User login
Thinking Inside the Box
A 65‐year‐old man was referred for evaluation of worsening ascites and end‐stage liver disease. The patient had been well until 1 year ago, when he developed lower extremity edema and abdominal distention. After evaluation by his primary care physician, he was given a diagnosis of cryptogenic cirrhosis. He underwent several paracenteses and was placed on furosemide and spironolactone. The patient had been stable on his diuretic regimen until 2 weeks previously, when he suddenly developed worsening edema and ascites, along with dizziness, nausea, and hypotension. His physician stopped the diuretics and referred him to the hospital.
Before diagnosing a patient with cryptogenic cirrhosis, it is necessary to exclude common etiologies of cirrhosis such as alcohol, viral hepatitis, and non‐alcoholic fatty liver disease and numerous uncommon causes, including Wilson's disease, hemochromatosis, Budd‐Chiari, and biliary cirrhosis. It is also important to remember that patients with liver disease are not immune to extrahepatic causes of ascites, such as peritoneal carcinomatosis and tuberculous ascites. Simultaneously, reasons for chronic liver disease decompensating acutely must be considered: medication nonadherence, excess salt intake, hepatotoxicity from acetaminophen or alcohol, and other acute insults, such as hepatocellular carcinoma, an intervening infection (especially spontaneous bacterial peritonitis), ascending cholangitis, or a flare of chronic viral hepatitis.
Past medical and surgical history included diabetes mellitus (diagnosed 10 years previously), obstructive sleep apnea, hypertension, hypothyroidism, and mild chronic kidney disease. Medications included levothyroxine, lactulose, sulfamethoxazole, pioglitazone (started 4 months prior), and ibuprofen. Furosemide and spironolactone had been discontinued 2 weeks previously. He currently resided in the Central Valley of California. He had lived in Thailand from age 7 to 17 and traveled to India more than 1 year ago. He did not smoke and had never used intravenous drugs or received a blood transfusion. He rarely drank alcohol. He worked as a chemist. There was no family history of liver disease.
There is no obvious explanation for the underlying liver disease or the acute decompensation. Sulfamethoxazole is a rare cause of allergic or granulomatous hepatitis. Pioglitazone is a thiazolinedione which in earlier formulations was linked to hepatitis but can be excluded as a cause of this patient's cirrhosis because it was started after liver disease was detected. As a chemist, he might have been exposed to carbon tetrachloride, a known hepatotoxin. Obstructive sleep apnea causes pulmonary hypertension, but severe ascites and acute hepatic decompensation would be unusual. Ibuprofen might precipitate worsening renal function and fluid accumulation. Time in Thailand and India raises the possibility of tuberculous ascites.
The patient had no headache, vision changes, abdominal pain, emesis, melena, hematochezia, chest pain, palpitations, dysuria, polyuria, pruritus, dark urine, or rashes. He reported difficulty with concentration when lactulose was decreased. He noted worsening exercise tolerance with dyspnea after 10 steps and reported a weight gain of 12 pounds in the past 2 weeks.
On examination, temperature was 36.8C; blood pressure, 129/87 mm Hg; heart rate, 85 beats per minute; respirations, 20 per minute; and oxygen saturation, 94% on room air. He was uncomfortable but alert. There was no scleral icterus or conjunctival pallor. Jugular venous pressure was elevated. The lungs were clear, and the heart was regular, with no murmur, rub, or gallops. The abdomen was massively distended with a fluid wave; the liver and spleen could not be palpated. There was pitting edema of the sacrum and lower extremities. There was no asterixis, palmar erythema, spider angiomata, or skin discoloration.
The additional history and physical exam suggest that the primary problem may lie outside the liver, especially as signs of advanced liver disease (other than ascites) are absent. Dyspnea on exertion is consistent with the physical stress of a large volume of ascites or could be secondary to several pulmonary complications associated with liver disease, including portopulmonary hypertension, hepatopulmonary syndrome, or hepatic hydrothorax. Alternatively, the dyspnea raises the possibility that the ascites is not related to a primary liver disorder but rather to anemia or to a cardiac disorder, such as chronic left ventricular failure, isolated right‐sided heart failure, or constrictive pericarditis. These diagnoses are suggested by the elevated jugular venous pressure, which is atypical in cirrhosis.
Although portal hypertension accounts for most cases of ascites, peritoneal fluid should be examined to exclude peritoneal carcinomatosis and tuberculous ascites. I am interested in the results of an echocardiogram.
Initial laboratory studies demonstrated a sodium concentration of 136 mEq/dL; potassium, 4.7 mEq/dL; chloride, 99 mEq/dL; bicarbonate, 24 mEq/dL; blood urea nitrogen, 54 mg/dL; creatinine, 3.3 mg/dL (increased from baseline of 1.6 mg/dL 4 months previously); white cell count, 7000/mm3; hemoglobin, 10.5 g/dL; MCV, 89 fL; platelet count, 205,000/mm3; bilirubin, 0.6 mg/dL; aspartate aminotransferase, 15 U/L; alanine aminotransferase, 8 U/L; alkaline phosphatase, 102 U/L; albumin, 4.2 g/dL; total protein, 8.2 g/dL; international normalized ratio, 1.2; and partial thromboplastin time, 31.8 seconds. A urine dipstick demonstrated 1+ protein. The chest radiograph was normal. Electrocardiogram had borderline low voltage with nonspecific T‐wave abnormalities. Additional studies showed a serum iron concentration of 49 mg/dL, transferrin saturation of 16%, total iron binding capacity of 310 mg/dL, and ferritin of 247 mg/mL. Hemoglobin A1c was 7.0%. Acute and chronic antibodies to hepatitis A, B, and C viruses were negative. The following study results were normal or negative: antinuclear antibody, alpha‐1‐antitrypsin, ceruloplasmin, alpha‐fetoprotein, carcinoembryonic antigen, and 24‐hour urinary copper. The thyroid function studies were normal. A purified protein derivative (PPD) skin test was nonreactive.
There continues to be a paucity of evidence of a primary liver disorder. The hepatic enzymes and tests of liver synthetic function are normal, and there is no pancytopenia, as might result from hypersplenism. I remain most suspicious of either a primary cardiac or pericardial disorder with secondary hepatic congestion or a disease that simultaneously affects the heart and liver.
The reasons for the low voltage on the electrocardiogram include processes that infiltrate the myocardium (amyloidosis, sarcoidosis, hemochromatosis, and myxedema fluid) and processes that increase the distance between the myocardium and surface electrodes, such as adipose tissue, air (from emphysema or pneumothorax), or pericardial effusion. Pericardial effusion may present subacutely with predominant features of right ventricular failure. Low voltage, liver disease, and possible heart failure raise the possibility of amyloidosis or hemochromatosis. The low transferrin saturation renders hemochromatosis unlikely. Although normal alkaline phosphatase and serum albumin are not characteristic when AL amyloid affects the liver and kidneys, serum and urine protein electrophoresis and immunofixation should be considered.
With paracentesis 3.5 L of ascitic fluid was removed. The red cell count was 4000/mm3, and white blood cell count was 505/mm3, of which 25% were polymorphonuclear cells, 22% were lymphocytes, and 53% were monocytes. Additional peritoneal fluid chemistries included albumin of 3.0 g/dL and total protein of 5.3 g/dL. Abdominal ultrasound with Doppler demonstrated a liver of normal size and echogenicity with patent hepatic arteries, hepatic veins, and portal vein. There was mild splenomegaly with normal kidneys. Evaluation for a possible liver transplant was initiated. Blood, urine, and peritoneal fluid cultures demonstrated no growth. Echocardiography demonstrated borderline concentric left ventricular hypertrophy, normal right and left ventricular function, dilated superior and inferior vena cavae, and no pericardial effusion or thickening.
