User login
Mortality Due to Elevated Troponin
Acute coronary syndromes (ACS) are potentially lethal and present with a wide variety of symptoms. As such, physicians frequently order cardiac biomarkers, such as cardiac troponin, for patients with acute complaints. Elevated troponin is associated with higher risk of mortality regardless of the causes, which can be myriad, both chronic and acute.[1] Among patients with an elevated troponin, distinguishing ACS from non‐ACS can be challenging.
Making the distinction between ACS and non‐ACS troponin elevation is crucial because the underlying pathophysiology and subsequent management strategies are markedly different.[2] According to evidence‐based practice guidelines, ACS is managed with antiplatelet drugs, statins, and percutaneous coronary intervention, improving clinical outcomes.[3] In contrast, care for patients with non‐ACS troponin elevations is usually supportive, with a focus on the underlying conditions. The lack of specific treatment options for such patients is concerning given that several series have suggested that non‐ACS troponin patients may have a higher mortality risk than ACS patients.[4, 5, 6] Non‐ACS troponin elevation can be the result of a multitude of conditions.[7, 8] What remains unclear at this point is whether the excess mortality observed with non‐ACS troponin elevation is due to myocardial damage or to the underlying conditions that predispose to troponin release.
Using data from a quality improvement (QI) project collected at our Veterans Affairs (VA) medical center, we investigated the mortality risk associated with ACS and non‐ACS troponin elevation including an analysis of factors associated with mortality. We hypothesized that non‐ACS troponin elevation will have a higher mortality risk than troponin elevation due to ACS, and that important contributors to this relationship could be identified to provide direction for future investigation directed at modifying this mortality risk.
METHODS
We analyzed data that were prospectively collected for a quality initiative between 2006 and 2007. The project was a collaborative endeavor between cardiology, hospital medicine, and emergency medicine with the process goal of better identifying patients with ACS to hopefully improve outcomes. The QI team was consulted in real time to assist with treatment recommendations; no retrospective decisions were made regarding whether or not ACS was present. As the goal of the project was to improve cardiovascular outcomes, consultative advice was freely provided, and no physicians or teams were subject to any adverse repercussions for their diagnoses or management decisions.
A cardiologist‐led team was created to improve quality of care for myocardial infarction patients by evaluating all patients at our facility with an elevated troponin. On a daily basis, a specialist clinical coordinator (nurse practitioner or physician assistant) received a list of all patients with elevated troponin from the chemistry lab. The coordinator reviewed the patients' medical records with a cardiologist. A positive troponin was defined as a troponin T level of greater than 0.03 ng/mL (99th percentile at our facility). Each attending cardiologist prospectively determined if troponin elevation was related to clinical findings consistent with an ACS based on review of the patients' symptoms (duration, quality, severity, chronicity, and alleviating/aggravating factors), medical history, and noninvasive cardiac testing including electrocardiograms, cardiac biomarkers, and any other available imaging tests.
We have previously demonstrated that the cardiologists at our facility have a similar rate of diagnosing ACS.[9] All cardiologists at our facility maintain current American Board of Internal Medicine certification in cardiovascular disease and have academic appointments at the University of Florida College of Medicine. All patients were followed prospectively, and data on their medical history, acute evaluation, and outcomes were tracked in an electronic database. Given the higher risk of mortality with ST‐elevation myocardial infarction, such patients were excluded from this investigation. By definition, patients with unstable angina do not have elevated biomarkers and thus would not have been included in the database to begin with. Prospectively recorded data elements included: age, gender, chief complaint, tobacco use, presence of hypertension, hyperlipidemia, prior coronary disease, chronic kidney disease, diabetes mellitus, cardiac troponin values, serum creatinine, electrocardiogram (ECG) variables, Thrombolysis in Myocardial Infarction (TIMI) score, and if the patient was placed under hospice care or an active do‐not‐resuscitate (DNR) order. Additional data elements gathered at a later date included maximum temperature, white blood cell count, N‐terminal pro‐brain natriuretic peptide (NT‐proBNP), administration of advanced cardiac life support (ACLS), and admission to an intensive care unit (ICU). All consecutive patients with elevated troponin were included in the database; if patients were included more than once, we used their index evaluation only. All patients with troponin elevation after revascularization (percutaneous coronary intervention or coronary bypass surgery) were excluded. Our investigational design was reviewed by our institutional review board, who waived the requirement for formal written informed consent and approved use of data from this QI project for research purposes.
We focused this investigation on an analysis of all‐cause mortality in February 2014. We analyzed mortality at 30 days, 1 year, and 6 years. As secondary outcomes we analyzed the likelihood of the patients' chief complaint for the diagnosis of ACS and evaluated predictors of mortality based on Cox proportional hazard modeling. Mortality within the VA system is reliably tracked and compares favorably to the Social Security National Death Index Master File for accuracy.[10, 11]
Categorical variables were compared by 2 test. The Student t test was used to compare normally distributed continuous variables, and nonparametric tests were used for non‐normal distributions as appropriate. Mortality data at 30 days, 1 year, and 6 years were compared by log‐rank test and Kaplan‐Meier graphs. A formal power analysis was not performed; the entire available population was included. A Cox proportional hazard model was created to estimate mortality risk at each time point. Variables included in our Cox regression model were age, gender, history of coronary artery disease (CAD), hypertension, diabetes mellitus or hyperlipidemia, ACS diagnosis, dynamic ECG changes, TIMI risk score, initial troponin level, creatinine level at time of initial troponin (per mg/dL), presence of fever, maximum white blood cell count, NT‐proBNP level (per 1000 pg/mL), if ACLS was performed, if the patient was under hospice care, if there was a DNR order, and if they required ICU admission. This model was also constructed independently for the ACS and non‐ACS cohorts for mortality at 1 year. A forward stepwise model was used. Statistical results were considered significant at P < 0.05. Statistical analyses were performed using SPSS version 21 (IBM, Armonk, NY).
RESULTS
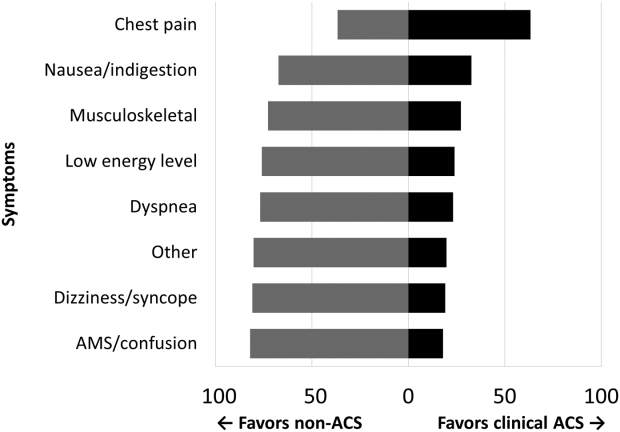
Among the 761 patients, 502 (66.0%) were classified as non‐ACS and 259 (34.0%) as ACS (Table 1). The mean age was higher in the non‐ACS group (71 years vs 69 years in the ACS group, P = 0.006). Hypertension, diabetes mellitus, and prior CAD were frequent in both groups and not significantly different. Median initial troponin T was higher in the ACS group (0.12 ng/mL vs 0.06 ng/mL, P < 0.001) as were the frequency of a TIMI risk score >2 (92.5% vs 74.3%, P < 0.001) and new ECG changes (29.7% vs 8.2%, P < 0.001). Hospice, DNR orders, and administration of ACLS were not different between groups; however, admission to the ICU was more frequent in the ACS group (44.8% vs 31.9%, P < 0.001). Chest pain was the symptom with the highest positive predictive value for the diagnosis of ACS (63.3%), whereas the least predictive was altered mental status or confusion (18.0%) (Figure 1).
| Non‐ACS, N = 502 | ACS, N = 259 | P Value | |
|---|---|---|---|
| |||
| Baseline characteristics, n (%) | |||
| Age, y | 71 11 | 69 11 | 0.006 |
| Female | 6 (1.2%) | 1 (0.4%) | 0.27 |
| Coronary artery disease | 244 (48.6%) | 141 (54.4%) | 0.13 |
| Hypertension | 381 (75.9%) | 203 (78.4%) | 0.44 |
| Diabetes mellitus | 220 (43.8%) | 119 (45.9%) | 0.58 |
| Hyperlipidemia | 268 (53.4%) | 170 (65.6%) | 0.001 |
| Current smoker | 24 (4.8%) | 49 (18.9%) | <0.001 |
| Clinical presentation | |||
| Initial troponin T, ng/mL, median [IQR] | 0.06 [0.040.11] | 0.12 [0.050.32] | <0.001 |
| White cell count, 109/L, median [IQR] | 10 [8.014.0] | 11 [8.015.0] | 0.005 |
| NT‐proBNP, pg/mL, median [IQR] | 3,531 [1,20110,519] | 1,932 [3199,100] | 0.001 |
| Creatinine, mg/dL, median [IQR] | 1.6 [1.12.4] | 1.1 [0.91.5] | <0.001 |
| New ECG changes, no. (%) | 41 (8.2%) | 77 (29.7%) | <0.001 |
| TIMI score over 2, no. (%) | 365 (74.3%) | 235 (92.5%) | <0.001 |
| Fever (over 100.4 F), no. (%) | 75 (15.0%) | 38 (14.7%) | 0.91 |
| Hospice, no. (%) | 8 (1.6%) | 5 (1.9%) | 0.73 |
| Do not resuscitate, no. (%) | 62 (12.4%) | 30 (11.6%) | 0.76 |
| Intensive care admission, no. (%) | 160 (31.9%) | 116 (44.8%) | <0.001 |
| ACLS administered, no. (%) | 38 (7.6%) | 17 (6.6%) | 0.6 |
| Outcomes, no. (%) | |||
| Death, 30 days | 67 (13.3%) | 30 (11.6%) | 0.49 |
| Death, 1 year | 211 (42.0%) | 75 (29.0%) | <0.001 |
| Death, 6 years | 390 (77.7%) | 152 (58.7%) | <0.001 |
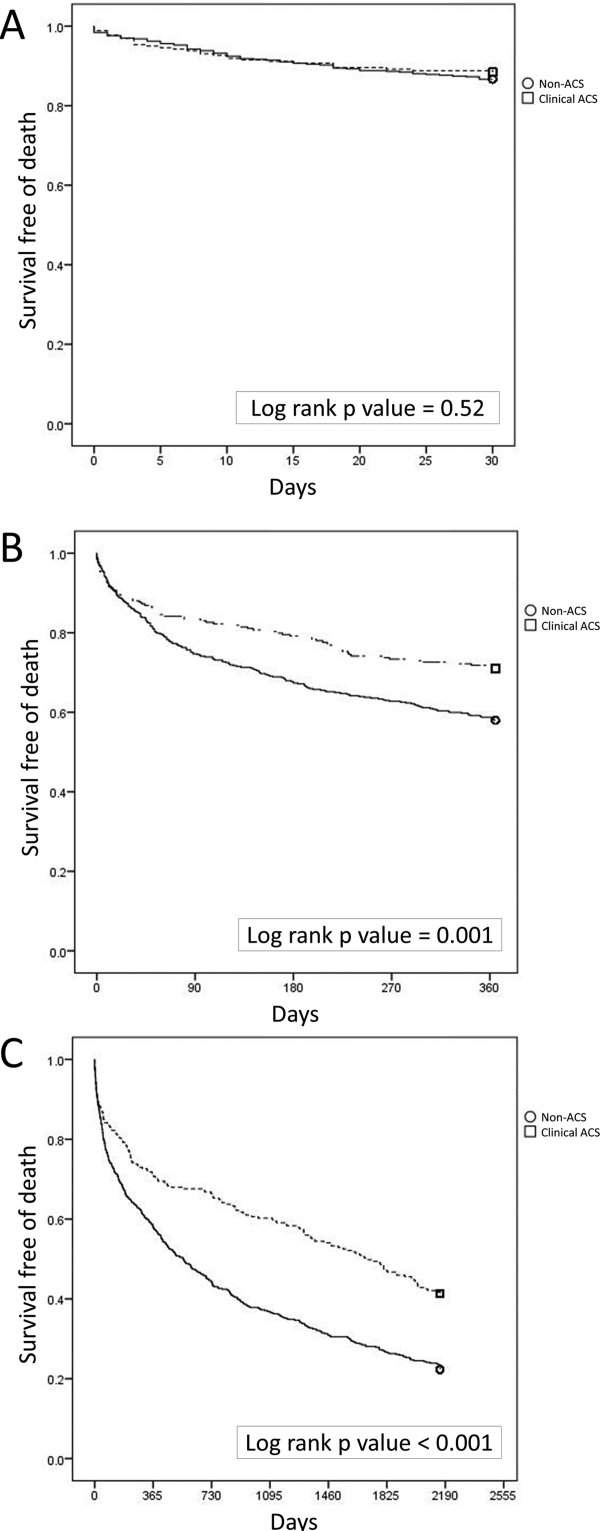
Mortality at 30 days was not different between the 2 groups, but mortality was higher for the non‐ACS cohort at 1 year and at 6 years (Table 1). Kaplan‐Meier curves demonstrate that mortality for the 2 cohorts begins to diverge between 30 and 60 days until approximately 2 years when the curves again are parallel (Figure 2).
In Cox proportional hazards models, 5 factors were associated with higher mortality at 30 days, 1 year, and at 6 years: age, hospice, DNR order, need for ACLS, and admission to the ICU (Table 2). Additionally, at 1 and 6 years, NT‐proBNP and non‐ACS were associated with higher mortality. At 6 years, creatinine was an additional significant factor. We separated the ACS and non‐ACS cohorts and performed the same model for 1‐year mortality (Table 3). The models yielded similar factors associated with higher mortality: hospice, DNR order, need for ACLS, age, and NT‐proBNP, with ICU admission being significant only in the non‐ACS cohort.
| P Value | Hazard Ratio | 95% CI | |
|---|---|---|---|
| |||
| 30 days | |||
| Intensive care unit admission | <0.0001 | 2.18 | 1.283.72 |
| Hospice | <0.0001 | 4.67 | 1.9111.40 |
| Do not resuscitate | <0.0001 | 3.19 | 1.945.24 |
| ACLS performed | <0.0001 | 10.17 | 6.0317.17 |
| Age, per year | <0.0001 | 1.04 | 1.021.06 |
| 1 year | |||
| Intensive care unit admission | <0.0001 | 1.66 | 1.262.20 |
| Hospice | <0.0001 | 4.98 | 2.699.21 |
| Do not resuscitate | <0.0001 | 2.52 | 1.833.47 |
| Non‐ACS | <0.0001 | 1.57 | 1.192.08 |
| ACLS performed | <0.0001 | 6.03 | 4.178.72 |
| Age, per year | <0.0001 | 1.03 | 1.021.04 |
| NT‐proBNP, per 1,000 pg/mL | <0.0001 | 1.02 | 1.011.03 |
| Extended follow‐up | |||
| Intensive care unit admission | <0.0001 | 1.35 | 1.111.65 |
| Hospice | <0.0001 | 3.81 | 2.136.81 |
| Do not resuscitate | <0.0001 | 2.11 | 1.622.74 |
| Non‐ACS | <0.0001 | 1.53 | 1.251.88 |
| ACLS performed | <0.0001 | 4.19 | 3.015.84 |
| Age, per year | <0.0001 | 1.03 | 1.031.04 |
| Creatinine, per mg/dL | 0.02 | 1.06 | 1.011.12 |
| NT‐proBNP, per 1,000 pg/mL | <0.0001 | 1.02 | 1.021.03 |
| P Value | Hazard Ratio | 95% CI | |
|---|---|---|---|
| |||
| Non‐ACS | |||
| Intensive care unit admission | <0.0001 | 1.86 | 1.352.58 |
| Hospice | <0.0001 | 7.55 | 3.5715.93 |
| Do not resuscitate | <0.0001 | 2.33 | 1.603.41 |
| ACLS performed | <0.0001 | 4.42 | 2.836.92 |
| Age, per year | <0.0001 | 1.03 | 1.011.04 |
| NT‐proBNP, per 1,000 pg/mL | 0.002 | 1.02 | 1.011.03 |
| Clinical ACS | |||
| Hospice | 0.036 | 3.17 | 1.089.32 |
| Do not resuscitate | 0.003 | 2.49 | 1.364.55 |
| ACLS performed | <0.0001 | 12.04 | 6.3322.91 |
| Age, per year | <0.0001 | 1.05 | 1.021.07 |
| NT‐proBNP, per 1,000 pg/mL | 0.001 | 1.04 | 1.011.06 |
DISCUSSION
Our findings confirm the important, but perhaps not well‐recognized, fact that an elevated troponin without ACS is associated with higher mortality than with ACS. This has been previously observed in veteran and nonveteran populations.[4, 6, 8, 12] The novel finding from our investigation is that mortality risk with troponin elevation is most strongly associated with unmodifiable clinical factors that are plausible explanations of risk. Furthermore, the distribution of these factors between our 2 cohorts does not sufficiently explain the difference in risk between ACS and non‐ACS patients.
At each time point we evaluated, ICU admission and need for ACLS were associated with mortality. These are indicators of a severely ill population and are not surprising to find associated with mortality. Many hospitals have instituted some form of pre‐code approach or rapid response team to identify patients before they need ACLS. These efforts, although well meaning, have not yielded convincing results of effectiveness.[13] Hospice and DNR patients were also, not surprisingly, associated with higher mortality. Although these factors were statistically significant, the low prevalence suggests that they are not clinically impactful on the primary questions of the investigation. These factors can be altered but are not intended as modifiable as they reflect the wishes of patients and their decision makers. The distribution of the factors in our model, however, did not adequately explain the higher risk of death with non‐ACS troponin elevation. For example, ACLS administration, hospice care, and DNR orders were strong predictors but were similar between the groups. ICU admission was actually more common with ACS patients, despite strong association with mortality. Age and NT‐proBNP were associated with mortality and higher in the non‐ACS group; however the magnitude of hazard was less than for the other factors. These findings lead us back to the possible explanation that non‐ACS troponin elevation stands as an independent risk factor, and that ACS patients have a distinct advantage in the myriad treatments available. If ACS patients were misdiagnosed as non‐ACS and failed to receive appropriate treatments, that might have contributed to higher mortality; however, we consider that unlikely given that the goal of the QI project was to minimize missed ACS diagnoses.
