User login
A Strong Diagnosis of Weakness
A 52-year-old man presented with bilateral weakness in all extremities. He noted the gradual onset of progressive muscle weakness 6 months prior to presentation. He reported generalized fatigue and difficulty with climbing stairs and carrying heavy objects.
Initial considerations of chronic weakness and fatigue are myopathy, polyneuropathy, medications, malignancy, endocrinopathies, human immunodeficiency virus (HIV), neuromuscular junction dysfunction, and central nervous system (CNS) disorders, such as amyotrophic lateral sclerosis (ALS) or multiple sclerosis (MS). Symmetrical muscle involvement and proximal weakness make myopathy most likely. Polyneuropathy, such as chronic inflammatory demyelinating polyneuropathy (CIDP), is less likely but still possible given the slowly progressive course. The use of medications that can cause myopathy should be explored, including colchicine, steroids, and statins. Gathering further history should focus on risk factors for HIV, as well as alcohol and illicit drug use. Malignancy can cause paraneoplastic myopathy. The review of systems should include symptoms of endocrinopathies, such as thyrotoxicosis and hypothyroidism. Fluctuations in weakness and dysphagia or ocular symptoms would suggest myasthenia gravis (MG). The time course and symmetrical weakness make a central disorder, such as ALS or MS, unlikely.
His past medical history was notable for pulmonary tuberculosis diagnosed at the age of 6 years, which was treated with hospitalization and an unknown medication regimen. He was not taking medications prior to this admission. His family history was significant for diabetes mellitus in both parents. He denied sick contacts. He was sexually active with his wife. He denied the use of tobacco and illicit drugs but endorsed alcohol consumption on a daily basis over the last 32 years. He reported no fluctuation in his symptoms, muscle or joint pains, rash, fevers, chills, diaphoresis, chest pain, dyspnea, abdominal pain, diarrhea, paresthesias, weight loss, or night sweats. He had never had a colonoscopy.
Painless progressive weakness of the limbs without sensory deficit is typical of a myopathy. Though CIDP can present with only motor weakness, the majority of patients have sensory symptoms, making this less likely. Although chronic alcohol abuse can cause myopathy, it seems less likely because other neurologic complications, such as sensory polyneuropathy or ataxia, would be expected. A review of systems does not suggest a thyroid disorder or malignancy, although this does not preclude an evaluation for both. The absence of fluctuations in weakness argues against MG. Though ALS, MG, MS, and CIDP are less likely, a neurologic exam is crucial in excluding them. The hallmark of ALS is upper motor neuron (UMN) and lower motor neuron signs in the absence of sensory symptoms and signs, while global hyporeflexia would be expected in CIDP, and fatigability on repeated power testing would be expected in MG. Neurologic findings disseminated in space (neuro-anatomically) would be expected in MS.
On physical examination, the patient had a temperature of 36.9°C, heart rate of 70 beats per minute, and regular respiratory rate of 10 breaths per minute, blood pressure 130/80 mmHg, and oxygen saturation 98% while breathing ambient air. Auscultation of the heart and lungs revealed normal findings. The abdomen was soft, nontender, and without masses or organomegaly. Neurologic examination disclosed bilateral symmetric upper and lower extremity weakness with positive Gower sign. Muscle strength scores of the bilateral biceps brachii, iliopsoas, and digitis extensor were between 4 and 5 without fatigability. Grasping power was impaired. Deep tendon reflexes were preserved, and there were no UMN signs. There was no tenderness to palpation in any muscle groups. Sensory testing was normal. Skin and lymph examinations were without abnormality. The rest of the physical examination was unremarkable.
Gower sign, characteristic of but not specific to muscular dystrophy, indicates proximal muscle weakness of lower extremities, wherein hands and arms are used to walk up the body into an upright position. The exam also reveals distal weakness as shown by reduced hand grasp. Symmetrical proximal weakness of all extremities without sensory deficits suggests a myopathic process, albeit one with some distal involvement. The absence of UMN signs argues against ALS, lack of fatigability argues against MG, and the absence of CNS or sensory deficits argues against MS.
Because myopathy is most likely, the next step would be to determine if this is an idiopathic inflammatory myopathy, such as polymyositis (PM) or dermatomyositis (DM), secondary inflammatory myopathy, or noninflammatory myopathy due to endocrinopathies. The time course is consistent with an inflammatory myopathy, such as PM or DM. Inclusion body myositis (IBM), another inflammatory myopathy, presents much more insidiously over years and tends to be asymmetric compared to PM. The absence of myalgia, arthralgia, rash, and gastrointestinal symptoms makes myopathy as a component of a connective tissue disease, such as systemic lupus erythematosus, or a mixed connective tissue disease unlikely. The next steps would be laboratory testing of muscle enzymes, complete blood count, biochemical profile, and antinuclear antibody (ANA).
Laboratory studies revealed a white blood cell count of 4460/mm3 with normal differential, hemoglobin 12.5 g/dL, and platelet count 345,000/mm3. Creatinine was 0.87 mg/dL, aspartate aminotransferase 61 IU/mL, alanine aminotransferase 45 IU/mL, and creatine kinase (CK) 529 U/L (normal range, 38-174 U/L). Other liver function enzymes were normal. Biochemistry studies disclosed normal sodium, potassium, glucose, calcium, and magnesium levels. Dipstick urinalysis revealed blood and protein, and the microscopic examination of urinary sediment was unremarkable without the presence of erythrocytes. Twenty-four-hour creatinine clearance was 106 mL/min (normal range, 97-137 mL/min). Chest radiography was unrevealing.
The modest increase in CK, evidence of myoglobinuria, and proteinuria can all occur with an inflammatory or metabolic myopathy. The combination of proximal and distal weakness, coupled with only a modestly elevated CK, makes IBM more likely than PM, as PM usually presents with proximal weakness and much higher CK values. Normal skin examination makes DM less likely, as skin manifestations are generally found at time of presentation. The onset of symptoms after age 50 and the patient being male also favor IBM, though a longer time course would be expected. Definitively distinguishing IBM from PM is important because treatment and prognosis differ.
Thyroid function and HIV testing should be obtained. ANA, more common in PM than in IBM, should be checked because these myopathies can be associated with other autoimmune diseases. Imaging is generally not essential, although magnetic resonance imaging (MRI) of the thighs may help to differentiate IBM from PM. Electromyography (EMG) should be done to determine the pattern of myopathy and select muscle biopsy sites.
Additional testing revealed a normal thyroid stimulating hormone level. HIV and ANA were negative. Serum aldolase level was 19 IU/L (normal range, 2.7-5.9 IU/L), myoglobin 277 ng/mL (normal range, 28-72 ng/mL), lactate dehydrogenase 416 IU/mL (normal range, 119-229 IU/mL), and C-reactive protein 0.32 mg/dL. An EMG revealed mild myogenic changes in all extremities. An MRI of the left brachial muscle revealed multiple scattered high-signal lesions.
