User login
Four Nephrology Myths Debunked
There are many controversial topics relating to renal disease in hospitalized patients. The aim of this review is to shed light on some important and often debated issues. We will first discuss topics related to electrolytes disorder commonly seen in hospitalized patients (hyponatremia, hyperkalemia, metabolic acidosis) then the use of diuretics in patients with allergy to sulfa containing antibiotics.
Hypothyroidism and Hyponatremia
Hyponatremia is common in hospitalized patients and is associated with worse outcomes.1 It can be seen with a variety of conditions ranging from congestive heart failure to volume depletion. Careful history and physical examination are paramount and the initial work‐up usually includes serum and urine osmolality and urine sodium concentration.
For euvolemic hyponatremia, the differential diagnosis includes the syndrome of inappropriate adenine dinucleotide (ADH) secretion (SIADH), hypoadrenalism, and beer potomania. Additionally, many authorities also include hypothyroidism.
Although the simultaneous finding of hypothyroidism and hyponatremia can occur in patients as both diseases are widely prevalent in the general population, causation has yet to be convincingly demonstrated.
ADH is released in response to effective volume depletion; consequently when hypothyroidism is encountered in the setting of complete pituitary failure there is often hyponatremia.2, 3 Alternatively, with myxedema, the ability of the kidney to handle a water load and concentrate urine can be impaired.4
However, the observation that thyroid hormone administration did not raise sodium values in newborns with congenital hypothyroidism or in adults supports the absence of causal effect.5, 6And in addition, large studies done in the hospital and outpatient setting showed no differences between the serum sodium values of hypothyroid patients and that of controls.7, 8 In the study of outpatients, among those with hypothyroidism, for every increase of 10 mU/L of thyroid‐stimulating hormone (TSH), there was a drop of only 0.14 mmol/L of Na concentration.8 Thus, the elevation of TSH required for a clinically meaningful drop in sodium to occur was considerable.
Hence in patients with hyponatremia, the hospitalist should look for etiologies other than hypothyroidism and should only consider thyroid hypofunction as a culprit in cases of myxedema, or panhypopituitarism.
Sodium Bicarbonate for Hyperkalemia
Hyperkalemia is one of the most feared electrolyte disorders encountered in hospitalized patients and can lead to dire outcomes.9, 10
Potassium (K+) homeostasis is maintained in the body by 2 complimentary systems: a short‐term system that regulates K+ variation by modifying translocation across the cellular membrane and a long‐term system that adjusts overall K+ balance. The translocation system is regulated primarily by insulin and ‐2 stimulation. Overall K+ balance is mainly controlled by the kidney (90‐95%) although the gastrointestinal (GI) tract can have a more preponderant role in anephric patients.
Hyperkalemia can ensue by either a dysregulation of the translocation system (as in diabetic Ketoacidosis secondary to insulin deficiency) or impairment of K+ elimination.
Acid‐base status was previously thought to have a prominent influence on K+ concentration, based on studies that demonstrated that. However, studies looking at metabolic acidosis revealed that contrary to the effect of mineral acidosis (excess of nonmetabolizable anions)11 where there is an inverse correlation between potassium concentration and pH; organic acidosis (excess of metabolizable anions)11 was not associated with hyperkalemia.1214 However, organic acidosis can be seen simultaneously and induced by a same underlying disease (such as organ ischemia with lactic acidosis or insulin deficiency complicated by ketoacidosis). Also, when changes in pH are induced by respiratory variations or with alkalosis, the impact on serum K+ concentration is less remarkable.15 Hence, it seems that it is the nature of the acid‐base disturbance that impacts K+ concentration more than the change in pH itself.
In the kidney, the main site for regulation of K+ balance is the collecting duct. Factors that affect elimination include urinary sodium delivery, urine flow, and aldosterone.16 In order to adequately eliminate K+ these factors must be optimized in conjunction.
Treatment of hyperkalemia includes the sequential administration of agents that stabilize the cardiac membrane (calcium gluconate), shift the potassium intracellularly (insulin, ‐2 agonists), and remove the potassium (diuretics, sodium polystyrene, or dialysis).
The use of sodium bicarbonate for treatment of hyperkalemia has been long advocated.17 It was thought to act by translocation of potassium hence could be used to quickly lower K+ concentration. However, this dogma has been challenged recently.
To assess the true impact of sodium bicarbonate on potassium translocation, studies have been conducted on anephric patients with hyperkalemia. Bicarbonate infusion failed to elicit a significant rapid change in serum K+ concentration despite increase in bicarbonate concentration, arguing against a translocation mechanism.1821 After 60 minutes of treatment, neither isotonic nor hypertonic bicarbonate infusion affected Serum K+ levels in end‐stage renal disease (ESRD) patients.19, 20 On the contrary, hypertonic sodium bicarbonate increased the K+ concentration after 180 minutes of treatment,20 and it took a prolonged infusion of 4 hours to see a significant decrease in K+ concentration (0.6 mmol/L); half of which could be accounted for solely by volume administration. Moreover, this reduction was highly variable.
Rather, sodium bicarbonate seems to enhance potassium elimination by increasing sodium delivery to the distal tubule, increasing urinary pH and negative luminal charge and potentiating the action of diuretics.23 In an elegant study on normovolemic patients, the induction of bicarbonaturia practically doubled potassium excretion.23 However, such an effect is heterogeneous and usually takes place over 4 hours to 6 hours.17
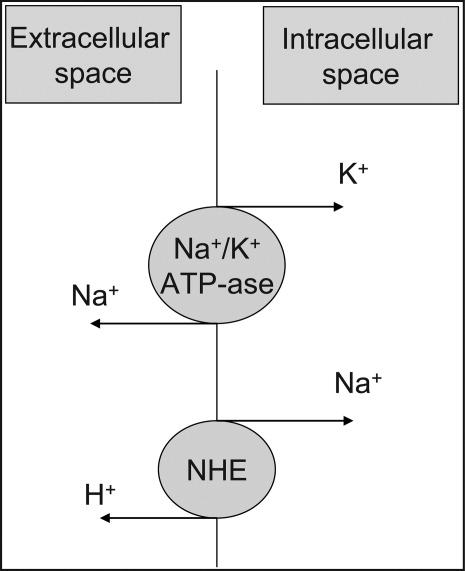
At the cellular level, 2 ion exchange pumps cooperate to handle Na/K/H movement across the cellular membrane: an Na+/H+ exchanger (NHE) and the Na+/K+ ATP‐ase pump (Figure 1). The NHE is normally inactive and is only upregulated in cases of severe intracellular acidosis.24 The infusion of sodium bicarbonate to patients with severe metabolic acidosis could possibly decrease the serum potassium concentration by translocation if the NHE was significantly upregulated. However, this treatment can be associated with a drop in the ionized calcium level, a worsening of the intracellular acidosis, and a decreased peripheral oxygen delivery.25 Thus, the benefits should be balanced with the potential adverse effects and, even in cases of severe metabolic acidosis with hyperkalemia, we would advise the clinician to restrictively administer sodium bicarbonate.
In addition, in ESRD patients, the administration of sodium bicarbonate can be problematic owing to the osmotic and volume burden it carries. It should also be avoided in patients who are volume overload or in those with decreased ability to eliminate potassium.
When treating hyperkalemic patients, hospitalists should use sodium bicarbonate to potentiate urinary elimination of potassium and should consider administering it either with acetazolamide or a loop diuretic, anticipating a lowering effect after a few hours.26 It should be avoided in patients with volume overload and anuria. Immediate translocation of potassium into cells is best achieved by insulin and ‐2 agonists.