The serum‐ascites albumin gradient (SAAG) of 1.2 is consistent with portal hypertension as the cause of the ascites. The Doppler findings exclude postsinusoidal causes of portal hypertension from hepatic vein obstruction or thrombosis. The combination of the elevated SAAG, elevated jugular venous pressure, borderline low voltage on ECG, and elevated peritoneal total protein make cardiac and pericardial disease the leading considerations. Given the normal ventricular function, I am concerned about elevated intracardiac pressures resulting from pericardial disease or restrictive cardiomyopathy. At this point, right heart catheterization would be useful for assessing intracardiac pressures.
On the fourth hospital day, paracentesis was repeated, and 15 L of fluid was removed. A transjugular liver biopsy demonstrated diffuse patchy fibrosis consistent with early cirrhosis and minor intralobular changes with minimal ballooning. There was no steatosis, active inflammation, granulomata, iron deposition, or evidence of viral hepatitis. Right heart catheterization revealed a right atrial pressure of 18 cm H20, right ventricular pressure of 34/20 cm H20, pulmonary artery pressure of 34/18 cm H20 (mean 25), pulmonary capillary wedge pressure of 20 cm H20, cardiac output of 5.8 L/min, and cardiac index of 2.5 L/min/m2.
The mild hepatic histologic abnormalities do not support an intrinsic liver disease as the cause of his massive ascites and end‐stage liver disease physiology. Cardiac catheterization demonstrates equalization of diastolic pressures, which suggests constrictive pericarditis or restrictive cardiomyopathy. Despite the normal chest radiograph and nonreactive PPD, tuberculosis would be my leading explanation for constrictive pericarditis given the time spent in areas endemic with TB. Although lateral chest radiography may demonstrate pericardial calcifications, magnetic resonance imaging (MRI) is the best imaging modality to detect constrictive pericarditis. Alternately, cardiac amyloidosis could cause restrictive cardiomyopathy and has not been definitively excluded. A cardiac MRI to assess the pericardium would be my next test, and I would request Congo red stains of the liver biopsy. If these tests are unrevealing, endomyocardial biopsy may be necessary.
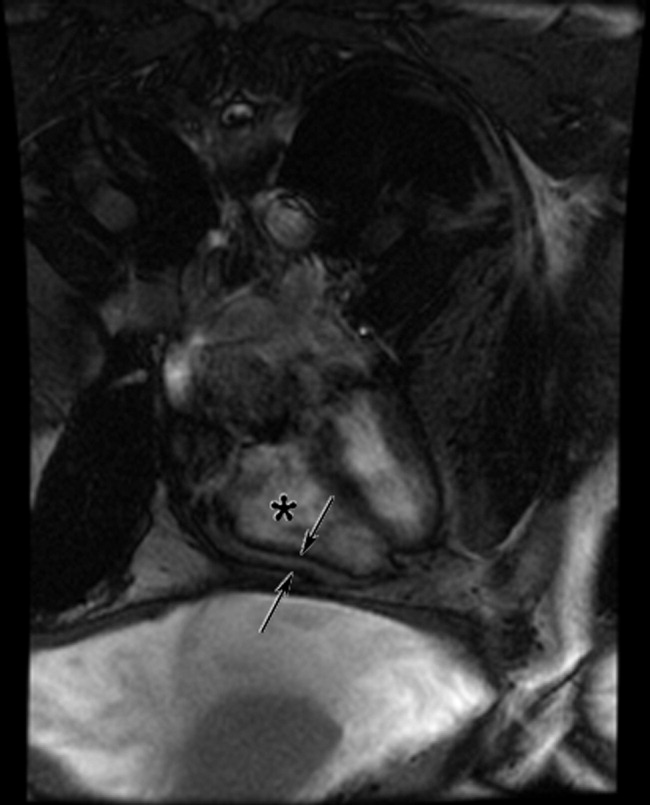
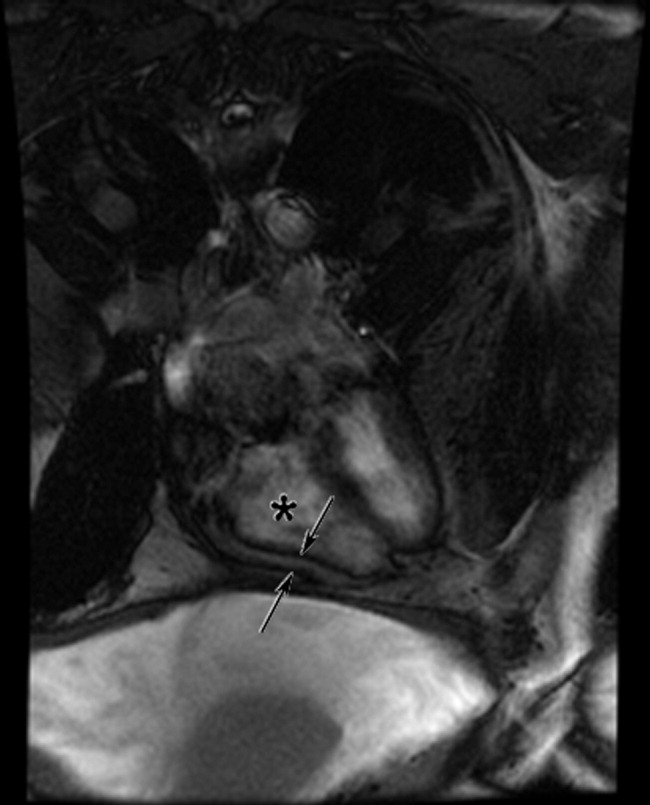
The cardiac MRI revealed a severely thickened 7‐mm pericardium (normal < 3 mm) most prominent over the right atrium and ventricle. The right ventricle was described as bullet‐shaped, suggesting constrictive pericardial disease (Fig. 1). Left heart catheterization to evaluate coronary anatomy and left ventricular pressures revealed no significant coronary arterial disease and demonstrated an elevated left ventricular end‐diastolic pressure consistent with constrictive pericarditis. Endomyocardial biopsy showed no evidence of infiltrative disease, granulomata, or other significant abnormality. The following day the patient underwent pericardiectomy. Postoperatively, his ascites was easily managed with low doses of diuretics. The pericardial tissue revealed chronic inflammatory cells and dense collagenous fibrosis characteristic of constrictive pericarditis without evidence of malignancy or granulomatous disease. Pericardial cultures were negative for bacteria, viruses, fungi, and mycobacteria.
