Article
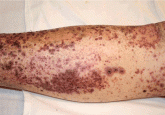
A 72-year-old man with a purpuric rash
He has multiple medical problems and a new rash that appeared 2 weeks ago. Now he presents to the emergency department confused and diaphoretic....
Article
Update on Wegener granulomatosis
Although Wegener granulomatosis is uncommon, it is relevant to internists because it is a multisystem disease that presents in a variety of ways....
Article
The diagnostic utility of c-ANCA in Wegener’s granulomatosis
A positive c-ANCA may suggest Wegener's granulomatosis, but in most cases it should not be used instead of biopsy to make the diagnosis.