5‐Oxoprolinuria: A Newly Recognized Cause of High Anion Gap Metabolic Acidosis
There are several causes of metabolic high anion gap acidosis in hospitalized patients. However, despite careful investigations, the cause of that disorder is not always apparent.27 Recently, 5‐oxoprolinuria (also called pyroglutamic acidosis) has become increasingly recognized as a potential etiology.2832
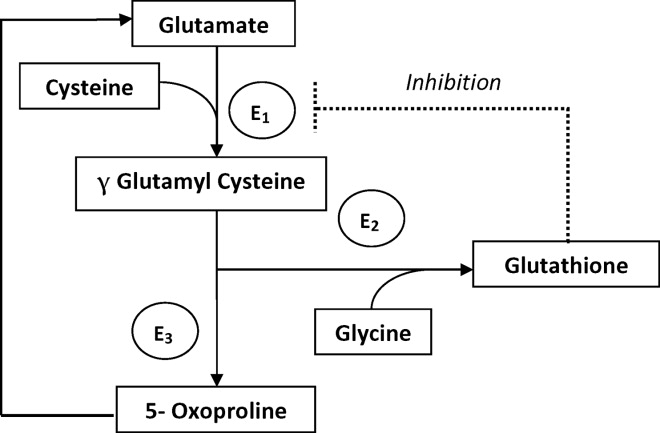
The metabolism of glutamate (though the ‐Glutamyl cycle) generates glutathione which provides negative feedback to the ‐Glutamyl‐cysteine synthetase enzyme. The depletion of glutathione increases 5‐oxoproline production owing to the loss of that inhibition (Figures 2 and 3). Low glutathione levels can be seen with liver disease,33 chronic alcohol intake,34 acetaminophen use,35 malnutrition,36 renal dysfunction,29 use of vigabatrim (an antiepileptic that received Food and Drug Administration [FDA] approval for use in April 2009)37 and sepsis.38 Most of the reported cases were female and had more than one risk factor.39
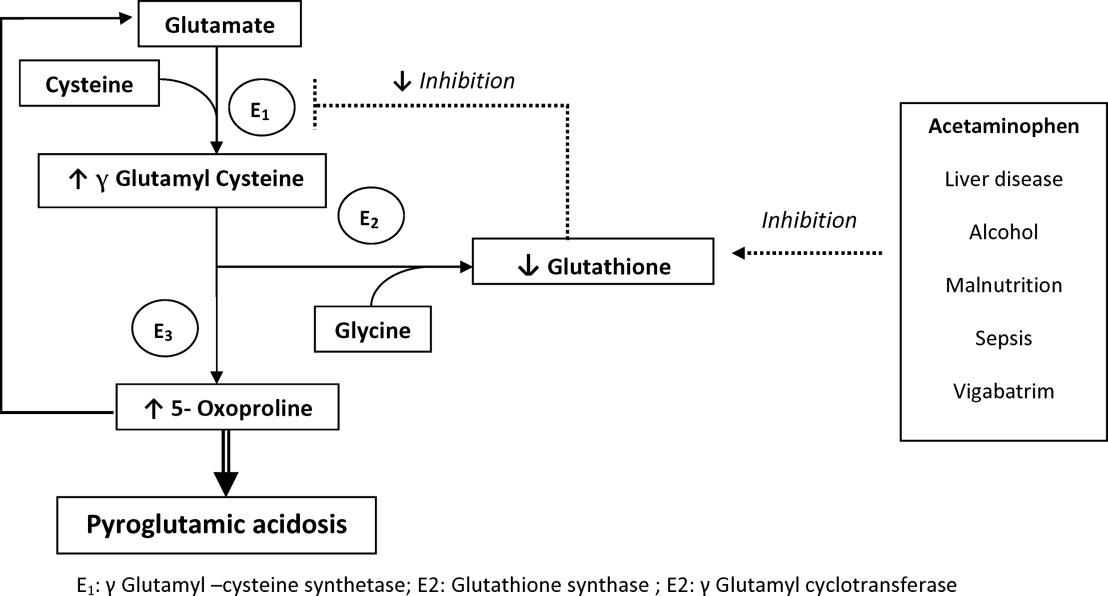
Typically, patients present with high anion gap acidosis (often more than 20)28 with normal acetaminophen levels and all usual tests being negative. A history of chronic acetaminophen use with or without other risk factors can frequently be found. The true independent impact of this type of acidosis on outcomes is difficult to determine as all of the reported cases had many confounding factors.
A urinary organic acid level is diagnostic and will reveal increased levels of pyroglutamic acid. Alternatively, the finding of a positive urinary anion gap (UNa +UK UCl) with a positive urinary osmolar gap (Uosmmeasured‐Uosmcalculated) in the appropriate clinical setting (unexplained high anion gap acidosis with negative workup and presence of risk factors for 5‐oxoproliniuria) can point towards the diagnosis.40
A study of patients with unexplained metabolic acidosis did not find any cases of 5‐oxoprolinuria.41 Although this might suggest that the incidence of this disease is low, very few of those patients were actually taking acetaminophen (therefore had a reduced propensity for developing pyroglutamic acidosis).41 Thus, the actual incidence of 5‐oxoprolinuria is hard to determine.
Once recognized, acetaminophen should be withheld and N‐acetylcysteine (NAC) can be used to replete glutathione levels although there is no convincing evidence for this use.42 It is important for hospitalists to be aware of this disorder as it can pose a diagnostic challenge (negative usual work‐up), is easy to treat by stopping acetaminophen, and can (possibly) negatively affect outcomes.
Furosemide and Patients With Sulfa Allergy
Allergic reactions are a common occurrence with sulfa‐containing antibiotics (SCA) and reports estimates the incidence to be approximately 3% to 5%.43, 44
One misbelief is that patients who are allergic to SCAs should not receive sulfa containing diuretics or other sulfa‐containing medications.45 This leads some physicians to substitute commonly used diuretics (such as furosemide or thiazides) for ethacrynic acid. The use of ethacrynic acid has several challenges: the limited supply of the intravenous form, the discontinuation of the oral form, the increased cost, and the risk of permanent ototoxicity.
The evidence for potential allergic cross‐reactivity among medications containing the sulfa moiety has been primarily derived from Case Reports.4649
The molecular structures of sulfamethoxazole and furosemide are shown below. The allergic antigen is most often the N1 component,45 and sometimes N4 but not the sulfa moiety. Both of the incriminated antigens are not present in the furosemide structure (as well as all other sulfa containing diuretics) (Figures 4 and 5).
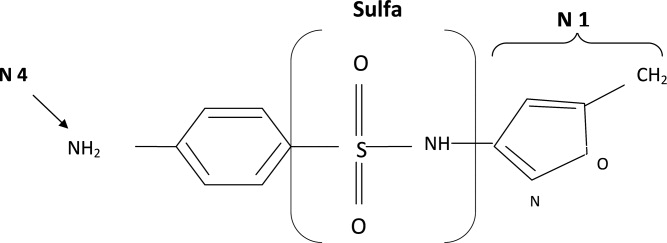
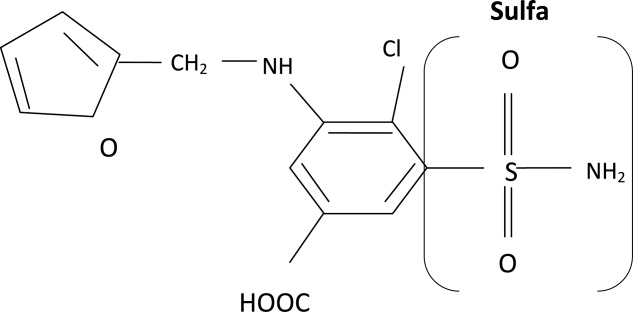
Experimental data showed that serum from patients allergic to SCAs did not bind to diuretics.50 In addition, clinical reports failed to demonstrate cross‐reactivity.5153 In a large clinical trial, Strom et al.53 showed that although there was a higher risk for allergic reaction to sulfa containing medications (SCM) in patients allergic to SCA (compared to those who were not), it was lower among patients with an allergy to sulfa antibiotic than among patients with a history of hypersensitivity to penicillins, suggesting this was due to a predisposition to allergic reactions in general rather than true cross‐reactivity. In another report, patients who were receiving ethacrynic acid for many years were successfully and uneventfully switched to furosemide.54
Taken together, these findings suggest that there is no evidence for withholding sulfa nonantibiotics in patients allergic to sulfa containing antibiotics.
