasia as the cause of primary hyperparathyroidism in patients with prolonged lithium therapy. World J Surg. 2003;27(4):486-488.
12. Farndon JR. Postoperative complications of parathyroidectomy. In: Holzheimer RG, Mannick JA, eds. Surgical Treatment: Evidence-Based and Problem-Oriented. Zuckschwerdt; 2001. Accessed October 25, 2022. https://www.ncbi.nlm.nih.gov/books/NBK6967
Hypercalcemia is a well-known but underrecognized adverse effect of lithium. Most patients with lithium-associated hypercalcemia (LAH) have either nonspecific symptoms (eg, persistent tiredness, constipation, polyuria, polydipsia) or no symptoms. Clinically, LAH differs from primary hyperparathyroidism, though the management protocol of these 2 conditions is almost the same. In this article, we discuss how lithium can affect calcium and parathyroid hormone (PTH) levels and how LAH and lithium-associated hyperparathyroidism (LAHP) differs from primary hyperparathyroidism. We also outline a suggested approach to monitoring and management.
An insidious problem
Due to the varying definitions and methods used to assess hypercalcemia, the reported prevalence of LAH varies from 4.3% to 80%.1 McKnight et al2 conducted a systematic review and meta-analysis of studies of the relationship between lithium and parathyroid function that included 14 case-control studies, 36 case reports, and 6 cross-sectional studies without a control group. They found that the levels of calcium and PTH were 10% higher in lithium-treated patients than in controls.2
Pathophysiology. Lithium is known to increase both calcium and PTH levels. PTH is responsible for calcium homeostasis. It is secreted in response to low calcium levels, which it increases by its action on bones, intestines, and kidneys. Vitamin D also plays a crucial role in calcium homeostasis. A deficiency of vitamin D triggers a compensatory increase in PTH to maintain calcium levels.3
Calcium and PTH levels increase soon after administration of lithium, but the rise is usually mild and insidious. In a small proportion of patients who receive long-term lithium treatment, calcium levels can exceed the normal range. Patients who develop LAH typically have serum calcium levels slightly above the normal range and PTH levels ranging from the higher side of the normal range to several times the upper limit of the normal range. Patients might also experience elevated PTH levels without any increase in calcium levels. Lithium can affect calcium and PTH levels in multiple ways. For instance, it increases the reabsorption of calcium in the kidney as well as the reset point of calcium-sensing receptors. Therefore, only higher levels of calcium can inhibit the release of PTH. Hence, in cases where the PTH level is within the normal range, it is generally higher than would be expected for a given serum calcium level. Lithium can also directly affect the parathyroid glands and can lead to either single-nodule or multimodule hyperplasia.4
Long-term lithium use can cause chronic kidney disease (CKD), which in turn leads to vitamin D deficiency and hyperparathyroidism. However, secondary hyperparathyroidism with CKD is usually seen in the more advanced stages of CKD, and is associated with low-to-normal calcium levels (as opposed to the high levels seen in LAH).3-5
Lithium-associated hyperparathyroidism
Primary hyperparathyroidism is the most common cause of hypercalcemia. Its prevalence ranges from 1 to 7 cases per 1,000 adults. The incidence of LAH/LAHP is 4- to 6-fold higher compared to the general population.6 Similar to LAH/LAHP, primary hyperparathyroidism is more common in older adults (age >60) and females. Hence, some researchers have suggested that lithium probably unmasks hyperparathyroidism in patients who are susceptible to primary hyperparathyroidism.3
Look for these clinical manifestations
Symptoms of primary hyperparathyroidism are related to high calcium and PTH levels. They are commonly described as “painful bones, renal stones, abdominal groans (due to hypercalcemia-induced ileus), and psychic moans (lethargy, poor concentration, depression).” Common adverse outcomes associated with primary hyperparathyroidism are renal stones, high risk of fracture, constipation, peptic ulcer, and pancreatitis.3,7
Continue: In contrast...
In contrast, LAHP is characterized by mild, intermittent, and/or persistent hypercalcemia and mildly increased PTH (Table 1).1,3,4 In some patients, it could improve without active intervention. Because lithium increases the absorption of urinary calcium, it is associated with hypocalciuria and a lower risk of renal stones. Additionally, lithium has osteoprotective effects and has not been associated with an increased risk of fracture. Some researchers have suggested that the presentation of LAHP is more like familial hypocalciuric hypercalcemia (FHC), which is also associated with hypocalciuria. FHC is a benign condition and does not require active intervention.3,4 Similar to those with FHC, many patients with LAHP may live with chronic asymptomatic hypercalcemia without any significant adverse outcome.
A suggested approach to monitoring
In most cases, LAH is an insidious adverse effect that is usually detected on blood tests after many years of lithium therapy.8 For patients starting lithium therapy, International Society of Bipolar Disorder guidelines recommend testing calcium levels at baseline, 6 months, and annually thereafter, or as clinically indicated, to detect and monitor hypercalcemia and hyperparathyroidism. However, these guidelines do not provide any recommendations regarding how to manage abnormal findings.9
Clinical laboratories report both total and adjusted calcium values. The adjusted calcium value takes into account albumin levels. This is a way to compensate for an abnormal concentration of albumin (establishing what a patient’s total calcium concentration would be if the albumin concentration was normal). Table 25 shows the categorization of adjusted calcium values.For patients receiving lithium, some researchers have suggested monitoring PTH as well as calcium.1
The Figure outlines our proposed approach to monitoring for LAH in patients receiving lithium. An isolated high value of calcium could be due to prolonged venous stasis if a tourniquet is used for phlebotomy. In such instances, the calcium level should be tested again without a tourniquet.10 If the repeat blood test shows elevated calcium levels, then both PTH and serum calcium should be tested.
If the PTH level is higher than the midpoint of the reference range, LAH should be suspected, though sometimes hypercalcemia can present without raised PTH. LAH has also been reported to cause a transient increase in calcium levels. If hypercalcemia frequently recurs, PTH levels should be monitored. If PTH is suppressed, then the raised calcium levels are probably secondary to something other than lithium; common reasons for this include the use of vitamin D supplements or thiazide diuretics, or malignancies such as multiple myeloma.3,5,8
Continue to: Treatment
Treatment: Continue lithium?
There are several options for treating LAH. Lithium may be continued or discontinued following close monitoring of calcium and PTH levels, with or without active interventions such as surgery or pharmacotherapy, and as deemed appropriate after consultation with an endocrinologist. The decision should be informed by evaluating the risks and benefits to the patient’s physical and mental health. LAH can be reversed by discontinuing lithium, but this might not be the case in patients receiving long-term lithium therapy, especially if their elevated calcium levels are associated with parathyroid adenomas or hyperplasia. Hence, close monitoring of calcium and PTH is required even after discontinuing lithium.3,8
Surgical treatment. The primary treatment of LAH and primary hyperparathyroidism is parathyroidectomy. The possibility of recovery after parathyroidectomy for primary hyperparathyroidism is 60% to 80%, though a small proportion of patients might experience recurrence. This figure might be higher for LAH, because it is more likely to affect multiple glands.1,11 Other potential complications of parathyroidectomy are recurrent laryngeal nerve injury causing paralysis of vocal cords leading to hoarseness of voice, stridor, or aspiration, and local hematoma and hypocalcemia (requiring vitamin D and/or calcium supplements).12
Pharmacotherapy. Cinacalcet is a calcimimetic drug that decreases the reset point of the calcium-sensing receptor. It can be used if a patient is not suitable for or apprehensive about surgical intervention.1,8
Bottom Line
Calcium levels should be regularly monitored in patients receiving lithium. If calcium levels are persistently high, parathyroid hormone levels should also be measured. Management of lithium-associated hypercalcemia includes watchful waiting, discontinuing lithium, parathyroidectomy, and pharmacotherapy with cinacalcet.
Related Resources
- Laski M, Foreman R, Hancock H, et al. Lithium: an underutilized element. Current Psychiatry. 2021;20(12):27-30,34. doi:10.12788/cp.0193
- Pelekanos M, Foo K. A resident’s guide to lithium. Current Psychiatry. 2021;20(4):e3-e7. doi:10.12788/cp.0113
Drug Brand Names
Cinacalcet • Sensipar
Hypercalcemia is a well-known but underrecognized adverse effect of lithium. Most patients with lithium-associated hypercalcemia (LAH) have either nonspecific symptoms (eg, persistent tiredness, constipation, polyuria, polydipsia) or no symptoms. Clinically, LAH differs from primary hyperparathyroidism, though the management protocol of these 2 conditions is almost the same. In this article, we discuss how lithium can affect calcium and parathyroid hormone (PTH) levels and how LAH and lithium-associated hyperparathyroidism (LAHP) differs from primary hyperparathyroidism. We also outline a suggested approach to monitoring and management.
An insidious problem
Due to the varying definitions and methods used to assess hypercalcemia, the reported prevalence of LAH varies from 4.3% to 80%.1 McKnight et al2 conducted a systematic review and meta-analysis of studies of the relationship between lithium and parathyroid function that included 14 case-control studies, 36 case reports, and 6 cross-sectional studies without a control group. They found that the levels of calcium and PTH were 10% higher in lithium-treated patients than in controls.2
Pathophysiology. Lithium is known to increase both calcium and PTH levels. PTH is responsible for calcium homeostasis. It is secreted in response to low calcium levels, which it increases by its action on bones, intestines, and kidneys. Vitamin D also plays a crucial role in calcium homeostasis. A deficiency of vitamin D triggers a compensatory increase in PTH to maintain calcium levels.3
Calcium and PTH levels increase soon after administration of lithium, but the rise is usually mild and insidious. In a small proportion of patients who receive long-term lithium treatment, calcium levels can exceed the normal range. Patients who develop LAH typically have serum calcium levels slightly above the normal range and PTH levels ranging from the higher side of the normal range to several times the upper limit of the normal range. Patients might also experience elevated PTH levels without any increase in calcium levels. Lithium can affect calcium and PTH levels in multiple ways. For instance, it increases the reabsorption of calcium in the kidney as well as the reset point of calcium-sensing receptors. Therefore, only higher levels of calcium can inhibit the release of PTH. Hence, in cases where the PTH level is within the normal range, it is generally higher than would be expected for a given serum calcium level. Lithium can also directly affect the parathyroid glands and can lead to either single-nodule or multimodule hyperplasia.4
Long-term lithium use can cause chronic kidney disease (CKD), which in turn leads to vitamin D deficiency and hyperparathyroidism. However, secondary hyperparathyroidism with CKD is usually seen in the more advanced stages of CKD, and is associated with low-to-normal calcium levels (as opposed to the high levels seen in LAH).3-5
Lithium-associated hyperparathyroidism
Primary hyperparathyroidism is the most common cause of hypercalcemia. Its prevalence ranges from 1 to 7 cases per 1,000 adults. The incidence of LAH/LAHP is 4- to 6-fold higher compared to the general population.6 Similar to LAH/LAHP, primary hyperparathyroidism is more common in older adults (age >60) and females. Hence, some researchers have suggested that lithium probably unmasks hyperparathyroidism in patients who are susceptible to primary hyperparathyroidism.3
Look for these clinical manifestations
Symptoms of primary hyperparathyroidism are related to high calcium and PTH levels. They are commonly described as “painful bones, renal stones, abdominal groans (due to hypercalcemia-induced ileus), and psychic moans (lethargy, poor concentration, depression).” Common adverse outcomes associated with primary hyperparathyroidism are renal stones, high risk of fracture, constipation, peptic ulcer, and pancreatitis.3,7
Continue: In contrast...
In contrast, LAHP is characterized by mild, intermittent, and/or persistent hypercalcemia and mildly increased PTH (Table 1).1,3,4 In some patients, it could improve without active intervention. Because lithium increases the absorption of urinary calcium, it is associated with hypocalciuria and a lower risk of renal stones. Additionally, lithium has osteoprotective effects and has not been associated with an increased risk of fracture. Some researchers have suggested that the presentation of LAHP is more like familial hypocalciuric hypercalcemia (FHC), which is also associated with hypocalciuria. FHC is a benign condition and does not require active intervention.3,4 Similar to those with FHC, many patients with LAHP may live with chronic asymptomatic hypercalcemia without any significant adverse outcome.
A suggested approach to monitoring
In most cases, LAH is an insidious adverse effect that is usually detected on blood tests after many years of lithium therapy.8 For patients starting lithium therapy, International Society of Bipolar Disorder guidelines recommend testing calcium levels at baseline, 6 months, and annually thereafter, or as clinically indicated, to detect and monitor hypercalcemia and hyperparathyroidism. However, these guidelines do not provide any recommendations regarding how to manage abnormal findings.9
Clinical laboratories report both total and adjusted calcium values. The adjusted calcium value takes into account albumin levels. This is a way to compensate for an abnormal concentration of albumin (establishing what a patient’s total calcium concentration would be if the albumin concentration was normal). Table 25 shows the categorization of adjusted calcium values.For patients receiving lithium, some researchers have suggested monitoring PTH as well as calcium.1
The Figure outlines our proposed approach to monitoring for LAH in patients receiving lithium. An isolated high value of calcium could be due to prolonged venous stasis if a tourniquet is used for phlebotomy. In such instances, the calcium level should be tested again without a tourniquet.10 If the repeat blood test shows elevated calcium levels, then both PTH and serum calcium should be tested.
If the PTH level is higher than the midpoint of the reference range, LAH should be suspected, though sometimes hypercalcemia can present without raised PTH. LAH has also been reported to cause a transient increase in calcium levels. If hypercalcemia frequently recurs, PTH levels should be monitored. If PTH is suppressed, then the raised calcium levels are probably secondary to something other than lithium; common reasons for this include the use of vitamin D supplements or thiazide diuretics, or malignancies such as multiple myeloma.3,5,8
Continue to: Treatment
Treatment: Continue lithium?
There are several options for treating LAH. Lithium may be continued or discontinued following close monitoring of calcium and PTH levels, with or without active interventions such as surgery or pharmacotherapy, and as deemed appropriate after consultation with an endocrinologist. The decision should be informed by evaluating the risks and benefits to the patient’s physical and mental health. LAH can be reversed by discontinuing lithium, but this might not be the case in patients receiving long-term lithium therapy, especially if their elevated calcium levels are associated with parathyroid adenomas or hyperplasia. Hence, close monitoring of calcium and PTH is required even after discontinuing lithium.3,8
Surgical treatment. The primary treatment of LAH and primary hyperparathyroidism is parathyroidectomy. The possibility of recovery after parathyroidectomy for primary hyperparathyroidism is 60% to 80%, though a small proportion of patients might experience recurrence. This figure might be higher for LAH, because it is more likely to affect multiple glands.1,11 Other potential complications of parathyroidectomy are recurrent laryngeal nerve injury causing paralysis of vocal cords leading to hoarseness of voice, stridor, or aspiration, and local hematoma and hypocalcemia (requiring vitamin D and/or calcium supplements).12
Pharmacotherapy. Cinacalcet is a calcimimetic drug that decreases the reset point of the calcium-sensing receptor. It can be used if a patient is not suitable for or apprehensive about surgical intervention.1,8
Bottom Line
Calcium levels should be regularly monitored in patients receiving lithium. If calcium levels are persistently high, parathyroid hormone levels should also be measured. Management of lithium-associated hypercalcemia includes watchful waiting, discontinuing lithium, parathyroidectomy, and pharmacotherapy with cinacalcet.
Related Resources
- Laski M, Foreman R, Hancock H, et al. Lithium: an underutilized element. Current Psychiatry. 2021;20(12):27-30,34. doi:10.12788/cp.0193
- Pelekanos M, Foo K. A resident’s guide to lithium. Current Psychiatry. 2021;20(4):e3-e7. doi:10.12788/cp.0113
Drug Brand Names
Cinacalcet • Sensipar
1. Meehan AD, Udumyan R, Kardell M, et al. Lithium-associated hypercalcemia: pathophysiology, prevalence, management. World J Surg. 2018;42(2):415-424.
2. McKnight RF, Adida M, Budge K, et al. Lithium toxicity profile: a systematic review and meta-analysis. Lancet. 2012;379(9817):721-728.
3. Shapiro HI, Davis KA. Hypercalcemia and “primary” hyperparathyroidism during lithium therapy. Am J Psychiatry. 2015;172(1):12-15.
4. Lerena VS, León NS, Sosa S, et al. Lithium and endocrine dysfunction. Medicina (B Aires). 2022;82(1):130-137.
5. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966.
6. Yeh MW, Ituarte PH, Zhou HC, et al. Incidence and prevalence of primary hyperparathyroidism in a racially mixed population. J Clin Endocrinol Metab. 2013;98(3):1122-1129.
7. Dandurand K, Ali DS, Khan AA. Primary hyperparathyroidism: a narrative review of diagnosis and medical management. J Clin Med. 2021;10(8):1604.
8. Mifsud S, Cilia K, Mifsud EL, et al. Lithium-associated hyperparathyroidism. Br J Hosp Med (Lond). 2020;81(11):1-9.
9. Yatham LN, Kennedy SH, Parikh SV, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) 2018 guidelines for the management of patients with bipolar disorder. Bipolar Disord. 2018;20(2):97-170.
10. Mieebi WM, Solomon AE, Wabote AP. The effect of tourniquet application on serum calcium and inorganic phosphorus determination. Journal of Health, Medicine and Nursing. 2019;65:51-54.
11. Awad SS, Miskulin J, Thompson N. Parathyroid adenomas versus four-gland hyperplasia as the cause of primary hyperparathyroidism in patients with prolonged lithium therapy. World J Surg. 2003;27(4):486-488.
12. Farndon JR. Postoperative complications of parathyroidectomy. In: Holzheimer RG, Mannick JA, eds. Surgical Treatment: Evidence-Based and Problem-Oriented. Zuckschwerdt; 2001. Accessed October 25, 2022. https://www.ncbi.nlm.nih.gov/books/NBK6967
1. Meehan AD, Udumyan R, Kardell M, et al. Lithium-associated hypercalcemia: pathophysiology, prevalence, management. World J Surg. 2018;42(2):415-424.
2. McKnight RF, Adida M, Budge K, et al. Lithium toxicity profile: a systematic review and meta-analysis. Lancet. 2012;379(9817):721-728.
3. Shapiro HI, Davis KA. Hypercalcemia and “primary” hyperparathyroidism during lithium therapy. Am J Psychiatry. 2015;172(1):12-15.
4. Lerena VS, León NS, Sosa S, et al. Lithium and endocrine dysfunction. Medicina (B Aires). 2022;82(1):130-137.
5. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966.
6. Yeh MW, Ituarte PH, Zhou HC, et al. Incidence and prevalence of primary hyperparathyroidism in a racially mixed population. J Clin Endocrinol Metab. 2013;98(3):1122-1129.
7. Dandurand K, Ali DS, Khan AA. Primary hyperparathyroidism: a narrative review of diagnosis and medical management. J Clin Med. 2021;10(8):1604.
8. Mifsud S, Cilia K, Mifsud EL, et al. Lithium-associated hyperparathyroidism. Br J Hosp Med (Lond). 2020;81(11):1-9.
9. Yatham LN, Kennedy SH, Parikh SV, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) 2018 guidelines for the management of patients with bipolar disorder. Bipolar Disord. 2018;20(2):97-170.
10. Mieebi WM, Solomon AE, Wabote AP. The effect of tourniquet application on serum calcium and inorganic phosphorus determination. Journal of Health, Medicine and Nursing. 2019;65:51-54.
11. Awad SS, Miskulin J, Thompson N. Parathyroid adenomas versus four-gland hyperplasia as the cause of primary hyperparathyroidism in patients with prolonged lithium therapy. World J Surg. 2003;27(4):486-488.
12. Farndon JR. Postoperative complications of parathyroidectomy. In: Holzheimer RG, Mannick JA, eds. Surgical Treatment: Evidence-Based and Problem-Oriented. Zuckschwerdt; 2001. Accessed October 25, 2022. https://www.ncbi.nlm.nih.gov/books/NBK6967

Is it psychosis, or an autoimmune encephalitis?
Hidden within routine presentations of first-episode psychosis is a rare subpopulation whose symptoms are mediated by an autoimmune process for which proper treatment differs significantly from standard care for typical psychotic illness. In this article, we present a hypothetical case and describe how to assess if a patient has an elevated probability of autoimmune encephalitis, determine what diagnostics or medication-induced effects to consider, and identify unresolved questions about best practices.
CASE REPORT
Bizarre behavior and isolation
Ms. L, age 21, is brought to the emergency department (ED) by her college roommate after exhibiting out-of-character behavior and gradual self-isolation over the last 2 months. Her roommate noticed that she had been spending more time isolated in her dorm room and remaining in bed into the early afternoon, though she does not appear to be asleep. Ms. L’s mother is concerned about her daughter’s uncharacteristic refusal to travel home for a family event. Ms. L expresses concern about the intentions of her research preceptor, and recalls messages from the association of colleges telling her to “change her future.” Ms. L hears voices telling her who she can and cannot trust. In the ED, she says she has a headache, experiences mild dizziness while standing, and reports having a brief upper respiratory illness at the end of last semester. Otherwise, a medical review of systems is negative.
Although the etiology of first-episode psychosis can be numerous or unknown, many psychiatrists feel comfortable with the initial diagnostic for this type of clinical presentation. However, for some clinicians, it may be challenging to feel confident in making a diagnosis of autoimmune encephalitis.
Autoimmune encephalitis is a family of syndromes caused by autoantibodies targeting either intracellular or extracellular neuronal antigens. Anti-N-methyl-
In this article, we focus on anti-NMDA receptor encephalitis and use the term interchangeably with autoimmune encephalitis for 2 reasons. First, anti-NMDA receptor encephalitis can present with psychotic symptoms as the only symptoms (prior to cognitive or neurologic manifestations) or can present with psychotic symptoms as the main indicator (with other symptoms that are more subtle and possibly missed). Second, anti-NMDA receptor encephalitis often occurs in young adults, which is when it is common to see the onset of a primary psychotic illness. These 2 factors make it likely that these cases will come into the evaluative sphere of psychiatrists. We give special attention to features of cases of anti-NMDA receptor encephalitis confirmed with antineuronal antibodies in the CSF, as it has emerged that antibodies in the serum can be nonspecific and nonpathogenic.2,3
What does anti-NMDA receptor encephalitis look like?
Symptoms of anti-NMDA receptor encephalitis resemble those of a primary psychotic disorder, which can make it challenging to differentiate between the 2 conditions, and might cause the correct diagnosis to be missed. Pollak et al4 proposed that psychiatrically confusing presentations that don’t clearly match an identifiable psychotic disorder should raise a red flag for an autoimmune etiology. However, studies often fail to describe the specific psychiatric features of anti-NMDA receptor encephalitis, and thus provide little practical evidence to guide diagnosis. In some of the largest studies of patients with anti-NMDA receptor encephalitis, psychiatric clinical findings are often combined into nonspecific headings such as “abnormal behavior” or “behavioral and cognitive” symptoms.5 Such groupings make this the most common clinical finding (95%)5 but make it difficult to discern particular clinical characteristics. Where available, specific symptoms identified across studies include agitation, aggression, changes in mood and/or irritability, insomnia, delusions, hallucinations, and occasionally catatonic features.6,7 Attempts to identify specific psychiatric phenotypes distinct from primary psychotic illnesses have fallen short due to contradictory findings and lack of clinical practicality.8 One exception is the presence of catatonic features, which have been found in CSF-confirmed studies.2 In contrast to the typical teaching that the hallucination modality (eg, visual or tactile) can be helpful in estimating the likelihood of a secondary psychosis (ie, drug-induced, neurodegenerative, or autoimmune), there does not appear to be a difference in hallucination modality between encephalitis and primary psychotic disorders.9
History and review of systems
Another red flag to consider is the rapidity of symptom presentation. Symptoms that progress within 3 months increase the likelihood that the patient has autoimmune encephalitis.10 Cases where collateral information indicates the psychotic episode was preceded by a long, subtle decline in school performance, social withdrawal, and attenuated psychotic symptoms typical of a schizophrenia prodrome are less likely to be an autoimmune psychosis.11 A more delayed presentation does not entirely exclude autoimmune encephalitis; however, a viral-like prodrome before the onset of psychosis increases the likelihood of autoimmune encephalitis. Such a prodrome may include fever, headache, nausea, vomiting, and diarrhea.7
Continue to: Another indication is the presence...
Another indication is the presence of new seizures within 1 year of presenting with psychotic symptoms.10 The possibility of undiagnosed seizures should be considered in a patient with psychosis who has episodes of unresponsiveness, dissociative episodes, or seizure-like activity that is thought to be psychogenic but has not been fully evaluated. Seizures in autoimmune encephalitis involve deep structures in the brain and can be present without overt epileptiform activity on EEG, but rather causing only bilateral slowing that is often described as nonspecific.12
In a young patient presenting with first-episode psychosis, a recent diagnosis of cancer or abnormal finding in the ovaries increases the likelihood of autoimmune encephalitis.4 Historically, however, this type of medical history has been irrelevant to psychosis. Although rare, any person presenting with first-episode psychosis and a history of herpes simplex virus (HSV) encephalitis should be evaluated for autoimmune encephalitis because anti-NMDA receptor antibodies have been reported to be present in approximately one-third of these patients.13 Finally, the report of focal neurologic symptoms, including neck stiffness or neck pain, should raise concern, although sensory, working memory, and cognitive deficits may be difficult to fully distinguish from common somatic and cognitive symptoms in a primary psychiatric presentation.
Table 1 lists 4 questions to ask patients who present with first-episode psychosis that may not usually be part of a typical evaluation.

CASE CONTINUED
Uncooperative with examination
In the ED, Ms. L’s heart rate is 101 beats per minute and her blood pressure is 102/72 mm Hg. Her body mass index (BMI) is 22, which suggests an approximate 8-pound weight loss since her BMI was last assessed. Ms. L responds to questions with 1- to 6-word sentences, without clear verbigeration. Though her speech is not pressured, it is of increased rate. Her gaze scans the room, occasionally becoming fixed for 5 to 10 seconds but is aborted by the interviewer’s comment on this behavior. Ms. L efficiently and accurately spells WORLD backwards, then asks “Why?” and refuses to engage in further cognitive testing, stating “Not doing that.” When the interviewer asks “Why not?” she responds “Not doing that.” Her cranial nerves are intact, and she refuses cerebellar testing or requests to assess tone. There are no observed stereotypies, posturing, or echopraxia.
While not necessary for a diagnosis of autoimmune encephalitis, short-term memory loss is a common cognitive finding across studies.5-7 A common clinical finding from a mental status exam is speech disorders, including (but not limited to) increased rates of speech or decreased verbal output.7 Autonomic instability—including tachycardia, markedly labile blood pressures, and orthostasis—all increase the likelihood of autoimmune encephalitis.14 Interpreting a patient’s vital sign changes can be confounded if they are agitated or anxious, or if they are taking an antipsychotic that produces adverse anticholinergic effects. However, vital sign abnormalities that precede medication administration or do not correlate with fluctuations in mental status increase suspicion for an autoimmune encephalitis.
Continue to: In the absence of the adverse effect...
In the absence of the adverse effect of a medication, orthostasis is uncommon in a well-hydrated young person. Some guidelines4 suggest that symptoms of catatonia should be considered a red flag for autoimmune encephalitis. According to the Bush-Francis Catatonia Rating Scale, commonly identified features include immobility, staring, mutism, posturing, withdrawal, rigidity, and gegenhalten.15 Catatonia is common among patients with anti-NDMA receptor encephalitis, though it may not be initially present and could emerge later.2 However, there are documented cases of autoimmune encephalitis where the patient had only isolated features of catatonia, such as echolalia or mutism.2
CASE CONTINUED
History helps narrow the diagnosis
Ms. L’s parents say their daughter has not had prior contact with a therapist or psychiatrist, previous psychiatric diagnoses, hospitalizations, suicide attempts, self-injury, or binging or purging behaviors. Ms. L’s paternal grandfather was diagnosed with schizophrenia, but he is currently employed, lives alone, and has not taken medication for many years. Her mother has hypothyroidism. Ms. L was born at full term via vaginal delivery without cardiac defects or a neonatal intensive care unit stay. Her mother said she did not have postpartum depression or anxiety, a complicated pregnancy, or exposure to tobacco, alcohol, or illicit drug use. Ms. L has no history of childhood seizures or head injury with loss of consciousness. She is an only child, born and raised in a house in a metropolitan area, walked at 13 months, did not require early intervention or speech therapy, and met normal language milestones.
She attended kindergarten at age 6 and progressed throughout public school without regressions in reading, writing, or behavioral manifestations, and did not require a 504 Plan or individualized education program. Ms. L graduated high school in the top 30% of her class, was socially active, and attended a local college. In college, she achieved honor roll, enrolled in a sorority, and was a part of a research lab. Her only medication is oral contraception. She consumes alcohol socially, and reports no cannabis, cigarette, or vaping use. Ms. L says she does not use hallucinogens, stimulants, opiates, or cocaine, and her roommate and family confirm this. She denies recent travel and is sexually active. Ms. L’s urinary and serum toxicology are unremarkable, human chorionic gonadotropin is undetectable, and her sodium level is 133 mEq/L. A measure of serum neutrophils is 6.8 x 109/L and serum lymphocytes is 1.7 x 109/L. Her parents adamantly request a Neurology consultation and further workup, including a lumbar puncture (LP), EEG, and brain imaging (MRI).
This information is useful in ruling out other potential causes of psychosis, such as substance-induced psychosis and neurodevelopmental disorders that can present with psychosis. Additionally, neurodevelopmental abnormalities and psychiatric prodromal symptoms are known precedents in individuals who develop a primary psychotic disorder such as schizophrenia.16 A family history that includes a psychotic illness may increase the likelihood of a primary psychotic disorder in offspring; however, clinicians must also consider the accuracy of diagnosis in the family, as this can often be inaccurate or influenced by historical cultural bias. We recommend further elucidating the likelihood of a genetic predisposition to a primary psychotic disorder by clarifying familial medication history and functionality.
For example, the fact that Ms. L’s grandfather has not taken medication for many years and has a high degree of functioning and/or absence of cognitive deficits would lower our suspicion for an accurate diagnosis of schizophrenia (given the typical cognitive decline with untreated illness). Another piece of family history relevant to autoimmune encephalitis includes the propensity for autoimmune disorders, but expert opinion on this matter is mixed.17 Ms. L’s mother has hypothyroidism, which is commonly caused by a prior episode of Hashimoto’s autoimmune thyroiditis. Some physicians advocate for measuring antithyroid antibodies and erythrocyte sedimentation rate or C-reactive protein to gauge the level of autoimmunity, but the usefulness of these measures for detecting autoimmune encephalitis is unclear. These serum markers can be useful in detecting additional important etiologies such as systemic infection or systemic inflammation, and there are conditions such as steroid-responsive encephalopathy with associated thyroiditis, which, as the name suggests, responds to steroids rather than other psychotropic medications. Other risk factors for autoimmune encephalitis include being female, being young, having viral infections (eg, HSV), prior tumor burden, and being in the postpartum period.18 Some experts also suggest the presence of neurologic symptoms 4 weeks after the first psychiatric or cognitive symptom presentation increases the likelihood of anti-NMDA receptor encephalitis, and a lack of neurologic symptoms would make this diagnosis less likely.6,19
Continue to: Another item of interest...
Another item of interest in Ms. L’s case is her parents’ request for a Neurology consultation and further workup, as there is an association between caregiver request for workup and eventual diagnosis.6 While the etiology of this phenomenon is unclear, the literature suggests individuals with autoimmune encephalitis who initially present to Psychiatry experience longer delays to the appropriate treatment with immunomodulatory therapy than those who first present to Neurology.20
Laboratory and diagnostic testing
Guasp et al2 recommend EEG, MRI, and serum autoimmune antibodies (ie, screening for anti-NMDA receptor antibodies) for patients who present with first-episode psychosis, even in the absence of some of the red flags previously discussed. A recent economic analysis suggested screening all patients with first-episode psychosis for serum antibodies may be cost-effective.21
For patients whose presentations include features concerning for anti-NMDA receptor encephalitis, an EEG and MRI are reasonable. In a review of EEG abnormalities in anti-NMDA receptor encephalitis, Gillinder et al23 noted that while 30% did not have initial findings, 83.6% of those with confirmed anti-NMDA receptor encephalitis demonstrated EEG abnormalities; the most common were generalized slowing, delta slowing, and focal abnormalities. Discovering an extreme delta-brush activity on EEG is specific for anti-NMDA receptor encephalitis, but its absence is not fully informative. Practically, slowing can be a nonspecific manifestation of encephalopathy or a medication effect, and many people who present with first-episode psychosis will have recently received antipsychotics, which alter EEG frequency. In a study of EEG changes with antipsychotics, Centorrino et al24 found that generalized background slowing into the theta range across all antipsychotics was not significantly different from control participants, while theta to delta range slowing occurred in 8.2% of those receiving antipsychotics vs 3.3% of controls. Clozapine and olanzapine may be associated with greater EEG abnormalities, while haloperidol and quetiapine contribute a lower risk.25 For young patients with first-episode psychosis without a clear alternative explanation, we advocate for further autoimmune encephalitis workup among all individuals with generalized theta or delta wave slowing.
Because these medication effects are most likely to decrease specificity but not sensitivity of EEG for autoimmune encephalitis, a normal EEG without slowing can be reassuring.26 Moreover, for patients who receive neuroimaging, an MRI may detect inflammation that is not visible on CT. The concerning findings for anti-NMDA receptor encephalitis are temporal or multifocal T2 hyperintensities, though the MRI is normal in most cases and thus should not be reassuring if other concerning features are present.27
The role of lumbar puncture
Another area of active debate surrounds the usefulness and timing of LP. Guasp et al2 proposed that all individuals with first-episode psychosis and focal neurologic findings should receive LP and CSF antineuronal antibody testing. They recommend that patients with first-episode psychosis without focal neurologic findings also should receive LP and CSF testing if ≥1 of the following is present:
- slowing on EEG
- temporal or multifocal T2 hyperintensities on MRI
- positive anti-NMDA receptor antibody in the serum.2
Continue to: Evidence suggests that basic CSF parameters...
Evidence suggests that basic CSF parameters, such as elevated protein and white blood cell counts, are some of the most sensitive and specific tests for autoimmune encephalitis.2 Thus, if the patient is amenable and logistical factors are in place, it may be reasonable to pursue LP earlier in some cases without waiting for serum antibody assays to return (these results can take several weeks). CSF inflammatory changes without neuronal antibodies should lead to other diagnostic considerations (eg, systemic inflammatory disease, psychosis attributed to systemic lupus erythematosus).7 While nonspecific, serum laboratory values that may increase suspicion of anti-NMDA receptor encephalitis include hyponatremia6 and an elevated neutrophil-to-lymphocyte ratio (NLR).28 An NLR >4 in conjunction with CSF albumin-to- serum albumin ratio >7 is associated with impaired blood brain barrier integrity and a worse prognosis for those with anti-NMDA receptor encephalitis.28
Additional clinical features that may sway decisions in favor of obtaining LP despite negative findings on EEG, MRI, and serum antibodies include increased adverse reactions to antipsychotics (eg, neuroleptic malignant syndrome), prodromal infectious symptoms, known tumor, or new-onset neurologic symptoms after initial evaluation.2,8
Table 2 summarizes key features of laboratory and diagnostic findings in anti-NMDA receptor encephalitis.

