User login
Diabetes management: Beyond hemoglobin A1c
When scientists discovered the band of hemoglobin A1c during electrophoresis in the 1950s and 1960s and discerned it was elevated in patients with diabetes, little did they know the important role it would play in the diagnosis and treatment of diabetes in the decades to come.1–3 Despite some caveats, a hemoglobin A1c level of 6.5% or higher is diagnostic of diabetes across most populations, and hemoglobin A1c goals ranging from 6.5% to 7.5% have been set for different subsets of patients depending on comorbidities, complications, risk of hypoglycemia, life expectancy, disease duration, patient preferences, and available resources.4

With a growing number of medications for diabetes—insulin in its various formulations and 11 other classes—hemoglobin A1c targets can now be tailored to fit individual patient profiles. Although helping patients attain their glycemic goals is paramount, other factors should be considered when prescribing or changing a drug treatment regimen, such as cardiovascular risk reduction, weight control, avoidance of hypoglycemia, and minimizing out-of-pocket drug costs (Table 1).
CARDIOVASCULAR BENEFIT
Patients with type 2 diabetes have a 2 to 3 times higher risk of clinical atherosclerotic disease, according to 20 years of surveillance data from the Framingham cohort.5
Mixed results with intensive treatment
Reducing cardiovascular risk remains an important goal in diabetes management, but unfortunately, data from the long-term clinical trials aimed at reducing macrovascular risk with intensive glycemic management have been conflicting.
The United Kingdom Prospective Diabetes Study (UKPDS),6 which enrolled more than 4,000 patients with newly diagnosed type 2 diabetes, did not initially show a statistically significant difference in the incidence of myocardial infarction with intensive control vs conventional control, although intensive treatment did reduce the incidence of microvascular disease. However, 10 years after the trial ended, the incidence was 15% lower in the intensive-treatment group than in the conventional-treatment group, and the difference was statistically significant.7
A 10-year follow-up analysis of the Veterans Affairs Diabetes Trial (VADT)8 showed that patients who had been randomly assigned to intensive glucose control for 5.6 years had 8.6 fewer major cardiovascular events per 1,000 person-years than those assigned to standard therapy, but no improvement in median overall survival. The hemoglobin A1c levels achieved during the trial were 6.9% and 8.4%, respectively.
In 2008, the US Food and Drug Administration (FDA)9 mandated that all new applications for diabetes drugs must include cardiovascular outcome studies. Therefore, we now have data on the cardiovascular benefits of two antihyperglycemic drug classes—incretins and sodium-glucose cotransporter 2 (SGLT2) inhibitors, making them attractive medications to target both cardiac and glucose concerns.
Incretins
The incretin drugs comprise 2 classes, glucagon-like peptide 1 (GLP-1) receptor agonists and dipeptidyl peptidase 4 (DPP-4) inhibitors.
Liraglutide. The Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results (LEADER) trial10 compared liraglutide (a GLP-1 receptor agonist) and placebo in 9,000 patients with diabetes who either had or were at high risk of cardiovascular disease. Patients in the liraglutide group had a lower risk of the primary composite end point of death from cardiovascular causes or the first episode of nonfatal (including silent) myocardial infarction or nonfatal stroke, and a lower risk of cardiovascular death, all-cause mortality, and microvascular events than those in the placebo group. The number of patients who would need to be treated to prevent 1 event in 3 years was 66 in the analysis of the primary outcome and 98 in the analysis of death from any cause.9
Lixisenatide. The Evaluation of Lixisenatide in Acute Coronary Syndrome (ELIXA) trial11 studied the effect of the once-daily GLP-1 receptor agonist lixisenatide on cardiovascular outcomes in 6,000 patients with type 2 diabetes with a recent coronary event. In contrast to LEADER, ELIXA did not show a cardiovascular benefit over placebo.
Exenatide. The Exenatide Study of Cardiovascular Event Lowering (EXSCEL)12 assessed another GLP-1 extended-release drug, exenatide, in 14,000 patients, 73% of whom had established cardiovascular disease. In those patients, the drug had a modest benefit in terms of first occurrence of any component of the composite outcome of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke (3-component major adverse cardiac event [MACE] outcome) in a time-to-event analysis, but the results were not statistically significant. However, the drug did significantly reduce all-cause mortality.