The EMG and MRI findings are consistent with an inflammatory myopathy. The modest elevation in muscle enzymes and negative ANA are more consistent with IBM since most patients with PM or DM are ANA positive. Muscle biopsy can be very helpful in establishing the etiology of myopathy.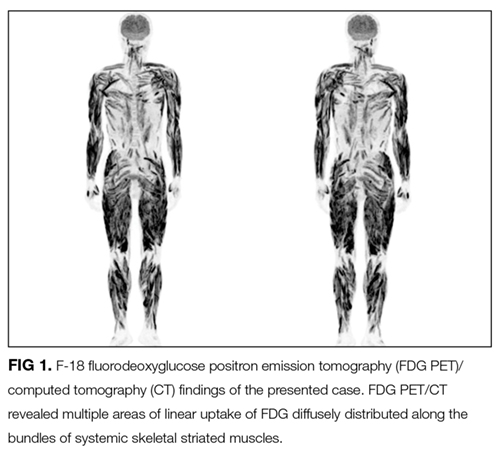
Malignancy is associated with DM and PM in about 9% and 4% of patients, respectively. The common cancers associated with these conditions are adenocarcinomas of the ovary, cervix, lung, pancreas, and stomach. Most cancers are diagnosed around the time of myositis diagnosis, although they can precede or follow by years. Idiopathic IBM is not associated with cancer.
Open surgical muscle biopsy of the left biceps brachii was performed. Light microscopic examination disclosed interstitial edema and noncaseating granulomas. Immunostaining revealed an increase in the number of cluster of differentiation (CD) 4+ T cells. Caseating granulomas and Langhans giant cells were not present (Figure 3).

Tuberculin reaction and interferon-γ-release assay were negative. Staining for AFB and fungi was negative. ANCA, rheumatoid factor (RF), anti-Ro/SSA, anti-La/SSB, anti-Sm, anti-RNP, and anti-Jo-1 were all negative or unremarkable. Serum angiotensin converting enzyme (ACE) level was 155.6 U/L (normal range, 7-25 U/L). Twenty-four-hour urine analysis revealed calcium excretion of 517.7 mg/day (normal range, 58-450 mg/day), β2-microglobulin 69,627 ug/day (normal range, <254 ug/day), and N-acetyl-D-glucosamine 95.3 U/day (normal range, <5.1 U/day) with a normal creatinine clearance. Serum intact parathyroid hormone level (PTH) was 5 pg/mL (normal range, 10-65 pg/mL), and 25-hydroxyvitamin D level was 51.1 ng/mL (normal range, 30-80 ng/mL). A CT of the thorax revealed a small ground-glass density lesion in the left lower lobe but no hilar or mediastinal lymphadenopathy.
Negative ANCA, RF, and autoantibodies exclude systemic vasculitis and connective tissue disease as causes of GM. Hypercalciuria is suggestive of granulomatous production of calcitriol, which, in turn, suppresses PTH. Hypercalcemia is not common in patients with sarcoidosis, but hypercalciuria occurs frequently. Serum ACE is a marker associated with sarcoidosis, but its diagnostic and prognostic utility is unclear.
Though there is a concern for sarcoidosis, this diagnosis can only be confidently made by finding noncaseating granulomas on a background of compatible clinical and radiologic findings after alternate possible etiologies are excluded. The chest CT reveals a small ground-glass density lesion without hilar adenopathy. These findings, though not incompatible, are not typical for pulmonary sarcoidosis. Therefore, finding noncaeseating granulomas in a second organ system would point toward systemic sarcoidosis as a unifying diagnosis. Bronchoscopy with bronchoalveolar lavage (BAL) and transbronchial biopsy has a reasonable yield even in the absence of hilar adenopathy or typical parenchymal findings. A CD4/CD8 T-cell ratio of 2 or more on BAL provides supportive evidence for sarcoidosis.
It is reasonable to start empiric glucocorticoids for GM given that the AFB and fungal stains on histopathology are negative and that there is no evidence of lymphoma.
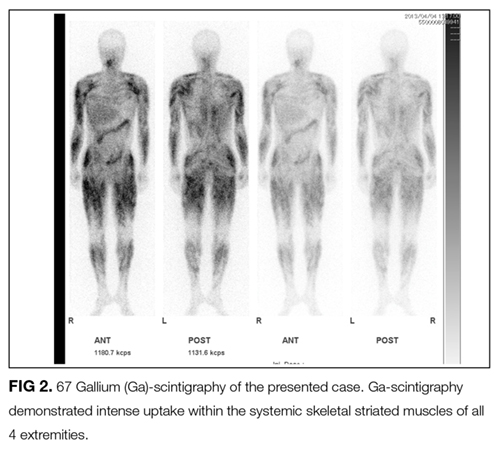
The CD4/CD8 T-cell ratio greater than 2, combined with the absence of neutrophils and eosinophils on BAL, is helpful in distinguishing sarcoidosis from other pulmonary diseases. This patient’s inflammatory myopathy was revealed to be a rare initial manifestation of systemic sarcoidosis.
DISCUSSION
Weakness is a common symptom of muscle disorders such as myopathies and muscular dystrophy. Idiopathic inflammatory myopathies include PM, DM, and others.1,2 These usually present with proximal-dominant muscle weakness, decreased endurance, and muscle inflammation. A diagnosis is made according to symptoms in combination with diagnostic examinations, including elevated serum CK levels, abnormal EMG findings, and histopathology of skeletal muscle biopsy specimens.
Sarcoidosis, a multisystem disorder of unknown etiology, is characterized histopathologically by noncaseating granulomas in affected organs.3 It typically affects young adults, with incidence peaking at 20 to 39 years of age. Although any organ may be involved, the disorder usually presents with 1 or more common abnormalities, including bilateral hilar lymphadenopathy, lung lesions, and skin and eye involvement. Musculoskeletal involvement is less common. It is estimated that skeletal muscle is involved in 50% to 80% of patients with sarcoidosis but is rarely symptomatic (0.5% to 2.5%).4-6
In this patient, weakness was distributed in both proximal and distal muscles, yet proximal weakness is the most characteristic feature in PM and DM. Therefore, sarcoidosis should be considered in the differential diagnosis of idiopathic inflammatory myopathies, especially when weakness accompanies abnormalities in other organs typically affected by sarcoidosis.
Myoglobinuria often is observed in rhabdomyolysis and inflammatory myopathies, conditions that produce high levels of serum CK and myoglobin. Myoglobinuria, often accompanied by the elevation of urinary β2-microglobulin and N-acetyl-D-glucosamine levels, can induce tubulointerstitial damage, which leads to acute kidney injury. In this case, however, these abnormal kidney findings were observed without high levels of serum CK or myoglobin. This suggests the potential for other causes of tubulointerstitial damage, such as granulomatous interstitial nephritis in renal sarcoidosis.3
Another characteristic abnormality was the elevation of urinary calcium excretion, which indicated an underlying granulomatous disorder, such as mycobacterial infection, granulomatosis with polyangiitis, or sarcoidosis. In sarcoidosis, hypercalciuria occurs in 40% of patients, hypercalcemia in 11%, and renal calculi in 10%.3,7 Hypercalciuria, for this patient, was important in arriving at the correct diagnosis after the gallium scan was obtained given the dearth of other typical features of sarcoidosis.