Conclusion
Hypothyroidism, unlike myxedema, is not a cause of hyponatremia (although it can be sometimes seen in conjunction with the latter) and additional investigations should be done to determine its etiology. Sodium bicarbonate is effective for treatment of hyperkalemia by enhancing renal potassium elimination, rather than from shifting potassium into cells. The 5‐oxoprolinuria is a newly recognized cause of high anion‐gap metabolic acidosis and should be considered in patients who have taken acetaminophen. Furosemide (and sulfa containing diuretics) can be used safely in patients with an allergy to SCA.
- ,,.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122(9):857–865.
- ,.Hyponatremia as the presenting manifestation of Sheehan's syndrome in elderly patients.Aging Clin Exp Res.2006;18(6):536–539.
- ,,.Sodium and water disturbances in patients with Sheehan's syndrome.Am J Kidney Dis.2001;38(3):E14.
- ,,,,,.Effect of acute water loading on plasma levels of antidiuretic hormone AVP aldosterone, ANP fractional excretion of sodium and plasma and urine osmolalities in myxedema.Chin Med J (Engl).1990;103(9):704–708.
- ,.Sodium handling in congenitally hypothyroid neonates.Acta Paediatr.2004;93(1):22–24.
- ,,.Prevalence and severity of hyponatremia and hypercreatininemia in short‐term uncomplicated hypothyroidism.J Endocrinol Invest.1999;22(1):35–39.
- ,,,,.Absence of relation between hyponatraemia and hypothyroidism.Lancet.1997;350(9088):1402.
- ,,.The effect of newly diagnosed hypothyroidism on serum sodium concentrations: a retrospective study.Clin Endocrinol (Oxf).2006;64(5):598–599.
- .Disorders of potassium homeostasis. Hypokalemia and hyperkalemia.Crit Care Clin.2002;18(2):273–288,vi.
- ,.Hyperkalemia in hospitalized patients.Int Urol Nephrol.2000;32(2):177–180.
- ,.Treatment of metabolic acidosis.Curr Opin Crit Care.2003;9(4):260–265.
- ,,,.Natural history of lactic acidosis after grand‐mal seizures. A model for the study of an anion‐gap acidosis not associated with hyperkalemia.N Engl J Med.1977;297(15):796–799.
- ,,,,.The plasma potassium concentration in metabolic acidosis: a re‐evaluation.Am J Kidney Dis.1988;11(3):220–224.
- ,,,.Determinants of plasma potassium levels in diabetic ketoacidosis.Medicine (Baltimore).1986;65(3):163–172.
- ,.Changes in plasma potassium concentration during acute acid‐base disturbances.Am J Med.1981;71(3):456–467.
- ,,.New aspects of renal potassium transport.Pflugers Arch.2003;446(3):289–297.
- ,.Correction of hyperkalemia by bicarbonate despite constant blood pH.Kidney Int.1977;12(5):354–360.
- ,,,.Effect of various therapeutic approaches on plasma potassium and major regulating factors in terminal renal failure.Am J Med.1988;85(4):507–512.
- ,.Effect of bicarbonate administration on plasma potassium in dialysis patients: interactions with insulin and albuterol.Am J Kidney Dis.1996;28(4):508–514.
- ,,,,.Effect of hypertonic versus isotonic sodium bicarbonate on plasma potassium concentration in patients with end‐stage renal disease.Miner Electrolyte Metab.1991;17(5):297–302.
- .Combined effect of bicarbonate and insulin with glucose in acute therapy of hyperkalemia in end‐stage renal disease patients.Nephron.1996;72(3):476–482.
- ,,.Effect of prolonged bicarbonate administration on plasma potassium in terminal renal failure.Kidney Int.1992;41(2):369–374.
- ,,, et al.Modulation of the secretion of potassium by accompanying anions in humans.Kidney Int.1991;39(6):1206–1212.
- ,.Controversial issues in the treatment of hyperkalaemia.Nephrol Dial Transplant.2003;18(11):2215–2218.
- ,.Sodium bicarbonate for the treatment of lactic acidosis.Chest.2000;117(1):260–267.
- .Management of severe hyperkalemia.Crit Care Med.2008;36(12):3246–3251.
- ,.Pyroglutamic acid and high anion gap: looking through the keyhole?Crit Care Med.2000;28(6):2140–2141.
- ,,,,.Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen.Clin J Am Soc Nephrol.2006;1(3):441–447.
- ,,,.Pyroglutamic acidemia: a cause of high anion gap metabolic acidosis.Crit Care Med.2000;28(6):1803–1807.
- ,,,,,.Acetaminophen‐induced anion gap metabolic acidosis and 5‐oxoprolinuria (pyroglutamic aciduria) acquired in hospital.Am J Kidney Dis.2005;46(1):143–146.
- ,.Anion gap acidosis associated with acetaminophen.Ann Intern Med.2000;133(9):752–753.
- ,,.Pyroglutamic acidosis in a renal transplant patient.Nephrol Dial Transplant.2005;20(12):2836–2838.
- ,,.Hepatic glutathione content in patients with alcoholic and non alcoholic liver diseases.Life Sci.1988;43(12):991–998.
- ,,,,,.Decreased hepatic glutathione in chronic alcoholic patients.J Hepatol.1986;3(1):1–6.
- ,.Intracellular signaling mechanisms of acetaminophen‐induced liver cell death.Toxicol Sci.2006;89(1):31–41.
- ,,.Urinary excretion of 5‐L‐oxoproline (pyroglutamic acid) is increased during recovery from severe childhood malnutrition and responds to supplemental glycine.J Nutr.1996;126(11):2823–2830.
- ,,,.Pyroglutamicaciduria from vigabatrin.Lancet.1989;1(8652):1452–1453.
- ,,, et al.Cysteine metabolism and whole blood glutathione synthesis in septic pediatric patients.Crit Care Med.2001;29(4):870–877.
- ,.Transient 5‐oxoprolinuria and high anion gap metabolic acidosis: clinical and biochemical findings in eleven subjects.Clin Chem.1998;44(7):1497–1503.
- ,,,.Guilty as charged: unmeasured urinary anions in a case of pyroglutamic acidosis.Neth J Med.2008;66(8):351–353.
- ,,.Unexplained metabolic acidosis in critically ill patients: the role of pyroglutamic acid.Intensive Care Med.2004;30(3):502–505.
- ,,.A therapeutic trial with N‐acetylcysteine in subjects with hereditary glutathione synthetase deficiency (5‐oxoprolinuria).J Inherit Metab Dis.1989;12(2):120–130.
- .Diagnosis of allergic reactions to sulfonamides.Allergy.1999;54Suppl 58:28–32.
- .Practical issues in the management of hypersensitivity reactions: sulfonamides.South Med J.2001;94(8):817–824.
- ,,.Should celecoxib be contraindicated in patients who are allergic to sulfonamides? Revisiting the meaning of ‘sulfa’ allergy.Drug Saf.2001;24(4):239–247.
- .Thrombocytopenia due to sulfonamide cross‐sensitivity.Wis Med J.1982;81(6):21–23.
- ,,.Leukocytoclastic vasculitis induced by use of glyburide: a case of possible cross‐reaction of a sulfonamide and a sulfonylurea.Cutis.2003;71(3):235–238.
- ,.Celecoxib‐induced erythema multiforme with glyburide cross‐reactivity.Pharmacotherapy.2002;22(5):637–640.