The overall mortality risk in our study was high: 12.7% at 30 days and 37.6% at 1 year. This reflects the high‐risk population with elevated troponin seen at our facility with ages nearly 70 years and high prevalence of multiple cardiovascular risk factors. Despite a high event rate, many clinically relevant risk factors were not retained in our Cox hazard model. Among sepsis patients, elevation in troponin is associated with mortality[14]; however, in our population neither fever or white blood cell count were significant mortality factors. The relationship between chronic kidney disease and troponin is complex. Renal dysfunction may result in troponin elevation and troponin elevation is a predictor of risk within kidney disease patients.[15] In our study, we did not evaluate chronic kidney disease as a predictor, instead opting to use the serum creatinine. This was not associated with mortality except at the 6‐year time point.
The TIMI score was not associated with mortality in either the overall population or the ACS cohort. The proportion of patients in our cohort with TIMI score under 3 was 16.5% as compared with 21.6% in the original derivation study.[16] The limited data on the prognostic value of the TIMI score within a veteran population suggest a modest predictive capacity.[17] Our data raises the possibility that TIMI is not an optimal choice; however, our analysis only includes all‐cause mortality, different from the original intended use of TIMI, predicting a variety of major cardiac events.
Our data confirm that ACS can be detected in a wide range of clinical presentations. Within our population of troponin positive patients, those with chest pain were most likely to be diagnosed with ACS, although one‐third of chest pain patients were felt to have a non‐ACS diagnosis. On the opposite end of the spectrum, an elevation in troponin with altered mental status or confusion was rarely diagnosed as ACSonly 18% of the time. Many symptoms were poor predictors of ACS; however, none were low enough to disregard. Our data would suggest that most patients with elevated troponin warrant evaluation by a cardiovascular expert.
Our study population came from a single VA hospital that is comprised of elderly and predominantly male patients limiting applicability to other populations. Despite this, other investigations in younger populations and with a higher proportion of women have found similar mortality trends.[4, 8, 12] We did not have sufficient data to determine the cause of death or to further classify as cardiac versus noncardiac; knowledge of the cause of the specific death may better inform future investigations into this important clinical question. Our investigation did not use a standardized definition to determine ACS, a notable limitation that could introduce bias or variation in care. Because all determinations about ACS were made prospectively as part of a QI project, we have little reason to suspect any systematic bias to the determination of ACS. With regard to variation in care, we have previously presented data demonstrating consistent rates of ACS diagnosis across the physicians at our facility.
Based on our investigation and others on this topic, non‐ACS troponin elevation is a common, high‐risk clinical scenario. In our cohort, non‐ACS troponin elevation is about twice as frequent as ACS, and the problem is likely to grow dramatically within the next few years as ultrasensitive troponin assays are eventually approved for use in the United States. These assays are much more sensitive than the current assays, and may make it challenging to distinguish between someone with an acute supply/demand mismatch from someone with an elevated troponin due to chronic, but stable, illness such as CAD, heart failure, or diabetes. Non‐ACS troponin elevation remains poorly understood, with no viable treatment options other than addressing the pathophysiology resulting in the troponin elevation. Due to the heterogeneity of the diagnoses and pathophysiological conditions that result in elevated troponin, a unifying treatment is not likely feasible.
In conclusion, in this elderly, male veteran population, the mortality impact associated with a cardiac troponin elevation was not limited to ACS, as mortality was high among those without ACS. Factors independently associated with this non‐ACS mortality risk were plausible, but did not elucidate the reasons why non‐ACS troponin elevation carries a higher risk. Attempting to better understand the biological basis for the troponin elevation in these non‐ACS patients is a critical unmet need.
Disclosure
Nothing to report.
- , , , , . Prognostic significance of elevated troponin in non‐cardiac hospitalized patients: a systematic review and meta‐analysis. Ann Med. 2014;46:653–663.
- , , , , , ; Joint ESC/ACCF/AHA/WHF Task Force for Universal Definition of Myocardial Infarction. Third universal definition of myocardial infarction. J Am Coll Cardiol. 2012;60:1581–1598.
- , , , et al.; ACC/AHA Task Force Members; Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. 2014 AHA/ACC guideline for the management of patients with non‐st‐elevation acute coronary syndromes: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;130:2354–2394.
- , , , , , . Acute coronary syndrome vs nonspecific troponin elevation: clinical predictors and survival analysis. Arch Intern Med. 2007;167:276–281.
- , , , , , , TOTAL‐AMI study group. Type 2 myocardial infarction in clinical practice. Heart. 2015;101:101–106.
- , , , et al. Outcomes of hospitalized patients with non‐acute coronary syndrome and elevated cardiac troponin level. Am J Med. 2011;124:630–635.
- , , , et al. Classification of myocardial infarction: frequency and features of type 2 myocardial infarction. Am J Med. 2013;126:789–797.
- , , , , . Cardiac troponin I elevation in hospitalized patients without acute coronary syndromes. Am J Cardiol. 2008;101:1384–1388.
- , , , . Inter‐provider variation in diagnoses and cardiac catheterization use (abstract). Cardiology. 2014;128:346.
- , , , . A primer and comparative review of major us mortality databases. Ann Epidemiol. 2002;12:462–468.
- , , . Assessment of vital status in department of veterans affairs national databases. Comparison with state death certificates. Ann Epidemiol. 2001;11:286–291.
- , , , , , . Raised cardiac troponin T levels in patients without acute coronary syndrome. Postgrad Med J. 2007;83:200–205.
- , , , , . Rapid response teams: a systematic review and meta‐analysis. Arch Intern Med. 2010;170:18–26.
- , , , , . Prognostic value of troponins in sepsis: a meta‐analysis. Intensive Care Med. 2013;39:1181–1189.
- , , , et al. Prognostic value of cardiac troponin in patients with chronic kidney disease without suspected acute coronary syndrome: a systematic review and meta‐analysis. Ann Intern Med. 2014;161:491–501.
- , , , et al. The TIMI risk score for unstable angina/non‐ST elevation MI: a method for prognostication and therapeutic decision making. JAMA. 2000;284:835–842.
- , , , , , . Usefulness of the TIMI risk score in predicting both short‐ and long‐term outcomes in the Veterans Affairs non‐Q‐wave myocardial infarction strategies in‐hospital (VANQWISH) trial. Am J Cardiol. 2002;90:922–926.
Acute coronary syndromes (ACS) are potentially lethal and present with a wide variety of symptoms. As such, physicians frequently order cardiac biomarkers, such as cardiac troponin, for patients with acute complaints. Elevated troponin is associated with higher risk of mortality regardless of the causes, which can be myriad, both chronic and acute.[1] Among patients with an elevated troponin, distinguishing ACS from non‐ACS can be challenging.
Making the distinction between ACS and non‐ACS troponin elevation is crucial because the underlying pathophysiology and subsequent management strategies are markedly different.[2] According to evidence‐based practice guidelines, ACS is managed with antiplatelet drugs, statins, and percutaneous coronary intervention, improving clinical outcomes.[3] In contrast, care for patients with non‐ACS troponin elevations is usually supportive, with a focus on the underlying conditions. The lack of specific treatment options for such patients is concerning given that several series have suggested that non‐ACS troponin patients may have a higher mortality risk than ACS patients.[4, 5, 6] Non‐ACS troponin elevation can be the result of a multitude of conditions.[7, 8] What remains unclear at this point is whether the excess mortality observed with non‐ACS troponin elevation is due to myocardial damage or to the underlying conditions that predispose to troponin release.
Using data from a quality improvement (QI) project collected at our Veterans Affairs (VA) medical center, we investigated the mortality risk associated with ACS and non‐ACS troponin elevation including an analysis of factors associated with mortality. We hypothesized that non‐ACS troponin elevation will have a higher mortality risk than troponin elevation due to ACS, and that important contributors to this relationship could be identified to provide direction for future investigation directed at modifying this mortality risk.
METHODS
We analyzed data that were prospectively collected for a quality initiative between 2006 and 2007. The project was a collaborative endeavor between cardiology, hospital medicine, and emergency medicine with the process goal of better identifying patients with ACS to hopefully improve outcomes. The QI team was consulted in real time to assist with treatment recommendations; no retrospective decisions were made regarding whether or not ACS was present. As the goal of the project was to improve cardiovascular outcomes, consultative advice was freely provided, and no physicians or teams were subject to any adverse repercussions for their diagnoses or management decisions.
A cardiologist‐led team was created to improve quality of care for myocardial infarction patients by evaluating all patients at our facility with an elevated troponin. On a daily basis, a specialist clinical coordinator (nurse practitioner or physician assistant) received a list of all patients with elevated troponin from the chemistry lab. The coordinator reviewed the patients' medical records with a cardiologist. A positive troponin was defined as a troponin T level of greater than 0.03 ng/mL (99th percentile at our facility). Each attending cardiologist prospectively determined if troponin elevation was related to clinical findings consistent with an ACS based on review of the patients' symptoms (duration, quality, severity, chronicity, and alleviating/aggravating factors), medical history, and noninvasive cardiac testing including electrocardiograms, cardiac biomarkers, and any other available imaging tests.
We have previously demonstrated that the cardiologists at our facility have a similar rate of diagnosing ACS.[9] All cardiologists at our facility maintain current American Board of Internal Medicine certification in cardiovascular disease and have academic appointments at the University of Florida College of Medicine. All patients were followed prospectively, and data on their medical history, acute evaluation, and outcomes were tracked in an electronic database. Given the higher risk of mortality with ST‐elevation myocardial infarction, such patients were excluded from this investigation. By definition, patients with unstable angina do not have elevated biomarkers and thus would not have been included in the database to begin with. Prospectively recorded data elements included: age, gender, chief complaint, tobacco use, presence of hypertension, hyperlipidemia, prior coronary disease, chronic kidney disease, diabetes mellitus, cardiac troponin values, serum creatinine, electrocardiogram (ECG) variables, Thrombolysis in Myocardial Infarction (TIMI) score, and if the patient was placed under hospice care or an active do‐not‐resuscitate (DNR) order. Additional data elements gathered at a later date included maximum temperature, white blood cell count, N‐terminal pro‐brain natriuretic peptide (NT‐proBNP), administration of advanced cardiac life support (ACLS), and admission to an intensive care unit (ICU). All consecutive patients with elevated troponin were included in the database; if patients were included more than once, we used their index evaluation only. All patients with troponin elevation after revascularization (percutaneous coronary intervention or coronary bypass surgery) were excluded. Our investigational design was reviewed by our institutional review board, who waived the requirement for formal written informed consent and approved use of data from this QI project for research purposes.
We focused this investigation on an analysis of all‐cause mortality in February 2014. We analyzed mortality at 30 days, 1 year, and 6 years. As secondary outcomes we analyzed the likelihood of the patients' chief complaint for the diagnosis of ACS and evaluated predictors of mortality based on Cox proportional hazard modeling. Mortality within the VA system is reliably tracked and compares favorably to the Social Security National Death Index Master File for accuracy.[10, 11]
Categorical variables were compared by 2 test. The Student t test was used to compare normally distributed continuous variables, and nonparametric tests were used for non‐normal distributions as appropriate. Mortality data at 30 days, 1 year, and 6 years were compared by log‐rank test and Kaplan‐Meier graphs. A formal power analysis was not performed; the entire available population was included. A Cox proportional hazard model was created to estimate mortality risk at each time point. Variables included in our Cox regression model were age, gender, history of coronary artery disease (CAD), hypertension, diabetes mellitus or hyperlipidemia, ACS diagnosis, dynamic ECG changes, TIMI risk score, initial troponin level, creatinine level at time of initial troponin (per mg/dL), presence of fever, maximum white blood cell count, NT‐proBNP level (per 1000 pg/mL), if ACLS was performed, if the patient was under hospice care, if there was a DNR order, and if they required ICU admission. This model was also constructed independently for the ACS and non‐ACS cohorts for mortality at 1 year. A forward stepwise model was used. Statistical results were considered significant at P < 0.05. Statistical analyses were performed using SPSS version 21 (IBM, Armonk, NY).
RESULTS
Among the 761 patients, 502 (66.0%) were classified as non‐ACS and 259 (34.0%) as ACS (Table 1). The mean age was higher in the non‐ACS group (71 years vs 69 years in the ACS group, P = 0.006). Hypertension, diabetes mellitus, and prior CAD were frequent in both groups and not significantly different. Median initial troponin T was higher in the ACS group (0.12 ng/mL vs 0.06 ng/mL, P < 0.001) as were the frequency of a TIMI risk score >2 (92.5% vs 74.3%, P < 0.001) and new ECG changes (29.7% vs 8.2%, P < 0.001). Hospice, DNR orders, and administration of ACLS were not different between groups; however, admission to the ICU was more frequent in the ACS group (44.8% vs 31.9%, P < 0.001). Chest pain was the symptom with the highest positive predictive value for the diagnosis of ACS (63.3%), whereas the least predictive was altered mental status or confusion (18.0%) (Figure 1).
| Non‐ACS, N = 502 | ACS, N = 259 | P Value | |
|---|---|---|---|
| |||
| Baseline characteristics, n (%) | |||
| Age, y | 71 11 | 69 11 | 0.006 |
| Female | 6 (1.2%) | 1 (0.4%) | 0.27 |
| Coronary artery disease | 244 (48.6%) | 141 (54.4%) | 0.13 |
| Hypertension | 381 (75.9%) | 203 (78.4%) | 0.44 |
| Diabetes mellitus | 220 (43.8%) | 119 (45.9%) | 0.58 |
| Hyperlipidemia | 268 (53.4%) | 170 (65.6%) | 0.001 |
| Current smoker | 24 (4.8%) | 49 (18.9%) | <0.001 |
| Clinical presentation | |||
| Initial troponin T, ng/mL, median [IQR] | 0.06 [0.040.11] | 0.12 [0.050.32] | <0.001 |
| White cell count, 109/L, median [IQR] | 10 [8.014.0] | 11 [8.015.0] | 0.005 |
| NT‐proBNP, pg/mL, median [IQR] | 3,531 [1,20110,519] | 1,932 [3199,100] | 0.001 |
| Creatinine, mg/dL, median [IQR] | 1.6 [1.12.4] | 1.1 [0.91.5] | <0.001 |
| New ECG changes, no. (%) | 41 (8.2%) | 77 (29.7%) | <0.001 |
| TIMI score over 2, no. (%) | 365 (74.3%) | 235 (92.5%) | <0.001 |
| Fever (over 100.4 F), no. (%) | 75 (15.0%) | 38 (14.7%) | 0.91 |
| Hospice, no. (%) | 8 (1.6%) | 5 (1.9%) | 0.73 |
| Do not resuscitate, no. (%) | 62 (12.4%) | 30 (11.6%) | 0.76 |
| Intensive care admission, no. (%) | 160 (31.9%) | 116 (44.8%) | <0.001 |
| ACLS administered, no. (%) | 38 (7.6%) | 17 (6.6%) | 0.6 |
| Outcomes, no. (%) | |||
| Death, 30 days | 67 (13.3%) | 30 (11.6%) | 0.49 |
| Death, 1 year | 211 (42.0%) | 75 (29.0%) | <0.001 |
| Death, 6 years | 390 (77.7%) | 152 (58.7%) | <0.001 |
Mortality at 30 days was not different between the 2 groups, but mortality was higher for the non‐ACS cohort at 1 year and at 6 years (Table 1). Kaplan‐Meier curves demonstrate that mortality for the 2 cohorts begins to diverge between 30 and 60 days until approximately 2 years when the curves again are parallel (Figure 2).
