User login
Ceftaroline fosamil: A super-cephalosporin?
Ceftaroline fosamil (Teflaro), introduced to the US market in October 2010, is the first beta-lactam agent with clinically useful activity against methicillin-resistant Staphylococcus aureus (MRSA). Currently, it is approved by the US Food and Drug Administration (FDA) to treat acute bacterial skin and skin-structure infections and community-acquired bacterial pneumonia caused by susceptible microorganisms.
In an era of increasing drug resistance and limited numbers of antimicrobials in the drug-production pipeline, ceftaroline is a step forward in fulfilling the Infectious Diseases Society of America’s “10 × ’20 Initiative” to increase support for drug research and manufacturing, with the goal of producing 10 new antimicrobial drugs by the year 2020.1 Ceftaroline was the first of several antibiotics to receive FDA approval in response to this initiative. It was followed by dalbavancin (May 2014), tedizolid phosphate (June 2014), oritavancin (August 2014), ceftolozane-tazobactam (December 2014), and ceftazidime-avibactam (February 2015). These antibiotic agents are aimed at treating infections caused by drug-resistant gram-positive and gram-negative microorganisms. It is important to understand and optimize the use of these new antibiotic agents in order to decrease the risk of emerging antibiotic resistance and superinfections (eg, Clostridium difficile infection) caused by antibiotic overuse or misuse.
This article provides an overview of ceftaroline’s mechanisms of action and resistance, spectrum of activity, pharmacokinetic properties, adverse effects, and current place in therapy.
AN ERA OF MULTIDRUG-RESISTANT MICROORGANISMS
Increasing rates of antimicrobial resistance threaten the efficacy of antimicrobial drugs in the daily practice of medicine. The World Health Organization has labeled antimicrobial resistance one of the three greatest threats to human health. Global efforts are under way to stimulate development of new antimicrobial agents and to decrease rates of antimicrobial resistance.
Staphylococcus aureus: A threat, even with vancomycin
Between 1998 and 2005, S aureus was one of the most common inpatient and outpatient isolates reported by clinical laboratories throughout the United States.2
Treatment of S aureus infection is complicated by a variety of resistance mechanisms that have evolved over time. In fact, the first resistant isolate of S aureus emerged not long after penicillin’s debut into clinical practice, and now the majority of strains are resistant to penicillin.
Methicillin was designed to overcome this beta-lactamase resistance and became the treatment of choice for penicillin-resistant S aureus isolates. However, MRSA isolates soon emerged because of the organism’s acquisition of penicillin-binding protein PBP2a via the mecA gene, leading to decreased binding affinity of methicillin.3
Since then, several agents active against MRSA (vancomycin, daptomycin, linezolid, tigecycline) have been introduced and continue to be widely used. While vancomycin is considered the first-line option for a variety of MRSA infections, its use has been threatened because of the emergence of vancomycin-intermediate-resistant S aureus (VISA), S aureus strains displaying vancomycin heteroresistance (hVISA), and vancomycin-resistant S aureus (VRSA) strains.4
VISA and hVISA isolates emerged through sequential mutations that lead to autolytic activity and cell-wall thickening. In contrast, the mechanism of resistance in VRSA is by acquisition of the vanA resistance gene, which alters the binding site of vancomycin from d-alanine-d-alanine to d-alanine-d-lactate.5
Streptococcus pneumoniae resistance: A continuing problem
The prevalence of drug resistance in S pneumoniae has risen since the late 1990s. A 2013 report from the SENTRY Antimicrobial Surveillance Program stated that almost 20% of S pneumoniae isolates were resistant to amoxicillin-clavulanate, and similar trends have been observed for penicillin (14.8%) and ceftriaxone (11.7%).6
S pneumoniae resistance is acquired through modifications of the penicillin-binding proteins, namely PBP1a, PBP2b, PBP2x, and, less frequently, PBP2a. These modifications lead to decreased binding affinity for most beta-lactams.7
Clinical impact of multidrug-resistant S aureus and S pneumoniae
In 2011, the US Centers for Disease Control and Prevention reported an estimated 80,000 severe MRSA infections and 11,000 MRSA-related deaths in the United States.8 In the same report, drug-resistant S pneumoniae was estimated to be responsible for almost 1.2 million illnesses and 7,000 deaths per year, leading to upwards of $96 million in related medical costs.
While invasive drug-resistant S pneumoniae infections usually affect patients at the extremes of age (under age 5 and over age 65), they have had a serious impact on patients of all ages.8
In light of the increasing prevalence of multidrug-resistant organisms, newer antimicrobial agents with novel mechanisms of action are needed.
CEFTAROLINE: A BETA-LACTAM WITH ANTI-MRSA ACTIVITY
The cephalosporins, a class of beta-lactam antibiotics, were originally derived from the fungus Cephalosporium (now called Acremonium). There are now many agents in this class, each containing a nucleus consisting of a beta-lactam ring fused to a six-member dihydrothiazine ring, and two side chains that can be modified to affect antibacterial activity and pharmacokinetic properties.
Cephalosporins are typically categorized into “generations.” With some exceptions, the first- and second-generation agents have good activity against gram-positive microorganisms, including methicillin-susceptible S aureus—but not against MRSA. The third- and fourth-generation cephalosporins have better gram-negative activity, with many agents having activity against the gram-negative bacterium Pseudomonas aeruginosa.
Enterococcal isolates are intrinsically resistant to cephalosporins. Additionally, cephalosporins are not active against anaerobic bacteria, except for a subset of structurally unique second-generation cephalosporins, ie, cefotetan and cefoxitin.
Ceftaroline was synthesized with specific manipulations of the side chains to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae isolates, making it the first available beta-lactam with this ability.
Mechanism of action
Ceftaroline binds to penicillin-binding proteins, inhibiting transpeptidation. This interaction blocks the final stage of peptidoglycan synthesis and inhibits bacterial cell wall formation, ultimately leading to cellular autolysis and microorganism death. Ceftaroline binds with high affinity to PBP2a and PBP2x, expanding its activity to encompass MRSA and penicillin-resistant S pneumoniae isolates.9
Spectrum of activity
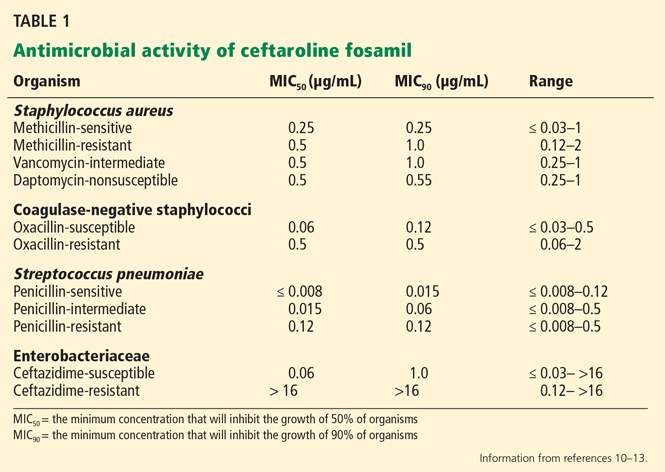
Ceftaroline has in vitro activity against many gram-positive and gram-negative bacteria,10–13 including (Table 1):
- Methicillin-susceptible and methicillin-resistant staphylococci
- VISA, VRSA, and hVISA
- Daptomycin-nonsusceptible S aureus
- Streptococcal species, including penicillin-resistant S pneumoniae
- Enterobacteriaceae, including Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Citrobacter koseri, Citrobacter freundii, Enterobacter cloacae, Enterobacter aerogenes, Moraxella catarrhalis, Morganella morganii, and Proteus mirabilis.
Of note, ceftaroline is not active against Pseudomonas species, Enterococcus species, or Bacteroides fragilis. In addition, it is not active against the “atypical” respiratory pathogens Mycoplasma pneumoniae, Chlamydophila pneumoniae, and Legionella pneumophila.
Ceftaroline resistance
Gram-negative organisms appear to develop resistance to ceftaroline at rates similar to those observed with the other oxyimino-cephalosporins (eg, ceftriaxone). Ceftaroline is inactive against gram-negative organisms producing extended-spectrum beta-lactamases, including K pneumoniae carbapenemase and metallo-beta-lactamases.14 In addition, it induces the expression of AmpC beta-lactamases.
Although currently uncommon, resistance to ceftaroline has also been reported in S aureus strains.15 The mechanism of resistance is decreased binding affinity for PBP2a due to amino acid substitutions on the nonpenicillin-binding domains.15
Pharmacokinetic profile
An understanding of pharmacokinetics is key in optimizing the dose of antimicrobials so that the drugs are used most effectively and pathogens do not develop resistance to them.

Ceftaroline fosamil is a prodrug that, upon intravenous administration, is rapidly converted by phosphatase enzymes to its active moiety, ceftaroline. Its pharmacokinetic profile is summarized in Table 2.16,17 Its volume of distribution is similar to that of the fourth-generation cephalosporin cefepime.
Ceftaroline is then hydrolyzed into its inactive metabolite, ceftaroline M-1. It undergoes little hepatic metabolism and lacks properties to make it a substrate, inhibitor, or inducer of the CYP450 enzyme system and therefore is not likely to cause notable CYP450-related drug-drug interactions.
Like most other beta-lactams, ceftaroline is primarily excreted by the kidneys. Furthermore, an estimated 21% of a dose is eliminated with each intermittent hemodialysis session. Therefore, renal and intermittent hemodialysis dose adjustments are necessary. The estimated elimination half-life is 2.6 hours, necessitating dosing two to three times daily, depending on the indication and infectious inoculum.
Ceftaroline dosing
Ceftaroline is available only in a parenteral preparation and is typically given at a dose of 600 mg every 12 hours.10 The intravenous infusion is given over 1 hour.
The current stability data require reconstituted ceftaroline to be used within 6 hours at room temperature and within 24 hours if refrigerated.10
Ceftaroline requires dosing adjustments for patients with renal insufficiency. Per the manufacturer, renal dosing adjustments are based on the creatinine clearance rate, as estimated by the Cockroft-Gault formula:
- Creatinine clearance > 50 mL/min: no dosage adjustment necessary
- Creatinine clearance > 30 to ≤ 50 mL/min: give 400 mg every 12 hours
- Creatinine clearance ≥ 15 to ≤ 30 mL/min: give 300 mg every 12 hours
- Creatinine clearance < 15 mL/min or on intermittent dialysis: give 200 mg every 12 hours.
Ongoing clinical trials are investigating a higher-dosing strategy of 600 mg every 8 hours for patients with community-acquired bacterial pneumonia at risk of MRSA bacteremia.18
CLINICAL TRIALS LEADING TO CEFTAROLINE’S APPROVAL
Ceftaroline was approved for the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections due to susceptible pathogens on the basis of phase 3 comparator trials.
Community-acquired bacterial pneumonia: The FOCUS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of community-acquired bacterial pneumonia was studied in two randomized, double-blind, noninferiority trials, known as Ceftaroline Community-acquired Pneumonia vs Ceftriaxone (FOCUS) 1 and FOCUS 2.19,20
Patients were adults and not critically ill, as was reflected by their being in Pneumonia Outcomes Research Team (PORT) risk class III or IV (with class V indicating the highest risk of death). Therefore, the results may not be completely applicable to critically ill patients or those not admitted to the hospital. Of note, patients were excluded from the trials if they had infections known or thought to be due to MRSA or to atypical organisms.21 Baseline characteristics and patient demographics were similar between study groups in both trials.
A bacterial pathogen was identified in 26.1% of the patients included in the modified intent-to-treat analysis of the pooled data of the trials; the most common pathogens were S pneumoniae, methicillin-sensitive S aureus, Haemophilus influenzae, K pneumoniae, and E coli.21
Treatment. Patients received either ceftaroline 600 mg every 12 hours (or a lower dose based on renal function) or ceftriaxone 1 g every 24 hours. In addition, in the FOCUS 1 trial, patients in both treatment groups received clarithromycin 500 mg every 12 hours for the first day.19

Results. In both trials and in the integrated analysis, ceftaroline was noninferior to ceftriaxone (Table 3).22 In the integrated analysis of both trials, compared with the ceftriaxone group, the ceftaroline group had a higher clinical cure rate among patients classified as PORT risk class III (86.8% vs 79.2%, weighted treatment difference 12.6%, 95% confidence interval [CI] 1.3–13.8) and among patients who had not received prior antibiotic treatment (85.5% vs 74.9%, weighted treatment difference 11.2%, 95% CI 4.5–18.0).21
Acute bacterial skin and skin-structure infections: The CANVAS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of complicated acute bacterial skin and skin-structure infections was studied in two randomized, double-blind trials: Ceftaroline Versus Vancomycin in Skin and Skin Structure Infections (CANVAS) 1 and CANVAS 2.23,24
Patients. Adult patients with a diagnosis of community-acquired skin and skin-structure infections warranting at least 5 days of intravenous antimicrobial therapy were included in the trials. Important protocol exclusions were patients with diabetic foot ulcers, decubitus ulcers, burns, ulcers associated with peripheral vascular disease accompanied by osteomyelitis, and suspected P aeruginosa infections.25 This limits the external validity of ceftaroline use in the aforementioned excluded patient populations.
Patients in each treatment group of the trials had similar demographic characteristics. The most common infections were cellulitis, major abscess requiring surgical intervention, wound infection, and infected ulcer. Bacteremia was present in 4.2% of patients in the ceftaroline group and in 3.8% of patients in the vancomycin-aztreonam group. The most common pathogen was S aureus. Methicillin resistance was present in 40% of the ceftaroline group and 34% of the control group.
Treatment. Patients received either ceftaroline 600 mg every 12 hours or the combination of vancomycin 1 g plus aztreonam 1 g given 12 hours, for 5 to 14 days.

Results. As assessed at a “test-of-cure” visit 8 to 15 days after the last dose of study medication, the efficacy of ceftaroline was similar to that of vancomycin-aztreonam, meeting the set noninferiority goal (Table 4).25 Moreover, if assessed on day 2 or 3 (a new end point recommended by the FDA), the rate of cessation of erythema spread and absence of fever was higher in the ceftaroline group than in the vancomycin-aztreonam group.26 However, this end point was not in the original trial protocol.
CEFTAROLINE FOR OTHER INDICATIONS
As noted, ceftaroline has been approved for treating community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections. In addition, it has been used in several studies in animals, and case reports of non-FDA approved indications including endocarditis and osteomyelitis have been published. Clinical trials are evaluating its use in pediatric patients, as well as for community-acquired bacterial pneumonia with risk for MRSA and for MRSA bacteremia.
Endocarditis
Animal studies have demonstrated ceftaroline to have bactericidal activity against MRSA and hVISA in endocarditis.27
A few case series have been published describing ceftaroline’s use as salvage therapy for persistent MRSA bacteremia and endocarditis. For example, Ho et al28 reported using it in three patients who had endocarditis as a source of their persistent bacteremia. All three patients had resolution of their MRSA bloodstream infection following ceftaroline therapy. The dosage was 600 mg every 8 hours, which is higher than in the manufacturer’s prescribing information.
Lin et al29 reported using ceftaroline in five patients with either possible or probable endocarditis. Three of the five patients had clinical cure as defined by resolution or improvement of all signs and symptoms of infection, and not requiring further antimicrobial therapy.29
More data from clinical trials would be beneficial in defining ceftaroline’s role in treating endocarditis caused by susceptible microorganisms.
Osteomyelitis
In animal studies of osteomyelitis, ceftaroline exhibited activity against MRSA in infected bone and joint fluid. Compared with vancomycin and linezolid, ceftaroline was associated with more significant decreases in bacterial load in the infected joint fluid, bone marrow, and bone.30
Lin et al29 gave ceftaroline to two patients with bone and joint infections, both of whom had received other therapies that had failed. The doses of ceftaroline were higher than those recommended in the prescribing information; clinical cure was noted in both cases following the switch.
These data come from case series, and more study of ceftaroline’s role in the treatment of osteomyelitis infections is warranted.
Meningitis
The use of ceftaroline in meningitis has been studied in rabbits. While ceftaroline penetrated into the cerebrospinal fluid in only negligible amounts in healthy rabbits (3% penetration), its penetration improved to 15% in animals with inflamed meninges. Ceftaroline cerebrospinal fluid levels in inflamed meninges were sufficient to provide bactericidal activity against penicillin-sensitive and resistant S pneumoniae strains as well as K pneumoniae and E coli strains.31,32
REPORTED ADVERSE EFFECTS OF CEFTAROLINE
Overall, ceftaroline was well tolerated in clinical trials, and its safety profile was similar to those of the comparator agents (ceftriaxone and vancomycin-aztreonam).
As with the other cephalosporins, hypersensitivity reactions have been reported with ceftaroline. In the clinical trials, 3% of patients developed a rash with ceftaroline.33,34 Patients with a history of beta-lactam allergy were excluded from the trials, so the rate of cross-reactivity with penicillins and with other cephalosporins is unknown.
In the phase 3 clinical trials, gastrointestinal side effects including diarrhea (5%), nausea (4%), and vomiting (2%) were reported with ceftaroline. C difficile-associated diarrhea has also been reported.33
As with other cephalosporins, ceftaroline can cause a false-positive result on the Coombs test. Approximately 11% of ceftaroline-treated patients in phase 3 clinical trials had a positive Coombs test, but hemolytic anemia did not occur in any patients.33,34
Discontinuation of ceftaroline due to an adverse reaction was reported in 2.7% of patients receiving the drug during phase 3 trials, compared with 3.7% with comparator agents.
WHEN SHOULD CEFTAROLINE BE USED IN DAILY PRACTICE?
Ceftaroline has been shown to be at least as effective as ceftriaxone in treating community-acquired bacterial pneumonia, and at least as effective as vancomycin-aztreonam in treating acute bacterial skin and skin-structure infections. The 2014 Infectious Diseases Society of America’s guidelines for the diagnosis and management of skin and soft-tissue infections recommend ceftaroline as an option for empiric therapy for purulent skin and soft-tissue infections.35
The guidelines on community-acquired pneumonia have not been updated since 2007, which was before ceftaroline was approved. However, these guidelines are currently undergoing revision and may provide insight on ceftaroline’s place in the treatment of community-acquired bacterial pneumonia.36
Currently, ceftaroline’s routine use for these indications should be balanced by its higher cost ($150 for a 600-mg dose) compared with ceftriaxone ($5 for a 1-g dose) or vancomycin ($25 for a 1-g dose). The drug’s in vitro activity against drug-resistant pneumococci and S aureus, including MRSA, hVISA, and VISA may help fill an unmet need or provide a safer and more tolerable alternative to currently available therapies.
However, ceftaroline’s lack of activity against P aeruginosa and carbapenem-resistant Enterobacteriaceae does not meet the public health threat needs stemming from these multidrug-resistant microorganisms. Ongoing clinical trials in patients with more serious MRSA infections will provide important information about ceftaroline’s role as an anti-MRSA agent.
While the discovery of antimicrobials has had one of the greatest impacts on medicine, continued antibiotic use is threatened by the emergence of drug-resistant pathogens. Therefore, it is as important as ever to be good stewards of our currently available antimicrobials. Developing usage and dosing criteria for antimicrobials based on available data and literature is a step forward in optimizing the use of antibiotics—a precious medical resource.
- Infectious Diseases Society of America. The 10 x ‘20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 2010; 50:1081–1083.
- Styers D, Sheehan DJ, Hogan P, Sahm DF. Laboratory-based surveillance of current antimicrobial resistance patterns and trends among Staphylococcus aureus: 2005 status in the United States. Ann Clin Microbiol Antimicrob 2006; 5:2.