Although muscle biopsy is essential, imaging studies for idiopathic inflammatory myopathy are considered useful tools to narrow the differential diagnosis. The use of MRI of the skeletal muscle is helpful to both identify an adequate muscle for biopsy and demonstrate the pattern of affected muscles beyond clinical appearance, which aids in excluding, for example, muscular dystrophies.8,9
FDG PET/CT is a very sensitive imaging modality used to detect neoplastic lesions and has been widely used to screen for occult neoplasms and detect metastases.10-12 It is also useful for detecting inflammation in patients with osteomyelitis, metastatic infectious diseases, rheumatoid arthritis, vasculitis, inflammatory bowel diseases, fever of unknown origin, and sarcoidosis.11,12 In PM and DM, however, the sensitivity of FDG PET/CT for detection of myositis is reportedly lower than that of EMG and MRI.13 Similarly, gallium scintigraphy is usually performed to examine the disease activity of interstitial pneumonia or to detect malignancy. Previous literature and this case show that the striking images of gallium scintigraphy and FDG PET/CT have utility, not only for detection of sarcoid myopathy but also for the evaluation of treatment efficacy.14-17 Characteristic imaging findings on FDG PET/CT have been described as a “tiger man” appearance.17
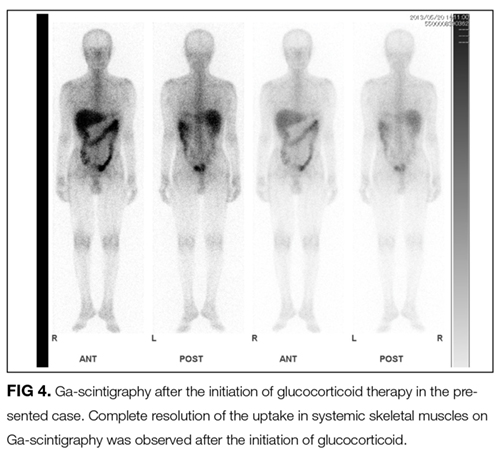
For the treatment of sarcoid myopathy, systemic glucocorticoids are used for patients with symptomatic acute or chronic forms. The standard doses of prednisolone used for other forms of idiopathic inflammatory myopathies are usually administered.3-6 In general, the response of acute sarcoid myopathy to glucocorticoid therapy is favorable, and the clinical course is usually benign. However, the course in chronic sarcoid myopathy can be unpredictable with exacerbations. Given the lack of randomized trials of this therapy and because glucocorticoids themselves can cause steroid-induced myopathy, they are not used for asymptomatic patients.
In the end, astute clinical thinking, deductive reasoning, and pattern recognition were all instrumental in making this strong diagnosis of weakness.
KEY TEACHING POINTS
- Proximal muscle–dominant weakness is the characteristic feature in inflammatory myopathies like PM and DM. Myopathy causing proximal and distal weakness is more characteristic of sarcoidosis, IBM, alcohol, and statins.
- Elevations of urinary Times New Romanβ2-microglobulin and N-acetyl-D-glucosamine are often observed in inflammatory muscle diseases because of myoglobin-induced tubulointerstitial damage. These findings may also be caused by other conditions that affect the tubules, such as lupus nephritis, Sjogren’s syndrome, or renal sarcoidosis.
- Hypercalciuria in a patient with myopathy could suggest an underlying granulomatous disorder, such as mycobacterial infection, granulomatosis with polyangiitis, or sarcoidosis.
- The striking uptake within systemic skeletal striated muscles on gallium scintigraphy and “tiger man” appearance on FDG PET/CT are characteristic features of acute sarcoid myopathy; these are not common in other inflammatory myopathies.
Disclosure
Drs. Sudo, Wada, Narita, Mba, and Houchens have no conflicts of interest to disclose.
1. Vincze M, Danko K. Idiopathic inflammatory myopathies. Best Pract Res Clin Rheumatol. 2012;26:25-45. PubMed
2. Carstens PO, Schmidt J. Diagnosis, pathogenesis, and treatment of myositis: recent advances. Clin Exp Immunol. 2014;175:425-438. PubMed
3. Lannuzzi MC, Rhbicki BA, Teirstein AS. Sarcoidosis. N Eng J Med. 2007;357:2153-2165. PubMed
4. Baydur A, Pandya K, Sharma OP, et al. Control of ventilation, respiratory muscle strength, and granulomatous involvement of skeletal muscle in patients with sarcoidosis. Chest. 1993;103:396-402. PubMed
5. Zisman DA, Biermann JS, Martinez FJ, et al. Sarcoidosis presenting as a tumorlike muscular lesion. Case report and review of the literature. Medicine (Baltimore). 1999;78:112-122. PubMed
6. Fayad F, Liote F, Berenbaum F, et al. Muscle involvement in sarcoidosis: a retrospective and followup studies. J Rheumatol. 2006;33:98-103. PubMed
7. Berliner AR, Haas M, Choi MJ. Sarcoidosis: the nephrologist’s perspective. Am J Kidney Dis. 2006;48:856-870. PubMed
8. Otake S, Ishigaki T. Musular sarcoidosis. Semin Musculoskelet Radiol. 2001;5:167-170. PubMed
9. Otake S, Imagumbai N, Suzuki M, et al. MR imaging of muscular sarcoidosis after steroid therapy. Eur Radiol. 1998;8:1651-1653. PubMed
10. Hoffman JM, Gambhir SS. Molecular imaging: The vision and opportunity for radiology in the future. Radiology. 2007;244:39-47. PubMed
11. Basu S, Zhuang H, Torigian DA, et al. Functional imaging of inflammatory diseases using nuclear medicine techniques. Semin Nucl Med. 2009;39:124-145. PubMed
12. Gotthardt M, Cleeker-Rovers CP, Boerman OC, et al. Imaging of inflammation by PET, conventional scintigraphy, and other imaging techniques. J Nucl Med. 2010;51:1937-1949. PubMed
13. Owada T, Maezawa R, Kurasawa K, et al. Detection of inflammatory lesions by F-18 fluorodeoxyglucose positron emission tomography in patients with polymyositis and dermatomyositis. J Rheumatol. 2012;39:1659-1665. PubMed
14. Liem IH, Drent M, Antevska E, et al. Intense muscle uptake of gallium-67 in a patient with sarcoidosis. J Nucl Med. 1998;39:1605-1607. PubMed
15. Suehiro S, Shiokawa S, Taniguchi S, et al. Gallium-67 scintigraphy in the diagnosis and management of chronic sarcoid myopathy. Clin Rheumatol. 2003;22:146-148. PubMed
16. Marie I, Josse S, Lahaxe L, et al. Clinical images: muscle sarcoidosis demonstrated on positron emission tomography. Arthritis Rheum. 2009;60:2847. PubMed
17. Wieers G, Lhommel R, Lecouvet F, et al. A tiger man. Lancet. 2012;380:1859. PubMed
A 52-year-old man presented with bilateral weakness in all extremities. He noted the gradual onset of progressive muscle weakness 6 months prior to presentation. He reported generalized fatigue and difficulty with climbing stairs and carrying heavy objects.
Initial considerations of chronic weakness and fatigue are myopathy, polyneuropathy, medications, malignancy, endocrinopathies, human immunodeficiency virus (HIV), neuromuscular junction dysfunction, and central nervous system (CNS) disorders, such as amyotrophic lateral sclerosis (ALS) or multiple sclerosis (MS). Symmetrical muscle involvement and proximal weakness make myopathy most likely. Polyneuropathy, such as chronic inflammatory demyelinating polyneuropathy (CIDP), is less likely but still possible given the slowly progressive course. The use of medications that can cause myopathy should be explored, including colchicine, steroids, and statins. Gathering further history should focus on risk factors for HIV, as well as alcohol and illicit drug use. Malignancy can cause paraneoplastic myopathy. The review of systems should include symptoms of endocrinopathies, such as thyrotoxicosis and hypothyroidism. Fluctuations in weakness and dysphagia or ocular symptoms would suggest myasthenia gravis (MG). The time course and symmetrical weakness make a central disorder, such as ALS or MS, unlikely.
