Article
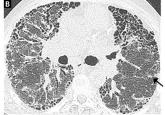
Idiopathic pulmonary fibrosis: What primary care physicians need to know
This devastating and fatal lung disease generally affects older adults, especially men, and can be mistaken for COPD.
Article
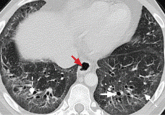
Managing interstitial lung disease detected on CT during lung cancer screening
How to distinguish among the most common forms, and the role of the primary care physician in managing them.