DISCUSSION
Constrictive pericarditis is characterized by chronic fibrous thickening of the once‐elastic pericardial sac and can occur following any disease process that affects the pericardium (Table 1).1, 2 The challenge in the diagnosis of constrictive pericarditis lies in the recognition of this slowly progressive and uncommon disease. In many cases, nonspecific symptoms of reduced cardiac output and insidious right‐sided heart failure are present for 12 months or longer before a diagnosis is established.1, 3 A typical presentation of constrictive pericarditis is peripheral edema, ascites, and hepatomegaly, a combination that may understandably lead to a misdiagnosis of chronic liver disease and even subject a patient to the unnecessary risk of a liver biopsy, as in this case.
|
Cryptogenic cirrhosis, the initial diagnosis of this patient, is a term used only after excluding the common and uncommon causes of cirrhosis (Table 2).46 With expanded knowledge of the causes of cirrhosis, especially nonalcoholic fatty liver disease, the number of cases of cirrhosis considered to be cryptogenic has decreased from nearly one‐third of all cases in 1960 to approximately 5% in a modern series.7, 8 Chronic or repetitive heart failure can lead to progressive hepatic fibrosis and cirrhosis. Distinguishing features compared to other causes of cirrhosis include an ascitic protein concentration greater than 2.5 g/dL, relatively preserved synthetic function, and infrequent stigmata of end‐stage liver disease such as spider angiomata or pronounced jaundice.9, 10
|
Most common
|
Less common
|
A key exam feature that distinguishes cardiac cirrhosis from other causes of liver failure is an elevated jugular venous pressure. Hepatic causes of cirrhosis induce increased nitric oxide production, which leads to splanchnic and peripheral arterial vasodilatation with a reduced effective circulating volume and normal or low jugular venous pressure.11, 12 Therefore, a patient with cirrhosis and ascites having an elevated jugular venous pressure should prompt echocardiographic evaluation.13 When echocardiography excludes ventricular dysfunction, valvular abnormalities, and pulmonary hypertension, constrictive pericarditis and restrictive cardiomyopathy remain important diagnostic considerations.
In both constrictive pericarditis and restrictive cardiomyopathy, ventricular filling is limited. Pressures in the chambers rise abruptly and rapidly during ventricular filling until equilibrium is reached in early diastole. This can be conceptualized as the cardiac chambers being constrained by the limitations of a rigid external box. In constrictive pericarditis, the rigid external box is the fibrosed and thickened pericardial sac, which loses its elasticity and impairs filling of the ventricles. In restrictive cardiomyopathy, the stiff myocardium limits ventricular filling.
There is considerable overlap in the clinical, echocardiographic, and hemodynamic findings of constrictive pericarditis and restrictive cardiomyopathy.14 Both may present insidiously with progressive heart failure. Echocardiography demonstrates impaired diastolic function. Cardiac hemodynamics demonstrate abrupt and rapid early diastolic filling, elevated and equal ventricular end‐diastolic pressures, and reduced stroke volume and cardiac output. A diagnosis of constrictive pericarditis is favored when a marked inspiratory increase in right ventricular pressures and decrease in left ventricular pressures are seen on heart catheterization or a similar inspiratory increase in transvalvular flow velocities across the tricuspid valve compared with the mitral valve is shown by echocardiography. This finding results from normal inspiratory increases in intrathoracic pressures, which are unable to be transmitted through the rigid pericardium but continue to augment venous return to the right side of the heart. As many as one‐third of patients with pericardial constriction lack these characteristic findings on echocardiogram.14
The results of pericardial imaging may suggest a diagnosis of constrictive pericarditis. Lateral chest radiography demonstrates pericardial calcifications in less than 30% of cases.15 Cardiac computed tomography (CT) and MRI are the best imaging modalities for detecting an increase in pericardial thickness (3 mm or greater).16 However, in as many as 20% of patients with surgically confirmed constrictive pericarditis, CT and MRI will demonstrate a pericardium of normal thickness.17
When faced with the diagnostic conundrum of constrictive pericarditis versus restrictive cardiomyopathy, strong clinical suspicion, thorough echocardiography, careful hemodynamic assessment with right and left heart catheterization,14, 18 pericardial imaging, and sometimes endomyocardial biopsy to exclude restrictive cardiomyopathy are often needed before proceeding to pericardiectomy, which carries a significant surgical risk but can also be curative.
This case highlights many of the features of constrictive pericarditis, the challenges and delay in its diagnosis, and its occasional misdiagnosis as chronic liver disease. Clinicians may recognize the typical combination of cirrhosis (or suspected cirrhosis), high SAAG ascites, and edema as characteristic of advanced intrinsic liver disease. However, they must not be seduced into immediate pattern recognition when contrary evidencesuch as elevated neck veins, elevated ascitic total protein, or relatively preserved hepatic synthetic functionaccompanies that picture. Under such circumstances, they must remember to think outside the box and bear in mind that the heart may be trapped inside a box.
Take‐Home Points
-
Constrictive pericarditis is often unrecognized initially, resulting in delayed diagnosis. Patients typically present with nonspecific signs and symptoms of low cardiac output and progressive right‐sided heart failure. Clinical suspicion is key to prompt diagnosis and pericardiectomy, which may be curative.
-
Distinguishing features in the presentation of cardiac or pericardial etiologies of ascites and cirrhosis include elevated neck veins, elevated ascitic protein content, relatively preserved hepatic synthetic function, and absence of the stigmata of end‐stage liver disease.
-
Constrictive pericarditis and restrictive cardiomyopathy can present with a similar clinical picture and hemodynamics showing impaired ventricular filling. Right and left heart catheterization, pericardial imaging, and endomyocardial biopsy may differentiate the 2 conditions. For constrictive pericarditis, surgical and pathological confirmation is the gold standard for diagnosis and the only definitive treatment.
- ,,, et al.Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy.Circulation.1999;100:1380–1386.
- ,,, et al.Constrictive pericarditis: etiology and cause‐specific survival after pericardiectomy.J Am Coll Cardiol.2004;43:1445–1452.
- .Chronic constrictive pericarditis.Am J Cardiol.1961;7:48–61.
- American Gastroenterological Association.AGA technical review on the evaluation of liver chemistry tests.Gastroenterology.2002;123:1367–1384.
- ,.AASLD practice guidelines: evaluation of the patient for liver transplantation.Hepatology.2005;41:1–26.
- Feldman M,Friedman LS,Brandt LJ, eds.Sleisenger and Fordtran's Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management.Philadelphia:Saunders Elsevier;2006.
- ,,, et al.Cirrhosis of the liver: a study of alcoholic and nonalcoholic patients in Boston and London.N Engl J Med.1960;261:1–9.
- ,,, et al.Liver transplantation for cryptogenic cirrhosis.Liver Transpl Surg.1997;3:359–364.
- ,,, et al.Heart diseases affecting the liver and liver disease affecting the heart.Am Heart J.2000;140:111–120.
- ,,.The liver in heart failure.Clin Liver Dis.2002;6:947–967.
- ,,, et al.Portal hypertension: from pathophysiology to clinical practice.Liver Int.2005;25:1079–1090.
- .Portal hypertension.Curr Opin Gastroenterol.2006;22:254–262.
- ,,, et al.Negative influences of ascites on the cardiac function of cirrhotic patients.Am J Med.1975;59:165–170.
- .Constrictive pericarditis in the modern era: a diagnostic dilemma.Heart.2001;86:619–623.
- ,,, et al.Calcific constrictive pericarditis: is it still with us?Ann Intern Med.2000;132:444–450.
- ,,,,,.CT and MR imaging of pericardial disease.Radiographics.2003;23:S167–S180.
- ,,, et al.Constrictive pericarditis in 26 patients with histologically normal pericardial thickness.Circulation.2003;108:1852–1857.
- ,,, et al.Value of dynamic respiratory changes in left and right ventricular pressures for the diagnosis of constrictive pericarditis.Circulation.1996;93:2007–2013.