His past medical history was notable for pulmonary tuberculosis diagnosed at the age of 6 years, which was treated with hospitalization and an unknown medication regimen. He was not taking medications prior to this admission. His family history was significant for diabetes mellitus in both parents. He denied sick contacts. He was sexually active with his wife. He denied the use of tobacco and illicit drugs but endorsed alcohol consumption on a daily basis over the last 32 years. He reported no fluctuation in his symptoms, muscle or joint pains, rash, fevers, chills, diaphoresis, chest pain, dyspnea, abdominal pain, diarrhea, paresthesias, weight loss, or night sweats. He had never had a colonoscopy.
Painless progressive weakness of the limbs without sensory deficit is typical of a myopathy. Though CIDP can present with only motor weakness, the majority of patients have sensory symptoms, making this less likely. Although chronic alcohol abuse can cause myopathy, it seems less likely because other neurologic complications, such as sensory polyneuropathy or ataxia, would be expected. A review of systems does not suggest a thyroid disorder or malignancy, although this does not preclude an evaluation for both. The absence of fluctuations in weakness argues against MG. Though ALS, MG, MS, and CIDP are less likely, a neurologic exam is crucial in excluding them. The hallmark of ALS is upper motor neuron (UMN) and lower motor neuron signs in the absence of sensory symptoms and signs, while global hyporeflexia would be expected in CIDP, and fatigability on repeated power testing would be expected in MG. Neurologic findings disseminated in space (neuro-anatomically) would be expected in MS.
On physical examination, the patient had a temperature of 36.9°C, heart rate of 70 beats per minute, and regular respiratory rate of 10 breaths per minute, blood pressure 130/80 mmHg, and oxygen saturation 98% while breathing ambient air. Auscultation of the heart and lungs revealed normal findings. The abdomen was soft, nontender, and without masses or organomegaly. Neurologic examination disclosed bilateral symmetric upper and lower extremity weakness with positive Gower sign. Muscle strength scores of the bilateral biceps brachii, iliopsoas, and digitis extensor were between 4 and 5 without fatigability. Grasping power was impaired. Deep tendon reflexes were preserved, and there were no UMN signs. There was no tenderness to palpation in any muscle groups. Sensory testing was normal. Skin and lymph examinations were without abnormality. The rest of the physical examination was unremarkable.
Gower sign, characteristic of but not specific to muscular dystrophy, indicates proximal muscle weakness of lower extremities, wherein hands and arms are used to walk up the body into an upright position. The exam also reveals distal weakness as shown by reduced hand grasp. Symmetrical proximal weakness of all extremities without sensory deficits suggests a myopathic process, albeit one with some distal involvement. The absence of UMN signs argues against ALS, lack of fatigability argues against MG, and the absence of CNS or sensory deficits argues against MS.
Because myopathy is most likely, the next step would be to determine if this is an idiopathic inflammatory myopathy, such as polymyositis (PM) or dermatomyositis (DM), secondary inflammatory myopathy, or noninflammatory myopathy due to endocrinopathies. The time course is consistent with an inflammatory myopathy, such as PM or DM. Inclusion body myositis (IBM), another inflammatory myopathy, presents much more insidiously over years and tends to be asymmetric compared to PM. The absence of myalgia, arthralgia, rash, and gastrointestinal symptoms makes myopathy as a component of a connective tissue disease, such as systemic lupus erythematosus, or a mixed connective tissue disease unlikely. The next steps would be laboratory testing of muscle enzymes, complete blood count, biochemical profile, and antinuclear antibody (ANA).
Laboratory studies revealed a white blood cell count of 4460/mm3 with normal differential, hemoglobin 12.5 g/dL, and platelet count 345,000/mm3. Creatinine was 0.87 mg/dL, aspartate aminotransferase 61 IU/mL, alanine aminotransferase 45 IU/mL, and creatine kinase (CK) 529 U/L (normal range, 38-174 U/L). Other liver function enzymes were normal. Biochemistry studies disclosed normal sodium, potassium, glucose, calcium, and magnesium levels. Dipstick urinalysis revealed blood and protein, and the microscopic examination of urinary sediment was unremarkable without the presence of erythrocytes. Twenty-four-hour creatinine clearance was 106 mL/min (normal range, 97-137 mL/min). Chest radiography was unrevealing.
The modest increase in CK, evidence of myoglobinuria, and proteinuria can all occur with an inflammatory or metabolic myopathy. The combination of proximal and distal weakness, coupled with only a modestly elevated CK, makes IBM more likely than PM, as PM usually presents with proximal weakness and much higher CK values. Normal skin examination makes DM less likely, as skin manifestations are generally found at time of presentation. The onset of symptoms after age 50 and the patient being male also favor IBM, though a longer time course would be expected. Definitively distinguishing IBM from PM is important because treatment and prognosis differ.
Thyroid function and HIV testing should be obtained. ANA, more common in PM than in IBM, should be checked because these myopathies can be associated with other autoimmune diseases. Imaging is generally not essential, although magnetic resonance imaging (MRI) of the thighs may help to differentiate IBM from PM. Electromyography (EMG) should be done to determine the pattern of myopathy and select muscle biopsy sites.
Additional testing revealed a normal thyroid stimulating hormone level. HIV and ANA were negative. Serum aldolase level was 19 IU/L (normal range, 2.7-5.9 IU/L), myoglobin 277 ng/mL (normal range, 28-72 ng/mL), lactate dehydrogenase 416 IU/mL (normal range, 119-229 IU/mL), and C-reactive protein 0.32 mg/dL. An EMG revealed mild myogenic changes in all extremities. An MRI of the left brachial muscle revealed multiple scattered high-signal lesions.
The EMG and MRI findings are consistent with an inflammatory myopathy. The modest elevation in muscle enzymes and negative ANA are more consistent with IBM since most patients with PM or DM are ANA positive. Muscle biopsy can be very helpful in establishing the etiology of myopathy.
Malignancy is associated with DM and PM in about 9% and 4% of patients, respectively. The common cancers associated with these conditions are adenocarcinomas of the ovary, cervix, lung, pancreas, and stomach. Most cancers are diagnosed around the time of myositis diagnosis, although they can precede or follow by years. Idiopathic IBM is not associated with cancer.
Open surgical muscle biopsy of the left biceps brachii was performed. Light microscopic examination disclosed interstitial edema and noncaseating granulomas. Immunostaining revealed an increase in the number of cluster of differentiation (CD) 4+ T cells. Caseating granulomas and Langhans giant cells were not present (Figure 3).