In Cox proportional hazards models, 5 factors were associated with higher mortality at 30 days, 1 year, and at 6 years: age, hospice, DNR order, need for ACLS, and admission to the ICU (Table 2). Additionally, at 1 and 6 years, NT‐proBNP and non‐ACS were associated with higher mortality. At 6 years, creatinine was an additional significant factor. We separated the ACS and non‐ACS cohorts and performed the same model for 1‐year mortality (Table 3). The models yielded similar factors associated with higher mortality: hospice, DNR order, need for ACLS, age, and NT‐proBNP, with ICU admission being significant only in the non‐ACS cohort.
| P Value | Hazard Ratio | 95% CI | |
|---|---|---|---|
| |||
| 30 days | |||
| Intensive care unit admission | <0.0001 | 2.18 | 1.283.72 |
| Hospice | <0.0001 | 4.67 | 1.9111.40 |
| Do not resuscitate | <0.0001 | 3.19 | 1.945.24 |
| ACLS performed | <0.0001 | 10.17 | 6.0317.17 |
| Age, per year | <0.0001 | 1.04 | 1.021.06 |
| 1 year | |||
| Intensive care unit admission | <0.0001 | 1.66 | 1.262.20 |
| Hospice | <0.0001 | 4.98 | 2.699.21 |
| Do not resuscitate | <0.0001 | 2.52 | 1.833.47 |
| Non‐ACS | <0.0001 | 1.57 | 1.192.08 |
| ACLS performed | <0.0001 | 6.03 | 4.178.72 |
| Age, per year | <0.0001 | 1.03 | 1.021.04 |
| NT‐proBNP, per 1,000 pg/mL | <0.0001 | 1.02 | 1.011.03 |
| Extended follow‐up | |||
| Intensive care unit admission | <0.0001 | 1.35 | 1.111.65 |
| Hospice | <0.0001 | 3.81 | 2.136.81 |
| Do not resuscitate | <0.0001 | 2.11 | 1.622.74 |
| Non‐ACS | <0.0001 | 1.53 | 1.251.88 |
| ACLS performed | <0.0001 | 4.19 | 3.015.84 |
| Age, per year | <0.0001 | 1.03 | 1.031.04 |
| Creatinine, per mg/dL | 0.02 | 1.06 | 1.011.12 |
| NT‐proBNP, per 1,000 pg/mL | <0.0001 | 1.02 | 1.021.03 |
| P Value | Hazard Ratio | 95% CI | |
|---|---|---|---|
| |||
| Non‐ACS | |||
| Intensive care unit admission | <0.0001 | 1.86 | 1.352.58 |
| Hospice | <0.0001 | 7.55 | 3.5715.93 |
| Do not resuscitate | <0.0001 | 2.33 | 1.603.41 |
| ACLS performed | <0.0001 | 4.42 | 2.836.92 |
| Age, per year | <0.0001 | 1.03 | 1.011.04 |
| NT‐proBNP, per 1,000 pg/mL | 0.002 | 1.02 | 1.011.03 |
| Clinical ACS | |||
| Hospice | 0.036 | 3.17 | 1.089.32 |
| Do not resuscitate | 0.003 | 2.49 | 1.364.55 |
| ACLS performed | <0.0001 | 12.04 | 6.3322.91 |
| Age, per year | <0.0001 | 1.05 | 1.021.07 |
| NT‐proBNP, per 1,000 pg/mL | 0.001 | 1.04 | 1.011.06 |
DISCUSSION
Our findings confirm the important, but perhaps not well‐recognized, fact that an elevated troponin without ACS is associated with higher mortality than with ACS. This has been previously observed in veteran and nonveteran populations.[4, 6, 8, 12] The novel finding from our investigation is that mortality risk with troponin elevation is most strongly associated with unmodifiable clinical factors that are plausible explanations of risk. Furthermore, the distribution of these factors between our 2 cohorts does not sufficiently explain the difference in risk between ACS and non‐ACS patients.
At each time point we evaluated, ICU admission and need for ACLS were associated with mortality. These are indicators of a severely ill population and are not surprising to find associated with mortality. Many hospitals have instituted some form of pre‐code approach or rapid response team to identify patients before they need ACLS. These efforts, although well meaning, have not yielded convincing results of effectiveness.[13] Hospice and DNR patients were also, not surprisingly, associated with higher mortality. Although these factors were statistically significant, the low prevalence suggests that they are not clinically impactful on the primary questions of the investigation. These factors can be altered but are not intended as modifiable as they reflect the wishes of patients and their decision makers. The distribution of the factors in our model, however, did not adequately explain the higher risk of death with non‐ACS troponin elevation. For example, ACLS administration, hospice care, and DNR orders were strong predictors but were similar between the groups. ICU admission was actually more common with ACS patients, despite strong association with mortality. Age and NT‐proBNP were associated with mortality and higher in the non‐ACS group; however the magnitude of hazard was less than for the other factors. These findings lead us back to the possible explanation that non‐ACS troponin elevation stands as an independent risk factor, and that ACS patients have a distinct advantage in the myriad treatments available. If ACS patients were misdiagnosed as non‐ACS and failed to receive appropriate treatments, that might have contributed to higher mortality; however, we consider that unlikely given that the goal of the QI project was to minimize missed ACS diagnoses.
The overall mortality risk in our study was high: 12.7% at 30 days and 37.6% at 1 year. This reflects the high‐risk population with elevated troponin seen at our facility with ages nearly 70 years and high prevalence of multiple cardiovascular risk factors. Despite a high event rate, many clinically relevant risk factors were not retained in our Cox hazard model. Among sepsis patients, elevation in troponin is associated with mortality[14]; however, in our population neither fever or white blood cell count were significant mortality factors. The relationship between chronic kidney disease and troponin is complex. Renal dysfunction may result in troponin elevation and troponin elevation is a predictor of risk within kidney disease patients.[15] In our study, we did not evaluate chronic kidney disease as a predictor, instead opting to use the serum creatinine. This was not associated with mortality except at the 6‐year time point.
The TIMI score was not associated with mortality in either the overall population or the ACS cohort. The proportion of patients in our cohort with TIMI score under 3 was 16.5% as compared with 21.6% in the original derivation study.[16] The limited data on the prognostic value of the TIMI score within a veteran population suggest a modest predictive capacity.[17] Our data raises the possibility that TIMI is not an optimal choice; however, our analysis only includes all‐cause mortality, different from the original intended use of TIMI, predicting a variety of major cardiac events.
Our data confirm that ACS can be detected in a wide range of clinical presentations. Within our population of troponin positive patients, those with chest pain were most likely to be diagnosed with ACS, although one‐third of chest pain patients were felt to have a non‐ACS diagnosis. On the opposite end of the spectrum, an elevation in troponin with altered mental status or confusion was rarely diagnosed as ACSonly 18% of the time. Many symptoms were poor predictors of ACS; however, none were low enough to disregard. Our data would suggest that most patients with elevated troponin warrant evaluation by a cardiovascular expert.
Our study population came from a single VA hospital that is comprised of elderly and predominantly male patients limiting applicability to other populations. Despite this, other investigations in younger populations and with a higher proportion of women have found similar mortality trends.[4, 8, 12] We did not have sufficient data to determine the cause of death or to further classify as cardiac versus noncardiac; knowledge of the cause of the specific death may better inform future investigations into this important clinical question. Our investigation did not use a standardized definition to determine ACS, a notable limitation that could introduce bias or variation in care. Because all determinations about ACS were made prospectively as part of a QI project, we have little reason to suspect any systematic bias to the determination of ACS. With regard to variation in care, we have previously presented data demonstrating consistent rates of ACS diagnosis across the physicians at our facility.
Based on our investigation and others on this topic, non‐ACS troponin elevation is a common, high‐risk clinical scenario. In our cohort, non‐ACS troponin elevation is about twice as frequent as ACS, and the problem is likely to grow dramatically within the next few years as ultrasensitive troponin assays are eventually approved for use in the United States. These assays are much more sensitive than the current assays, and may make it challenging to distinguish between someone with an acute supply/demand mismatch from someone with an elevated troponin due to chronic, but stable, illness such as CAD, heart failure, or diabetes. Non‐ACS troponin elevation remains poorly understood, with no viable treatment options other than addressing the pathophysiology resulting in the troponin elevation. Due to the heterogeneity of the diagnoses and pathophysiological conditions that result in elevated troponin, a unifying treatment is not likely feasible.
In conclusion, in this elderly, male veteran population, the mortality impact associated with a cardiac troponin elevation was not limited to ACS, as mortality was high among those without ACS. Factors independently associated with this non‐ACS mortality risk were plausible, but did not elucidate the reasons why non‐ACS troponin elevation carries a higher risk. Attempting to better understand the biological basis for the troponin elevation in these non‐ACS patients is a critical unmet need.
Disclosure
Nothing to report.
Acute coronary syndromes (ACS) are potentially lethal and present with a wide variety of symptoms. As such, physicians frequently order cardiac biomarkers, such as cardiac troponin, for patients with acute complaints. Elevated troponin is associated with higher risk of mortality regardless of the causes, which can be myriad, both chronic and acute.[1] Among patients with an elevated troponin, distinguishing ACS from non‐ACS can be challenging.
Making the distinction between ACS and non‐ACS troponin elevation is crucial because the underlying pathophysiology and subsequent management strategies are markedly different.[2] According to evidence‐based practice guidelines, ACS is managed with antiplatelet drugs, statins, and percutaneous coronary intervention, improving clinical outcomes.[3] In contrast, care for patients with non‐ACS troponin elevations is usually supportive, with a focus on the underlying conditions. The lack of specific treatment options for such patients is concerning given that several series have suggested that non‐ACS troponin patients may have a higher mortality risk than ACS patients.[4, 5, 6] Non‐ACS troponin elevation can be the result of a multitude of conditions.[7, 8] What remains unclear at this point is whether the excess mortality observed with non‐ACS troponin elevation is due to myocardial damage or to the underlying conditions that predispose to troponin release.
Using data from a quality improvement (QI) project collected at our Veterans Affairs (VA) medical center, we investigated the mortality risk associated with ACS and non‐ACS troponin elevation including an analysis of factors associated with mortality. We hypothesized that non‐ACS troponin elevation will have a higher mortality risk than troponin elevation due to ACS, and that important contributors to this relationship could be identified to provide direction for future investigation directed at modifying this mortality risk.
METHODS
We analyzed data that were prospectively collected for a quality initiative between 2006 and 2007. The project was a collaborative endeavor between cardiology, hospital medicine, and emergency medicine with the process goal of better identifying patients with ACS to hopefully improve outcomes. The QI team was consulted in real time to assist with treatment recommendations; no retrospective decisions were made regarding whether or not ACS was present. As the goal of the project was to improve cardiovascular outcomes, consultative advice was freely provided, and no physicians or teams were subject to any adverse repercussions for their diagnoses or management decisions.
A cardiologist‐led team was created to improve quality of care for myocardial infarction patients by evaluating all patients at our facility with an elevated troponin. On a daily basis, a specialist clinical coordinator (nurse practitioner or physician assistant) received a list of all patients with elevated troponin from the chemistry lab. The coordinator reviewed the patients' medical records with a cardiologist. A positive troponin was defined as a troponin T level of greater than 0.03 ng/mL (99th percentile at our facility). Each attending cardiologist prospectively determined if troponin elevation was related to clinical findings consistent with an ACS based on review of the patients' symptoms (duration, quality, severity, chronicity, and alleviating/aggravating factors), medical history, and noninvasive cardiac testing including electrocardiograms, cardiac biomarkers, and any other available imaging tests.
We have previously demonstrated that the cardiologists at our facility have a similar rate of diagnosing ACS.[9] All cardiologists at our facility maintain current American Board of Internal Medicine certification in cardiovascular disease and have academic appointments at the University of Florida College of Medicine. All patients were followed prospectively, and data on their medical history, acute evaluation, and outcomes were tracked in an electronic database. Given the higher risk of mortality with ST‐elevation myocardial infarction, such patients were excluded from this investigation. By definition, patients with unstable angina do not have elevated biomarkers and thus would not have been included in the database to begin with. Prospectively recorded data elements included: age, gender, chief complaint, tobacco use, presence of hypertension, hyperlipidemia, prior coronary disease, chronic kidney disease, diabetes mellitus, cardiac troponin values, serum creatinine, electrocardiogram (ECG) variables, Thrombolysis in Myocardial Infarction (TIMI) score, and if the patient was placed under hospice care or an active do‐not‐resuscitate (DNR) order. Additional data elements gathered at a later date included maximum temperature, white blood cell count, N‐terminal pro‐brain natriuretic peptide (NT‐proBNP), administration of advanced cardiac life support (ACLS), and admission to an intensive care unit (ICU). All consecutive patients with elevated troponin were included in the database; if patients were included more than once, we used their index evaluation only. All patients with troponin elevation after revascularization (percutaneous coronary intervention or coronary bypass surgery) were excluded. Our investigational design was reviewed by our institutional review board, who waived the requirement for formal written informed consent and approved use of data from this QI project for research purposes.
We focused this investigation on an analysis of all‐cause mortality in February 2014. We analyzed mortality at 30 days, 1 year, and 6 years. As secondary outcomes we analyzed the likelihood of the patients' chief complaint for the diagnosis of ACS and evaluated predictors of mortality based on Cox proportional hazard modeling. Mortality within the VA system is reliably tracked and compares favorably to the Social Security National Death Index Master File for accuracy.[10, 11]
Categorical variables were compared by 2 test. The Student t test was used to compare normally distributed continuous variables, and nonparametric tests were used for non‐normal distributions as appropriate. Mortality data at 30 days, 1 year, and 6 years were compared by log‐rank test and Kaplan‐Meier graphs. A formal power analysis was not performed; the entire available population was included. A Cox proportional hazard model was created to estimate mortality risk at each time point. Variables included in our Cox regression model were age, gender, history of coronary artery disease (CAD), hypertension, diabetes mellitus or hyperlipidemia, ACS diagnosis, dynamic ECG changes, TIMI risk score, initial troponin level, creatinine level at time of initial troponin (per mg/dL), presence of fever, maximum white blood cell count, NT‐proBNP level (per 1000 pg/mL), if ACLS was performed, if the patient was under hospice care, if there was a DNR order, and if they required ICU admission. This model was also constructed independently for the ACS and non‐ACS cohorts for mortality at 1 year. A forward stepwise model was used. Statistical results were considered significant at P < 0.05. Statistical analyses were performed using SPSS version 21 (IBM, Armonk, NY).
RESULTS
Among the 761 patients, 502 (66.0%) were classified as non‐ACS and 259 (34.0%) as ACS (Table 1). The mean age was higher in the non‐ACS group (71 years vs 69 years in the ACS group, P = 0.006). Hypertension, diabetes mellitus, and prior CAD were frequent in both groups and not significantly different. Median initial troponin T was higher in the ACS group (0.12 ng/mL vs 0.06 ng/mL, P < 0.001) as were the frequency of a TIMI risk score >2 (92.5% vs 74.3%, P < 0.001) and new ECG changes (29.7% vs 8.2%, P < 0.001). Hospice, DNR orders, and administration of ACLS were not different between groups; however, admission to the ICU was more frequent in the ACS group (44.8% vs 31.9%, P < 0.001). Chest pain was the symptom with the highest positive predictive value for the diagnosis of ACS (63.3%), whereas the least predictive was altered mental status or confusion (18.0%) (Figure 1).
| Non‐ACS, N = 502 | ACS, N = 259 | P Value | |
|---|---|---|---|
| |||
| Baseline characteristics, n (%) | |||
| Age, y | 71 11 | 69 11 | 0.006 |
| Female | 6 (1.2%) | 1 (0.4%) | 0.27 |
| Coronary artery disease | 244 (48.6%) | 141 (54.4%) | 0.13 |
| Hypertension | 381 (75.9%) | 203 (78.4%) | 0.44 |
| Diabetes mellitus | 220 (43.8%) | 119 (45.9%) | 0.58 |
| Hyperlipidemia | 268 (53.4%) | 170 (65.6%) | 0.001 |
| Current smoker | 24 (4.8%) | 49 (18.9%) | <0.001 |
| Clinical presentation | |||
| Initial troponin T, ng/mL, median [IQR] | 0.06 [0.040.11] | 0.12 [0.050.32] | <0.001 |
| White cell count, 109/L, median [IQR] | 10 [8.014.0] | 11 [8.015.0] | 0.005 |
| NT‐proBNP, pg/mL, median [IQR] | 3,531 [1,20110,519] | 1,932 [3199,100] | 0.001 |
| Creatinine, mg/dL, median [IQR] | 1.6 [1.12.4] | 1.1 [0.91.5] | <0.001 |
| New ECG changes, no. (%) | 41 (8.2%) | 77 (29.7%) | <0.001 |
| TIMI score over 2, no. (%) | 365 (74.3%) | 235 (92.5%) | <0.001 |
| Fever (over 100.4 F), no. (%) | 75 (15.0%) | 38 (14.7%) | 0.91 |
| Hospice, no. (%) | 8 (1.6%) | 5 (1.9%) | 0.73 |
| Do not resuscitate, no. (%) | 62 (12.4%) | 30 (11.6%) | 0.76 |
| Intensive care admission, no. (%) | 160 (31.9%) | 116 (44.8%) | <0.001 |
| ACLS administered, no. (%) | 38 (7.6%) | 17 (6.6%) | 0.6 |
| Outcomes, no. (%) | |||
| Death, 30 days | 67 (13.3%) | 30 (11.6%) | 0.49 |
| Death, 1 year | 211 (42.0%) | 75 (29.0%) | <0.001 |
| Death, 6 years | 390 (77.7%) | 152 (58.7%) | <0.001 |
Mortality at 30 days was not different between the 2 groups, but mortality was higher for the non‐ACS cohort at 1 year and at 6 years (Table 1). Kaplan‐Meier curves demonstrate that mortality for the 2 cohorts begins to diverge between 30 and 60 days until approximately 2 years when the curves again are parallel (Figure 2).