- Farrell DJ, Castanheira M, Mendes RE, Sader HS, Jones RN. In vitro activity of ceftaroline against multidrug-resistant Staphylococcus aureus and Streptococcus pneumoniae: a review of published studies and the AWARE Surveillance Program (2008-2010). Clin Infect Dis 2012; 55(suppl 3):S206–S214.
- Holmes NE, Johnson PD, Howden BP. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J Clin Microbiol 2012; 50:2548–2552.
- Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 2003; 111:1265–1273.
- Jones RN, Sader HS, Mendes RE, Flamm RK. Update on antimicrobial susceptibility trends among Streptococcus pneumoniae in the United States: report of ceftaroline activity from the SENTRY Antimicrobial Surveillance Program (1998-2011). Diag Microbiol Infect Dis 2013; 75:107–109.
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 2008; 32:361–385.
- Centers for Disease Control and Prevention (CDC). Antibiotic resistance threats in the United States 2013. cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. Accessed June 1, 2015.
- Moisan H, Pruneau M, Malouin F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J Antimicrob Chemother 2010; 65:713–716.
- Forest Laboratories, Inc. Teflaro® (ceftaroline fosamil): prescribing information. www.frx.com/pi/teflaro_pi.pdf. Accessed June 1, 2015.
- Richter SS, Heilmann KP, Dohrn CL, et al. Activity of ceftaroline and epidemiologic trends in Staphylococcus aureus isolates collected from 43 medical centers in the United States in 2009. Antimicrob Agents Chemother 2011; 55:4154–4160.
- Ge Y, Biek D, Talbot GH, Sahm DF. In vitro profiling of ceftaroline against a collection of recent bacterial clinical isolates from across the United States. Antimicrob Agents Chemother 2008; 52:3398–3407.
- Saravolatz L, Pawlak J, Johnson L. In vitro activity of ceftaroline against community-associated methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, and daptomycin-nonsusceptible Staphylococcus aureus isolates. Antimicrob Agents Chemother 2010; 54:3027–3030.
- Mushtaq S, Livermore DM. AmpC induction by ceftaroline. J Antimicrob Chemother 2010; 65:586–588.
- Mendes RE, Tsakris A, Sader HS, et al. Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J Antimicrob Chemother 2012; 67:1321–1324.
- Lodise TP, Low DE. Ceftaroline fosamil in the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections. Drugs 2012; 72:1473–1493.
- Riccobene TA, Su SF, Rank D. Single- and multiple-dose study to determine the safety, tolerability, and pharmacokinetics of ceftaroline fosamil in combination with avibactam in healthy subjects. Antimicrob Agents Chemother 2013; 57:1496–1504.
- US National Institutes of Health. ClinicalTrials.gov. Evaluation of ceftaroline fosamil versus a comparator in adult subjects with community-acquired bacterial pneumonia (CABP) with risk for methicillin-resistant Staphylococcus aureus. http://clinicaltrials.gov/ct2/show/NCT01645735. Accessed June 1, 2015.
- File TM Jr, Low DE, Eckburg PB, et al; FOCUS 1 investigators. FOCUS 1: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii19–iii32.
- Low DE, File TM Jr, Eckburg PB, et al; FOCUS 2 investigators. FOCUS 2: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii33–iii44.
- File TM Jr, Low DE, Eckburg PB, et al. Integrated analysis of FOCUS 1 and FOCUS 2: randomized, doubled-blinded, multicenter phase 3 trials of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in patients with community-acquired pneumonia. Clin Infect Dis 2010; 51:1395–1405.
- Food and Drug Administration (FDA). Ceftaroline fosamil for the treatment of community-acquired bacterial pneumonia and complicated skin and skin structure infections. www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/anti-infectivedrugsadvisorycommittee/ucm224656.pdf. Accessed June 1, 2015.
- Corey GR, Wilcox MH, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 1 investigators. CANVAS 1: the first phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv41–iv51.
- Wilcox MH, Corey GR, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 2 investigators. CANVAS 2: the second phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv53-iv65.
- Corey GR, Wilcox M, Talbot GH, et al. Integrated analysis of CANVAS 1 and 2: phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin Infect Dis 2010; 51:641–650.
- Friedland HD, O’Neal T, Biek D, et al. CANVAS 1 and 2: analysis of clinical response at day 3 in two phase 3 trials of ceftaroline fosamil versus vancomycin plus aztreonam in treatment of acute bacterial skin and skin structure infections. Antimicrob Agents Chemother 2012; 56:2231–2236.
- Jacqueline C, Caillon J, Le Mabecque V, et al. In vivo efficacy of ceftaroline (PPI-0903), a new broad-spectrum cephalosporin, compared with linezolid and vancomycin against methicillin-resistant and vancomycin-intermediate Staphylococcus aureus in a rabbit endocarditis model. Antimicrob Agents Chemother 2007; 51:3397–3400.
- Ho TT, Cadena J, Childs LM, Gonzalez-Velez M, Lewis JS 2nd. Methicillin-resistant Staphylococcus aureus bacteraemia and endocarditis treated with ceftaroline salvage therapy. J Antimicrob Chemother 2012; 67:1267–1270.
- Lin JC, Aung G, Thomas A, Jahng M, Johns S, Fierer J. The use of ceftaroline fosamil in methicillin-resistant Staphylococcus aureus endocarditis and deep-seated MRSA infections: a retrospective case series of 10 patients. J Infect Chemother 2013; 19:42–49.
- Jacqueline C, Amador G, Caillon J, et al. Efficacy of the new cephalosporin ceftaroline in the treatment of experimental methicillin-resistant Staphylococcus aureus acute osteomyelitis. J Antimicrob Chemother 2010; 65:1749–1752.
- Stucki A, Acosta F, Cottagnoud M, Cottagnoud P. Efficacy of ceftaroline fosamil against Escherichia coli and Klebsiella pneumoniae strains in a rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:5808–5810.
- Cottagnoud P, Cottagnoud M, Acosta F, Stucki A. Efficacy of ceftaroline fosamil against penicillin-sensitive and -resistant Streptococcus pneumoniae in an experimental rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:4653–4655.
- Corrado ML. Integrated safety summary of CANVAS 1 and 2 trials: phase III, randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv67–iv71.
- Rank DR, Friedland HD, Laudano JB. Integrated safety summary of FOCUS 1 and FOCUS 2 trials: phase III randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii53–iii59.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis 2014; 59:147–159.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44:S27–S72.
Ceftaroline fosamil (Teflaro), introduced to the US market in October 2010, is the first beta-lactam agent with clinically useful activity against methicillin-resistant Staphylococcus aureus (MRSA). Currently, it is approved by the US Food and Drug Administration (FDA) to treat acute bacterial skin and skin-structure infections and community-acquired bacterial pneumonia caused by susceptible microorganisms.
In an era of increasing drug resistance and limited numbers of antimicrobials in the drug-production pipeline, ceftaroline is a step forward in fulfilling the Infectious Diseases Society of America’s “10 × ’20 Initiative” to increase support for drug research and manufacturing, with the goal of producing 10 new antimicrobial drugs by the year 2020.1 Ceftaroline was the first of several antibiotics to receive FDA approval in response to this initiative. It was followed by dalbavancin (May 2014), tedizolid phosphate (June 2014), oritavancin (August 2014), ceftolozane-tazobactam (December 2014), and ceftazidime-avibactam (February 2015). These antibiotic agents are aimed at treating infections caused by drug-resistant gram-positive and gram-negative microorganisms. It is important to understand and optimize the use of these new antibiotic agents in order to decrease the risk of emerging antibiotic resistance and superinfections (eg, Clostridium difficile infection) caused by antibiotic overuse or misuse.
This article provides an overview of ceftaroline’s mechanisms of action and resistance, spectrum of activity, pharmacokinetic properties, adverse effects, and current place in therapy.
AN ERA OF MULTIDRUG-RESISTANT MICROORGANISMS
Increasing rates of antimicrobial resistance threaten the efficacy of antimicrobial drugs in the daily practice of medicine. The World Health Organization has labeled antimicrobial resistance one of the three greatest threats to human health. Global efforts are under way to stimulate development of new antimicrobial agents and to decrease rates of antimicrobial resistance.
Staphylococcus aureus: A threat, even with vancomycin
Between 1998 and 2005, S aureus was one of the most common inpatient and outpatient isolates reported by clinical laboratories throughout the United States.2
Treatment of S aureus infection is complicated by a variety of resistance mechanisms that have evolved over time. In fact, the first resistant isolate of S aureus emerged not long after penicillin’s debut into clinical practice, and now the majority of strains are resistant to penicillin.
Methicillin was designed to overcome this beta-lactamase resistance and became the treatment of choice for penicillin-resistant S aureus isolates. However, MRSA isolates soon emerged because of the organism’s acquisition of penicillin-binding protein PBP2a via the mecA gene, leading to decreased binding affinity of methicillin.3
Since then, several agents active against MRSA (vancomycin, daptomycin, linezolid, tigecycline) have been introduced and continue to be widely used. While vancomycin is considered the first-line option for a variety of MRSA infections, its use has been threatened because of the emergence of vancomycin-intermediate-resistant S aureus (VISA), S aureus strains displaying vancomycin heteroresistance (hVISA), and vancomycin-resistant S aureus (VRSA) strains.4
VISA and hVISA isolates emerged through sequential mutations that lead to autolytic activity and cell-wall thickening. In contrast, the mechanism of resistance in VRSA is by acquisition of the vanA resistance gene, which alters the binding site of vancomycin from d-alanine-d-alanine to d-alanine-d-lactate.5
Streptococcus pneumoniae resistance: A continuing problem
The prevalence of drug resistance in S pneumoniae has risen since the late 1990s. A 2013 report from the SENTRY Antimicrobial Surveillance Program stated that almost 20% of S pneumoniae isolates were resistant to amoxicillin-clavulanate, and similar trends have been observed for penicillin (14.8%) and ceftriaxone (11.7%).6
S pneumoniae resistance is acquired through modifications of the penicillin-binding proteins, namely PBP1a, PBP2b, PBP2x, and, less frequently, PBP2a. These modifications lead to decreased binding affinity for most beta-lactams.7
Clinical impact of multidrug-resistant S aureus and S pneumoniae
In 2011, the US Centers for Disease Control and Prevention reported an estimated 80,000 severe MRSA infections and 11,000 MRSA-related deaths in the United States.8 In the same report, drug-resistant S pneumoniae was estimated to be responsible for almost 1.2 million illnesses and 7,000 deaths per year, leading to upwards of $96 million in related medical costs.
While invasive drug-resistant S pneumoniae infections usually affect patients at the extremes of age (under age 5 and over age 65), they have had a serious impact on patients of all ages.8
In light of the increasing prevalence of multidrug-resistant organisms, newer antimicrobial agents with novel mechanisms of action are needed.
CEFTAROLINE: A BETA-LACTAM WITH ANTI-MRSA ACTIVITY
The cephalosporins, a class of beta-lactam antibiotics, were originally derived from the fungus Cephalosporium (now called Acremonium). There are now many agents in this class, each containing a nucleus consisting of a beta-lactam ring fused to a six-member dihydrothiazine ring, and two side chains that can be modified to affect antibacterial activity and pharmacokinetic properties.
Cephalosporins are typically categorized into “generations.” With some exceptions, the first- and second-generation agents have good activity against gram-positive microorganisms, including methicillin-susceptible S aureus—but not against MRSA. The third- and fourth-generation cephalosporins have better gram-negative activity, with many agents having activity against the gram-negative bacterium Pseudomonas aeruginosa.
Enterococcal isolates are intrinsically resistant to cephalosporins. Additionally, cephalosporins are not active against anaerobic bacteria, except for a subset of structurally unique second-generation cephalosporins, ie, cefotetan and cefoxitin.
Ceftaroline was synthesized with specific manipulations of the side chains to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae isolates, making it the first available beta-lactam with this ability.
Mechanism of action
Ceftaroline binds to penicillin-binding proteins, inhibiting transpeptidation. This interaction blocks the final stage of peptidoglycan synthesis and inhibits bacterial cell wall formation, ultimately leading to cellular autolysis and microorganism death. Ceftaroline binds with high affinity to PBP2a and PBP2x, expanding its activity to encompass MRSA and penicillin-resistant S pneumoniae isolates.9
Spectrum of activity
Ceftaroline has in vitro activity against many gram-positive and gram-negative bacteria,10–13 including (Table 1):
- Methicillin-susceptible and methicillin-resistant staphylococci
- VISA, VRSA, and hVISA
- Daptomycin-nonsusceptible S aureus
- Streptococcal species, including penicillin-resistant S pneumoniae
- Enterobacteriaceae, including Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Citrobacter koseri, Citrobacter freundii, Enterobacter cloacae, Enterobacter aerogenes, Moraxella catarrhalis, Morganella morganii, and Proteus mirabilis.
Of note, ceftaroline is not active against Pseudomonas species, Enterococcus species, or Bacteroides fragilis. In addition, it is not active against the “atypical” respiratory pathogens Mycoplasma pneumoniae, Chlamydophila pneumoniae, and Legionella pneumophila.
Ceftaroline resistance
Gram-negative organisms appear to develop resistance to ceftaroline at rates similar to those observed with the other oxyimino-cephalosporins (eg, ceftriaxone). Ceftaroline is inactive against gram-negative organisms producing extended-spectrum beta-lactamases, including K pneumoniae carbapenemase and metallo-beta-lactamases.14 In addition, it induces the expression of AmpC beta-lactamases.
Although currently uncommon, resistance to ceftaroline has also been reported in S aureus strains.15 The mechanism of resistance is decreased binding affinity for PBP2a due to amino acid substitutions on the nonpenicillin-binding domains.15
Pharmacokinetic profile
An understanding of pharmacokinetics is key in optimizing the dose of antimicrobials so that the drugs are used most effectively and pathogens do not develop resistance to them.
Ceftaroline fosamil is a prodrug that, upon intravenous administration, is rapidly converted by phosphatase enzymes to its active moiety, ceftaroline. Its pharmacokinetic profile is summarized in Table 2.16,17 Its volume of distribution is similar to that of the fourth-generation cephalosporin cefepime.
Ceftaroline is then hydrolyzed into its inactive metabolite, ceftaroline M-1. It undergoes little hepatic metabolism and lacks properties to make it a substrate, inhibitor, or inducer of the CYP450 enzyme system and therefore is not likely to cause notable CYP450-related drug-drug interactions.
Like most other beta-lactams, ceftaroline is primarily excreted by the kidneys. Furthermore, an estimated 21% of a dose is eliminated with each intermittent hemodialysis session. Therefore, renal and intermittent hemodialysis dose adjustments are necessary. The estimated elimination half-life is 2.6 hours, necessitating dosing two to three times daily, depending on the indication and infectious inoculum.
Ceftaroline dosing
Ceftaroline is available only in a parenteral preparation and is typically given at a dose of 600 mg every 12 hours.10 The intravenous infusion is given over 1 hour.
The current stability data require reconstituted ceftaroline to be used within 6 hours at room temperature and within 24 hours if refrigerated.10
Ceftaroline requires dosing adjustments for patients with renal insufficiency. Per the manufacturer, renal dosing adjustments are based on the creatinine clearance rate, as estimated by the Cockroft-Gault formula:
- Creatinine clearance > 50 mL/min: no dosage adjustment necessary
- Creatinine clearance > 30 to ≤ 50 mL/min: give 400 mg every 12 hours
- Creatinine clearance ≥ 15 to ≤ 30 mL/min: give 300 mg every 12 hours
- Creatinine clearance < 15 mL/min or on intermittent dialysis: give 200 mg every 12 hours.
Ongoing clinical trials are investigating a higher-dosing strategy of 600 mg every 8 hours for patients with community-acquired bacterial pneumonia at risk of MRSA bacteremia.18
CLINICAL TRIALS LEADING TO CEFTAROLINE’S APPROVAL
Ceftaroline was approved for the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections due to susceptible pathogens on the basis of phase 3 comparator trials.
Community-acquired bacterial pneumonia: The FOCUS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of community-acquired bacterial pneumonia was studied in two randomized, double-blind, noninferiority trials, known as Ceftaroline Community-acquired Pneumonia vs Ceftriaxone (FOCUS) 1 and FOCUS 2.19,20
Patients were adults and not critically ill, as was reflected by their being in Pneumonia Outcomes Research Team (PORT) risk class III or IV (with class V indicating the highest risk of death). Therefore, the results may not be completely applicable to critically ill patients or those not admitted to the hospital. Of note, patients were excluded from the trials if they had infections known or thought to be due to MRSA or to atypical organisms.21 Baseline characteristics and patient demographics were similar between study groups in both trials.
A bacterial pathogen was identified in 26.1% of the patients included in the modified intent-to-treat analysis of the pooled data of the trials; the most common pathogens were S pneumoniae, methicillin-sensitive S aureus, Haemophilus influenzae, K pneumoniae, and E coli.21
Treatment. Patients received either ceftaroline 600 mg every 12 hours (or a lower dose based on renal function) or ceftriaxone 1 g every 24 hours. In addition, in the FOCUS 1 trial, patients in both treatment groups received clarithromycin 500 mg every 12 hours for the first day.19
Results. In both trials and in the integrated analysis, ceftaroline was noninferior to ceftriaxone (Table 3).22 In the integrated analysis of both trials, compared with the ceftriaxone group, the ceftaroline group had a higher clinical cure rate among patients classified as PORT risk class III (86.8% vs 79.2%, weighted treatment difference 12.6%, 95% confidence interval [CI] 1.3–13.8) and among patients who had not received prior antibiotic treatment (85.5% vs 74.9%, weighted treatment difference 11.2%, 95% CI 4.5–18.0).21
Acute bacterial skin and skin-structure infections: The CANVAS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of complicated acute bacterial skin and skin-structure infections was studied in two randomized, double-blind trials: Ceftaroline Versus Vancomycin in Skin and Skin Structure Infections (CANVAS) 1 and CANVAS 2.23,24
Patients. Adult patients with a diagnosis of community-acquired skin and skin-structure infections warranting at least 5 days of intravenous antimicrobial therapy were included in the trials. Important protocol exclusions were patients with diabetic foot ulcers, decubitus ulcers, burns, ulcers associated with peripheral vascular disease accompanied by osteomyelitis, and suspected P aeruginosa infections.25 This limits the external validity of ceftaroline use in the aforementioned excluded patient populations.
Patients in each treatment group of the trials had similar demographic characteristics. The most common infections were cellulitis, major abscess requiring surgical intervention, wound infection, and infected ulcer. Bacteremia was present in 4.2% of patients in the ceftaroline group and in 3.8% of patients in the vancomycin-aztreonam group. The most common pathogen was S aureus. Methicillin resistance was present in 40% of the ceftaroline group and 34% of the control group.
Treatment. Patients received either ceftaroline 600 mg every 12 hours or the combination of vancomycin 1 g plus aztreonam 1 g given 12 hours, for 5 to 14 days.
Results. As assessed at a “test-of-cure” visit 8 to 15 days after the last dose of study medication, the efficacy of ceftaroline was similar to that of vancomycin-aztreonam, meeting the set noninferiority goal (Table 4).25 Moreover, if assessed on day 2 or 3 (a new end point recommended by the FDA), the rate of cessation of erythema spread and absence of fever was higher in the ceftaroline group than in the vancomycin-aztreonam group.26 However, this end point was not in the original trial protocol.
CEFTAROLINE FOR OTHER INDICATIONS
As noted, ceftaroline has been approved for treating community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections. In addition, it has been used in several studies in animals, and case reports of non-FDA approved indications including endocarditis and osteomyelitis have been published. Clinical trials are evaluating its use in pediatric patients, as well as for community-acquired bacterial pneumonia with risk for MRSA and for MRSA bacteremia.