Tuberculin reaction and interferon-γ-release assay were negative. Staining for AFB and fungi was negative. ANCA, rheumatoid factor (RF), anti-Ro/SSA, anti-La/SSB, anti-Sm, anti-RNP, and anti-Jo-1 were all negative or unremarkable. Serum angiotensin converting enzyme (ACE) level was 155.6 U/L (normal range, 7-25 U/L). Twenty-four-hour urine analysis revealed calcium excretion of 517.7 mg/day (normal range, 58-450 mg/day), β2-microglobulin 69,627 ug/day (normal range, <254 ug/day), and N-acetyl-D-glucosamine 95.3 U/day (normal range, <5.1 U/day) with a normal creatinine clearance. Serum intact parathyroid hormone level (PTH) was 5 pg/mL (normal range, 10-65 pg/mL), and 25-hydroxyvitamin D level was 51.1 ng/mL (normal range, 30-80 ng/mL). A CT of the thorax revealed a small ground-glass density lesion in the left lower lobe but no hilar or mediastinal lymphadenopathy.
Negative ANCA, RF, and autoantibodies exclude systemic vasculitis and connective tissue disease as causes of GM. Hypercalciuria is suggestive of granulomatous production of calcitriol, which, in turn, suppresses PTH. Hypercalcemia is not common in patients with sarcoidosis, but hypercalciuria occurs frequently. Serum ACE is a marker associated with sarcoidosis, but its diagnostic and prognostic utility is unclear.
Though there is a concern for sarcoidosis, this diagnosis can only be confidently made by finding noncaseating granulomas on a background of compatible clinical and radiologic findings after alternate possible etiologies are excluded. The chest CT reveals a small ground-glass density lesion without hilar adenopathy. These findings, though not incompatible, are not typical for pulmonary sarcoidosis. Therefore, finding noncaeseating granulomas in a second organ system would point toward systemic sarcoidosis as a unifying diagnosis. Bronchoscopy with bronchoalveolar lavage (BAL) and transbronchial biopsy has a reasonable yield even in the absence of hilar adenopathy or typical parenchymal findings. A CD4/CD8 T-cell ratio of 2 or more on BAL provides supportive evidence for sarcoidosis.
It is reasonable to start empiric glucocorticoids for GM given that the AFB and fungal stains on histopathology are negative and that there is no evidence of lymphoma.
The CD4/CD8 T-cell ratio greater than 2, combined with the absence of neutrophils and eosinophils on BAL, is helpful in distinguishing sarcoidosis from other pulmonary diseases. This patient’s inflammatory myopathy was revealed to be a rare initial manifestation of systemic sarcoidosis.
DISCUSSION
Weakness is a common symptom of muscle disorders such as myopathies and muscular dystrophy. Idiopathic inflammatory myopathies include PM, DM, and others.1,2 These usually present with proximal-dominant muscle weakness, decreased endurance, and muscle inflammation. A diagnosis is made according to symptoms in combination with diagnostic examinations, including elevated serum CK levels, abnormal EMG findings, and histopathology of skeletal muscle biopsy specimens.
Sarcoidosis, a multisystem disorder of unknown etiology, is characterized histopathologically by noncaseating granulomas in affected organs.3 It typically affects young adults, with incidence peaking at 20 to 39 years of age. Although any organ may be involved, the disorder usually presents with 1 or more common abnormalities, including bilateral hilar lymphadenopathy, lung lesions, and skin and eye involvement. Musculoskeletal involvement is less common. It is estimated that skeletal muscle is involved in 50% to 80% of patients with sarcoidosis but is rarely symptomatic (0.5% to 2.5%).4-6
In this patient, weakness was distributed in both proximal and distal muscles, yet proximal weakness is the most characteristic feature in PM and DM. Therefore, sarcoidosis should be considered in the differential diagnosis of idiopathic inflammatory myopathies, especially when weakness accompanies abnormalities in other organs typically affected by sarcoidosis.
Myoglobinuria often is observed in rhabdomyolysis and inflammatory myopathies, conditions that produce high levels of serum CK and myoglobin. Myoglobinuria, often accompanied by the elevation of urinary β2-microglobulin and N-acetyl-D-glucosamine levels, can induce tubulointerstitial damage, which leads to acute kidney injury. In this case, however, these abnormal kidney findings were observed without high levels of serum CK or myoglobin. This suggests the potential for other causes of tubulointerstitial damage, such as granulomatous interstitial nephritis in renal sarcoidosis.3
Another characteristic abnormality was the elevation of urinary calcium excretion, which indicated an underlying granulomatous disorder, such as mycobacterial infection, granulomatosis with polyangiitis, or sarcoidosis. In sarcoidosis, hypercalciuria occurs in 40% of patients, hypercalcemia in 11%, and renal calculi in 10%.3,7 Hypercalciuria, for this patient, was important in arriving at the correct diagnosis after the gallium scan was obtained given the dearth of other typical features of sarcoidosis.
Although muscle biopsy is essential, imaging studies for idiopathic inflammatory myopathy are considered useful tools to narrow the differential diagnosis. The use of MRI of the skeletal muscle is helpful to both identify an adequate muscle for biopsy and demonstrate the pattern of affected muscles beyond clinical appearance, which aids in excluding, for example, muscular dystrophies.8,9
FDG PET/CT is a very sensitive imaging modality used to detect neoplastic lesions and has been widely used to screen for occult neoplasms and detect metastases.10-12 It is also useful for detecting inflammation in patients with osteomyelitis, metastatic infectious diseases, rheumatoid arthritis, vasculitis, inflammatory bowel diseases, fever of unknown origin, and sarcoidosis.11,12 In PM and DM, however, the sensitivity of FDG PET/CT for detection of myositis is reportedly lower than that of EMG and MRI.13 Similarly, gallium scintigraphy is usually performed to examine the disease activity of interstitial pneumonia or to detect malignancy. Previous literature and this case show that the striking images of gallium scintigraphy and FDG PET/CT have utility, not only for detection of sarcoid myopathy but also for the evaluation of treatment efficacy.14-17 Characteristic imaging findings on FDG PET/CT have been described as a “tiger man” appearance.17
For the treatment of sarcoid myopathy, systemic glucocorticoids are used for patients with symptomatic acute or chronic forms. The standard doses of prednisolone used for other forms of idiopathic inflammatory myopathies are usually administered.3-6 In general, the response of acute sarcoid myopathy to glucocorticoid therapy is favorable, and the clinical course is usually benign. However, the course in chronic sarcoid myopathy can be unpredictable with exacerbations. Given the lack of randomized trials of this therapy and because glucocorticoids themselves can cause steroid-induced myopathy, they are not used for asymptomatic patients.
In the end, astute clinical thinking, deductive reasoning, and pattern recognition were all instrumental in making this strong diagnosis of weakness.
KEY TEACHING POINTS
- Proximal muscle–dominant weakness is the characteristic feature in inflammatory myopathies like PM and DM. Myopathy causing proximal and distal weakness is more characteristic of sarcoidosis, IBM, alcohol, and statins.
- Elevations of urinary Times New Romanβ2-microglobulin and N-acetyl-D-glucosamine are often observed in inflammatory muscle diseases because of myoglobin-induced tubulointerstitial damage. These findings may also be caused by other conditions that affect the tubules, such as lupus nephritis, Sjogren’s syndrome, or renal sarcoidosis.
- Hypercalciuria in a patient with myopathy could suggest an underlying granulomatous disorder, such as mycobacterial infection, granulomatosis with polyangiitis, or sarcoidosis.
- The striking uptake within systemic skeletal striated muscles on gallium scintigraphy and “tiger man” appearance on FDG PET/CT are characteristic features of acute sarcoid myopathy; these are not common in other inflammatory myopathies.
Disclosure
Drs. Sudo, Wada, Narita, Mba, and Houchens have no conflicts of interest to disclose.