A 65‐year‐old man was referred for evaluation of worsening ascites and end‐stage liver disease. The patient had been well until 1 year ago, when he developed lower extremity edema and abdominal distention. After evaluation by his primary care physician, he was given a diagnosis of cryptogenic cirrhosis. He underwent several paracenteses and was placed on furosemide and spironolactone. The patient had been stable on his diuretic regimen until 2 weeks previously, when he suddenly developed worsening edema and ascites, along with dizziness, nausea, and hypotension. His physician stopped the diuretics and referred him to the hospital.
Before diagnosing a patient with cryptogenic cirrhosis, it is necessary to exclude common etiologies of cirrhosis such as alcohol, viral hepatitis, and non‐alcoholic fatty liver disease and numerous uncommon causes, including Wilson's disease, hemochromatosis, Budd‐Chiari, and biliary cirrhosis. It is also important to remember that patients with liver disease are not immune to extrahepatic causes of ascites, such as peritoneal carcinomatosis and tuberculous ascites. Simultaneously, reasons for chronic liver disease decompensating acutely must be considered: medication nonadherence, excess salt intake, hepatotoxicity from acetaminophen or alcohol, and other acute insults, such as hepatocellular carcinoma, an intervening infection (especially spontaneous bacterial peritonitis), ascending cholangitis, or a flare of chronic viral hepatitis.
Past medical and surgical history included diabetes mellitus (diagnosed 10 years previously), obstructive sleep apnea, hypertension, hypothyroidism, and mild chronic kidney disease. Medications included levothyroxine, lactulose, sulfamethoxazole, pioglitazone (started 4 months prior), and ibuprofen. Furosemide and spironolactone had been discontinued 2 weeks previously. He currently resided in the Central Valley of California. He had lived in Thailand from age 7 to 17 and traveled to India more than 1 year ago. He did not smoke and had never used intravenous drugs or received a blood transfusion. He rarely drank alcohol. He worked as a chemist. There was no family history of liver disease.
There is no obvious explanation for the underlying liver disease or the acute decompensation. Sulfamethoxazole is a rare cause of allergic or granulomatous hepatitis. Pioglitazone is a thiazolinedione which in earlier formulations was linked to hepatitis but can be excluded as a cause of this patient's cirrhosis because it was started after liver disease was detected. As a chemist, he might have been exposed to carbon tetrachloride, a known hepatotoxin. Obstructive sleep apnea causes pulmonary hypertension, but severe ascites and acute hepatic decompensation would be unusual. Ibuprofen might precipitate worsening renal function and fluid accumulation. Time in Thailand and India raises the possibility of tuberculous ascites.
The patient had no headache, vision changes, abdominal pain, emesis, melena, hematochezia, chest pain, palpitations, dysuria, polyuria, pruritus, dark urine, or rashes. He reported difficulty with concentration when lactulose was decreased. He noted worsening exercise tolerance with dyspnea after 10 steps and reported a weight gain of 12 pounds in the past 2 weeks.
On examination, temperature was 36.8C; blood pressure, 129/87 mm Hg; heart rate, 85 beats per minute; respirations, 20 per minute; and oxygen saturation, 94% on room air. He was uncomfortable but alert. There was no scleral icterus or conjunctival pallor. Jugular venous pressure was elevated. The lungs were clear, and the heart was regular, with no murmur, rub, or gallops. The abdomen was massively distended with a fluid wave; the liver and spleen could not be palpated. There was pitting edema of the sacrum and lower extremities. There was no asterixis, palmar erythema, spider angiomata, or skin discoloration.
The additional history and physical exam suggest that the primary problem may lie outside the liver, especially as signs of advanced liver disease (other than ascites) are absent. Dyspnea on exertion is consistent with the physical stress of a large volume of ascites or could be secondary to several pulmonary complications associated with liver disease, including portopulmonary hypertension, hepatopulmonary syndrome, or hepatic hydrothorax. Alternatively, the dyspnea raises the possibility that the ascites is not related to a primary liver disorder but rather to anemia or to a cardiac disorder, such as chronic left ventricular failure, isolated right‐sided heart failure, or constrictive pericarditis. These diagnoses are suggested by the elevated jugular venous pressure, which is atypical in cirrhosis.
Although portal hypertension accounts for most cases of ascites, peritoneal fluid should be examined to exclude peritoneal carcinomatosis and tuberculous ascites. I am interested in the results of an echocardiogram.
Initial laboratory studies demonstrated a sodium concentration of 136 mEq/dL; potassium, 4.7 mEq/dL; chloride, 99 mEq/dL; bicarbonate, 24 mEq/dL; blood urea nitrogen, 54 mg/dL; creatinine, 3.3 mg/dL (increased from baseline of 1.6 mg/dL 4 months previously); white cell count, 7000/mm3; hemoglobin, 10.5 g/dL; MCV, 89 fL; platelet count, 205,000/mm3; bilirubin, 0.6 mg/dL; aspartate aminotransferase, 15 U/L; alanine aminotransferase, 8 U/L; alkaline phosphatase, 102 U/L; albumin, 4.2 g/dL; total protein, 8.2 g/dL; international normalized ratio, 1.2; and partial thromboplastin time, 31.8 seconds. A urine dipstick demonstrated 1+ protein. The chest radiograph was normal. Electrocardiogram had borderline low voltage with nonspecific T‐wave abnormalities. Additional studies showed a serum iron concentration of 49 mg/dL, transferrin saturation of 16%, total iron binding capacity of 310 mg/dL, and ferritin of 247 mg/mL. Hemoglobin A1c was 7.0%. Acute and chronic antibodies to hepatitis A, B, and C viruses were negative. The following study results were normal or negative: antinuclear antibody, alpha‐1‐antitrypsin, ceruloplasmin, alpha‐fetoprotein, carcinoembryonic antigen, and 24‐hour urinary copper. The thyroid function studies were normal. A purified protein derivative (PPD) skin test was nonreactive.
There continues to be a paucity of evidence of a primary liver disorder. The hepatic enzymes and tests of liver synthetic function are normal, and there is no pancytopenia, as might result from hypersplenism. I remain most suspicious of either a primary cardiac or pericardial disorder with secondary hepatic congestion or a disease that simultaneously affects the heart and liver.
The reasons for the low voltage on the electrocardiogram include processes that infiltrate the myocardium (amyloidosis, sarcoidosis, hemochromatosis, and myxedema fluid) and processes that increase the distance between the myocardium and surface electrodes, such as adipose tissue, air (from emphysema or pneumothorax), or pericardial effusion. Pericardial effusion may present subacutely with predominant features of right ventricular failure. Low voltage, liver disease, and possible heart failure raise the possibility of amyloidosis or hemochromatosis. The low transferrin saturation renders hemochromatosis unlikely. Although normal alkaline phosphatase and serum albumin are not characteristic when AL amyloid affects the liver and kidneys, serum and urine protein electrophoresis and immunofixation should be considered.
With paracentesis 3.5 L of ascitic fluid was removed. The red cell count was 4000/mm3, and white blood cell count was 505/mm3, of which 25% were polymorphonuclear cells, 22% were lymphocytes, and 53% were monocytes. Additional peritoneal fluid chemistries included albumin of 3.0 g/dL and total protein of 5.3 g/dL. Abdominal ultrasound with Doppler demonstrated a liver of normal size and echogenicity with patent hepatic arteries, hepatic veins, and portal vein. There was mild splenomegaly with normal kidneys. Evaluation for a possible liver transplant was initiated. Blood, urine, and peritoneal fluid cultures demonstrated no growth. Echocardiography demonstrated borderline concentric left ventricular hypertrophy, normal right and left ventricular function, dilated superior and inferior vena cavae, and no pericardial effusion or thickening.