- ,.Vesiculobullous rash in a patient with systemic lupus erythematosus.Ann Allergy.1993;70(3):196–203.
- ,,, et al.Use of optical biosensor technology to study immunological cross‐reactivity between different sulfonamide drugs.Anal Biochem.2002;300(2):177–184.
- ,,,.Adverse reactions to sulphonamide and sulphonamide‐trimethoprim antimicrobials: clinical syndromes and pathogenesis.Adverse Drug React Toxicol Rev.1996;15(1):9–50.
- ,,.Cross‐reactivity in HIV‐infected patients switched from trimethoprim‐sulfamethoxazole to dapsone.Pharmacotherapy.1998;18(4):831–835.
- ,,, et al.Absence of cross‐reactivity between sulfonamide antibiotics and sulfonamide nonantibiotics.N Engl J Med.2003;349(17):1628–1635.
- ,,.Furosemide challenge in patients with heart failure and adverse reactions to sulfa‐containing diuretics.Ann Intern Med.2003;138(4):358–359.
There are many controversial topics relating to renal disease in hospitalized patients. The aim of this review is to shed light on some important and often debated issues. We will first discuss topics related to electrolytes disorder commonly seen in hospitalized patients (hyponatremia, hyperkalemia, metabolic acidosis) then the use of diuretics in patients with allergy to sulfa containing antibiotics.
Hypothyroidism and Hyponatremia
Hyponatremia is common in hospitalized patients and is associated with worse outcomes.1 It can be seen with a variety of conditions ranging from congestive heart failure to volume depletion. Careful history and physical examination are paramount and the initial work‐up usually includes serum and urine osmolality and urine sodium concentration.
For euvolemic hyponatremia, the differential diagnosis includes the syndrome of inappropriate adenine dinucleotide (ADH) secretion (SIADH), hypoadrenalism, and beer potomania. Additionally, many authorities also include hypothyroidism.
Although the simultaneous finding of hypothyroidism and hyponatremia can occur in patients as both diseases are widely prevalent in the general population, causation has yet to be convincingly demonstrated.
ADH is released in response to effective volume depletion; consequently when hypothyroidism is encountered in the setting of complete pituitary failure there is often hyponatremia.2, 3 Alternatively, with myxedema, the ability of the kidney to handle a water load and concentrate urine can be impaired.4
However, the observation that thyroid hormone administration did not raise sodium values in newborns with congenital hypothyroidism or in adults supports the absence of causal effect.5, 6And in addition, large studies done in the hospital and outpatient setting showed no differences between the serum sodium values of hypothyroid patients and that of controls.7, 8 In the study of outpatients, among those with hypothyroidism, for every increase of 10 mU/L of thyroid‐stimulating hormone (TSH), there was a drop of only 0.14 mmol/L of Na concentration.8 Thus, the elevation of TSH required for a clinically meaningful drop in sodium to occur was considerable.
Hence in patients with hyponatremia, the hospitalist should look for etiologies other than hypothyroidism and should only consider thyroid hypofunction as a culprit in cases of myxedema, or panhypopituitarism.
Sodium Bicarbonate for Hyperkalemia
Hyperkalemia is one of the most feared electrolyte disorders encountered in hospitalized patients and can lead to dire outcomes.9, 10
Potassium (K+) homeostasis is maintained in the body by 2 complimentary systems: a short‐term system that regulates K+ variation by modifying translocation across the cellular membrane and a long‐term system that adjusts overall K+ balance. The translocation system is regulated primarily by insulin and ‐2 stimulation. Overall K+ balance is mainly controlled by the kidney (90‐95%) although the gastrointestinal (GI) tract can have a more preponderant role in anephric patients.
Hyperkalemia can ensue by either a dysregulation of the translocation system (as in diabetic Ketoacidosis secondary to insulin deficiency) or impairment of K+ elimination.
Acid‐base status was previously thought to have a prominent influence on K+ concentration, based on studies that demonstrated that. However, studies looking at metabolic acidosis revealed that contrary to the effect of mineral acidosis (excess of nonmetabolizable anions)11 where there is an inverse correlation between potassium concentration and pH; organic acidosis (excess of metabolizable anions)11 was not associated with hyperkalemia.1214 However, organic acidosis can be seen simultaneously and induced by a same underlying disease (such as organ ischemia with lactic acidosis or insulin deficiency complicated by ketoacidosis). Also, when changes in pH are induced by respiratory variations or with alkalosis, the impact on serum K+ concentration is less remarkable.15 Hence, it seems that it is the nature of the acid‐base disturbance that impacts K+ concentration more than the change in pH itself.
In the kidney, the main site for regulation of K+ balance is the collecting duct. Factors that affect elimination include urinary sodium delivery, urine flow, and aldosterone.16 In order to adequately eliminate K+ these factors must be optimized in conjunction.
Treatment of hyperkalemia includes the sequential administration of agents that stabilize the cardiac membrane (calcium gluconate), shift the potassium intracellularly (insulin, ‐2 agonists), and remove the potassium (diuretics, sodium polystyrene, or dialysis).
The use of sodium bicarbonate for treatment of hyperkalemia has been long advocated.17 It was thought to act by translocation of potassium hence could be used to quickly lower K+ concentration. However, this dogma has been challenged recently.
To assess the true impact of sodium bicarbonate on potassium translocation, studies have been conducted on anephric patients with hyperkalemia. Bicarbonate infusion failed to elicit a significant rapid change in serum K+ concentration despite increase in bicarbonate concentration, arguing against a translocation mechanism.1821 After 60 minutes of treatment, neither isotonic nor hypertonic bicarbonate infusion affected Serum K+ levels in end‐stage renal disease (ESRD) patients.19, 20 On the contrary, hypertonic sodium bicarbonate increased the K+ concentration after 180 minutes of treatment,20 and it took a prolonged infusion of 4 hours to see a significant decrease in K+ concentration (0.6 mmol/L); half of which could be accounted for solely by volume administration. Moreover, this reduction was highly variable.
Rather, sodium bicarbonate seems to enhance potassium elimination by increasing sodium delivery to the distal tubule, increasing urinary pH and negative luminal charge and potentiating the action of diuretics.23 In an elegant study on normovolemic patients, the induction of bicarbonaturia practically doubled potassium excretion.23 However, such an effect is heterogeneous and usually takes place over 4 hours to 6 hours.17
At the cellular level, 2 ion exchange pumps cooperate to handle Na/K/H movement across the cellular membrane: an Na+/H+ exchanger (NHE) and the Na+/K+ ATP‐ase pump (Figure 1). The NHE is normally inactive and is only upregulated in cases of severe intracellular acidosis.24 The infusion of sodium bicarbonate to patients with severe metabolic acidosis could possibly decrease the serum potassium concentration by translocation if the NHE was significantly upregulated. However, this treatment can be associated with a drop in the ionized calcium level, a worsening of the intracellular acidosis, and a decreased peripheral oxygen delivery.25 Thus, the benefits should be balanced with the potential adverse effects and, even in cases of severe metabolic acidosis with hyperkalemia, we would advise the clinician to restrictively administer sodium bicarbonate.
In addition, in ESRD patients, the administration of sodium bicarbonate can be problematic owing to the osmotic and volume burden it carries. It should also be avoided in patients who are volume overload or in those with decreased ability to eliminate potassium.
When treating hyperkalemic patients, hospitalists should use sodium bicarbonate to potentiate urinary elimination of potassium and should consider administering it either with acetazolamide or a loop diuretic, anticipating a lowering effect after a few hours.26 It should be avoided in patients with volume overload and anuria. Immediate translocation of potassium into cells is best achieved by insulin and ‐2 agonists.