In Cox proportional hazards models, 5 factors were associated with higher mortality at 30 days, 1 year, and at 6 years: age, hospice, DNR order, need for ACLS, and admission to the ICU (Table 2). Additionally, at 1 and 6 years, NT‐proBNP and non‐ACS were associated with higher mortality. At 6 years, creatinine was an additional significant factor. We separated the ACS and non‐ACS cohorts and performed the same model for 1‐year mortality (Table 3). The models yielded similar factors associated with higher mortality: hospice, DNR order, need for ACLS, age, and NT‐proBNP, with ICU admission being significant only in the non‐ACS cohort.
| P Value | Hazard Ratio | 95% CI | |
|---|---|---|---|
| |||
| 30 days | |||
| Intensive care unit admission | <0.0001 | 2.18 | 1.283.72 |
| Hospice | <0.0001 | 4.67 | 1.9111.40 |
| Do not resuscitate | <0.0001 | 3.19 | 1.945.24 |
| ACLS performed | <0.0001 | 10.17 | 6.0317.17 |
| Age, per year | <0.0001 | 1.04 | 1.021.06 |
| 1 year | |||
| Intensive care unit admission | <0.0001 | 1.66 | 1.262.20 |
| Hospice | <0.0001 | 4.98 | 2.699.21 |
| Do not resuscitate | <0.0001 | 2.52 | 1.833.47 |
| Non‐ACS | <0.0001 | 1.57 | 1.192.08 |
| ACLS performed | <0.0001 | 6.03 | 4.178.72 |
| Age, per year | <0.0001 | 1.03 | 1.021.04 |
| NT‐proBNP, per 1,000 pg/mL | <0.0001 | 1.02 | 1.011.03 |
| Extended follow‐up | |||
| Intensive care unit admission | <0.0001 | 1.35 | 1.111.65 |
| Hospice | <0.0001 | 3.81 | 2.136.81 |
| Do not resuscitate | <0.0001 | 2.11 | 1.622.74 |
| Non‐ACS | <0.0001 | 1.53 | 1.251.88 |
| ACLS performed | <0.0001 | 4.19 | 3.015.84 |
| Age, per year | <0.0001 | 1.03 | 1.031.04 |
| Creatinine, per mg/dL | 0.02 | 1.06 | 1.011.12 |
| NT‐proBNP, per 1,000 pg/mL | <0.0001 | 1.02 | 1.021.03 |
| P Value | Hazard Ratio | 95% CI | |
|---|---|---|---|
| |||
| Non‐ACS | |||
| Intensive care unit admission | <0.0001 | 1.86 | 1.352.58 |
| Hospice | <0.0001 | 7.55 | 3.5715.93 |
| Do not resuscitate | <0.0001 | 2.33 | 1.603.41 |
| ACLS performed | <0.0001 | 4.42 | 2.836.92 |
| Age, per year | <0.0001 | 1.03 | 1.011.04 |
| NT‐proBNP, per 1,000 pg/mL | 0.002 | 1.02 | 1.011.03 |
| Clinical ACS | |||
| Hospice | 0.036 | 3.17 | 1.089.32 |
| Do not resuscitate | 0.003 | 2.49 | 1.364.55 |
| ACLS performed | <0.0001 | 12.04 | 6.3322.91 |
| Age, per year | <0.0001 | 1.05 | 1.021.07 |
| NT‐proBNP, per 1,000 pg/mL | 0.001 | 1.04 | 1.011.06 |
DISCUSSION
Our findings confirm the important, but perhaps not well‐recognized, fact that an elevated troponin without ACS is associated with higher mortality than with ACS. This has been previously observed in veteran and nonveteran populations.[4, 6, 8, 12] The novel finding from our investigation is that mortality risk with troponin elevation is most strongly associated with unmodifiable clinical factors that are plausible explanations of risk. Furthermore, the distribution of these factors between our 2 cohorts does not sufficiently explain the difference in risk between ACS and non‐ACS patients.
At each time point we evaluated, ICU admission and need for ACLS were associated with mortality. These are indicators of a severely ill population and are not surprising to find associated with mortality. Many hospitals have instituted some form of pre‐code approach or rapid response team to identify patients before they need ACLS. These efforts, although well meaning, have not yielded convincing results of effectiveness.[13] Hospice and DNR patients were also, not surprisingly, associated with higher mortality. Although these factors were statistically significant, the low prevalence suggests that they are not clinically impactful on the primary questions of the investigation. These factors can be altered but are not intended as modifiable as they reflect the wishes of patients and their decision makers. The distribution of the factors in our model, however, did not adequately explain the higher risk of death with non‐ACS troponin elevation. For example, ACLS administration, hospice care, and DNR orders were strong predictors but were similar between the groups. ICU admission was actually more common with ACS patients, despite strong association with mortality. Age and NT‐proBNP were associated with mortality and higher in the non‐ACS group; however the magnitude of hazard was less than for the other factors. These findings lead us back to the possible explanation that non‐ACS troponin elevation stands as an independent risk factor, and that ACS patients have a distinct advantage in the myriad treatments available. If ACS patients were misdiagnosed as non‐ACS and failed to receive appropriate treatments, that might have contributed to higher mortality; however, we consider that unlikely given that the goal of the QI project was to minimize missed ACS diagnoses.
The overall mortality risk in our study was high: 12.7% at 30 days and 37.6% at 1 year. This reflects the high‐risk population with elevated troponin seen at our facility with ages nearly 70 years and high prevalence of multiple cardiovascular risk factors. Despite a high event rate, many clinically relevant risk factors were not retained in our Cox hazard model. Among sepsis patients, elevation in troponin is associated with mortality[14]; however, in our population neither fever or white blood cell count were significant mortality factors. The relationship between chronic kidney disease and troponin is complex. Renal dysfunction may result in troponin elevation and troponin elevation is a predictor of risk within kidney disease patients.[15] In our study, we did not evaluate chronic kidney disease as a predictor, instead opting to use the serum creatinine. This was not associated with mortality except at the 6‐year time point.
The TIMI score was not associated with mortality in either the overall population or the ACS cohort. The proportion of patients in our cohort with TIMI score under 3 was 16.5% as compared with 21.6% in the original derivation study.[16] The limited data on the prognostic value of the TIMI score within a veteran population suggest a modest predictive capacity.[17] Our data raises the possibility that TIMI is not an optimal choice; however, our analysis only includes all‐cause mortality, different from the original intended use of TIMI, predicting a variety of major cardiac events.
Our data confirm that ACS can be detected in a wide range of clinical presentations. Within our population of troponin positive patients, those with chest pain were most likely to be diagnosed with ACS, although one‐third of chest pain patients were felt to have a non‐ACS diagnosis. On the opposite end of the spectrum, an elevation in troponin with altered mental status or confusion was rarely diagnosed as ACSonly 18% of the time. Many symptoms were poor predictors of ACS; however, none were low enough to disregard. Our data would suggest that most patients with elevated troponin warrant evaluation by a cardiovascular expert.
Our study population came from a single VA hospital that is comprised of elderly and predominantly male patients limiting applicability to other populations. Despite this, other investigations in younger populations and with a higher proportion of women have found similar mortality trends.[4, 8, 12] We did not have sufficient data to determine the cause of death or to further classify as cardiac versus noncardiac; knowledge of the cause of the specific death may better inform future investigations into this important clinical question. Our investigation did not use a standardized definition to determine ACS, a notable limitation that could introduce bias or variation in care. Because all determinations about ACS were made prospectively as part of a QI project, we have little reason to suspect any systematic bias to the determination of ACS. With regard to variation in care, we have previously presented data demonstrating consistent rates of ACS diagnosis across the physicians at our facility.
Based on our investigation and others on this topic, non‐ACS troponin elevation is a common, high‐risk clinical scenario. In our cohort, non‐ACS troponin elevation is about twice as frequent as ACS, and the problem is likely to grow dramatically within the next few years as ultrasensitive troponin assays are eventually approved for use in the United States. These assays are much more sensitive than the current assays, and may make it challenging to distinguish between someone with an acute supply/demand mismatch from someone with an elevated troponin due to chronic, but stable, illness such as CAD, heart failure, or diabetes. Non‐ACS troponin elevation remains poorly understood, with no viable treatment options other than addressing the pathophysiology resulting in the troponin elevation. Due to the heterogeneity of the diagnoses and pathophysiological conditions that result in elevated troponin, a unifying treatment is not likely feasible.
In conclusion, in this elderly, male veteran population, the mortality impact associated with a cardiac troponin elevation was not limited to ACS, as mortality was high among those without ACS. Factors independently associated with this non‐ACS mortality risk were plausible, but did not elucidate the reasons why non‐ACS troponin elevation carries a higher risk. Attempting to better understand the biological basis for the troponin elevation in these non‐ACS patients is a critical unmet need.
Disclosure
Nothing to report.
- , , , , . Prognostic significance of elevated troponin in non‐cardiac hospitalized patients: a systematic review and meta‐analysis. Ann Med. 2014;46:653–663.
- , , , , , ; Joint ESC/ACCF/AHA/WHF Task Force for Universal Definition of Myocardial Infarction. Third universal definition of myocardial infarction. J Am Coll Cardiol. 2012;60:1581–1598.
- , , , et al.; ACC/AHA Task Force Members; Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. 2014 AHA/ACC guideline for the management of patients with non‐st‐elevation acute coronary syndromes: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;130:2354–2394.
- , , , , , . Acute coronary syndrome vs nonspecific troponin elevation: clinical predictors and survival analysis. Arch Intern Med. 2007;167:276–281.
- , , , , , , TOTAL‐AMI study group. Type 2 myocardial infarction in clinical practice. Heart. 2015;101:101–106.
- , , , et al. Outcomes of hospitalized patients with non‐acute coronary syndrome and elevated cardiac troponin level. Am J Med. 2011;124:630–635.
- , , , et al. Classification of myocardial infarction: frequency and features of type 2 myocardial infarction. Am J Med. 2013;126:789–797.
- , , , , . Cardiac troponin I elevation in hospitalized patients without acute coronary syndromes. Am J Cardiol. 2008;101:1384–1388.
- , , , . Inter‐provider variation in diagnoses and cardiac catheterization use (abstract). Cardiology. 2014;128:346.
- , , , . A primer and comparative review of major us mortality databases. Ann Epidemiol. 2002;12:462–468.
- , , . Assessment of vital status in department of veterans affairs national databases. Comparison with state death certificates. Ann Epidemiol. 2001;11:286–291.
- , , , , , . Raised cardiac troponin T levels in patients without acute coronary syndrome. Postgrad Med J. 2007;83:200–205.
- , , , , . Rapid response teams: a systematic review and meta‐analysis. Arch Intern Med. 2010;170:18–26.
- , , , , . Prognostic value of troponins in sepsis: a meta‐analysis. Intensive Care Med. 2013;39:1181–1189.
- , , , et al. Prognostic value of cardiac troponin in patients with chronic kidney disease without suspected acute coronary syndrome: a systematic review and meta‐analysis. Ann Intern Med. 2014;161:491–501.
- , , , et al. The TIMI risk score for unstable angina/non‐ST elevation MI: a method for prognostication and therapeutic decision making. JAMA. 2000;284:835–842.
- , , , , , . Usefulness of the TIMI risk score in predicting both short‐ and long‐term outcomes in the Veterans Affairs non‐Q‐wave myocardial infarction strategies in‐hospital (VANQWISH) trial. Am J Cardiol. 2002;90:922–926.
- , , , , . Prognostic significance of elevated troponin in non‐cardiac hospitalized patients: a systematic review and meta‐analysis. Ann Med. 2014;46:653–663.
- , , , , , ; Joint ESC/ACCF/AHA/WHF Task Force for Universal Definition of Myocardial Infarction. Third universal definition of myocardial infarction. J Am Coll Cardiol. 2012;60:1581–1598.
- , , , et al.; ACC/AHA Task Force Members; Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. 2014 AHA/ACC guideline for the management of patients with non‐st‐elevation acute coronary syndromes: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;130:2354–2394.
- , , , , , . Acute coronary syndrome vs nonspecific troponin elevation: clinical predictors and survival analysis. Arch Intern Med. 2007;167:276–281.
- , , , , , , TOTAL‐AMI study group. Type 2 myocardial infarction in clinical practice. Heart. 2015;101:101–106.
- , , , et al. Outcomes of hospitalized patients with non‐acute coronary syndrome and elevated cardiac troponin level. Am J Med. 2011;124:630–635.
- , , , et al. Classification of myocardial infarction: frequency and features of type 2 myocardial infarction. Am J Med. 2013;126:789–797.
- , , , , . Cardiac troponin I elevation in hospitalized patients without acute coronary syndromes. Am J Cardiol. 2008;101:1384–1388.
- , , , . Inter‐provider variation in diagnoses and cardiac catheterization use (abstract). Cardiology. 2014;128:346.
- , , , . A primer and comparative review of major us mortality databases. Ann Epidemiol. 2002;12:462–468.
- , , . Assessment of vital status in department of veterans affairs national databases. Comparison with state death certificates. Ann Epidemiol. 2001;11:286–291.
- , , , , , . Raised cardiac troponin T levels in patients without acute coronary syndrome. Postgrad Med J. 2007;83:200–205.
- , , , , . Rapid response teams: a systematic review and meta‐analysis. Arch Intern Med. 2010;170:18–26.
- , , , , . Prognostic value of troponins in sepsis: a meta‐analysis. Intensive Care Med. 2013;39:1181–1189.
- , , , et al. Prognostic value of cardiac troponin in patients with chronic kidney disease without suspected acute coronary syndrome: a systematic review and meta‐analysis. Ann Intern Med. 2014;161:491–501.
- , , , et al. The TIMI risk score for unstable angina/non‐ST elevation MI: a method for prognostication and therapeutic decision making. JAMA. 2000;284:835–842.
- , , , , , . Usefulness of the TIMI risk score in predicting both short‐ and long‐term outcomes in the Veterans Affairs non‐Q‐wave myocardial infarction strategies in‐hospital (VANQWISH) trial. Am J Cardiol. 2002;90:922–926.
Pathophysiologic mechanisms linking impaired cardiovascular health and neurologic dysfunction: The year in review
The complex interaction between the nervous system and cardiovascular (CV) health is well recognized. In addition to stroke, migraine, and other disorders, the focus has shifted to depression and related negative-affect states including anxiety, chronic stress, posttraumatic stress disorder, and social isolation. Although all of these conditions have been linked to poor CV health, the mechanisms responsible for this brain-cardiovascular interaction have been poorly understood until recently.
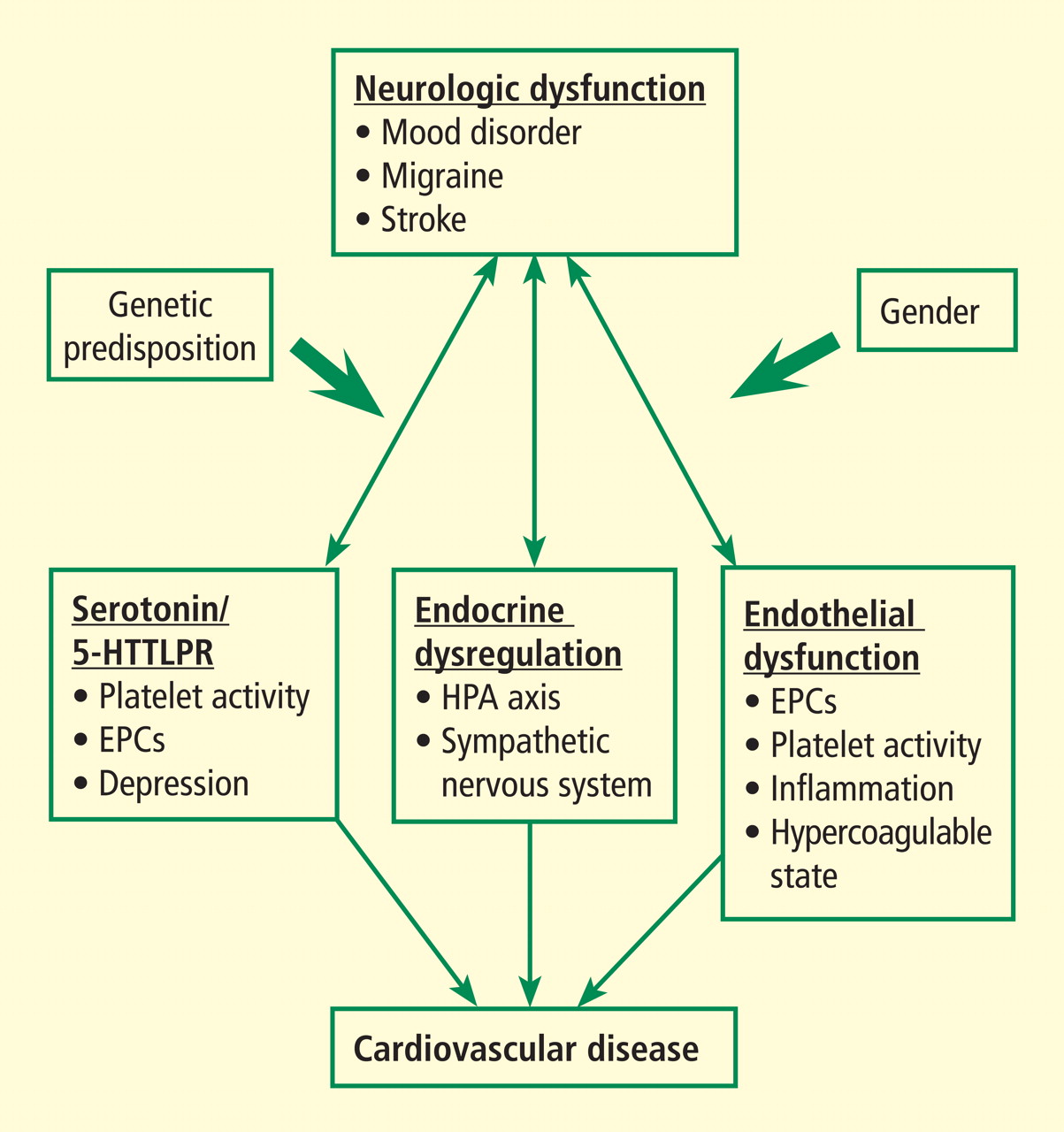
This paper reviews a sample of the more recent advances in our understanding the pathophysiologic mechanisms underlying this complex brain-vascular interaction—including the roles of genetic predisposition, endothelial cell dysfunction, and endocrine dysregulation—and discusses future directions for research in this area.
GENETIC PREDISPOSITION
Increasing evidence highlights the importance of genetic predisposition in both psychological dysfunction and CV disease. The importance of common genetic variability in these two conditions originated from studies of twins that established that both depression and coronary artery disease (CAD) tended to run in families. However, recent studies focused on identifying specific genes that may link depression/negative affect and CAD through various pathways related to platelet reactivity, inflammation, and autonomic nervous system regulation, among many others. A candidate gene study by McCaffery et al1 sought to identify specific genes influencing depressive symptoms in CAD patients. Genes were selected based on their role in biologic pathways involved in inflammation, platelet activation, and the HPA axis and sympathetic nervous system. Among the 59 genes analyzed, the strongest associations were between markers involved in endothelial dysfunction and platelet aggregation—specifically, the involvement of von Willebrand factor in platelet recruitment and of vascular cell adhesion molecule–1 in recruitment and adhesion of inflammatory cells to injured endothelium. Other recent studies have analyzed the role of various genes in mediating neurologic and CV dysfunction.