Semaglutide, another GLP-1 receptor agonist recently approved by the FDA, also showed benefit in patients who had cardiovascular disease or were at high risk, with significant reduction in the primary composite end point of death from cardiovascular causes or the first occurrence of nonfatal myocardial infarction (including silent) or nonfatal stroke.13
Dulaglutide, a newer GLP-1 drug, was associated with significantly reduced major adverse cardiovascular events (a composite end point of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke) in about 9,900 patients with diabetes, with a median follow-up of more than 5 years. Only 31% of the patients in the trial had established cardiovascular disease.14
Comment. GLP-1 drugs as a class are a good option for patients with diabetes who require weight loss, and liraglutide is now FDA-approved for reduction of cardiovascular events in patients with type 2 diabetes with established cardiovascular disease. However, other factors should be considered when prescribing these drugs: they have adverse gastrointestinal effects, the cardiovascular benefit was not a class effect, they are relatively expensive, and they must be injected. Also, they should not be prescribed concurrently with a DPP-4 inhibitor because they target the same pathway.
SGLT2 inhibitors
The other class of diabetes drugs that have shown cardiovascular benefit are the SGLT2 inhibitors.
Empagliflozin. The Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients (EMPA-REG)15 compared the efficacy of empagliflozin vs placebo in 7,000 patients with diabetes and cardiovascular disease and showed relative risk reductions of 38% in death from cardiovascular death, 31% in sudden death, and 35% in heart failure hospitalizations. Empagliflozin also showed benefit in terms of progression of kidney disease and occurrence of clinically relevant renal events in this population.16
Canagliflozin also has cardiovascular outcome data and showed significant benefit when compared with placebo in the primary outcome of the composite of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke, but no significant effects on cardiovascular death or all-cause mortality.17 Data from this trial also suggested a nonsignificant benefit of canagliflozin in decreasing progression of albuminuria and in the composite outcome of a sustained 40% reduction in the estimated glomerular filtration rate (eGFR), the need for renal replacement therapy, or death from renal causes.
The above data led to an additional indication from the FDA for empagliflozin—and recently, canagliflozin—to prevent cardiovascular death in patients with diabetes with established disease, but other factors should be considered when prescribing them. Patients taking canagliflozin showed a significantly increased risk of amputation. SGLT2 inhibitors as a class also increase the risk of genital infections in men and women; this is an important consideration since patients with diabetes complain of vaginal fungal and urinary tract infections even without the use of these drugs. A higher incidence of fractures with canagliflozin should also be considered when using these medications in elderly and osteoporosis-prone patients at high risk of falling.
Dapagliflozin, the third drug in this class, was associated with a lower rate of hospitalization for heart failure in about 17,160 patients—including 10,186 without atherosclerotic cardiovascular disease—who were followed for a median of 4.2 years.18 It did not show benefit for the primary safety outcome, a composite of major adverse cardiovascular events defined as cardiovascular death, myocardial infarction, or ischemic stroke.
WEIGHT MANAGEMENT
Weight loss can help overweight patients reach their hemoglobin A1c target.
Metformin should be continued as other drugs are added because it does not induce weight gain and may help with weight loss of up to 2 kg as shown in the Diabetes Prevention Program Outcomes Study.19
GLP-1 receptor agonists and SGLT2 inhibitors help with weight loss and are good additions to a basal insulin regimen to minimize weight gain.
Liraglutide was associated with a mean weight loss of 2.3 kg over 36 months of treatment compared with placebo in the LEADER trial.10
In the Trial to Evaluate Cardiovascular and Other Long-term Outcomes With Semaglutide in Subjects With Type 2 Diabetes (SUSTAIN-6),20 the mean body weight in the semaglutide group, compared with the placebo group, was 2.9 kg lower in the group receiving a lower dose and 4.3 kg lower in the group receiving a higher dose of the drug.
In a 24-week trial in 182 patients with type 2 diabetes inadequately controlled on metformin, dapagliflozin produced a statistically significant weight reduction of 2.08 kg (95% confidence interval 2.84–1.31; P < .0001) compared with placebo.21
Lifestyle changes aimed at weight management should be emphasized and discussed at every visit.