When should you pursue a more extensive workup?
There are some practical tools and rating scales to help clinicians conceptualize risk for autoimmune encephalitis. For psychiatric purposes, however, many of these scales assume that LP, MRI, and EEG have already been completed, and thus it is challenging to incorporate them into psychiatric practice. One such tool is the Antibody Prevalence in Epilepsy and Encephalopathy scale; a score ≥4 is 98% sensitive and 78% to 84% specific for predicting antineural autoantibody positivity.10 Table 3 describes warning signs that may be useful in helping clinicians decide how urgently to pursue a more extensive workup in the possibility of autoimmune encephalitis.

The importance of catching anti-NMDA receptor encephalitis is underscored by the fact that appropriate treatment is very different than for primary psychosis, and outcomes worsen with delay to appropriate treatment.20 Without treatment, severe cases may progress to autonomic instability, altered consciousness, and respiratory compromise warranting admission to an intensive care unit. While the details are beyond the scope of this review, the recommended treatment for confirmed cases of anti-NMDA receptor encephalitis includes tumor removal (if indicated), reducing inflammation (steroids), removing antibodies via IV immunoglobulins, or plasma exchange.8,29 Progression of the disease may warrant consideration of rituximab or cyclophosphamide. In nonresponsive cases, third-line treatments include proteasome inhibitors or interleukin-6 receptor antagonists.8 For patients with severe catatonia, some studies have investigated the utility of electroconvulsive therapy.30 Conceptually, clinicians may consider the utility of antipsychotics as similar to recommendations for hyperactive delirium for the management of psychotic symptoms, agitation, or insomnia. However, given the risk for antipsychotic intolerance, using the lowest effective dose and vigilant screening for the emergence of extrapyramidal symptoms, fever, and autonomic instability is recommended.
CASE CONTINUED
Finally, something objective
Ms. L receives haloperidol 2 mg and undergoes an MRI without contrast. Findings are unremarkable. A spot EEG notes diffuse background slowing in the theta range, prompting lumbar puncture. Findings note 0.40 g/L, 0.2 g/L, and 3.5 for the total protein, albumin, and albumin/CSF-serum quotient (QAlb), respectively; all values are within normal limits. A mild lymphocytic pleocytosis is present as evidenced by a cell count of 35 cells/µL. The CSF is sent for qualitative examination of immunoglobulin G and electrophoresis of proteins in the CSF and serum, of which an increased concentration of restricted bands (oligoclonal bands) in the CSF but not the serum would indicate findings of oligoclonal bands. CSF is sent for detection of anti-NMDA receptor antibodies by indirect immunofluorescence, with a plan to involve an interdisciplinary team for treatment if the antibodies return positive and to manage the case symptomatically in the interim.
Bottom Line
A small subpopulation of patients who present with apparent first-episode psychosis will have symptoms caused by autoimmune encephalitis (specifically, anti-NMDA receptor encephalitis). We provide 4 screening questions to determine when to pursue a workup for an autoimmune encephalitis, and describe relevant clinical symptoms and warning signs to help differentiate the 2 conditions.
Related Resources
- Askandaryan AS, Naqvi A, Varughese A, et al. Anti-N-methyl-D-aspartate receptor encephalitis: neuropsychiatric and multidisciplinary approach to a patient not responding to first-line treatment. Cureus. 2022;14(6):e25751.
- Kayser MS, Titulaer MJ, Gresa-Arribas N, et al. Frequency and characteristics of isolated psychiatric episodes in anti-NMDA receptor encephalitis. JAMA Neurol. 2013;70(9):1133-1139.
Drug Brand Names
Clozapine • Clozaril
Haloperidol • Haldol
Olanzapine • Zyprexa
Quetiapine • Seroquel
Rituximab • Rituxan
1. Granerod J, Ambrose HE, Davies NW, et al; UK Health Protection Agency (HPA) Aetiology of Encephalitis Study Group. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis. 2010;10(12):835-44. doi:10.1016/S1473-3099(10)70222-X
2. Guasp M, Giné-Servén E, Maudes E, et al. Clinical, neuroimmunologic, and CSF investigations in first episode psychosis. Neurology. 2021;97(1):e61-e75.
3. From the American Association of Neurological Surgeons (AANS), American Society of Neuroradiology (ASNR), Cardiovascular and Interventional Radiology Society of Europe (CIRSE), Canadian Interventional Radiology Association (CIRA), Congress of Neurological Surgeons (CNS), European Society of Minimally Invasive Neurological Therapy (ESMINT), European Society of Neuroradiology (ESNR), European Stroke Organization (ESO), Society for Cardiovascular Angiography and Interventions (SCAI), Society of Interventional Radiology (SIR), Society of NeuroInterventional Surgery (SNIS), and World Stroke Organization (WSO), Sacks D, Baxter B, Campbell BCV, et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int J Stroke. 2018;13(6):612-632. doi:10.1177/1747493018778713
4. Pollak TA, Lennox BR, Muller S, et al. Autoimmune psychosis: an international consensus on an approach to the diagnosis and management of psychosis of suspected autoimmune origin. Lancet Psychiatry. 2020;7(1):93-108.
5. Guasp M, Módena Y, Armangue T, et al. Clinical features of seronegative, but CSF antibody-positive, anti-NMDA receptor encephalitis. Neurol Neuroimmunol Neuroinflamm. 2020;7(2):e659.
6. Herken J, Prüss H. Red flags: clinical signs for identifying autoimmune encephalitis in psychiatric patients. Front Psychiatry. 2017;8:25. doi:10.3389/fpsyt.2017.00025
7. Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404.
8. Dalmau J, Armangue T, Planaguma J, et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: mechanisms and models. Lancet Neurol. 2019;18(11):1045-1057.
9. Rattay TW, Martin P, Vittore D, et al. Cerebrospinal fluid findings in patients with psychotic symptoms—a retrospective analysis. Sci Rep. 2021;11(1):7169.
10. Dubey D, Pittock SJ, McKeon A. Antibody prevalence in epilepsy and encephalopathy score: increased specificity and applicability. Epilepsia. 2019;60(2):367-369.
11. Maj M, van Os J, De Hert M, et al. The clinical characterization of the patient with primary psychosis aimed at personalization of management. World Psychiatry. 2021;20(1):4-33. doi:10.1002/wps.20809
12. Caplan JP, Binius T, Lennon VA, et al. Pseudopseudoseizures: conditions that may mimic psychogenic non-epileptic seizures. Psychosomatics. 2011;52(6):501-506.
13. Armangue T, Spatola M, Vlagea A, et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis. Lancet Neurol. 2018;17(9):760-772.
14. Takamatsu K, Nakane S. Autonomic manifestations in autoimmune encephalitis. Neurol Clin Neurosci. 2022;10:130-136. doi:10.1111/ncn3.12557
15. Espinola-Nadurille M, Flores-Rivera J, Rivas-Alonso V, et al. Catatonia in patients with anti-NMDA receptor encephalitis. Psychiatry Clin Neurosci. 2019;73(9):574-580.
16. Keshavan M, Montrose DM, Rajarethinam R, et al. Psychopathology among offspring of parents with schizophrenia: relationship to premorbid impairments. Schizophr Res. 2008;103(1-3):114-120.
17. Jeppesen R, Benros ME. Autoimmune diseases and psychotic disorders. Front Psychiatry. 2019;10:131.
18. Bergink V, Armangue T, Titulaer MJ, et al. Autoimmune encephalitis in postpartum psychosis. Am J Psychiatry. 2015;172(9):901-908.
19. Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091-8. doi: 10.1016/S1474-4422(08)70224-2
20. Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12(2):157-165.
21. Ross EL, Becker JE, Linnoila JJ, et al. Cost-effectiveness of routine screening for autoimmune encephalitis in patients with first-episode psychosis in the United States. J Clin Psychiatry. 2020;82(1):19m13168.
22. Sonderen AV, Arends S, Tavy DLJ, et al. Predictive value of electroencephalography in anti-NMDA receptor encephalitis. J Neurol Neurosurg Psychiatry. 2018;89(10):1101-1106.
23. Gillinder L, Warren N, Hartel G, et al. EEG findings in NMDA encephalitis--a systematic review. Seizure. 2019;65:20-24.
24. Centorrino F, Price BH, Tuttle M, et al. EEG abnormalities during treatment with typical and atypical antipsychotics. Am J Psychiatry. 2002;159(1):109-115.
25. Raymond N, Lizano P, Kelly S, et al. What can clozapine’s effect on neural oscillations tell us about its therapeutic effects? A scoping review and synthesis. Biomarkers in Neuropsychiatry. 2022;6:100048.
26. Kaufman DM, Geyer H, Milstein MJ. Kaufman’s Clinical Neurology for Psychiatrists. 8th ed. Elsevier Inc; 2016.
27. Kelley BP, Patel SC, Marin HL, et al. Autoimmune encephalitis: pathophysiology and imaging review of an overlooked diagnosis. AJNR Am J Neuroradiol. 2017;38(6):1070-1078.
28. Yu Y, Wu Y, Cao X, et al. The clinical features and prognosis of anti-NMDAR encephalitis depends on blood brain barrier integrity. Mult Scler Relat Disord. 2021;47:102604.
29. Dalmau J, Graus F. Antibody-mediated neuropsychiatric disorders. J Allergy Clin Immunol. 2022;149(1):37-40.
30. Warren N, Grote V, O’Gorman C, et al. Electroconvulsive therapy for anti-N-methyl-daspartate (NMDA) receptor encephalitis: a systematic review of cases. Brain Stimul. 2019;12(2):329-334.
Hidden within routine presentations of first-episode psychosis is a rare subpopulation whose symptoms are mediated by an autoimmune process for which proper treatment differs significantly from standard care for typical psychotic illness. In this article, we present a hypothetical case and describe how to assess if a patient has an elevated probability of autoimmune encephalitis, determine what diagnostics or medication-induced effects to consider, and identify unresolved questions about best practices.
CASE REPORT
Bizarre behavior and isolation
Ms. L, age 21, is brought to the emergency department (ED) by her college roommate after exhibiting out-of-character behavior and gradual self-isolation over the last 2 months. Her roommate noticed that she had been spending more time isolated in her dorm room and remaining in bed into the early afternoon, though she does not appear to be asleep. Ms. L’s mother is concerned about her daughter’s uncharacteristic refusal to travel home for a family event. Ms. L expresses concern about the intentions of her research preceptor, and recalls messages from the association of colleges telling her to “change her future.” Ms. L hears voices telling her who she can and cannot trust. In the ED, she says she has a headache, experiences mild dizziness while standing, and reports having a brief upper respiratory illness at the end of last semester. Otherwise, a medical review of systems is negative.
Although the etiology of first-episode psychosis can be numerous or unknown, many psychiatrists feel comfortable with the initial diagnostic for this type of clinical presentation. However, for some clinicians, it may be challenging to feel confident in making a diagnosis of autoimmune encephalitis.
Autoimmune encephalitis is a family of syndromes caused by autoantibodies targeting either intracellular or extracellular neuronal antigens. Anti-N-methyl-
In this article, we focus on anti-NMDA receptor encephalitis and use the term interchangeably with autoimmune encephalitis for 2 reasons. First, anti-NMDA receptor encephalitis can present with psychotic symptoms as the only symptoms (prior to cognitive or neurologic manifestations) or can present with psychotic symptoms as the main indicator (with other symptoms that are more subtle and possibly missed). Second, anti-NMDA receptor encephalitis often occurs in young adults, which is when it is common to see the onset of a primary psychotic illness. These 2 factors make it likely that these cases will come into the evaluative sphere of psychiatrists. We give special attention to features of cases of anti-NMDA receptor encephalitis confirmed with antineuronal antibodies in the CSF, as it has emerged that antibodies in the serum can be nonspecific and nonpathogenic.2,3
What does anti-NMDA receptor encephalitis look like?
Symptoms of anti-NMDA receptor encephalitis resemble those of a primary psychotic disorder, which can make it challenging to differentiate between the 2 conditions, and might cause the correct diagnosis to be missed. Pollak et al4 proposed that psychiatrically confusing presentations that don’t clearly match an identifiable psychotic disorder should raise a red flag for an autoimmune etiology. However, studies often fail to describe the specific psychiatric features of anti-NMDA receptor encephalitis, and thus provide little practical evidence to guide diagnosis. In some of the largest studies of patients with anti-NMDA receptor encephalitis, psychiatric clinical findings are often combined into nonspecific headings such as “abnormal behavior” or “behavioral and cognitive” symptoms.5 Such groupings make this the most common clinical finding (95%)5 but make it difficult to discern particular clinical characteristics. Where available, specific symptoms identified across studies include agitation, aggression, changes in mood and/or irritability, insomnia, delusions, hallucinations, and occasionally catatonic features.6,7 Attempts to identify specific psychiatric phenotypes distinct from primary psychotic illnesses have fallen short due to contradictory findings and lack of clinical practicality.8 One exception is the presence of catatonic features, which have been found in CSF-confirmed studies.2 In contrast to the typical teaching that the hallucination modality (eg, visual or tactile) can be helpful in estimating the likelihood of a secondary psychosis (ie, drug-induced, neurodegenerative, or autoimmune), there does not appear to be a difference in hallucination modality between encephalitis and primary psychotic disorders.9
History and review of systems
Another red flag to consider is the rapidity of symptom presentation. Symptoms that progress within 3 months increase the likelihood that the patient has autoimmune encephalitis.10 Cases where collateral information indicates the psychotic episode was preceded by a long, subtle decline in school performance, social withdrawal, and attenuated psychotic symptoms typical of a schizophrenia prodrome are less likely to be an autoimmune psychosis.11 A more delayed presentation does not entirely exclude autoimmune encephalitis; however, a viral-like prodrome before the onset of psychosis increases the likelihood of autoimmune encephalitis. Such a prodrome may include fever, headache, nausea, vomiting, and diarrhea.7
Continue to: Another indication is the presence...
Another indication is the presence of new seizures within 1 year of presenting with psychotic symptoms.10 The possibility of undiagnosed seizures should be considered in a patient with psychosis who has episodes of unresponsiveness, dissociative episodes, or seizure-like activity that is thought to be psychogenic but has not been fully evaluated. Seizures in autoimmune encephalitis involve deep structures in the brain and can be present without overt epileptiform activity on EEG, but rather causing only bilateral slowing that is often described as nonspecific.12
In a young patient presenting with first-episode psychosis, a recent diagnosis of cancer or abnormal finding in the ovaries increases the likelihood of autoimmune encephalitis.4 Historically, however, this type of medical history has been irrelevant to psychosis. Although rare, any person presenting with first-episode psychosis and a history of herpes simplex virus (HSV) encephalitis should be evaluated for autoimmune encephalitis because anti-NMDA receptor antibodies have been reported to be present in approximately one-third of these patients.13 Finally, the report of focal neurologic symptoms, including neck stiffness or neck pain, should raise concern, although sensory, working memory, and cognitive deficits may be difficult to fully distinguish from common somatic and cognitive symptoms in a primary psychiatric presentation.
Table 1 lists 4 questions to ask patients who present with first-episode psychosis that may not usually be part of a typical evaluation.
CASE CONTINUED
Uncooperative with examination
In the ED, Ms. L’s heart rate is 101 beats per minute and her blood pressure is 102/72 mm Hg. Her body mass index (BMI) is 22, which suggests an approximate 8-pound weight loss since her BMI was last assessed. Ms. L responds to questions with 1- to 6-word sentences, without clear verbigeration. Though her speech is not pressured, it is of increased rate. Her gaze scans the room, occasionally becoming fixed for 5 to 10 seconds but is aborted by the interviewer’s comment on this behavior. Ms. L efficiently and accurately spells WORLD backwards, then asks “Why?” and refuses to engage in further cognitive testing, stating “Not doing that.” When the interviewer asks “Why not?” she responds “Not doing that.” Her cranial nerves are intact, and she refuses cerebellar testing or requests to assess tone. There are no observed stereotypies, posturing, or echopraxia.
While not necessary for a diagnosis of autoimmune encephalitis, short-term memory loss is a common cognitive finding across studies.5-7 A common clinical finding from a mental status exam is speech disorders, including (but not limited to) increased rates of speech or decreased verbal output.7 Autonomic instability—including tachycardia, markedly labile blood pressures, and orthostasis—all increase the likelihood of autoimmune encephalitis.14 Interpreting a patient’s vital sign changes can be confounded if they are agitated or anxious, or if they are taking an antipsychotic that produces adverse anticholinergic effects. However, vital sign abnormalities that precede medication administration or do not correlate with fluctuations in mental status increase suspicion for an autoimmune encephalitis.
Continue to: In the absence of the adverse effect...
In the absence of the adverse effect of a medication, orthostasis is uncommon in a well-hydrated young person. Some guidelines4 suggest that symptoms of catatonia should be considered a red flag for autoimmune encephalitis. According to the Bush-Francis Catatonia Rating Scale, commonly identified features include immobility, staring, mutism, posturing, withdrawal, rigidity, and gegenhalten.15 Catatonia is common among patients with anti-NDMA receptor encephalitis, though it may not be initially present and could emerge later.2 However, there are documented cases of autoimmune encephalitis where the patient had only isolated features of catatonia, such as echolalia or mutism.2
CASE CONTINUED
History helps narrow the diagnosis
Ms. L’s parents say their daughter has not had prior contact with a therapist or psychiatrist, previous psychiatric diagnoses, hospitalizations, suicide attempts, self-injury, or binging or purging behaviors. Ms. L’s paternal grandfather was diagnosed with schizophrenia, but he is currently employed, lives alone, and has not taken medication for many years. Her mother has hypothyroidism. Ms. L was born at full term via vaginal delivery without cardiac defects or a neonatal intensive care unit stay. Her mother said she did not have postpartum depression or anxiety, a complicated pregnancy, or exposure to tobacco, alcohol, or illicit drug use. Ms. L has no history of childhood seizures or head injury with loss of consciousness. She is an only child, born and raised in a house in a metropolitan area, walked at 13 months, did not require early intervention or speech therapy, and met normal language milestones.
She attended kindergarten at age 6 and progressed throughout public school without regressions in reading, writing, or behavioral manifestations, and did not require a 504 Plan or individualized education program. Ms. L graduated high school in the top 30% of her class, was socially active, and attended a local college. In college, she achieved honor roll, enrolled in a sorority, and was a part of a research lab. Her only medication is oral contraception. She consumes alcohol socially, and reports no cannabis, cigarette, or vaping use. Ms. L says she does not use hallucinogens, stimulants, opiates, or cocaine, and her roommate and family confirm this. She denies recent travel and is sexually active. Ms. L’s urinary and serum toxicology are unremarkable, human chorionic gonadotropin is undetectable, and her sodium level is 133 mEq/L. A measure of serum neutrophils is 6.8 x 109/L and serum lymphocytes is 1.7 x 109/L. Her parents adamantly request a Neurology consultation and further workup, including a lumbar puncture (LP), EEG, and brain imaging (MRI).
This information is useful in ruling out other potential causes of psychosis, such as substance-induced psychosis and neurodevelopmental disorders that can present with psychosis. Additionally, neurodevelopmental abnormalities and psychiatric prodromal symptoms are known precedents in individuals who develop a primary psychotic disorder such as schizophrenia.16 A family history that includes a psychotic illness may increase the likelihood of a primary psychotic disorder in offspring; however, clinicians must also consider the accuracy of diagnosis in the family, as this can often be inaccurate or influenced by historical cultural bias. We recommend further elucidating the likelihood of a genetic predisposition to a primary psychotic disorder by clarifying familial medication history and functionality.
For example, the fact that Ms. L’s grandfather has not taken medication for many years and has a high degree of functioning and/or absence of cognitive deficits would lower our suspicion for an accurate diagnosis of schizophrenia (given the typical cognitive decline with untreated illness). Another piece of family history relevant to autoimmune encephalitis includes the propensity for autoimmune disorders, but expert opinion on this matter is mixed.17 Ms. L’s mother has hypothyroidism, which is commonly caused by a prior episode of Hashimoto’s autoimmune thyroiditis. Some physicians advocate for measuring antithyroid antibodies and erythrocyte sedimentation rate or C-reactive protein to gauge the level of autoimmunity, but the usefulness of these measures for detecting autoimmune encephalitis is unclear. These serum markers can be useful in detecting additional important etiologies such as systemic infection or systemic inflammation, and there are conditions such as steroid-responsive encephalopathy with associated thyroiditis, which, as the name suggests, responds to steroids rather than other psychotropic medications. Other risk factors for autoimmune encephalitis include being female, being young, having viral infections (eg, HSV), prior tumor burden, and being in the postpartum period.18 Some experts also suggest the presence of neurologic symptoms 4 weeks after the first psychiatric or cognitive symptom presentation increases the likelihood of anti-NMDA receptor encephalitis, and a lack of neurologic symptoms would make this diagnosis less likely.6,19
Continue to: Another item of interest...
Another item of interest in Ms. L’s case is her parents’ request for a Neurology consultation and further workup, as there is an association between caregiver request for workup and eventual diagnosis.6 While the etiology of this phenomenon is unclear, the literature suggests individuals with autoimmune encephalitis who initially present to Psychiatry experience longer delays to the appropriate treatment with immunomodulatory therapy than those who first present to Neurology.20
Laboratory and diagnostic testing
Guasp et al2 recommend EEG, MRI, and serum autoimmune antibodies (ie, screening for anti-NMDA receptor antibodies) for patients who present with first-episode psychosis, even in the absence of some of the red flags previously discussed. A recent economic analysis suggested screening all patients with first-episode psychosis for serum antibodies may be cost-effective.21
For patients whose presentations include features concerning for anti-NMDA receptor encephalitis, an EEG and MRI are reasonable. In a review of EEG abnormalities in anti-NMDA receptor encephalitis, Gillinder et al23 noted that while 30% did not have initial findings, 83.6% of those with confirmed anti-NMDA receptor encephalitis demonstrated EEG abnormalities; the most common were generalized slowing, delta slowing, and focal abnormalities. Discovering an extreme delta-brush activity on EEG is specific for anti-NMDA receptor encephalitis, but its absence is not fully informative. Practically, slowing can be a nonspecific manifestation of encephalopathy or a medication effect, and many people who present with first-episode psychosis will have recently received antipsychotics, which alter EEG frequency. In a study of EEG changes with antipsychotics, Centorrino et al24 found that generalized background slowing into the theta range across all antipsychotics was not significantly different from control participants, while theta to delta range slowing occurred in 8.2% of those receiving antipsychotics vs 3.3% of controls. Clozapine and olanzapine may be associated with greater EEG abnormalities, while haloperidol and quetiapine contribute a lower risk.25 For young patients with first-episode psychosis without a clear alternative explanation, we advocate for further autoimmune encephalitis workup among all individuals with generalized theta or delta wave slowing.
Because these medication effects are most likely to decrease specificity but not sensitivity of EEG for autoimmune encephalitis, a normal EEG without slowing can be reassuring.26 Moreover, for patients who receive neuroimaging, an MRI may detect inflammation that is not visible on CT. The concerning findings for anti-NMDA receptor encephalitis are temporal or multifocal T2 hyperintensities, though the MRI is normal in most cases and thus should not be reassuring if other concerning features are present.27
The role of lumbar puncture
Another area of active debate surrounds the usefulness and timing of LP. Guasp et al2 proposed that all individuals with first-episode psychosis and focal neurologic findings should receive LP and CSF antineuronal antibody testing. They recommend that patients with first-episode psychosis without focal neurologic findings also should receive LP and CSF testing if ≥1 of the following is present:
- slowing on EEG
- temporal or multifocal T2 hyperintensities on MRI
- positive anti-NMDA receptor antibody in the serum.2
Continue to: Evidence suggests that basic CSF parameters...
Evidence suggests that basic CSF parameters, such as elevated protein and white blood cell counts, are some of the most sensitive and specific tests for autoimmune encephalitis.2 Thus, if the patient is amenable and logistical factors are in place, it may be reasonable to pursue LP earlier in some cases without waiting for serum antibody assays to return (these results can take several weeks). CSF inflammatory changes without neuronal antibodies should lead to other diagnostic considerations (eg, systemic inflammatory disease, psychosis attributed to systemic lupus erythematosus).7 While nonspecific, serum laboratory values that may increase suspicion of anti-NMDA receptor encephalitis include hyponatremia6 and an elevated neutrophil-to-lymphocyte ratio (NLR).28 An NLR >4 in conjunction with CSF albumin-to- serum albumin ratio >7 is associated with impaired blood brain barrier integrity and a worse prognosis for those with anti-NMDA receptor encephalitis.28
Additional clinical features that may sway decisions in favor of obtaining LP despite negative findings on EEG, MRI, and serum antibodies include increased adverse reactions to antipsychotics (eg, neuroleptic malignant syndrome), prodromal infectious symptoms, known tumor, or new-onset neurologic symptoms after initial evaluation.2,8
Table 2 summarizes key features of laboratory and diagnostic findings in anti-NMDA receptor encephalitis.
When should you pursue a more extensive workup?
There are some practical tools and rating scales to help clinicians conceptualize risk for autoimmune encephalitis. For psychiatric purposes, however, many of these scales assume that LP, MRI, and EEG have already been completed, and thus it is challenging to incorporate them into psychiatric practice. One such tool is the Antibody Prevalence in Epilepsy and Encephalopathy scale; a score ≥4 is 98% sensitive and 78% to 84% specific for predicting antineural autoantibody positivity.10 Table 3 describes warning signs that may be useful in helping clinicians decide how urgently to pursue a more extensive workup in the possibility of autoimmune encephalitis.
The importance of catching anti-NMDA receptor encephalitis is underscored by the fact that appropriate treatment is very different than for primary psychosis, and outcomes worsen with delay to appropriate treatment.20 Without treatment, severe cases may progress to autonomic instability, altered consciousness, and respiratory compromise warranting admission to an intensive care unit. While the details are beyond the scope of this review, the recommended treatment for confirmed cases of anti-NMDA receptor encephalitis includes tumor removal (if indicated), reducing inflammation (steroids), removing antibodies via IV immunoglobulins, or plasma exchange.8,29 Progression of the disease may warrant consideration of rituximab or cyclophosphamide. In nonresponsive cases, third-line treatments include proteasome inhibitors or interleukin-6 receptor antagonists.8 For patients with severe catatonia, some studies have investigated the utility of electroconvulsive therapy.30 Conceptually, clinicians may consider the utility of antipsychotics as similar to recommendations for hyperactive delirium for the management of psychotic symptoms, agitation, or insomnia. However, given the risk for antipsychotic intolerance, using the lowest effective dose and vigilant screening for the emergence of extrapyramidal symptoms, fever, and autonomic instability is recommended.
CASE CONTINUED
Finally, something objective
Ms. L receives haloperidol 2 mg and undergoes an MRI without contrast. Findings are unremarkable. A spot EEG notes diffuse background slowing in the theta range, prompting lumbar puncture. Findings note 0.40 g/L, 0.2 g/L, and 3.5 for the total protein, albumin, and albumin/CSF-serum quotient (QAlb), respectively; all values are within normal limits. A mild lymphocytic pleocytosis is present as evidenced by a cell count of 35 cells/µL. The CSF is sent for qualitative examination of immunoglobulin G and electrophoresis of proteins in the CSF and serum, of which an increased concentration of restricted bands (oligoclonal bands) in the CSF but not the serum would indicate findings of oligoclonal bands. CSF is sent for detection of anti-NMDA receptor antibodies by indirect immunofluorescence, with a plan to involve an interdisciplinary team for treatment if the antibodies return positive and to manage the case symptomatically in the interim.
Bottom Line
A small subpopulation of patients who present with apparent first-episode psychosis will have symptoms caused by autoimmune encephalitis (specifically, anti-NMDA receptor encephalitis). We provide 4 screening questions to determine when to pursue a workup for an autoimmune encephalitis, and describe relevant clinical symptoms and warning signs to help differentiate the 2 conditions.
Related Resources
- Askandaryan AS, Naqvi A, Varughese A, et al. Anti-N-methyl-D-aspartate receptor encephalitis: neuropsychiatric and multidisciplinary approach to a patient not responding to first-line treatment. Cureus. 2022;14(6):e25751.
- Kayser MS, Titulaer MJ, Gresa-Arribas N, et al. Frequency and characteristics of isolated psychiatric episodes in anti-NMDA receptor encephalitis. JAMA Neurol. 2013;70(9):1133-1139.
Drug Brand Names
Clozapine • Clozaril
Haloperidol • Haldol
Olanzapine • Zyprexa
Quetiapine • Seroquel
Rituximab • Rituxan
Hidden within routine presentations of first-episode psychosis is a rare subpopulation whose symptoms are mediated by an autoimmune process for which proper treatment differs significantly from standard care for typical psychotic illness. In this article, we present a hypothetical case and describe how to assess if a patient has an elevated probability of autoimmune encephalitis, determine what diagnostics or medication-induced effects to consider, and identify unresolved questions about best practices.
CASE REPORT
Bizarre behavior and isolation
Ms. L, age 21, is brought to the emergency department (ED) by her college roommate after exhibiting out-of-character behavior and gradual self-isolation over the last 2 months. Her roommate noticed that she had been spending more time isolated in her dorm room and remaining in bed into the early afternoon, though she does not appear to be asleep. Ms. L’s mother is concerned about her daughter’s uncharacteristic refusal to travel home for a family event. Ms. L expresses concern about the intentions of her research preceptor, and recalls messages from the association of colleges telling her to “change her future.” Ms. L hears voices telling her who she can and cannot trust. In the ED, she says she has a headache, experiences mild dizziness while standing, and reports having a brief upper respiratory illness at the end of last semester. Otherwise, a medical review of systems is negative.
Although the etiology of first-episode psychosis can be numerous or unknown, many psychiatrists feel comfortable with the initial diagnostic for this type of clinical presentation. However, for some clinicians, it may be challenging to feel confident in making a diagnosis of autoimmune encephalitis.
Autoimmune encephalitis is a family of syndromes caused by autoantibodies targeting either intracellular or extracellular neuronal antigens. Anti-N-methyl-
In this article, we focus on anti-NMDA receptor encephalitis and use the term interchangeably with autoimmune encephalitis for 2 reasons. First, anti-NMDA receptor encephalitis can present with psychotic symptoms as the only symptoms (prior to cognitive or neurologic manifestations) or can present with psychotic symptoms as the main indicator (with other symptoms that are more subtle and possibly missed). Second, anti-NMDA receptor encephalitis often occurs in young adults, which is when it is common to see the onset of a primary psychotic illness. These 2 factors make it likely that these cases will come into the evaluative sphere of psychiatrists. We give special attention to features of cases of anti-NMDA receptor encephalitis confirmed with antineuronal antibodies in the CSF, as it has emerged that antibodies in the serum can be nonspecific and nonpathogenic.2,3
What does anti-NMDA receptor encephalitis look like?
Symptoms of anti-NMDA receptor encephalitis resemble those of a primary psychotic disorder, which can make it challenging to differentiate between the 2 conditions, and might cause the correct diagnosis to be missed. Pollak et al4 proposed that psychiatrically confusing presentations that don’t clearly match an identifiable psychotic disorder should raise a red flag for an autoimmune etiology. However, studies often fail to describe the specific psychiatric features of anti-NMDA receptor encephalitis, and thus provide little practical evidence to guide diagnosis. In some of the largest studies of patients with anti-NMDA receptor encephalitis, psychiatric clinical findings are often combined into nonspecific headings such as “abnormal behavior” or “behavioral and cognitive” symptoms.5 Such groupings make this the most common clinical finding (95%)5 but make it difficult to discern particular clinical characteristics. Where available, specific symptoms identified across studies include agitation, aggression, changes in mood and/or irritability, insomnia, delusions, hallucinations, and occasionally catatonic features.6,7 Attempts to identify specific psychiatric phenotypes distinct from primary psychotic illnesses have fallen short due to contradictory findings and lack of clinical practicality.8 One exception is the presence of catatonic features, which have been found in CSF-confirmed studies.2 In contrast to the typical teaching that the hallucination modality (eg, visual or tactile) can be helpful in estimating the likelihood of a secondary psychosis (ie, drug-induced, neurodegenerative, or autoimmune), there does not appear to be a difference in hallucination modality between encephalitis and primary psychotic disorders.9
History and review of systems
Another red flag to consider is the rapidity of symptom presentation. Symptoms that progress within 3 months increase the likelihood that the patient has autoimmune encephalitis.10 Cases where collateral information indicates the psychotic episode was preceded by a long, subtle decline in school performance, social withdrawal, and attenuated psychotic symptoms typical of a schizophrenia prodrome are less likely to be an autoimmune psychosis.11 A more delayed presentation does not entirely exclude autoimmune encephalitis; however, a viral-like prodrome before the onset of psychosis increases the likelihood of autoimmune encephalitis. Such a prodrome may include fever, headache, nausea, vomiting, and diarrhea.7
Continue to: Another indication is the presence...
Another indication is the presence of new seizures within 1 year of presenting with psychotic symptoms.10 The possibility of undiagnosed seizures should be considered in a patient with psychosis who has episodes of unresponsiveness, dissociative episodes, or seizure-like activity that is thought to be psychogenic but has not been fully evaluated. Seizures in autoimmune encephalitis involve deep structures in the brain and can be present without overt epileptiform activity on EEG, but rather causing only bilateral slowing that is often described as nonspecific.12
In a young patient presenting with first-episode psychosis, a recent diagnosis of cancer or abnormal finding in the ovaries increases the likelihood of autoimmune encephalitis.4 Historically, however, this type of medical history has been irrelevant to psychosis. Although rare, any person presenting with first-episode psychosis and a history of herpes simplex virus (HSV) encephalitis should be evaluated for autoimmune encephalitis because anti-NMDA receptor antibodies have been reported to be present in approximately one-third of these patients.13 Finally, the report of focal neurologic symptoms, including neck stiffness or neck pain, should raise concern, although sensory, working memory, and cognitive deficits may be difficult to fully distinguish from common somatic and cognitive symptoms in a primary psychiatric presentation.
Table 1 lists 4 questions to ask patients who present with first-episode psychosis that may not usually be part of a typical evaluation.
CASE CONTINUED
Uncooperative with examination
In the ED, Ms. L’s heart rate is 101 beats per minute and her blood pressure is 102/72 mm Hg. Her body mass index (BMI) is 22, which suggests an approximate 8-pound weight loss since her BMI was last assessed. Ms. L responds to questions with 1- to 6-word sentences, without clear verbigeration. Though her speech is not pressured, it is of increased rate. Her gaze scans the room, occasionally becoming fixed for 5 to 10 seconds but is aborted by the interviewer’s comment on this behavior. Ms. L efficiently and accurately spells WORLD backwards, then asks “Why?” and refuses to engage in further cognitive testing, stating “Not doing that.” When the interviewer asks “Why not?” she responds “Not doing that.” Her cranial nerves are intact, and she refuses cerebellar testing or requests to assess tone. There are no observed stereotypies, posturing, or echopraxia.
While not necessary for a diagnosis of autoimmune encephalitis, short-term memory loss is a common cognitive finding across studies.5-7 A common clinical finding from a mental status exam is speech disorders, including (but not limited to) increased rates of speech or decreased verbal output.7 Autonomic instability—including tachycardia, markedly labile blood pressures, and orthostasis—all increase the likelihood of autoimmune encephalitis.14 Interpreting a patient’s vital sign changes can be confounded if they are agitated or anxious, or if they are taking an antipsychotic that produces adverse anticholinergic effects. However, vital sign abnormalities that precede medication administration or do not correlate with fluctuations in mental status increase suspicion for an autoimmune encephalitis.
Continue to: In the absence of the adverse effect...
In the absence of the adverse effect of a medication, orthostasis is uncommon in a well-hydrated young person. Some guidelines4 suggest that symptoms of catatonia should be considered a red flag for autoimmune encephalitis. According to the Bush-Francis Catatonia Rating Scale, commonly identified features include immobility, staring, mutism, posturing, withdrawal, rigidity, and gegenhalten.15 Catatonia is common among patients with anti-NDMA receptor encephalitis, though it may not be initially present and could emerge later.2 However, there are documented cases of autoimmune encephalitis where the patient had only isolated features of catatonia, such as echolalia or mutism.2
CASE CONTINUED
History helps narrow the diagnosis
Ms. L’s parents say their daughter has not had prior contact with a therapist or psychiatrist, previous psychiatric diagnoses, hospitalizations, suicide attempts, self-injury, or binging or purging behaviors. Ms. L’s paternal grandfather was diagnosed with schizophrenia, but he is currently employed, lives alone, and has not taken medication for many years. Her mother has hypothyroidism. Ms. L was born at full term via vaginal delivery without cardiac defects or a neonatal intensive care unit stay. Her mother said she did not have postpartum depression or anxiety, a complicated pregnancy, or exposure to tobacco, alcohol, or illicit drug use. Ms. L has no history of childhood seizures or head injury with loss of consciousness. She is an only child, born and raised in a house in a metropolitan area, walked at 13 months, did not require early intervention or speech therapy, and met normal language milestones.
She attended kindergarten at age 6 and progressed throughout public school without regressions in reading, writing, or behavioral manifestations, and did not require a 504 Plan or individualized education program. Ms. L graduated high school in the top 30% of her class, was socially active, and attended a local college. In college, she achieved honor roll, enrolled in a sorority, and was a part of a research lab. Her only medication is oral contraception. She consumes alcohol socially, and reports no cannabis, cigarette, or vaping use. Ms. L says she does not use hallucinogens, stimulants, opiates, or cocaine, and her roommate and family confirm this. She denies recent travel and is sexually active. Ms. L’s urinary and serum toxicology are unremarkable, human chorionic gonadotropin is undetectable, and her sodium level is 133 mEq/L. A measure of serum neutrophils is 6.8 x 109/L and serum lymphocytes is 1.7 x 109/L. Her parents adamantly request a Neurology consultation and further workup, including a lumbar puncture (LP), EEG, and brain imaging (MRI).
This information is useful in ruling out other potential causes of psychosis, such as substance-induced psychosis and neurodevelopmental disorders that can present with psychosis. Additionally, neurodevelopmental abnormalities and psychiatric prodromal symptoms are known precedents in individuals who develop a primary psychotic disorder such as schizophrenia.16 A family history that includes a psychotic illness may increase the likelihood of a primary psychotic disorder in offspring; however, clinicians must also consider the accuracy of diagnosis in the family, as this can often be inaccurate or influenced by historical cultural bias. We recommend further elucidating the likelihood of a genetic predisposition to a primary psychotic disorder by clarifying familial medication history and functionality.
For example, the fact that Ms. L’s grandfather has not taken medication for many years and has a high degree of functioning and/or absence of cognitive deficits would lower our suspicion for an accurate diagnosis of schizophrenia (given the typical cognitive decline with untreated illness). Another piece of family history relevant to autoimmune encephalitis includes the propensity for autoimmune disorders, but expert opinion on this matter is mixed.17 Ms. L’s mother has hypothyroidism, which is commonly caused by a prior episode of Hashimoto’s autoimmune thyroiditis. Some physicians advocate for measuring antithyroid antibodies and erythrocyte sedimentation rate or C-reactive protein to gauge the level of autoimmunity, but the usefulness of these measures for detecting autoimmune encephalitis is unclear. These serum markers can be useful in detecting additional important etiologies such as systemic infection or systemic inflammation, and there are conditions such as steroid-responsive encephalopathy with associated thyroiditis, which, as the name suggests, responds to steroids rather than other psychotropic medications. Other risk factors for autoimmune encephalitis include being female, being young, having viral infections (eg, HSV), prior tumor burden, and being in the postpartum period.18 Some experts also suggest the presence of neurologic symptoms 4 weeks after the first psychiatric or cognitive symptom presentation increases the likelihood of anti-NMDA receptor encephalitis, and a lack of neurologic symptoms would make this diagnosis less likely.6,19
Continue to: Another item of interest...
Another item of interest in Ms. L’s case is her parents’ request for a Neurology consultation and further workup, as there is an association between caregiver request for workup and eventual diagnosis.6 While the etiology of this phenomenon is unclear, the literature suggests individuals with autoimmune encephalitis who initially present to Psychiatry experience longer delays to the appropriate treatment with immunomodulatory therapy than those who first present to Neurology.20
Laboratory and diagnostic testing
Guasp et al2 recommend EEG, MRI, and serum autoimmune antibodies (ie, screening for anti-NMDA receptor antibodies) for patients who present with first-episode psychosis, even in the absence of some of the red flags previously discussed. A recent economic analysis suggested screening all patients with first-episode psychosis for serum antibodies may be cost-effective.21
For patients whose presentations include features concerning for anti-NMDA receptor encephalitis, an EEG and MRI are reasonable. In a review of EEG abnormalities in anti-NMDA receptor encephalitis, Gillinder et al23 noted that while 30% did not have initial findings, 83.6% of those with confirmed anti-NMDA receptor encephalitis demonstrated EEG abnormalities; the most common were generalized slowing, delta slowing, and focal abnormalities. Discovering an extreme delta-brush activity on EEG is specific for anti-NMDA receptor encephalitis, but its absence is not fully informative. Practically, slowing can be a nonspecific manifestation of encephalopathy or a medication effect, and many people who present with first-episode psychosis will have recently received antipsychotics, which alter EEG frequency. In a study of EEG changes with antipsychotics, Centorrino et al24 found that generalized background slowing into the theta range across all antipsychotics was not significantly different from control participants, while theta to delta range slowing occurred in 8.2% of those receiving antipsychotics vs 3.3% of controls. Clozapine and olanzapine may be associated with greater EEG abnormalities, while haloperidol and quetiapine contribute a lower risk.25 For young patients with first-episode psychosis without a clear alternative explanation, we advocate for further autoimmune encephalitis workup among all individuals with generalized theta or delta wave slowing.
Because these medication effects are most likely to decrease specificity but not sensitivity of EEG for autoimmune encephalitis, a normal EEG without slowing can be reassuring.26 Moreover, for patients who receive neuroimaging, an MRI may detect inflammation that is not visible on CT. The concerning findings for anti-NMDA receptor encephalitis are temporal or multifocal T2 hyperintensities, though the MRI is normal in most cases and thus should not be reassuring if other concerning features are present.27
The role of lumbar puncture
Another area of active debate surrounds the usefulness and timing of LP. Guasp et al2 proposed that all individuals with first-episode psychosis and focal neurologic findings should receive LP and CSF antineuronal antibody testing. They recommend that patients with first-episode psychosis without focal neurologic findings also should receive LP and CSF testing if ≥1 of the following is present:
- slowing on EEG
- temporal or multifocal T2 hyperintensities on MRI
- positive anti-NMDA receptor antibody in the serum.2
Continue to: Evidence suggests that basic CSF parameters...
Evidence suggests that basic CSF parameters, such as elevated protein and white blood cell counts, are some of the most sensitive and specific tests for autoimmune encephalitis.2 Thus, if the patient is amenable and logistical factors are in place, it may be reasonable to pursue LP earlier in some cases without waiting for serum antibody assays to return (these results can take several weeks). CSF inflammatory changes without neuronal antibodies should lead to other diagnostic considerations (eg, systemic inflammatory disease, psychosis attributed to systemic lupus erythematosus).7 While nonspecific, serum laboratory values that may increase suspicion of anti-NMDA receptor encephalitis include hyponatremia6 and an elevated neutrophil-to-lymphocyte ratio (NLR).28 An NLR >4 in conjunction with CSF albumin-to- serum albumin ratio >7 is associated with impaired blood brain barrier integrity and a worse prognosis for those with anti-NMDA receptor encephalitis.28
Additional clinical features that may sway decisions in favor of obtaining LP despite negative findings on EEG, MRI, and serum antibodies include increased adverse reactions to antipsychotics (eg, neuroleptic malignant syndrome), prodromal infectious symptoms, known tumor, or new-onset neurologic symptoms after initial evaluation.2,8
Table 2 summarizes key features of laboratory and diagnostic findings in anti-NMDA receptor encephalitis.
When should you pursue a more extensive workup?
There are some practical tools and rating scales to help clinicians conceptualize risk for autoimmune encephalitis. For psychiatric purposes, however, many of these scales assume that LP, MRI, and EEG have already been completed, and thus it is challenging to incorporate them into psychiatric practice. One such tool is the Antibody Prevalence in Epilepsy and Encephalopathy scale; a score ≥4 is 98% sensitive and 78% to 84% specific for predicting antineural autoantibody positivity.10 Table 3 describes warning signs that may be useful in helping clinicians decide how urgently to pursue a more extensive workup in the possibility of autoimmune encephalitis.
The importance of catching anti-NMDA receptor encephalitis is underscored by the fact that appropriate treatment is very different than for primary psychosis, and outcomes worsen with delay to appropriate treatment.20 Without treatment, severe cases may progress to autonomic instability, altered consciousness, and respiratory compromise warranting admission to an intensive care unit. While the details are beyond the scope of this review, the recommended treatment for confirmed cases of anti-NMDA receptor encephalitis includes tumor removal (if indicated), reducing inflammation (steroids), removing antibodies via IV immunoglobulins, or plasma exchange.8,29 Progression of the disease may warrant consideration of rituximab or cyclophosphamide. In nonresponsive cases, third-line treatments include proteasome inhibitors or interleukin-6 receptor antagonists.8 For patients with severe catatonia, some studies have investigated the utility of electroconvulsive therapy.30 Conceptually, clinicians may consider the utility of antipsychotics as similar to recommendations for hyperactive delirium for the management of psychotic symptoms, agitation, or insomnia. However, given the risk for antipsychotic intolerance, using the lowest effective dose and vigilant screening for the emergence of extrapyramidal symptoms, fever, and autonomic instability is recommended.
CASE CONTINUED
Finally, something objective
Ms. L receives haloperidol 2 mg and undergoes an MRI without contrast. Findings are unremarkable. A spot EEG notes diffuse background slowing in the theta range, prompting lumbar puncture. Findings note 0.40 g/L, 0.2 g/L, and 3.5 for the total protein, albumin, and albumin/CSF-serum quotient (QAlb), respectively; all values are within normal limits. A mild lymphocytic pleocytosis is present as evidenced by a cell count of 35 cells/µL. The CSF is sent for qualitative examination of immunoglobulin G and electrophoresis of proteins in the CSF and serum, of which an increased concentration of restricted bands (oligoclonal bands) in the CSF but not the serum would indicate findings of oligoclonal bands. CSF is sent for detection of anti-NMDA receptor antibodies by indirect immunofluorescence, with a plan to involve an interdisciplinary team for treatment if the antibodies return positive and to manage the case symptomatically in the interim.
Bottom Line
A small subpopulation of patients who present with apparent first-episode psychosis will have symptoms caused by autoimmune encephalitis (specifically, anti-NMDA receptor encephalitis). We provide 4 screening questions to determine when to pursue a workup for an autoimmune encephalitis, and describe relevant clinical symptoms and warning signs to help differentiate the 2 conditions.
Related Resources
- Askandaryan AS, Naqvi A, Varughese A, et al. Anti-N-methyl-D-aspartate receptor encephalitis: neuropsychiatric and multidisciplinary approach to a patient not responding to first-line treatment. Cureus. 2022;14(6):e25751.
- Kayser MS, Titulaer MJ, Gresa-Arribas N, et al. Frequency and characteristics of isolated psychiatric episodes in anti-NMDA receptor encephalitis. JAMA Neurol. 2013;70(9):1133-1139.
Drug Brand Names
Clozapine • Clozaril
Haloperidol • Haldol
Olanzapine • Zyprexa
Quetiapine • Seroquel
Rituximab • Rituxan
1. Granerod J, Ambrose HE, Davies NW, et al; UK Health Protection Agency (HPA) Aetiology of Encephalitis Study Group. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis. 2010;10(12):835-44. doi:10.1016/S1473-3099(10)70222-X
2. Guasp M, Giné-Servén E, Maudes E, et al. Clinical, neuroimmunologic, and CSF investigations in first episode psychosis. Neurology. 2021;97(1):e61-e75.
3. From the American Association of Neurological Surgeons (AANS), American Society of Neuroradiology (ASNR), Cardiovascular and Interventional Radiology Society of Europe (CIRSE), Canadian Interventional Radiology Association (CIRA), Congress of Neurological Surgeons (CNS), European Society of Minimally Invasive Neurological Therapy (ESMINT), European Society of Neuroradiology (ESNR), European Stroke Organization (ESO), Society for Cardiovascular Angiography and Interventions (SCAI), Society of Interventional Radiology (SIR), Society of NeuroInterventional Surgery (SNIS), and World Stroke Organization (WSO), Sacks D, Baxter B, Campbell BCV, et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int J Stroke. 2018;13(6):612-632. doi:10.1177/1747493018778713
4. Pollak TA, Lennox BR, Muller S, et al. Autoimmune psychosis: an international consensus on an approach to the diagnosis and management of psychosis of suspected autoimmune origin. Lancet Psychiatry. 2020;7(1):93-108.
5. Guasp M, Módena Y, Armangue T, et al. Clinical features of seronegative, but CSF antibody-positive, anti-NMDA receptor encephalitis. Neurol Neuroimmunol Neuroinflamm. 2020;7(2):e659.
6. Herken J, Prüss H. Red flags: clinical signs for identifying autoimmune encephalitis in psychiatric patients. Front Psychiatry. 2017;8:25. doi:10.3389/fpsyt.2017.00025
7. Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404.
8. Dalmau J, Armangue T, Planaguma J, et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: mechanisms and models. Lancet Neurol. 2019;18(11):1045-1057.
9. Rattay TW, Martin P, Vittore D, et al. Cerebrospinal fluid findings in patients with psychotic symptoms—a retrospective analysis. Sci Rep. 2021;11(1):7169.
10. Dubey D, Pittock SJ, McKeon A. Antibody prevalence in epilepsy and encephalopathy score: increased specificity and applicability. Epilepsia. 2019;60(2):367-369.
11. Maj M, van Os J, De Hert M, et al. The clinical characterization of the patient with primary psychosis aimed at personalization of management. World Psychiatry. 2021;20(1):4-33. doi:10.1002/wps.20809
12. Caplan JP, Binius T, Lennon VA, et al. Pseudopseudoseizures: conditions that may mimic psychogenic non-epileptic seizures. Psychosomatics. 2011;52(6):501-506.
13. Armangue T, Spatola M, Vlagea A, et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis. Lancet Neurol. 2018;17(9):760-772.
14. Takamatsu K, Nakane S. Autonomic manifestations in autoimmune encephalitis. Neurol Clin Neurosci. 2022;10:130-136. doi:10.1111/ncn3.12557
15. Espinola-Nadurille M, Flores-Rivera J, Rivas-Alonso V, et al. Catatonia in patients with anti-NMDA receptor encephalitis. Psychiatry Clin Neurosci. 2019;73(9):574-580.
16. Keshavan M, Montrose DM, Rajarethinam R, et al. Psychopathology among offspring of parents with schizophrenia: relationship to premorbid impairments. Schizophr Res. 2008;103(1-3):114-120.
17. Jeppesen R, Benros ME. Autoimmune diseases and psychotic disorders. Front Psychiatry. 2019;10:131.
18. Bergink V, Armangue T, Titulaer MJ, et al. Autoimmune encephalitis in postpartum psychosis. Am J Psychiatry. 2015;172(9):901-908.
19. Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091-8. doi: 10.1016/S1474-4422(08)70224-2
20. Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12(2):157-165.
21. Ross EL, Becker JE, Linnoila JJ, et al. Cost-effectiveness of routine screening for autoimmune encephalitis in patients with first-episode psychosis in the United States. J Clin Psychiatry. 2020;82(1):19m13168.
22. Sonderen AV, Arends S, Tavy DLJ, et al. Predictive value of electroencephalography in anti-NMDA receptor encephalitis. J Neurol Neurosurg Psychiatry. 2018;89(10):1101-1106.
23. Gillinder L, Warren N, Hartel G, et al. EEG findings in NMDA encephalitis--a systematic review. Seizure. 2019;65:20-24.
24. Centorrino F, Price BH, Tuttle M, et al. EEG abnormalities during treatment with typical and atypical antipsychotics. Am J Psychiatry. 2002;159(1):109-115.
25. Raymond N, Lizano P, Kelly S, et al. What can clozapine’s effect on neural oscillations tell us about its therapeutic effects? A scoping review and synthesis. Biomarkers in Neuropsychiatry. 2022;6:100048.
26. Kaufman DM, Geyer H, Milstein MJ. Kaufman’s Clinical Neurology for Psychiatrists. 8th ed. Elsevier Inc; 2016.
27. Kelley BP, Patel SC, Marin HL, et al. Autoimmune encephalitis: pathophysiology and imaging review of an overlooked diagnosis. AJNR Am J Neuroradiol. 2017;38(6):1070-1078.
28. Yu Y, Wu Y, Cao X, et al. The clinical features and prognosis of anti-NMDAR encephalitis depends on blood brain barrier integrity. Mult Scler Relat Disord. 2021;47:102604.
29. Dalmau J, Graus F. Antibody-mediated neuropsychiatric disorders. J Allergy Clin Immunol. 2022;149(1):37-40.
30. Warren N, Grote V, O’Gorman C, et al. Electroconvulsive therapy for anti-N-methyl-daspartate (NMDA) receptor encephalitis: a systematic review of cases. Brain Stimul. 2019;12(2):329-334.
1. Granerod J, Ambrose HE, Davies NW, et al; UK Health Protection Agency (HPA) Aetiology of Encephalitis Study Group. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis. 2010;10(12):835-44. doi:10.1016/S1473-3099(10)70222-X
2. Guasp M, Giné-Servén E, Maudes E, et al. Clinical, neuroimmunologic, and CSF investigations in first episode psychosis. Neurology. 2021;97(1):e61-e75.
3. From the American Association of Neurological Surgeons (AANS), American Society of Neuroradiology (ASNR), Cardiovascular and Interventional Radiology Society of Europe (CIRSE), Canadian Interventional Radiology Association (CIRA), Congress of Neurological Surgeons (CNS), European Society of Minimally Invasive Neurological Therapy (ESMINT), European Society of Neuroradiology (ESNR), European Stroke Organization (ESO), Society for Cardiovascular Angiography and Interventions (SCAI), Society of Interventional Radiology (SIR), Society of NeuroInterventional Surgery (SNIS), and World Stroke Organization (WSO), Sacks D, Baxter B, Campbell BCV, et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int J Stroke. 2018;13(6):612-632. doi:10.1177/1747493018778713
4. Pollak TA, Lennox BR, Muller S, et al. Autoimmune psychosis: an international consensus on an approach to the diagnosis and management of psychosis of suspected autoimmune origin. Lancet Psychiatry. 2020;7(1):93-108.
5. Guasp M, Módena Y, Armangue T, et al. Clinical features of seronegative, but CSF antibody-positive, anti-NMDA receptor encephalitis. Neurol Neuroimmunol Neuroinflamm. 2020;7(2):e659.
6. Herken J, Prüss H. Red flags: clinical signs for identifying autoimmune encephalitis in psychiatric patients. Front Psychiatry. 2017;8:25. doi:10.3389/fpsyt.2017.00025
7. Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404.
8. Dalmau J, Armangue T, Planaguma J, et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: mechanisms and models. Lancet Neurol. 2019;18(11):1045-1057.
9. Rattay TW, Martin P, Vittore D, et al. Cerebrospinal fluid findings in patients with psychotic symptoms—a retrospective analysis. Sci Rep. 2021;11(1):7169.
10. Dubey D, Pittock SJ, McKeon A. Antibody prevalence in epilepsy and encephalopathy score: increased specificity and applicability. Epilepsia. 2019;60(2):367-369.
11. Maj M, van Os J, De Hert M, et al. The clinical characterization of the patient with primary psychosis aimed at personalization of management. World Psychiatry. 2021;20(1):4-33. doi:10.1002/wps.20809
12. Caplan JP, Binius T, Lennon VA, et al. Pseudopseudoseizures: conditions that may mimic psychogenic non-epileptic seizures. Psychosomatics. 2011;52(6):501-506.
13. Armangue T, Spatola M, Vlagea A, et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis. Lancet Neurol. 2018;17(9):760-772.
14. Takamatsu K, Nakane S. Autonomic manifestations in autoimmune encephalitis. Neurol Clin Neurosci. 2022;10:130-136. doi:10.1111/ncn3.12557
15. Espinola-Nadurille M, Flores-Rivera J, Rivas-Alonso V, et al. Catatonia in patients with anti-NMDA receptor encephalitis. Psychiatry Clin Neurosci. 2019;73(9):574-580.
16. Keshavan M, Montrose DM, Rajarethinam R, et al. Psychopathology among offspring of parents with schizophrenia: relationship to premorbid impairments. Schizophr Res. 2008;103(1-3):114-120.
17. Jeppesen R, Benros ME. Autoimmune diseases and psychotic disorders. Front Psychiatry. 2019;10:131.
18. Bergink V, Armangue T, Titulaer MJ, et al. Autoimmune encephalitis in postpartum psychosis. Am J Psychiatry. 2015;172(9):901-908.
19. Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091-8. doi: 10.1016/S1474-4422(08)70224-2
20. Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12(2):157-165.
21. Ross EL, Becker JE, Linnoila JJ, et al. Cost-effectiveness of routine screening for autoimmune encephalitis in patients with first-episode psychosis in the United States. J Clin Psychiatry. 2020;82(1):19m13168.
22. Sonderen AV, Arends S, Tavy DLJ, et al. Predictive value of electroencephalography in anti-NMDA receptor encephalitis. J Neurol Neurosurg Psychiatry. 2018;89(10):1101-1106.
23. Gillinder L, Warren N, Hartel G, et al. EEG findings in NMDA encephalitis--a systematic review. Seizure. 2019;65:20-24.
24. Centorrino F, Price BH, Tuttle M, et al. EEG abnormalities during treatment with typical and atypical antipsychotics. Am J Psychiatry. 2002;159(1):109-115.
25. Raymond N, Lizano P, Kelly S, et al. What can clozapine’s effect on neural oscillations tell us about its therapeutic effects? A scoping review and synthesis. Biomarkers in Neuropsychiatry. 2022;6:100048.
26. Kaufman DM, Geyer H, Milstein MJ. Kaufman’s Clinical Neurology for Psychiatrists. 8th ed. Elsevier Inc; 2016.
27. Kelley BP, Patel SC, Marin HL, et al. Autoimmune encephalitis: pathophysiology and imaging review of an overlooked diagnosis. AJNR Am J Neuroradiol. 2017;38(6):1070-1078.
28. Yu Y, Wu Y, Cao X, et al. The clinical features and prognosis of anti-NMDAR encephalitis depends on blood brain barrier integrity. Mult Scler Relat Disord. 2021;47:102604.
29. Dalmau J, Graus F. Antibody-mediated neuropsychiatric disorders. J Allergy Clin Immunol. 2022;149(1):37-40.
30. Warren N, Grote V, O’Gorman C, et al. Electroconvulsive therapy for anti-N-methyl-daspartate (NMDA) receptor encephalitis: a systematic review of cases. Brain Stimul. 2019;12(2):329-334.