Serotonin: A multitude of cardiovascular effects
Serotonin (5-HT) is best known as a neurotransmitter involved in mood regulation, but it also directly affects endothelial cells and vascular smooth muscle. Human brain microvascular endothelial cells have 5-HT2A receptors, which are thought to be involved in blood-brain trafficking.2 Polymorphisms in this receptor have been associated with impaired glucose tolerance and type 2 diabetes.3
However, the role of serotonin within the CV system has only recently been realized. Along with its associated serotonin transporter (SERT), serotonin has been best studied within the CV system for its role in development of pulmonary hypertension via vasoconstricton. However, serotonin and SERT exhibit other effects on the CV system, including regulation of factors involved in vascular integrity, such as platelet activity, endothelial dysfunction, and smooth muscle cell and endothelial cell mitogenesis. Serotonin is stored peripherally in platelets and, when released at sites of endothelial damage, promotes platelet aggregation. Cardiomyocytes also are a source of serotonin within the heart, resulting in positive chronotropy, positive inotropy, and activation of mitogen activity.4
Serotonin also affects endothelium through effects on bone marrow production of EPCs. It has been shown in mice to increase the proliferative activity of hematopoietic stem cells in the bone marrow via 5-HT2 receptors.5 Serotonin is a mitogen for canine and bovine endothelial cells6 and has also been shown to enhance ex vivo expansion of CD34+ hematopoietic stem cells in mice.7 It also seems to have an effect on regulation of inflammatory cytokines by regulating different secretory pathways. Activity of several different serotonin receptors, including 5-HT2A, 5-HT4, and 5-HT7, inhibits tumor necrosis factor production in peripheral cells.8
Insights from genetic analysis of SERT
These areas of recent research continue to highlight the importance of serotonin within the CV system, and its overlapping role in depression and other forms of psychological dysfunction underscores the importance of understanding its regulation.
The best-studied component of serotonin regulation is SERT, a sodium-dependent transporter that removes serotonin from outside the cell, bringing it back into the cell for repackaging. It is recognized as the site of action of the selective serotonin reuptake inhibitors (SSRIs).
Analysis of the serotonin transporter gene has traditionally focused on polymorphisms within the promoter region (5-HTTLPR). Two common alleles, the long (L) and short (S) variants, are the best characterized. The S variant is associated with a lower expression of SERT, leading to reduced uptake and release of serotonin. The SS genotype of SERT has been linked to major depressive disorder9 and to increased risk of subsequent cardiac events after myocardial infarction (MI).10
A study of the effects of environmental stress and gender on associations between depression and 5-HTTLPR found that this link varied according to gender and stressful life events.11 Specifically, women with the SS genotype, which leads to less transcriptional activity of 5-HTTLPR, tended to be more susceptible to depression under stressful life conditions. However, in men the LL genotype was associated with increased transcriptional activity and the same increased susceptibility to depression. 11 Expression of the LL genotype also resulted in upregulation of SERT, leading to higher MI risk.12
Another study investigated the relationship between genetic variability in two serotonin-related gene polymorphisms and found that the 5-HTTLPR gene polymorphism was associated with adverse cardiac events after coronary artery bypass graft surgery in combination with depression, specifically in patients with the L allele compared with SS.13
Humans who possess the L variant of the SERT protein have more rapid platelet serotonin uptake.14 Inhibitors of SERT have been shown to reduce platelet serotonin content, leading to disruption of thrombosis and increased bleeding due to inhibition of serotonin reuptake. A study in middle-aged men also found that genetic variability within SERT was associated with increased depressive symptoms and elevated levels of interleukin-6, a marker of inflammation.15 This indicates a common source of genetic vulnerability accounting for both depression and inflammation, and could help to explain the increased risk of CV disease in patients with depression.15
Mental stress–induced myocardial ischemia
The association between mental stress and activation of the sympathetic nervous system is well established. Stress leads to increases in blood pressure and heart rate. In patients with CAD, mental stress–induced myocardial ischemia (MSIMI) is a well-characterized phenomenon whereby ischemia is provoked by psychological stress. Sympathetic nervous system activation from stressful life events can increase vulnerability to CV outcomes such as MI, arrhythmias, and sudden cardiac death. MSIMI has been identified as a risk factor for poor outcomes in patients with known CAD. Proposed mechanisms include mismatch of myocardial blood supply and demand, as well as coronary spasm.
A recent study by Hassan et al evaluated associations of beta1-adrenergic receptor gene (ADBR1) polymorphisms with MSIMI in CAD patients.16 The researchers found a threefold increase in MSIMI in patients with a particular allele of the ADBR1 gene. This is the first report to highlight a specific genetic predisposition to susceptibility to MSIMI; it could help explain why some patients are at increased risk and also suggest targeting specific behavioral or pharmacologic therapies to reduce MSIMI.
ENDOTHELIAL DYSFUNCTION
Dysfunctional endothelium is important in the link between neurologic dysfunction and CV disease. Circulating EPCs have been recognized as playing an important role in maintaining vascular health. These cells originate in the bone marrow and may be identified by their surface markers and various functional characteristics. They have been shown to be a key part of the process involved in repair of biologic risk factor–mediated damage to endothelium and are reduced and/or dysfunctional in patients with CV risk factors such as tobacco use, diabetes, and hypertension. Low EPC levels have also been found in patients with cerebrovascular disease and are key in vascular neogenesis after ischemic insult.
Genetic effects on endothelium in migraine and stroke
A more recent area of interest in brain and cardiovascular health has been the role of EPCs in migraine, which is often debilitating. The association between migraine and increased risk of depression,17 as well as stroke and MI, has been well recognized. A recent study by MacClellan et al evaluated whether polymorphisms in genes regulating endothelial function and vascular tone influenced susceptibility to migraine and stroke in a large subset of young women.18 Analyzed genes included endothelin-1 (EDN), endothelin receptor type B (EDNRB), and nitric oxide synthase-3 (NOS3). Several polymorphisms within these genes were associated with stroke in white women but not in black women. However, the study did not show whether the association between migraine and stroke was mediated by the polymorphisms studied from the candidate genes. Others have found no association with EDNRB but found that the homozygous minor genotype (present in 5% of cases) of the EDNRA SNP rs2048894 showed association with migraine with aura (odds ratio [OR] = 1.61, 95% confidence interval [CI], 1.12–2.32; P = .010] when adjusted for gender and sample origin.19 Early age at onset (P = .011) in the pooled sample.19 Studies on larger samples will be needed to confirm these findings.
Depression may alter endothelial homeostasis
One mechanism by which depression may lead to increased CV disease risk is through effects on endothelial homeostasis. Several studies have found an association between depression and attenuated flow-mediated vasodilation. However, it has been postulated that this effect was mediated by atherosclerosis, as these studies were performed in older cohorts. To evaluate this association without the possibility of confounding severe atherosclerosis, a recent study evaluated this association in adolescent women with no known health problems.20 Regression analysis demonstrated a significant inverse relationship between depression and endothelial function as measured by pulse-wave amplitude. Most patients in this study had evidence of subclinical depression, suggesting that even those with mild symptoms can have endothelial dysfunction.20 These findings provide evidence to suggest that there may be some factors that underlie the vascular dysfunction associated with both depression and early subclinical atherosclerosis.
Focus on endothelial progenitor cells
EPCs from bone marrow and likely other sites (eg, spleen, perivascular omentum, liver, mesentery) play an important role in maintaining vascular integrity. The interaction between the bone marrow, nervous system, HPA axis, and progenitor cells, together with the effect of this interaction on vascular integrity, has become an area of increasing research. Specifically, the sympathetic nervous system has been identified as playing an important role in progenitor cell egress from the bone marrow. Mice with abnormal nerve conduction produce virtually no progenitor cells when treated with granulocyte colony-stimulating factor.21
Psychosocial factors influence activation of the sympathetic nervous system. An innovative recent study examined the effect of psychosocial determinants on bone marrow–derived progenitor cells; the psychosocial variables and progenitor cell counts were associated independently from traditional biological and behavioral risk factors.22 This is one of the first studies to examine the effect of psychosocial stressors on progenitor cells and should open the way for further research exploring this association and its effects on CV health.
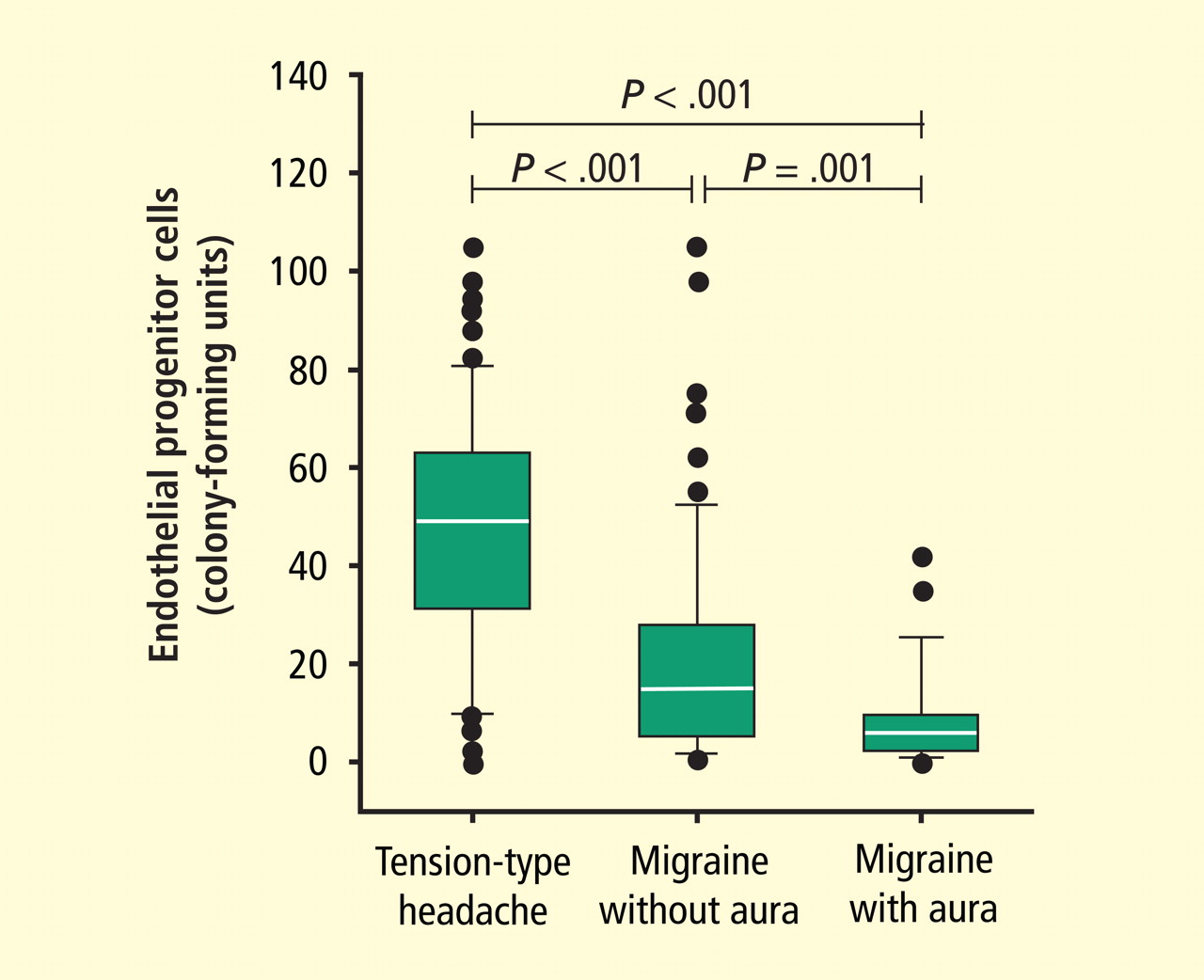
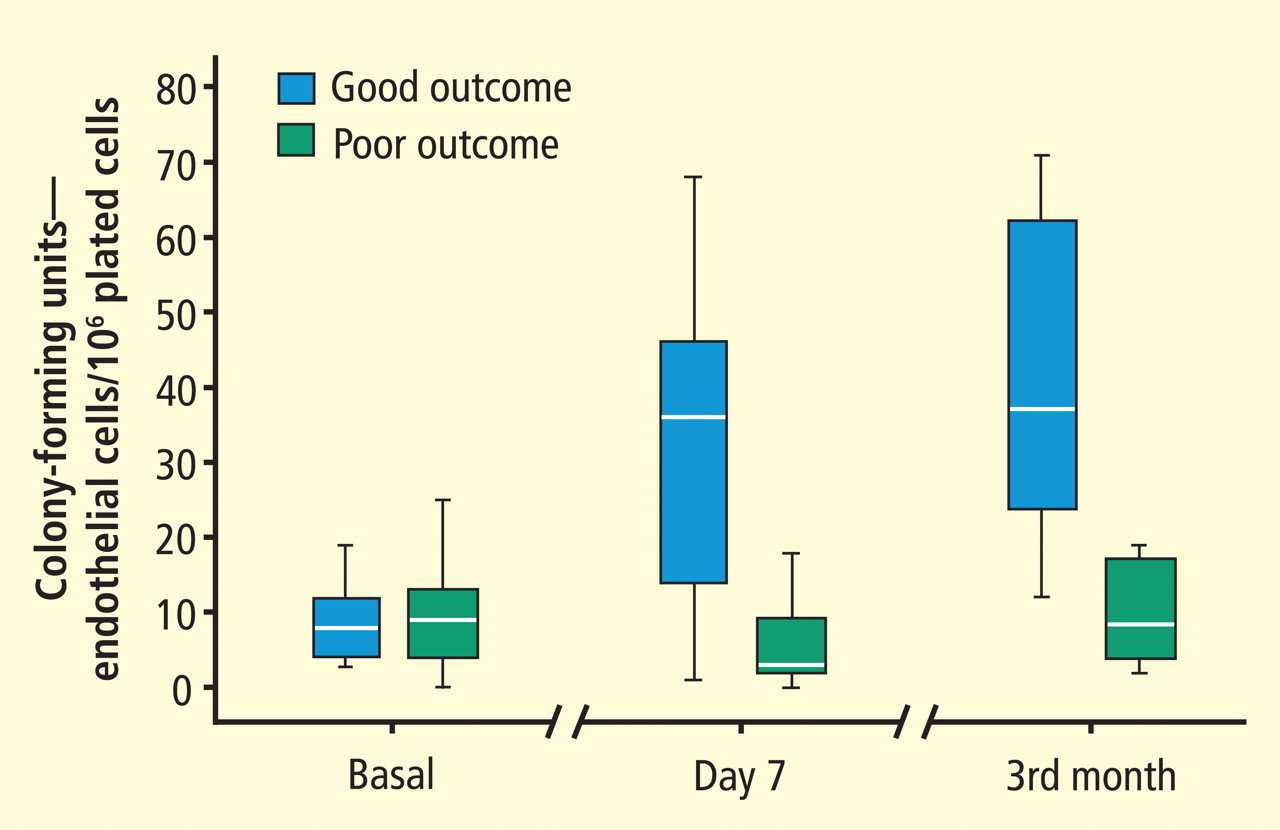
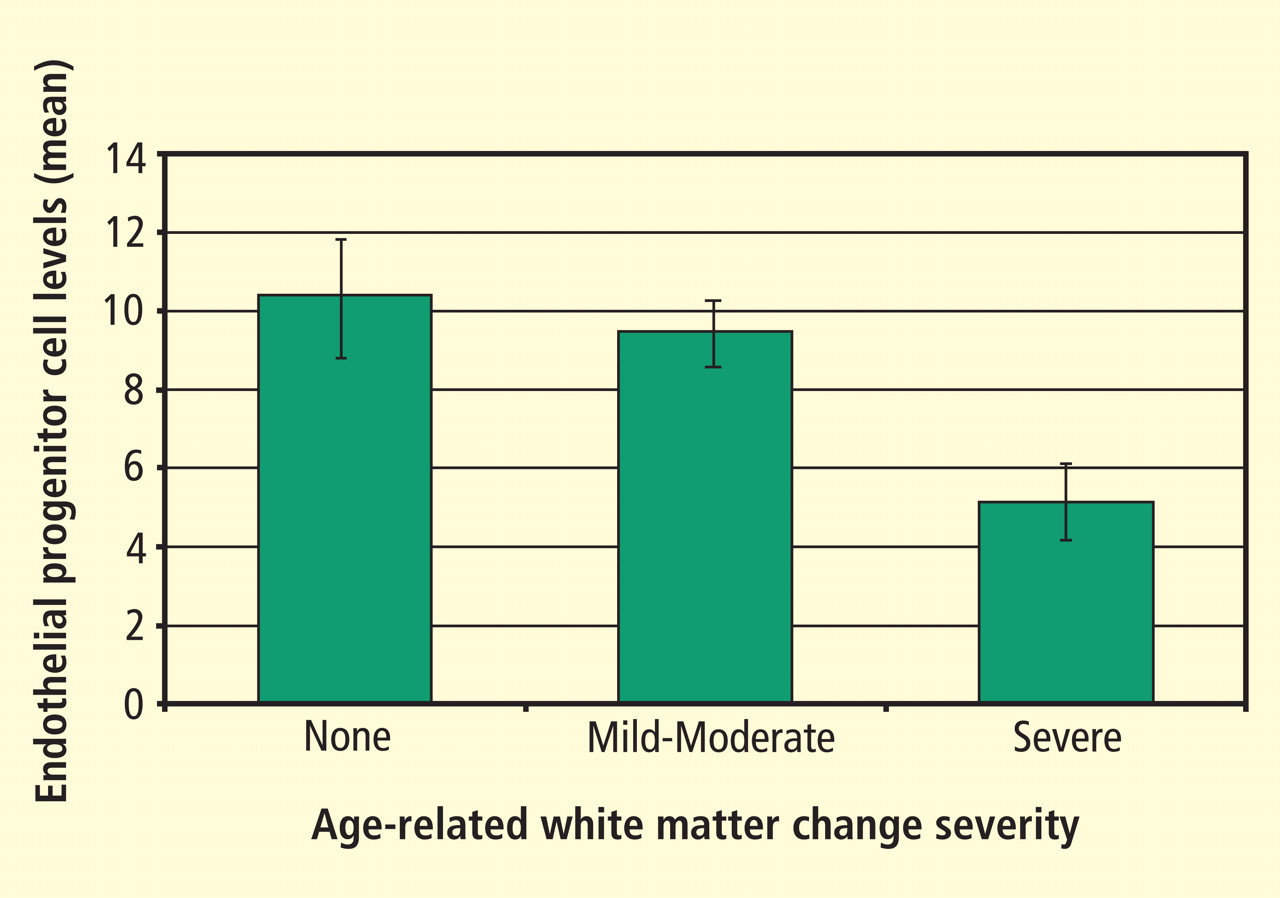
Platelet activity
Platelets play a critical role in endothelial hemostasis. Alterations in platelet function have been hypothesized as a mechanism by which depression may lead to CV disease.27 A recent study analyzed the effects of persistent depressive symptoms on platelet activation in a cohort of spousal dementia caregivers.28 P-selectin, measured as an index of platelet activation, and depression, mainly in the subclinical range, were associated with elevated platelet reactivity and recovery. This may be one mechanism by which elderly caregivers are at risk of CV disease even without evidence of clinical depression.
