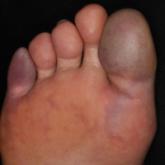
Article
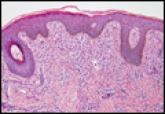
Brown Macule on the Waist
Article
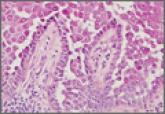
Hailey-Hailey Disease
Hailey-Hailey disease typically presents as suprabasal blisters with a perivascular and interstitial lymphocytic infiltrate. The differential...
Hailey-Hailey disease typically presents as suprabasal blisters with a perivascular and interstitial lymphocytic infiltrate. The differential...