Ketamine for acute catatonia: A case report
Ms. C, age 44, who has major depressive disorder (MDD), anxiety, obsessive-compulsive disorder (OCD) (religious subtype), and has experienced multiple episodes of treatment-resistant catatonia, is brought to the emergency department (ED) by her parents. She has immobility, mutism, rigidity, and decreased oral intake that she has experienced for 1 day.
The night before, Ms. C had been stressed about an upcoming job interview. She cancelled the interview and went to her bedroom. Later that night her parents found her lying on the floor, immobile.
Before the onset of her psychiatric symptoms, Ms. C had been high functioning. She had been an athlete in college and had a career as a school psychologist. The Sidebar summarizes Ms. C’s psychiatric history, which includes similar complex episodes and multiple hospitalizations. She also has a history of hypothyroidism.
SIDEBAR
In 2013, Ms. C experienced severe social stress from both her work as a psychologist and a divorce. She sold all of her possessions and was living in motels and hotels searching for the “truth of God.” In February 2016, she was hospitalized after refusing to eat and self-discontinuing all medications, including her thyroid medications. She was then placed under the conservatorship of her parents.
In July 2017, Ms. C was hospitalized again for refusing to eat or take her medications; this time she also exhibited selective mutism. Catatonia was suspected and she was started on oral lorazepam, 2 mg 3 times a day. Duloxetine and ziprasidone were also trialed but were stopped due to noncompliance and adverse effects. Ms. C showed little improvement on these regimens. In the hospital, IV lorazepam, 4 mg, was trialed with good effect, and she began to respond to questioning. She was transitioned to oral lorazepam, 4 mg 5 times per day, and mirtazapine, 15 mg/d. With this regimen, Ms. C became progressively more interactive; however, she still refused to eat. Throughout her hospitalization, multiple medications were prescribed, including divalproex sodium, memantine, zolpidem, olanzapine, and dextroamphetamine/levoamphetamine, all of which were not effective in stimulating her appetite. Due to malnutrition, Ms. C was placed on total parenteral nutrition. During this time, the highest dose of IM lorazepam was 20 mg/d in divided doses.
Some improvement with ECT
Four months into her hospitalization, Ms. C’s lorazepam was titrated down to 4 mg 4 times a day, and she underwent a trial of electroconvulsive therapy (ECT). Following the fourth ECT session, she displayed significant improvement. Ms. C engaged with her clinicians, displayed bright mood and affect, began eating again, and was able to recount her depressive symptoms following her divorce. At this time, she received a total of 8 ECT treatments and was started on fluoxetine. At the end of January 2018, after 19 days of hospitalization, she was transitioned to a partial hospitalization program (PHP) on a regimen of lorazepam, 2 mg 3 times daily; fluoxetine, 40 mg/d; midodrine, 10 mg 3 times daily; fludrocortisone; and levothyroxine. Her discharge diagnosis was major depressive disorder with psychotic features and catatonia.
Between her first hospitalization and her current presentation to the emergency department (ED), Ms. C presented several times to the ED with similar symptoms of decreased speech, movement, and oral intake. In February 2018, she was hospitalized and responded after 4 sessions of ECT. She returned to work as a substitute teacher and was stable for >1 year on a regimen of lorazepam, olanzapine, and risperidone. In June 2019, her symptoms returned. She was hospitalized and required a nasogastric tube to address malnutrition. She was eventually stabilized on a regimen of risperidone and lorazepam, which she continued as an outpatient until she was hospitalized again in August 2019. During this hospitalization, Ms. C failed to respond to risperidone or lorazepam, up to 2 mg 3 times a day. After several changes to her regimen, she began to respond to olanzapine, 30 mg/d; mirtazapine, 15 mg/d; and lorazepam, 2 mg 3 times a day.
Throughout her hospitalizations, once she became verbal, Ms. C demonstrated hyper-religiosity. She would ask to read the Bible, and state that her purpose was to find the truth of God. As an outpatient, she would compulsively go to church in the middle of the night and read the Bible for hours. A preliminary diagnosis of obsessive-compulsive disorder was made based on her scrupulosity, and mirtazapine was cross-titrated to fluvoxamine prior to discharge.
Shortly after discharge, she was readmitted to a PHP, and did well on fluvoxamine, 100 mg twice a day; olanzapine, 5 mg every night; levothyroxine, 100 mcg/d; and oral lorazepam, 1 mg 4 times a day. Ms. C displayed full mood, appropriate affect, and began working part-time as a substitute teacher. She had begun to interview for full-time jobs before her most recent ED presentation.
In the ED, the psychiatry team evaluates Ms. C. She displays a similar pattern of mutism, immobility, and rigidity as she did upon her initial presentation. Her father reports that she had been compliant with her medications but had not taken them the previous night. Ms. C screens positive for catatonia on the Bush-Francis Catatonia Rating Scale (BFCRS). Her severity score of 10/69 indicates a mild presentation. She is diagnosed with catatonia and is administered IV
Because Ms. C has been hospitalized many times for similar presentations, the treatment team decides to initiate a trial of IV ketamine.
Catatonia can manifest in many different ways in patients with psychiatric illness. If left untreated, it is associated with a high rate of mortality.1 Catatonia often is described along a continuum from retarded/stuporous to excited, and presentations can vary substantially. The physiologic and psychological mechanisms of catatonia are poorly understood.
Traditionally, most patients respond well to low-dose benzodiazepines, with electroconvulsive therapy as a second-line intervention for refractory and malignant cases. However, these interventions are not always successful or readily available.
Continue to: Research into the anesthetic ketamine...
Research into the anesthetic ketamine is gradually expanding, and the use of this agent for treating various psychiatric illnesses, including both unipolar and bipolar depression, has been increasing.2 Empiric evidence suggests ketamine is effective for certain psychiatric disorders, but the mechanism of action remains unclear. Although the evidence base is small, additional cases demonstrating the effectiveness of ketamine in the treatment of acute catatonia might make it a therapeutic option for use by psychiatrists and emergency medicine clinicians.
In this article, we discuss ketamine’s possible role in the treatment of catatonia, possible adverse effects, dosing strategies, and theories about ketamine’s mechanism of action.
Ketamine’s utility in psychiatry
Ketamine is a rapid-acting anesthetic that acts primarily by antagonizing N-methyl-
Previously, ketamine had been thought to induce a catatonic state, which was supported by a neurophysiologic model of catatonia that suggested the condition was caused in part by glutamate hypoactivity at the NMDA receptor.9 However, recent studies have shown that the NMDA receptor antagonists
Ketamine originally was used for sedation, and much of its safety and risk profile has been developed from decades of administration as an anesthetic. Studies have found that ketamine has a large therapeutic window in children and adults.15,16 Moreover, it does not depress the respiratory system. As an anesthetic, ketamine has a rapid onset and a quick resolution, with its sedative and disorienting effects resolving within 30 to 120 minutes.17 Ketamine’s rapid onset of action extends beyond its sedating effects. Trials with the intranasal spray esketamine for treatment-resistant depression have demonstrated an onset antidepressant effects within 2 days.18 This is much faster than that of traditional antidepressants, such as selective serotonin reuptake inhibitors.18 Based on these features, ketamine has the potential to be a useful medication in the emergency psychiatric setting, particularly for acute presentations such as catatonia.
Continue to: Beware of the potential risks
Beware of the potential risks
Although ketamine may be clinically useful, it also carries some risks. Adverse effects associated with ketamine include sedation, dissociation, hallucinations, elevated blood pressure, nausea, increased heart rate, vomiting, dizziness, fatigue, blurred vision, itching, and emesis. Clinicians also should be aware that some patients may use illicit ketamine, either as self-treatment to control depressive symptoms or for recreational purposes. When misused/abused, long-term use of ketamine can cause neurologic damage.19 Studies also have reported rare occurrences of recurrent hallucinations even after discontinuation of ketamine.20 Animal studies have demonstrated addiction and cognitive deficits with repeated use of ketamine in rodents.21 This research has led to concerns that chronic use of ketamine to treat illnesses such as depression might lead to similar long-term adverse outcomes.
Dosing
As a sedative, IV ketamine dosing is generally 1 to 2 mg/kg, and IM ketamine dosing is 3 to 5 mg/kg.16 As an antidepressant, small clinical trials have suggested that the preferred dose of IV ketamine may be 0.5 to 1 mg/kg, with dose-dependent increases in dissociation and blood pressure.21 Studies have also demonstrated that once-daily IV ketamine, 0.5 mg/kg administered over 40 minutes, led to greater improvements in patients with MDD than placebo, whereas once-daily IV ketamine, 0.2 mg/kg, did not.20
CASE CONTINUED
The team begins to treat Ms. C with IV ketamine. Ketamine, 0.2 mg/kg, is used to calculate the initial dose, and a total of 10 mg is administered over 10 minutes. Fifteen minutes after administration, Ms. C is able to move around in her bed, make eye contact, and nod to questions. She has purposeful movements, such as examining her IV line, scratching her head, and repositioning herself in the bed. After a few more minutes, she makes eye contact with her father, and nods to him during conversation. She is able to make a few noises but does not speak.
Later that day, Ms. C is discharged home (in a wheelchair) with her parents, on a medication regimen of fluvoxamine, 100 mg/d; lorazepam, 1 mg 4 times a day; and olanzapine, 5 mg/d. She is scheduled for an outpatient follow-up appointment 5 days later. Her parents are given instructions and several precautions to ensure that Ms. C receives proper nutrition until her appointment. That evening, Ms. C is able to eat voluntarily.
Five days later, Ms. C visits the outpatient psychiatric clinic and is verbal and ambulatory. Her father reports that she has become more verbal. During her follow-up interview, she is observed to be more subdued and less verbal than her baseline, but is vocal and able to voice her understanding of the treatment plan.
Continue to: After 3 months of being stable...
After 3 months of being stable on her outpatient regimen, Ms. C’s catatonic symptoms return, including refusing to eat and mutism. She is administered IV lorazepam, 4 mg, with no response and is admitted to the hospital for placement of a nasogastric feeding tube to address malnutrition. After several days, Ms. C responds to lorazepam, 4 mg every 6 hours. Six days later, after she begins eating and taking her medications voluntarily and the nasogastric tube is removed, Ms. C is discharged to home.
Findings need to be replicated in larger studies
Although some research has indicated that ketamine may be pro-catatonic, Ms. C’s improvement after receiving ketamine suggests that perhaps the situation is more complex.12,22 The exact mechanisms underlying catatonia remain uncertain. Carroll et al9 described 4 theories, and only 1 of them involved glutamate. Additionally, ketamine’s mechanism of action may extend beyond NMDA antagonism. In our case, Ms. C’s low BFCRS score during her most recent visit to the ED suggests she may have had a milder or less typical form of catatonia compared with her previous presentations (Sidebar). However, Ms. C’s clinical improvement after receiving ketamine is noteworthy.
A review of the literature yielded only 1 other case report that described using ketamine to treat catatonia.23 Iserson et al23 reported that their patient’s catatonic symptoms resolved after a total of 12.5 mg of ketamine was administered in 0.03 mg/kg boluses every 3 minutes. Compared with our own protocol, ketamine was administered at a much slower rate in this case, although both total doses of ketamine were comparable and well below the dose used for sedation. Additionally, in Iserson et al,23 lorazepam was not administered before ketamine because lorazepam was not readily available in the treatment setting. In our case, Ms. C may have had a delayed response to the IV lorazepam she received an hour before the ketamine dose; however, she exhibited a distinct clinical improvement 10 to 15 minutes after IV ketamine was administered. Nevertheless, both cases demonstrated rapid resolution of catatonic symptoms following administration of ketamine.
The marked improvement after the ketamine infusion allowed Ms. C to be discharged from the ED the same day, which was never possible after her previous catatonic episodes. Five days after discharge, she was walking, eating, talking, and able to attend to her activities of daily living without any change to her other medications. Moreover, these effects outlasted the duration of ketamine. Ms. C remained stable for 5 months until she destabilized in June 2020. At that time, she did not respond to lorazepam in the ED, needed to be hospitalized, and required a nasogastric feeding tube. Ketamine was not trialed during this presentation, so it remains to be seen if the patient’s response to ketamine was an isolated incident, or whether it could potentially spare her from future hospitalizations.
Bottom Line
In our case report, a woman with a long history of catatonia responded to a single infusion of IV ketamine, and the beneficial effects lasted for months. More research evaluating the efficacy of ketamine is needed to determine if this agent has a place in the treatment of catatonia.
Continue to: Related Resources
Related Resources
- Dubovsky SL, Dubovsky AN. Catatonia: How to identify and treat it. Current Psychiatry. 2018;17(8):16-26.
- Iserson KV, Durga D. Catatonia-like syndrome treated with low-dose ketamine. J Emerg Med. 2020;58(5):771-774.
Drug Brand Names
Amantadine • Gocovri
Dextroamphetamine sulfate/levoamphetamine sulfate • Evekeo
Divalproex sodium • Depakote
Duloxetine • Cymbalta
Esketamine • Spravato
Fluoxetine • Prozac
Fludrocortisone • Florinef
Fluvoxamine • Luvox
Ketamine • Ketalar
Levothyroxine • Synthroid
Lorazepam • Ativan
Memantine • Namenda
Mirtazapine • Remeron
Olanzapine • Zyprexa
Risperidone • Risperdal
Ziprasidone • Geodon
Zolpidem • Ambien
1. Rasmussen SA, Mazurek MF, Rosebush PI. Catatonia: our current understanding of its diagnosis, treatment and pathophysiology. World J Psychiatry. 2016;6(4):391-398.
2. Grady SE, Marsh TA, Tenhouse A, et al. Ketamine for the treatment of major depressive disorder and bipolar depression: a review of the literature. Mental Health Clin. 2017;7(1):16-23.
3. KETALAR (ketamine hydrochloride) injection. (n.d.). Accessed April 29, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/016812s043lbl.pdf
4. Williams NR, Schatzberg AF. NMDA antagonist treatment of depression. Curr Opin Neurobiol. 2016;36:112-117.
5. Parashchanka A, Schelfout S, Coppens M. Role of novel drugs in sedation outside the operating room: dexmedetomidine, ketamine and remifentanil. Curr Opin Anaesthesiol. 2014;27(4):442-447.
6. Radvansky BM, Puri S, Sifonios AN, et al. Ketamine—a narrative review of its uses in medicine. Am J Ther. 2016;23(6):e1414-e1426. doi: 10.1097/MJT.0000000000000257
7. O’Brien SL, Pangarkar S, Prager J. The use of ketamine in neuropathic pain. Current Physical Medicine and Rehabilitation Reports. 2014;2(2):128-145.
8. Swainson J, Thomas RK, Archer S, et al. Esketamine for treatment resistant depression. Expert Rev Neurother. 2019;19(10):899-911.
9. Carroll BT. The universal field hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr. 2000;5(7):26-33.
10. Carroll BT, Goforth HW, Thomas C, et al. Review of adjunctive glutamate antagonist therapy in the treatment of catatonic syndromes. J Neuropsychiatry Clin Neurosci. 2007;19(4):406-412.
11. Northoff G, Eckert J, Fritze J. Glutamatergic dysfunction in catatonia? Successful treatment of three acute akinetic catatonic patients with the NMDA antagonist amantadine. J Neurol Neurosurg Psychiatry. 1997;62(4):404-406.
12. Denysenko L, Sica N, Penders TM, et al. Catatonia in the medically ill: etiology, diagnosis, and treatment. The Academy of Consultation-Liaison Psychiatry Evidence-Based Medicine Subcommittee Monograph. Ann Clin Psychiatry. 2018;30(2):140-155.
13. Gideons ES, Kavalali ET, Monteggia LM. Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc Natl Acad Sci U S A. 2014;111(23):8649-8654.
14. de Bartolomeis A, Sarappa C, Buonaguro EF, et al. Different effects of the NMDA receptor antagonists ketamine, MK-801, and memantine on postsynaptic density transcripts and their topography: role of Homer signaling, and implications for novel antipsychotic and pro-cognitive targets in psychosis. Prog Neuropsychopharmacol Biol Psychiatry. 2013;46:1-12.
15. Green SM, Johnson NE. Ketamine sedation for pediatric procedures: part 2, review and implications. Ann Emerg Med. 1990;19(9):1033-1046.
16. Kurdi MS, Theerth KA, Deva RS. Ketamine: current applications in anesthesia, pain, and critical care. Anesth Essays Res. 2014;8(3):283-290.
17. Majidi S, Parna A, Zamani M, et al. Onset and effect duration of intrabuccal space and intramuscular ketamine in pediatrics. Adv Biomed Res. 2018;7:91.
18. Bahr R, Lopez A, Rey JA. Intranasal esketamine (SpravatoTM) for use in treatment-resistant depression in conjunction with an oral antidepressant. P T. 2019;44(6):340-342,344-346,375.
19. Strong CE, Kabbaj M. On the safety of repeated ketamine infusions for the treatment of depression: effects of sex and developmental periods. Neurobiol Stress. 2018;9:166-175.
20. Su TP, Chen MH, Li CT, et al. Dose-related effects of adjunctive ketamine in Taiwanese patients with treatment-resistant depression. Neuropsychopharmacology. 2017;42(13):2482-2492.
21. Fava M, Freeman MP, Flynn M, et al. Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD). Mol Psychiatry. 2020;25(7):1592-1603.
22. Wong DH, Jenkins LC. An experimental study of the mechanism of action of ketamine on the central nervous system. Can Anaesth Soc J. 1974;21(1):57-67.
23. Iserson KV, Durga D. Catatonia-like syndrome treated with low-dose ketamine. J Emerg Med. 2020;58(5):771-774.
Ms. C, age 44, who has major depressive disorder (MDD), anxiety, obsessive-compulsive disorder (OCD) (religious subtype), and has experienced multiple episodes of treatment-resistant catatonia, is brought to the emergency department (ED) by her parents. She has immobility, mutism, rigidity, and decreased oral intake that she has experienced for 1 day.
The night before, Ms. C had been stressed about an upcoming job interview. She cancelled the interview and went to her bedroom. Later that night her parents found her lying on the floor, immobile.
Before the onset of her psychiatric symptoms, Ms. C had been high functioning. She had been an athlete in college and had a career as a school psychologist. The Sidebar summarizes Ms. C’s psychiatric history, which includes similar complex episodes and multiple hospitalizations. She also has a history of hypothyroidism.
SIDEBAR
In 2013, Ms. C experienced severe social stress from both her work as a psychologist and a divorce. She sold all of her possessions and was living in motels and hotels searching for the “truth of God.” In February 2016, she was hospitalized after refusing to eat and self-discontinuing all medications, including her thyroid medications. She was then placed under the conservatorship of her parents.
In July 2017, Ms. C was hospitalized again for refusing to eat or take her medications; this time she also exhibited selective mutism. Catatonia was suspected and she was started on oral lorazepam, 2 mg 3 times a day. Duloxetine and ziprasidone were also trialed but were stopped due to noncompliance and adverse effects. Ms. C showed little improvement on these regimens. In the hospital, IV lorazepam, 4 mg, was trialed with good effect, and she began to respond to questioning. She was transitioned to oral lorazepam, 4 mg 5 times per day, and mirtazapine, 15 mg/d. With this regimen, Ms. C became progressively more interactive; however, she still refused to eat. Throughout her hospitalization, multiple medications were prescribed, including divalproex sodium, memantine, zolpidem, olanzapine, and dextroamphetamine/levoamphetamine, all of which were not effective in stimulating her appetite. Due to malnutrition, Ms. C was placed on total parenteral nutrition. During this time, the highest dose of IM lorazepam was 20 mg/d in divided doses.
Some improvement with ECT
Four months into her hospitalization, Ms. C’s lorazepam was titrated down to 4 mg 4 times a day, and she underwent a trial of electroconvulsive therapy (ECT). Following the fourth ECT session, she displayed significant improvement. Ms. C engaged with her clinicians, displayed bright mood and affect, began eating again, and was able to recount her depressive symptoms following her divorce. At this time, she received a total of 8 ECT treatments and was started on fluoxetine. At the end of January 2018, after 19 days of hospitalization, she was transitioned to a partial hospitalization program (PHP) on a regimen of lorazepam, 2 mg 3 times daily; fluoxetine, 40 mg/d; midodrine, 10 mg 3 times daily; fludrocortisone; and levothyroxine. Her discharge diagnosis was major depressive disorder with psychotic features and catatonia.
Between her first hospitalization and her current presentation to the emergency department (ED), Ms. C presented several times to the ED with similar symptoms of decreased speech, movement, and oral intake. In February 2018, she was hospitalized and responded after 4 sessions of ECT. She returned to work as a substitute teacher and was stable for >1 year on a regimen of lorazepam, olanzapine, and risperidone. In June 2019, her symptoms returned. She was hospitalized and required a nasogastric tube to address malnutrition. She was eventually stabilized on a regimen of risperidone and lorazepam, which she continued as an outpatient until she was hospitalized again in August 2019. During this hospitalization, Ms. C failed to respond to risperidone or lorazepam, up to 2 mg 3 times a day. After several changes to her regimen, she began to respond to olanzapine, 30 mg/d; mirtazapine, 15 mg/d; and lorazepam, 2 mg 3 times a day.
Throughout her hospitalizations, once she became verbal, Ms. C demonstrated hyper-religiosity. She would ask to read the Bible, and state that her purpose was to find the truth of God. As an outpatient, she would compulsively go to church in the middle of the night and read the Bible for hours. A preliminary diagnosis of obsessive-compulsive disorder was made based on her scrupulosity, and mirtazapine was cross-titrated to fluvoxamine prior to discharge.
Shortly after discharge, she was readmitted to a PHP, and did well on fluvoxamine, 100 mg twice a day; olanzapine, 5 mg every night; levothyroxine, 100 mcg/d; and oral lorazepam, 1 mg 4 times a day. Ms. C displayed full mood, appropriate affect, and began working part-time as a substitute teacher. She had begun to interview for full-time jobs before her most recent ED presentation.
In the ED, the psychiatry team evaluates Ms. C. She displays a similar pattern of mutism, immobility, and rigidity as she did upon her initial presentation. Her father reports that she had been compliant with her medications but had not taken them the previous night. Ms. C screens positive for catatonia on the Bush-Francis Catatonia Rating Scale (BFCRS). Her severity score of 10/69 indicates a mild presentation. She is diagnosed with catatonia and is administered IV
Because Ms. C has been hospitalized many times for similar presentations, the treatment team decides to initiate a trial of IV ketamine.
Catatonia can manifest in many different ways in patients with psychiatric illness. If left untreated, it is associated with a high rate of mortality.1 Catatonia often is described along a continuum from retarded/stuporous to excited, and presentations can vary substantially. The physiologic and psychological mechanisms of catatonia are poorly understood.
Traditionally, most patients respond well to low-dose benzodiazepines, with electroconvulsive therapy as a second-line intervention for refractory and malignant cases. However, these interventions are not always successful or readily available.
Continue to: Research into the anesthetic ketamine...
Research into the anesthetic ketamine is gradually expanding, and the use of this agent for treating various psychiatric illnesses, including both unipolar and bipolar depression, has been increasing.2 Empiric evidence suggests ketamine is effective for certain psychiatric disorders, but the mechanism of action remains unclear. Although the evidence base is small, additional cases demonstrating the effectiveness of ketamine in the treatment of acute catatonia might make it a therapeutic option for use by psychiatrists and emergency medicine clinicians.
In this article, we discuss ketamine’s possible role in the treatment of catatonia, possible adverse effects, dosing strategies, and theories about ketamine’s mechanism of action.
Ketamine’s utility in psychiatry
Ketamine is a rapid-acting anesthetic that acts primarily by antagonizing N-methyl-
Previously, ketamine had been thought to induce a catatonic state, which was supported by a neurophysiologic model of catatonia that suggested the condition was caused in part by glutamate hypoactivity at the NMDA receptor.9 However, recent studies have shown that the NMDA receptor antagonists
Ketamine originally was used for sedation, and much of its safety and risk profile has been developed from decades of administration as an anesthetic. Studies have found that ketamine has a large therapeutic window in children and adults.15,16 Moreover, it does not depress the respiratory system. As an anesthetic, ketamine has a rapid onset and a quick resolution, with its sedative and disorienting effects resolving within 30 to 120 minutes.17 Ketamine’s rapid onset of action extends beyond its sedating effects. Trials with the intranasal spray esketamine for treatment-resistant depression have demonstrated an onset antidepressant effects within 2 days.18 This is much faster than that of traditional antidepressants, such as selective serotonin reuptake inhibitors.18 Based on these features, ketamine has the potential to be a useful medication in the emergency psychiatric setting, particularly for acute presentations such as catatonia.
Continue to: Beware of the potential risks
Beware of the potential risks
Although ketamine may be clinically useful, it also carries some risks. Adverse effects associated with ketamine include sedation, dissociation, hallucinations, elevated blood pressure, nausea, increased heart rate, vomiting, dizziness, fatigue, blurred vision, itching, and emesis. Clinicians also should be aware that some patients may use illicit ketamine, either as self-treatment to control depressive symptoms or for recreational purposes. When misused/abused, long-term use of ketamine can cause neurologic damage.19 Studies also have reported rare occurrences of recurrent hallucinations even after discontinuation of ketamine.20 Animal studies have demonstrated addiction and cognitive deficits with repeated use of ketamine in rodents.21 This research has led to concerns that chronic use of ketamine to treat illnesses such as depression might lead to similar long-term adverse outcomes.
Dosing
As a sedative, IV ketamine dosing is generally 1 to 2 mg/kg, and IM ketamine dosing is 3 to 5 mg/kg.16 As an antidepressant, small clinical trials have suggested that the preferred dose of IV ketamine may be 0.5 to 1 mg/kg, with dose-dependent increases in dissociation and blood pressure.21 Studies have also demonstrated that once-daily IV ketamine, 0.5 mg/kg administered over 40 minutes, led to greater improvements in patients with MDD than placebo, whereas once-daily IV ketamine, 0.2 mg/kg, did not.20
CASE CONTINUED
The team begins to treat Ms. C with IV ketamine. Ketamine, 0.2 mg/kg, is used to calculate the initial dose, and a total of 10 mg is administered over 10 minutes. Fifteen minutes after administration, Ms. C is able to move around in her bed, make eye contact, and nod to questions. She has purposeful movements, such as examining her IV line, scratching her head, and repositioning herself in the bed. After a few more minutes, she makes eye contact with her father, and nods to him during conversation. She is able to make a few noises but does not speak.
Later that day, Ms. C is discharged home (in a wheelchair) with her parents, on a medication regimen of fluvoxamine, 100 mg/d; lorazepam, 1 mg 4 times a day; and olanzapine, 5 mg/d. She is scheduled for an outpatient follow-up appointment 5 days later. Her parents are given instructions and several precautions to ensure that Ms. C receives proper nutrition until her appointment. That evening, Ms. C is able to eat voluntarily.
Five days later, Ms. C visits the outpatient psychiatric clinic and is verbal and ambulatory. Her father reports that she has become more verbal. During her follow-up interview, she is observed to be more subdued and less verbal than her baseline, but is vocal and able to voice her understanding of the treatment plan.
Continue to: After 3 months of being stable...
After 3 months of being stable on her outpatient regimen, Ms. C’s catatonic symptoms return, including refusing to eat and mutism. She is administered IV lorazepam, 4 mg, with no response and is admitted to the hospital for placement of a nasogastric feeding tube to address malnutrition. After several days, Ms. C responds to lorazepam, 4 mg every 6 hours. Six days later, after she begins eating and taking her medications voluntarily and the nasogastric tube is removed, Ms. C is discharged to home.
Findings need to be replicated in larger studies
Although some research has indicated that ketamine may be pro-catatonic, Ms. C’s improvement after receiving ketamine suggests that perhaps the situation is more complex.12,22 The exact mechanisms underlying catatonia remain uncertain. Carroll et al9 described 4 theories, and only 1 of them involved glutamate. Additionally, ketamine’s mechanism of action may extend beyond NMDA antagonism. In our case, Ms. C’s low BFCRS score during her most recent visit to the ED suggests she may have had a milder or less typical form of catatonia compared with her previous presentations (Sidebar). However, Ms. C’s clinical improvement after receiving ketamine is noteworthy.
A review of the literature yielded only 1 other case report that described using ketamine to treat catatonia.23 Iserson et al23 reported that their patient’s catatonic symptoms resolved after a total of 12.5 mg of ketamine was administered in 0.03 mg/kg boluses every 3 minutes. Compared with our own protocol, ketamine was administered at a much slower rate in this case, although both total doses of ketamine were comparable and well below the dose used for sedation. Additionally, in Iserson et al,23 lorazepam was not administered before ketamine because lorazepam was not readily available in the treatment setting. In our case, Ms. C may have had a delayed response to the IV lorazepam she received an hour before the ketamine dose; however, she exhibited a distinct clinical improvement 10 to 15 minutes after IV ketamine was administered. Nevertheless, both cases demonstrated rapid resolution of catatonic symptoms following administration of ketamine.
The marked improvement after the ketamine infusion allowed Ms. C to be discharged from the ED the same day, which was never possible after her previous catatonic episodes. Five days after discharge, she was walking, eating, talking, and able to attend to her activities of daily living without any change to her other medications. Moreover, these effects outlasted the duration of ketamine. Ms. C remained stable for 5 months until she destabilized in June 2020. At that time, she did not respond to lorazepam in the ED, needed to be hospitalized, and required a nasogastric feeding tube. Ketamine was not trialed during this presentation, so it remains to be seen if the patient’s response to ketamine was an isolated incident, or whether it could potentially spare her from future hospitalizations.
Bottom Line
In our case report, a woman with a long history of catatonia responded to a single infusion of IV ketamine, and the beneficial effects lasted for months. More research evaluating the efficacy of ketamine is needed to determine if this agent has a place in the treatment of catatonia.
Continue to: Related Resources
Related Resources
- Dubovsky SL, Dubovsky AN. Catatonia: How to identify and treat it. Current Psychiatry. 2018;17(8):16-26.
- Iserson KV, Durga D. Catatonia-like syndrome treated with low-dose ketamine. J Emerg Med. 2020;58(5):771-774.
Drug Brand Names
Amantadine • Gocovri
Dextroamphetamine sulfate/levoamphetamine sulfate • Evekeo
Divalproex sodium • Depakote
Duloxetine • Cymbalta
Esketamine • Spravato
Fluoxetine • Prozac
Fludrocortisone • Florinef
Fluvoxamine • Luvox
Ketamine • Ketalar
Levothyroxine • Synthroid
Lorazepam • Ativan
Memantine • Namenda
Mirtazapine • Remeron
Olanzapine • Zyprexa
Risperidone • Risperdal
Ziprasidone • Geodon
Zolpidem • Ambien
Ms. C, age 44, who has major depressive disorder (MDD), anxiety, obsessive-compulsive disorder (OCD) (religious subtype), and has experienced multiple episodes of treatment-resistant catatonia, is brought to the emergency department (ED) by her parents. She has immobility, mutism, rigidity, and decreased oral intake that she has experienced for 1 day.
The night before, Ms. C had been stressed about an upcoming job interview. She cancelled the interview and went to her bedroom. Later that night her parents found her lying on the floor, immobile.
Before the onset of her psychiatric symptoms, Ms. C had been high functioning. She had been an athlete in college and had a career as a school psychologist. The Sidebar summarizes Ms. C’s psychiatric history, which includes similar complex episodes and multiple hospitalizations. She also has a history of hypothyroidism.
SIDEBAR
In 2013, Ms. C experienced severe social stress from both her work as a psychologist and a divorce. She sold all of her possessions and was living in motels and hotels searching for the “truth of God.” In February 2016, she was hospitalized after refusing to eat and self-discontinuing all medications, including her thyroid medications. She was then placed under the conservatorship of her parents.
In July 2017, Ms. C was hospitalized again for refusing to eat or take her medications; this time she also exhibited selective mutism. Catatonia was suspected and she was started on oral lorazepam, 2 mg 3 times a day. Duloxetine and ziprasidone were also trialed but were stopped due to noncompliance and adverse effects. Ms. C showed little improvement on these regimens. In the hospital, IV lorazepam, 4 mg, was trialed with good effect, and she began to respond to questioning. She was transitioned to oral lorazepam, 4 mg 5 times per day, and mirtazapine, 15 mg/d. With this regimen, Ms. C became progressively more interactive; however, she still refused to eat. Throughout her hospitalization, multiple medications were prescribed, including divalproex sodium, memantine, zolpidem, olanzapine, and dextroamphetamine/levoamphetamine, all of which were not effective in stimulating her appetite. Due to malnutrition, Ms. C was placed on total parenteral nutrition. During this time, the highest dose of IM lorazepam was 20 mg/d in divided doses.
Some improvement with ECT
Four months into her hospitalization, Ms. C’s lorazepam was titrated down to 4 mg 4 times a day, and she underwent a trial of electroconvulsive therapy (ECT). Following the fourth ECT session, she displayed significant improvement. Ms. C engaged with her clinicians, displayed bright mood and affect, began eating again, and was able to recount her depressive symptoms following her divorce. At this time, she received a total of 8 ECT treatments and was started on fluoxetine. At the end of January 2018, after 19 days of hospitalization, she was transitioned to a partial hospitalization program (PHP) on a regimen of lorazepam, 2 mg 3 times daily; fluoxetine, 40 mg/d; midodrine, 10 mg 3 times daily; fludrocortisone; and levothyroxine. Her discharge diagnosis was major depressive disorder with psychotic features and catatonia.
Between her first hospitalization and her current presentation to the emergency department (ED), Ms. C presented several times to the ED with similar symptoms of decreased speech, movement, and oral intake. In February 2018, she was hospitalized and responded after 4 sessions of ECT. She returned to work as a substitute teacher and was stable for >1 year on a regimen of lorazepam, olanzapine, and risperidone. In June 2019, her symptoms returned. She was hospitalized and required a nasogastric tube to address malnutrition. She was eventually stabilized on a regimen of risperidone and lorazepam, which she continued as an outpatient until she was hospitalized again in August 2019. During this hospitalization, Ms. C failed to respond to risperidone or lorazepam, up to 2 mg 3 times a day. After several changes to her regimen, she began to respond to olanzapine, 30 mg/d; mirtazapine, 15 mg/d; and lorazepam, 2 mg 3 times a day.
Throughout her hospitalizations, once she became verbal, Ms. C demonstrated hyper-religiosity. She would ask to read the Bible, and state that her purpose was to find the truth of God. As an outpatient, she would compulsively go to church in the middle of the night and read the Bible for hours. A preliminary diagnosis of obsessive-compulsive disorder was made based on her scrupulosity, and mirtazapine was cross-titrated to fluvoxamine prior to discharge.
Shortly after discharge, she was readmitted to a PHP, and did well on fluvoxamine, 100 mg twice a day; olanzapine, 5 mg every night; levothyroxine, 100 mcg/d; and oral lorazepam, 1 mg 4 times a day. Ms. C displayed full mood, appropriate affect, and began working part-time as a substitute teacher. She had begun to interview for full-time jobs before her most recent ED presentation.
In the ED, the psychiatry team evaluates Ms. C. She displays a similar pattern of mutism, immobility, and rigidity as she did upon her initial presentation. Her father reports that she had been compliant with her medications but had not taken them the previous night. Ms. C screens positive for catatonia on the Bush-Francis Catatonia Rating Scale (BFCRS). Her severity score of 10/69 indicates a mild presentation. She is diagnosed with catatonia and is administered IV
Because Ms. C has been hospitalized many times for similar presentations, the treatment team decides to initiate a trial of IV ketamine.
Catatonia can manifest in many different ways in patients with psychiatric illness. If left untreated, it is associated with a high rate of mortality.1 Catatonia often is described along a continuum from retarded/stuporous to excited, and presentations can vary substantially. The physiologic and psychological mechanisms of catatonia are poorly understood.
Traditionally, most patients respond well to low-dose benzodiazepines, with electroconvulsive therapy as a second-line intervention for refractory and malignant cases. However, these interventions are not always successful or readily available.
Continue to: Research into the anesthetic ketamine...
Research into the anesthetic ketamine is gradually expanding, and the use of this agent for treating various psychiatric illnesses, including both unipolar and bipolar depression, has been increasing.2 Empiric evidence suggests ketamine is effective for certain psychiatric disorders, but the mechanism of action remains unclear. Although the evidence base is small, additional cases demonstrating the effectiveness of ketamine in the treatment of acute catatonia might make it a therapeutic option for use by psychiatrists and emergency medicine clinicians.
In this article, we discuss ketamine’s possible role in the treatment of catatonia, possible adverse effects, dosing strategies, and theories about ketamine’s mechanism of action.
Ketamine’s utility in psychiatry
Ketamine is a rapid-acting anesthetic that acts primarily by antagonizing N-methyl-
Previously, ketamine had been thought to induce a catatonic state, which was supported by a neurophysiologic model of catatonia that suggested the condition was caused in part by glutamate hypoactivity at the NMDA receptor.9 However, recent studies have shown that the NMDA receptor antagonists
Ketamine originally was used for sedation, and much of its safety and risk profile has been developed from decades of administration as an anesthetic. Studies have found that ketamine has a large therapeutic window in children and adults.15,16 Moreover, it does not depress the respiratory system. As an anesthetic, ketamine has a rapid onset and a quick resolution, with its sedative and disorienting effects resolving within 30 to 120 minutes.17 Ketamine’s rapid onset of action extends beyond its sedating effects. Trials with the intranasal spray esketamine for treatment-resistant depression have demonstrated an onset antidepressant effects within 2 days.18 This is much faster than that of traditional antidepressants, such as selective serotonin reuptake inhibitors.18 Based on these features, ketamine has the potential to be a useful medication in the emergency psychiatric setting, particularly for acute presentations such as catatonia.
Continue to: Beware of the potential risks
Beware of the potential risks
Although ketamine may be clinically useful, it also carries some risks. Adverse effects associated with ketamine include sedation, dissociation, hallucinations, elevated blood pressure, nausea, increased heart rate, vomiting, dizziness, fatigue, blurred vision, itching, and emesis. Clinicians also should be aware that some patients may use illicit ketamine, either as self-treatment to control depressive symptoms or for recreational purposes. When misused/abused, long-term use of ketamine can cause neurologic damage.19 Studies also have reported rare occurrences of recurrent hallucinations even after discontinuation of ketamine.20 Animal studies have demonstrated addiction and cognitive deficits with repeated use of ketamine in rodents.21 This research has led to concerns that chronic use of ketamine to treat illnesses such as depression might lead to similar long-term adverse outcomes.
Dosing
As a sedative, IV ketamine dosing is generally 1 to 2 mg/kg, and IM ketamine dosing is 3 to 5 mg/kg.16 As an antidepressant, small clinical trials have suggested that the preferred dose of IV ketamine may be 0.5 to 1 mg/kg, with dose-dependent increases in dissociation and blood pressure.21 Studies have also demonstrated that once-daily IV ketamine, 0.5 mg/kg administered over 40 minutes, led to greater improvements in patients with MDD than placebo, whereas once-daily IV ketamine, 0.2 mg/kg, did not.20
CASE CONTINUED
The team begins to treat Ms. C with IV ketamine. Ketamine, 0.2 mg/kg, is used to calculate the initial dose, and a total of 10 mg is administered over 10 minutes. Fifteen minutes after administration, Ms. C is able to move around in her bed, make eye contact, and nod to questions. She has purposeful movements, such as examining her IV line, scratching her head, and repositioning herself in the bed. After a few more minutes, she makes eye contact with her father, and nods to him during conversation. She is able to make a few noises but does not speak.
Later that day, Ms. C is discharged home (in a wheelchair) with her parents, on a medication regimen of fluvoxamine, 100 mg/d; lorazepam, 1 mg 4 times a day; and olanzapine, 5 mg/d. She is scheduled for an outpatient follow-up appointment 5 days later. Her parents are given instructions and several precautions to ensure that Ms. C receives proper nutrition until her appointment. That evening, Ms. C is able to eat voluntarily.
Five days later, Ms. C visits the outpatient psychiatric clinic and is verbal and ambulatory. Her father reports that she has become more verbal. During her follow-up interview, she is observed to be more subdued and less verbal than her baseline, but is vocal and able to voice her understanding of the treatment plan.
Continue to: After 3 months of being stable...
After 3 months of being stable on her outpatient regimen, Ms. C’s catatonic symptoms return, including refusing to eat and mutism. She is administered IV lorazepam, 4 mg, with no response and is admitted to the hospital for placement of a nasogastric feeding tube to address malnutrition. After several days, Ms. C responds to lorazepam, 4 mg every 6 hours. Six days later, after she begins eating and taking her medications voluntarily and the nasogastric tube is removed, Ms. C is discharged to home.
Findings need to be replicated in larger studies
Although some research has indicated that ketamine may be pro-catatonic, Ms. C’s improvement after receiving ketamine suggests that perhaps the situation is more complex.12,22 The exact mechanisms underlying catatonia remain uncertain. Carroll et al9 described 4 theories, and only 1 of them involved glutamate. Additionally, ketamine’s mechanism of action may extend beyond NMDA antagonism. In our case, Ms. C’s low BFCRS score during her most recent visit to the ED suggests she may have had a milder or less typical form of catatonia compared with her previous presentations (Sidebar). However, Ms. C’s clinical improvement after receiving ketamine is noteworthy.
A review of the literature yielded only 1 other case report that described using ketamine to treat catatonia.23 Iserson et al23 reported that their patient’s catatonic symptoms resolved after a total of 12.5 mg of ketamine was administered in 0.03 mg/kg boluses every 3 minutes. Compared with our own protocol, ketamine was administered at a much slower rate in this case, although both total doses of ketamine were comparable and well below the dose used for sedation. Additionally, in Iserson et al,23 lorazepam was not administered before ketamine because lorazepam was not readily available in the treatment setting. In our case, Ms. C may have had a delayed response to the IV lorazepam she received an hour before the ketamine dose; however, she exhibited a distinct clinical improvement 10 to 15 minutes after IV ketamine was administered. Nevertheless, both cases demonstrated rapid resolution of catatonic symptoms following administration of ketamine.
The marked improvement after the ketamine infusion allowed Ms. C to be discharged from the ED the same day, which was never possible after her previous catatonic episodes. Five days after discharge, she was walking, eating, talking, and able to attend to her activities of daily living without any change to her other medications. Moreover, these effects outlasted the duration of ketamine. Ms. C remained stable for 5 months until she destabilized in June 2020. At that time, she did not respond to lorazepam in the ED, needed to be hospitalized, and required a nasogastric feeding tube. Ketamine was not trialed during this presentation, so it remains to be seen if the patient’s response to ketamine was an isolated incident, or whether it could potentially spare her from future hospitalizations.
Bottom Line
In our case report, a woman with a long history of catatonia responded to a single infusion of IV ketamine, and the beneficial effects lasted for months. More research evaluating the efficacy of ketamine is needed to determine if this agent has a place in the treatment of catatonia.
Continue to: Related Resources
Related Resources
- Dubovsky SL, Dubovsky AN. Catatonia: How to identify and treat it. Current Psychiatry. 2018;17(8):16-26.
- Iserson KV, Durga D. Catatonia-like syndrome treated with low-dose ketamine. J Emerg Med. 2020;58(5):771-774.
Drug Brand Names
Amantadine • Gocovri
Dextroamphetamine sulfate/levoamphetamine sulfate • Evekeo
Divalproex sodium • Depakote
Duloxetine • Cymbalta
Esketamine • Spravato
Fluoxetine • Prozac
Fludrocortisone • Florinef
Fluvoxamine • Luvox
Ketamine • Ketalar
Levothyroxine • Synthroid
Lorazepam • Ativan
Memantine • Namenda
Mirtazapine • Remeron
Olanzapine • Zyprexa
Risperidone • Risperdal
Ziprasidone • Geodon
Zolpidem • Ambien
1. Rasmussen SA, Mazurek MF, Rosebush PI. Catatonia: our current understanding of its diagnosis, treatment and pathophysiology. World J Psychiatry. 2016;6(4):391-398.
2. Grady SE, Marsh TA, Tenhouse A, et al. Ketamine for the treatment of major depressive disorder and bipolar depression: a review of the literature. Mental Health Clin. 2017;7(1):16-23.
3. KETALAR (ketamine hydrochloride) injection. (n.d.). Accessed April 29, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/016812s043lbl.pdf
4. Williams NR, Schatzberg AF. NMDA antagonist treatment of depression. Curr Opin Neurobiol. 2016;36:112-117.
5. Parashchanka A, Schelfout S, Coppens M. Role of novel drugs in sedation outside the operating room: dexmedetomidine, ketamine and remifentanil. Curr Opin Anaesthesiol. 2014;27(4):442-447.
6. Radvansky BM, Puri S, Sifonios AN, et al. Ketamine—a narrative review of its uses in medicine. Am J Ther. 2016;23(6):e1414-e1426. doi: 10.1097/MJT.0000000000000257
7. O’Brien SL, Pangarkar S, Prager J. The use of ketamine in neuropathic pain. Current Physical Medicine and Rehabilitation Reports. 2014;2(2):128-145.
8. Swainson J, Thomas RK, Archer S, et al. Esketamine for treatment resistant depression. Expert Rev Neurother. 2019;19(10):899-911.
9. Carroll BT. The universal field hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr. 2000;5(7):26-33.
10. Carroll BT, Goforth HW, Thomas C, et al. Review of adjunctive glutamate antagonist therapy in the treatment of catatonic syndromes. J Neuropsychiatry Clin Neurosci. 2007;19(4):406-412.
11. Northoff G, Eckert J, Fritze J. Glutamatergic dysfunction in catatonia? Successful treatment of three acute akinetic catatonic patients with the NMDA antagonist amantadine. J Neurol Neurosurg Psychiatry. 1997;62(4):404-406.
12. Denysenko L, Sica N, Penders TM, et al. Catatonia in the medically ill: etiology, diagnosis, and treatment. The Academy of Consultation-Liaison Psychiatry Evidence-Based Medicine Subcommittee Monograph. Ann Clin Psychiatry. 2018;30(2):140-155.
13. Gideons ES, Kavalali ET, Monteggia LM. Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc Natl Acad Sci U S A. 2014;111(23):8649-8654.
14. de Bartolomeis A, Sarappa C, Buonaguro EF, et al. Different effects of the NMDA receptor antagonists ketamine, MK-801, and memantine on postsynaptic density transcripts and their topography: role of Homer signaling, and implications for novel antipsychotic and pro-cognitive targets in psychosis. Prog Neuropsychopharmacol Biol Psychiatry. 2013;46:1-12.
15. Green SM, Johnson NE. Ketamine sedation for pediatric procedures: part 2, review and implications. Ann Emerg Med. 1990;19(9):1033-1046.
16. Kurdi MS, Theerth KA, Deva RS. Ketamine: current applications in anesthesia, pain, and critical care. Anesth Essays Res. 2014;8(3):283-290.
17. Majidi S, Parna A, Zamani M, et al. Onset and effect duration of intrabuccal space and intramuscular ketamine in pediatrics. Adv Biomed Res. 2018;7:91.
18. Bahr R, Lopez A, Rey JA. Intranasal esketamine (SpravatoTM) for use in treatment-resistant depression in conjunction with an oral antidepressant. P T. 2019;44(6):340-342,344-346,375.
19. Strong CE, Kabbaj M. On the safety of repeated ketamine infusions for the treatment of depression: effects of sex and developmental periods. Neurobiol Stress. 2018;9:166-175.
20. Su TP, Chen MH, Li CT, et al. Dose-related effects of adjunctive ketamine in Taiwanese patients with treatment-resistant depression. Neuropsychopharmacology. 2017;42(13):2482-2492.
21. Fava M, Freeman MP, Flynn M, et al. Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD). Mol Psychiatry. 2020;25(7):1592-1603.
22. Wong DH, Jenkins LC. An experimental study of the mechanism of action of ketamine on the central nervous system. Can Anaesth Soc J. 1974;21(1):57-67.
23. Iserson KV, Durga D. Catatonia-like syndrome treated with low-dose ketamine. J Emerg Med. 2020;58(5):771-774.
1. Rasmussen SA, Mazurek MF, Rosebush PI. Catatonia: our current understanding of its diagnosis, treatment and pathophysiology. World J Psychiatry. 2016;6(4):391-398.
2. Grady SE, Marsh TA, Tenhouse A, et al. Ketamine for the treatment of major depressive disorder and bipolar depression: a review of the literature. Mental Health Clin. 2017;7(1):16-23.
3. KETALAR (ketamine hydrochloride) injection. (n.d.). Accessed April 29, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/016812s043lbl.pdf
4. Williams NR, Schatzberg AF. NMDA antagonist treatment of depression. Curr Opin Neurobiol. 2016;36:112-117.
5. Parashchanka A, Schelfout S, Coppens M. Role of novel drugs in sedation outside the operating room: dexmedetomidine, ketamine and remifentanil. Curr Opin Anaesthesiol. 2014;27(4):442-447.
6. Radvansky BM, Puri S, Sifonios AN, et al. Ketamine—a narrative review of its uses in medicine. Am J Ther. 2016;23(6):e1414-e1426. doi: 10.1097/MJT.0000000000000257
7. O’Brien SL, Pangarkar S, Prager J. The use of ketamine in neuropathic pain. Current Physical Medicine and Rehabilitation Reports. 2014;2(2):128-145.
8. Swainson J, Thomas RK, Archer S, et al. Esketamine for treatment resistant depression. Expert Rev Neurother. 2019;19(10):899-911.
9. Carroll BT. The universal field hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr. 2000;5(7):26-33.
10. Carroll BT, Goforth HW, Thomas C, et al. Review of adjunctive glutamate antagonist therapy in the treatment of catatonic syndromes. J Neuropsychiatry Clin Neurosci. 2007;19(4):406-412.
11. Northoff G, Eckert J, Fritze J. Glutamatergic dysfunction in catatonia? Successful treatment of three acute akinetic catatonic patients with the NMDA antagonist amantadine. J Neurol Neurosurg Psychiatry. 1997;62(4):404-406.
12. Denysenko L, Sica N, Penders TM, et al. Catatonia in the medically ill: etiology, diagnosis, and treatment. The Academy of Consultation-Liaison Psychiatry Evidence-Based Medicine Subcommittee Monograph. Ann Clin Psychiatry. 2018;30(2):140-155.
13. Gideons ES, Kavalali ET, Monteggia LM. Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc Natl Acad Sci U S A. 2014;111(23):8649-8654.
14. de Bartolomeis A, Sarappa C, Buonaguro EF, et al. Different effects of the NMDA receptor antagonists ketamine, MK-801, and memantine on postsynaptic density transcripts and their topography: role of Homer signaling, and implications for novel antipsychotic and pro-cognitive targets in psychosis. Prog Neuropsychopharmacol Biol Psychiatry. 2013;46:1-12.
15. Green SM, Johnson NE. Ketamine sedation for pediatric procedures: part 2, review and implications. Ann Emerg Med. 1990;19(9):1033-1046.
16. Kurdi MS, Theerth KA, Deva RS. Ketamine: current applications in anesthesia, pain, and critical care. Anesth Essays Res. 2014;8(3):283-290.
17. Majidi S, Parna A, Zamani M, et al. Onset and effect duration of intrabuccal space and intramuscular ketamine in pediatrics. Adv Biomed Res. 2018;7:91.
18. Bahr R, Lopez A, Rey JA. Intranasal esketamine (SpravatoTM) for use in treatment-resistant depression in conjunction with an oral antidepressant. P T. 2019;44(6):340-342,344-346,375.
19. Strong CE, Kabbaj M. On the safety of repeated ketamine infusions for the treatment of depression: effects of sex and developmental periods. Neurobiol Stress. 2018;9:166-175.
20. Su TP, Chen MH, Li CT, et al. Dose-related effects of adjunctive ketamine in Taiwanese patients with treatment-resistant depression. Neuropsychopharmacology. 2017;42(13):2482-2492.
21. Fava M, Freeman MP, Flynn M, et al. Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD). Mol Psychiatry. 2020;25(7):1592-1603.
22. Wong DH, Jenkins LC. An experimental study of the mechanism of action of ketamine on the central nervous system. Can Anaesth Soc J. 1974;21(1):57-67.
23. Iserson KV, Durga D. Catatonia-like syndrome treated with low-dose ketamine. J Emerg Med. 2020;58(5):771-774.