Serotonin is also known for its effect on platelet function and vascular tone and is one of the main targets of antidepressant therapy. Several studies have analyzed the effect of depression treatment with SSRIs on platelet function. An initial analysis from the SADHART trial in 2003 found that in depressed patients treated with sertraline after acute coronary syndrome, reductions in platelet/endothelial activation occurred despite concurrent treatment with antiplatelet drugs, including aspirin and clopidogrel.29 This suggests that the antiplatelet properties of sertraline differ from those of aspirin and other antiplatelet therapies. A study by van Zyl et al30 yielded similar results with a different SSRI, citalopram, along with enhanced production of nitric oxide.
HORMONES, NEGATIVE AFFECT, AND CV DISEASE
Patients with CAD have different patterns of cortisol excretion compared with controls. A dysfunctional HPA axis has been implicated, leading to a failure in containing inflammatory activity.31 In healthy older patients without known CAD, heightened cortisol reactivity is associated with a greater extent of coronary artery calcification.32 Cynical hostility is also associated with CAD, but the mechanism is unclear. Higher levels of cynical hostility were associated with attenuation of the decreasing phase of the cortisol awakening response.33 Another recent study also found a higher cortisol awakening response and a larger ratio of total cholesterol to high-density lipoprotein cholesterol in response to stress in socially isolated men.34
Although cortisol is the most studied end product of the HPA axis, aldosterone is also released. Recent research has focused on the role of aldosterone in CV injury through its increase in superoxide generation and its upregulation of genes involved in inflammation, fibrosis, and atherosclerosis.35,36 Several studies have demonstrated the increased incidence of CV disease, including atrial fibrillation, stroke, and MI, among patients with primary hyperaldosteronism compared with patients with similar blood pressure elevations.37,38 Continued evidence supports the importance of the endocrine pathways in modulation of CV disease, and future research should continue to explore the role of various hormones in this interaction.
DIRECTIONS FOR FUTURE RESEARCH
Although advances have been made in understanding the pathophysiologic mechanisms involved in interactions between the nervous system and CV disease, many factors have yet to be completely understood. For example, the role of gender in the interaction between psychological distress and heart disease is an area of increasing discussion. CV disease is a significant cause of morbidity and mortality among women, who are also at increased risk of depression and anxiety as well as stroke and migraine. Recent literature supports the concept that significant biologic differences exist in the effect of stress and depression on men and women. A recent study in mice found that a lifelong increase in SERT function decreased constitutive cerebral metabolism in a number of brain regions, and that this effect was significant only in females.39 Gender differences also appear to exist in the efficacy of antidepressant therapy.11 Although these differences have been observed, it remains unclear whether they primarily are due to obvious hormonal differences or to differences in genetic predisposition.
Stem cell therapy—for stroke and perhaps even for migraine and other disorders—is another area of potential future research. In CV disease, bone marrow–derived EPCs have been used in the post-MI setting and for heart failure. Injured endothelium presents a similar target for endothelial repair with cell therapy in stroke and migraine as well. Continued research in these areas will provide new insights into this brain-vascular interaction and could help provide new directions for treatment in the future.
- McCaffery JM, Duan QL, Frasure-Smith N, et al Genetic predictors of depressive symptoms in cardiac patients. Am J Med Genet B Neuropsychiatr Genet 2009; 150B:381–388.
- Cohen Z, Bouchelet I, Olivier A, et al Multiple microvascular and astroglial 5-hydroxytryptamine receptor subtypes in human brain: molecular and pharmacologic characterization. J Cereb Blood Flow Metab 1999; 19:908–917.
- Kring SI, Werge T, Holst C, et al Polymorphisms of serotonin receptor 2A and 2C genes and COMT in relation to obesity and type 2 diabetes. PLoS One 2009; 4:e6696.
- Ni W, Watts SW. 5-Hydroxytryptamine in the cardiovascular system: focus on the serotonin transporter (SERT). Clin Exp Pharmacol Physiol 2006; 33:575–583.
- Nefedov VP, Nefedova VV, Kononykhina VS, et al The effect of serotonin on the hematopoietic stem cells of the bone marrow [in Russian]. Biull Eksp Biol Med 1991; 112:488–489.
- Pakala R, Willerson JT, Benedict CR. Mitogenic effect of serotonin on vascular endothelial cells. Circulation 1994; 90:1919–1926.
- Yang M, Li K, Ng PC, et al Promoting effects of serotonin on hematopoiesis: ex vivo expansion of cord blood CD34+ stem/progenitor cells, proliferation of bone marrow stromal cells, and antiapoptosis. Stem Cells 2007; 25:1800–1806.
- Cloez-Tayarani I, Petit-Bertron AF, Venters HD, et al Differential effect of serotonin on cytokine production in lipopolysaccharide-stimulated human peripheral blood mononuclear cells: involvement of 5-hydroxytryptamine2A receptors. Int Immunol 2003; 15:233–240.
- Hoefgen B, Schulze TG, Ohlraun S, et al The power of sample size and homogenous sampling: association between the 5-HTTLPR serotonin transporter polymorphism and major depressive disorder. Biol Psychiatry 2005; 57:247–251.
- Nakatani D, Sato H, Sakata Y, et al Influence of serotonin transporter gene polymorphism on depressive symptoms and new cardiac events after acute myocardial infarction. Am Heart J 2005; 150:652–658.
- Brummett BH, Boyle SH, Siegler IC, et al Effects of environmental stress and gender on associations among symptoms of depression and the serotonin transporter gene linked polymorphic region (5-HTTLPR). Behav Genet 2008; 38:34–43.
- Fumeron F, Betoulle D, Nicaud V, et al Serotonin transporter gene polymorphism and myocardial infarction: Etude Cas-Témoins de l’Infarctus du Myocarde (ECTIM). Circulation 2002; 105:2943–2945.
- Phillips-Bute B, Mathew JP, Blumenthal JA, et al Relationship of genetic variability and depressive symptoms to adverse events after coronary artery bypass graft surgery. Psychosom Med 2008; 70:953–959.
- Greenberg BD, Tolliver TJ, Huang SJ, et al Genetic variation in the serotonin transporter promoter region affects serotonin uptake in human blood platelets. Am J Med Genet 1999; 88:83–87.
- Su S, Zhao J, Bremner JD, et al Serotonin transporter gene, depressive symptoms, and interleukin-6. Circ Cardiovasc Genet 2009; 2:614–620.
- Hassan M, York KM, Li H, et al Association of beta1-adrenergic receptor genetic polymorphism with mental stress-induced myocardial ischemia in patients with coronary artery disease. Arch Intern Med 2008; 168:763–770.
- Victor TW, Hu X, Campbell J, et al Association between migraine, anxiety and depression. Cephalalgia 2009; Jul 9[Epub ahead of print]
- MacClellan LR, Howard TD, Cole JW, et al Relation of candidate genes that encode for endothelial function to migraine and stroke: the Stroke Prevention in Young Women study. Stroke 2009; 40:e550–e557.
- Tikka-Kleemola P, Kaunisto MA, Hamalainen E, et al Genetic association study of endothelin-1 and its receptors EDNRA and EDNRB in migraine with aura. Cephalalgia 2009; 29:1224–1231.
- Tomfohr LM, Martin TM, Miller GE. Symptoms of depression and impaired endothelial function in healthy adolescent women. J Behav Med 2008; 31:137–143.
- Katayama Y, Battista M, Kao WM, et al Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006; 124:407–421.
- Fischer JC, Kudielka BM, von Kanel R, et al Bone-marrow derived progenitor cells are associated with psychosocial determinants of health after controlling for classical biological and behavioral cardiovascular risk factors. Brain Behav Immun 2009; 23:419–426.
- Baskin SM, Smitherman TA. Migraine and psychiatric disorders: comorbidities, mechanisms, and clinical applications. Neurol Sci 2009; 30( suppl 1):S61–S65.
- Lee S-T, Chu K, Jung KH, et al Decreased number and function of endothelial progenitor cells in patients with migraine. Neurology 2008; 70:1510–1517.
- Sobrino T, Hurtado O, Moro MA, et al The increase of circulating endothelial progenitor cells after acute ischemic stroke is associated with good outcome. Stroke 2007; 38:2759–2764.
- Jickling G, Salam A, Mohammad A, et al Circulating endothelial progenitor cells and age-related white matter changes. Stroke 2009; 40:3191–3196.
- Parakh K, Sakhuja A, Bhat U, et al Platelet function in patients with depression. South Med J 2008; 101:612–617.
- Aschbacher K, Mills PJ, von Känel R, et al Effects of depressive and anxious symptoms on norepinephrine and platelet P-selectin responses to acute psychological stress among elderly caregivers. Brain Behav Immun 2008; 22:493–502.
- Serebruany VL, Glassman AH, Malinin AI, et al Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events: the Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART) platelet substudy. Circulation 2003; 108:939–944.
- van Zyl LT, Lesperance F, Frasure-Smith N, et al Platelet and endothelial activity in comorbid major depression and coronary artery disease patients treated with citalopram: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy Trial (CREATE) biomarker sub-study. J Thromb Thrombolysis 2009; 27:48–56.
- Nijm J, Kristenson M, Olsson AG, et al Impaired cortisol response to acute stressors in patients with coronary disease: implications for inflammatory activity. J Intern Med 2007; 262:375–384.
- Hamer M, O’Donnell K, Lahiri A, Steptoe A. Salivary cortisol responses to mental stress are associated with coronary artery calcification in healthy men and women. Eur Heart J 2010; 31:424–429.
- Ranjit N, Diez-Roux AV, Sanchez B, et al Association of salivary cortisol circadian pattern with cynical hostility: multi-ethnic study of atherosclerosis. Psychosom Med 2009; 71:748–755.
- Grant N, Hamer M, Steptoe A. Social isolation and stress-related cardiovascular, lipid, and cortisol responses. Ann Behav Med 2009; 37:29–37.
- Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res 2005; 96:643–650.
- Kubzansky LD, Adler GK. Aldosterone: a forgotten mediator of the relationship between psychological stress and heart disease. Neurosci Biobehav Rev 2010; 34:80–86.
- Milliez P, Girerd X, Plouin PF, et al Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol 2005; 45:1243–1248.
- Duprez DA, Bauwens FR, De Buyzere ML, et al Influence of arterial blood pressure and aldosterone on left ventricular hypertrophy in moderate essential hypertension. Am J Cardiol 1993; 71:17A–20A.
- Dawson N, Ferrington L, Olverman HJ, et al Sex influences the effect of a lifelong increase in serotonin transporter function on cerebral metabolism. J Neurosci Res 2009; 87:2375–2385.
The complex interaction between the nervous system and cardiovascular (CV) health is well recognized. In addition to stroke, migraine, and other disorders, the focus has shifted to depression and related negative-affect states including anxiety, chronic stress, posttraumatic stress disorder, and social isolation. Although all of these conditions have been linked to poor CV health, the mechanisms responsible for this brain-cardiovascular interaction have been poorly understood until recently.
This paper reviews a sample of the more recent advances in our understanding the pathophysiologic mechanisms underlying this complex brain-vascular interaction—including the roles of genetic predisposition, endothelial cell dysfunction, and endocrine dysregulation—and discusses future directions for research in this area.
GENETIC PREDISPOSITION
Increasing evidence highlights the importance of genetic predisposition in both psychological dysfunction and CV disease. The importance of common genetic variability in these two conditions originated from studies of twins that established that both depression and coronary artery disease (CAD) tended to run in families. However, recent studies focused on identifying specific genes that may link depression/negative affect and CAD through various pathways related to platelet reactivity, inflammation, and autonomic nervous system regulation, among many others. A candidate gene study by McCaffery et al1 sought to identify specific genes influencing depressive symptoms in CAD patients. Genes were selected based on their role in biologic pathways involved in inflammation, platelet activation, and the HPA axis and sympathetic nervous system. Among the 59 genes analyzed, the strongest associations were between markers involved in endothelial dysfunction and platelet aggregation—specifically, the involvement of von Willebrand factor in platelet recruitment and of vascular cell adhesion molecule–1 in recruitment and adhesion of inflammatory cells to injured endothelium. Other recent studies have analyzed the role of various genes in mediating neurologic and CV dysfunction.
Serotonin: A multitude of cardiovascular effects
Serotonin (5-HT) is best known as a neurotransmitter involved in mood regulation, but it also directly affects endothelial cells and vascular smooth muscle. Human brain microvascular endothelial cells have 5-HT2A receptors, which are thought to be involved in blood-brain trafficking.2 Polymorphisms in this receptor have been associated with impaired glucose tolerance and type 2 diabetes.3
However, the role of serotonin within the CV system has only recently been realized. Along with its associated serotonin transporter (SERT), serotonin has been best studied within the CV system for its role in development of pulmonary hypertension via vasoconstricton. However, serotonin and SERT exhibit other effects on the CV system, including regulation of factors involved in vascular integrity, such as platelet activity, endothelial dysfunction, and smooth muscle cell and endothelial cell mitogenesis. Serotonin is stored peripherally in platelets and, when released at sites of endothelial damage, promotes platelet aggregation. Cardiomyocytes also are a source of serotonin within the heart, resulting in positive chronotropy, positive inotropy, and activation of mitogen activity.4
Serotonin also affects endothelium through effects on bone marrow production of EPCs. It has been shown in mice to increase the proliferative activity of hematopoietic stem cells in the bone marrow via 5-HT2 receptors.5 Serotonin is a mitogen for canine and bovine endothelial cells6 and has also been shown to enhance ex vivo expansion of CD34+ hematopoietic stem cells in mice.7 It also seems to have an effect on regulation of inflammatory cytokines by regulating different secretory pathways. Activity of several different serotonin receptors, including 5-HT2A, 5-HT4, and 5-HT7, inhibits tumor necrosis factor production in peripheral cells.8
Insights from genetic analysis of SERT
These areas of recent research continue to highlight the importance of serotonin within the CV system, and its overlapping role in depression and other forms of psychological dysfunction underscores the importance of understanding its regulation.
The best-studied component of serotonin regulation is SERT, a sodium-dependent transporter that removes serotonin from outside the cell, bringing it back into the cell for repackaging. It is recognized as the site of action of the selective serotonin reuptake inhibitors (SSRIs).
Analysis of the serotonin transporter gene has traditionally focused on polymorphisms within the promoter region (5-HTTLPR). Two common alleles, the long (L) and short (S) variants, are the best characterized. The S variant is associated with a lower expression of SERT, leading to reduced uptake and release of serotonin. The SS genotype of SERT has been linked to major depressive disorder9 and to increased risk of subsequent cardiac events after myocardial infarction (MI).10
A study of the effects of environmental stress and gender on associations between depression and 5-HTTLPR found that this link varied according to gender and stressful life events.11 Specifically, women with the SS genotype, which leads to less transcriptional activity of 5-HTTLPR, tended to be more susceptible to depression under stressful life conditions. However, in men the LL genotype was associated with increased transcriptional activity and the same increased susceptibility to depression. 11 Expression of the LL genotype also resulted in upregulation of SERT, leading to higher MI risk.12
Another study investigated the relationship between genetic variability in two serotonin-related gene polymorphisms and found that the 5-HTTLPR gene polymorphism was associated with adverse cardiac events after coronary artery bypass graft surgery in combination with depression, specifically in patients with the L allele compared with SS.13
Humans who possess the L variant of the SERT protein have more rapid platelet serotonin uptake.14 Inhibitors of SERT have been shown to reduce platelet serotonin content, leading to disruption of thrombosis and increased bleeding due to inhibition of serotonin reuptake. A study in middle-aged men also found that genetic variability within SERT was associated with increased depressive symptoms and elevated levels of interleukin-6, a marker of inflammation.15 This indicates a common source of genetic vulnerability accounting for both depression and inflammation, and could help to explain the increased risk of CV disease in patients with depression.15
Mental stress–induced myocardial ischemia
The association between mental stress and activation of the sympathetic nervous system is well established. Stress leads to increases in blood pressure and heart rate. In patients with CAD, mental stress–induced myocardial ischemia (MSIMI) is a well-characterized phenomenon whereby ischemia is provoked by psychological stress. Sympathetic nervous system activation from stressful life events can increase vulnerability to CV outcomes such as MI, arrhythmias, and sudden cardiac death. MSIMI has been identified as a risk factor for poor outcomes in patients with known CAD. Proposed mechanisms include mismatch of myocardial blood supply and demand, as well as coronary spasm.