Endocarditis
Animal studies have demonstrated ceftaroline to have bactericidal activity against MRSA and hVISA in endocarditis.27
A few case series have been published describing ceftaroline’s use as salvage therapy for persistent MRSA bacteremia and endocarditis. For example, Ho et al28 reported using it in three patients who had endocarditis as a source of their persistent bacteremia. All three patients had resolution of their MRSA bloodstream infection following ceftaroline therapy. The dosage was 600 mg every 8 hours, which is higher than in the manufacturer’s prescribing information.
Lin et al29 reported using ceftaroline in five patients with either possible or probable endocarditis. Three of the five patients had clinical cure as defined by resolution or improvement of all signs and symptoms of infection, and not requiring further antimicrobial therapy.29
More data from clinical trials would be beneficial in defining ceftaroline’s role in treating endocarditis caused by susceptible microorganisms.
Osteomyelitis
In animal studies of osteomyelitis, ceftaroline exhibited activity against MRSA in infected bone and joint fluid. Compared with vancomycin and linezolid, ceftaroline was associated with more significant decreases in bacterial load in the infected joint fluid, bone marrow, and bone.30
Lin et al29 gave ceftaroline to two patients with bone and joint infections, both of whom had received other therapies that had failed. The doses of ceftaroline were higher than those recommended in the prescribing information; clinical cure was noted in both cases following the switch.
These data come from case series, and more study of ceftaroline’s role in the treatment of osteomyelitis infections is warranted.
Meningitis
The use of ceftaroline in meningitis has been studied in rabbits. While ceftaroline penetrated into the cerebrospinal fluid in only negligible amounts in healthy rabbits (3% penetration), its penetration improved to 15% in animals with inflamed meninges. Ceftaroline cerebrospinal fluid levels in inflamed meninges were sufficient to provide bactericidal activity against penicillin-sensitive and resistant S pneumoniae strains as well as K pneumoniae and E coli strains.31,32
REPORTED ADVERSE EFFECTS OF CEFTAROLINE
Overall, ceftaroline was well tolerated in clinical trials, and its safety profile was similar to those of the comparator agents (ceftriaxone and vancomycin-aztreonam).
As with the other cephalosporins, hypersensitivity reactions have been reported with ceftaroline. In the clinical trials, 3% of patients developed a rash with ceftaroline.33,34 Patients with a history of beta-lactam allergy were excluded from the trials, so the rate of cross-reactivity with penicillins and with other cephalosporins is unknown.
In the phase 3 clinical trials, gastrointestinal side effects including diarrhea (5%), nausea (4%), and vomiting (2%) were reported with ceftaroline. C difficile-associated diarrhea has also been reported.33
As with other cephalosporins, ceftaroline can cause a false-positive result on the Coombs test. Approximately 11% of ceftaroline-treated patients in phase 3 clinical trials had a positive Coombs test, but hemolytic anemia did not occur in any patients.33,34
Discontinuation of ceftaroline due to an adverse reaction was reported in 2.7% of patients receiving the drug during phase 3 trials, compared with 3.7% with comparator agents.
WHEN SHOULD CEFTAROLINE BE USED IN DAILY PRACTICE?
Ceftaroline has been shown to be at least as effective as ceftriaxone in treating community-acquired bacterial pneumonia, and at least as effective as vancomycin-aztreonam in treating acute bacterial skin and skin-structure infections. The 2014 Infectious Diseases Society of America’s guidelines for the diagnosis and management of skin and soft-tissue infections recommend ceftaroline as an option for empiric therapy for purulent skin and soft-tissue infections.35
The guidelines on community-acquired pneumonia have not been updated since 2007, which was before ceftaroline was approved. However, these guidelines are currently undergoing revision and may provide insight on ceftaroline’s place in the treatment of community-acquired bacterial pneumonia.36
Currently, ceftaroline’s routine use for these indications should be balanced by its higher cost ($150 for a 600-mg dose) compared with ceftriaxone ($5 for a 1-g dose) or vancomycin ($25 for a 1-g dose). The drug’s in vitro activity against drug-resistant pneumococci and S aureus, including MRSA, hVISA, and VISA may help fill an unmet need or provide a safer and more tolerable alternative to currently available therapies.
However, ceftaroline’s lack of activity against P aeruginosa and carbapenem-resistant Enterobacteriaceae does not meet the public health threat needs stemming from these multidrug-resistant microorganisms. Ongoing clinical trials in patients with more serious MRSA infections will provide important information about ceftaroline’s role as an anti-MRSA agent.
While the discovery of antimicrobials has had one of the greatest impacts on medicine, continued antibiotic use is threatened by the emergence of drug-resistant pathogens. Therefore, it is as important as ever to be good stewards of our currently available antimicrobials. Developing usage and dosing criteria for antimicrobials based on available data and literature is a step forward in optimizing the use of antibiotics—a precious medical resource.
Ceftaroline fosamil (Teflaro), introduced to the US market in October 2010, is the first beta-lactam agent with clinically useful activity against methicillin-resistant Staphylococcus aureus (MRSA). Currently, it is approved by the US Food and Drug Administration (FDA) to treat acute bacterial skin and skin-structure infections and community-acquired bacterial pneumonia caused by susceptible microorganisms.
In an era of increasing drug resistance and limited numbers of antimicrobials in the drug-production pipeline, ceftaroline is a step forward in fulfilling the Infectious Diseases Society of America’s “10 × ’20 Initiative” to increase support for drug research and manufacturing, with the goal of producing 10 new antimicrobial drugs by the year 2020.1 Ceftaroline was the first of several antibiotics to receive FDA approval in response to this initiative. It was followed by dalbavancin (May 2014), tedizolid phosphate (June 2014), oritavancin (August 2014), ceftolozane-tazobactam (December 2014), and ceftazidime-avibactam (February 2015). These antibiotic agents are aimed at treating infections caused by drug-resistant gram-positive and gram-negative microorganisms. It is important to understand and optimize the use of these new antibiotic agents in order to decrease the risk of emerging antibiotic resistance and superinfections (eg, Clostridium difficile infection) caused by antibiotic overuse or misuse.
This article provides an overview of ceftaroline’s mechanisms of action and resistance, spectrum of activity, pharmacokinetic properties, adverse effects, and current place in therapy.
AN ERA OF MULTIDRUG-RESISTANT MICROORGANISMS
Increasing rates of antimicrobial resistance threaten the efficacy of antimicrobial drugs in the daily practice of medicine. The World Health Organization has labeled antimicrobial resistance one of the three greatest threats to human health. Global efforts are under way to stimulate development of new antimicrobial agents and to decrease rates of antimicrobial resistance.
Staphylococcus aureus: A threat, even with vancomycin
Between 1998 and 2005, S aureus was one of the most common inpatient and outpatient isolates reported by clinical laboratories throughout the United States.2
Treatment of S aureus infection is complicated by a variety of resistance mechanisms that have evolved over time. In fact, the first resistant isolate of S aureus emerged not long after penicillin’s debut into clinical practice, and now the majority of strains are resistant to penicillin.
Methicillin was designed to overcome this beta-lactamase resistance and became the treatment of choice for penicillin-resistant S aureus isolates. However, MRSA isolates soon emerged because of the organism’s acquisition of penicillin-binding protein PBP2a via the mecA gene, leading to decreased binding affinity of methicillin.3
Since then, several agents active against MRSA (vancomycin, daptomycin, linezolid, tigecycline) have been introduced and continue to be widely used. While vancomycin is considered the first-line option for a variety of MRSA infections, its use has been threatened because of the emergence of vancomycin-intermediate-resistant S aureus (VISA), S aureus strains displaying vancomycin heteroresistance (hVISA), and vancomycin-resistant S aureus (VRSA) strains.4
VISA and hVISA isolates emerged through sequential mutations that lead to autolytic activity and cell-wall thickening. In contrast, the mechanism of resistance in VRSA is by acquisition of the vanA resistance gene, which alters the binding site of vancomycin from d-alanine-d-alanine to d-alanine-d-lactate.5
Streptococcus pneumoniae resistance: A continuing problem
The prevalence of drug resistance in S pneumoniae has risen since the late 1990s. A 2013 report from the SENTRY Antimicrobial Surveillance Program stated that almost 20% of S pneumoniae isolates were resistant to amoxicillin-clavulanate, and similar trends have been observed for penicillin (14.8%) and ceftriaxone (11.7%).6
S pneumoniae resistance is acquired through modifications of the penicillin-binding proteins, namely PBP1a, PBP2b, PBP2x, and, less frequently, PBP2a. These modifications lead to decreased binding affinity for most beta-lactams.7
Clinical impact of multidrug-resistant S aureus and S pneumoniae
In 2011, the US Centers for Disease Control and Prevention reported an estimated 80,000 severe MRSA infections and 11,000 MRSA-related deaths in the United States.8 In the same report, drug-resistant S pneumoniae was estimated to be responsible for almost 1.2 million illnesses and 7,000 deaths per year, leading to upwards of $96 million in related medical costs.
While invasive drug-resistant S pneumoniae infections usually affect patients at the extremes of age (under age 5 and over age 65), they have had a serious impact on patients of all ages.8
In light of the increasing prevalence of multidrug-resistant organisms, newer antimicrobial agents with novel mechanisms of action are needed.
CEFTAROLINE: A BETA-LACTAM WITH ANTI-MRSA ACTIVITY
The cephalosporins, a class of beta-lactam antibiotics, were originally derived from the fungus Cephalosporium (now called Acremonium). There are now many agents in this class, each containing a nucleus consisting of a beta-lactam ring fused to a six-member dihydrothiazine ring, and two side chains that can be modified to affect antibacterial activity and pharmacokinetic properties.
Cephalosporins are typically categorized into “generations.” With some exceptions, the first- and second-generation agents have good activity against gram-positive microorganisms, including methicillin-susceptible S aureus—but not against MRSA. The third- and fourth-generation cephalosporins have better gram-negative activity, with many agents having activity against the gram-negative bacterium Pseudomonas aeruginosa.
Enterococcal isolates are intrinsically resistant to cephalosporins. Additionally, cephalosporins are not active against anaerobic bacteria, except for a subset of structurally unique second-generation cephalosporins, ie, cefotetan and cefoxitin.
Ceftaroline was synthesized with specific manipulations of the side chains to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae isolates, making it the first available beta-lactam with this ability.
Mechanism of action
Ceftaroline binds to penicillin-binding proteins, inhibiting transpeptidation. This interaction blocks the final stage of peptidoglycan synthesis and inhibits bacterial cell wall formation, ultimately leading to cellular autolysis and microorganism death. Ceftaroline binds with high affinity to PBP2a and PBP2x, expanding its activity to encompass MRSA and penicillin-resistant S pneumoniae isolates.9
Spectrum of activity
Ceftaroline has in vitro activity against many gram-positive and gram-negative bacteria,10–13 including (Table 1):
- Methicillin-susceptible and methicillin-resistant staphylococci
- VISA, VRSA, and hVISA
- Daptomycin-nonsusceptible S aureus
- Streptococcal species, including penicillin-resistant S pneumoniae
- Enterobacteriaceae, including Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Citrobacter koseri, Citrobacter freundii, Enterobacter cloacae, Enterobacter aerogenes, Moraxella catarrhalis, Morganella morganii, and Proteus mirabilis.
Of note, ceftaroline is not active against Pseudomonas species, Enterococcus species, or Bacteroides fragilis. In addition, it is not active against the “atypical” respiratory pathogens Mycoplasma pneumoniae, Chlamydophila pneumoniae, and Legionella pneumophila.
Ceftaroline resistance
Gram-negative organisms appear to develop resistance to ceftaroline at rates similar to those observed with the other oxyimino-cephalosporins (eg, ceftriaxone). Ceftaroline is inactive against gram-negative organisms producing extended-spectrum beta-lactamases, including K pneumoniae carbapenemase and metallo-beta-lactamases.14 In addition, it induces the expression of AmpC beta-lactamases.
Although currently uncommon, resistance to ceftaroline has also been reported in S aureus strains.15 The mechanism of resistance is decreased binding affinity for PBP2a due to amino acid substitutions on the nonpenicillin-binding domains.15
Pharmacokinetic profile
An understanding of pharmacokinetics is key in optimizing the dose of antimicrobials so that the drugs are used most effectively and pathogens do not develop resistance to them.
Ceftaroline fosamil is a prodrug that, upon intravenous administration, is rapidly converted by phosphatase enzymes to its active moiety, ceftaroline. Its pharmacokinetic profile is summarized in Table 2.16,17 Its volume of distribution is similar to that of the fourth-generation cephalosporin cefepime.
Ceftaroline is then hydrolyzed into its inactive metabolite, ceftaroline M-1. It undergoes little hepatic metabolism and lacks properties to make it a substrate, inhibitor, or inducer of the CYP450 enzyme system and therefore is not likely to cause notable CYP450-related drug-drug interactions.
Like most other beta-lactams, ceftaroline is primarily excreted by the kidneys. Furthermore, an estimated 21% of a dose is eliminated with each intermittent hemodialysis session. Therefore, renal and intermittent hemodialysis dose adjustments are necessary. The estimated elimination half-life is 2.6 hours, necessitating dosing two to three times daily, depending on the indication and infectious inoculum.
Ceftaroline dosing
Ceftaroline is available only in a parenteral preparation and is typically given at a dose of 600 mg every 12 hours.10 The intravenous infusion is given over 1 hour.
The current stability data require reconstituted ceftaroline to be used within 6 hours at room temperature and within 24 hours if refrigerated.10
Ceftaroline requires dosing adjustments for patients with renal insufficiency. Per the manufacturer, renal dosing adjustments are based on the creatinine clearance rate, as estimated by the Cockroft-Gault formula:
- Creatinine clearance > 50 mL/min: no dosage adjustment necessary
- Creatinine clearance > 30 to ≤ 50 mL/min: give 400 mg every 12 hours
- Creatinine clearance ≥ 15 to ≤ 30 mL/min: give 300 mg every 12 hours
- Creatinine clearance < 15 mL/min or on intermittent dialysis: give 200 mg every 12 hours.
Ongoing clinical trials are investigating a higher-dosing strategy of 600 mg every 8 hours for patients with community-acquired bacterial pneumonia at risk of MRSA bacteremia.18
CLINICAL TRIALS LEADING TO CEFTAROLINE’S APPROVAL
Ceftaroline was approved for the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections due to susceptible pathogens on the basis of phase 3 comparator trials.
Community-acquired bacterial pneumonia: The FOCUS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of community-acquired bacterial pneumonia was studied in two randomized, double-blind, noninferiority trials, known as Ceftaroline Community-acquired Pneumonia vs Ceftriaxone (FOCUS) 1 and FOCUS 2.19,20
Patients were adults and not critically ill, as was reflected by their being in Pneumonia Outcomes Research Team (PORT) risk class III or IV (with class V indicating the highest risk of death). Therefore, the results may not be completely applicable to critically ill patients or those not admitted to the hospital. Of note, patients were excluded from the trials if they had infections known or thought to be due to MRSA or to atypical organisms.21 Baseline characteristics and patient demographics were similar between study groups in both trials.
A bacterial pathogen was identified in 26.1% of the patients included in the modified intent-to-treat analysis of the pooled data of the trials; the most common pathogens were S pneumoniae, methicillin-sensitive S aureus, Haemophilus influenzae, K pneumoniae, and E coli.21
Treatment. Patients received either ceftaroline 600 mg every 12 hours (or a lower dose based on renal function) or ceftriaxone 1 g every 24 hours. In addition, in the FOCUS 1 trial, patients in both treatment groups received clarithromycin 500 mg every 12 hours for the first day.19
Results. In both trials and in the integrated analysis, ceftaroline was noninferior to ceftriaxone (Table 3).22 In the integrated analysis of both trials, compared with the ceftriaxone group, the ceftaroline group had a higher clinical cure rate among patients classified as PORT risk class III (86.8% vs 79.2%, weighted treatment difference 12.6%, 95% confidence interval [CI] 1.3–13.8) and among patients who had not received prior antibiotic treatment (85.5% vs 74.9%, weighted treatment difference 11.2%, 95% CI 4.5–18.0).21
Acute bacterial skin and skin-structure infections: The CANVAS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of complicated acute bacterial skin and skin-structure infections was studied in two randomized, double-blind trials: Ceftaroline Versus Vancomycin in Skin and Skin Structure Infections (CANVAS) 1 and CANVAS 2.23,24
Patients. Adult patients with a diagnosis of community-acquired skin and skin-structure infections warranting at least 5 days of intravenous antimicrobial therapy were included in the trials. Important protocol exclusions were patients with diabetic foot ulcers, decubitus ulcers, burns, ulcers associated with peripheral vascular disease accompanied by osteomyelitis, and suspected P aeruginosa infections.25 This limits the external validity of ceftaroline use in the aforementioned excluded patient populations.
Patients in each treatment group of the trials had similar demographic characteristics. The most common infections were cellulitis, major abscess requiring surgical intervention, wound infection, and infected ulcer. Bacteremia was present in 4.2% of patients in the ceftaroline group and in 3.8% of patients in the vancomycin-aztreonam group. The most common pathogen was S aureus. Methicillin resistance was present in 40% of the ceftaroline group and 34% of the control group.
Treatment. Patients received either ceftaroline 600 mg every 12 hours or the combination of vancomycin 1 g plus aztreonam 1 g given 12 hours, for 5 to 14 days.
Results. As assessed at a “test-of-cure” visit 8 to 15 days after the last dose of study medication, the efficacy of ceftaroline was similar to that of vancomycin-aztreonam, meeting the set noninferiority goal (Table 4).25 Moreover, if assessed on day 2 or 3 (a new end point recommended by the FDA), the rate of cessation of erythema spread and absence of fever was higher in the ceftaroline group than in the vancomycin-aztreonam group.26 However, this end point was not in the original trial protocol.
CEFTAROLINE FOR OTHER INDICATIONS
As noted, ceftaroline has been approved for treating community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections. In addition, it has been used in several studies in animals, and case reports of non-FDA approved indications including endocarditis and osteomyelitis have been published. Clinical trials are evaluating its use in pediatric patients, as well as for community-acquired bacterial pneumonia with risk for MRSA and for MRSA bacteremia.
Endocarditis
Animal studies have demonstrated ceftaroline to have bactericidal activity against MRSA and hVISA in endocarditis.27
A few case series have been published describing ceftaroline’s use as salvage therapy for persistent MRSA bacteremia and endocarditis. For example, Ho et al28 reported using it in three patients who had endocarditis as a source of their persistent bacteremia. All three patients had resolution of their MRSA bloodstream infection following ceftaroline therapy. The dosage was 600 mg every 8 hours, which is higher than in the manufacturer’s prescribing information.
Lin et al29 reported using ceftaroline in five patients with either possible or probable endocarditis. Three of the five patients had clinical cure as defined by resolution or improvement of all signs and symptoms of infection, and not requiring further antimicrobial therapy.29
More data from clinical trials would be beneficial in defining ceftaroline’s role in treating endocarditis caused by susceptible microorganisms.
Osteomyelitis
In animal studies of osteomyelitis, ceftaroline exhibited activity against MRSA in infected bone and joint fluid. Compared with vancomycin and linezolid, ceftaroline was associated with more significant decreases in bacterial load in the infected joint fluid, bone marrow, and bone.30
Lin et al29 gave ceftaroline to two patients with bone and joint infections, both of whom had received other therapies that had failed. The doses of ceftaroline were higher than those recommended in the prescribing information; clinical cure was noted in both cases following the switch.
These data come from case series, and more study of ceftaroline’s role in the treatment of osteomyelitis infections is warranted.