A 52-year-old man presented with bilateral weakness in all extremities. He noted the gradual onset of progressive muscle weakness 6 months prior to presentation. He reported generalized fatigue and difficulty with climbing stairs and carrying heavy objects.
Initial considerations of chronic weakness and fatigue are myopathy, polyneuropathy, medications, malignancy, endocrinopathies, human immunodeficiency virus (HIV), neuromuscular junction dysfunction, and central nervous system (CNS) disorders, such as amyotrophic lateral sclerosis (ALS) or multiple sclerosis (MS). Symmetrical muscle involvement and proximal weakness make myopathy most likely. Polyneuropathy, such as chronic inflammatory demyelinating polyneuropathy (CIDP), is less likely but still possible given the slowly progressive course. The use of medications that can cause myopathy should be explored, including colchicine, steroids, and statins. Gathering further history should focus on risk factors for HIV, as well as alcohol and illicit drug use. Malignancy can cause paraneoplastic myopathy. The review of systems should include symptoms of endocrinopathies, such as thyrotoxicosis and hypothyroidism. Fluctuations in weakness and dysphagia or ocular symptoms would suggest myasthenia gravis (MG). The time course and symmetrical weakness make a central disorder, such as ALS or MS, unlikely.
His past medical history was notable for pulmonary tuberculosis diagnosed at the age of 6 years, which was treated with hospitalization and an unknown medication regimen. He was not taking medications prior to this admission. His family history was significant for diabetes mellitus in both parents. He denied sick contacts. He was sexually active with his wife. He denied the use of tobacco and illicit drugs but endorsed alcohol consumption on a daily basis over the last 32 years. He reported no fluctuation in his symptoms, muscle or joint pains, rash, fevers, chills, diaphoresis, chest pain, dyspnea, abdominal pain, diarrhea, paresthesias, weight loss, or night sweats. He had never had a colonoscopy.
Painless progressive weakness of the limbs without sensory deficit is typical of a myopathy. Though CIDP can present with only motor weakness, the majority of patients have sensory symptoms, making this less likely. Although chronic alcohol abuse can cause myopathy, it seems less likely because other neurologic complications, such as sensory polyneuropathy or ataxia, would be expected. A review of systems does not suggest a thyroid disorder or malignancy, although this does not preclude an evaluation for both. The absence of fluctuations in weakness argues against MG. Though ALS, MG, MS, and CIDP are less likely, a neurologic exam is crucial in excluding them. The hallmark of ALS is upper motor neuron (UMN) and lower motor neuron signs in the absence of sensory symptoms and signs, while global hyporeflexia would be expected in CIDP, and fatigability on repeated power testing would be expected in MG. Neurologic findings disseminated in space (neuro-anatomically) would be expected in MS.
On physical examination, the patient had a temperature of 36.9°C, heart rate of 70 beats per minute, and regular respiratory rate of 10 breaths per minute, blood pressure 130/80 mmHg, and oxygen saturation 98% while breathing ambient air. Auscultation of the heart and lungs revealed normal findings. The abdomen was soft, nontender, and without masses or organomegaly. Neurologic examination disclosed bilateral symmetric upper and lower extremity weakness with positive Gower sign. Muscle strength scores of the bilateral biceps brachii, iliopsoas, and digitis extensor were between 4 and 5 without fatigability. Grasping power was impaired. Deep tendon reflexes were preserved, and there were no UMN signs. There was no tenderness to palpation in any muscle groups. Sensory testing was normal. Skin and lymph examinations were without abnormality. The rest of the physical examination was unremarkable.
Gower sign, characteristic of but not specific to muscular dystrophy, indicates proximal muscle weakness of lower extremities, wherein hands and arms are used to walk up the body into an upright position. The exam also reveals distal weakness as shown by reduced hand grasp. Symmetrical proximal weakness of all extremities without sensory deficits suggests a myopathic process, albeit one with some distal involvement. The absence of UMN signs argues against ALS, lack of fatigability argues against MG, and the absence of CNS or sensory deficits argues against MS.
Because myopathy is most likely, the next step would be to determine if this is an idiopathic inflammatory myopathy, such as polymyositis (PM) or dermatomyositis (DM), secondary inflammatory myopathy, or noninflammatory myopathy due to endocrinopathies. The time course is consistent with an inflammatory myopathy, such as PM or DM. Inclusion body myositis (IBM), another inflammatory myopathy, presents much more insidiously over years and tends to be asymmetric compared to PM. The absence of myalgia, arthralgia, rash, and gastrointestinal symptoms makes myopathy as a component of a connective tissue disease, such as systemic lupus erythematosus, or a mixed connective tissue disease unlikely. The next steps would be laboratory testing of muscle enzymes, complete blood count, biochemical profile, and antinuclear antibody (ANA).
Laboratory studies revealed a white blood cell count of 4460/mm3 with normal differential, hemoglobin 12.5 g/dL, and platelet count 345,000/mm3. Creatinine was 0.87 mg/dL, aspartate aminotransferase 61 IU/mL, alanine aminotransferase 45 IU/mL, and creatine kinase (CK) 529 U/L (normal range, 38-174 U/L). Other liver function enzymes were normal. Biochemistry studies disclosed normal sodium, potassium, glucose, calcium, and magnesium levels. Dipstick urinalysis revealed blood and protein, and the microscopic examination of urinary sediment was unremarkable without the presence of erythrocytes. Twenty-four-hour creatinine clearance was 106 mL/min (normal range, 97-137 mL/min). Chest radiography was unrevealing.
The modest increase in CK, evidence of myoglobinuria, and proteinuria can all occur with an inflammatory or metabolic myopathy. The combination of proximal and distal weakness, coupled with only a modestly elevated CK, makes IBM more likely than PM, as PM usually presents with proximal weakness and much higher CK values. Normal skin examination makes DM less likely, as skin manifestations are generally found at time of presentation. The onset of symptoms after age 50 and the patient being male also favor IBM, though a longer time course would be expected. Definitively distinguishing IBM from PM is important because treatment and prognosis differ.
Thyroid function and HIV testing should be obtained. ANA, more common in PM than in IBM, should be checked because these myopathies can be associated with other autoimmune diseases. Imaging is generally not essential, although magnetic resonance imaging (MRI) of the thighs may help to differentiate IBM from PM. Electromyography (EMG) should be done to determine the pattern of myopathy and select muscle biopsy sites.
Additional testing revealed a normal thyroid stimulating hormone level. HIV and ANA were negative. Serum aldolase level was 19 IU/L (normal range, 2.7-5.9 IU/L), myoglobin 277 ng/mL (normal range, 28-72 ng/mL), lactate dehydrogenase 416 IU/mL (normal range, 119-229 IU/mL), and C-reactive protein 0.32 mg/dL. An EMG revealed mild myogenic changes in all extremities. An MRI of the left brachial muscle revealed multiple scattered high-signal lesions.
The EMG and MRI findings are consistent with an inflammatory myopathy. The modest elevation in muscle enzymes and negative ANA are more consistent with IBM since most patients with PM or DM are ANA positive. Muscle biopsy can be very helpful in establishing the etiology of myopathy.
Malignancy is associated with DM and PM in about 9% and 4% of patients, respectively. The common cancers associated with these conditions are adenocarcinomas of the ovary, cervix, lung, pancreas, and stomach. Most cancers are diagnosed around the time of myositis diagnosis, although they can precede or follow by years. Idiopathic IBM is not associated with cancer.