The serum‐ascites albumin gradient (SAAG) of 1.2 is consistent with portal hypertension as the cause of the ascites. The Doppler findings exclude postsinusoidal causes of portal hypertension from hepatic vein obstruction or thrombosis. The combination of the elevated SAAG, elevated jugular venous pressure, borderline low voltage on ECG, and elevated peritoneal total protein make cardiac and pericardial disease the leading considerations. Given the normal ventricular function, I am concerned about elevated intracardiac pressures resulting from pericardial disease or restrictive cardiomyopathy. At this point, right heart catheterization would be useful for assessing intracardiac pressures.
On the fourth hospital day, paracentesis was repeated, and 15 L of fluid was removed. A transjugular liver biopsy demonstrated diffuse patchy fibrosis consistent with early cirrhosis and minor intralobular changes with minimal ballooning. There was no steatosis, active inflammation, granulomata, iron deposition, or evidence of viral hepatitis. Right heart catheterization revealed a right atrial pressure of 18 cm H20, right ventricular pressure of 34/20 cm H20, pulmonary artery pressure of 34/18 cm H20 (mean 25), pulmonary capillary wedge pressure of 20 cm H20, cardiac output of 5.8 L/min, and cardiac index of 2.5 L/min/m2.
The mild hepatic histologic abnormalities do not support an intrinsic liver disease as the cause of his massive ascites and end‐stage liver disease physiology. Cardiac catheterization demonstrates equalization of diastolic pressures, which suggests constrictive pericarditis or restrictive cardiomyopathy. Despite the normal chest radiograph and nonreactive PPD, tuberculosis would be my leading explanation for constrictive pericarditis given the time spent in areas endemic with TB. Although lateral chest radiography may demonstrate pericardial calcifications, magnetic resonance imaging (MRI) is the best imaging modality to detect constrictive pericarditis. Alternately, cardiac amyloidosis could cause restrictive cardiomyopathy and has not been definitively excluded. A cardiac MRI to assess the pericardium would be my next test, and I would request Congo red stains of the liver biopsy. If these tests are unrevealing, endomyocardial biopsy may be necessary.
The cardiac MRI revealed a severely thickened 7‐mm pericardium (normal < 3 mm) most prominent over the right atrium and ventricle. The right ventricle was described as bullet‐shaped, suggesting constrictive pericardial disease (Fig. 1). Left heart catheterization to evaluate coronary anatomy and left ventricular pressures revealed no significant coronary arterial disease and demonstrated an elevated left ventricular end‐diastolic pressure consistent with constrictive pericarditis. Endomyocardial biopsy showed no evidence of infiltrative disease, granulomata, or other significant abnormality. The following day the patient underwent pericardiectomy. Postoperatively, his ascites was easily managed with low doses of diuretics. The pericardial tissue revealed chronic inflammatory cells and dense collagenous fibrosis characteristic of constrictive pericarditis without evidence of malignancy or granulomatous disease. Pericardial cultures were negative for bacteria, viruses, fungi, and mycobacteria.
DISCUSSION
Constrictive pericarditis is characterized by chronic fibrous thickening of the once‐elastic pericardial sac and can occur following any disease process that affects the pericardium (Table 1).1, 2 The challenge in the diagnosis of constrictive pericarditis lies in the recognition of this slowly progressive and uncommon disease. In many cases, nonspecific symptoms of reduced cardiac output and insidious right‐sided heart failure are present for 12 months or longer before a diagnosis is established.1, 3 A typical presentation of constrictive pericarditis is peripheral edema, ascites, and hepatomegaly, a combination that may understandably lead to a misdiagnosis of chronic liver disease and even subject a patient to the unnecessary risk of a liver biopsy, as in this case.
|
Cryptogenic cirrhosis, the initial diagnosis of this patient, is a term used only after excluding the common and uncommon causes of cirrhosis (Table 2).46 With expanded knowledge of the causes of cirrhosis, especially nonalcoholic fatty liver disease, the number of cases of cirrhosis considered to be cryptogenic has decreased from nearly one‐third of all cases in 1960 to approximately 5% in a modern series.7, 8 Chronic or repetitive heart failure can lead to progressive hepatic fibrosis and cirrhosis. Distinguishing features compared to other causes of cirrhosis include an ascitic protein concentration greater than 2.5 g/dL, relatively preserved synthetic function, and infrequent stigmata of end‐stage liver disease such as spider angiomata or pronounced jaundice.9, 10
|
Most common
|
Less common
|
A key exam feature that distinguishes cardiac cirrhosis from other causes of liver failure is an elevated jugular venous pressure. Hepatic causes of cirrhosis induce increased nitric oxide production, which leads to splanchnic and peripheral arterial vasodilatation with a reduced effective circulating volume and normal or low jugular venous pressure.11, 12 Therefore, a patient with cirrhosis and ascites having an elevated jugular venous pressure should prompt echocardiographic evaluation.13 When echocardiography excludes ventricular dysfunction, valvular abnormalities, and pulmonary hypertension, constrictive pericarditis and restrictive cardiomyopathy remain important diagnostic considerations.
In both constrictive pericarditis and restrictive cardiomyopathy, ventricular filling is limited. Pressures in the chambers rise abruptly and rapidly during ventricular filling until equilibrium is reached in early diastole. This can be conceptualized as the cardiac chambers being constrained by the limitations of a rigid external box. In constrictive pericarditis, the rigid external box is the fibrosed and thickened pericardial sac, which loses its elasticity and impairs filling of the ventricles. In restrictive cardiomyopathy, the stiff myocardium limits ventricular filling.
There is considerable overlap in the clinical, echocardiographic, and hemodynamic findings of constrictive pericarditis and restrictive cardiomyopathy.14 Both may present insidiously with progressive heart failure. Echocardiography demonstrates impaired diastolic function. Cardiac hemodynamics demonstrate abrupt and rapid early diastolic filling, elevated and equal ventricular end‐diastolic pressures, and reduced stroke volume and cardiac output. A diagnosis of constrictive pericarditis is favored when a marked inspiratory increase in right ventricular pressures and decrease in left ventricular pressures are seen on heart catheterization or a similar inspiratory increase in transvalvular flow velocities across the tricuspid valve compared with the mitral valve is shown by echocardiography. This finding results from normal inspiratory increases in intrathoracic pressures, which are unable to be transmitted through the rigid pericardium but continue to augment venous return to the right side of the heart. As many as one‐third of patients with pericardial constriction lack these characteristic findings on echocardiogram.14
The results of pericardial imaging may suggest a diagnosis of constrictive pericarditis. Lateral chest radiography demonstrates pericardial calcifications in less than 30% of cases.15 Cardiac computed tomography (CT) and MRI are the best imaging modalities for detecting an increase in pericardial thickness (3 mm or greater).16 However, in as many as 20% of patients with surgically confirmed constrictive pericarditis, CT and MRI will demonstrate a pericardium of normal thickness.17
When faced with the diagnostic conundrum of constrictive pericarditis versus restrictive cardiomyopathy, strong clinical suspicion, thorough echocardiography, careful hemodynamic assessment with right and left heart catheterization,14, 18 pericardial imaging, and sometimes endomyocardial biopsy to exclude restrictive cardiomyopathy are often needed before proceeding to pericardiectomy, which carries a significant surgical risk but can also be curative.