5‐Oxoprolinuria: A Newly Recognized Cause of High Anion Gap Metabolic Acidosis
There are several causes of metabolic high anion gap acidosis in hospitalized patients. However, despite careful investigations, the cause of that disorder is not always apparent.27 Recently, 5‐oxoprolinuria (also called pyroglutamic acidosis) has become increasingly recognized as a potential etiology.2832
The metabolism of glutamate (though the ‐Glutamyl cycle) generates glutathione which provides negative feedback to the ‐Glutamyl‐cysteine synthetase enzyme. The depletion of glutathione increases 5‐oxoproline production owing to the loss of that inhibition (Figures 2 and 3). Low glutathione levels can be seen with liver disease,33 chronic alcohol intake,34 acetaminophen use,35 malnutrition,36 renal dysfunction,29 use of vigabatrim (an antiepileptic that received Food and Drug Administration [FDA] approval for use in April 2009)37 and sepsis.38 Most of the reported cases were female and had more than one risk factor.39
Typically, patients present with high anion gap acidosis (often more than 20)28 with normal acetaminophen levels and all usual tests being negative. A history of chronic acetaminophen use with or without other risk factors can frequently be found. The true independent impact of this type of acidosis on outcomes is difficult to determine as all of the reported cases had many confounding factors.
A urinary organic acid level is diagnostic and will reveal increased levels of pyroglutamic acid. Alternatively, the finding of a positive urinary anion gap (UNa +UK UCl) with a positive urinary osmolar gap (Uosmmeasured‐Uosmcalculated) in the appropriate clinical setting (unexplained high anion gap acidosis with negative workup and presence of risk factors for 5‐oxoproliniuria) can point towards the diagnosis.40
A study of patients with unexplained metabolic acidosis did not find any cases of 5‐oxoprolinuria.41 Although this might suggest that the incidence of this disease is low, very few of those patients were actually taking acetaminophen (therefore had a reduced propensity for developing pyroglutamic acidosis).41 Thus, the actual incidence of 5‐oxoprolinuria is hard to determine.
Once recognized, acetaminophen should be withheld and N‐acetylcysteine (NAC) can be used to replete glutathione levels although there is no convincing evidence for this use.42 It is important for hospitalists to be aware of this disorder as it can pose a diagnostic challenge (negative usual work‐up), is easy to treat by stopping acetaminophen, and can (possibly) negatively affect outcomes.
Furosemide and Patients With Sulfa Allergy
Allergic reactions are a common occurrence with sulfa‐containing antibiotics (SCA) and reports estimates the incidence to be approximately 3% to 5%.43, 44
One misbelief is that patients who are allergic to SCAs should not receive sulfa containing diuretics or other sulfa‐containing medications.45 This leads some physicians to substitute commonly used diuretics (such as furosemide or thiazides) for ethacrynic acid. The use of ethacrynic acid has several challenges: the limited supply of the intravenous form, the discontinuation of the oral form, the increased cost, and the risk of permanent ototoxicity.
The evidence for potential allergic cross‐reactivity among medications containing the sulfa moiety has been primarily derived from Case Reports.4649
The molecular structures of sulfamethoxazole and furosemide are shown below. The allergic antigen is most often the N1 component,45 and sometimes N4 but not the sulfa moiety. Both of the incriminated antigens are not present in the furosemide structure (as well as all other sulfa containing diuretics) (Figures 4 and 5).
Experimental data showed that serum from patients allergic to SCAs did not bind to diuretics.50 In addition, clinical reports failed to demonstrate cross‐reactivity.5153 In a large clinical trial, Strom et al.53 showed that although there was a higher risk for allergic reaction to sulfa containing medications (SCM) in patients allergic to SCA (compared to those who were not), it was lower among patients with an allergy to sulfa antibiotic than among patients with a history of hypersensitivity to penicillins, suggesting this was due to a predisposition to allergic reactions in general rather than true cross‐reactivity. In another report, patients who were receiving ethacrynic acid for many years were successfully and uneventfully switched to furosemide.54
Taken together, these findings suggest that there is no evidence for withholding sulfa nonantibiotics in patients allergic to sulfa containing antibiotics.
Conclusion
Hypothyroidism, unlike myxedema, is not a cause of hyponatremia (although it can be sometimes seen in conjunction with the latter) and additional investigations should be done to determine its etiology. Sodium bicarbonate is effective for treatment of hyperkalemia by enhancing renal potassium elimination, rather than from shifting potassium into cells. The 5‐oxoprolinuria is a newly recognized cause of high anion‐gap metabolic acidosis and should be considered in patients who have taken acetaminophen. Furosemide (and sulfa containing diuretics) can be used safely in patients with an allergy to SCA.
There are many controversial topics relating to renal disease in hospitalized patients. The aim of this review is to shed light on some important and often debated issues. We will first discuss topics related to electrolytes disorder commonly seen in hospitalized patients (hyponatremia, hyperkalemia, metabolic acidosis) then the use of diuretics in patients with allergy to sulfa containing antibiotics.
Hypothyroidism and Hyponatremia
Hyponatremia is common in hospitalized patients and is associated with worse outcomes.1 It can be seen with a variety of conditions ranging from congestive heart failure to volume depletion. Careful history and physical examination are paramount and the initial work‐up usually includes serum and urine osmolality and urine sodium concentration.
For euvolemic hyponatremia, the differential diagnosis includes the syndrome of inappropriate adenine dinucleotide (ADH) secretion (SIADH), hypoadrenalism, and beer potomania. Additionally, many authorities also include hypothyroidism.
Although the simultaneous finding of hypothyroidism and hyponatremia can occur in patients as both diseases are widely prevalent in the general population, causation has yet to be convincingly demonstrated.
ADH is released in response to effective volume depletion; consequently when hypothyroidism is encountered in the setting of complete pituitary failure there is often hyponatremia.2, 3 Alternatively, with myxedema, the ability of the kidney to handle a water load and concentrate urine can be impaired.4
However, the observation that thyroid hormone administration did not raise sodium values in newborns with congenital hypothyroidism or in adults supports the absence of causal effect.5, 6And in addition, large studies done in the hospital and outpatient setting showed no differences between the serum sodium values of hypothyroid patients and that of controls.7, 8 In the study of outpatients, among those with hypothyroidism, for every increase of 10 mU/L of thyroid‐stimulating hormone (TSH), there was a drop of only 0.14 mmol/L of Na concentration.8 Thus, the elevation of TSH required for a clinically meaningful drop in sodium to occur was considerable.
Hence in patients with hyponatremia, the hospitalist should look for etiologies other than hypothyroidism and should only consider thyroid hypofunction as a culprit in cases of myxedema, or panhypopituitarism.
Sodium Bicarbonate for Hyperkalemia
Hyperkalemia is one of the most feared electrolyte disorders encountered in hospitalized patients and can lead to dire outcomes.9, 10
Potassium (K+) homeostasis is maintained in the body by 2 complimentary systems: a short‐term system that regulates K+ variation by modifying translocation across the cellular membrane and a long‐term system that adjusts overall K+ balance. The translocation system is regulated primarily by insulin and ‐2 stimulation. Overall K+ balance is mainly controlled by the kidney (90‐95%) although the gastrointestinal (GI) tract can have a more preponderant role in anephric patients.
Hyperkalemia can ensue by either a dysregulation of the translocation system (as in diabetic Ketoacidosis secondary to insulin deficiency) or impairment of K+ elimination.
Acid‐base status was previously thought to have a prominent influence on K+ concentration, based on studies that demonstrated that. However, studies looking at metabolic acidosis revealed that contrary to the effect of mineral acidosis (excess of nonmetabolizable anions)11 where there is an inverse correlation between potassium concentration and pH; organic acidosis (excess of metabolizable anions)11 was not associated with hyperkalemia.1214 However, organic acidosis can be seen simultaneously and induced by a same underlying disease (such as organ ischemia with lactic acidosis or insulin deficiency complicated by ketoacidosis). Also, when changes in pH are induced by respiratory variations or with alkalosis, the impact on serum K+ concentration is less remarkable.15 Hence, it seems that it is the nature of the acid‐base disturbance that impacts K+ concentration more than the change in pH itself.
