Opinion
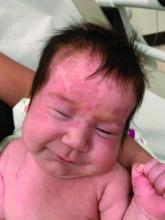
An otherwise healthy 1-month-old female presents with lesions on the face, scalp, and chest
A 1-month-old female infant presented to the dermatology clinic with a 3-week history of recurrent skin lesions on the scalp, face, and chest.
Opinion
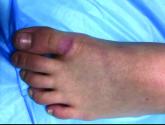
Pediatric Dermatology Consult - June 2018
If unusual features make you suspect tinea, leprosy, mycosis fungoides, or other annular lesions, then biopsy can reveal the correct diagnosis.
Opinion
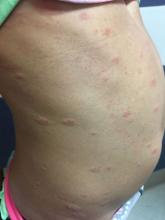
Pediatric Dermatology Consult - February 2018
What benign, self-limited condition is heralded with the eruption of a large, scaly, pink- to salmon-colored lesion?
Opinion
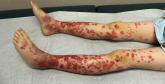
Pediatric Dermatology Consult - November 2017
What presents with palpable purpura, abdominal pain, arthritis, and hematuria 1-2 weeks after an upper respiratory infection?