This case highlights many of the features of constrictive pericarditis, the challenges and delay in its diagnosis, and its occasional misdiagnosis as chronic liver disease. Clinicians may recognize the typical combination of cirrhosis (or suspected cirrhosis), high SAAG ascites, and edema as characteristic of advanced intrinsic liver disease. However, they must not be seduced into immediate pattern recognition when contrary evidencesuch as elevated neck veins, elevated ascitic total protein, or relatively preserved hepatic synthetic functionaccompanies that picture. Under such circumstances, they must remember to think outside the box and bear in mind that the heart may be trapped inside a box.
Take‐Home Points
-
Constrictive pericarditis is often unrecognized initially, resulting in delayed diagnosis. Patients typically present with nonspecific signs and symptoms of low cardiac output and progressive right‐sided heart failure. Clinical suspicion is key to prompt diagnosis and pericardiectomy, which may be curative.
-
Distinguishing features in the presentation of cardiac or pericardial etiologies of ascites and cirrhosis include elevated neck veins, elevated ascitic protein content, relatively preserved hepatic synthetic function, and absence of the stigmata of end‐stage liver disease.
-
Constrictive pericarditis and restrictive cardiomyopathy can present with a similar clinical picture and hemodynamics showing impaired ventricular filling. Right and left heart catheterization, pericardial imaging, and endomyocardial biopsy may differentiate the 2 conditions. For constrictive pericarditis, surgical and pathological confirmation is the gold standard for diagnosis and the only definitive treatment.
A 65‐year‐old man was referred for evaluation of worsening ascites and end‐stage liver disease. The patient had been well until 1 year ago, when he developed lower extremity edema and abdominal distention. After evaluation by his primary care physician, he was given a diagnosis of cryptogenic cirrhosis. He underwent several paracenteses and was placed on furosemide and spironolactone. The patient had been stable on his diuretic regimen until 2 weeks previously, when he suddenly developed worsening edema and ascites, along with dizziness, nausea, and hypotension. His physician stopped the diuretics and referred him to the hospital.
Before diagnosing a patient with cryptogenic cirrhosis, it is necessary to exclude common etiologies of cirrhosis such as alcohol, viral hepatitis, and non‐alcoholic fatty liver disease and numerous uncommon causes, including Wilson's disease, hemochromatosis, Budd‐Chiari, and biliary cirrhosis. It is also important to remember that patients with liver disease are not immune to extrahepatic causes of ascites, such as peritoneal carcinomatosis and tuberculous ascites. Simultaneously, reasons for chronic liver disease decompensating acutely must be considered: medication nonadherence, excess salt intake, hepatotoxicity from acetaminophen or alcohol, and other acute insults, such as hepatocellular carcinoma, an intervening infection (especially spontaneous bacterial peritonitis), ascending cholangitis, or a flare of chronic viral hepatitis.
Past medical and surgical history included diabetes mellitus (diagnosed 10 years previously), obstructive sleep apnea, hypertension, hypothyroidism, and mild chronic kidney disease. Medications included levothyroxine, lactulose, sulfamethoxazole, pioglitazone (started 4 months prior), and ibuprofen. Furosemide and spironolactone had been discontinued 2 weeks previously. He currently resided in the Central Valley of California. He had lived in Thailand from age 7 to 17 and traveled to India more than 1 year ago. He did not smoke and had never used intravenous drugs or received a blood transfusion. He rarely drank alcohol. He worked as a chemist. There was no family history of liver disease.
There is no obvious explanation for the underlying liver disease or the acute decompensation. Sulfamethoxazole is a rare cause of allergic or granulomatous hepatitis. Pioglitazone is a thiazolinedione which in earlier formulations was linked to hepatitis but can be excluded as a cause of this patient's cirrhosis because it was started after liver disease was detected. As a chemist, he might have been exposed to carbon tetrachloride, a known hepatotoxin. Obstructive sleep apnea causes pulmonary hypertension, but severe ascites and acute hepatic decompensation would be unusual. Ibuprofen might precipitate worsening renal function and fluid accumulation. Time in Thailand and India raises the possibility of tuberculous ascites.
The patient had no headache, vision changes, abdominal pain, emesis, melena, hematochezia, chest pain, palpitations, dysuria, polyuria, pruritus, dark urine, or rashes. He reported difficulty with concentration when lactulose was decreased. He noted worsening exercise tolerance with dyspnea after 10 steps and reported a weight gain of 12 pounds in the past 2 weeks.
On examination, temperature was 36.8C; blood pressure, 129/87 mm Hg; heart rate, 85 beats per minute; respirations, 20 per minute; and oxygen saturation, 94% on room air. He was uncomfortable but alert. There was no scleral icterus or conjunctival pallor. Jugular venous pressure was elevated. The lungs were clear, and the heart was regular, with no murmur, rub, or gallops. The abdomen was massively distended with a fluid wave; the liver and spleen could not be palpated. There was pitting edema of the sacrum and lower extremities. There was no asterixis, palmar erythema, spider angiomata, or skin discoloration.
The additional history and physical exam suggest that the primary problem may lie outside the liver, especially as signs of advanced liver disease (other than ascites) are absent. Dyspnea on exertion is consistent with the physical stress of a large volume of ascites or could be secondary to several pulmonary complications associated with liver disease, including portopulmonary hypertension, hepatopulmonary syndrome, or hepatic hydrothorax. Alternatively, the dyspnea raises the possibility that the ascites is not related to a primary liver disorder but rather to anemia or to a cardiac disorder, such as chronic left ventricular failure, isolated right‐sided heart failure, or constrictive pericarditis. These diagnoses are suggested by the elevated jugular venous pressure, which is atypical in cirrhosis.
Although portal hypertension accounts for most cases of ascites, peritoneal fluid should be examined to exclude peritoneal carcinomatosis and tuberculous ascites. I am interested in the results of an echocardiogram.
Initial laboratory studies demonstrated a sodium concentration of 136 mEq/dL; potassium, 4.7 mEq/dL; chloride, 99 mEq/dL; bicarbonate, 24 mEq/dL; blood urea nitrogen, 54 mg/dL; creatinine, 3.3 mg/dL (increased from baseline of 1.6 mg/dL 4 months previously); white cell count, 7000/mm3; hemoglobin, 10.5 g/dL; MCV, 89 fL; platelet count, 205,000/mm3; bilirubin, 0.6 mg/dL; aspartate aminotransferase, 15 U/L; alanine aminotransferase, 8 U/L; alkaline phosphatase, 102 U/L; albumin, 4.2 g/dL; total protein, 8.2 g/dL; international normalized ratio, 1.2; and partial thromboplastin time, 31.8 seconds. A urine dipstick demonstrated 1+ protein. The chest radiograph was normal. Electrocardiogram had borderline low voltage with nonspecific T‐wave abnormalities. Additional studies showed a serum iron concentration of 49 mg/dL, transferrin saturation of 16%, total iron binding capacity of 310 mg/dL, and ferritin of 247 mg/mL. Hemoglobin A1c was 7.0%. Acute and chronic antibodies to hepatitis A, B, and C viruses were negative. The following study results were normal or negative: antinuclear antibody, alpha‐1‐antitrypsin, ceruloplasmin, alpha‐fetoprotein, carcinoembryonic antigen, and 24‐hour urinary copper. The thyroid function studies were normal. A purified protein derivative (PPD) skin test was nonreactive.