A recent study by Hassan et al evaluated associations of beta1-adrenergic receptor gene (ADBR1) polymorphisms with MSIMI in CAD patients.16 The researchers found a threefold increase in MSIMI in patients with a particular allele of the ADBR1 gene. This is the first report to highlight a specific genetic predisposition to susceptibility to MSIMI; it could help explain why some patients are at increased risk and also suggest targeting specific behavioral or pharmacologic therapies to reduce MSIMI.
ENDOTHELIAL DYSFUNCTION
Dysfunctional endothelium is important in the link between neurologic dysfunction and CV disease. Circulating EPCs have been recognized as playing an important role in maintaining vascular health. These cells originate in the bone marrow and may be identified by their surface markers and various functional characteristics. They have been shown to be a key part of the process involved in repair of biologic risk factor–mediated damage to endothelium and are reduced and/or dysfunctional in patients with CV risk factors such as tobacco use, diabetes, and hypertension. Low EPC levels have also been found in patients with cerebrovascular disease and are key in vascular neogenesis after ischemic insult.
Genetic effects on endothelium in migraine and stroke
A more recent area of interest in brain and cardiovascular health has been the role of EPCs in migraine, which is often debilitating. The association between migraine and increased risk of depression,17 as well as stroke and MI, has been well recognized. A recent study by MacClellan et al evaluated whether polymorphisms in genes regulating endothelial function and vascular tone influenced susceptibility to migraine and stroke in a large subset of young women.18 Analyzed genes included endothelin-1 (EDN), endothelin receptor type B (EDNRB), and nitric oxide synthase-3 (NOS3). Several polymorphisms within these genes were associated with stroke in white women but not in black women. However, the study did not show whether the association between migraine and stroke was mediated by the polymorphisms studied from the candidate genes. Others have found no association with EDNRB but found that the homozygous minor genotype (present in 5% of cases) of the EDNRA SNP rs2048894 showed association with migraine with aura (odds ratio [OR] = 1.61, 95% confidence interval [CI], 1.12–2.32; P = .010] when adjusted for gender and sample origin.19 Early age at onset (P = .011) in the pooled sample.19 Studies on larger samples will be needed to confirm these findings.
Depression may alter endothelial homeostasis
One mechanism by which depression may lead to increased CV disease risk is through effects on endothelial homeostasis. Several studies have found an association between depression and attenuated flow-mediated vasodilation. However, it has been postulated that this effect was mediated by atherosclerosis, as these studies were performed in older cohorts. To evaluate this association without the possibility of confounding severe atherosclerosis, a recent study evaluated this association in adolescent women with no known health problems.20 Regression analysis demonstrated a significant inverse relationship between depression and endothelial function as measured by pulse-wave amplitude. Most patients in this study had evidence of subclinical depression, suggesting that even those with mild symptoms can have endothelial dysfunction.20 These findings provide evidence to suggest that there may be some factors that underlie the vascular dysfunction associated with both depression and early subclinical atherosclerosis.
Focus on endothelial progenitor cells
EPCs from bone marrow and likely other sites (eg, spleen, perivascular omentum, liver, mesentery) play an important role in maintaining vascular integrity. The interaction between the bone marrow, nervous system, HPA axis, and progenitor cells, together with the effect of this interaction on vascular integrity, has become an area of increasing research. Specifically, the sympathetic nervous system has been identified as playing an important role in progenitor cell egress from the bone marrow. Mice with abnormal nerve conduction produce virtually no progenitor cells when treated with granulocyte colony-stimulating factor.21
Psychosocial factors influence activation of the sympathetic nervous system. An innovative recent study examined the effect of psychosocial determinants on bone marrow–derived progenitor cells; the psychosocial variables and progenitor cell counts were associated independently from traditional biological and behavioral risk factors.22 This is one of the first studies to examine the effect of psychosocial stressors on progenitor cells and should open the way for further research exploring this association and its effects on CV health.
Platelet activity
Platelets play a critical role in endothelial hemostasis. Alterations in platelet function have been hypothesized as a mechanism by which depression may lead to CV disease.27 A recent study analyzed the effects of persistent depressive symptoms on platelet activation in a cohort of spousal dementia caregivers.28 P-selectin, measured as an index of platelet activation, and depression, mainly in the subclinical range, were associated with elevated platelet reactivity and recovery. This may be one mechanism by which elderly caregivers are at risk of CV disease even without evidence of clinical depression.
Serotonin is also known for its effect on platelet function and vascular tone and is one of the main targets of antidepressant therapy. Several studies have analyzed the effect of depression treatment with SSRIs on platelet function. An initial analysis from the SADHART trial in 2003 found that in depressed patients treated with sertraline after acute coronary syndrome, reductions in platelet/endothelial activation occurred despite concurrent treatment with antiplatelet drugs, including aspirin and clopidogrel.29 This suggests that the antiplatelet properties of sertraline differ from those of aspirin and other antiplatelet therapies. A study by van Zyl et al30 yielded similar results with a different SSRI, citalopram, along with enhanced production of nitric oxide.
HORMONES, NEGATIVE AFFECT, AND CV DISEASE
Patients with CAD have different patterns of cortisol excretion compared with controls. A dysfunctional HPA axis has been implicated, leading to a failure in containing inflammatory activity.31 In healthy older patients without known CAD, heightened cortisol reactivity is associated with a greater extent of coronary artery calcification.32 Cynical hostility is also associated with CAD, but the mechanism is unclear. Higher levels of cynical hostility were associated with attenuation of the decreasing phase of the cortisol awakening response.33 Another recent study also found a higher cortisol awakening response and a larger ratio of total cholesterol to high-density lipoprotein cholesterol in response to stress in socially isolated men.34
Although cortisol is the most studied end product of the HPA axis, aldosterone is also released. Recent research has focused on the role of aldosterone in CV injury through its increase in superoxide generation and its upregulation of genes involved in inflammation, fibrosis, and atherosclerosis.35,36 Several studies have demonstrated the increased incidence of CV disease, including atrial fibrillation, stroke, and MI, among patients with primary hyperaldosteronism compared with patients with similar blood pressure elevations.37,38 Continued evidence supports the importance of the endocrine pathways in modulation of CV disease, and future research should continue to explore the role of various hormones in this interaction.
DIRECTIONS FOR FUTURE RESEARCH
Although advances have been made in understanding the pathophysiologic mechanisms involved in interactions between the nervous system and CV disease, many factors have yet to be completely understood. For example, the role of gender in the interaction between psychological distress and heart disease is an area of increasing discussion. CV disease is a significant cause of morbidity and mortality among women, who are also at increased risk of depression and anxiety as well as stroke and migraine. Recent literature supports the concept that significant biologic differences exist in the effect of stress and depression on men and women. A recent study in mice found that a lifelong increase in SERT function decreased constitutive cerebral metabolism in a number of brain regions, and that this effect was significant only in females.39 Gender differences also appear to exist in the efficacy of antidepressant therapy.11 Although these differences have been observed, it remains unclear whether they primarily are due to obvious hormonal differences or to differences in genetic predisposition.
Stem cell therapy—for stroke and perhaps even for migraine and other disorders—is another area of potential future research. In CV disease, bone marrow–derived EPCs have been used in the post-MI setting and for heart failure. Injured endothelium presents a similar target for endothelial repair with cell therapy in stroke and migraine as well. Continued research in these areas will provide new insights into this brain-vascular interaction and could help provide new directions for treatment in the future.
The complex interaction between the nervous system and cardiovascular (CV) health is well recognized. In addition to stroke, migraine, and other disorders, the focus has shifted to depression and related negative-affect states including anxiety, chronic stress, posttraumatic stress disorder, and social isolation. Although all of these conditions have been linked to poor CV health, the mechanisms responsible for this brain-cardiovascular interaction have been poorly understood until recently.
This paper reviews a sample of the more recent advances in our understanding the pathophysiologic mechanisms underlying this complex brain-vascular interaction—including the roles of genetic predisposition, endothelial cell dysfunction, and endocrine dysregulation—and discusses future directions for research in this area.
GENETIC PREDISPOSITION
Increasing evidence highlights the importance of genetic predisposition in both psychological dysfunction and CV disease. The importance of common genetic variability in these two conditions originated from studies of twins that established that both depression and coronary artery disease (CAD) tended to run in families. However, recent studies focused on identifying specific genes that may link depression/negative affect and CAD through various pathways related to platelet reactivity, inflammation, and autonomic nervous system regulation, among many others. A candidate gene study by McCaffery et al1 sought to identify specific genes influencing depressive symptoms in CAD patients. Genes were selected based on their role in biologic pathways involved in inflammation, platelet activation, and the HPA axis and sympathetic nervous system. Among the 59 genes analyzed, the strongest associations were between markers involved in endothelial dysfunction and platelet aggregation—specifically, the involvement of von Willebrand factor in platelet recruitment and of vascular cell adhesion molecule–1 in recruitment and adhesion of inflammatory cells to injured endothelium. Other recent studies have analyzed the role of various genes in mediating neurologic and CV dysfunction.
Serotonin: A multitude of cardiovascular effects
Serotonin (5-HT) is best known as a neurotransmitter involved in mood regulation, but it also directly affects endothelial cells and vascular smooth muscle. Human brain microvascular endothelial cells have 5-HT2A receptors, which are thought to be involved in blood-brain trafficking.2 Polymorphisms in this receptor have been associated with impaired glucose tolerance and type 2 diabetes.3
However, the role of serotonin within the CV system has only recently been realized. Along with its associated serotonin transporter (SERT), serotonin has been best studied within the CV system for its role in development of pulmonary hypertension via vasoconstricton. However, serotonin and SERT exhibit other effects on the CV system, including regulation of factors involved in vascular integrity, such as platelet activity, endothelial dysfunction, and smooth muscle cell and endothelial cell mitogenesis. Serotonin is stored peripherally in platelets and, when released at sites of endothelial damage, promotes platelet aggregation. Cardiomyocytes also are a source of serotonin within the heart, resulting in positive chronotropy, positive inotropy, and activation of mitogen activity.4
Serotonin also affects endothelium through effects on bone marrow production of EPCs. It has been shown in mice to increase the proliferative activity of hematopoietic stem cells in the bone marrow via 5-HT2 receptors.5 Serotonin is a mitogen for canine and bovine endothelial cells6 and has also been shown to enhance ex vivo expansion of CD34+ hematopoietic stem cells in mice.7 It also seems to have an effect on regulation of inflammatory cytokines by regulating different secretory pathways. Activity of several different serotonin receptors, including 5-HT2A, 5-HT4, and 5-HT7, inhibits tumor necrosis factor production in peripheral cells.8
Insights from genetic analysis of SERT
These areas of recent research continue to highlight the importance of serotonin within the CV system, and its overlapping role in depression and other forms of psychological dysfunction underscores the importance of understanding its regulation.
The best-studied component of serotonin regulation is SERT, a sodium-dependent transporter that removes serotonin from outside the cell, bringing it back into the cell for repackaging. It is recognized as the site of action of the selective serotonin reuptake inhibitors (SSRIs).
Analysis of the serotonin transporter gene has traditionally focused on polymorphisms within the promoter region (5-HTTLPR). Two common alleles, the long (L) and short (S) variants, are the best characterized. The S variant is associated with a lower expression of SERT, leading to reduced uptake and release of serotonin. The SS genotype of SERT has been linked to major depressive disorder9 and to increased risk of subsequent cardiac events after myocardial infarction (MI).10
A study of the effects of environmental stress and gender on associations between depression and 5-HTTLPR found that this link varied according to gender and stressful life events.11 Specifically, women with the SS genotype, which leads to less transcriptional activity of 5-HTTLPR, tended to be more susceptible to depression under stressful life conditions. However, in men the LL genotype was associated with increased transcriptional activity and the same increased susceptibility to depression. 11 Expression of the LL genotype also resulted in upregulation of SERT, leading to higher MI risk.12
Another study investigated the relationship between genetic variability in two serotonin-related gene polymorphisms and found that the 5-HTTLPR gene polymorphism was associated with adverse cardiac events after coronary artery bypass graft surgery in combination with depression, specifically in patients with the L allele compared with SS.13
Humans who possess the L variant of the SERT protein have more rapid platelet serotonin uptake.14 Inhibitors of SERT have been shown to reduce platelet serotonin content, leading to disruption of thrombosis and increased bleeding due to inhibition of serotonin reuptake. A study in middle-aged men also found that genetic variability within SERT was associated with increased depressive symptoms and elevated levels of interleukin-6, a marker of inflammation.15 This indicates a common source of genetic vulnerability accounting for both depression and inflammation, and could help to explain the increased risk of CV disease in patients with depression.15
Mental stress–induced myocardial ischemia
The association between mental stress and activation of the sympathetic nervous system is well established. Stress leads to increases in blood pressure and heart rate. In patients with CAD, mental stress–induced myocardial ischemia (MSIMI) is a well-characterized phenomenon whereby ischemia is provoked by psychological stress. Sympathetic nervous system activation from stressful life events can increase vulnerability to CV outcomes such as MI, arrhythmias, and sudden cardiac death. MSIMI has been identified as a risk factor for poor outcomes in patients with known CAD. Proposed mechanisms include mismatch of myocardial blood supply and demand, as well as coronary spasm.
A recent study by Hassan et al evaluated associations of beta1-adrenergic receptor gene (ADBR1) polymorphisms with MSIMI in CAD patients.16 The researchers found a threefold increase in MSIMI in patients with a particular allele of the ADBR1 gene. This is the first report to highlight a specific genetic predisposition to susceptibility to MSIMI; it could help explain why some patients are at increased risk and also suggest targeting specific behavioral or pharmacologic therapies to reduce MSIMI.
ENDOTHELIAL DYSFUNCTION
Dysfunctional endothelium is important in the link between neurologic dysfunction and CV disease. Circulating EPCs have been recognized as playing an important role in maintaining vascular health. These cells originate in the bone marrow and may be identified by their surface markers and various functional characteristics. They have been shown to be a key part of the process involved in repair of biologic risk factor–mediated damage to endothelium and are reduced and/or dysfunctional in patients with CV risk factors such as tobacco use, diabetes, and hypertension. Low EPC levels have also been found in patients with cerebrovascular disease and are key in vascular neogenesis after ischemic insult.
Genetic effects on endothelium in migraine and stroke
A more recent area of interest in brain and cardiovascular health has been the role of EPCs in migraine, which is often debilitating. The association between migraine and increased risk of depression,17 as well as stroke and MI, has been well recognized. A recent study by MacClellan et al evaluated whether polymorphisms in genes regulating endothelial function and vascular tone influenced susceptibility to migraine and stroke in a large subset of young women.18 Analyzed genes included endothelin-1 (EDN), endothelin receptor type B (EDNRB), and nitric oxide synthase-3 (NOS3). Several polymorphisms within these genes were associated with stroke in white women but not in black women. However, the study did not show whether the association between migraine and stroke was mediated by the polymorphisms studied from the candidate genes. Others have found no association with EDNRB but found that the homozygous minor genotype (present in 5% of cases) of the EDNRA SNP rs2048894 showed association with migraine with aura (odds ratio [OR] = 1.61, 95% confidence interval [CI], 1.12–2.32; P = .010] when adjusted for gender and sample origin.19 Early age at onset (P = .011) in the pooled sample.19 Studies on larger samples will be needed to confirm these findings.
Depression may alter endothelial homeostasis
One mechanism by which depression may lead to increased CV disease risk is through effects on endothelial homeostasis. Several studies have found an association between depression and attenuated flow-mediated vasodilation. However, it has been postulated that this effect was mediated by atherosclerosis, as these studies were performed in older cohorts. To evaluate this association without the possibility of confounding severe atherosclerosis, a recent study evaluated this association in adolescent women with no known health problems.20 Regression analysis demonstrated a significant inverse relationship between depression and endothelial function as measured by pulse-wave amplitude. Most patients in this study had evidence of subclinical depression, suggesting that even those with mild symptoms can have endothelial dysfunction.20 These findings provide evidence to suggest that there may be some factors that underlie the vascular dysfunction associated with both depression and early subclinical atherosclerosis.
Focus on endothelial progenitor cells
EPCs from bone marrow and likely other sites (eg, spleen, perivascular omentum, liver, mesentery) play an important role in maintaining vascular integrity. The interaction between the bone marrow, nervous system, HPA axis, and progenitor cells, together with the effect of this interaction on vascular integrity, has become an area of increasing research. Specifically, the sympathetic nervous system has been identified as playing an important role in progenitor cell egress from the bone marrow. Mice with abnormal nerve conduction produce virtually no progenitor cells when treated with granulocyte colony-stimulating factor.21
Psychosocial factors influence activation of the sympathetic nervous system. An innovative recent study examined the effect of psychosocial determinants on bone marrow–derived progenitor cells; the psychosocial variables and progenitor cell counts were associated independently from traditional biological and behavioral risk factors.22 This is one of the first studies to examine the effect of psychosocial stressors on progenitor cells and should open the way for further research exploring this association and its effects on CV health.
Platelet activity
Platelets play a critical role in endothelial hemostasis. Alterations in platelet function have been hypothesized as a mechanism by which depression may lead to CV disease.27 A recent study analyzed the effects of persistent depressive symptoms on platelet activation in a cohort of spousal dementia caregivers.28 P-selectin, measured as an index of platelet activation, and depression, mainly in the subclinical range, were associated with elevated platelet reactivity and recovery. This may be one mechanism by which elderly caregivers are at risk of CV disease even without evidence of clinical depression.