In the kidney, the main site for regulation of K+ balance is the collecting duct. Factors that affect elimination include urinary sodium delivery, urine flow, and aldosterone.16 In order to adequately eliminate K+ these factors must be optimized in conjunction.
Treatment of hyperkalemia includes the sequential administration of agents that stabilize the cardiac membrane (calcium gluconate), shift the potassium intracellularly (insulin, ‐2 agonists), and remove the potassium (diuretics, sodium polystyrene, or dialysis).
The use of sodium bicarbonate for treatment of hyperkalemia has been long advocated.17 It was thought to act by translocation of potassium hence could be used to quickly lower K+ concentration. However, this dogma has been challenged recently.
To assess the true impact of sodium bicarbonate on potassium translocation, studies have been conducted on anephric patients with hyperkalemia. Bicarbonate infusion failed to elicit a significant rapid change in serum K+ concentration despite increase in bicarbonate concentration, arguing against a translocation mechanism.1821 After 60 minutes of treatment, neither isotonic nor hypertonic bicarbonate infusion affected Serum K+ levels in end‐stage renal disease (ESRD) patients.19, 20 On the contrary, hypertonic sodium bicarbonate increased the K+ concentration after 180 minutes of treatment,20 and it took a prolonged infusion of 4 hours to see a significant decrease in K+ concentration (0.6 mmol/L); half of which could be accounted for solely by volume administration. Moreover, this reduction was highly variable.
Rather, sodium bicarbonate seems to enhance potassium elimination by increasing sodium delivery to the distal tubule, increasing urinary pH and negative luminal charge and potentiating the action of diuretics.23 In an elegant study on normovolemic patients, the induction of bicarbonaturia practically doubled potassium excretion.23 However, such an effect is heterogeneous and usually takes place over 4 hours to 6 hours.17
At the cellular level, 2 ion exchange pumps cooperate to handle Na/K/H movement across the cellular membrane: an Na+/H+ exchanger (NHE) and the Na+/K+ ATP‐ase pump (Figure 1). The NHE is normally inactive and is only upregulated in cases of severe intracellular acidosis.24 The infusion of sodium bicarbonate to patients with severe metabolic acidosis could possibly decrease the serum potassium concentration by translocation if the NHE was significantly upregulated. However, this treatment can be associated with a drop in the ionized calcium level, a worsening of the intracellular acidosis, and a decreased peripheral oxygen delivery.25 Thus, the benefits should be balanced with the potential adverse effects and, even in cases of severe metabolic acidosis with hyperkalemia, we would advise the clinician to restrictively administer sodium bicarbonate.
In addition, in ESRD patients, the administration of sodium bicarbonate can be problematic owing to the osmotic and volume burden it carries. It should also be avoided in patients who are volume overload or in those with decreased ability to eliminate potassium.
When treating hyperkalemic patients, hospitalists should use sodium bicarbonate to potentiate urinary elimination of potassium and should consider administering it either with acetazolamide or a loop diuretic, anticipating a lowering effect after a few hours.26 It should be avoided in patients with volume overload and anuria. Immediate translocation of potassium into cells is best achieved by insulin and ‐2 agonists.
5‐Oxoprolinuria: A Newly Recognized Cause of High Anion Gap Metabolic Acidosis
There are several causes of metabolic high anion gap acidosis in hospitalized patients. However, despite careful investigations, the cause of that disorder is not always apparent.27 Recently, 5‐oxoprolinuria (also called pyroglutamic acidosis) has become increasingly recognized as a potential etiology.2832
The metabolism of glutamate (though the ‐Glutamyl cycle) generates glutathione which provides negative feedback to the ‐Glutamyl‐cysteine synthetase enzyme. The depletion of glutathione increases 5‐oxoproline production owing to the loss of that inhibition (Figures 2 and 3). Low glutathione levels can be seen with liver disease,33 chronic alcohol intake,34 acetaminophen use,35 malnutrition,36 renal dysfunction,29 use of vigabatrim (an antiepileptic that received Food and Drug Administration [FDA] approval for use in April 2009)37 and sepsis.38 Most of the reported cases were female and had more than one risk factor.39
Typically, patients present with high anion gap acidosis (often more than 20)28 with normal acetaminophen levels and all usual tests being negative. A history of chronic acetaminophen use with or without other risk factors can frequently be found. The true independent impact of this type of acidosis on outcomes is difficult to determine as all of the reported cases had many confounding factors.
A urinary organic acid level is diagnostic and will reveal increased levels of pyroglutamic acid. Alternatively, the finding of a positive urinary anion gap (UNa +UK UCl) with a positive urinary osmolar gap (Uosmmeasured‐Uosmcalculated) in the appropriate clinical setting (unexplained high anion gap acidosis with negative workup and presence of risk factors for 5‐oxoproliniuria) can point towards the diagnosis.40
A study of patients with unexplained metabolic acidosis did not find any cases of 5‐oxoprolinuria.41 Although this might suggest that the incidence of this disease is low, very few of those patients were actually taking acetaminophen (therefore had a reduced propensity for developing pyroglutamic acidosis).41 Thus, the actual incidence of 5‐oxoprolinuria is hard to determine.
Once recognized, acetaminophen should be withheld and N‐acetylcysteine (NAC) can be used to replete glutathione levels although there is no convincing evidence for this use.42 It is important for hospitalists to be aware of this disorder as it can pose a diagnostic challenge (negative usual work‐up), is easy to treat by stopping acetaminophen, and can (possibly) negatively affect outcomes.
Furosemide and Patients With Sulfa Allergy
Allergic reactions are a common occurrence with sulfa‐containing antibiotics (SCA) and reports estimates the incidence to be approximately 3% to 5%.43, 44
One misbelief is that patients who are allergic to SCAs should not receive sulfa containing diuretics or other sulfa‐containing medications.45 This leads some physicians to substitute commonly used diuretics (such as furosemide or thiazides) for ethacrynic acid. The use of ethacrynic acid has several challenges: the limited supply of the intravenous form, the discontinuation of the oral form, the increased cost, and the risk of permanent ototoxicity.
The evidence for potential allergic cross‐reactivity among medications containing the sulfa moiety has been primarily derived from Case Reports.4649
The molecular structures of sulfamethoxazole and furosemide are shown below. The allergic antigen is most often the N1 component,45 and sometimes N4 but not the sulfa moiety. Both of the incriminated antigens are not present in the furosemide structure (as well as all other sulfa containing diuretics) (Figures 4 and 5).
Experimental data showed that serum from patients allergic to SCAs did not bind to diuretics.50 In addition, clinical reports failed to demonstrate cross‐reactivity.5153 In a large clinical trial, Strom et al.53 showed that although there was a higher risk for allergic reaction to sulfa containing medications (SCM) in patients allergic to SCA (compared to those who were not), it was lower among patients with an allergy to sulfa antibiotic than among patients with a history of hypersensitivity to penicillins, suggesting this was due to a predisposition to allergic reactions in general rather than true cross‐reactivity. In another report, patients who were receiving ethacrynic acid for many years were successfully and uneventfully switched to furosemide.54
Taken together, these findings suggest that there is no evidence for withholding sulfa nonantibiotics in patients allergic to sulfa containing antibiotics.
Conclusion
Hypothyroidism, unlike myxedema, is not a cause of hyponatremia (although it can be sometimes seen in conjunction with the latter) and additional investigations should be done to determine its etiology. Sodium bicarbonate is effective for treatment of hyperkalemia by enhancing renal potassium elimination, rather than from shifting potassium into cells. The 5‐oxoprolinuria is a newly recognized cause of high anion‐gap metabolic acidosis and should be considered in patients who have taken acetaminophen. Furosemide (and sulfa containing diuretics) can be used safely in patients with an allergy to SCA.