Open surgical muscle biopsy of the left biceps brachii was performed. Light microscopic examination disclosed interstitial edema and noncaseating granulomas. Immunostaining revealed an increase in the number of cluster of differentiation (CD) 4+ T cells. Caseating granulomas and Langhans giant cells were not present (Figure 3).
Tuberculin reaction and interferon-γ-release assay were negative. Staining for AFB and fungi was negative. ANCA, rheumatoid factor (RF), anti-Ro/SSA, anti-La/SSB, anti-Sm, anti-RNP, and anti-Jo-1 were all negative or unremarkable. Serum angiotensin converting enzyme (ACE) level was 155.6 U/L (normal range, 7-25 U/L). Twenty-four-hour urine analysis revealed calcium excretion of 517.7 mg/day (normal range, 58-450 mg/day), β2-microglobulin 69,627 ug/day (normal range, <254 ug/day), and N-acetyl-D-glucosamine 95.3 U/day (normal range, <5.1 U/day) with a normal creatinine clearance. Serum intact parathyroid hormone level (PTH) was 5 pg/mL (normal range, 10-65 pg/mL), and 25-hydroxyvitamin D level was 51.1 ng/mL (normal range, 30-80 ng/mL). A CT of the thorax revealed a small ground-glass density lesion in the left lower lobe but no hilar or mediastinal lymphadenopathy.
Negative ANCA, RF, and autoantibodies exclude systemic vasculitis and connective tissue disease as causes of GM. Hypercalciuria is suggestive of granulomatous production of calcitriol, which, in turn, suppresses PTH. Hypercalcemia is not common in patients with sarcoidosis, but hypercalciuria occurs frequently. Serum ACE is a marker associated with sarcoidosis, but its diagnostic and prognostic utility is unclear.
Though there is a concern for sarcoidosis, this diagnosis can only be confidently made by finding noncaseating granulomas on a background of compatible clinical and radiologic findings after alternate possible etiologies are excluded. The chest CT reveals a small ground-glass density lesion without hilar adenopathy. These findings, though not incompatible, are not typical for pulmonary sarcoidosis. Therefore, finding noncaeseating granulomas in a second organ system would point toward systemic sarcoidosis as a unifying diagnosis. Bronchoscopy with bronchoalveolar lavage (BAL) and transbronchial biopsy has a reasonable yield even in the absence of hilar adenopathy or typical parenchymal findings. A CD4/CD8 T-cell ratio of 2 or more on BAL provides supportive evidence for sarcoidosis.
It is reasonable to start empiric glucocorticoids for GM given that the AFB and fungal stains on histopathology are negative and that there is no evidence of lymphoma.
The CD4/CD8 T-cell ratio greater than 2, combined with the absence of neutrophils and eosinophils on BAL, is helpful in distinguishing sarcoidosis from other pulmonary diseases. This patient’s inflammatory myopathy was revealed to be a rare initial manifestation of systemic sarcoidosis.
DISCUSSION
Weakness is a common symptom of muscle disorders such as myopathies and muscular dystrophy. Idiopathic inflammatory myopathies include PM, DM, and others.1,2 These usually present with proximal-dominant muscle weakness, decreased endurance, and muscle inflammation. A diagnosis is made according to symptoms in combination with diagnostic examinations, including elevated serum CK levels, abnormal EMG findings, and histopathology of skeletal muscle biopsy specimens.
Sarcoidosis, a multisystem disorder of unknown etiology, is characterized histopathologically by noncaseating granulomas in affected organs.3 It typically affects young adults, with incidence peaking at 20 to 39 years of age. Although any organ may be involved, the disorder usually presents with 1 or more common abnormalities, including bilateral hilar lymphadenopathy, lung lesions, and skin and eye involvement. Musculoskeletal involvement is less common. It is estimated that skeletal muscle is involved in 50% to 80% of patients with sarcoidosis but is rarely symptomatic (0.5% to 2.5%).4-6
In this patient, weakness was distributed in both proximal and distal muscles, yet proximal weakness is the most characteristic feature in PM and DM. Therefore, sarcoidosis should be considered in the differential diagnosis of idiopathic inflammatory myopathies, especially when weakness accompanies abnormalities in other organs typically affected by sarcoidosis.
Myoglobinuria often is observed in rhabdomyolysis and inflammatory myopathies, conditions that produce high levels of serum CK and myoglobin. Myoglobinuria, often accompanied by the elevation of urinary β2-microglobulin and N-acetyl-D-glucosamine levels, can induce tubulointerstitial damage, which leads to acute kidney injury. In this case, however, these abnormal kidney findings were observed without high levels of serum CK or myoglobin. This suggests the potential for other causes of tubulointerstitial damage, such as granulomatous interstitial nephritis in renal sarcoidosis.3
Another characteristic abnormality was the elevation of urinary calcium excretion, which indicated an underlying granulomatous disorder, such as mycobacterial infection, granulomatosis with polyangiitis, or sarcoidosis. In sarcoidosis, hypercalciuria occurs in 40% of patients, hypercalcemia in 11%, and renal calculi in 10%.3,7 Hypercalciuria, for this patient, was important in arriving at the correct diagnosis after the gallium scan was obtained given the dearth of other typical features of sarcoidosis.
Although muscle biopsy is essential, imaging studies for idiopathic inflammatory myopathy are considered useful tools to narrow the differential diagnosis. The use of MRI of the skeletal muscle is helpful to both identify an adequate muscle for biopsy and demonstrate the pattern of affected muscles beyond clinical appearance, which aids in excluding, for example, muscular dystrophies.8,9
FDG PET/CT is a very sensitive imaging modality used to detect neoplastic lesions and has been widely used to screen for occult neoplasms and detect metastases.10-12 It is also useful for detecting inflammation in patients with osteomyelitis, metastatic infectious diseases, rheumatoid arthritis, vasculitis, inflammatory bowel diseases, fever of unknown origin, and sarcoidosis.11,12 In PM and DM, however, the sensitivity of FDG PET/CT for detection of myositis is reportedly lower than that of EMG and MRI.13 Similarly, gallium scintigraphy is usually performed to examine the disease activity of interstitial pneumonia or to detect malignancy. Previous literature and this case show that the striking images of gallium scintigraphy and FDG PET/CT have utility, not only for detection of sarcoid myopathy but also for the evaluation of treatment efficacy.14-17 Characteristic imaging findings on FDG PET/CT have been described as a “tiger man” appearance.17
For the treatment of sarcoid myopathy, systemic glucocorticoids are used for patients with symptomatic acute or chronic forms. The standard doses of prednisolone used for other forms of idiopathic inflammatory myopathies are usually administered.3-6 In general, the response of acute sarcoid myopathy to glucocorticoid therapy is favorable, and the clinical course is usually benign. However, the course in chronic sarcoid myopathy can be unpredictable with exacerbations. Given the lack of randomized trials of this therapy and because glucocorticoids themselves can cause steroid-induced myopathy, they are not used for asymptomatic patients.
In the end, astute clinical thinking, deductive reasoning, and pattern recognition were all instrumental in making this strong diagnosis of weakness.