Meningitis
The use of ceftaroline in meningitis has been studied in rabbits. While ceftaroline penetrated into the cerebrospinal fluid in only negligible amounts in healthy rabbits (3% penetration), its penetration improved to 15% in animals with inflamed meninges. Ceftaroline cerebrospinal fluid levels in inflamed meninges were sufficient to provide bactericidal activity against penicillin-sensitive and resistant S pneumoniae strains as well as K pneumoniae and E coli strains.31,32
REPORTED ADVERSE EFFECTS OF CEFTAROLINE
Overall, ceftaroline was well tolerated in clinical trials, and its safety profile was similar to those of the comparator agents (ceftriaxone and vancomycin-aztreonam).
As with the other cephalosporins, hypersensitivity reactions have been reported with ceftaroline. In the clinical trials, 3% of patients developed a rash with ceftaroline.33,34 Patients with a history of beta-lactam allergy were excluded from the trials, so the rate of cross-reactivity with penicillins and with other cephalosporins is unknown.
In the phase 3 clinical trials, gastrointestinal side effects including diarrhea (5%), nausea (4%), and vomiting (2%) were reported with ceftaroline. C difficile-associated diarrhea has also been reported.33
As with other cephalosporins, ceftaroline can cause a false-positive result on the Coombs test. Approximately 11% of ceftaroline-treated patients in phase 3 clinical trials had a positive Coombs test, but hemolytic anemia did not occur in any patients.33,34
Discontinuation of ceftaroline due to an adverse reaction was reported in 2.7% of patients receiving the drug during phase 3 trials, compared with 3.7% with comparator agents.
WHEN SHOULD CEFTAROLINE BE USED IN DAILY PRACTICE?
Ceftaroline has been shown to be at least as effective as ceftriaxone in treating community-acquired bacterial pneumonia, and at least as effective as vancomycin-aztreonam in treating acute bacterial skin and skin-structure infections. The 2014 Infectious Diseases Society of America’s guidelines for the diagnosis and management of skin and soft-tissue infections recommend ceftaroline as an option for empiric therapy for purulent skin and soft-tissue infections.35
The guidelines on community-acquired pneumonia have not been updated since 2007, which was before ceftaroline was approved. However, these guidelines are currently undergoing revision and may provide insight on ceftaroline’s place in the treatment of community-acquired bacterial pneumonia.36
Currently, ceftaroline’s routine use for these indications should be balanced by its higher cost ($150 for a 600-mg dose) compared with ceftriaxone ($5 for a 1-g dose) or vancomycin ($25 for a 1-g dose). The drug’s in vitro activity against drug-resistant pneumococci and S aureus, including MRSA, hVISA, and VISA may help fill an unmet need or provide a safer and more tolerable alternative to currently available therapies.
However, ceftaroline’s lack of activity against P aeruginosa and carbapenem-resistant Enterobacteriaceae does not meet the public health threat needs stemming from these multidrug-resistant microorganisms. Ongoing clinical trials in patients with more serious MRSA infections will provide important information about ceftaroline’s role as an anti-MRSA agent.
While the discovery of antimicrobials has had one of the greatest impacts on medicine, continued antibiotic use is threatened by the emergence of drug-resistant pathogens. Therefore, it is as important as ever to be good stewards of our currently available antimicrobials. Developing usage and dosing criteria for antimicrobials based on available data and literature is a step forward in optimizing the use of antibiotics—a precious medical resource.
- Infectious Diseases Society of America. The 10 x ‘20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 2010; 50:1081–1083.
- Styers D, Sheehan DJ, Hogan P, Sahm DF. Laboratory-based surveillance of current antimicrobial resistance patterns and trends among Staphylococcus aureus: 2005 status in the United States. Ann Clin Microbiol Antimicrob 2006; 5:2.
- Farrell DJ, Castanheira M, Mendes RE, Sader HS, Jones RN. In vitro activity of ceftaroline against multidrug-resistant Staphylococcus aureus and Streptococcus pneumoniae: a review of published studies and the AWARE Surveillance Program (2008-2010). Clin Infect Dis 2012; 55(suppl 3):S206–S214.
- Holmes NE, Johnson PD, Howden BP. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J Clin Microbiol 2012; 50:2548–2552.
- Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 2003; 111:1265–1273.
- Jones RN, Sader HS, Mendes RE, Flamm RK. Update on antimicrobial susceptibility trends among Streptococcus pneumoniae in the United States: report of ceftaroline activity from the SENTRY Antimicrobial Surveillance Program (1998-2011). Diag Microbiol Infect Dis 2013; 75:107–109.
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 2008; 32:361–385.
- Centers for Disease Control and Prevention (CDC). Antibiotic resistance threats in the United States 2013. cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. Accessed June 1, 2015.
- Moisan H, Pruneau M, Malouin F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J Antimicrob Chemother 2010; 65:713–716.
- Forest Laboratories, Inc. Teflaro® (ceftaroline fosamil): prescribing information. www.frx.com/pi/teflaro_pi.pdf. Accessed June 1, 2015.
- Richter SS, Heilmann KP, Dohrn CL, et al. Activity of ceftaroline and epidemiologic trends in Staphylococcus aureus isolates collected from 43 medical centers in the United States in 2009. Antimicrob Agents Chemother 2011; 55:4154–4160.
- Ge Y, Biek D, Talbot GH, Sahm DF. In vitro profiling of ceftaroline against a collection of recent bacterial clinical isolates from across the United States. Antimicrob Agents Chemother 2008; 52:3398–3407.
- Saravolatz L, Pawlak J, Johnson L. In vitro activity of ceftaroline against community-associated methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, and daptomycin-nonsusceptible Staphylococcus aureus isolates. Antimicrob Agents Chemother 2010; 54:3027–3030.
- Mushtaq S, Livermore DM. AmpC induction by ceftaroline. J Antimicrob Chemother 2010; 65:586–588.
- Mendes RE, Tsakris A, Sader HS, et al. Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J Antimicrob Chemother 2012; 67:1321–1324.
- Lodise TP, Low DE. Ceftaroline fosamil in the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections. Drugs 2012; 72:1473–1493.
- Riccobene TA, Su SF, Rank D. Single- and multiple-dose study to determine the safety, tolerability, and pharmacokinetics of ceftaroline fosamil in combination with avibactam in healthy subjects. Antimicrob Agents Chemother 2013; 57:1496–1504.
- US National Institutes of Health. ClinicalTrials.gov. Evaluation of ceftaroline fosamil versus a comparator in adult subjects with community-acquired bacterial pneumonia (CABP) with risk for methicillin-resistant Staphylococcus aureus. http://clinicaltrials.gov/ct2/show/NCT01645735. Accessed June 1, 2015.
- File TM Jr, Low DE, Eckburg PB, et al; FOCUS 1 investigators. FOCUS 1: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii19–iii32.
- Low DE, File TM Jr, Eckburg PB, et al; FOCUS 2 investigators. FOCUS 2: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii33–iii44.
- File TM Jr, Low DE, Eckburg PB, et al. Integrated analysis of FOCUS 1 and FOCUS 2: randomized, doubled-blinded, multicenter phase 3 trials of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in patients with community-acquired pneumonia. Clin Infect Dis 2010; 51:1395–1405.
- Food and Drug Administration (FDA). Ceftaroline fosamil for the treatment of community-acquired bacterial pneumonia and complicated skin and skin structure infections. www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/anti-infectivedrugsadvisorycommittee/ucm224656.pdf. Accessed June 1, 2015.
- Corey GR, Wilcox MH, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 1 investigators. CANVAS 1: the first phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv41–iv51.
- Wilcox MH, Corey GR, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 2 investigators. CANVAS 2: the second phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv53-iv65.
- Corey GR, Wilcox M, Talbot GH, et al. Integrated analysis of CANVAS 1 and 2: phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin Infect Dis 2010; 51:641–650.
- Friedland HD, O’Neal T, Biek D, et al. CANVAS 1 and 2: analysis of clinical response at day 3 in two phase 3 trials of ceftaroline fosamil versus vancomycin plus aztreonam in treatment of acute bacterial skin and skin structure infections. Antimicrob Agents Chemother 2012; 56:2231–2236.
- Jacqueline C, Caillon J, Le Mabecque V, et al. In vivo efficacy of ceftaroline (PPI-0903), a new broad-spectrum cephalosporin, compared with linezolid and vancomycin against methicillin-resistant and vancomycin-intermediate Staphylococcus aureus in a rabbit endocarditis model. Antimicrob Agents Chemother 2007; 51:3397–3400.
- Ho TT, Cadena J, Childs LM, Gonzalez-Velez M, Lewis JS 2nd. Methicillin-resistant Staphylococcus aureus bacteraemia and endocarditis treated with ceftaroline salvage therapy. J Antimicrob Chemother 2012; 67:1267–1270.
- Lin JC, Aung G, Thomas A, Jahng M, Johns S, Fierer J. The use of ceftaroline fosamil in methicillin-resistant Staphylococcus aureus endocarditis and deep-seated MRSA infections: a retrospective case series of 10 patients. J Infect Chemother 2013; 19:42–49.
- Jacqueline C, Amador G, Caillon J, et al. Efficacy of the new cephalosporin ceftaroline in the treatment of experimental methicillin-resistant Staphylococcus aureus acute osteomyelitis. J Antimicrob Chemother 2010; 65:1749–1752.
- Stucki A, Acosta F, Cottagnoud M, Cottagnoud P. Efficacy of ceftaroline fosamil against Escherichia coli and Klebsiella pneumoniae strains in a rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:5808–5810.
- Cottagnoud P, Cottagnoud M, Acosta F, Stucki A. Efficacy of ceftaroline fosamil against penicillin-sensitive and -resistant Streptococcus pneumoniae in an experimental rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:4653–4655.
- Corrado ML. Integrated safety summary of CANVAS 1 and 2 trials: phase III, randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv67–iv71.
- Rank DR, Friedland HD, Laudano JB. Integrated safety summary of FOCUS 1 and FOCUS 2 trials: phase III randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii53–iii59.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis 2014; 59:147–159.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44:S27–S72.
- Infectious Diseases Society of America. The 10 x ‘20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 2010; 50:1081–1083.
- Styers D, Sheehan DJ, Hogan P, Sahm DF. Laboratory-based surveillance of current antimicrobial resistance patterns and trends among Staphylococcus aureus: 2005 status in the United States. Ann Clin Microbiol Antimicrob 2006; 5:2.
- Farrell DJ, Castanheira M, Mendes RE, Sader HS, Jones RN. In vitro activity of ceftaroline against multidrug-resistant Staphylococcus aureus and Streptococcus pneumoniae: a review of published studies and the AWARE Surveillance Program (2008-2010). Clin Infect Dis 2012; 55(suppl 3):S206–S214.
- Holmes NE, Johnson PD, Howden BP. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J Clin Microbiol 2012; 50:2548–2552.
- Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 2003; 111:1265–1273.
- Jones RN, Sader HS, Mendes RE, Flamm RK. Update on antimicrobial susceptibility trends among Streptococcus pneumoniae in the United States: report of ceftaroline activity from the SENTRY Antimicrobial Surveillance Program (1998-2011). Diag Microbiol Infect Dis 2013; 75:107–109.
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 2008; 32:361–385.
- Centers for Disease Control and Prevention (CDC). Antibiotic resistance threats in the United States 2013. cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. Accessed June 1, 2015.
- Moisan H, Pruneau M, Malouin F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J Antimicrob Chemother 2010; 65:713–716.
- Forest Laboratories, Inc. Teflaro® (ceftaroline fosamil): prescribing information. www.frx.com/pi/teflaro_pi.pdf. Accessed June 1, 2015.
- Richter SS, Heilmann KP, Dohrn CL, et al. Activity of ceftaroline and epidemiologic trends in Staphylococcus aureus isolates collected from 43 medical centers in the United States in 2009. Antimicrob Agents Chemother 2011; 55:4154–4160.
- Ge Y, Biek D, Talbot GH, Sahm DF. In vitro profiling of ceftaroline against a collection of recent bacterial clinical isolates from across the United States. Antimicrob Agents Chemother 2008; 52:3398–3407.
- Saravolatz L, Pawlak J, Johnson L. In vitro activity of ceftaroline against community-associated methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, and daptomycin-nonsusceptible Staphylococcus aureus isolates. Antimicrob Agents Chemother 2010; 54:3027–3030.
- Mushtaq S, Livermore DM. AmpC induction by ceftaroline. J Antimicrob Chemother 2010; 65:586–588.
- Mendes RE, Tsakris A, Sader HS, et al. Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J Antimicrob Chemother 2012; 67:1321–1324.
- Lodise TP, Low DE. Ceftaroline fosamil in the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections. Drugs 2012; 72:1473–1493.
- Riccobene TA, Su SF, Rank D. Single- and multiple-dose study to determine the safety, tolerability, and pharmacokinetics of ceftaroline fosamil in combination with avibactam in healthy subjects. Antimicrob Agents Chemother 2013; 57:1496–1504.
- US National Institutes of Health. ClinicalTrials.gov. Evaluation of ceftaroline fosamil versus a comparator in adult subjects with community-acquired bacterial pneumonia (CABP) with risk for methicillin-resistant Staphylococcus aureus. http://clinicaltrials.gov/ct2/show/NCT01645735. Accessed June 1, 2015.
- File TM Jr, Low DE, Eckburg PB, et al; FOCUS 1 investigators. FOCUS 1: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii19–iii32.
- Low DE, File TM Jr, Eckburg PB, et al; FOCUS 2 investigators. FOCUS 2: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii33–iii44.
- File TM Jr, Low DE, Eckburg PB, et al. Integrated analysis of FOCUS 1 and FOCUS 2: randomized, doubled-blinded, multicenter phase 3 trials of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in patients with community-acquired pneumonia. Clin Infect Dis 2010; 51:1395–1405.
- Food and Drug Administration (FDA). Ceftaroline fosamil for the treatment of community-acquired bacterial pneumonia and complicated skin and skin structure infections. www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/anti-infectivedrugsadvisorycommittee/ucm224656.pdf. Accessed June 1, 2015.
- Corey GR, Wilcox MH, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 1 investigators. CANVAS 1: the first phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv41–iv51.
- Wilcox MH, Corey GR, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 2 investigators. CANVAS 2: the second phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv53-iv65.
- Corey GR, Wilcox M, Talbot GH, et al. Integrated analysis of CANVAS 1 and 2: phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin Infect Dis 2010; 51:641–650.
- Friedland HD, O’Neal T, Biek D, et al. CANVAS 1 and 2: analysis of clinical response at day 3 in two phase 3 trials of ceftaroline fosamil versus vancomycin plus aztreonam in treatment of acute bacterial skin and skin structure infections. Antimicrob Agents Chemother 2012; 56:2231–2236.
- Jacqueline C, Caillon J, Le Mabecque V, et al. In vivo efficacy of ceftaroline (PPI-0903), a new broad-spectrum cephalosporin, compared with linezolid and vancomycin against methicillin-resistant and vancomycin-intermediate Staphylococcus aureus in a rabbit endocarditis model. Antimicrob Agents Chemother 2007; 51:3397–3400.
- Ho TT, Cadena J, Childs LM, Gonzalez-Velez M, Lewis JS 2nd. Methicillin-resistant Staphylococcus aureus bacteraemia and endocarditis treated with ceftaroline salvage therapy. J Antimicrob Chemother 2012; 67:1267–1270.
- Lin JC, Aung G, Thomas A, Jahng M, Johns S, Fierer J. The use of ceftaroline fosamil in methicillin-resistant Staphylococcus aureus endocarditis and deep-seated MRSA infections: a retrospective case series of 10 patients. J Infect Chemother 2013; 19:42–49.
- Jacqueline C, Amador G, Caillon J, et al. Efficacy of the new cephalosporin ceftaroline in the treatment of experimental methicillin-resistant Staphylococcus aureus acute osteomyelitis. J Antimicrob Chemother 2010; 65:1749–1752.
- Stucki A, Acosta F, Cottagnoud M, Cottagnoud P. Efficacy of ceftaroline fosamil against Escherichia coli and Klebsiella pneumoniae strains in a rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:5808–5810.
- Cottagnoud P, Cottagnoud M, Acosta F, Stucki A. Efficacy of ceftaroline fosamil against penicillin-sensitive and -resistant Streptococcus pneumoniae in an experimental rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:4653–4655.
- Corrado ML. Integrated safety summary of CANVAS 1 and 2 trials: phase III, randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv67–iv71.
- Rank DR, Friedland HD, Laudano JB. Integrated safety summary of FOCUS 1 and FOCUS 2 trials: phase III randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii53–iii59.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis 2014; 59:147–159.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44:S27–S72.
KEY POINTS
- Resistance of S aureus and S pneumoniae to multiple antimicrobial drugs is on the rise, and new agents are urgently needed.
- Ceftaroline’s molecular structure was designed to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae.
- In clinical trials leading to its approval, ceftaroline was found to be at least as effective as ceftriaxone in treating community-acquired pneumonia and at least as effective as vancomycin plus aztreonam in treating acute bacterial skin and skin-structure infections.
- The routine use of ceftaroline for these indications should be balanced by its higher cost compared with ceftriaxone or vancomycin. Ongoing studies should shed more light on its role in treatment.
Vancomycin: A 50-something-year-old antibiotic we still don’t understand
In the past half-century, vancomycin has gone from near-orphan status to being one of the most often used antibiotics in our formulary. The driving force for its use is clear: the evolution of Staphylococcus aureus. At first, vancomycin was used to treat infections caused by penicillin-resistant strains. However, the discovery of methicillin curbed its use for more than 2 decades.1
Then, as methicillin-resistant S aureus (MRSA) began to spread in the 1980s, the use of vancomycin began to increase, and with the rise in community-associated MRSA infections in the 1990s, it became even more widely prescribed. The recent Infectious Diseases Society of America (IDSA) guidelines for treatment of infections due to MRSA are replete with references to the use of vancomycin.2
Another factor driving the use of vancomycin is the increased prevalence of device-associated infections, many of which are caused by coagulase-negative staphylococci and other organisms that colonize the skin.3 Many of these bacteria are susceptible only to vancomycin; they may be associated with infections of vascular catheters, cardiac valves, pacemakers, implantable cardioverter-defibrillators, orthopedic implants, neurosurgical devices, and other devices.
To use vancomycin appropriately, we need to recognize the changing minimum inhibitory concentrations (MICs), to select proper doses and dosing intervals, and to know how to monitor its use. Despite more than 50 years of experience with vancomycin, we sometimes find ourselves with more questions than answers about its optimal use.
WHAT IS VANCOMYCIN?
Vancomycin is a glycopeptide antibiotic isolated from a strain of Streptomyces orientalis discovered in a soil sample from Borneo in the mid-1950s.1 It exerts its action by binding to a d-alanyl-d-alanine cell wall precursor necessary for peptidoglycan cross-linking and, therefore, for inhibiting bacterial cell wall synthesis.
Vancomycin is bactericidal against most gram-positive species, including streptococci and staphylococci, with the exception of Enterococcus species, for which it is bacteriostatic. Though it is bactericidal, it appears to kill bacteria more slowly than beta-lactam antibiotics, and therefore it may take longer to clear bacteremia.4
WHAT IS THE BEST WAY TO DOSE VANCOMYCIN?
Vancomycin is widely distributed to most tissues, with an approximate volume of distribution of 0.4 to 1 L/kg; 50% to 55% is protein-bound. Because of this large volume of distribution, vancomycin’s dosing is based on actual body weight.
Vancomycin is not metabolized and is primarily excreted unchanged in the urine via glomerular filtration. It therefore requires dosage adjustments for renal insufficiency.
Vancomycin’s molecular weight is 1,485.73 Da, making it less susceptible to removal by dialysis than smaller molecules. Dosing of vancomycin in patients on hemodialysis depends on many factors specific to the dialysis center, including but not limited to the type of filter used, the duration of filtration, and whether high-flux filtration is used.