- ,,.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122(9):857–865.
- ,.Hyponatremia as the presenting manifestation of Sheehan's syndrome in elderly patients.Aging Clin Exp Res.2006;18(6):536–539.
- ,,.Sodium and water disturbances in patients with Sheehan's syndrome.Am J Kidney Dis.2001;38(3):E14.
- ,,,,,.Effect of acute water loading on plasma levels of antidiuretic hormone AVP aldosterone, ANP fractional excretion of sodium and plasma and urine osmolalities in myxedema.Chin Med J (Engl).1990;103(9):704–708.
- ,.Sodium handling in congenitally hypothyroid neonates.Acta Paediatr.2004;93(1):22–24.
- ,,.Prevalence and severity of hyponatremia and hypercreatininemia in short‐term uncomplicated hypothyroidism.J Endocrinol Invest.1999;22(1):35–39.
- ,,,,.Absence of relation between hyponatraemia and hypothyroidism.Lancet.1997;350(9088):1402.
- ,,.The effect of newly diagnosed hypothyroidism on serum sodium concentrations: a retrospective study.Clin Endocrinol (Oxf).2006;64(5):598–599.
- .Disorders of potassium homeostasis. Hypokalemia and hyperkalemia.Crit Care Clin.2002;18(2):273–288,vi.
- ,.Hyperkalemia in hospitalized patients.Int Urol Nephrol.2000;32(2):177–180.
- ,.Treatment of metabolic acidosis.Curr Opin Crit Care.2003;9(4):260–265.
- ,,,.Natural history of lactic acidosis after grand‐mal seizures. A model for the study of an anion‐gap acidosis not associated with hyperkalemia.N Engl J Med.1977;297(15):796–799.
- ,,,,.The plasma potassium concentration in metabolic acidosis: a re‐evaluation.Am J Kidney Dis.1988;11(3):220–224.
- ,,,.Determinants of plasma potassium levels in diabetic ketoacidosis.Medicine (Baltimore).1986;65(3):163–172.
- ,.Changes in plasma potassium concentration during acute acid‐base disturbances.Am J Med.1981;71(3):456–467.
- ,,.New aspects of renal potassium transport.Pflugers Arch.2003;446(3):289–297.
- ,.Correction of hyperkalemia by bicarbonate despite constant blood pH.Kidney Int.1977;12(5):354–360.
- ,,,.Effect of various therapeutic approaches on plasma potassium and major regulating factors in terminal renal failure.Am J Med.1988;85(4):507–512.
- ,.Effect of bicarbonate administration on plasma potassium in dialysis patients: interactions with insulin and albuterol.Am J Kidney Dis.1996;28(4):508–514.
- ,,,,.Effect of hypertonic versus isotonic sodium bicarbonate on plasma potassium concentration in patients with end‐stage renal disease.Miner Electrolyte Metab.1991;17(5):297–302.
- .Combined effect of bicarbonate and insulin with glucose in acute therapy of hyperkalemia in end‐stage renal disease patients.Nephron.1996;72(3):476–482.
- ,,.Effect of prolonged bicarbonate administration on plasma potassium in terminal renal failure.Kidney Int.1992;41(2):369–374.
- ,,, et al.Modulation of the secretion of potassium by accompanying anions in humans.Kidney Int.1991;39(6):1206–1212.
- ,.Controversial issues in the treatment of hyperkalaemia.Nephrol Dial Transplant.2003;18(11):2215–2218.
- ,.Sodium bicarbonate for the treatment of lactic acidosis.Chest.2000;117(1):260–267.
- .Management of severe hyperkalemia.Crit Care Med.2008;36(12):3246–3251.
- ,.Pyroglutamic acid and high anion gap: looking through the keyhole?Crit Care Med.2000;28(6):2140–2141.
- ,,,,.Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen.Clin J Am Soc Nephrol.2006;1(3):441–447.
- ,,,.Pyroglutamic acidemia: a cause of high anion gap metabolic acidosis.Crit Care Med.2000;28(6):1803–1807.
- ,,,,,.Acetaminophen‐induced anion gap metabolic acidosis and 5‐oxoprolinuria (pyroglutamic aciduria) acquired in hospital.Am J Kidney Dis.2005;46(1):143–146.
- ,.Anion gap acidosis associated with acetaminophen.Ann Intern Med.2000;133(9):752–753.
- ,,.Pyroglutamic acidosis in a renal transplant patient.Nephrol Dial Transplant.2005;20(12):2836–2838.
- ,,.Hepatic glutathione content in patients with alcoholic and non alcoholic liver diseases.Life Sci.1988;43(12):991–998.
- ,,,,,.Decreased hepatic glutathione in chronic alcoholic patients.J Hepatol.1986;3(1):1–6.
- ,.Intracellular signaling mechanisms of acetaminophen‐induced liver cell death.Toxicol Sci.2006;89(1):31–41.
- ,,.Urinary excretion of 5‐L‐oxoproline (pyroglutamic acid) is increased during recovery from severe childhood malnutrition and responds to supplemental glycine.J Nutr.1996;126(11):2823–2830.
- ,,,.Pyroglutamicaciduria from vigabatrin.Lancet.1989;1(8652):1452–1453.
- ,,, et al.Cysteine metabolism and whole blood glutathione synthesis in septic pediatric patients.Crit Care Med.2001;29(4):870–877.
- ,.Transient 5‐oxoprolinuria and high anion gap metabolic acidosis: clinical and biochemical findings in eleven subjects.Clin Chem.1998;44(7):1497–1503.
- ,,,.Guilty as charged: unmeasured urinary anions in a case of pyroglutamic acidosis.Neth J Med.2008;66(8):351–353.
- ,,.Unexplained metabolic acidosis in critically ill patients: the role of pyroglutamic acid.Intensive Care Med.2004;30(3):502–505.
- ,,.A therapeutic trial with N‐acetylcysteine in subjects with hereditary glutathione synthetase deficiency (5‐oxoprolinuria).J Inherit Metab Dis.1989;12(2):120–130.
- .Diagnosis of allergic reactions to sulfonamides.Allergy.1999;54Suppl 58:28–32.
- .Practical issues in the management of hypersensitivity reactions: sulfonamides.South Med J.2001;94(8):817–824.
- ,,.Should celecoxib be contraindicated in patients who are allergic to sulfonamides? Revisiting the meaning of ‘sulfa’ allergy.Drug Saf.2001;24(4):239–247.
- .Thrombocytopenia due to sulfonamide cross‐sensitivity.Wis Med J.1982;81(6):21–23.
- ,,.Leukocytoclastic vasculitis induced by use of glyburide: a case of possible cross‐reaction of a sulfonamide and a sulfonylurea.Cutis.2003;71(3):235–238.
- ,.Celecoxib‐induced erythema multiforme with glyburide cross‐reactivity.Pharmacotherapy.2002;22(5):637–640.
- ,.Vesiculobullous rash in a patient with systemic lupus erythematosus.Ann Allergy.1993;70(3):196–203.
- ,,, et al.Use of optical biosensor technology to study immunological cross‐reactivity between different sulfonamide drugs.Anal Biochem.2002;300(2):177–184.
- ,,,.Adverse reactions to sulphonamide and sulphonamide‐trimethoprim antimicrobials: clinical syndromes and pathogenesis.Adverse Drug React Toxicol Rev.1996;15(1):9–50.
- ,,.Cross‐reactivity in HIV‐infected patients switched from trimethoprim‐sulfamethoxazole to dapsone.Pharmacotherapy.1998;18(4):831–835.
- ,,, et al.Absence of cross‐reactivity between sulfonamide antibiotics and sulfonamide nonantibiotics.N Engl J Med.2003;349(17):1628–1635.
- ,,.Furosemide challenge in patients with heart failure and adverse reactions to sulfa‐containing diuretics.Ann Intern Med.2003;138(4):358–359.