Steroid-induced psychiatric symptoms: What you need to know
Ms. N, age 30, presents to the emergency department for altered mental status, insomnia, and behavioral changes, which she has experienced for 1 week. On evaluation, she grabs a clinician’s hand and details her business ideas and life story with no prompting. Ms. N’s mental status examination is significant for hyperverbal speech with increased rate and volume; tangential thought process; and bright, expanded affect.
One week earlier, Ms. N was hospitalized for sudden-onset chest pain, weakness, and dizziness. She received 45 minutes of cardiopulmonary resuscitation prior to presentation and was found to have a ST-segment elevation myocardial infarction that required emergent left anterior descending coronary artery and right coronary artery percutaneous coronary intervention to place drug-eluting stents. Her recovery was complicated by acute cardiogenic shock, pulmonary edema, and hypoxic respiratory failure. Subsequently, she was intubated, admitted to the ICU, and received high-dose corticosteroids, including IV methylprednisolone, 40 mg every 12 hours, which was tapered prior to discharge. Her husband reports that since Ms. N came home, she has been more talkative and irritable, ruminating about past events, unable to sleep (<1 hour/night), and crying frequently. She has also been endorsing visual and auditory hallucinations, with increased praying and listening to religious music.
The frequent clinical use of steroids necessitates an understanding of these medications’ various adverse effects. The manifestations of steroid-induced psychiatric symptoms are broad and can involve affective, behavioral, and cognitive domains. While the current mechanism is unknown, this phenomenon may be related to decreased levels of corticotropin, norepinephrine, and beta-endorphin immunoreactivity, as well as effects on brain regions such as the hippocampus and amygdala. The best interventions for steroid-induced psychiatric symptoms are awareness and early diagnosis. There are no FDA-approved treatments for steroid-induced psychiatric symptoms; initial measures should include tapering or discontinuing corticosteroids.
In this article, we review the literature on the incidence, characteristics, differential diagnoses, proposed mechanism, risk factors, and proposed treatments of steroid-induced psychiatric symptoms.
A wide range of presentations
Steroid use has increased over the past 2 decades, with 10% of medical and surgical inpatients and 1% to 3% of the general population taking long-term glucocorticoids.1 Even with topical application, steroid therapy is often systemically absorbed, and thus may lead to steroid-induced psychiatric symptoms. The incidence of steroid-induced psychiatric symptoms is difficult to assess because there can be a wide range of reactions that are dose- and time-related. Three reviews of a total of 122 cases reports found that an estimated 5% of patients treated with steroids experience severe psychiatric reactions.1-3
Steroid-induced psychopathology can include mood, behavioral, and/or cognitive impairments. Mania/hypomania is the most common overall psychiatric symptom; the most common mood manifestations are anxiety and depression.4,5 Other possible steroid-induced symptoms include psychosis, dementia, panic disorder, delirium, suicidal thinking and behavior, aggressive behavior, insomnia, agitation, depersonalization, and euphoria.5 The most common cognitive impairment is verbal or declarative memory deficit; others include distractibility and deficits in attention and psychomotor speed.5 These psychiatric symptoms can have a rapid onset, possibly within hours of starting steroids.1 However, studies have reported a median time to onset of 11.5 days; 39% of cases had onset during the first week and 62% within 2 weeks.3,6 After reducing or stopping the steroid, it may take days to weeks before symptoms start to subside.2
What to consider in the differential Dx
Psychiatric symptoms that are induced by steroids can mimic metabolic, neurologic, or toxic disorders. Other factors to consider include drug withdrawal/intoxication, infections, and paraneoplastic syndromes.4,5 Although there is no reported correlation between the location of neurologic lesions and the development of specific psychiatric symptoms, manic symptoms appear most commonly with lesions in the right frontal lobe. 4 Other factors to note include the presence of new-onset psychiatric illnesses such as bipolar, mood, or thought disorders,4 as well as psychosocial stressors that might be contributing to the patient’s presentation.5
Continue to: Proposed mechanisms
Proposed mechanisms
Although the exact mechanism by which steroids induce psychiatric symptoms is unknown, several mechanisms have been proposed. One hypothesis is that steroid-induced psychopathology is related to decreased levels of corticotropin, norepinephrine, and beta-endorphin immunoreactivity.4,5,7 This may explain why many patients with major depressive disorder have elevated cortisol production and/or lack of suppression of cortisol secretion during a dexamethasone stimulation test, and why approximately one-half of patients with Cushing’s disease experience depressive symptoms.8 This is also likely why antipsychotics, which typically reduce cortisol, are efficacious treatments for some steroid-induced psychiatric symptoms.9
Cognitive impairments from steroid use may be related to these agents’ effects on certain brain regions. One such area is the hippocampus, an important mediator in the creation and maintenance of episodic and declarative memories.5,8,9 Acute glucocorticoid use is associated with decreased activity in the left hippocampus, reduced hippocampal glucose metabolism, and reduced cerebral blood flow in the posterior medial temporal lobe.10 Long-term glucocorticoid exposure is associated with smaller hippocampal volume and lower levels of temporal lobe N-acetylaspartate, a marker of neuronal viability.10 Because working memory depends on the prefrontal cortex and declarative memory relies on the hippocampus, deficits in these functions can be attributed to the effect of prolonged glucocorticoid exposure on glucocorticoid or mineralocorticoid receptors in the hippocampus, reduction of hippocampal volume, or elevated glutamate accumulation in that area.11 In addition, high cortisol levels inhibit brain-derived neurotrophic factor, which plays a crucial role in maintaining neural architecture in key brain regions such as the hippocampus and prefrontal cortex.11 There is also a correlation between the duration of prednisone treatment and atrophy of the right amygdala, which is an important regulator of mood and anxiety.11 Both the hippocampus and amygdala have dense collections of glucocorticoid receptors. This may explain why patients who receive high-dose corticosteroids can have reversible atrophy in the hypothalamus and amygdala, leading to deficits in emotional learning and the stress response.
Factors that increase risk
Several factors can increase the risk of steroid-induced psychopathology. The most significant is the dose; higher doses are more likely to produce psychiatric symptoms.1,5 Concurrent use of drugs that increase circulating levels of corticosteroids, such as inhibitors of the cytochrome P450 (CYP) enzyme (eg, clarithromycin), also increases the likelihood of developing psychiatric symptoms.1,5 Risk is also increased in patients with liver or renal dysfunction.1 Cerebral spinal fluid/serum albumin ratio, a marker of blood-brain barrier damage, and low serum complement levels were also reported to be independent risk factors,12 with the thought that increased permeability of the blood-brain barrier may allow hydrophobic steroid molecules to more easily penetrate the CNS, leading to increased neuropsychiatric effects. Hypoalbuminemia is another reported risk factor, perhaps because lower levels of serum albumin are related to higher levels of free and active glucocorticoids, which are normally inactive when bound to albumin.13 There also appears to be an increased prevalence of steroid-induced psychopathology in women, perhaps due to greater propensity in women to seek medical care or a higher prevalence of women with medical disorders that are treated with steroids.5 A previous history of psychiatric disorders may not increase risk.5
Several methods for reducing risk have been proposed, including using a divided-dosing regimens that may lower peak steroid plasma concentrations.13,14 However, the best prevention of steroid-induced psychiatric symptoms are awareness, early diagnosis, and intervention. Studies have suggested that N-methyl-
Treatment options
There are no FDA-approved medications for managing steroid-induced psychiatric symptoms.1,16 Treatment is based on evidence from case reports and a few small case series (Table2-5,17,18).