Is continuous intravenous infusion better than standard dosing?
Giving vancomycin by continuous infusion has been suggested as a way to optimize its serum concentration and improve its clinical effectiveness.
Wysocki et al5 conducted a multicenter, prospective, randomized study comparing continuous and intermittent intravenous infusions of vancomycin (the latter every 12 hours) to treat severe hospital-acquired MRSA infections, including bloodstream infections and pneumonia. Although blood concentrations above 10 μg/mL were reached more than 30 hours faster with continuous infusions than with intermittent ones, the microbiologic and clinical outcomes were similar with either method.
James et al6 compared the pharmacodynamics of conventional dosing of vancomycin (ie, 1 g every 12 hours) and continuous infusion in 10 patients with suspected or documented gram-positive infections in a prospective, randomized, crossover study. While no adverse effects were observed, the authors also found no statistically significant difference between the treatment groups in the pharmacodynamic variables investigated, including the area under the curve (AUC) divided by the MIC (the AUC-MIC ratio).
In view of the currently available data, the guidelines for monitoring vancomycin therapy note that there does not appear to be any difference in patient outcomes with continuous infusion vs intermittent dosing.7
Should a loading dose be given?
Another proposed strategy for optimizing vancomycin’s effectiveness is to give a higher initial dose, ie, a loading dose.
Wang et al8 performed a single-center study in 28 patients who received a 25 mg/kg loading dose at a rate of 500 mg/hour. This loading dose was safe, but the authors did not evaluate its efficacy.
Mohammedi et al9 compared loading doses of 500 mg and 15 mg/kg in critically ill patients receiving vancomycin by continuous infusion. The weight-based loading dose produced higher post-dose levels and a significantly higher rate of clinical cure, but there was no significant difference in the rate of survival to discharge from the intensive care unit.
While the use of a loading dose appears to be safe and likely leads to more rapid attainment of therapeutic blood levels, we lack data on whether it improves clinical outcomes, and further study is needed to determine its role.
WHAT IS THE BEST WAY TO MONITOR VANCOMYCIN THERAPY?
Whether and how to use the serum vancomycin concentration to adjust the dosing has been a matter of debate for many years. Convincing evidence that vancomycin levels predict clinical outcomes or that measuring them prevents toxicity is lacking.7
A consensus statement from the American Society of Health-System Pharmacists, the IDSA, and the Society of Infectious Diseases Pharmacists7 contains recommendations for monitoring vancomycin therapy, based on a critical evaluation of the available scientific evidence. Their recommendations:
- Vancomycin serum concentrations should be checked to optimize therapy and used as a surrogate marker of effectiveness.
- Trough, rather than peak, levels should be monitored.
- Trough levels should be checked just before the fourth dose, when steady-state levels are likely to have been achieved. More frequent monitoring may be considered in patients with fluctuating renal function.
- Trough levels should be higher than 10 mg/L to prevent the development of resistance.
- To improve antibiotic penetration and optimize the likelihood of achieving pharmacokinetic and pharmacodynamic targets, trough levels of 15 to 20 mg/L are recommended for pathogens with a vancomycin MIC of 1 mg/L or higher and for complicated infections such as endocarditis, osteomyelitis, meningitis, and hospital-acquired pneumonia.
- For prolonged courses, it is appropriate to check vancomycin levels weekly in hemodynamically stable patients and more often in those who are not hemodynamically stable.
IS VANCOMYCIN NEPHROTOXIC?
In the 1950s, vancomycin formulations were sometimes called “Mississippi mud” because of the many impurities they contained.1 These impurities were associated with significant nephrotoxicity. Better purification methods used in the manufacture of current formulations mitigate this problem, resulting in a lower incidence of nephrotoxicity.
Over the last several years, organizations such as the American Thoracic Society and the IDSA have recommended targeting higher vancomycin trough concentrations.10 The consequent widespread use of higher doses has renewed interest in vancomycin’s potential nephrotoxicity.
Lodise et al,11 in a cohort study, examined the incidence of nephrotoxicity with higher daily doses of vancomycin (≥ 4 g/day), lower daily doses (< 4 g/day), and linezolid (Zyvox). They defined nephrotoxicity as an increase in serum creatinine of 0.5 mg/dL or a decrease in calculated creatinine clearance of 50% from baseline on 2 consecutive days.
The incidence of nephrotoxicity was significantly higher in the high-dose vancomycin group (34.6%) than in the low-dose vancomycin group (10.9%) and in the linezolid group (6.7%) (P = .001). Additional factors associated with nephrotoxicity in this study included baseline creatinine clearance less than 86.6 mL/minute, weight greater than 101.4 kg (223.5 lb), and being in an intensive care unit.
Hidayat et al12 investigated outcomes in patients with high vs low vancomycin trough levels (≥ 15 mg/L vs < 15 mg/L) in a prospective cohort study. Sixty-three patients achieved an average vancomycin trough of 15 to 20 mg/L, and of these, 11 developed nephrotoxicity, compared with no patients in the low-trough group (P = .01). Of the 11 who developed nephrotoxicity, 10 were concomitantly taking other potentially nephrotoxic agents.
Comment. The data on vancomycin and nephrotoxicity are mostly from studies that had limitations such as small numbers of patients, retrospective design, and variable definitions of nephrotoxicity. Many of the patients in these studies had additional factors contributing to nephrotoxicity, including hemodynamic instability and concomitant exposure to other nephrotoxins. Additionally, the sequence of events (nephrotoxicity leading to elevated vancomycin levels vs elevated vancomycin levels causing nephrotoxicity) is still debatable.
The incidence of nephrotoxicity associated with vancomycin therapy is difficult to determine. However, based on current information, the incidence of nephrotoxicity appears to be low when vancomycin is used as monotherapy.
IS S AUREUS BECOMING RESISTANT TO VANCOMYCIN?
An issue of increasing importance in health care settings is the emergence of vancomycin-intermediate S aureus (VISA) and vancomycin-resistant S aureus (VRSA). Eleven cases of VRSA were identified in the United States from 2002 to 2005.13 All cases of VRSA in the United States have involved the incorporation of enterococcal vanA cassette into the S aureus genome.14 While true VRSA isolates remain rare, VISA isolates are becoming more common.
Heteroresistant VISA: An emerging subpopulation of MRSA
Another population of S aureus that has emerged is heteroresistant vancomycin-intermediate S aureus (hVISA). It is defined as the presence of subpopulations of VISA within a population of MRSA at a rate of one organism per 105 to 106 organisms. With traditional testing methods, the vancomycin MIC for the entire population of the strain is within the susceptible range.15 These hVISA populations are thought to be precursors to the development of VISA.16
The resistance to vancomycin in hVISA and VISA populations is due to increased cell wall thickness, altered penicillin-binding protein profiles, and decreased cell wall autolysis.
While the true prevalence of hVISA is difficult to predict because of challenges in microbiological detection and probably varies between geographic regions and individual institutions, different studies have reported hVISA rates between 2% and 13% of all MRSA isolates.15–17
Reduced vancomycin susceptibility can develop regardless of methicillin susceptibility.18
While hVISA is not common, its presence is thought to be a predictor of failing vancomycin therapy.15
Factors associated with hVISA bacteremia include high-bacterial-load infections, treatment failure (including persistent bacteremia for more than 7 days), and initially low serum vancomycin levels.15
‘MIC creep’: Is it real?
Also worrisome, the average vancomycin MIC for S aureus has been shifting upward, based on reports from several institutions, although it is still within the susceptible range.19,20 However, this “MIC creep” likely reflects, at least in part, differences in MIC testing and varying methods used to analyze the data.19,20
Holmes and Jorgensen,21 in a single-institution study of MRSA isolates recovered from bacteremic patients from 1999 to 2006, determined that no MIC creep existed when they tested vancomycin MICs using the broth microdilution method. The authors found the MIC90 (ie, the MIC in at least 90% of the isolates) remained less than 1 mg/L during each year of the study.
Sader et al,22 in a multicenter study, evaluated 1,800 MRSA bloodstream isolates from nine hospitals across the United States from 2002 to 2006. Vancomycin MICs were again measured by broth microdilution methods. The mode MIC remained stable at 0.625 mg/L during the study period, and the authors did not detect a trend of rising MICs.
The inconsistency between reports of MIC creep at single institutions and the absence of this phenomenon in large, multicenter studies seems to imply that vancomycin MIC creep is not occurring on a grand scale.
Vancomycin tolerance
Another troubling matter with S aureus and vancomycin is the issue of tolerance. Vancomycin tolerance, defined in terms of increased minimum bactericidal concentration, represents a loss of bactericidal activity. Tolerance to vancomycin can occur even if the MIC remains in the susceptible range.23
Safdar and Rolston,24 in an observational study from a cancer center, reported that of eight cases of bacteremia that was resistant to vancomycin therapy, three were caused by S aureus.
Sakoulas et al25 found that higher levels of vancomycin bactericidal activity were associated with higher rates of clinical success; however, they found no effect on the mortality rate.
The issue of vancomycin tolerance remains controversial, and because testing for it is impractical in clinical microbiology laboratories, its implications outside the research arena are difficult to ascertain at present.
IS VANCOMYCIN STILL THE BEST DRUG FOR S AUREUS?
MIC break points have been lowered
In 2006, the Clinical Laboratories and Standards Institute lowered its break points for vancomycin MIC categories for S aureus:
- Susceptible: ≤ 2 mg/L (formerly ≤ 4 mg/L)
- Intermediate: 4–8 mg/L (formerly 8–16 mg/L)
- Resistant: ≥ 16 mg/L (formerly ≥ 32 mg/L).
The rationales for these changes were that the lower break points would better detect hVISA, and that cases have been reported of clinical treatment failure of S aureus infections in which the MICs for vancomycin were 4 mg/L.26
Since 2006, the question has been raised whether to lower the break points even further. A reason for this proposal comes from an enhanced understanding of the pharmacokinetics and pharmacodynamics of vancomycin.
The variable most closely associated with clinical response to vancomycin is the AUC-MIC ratio. An AUC-MIC ratio of 400 or higher may be associated with better outcomes in patients with serious S aureus infection. A study of 108 patients with S aureus infection of the lower respiratory tract indicated that organism eradication was more likely if the AUC-MIC ratio was 400 or greater compared with values less than 400, and this was statistically significant.27 However, in cases of S aureus infection with a vancomycin MIC of 2 mg/L or higher, this ratio may not be achievable.
A prospective study of 414 MRSA bacteremia episodes found a vancomycin MIC of 2 mg/L to be a predictor of death.28 The authors concluded that vancomycin may not be the optimal treatment for MRSA with a vancomycin MIC of 2 mg/L.28 Additional studies have also suggested a possible decrease in response to vancomycin in MRSA isolates with elevated MICs within the susceptible range.25,29
Recent guidelines from the IDSA recommend using the clinical response, regardless of the MIC, to guide antimicrobial selection for isolates with MICs in the susceptible range.2
Combination therapy with vancomycin
As vancomycin use has increased, therapeutic failures with vancomycin have become apparent. Combination therapy has been suggested as an option to increase the efficacy of vancomycin when treating complicated infections.
Rifampin plus vancomycin is controversial.30 The combination is theoretically beneficial, especially in infections associated with prosthetic devices. However, clinical studies have failed to convincingly support its use, and some have suggested that it might prolong bacteremia. In addition, it has numerous drug interactions to consider and adverse effects.31
Gentamicin plus vancomycin. The evidence supporting the use of this combination is weak at best. It appears that clinicians may have extrapolated from the success reported by Korzeniowski and Sande,32 who found that methicillin-susceptible S aureus bacteremia was cleared faster if gentamicin was added to nafcillin. A more recent study33 that compared daptomycin (Cubicin) monotherapy with combined vancomycin and gentamicin to treat MRSA bacteremia and endocarditis showed a better overall success rate with daptomycin (44% vs 32.6%), but the difference was not statistically significant.
Gentamicin has some toxicity. Even short-term use (for the first 4 days of therapy) at low doses for bacteremia and endocarditis due to staphylococci has been associated with a higher rate of renal adverse events, including a significant decrease in creatinine clearance.34
Clindamycin or linezolid plus vancomycin is used to decrease toxin production by S aureus.30
While combination therapy with vancomycin is recommended in specific clinical situations, and the combinations are synergistic in vitro, information is lacking about clinical outcomes to support their use.
Don’t use vancomycin when another drug would be better
Vancomycin continues to be the drug of choice in many circumstances, but in some instances its role is under scrutiny and another drug might be better.
Beta-lactams. In patients with infection due to methicillin-susceptible S aureus, failure rates are higher with vancomycin than with beta-lactam therapy, specifically nafcillin.35–37 Beta-lactam antibiotics are thus the drugs of choice for treating infection with beta-lactam-susceptible strains of S aureus.
Linezolid. In theory, linezolid’s ability to decrease production of the S aureus Panton-Valentine leukocidin (PVL) toxin may be an advantage over vancomycin for treating necrotizing pneumonias. For the treatment of MRSA pneumonia, however, controversy exists as to whether linezolid is superior to vancomycin. An analysis of two prospective, randomized, double-blind studies of patients with MRSA pneumonia suggested that initial therapy with linezolid was associated with better survival and clinical cure rates,38 but a subsequent meta-analysis did not substantiate this finding.39 An additional comparative study has been completed, and analysis of the results is in progress.
Daptomycin, approved for skin and soft-tissue infections and bacteremias, including those with right-sided endocarditis, is a lipopeptide antibiotic with a spectrum of action similar to that of vancomycin.40 Daptomycin is also active against many strains of vancomycin-resistant enterococci. As noted above, in the MRSA subgroup of the pivotal comparative study of treatment for S aureus bacteremia and endocarditis, the success rate for daptomycin-treated patients (44.4%) was better than that for patients treated with vancomycin plus gentamicin (32.6%), but the difference was not statistically significant.33,41
The creatine phosphokinase concentration should be monitored weekly in patients on daptomycin.42 Daptomycin is inactivated by lung surfactant and should not be used to treat pneumonia.
Other treatment options approved by the US Food and Drug Administration (FDA) for MRSA infections include tigecycline (Tygacil), quinupristin-dalfopristin (Synercid), telavancin (Vibativ), and ceftaroline (Teflaro).
Tigecycline is a glycylcycline with bacteriostatic activity against S aureus and wide distribution to the tissues.43
Quinupristin-dalfopristin, a streptogramin antibiotic, has activity against S aureus. Its use may be associated with severe myalgias, sometimes leading patients to stop taking it.
Telavancin, recently approved by the FDA, is a lipoglycopeptide antibiotic.44 It is currently approved to treat complicated skin and skin structure infections and was found to be not inferior to vancomycin. An important side effect of this agent is nephrotoxicity. A negative pregnancy test is required before using this agent in women of childbearing potential.
Ceftaroline, a fifth-generation cephalosporin active against MRSA, has been approved by the FDA for the treatment of skin and skin structure infections and community-acquired pneumonia.45
- Murray BE, Nannini EC. Glycopeptides (vancomycin and teicoplanin), streptogramins (quinupristin-dalfopristin), and lipopeptides (daptomycin). In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2010:449–468.
- Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis 2011; 52:285–292.
- Baddour LM, Epstein AE, Erickson CC, et al; American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee. Update on cardiovascular implantable electronic device infections and their management: a scientific statement from the American Heart Association. Circulation 2010; 121:458–477.
- Chang FY, Peacock JE, Musher DM, et al. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore) 2003; 82:333–339.
- Wysocki M, Delatour F, Faurisson F, et al. Continuous versus intermittent infusion of vancomycin in severe staphylococcal infections: prospective multicenter randomized study. Antimicrob Agents Chemother 2001; 45:2460–2467.
- James JK, Palmer SM, Levine DP, Rybak MJ. Comparison of conventional dosing versus continuous-infusion vancomycin therapy for patients with suspected or documented gram-positive infections. Antimicrob Agents Chemother 1996; 40:696–700.
- Rybak M, Lomaestro B, Rotschafer JC, et al. Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am J Health Syst Pharm 2009; 66:82–98.
- Wang JT, Fang CT, Chen YC, Chang SC. Necessity of a loading dose when using vancomycin in critically ill patients (letter). J Antimicrob Chemother 2001; 47:246.
- Mohammedi I, Descloux E, Argaud L, Le Scanff J, Robert D. Loading dose of vancomycin in critically ill patients: 15 mg/kg is a better choice than 500 mg. Int J Antimicrob Agents 2006; 27:259–262.
- American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 2005; 171:388–416.
- Lodise TP, Lomaestro B, Graves J, Drusano GL. Larger vancomycin doses (at least four grams per day) are associated with an increased incidence of nephrotoxicity. Antimicrob Agents Chemother 2008; 52:1330–1336.
- Hidayat LK, Hsu DI, Quist R, Shriner KA, Wong-Beringer A. High-dose vancomycin therapy for methicillin-resistant Staphylococcus aureus infections: efficacy and toxicity. Arch Intern Med 2006; 166:2138–2144.
- Centers for Disease Control and Prevention. CDC reminds clinical laboratories and healthcare infection preventionists of their role in the search and containment of vancomycin-resistant Staphylococcus aureus (VRSA), May 2010. http://emergency.cdc.gov/coca/reminders/2010/2010may06.asp. Accessed June 7, 2011.
- Sievert DM, Rudrik JT, Patel JB, McDonald LC, Wilkins MJ, Hageman JC. Vancomycin-resistant Staphylococcus aureus in the United States, 2002–2006. Clin Infect Dis 2008; 46:668–674.
- Charles PG, Ward PB, Johnson PD, Howden BP, Grayson ML. Clinical features associated with bacteremia due to heterogeneous vancomycin-intermediate Staphylococcus aureus. Clin Infect Dis 2004; 38:448–451.
- Liu C, Chambers HF. Staphylococcus aureus with heterogeneous resistance to vancomycin: epidemiology, clinical significance, and critical assessment of diagnostic methods. Antimicrob Agents Chemother 2003; 47:3040–3045.
- Sader HS, Jones RN, Rossi KL, Rybak MJ. Occurrence of vancomycin-tolerant and heterogeneous vancomycin-intermediate strains (hVISA) among Staphylococcus aureus causing bloodstream infections in nine USA hospitals. J Antimicrob Chemother 2009; 64:1024–1028.
- Pillai SK, Wennersten C, Venkataraman L, Eliopoulos GM, Moellering RC, Karchmer AW. Development of reduced vancomycin susceptibility in methicillin-susceptible Staphylococcus aureus. Clin Infect Dis 2009; 49:1169–1174.
- Wang G, Hindler JF, Ward KW, Bruckner DA. Increased vancomycin MICs for Staphylococcus aureus clinical isolates from a university hospital during a 5-year period. J Clin Microbiol 2006; 44:3883–3886.
- Steinkraus G, White R, Friedrich L. Vancomycin MIC creep in nonvancomycin-intermediate Staphylococcus aureus (VISA), vancomycin-susceptible clinical methicillin-resistant S. aureus (MRSA) blood isolates from 2001–05. J Antimicrob Chemother 2007; 60:788–794.
- Holmes RL, Jorgensen JH. Inhibitory activities of 11 antimicrobial agents and bactericidal activities of vancomycin and daptomycin against invasive methicillin-resistant Staphylococcus aureus isolates obtained from 1999 through 2006. Antimicrob Agents Chemother 2008; 52:757–760.
- Sader HS, Fey PD, Limaye AP, et al. Evaluation of vancomycin and daptomycin potency trends (MIC creep) against methicillin-resistant Staphylococcus aureus isolates collected in nine U.S. medical centers from 2002 to 2006. Antimicrob Agents Chemother 2009; 53:4127–4132.