- ,,.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122(9):857–865.
- ,.Hyponatremia as the presenting manifestation of Sheehan's syndrome in elderly patients.Aging Clin Exp Res.2006;18(6):536–539.
- ,,.Sodium and water disturbances in patients with Sheehan's syndrome.Am J Kidney Dis.2001;38(3):E14.
- ,,,,,.Effect of acute water loading on plasma levels of antidiuretic hormone AVP aldosterone, ANP fractional excretion of sodium and plasma and urine osmolalities in myxedema.Chin Med J (Engl).1990;103(9):704–708.
- ,.Sodium handling in congenitally hypothyroid neonates.Acta Paediatr.2004;93(1):22–24.
- ,,.Prevalence and severity of hyponatremia and hypercreatininemia in short‐term uncomplicated hypothyroidism.J Endocrinol Invest.1999;22(1):35–39.
- ,,,,.Absence of relation between hyponatraemia and hypothyroidism.Lancet.1997;350(9088):1402.
- ,,.The effect of newly diagnosed hypothyroidism on serum sodium concentrations: a retrospective study.Clin Endocrinol (Oxf).2006;64(5):598–599.
- .Disorders of potassium homeostasis. Hypokalemia and hyperkalemia.Crit Care Clin.2002;18(2):273–288,vi.
- ,.Hyperkalemia in hospitalized patients.Int Urol Nephrol.2000;32(2):177–180.
- ,.Treatment of metabolic acidosis.Curr Opin Crit Care.2003;9(4):260–265.
- ,,,.Natural history of lactic acidosis after grand‐mal seizures. A model for the study of an anion‐gap acidosis not associated with hyperkalemia.N Engl J Med.1977;297(15):796–799.
- ,,,,.The plasma potassium concentration in metabolic acidosis: a re‐evaluation.Am J Kidney Dis.1988;11(3):220–224.
- ,,,.Determinants of plasma potassium levels in diabetic ketoacidosis.Medicine (Baltimore).1986;65(3):163–172.
- ,.Changes in plasma potassium concentration during acute acid‐base disturbances.Am J Med.1981;71(3):456–467.
- ,,.New aspects of renal potassium transport.Pflugers Arch.2003;446(3):289–297.
- ,.Correction of hyperkalemia by bicarbonate despite constant blood pH.Kidney Int.1977;12(5):354–360.
- ,,,.Effect of various therapeutic approaches on plasma potassium and major regulating factors in terminal renal failure.Am J Med.1988;85(4):507–512.
- ,.Effect of bicarbonate administration on plasma potassium in dialysis patients: interactions with insulin and albuterol.Am J Kidney Dis.1996;28(4):508–514.
- ,,,,.Effect of hypertonic versus isotonic sodium bicarbonate on plasma potassium concentration in patients with end‐stage renal disease.Miner Electrolyte Metab.1991;17(5):297–302.
- .Combined effect of bicarbonate and insulin with glucose in acute therapy of hyperkalemia in end‐stage renal disease patients.Nephron.1996;72(3):476–482.
- ,,.Effect of prolonged bicarbonate administration on plasma potassium in terminal renal failure.Kidney Int.1992;41(2):369–374.
- ,,, et al.Modulation of the secretion of potassium by accompanying anions in humans.Kidney Int.1991;39(6):1206–1212.
- ,.Controversial issues in the treatment of hyperkalaemia.Nephrol Dial Transplant.2003;18(11):2215–2218.
- ,.Sodium bicarbonate for the treatment of lactic acidosis.Chest.2000;117(1):260–267.
- .Management of severe hyperkalemia.Crit Care Med.2008;36(12):3246–3251.
- ,.Pyroglutamic acid and high anion gap: looking through the keyhole?Crit Care Med.2000;28(6):2140–2141.
- ,,,,.Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen.Clin J Am Soc Nephrol.2006;1(3):441–447.
- ,,,.Pyroglutamic acidemia: a cause of high anion gap metabolic acidosis.Crit Care Med.2000;28(6):1803–1807.
- ,,,,,.Acetaminophen‐induced anion gap metabolic acidosis and 5‐oxoprolinuria (pyroglutamic aciduria) acquired in hospital.Am J Kidney Dis.2005;46(1):143–146.
- ,.Anion gap acidosis associated with acetaminophen.Ann Intern Med.2000;133(9):752–753.
- ,,.Pyroglutamic acidosis in a renal transplant patient.Nephrol Dial Transplant.2005;20(12):2836–2838.
- ,,.Hepatic glutathione content in patients with alcoholic and non alcoholic liver diseases.Life Sci.1988;43(12):991–998.
- ,,,,,.Decreased hepatic glutathione in chronic alcoholic patients.J Hepatol.1986;3(1):1–6.
- ,.Intracellular signaling mechanisms of acetaminophen‐induced liver cell death.Toxicol Sci.2006;89(1):31–41.
- ,,.Urinary excretion of 5‐L‐oxoproline (pyroglutamic acid) is increased during recovery from severe childhood malnutrition and responds to supplemental glycine.J Nutr.1996;126(11):2823–2830.
- ,,,.Pyroglutamicaciduria from vigabatrin.Lancet.1989;1(8652):1452–1453.
- ,,, et al.Cysteine metabolism and whole blood glutathione synthesis in septic pediatric patients.Crit Care Med.2001;29(4):870–877.
- ,.Transient 5‐oxoprolinuria and high anion gap metabolic acidosis: clinical and biochemical findings in eleven subjects.Clin Chem.1998;44(7):1497–1503.
- ,,,.Guilty as charged: unmeasured urinary anions in a case of pyroglutamic acidosis.Neth J Med.2008;66(8):351–353.
- ,,.Unexplained metabolic acidosis in critically ill patients: the role of pyroglutamic acid.Intensive Care Med.2004;30(3):502–505.
- ,,.A therapeutic trial with N‐acetylcysteine in subjects with hereditary glutathione synthetase deficiency (5‐oxoprolinuria).J Inherit Metab Dis.1989;12(2):120–130.
- .Diagnosis of allergic reactions to sulfonamides.Allergy.1999;54Suppl 58:28–32.
- .Practical issues in the management of hypersensitivity reactions: sulfonamides.South Med J.2001;94(8):817–824.
- ,,.Should celecoxib be contraindicated in patients who are allergic to sulfonamides? Revisiting the meaning of ‘sulfa’ allergy.Drug Saf.2001;24(4):239–247.
- .Thrombocytopenia due to sulfonamide cross‐sensitivity.Wis Med J.1982;81(6):21–23.
- ,,.Leukocytoclastic vasculitis induced by use of glyburide: a case of possible cross‐reaction of a sulfonamide and a sulfonylurea.Cutis.2003;71(3):235–238.
- ,.Celecoxib‐induced erythema multiforme with glyburide cross‐reactivity.Pharmacotherapy.2002;22(5):637–640.
- ,.Vesiculobullous rash in a patient with systemic lupus erythematosus.Ann Allergy.1993;70(3):196–203.
- ,,, et al.Use of optical biosensor technology to study immunological cross‐reactivity between different sulfonamide drugs.Anal Biochem.2002;300(2):177–184.
- ,,,.Adverse reactions to sulphonamide and sulphonamide‐trimethoprim antimicrobials: clinical syndromes and pathogenesis.Adverse Drug React Toxicol Rev.1996;15(1):9–50.
- ,,.Cross‐reactivity in HIV‐infected patients switched from trimethoprim‐sulfamethoxazole to dapsone.Pharmacotherapy.1998;18(4):831–835.
- ,,, et al.Absence of cross‐reactivity between sulfonamide antibiotics and sulfonamide nonantibiotics.N Engl J Med.2003;349(17):1628–1635.
- ,,.Furosemide challenge in patients with heart failure and adverse reactions to sulfa‐containing diuretics.Ann Intern Med.2003;138(4):358–359.