There continues to be a paucity of evidence of a primary liver disorder. The hepatic enzymes and tests of liver synthetic function are normal, and there is no pancytopenia, as might result from hypersplenism. I remain most suspicious of either a primary cardiac or pericardial disorder with secondary hepatic congestion or a disease that simultaneously affects the heart and liver.
The reasons for the low voltage on the electrocardiogram include processes that infiltrate the myocardium (amyloidosis, sarcoidosis, hemochromatosis, and myxedema fluid) and processes that increase the distance between the myocardium and surface electrodes, such as adipose tissue, air (from emphysema or pneumothorax), or pericardial effusion. Pericardial effusion may present subacutely with predominant features of right ventricular failure. Low voltage, liver disease, and possible heart failure raise the possibility of amyloidosis or hemochromatosis. The low transferrin saturation renders hemochromatosis unlikely. Although normal alkaline phosphatase and serum albumin are not characteristic when AL amyloid affects the liver and kidneys, serum and urine protein electrophoresis and immunofixation should be considered.
With paracentesis 3.5 L of ascitic fluid was removed. The red cell count was 4000/mm3, and white blood cell count was 505/mm3, of which 25% were polymorphonuclear cells, 22% were lymphocytes, and 53% were monocytes. Additional peritoneal fluid chemistries included albumin of 3.0 g/dL and total protein of 5.3 g/dL. Abdominal ultrasound with Doppler demonstrated a liver of normal size and echogenicity with patent hepatic arteries, hepatic veins, and portal vein. There was mild splenomegaly with normal kidneys. Evaluation for a possible liver transplant was initiated. Blood, urine, and peritoneal fluid cultures demonstrated no growth. Echocardiography demonstrated borderline concentric left ventricular hypertrophy, normal right and left ventricular function, dilated superior and inferior vena cavae, and no pericardial effusion or thickening.
The serum‐ascites albumin gradient (SAAG) of 1.2 is consistent with portal hypertension as the cause of the ascites. The Doppler findings exclude postsinusoidal causes of portal hypertension from hepatic vein obstruction or thrombosis. The combination of the elevated SAAG, elevated jugular venous pressure, borderline low voltage on ECG, and elevated peritoneal total protein make cardiac and pericardial disease the leading considerations. Given the normal ventricular function, I am concerned about elevated intracardiac pressures resulting from pericardial disease or restrictive cardiomyopathy. At this point, right heart catheterization would be useful for assessing intracardiac pressures.
On the fourth hospital day, paracentesis was repeated, and 15 L of fluid was removed. A transjugular liver biopsy demonstrated diffuse patchy fibrosis consistent with early cirrhosis and minor intralobular changes with minimal ballooning. There was no steatosis, active inflammation, granulomata, iron deposition, or evidence of viral hepatitis. Right heart catheterization revealed a right atrial pressure of 18 cm H20, right ventricular pressure of 34/20 cm H20, pulmonary artery pressure of 34/18 cm H20 (mean 25), pulmonary capillary wedge pressure of 20 cm H20, cardiac output of 5.8 L/min, and cardiac index of 2.5 L/min/m2.
The mild hepatic histologic abnormalities do not support an intrinsic liver disease as the cause of his massive ascites and end‐stage liver disease physiology. Cardiac catheterization demonstrates equalization of diastolic pressures, which suggests constrictive pericarditis or restrictive cardiomyopathy. Despite the normal chest radiograph and nonreactive PPD, tuberculosis would be my leading explanation for constrictive pericarditis given the time spent in areas endemic with TB. Although lateral chest radiography may demonstrate pericardial calcifications, magnetic resonance imaging (MRI) is the best imaging modality to detect constrictive pericarditis. Alternately, cardiac amyloidosis could cause restrictive cardiomyopathy and has not been definitively excluded. A cardiac MRI to assess the pericardium would be my next test, and I would request Congo red stains of the liver biopsy. If these tests are unrevealing, endomyocardial biopsy may be necessary.
The cardiac MRI revealed a severely thickened 7‐mm pericardium (normal < 3 mm) most prominent over the right atrium and ventricle. The right ventricle was described as bullet‐shaped, suggesting constrictive pericardial disease (Fig. 1). Left heart catheterization to evaluate coronary anatomy and left ventricular pressures revealed no significant coronary arterial disease and demonstrated an elevated left ventricular end‐diastolic pressure consistent with constrictive pericarditis. Endomyocardial biopsy showed no evidence of infiltrative disease, granulomata, or other significant abnormality. The following day the patient underwent pericardiectomy. Postoperatively, his ascites was easily managed with low doses of diuretics. The pericardial tissue revealed chronic inflammatory cells and dense collagenous fibrosis characteristic of constrictive pericarditis without evidence of malignancy or granulomatous disease. Pericardial cultures were negative for bacteria, viruses, fungi, and mycobacteria.
DISCUSSION
Constrictive pericarditis is characterized by chronic fibrous thickening of the once‐elastic pericardial sac and can occur following any disease process that affects the pericardium (Table 1).1, 2 The challenge in the diagnosis of constrictive pericarditis lies in the recognition of this slowly progressive and uncommon disease. In many cases, nonspecific symptoms of reduced cardiac output and insidious right‐sided heart failure are present for 12 months or longer before a diagnosis is established.1, 3 A typical presentation of constrictive pericarditis is peripheral edema, ascites, and hepatomegaly, a combination that may understandably lead to a misdiagnosis of chronic liver disease and even subject a patient to the unnecessary risk of a liver biopsy, as in this case.
|
Cryptogenic cirrhosis, the initial diagnosis of this patient, is a term used only after excluding the common and uncommon causes of cirrhosis (Table 2).46 With expanded knowledge of the causes of cirrhosis, especially nonalcoholic fatty liver disease, the number of cases of cirrhosis considered to be cryptogenic has decreased from nearly one‐third of all cases in 1960 to approximately 5% in a modern series.7, 8 Chronic or repetitive heart failure can lead to progressive hepatic fibrosis and cirrhosis. Distinguishing features compared to other causes of cirrhosis include an ascitic protein concentration greater than 2.5 g/dL, relatively preserved synthetic function, and infrequent stigmata of end‐stage liver disease such as spider angiomata or pronounced jaundice.9, 10
|
Most common
|
Less common
|
A key exam feature that distinguishes cardiac cirrhosis from other causes of liver failure is an elevated jugular venous pressure. Hepatic causes of cirrhosis induce increased nitric oxide production, which leads to splanchnic and peripheral arterial vasodilatation with a reduced effective circulating volume and normal or low jugular venous pressure.11, 12 Therefore, a patient with cirrhosis and ascites having an elevated jugular venous pressure should prompt echocardiographic evaluation.13 When echocardiography excludes ventricular dysfunction, valvular abnormalities, and pulmonary hypertension, constrictive pericarditis and restrictive cardiomyopathy remain important diagnostic considerations.
In both constrictive pericarditis and restrictive cardiomyopathy, ventricular filling is limited. Pressures in the chambers rise abruptly and rapidly during ventricular filling until equilibrium is reached in early diastole. This can be conceptualized as the cardiac chambers being constrained by the limitations of a rigid external box. In constrictive pericarditis, the rigid external box is the fibrosed and thickened pericardial sac, which loses its elasticity and impairs filling of the ventricles. In restrictive cardiomyopathy, the stiff myocardium limits ventricular filling.