- May J, Shannon K, King A, French G. Glycopeptide tolerance in Staphylococcus aureus. J Antimicrob Chemother 1998; 42:189–197.
- Safdar A, Rolston KV. Vancomycin tolerance, a potential mechanism for refractory gram-positive bacteremia observational study in patients with cancer. Cancer 2006; 106:1815–1820.
- Sakoulas G, Moise-Broder PA, Schentag J, Forrest A, Moellering RC, Eliopoulos GM. Relationship of MIC and bactericidal activity to efficacy of vancomycin for treatment of methicillin-resistant Staphylococcus aureus bacteremia. J Clin Microbiol 2004; 42:2398–2402.
- Tenover FC, Moellering RC. The rationale for revising the Clinical and Laboratory Standards Institute vancomycin minimal inhibitory concentration interpretive criteria for Staphylococcus aureus. Clin Infect Dis 2007; 44:1208–1215.
- Moise-Broder PA, Forrest A, Birmingham MC, Schentag JJ. Pharmacodynamics of vancomycin and other antimicrobials in patients with Staphylococcus aureus lower respiratory tract infections. Clin Pharmacokinet 2004; 43:925–942.
- Soriano A, Marco F, Martínez JA, et al. Influence of vancomycin minimum inhibitory concentration on the treatment of methicillin-resistant Staphylococcus aureus bacteremia. Clin Infect Dis 2008; 46:193–200.
- Lodise TP, Graves J, Evans A, et al. Relationship between vancomycin MIC and failure among patients with methicillin-resistant Staphylococcus aureus bacteremia treated with vancomycin. Antimicrob Agents Chemother 2008; 52:3315–3320.
- Deresinski S. Vancomycin in combination with other antibiotics for the treatment of serious methicillin-resistant Staphylococcus aureus infections. Clin Infect Dis 2009; 49:1072–1079.
- Levine DP, Fromm BS, Reddy BR. Slow response to vancomycin or vancomycin plus rifampin in methicillin-resistant Staphylococcus aureus endocarditis. Ann Intern Med 1991; 115:674–680.
- Korzeniowski O, Sande MA. Combination antimicrobial therapy for Staphylococcus aureus endocarditis in patients addicted to parenteral drugs and in nonaddicts: a prospective study. Ann Intern Med 1982; 97:496–503.
- Rehm SJ, Boucher H, Levine D, et al. Daptomycin versus vancomycin plus gentamicin for treatment of bacteraemia and endocarditis due to Staphylococcus aureus: subset analysis of patients infected with methicillin-resistant isolates. J Antimicrob Chemother 2008; 62:1413–1421.
- Cosgrove SE, Vigliani GA, Fowler VG, et al. Initial low-dose gentamicin for Staphylococcus aureus bacteremia and endocarditis is nephrotoxic. Clin Infect Dis 2009; 48:713–721.
- Small PM, Chambers HF. Vancomycin for Staphylococcus aureus endocarditis in intravenous drug users. Antimicrob Agents Chemother 1990; 34:1227–1231.
- Gentry CA, Rodvold KA, Novak RM, Hershow RC, Naderer OJ. Retrospective evaluation of therapies for Staphylococcus aureus endocarditis. Pharmacotherapy 1997; 17:990–997.
- Chang FY, Peacock JE, Musher DM, et al. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore) 2003; 82:333–339.
- Wunderink RG, Rello J, Cammarata SK, Croos-Dabrera RV, Kollef MH. Linezolid vs vancomycin: analysis of two double-blind studies of patients with methicillin-resistant Staphylococcus aureus nosocomial pneumonia. Chest 2003; 124:1789–1797.
- Kalil AC, Murthy MH, Hermsen ED, Neto FK, Sun J, Rupp ME. Linezolid versus vancomycin or teicoplanin for nosocomial pneumonia: a systematic review and meta-analysis. Crit Care Med 2010; 38:1802–1808.
- Kosmidis C, Levine DP. Daptomycin: pharmacology and clinical use. Expert Opin Pharmacother 2010; 11:615–625.
- Fowler VG, Boucher HW, Corey GR, et al; S aureus Endocarditis and Bacteremia Study Group. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N Engl J Med 2006; 355:653–665.
- Daptomycin package insert. Lexington, MA. Cubist Pharmaceuticals, Inc. November 2010. www.cubicin.com/pdf/PrescribingInformation.pdf. Accessed June 7, 2011.
- Peterson LR. A review of tigecycline—the first glycylcycline. Int J Antimicrob Agents 2008; 32(suppl 4):S215–S222.
- Saravolatz LD, Stein GE, Johnson LB. Telavancin: a novel lipoglycopeptide. Clin Infect Dis 2009; 49:1908–1914.
- Ceftaroline package insert. St. Louis, MO. Forest Pharmaceuticals. October 2010.
In the past half-century, vancomycin has gone from near-orphan status to being one of the most often used antibiotics in our formulary. The driving force for its use is clear: the evolution of Staphylococcus aureus. At first, vancomycin was used to treat infections caused by penicillin-resistant strains. However, the discovery of methicillin curbed its use for more than 2 decades.1
Then, as methicillin-resistant S aureus (MRSA) began to spread in the 1980s, the use of vancomycin began to increase, and with the rise in community-associated MRSA infections in the 1990s, it became even more widely prescribed. The recent Infectious Diseases Society of America (IDSA) guidelines for treatment of infections due to MRSA are replete with references to the use of vancomycin.2
Another factor driving the use of vancomycin is the increased prevalence of device-associated infections, many of which are caused by coagulase-negative staphylococci and other organisms that colonize the skin.3 Many of these bacteria are susceptible only to vancomycin; they may be associated with infections of vascular catheters, cardiac valves, pacemakers, implantable cardioverter-defibrillators, orthopedic implants, neurosurgical devices, and other devices.
To use vancomycin appropriately, we need to recognize the changing minimum inhibitory concentrations (MICs), to select proper doses and dosing intervals, and to know how to monitor its use. Despite more than 50 years of experience with vancomycin, we sometimes find ourselves with more questions than answers about its optimal use.
WHAT IS VANCOMYCIN?
Vancomycin is a glycopeptide antibiotic isolated from a strain of Streptomyces orientalis discovered in a soil sample from Borneo in the mid-1950s.1 It exerts its action by binding to a d-alanyl-d-alanine cell wall precursor necessary for peptidoglycan cross-linking and, therefore, for inhibiting bacterial cell wall synthesis.
Vancomycin is bactericidal against most gram-positive species, including streptococci and staphylococci, with the exception of Enterococcus species, for which it is bacteriostatic. Though it is bactericidal, it appears to kill bacteria more slowly than beta-lactam antibiotics, and therefore it may take longer to clear bacteremia.4
WHAT IS THE BEST WAY TO DOSE VANCOMYCIN?
Vancomycin is widely distributed to most tissues, with an approximate volume of distribution of 0.4 to 1 L/kg; 50% to 55% is protein-bound. Because of this large volume of distribution, vancomycin’s dosing is based on actual body weight.
Vancomycin is not metabolized and is primarily excreted unchanged in the urine via glomerular filtration. It therefore requires dosage adjustments for renal insufficiency.
Vancomycin’s molecular weight is 1,485.73 Da, making it less susceptible to removal by dialysis than smaller molecules. Dosing of vancomycin in patients on hemodialysis depends on many factors specific to the dialysis center, including but not limited to the type of filter used, the duration of filtration, and whether high-flux filtration is used.
Is continuous intravenous infusion better than standard dosing?
Giving vancomycin by continuous infusion has been suggested as a way to optimize its serum concentration and improve its clinical effectiveness.
Wysocki et al5 conducted a multicenter, prospective, randomized study comparing continuous and intermittent intravenous infusions of vancomycin (the latter every 12 hours) to treat severe hospital-acquired MRSA infections, including bloodstream infections and pneumonia. Although blood concentrations above 10 μg/mL were reached more than 30 hours faster with continuous infusions than with intermittent ones, the microbiologic and clinical outcomes were similar with either method.
James et al6 compared the pharmacodynamics of conventional dosing of vancomycin (ie, 1 g every 12 hours) and continuous infusion in 10 patients with suspected or documented gram-positive infections in a prospective, randomized, crossover study. While no adverse effects were observed, the authors also found no statistically significant difference between the treatment groups in the pharmacodynamic variables investigated, including the area under the curve (AUC) divided by the MIC (the AUC-MIC ratio).
In view of the currently available data, the guidelines for monitoring vancomycin therapy note that there does not appear to be any difference in patient outcomes with continuous infusion vs intermittent dosing.7
Should a loading dose be given?
Another proposed strategy for optimizing vancomycin’s effectiveness is to give a higher initial dose, ie, a loading dose.
Wang et al8 performed a single-center study in 28 patients who received a 25 mg/kg loading dose at a rate of 500 mg/hour. This loading dose was safe, but the authors did not evaluate its efficacy.
Mohammedi et al9 compared loading doses of 500 mg and 15 mg/kg in critically ill patients receiving vancomycin by continuous infusion. The weight-based loading dose produced higher post-dose levels and a significantly higher rate of clinical cure, but there was no significant difference in the rate of survival to discharge from the intensive care unit.
While the use of a loading dose appears to be safe and likely leads to more rapid attainment of therapeutic blood levels, we lack data on whether it improves clinical outcomes, and further study is needed to determine its role.
WHAT IS THE BEST WAY TO MONITOR VANCOMYCIN THERAPY?
Whether and how to use the serum vancomycin concentration to adjust the dosing has been a matter of debate for many years. Convincing evidence that vancomycin levels predict clinical outcomes or that measuring them prevents toxicity is lacking.7
A consensus statement from the American Society of Health-System Pharmacists, the IDSA, and the Society of Infectious Diseases Pharmacists7 contains recommendations for monitoring vancomycin therapy, based on a critical evaluation of the available scientific evidence. Their recommendations:
- Vancomycin serum concentrations should be checked to optimize therapy and used as a surrogate marker of effectiveness.
- Trough, rather than peak, levels should be monitored.
- Trough levels should be checked just before the fourth dose, when steady-state levels are likely to have been achieved. More frequent monitoring may be considered in patients with fluctuating renal function.
- Trough levels should be higher than 10 mg/L to prevent the development of resistance.
- To improve antibiotic penetration and optimize the likelihood of achieving pharmacokinetic and pharmacodynamic targets, trough levels of 15 to 20 mg/L are recommended for pathogens with a vancomycin MIC of 1 mg/L or higher and for complicated infections such as endocarditis, osteomyelitis, meningitis, and hospital-acquired pneumonia.
- For prolonged courses, it is appropriate to check vancomycin levels weekly in hemodynamically stable patients and more often in those who are not hemodynamically stable.
IS VANCOMYCIN NEPHROTOXIC?
In the 1950s, vancomycin formulations were sometimes called “Mississippi mud” because of the many impurities they contained.1 These impurities were associated with significant nephrotoxicity. Better purification methods used in the manufacture of current formulations mitigate this problem, resulting in a lower incidence of nephrotoxicity.
Over the last several years, organizations such as the American Thoracic Society and the IDSA have recommended targeting higher vancomycin trough concentrations.10 The consequent widespread use of higher doses has renewed interest in vancomycin’s potential nephrotoxicity.
Lodise et al,11 in a cohort study, examined the incidence of nephrotoxicity with higher daily doses of vancomycin (≥ 4 g/day), lower daily doses (< 4 g/day), and linezolid (Zyvox). They defined nephrotoxicity as an increase in serum creatinine of 0.5 mg/dL or a decrease in calculated creatinine clearance of 50% from baseline on 2 consecutive days.
The incidence of nephrotoxicity was significantly higher in the high-dose vancomycin group (34.6%) than in the low-dose vancomycin group (10.9%) and in the linezolid group (6.7%) (P = .001). Additional factors associated with nephrotoxicity in this study included baseline creatinine clearance less than 86.6 mL/minute, weight greater than 101.4 kg (223.5 lb), and being in an intensive care unit.
Hidayat et al12 investigated outcomes in patients with high vs low vancomycin trough levels (≥ 15 mg/L vs < 15 mg/L) in a prospective cohort study. Sixty-three patients achieved an average vancomycin trough of 15 to 20 mg/L, and of these, 11 developed nephrotoxicity, compared with no patients in the low-trough group (P = .01). Of the 11 who developed nephrotoxicity, 10 were concomitantly taking other potentially nephrotoxic agents.
Comment. The data on vancomycin and nephrotoxicity are mostly from studies that had limitations such as small numbers of patients, retrospective design, and variable definitions of nephrotoxicity. Many of the patients in these studies had additional factors contributing to nephrotoxicity, including hemodynamic instability and concomitant exposure to other nephrotoxins. Additionally, the sequence of events (nephrotoxicity leading to elevated vancomycin levels vs elevated vancomycin levels causing nephrotoxicity) is still debatable.
The incidence of nephrotoxicity associated with vancomycin therapy is difficult to determine. However, based on current information, the incidence of nephrotoxicity appears to be low when vancomycin is used as monotherapy.
IS S AUREUS BECOMING RESISTANT TO VANCOMYCIN?
An issue of increasing importance in health care settings is the emergence of vancomycin-intermediate S aureus (VISA) and vancomycin-resistant S aureus (VRSA). Eleven cases of VRSA were identified in the United States from 2002 to 2005.13 All cases of VRSA in the United States have involved the incorporation of enterococcal vanA cassette into the S aureus genome.14 While true VRSA isolates remain rare, VISA isolates are becoming more common.
Heteroresistant VISA: An emerging subpopulation of MRSA
Another population of S aureus that has emerged is heteroresistant vancomycin-intermediate S aureus (hVISA). It is defined as the presence of subpopulations of VISA within a population of MRSA at a rate of one organism per 105 to 106 organisms. With traditional testing methods, the vancomycin MIC for the entire population of the strain is within the susceptible range.15 These hVISA populations are thought to be precursors to the development of VISA.16
The resistance to vancomycin in hVISA and VISA populations is due to increased cell wall thickness, altered penicillin-binding protein profiles, and decreased cell wall autolysis.
While the true prevalence of hVISA is difficult to predict because of challenges in microbiological detection and probably varies between geographic regions and individual institutions, different studies have reported hVISA rates between 2% and 13% of all MRSA isolates.15–17
Reduced vancomycin susceptibility can develop regardless of methicillin susceptibility.18
While hVISA is not common, its presence is thought to be a predictor of failing vancomycin therapy.15
Factors associated with hVISA bacteremia include high-bacterial-load infections, treatment failure (including persistent bacteremia for more than 7 days), and initially low serum vancomycin levels.15
‘MIC creep’: Is it real?
Also worrisome, the average vancomycin MIC for S aureus has been shifting upward, based on reports from several institutions, although it is still within the susceptible range.19,20 However, this “MIC creep” likely reflects, at least in part, differences in MIC testing and varying methods used to analyze the data.19,20
Holmes and Jorgensen,21 in a single-institution study of MRSA isolates recovered from bacteremic patients from 1999 to 2006, determined that no MIC creep existed when they tested vancomycin MICs using the broth microdilution method. The authors found the MIC90 (ie, the MIC in at least 90% of the isolates) remained less than 1 mg/L during each year of the study.
Sader et al,22 in a multicenter study, evaluated 1,800 MRSA bloodstream isolates from nine hospitals across the United States from 2002 to 2006. Vancomycin MICs were again measured by broth microdilution methods. The mode MIC remained stable at 0.625 mg/L during the study period, and the authors did not detect a trend of rising MICs.
The inconsistency between reports of MIC creep at single institutions and the absence of this phenomenon in large, multicenter studies seems to imply that vancomycin MIC creep is not occurring on a grand scale.
Vancomycin tolerance
Another troubling matter with S aureus and vancomycin is the issue of tolerance. Vancomycin tolerance, defined in terms of increased minimum bactericidal concentration, represents a loss of bactericidal activity. Tolerance to vancomycin can occur even if the MIC remains in the susceptible range.23
Safdar and Rolston,24 in an observational study from a cancer center, reported that of eight cases of bacteremia that was resistant to vancomycin therapy, three were caused by S aureus.
Sakoulas et al25 found that higher levels of vancomycin bactericidal activity were associated with higher rates of clinical success; however, they found no effect on the mortality rate.
The issue of vancomycin tolerance remains controversial, and because testing for it is impractical in clinical microbiology laboratories, its implications outside the research arena are difficult to ascertain at present.
IS VANCOMYCIN STILL THE BEST DRUG FOR S AUREUS?
MIC break points have been lowered
In 2006, the Clinical Laboratories and Standards Institute lowered its break points for vancomycin MIC categories for S aureus:
- Susceptible: ≤ 2 mg/L (formerly ≤ 4 mg/L)
- Intermediate: 4–8 mg/L (formerly 8–16 mg/L)
- Resistant: ≥ 16 mg/L (formerly ≥ 32 mg/L).
The rationales for these changes were that the lower break points would better detect hVISA, and that cases have been reported of clinical treatment failure of S aureus infections in which the MICs for vancomycin were 4 mg/L.26
Since 2006, the question has been raised whether to lower the break points even further. A reason for this proposal comes from an enhanced understanding of the pharmacokinetics and pharmacodynamics of vancomycin.
The variable most closely associated with clinical response to vancomycin is the AUC-MIC ratio. An AUC-MIC ratio of 400 or higher may be associated with better outcomes in patients with serious S aureus infection. A study of 108 patients with S aureus infection of the lower respiratory tract indicated that organism eradication was more likely if the AUC-MIC ratio was 400 or greater compared with values less than 400, and this was statistically significant.27 However, in cases of S aureus infection with a vancomycin MIC of 2 mg/L or higher, this ratio may not be achievable.
A prospective study of 414 MRSA bacteremia episodes found a vancomycin MIC of 2 mg/L to be a predictor of death.28 The authors concluded that vancomycin may not be the optimal treatment for MRSA with a vancomycin MIC of 2 mg/L.28 Additional studies have also suggested a possible decrease in response to vancomycin in MRSA isolates with elevated MICs within the susceptible range.25,29
Recent guidelines from the IDSA recommend using the clinical response, regardless of the MIC, to guide antimicrobial selection for isolates with MICs in the susceptible range.2
Combination therapy with vancomycin
As vancomycin use has increased, therapeutic failures with vancomycin have become apparent. Combination therapy has been suggested as an option to increase the efficacy of vancomycin when treating complicated infections.
Rifampin plus vancomycin is controversial.30 The combination is theoretically beneficial, especially in infections associated with prosthetic devices. However, clinical studies have failed to convincingly support its use, and some have suggested that it might prolong bacteremia. In addition, it has numerous drug interactions to consider and adverse effects.31
Gentamicin plus vancomycin. The evidence supporting the use of this combination is weak at best. It appears that clinicians may have extrapolated from the success reported by Korzeniowski and Sande,32 who found that methicillin-susceptible S aureus bacteremia was cleared faster if gentamicin was added to nafcillin. A more recent study33 that compared daptomycin (Cubicin) monotherapy with combined vancomycin and gentamicin to treat MRSA bacteremia and endocarditis showed a better overall success rate with daptomycin (44% vs 32.6%), but the difference was not statistically significant.
Gentamicin has some toxicity. Even short-term use (for the first 4 days of therapy) at low doses for bacteremia and endocarditis due to staphylococci has been associated with a higher rate of renal adverse events, including a significant decrease in creatinine clearance.34
Clindamycin or linezolid plus vancomycin is used to decrease toxin production by S aureus.30
While combination therapy with vancomycin is recommended in specific clinical situations, and the combinations are synergistic in vitro, information is lacking about clinical outcomes to support their use.
Don’t use vancomycin when another drug would be better
Vancomycin continues to be the drug of choice in many circumstances, but in some instances its role is under scrutiny and another drug might be better.