Continue to: When possible, initial treatment...
When possible, initial treatment should include discontinuing or tapering corticosteroids to <40 mg/d of prednisone-equivalent.1,4,10,18 Most studies have reported rapid reversal of deficits in declarative memory and of hippocampal volume loss once corticosteroids were tapered and discontinued.1,18 One study reported that >90% of patients recovered within 6 weeks, with patients with delirium recovering more quickly (mean: 5.4 days) than those with depression, mania, or psychosis (mean: 19.3 days).3 Another found that the vast majority (92%) of patients treated only with a steroid taper achieved clinical recovery, and 84% recovered with administration of antipsychotics without a steroid taper.3 In this study, all patients who received electroconvulsive therapy (ECT) recovered, as did those who received a steroid taper plus lithium or antipsychotics. Steroid tapering regimens are especially important for patients who have received long-term glucocorticoid treatment. Patients need to be closely monitored for signs of new or increased depression, delirium, or confusion during the taper. If these symptoms occur, the patient should be checked for adrenocortical insufficiency, which can be resolved by re-administering or increasing the dosage of the glucocorticoid.10
Mania. The treatment of mania/hypomania includes mood stabilizers (valproate, lithium, lamotrigine) and antipsychotics (quetiapine, olanzapine, haloperidol).2,4,5,10,14,18 Valproate has been reported to be an effective prophylactic of corticosteroid-induced mania,2 perhaps because it dampens neuronal hyperexcitability by attenuating NMDA receptors, blocking voltage-dependent sodium channels, and inhibiting the synthesis of cortical GABAergic steroids. Starting valproate while continuing corticosteroids (if necessary) may help lessen mania.2 Benzodiazepines also may be useful on a short-term basis.
Depression. Steroid-induced depression may be treated with sertraline or other first-line antidepressants.5,14 Consider ECT for patients with severe depression. Support for the use of antipsychotic medications stems from studies that reported steroids’ role in disrupting dopamine and 5HT2 activity. Lithium also has been used successfully to manage and prevent glucocorticoid-associated affective disorder.10,18 It can be used alone or in combination with selective serotonin reuptake inhibitors to alleviate depressive symptoms.10 Tricyclic antidepressants are generally avoided because their anticholinergic effects can exacerbate or worsen delirium.18 In general, ECT is an effective treatment for persistent and/or unresponsive steroid-induced depression,2,10 but may be difficult to use in patients with serious medical illnesses.
Agitation. Medications that have been proposed for treating steroid-induced agitation include benzodiazepines, haloperidol, and second-generation antipsychotics.5,17
Other considerations. Clinicians, patients, and families should discuss in detail the risks of steroid-induced psychiatric symptoms so an early diagnosis and appropriate intervention can be implemented. Before starting steroids, it is important to review the patient’s current medication list to ensure that steroid treatment is indicated, and to check for potential drug–drug interactions. In addition, the medical condition that is being treated with steroids also needs to be carefully reviewed, because certain illnesses are associated with the development of psychiatric symptoms. 5,10
Continue to: Young children...
Young children (age <6) and older adults appear to be at greater risk for cognitive and memory disturbances from steroid use.10 In addition, patients have individual levels of susceptibility to steroid-induced psychiatric symptoms that can vary over time. The risk for adverse effects may be elevated based on response to previous courses of glucocorticoid treatment.10 While gender, age, dosage, and duration of treatment influence risk, it is not possible to predict which patients will experience psychiatric effects during a given course of glucocorticoid therapy. Therefore, all patients should be considered to have the potential of developing such effects, and should be monitored during glucocorticoid treatment and withdrawal.
Goals for future research
To help reduce the severity of and cost associated with steroid-induced psychiatric symptoms,5,14 future studies should focus on controlled trials of preventative strategies. In particular, recent advances in genetic mapping may help identify involvement of certain genes or polymorphisms.5 Because current guidelines for the prevention and treatment of steroid-induced psychiatric symptoms are not evidence-based, controlled clinical trials are needed to elucidate the optimal management of such symptoms. There is much interplay between many of the proposed mechanisms of steroid-induced psychiatric symptoms, and future studies can help uncover a deeper understanding of the intricacies of this phenomenon.
CASE CONTINUED
Mrs. N is admitted for altered mental status. Medical workup includes MRI of the brain, MRI of the neck, cardiac echocardiogram, and EEG. There is no evidence of acute structural pathology. She is started on olanzapine, 10 mg/d at bedtime for manic and psychotic symptoms, and is discharged after 5 days. After 1 month, the outpatient psychiatrist gradually decreases and discontinues olanzapine as Mrs. N steadily returns to baseline. One year after discharge, Mrs. N continues to report resolution of her manic and psychotic symptoms.
Bottom Line
Steroids can induce a wide range of psychiatric symptoms, including mania/ hypomania, anxiety, and depression. Initial treatment typically includes tapering or discontinuing the steroid when possible. Other proposed treatments include certain antipsychotics, antidepressants, and other psychotropics, but the supporting evidence is largely anecdotal or based on case studies. Additional research is needed to elucidate the mechanism and treatment recommendations.
Related Resources
- Janes M, Kuster S, Goldson TM, et al. Steroid-induced psychosis. Proc (Bayl Univ Med Cent). 2019;32(4):614-615.
- Mayo Clinic. Prednisone and other corticosteroids. https://www.mayoclinic.org/steroids/art-20045692
Drug Brand Names
Haloperidol • Haldol
Lamotrigine • Lamictal
Lithium • Eskalith, Lithobid
Methylprednisolone injection • Solu-Medrol
Olanzapine • Zyprexa
Paroxetine • Paxil
Phenytoin • Dilantin
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Valproate • Depakote
1. Dubovsky AN, Arvikar S, Stern TA, et al. The neuropsychiatric complications of glucocorticoid use: steroid psychosis revisited. Psychosomatics. 2012;53(2):103-115.
2. Roxanas MG, Hunt GE. Rapid reversal of corticosteroid-induced mania with sodium valproate: a case series of 20 patients. Psychosomatics. 2012;53(6):575-581.
3. Lewis DA, Smith RE. Steroid‐induced psychiatric syndromes. A report of 14 cases and a review of the literature. J Affect Disord. 1983;5(4):319-332.
4. Warren KN, Katakam J, Espiridion ED. Acute-onset mania in a patient with non-small cell lung cancer. Cureus. 2019;11(8):e5436.
5. Kenna HA, Poon AW, de los Angeles CP, et al. Psychiatric complications of treatment with corticosteroids: review with case report. Psychiatry Clin Neurosci. 2011;65(6):549-560.
6. Ling MH, Perry PJ, Tsuang MT. Side effects of corticosteroid therapy. Psychiatric aspects. Arch Gen. Psychiatry. 1981;38(4):471-477.
7. Ularntinon S, Tzuang D, Dahl G, et al. Concurrent treatment of steroid-related mood and psychotic symptoms with risperidone. Pediatrics. 2010;125(5):e1241-e1245.
8. Pokladinkova J, Meyboom RH, Vlcek J, et al. Intranasally administered corticosteroids and neuropsychiatric disturbances: a review of the international pharmacovigilance programme of the World Health Organization. Ann Allergy Asthma Immunol. 2008;101(1):67-73.
9. Walker EF, Trotman HD, Pearce BD, et al. Cortisol levels and risk for psychosis: initial findings from the North American prodrome longitudinal study. Biol Psychiatry. 2013;74(6):410-417.
10. Wolkowitz OM, Reus UI. Treatment of depression with antiglucocorticoid drugs. Psychosom Med. 1999;61(5):698-711.
11. Judd LL, Schettler PJ, Brown ES, et al. Adverse consequences of glucocorticoid medication: psychological, cognitive, and behavioral effects. Am J Psychiatry. 2014;171(10):1045-1051.
12. Appenzeller S, Cendes F, Costallat LT. Acute psychosis in systemic lupus erythematosus. Rheumatol Int. 2008;28(3):237-243.
13. Glynne-Jones R, Vernon CC, Bell G. Is steroid psychosis preventable by divided doses? Lancet. 1986;2(8520):1404.
14. Ismail MF, Lavelle C, Cassidy EM. Steroid-induced mental disorders in cancer patients: a systematic review. Future Oncol. 2017;13(29):2719-2731.
15. Magariños AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience. 1995;69(1):89-98.
16. Brown BS, Stuard G, Liggin JDM, et al. Effect of phenytoin on mood and declarative memory during prescription corticosteroid therapy. Biol Psychiatry. 2005;57(5):543-548.
17. Desai S, Khanani S, Shad MU, et al. Attenutation of amygdala atrophy with lamotrigine in patients receiving corticosteroid therapy. J Clin Psychopharmacol. 2009;29(3):284-287.
18. Gable M, Depry D. Sustained corticosteroid-induced mania and psychosis despite cessation: a case study and brief literature review. Int J Psychiatry Med. 2015;50(4):398-404.
Ms. N, age 30, presents to the emergency department for altered mental status, insomnia, and behavioral changes, which she has experienced for 1 week. On evaluation, she grabs a clinician’s hand and details her business ideas and life story with no prompting. Ms. N’s mental status examination is significant for hyperverbal speech with increased rate and volume; tangential thought process; and bright, expanded affect.
One week earlier, Ms. N was hospitalized for sudden-onset chest pain, weakness, and dizziness. She received 45 minutes of cardiopulmonary resuscitation prior to presentation and was found to have a ST-segment elevation myocardial infarction that required emergent left anterior descending coronary artery and right coronary artery percutaneous coronary intervention to place drug-eluting stents. Her recovery was complicated by acute cardiogenic shock, pulmonary edema, and hypoxic respiratory failure. Subsequently, she was intubated, admitted to the ICU, and received high-dose corticosteroids, including IV methylprednisolone, 40 mg every 12 hours, which was tapered prior to discharge. Her husband reports that since Ms. N came home, she has been more talkative and irritable, ruminating about past events, unable to sleep (<1 hour/night), and crying frequently. She has also been endorsing visual and auditory hallucinations, with increased praying and listening to religious music.
The frequent clinical use of steroids necessitates an understanding of these medications’ various adverse effects. The manifestations of steroid-induced psychiatric symptoms are broad and can involve affective, behavioral, and cognitive domains. While the current mechanism is unknown, this phenomenon may be related to decreased levels of corticotropin, norepinephrine, and beta-endorphin immunoreactivity, as well as effects on brain regions such as the hippocampus and amygdala. The best interventions for steroid-induced psychiatric symptoms are awareness and early diagnosis. There are no FDA-approved treatments for steroid-induced psychiatric symptoms; initial measures should include tapering or discontinuing corticosteroids.
In this article, we review the literature on the incidence, characteristics, differential diagnoses, proposed mechanism, risk factors, and proposed treatments of steroid-induced psychiatric symptoms.
A wide range of presentations
Steroid use has increased over the past 2 decades, with 10% of medical and surgical inpatients and 1% to 3% of the general population taking long-term glucocorticoids.1 Even with topical application, steroid therapy is often systemically absorbed, and thus may lead to steroid-induced psychiatric symptoms. The incidence of steroid-induced psychiatric symptoms is difficult to assess because there can be a wide range of reactions that are dose- and time-related. Three reviews of a total of 122 cases reports found that an estimated 5% of patients treated with steroids experience severe psychiatric reactions.1-3
Steroid-induced psychopathology can include mood, behavioral, and/or cognitive impairments. Mania/hypomania is the most common overall psychiatric symptom; the most common mood manifestations are anxiety and depression.4,5 Other possible steroid-induced symptoms include psychosis, dementia, panic disorder, delirium, suicidal thinking and behavior, aggressive behavior, insomnia, agitation, depersonalization, and euphoria.5 The most common cognitive impairment is verbal or declarative memory deficit; others include distractibility and deficits in attention and psychomotor speed.5 These psychiatric symptoms can have a rapid onset, possibly within hours of starting steroids.1 However, studies have reported a median time to onset of 11.5 days; 39% of cases had onset during the first week and 62% within 2 weeks.3,6 After reducing or stopping the steroid, it may take days to weeks before symptoms start to subside.2
What to consider in the differential Dx
Psychiatric symptoms that are induced by steroids can mimic metabolic, neurologic, or toxic disorders. Other factors to consider include drug withdrawal/intoxication, infections, and paraneoplastic syndromes.4,5 Although there is no reported correlation between the location of neurologic lesions and the development of specific psychiatric symptoms, manic symptoms appear most commonly with lesions in the right frontal lobe. 4 Other factors to note include the presence of new-onset psychiatric illnesses such as bipolar, mood, or thought disorders,4 as well as psychosocial stressors that might be contributing to the patient’s presentation.5
Continue to: Proposed mechanisms
Proposed mechanisms
Although the exact mechanism by which steroids induce psychiatric symptoms is unknown, several mechanisms have been proposed. One hypothesis is that steroid-induced psychopathology is related to decreased levels of corticotropin, norepinephrine, and beta-endorphin immunoreactivity.4,5,7 This may explain why many patients with major depressive disorder have elevated cortisol production and/or lack of suppression of cortisol secretion during a dexamethasone stimulation test, and why approximately one-half of patients with Cushing’s disease experience depressive symptoms.8 This is also likely why antipsychotics, which typically reduce cortisol, are efficacious treatments for some steroid-induced psychiatric symptoms.9
Cognitive impairments from steroid use may be related to these agents’ effects on certain brain regions. One such area is the hippocampus, an important mediator in the creation and maintenance of episodic and declarative memories.5,8,9 Acute glucocorticoid use is associated with decreased activity in the left hippocampus, reduced hippocampal glucose metabolism, and reduced cerebral blood flow in the posterior medial temporal lobe.10 Long-term glucocorticoid exposure is associated with smaller hippocampal volume and lower levels of temporal lobe N-acetylaspartate, a marker of neuronal viability.10 Because working memory depends on the prefrontal cortex and declarative memory relies on the hippocampus, deficits in these functions can be attributed to the effect of prolonged glucocorticoid exposure on glucocorticoid or mineralocorticoid receptors in the hippocampus, reduction of hippocampal volume, or elevated glutamate accumulation in that area.11 In addition, high cortisol levels inhibit brain-derived neurotrophic factor, which plays a crucial role in maintaining neural architecture in key brain regions such as the hippocampus and prefrontal cortex.11 There is also a correlation between the duration of prednisone treatment and atrophy of the right amygdala, which is an important regulator of mood and anxiety.11 Both the hippocampus and amygdala have dense collections of glucocorticoid receptors. This may explain why patients who receive high-dose corticosteroids can have reversible atrophy in the hypothalamus and amygdala, leading to deficits in emotional learning and the stress response.
Factors that increase risk
Several factors can increase the risk of steroid-induced psychopathology. The most significant is the dose; higher doses are more likely to produce psychiatric symptoms.1,5 Concurrent use of drugs that increase circulating levels of corticosteroids, such as inhibitors of the cytochrome P450 (CYP) enzyme (eg, clarithromycin), also increases the likelihood of developing psychiatric symptoms.1,5 Risk is also increased in patients with liver or renal dysfunction.1 Cerebral spinal fluid/serum albumin ratio, a marker of blood-brain barrier damage, and low serum complement levels were also reported to be independent risk factors,12 with the thought that increased permeability of the blood-brain barrier may allow hydrophobic steroid molecules to more easily penetrate the CNS, leading to increased neuropsychiatric effects. Hypoalbuminemia is another reported risk factor, perhaps because lower levels of serum albumin are related to higher levels of free and active glucocorticoids, which are normally inactive when bound to albumin.13 There also appears to be an increased prevalence of steroid-induced psychopathology in women, perhaps due to greater propensity in women to seek medical care or a higher prevalence of women with medical disorders that are treated with steroids.5 A previous history of psychiatric disorders may not increase risk.5
Several methods for reducing risk have been proposed, including using a divided-dosing regimens that may lower peak steroid plasma concentrations.13,14 However, the best prevention of steroid-induced psychiatric symptoms are awareness, early diagnosis, and intervention. Studies have suggested that N-methyl-
Treatment options
There are no FDA-approved medications for managing steroid-induced psychiatric symptoms.1,16 Treatment is based on evidence from case reports and a few small case series (Table2-5,17,18).
Continue to: When possible, initial treatment...
When possible, initial treatment should include discontinuing or tapering corticosteroids to <40 mg/d of prednisone-equivalent.1,4,10,18 Most studies have reported rapid reversal of deficits in declarative memory and of hippocampal volume loss once corticosteroids were tapered and discontinued.1,18 One study reported that >90% of patients recovered within 6 weeks, with patients with delirium recovering more quickly (mean: 5.4 days) than those with depression, mania, or psychosis (mean: 19.3 days).3 Another found that the vast majority (92%) of patients treated only with a steroid taper achieved clinical recovery, and 84% recovered with administration of antipsychotics without a steroid taper.3 In this study, all patients who received electroconvulsive therapy (ECT) recovered, as did those who received a steroid taper plus lithium or antipsychotics. Steroid tapering regimens are especially important for patients who have received long-term glucocorticoid treatment. Patients need to be closely monitored for signs of new or increased depression, delirium, or confusion during the taper. If these symptoms occur, the patient should be checked for adrenocortical insufficiency, which can be resolved by re-administering or increasing the dosage of the glucocorticoid.10
Mania. The treatment of mania/hypomania includes mood stabilizers (valproate, lithium, lamotrigine) and antipsychotics (quetiapine, olanzapine, haloperidol).2,4,5,10,14,18 Valproate has been reported to be an effective prophylactic of corticosteroid-induced mania,2 perhaps because it dampens neuronal hyperexcitability by attenuating NMDA receptors, blocking voltage-dependent sodium channels, and inhibiting the synthesis of cortical GABAergic steroids. Starting valproate while continuing corticosteroids (if necessary) may help lessen mania.2 Benzodiazepines also may be useful on a short-term basis.
Depression. Steroid-induced depression may be treated with sertraline or other first-line antidepressants.5,14 Consider ECT for patients with severe depression. Support for the use of antipsychotic medications stems from studies that reported steroids’ role in disrupting dopamine and 5HT2 activity. Lithium also has been used successfully to manage and prevent glucocorticoid-associated affective disorder.10,18 It can be used alone or in combination with selective serotonin reuptake inhibitors to alleviate depressive symptoms.10 Tricyclic antidepressants are generally avoided because their anticholinergic effects can exacerbate or worsen delirium.18 In general, ECT is an effective treatment for persistent and/or unresponsive steroid-induced depression,2,10 but may be difficult to use in patients with serious medical illnesses.
Agitation. Medications that have been proposed for treating steroid-induced agitation include benzodiazepines, haloperidol, and second-generation antipsychotics.5,17
Other considerations. Clinicians, patients, and families should discuss in detail the risks of steroid-induced psychiatric symptoms so an early diagnosis and appropriate intervention can be implemented. Before starting steroids, it is important to review the patient’s current medication list to ensure that steroid treatment is indicated, and to check for potential drug–drug interactions. In addition, the medical condition that is being treated with steroids also needs to be carefully reviewed, because certain illnesses are associated with the development of psychiatric symptoms. 5,10
Continue to: Young children...
Young children (age <6) and older adults appear to be at greater risk for cognitive and memory disturbances from steroid use.10 In addition, patients have individual levels of susceptibility to steroid-induced psychiatric symptoms that can vary over time. The risk for adverse effects may be elevated based on response to previous courses of glucocorticoid treatment.10 While gender, age, dosage, and duration of treatment influence risk, it is not possible to predict which patients will experience psychiatric effects during a given course of glucocorticoid therapy. Therefore, all patients should be considered to have the potential of developing such effects, and should be monitored during glucocorticoid treatment and withdrawal.
Goals for future research
To help reduce the severity of and cost associated with steroid-induced psychiatric symptoms,5,14 future studies should focus on controlled trials of preventative strategies. In particular, recent advances in genetic mapping may help identify involvement of certain genes or polymorphisms.5 Because current guidelines for the prevention and treatment of steroid-induced psychiatric symptoms are not evidence-based, controlled clinical trials are needed to elucidate the optimal management of such symptoms. There is much interplay between many of the proposed mechanisms of steroid-induced psychiatric symptoms, and future studies can help uncover a deeper understanding of the intricacies of this phenomenon.
CASE CONTINUED
Mrs. N is admitted for altered mental status. Medical workup includes MRI of the brain, MRI of the neck, cardiac echocardiogram, and EEG. There is no evidence of acute structural pathology. She is started on olanzapine, 10 mg/d at bedtime for manic and psychotic symptoms, and is discharged after 5 days. After 1 month, the outpatient psychiatrist gradually decreases and discontinues olanzapine as Mrs. N steadily returns to baseline. One year after discharge, Mrs. N continues to report resolution of her manic and psychotic symptoms.
Bottom Line
Steroids can induce a wide range of psychiatric symptoms, including mania/ hypomania, anxiety, and depression. Initial treatment typically includes tapering or discontinuing the steroid when possible. Other proposed treatments include certain antipsychotics, antidepressants, and other psychotropics, but the supporting evidence is largely anecdotal or based on case studies. Additional research is needed to elucidate the mechanism and treatment recommendations.
Related Resources
- Janes M, Kuster S, Goldson TM, et al. Steroid-induced psychosis. Proc (Bayl Univ Med Cent). 2019;32(4):614-615.
- Mayo Clinic. Prednisone and other corticosteroids. https://www.mayoclinic.org/steroids/art-20045692
Drug Brand Names
Haloperidol • Haldol
Lamotrigine • Lamictal
Lithium • Eskalith, Lithobid
Methylprednisolone injection • Solu-Medrol
Olanzapine • Zyprexa
Paroxetine • Paxil
Phenytoin • Dilantin
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Valproate • Depakote
Ms. N, age 30, presents to the emergency department for altered mental status, insomnia, and behavioral changes, which she has experienced for 1 week. On evaluation, she grabs a clinician’s hand and details her business ideas and life story with no prompting. Ms. N’s mental status examination is significant for hyperverbal speech with increased rate and volume; tangential thought process; and bright, expanded affect.
One week earlier, Ms. N was hospitalized for sudden-onset chest pain, weakness, and dizziness. She received 45 minutes of cardiopulmonary resuscitation prior to presentation and was found to have a ST-segment elevation myocardial infarction that required emergent left anterior descending coronary artery and right coronary artery percutaneous coronary intervention to place drug-eluting stents. Her recovery was complicated by acute cardiogenic shock, pulmonary edema, and hypoxic respiratory failure. Subsequently, she was intubated, admitted to the ICU, and received high-dose corticosteroids, including IV methylprednisolone, 40 mg every 12 hours, which was tapered prior to discharge. Her husband reports that since Ms. N came home, she has been more talkative and irritable, ruminating about past events, unable to sleep (<1 hour/night), and crying frequently. She has also been endorsing visual and auditory hallucinations, with increased praying and listening to religious music.
The frequent clinical use of steroids necessitates an understanding of these medications’ various adverse effects. The manifestations of steroid-induced psychiatric symptoms are broad and can involve affective, behavioral, and cognitive domains. While the current mechanism is unknown, this phenomenon may be related to decreased levels of corticotropin, norepinephrine, and beta-endorphin immunoreactivity, as well as effects on brain regions such as the hippocampus and amygdala. The best interventions for steroid-induced psychiatric symptoms are awareness and early diagnosis. There are no FDA-approved treatments for steroid-induced psychiatric symptoms; initial measures should include tapering or discontinuing corticosteroids.
In this article, we review the literature on the incidence, characteristics, differential diagnoses, proposed mechanism, risk factors, and proposed treatments of steroid-induced psychiatric symptoms.
A wide range of presentations
Steroid use has increased over the past 2 decades, with 10% of medical and surgical inpatients and 1% to 3% of the general population taking long-term glucocorticoids.1 Even with topical application, steroid therapy is often systemically absorbed, and thus may lead to steroid-induced psychiatric symptoms. The incidence of steroid-induced psychiatric symptoms is difficult to assess because there can be a wide range of reactions that are dose- and time-related. Three reviews of a total of 122 cases reports found that an estimated 5% of patients treated with steroids experience severe psychiatric reactions.1-3
Steroid-induced psychopathology can include mood, behavioral, and/or cognitive impairments. Mania/hypomania is the most common overall psychiatric symptom; the most common mood manifestations are anxiety and depression.4,5 Other possible steroid-induced symptoms include psychosis, dementia, panic disorder, delirium, suicidal thinking and behavior, aggressive behavior, insomnia, agitation, depersonalization, and euphoria.5 The most common cognitive impairment is verbal or declarative memory deficit; others include distractibility and deficits in attention and psychomotor speed.5 These psychiatric symptoms can have a rapid onset, possibly within hours of starting steroids.1 However, studies have reported a median time to onset of 11.5 days; 39% of cases had onset during the first week and 62% within 2 weeks.3,6 After reducing or stopping the steroid, it may take days to weeks before symptoms start to subside.2
What to consider in the differential Dx
Psychiatric symptoms that are induced by steroids can mimic metabolic, neurologic, or toxic disorders. Other factors to consider include drug withdrawal/intoxication, infections, and paraneoplastic syndromes.4,5 Although there is no reported correlation between the location of neurologic lesions and the development of specific psychiatric symptoms, manic symptoms appear most commonly with lesions in the right frontal lobe. 4 Other factors to note include the presence of new-onset psychiatric illnesses such as bipolar, mood, or thought disorders,4 as well as psychosocial stressors that might be contributing to the patient’s presentation.5
Continue to: Proposed mechanisms
Proposed mechanisms
Although the exact mechanism by which steroids induce psychiatric symptoms is unknown, several mechanisms have been proposed. One hypothesis is that steroid-induced psychopathology is related to decreased levels of corticotropin, norepinephrine, and beta-endorphin immunoreactivity.4,5,7 This may explain why many patients with major depressive disorder have elevated cortisol production and/or lack of suppression of cortisol secretion during a dexamethasone stimulation test, and why approximately one-half of patients with Cushing’s disease experience depressive symptoms.8 This is also likely why antipsychotics, which typically reduce cortisol, are efficacious treatments for some steroid-induced psychiatric symptoms.9
Cognitive impairments from steroid use may be related to these agents’ effects on certain brain regions. One such area is the hippocampus, an important mediator in the creation and maintenance of episodic and declarative memories.5,8,9 Acute glucocorticoid use is associated with decreased activity in the left hippocampus, reduced hippocampal glucose metabolism, and reduced cerebral blood flow in the posterior medial temporal lobe.10 Long-term glucocorticoid exposure is associated with smaller hippocampal volume and lower levels of temporal lobe N-acetylaspartate, a marker of neuronal viability.10 Because working memory depends on the prefrontal cortex and declarative memory relies on the hippocampus, deficits in these functions can be attributed to the effect of prolonged glucocorticoid exposure on glucocorticoid or mineralocorticoid receptors in the hippocampus, reduction of hippocampal volume, or elevated glutamate accumulation in that area.11 In addition, high cortisol levels inhibit brain-derived neurotrophic factor, which plays a crucial role in maintaining neural architecture in key brain regions such as the hippocampus and prefrontal cortex.11 There is also a correlation between the duration of prednisone treatment and atrophy of the right amygdala, which is an important regulator of mood and anxiety.11 Both the hippocampus and amygdala have dense collections of glucocorticoid receptors. This may explain why patients who receive high-dose corticosteroids can have reversible atrophy in the hypothalamus and amygdala, leading to deficits in emotional learning and the stress response.
Factors that increase risk
Several factors can increase the risk of steroid-induced psychopathology. The most significant is the dose; higher doses are more likely to produce psychiatric symptoms.1,5 Concurrent use of drugs that increase circulating levels of corticosteroids, such as inhibitors of the cytochrome P450 (CYP) enzyme (eg, clarithromycin), also increases the likelihood of developing psychiatric symptoms.1,5 Risk is also increased in patients with liver or renal dysfunction.1 Cerebral spinal fluid/serum albumin ratio, a marker of blood-brain barrier damage, and low serum complement levels were also reported to be independent risk factors,12 with the thought that increased permeability of the blood-brain barrier may allow hydrophobic steroid molecules to more easily penetrate the CNS, leading to increased neuropsychiatric effects. Hypoalbuminemia is another reported risk factor, perhaps because lower levels of serum albumin are related to higher levels of free and active glucocorticoids, which are normally inactive when bound to albumin.13 There also appears to be an increased prevalence of steroid-induced psychopathology in women, perhaps due to greater propensity in women to seek medical care or a higher prevalence of women with medical disorders that are treated with steroids.5 A previous history of psychiatric disorders may not increase risk.5
Several methods for reducing risk have been proposed, including using a divided-dosing regimens that may lower peak steroid plasma concentrations.13,14 However, the best prevention of steroid-induced psychiatric symptoms are awareness, early diagnosis, and intervention. Studies have suggested that N-methyl-
Treatment options
There are no FDA-approved medications for managing steroid-induced psychiatric symptoms.1,16 Treatment is based on evidence from case reports and a few small case series (Table2-5,17,18).
Continue to: When possible, initial treatment...
When possible, initial treatment should include discontinuing or tapering corticosteroids to <40 mg/d of prednisone-equivalent.1,4,10,18 Most studies have reported rapid reversal of deficits in declarative memory and of hippocampal volume loss once corticosteroids were tapered and discontinued.1,18 One study reported that >90% of patients recovered within 6 weeks, with patients with delirium recovering more quickly (mean: 5.4 days) than those with depression, mania, or psychosis (mean: 19.3 days).3 Another found that the vast majority (92%) of patients treated only with a steroid taper achieved clinical recovery, and 84% recovered with administration of antipsychotics without a steroid taper.3 In this study, all patients who received electroconvulsive therapy (ECT) recovered, as did those who received a steroid taper plus lithium or antipsychotics. Steroid tapering regimens are especially important for patients who have received long-term glucocorticoid treatment. Patients need to be closely monitored for signs of new or increased depression, delirium, or confusion during the taper. If these symptoms occur, the patient should be checked for adrenocortical insufficiency, which can be resolved by re-administering or increasing the dosage of the glucocorticoid.10
Mania. The treatment of mania/hypomania includes mood stabilizers (valproate, lithium, lamotrigine) and antipsychotics (quetiapine, olanzapine, haloperidol).2,4,5,10,14,18 Valproate has been reported to be an effective prophylactic of corticosteroid-induced mania,2 perhaps because it dampens neuronal hyperexcitability by attenuating NMDA receptors, blocking voltage-dependent sodium channels, and inhibiting the synthesis of cortical GABAergic steroids. Starting valproate while continuing corticosteroids (if necessary) may help lessen mania.2 Benzodiazepines also may be useful on a short-term basis.
Depression. Steroid-induced depression may be treated with sertraline or other first-line antidepressants.5,14 Consider ECT for patients with severe depression. Support for the use of antipsychotic medications stems from studies that reported steroids’ role in disrupting dopamine and 5HT2 activity. Lithium also has been used successfully to manage and prevent glucocorticoid-associated affective disorder.10,18 It can be used alone or in combination with selective serotonin reuptake inhibitors to alleviate depressive symptoms.10 Tricyclic antidepressants are generally avoided because their anticholinergic effects can exacerbate or worsen delirium.18 In general, ECT is an effective treatment for persistent and/or unresponsive steroid-induced depression,2,10 but may be difficult to use in patients with serious medical illnesses.
Agitation. Medications that have been proposed for treating steroid-induced agitation include benzodiazepines, haloperidol, and second-generation antipsychotics.5,17
Other considerations. Clinicians, patients, and families should discuss in detail the risks of steroid-induced psychiatric symptoms so an early diagnosis and appropriate intervention can be implemented. Before starting steroids, it is important to review the patient’s current medication list to ensure that steroid treatment is indicated, and to check for potential drug–drug interactions. In addition, the medical condition that is being treated with steroids also needs to be carefully reviewed, because certain illnesses are associated with the development of psychiatric symptoms. 5,10
Continue to: Young children...
Young children (age <6) and older adults appear to be at greater risk for cognitive and memory disturbances from steroid use.10 In addition, patients have individual levels of susceptibility to steroid-induced psychiatric symptoms that can vary over time. The risk for adverse effects may be elevated based on response to previous courses of glucocorticoid treatment.10 While gender, age, dosage, and duration of treatment influence risk, it is not possible to predict which patients will experience psychiatric effects during a given course of glucocorticoid therapy. Therefore, all patients should be considered to have the potential of developing such effects, and should be monitored during glucocorticoid treatment and withdrawal.
Goals for future research
To help reduce the severity of and cost associated with steroid-induced psychiatric symptoms,5,14 future studies should focus on controlled trials of preventative strategies. In particular, recent advances in genetic mapping may help identify involvement of certain genes or polymorphisms.5 Because current guidelines for the prevention and treatment of steroid-induced psychiatric symptoms are not evidence-based, controlled clinical trials are needed to elucidate the optimal management of such symptoms. There is much interplay between many of the proposed mechanisms of steroid-induced psychiatric symptoms, and future studies can help uncover a deeper understanding of the intricacies of this phenomenon.
CASE CONTINUED
Mrs. N is admitted for altered mental status. Medical workup includes MRI of the brain, MRI of the neck, cardiac echocardiogram, and EEG. There is no evidence of acute structural pathology. She is started on olanzapine, 10 mg/d at bedtime for manic and psychotic symptoms, and is discharged after 5 days. After 1 month, the outpatient psychiatrist gradually decreases and discontinues olanzapine as Mrs. N steadily returns to baseline. One year after discharge, Mrs. N continues to report resolution of her manic and psychotic symptoms.
Bottom Line
Steroids can induce a wide range of psychiatric symptoms, including mania/ hypomania, anxiety, and depression. Initial treatment typically includes tapering or discontinuing the steroid when possible. Other proposed treatments include certain antipsychotics, antidepressants, and other psychotropics, but the supporting evidence is largely anecdotal or based on case studies. Additional research is needed to elucidate the mechanism and treatment recommendations.
Related Resources
- Janes M, Kuster S, Goldson TM, et al. Steroid-induced psychosis. Proc (Bayl Univ Med Cent). 2019;32(4):614-615.
- Mayo Clinic. Prednisone and other corticosteroids. https://www.mayoclinic.org/steroids/art-20045692
Drug Brand Names
Haloperidol • Haldol
Lamotrigine • Lamictal
Lithium • Eskalith, Lithobid
Methylprednisolone injection • Solu-Medrol
Olanzapine • Zyprexa
Paroxetine • Paxil
Phenytoin • Dilantin
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Valproate • Depakote
1. Dubovsky AN, Arvikar S, Stern TA, et al. The neuropsychiatric complications of glucocorticoid use: steroid psychosis revisited. Psychosomatics. 2012;53(2):103-115.
2. Roxanas MG, Hunt GE. Rapid reversal of corticosteroid-induced mania with sodium valproate: a case series of 20 patients. Psychosomatics. 2012;53(6):575-581.
3. Lewis DA, Smith RE. Steroid‐induced psychiatric syndromes. A report of 14 cases and a review of the literature. J Affect Disord. 1983;5(4):319-332.
4. Warren KN, Katakam J, Espiridion ED. Acute-onset mania in a patient with non-small cell lung cancer. Cureus. 2019;11(8):e5436.
5. Kenna HA, Poon AW, de los Angeles CP, et al. Psychiatric complications of treatment with corticosteroids: review with case report. Psychiatry Clin Neurosci. 2011;65(6):549-560.
6. Ling MH, Perry PJ, Tsuang MT. Side effects of corticosteroid therapy. Psychiatric aspects. Arch Gen. Psychiatry. 1981;38(4):471-477.
7. Ularntinon S, Tzuang D, Dahl G, et al. Concurrent treatment of steroid-related mood and psychotic symptoms with risperidone. Pediatrics. 2010;125(5):e1241-e1245.
8. Pokladinkova J, Meyboom RH, Vlcek J, et al. Intranasally administered corticosteroids and neuropsychiatric disturbances: a review of the international pharmacovigilance programme of the World Health Organization. Ann Allergy Asthma Immunol. 2008;101(1):67-73.
9. Walker EF, Trotman HD, Pearce BD, et al. Cortisol levels and risk for psychosis: initial findings from the North American prodrome longitudinal study. Biol Psychiatry. 2013;74(6):410-417.
10. Wolkowitz OM, Reus UI. Treatment of depression with antiglucocorticoid drugs. Psychosom Med. 1999;61(5):698-711.
11. Judd LL, Schettler PJ, Brown ES, et al. Adverse consequences of glucocorticoid medication: psychological, cognitive, and behavioral effects. Am J Psychiatry. 2014;171(10):1045-1051.
12. Appenzeller S, Cendes F, Costallat LT. Acute psychosis in systemic lupus erythematosus. Rheumatol Int. 2008;28(3):237-243.
13. Glynne-Jones R, Vernon CC, Bell G. Is steroid psychosis preventable by divided doses? Lancet. 1986;2(8520):1404.
14. Ismail MF, Lavelle C, Cassidy EM. Steroid-induced mental disorders in cancer patients: a systematic review. Future Oncol. 2017;13(29):2719-2731.
15. Magariños AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience. 1995;69(1):89-98.
16. Brown BS, Stuard G, Liggin JDM, et al. Effect of phenytoin on mood and declarative memory during prescription corticosteroid therapy. Biol Psychiatry. 2005;57(5):543-548.
17. Desai S, Khanani S, Shad MU, et al. Attenutation of amygdala atrophy with lamotrigine in patients receiving corticosteroid therapy. J Clin Psychopharmacol. 2009;29(3):284-287.
18. Gable M, Depry D. Sustained corticosteroid-induced mania and psychosis despite cessation: a case study and brief literature review. Int J Psychiatry Med. 2015;50(4):398-404.
1. Dubovsky AN, Arvikar S, Stern TA, et al. The neuropsychiatric complications of glucocorticoid use: steroid psychosis revisited. Psychosomatics. 2012;53(2):103-115.
2. Roxanas MG, Hunt GE. Rapid reversal of corticosteroid-induced mania with sodium valproate: a case series of 20 patients. Psychosomatics. 2012;53(6):575-581.
3. Lewis DA, Smith RE. Steroid‐induced psychiatric syndromes. A report of 14 cases and a review of the literature. J Affect Disord. 1983;5(4):319-332.
4. Warren KN, Katakam J, Espiridion ED. Acute-onset mania in a patient with non-small cell lung cancer. Cureus. 2019;11(8):e5436.
5. Kenna HA, Poon AW, de los Angeles CP, et al. Psychiatric complications of treatment with corticosteroids: review with case report. Psychiatry Clin Neurosci. 2011;65(6):549-560.
6. Ling MH, Perry PJ, Tsuang MT. Side effects of corticosteroid therapy. Psychiatric aspects. Arch Gen. Psychiatry. 1981;38(4):471-477.
7. Ularntinon S, Tzuang D, Dahl G, et al. Concurrent treatment of steroid-related mood and psychotic symptoms with risperidone. Pediatrics. 2010;125(5):e1241-e1245.
8. Pokladinkova J, Meyboom RH, Vlcek J, et al. Intranasally administered corticosteroids and neuropsychiatric disturbances: a review of the international pharmacovigilance programme of the World Health Organization. Ann Allergy Asthma Immunol. 2008;101(1):67-73.
9. Walker EF, Trotman HD, Pearce BD, et al. Cortisol levels and risk for psychosis: initial findings from the North American prodrome longitudinal study. Biol Psychiatry. 2013;74(6):410-417.
10. Wolkowitz OM, Reus UI. Treatment of depression with antiglucocorticoid drugs. Psychosom Med. 1999;61(5):698-711.
11. Judd LL, Schettler PJ, Brown ES, et al. Adverse consequences of glucocorticoid medication: psychological, cognitive, and behavioral effects. Am J Psychiatry. 2014;171(10):1045-1051.
12. Appenzeller S, Cendes F, Costallat LT. Acute psychosis in systemic lupus erythematosus. Rheumatol Int. 2008;28(3):237-243.
13. Glynne-Jones R, Vernon CC, Bell G. Is steroid psychosis preventable by divided doses? Lancet. 1986;2(8520):1404.
14. Ismail MF, Lavelle C, Cassidy EM. Steroid-induced mental disorders in cancer patients: a systematic review. Future Oncol. 2017;13(29):2719-2731.
15. Magariños AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience. 1995;69(1):89-98.
16. Brown BS, Stuard G, Liggin JDM, et al. Effect of phenytoin on mood and declarative memory during prescription corticosteroid therapy. Biol Psychiatry. 2005;57(5):543-548.
17. Desai S, Khanani S, Shad MU, et al. Attenutation of amygdala atrophy with lamotrigine in patients receiving corticosteroid therapy. J Clin Psychopharmacol. 2009;29(3):284-287.
18. Gable M, Depry D. Sustained corticosteroid-induced mania and psychosis despite cessation: a case study and brief literature review. Int J Psychiatry Med. 2015;50(4):398-404.