Serotonin is also known for its effect on platelet function and vascular tone and is one of the main targets of antidepressant therapy. Several studies have analyzed the effect of depression treatment with SSRIs on platelet function. An initial analysis from the SADHART trial in 2003 found that in depressed patients treated with sertraline after acute coronary syndrome, reductions in platelet/endothelial activation occurred despite concurrent treatment with antiplatelet drugs, including aspirin and clopidogrel.29 This suggests that the antiplatelet properties of sertraline differ from those of aspirin and other antiplatelet therapies. A study by van Zyl et al30 yielded similar results with a different SSRI, citalopram, along with enhanced production of nitric oxide.
HORMONES, NEGATIVE AFFECT, AND CV DISEASE
Patients with CAD have different patterns of cortisol excretion compared with controls. A dysfunctional HPA axis has been implicated, leading to a failure in containing inflammatory activity.31 In healthy older patients without known CAD, heightened cortisol reactivity is associated with a greater extent of coronary artery calcification.32 Cynical hostility is also associated with CAD, but the mechanism is unclear. Higher levels of cynical hostility were associated with attenuation of the decreasing phase of the cortisol awakening response.33 Another recent study also found a higher cortisol awakening response and a larger ratio of total cholesterol to high-density lipoprotein cholesterol in response to stress in socially isolated men.34
Although cortisol is the most studied end product of the HPA axis, aldosterone is also released. Recent research has focused on the role of aldosterone in CV injury through its increase in superoxide generation and its upregulation of genes involved in inflammation, fibrosis, and atherosclerosis.35,36 Several studies have demonstrated the increased incidence of CV disease, including atrial fibrillation, stroke, and MI, among patients with primary hyperaldosteronism compared with patients with similar blood pressure elevations.37,38 Continued evidence supports the importance of the endocrine pathways in modulation of CV disease, and future research should continue to explore the role of various hormones in this interaction.
DIRECTIONS FOR FUTURE RESEARCH
Although advances have been made in understanding the pathophysiologic mechanisms involved in interactions between the nervous system and CV disease, many factors have yet to be completely understood. For example, the role of gender in the interaction between psychological distress and heart disease is an area of increasing discussion. CV disease is a significant cause of morbidity and mortality among women, who are also at increased risk of depression and anxiety as well as stroke and migraine. Recent literature supports the concept that significant biologic differences exist in the effect of stress and depression on men and women. A recent study in mice found that a lifelong increase in SERT function decreased constitutive cerebral metabolism in a number of brain regions, and that this effect was significant only in females.39 Gender differences also appear to exist in the efficacy of antidepressant therapy.11 Although these differences have been observed, it remains unclear whether they primarily are due to obvious hormonal differences or to differences in genetic predisposition.
Stem cell therapy—for stroke and perhaps even for migraine and other disorders—is another area of potential future research. In CV disease, bone marrow–derived EPCs have been used in the post-MI setting and for heart failure. Injured endothelium presents a similar target for endothelial repair with cell therapy in stroke and migraine as well. Continued research in these areas will provide new insights into this brain-vascular interaction and could help provide new directions for treatment in the future.
- McCaffery JM, Duan QL, Frasure-Smith N, et al Genetic predictors of depressive symptoms in cardiac patients. Am J Med Genet B Neuropsychiatr Genet 2009; 150B:381–388.
- Cohen Z, Bouchelet I, Olivier A, et al Multiple microvascular and astroglial 5-hydroxytryptamine receptor subtypes in human brain: molecular and pharmacologic characterization. J Cereb Blood Flow Metab 1999; 19:908–917.
- Kring SI, Werge T, Holst C, et al Polymorphisms of serotonin receptor 2A and 2C genes and COMT in relation to obesity and type 2 diabetes. PLoS One 2009; 4:e6696.
- Ni W, Watts SW. 5-Hydroxytryptamine in the cardiovascular system: focus on the serotonin transporter (SERT). Clin Exp Pharmacol Physiol 2006; 33:575–583.
- Nefedov VP, Nefedova VV, Kononykhina VS, et al The effect of serotonin on the hematopoietic stem cells of the bone marrow [in Russian]. Biull Eksp Biol Med 1991; 112:488–489.
- Pakala R, Willerson JT, Benedict CR. Mitogenic effect of serotonin on vascular endothelial cells. Circulation 1994; 90:1919–1926.
- Yang M, Li K, Ng PC, et al Promoting effects of serotonin on hematopoiesis: ex vivo expansion of cord blood CD34+ stem/progenitor cells, proliferation of bone marrow stromal cells, and antiapoptosis. Stem Cells 2007; 25:1800–1806.
- Cloez-Tayarani I, Petit-Bertron AF, Venters HD, et al Differential effect of serotonin on cytokine production in lipopolysaccharide-stimulated human peripheral blood mononuclear cells: involvement of 5-hydroxytryptamine2A receptors. Int Immunol 2003; 15:233–240.
- Hoefgen B, Schulze TG, Ohlraun S, et al The power of sample size and homogenous sampling: association between the 5-HTTLPR serotonin transporter polymorphism and major depressive disorder. Biol Psychiatry 2005; 57:247–251.
- Nakatani D, Sato H, Sakata Y, et al Influence of serotonin transporter gene polymorphism on depressive symptoms and new cardiac events after acute myocardial infarction. Am Heart J 2005; 150:652–658.
- Brummett BH, Boyle SH, Siegler IC, et al Effects of environmental stress and gender on associations among symptoms of depression and the serotonin transporter gene linked polymorphic region (5-HTTLPR). Behav Genet 2008; 38:34–43.
- Fumeron F, Betoulle D, Nicaud V, et al Serotonin transporter gene polymorphism and myocardial infarction: Etude Cas-Témoins de l’Infarctus du Myocarde (ECTIM). Circulation 2002; 105:2943–2945.
- Phillips-Bute B, Mathew JP, Blumenthal JA, et al Relationship of genetic variability and depressive symptoms to adverse events after coronary artery bypass graft surgery. Psychosom Med 2008; 70:953–959.
- Greenberg BD, Tolliver TJ, Huang SJ, et al Genetic variation in the serotonin transporter promoter region affects serotonin uptake in human blood platelets. Am J Med Genet 1999; 88:83–87.
- Su S, Zhao J, Bremner JD, et al Serotonin transporter gene, depressive symptoms, and interleukin-6. Circ Cardiovasc Genet 2009; 2:614–620.
- Hassan M, York KM, Li H, et al Association of beta1-adrenergic receptor genetic polymorphism with mental stress-induced myocardial ischemia in patients with coronary artery disease. Arch Intern Med 2008; 168:763–770.
- Victor TW, Hu X, Campbell J, et al Association between migraine, anxiety and depression. Cephalalgia 2009; Jul 9[Epub ahead of print]
- MacClellan LR, Howard TD, Cole JW, et al Relation of candidate genes that encode for endothelial function to migraine and stroke: the Stroke Prevention in Young Women study. Stroke 2009; 40:e550–e557.
- Tikka-Kleemola P, Kaunisto MA, Hamalainen E, et al Genetic association study of endothelin-1 and its receptors EDNRA and EDNRB in migraine with aura. Cephalalgia 2009; 29:1224–1231.
- Tomfohr LM, Martin TM, Miller GE. Symptoms of depression and impaired endothelial function in healthy adolescent women. J Behav Med 2008; 31:137–143.
- Katayama Y, Battista M, Kao WM, et al Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006; 124:407–421.
- Fischer JC, Kudielka BM, von Kanel R, et al Bone-marrow derived progenitor cells are associated with psychosocial determinants of health after controlling for classical biological and behavioral cardiovascular risk factors. Brain Behav Immun 2009; 23:419–426.
- Baskin SM, Smitherman TA. Migraine and psychiatric disorders: comorbidities, mechanisms, and clinical applications. Neurol Sci 2009; 30( suppl 1):S61–S65.
- Lee S-T, Chu K, Jung KH, et al Decreased number and function of endothelial progenitor cells in patients with migraine. Neurology 2008; 70:1510–1517.
- Sobrino T, Hurtado O, Moro MA, et al The increase of circulating endothelial progenitor cells after acute ischemic stroke is associated with good outcome. Stroke 2007; 38:2759–2764.
- Jickling G, Salam A, Mohammad A, et al Circulating endothelial progenitor cells and age-related white matter changes. Stroke 2009; 40:3191–3196.
- Parakh K, Sakhuja A, Bhat U, et al Platelet function in patients with depression. South Med J 2008; 101:612–617.
- Aschbacher K, Mills PJ, von Känel R, et al Effects of depressive and anxious symptoms on norepinephrine and platelet P-selectin responses to acute psychological stress among elderly caregivers. Brain Behav Immun 2008; 22:493–502.
- Serebruany VL, Glassman AH, Malinin AI, et al Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events: the Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART) platelet substudy. Circulation 2003; 108:939–944.
- van Zyl LT, Lesperance F, Frasure-Smith N, et al Platelet and endothelial activity in comorbid major depression and coronary artery disease patients treated with citalopram: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy Trial (CREATE) biomarker sub-study. J Thromb Thrombolysis 2009; 27:48–56.
- Nijm J, Kristenson M, Olsson AG, et al Impaired cortisol response to acute stressors in patients with coronary disease: implications for inflammatory activity. J Intern Med 2007; 262:375–384.
- Hamer M, O’Donnell K, Lahiri A, Steptoe A. Salivary cortisol responses to mental stress are associated with coronary artery calcification in healthy men and women. Eur Heart J 2010; 31:424–429.
- Ranjit N, Diez-Roux AV, Sanchez B, et al Association of salivary cortisol circadian pattern with cynical hostility: multi-ethnic study of atherosclerosis. Psychosom Med 2009; 71:748–755.
- Grant N, Hamer M, Steptoe A. Social isolation and stress-related cardiovascular, lipid, and cortisol responses. Ann Behav Med 2009; 37:29–37.
- Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res 2005; 96:643–650.
- Kubzansky LD, Adler GK. Aldosterone: a forgotten mediator of the relationship between psychological stress and heart disease. Neurosci Biobehav Rev 2010; 34:80–86.
- Milliez P, Girerd X, Plouin PF, et al Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol 2005; 45:1243–1248.
- Duprez DA, Bauwens FR, De Buyzere ML, et al Influence of arterial blood pressure and aldosterone on left ventricular hypertrophy in moderate essential hypertension. Am J Cardiol 1993; 71:17A–20A.
- Dawson N, Ferrington L, Olverman HJ, et al Sex influences the effect of a lifelong increase in serotonin transporter function on cerebral metabolism. J Neurosci Res 2009; 87:2375–2385.
- McCaffery JM, Duan QL, Frasure-Smith N, et al Genetic predictors of depressive symptoms in cardiac patients. Am J Med Genet B Neuropsychiatr Genet 2009; 150B:381–388.
- Cohen Z, Bouchelet I, Olivier A, et al Multiple microvascular and astroglial 5-hydroxytryptamine receptor subtypes in human brain: molecular and pharmacologic characterization. J Cereb Blood Flow Metab 1999; 19:908–917.
- Kring SI, Werge T, Holst C, et al Polymorphisms of serotonin receptor 2A and 2C genes and COMT in relation to obesity and type 2 diabetes. PLoS One 2009; 4:e6696.
- Ni W, Watts SW. 5-Hydroxytryptamine in the cardiovascular system: focus on the serotonin transporter (SERT). Clin Exp Pharmacol Physiol 2006; 33:575–583.
- Nefedov VP, Nefedova VV, Kononykhina VS, et al The effect of serotonin on the hematopoietic stem cells of the bone marrow [in Russian]. Biull Eksp Biol Med 1991; 112:488–489.
- Pakala R, Willerson JT, Benedict CR. Mitogenic effect of serotonin on vascular endothelial cells. Circulation 1994; 90:1919–1926.
- Yang M, Li K, Ng PC, et al Promoting effects of serotonin on hematopoiesis: ex vivo expansion of cord blood CD34+ stem/progenitor cells, proliferation of bone marrow stromal cells, and antiapoptosis. Stem Cells 2007; 25:1800–1806.
- Cloez-Tayarani I, Petit-Bertron AF, Venters HD, et al Differential effect of serotonin on cytokine production in lipopolysaccharide-stimulated human peripheral blood mononuclear cells: involvement of 5-hydroxytryptamine2A receptors. Int Immunol 2003; 15:233–240.
- Hoefgen B, Schulze TG, Ohlraun S, et al The power of sample size and homogenous sampling: association between the 5-HTTLPR serotonin transporter polymorphism and major depressive disorder. Biol Psychiatry 2005; 57:247–251.
- Nakatani D, Sato H, Sakata Y, et al Influence of serotonin transporter gene polymorphism on depressive symptoms and new cardiac events after acute myocardial infarction. Am Heart J 2005; 150:652–658.
- Brummett BH, Boyle SH, Siegler IC, et al Effects of environmental stress and gender on associations among symptoms of depression and the serotonin transporter gene linked polymorphic region (5-HTTLPR). Behav Genet 2008; 38:34–43.
- Fumeron F, Betoulle D, Nicaud V, et al Serotonin transporter gene polymorphism and myocardial infarction: Etude Cas-Témoins de l’Infarctus du Myocarde (ECTIM). Circulation 2002; 105:2943–2945.
- Phillips-Bute B, Mathew JP, Blumenthal JA, et al Relationship of genetic variability and depressive symptoms to adverse events after coronary artery bypass graft surgery. Psychosom Med 2008; 70:953–959.
- Greenberg BD, Tolliver TJ, Huang SJ, et al Genetic variation in the serotonin transporter promoter region affects serotonin uptake in human blood platelets. Am J Med Genet 1999; 88:83–87.
- Su S, Zhao J, Bremner JD, et al Serotonin transporter gene, depressive symptoms, and interleukin-6. Circ Cardiovasc Genet 2009; 2:614–620.
- Hassan M, York KM, Li H, et al Association of beta1-adrenergic receptor genetic polymorphism with mental stress-induced myocardial ischemia in patients with coronary artery disease. Arch Intern Med 2008; 168:763–770.
- Victor TW, Hu X, Campbell J, et al Association between migraine, anxiety and depression. Cephalalgia 2009; Jul 9[Epub ahead of print]
- MacClellan LR, Howard TD, Cole JW, et al Relation of candidate genes that encode for endothelial function to migraine and stroke: the Stroke Prevention in Young Women study. Stroke 2009; 40:e550–e557.
- Tikka-Kleemola P, Kaunisto MA, Hamalainen E, et al Genetic association study of endothelin-1 and its receptors EDNRA and EDNRB in migraine with aura. Cephalalgia 2009; 29:1224–1231.
- Tomfohr LM, Martin TM, Miller GE. Symptoms of depression and impaired endothelial function in healthy adolescent women. J Behav Med 2008; 31:137–143.
- Katayama Y, Battista M, Kao WM, et al Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006; 124:407–421.
- Fischer JC, Kudielka BM, von Kanel R, et al Bone-marrow derived progenitor cells are associated with psychosocial determinants of health after controlling for classical biological and behavioral cardiovascular risk factors. Brain Behav Immun 2009; 23:419–426.
- Baskin SM, Smitherman TA. Migraine and psychiatric disorders: comorbidities, mechanisms, and clinical applications. Neurol Sci 2009; 30( suppl 1):S61–S65.
- Lee S-T, Chu K, Jung KH, et al Decreased number and function of endothelial progenitor cells in patients with migraine. Neurology 2008; 70:1510–1517.
- Sobrino T, Hurtado O, Moro MA, et al The increase of circulating endothelial progenitor cells after acute ischemic stroke is associated with good outcome. Stroke 2007; 38:2759–2764.
- Jickling G, Salam A, Mohammad A, et al Circulating endothelial progenitor cells and age-related white matter changes. Stroke 2009; 40:3191–3196.
- Parakh K, Sakhuja A, Bhat U, et al Platelet function in patients with depression. South Med J 2008; 101:612–617.
- Aschbacher K, Mills PJ, von Känel R, et al Effects of depressive and anxious symptoms on norepinephrine and platelet P-selectin responses to acute psychological stress among elderly caregivers. Brain Behav Immun 2008; 22:493–502.
- Serebruany VL, Glassman AH, Malinin AI, et al Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events: the Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART) platelet substudy. Circulation 2003; 108:939–944.
- van Zyl LT, Lesperance F, Frasure-Smith N, et al Platelet and endothelial activity in comorbid major depression and coronary artery disease patients treated with citalopram: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy Trial (CREATE) biomarker sub-study. J Thromb Thrombolysis 2009; 27:48–56.
- Nijm J, Kristenson M, Olsson AG, et al Impaired cortisol response to acute stressors in patients with coronary disease: implications for inflammatory activity. J Intern Med 2007; 262:375–384.
- Hamer M, O’Donnell K, Lahiri A, Steptoe A. Salivary cortisol responses to mental stress are associated with coronary artery calcification in healthy men and women. Eur Heart J 2010; 31:424–429.
- Ranjit N, Diez-Roux AV, Sanchez B, et al Association of salivary cortisol circadian pattern with cynical hostility: multi-ethnic study of atherosclerosis. Psychosom Med 2009; 71:748–755.
- Grant N, Hamer M, Steptoe A. Social isolation and stress-related cardiovascular, lipid, and cortisol responses. Ann Behav Med 2009; 37:29–37.
- Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res 2005; 96:643–650.
- Kubzansky LD, Adler GK. Aldosterone: a forgotten mediator of the relationship between psychological stress and heart disease. Neurosci Biobehav Rev 2010; 34:80–86.
- Milliez P, Girerd X, Plouin PF, et al Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol 2005; 45:1243–1248.
- Duprez DA, Bauwens FR, De Buyzere ML, et al Influence of arterial blood pressure and aldosterone on left ventricular hypertrophy in moderate essential hypertension. Am J Cardiol 1993; 71:17A–20A.
- Dawson N, Ferrington L, Olverman HJ, et al Sex influences the effect of a lifelong increase in serotonin transporter function on cerebral metabolism. J Neurosci Res 2009; 87:2375–2385.