Beta-lactams. In patients with infection due to methicillin-susceptible S aureus, failure rates are higher with vancomycin than with beta-lactam therapy, specifically nafcillin.35–37 Beta-lactam antibiotics are thus the drugs of choice for treating infection with beta-lactam-susceptible strains of S aureus.
Linezolid. In theory, linezolid’s ability to decrease production of the S aureus Panton-Valentine leukocidin (PVL) toxin may be an advantage over vancomycin for treating necrotizing pneumonias. For the treatment of MRSA pneumonia, however, controversy exists as to whether linezolid is superior to vancomycin. An analysis of two prospective, randomized, double-blind studies of patients with MRSA pneumonia suggested that initial therapy with linezolid was associated with better survival and clinical cure rates,38 but a subsequent meta-analysis did not substantiate this finding.39 An additional comparative study has been completed, and analysis of the results is in progress.
Daptomycin, approved for skin and soft-tissue infections and bacteremias, including those with right-sided endocarditis, is a lipopeptide antibiotic with a spectrum of action similar to that of vancomycin.40 Daptomycin is also active against many strains of vancomycin-resistant enterococci. As noted above, in the MRSA subgroup of the pivotal comparative study of treatment for S aureus bacteremia and endocarditis, the success rate for daptomycin-treated patients (44.4%) was better than that for patients treated with vancomycin plus gentamicin (32.6%), but the difference was not statistically significant.33,41
The creatine phosphokinase concentration should be monitored weekly in patients on daptomycin.42 Daptomycin is inactivated by lung surfactant and should not be used to treat pneumonia.
Other treatment options approved by the US Food and Drug Administration (FDA) for MRSA infections include tigecycline (Tygacil), quinupristin-dalfopristin (Synercid), telavancin (Vibativ), and ceftaroline (Teflaro).
Tigecycline is a glycylcycline with bacteriostatic activity against S aureus and wide distribution to the tissues.43
Quinupristin-dalfopristin, a streptogramin antibiotic, has activity against S aureus. Its use may be associated with severe myalgias, sometimes leading patients to stop taking it.
Telavancin, recently approved by the FDA, is a lipoglycopeptide antibiotic.44 It is currently approved to treat complicated skin and skin structure infections and was found to be not inferior to vancomycin. An important side effect of this agent is nephrotoxicity. A negative pregnancy test is required before using this agent in women of childbearing potential.
Ceftaroline, a fifth-generation cephalosporin active against MRSA, has been approved by the FDA for the treatment of skin and skin structure infections and community-acquired pneumonia.45
In the past half-century, vancomycin has gone from near-orphan status to being one of the most often used antibiotics in our formulary. The driving force for its use is clear: the evolution of Staphylococcus aureus. At first, vancomycin was used to treat infections caused by penicillin-resistant strains. However, the discovery of methicillin curbed its use for more than 2 decades.1
Then, as methicillin-resistant S aureus (MRSA) began to spread in the 1980s, the use of vancomycin began to increase, and with the rise in community-associated MRSA infections in the 1990s, it became even more widely prescribed. The recent Infectious Diseases Society of America (IDSA) guidelines for treatment of infections due to MRSA are replete with references to the use of vancomycin.2
Another factor driving the use of vancomycin is the increased prevalence of device-associated infections, many of which are caused by coagulase-negative staphylococci and other organisms that colonize the skin.3 Many of these bacteria are susceptible only to vancomycin; they may be associated with infections of vascular catheters, cardiac valves, pacemakers, implantable cardioverter-defibrillators, orthopedic implants, neurosurgical devices, and other devices.
To use vancomycin appropriately, we need to recognize the changing minimum inhibitory concentrations (MICs), to select proper doses and dosing intervals, and to know how to monitor its use. Despite more than 50 years of experience with vancomycin, we sometimes find ourselves with more questions than answers about its optimal use.
WHAT IS VANCOMYCIN?
Vancomycin is a glycopeptide antibiotic isolated from a strain of Streptomyces orientalis discovered in a soil sample from Borneo in the mid-1950s.1 It exerts its action by binding to a d-alanyl-d-alanine cell wall precursor necessary for peptidoglycan cross-linking and, therefore, for inhibiting bacterial cell wall synthesis.
Vancomycin is bactericidal against most gram-positive species, including streptococci and staphylococci, with the exception of Enterococcus species, for which it is bacteriostatic. Though it is bactericidal, it appears to kill bacteria more slowly than beta-lactam antibiotics, and therefore it may take longer to clear bacteremia.4
WHAT IS THE BEST WAY TO DOSE VANCOMYCIN?
Vancomycin is widely distributed to most tissues, with an approximate volume of distribution of 0.4 to 1 L/kg; 50% to 55% is protein-bound. Because of this large volume of distribution, vancomycin’s dosing is based on actual body weight.
Vancomycin is not metabolized and is primarily excreted unchanged in the urine via glomerular filtration. It therefore requires dosage adjustments for renal insufficiency.
Vancomycin’s molecular weight is 1,485.73 Da, making it less susceptible to removal by dialysis than smaller molecules. Dosing of vancomycin in patients on hemodialysis depends on many factors specific to the dialysis center, including but not limited to the type of filter used, the duration of filtration, and whether high-flux filtration is used.
Is continuous intravenous infusion better than standard dosing?
Giving vancomycin by continuous infusion has been suggested as a way to optimize its serum concentration and improve its clinical effectiveness.
Wysocki et al5 conducted a multicenter, prospective, randomized study comparing continuous and intermittent intravenous infusions of vancomycin (the latter every 12 hours) to treat severe hospital-acquired MRSA infections, including bloodstream infections and pneumonia. Although blood concentrations above 10 μg/mL were reached more than 30 hours faster with continuous infusions than with intermittent ones, the microbiologic and clinical outcomes were similar with either method.
James et al6 compared the pharmacodynamics of conventional dosing of vancomycin (ie, 1 g every 12 hours) and continuous infusion in 10 patients with suspected or documented gram-positive infections in a prospective, randomized, crossover study. While no adverse effects were observed, the authors also found no statistically significant difference between the treatment groups in the pharmacodynamic variables investigated, including the area under the curve (AUC) divided by the MIC (the AUC-MIC ratio).
In view of the currently available data, the guidelines for monitoring vancomycin therapy note that there does not appear to be any difference in patient outcomes with continuous infusion vs intermittent dosing.7
Should a loading dose be given?
Another proposed strategy for optimizing vancomycin’s effectiveness is to give a higher initial dose, ie, a loading dose.
Wang et al8 performed a single-center study in 28 patients who received a 25 mg/kg loading dose at a rate of 500 mg/hour. This loading dose was safe, but the authors did not evaluate its efficacy.
Mohammedi et al9 compared loading doses of 500 mg and 15 mg/kg in critically ill patients receiving vancomycin by continuous infusion. The weight-based loading dose produced higher post-dose levels and a significantly higher rate of clinical cure, but there was no significant difference in the rate of survival to discharge from the intensive care unit.
While the use of a loading dose appears to be safe and likely leads to more rapid attainment of therapeutic blood levels, we lack data on whether it improves clinical outcomes, and further study is needed to determine its role.
WHAT IS THE BEST WAY TO MONITOR VANCOMYCIN THERAPY?
Whether and how to use the serum vancomycin concentration to adjust the dosing has been a matter of debate for many years. Convincing evidence that vancomycin levels predict clinical outcomes or that measuring them prevents toxicity is lacking.7
A consensus statement from the American Society of Health-System Pharmacists, the IDSA, and the Society of Infectious Diseases Pharmacists7 contains recommendations for monitoring vancomycin therapy, based on a critical evaluation of the available scientific evidence. Their recommendations:
- Vancomycin serum concentrations should be checked to optimize therapy and used as a surrogate marker of effectiveness.
- Trough, rather than peak, levels should be monitored.
- Trough levels should be checked just before the fourth dose, when steady-state levels are likely to have been achieved. More frequent monitoring may be considered in patients with fluctuating renal function.
- Trough levels should be higher than 10 mg/L to prevent the development of resistance.
- To improve antibiotic penetration and optimize the likelihood of achieving pharmacokinetic and pharmacodynamic targets, trough levels of 15 to 20 mg/L are recommended for pathogens with a vancomycin MIC of 1 mg/L or higher and for complicated infections such as endocarditis, osteomyelitis, meningitis, and hospital-acquired pneumonia.
- For prolonged courses, it is appropriate to check vancomycin levels weekly in hemodynamically stable patients and more often in those who are not hemodynamically stable.
IS VANCOMYCIN NEPHROTOXIC?
In the 1950s, vancomycin formulations were sometimes called “Mississippi mud” because of the many impurities they contained.1 These impurities were associated with significant nephrotoxicity. Better purification methods used in the manufacture of current formulations mitigate this problem, resulting in a lower incidence of nephrotoxicity.
Over the last several years, organizations such as the American Thoracic Society and the IDSA have recommended targeting higher vancomycin trough concentrations.10 The consequent widespread use of higher doses has renewed interest in vancomycin’s potential nephrotoxicity.
Lodise et al,11 in a cohort study, examined the incidence of nephrotoxicity with higher daily doses of vancomycin (≥ 4 g/day), lower daily doses (< 4 g/day), and linezolid (Zyvox). They defined nephrotoxicity as an increase in serum creatinine of 0.5 mg/dL or a decrease in calculated creatinine clearance of 50% from baseline on 2 consecutive days.
The incidence of nephrotoxicity was significantly higher in the high-dose vancomycin group (34.6%) than in the low-dose vancomycin group (10.9%) and in the linezolid group (6.7%) (P = .001). Additional factors associated with nephrotoxicity in this study included baseline creatinine clearance less than 86.6 mL/minute, weight greater than 101.4 kg (223.5 lb), and being in an intensive care unit.
Hidayat et al12 investigated outcomes in patients with high vs low vancomycin trough levels (≥ 15 mg/L vs < 15 mg/L) in a prospective cohort study. Sixty-three patients achieved an average vancomycin trough of 15 to 20 mg/L, and of these, 11 developed nephrotoxicity, compared with no patients in the low-trough group (P = .01). Of the 11 who developed nephrotoxicity, 10 were concomitantly taking other potentially nephrotoxic agents.
Comment. The data on vancomycin and nephrotoxicity are mostly from studies that had limitations such as small numbers of patients, retrospective design, and variable definitions of nephrotoxicity. Many of the patients in these studies had additional factors contributing to nephrotoxicity, including hemodynamic instability and concomitant exposure to other nephrotoxins. Additionally, the sequence of events (nephrotoxicity leading to elevated vancomycin levels vs elevated vancomycin levels causing nephrotoxicity) is still debatable.
The incidence of nephrotoxicity associated with vancomycin therapy is difficult to determine. However, based on current information, the incidence of nephrotoxicity appears to be low when vancomycin is used as monotherapy.
IS S AUREUS BECOMING RESISTANT TO VANCOMYCIN?
An issue of increasing importance in health care settings is the emergence of vancomycin-intermediate S aureus (VISA) and vancomycin-resistant S aureus (VRSA). Eleven cases of VRSA were identified in the United States from 2002 to 2005.13 All cases of VRSA in the United States have involved the incorporation of enterococcal vanA cassette into the S aureus genome.14 While true VRSA isolates remain rare, VISA isolates are becoming more common.
Heteroresistant VISA: An emerging subpopulation of MRSA
Another population of S aureus that has emerged is heteroresistant vancomycin-intermediate S aureus (hVISA). It is defined as the presence of subpopulations of VISA within a population of MRSA at a rate of one organism per 105 to 106 organisms. With traditional testing methods, the vancomycin MIC for the entire population of the strain is within the susceptible range.15 These hVISA populations are thought to be precursors to the development of VISA.16
The resistance to vancomycin in hVISA and VISA populations is due to increased cell wall thickness, altered penicillin-binding protein profiles, and decreased cell wall autolysis.
While the true prevalence of hVISA is difficult to predict because of challenges in microbiological detection and probably varies between geographic regions and individual institutions, different studies have reported hVISA rates between 2% and 13% of all MRSA isolates.15–17
Reduced vancomycin susceptibility can develop regardless of methicillin susceptibility.18
While hVISA is not common, its presence is thought to be a predictor of failing vancomycin therapy.15
Factors associated with hVISA bacteremia include high-bacterial-load infections, treatment failure (including persistent bacteremia for more than 7 days), and initially low serum vancomycin levels.15
‘MIC creep’: Is it real?
Also worrisome, the average vancomycin MIC for S aureus has been shifting upward, based on reports from several institutions, although it is still within the susceptible range.19,20 However, this “MIC creep” likely reflects, at least in part, differences in MIC testing and varying methods used to analyze the data.19,20
Holmes and Jorgensen,21 in a single-institution study of MRSA isolates recovered from bacteremic patients from 1999 to 2006, determined that no MIC creep existed when they tested vancomycin MICs using the broth microdilution method. The authors found the MIC90 (ie, the MIC in at least 90% of the isolates) remained less than 1 mg/L during each year of the study.
Sader et al,22 in a multicenter study, evaluated 1,800 MRSA bloodstream isolates from nine hospitals across the United States from 2002 to 2006. Vancomycin MICs were again measured by broth microdilution methods. The mode MIC remained stable at 0.625 mg/L during the study period, and the authors did not detect a trend of rising MICs.
The inconsistency between reports of MIC creep at single institutions and the absence of this phenomenon in large, multicenter studies seems to imply that vancomycin MIC creep is not occurring on a grand scale.
Vancomycin tolerance
Another troubling matter with S aureus and vancomycin is the issue of tolerance. Vancomycin tolerance, defined in terms of increased minimum bactericidal concentration, represents a loss of bactericidal activity. Tolerance to vancomycin can occur even if the MIC remains in the susceptible range.23
Safdar and Rolston,24 in an observational study from a cancer center, reported that of eight cases of bacteremia that was resistant to vancomycin therapy, three were caused by S aureus.
Sakoulas et al25 found that higher levels of vancomycin bactericidal activity were associated with higher rates of clinical success; however, they found no effect on the mortality rate.
The issue of vancomycin tolerance remains controversial, and because testing for it is impractical in clinical microbiology laboratories, its implications outside the research arena are difficult to ascertain at present.
IS VANCOMYCIN STILL THE BEST DRUG FOR S AUREUS?
MIC break points have been lowered
In 2006, the Clinical Laboratories and Standards Institute lowered its break points for vancomycin MIC categories for S aureus:
- Susceptible: ≤ 2 mg/L (formerly ≤ 4 mg/L)
- Intermediate: 4–8 mg/L (formerly 8–16 mg/L)
- Resistant: ≥ 16 mg/L (formerly ≥ 32 mg/L).
The rationales for these changes were that the lower break points would better detect hVISA, and that cases have been reported of clinical treatment failure of S aureus infections in which the MICs for vancomycin were 4 mg/L.26
Since 2006, the question has been raised whether to lower the break points even further. A reason for this proposal comes from an enhanced understanding of the pharmacokinetics and pharmacodynamics of vancomycin.
The variable most closely associated with clinical response to vancomycin is the AUC-MIC ratio. An AUC-MIC ratio of 400 or higher may be associated with better outcomes in patients with serious S aureus infection. A study of 108 patients with S aureus infection of the lower respiratory tract indicated that organism eradication was more likely if the AUC-MIC ratio was 400 or greater compared with values less than 400, and this was statistically significant.27 However, in cases of S aureus infection with a vancomycin MIC of 2 mg/L or higher, this ratio may not be achievable.
A prospective study of 414 MRSA bacteremia episodes found a vancomycin MIC of 2 mg/L to be a predictor of death.28 The authors concluded that vancomycin may not be the optimal treatment for MRSA with a vancomycin MIC of 2 mg/L.28 Additional studies have also suggested a possible decrease in response to vancomycin in MRSA isolates with elevated MICs within the susceptible range.25,29
Recent guidelines from the IDSA recommend using the clinical response, regardless of the MIC, to guide antimicrobial selection for isolates with MICs in the susceptible range.2
Combination therapy with vancomycin
As vancomycin use has increased, therapeutic failures with vancomycin have become apparent. Combination therapy has been suggested as an option to increase the efficacy of vancomycin when treating complicated infections.
Rifampin plus vancomycin is controversial.30 The combination is theoretically beneficial, especially in infections associated with prosthetic devices. However, clinical studies have failed to convincingly support its use, and some have suggested that it might prolong bacteremia. In addition, it has numerous drug interactions to consider and adverse effects.31
Gentamicin plus vancomycin. The evidence supporting the use of this combination is weak at best. It appears that clinicians may have extrapolated from the success reported by Korzeniowski and Sande,32 who found that methicillin-susceptible S aureus bacteremia was cleared faster if gentamicin was added to nafcillin. A more recent study33 that compared daptomycin (Cubicin) monotherapy with combined vancomycin and gentamicin to treat MRSA bacteremia and endocarditis showed a better overall success rate with daptomycin (44% vs 32.6%), but the difference was not statistically significant.
Gentamicin has some toxicity. Even short-term use (for the first 4 days of therapy) at low doses for bacteremia and endocarditis due to staphylococci has been associated with a higher rate of renal adverse events, including a significant decrease in creatinine clearance.34
Clindamycin or linezolid plus vancomycin is used to decrease toxin production by S aureus.30
While combination therapy with vancomycin is recommended in specific clinical situations, and the combinations are synergistic in vitro, information is lacking about clinical outcomes to support their use.
Don’t use vancomycin when another drug would be better
Vancomycin continues to be the drug of choice in many circumstances, but in some instances its role is under scrutiny and another drug might be better.
Beta-lactams. In patients with infection due to methicillin-susceptible S aureus, failure rates are higher with vancomycin than with beta-lactam therapy, specifically nafcillin.35–37 Beta-lactam antibiotics are thus the drugs of choice for treating infection with beta-lactam-susceptible strains of S aureus.
Linezolid. In theory, linezolid’s ability to decrease production of the S aureus Panton-Valentine leukocidin (PVL) toxin may be an advantage over vancomycin for treating necrotizing pneumonias. For the treatment of MRSA pneumonia, however, controversy exists as to whether linezolid is superior to vancomycin. An analysis of two prospective, randomized, double-blind studies of patients with MRSA pneumonia suggested that initial therapy with linezolid was associated with better survival and clinical cure rates,38 but a subsequent meta-analysis did not substantiate this finding.39 An additional comparative study has been completed, and analysis of the results is in progress.
Daptomycin, approved for skin and soft-tissue infections and bacteremias, including those with right-sided endocarditis, is a lipopeptide antibiotic with a spectrum of action similar to that of vancomycin.40 Daptomycin is also active against many strains of vancomycin-resistant enterococci. As noted above, in the MRSA subgroup of the pivotal comparative study of treatment for S aureus bacteremia and endocarditis, the success rate for daptomycin-treated patients (44.4%) was better than that for patients treated with vancomycin plus gentamicin (32.6%), but the difference was not statistically significant.33,41
The creatine phosphokinase concentration should be monitored weekly in patients on daptomycin.42 Daptomycin is inactivated by lung surfactant and should not be used to treat pneumonia.
Other treatment options approved by the US Food and Drug Administration (FDA) for MRSA infections include tigecycline (Tygacil), quinupristin-dalfopristin (Synercid), telavancin (Vibativ), and ceftaroline (Teflaro).
Tigecycline is a glycylcycline with bacteriostatic activity against S aureus and wide distribution to the tissues.43
Quinupristin-dalfopristin, a streptogramin antibiotic, has activity against S aureus. Its use may be associated with severe myalgias, sometimes leading patients to stop taking it.
Telavancin, recently approved by the FDA, is a lipoglycopeptide antibiotic.44 It is currently approved to treat complicated skin and skin structure infections and was found to be not inferior to vancomycin. An important side effect of this agent is nephrotoxicity. A negative pregnancy test is required before using this agent in women of childbearing potential.