KEY TEACHING POINTS
- Proximal muscle–dominant weakness is the characteristic feature in inflammatory myopathies like PM and DM. Myopathy causing proximal and distal weakness is more characteristic of sarcoidosis, IBM, alcohol, and statins.
- Elevations of urinary Times New Romanβ2-microglobulin and N-acetyl-D-glucosamine are often observed in inflammatory muscle diseases because of myoglobin-induced tubulointerstitial damage. These findings may also be caused by other conditions that affect the tubules, such as lupus nephritis, Sjogren’s syndrome, or renal sarcoidosis.
- Hypercalciuria in a patient with myopathy could suggest an underlying granulomatous disorder, such as mycobacterial infection, granulomatosis with polyangiitis, or sarcoidosis.
- The striking uptake within systemic skeletal striated muscles on gallium scintigraphy and “tiger man” appearance on FDG PET/CT are characteristic features of acute sarcoid myopathy; these are not common in other inflammatory myopathies.
Disclosure
Drs. Sudo, Wada, Narita, Mba, and Houchens have no conflicts of interest to disclose.
1. Vincze M, Danko K. Idiopathic inflammatory myopathies. Best Pract Res Clin Rheumatol. 2012;26:25-45. PubMed
2. Carstens PO, Schmidt J. Diagnosis, pathogenesis, and treatment of myositis: recent advances. Clin Exp Immunol. 2014;175:425-438. PubMed
3. Lannuzzi MC, Rhbicki BA, Teirstein AS. Sarcoidosis. N Eng J Med. 2007;357:2153-2165. PubMed
4. Baydur A, Pandya K, Sharma OP, et al. Control of ventilation, respiratory muscle strength, and granulomatous involvement of skeletal muscle in patients with sarcoidosis. Chest. 1993;103:396-402. PubMed
5. Zisman DA, Biermann JS, Martinez FJ, et al. Sarcoidosis presenting as a tumorlike muscular lesion. Case report and review of the literature. Medicine (Baltimore). 1999;78:112-122. PubMed
6. Fayad F, Liote F, Berenbaum F, et al. Muscle involvement in sarcoidosis: a retrospective and followup studies. J Rheumatol. 2006;33:98-103. PubMed
7. Berliner AR, Haas M, Choi MJ. Sarcoidosis: the nephrologist’s perspective. Am J Kidney Dis. 2006;48:856-870. PubMed
8. Otake S, Ishigaki T. Musular sarcoidosis. Semin Musculoskelet Radiol. 2001;5:167-170. PubMed
9. Otake S, Imagumbai N, Suzuki M, et al. MR imaging of muscular sarcoidosis after steroid therapy. Eur Radiol. 1998;8:1651-1653. PubMed
10. Hoffman JM, Gambhir SS. Molecular imaging: The vision and opportunity for radiology in the future. Radiology. 2007;244:39-47. PubMed
11. Basu S, Zhuang H, Torigian DA, et al. Functional imaging of inflammatory diseases using nuclear medicine techniques. Semin Nucl Med. 2009;39:124-145. PubMed
12. Gotthardt M, Cleeker-Rovers CP, Boerman OC, et al. Imaging of inflammation by PET, conventional scintigraphy, and other imaging techniques. J Nucl Med. 2010;51:1937-1949. PubMed
13. Owada T, Maezawa R, Kurasawa K, et al. Detection of inflammatory lesions by F-18 fluorodeoxyglucose positron emission tomography in patients with polymyositis and dermatomyositis. J Rheumatol. 2012;39:1659-1665. PubMed
14. Liem IH, Drent M, Antevska E, et al. Intense muscle uptake of gallium-67 in a patient with sarcoidosis. J Nucl Med. 1998;39:1605-1607. PubMed
15. Suehiro S, Shiokawa S, Taniguchi S, et al. Gallium-67 scintigraphy in the diagnosis and management of chronic sarcoid myopathy. Clin Rheumatol. 2003;22:146-148. PubMed
16. Marie I, Josse S, Lahaxe L, et al. Clinical images: muscle sarcoidosis demonstrated on positron emission tomography. Arthritis Rheum. 2009;60:2847. PubMed
17. Wieers G, Lhommel R, Lecouvet F, et al. A tiger man. Lancet. 2012;380:1859. PubMed
1. Vincze M, Danko K. Idiopathic inflammatory myopathies. Best Pract Res Clin Rheumatol. 2012;26:25-45. PubMed
2. Carstens PO, Schmidt J. Diagnosis, pathogenesis, and treatment of myositis: recent advances. Clin Exp Immunol. 2014;175:425-438. PubMed
3. Lannuzzi MC, Rhbicki BA, Teirstein AS. Sarcoidosis. N Eng J Med. 2007;357:2153-2165. PubMed
4. Baydur A, Pandya K, Sharma OP, et al. Control of ventilation, respiratory muscle strength, and granulomatous involvement of skeletal muscle in patients with sarcoidosis. Chest. 1993;103:396-402. PubMed
5. Zisman DA, Biermann JS, Martinez FJ, et al. Sarcoidosis presenting as a tumorlike muscular lesion. Case report and review of the literature. Medicine (Baltimore). 1999;78:112-122. PubMed
6. Fayad F, Liote F, Berenbaum F, et al. Muscle involvement in sarcoidosis: a retrospective and followup studies. J Rheumatol. 2006;33:98-103. PubMed
7. Berliner AR, Haas M, Choi MJ. Sarcoidosis: the nephrologist’s perspective. Am J Kidney Dis. 2006;48:856-870. PubMed
8. Otake S, Ishigaki T. Musular sarcoidosis. Semin Musculoskelet Radiol. 2001;5:167-170. PubMed
9. Otake S, Imagumbai N, Suzuki M, et al. MR imaging of muscular sarcoidosis after steroid therapy. Eur Radiol. 1998;8:1651-1653. PubMed
10. Hoffman JM, Gambhir SS. Molecular imaging: The vision and opportunity for radiology in the future. Radiology. 2007;244:39-47. PubMed
11. Basu S, Zhuang H, Torigian DA, et al. Functional imaging of inflammatory diseases using nuclear medicine techniques. Semin Nucl Med. 2009;39:124-145. PubMed
12. Gotthardt M, Cleeker-Rovers CP, Boerman OC, et al. Imaging of inflammation by PET, conventional scintigraphy, and other imaging techniques. J Nucl Med. 2010;51:1937-1949. PubMed
13. Owada T, Maezawa R, Kurasawa K, et al. Detection of inflammatory lesions by F-18 fluorodeoxyglucose positron emission tomography in patients with polymyositis and dermatomyositis. J Rheumatol. 2012;39:1659-1665. PubMed
14. Liem IH, Drent M, Antevska E, et al. Intense muscle uptake of gallium-67 in a patient with sarcoidosis. J Nucl Med. 1998;39:1605-1607. PubMed
15. Suehiro S, Shiokawa S, Taniguchi S, et al. Gallium-67 scintigraphy in the diagnosis and management of chronic sarcoid myopathy. Clin Rheumatol. 2003;22:146-148. PubMed
16. Marie I, Josse S, Lahaxe L, et al. Clinical images: muscle sarcoidosis demonstrated on positron emission tomography. Arthritis Rheum. 2009;60:2847. PubMed
17. Wieers G, Lhommel R, Lecouvet F, et al. A tiger man. Lancet. 2012;380:1859. PubMed
© 2017 Society of Hospital Medicine