Differentiating serotonin syndrome and neuroleptic malignant syndrome
Serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) are each rare psychiatric emergencies that can lead to fatal outcomes. Their clinical presentations can overlap, which can make it difficult to differentiate between the 2 syndromes; however, their treatments are distinct, and it is imperative to know how to identify symptoms and accurately diagnose each of them to provide appropriate intervention. This article summarizes the 2 syndromes and their treatments, with a focus on how clinicians can distinguish them, provide prompt intervention, and prevent occurrence.
Serotonin syndrome
Mechanism. The decarboxylation and hydroxylation of tryptophan forms serotonin, also known as 5-hydroxytryptamine (5-HT), which can then be metabolized by monoamine oxidase-A (MAO-A) into 5-hydroxyindoleacetic acid (5-HIAA).1Medications can disrupt this pathway of serotonin production or its metabolism, and result in excessive levels of serotonin, which subsequently leads to an overactivation of central and peripheral serotonin receptors.1 Increased receptor activation leads to further upregulation, and ultimately more serotonin transmission. This can be caused by monotherapy or use of multiple serotonergic agents, polypharmacy with a combination of medication classes, drug interactions, or overdose. The wide variety of medications often prescribed by different clinicians can make identification of excessive serotonergic activity difficult, especially because mood stabilizers such as lithium,2 and non-psychiatric medications such as
- inhibition of serotonin uptake (seen with selective serotonin reuptake inhibitors [SSRIs], serotonin-norepinephrine reuptake inhibitors [SNRIs], and tricyclic antidepressants [TCAs])
- inhibition of serotonin metabolism (seen with monoamine oxidase inhibitors [MAOIs])
- increased serotonin synthesis (seen with stimulants)
- increased serotonin release (seen with stimulants and opiates)
- activation of serotonin receptors (seen with lithium)
- inhibition of certain cytochrome P450 (CYP450) enzymes (seen with ciprofloxacin, fluconazole, etc.).

It is important to recognize that various serotonergic agents are involved in the CYP450 system. Inhibition of the CYP450 pathway by common antibiotics such as ciprofloxacin, or antifungals such as fluconazole, may result in an accumulation of serotonergic agents and place patients at increased risk for developing SS.
Clinical presentation. The clinical presentation of SS can range from mild to fatal. There is no specific laboratory test for diagnosis, although an elevation of the total creatine kinase (CK) and leukocyte count, as well as increased transaminase levels or lower bicarbonate levels, have been reported in the literature.4
Symptoms of SS generally present within 24 hours of starting/changing therapy and include a triad of mental status changes (altered mental status [AMS]), autonomic instability, and abnormalities of neuromuscular tone. Examples of AMS include agitation, anxiety, disorientation, and restlessness. Symptoms of autonomic instability include hypertension, tachycardia, tachypnea, hyperthermia, diaphoresis, flushed skin, vomiting, diarrhea, and arrhythmias. Symptoms stemming from changes in neuromuscular tone include tremors, clonus, hyperreflexia, and muscle rigidity.1 The multiple possible clinical presentations, as well as symptoms that overlap with those of other syndromes, can make SS difficult to recognize quickly in a clinical setting.

Diagnostic criteria. Sternbach’s diagnostic criteria for SS are defined as the presence of 3 or more of the 10 most common clinical features (Table 25). Due to concerns that Sternbach’s diagnostic criteria overemphasized an abnormal mental state (leading to possible confusion of SS with other AMS syndromes), the Hunter serotonin toxicity criteria6 (Figure6) were developed in 2003, and were found to be more sensitive and specific than Sternbach’s criteria. Both tools are often used in clinical practice.

Treatment. Treatment of SS begins with prompt discontinuation of all serotonergic agents. The intensity of treatment depends on the severity of the symptoms. Mild symptoms can be managed with supportive care,3 and in such cases, the syndrome generally resolves within 24 hours.7 Clinicians may use supportive care to normalize vital signs (oxygenation to maintain SpO2 >94%, IV fluids for volume depletion, cooling agents, antihypertensives, benzodiazepines for sedation or control of agitation, etc.). Patients who are more ill may require more aggressive treatment, such as the use of a serotonergic antagonist (ie, cyproheptadine) and those who are severely hyperthermic (temperature >41.1ºC) may require neuromuscular sedation, paralysis, and possibly endotracheal intubation.3
Continue to: Management pitfalls include...
Management pitfalls include misdiagnosis of SS, failure to recognize its rapid rate of progression, and adverse effects of pharmacologic therapy.3 The most effective treatment for SS is prevention. SS can be prevented by astute pharmacologic understanding, avoidance of polypharmacy, and physician education.3
Neuroleptic malignant syndrome
Possible mechanisms. Neuromuscular malignant syndrome is thought to result from dopamine receptor antagonism leading to a hypodopaminergic state in the striatum and hypothalamus.8 The pathophysiology behind NMS has not fully been elucidated; however, several hypotheses attempt to explain this life-threatening reaction. The first focuses on dopamine D2 receptor antagonism, because many of the neuroleptic (antipsychotic) medications that can precipitate NMS are involved in dopamine blockade. In this theory, blocking dopamine D2 receptors in the anterior hypothalamus explains the hyperthermia seen in NMS, while blockade in the corpus striatum is believed to lead to muscle rigidity.9
The second hypothesis suggests that neuroleptics may have a direct toxic effect to muscle cells. Neuroleptics influence calcium transport across the sarcoplasmic reticulum and can lead to increased calcium release, which may contribute to the muscle rigidity and hyperthermia seen in NMS.9
The third hypothesis involves hyperactivity of the sympathetic nervous system; it is thought that psychologic stressors alter frontal lobe function, with neuroleptics disrupting the inhibitory pathways of the sympathetic nervous system. The autonomic nervous system innervates multiple organ systems, so this excessively dysregulated sympathetic nervous system may be responsible for multiple NMS symptoms (hyperthermia, muscle rigidity, hypertension, diaphoresis, tachycardia, elevated CK.10
NMS can be caused by neuroleptic agents (both first- and second-generation antipsychotics) as well as antiemetics (Table 31). The time between use of these medications and onset of symptoms is highly variable. NMS can occur after a single dose, after a dose adjustment, or possibly after years of treatment with the same medication. It is not dose-dependent.11 In certain individuals, NMS may occur at therapeutic doses.

Continue to: Clinical presentation
Clinical presentation. Patients with NMS typically present with a tetrad of symptoms: mental status changes, muscular rigidity, hyperthermia, and autonomic instability.12 Mental status changes can include confusion and agitation, as well as catatonic signs and mutism. The muscular rigidity of NMS is characterized by “lead pipe rigidity” and may be accompanied by tremor, dystonia, or dyskinesias. Laboratory findings include elevated serum CK (from severe rigidity), often >1,000 U/L, although normal levels can be observed if rigidity has not yet developed.13
Treatment. The first step for treatment is to discontinue the causative medication.14 Initiate supportive therapy immediately to restrict the progression of symptoms. Interventions include cooling blankets, fluid resuscitation, and antihypertensives to maintain autonomic stability15 or benzodiazepines to control agitation. In severe cases, muscular rigidity may extend to the airways and intubation may be required. The severity of these symptoms may warrant admission to the ICU for close monitoring. Pharmacologic treatment with
Differentiating between SS and NMS
Differentiating between these 2 syndromes (Table 417) is critical to direct appropriate intervention. Table 517 outlines the treatment overview for SS and NMS.

Detailed history. A detailed history is imperative in making accurate diagnoses. Useful components of the history include a patient’s duration of symptoms and medication history (prescription medications as well as over-the-counter medications, supplements, and illicit drugs). Also assess for medical comorbidities, because certain medical diagnoses may alert the clinician that it is likely the patient had been prescribed serotonergic agents or neuroleptics, and renal or liver impairment may alert the clinician of decreased metabolism rates. Medication history is arguably the most useful piece of the interview, because serotonergic agents can cause SS, whereas dopamine blockers cause NMS. It should be noted that excess serotonin acts as a true toxidrome and is concentration-dependent in causing SS, whereas NMS is an idiosyncratic reaction to a drug.