There is considerable overlap in the clinical, echocardiographic, and hemodynamic findings of constrictive pericarditis and restrictive cardiomyopathy.14 Both may present insidiously with progressive heart failure. Echocardiography demonstrates impaired diastolic function. Cardiac hemodynamics demonstrate abrupt and rapid early diastolic filling, elevated and equal ventricular end‐diastolic pressures, and reduced stroke volume and cardiac output. A diagnosis of constrictive pericarditis is favored when a marked inspiratory increase in right ventricular pressures and decrease in left ventricular pressures are seen on heart catheterization or a similar inspiratory increase in transvalvular flow velocities across the tricuspid valve compared with the mitral valve is shown by echocardiography. This finding results from normal inspiratory increases in intrathoracic pressures, which are unable to be transmitted through the rigid pericardium but continue to augment venous return to the right side of the heart. As many as one‐third of patients with pericardial constriction lack these characteristic findings on echocardiogram.14
The results of pericardial imaging may suggest a diagnosis of constrictive pericarditis. Lateral chest radiography demonstrates pericardial calcifications in less than 30% of cases.15 Cardiac computed tomography (CT) and MRI are the best imaging modalities for detecting an increase in pericardial thickness (3 mm or greater).16 However, in as many as 20% of patients with surgically confirmed constrictive pericarditis, CT and MRI will demonstrate a pericardium of normal thickness.17
When faced with the diagnostic conundrum of constrictive pericarditis versus restrictive cardiomyopathy, strong clinical suspicion, thorough echocardiography, careful hemodynamic assessment with right and left heart catheterization,14, 18 pericardial imaging, and sometimes endomyocardial biopsy to exclude restrictive cardiomyopathy are often needed before proceeding to pericardiectomy, which carries a significant surgical risk but can also be curative.
This case highlights many of the features of constrictive pericarditis, the challenges and delay in its diagnosis, and its occasional misdiagnosis as chronic liver disease. Clinicians may recognize the typical combination of cirrhosis (or suspected cirrhosis), high SAAG ascites, and edema as characteristic of advanced intrinsic liver disease. However, they must not be seduced into immediate pattern recognition when contrary evidencesuch as elevated neck veins, elevated ascitic total protein, or relatively preserved hepatic synthetic functionaccompanies that picture. Under such circumstances, they must remember to think outside the box and bear in mind that the heart may be trapped inside a box.
Take‐Home Points
-
Constrictive pericarditis is often unrecognized initially, resulting in delayed diagnosis. Patients typically present with nonspecific signs and symptoms of low cardiac output and progressive right‐sided heart failure. Clinical suspicion is key to prompt diagnosis and pericardiectomy, which may be curative.
-
Distinguishing features in the presentation of cardiac or pericardial etiologies of ascites and cirrhosis include elevated neck veins, elevated ascitic protein content, relatively preserved hepatic synthetic function, and absence of the stigmata of end‐stage liver disease.
-
Constrictive pericarditis and restrictive cardiomyopathy can present with a similar clinical picture and hemodynamics showing impaired ventricular filling. Right and left heart catheterization, pericardial imaging, and endomyocardial biopsy may differentiate the 2 conditions. For constrictive pericarditis, surgical and pathological confirmation is the gold standard for diagnosis and the only definitive treatment.
- ,,, et al.Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy.Circulation.1999;100:1380–1386.
- ,,, et al.Constrictive pericarditis: etiology and cause‐specific survival after pericardiectomy.J Am Coll Cardiol.2004;43:1445–1452.
- .Chronic constrictive pericarditis.Am J Cardiol.1961;7:48–61.
- American Gastroenterological Association.AGA technical review on the evaluation of liver chemistry tests.Gastroenterology.2002;123:1367–1384.
- ,.AASLD practice guidelines: evaluation of the patient for liver transplantation.Hepatology.2005;41:1–26.
- Feldman M,Friedman LS,Brandt LJ, eds.Sleisenger and Fordtran's Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management.Philadelphia:Saunders Elsevier;2006.
- ,,, et al.Cirrhosis of the liver: a study of alcoholic and nonalcoholic patients in Boston and London.N Engl J Med.1960;261:1–9.
- ,,, et al.Liver transplantation for cryptogenic cirrhosis.Liver Transpl Surg.1997;3:359–364.
- ,,, et al.Heart diseases affecting the liver and liver disease affecting the heart.Am Heart J.2000;140:111–120.
- ,,.The liver in heart failure.Clin Liver Dis.2002;6:947–967.
- ,,, et al.Portal hypertension: from pathophysiology to clinical practice.Liver Int.2005;25:1079–1090.
- .Portal hypertension.Curr Opin Gastroenterol.2006;22:254–262.
- ,,, et al.Negative influences of ascites on the cardiac function of cirrhotic patients.Am J Med.1975;59:165–170.
- .Constrictive pericarditis in the modern era: a diagnostic dilemma.Heart.2001;86:619–623.
- ,,, et al.Calcific constrictive pericarditis: is it still with us?Ann Intern Med.2000;132:444–450.
- ,,,,,.CT and MR imaging of pericardial disease.Radiographics.2003;23:S167–S180.
- ,,, et al.Constrictive pericarditis in 26 patients with histologically normal pericardial thickness.Circulation.2003;108:1852–1857.
- ,,, et al.Value of dynamic respiratory changes in left and right ventricular pressures for the diagnosis of constrictive pericarditis.Circulation.1996;93:2007–2013.
- ,,, et al.Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy.Circulation.1999;100:1380–1386.
- ,,, et al.Constrictive pericarditis: etiology and cause‐specific survival after pericardiectomy.J Am Coll Cardiol.2004;43:1445–1452.
- .Chronic constrictive pericarditis.Am J Cardiol.1961;7:48–61.
- American Gastroenterological Association.AGA technical review on the evaluation of liver chemistry tests.Gastroenterology.2002;123:1367–1384.
- ,.AASLD practice guidelines: evaluation of the patient for liver transplantation.Hepatology.2005;41:1–26.
- Feldman M,Friedman LS,Brandt LJ, eds.Sleisenger and Fordtran's Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management.Philadelphia:Saunders Elsevier;2006.
- ,,, et al.Cirrhosis of the liver: a study of alcoholic and nonalcoholic patients in Boston and London.N Engl J Med.1960;261:1–9.
- ,,, et al.Liver transplantation for cryptogenic cirrhosis.Liver Transpl Surg.1997;3:359–364.
- ,,, et al.Heart diseases affecting the liver and liver disease affecting the heart.Am Heart J.2000;140:111–120.
- ,,.The liver in heart failure.Clin Liver Dis.2002;6:947–967.
- ,,, et al.Portal hypertension: from pathophysiology to clinical practice.Liver Int.2005;25:1079–1090.
- .Portal hypertension.Curr Opin Gastroenterol.2006;22:254–262.
- ,,, et al.Negative influences of ascites on the cardiac function of cirrhotic patients.Am J Med.1975;59:165–170.
- .Constrictive pericarditis in the modern era: a diagnostic dilemma.Heart.2001;86:619–623.
- ,,, et al.Calcific constrictive pericarditis: is it still with us?Ann Intern Med.2000;132:444–450.
- ,,,,,.CT and MR imaging of pericardial disease.Radiographics.2003;23:S167–S180.
- ,,, et al.Constrictive pericarditis in 26 patients with histologically normal pericardial thickness.Circulation.2003;108:1852–1857.
- ,,, et al.Value of dynamic respiratory changes in left and right ventricular pressures for the diagnosis of constrictive pericarditis.Circulation.1996;93:2007–2013.