Ceftaroline, a fifth-generation cephalosporin active against MRSA, has been approved by the FDA for the treatment of skin and skin structure infections and community-acquired pneumonia.45
- Murray BE, Nannini EC. Glycopeptides (vancomycin and teicoplanin), streptogramins (quinupristin-dalfopristin), and lipopeptides (daptomycin). In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2010:449–468.
- Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis 2011; 52:285–292.
- Baddour LM, Epstein AE, Erickson CC, et al; American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee. Update on cardiovascular implantable electronic device infections and their management: a scientific statement from the American Heart Association. Circulation 2010; 121:458–477.
- Chang FY, Peacock JE, Musher DM, et al. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore) 2003; 82:333–339.
- Wysocki M, Delatour F, Faurisson F, et al. Continuous versus intermittent infusion of vancomycin in severe staphylococcal infections: prospective multicenter randomized study. Antimicrob Agents Chemother 2001; 45:2460–2467.
- James JK, Palmer SM, Levine DP, Rybak MJ. Comparison of conventional dosing versus continuous-infusion vancomycin therapy for patients with suspected or documented gram-positive infections. Antimicrob Agents Chemother 1996; 40:696–700.
- Rybak M, Lomaestro B, Rotschafer JC, et al. Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am J Health Syst Pharm 2009; 66:82–98.
- Wang JT, Fang CT, Chen YC, Chang SC. Necessity of a loading dose when using vancomycin in critically ill patients (letter). J Antimicrob Chemother 2001; 47:246.
- Mohammedi I, Descloux E, Argaud L, Le Scanff J, Robert D. Loading dose of vancomycin in critically ill patients: 15 mg/kg is a better choice than 500 mg. Int J Antimicrob Agents 2006; 27:259–262.
- American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 2005; 171:388–416.
- Lodise TP, Lomaestro B, Graves J, Drusano GL. Larger vancomycin doses (at least four grams per day) are associated with an increased incidence of nephrotoxicity. Antimicrob Agents Chemother 2008; 52:1330–1336.
- Hidayat LK, Hsu DI, Quist R, Shriner KA, Wong-Beringer A. High-dose vancomycin therapy for methicillin-resistant Staphylococcus aureus infections: efficacy and toxicity. Arch Intern Med 2006; 166:2138–2144.
- Centers for Disease Control and Prevention. CDC reminds clinical laboratories and healthcare infection preventionists of their role in the search and containment of vancomycin-resistant Staphylococcus aureus (VRSA), May 2010. http://emergency.cdc.gov/coca/reminders/2010/2010may06.asp. Accessed June 7, 2011.
- Sievert DM, Rudrik JT, Patel JB, McDonald LC, Wilkins MJ, Hageman JC. Vancomycin-resistant Staphylococcus aureus in the United States, 2002–2006. Clin Infect Dis 2008; 46:668–674.
- Charles PG, Ward PB, Johnson PD, Howden BP, Grayson ML. Clinical features associated with bacteremia due to heterogeneous vancomycin-intermediate Staphylococcus aureus. Clin Infect Dis 2004; 38:448–451.
- Liu C, Chambers HF. Staphylococcus aureus with heterogeneous resistance to vancomycin: epidemiology, clinical significance, and critical assessment of diagnostic methods. Antimicrob Agents Chemother 2003; 47:3040–3045.
- Sader HS, Jones RN, Rossi KL, Rybak MJ. Occurrence of vancomycin-tolerant and heterogeneous vancomycin-intermediate strains (hVISA) among Staphylococcus aureus causing bloodstream infections in nine USA hospitals. J Antimicrob Chemother 2009; 64:1024–1028.
- Pillai SK, Wennersten C, Venkataraman L, Eliopoulos GM, Moellering RC, Karchmer AW. Development of reduced vancomycin susceptibility in methicillin-susceptible Staphylococcus aureus. Clin Infect Dis 2009; 49:1169–1174.
- Wang G, Hindler JF, Ward KW, Bruckner DA. Increased vancomycin MICs for Staphylococcus aureus clinical isolates from a university hospital during a 5-year period. J Clin Microbiol 2006; 44:3883–3886.
- Steinkraus G, White R, Friedrich L. Vancomycin MIC creep in nonvancomycin-intermediate Staphylococcus aureus (VISA), vancomycin-susceptible clinical methicillin-resistant S. aureus (MRSA) blood isolates from 2001–05. J Antimicrob Chemother 2007; 60:788–794.
- Holmes RL, Jorgensen JH. Inhibitory activities of 11 antimicrobial agents and bactericidal activities of vancomycin and daptomycin against invasive methicillin-resistant Staphylococcus aureus isolates obtained from 1999 through 2006. Antimicrob Agents Chemother 2008; 52:757–760.
- Sader HS, Fey PD, Limaye AP, et al. Evaluation of vancomycin and daptomycin potency trends (MIC creep) against methicillin-resistant Staphylococcus aureus isolates collected in nine U.S. medical centers from 2002 to 2006. Antimicrob Agents Chemother 2009; 53:4127–4132.
- May J, Shannon K, King A, French G. Glycopeptide tolerance in Staphylococcus aureus. J Antimicrob Chemother 1998; 42:189–197.
- Safdar A, Rolston KV. Vancomycin tolerance, a potential mechanism for refractory gram-positive bacteremia observational study in patients with cancer. Cancer 2006; 106:1815–1820.
- Sakoulas G, Moise-Broder PA, Schentag J, Forrest A, Moellering RC, Eliopoulos GM. Relationship of MIC and bactericidal activity to efficacy of vancomycin for treatment of methicillin-resistant Staphylococcus aureus bacteremia. J Clin Microbiol 2004; 42:2398–2402.
- Tenover FC, Moellering RC. The rationale for revising the Clinical and Laboratory Standards Institute vancomycin minimal inhibitory concentration interpretive criteria for Staphylococcus aureus. Clin Infect Dis 2007; 44:1208–1215.
- Moise-Broder PA, Forrest A, Birmingham MC, Schentag JJ. Pharmacodynamics of vancomycin and other antimicrobials in patients with Staphylococcus aureus lower respiratory tract infections. Clin Pharmacokinet 2004; 43:925–942.
- Soriano A, Marco F, Martínez JA, et al. Influence of vancomycin minimum inhibitory concentration on the treatment of methicillin-resistant Staphylococcus aureus bacteremia. Clin Infect Dis 2008; 46:193–200.
- Lodise TP, Graves J, Evans A, et al. Relationship between vancomycin MIC and failure among patients with methicillin-resistant Staphylococcus aureus bacteremia treated with vancomycin. Antimicrob Agents Chemother 2008; 52:3315–3320.
- Deresinski S. Vancomycin in combination with other antibiotics for the treatment of serious methicillin-resistant Staphylococcus aureus infections. Clin Infect Dis 2009; 49:1072–1079.
- Levine DP, Fromm BS, Reddy BR. Slow response to vancomycin or vancomycin plus rifampin in methicillin-resistant Staphylococcus aureus endocarditis. Ann Intern Med 1991; 115:674–680.
- Korzeniowski O, Sande MA. Combination antimicrobial therapy for Staphylococcus aureus endocarditis in patients addicted to parenteral drugs and in nonaddicts: a prospective study. Ann Intern Med 1982; 97:496–503.
- Rehm SJ, Boucher H, Levine D, et al. Daptomycin versus vancomycin plus gentamicin for treatment of bacteraemia and endocarditis due to Staphylococcus aureus: subset analysis of patients infected with methicillin-resistant isolates. J Antimicrob Chemother 2008; 62:1413–1421.
- Cosgrove SE, Vigliani GA, Fowler VG, et al. Initial low-dose gentamicin for Staphylococcus aureus bacteremia and endocarditis is nephrotoxic. Clin Infect Dis 2009; 48:713–721.
- Small PM, Chambers HF. Vancomycin for Staphylococcus aureus endocarditis in intravenous drug users. Antimicrob Agents Chemother 1990; 34:1227–1231.
- Gentry CA, Rodvold KA, Novak RM, Hershow RC, Naderer OJ. Retrospective evaluation of therapies for Staphylococcus aureus endocarditis. Pharmacotherapy 1997; 17:990–997.
- Chang FY, Peacock JE, Musher DM, et al. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore) 2003; 82:333–339.
- Wunderink RG, Rello J, Cammarata SK, Croos-Dabrera RV, Kollef MH. Linezolid vs vancomycin: analysis of two double-blind studies of patients with methicillin-resistant Staphylococcus aureus nosocomial pneumonia. Chest 2003; 124:1789–1797.
- Kalil AC, Murthy MH, Hermsen ED, Neto FK, Sun J, Rupp ME. Linezolid versus vancomycin or teicoplanin for nosocomial pneumonia: a systematic review and meta-analysis. Crit Care Med 2010; 38:1802–1808.
- Kosmidis C, Levine DP. Daptomycin: pharmacology and clinical use. Expert Opin Pharmacother 2010; 11:615–625.
- Fowler VG, Boucher HW, Corey GR, et al; S aureus Endocarditis and Bacteremia Study Group. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N Engl J Med 2006; 355:653–665.
- Daptomycin package insert. Lexington, MA. Cubist Pharmaceuticals, Inc. November 2010. www.cubicin.com/pdf/PrescribingInformation.pdf. Accessed June 7, 2011.
- Peterson LR. A review of tigecycline—the first glycylcycline. Int J Antimicrob Agents 2008; 32(suppl 4):S215–S222.
- Saravolatz LD, Stein GE, Johnson LB. Telavancin: a novel lipoglycopeptide. Clin Infect Dis 2009; 49:1908–1914.
- Ceftaroline package insert. St. Louis, MO. Forest Pharmaceuticals. October 2010.
- Murray BE, Nannini EC. Glycopeptides (vancomycin and teicoplanin), streptogramins (quinupristin-dalfopristin), and lipopeptides (daptomycin). In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2010:449–468.
- Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis 2011; 52:285–292.
- Baddour LM, Epstein AE, Erickson CC, et al; American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee. Update on cardiovascular implantable electronic device infections and their management: a scientific statement from the American Heart Association. Circulation 2010; 121:458–477.
- Chang FY, Peacock JE, Musher DM, et al. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore) 2003; 82:333–339.
- Wysocki M, Delatour F, Faurisson F, et al. Continuous versus intermittent infusion of vancomycin in severe staphylococcal infections: prospective multicenter randomized study. Antimicrob Agents Chemother 2001; 45:2460–2467.
- James JK, Palmer SM, Levine DP, Rybak MJ. Comparison of conventional dosing versus continuous-infusion vancomycin therapy for patients with suspected or documented gram-positive infections. Antimicrob Agents Chemother 1996; 40:696–700.
- Rybak M, Lomaestro B, Rotschafer JC, et al. Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am J Health Syst Pharm 2009; 66:82–98.
- Wang JT, Fang CT, Chen YC, Chang SC. Necessity of a loading dose when using vancomycin in critically ill patients (letter). J Antimicrob Chemother 2001; 47:246.
- Mohammedi I, Descloux E, Argaud L, Le Scanff J, Robert D. Loading dose of vancomycin in critically ill patients: 15 mg/kg is a better choice than 500 mg. Int J Antimicrob Agents 2006; 27:259–262.
- American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 2005; 171:388–416.
- Lodise TP, Lomaestro B, Graves J, Drusano GL. Larger vancomycin doses (at least four grams per day) are associated with an increased incidence of nephrotoxicity. Antimicrob Agents Chemother 2008; 52:1330–1336.
- Hidayat LK, Hsu DI, Quist R, Shriner KA, Wong-Beringer A. High-dose vancomycin therapy for methicillin-resistant Staphylococcus aureus infections: efficacy and toxicity. Arch Intern Med 2006; 166:2138–2144.
- Centers for Disease Control and Prevention. CDC reminds clinical laboratories and healthcare infection preventionists of their role in the search and containment of vancomycin-resistant Staphylococcus aureus (VRSA), May 2010. http://emergency.cdc.gov/coca/reminders/2010/2010may06.asp. Accessed June 7, 2011.
- Sievert DM, Rudrik JT, Patel JB, McDonald LC, Wilkins MJ, Hageman JC. Vancomycin-resistant Staphylococcus aureus in the United States, 2002–2006. Clin Infect Dis 2008; 46:668–674.
- Charles PG, Ward PB, Johnson PD, Howden BP, Grayson ML. Clinical features associated with bacteremia due to heterogeneous vancomycin-intermediate Staphylococcus aureus. Clin Infect Dis 2004; 38:448–451.
- Liu C, Chambers HF. Staphylococcus aureus with heterogeneous resistance to vancomycin: epidemiology, clinical significance, and critical assessment of diagnostic methods. Antimicrob Agents Chemother 2003; 47:3040–3045.
- Sader HS, Jones RN, Rossi KL, Rybak MJ. Occurrence of vancomycin-tolerant and heterogeneous vancomycin-intermediate strains (hVISA) among Staphylococcus aureus causing bloodstream infections in nine USA hospitals. J Antimicrob Chemother 2009; 64:1024–1028.
- Pillai SK, Wennersten C, Venkataraman L, Eliopoulos GM, Moellering RC, Karchmer AW. Development of reduced vancomycin susceptibility in methicillin-susceptible Staphylococcus aureus. Clin Infect Dis 2009; 49:1169–1174.
- Wang G, Hindler JF, Ward KW, Bruckner DA. Increased vancomycin MICs for Staphylococcus aureus clinical isolates from a university hospital during a 5-year period. J Clin Microbiol 2006; 44:3883–3886.
- Steinkraus G, White R, Friedrich L. Vancomycin MIC creep in nonvancomycin-intermediate Staphylococcus aureus (VISA), vancomycin-susceptible clinical methicillin-resistant S. aureus (MRSA) blood isolates from 2001–05. J Antimicrob Chemother 2007; 60:788–794.
- Holmes RL, Jorgensen JH. Inhibitory activities of 11 antimicrobial agents and bactericidal activities of vancomycin and daptomycin against invasive methicillin-resistant Staphylococcus aureus isolates obtained from 1999 through 2006. Antimicrob Agents Chemother 2008; 52:757–760.
- Sader HS, Fey PD, Limaye AP, et al. Evaluation of vancomycin and daptomycin potency trends (MIC creep) against methicillin-resistant Staphylococcus aureus isolates collected in nine U.S. medical centers from 2002 to 2006. Antimicrob Agents Chemother 2009; 53:4127–4132.
- May J, Shannon K, King A, French G. Glycopeptide tolerance in Staphylococcus aureus. J Antimicrob Chemother 1998; 42:189–197.
- Safdar A, Rolston KV. Vancomycin tolerance, a potential mechanism for refractory gram-positive bacteremia observational study in patients with cancer. Cancer 2006; 106:1815–1820.
- Sakoulas G, Moise-Broder PA, Schentag J, Forrest A, Moellering RC, Eliopoulos GM. Relationship of MIC and bactericidal activity to efficacy of vancomycin for treatment of methicillin-resistant Staphylococcus aureus bacteremia. J Clin Microbiol 2004; 42:2398–2402.
- Tenover FC, Moellering RC. The rationale for revising the Clinical and Laboratory Standards Institute vancomycin minimal inhibitory concentration interpretive criteria for Staphylococcus aureus. Clin Infect Dis 2007; 44:1208–1215.
- Moise-Broder PA, Forrest A, Birmingham MC, Schentag JJ. Pharmacodynamics of vancomycin and other antimicrobials in patients with Staphylococcus aureus lower respiratory tract infections. Clin Pharmacokinet 2004; 43:925–942.
- Soriano A, Marco F, Martínez JA, et al. Influence of vancomycin minimum inhibitory concentration on the treatment of methicillin-resistant Staphylococcus aureus bacteremia. Clin Infect Dis 2008; 46:193–200.
- Lodise TP, Graves J, Evans A, et al. Relationship between vancomycin MIC and failure among patients with methicillin-resistant Staphylococcus aureus bacteremia treated with vancomycin. Antimicrob Agents Chemother 2008; 52:3315–3320.
- Deresinski S. Vancomycin in combination with other antibiotics for the treatment of serious methicillin-resistant Staphylococcus aureus infections. Clin Infect Dis 2009; 49:1072–1079.
- Levine DP, Fromm BS, Reddy BR. Slow response to vancomycin or vancomycin plus rifampin in methicillin-resistant Staphylococcus aureus endocarditis. Ann Intern Med 1991; 115:674–680.
- Korzeniowski O, Sande MA. Combination antimicrobial therapy for Staphylococcus aureus endocarditis in patients addicted to parenteral drugs and in nonaddicts: a prospective study. Ann Intern Med 1982; 97:496–503.
- Rehm SJ, Boucher H, Levine D, et al. Daptomycin versus vancomycin plus gentamicin for treatment of bacteraemia and endocarditis due to Staphylococcus aureus: subset analysis of patients infected with methicillin-resistant isolates. J Antimicrob Chemother 2008; 62:1413–1421.
- Cosgrove SE, Vigliani GA, Fowler VG, et al. Initial low-dose gentamicin for Staphylococcus aureus bacteremia and endocarditis is nephrotoxic. Clin Infect Dis 2009; 48:713–721.
- Small PM, Chambers HF. Vancomycin for Staphylococcus aureus endocarditis in intravenous drug users. Antimicrob Agents Chemother 1990; 34:1227–1231.
- Gentry CA, Rodvold KA, Novak RM, Hershow RC, Naderer OJ. Retrospective evaluation of therapies for Staphylococcus aureus endocarditis. Pharmacotherapy 1997; 17:990–997.
- Chang FY, Peacock JE, Musher DM, et al. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore) 2003; 82:333–339.
- Wunderink RG, Rello J, Cammarata SK, Croos-Dabrera RV, Kollef MH. Linezolid vs vancomycin: analysis of two double-blind studies of patients with methicillin-resistant Staphylococcus aureus nosocomial pneumonia. Chest 2003; 124:1789–1797.
- Kalil AC, Murthy MH, Hermsen ED, Neto FK, Sun J, Rupp ME. Linezolid versus vancomycin or teicoplanin for nosocomial pneumonia: a systematic review and meta-analysis. Crit Care Med 2010; 38:1802–1808.
- Kosmidis C, Levine DP. Daptomycin: pharmacology and clinical use. Expert Opin Pharmacother 2010; 11:615–625.
- Fowler VG, Boucher HW, Corey GR, et al; S aureus Endocarditis and Bacteremia Study Group. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N Engl J Med 2006; 355:653–665.
- Daptomycin package insert. Lexington, MA. Cubist Pharmaceuticals, Inc. November 2010. www.cubicin.com/pdf/PrescribingInformation.pdf. Accessed June 7, 2011.
- Peterson LR. A review of tigecycline—the first glycylcycline. Int J Antimicrob Agents 2008; 32(suppl 4):S215–S222.
- Saravolatz LD, Stein GE, Johnson LB. Telavancin: a novel lipoglycopeptide. Clin Infect Dis 2009; 49:1908–1914.
- Ceftaroline package insert. St. Louis, MO. Forest Pharmaceuticals. October 2010.
KEY POINTS
- Giving vancomycin by continuous infusion appears to offer no advantage over giving it every 12 hours.
- Therapeutic blood levels can be reached more quickly if a loading dose is given, but whether this offers a clinical advantage is unclear.
- The trough vancomycin serum concentration should be greater than 10 mg/L to prevent the development of resistance, and trough levels of 15 to 20 mg/L are recommended if the minimum inhibitory concentration (MIC) is 1 mg/L or higher.
- Whether S aureus is becoming resistant to vancomycin is not clear.
- The variable most closely associated with clinical response to vancomycin is the area under the curve (AUC) divided by the MIC (the AUC-MIC ratio), which should be greater than 400.