Physical exam. Although there are many overlapping clinical manifestations, SS produces neuromuscular hyperactivity (ie, clonus, hyperreflexia), whereas NMS is characterized by more sluggish responses (ie, rigidity, bradyreflexia).18
Continue to: Laboratory findings
Laboratory findings. Overlap between NMS and SS also occurs with lab findings; both syndromes can result in leukocytosis, elevated CK from muscle damage, and low serum iron levels. However, these findings are more commonly associated with NMS and are seen in 75% of cases.17,19
Course of illness. Duration of symptoms can also help differentiate the 2 syndromes. SS typically develops within 24 hours of starting/changing therapy, whereas NMS symptoms can be present for days to weeks. Resolution of symptoms may also be helpful in differentiation because SS typically resolves within a few days of initiating treatment, whereas NMS resolves within 9 to 14 days of starting treatment.19
Bottom Line
The clinical presentations of serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) overlap, which can make them difficult to differentiate; however, they each have distinct approaches to treatment. Features in SS that are distinct from NMS include a history of serotonergic agents, rapid onset of symptoms, hyperreflexia, and clonus. NMS is slower in onset and can be found in patients who are prescribed dopamine antagonists, with distinct symptoms of rigidity and hyporeflexia.
Related Resources
- Kimmel R. Serotonin syndrome or NMS? Clues to diagnosis. Current Psychiatry. 2010;9(2):92.
- Strawn JR, Keck Jr PE, Caroff SN. Neuroleptic malignant syndrome: Answers to 6 tough questions. Current Psychiatry. 2008;7(1):95-101.
Drug Brand Names
Amantadine • Symmetrel
Amitriptyline • Elavil, Endep
Aripiprazole • Abilify
Bromocriptine • Cycloset, Parlodel
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Ciprofloxacin • Cipro
Citalopram • Celexa
Clomipramine • Anafranil
Clozapine • Clozaril
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Dantrolene • Dantrium
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Dextromethorphan • Benylin, Dexalone
Dolasetron • Anzemet
Doxepin • Silenor
Droperidol • Inapsine
Duloxetine • Cymbalt
Escitalopram • Lexapro
Fentanyl • Actiq, Duragesic
Fluconazole • Diflucan
Fluoxetine • Prozac
Fluphenazine • Prolixin
Fluvoxamine • Luvox
Granisetron • Kytril
Haloperidol • Haldol
Isocarboxazid • Marplan
Levomilnacipran • Fetzima
Linezolid • Zyvox
Lithium • Eskalith, Lithobid
Meperidone • Demerol
Metoclopramide • Reglan
Milnacipran • Savella
Nefazodone • Serzone
Olanzapine • Zyprexa
Ondansetron • Zofran
Paliperidone • Invega
Palonosetron • Aloxi
Paroxetine • Paxil
Pentazocine • Talwin, Talacen
Perphenazine • Trilafon
Phenelzine • Nardil
Procarbazine • Matulane
Prochlorperazine • Compazine
Promethazine • Phenergan
Quetiapine • Seroquel
Rasagiline • Azilect
Risperidone • Risperdal
Safinamide • Xadago
Selegiline • Eldepryl, Zelapar
Sertraline • Zoloft
Sibutramine • Meridia
Tedizolid • Sivextro
Thioridazine • Mellaril
Tranylcypromine • Parnate
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Vilazodone • Viibryd
Vortioxetine • Trintellix
Valproate • Depacon
Ziprasidone • Geodon
1. Volpi-Abadie J, Kaye AM, Kaye AD. Serotonin syndrome. Ochsner J. 2013;13(4):533-540.
2. Werneke U, Jamshidi F, Taylor D, et al. Conundrums in neurology: diagnosing serotonin syndrome – a meta-analysis of cases. BMC Neurol. 2016;16:97.
3. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
4. Birmes P, Coppin D, Schmitt L, et al. Serotonin syndrome: a brief review. CMAJ. 2003;168(11):1439-1442.
5. Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148:705-713.
6. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003; 96(9):635-642.
7. Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med. 1994;331(15):1021-1022.
8. Nisijima K. Serotonin syndrome overlapping with neuroleptic malignant syndrome: A case report and approaches for differentially diagnosing the two syndromes. Asian J Psychiatr. 2015;18:100-101.
9. Adnet P, Lestavel P, Krivosic-Horber R. Neuroleptic malignant syndrome. Br J Anaesth. 2000;85(1):129-135.
10. Gurrera R. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
11. Pope HG Jr, Aizley HG, Keck PE Jr, et al. Neuroleptic malignant syndrome: long-term follow-up of 20 cases. J Clin Psychiatry. 1991;52(5):208-212.
12. Velamoor VR, Norman RM, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis. 1994;182(3):168-173.
13. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77(1):185-202.
14. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome. Ann Pharmacother. 2016;50(11):973-981.
15. San Gabriel MC, Eddula-Changala B, Tan Y, et al. Electroconvulsive in a schizophrenic patient with neuroleptic malignant syndrome and rhabdomyolysis. J ECT. 2015;31(3):197-200.
16. Buggenhout S, Vandenberghe J, Sienaert P. Electroconvulsion therapy for neuroleptic malignant syndrome. Tijdschr Psychiatr. 2014;56(9):612-615.
17. Perry PJ, Wilborn CA. Serotonin syndrome vs neuroleptic malignant syndrome: a contrast of causes, diagnoses, and management. Ann Clin Psychiatry. 2012;24(2):155-162.
18. Mills KC. Serotonin syndrome. A clinical update. Crit Care Clin. 1997;13(4):763-783.
19. Dosi R, Ambaliya A, Joshi H, et al. Serotonin syndrome versus neuroleptic malignant syndrome: a challenge clinical quandary. BMJ Case Rep. 2014;2014:bcr201404154. doi:10.1136/bcr-2014-204154.
Serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) are each rare psychiatric emergencies that can lead to fatal outcomes. Their clinical presentations can overlap, which can make it difficult to differentiate between the 2 syndromes; however, their treatments are distinct, and it is imperative to know how to identify symptoms and accurately diagnose each of them to provide appropriate intervention. This article summarizes the 2 syndromes and their treatments, with a focus on how clinicians can distinguish them, provide prompt intervention, and prevent occurrence.
Serotonin syndrome
Mechanism. The decarboxylation and hydroxylation of tryptophan forms serotonin, also known as 5-hydroxytryptamine (5-HT), which can then be metabolized by monoamine oxidase-A (MAO-A) into 5-hydroxyindoleacetic acid (5-HIAA).1Medications can disrupt this pathway of serotonin production or its metabolism, and result in excessive levels of serotonin, which subsequently leads to an overactivation of central and peripheral serotonin receptors.1 Increased receptor activation leads to further upregulation, and ultimately more serotonin transmission. This can be caused by monotherapy or use of multiple serotonergic agents, polypharmacy with a combination of medication classes, drug interactions, or overdose. The wide variety of medications often prescribed by different clinicians can make identification of excessive serotonergic activity difficult, especially because mood stabilizers such as lithium,2 and non-psychiatric medications such as
- inhibition of serotonin uptake (seen with selective serotonin reuptake inhibitors [SSRIs], serotonin-norepinephrine reuptake inhibitors [SNRIs], and tricyclic antidepressants [TCAs])
- inhibition of serotonin metabolism (seen with monoamine oxidase inhibitors [MAOIs])
- increased serotonin synthesis (seen with stimulants)
- increased serotonin release (seen with stimulants and opiates)
- activation of serotonin receptors (seen with lithium)
- inhibition of certain cytochrome P450 (CYP450) enzymes (seen with ciprofloxacin, fluconazole, etc.).
It is important to recognize that various serotonergic agents are involved in the CYP450 system. Inhibition of the CYP450 pathway by common antibiotics such as ciprofloxacin, or antifungals such as fluconazole, may result in an accumulation of serotonergic agents and place patients at increased risk for developing SS.
Clinical presentation. The clinical presentation of SS can range from mild to fatal. There is no specific laboratory test for diagnosis, although an elevation of the total creatine kinase (CK) and leukocyte count, as well as increased transaminase levels or lower bicarbonate levels, have been reported in the literature.4
Symptoms of SS generally present within 24 hours of starting/changing therapy and include a triad of mental status changes (altered mental status [AMS]), autonomic instability, and abnormalities of neuromuscular tone. Examples of AMS include agitation, anxiety, disorientation, and restlessness. Symptoms of autonomic instability include hypertension, tachycardia, tachypnea, hyperthermia, diaphoresis, flushed skin, vomiting, diarrhea, and arrhythmias. Symptoms stemming from changes in neuromuscular tone include tremors, clonus, hyperreflexia, and muscle rigidity.1 The multiple possible clinical presentations, as well as symptoms that overlap with those of other syndromes, can make SS difficult to recognize quickly in a clinical setting.
Diagnostic criteria. Sternbach’s diagnostic criteria for SS are defined as the presence of 3 or more of the 10 most common clinical features (Table 25). Due to concerns that Sternbach’s diagnostic criteria overemphasized an abnormal mental state (leading to possible confusion of SS with other AMS syndromes), the Hunter serotonin toxicity criteria6 (Figure6) were developed in 2003, and were found to be more sensitive and specific than Sternbach’s criteria. Both tools are often used in clinical practice.
Treatment. Treatment of SS begins with prompt discontinuation of all serotonergic agents. The intensity of treatment depends on the severity of the symptoms. Mild symptoms can be managed with supportive care,3 and in such cases, the syndrome generally resolves within 24 hours.7 Clinicians may use supportive care to normalize vital signs (oxygenation to maintain SpO2 >94%, IV fluids for volume depletion, cooling agents, antihypertensives, benzodiazepines for sedation or control of agitation, etc.). Patients who are more ill may require more aggressive treatment, such as the use of a serotonergic antagonist (ie, cyproheptadine) and those who are severely hyperthermic (temperature >41.1ºC) may require neuromuscular sedation, paralysis, and possibly endotracheal intubation.3
Continue to: Management pitfalls include...
Management pitfalls include misdiagnosis of SS, failure to recognize its rapid rate of progression, and adverse effects of pharmacologic therapy.3 The most effective treatment for SS is prevention. SS can be prevented by astute pharmacologic understanding, avoidance of polypharmacy, and physician education.3
Neuroleptic malignant syndrome
Possible mechanisms. Neuromuscular malignant syndrome is thought to result from dopamine receptor antagonism leading to a hypodopaminergic state in the striatum and hypothalamus.8 The pathophysiology behind NMS has not fully been elucidated; however, several hypotheses attempt to explain this life-threatening reaction. The first focuses on dopamine D2 receptor antagonism, because many of the neuroleptic (antipsychotic) medications that can precipitate NMS are involved in dopamine blockade. In this theory, blocking dopamine D2 receptors in the anterior hypothalamus explains the hyperthermia seen in NMS, while blockade in the corpus striatum is believed to lead to muscle rigidity.9
The second hypothesis suggests that neuroleptics may have a direct toxic effect to muscle cells. Neuroleptics influence calcium transport across the sarcoplasmic reticulum and can lead to increased calcium release, which may contribute to the muscle rigidity and hyperthermia seen in NMS.9
The third hypothesis involves hyperactivity of the sympathetic nervous system; it is thought that psychologic stressors alter frontal lobe function, with neuroleptics disrupting the inhibitory pathways of the sympathetic nervous system. The autonomic nervous system innervates multiple organ systems, so this excessively dysregulated sympathetic nervous system may be responsible for multiple NMS symptoms (hyperthermia, muscle rigidity, hypertension, diaphoresis, tachycardia, elevated CK.10
NMS can be caused by neuroleptic agents (both first- and second-generation antipsychotics) as well as antiemetics (Table 31). The time between use of these medications and onset of symptoms is highly variable. NMS can occur after a single dose, after a dose adjustment, or possibly after years of treatment with the same medication. It is not dose-dependent.11 In certain individuals, NMS may occur at therapeutic doses.
Continue to: Clinical presentation
Clinical presentation. Patients with NMS typically present with a tetrad of symptoms: mental status changes, muscular rigidity, hyperthermia, and autonomic instability.12 Mental status changes can include confusion and agitation, as well as catatonic signs and mutism. The muscular rigidity of NMS is characterized by “lead pipe rigidity” and may be accompanied by tremor, dystonia, or dyskinesias. Laboratory findings include elevated serum CK (from severe rigidity), often >1,000 U/L, although normal levels can be observed if rigidity has not yet developed.13
Treatment. The first step for treatment is to discontinue the causative medication.14 Initiate supportive therapy immediately to restrict the progression of symptoms. Interventions include cooling blankets, fluid resuscitation, and antihypertensives to maintain autonomic stability15 or benzodiazepines to control agitation. In severe cases, muscular rigidity may extend to the airways and intubation may be required. The severity of these symptoms may warrant admission to the ICU for close monitoring. Pharmacologic treatment with
Differentiating between SS and NMS
Differentiating between these 2 syndromes (Table 417) is critical to direct appropriate intervention. Table 517 outlines the treatment overview for SS and NMS.
Detailed history. A detailed history is imperative in making accurate diagnoses. Useful components of the history include a patient’s duration of symptoms and medication history (prescription medications as well as over-the-counter medications, supplements, and illicit drugs). Also assess for medical comorbidities, because certain medical diagnoses may alert the clinician that it is likely the patient had been prescribed serotonergic agents or neuroleptics, and renal or liver impairment may alert the clinician of decreased metabolism rates. Medication history is arguably the most useful piece of the interview, because serotonergic agents can cause SS, whereas dopamine blockers cause NMS. It should be noted that excess serotonin acts as a true toxidrome and is concentration-dependent in causing SS, whereas NMS is an idiosyncratic reaction to a drug.
Physical exam. Although there are many overlapping clinical manifestations, SS produces neuromuscular hyperactivity (ie, clonus, hyperreflexia), whereas NMS is characterized by more sluggish responses (ie, rigidity, bradyreflexia).18
Continue to: Laboratory findings
Laboratory findings. Overlap between NMS and SS also occurs with lab findings; both syndromes can result in leukocytosis, elevated CK from muscle damage, and low serum iron levels. However, these findings are more commonly associated with NMS and are seen in 75% of cases.17,19
Course of illness. Duration of symptoms can also help differentiate the 2 syndromes. SS typically develops within 24 hours of starting/changing therapy, whereas NMS symptoms can be present for days to weeks. Resolution of symptoms may also be helpful in differentiation because SS typically resolves within a few days of initiating treatment, whereas NMS resolves within 9 to 14 days of starting treatment.19
Bottom Line
The clinical presentations of serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) overlap, which can make them difficult to differentiate; however, they each have distinct approaches to treatment. Features in SS that are distinct from NMS include a history of serotonergic agents, rapid onset of symptoms, hyperreflexia, and clonus. NMS is slower in onset and can be found in patients who are prescribed dopamine antagonists, with distinct symptoms of rigidity and hyporeflexia.
Related Resources
- Kimmel R. Serotonin syndrome or NMS? Clues to diagnosis. Current Psychiatry. 2010;9(2):92.
- Strawn JR, Keck Jr PE, Caroff SN. Neuroleptic malignant syndrome: Answers to 6 tough questions. Current Psychiatry. 2008;7(1):95-101.
Drug Brand Names
Amantadine • Symmetrel
Amitriptyline • Elavil, Endep
Aripiprazole • Abilify
Bromocriptine • Cycloset, Parlodel
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Ciprofloxacin • Cipro
Citalopram • Celexa
Clomipramine • Anafranil
Clozapine • Clozaril
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Dantrolene • Dantrium
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Dextromethorphan • Benylin, Dexalone
Dolasetron • Anzemet
Doxepin • Silenor
Droperidol • Inapsine
Duloxetine • Cymbalt
Escitalopram • Lexapro
Fentanyl • Actiq, Duragesic
Fluconazole • Diflucan
Fluoxetine • Prozac
Fluphenazine • Prolixin
Fluvoxamine • Luvox
Granisetron • Kytril
Haloperidol • Haldol
Isocarboxazid • Marplan
Levomilnacipran • Fetzima
Linezolid • Zyvox
Lithium • Eskalith, Lithobid
Meperidone • Demerol
Metoclopramide • Reglan
Milnacipran • Savella
Nefazodone • Serzone
Olanzapine • Zyprexa
Ondansetron • Zofran
Paliperidone • Invega
Palonosetron • Aloxi
Paroxetine • Paxil
Pentazocine • Talwin, Talacen
Perphenazine • Trilafon
Phenelzine • Nardil
Procarbazine • Matulane
Prochlorperazine • Compazine
Promethazine • Phenergan
Quetiapine • Seroquel
Rasagiline • Azilect
Risperidone • Risperdal
Safinamide • Xadago
Selegiline • Eldepryl, Zelapar
Sertraline • Zoloft
Sibutramine • Meridia
Tedizolid • Sivextro
Thioridazine • Mellaril
Tranylcypromine • Parnate
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Vilazodone • Viibryd
Vortioxetine • Trintellix
Valproate • Depacon
Ziprasidone • Geodon
Serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) are each rare psychiatric emergencies that can lead to fatal outcomes. Their clinical presentations can overlap, which can make it difficult to differentiate between the 2 syndromes; however, their treatments are distinct, and it is imperative to know how to identify symptoms and accurately diagnose each of them to provide appropriate intervention. This article summarizes the 2 syndromes and their treatments, with a focus on how clinicians can distinguish them, provide prompt intervention, and prevent occurrence.
Serotonin syndrome
Mechanism. The decarboxylation and hydroxylation of tryptophan forms serotonin, also known as 5-hydroxytryptamine (5-HT), which can then be metabolized by monoamine oxidase-A (MAO-A) into 5-hydroxyindoleacetic acid (5-HIAA).1Medications can disrupt this pathway of serotonin production or its metabolism, and result in excessive levels of serotonin, which subsequently leads to an overactivation of central and peripheral serotonin receptors.1 Increased receptor activation leads to further upregulation, and ultimately more serotonin transmission. This can be caused by monotherapy or use of multiple serotonergic agents, polypharmacy with a combination of medication classes, drug interactions, or overdose. The wide variety of medications often prescribed by different clinicians can make identification of excessive serotonergic activity difficult, especially because mood stabilizers such as lithium,2 and non-psychiatric medications such as
- inhibition of serotonin uptake (seen with selective serotonin reuptake inhibitors [SSRIs], serotonin-norepinephrine reuptake inhibitors [SNRIs], and tricyclic antidepressants [TCAs])
- inhibition of serotonin metabolism (seen with monoamine oxidase inhibitors [MAOIs])
- increased serotonin synthesis (seen with stimulants)
- increased serotonin release (seen with stimulants and opiates)
- activation of serotonin receptors (seen with lithium)
- inhibition of certain cytochrome P450 (CYP450) enzymes (seen with ciprofloxacin, fluconazole, etc.).
It is important to recognize that various serotonergic agents are involved in the CYP450 system. Inhibition of the CYP450 pathway by common antibiotics such as ciprofloxacin, or antifungals such as fluconazole, may result in an accumulation of serotonergic agents and place patients at increased risk for developing SS.
Clinical presentation. The clinical presentation of SS can range from mild to fatal. There is no specific laboratory test for diagnosis, although an elevation of the total creatine kinase (CK) and leukocyte count, as well as increased transaminase levels or lower bicarbonate levels, have been reported in the literature.4
Symptoms of SS generally present within 24 hours of starting/changing therapy and include a triad of mental status changes (altered mental status [AMS]), autonomic instability, and abnormalities of neuromuscular tone. Examples of AMS include agitation, anxiety, disorientation, and restlessness. Symptoms of autonomic instability include hypertension, tachycardia, tachypnea, hyperthermia, diaphoresis, flushed skin, vomiting, diarrhea, and arrhythmias. Symptoms stemming from changes in neuromuscular tone include tremors, clonus, hyperreflexia, and muscle rigidity.1 The multiple possible clinical presentations, as well as symptoms that overlap with those of other syndromes, can make SS difficult to recognize quickly in a clinical setting.
Diagnostic criteria. Sternbach’s diagnostic criteria for SS are defined as the presence of 3 or more of the 10 most common clinical features (Table 25). Due to concerns that Sternbach’s diagnostic criteria overemphasized an abnormal mental state (leading to possible confusion of SS with other AMS syndromes), the Hunter serotonin toxicity criteria6 (Figure6) were developed in 2003, and were found to be more sensitive and specific than Sternbach’s criteria. Both tools are often used in clinical practice.
Treatment. Treatment of SS begins with prompt discontinuation of all serotonergic agents. The intensity of treatment depends on the severity of the symptoms. Mild symptoms can be managed with supportive care,3 and in such cases, the syndrome generally resolves within 24 hours.7 Clinicians may use supportive care to normalize vital signs (oxygenation to maintain SpO2 >94%, IV fluids for volume depletion, cooling agents, antihypertensives, benzodiazepines for sedation or control of agitation, etc.). Patients who are more ill may require more aggressive treatment, such as the use of a serotonergic antagonist (ie, cyproheptadine) and those who are severely hyperthermic (temperature >41.1ºC) may require neuromuscular sedation, paralysis, and possibly endotracheal intubation.3
Continue to: Management pitfalls include...
Management pitfalls include misdiagnosis of SS, failure to recognize its rapid rate of progression, and adverse effects of pharmacologic therapy.3 The most effective treatment for SS is prevention. SS can be prevented by astute pharmacologic understanding, avoidance of polypharmacy, and physician education.3
Neuroleptic malignant syndrome
Possible mechanisms. Neuromuscular malignant syndrome is thought to result from dopamine receptor antagonism leading to a hypodopaminergic state in the striatum and hypothalamus.8 The pathophysiology behind NMS has not fully been elucidated; however, several hypotheses attempt to explain this life-threatening reaction. The first focuses on dopamine D2 receptor antagonism, because many of the neuroleptic (antipsychotic) medications that can precipitate NMS are involved in dopamine blockade. In this theory, blocking dopamine D2 receptors in the anterior hypothalamus explains the hyperthermia seen in NMS, while blockade in the corpus striatum is believed to lead to muscle rigidity.9
The second hypothesis suggests that neuroleptics may have a direct toxic effect to muscle cells. Neuroleptics influence calcium transport across the sarcoplasmic reticulum and can lead to increased calcium release, which may contribute to the muscle rigidity and hyperthermia seen in NMS.9
The third hypothesis involves hyperactivity of the sympathetic nervous system; it is thought that psychologic stressors alter frontal lobe function, with neuroleptics disrupting the inhibitory pathways of the sympathetic nervous system. The autonomic nervous system innervates multiple organ systems, so this excessively dysregulated sympathetic nervous system may be responsible for multiple NMS symptoms (hyperthermia, muscle rigidity, hypertension, diaphoresis, tachycardia, elevated CK.10
NMS can be caused by neuroleptic agents (both first- and second-generation antipsychotics) as well as antiemetics (Table 31). The time between use of these medications and onset of symptoms is highly variable. NMS can occur after a single dose, after a dose adjustment, or possibly after years of treatment with the same medication. It is not dose-dependent.11 In certain individuals, NMS may occur at therapeutic doses.
Continue to: Clinical presentation
Clinical presentation. Patients with NMS typically present with a tetrad of symptoms: mental status changes, muscular rigidity, hyperthermia, and autonomic instability.12 Mental status changes can include confusion and agitation, as well as catatonic signs and mutism. The muscular rigidity of NMS is characterized by “lead pipe rigidity” and may be accompanied by tremor, dystonia, or dyskinesias. Laboratory findings include elevated serum CK (from severe rigidity), often >1,000 U/L, although normal levels can be observed if rigidity has not yet developed.13
Treatment. The first step for treatment is to discontinue the causative medication.14 Initiate supportive therapy immediately to restrict the progression of symptoms. Interventions include cooling blankets, fluid resuscitation, and antihypertensives to maintain autonomic stability15 or benzodiazepines to control agitation. In severe cases, muscular rigidity may extend to the airways and intubation may be required. The severity of these symptoms may warrant admission to the ICU for close monitoring. Pharmacologic treatment with
Differentiating between SS and NMS
Differentiating between these 2 syndromes (Table 417) is critical to direct appropriate intervention. Table 517 outlines the treatment overview for SS and NMS.
Detailed history. A detailed history is imperative in making accurate diagnoses. Useful components of the history include a patient’s duration of symptoms and medication history (prescription medications as well as over-the-counter medications, supplements, and illicit drugs). Also assess for medical comorbidities, because certain medical diagnoses may alert the clinician that it is likely the patient had been prescribed serotonergic agents or neuroleptics, and renal or liver impairment may alert the clinician of decreased metabolism rates. Medication history is arguably the most useful piece of the interview, because serotonergic agents can cause SS, whereas dopamine blockers cause NMS. It should be noted that excess serotonin acts as a true toxidrome and is concentration-dependent in causing SS, whereas NMS is an idiosyncratic reaction to a drug.
Physical exam. Although there are many overlapping clinical manifestations, SS produces neuromuscular hyperactivity (ie, clonus, hyperreflexia), whereas NMS is characterized by more sluggish responses (ie, rigidity, bradyreflexia).18
Continue to: Laboratory findings
Laboratory findings. Overlap between NMS and SS also occurs with lab findings; both syndromes can result in leukocytosis, elevated CK from muscle damage, and low serum iron levels. However, these findings are more commonly associated with NMS and are seen in 75% of cases.17,19
Course of illness. Duration of symptoms can also help differentiate the 2 syndromes. SS typically develops within 24 hours of starting/changing therapy, whereas NMS symptoms can be present for days to weeks. Resolution of symptoms may also be helpful in differentiation because SS typically resolves within a few days of initiating treatment, whereas NMS resolves within 9 to 14 days of starting treatment.19
Bottom Line
The clinical presentations of serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) overlap, which can make them difficult to differentiate; however, they each have distinct approaches to treatment. Features in SS that are distinct from NMS include a history of serotonergic agents, rapid onset of symptoms, hyperreflexia, and clonus. NMS is slower in onset and can be found in patients who are prescribed dopamine antagonists, with distinct symptoms of rigidity and hyporeflexia.
Related Resources
- Kimmel R. Serotonin syndrome or NMS? Clues to diagnosis. Current Psychiatry. 2010;9(2):92.
- Strawn JR, Keck Jr PE, Caroff SN. Neuroleptic malignant syndrome: Answers to 6 tough questions. Current Psychiatry. 2008;7(1):95-101.
Drug Brand Names
Amantadine • Symmetrel
Amitriptyline • Elavil, Endep
Aripiprazole • Abilify
Bromocriptine • Cycloset, Parlodel
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Ciprofloxacin • Cipro
Citalopram • Celexa
Clomipramine • Anafranil
Clozapine • Clozaril
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Dantrolene • Dantrium
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Dextromethorphan • Benylin, Dexalone
Dolasetron • Anzemet
Doxepin • Silenor
Droperidol • Inapsine
Duloxetine • Cymbalt
Escitalopram • Lexapro
Fentanyl • Actiq, Duragesic
Fluconazole • Diflucan
Fluoxetine • Prozac
Fluphenazine • Prolixin
Fluvoxamine • Luvox
Granisetron • Kytril
Haloperidol • Haldol
Isocarboxazid • Marplan
Levomilnacipran • Fetzima
Linezolid • Zyvox
Lithium • Eskalith, Lithobid
Meperidone • Demerol
Metoclopramide • Reglan
Milnacipran • Savella
Nefazodone • Serzone
Olanzapine • Zyprexa
Ondansetron • Zofran
Paliperidone • Invega
Palonosetron • Aloxi
Paroxetine • Paxil
Pentazocine • Talwin, Talacen
Perphenazine • Trilafon
Phenelzine • Nardil
Procarbazine • Matulane
Prochlorperazine • Compazine
Promethazine • Phenergan
Quetiapine • Seroquel
Rasagiline • Azilect
Risperidone • Risperdal
Safinamide • Xadago
Selegiline • Eldepryl, Zelapar
Sertraline • Zoloft
Sibutramine • Meridia
Tedizolid • Sivextro
Thioridazine • Mellaril
Tranylcypromine • Parnate
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Vilazodone • Viibryd
Vortioxetine • Trintellix
Valproate • Depacon
Ziprasidone • Geodon
1. Volpi-Abadie J, Kaye AM, Kaye AD. Serotonin syndrome. Ochsner J. 2013;13(4):533-540.
2. Werneke U, Jamshidi F, Taylor D, et al. Conundrums in neurology: diagnosing serotonin syndrome – a meta-analysis of cases. BMC Neurol. 2016;16:97.
3. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
4. Birmes P, Coppin D, Schmitt L, et al. Serotonin syndrome: a brief review. CMAJ. 2003;168(11):1439-1442.
5. Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148:705-713.
6. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003; 96(9):635-642.
7. Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med. 1994;331(15):1021-1022.
8. Nisijima K. Serotonin syndrome overlapping with neuroleptic malignant syndrome: A case report and approaches for differentially diagnosing the two syndromes. Asian J Psychiatr. 2015;18:100-101.
9. Adnet P, Lestavel P, Krivosic-Horber R. Neuroleptic malignant syndrome. Br J Anaesth. 2000;85(1):129-135.
10. Gurrera R. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
11. Pope HG Jr, Aizley HG, Keck PE Jr, et al. Neuroleptic malignant syndrome: long-term follow-up of 20 cases. J Clin Psychiatry. 1991;52(5):208-212.
12. Velamoor VR, Norman RM, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis. 1994;182(3):168-173.
13. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77(1):185-202.
14. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome. Ann Pharmacother. 2016;50(11):973-981.
15. San Gabriel MC, Eddula-Changala B, Tan Y, et al. Electroconvulsive in a schizophrenic patient with neuroleptic malignant syndrome and rhabdomyolysis. J ECT. 2015;31(3):197-200.
16. Buggenhout S, Vandenberghe J, Sienaert P. Electroconvulsion therapy for neuroleptic malignant syndrome. Tijdschr Psychiatr. 2014;56(9):612-615.
17. Perry PJ, Wilborn CA. Serotonin syndrome vs neuroleptic malignant syndrome: a contrast of causes, diagnoses, and management. Ann Clin Psychiatry. 2012;24(2):155-162.
18. Mills KC. Serotonin syndrome. A clinical update. Crit Care Clin. 1997;13(4):763-783.
19. Dosi R, Ambaliya A, Joshi H, et al. Serotonin syndrome versus neuroleptic malignant syndrome: a challenge clinical quandary. BMJ Case Rep. 2014;2014:bcr201404154. doi:10.1136/bcr-2014-204154.
1. Volpi-Abadie J, Kaye AM, Kaye AD. Serotonin syndrome. Ochsner J. 2013;13(4):533-540.
2. Werneke U, Jamshidi F, Taylor D, et al. Conundrums in neurology: diagnosing serotonin syndrome – a meta-analysis of cases. BMC Neurol. 2016;16:97.
3. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
4. Birmes P, Coppin D, Schmitt L, et al. Serotonin syndrome: a brief review. CMAJ. 2003;168(11):1439-1442.
5. Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148:705-713.
6. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003; 96(9):635-642.
7. Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med. 1994;331(15):1021-1022.
8. Nisijima K. Serotonin syndrome overlapping with neuroleptic malignant syndrome: A case report and approaches for differentially diagnosing the two syndromes. Asian J Psychiatr. 2015;18:100-101.
9. Adnet P, Lestavel P, Krivosic-Horber R. Neuroleptic malignant syndrome. Br J Anaesth. 2000;85(1):129-135.
10. Gurrera R. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
11. Pope HG Jr, Aizley HG, Keck PE Jr, et al. Neuroleptic malignant syndrome: long-term follow-up of 20 cases. J Clin Psychiatry. 1991;52(5):208-212.
12. Velamoor VR, Norman RM, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis. 1994;182(3):168-173.
13. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77(1):185-202.
14. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome. Ann Pharmacother. 2016;50(11):973-981.
15. San Gabriel MC, Eddula-Changala B, Tan Y, et al. Electroconvulsive in a schizophrenic patient with neuroleptic malignant syndrome and rhabdomyolysis. J ECT. 2015;31(3):197-200.
16. Buggenhout S, Vandenberghe J, Sienaert P. Electroconvulsion therapy for neuroleptic malignant syndrome. Tijdschr Psychiatr. 2014;56(9):612-615.
17. Perry PJ, Wilborn CA. Serotonin syndrome vs neuroleptic malignant syndrome: a contrast of causes, diagnoses, and management. Ann Clin Psychiatry. 2012;24(2):155-162.
18. Mills KC. Serotonin syndrome. A clinical update. Crit Care Clin. 1997;13(4):763-783.
19. Dosi R, Ambaliya A, Joshi H, et al. Serotonin syndrome versus neuroleptic malignant syndrome: a challenge clinical quandary. BMJ Case Rep. 2014;2014:bcr201404154. doi:10.1136/bcr-2014-204154.

How to diagnose and manage hypertension in a psychiatric patient
Hypertension is a widespread, under-recognized, and undertreated cause of morbidity and mortality in the United States and is associated with several psychiatric illnesses. Left untreated, hypertension can have significant consequences, including increased risk of stroke, coronary heart disease, heart failure, chronic kidney failure, and death. Approximately 70 million adults in the United States have hypertension, but only 60% of them have been diagnosed, and of those only 50% have their blood pressure under control.1 In 2013, 360,000 deaths in the United States were attributed to hypertension.2
Hypertension is associated with major depressive disorder, generalized anxiety disorder, bipolar disorder, and schizophrenia.3-5 Additionally, impulsive eating disorders, substance abuse, anxiety, and depression are associated with a hypertension diagnosis, although patients with panic disorder develop hypertension at a younger age.6 A 2007 study found a 61% prevalence of hypertension in those with bipolar disorder compared with 41% among the general population.7 The strong link between bipolar disorder and hypertension might be because of a common disease mechanism; both are associated with hyperactive cellular calcium signaling and increased platelet intracellular calcium ion concentrations.8
Hypertension not only is common among patients with psychiatric illness, it likely contributes to worse clinical outcomes. Studies across different cultures have found higher mortality rates in individuals with mental illness.9-11 Persons with schizophrenia and other severe mental illnesses may lose ≥25 years of life expectancy, with the primary cause of death being cardiovascular disease, not suicide.12 Patients with depression have a 50% greater risk of cardiovascular disease, which is equivalent to the risk of smoking.13
Schizophrenia is strongly associated with numerous comorbidities and has been linked significantly to an elevated 10-year cardiac risk after controlling for body mass index.5 The high rate of non-treatment of hypertension for patients with schizophrenia (62.4%) is especially concerning.14
Because of the well-documented morbidity and mortality of hypertension and its increased prevalence and undertreatment in the psychiatric population, mental health providers are in an important position to recognize hypertension and evaluate its inherent risks to direct their patients toward proper treatment. This article reviews:
- the signs and symptoms of hypertension
- the mental health provider’s role in the evaluation and diagnosis
- how psychotropic drugs influence blood pressure and drug–drug interactions
- the management of hypertension in psychiatric patients, including strategies for counseling and lifestyle management.
Diagnosing hypertension
Hypertension is defined as a blood pressure >140/90 mm Hg, the average of ≥2 properly measured readings at ≥2 visits in a medical setting.15 The proper equipment, including a well-fitting blood pressure cuff, and technique to measure blood pressure are essential to avoid misdiagnosis. The patient should be at rest for ≥5 minutes, without active pain or emotional distress.
Most cases of hypertension (90% to 95%) are primary, commonly called essential hypertension. However, the differential diagnosis also should consider secondary causes, which may include:
- obesity
- medications
- chronic alcohol use
- methamphetamine or cocaine use
- primary kidney disease
- atherosclerotic renal artery stenosis
- obstructive sleep apnea
- hypothyroidism
- primary hyperaldosteronism
- narrowing of the aorta
- Cushing syndrome
- primary hyperparathyroidism
- polycythemia
- pheochromocytoma.