HYPOGLYCEMIA RISK
Hypoglycemia is a major consideration when tailoring hemoglobin A1c targets. In the Action to Control Cardiovascular Risk (ACCORD) trial,22 severe, symptomatic hypoglycemia increased the risk of death in both the intensive and conventional treatment groups. In VADT, the occurrence of a recent severe hypoglycemic event was the strongest independent predictor of death within 90 days. Further analysis showed that even though serious hypoglycemia occurred more often in the intensive therapy group, it was associated with progression of coronary artery calcification in the standard therapy group.23 Hence, it is imperative that tight glycemic control not be achieved at the cost of severe or recurrent hypoglycemia.
In terms of hypoglycemia, metformin is an excellent medication. The American Diabetes Association24 recommends metformin as the first-line therapy for newly diagnosed diabetes. Long-term follow-up data from UKPDS showed that metformin decreased mortality and the incidence of myocardial infarction and lowered treatment costs as well as the overall risk of hypoglycemia.25 When prescribed, it should be titrated to the highest dose.
The FDA26 has changed the prescribing information for metformin in patients with renal impairment. Metformin should not be started if the eGFR is less than 45 mL/min/1.73 m2, but it can be continued if the patient is already receiving it and the eGFR is between 30 and 45. Previously, creatinine levels were used to define renal impairment and suitability for metformin. This change has increased the number of patients who can benefit from this medication.
In patients who have a contraindication to metformin, DPP-4 inhibitors can be considered, as they carry a low risk of hypoglycemia as well. Sulfonylureas should be used with caution in these patients, especially if their oral intake is variable. When sulfonylureas were compared to the DPP-4 inhibitor sitagliptin as an add-on to metformin, the rate of hypoglycemia was 32% in the sulfonylurea group vs 5% in the sitagliptin group.27
Of the sulfonylureas, glipizide and glimepiride are better than glyburide because of a comparatively lower risk of hypoglycemia and a higher selectivity for binding the KATP channel on the pancreatic beta cell.28
Meglitinides can be a good option for patients who skip meals, but they are more expensive than other generic oral hypoglycemic agents and require multiple daily dosing.
GLP-1 analogues also have a low risk of hypoglycemia but are only available in injectable formulations. Patients must be willing and able to perform the injections themselves.29
LOOSER TARGETS FOR OLDER PATIENTS
In 2010, among US residents age 65 and older, 10.9 million (about 27%) had diabetes,30 and this number is projected to increase to 26.7 million by 2050.31 This population is prone to hypoglycemia when treated with insulin and sulfonylureas. An injury sustained by a fall induced by hypoglycemia can be life-altering. In addition, no randomized clinical trials show the effect of tight glycemic control on complications in older patients with diabetes because patients older than 80 are often excluded.
A reasonable goal suggested by the European Diabetes Working Party for Older People 201132 and reiterated by the American Geriatrics Society in 201333 is a hemoglobin A1c between 7% and 7.5% for relatively healthy older patients and 7.5% to 8% or 8.5% in frail elderly patients with diabetes.
Consider prescribing medications that carry a low risk of hypoglycemia, can be dose-adjusted for kidney function, and do not rely on manual dexterity for administration (ie, do not require patients to give themselves injections). These include metformin and DPP-4 inhibitors.
DRUG COMBINATIONS
Polypharmacy is a concern for all patients with diabetes, especially since it increases the risk of drug interactions and adverse effects, increases out-of-pocket costs, and decreases the likelihood that patients will remain adherent to their treatment regimen. The use of combination medications can reduce the number of pills or injections required, as well as copayments.
Due to concern for multiple drug-drug interactions (and also due to the progressive nature of diabetes), many people with type 2 diabetes are given insulin in lieu of pills to lower their blood glucose. In addition to premixed insulin combinations (such as combinations of neutral protamine Hagedorn and regular insulin or combinations of insulin analogues), long-acting basal insulins can now be prescribed with a GLP-1 drug in fixed-dose combinations such as insulin glargine plus lixisenatide and insulin degludec plus liraglutide.