Medical evaluation. Once the diagnosis of hypertension is made, a medical evaluation is indicated to determine if the patient has end-organ damage from the elevated pressures, such as renal disease or heart disease, to identify other modifiable cardiovascular risk factors, such as hyperlipidemia, and to screen for secondary causes of hypertension. This evaluation includes15:
- a physical exam
- review of medications
- lipid profile
- urinalysis to screen for proteinuria
- serum electrolytes and creatinine
- electrocardiogram to screen for left ventricular hypertrophy or prior infarction
- fasting glucose or hemoglobin A1c to screen for type 2 diabetes mellitus.
Psychotropic drugs. In psychiatric patients, the evaluation must consider the potential impact psychotropic drug effects and drug–drug interactions can have on blood pressure (Table 2). For example, patients taking both diuretics and lithium are at increased risk for dehydration and increased serum lithium levels, which could cause severe neurologic symptoms and renal insufficiency.16 Several antihypertensives when taken with venlafaxine can increase blood pressure, but antihypertensives with α-1 blocking psychotropics can decrease blood pressure. Monoamine oxidase inhibitors can cause hypotension or hypertension with various classes of antihypertensives. Stimulants, such as methylphenidate, atomoxetine, dextroamphetamine, armodafinil, or modafinil, alone or combined with antihypertensives, can cause hypertension.17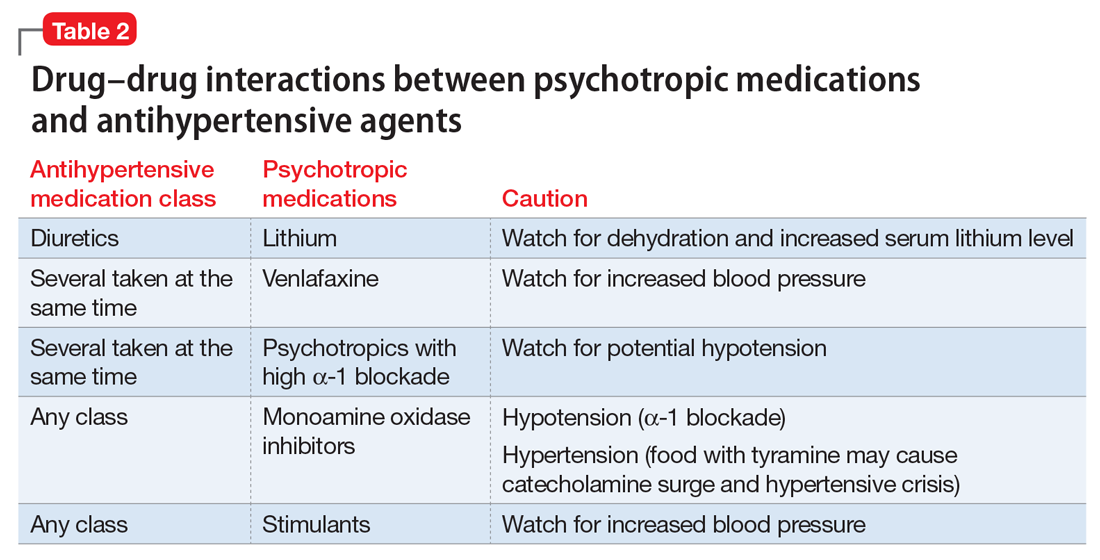
Substance abuse, particularly alcohol, methamphetamine, and cocaine, can cause difficulty controlling blood pressure. Patients with refractory hypertension should have a reassessment of substance abuse as a potential cause.
Screening guidelines for mental health providers
For many patients with severe mental illness, visits to their mental health providers might be their only contact with the medical system. Therefore, screening in the mental health settings could detect cases that otherwise would be missed.
Screening recommendations. The U.S. Preventive Services Task Force recommends screening for hypertension in the general population beginning at age 18.18 Adults age 18 to 39 with normal blood pressure (<130/85 mm Hg) and no other risk factors (eg, overweight, obese, or African American) can be screened every 3 years. Those with risk factors or a blood pressure of 130/85 to 139/89 mm Hg and adults age ≥40 should have annual screenings.
Ideally, psychiatrists and other mental health providers should monitor blood pressure at each visit, especially in patients taking psychotropics because of their higher risk for hypertension.
Optimizing treatment. Once the diagnosis of essential hypertension is established, identifying psychiatric comorbidities and the severity of psychiatric symptoms are important to optimize treatment adherence. Patients with increased depressive symptoms are less likely to comply with antihypertensive medication,19 and patients with confirmed depression are 3 times more likely to not adhere to medical treatment recommendations than non-depressed patients.20
Physicians’ attitudes toward hypertension also can affect patients’ compliance and blood pressure control.21 Psychiatrists should be empathetic and motivational toward patients attempting to control their blood pressure. The Seventh Joint National Committee on the Prevention, Detection, Evaluation, and Treatment of High Blood Pressure states, “Motivation improves when patients have positive experiences with, and trust in, the clinician. Empathy builds trust and is a potent motivator.”22
Treatment and management
Treatment of hypertension significantly reduces the risk of stroke, myocardial infarction, renal injury, heart failure, and premature death. Studies show that treatment that reduces systolic blood pressure by 12 mm Hg over 10 years will prevent 1 death for every 11 patients with essential hypertension. In those with concomitant cardiovascular disease or target organ damage, such a reduction would prevent death in 1 of every 9 patients treated.15Blood pressure goals. The 2014 Eighth Joint National Committee Guideline for Management of High Blood Pressure in Adults provides guidance on blood pressure goals depending on patients’ underlying medical history (Figure).23 Based on expert opinion and randomized controlled studies, blood pressure goals for patients without diabetes or chronic kidney disease (CKD)—an estimated or measured glomerular filtration rate (GFR) of ≤60 mL/min/1.73 m2—depend on age: <140/90 mm Hg for age 18 to 59 and <150/90 mm Hg for age ≥60. For patients with diabetes or CKD, the blood pressure goal is <140/90 mm Hg, regardless of age.
However, not all experts agree on these specific blood pressure goals. A major trial (SPRINT) published in 2015 found that intensive blood pressure goals do benefit higher-risk, non-diabetic patients.24 Specifically, the study randomized patients age ≥50 with systolic blood pressure of 130 to 180 mm Hg and increased cardiovascular risk to systolic blood pressure targets of <140 mm Hg (standard) or <120 mm Hg (intensive). Characteristics of increased cardiovascular risk were clinical or subclinical cardiovascular disease other than stroke, CKD with GFR of 20 to 60 mL/min/1.73 m2, age ≥75, or Framingham 10-year coronary heart disease risk score ≥15%. Intensive treatment significantly reduced overall mortality and the rate of acute coronary syndrome, myocardial infarction, heart failure, stroke, or cardiovascular death. However, the results of this study have not been assimilated into any recent guidelines. Therefore, consider a goal of <120 mm Hg for non-diabetic patients age ≥50 with any of these factors.
Lifestyle modifications. Psychiatrists are well equipped to motivate and encourage behavioral modification in patients with hypertension. Counseling and structured training courses could help to effectively lower blood pressure.25 Patients should receive education on lifestyle modifications including:
- weight reduction
- physical activity
- moderate alcohol consumption
- decreased sodium consumption
- implementation of the Dietary Approaches to Stop Hypertension (DASH) or Mediterranean diets.15
Maintaining a normal body weight is ideal, but weight reduction of 10 lb can reduce blood pressure in overweight patients. The DASH diet, consisting of fruits, vegetables, low-fat dairy products, high calcium and potassium intake, and reduced saturated and total fat intake can decrease systolic blood pressure from 8 to 14 mm Hg. Reduction of sodium intake to ≤2,400 mg/d can reduce systolic blood pressure from 2 to 8 mm Hg. Regular aerobic exercise of 30 minutes a day most days of the week can reduce systolic blood pressure up to 9 mm Hg. Patients also should be encouraged to quit smoking. Patients who implement ≥2 these modifications get better results.
Antihypertensive medications. Patients who do not reach their goals with lifestyle measures alone should receive antihypertensive medications. Most patients will require ≥2 agents to control their blood pressure. Clinical trials show that some patient subgroups have better outcomes with different first-line agents.
For example, in non-African American patients, thiazide diuretics, calcium channel blockers, angiotensin receptor blockers, and angiotensin-converting enzyme inhibitors are first-line treatments (Table 3). For African American patients without CKD, first-line treatments should be thiazide diuretics and calcium channel blockers, because angiotensin-converting enzyme inhibitors and angiotensin receptor blockers do not reduce cardiovascular events as effectively. African American patients with CKD and proteinuria, however, benefit from angiotensin-converting enzyme inhibitors or angiotensin receptor blockers and are preferred first-line agents. However, blood pressure control is a more important factor in improving outcomes than the choice of medication.
Psychiatrists’ role. Psychiatrists should aim to collaborate with the primary care provider when treating hypertension. However, when integrative care is not possible, they should start a first-line medication with follow-up in 1 month or sooner for patients with severe hypertension (>160/100 mm Hg) or significant comorbidities (eg, CKD, congestive heart failure, coronary disease). Patients with blood pressure >160/100 mm Hg often are started on a thiazide diuretic with one other medication because a single agent usually does not achieve goal blood pressure. Patients with CKD need close monitoring of potassium and creatinine when starting angiotensin-converting enzyme inhibitor or angiotensin receptor blocker therapy, usually within 1 to 2 days of starting or adjusting their medication. Adjust or add medication dosages monthly until blood pressure goals are reached.
A general internist, cardiologist, or nephrologist who has expertise in managing complex cases should oversee care of a psychiatric patient in any of the following scenarios:
- suspected secondary cause of hypertension
- adverse reaction to antihypertensive medications
- complicated comorbid conditions (ie, creatinine >1.8 mg/dL, worsening renal failure, hyperkalemia, heart failure, coronary disease)
- blood pressure >180/120 mm Hg
- requires ≥3 antihypertensive medications.
Summing up
Hypertension is a significant comorbidity in many psychiatric patients, but usually is asymptomatic. Often the psychiatrist or other mental health provider will diagnose hypertension because of their frequent contact with these patients. Once the diagnosis is made, an initial evaluation can direct lifestyle modifications. Patients who continue to have significant elevation of blood pressure should start pharmacotherapy, either by the psychiatrist or by ensuring follow-up with a primary care physician. The psychiatrist may be able to manage cases of essential hypertension, but always must be vigilant for potential drug–disease or drug–drug interactions during treatment. A team-based approach may improve health outcomes in psychiatric patients.
1. Centers for Disease Control and Prevention (CDC). Vital signs: awareness and treatment of uncontrolled hypertension among adults—United States, 2003-2010. MMWR Morb Mortal Wkly Rep. 2012;61:703-709.
2. Mozzafarian D, Benjamin EJ, Go AS, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29-e322.
3. Carroll D, Phillips AC, Gale CR, et al. Generalized anxiety and major depressive disorders, their comorbidity and hypertension in middle-aged men. Psychosom Med. 2010;72(1):16-19.
4. Leboyer M, Soreca I, Scott J, et al. Can bipolar disorder be viewed as a multi-system inflammatory disease? J Affect Disord. 2012;141(1):1-10.
5. Goff DC, Sullivan LM, McEvoy JP, et al. A comparison of ten-year cardiac risk estimates in schizophrenia patients from the CATIE study and matched controls. Schizophr Res. 2005;80(1):45-53.
6. Stein DJ, Aguilar-Gaxiola S, Alonso J, et al. Associations between mental disorders and subsequent onset of hypertension. Gen Hosp Psychiatry. 2014;36(2):142-149.
7. Birkenaes AB, Opjordsmoen S, Brunborg C, et al. The level of cardiovascular risk factors in bipolar disorder equals that of schizophrenia: a comparative study. J Clin Psychiatry. 2007;68(6):917-923.
8. Izzo JL, Black HR, Goodfriend TL. Hypertension primer: the essentials of high blood pressure. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.
9. Osby U, Correia N, Brandt L, et al. Mortality and causes of death in schizophrenia in Stockholm County, Sweden. Schizophr Res. 2000;45(1-2):21-28.
10. Brown S, Inskip H, Barraclough B. Causes of the excess mortality of schizophrenia. Br J Psychiatry. 2000;177:212-217.
11. Auquier P, Lançon C, Rouillon F, et al. Mortality in schizophrenia. Pharmacoepidemiol Drug Saf. 2007;16(12):1308-1312.
12. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
13. Bowis J, Parvanova A, McDaid D, et al. Mental and Physical Health Charter: bridging the gap between mental and physical health. https://www.idf.org/sites/default/files/Mental%2520and%2520Physical%2520Health%2520Charter%2520-%2520FINAL.pdf. Published October 7, 2009. Accessed March 6, 2017.
14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
15. Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289(19):2560-2571.
16. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
17. National Collaborating Centre for Mental Health (UK). Depression in adults with a chronic physical health problem: treatment and Management. Appendix 16: table of drug interactions. http://www.ncbi.nlm.nih.gov/books/NBK82914. Published 2010. Accessed March 6, 2017.
18. Siu AL; U.S. Preventive Services Task Force. Screening for high blood pressure in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2015:163(10):778-786.
19. Wang PS, Bohn RL, Knight E, et al. Noncompliance with antihypertensive medications: the impact of depressive symptoms and psychosocial factors. J Gen Intern Med. 2002;17(7):504-511.
20. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
21. Consoli SM, Lemogne C, Levy A, et al. Physicians’ degree of motivation regarding their perception of hypertension, and blood pressure control. J Hypertens. 2010;28(6):1330-1339.
22. National High Blood Pressure Education Program. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Improving Hypertension Control. Bethesda, MD: U.S. Department of Health and Human Services; 2004:61-64.
23. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520.
24. The SPRINT Research Group; Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373(22):2103-2016.
25. Boulware LE, Daumit GL, Frick KD, et al. An evidence-based review of patient-centered behavioral interventions for hypertension. Am J Prev Med. 2001;21(3):221-232.
Hypertension is a widespread, under-recognized, and undertreated cause of morbidity and mortality in the United States and is associated with several psychiatric illnesses. Left untreated, hypertension can have significant consequences, including increased risk of stroke, coronary heart disease, heart failure, chronic kidney failure, and death. Approximately 70 million adults in the United States have hypertension, but only 60% of them have been diagnosed, and of those only 50% have their blood pressure under control.1 In 2013, 360,000 deaths in the United States were attributed to hypertension.2
Hypertension is associated with major depressive disorder, generalized anxiety disorder, bipolar disorder, and schizophrenia.3-5 Additionally, impulsive eating disorders, substance abuse, anxiety, and depression are associated with a hypertension diagnosis, although patients with panic disorder develop hypertension at a younger age.6 A 2007 study found a 61% prevalence of hypertension in those with bipolar disorder compared with 41% among the general population.7 The strong link between bipolar disorder and hypertension might be because of a common disease mechanism; both are associated with hyperactive cellular calcium signaling and increased platelet intracellular calcium ion concentrations.8
Hypertension not only is common among patients with psychiatric illness, it likely contributes to worse clinical outcomes. Studies across different cultures have found higher mortality rates in individuals with mental illness.9-11 Persons with schizophrenia and other severe mental illnesses may lose ≥25 years of life expectancy, with the primary cause of death being cardiovascular disease, not suicide.12 Patients with depression have a 50% greater risk of cardiovascular disease, which is equivalent to the risk of smoking.13
Schizophrenia is strongly associated with numerous comorbidities and has been linked significantly to an elevated 10-year cardiac risk after controlling for body mass index.5 The high rate of non-treatment of hypertension for patients with schizophrenia (62.4%) is especially concerning.14
Because of the well-documented morbidity and mortality of hypertension and its increased prevalence and undertreatment in the psychiatric population, mental health providers are in an important position to recognize hypertension and evaluate its inherent risks to direct their patients toward proper treatment. This article reviews:
- the signs and symptoms of hypertension
- the mental health provider’s role in the evaluation and diagnosis
- how psychotropic drugs influence blood pressure and drug–drug interactions
- the management of hypertension in psychiatric patients, including strategies for counseling and lifestyle management.
Diagnosing hypertension
Hypertension is defined as a blood pressure >140/90 mm Hg, the average of ≥2 properly measured readings at ≥2 visits in a medical setting.15 The proper equipment, including a well-fitting blood pressure cuff, and technique to measure blood pressure are essential to avoid misdiagnosis. The patient should be at rest for ≥5 minutes, without active pain or emotional distress.
Most cases of hypertension (90% to 95%) are primary, commonly called essential hypertension. However, the differential diagnosis also should consider secondary causes, which may include:
- obesity
- medications
- chronic alcohol use
- methamphetamine or cocaine use
- primary kidney disease
- atherosclerotic renal artery stenosis
- obstructive sleep apnea
- hypothyroidism
- primary hyperaldosteronism
- narrowing of the aorta
- Cushing syndrome
- primary hyperparathyroidism
- polycythemia
- pheochromocytoma.
Medical evaluation. Once the diagnosis of hypertension is made, a medical evaluation is indicated to determine if the patient has end-organ damage from the elevated pressures, such as renal disease or heart disease, to identify other modifiable cardiovascular risk factors, such as hyperlipidemia, and to screen for secondary causes of hypertension. This evaluation includes15:
- a physical exam
- review of medications
- lipid profile
- urinalysis to screen for proteinuria
- serum electrolytes and creatinine
- electrocardiogram to screen for left ventricular hypertrophy or prior infarction
- fasting glucose or hemoglobin A1c to screen for type 2 diabetes mellitus.
Psychotropic drugs. In psychiatric patients, the evaluation must consider the potential impact psychotropic drug effects and drug–drug interactions can have on blood pressure (Table 2). For example, patients taking both diuretics and lithium are at increased risk for dehydration and increased serum lithium levels, which could cause severe neurologic symptoms and renal insufficiency.16 Several antihypertensives when taken with venlafaxine can increase blood pressure, but antihypertensives with α-1 blocking psychotropics can decrease blood pressure. Monoamine oxidase inhibitors can cause hypotension or hypertension with various classes of antihypertensives. Stimulants, such as methylphenidate, atomoxetine, dextroamphetamine, armodafinil, or modafinil, alone or combined with antihypertensives, can cause hypertension.17
Substance abuse, particularly alcohol, methamphetamine, and cocaine, can cause difficulty controlling blood pressure. Patients with refractory hypertension should have a reassessment of substance abuse as a potential cause.
Screening guidelines for mental health providers
For many patients with severe mental illness, visits to their mental health providers might be their only contact with the medical system. Therefore, screening in the mental health settings could detect cases that otherwise would be missed.
Screening recommendations. The U.S. Preventive Services Task Force recommends screening for hypertension in the general population beginning at age 18.18 Adults age 18 to 39 with normal blood pressure (<130/85 mm Hg) and no other risk factors (eg, overweight, obese, or African American) can be screened every 3 years. Those with risk factors or a blood pressure of 130/85 to 139/89 mm Hg and adults age ≥40 should have annual screenings.
Ideally, psychiatrists and other mental health providers should monitor blood pressure at each visit, especially in patients taking psychotropics because of their higher risk for hypertension.
Optimizing treatment. Once the diagnosis of essential hypertension is established, identifying psychiatric comorbidities and the severity of psychiatric symptoms are important to optimize treatment adherence. Patients with increased depressive symptoms are less likely to comply with antihypertensive medication,19 and patients with confirmed depression are 3 times more likely to not adhere to medical treatment recommendations than non-depressed patients.20
Physicians’ attitudes toward hypertension also can affect patients’ compliance and blood pressure control.21 Psychiatrists should be empathetic and motivational toward patients attempting to control their blood pressure. The Seventh Joint National Committee on the Prevention, Detection, Evaluation, and Treatment of High Blood Pressure states, “Motivation improves when patients have positive experiences with, and trust in, the clinician. Empathy builds trust and is a potent motivator.”22
Treatment and management
Treatment of hypertension significantly reduces the risk of stroke, myocardial infarction, renal injury, heart failure, and premature death. Studies show that treatment that reduces systolic blood pressure by 12 mm Hg over 10 years will prevent 1 death for every 11 patients with essential hypertension. In those with concomitant cardiovascular disease or target organ damage, such a reduction would prevent death in 1 of every 9 patients treated.15Blood pressure goals. The 2014 Eighth Joint National Committee Guideline for Management of High Blood Pressure in Adults provides guidance on blood pressure goals depending on patients’ underlying medical history (Figure).23 Based on expert opinion and randomized controlled studies, blood pressure goals for patients without diabetes or chronic kidney disease (CKD)—an estimated or measured glomerular filtration rate (GFR) of ≤60 mL/min/1.73 m2—depend on age: <140/90 mm Hg for age 18 to 59 and <150/90 mm Hg for age ≥60. For patients with diabetes or CKD, the blood pressure goal is <140/90 mm Hg, regardless of age.
However, not all experts agree on these specific blood pressure goals. A major trial (SPRINT) published in 2015 found that intensive blood pressure goals do benefit higher-risk, non-diabetic patients.24 Specifically, the study randomized patients age ≥50 with systolic blood pressure of 130 to 180 mm Hg and increased cardiovascular risk to systolic blood pressure targets of <140 mm Hg (standard) or <120 mm Hg (intensive). Characteristics of increased cardiovascular risk were clinical or subclinical cardiovascular disease other than stroke, CKD with GFR of 20 to 60 mL/min/1.73 m2, age ≥75, or Framingham 10-year coronary heart disease risk score ≥15%. Intensive treatment significantly reduced overall mortality and the rate of acute coronary syndrome, myocardial infarction, heart failure, stroke, or cardiovascular death. However, the results of this study have not been assimilated into any recent guidelines. Therefore, consider a goal of <120 mm Hg for non-diabetic patients age ≥50 with any of these factors.
Lifestyle modifications. Psychiatrists are well equipped to motivate and encourage behavioral modification in patients with hypertension. Counseling and structured training courses could help to effectively lower blood pressure.25 Patients should receive education on lifestyle modifications including:
- weight reduction
- physical activity
- moderate alcohol consumption
- decreased sodium consumption
- implementation of the Dietary Approaches to Stop Hypertension (DASH) or Mediterranean diets.15
Maintaining a normal body weight is ideal, but weight reduction of 10 lb can reduce blood pressure in overweight patients. The DASH diet, consisting of fruits, vegetables, low-fat dairy products, high calcium and potassium intake, and reduced saturated and total fat intake can decrease systolic blood pressure from 8 to 14 mm Hg. Reduction of sodium intake to ≤2,400 mg/d can reduce systolic blood pressure from 2 to 8 mm Hg. Regular aerobic exercise of 30 minutes a day most days of the week can reduce systolic blood pressure up to 9 mm Hg. Patients also should be encouraged to quit smoking. Patients who implement ≥2 these modifications get better results.
Antihypertensive medications. Patients who do not reach their goals with lifestyle measures alone should receive antihypertensive medications. Most patients will require ≥2 agents to control their blood pressure. Clinical trials show that some patient subgroups have better outcomes with different first-line agents.
For example, in non-African American patients, thiazide diuretics, calcium channel blockers, angiotensin receptor blockers, and angiotensin-converting enzyme inhibitors are first-line treatments (Table 3). For African American patients without CKD, first-line treatments should be thiazide diuretics and calcium channel blockers, because angiotensin-converting enzyme inhibitors and angiotensin receptor blockers do not reduce cardiovascular events as effectively. African American patients with CKD and proteinuria, however, benefit from angiotensin-converting enzyme inhibitors or angiotensin receptor blockers and are preferred first-line agents. However, blood pressure control is a more important factor in improving outcomes than the choice of medication.
Psychiatrists’ role. Psychiatrists should aim to collaborate with the primary care provider when treating hypertension. However, when integrative care is not possible, they should start a first-line medication with follow-up in 1 month or sooner for patients with severe hypertension (>160/100 mm Hg) or significant comorbidities (eg, CKD, congestive heart failure, coronary disease). Patients with blood pressure >160/100 mm Hg often are started on a thiazide diuretic with one other medication because a single agent usually does not achieve goal blood pressure. Patients with CKD need close monitoring of potassium and creatinine when starting angiotensin-converting enzyme inhibitor or angiotensin receptor blocker therapy, usually within 1 to 2 days of starting or adjusting their medication. Adjust or add medication dosages monthly until blood pressure goals are reached.
A general internist, cardiologist, or nephrologist who has expertise in managing complex cases should oversee care of a psychiatric patient in any of the following scenarios:
- suspected secondary cause of hypertension
- adverse reaction to antihypertensive medications
- complicated comorbid conditions (ie, creatinine >1.8 mg/dL, worsening renal failure, hyperkalemia, heart failure, coronary disease)
- blood pressure >180/120 mm Hg
- requires ≥3 antihypertensive medications.
Summing up
Hypertension is a significant comorbidity in many psychiatric patients, but usually is asymptomatic. Often the psychiatrist or other mental health provider will diagnose hypertension because of their frequent contact with these patients. Once the diagnosis is made, an initial evaluation can direct lifestyle modifications. Patients who continue to have significant elevation of blood pressure should start pharmacotherapy, either by the psychiatrist or by ensuring follow-up with a primary care physician. The psychiatrist may be able to manage cases of essential hypertension, but always must be vigilant for potential drug–disease or drug–drug interactions during treatment. A team-based approach may improve health outcomes in psychiatric patients.
Hypertension is a widespread, under-recognized, and undertreated cause of morbidity and mortality in the United States and is associated with several psychiatric illnesses. Left untreated, hypertension can have significant consequences, including increased risk of stroke, coronary heart disease, heart failure, chronic kidney failure, and death. Approximately 70 million adults in the United States have hypertension, but only 60% of them have been diagnosed, and of those only 50% have their blood pressure under control.1 In 2013, 360,000 deaths in the United States were attributed to hypertension.2
Hypertension is associated with major depressive disorder, generalized anxiety disorder, bipolar disorder, and schizophrenia.3-5 Additionally, impulsive eating disorders, substance abuse, anxiety, and depression are associated with a hypertension diagnosis, although patients with panic disorder develop hypertension at a younger age.6 A 2007 study found a 61% prevalence of hypertension in those with bipolar disorder compared with 41% among the general population.7 The strong link between bipolar disorder and hypertension might be because of a common disease mechanism; both are associated with hyperactive cellular calcium signaling and increased platelet intracellular calcium ion concentrations.8
Hypertension not only is common among patients with psychiatric illness, it likely contributes to worse clinical outcomes. Studies across different cultures have found higher mortality rates in individuals with mental illness.9-11 Persons with schizophrenia and other severe mental illnesses may lose ≥25 years of life expectancy, with the primary cause of death being cardiovascular disease, not suicide.12 Patients with depression have a 50% greater risk of cardiovascular disease, which is equivalent to the risk of smoking.13
Schizophrenia is strongly associated with numerous comorbidities and has been linked significantly to an elevated 10-year cardiac risk after controlling for body mass index.5 The high rate of non-treatment of hypertension for patients with schizophrenia (62.4%) is especially concerning.14
Because of the well-documented morbidity and mortality of hypertension and its increased prevalence and undertreatment in the psychiatric population, mental health providers are in an important position to recognize hypertension and evaluate its inherent risks to direct their patients toward proper treatment. This article reviews:
- the signs and symptoms of hypertension
- the mental health provider’s role in the evaluation and diagnosis
- how psychotropic drugs influence blood pressure and drug–drug interactions
- the management of hypertension in psychiatric patients, including strategies for counseling and lifestyle management.
Diagnosing hypertension
Hypertension is defined as a blood pressure >140/90 mm Hg, the average of ≥2 properly measured readings at ≥2 visits in a medical setting.15 The proper equipment, including a well-fitting blood pressure cuff, and technique to measure blood pressure are essential to avoid misdiagnosis. The patient should be at rest for ≥5 minutes, without active pain or emotional distress.
Most cases of hypertension (90% to 95%) are primary, commonly called essential hypertension. However, the differential diagnosis also should consider secondary causes, which may include:
- obesity
- medications
- chronic alcohol use
- methamphetamine or cocaine use
- primary kidney disease
- atherosclerotic renal artery stenosis
- obstructive sleep apnea
- hypothyroidism
- primary hyperaldosteronism
- narrowing of the aorta
- Cushing syndrome
- primary hyperparathyroidism
- polycythemia
- pheochromocytoma.
Medical evaluation. Once the diagnosis of hypertension is made, a medical evaluation is indicated to determine if the patient has end-organ damage from the elevated pressures, such as renal disease or heart disease, to identify other modifiable cardiovascular risk factors, such as hyperlipidemia, and to screen for secondary causes of hypertension. This evaluation includes15:
- a physical exam
- review of medications
- lipid profile
- urinalysis to screen for proteinuria
- serum electrolytes and creatinine
- electrocardiogram to screen for left ventricular hypertrophy or prior infarction
- fasting glucose or hemoglobin A1c to screen for type 2 diabetes mellitus.
Psychotropic drugs. In psychiatric patients, the evaluation must consider the potential impact psychotropic drug effects and drug–drug interactions can have on blood pressure (Table 2). For example, patients taking both diuretics and lithium are at increased risk for dehydration and increased serum lithium levels, which could cause severe neurologic symptoms and renal insufficiency.16 Several antihypertensives when taken with venlafaxine can increase blood pressure, but antihypertensives with α-1 blocking psychotropics can decrease blood pressure. Monoamine oxidase inhibitors can cause hypotension or hypertension with various classes of antihypertensives. Stimulants, such as methylphenidate, atomoxetine, dextroamphetamine, armodafinil, or modafinil, alone or combined with antihypertensives, can cause hypertension.17
Substance abuse, particularly alcohol, methamphetamine, and cocaine, can cause difficulty controlling blood pressure. Patients with refractory hypertension should have a reassessment of substance abuse as a potential cause.
Screening guidelines for mental health providers
For many patients with severe mental illness, visits to their mental health providers might be their only contact with the medical system. Therefore, screening in the mental health settings could detect cases that otherwise would be missed.
Screening recommendations. The U.S. Preventive Services Task Force recommends screening for hypertension in the general population beginning at age 18.18 Adults age 18 to 39 with normal blood pressure (<130/85 mm Hg) and no other risk factors (eg, overweight, obese, or African American) can be screened every 3 years. Those with risk factors or a blood pressure of 130/85 to 139/89 mm Hg and adults age ≥40 should have annual screenings.
Ideally, psychiatrists and other mental health providers should monitor blood pressure at each visit, especially in patients taking psychotropics because of their higher risk for hypertension.
Optimizing treatment. Once the diagnosis of essential hypertension is established, identifying psychiatric comorbidities and the severity of psychiatric symptoms are important to optimize treatment adherence. Patients with increased depressive symptoms are less likely to comply with antihypertensive medication,19 and patients with confirmed depression are 3 times more likely to not adhere to medical treatment recommendations than non-depressed patients.20
Physicians’ attitudes toward hypertension also can affect patients’ compliance and blood pressure control.21 Psychiatrists should be empathetic and motivational toward patients attempting to control their blood pressure. The Seventh Joint National Committee on the Prevention, Detection, Evaluation, and Treatment of High Blood Pressure states, “Motivation improves when patients have positive experiences with, and trust in, the clinician. Empathy builds trust and is a potent motivator.”22
Treatment and management
Treatment of hypertension significantly reduces the risk of stroke, myocardial infarction, renal injury, heart failure, and premature death. Studies show that treatment that reduces systolic blood pressure by 12 mm Hg over 10 years will prevent 1 death for every 11 patients with essential hypertension. In those with concomitant cardiovascular disease or target organ damage, such a reduction would prevent death in 1 of every 9 patients treated.15Blood pressure goals. The 2014 Eighth Joint National Committee Guideline for Management of High Blood Pressure in Adults provides guidance on blood pressure goals depending on patients’ underlying medical history (Figure).23 Based on expert opinion and randomized controlled studies, blood pressure goals for patients without diabetes or chronic kidney disease (CKD)—an estimated or measured glomerular filtration rate (GFR) of ≤60 mL/min/1.73 m2—depend on age: <140/90 mm Hg for age 18 to 59 and <150/90 mm Hg for age ≥60. For patients with diabetes or CKD, the blood pressure goal is <140/90 mm Hg, regardless of age.
However, not all experts agree on these specific blood pressure goals. A major trial (SPRINT) published in 2015 found that intensive blood pressure goals do benefit higher-risk, non-diabetic patients.24 Specifically, the study randomized patients age ≥50 with systolic blood pressure of 130 to 180 mm Hg and increased cardiovascular risk to systolic blood pressure targets of <140 mm Hg (standard) or <120 mm Hg (intensive). Characteristics of increased cardiovascular risk were clinical or subclinical cardiovascular disease other than stroke, CKD with GFR of 20 to 60 mL/min/1.73 m2, age ≥75, or Framingham 10-year coronary heart disease risk score ≥15%. Intensive treatment significantly reduced overall mortality and the rate of acute coronary syndrome, myocardial infarction, heart failure, stroke, or cardiovascular death. However, the results of this study have not been assimilated into any recent guidelines. Therefore, consider a goal of <120 mm Hg for non-diabetic patients age ≥50 with any of these factors.
Lifestyle modifications. Psychiatrists are well equipped to motivate and encourage behavioral modification in patients with hypertension. Counseling and structured training courses could help to effectively lower blood pressure.25 Patients should receive education on lifestyle modifications including:
- weight reduction
- physical activity
- moderate alcohol consumption
- decreased sodium consumption
- implementation of the Dietary Approaches to Stop Hypertension (DASH) or Mediterranean diets.15
Maintaining a normal body weight is ideal, but weight reduction of 10 lb can reduce blood pressure in overweight patients. The DASH diet, consisting of fruits, vegetables, low-fat dairy products, high calcium and potassium intake, and reduced saturated and total fat intake can decrease systolic blood pressure from 8 to 14 mm Hg. Reduction of sodium intake to ≤2,400 mg/d can reduce systolic blood pressure from 2 to 8 mm Hg. Regular aerobic exercise of 30 minutes a day most days of the week can reduce systolic blood pressure up to 9 mm Hg. Patients also should be encouraged to quit smoking. Patients who implement ≥2 these modifications get better results.
Antihypertensive medications. Patients who do not reach their goals with lifestyle measures alone should receive antihypertensive medications. Most patients will require ≥2 agents to control their blood pressure. Clinical trials show that some patient subgroups have better outcomes with different first-line agents.
For example, in non-African American patients, thiazide diuretics, calcium channel blockers, angiotensin receptor blockers, and angiotensin-converting enzyme inhibitors are first-line treatments (Table 3). For African American patients without CKD, first-line treatments should be thiazide diuretics and calcium channel blockers, because angiotensin-converting enzyme inhibitors and angiotensin receptor blockers do not reduce cardiovascular events as effectively. African American patients with CKD and proteinuria, however, benefit from angiotensin-converting enzyme inhibitors or angiotensin receptor blockers and are preferred first-line agents. However, blood pressure control is a more important factor in improving outcomes than the choice of medication.
Psychiatrists’ role. Psychiatrists should aim to collaborate with the primary care provider when treating hypertension. However, when integrative care is not possible, they should start a first-line medication with follow-up in 1 month or sooner for patients with severe hypertension (>160/100 mm Hg) or significant comorbidities (eg, CKD, congestive heart failure, coronary disease). Patients with blood pressure >160/100 mm Hg often are started on a thiazide diuretic with one other medication because a single agent usually does not achieve goal blood pressure. Patients with CKD need close monitoring of potassium and creatinine when starting angiotensin-converting enzyme inhibitor or angiotensin receptor blocker therapy, usually within 1 to 2 days of starting or adjusting their medication. Adjust or add medication dosages monthly until blood pressure goals are reached.
A general internist, cardiologist, or nephrologist who has expertise in managing complex cases should oversee care of a psychiatric patient in any of the following scenarios:
- suspected secondary cause of hypertension
- adverse reaction to antihypertensive medications
- complicated comorbid conditions (ie, creatinine >1.8 mg/dL, worsening renal failure, hyperkalemia, heart failure, coronary disease)
- blood pressure >180/120 mm Hg
- requires ≥3 antihypertensive medications.
Summing up
Hypertension is a significant comorbidity in many psychiatric patients, but usually is asymptomatic. Often the psychiatrist or other mental health provider will diagnose hypertension because of their frequent contact with these patients. Once the diagnosis is made, an initial evaluation can direct lifestyle modifications. Patients who continue to have significant elevation of blood pressure should start pharmacotherapy, either by the psychiatrist or by ensuring follow-up with a primary care physician. The psychiatrist may be able to manage cases of essential hypertension, but always must be vigilant for potential drug–disease or drug–drug interactions during treatment. A team-based approach may improve health outcomes in psychiatric patients.
1. Centers for Disease Control and Prevention (CDC). Vital signs: awareness and treatment of uncontrolled hypertension among adults—United States, 2003-2010. MMWR Morb Mortal Wkly Rep. 2012;61:703-709.
2. Mozzafarian D, Benjamin EJ, Go AS, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29-e322.
3. Carroll D, Phillips AC, Gale CR, et al. Generalized anxiety and major depressive disorders, their comorbidity and hypertension in middle-aged men. Psychosom Med. 2010;72(1):16-19.
4. Leboyer M, Soreca I, Scott J, et al. Can bipolar disorder be viewed as a multi-system inflammatory disease? J Affect Disord. 2012;141(1):1-10.
5. Goff DC, Sullivan LM, McEvoy JP, et al. A comparison of ten-year cardiac risk estimates in schizophrenia patients from the CATIE study and matched controls. Schizophr Res. 2005;80(1):45-53.
6. Stein DJ, Aguilar-Gaxiola S, Alonso J, et al. Associations between mental disorders and subsequent onset of hypertension. Gen Hosp Psychiatry. 2014;36(2):142-149.
7. Birkenaes AB, Opjordsmoen S, Brunborg C, et al. The level of cardiovascular risk factors in bipolar disorder equals that of schizophrenia: a comparative study. J Clin Psychiatry. 2007;68(6):917-923.
8. Izzo JL, Black HR, Goodfriend TL. Hypertension primer: the essentials of high blood pressure. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.
9. Osby U, Correia N, Brandt L, et al. Mortality and causes of death in schizophrenia in Stockholm County, Sweden. Schizophr Res. 2000;45(1-2):21-28.
10. Brown S, Inskip H, Barraclough B. Causes of the excess mortality of schizophrenia. Br J Psychiatry. 2000;177:212-217.
11. Auquier P, Lançon C, Rouillon F, et al. Mortality in schizophrenia. Pharmacoepidemiol Drug Saf. 2007;16(12):1308-1312.
12. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
13. Bowis J, Parvanova A, McDaid D, et al. Mental and Physical Health Charter: bridging the gap between mental and physical health. https://www.idf.org/sites/default/files/Mental%2520and%2520Physical%2520Health%2520Charter%2520-%2520FINAL.pdf. Published October 7, 2009. Accessed March 6, 2017.
14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
15. Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289(19):2560-2571.
16. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
17. National Collaborating Centre for Mental Health (UK). Depression in adults with a chronic physical health problem: treatment and Management. Appendix 16: table of drug interactions. http://www.ncbi.nlm.nih.gov/books/NBK82914. Published 2010. Accessed March 6, 2017.
18. Siu AL; U.S. Preventive Services Task Force. Screening for high blood pressure in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2015:163(10):778-786.
19. Wang PS, Bohn RL, Knight E, et al. Noncompliance with antihypertensive medications: the impact of depressive symptoms and psychosocial factors. J Gen Intern Med. 2002;17(7):504-511.
20. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
21. Consoli SM, Lemogne C, Levy A, et al. Physicians’ degree of motivation regarding their perception of hypertension, and blood pressure control. J Hypertens. 2010;28(6):1330-1339.
22. National High Blood Pressure Education Program. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Improving Hypertension Control. Bethesda, MD: U.S. Department of Health and Human Services; 2004:61-64.
23. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520.
24. The SPRINT Research Group; Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373(22):2103-2016.
25. Boulware LE, Daumit GL, Frick KD, et al. An evidence-based review of patient-centered behavioral interventions for hypertension. Am J Prev Med. 2001;21(3):221-232.
1. Centers for Disease Control and Prevention (CDC). Vital signs: awareness and treatment of uncontrolled hypertension among adults—United States, 2003-2010. MMWR Morb Mortal Wkly Rep. 2012;61:703-709.
2. Mozzafarian D, Benjamin EJ, Go AS, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29-e322.
3. Carroll D, Phillips AC, Gale CR, et al. Generalized anxiety and major depressive disorders, their comorbidity and hypertension in middle-aged men. Psychosom Med. 2010;72(1):16-19.
4. Leboyer M, Soreca I, Scott J, et al. Can bipolar disorder be viewed as a multi-system inflammatory disease? J Affect Disord. 2012;141(1):1-10.
5. Goff DC, Sullivan LM, McEvoy JP, et al. A comparison of ten-year cardiac risk estimates in schizophrenia patients from the CATIE study and matched controls. Schizophr Res. 2005;80(1):45-53.
6. Stein DJ, Aguilar-Gaxiola S, Alonso J, et al. Associations between mental disorders and subsequent onset of hypertension. Gen Hosp Psychiatry. 2014;36(2):142-149.
7. Birkenaes AB, Opjordsmoen S, Brunborg C, et al. The level of cardiovascular risk factors in bipolar disorder equals that of schizophrenia: a comparative study. J Clin Psychiatry. 2007;68(6):917-923.
8. Izzo JL, Black HR, Goodfriend TL. Hypertension primer: the essentials of high blood pressure. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.
9. Osby U, Correia N, Brandt L, et al. Mortality and causes of death in schizophrenia in Stockholm County, Sweden. Schizophr Res. 2000;45(1-2):21-28.
10. Brown S, Inskip H, Barraclough B. Causes of the excess mortality of schizophrenia. Br J Psychiatry. 2000;177:212-217.
11. Auquier P, Lançon C, Rouillon F, et al. Mortality in schizophrenia. Pharmacoepidemiol Drug Saf. 2007;16(12):1308-1312.
12. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
13. Bowis J, Parvanova A, McDaid D, et al. Mental and Physical Health Charter: bridging the gap between mental and physical health. https://www.idf.org/sites/default/files/Mental%2520and%2520Physical%2520Health%2520Charter%2520-%2520FINAL.pdf. Published October 7, 2009. Accessed March 6, 2017.
14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
15. Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289(19):2560-2571.
16. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
17. National Collaborating Centre for Mental Health (UK). Depression in adults with a chronic physical health problem: treatment and Management. Appendix 16: table of drug interactions. http://www.ncbi.nlm.nih.gov/books/NBK82914. Published 2010. Accessed March 6, 2017.
18. Siu AL; U.S. Preventive Services Task Force. Screening for high blood pressure in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2015:163(10):778-786.
19. Wang PS, Bohn RL, Knight E, et al. Noncompliance with antihypertensive medications: the impact of depressive symptoms and psychosocial factors. J Gen Intern Med. 2002;17(7):504-511.
20. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
21. Consoli SM, Lemogne C, Levy A, et al. Physicians’ degree of motivation regarding their perception of hypertension, and blood pressure control. J Hypertens. 2010;28(6):1330-1339.
22. National High Blood Pressure Education Program. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Improving Hypertension Control. Bethesda, MD: U.S. Department of Health and Human Services; 2004:61-64.
23. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520.
24. The SPRINT Research Group; Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373(22):2103-2016.
25. Boulware LE, Daumit GL, Frick KD, et al. An evidence-based review of patient-centered behavioral interventions for hypertension. Am J Prev Med. 2001;21(3):221-232.