COST CONSIDERATIONS
It is important to discuss medication cost with patients, because many newer diabetic drugs are expensive and add to the financial burden of patients already paying for multiple medications, such as antihypertensives and statins.
Metformin and sulfonylureas are less expensive alternatives for patients who cannot afford GLP-1 analogues or SGLT2 inhibitors. Even within the same drug class, the formulary-preferred drug may be cheaper than the nonformulary alternative. Thus, it is helpful to research formulary alternatives before discussing treatment regimens with patients.
- Allen DW, Schroeder WA, Balog J. Observations on the chromatographic heterogeneity of normal adult and fetal human hemoglobin: a study of the effects of crystallization and chromatography on the heterogeneity and isoleucine content. J Amer Chem Soc 1958; 80(7):1628–1634. doi:10.1021/ja01540a030
- Huisman TH, Dozy AM. Studies on the heterogeneity of hemoglobin. V. Binding of hemoglobin with oxidized glutathione. J Lab Clin Med 1962; 60:302–319. pmid:14449875
- Rahbar S, Blumenfeld O, Ranney HM. Studies of an unusual hemoglobin in patients with diabetes mellitus. Biochem Biophys Res Commun 1969; 36(5):838–843. pmid:5808299
- American Diabetes Association. 6. Glycemic targets: standards of medical care in diabetes—2018. Diabetes Care 2018; 41(suppl 1):S55–S64. doi:10.2337/dc18-S006
- Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA 1979; 241(19):2035–2038. pmid:430798
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352(9131):837–853. [Erratum in Lancet 1999; 354:602.] pmid:9742976
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359(15):1577–1589. doi:10.1056/NEJMoa0806470
- Hayward RA, Reaven PD, Wiitala WL, et al; VADT Investigators. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015; 372(23):2197–2206. doi:10.1056/NEJMoa1414266
- US Food and Drug Administration. Guidance for industry: diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. https://www.govinfo.gov/content/pkg/FR-2008-12-19/pdf/E8-30086.pdf. Accessed August 6, 2019.
- Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Steering Committee; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016; 375(4):311–322. doi:10.1056/NEJMoa1603827
- Pfeffer MA, Claggett B, Diaz R, et al; ELIXA Investigators. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med 2015; 373(23):2247–2257. doi:10.1056/NEJMoa1509225
- Holman RR, Bethel MA, Mentz RJ, et al; EXSCEL Study Group. Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2017; 377(13):1228–1239. doi:10.1056/NEJMoa1612917
- Cosmi F, Laini R, Nicolucci A. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2017; 376(9):890. doi:10.1056/NEJMc1615712
- Gerstein HC, Colhoun HM, Dagenais GR, et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet 2019; 394(10193):121–130. doi:10.1016/S0140-6736(19)31149-3
- Zinman B, Wanner C, Lachin JM, et al; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015; 373(22):2117–2128. doi:10.1056/NEJMoa1504720
- Wanner C, Inzucchi SE, Lachin JM, et al; EMPA-REG OUTCOME Investigators. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 2016; 375(4):323–334. doi:10.1056/NEJMoa1515920
- Neal B, Perkovic V, Mahaffey KW, et al; CANVAS Program Collaborative Group. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 2017; 377(7):644–657. doi:10.1056/NEJMoa1611925
- Wiviott SD, Raz I, Bonaca MP, et al; DECLARE–TIMI 58 Investigators. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2018. [Epub ahead of print] doi:10.1056/NEJMoa1812389
- Diabetes Prevention Program Research Group; Knowler WC, Fowler SE, Hamman RF, et al. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009; 374(9702):1677–1686. doi:10.1016/S0140-6736(09)61457-4
- Marso SP, Bain SC, Consoli A, et al, for the SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2016; 375:1834–1844. doi:10.1056/NEJMoa1607141
- Bolinder J, Ljunggren Ö, Kullberg J, et al. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab 2012; 97(3):1020–1031. doi:10.1210/jc.2011-2260
- Bonds DE, Miller ME, Bergenstal RM, et al. The association between symptomatic, severe hypoglycaemia and mortality in type 2 diabetes: retrospective epidemiological analysis of the ACCORD study. BMJ 2010; 340:b4909. doi:10.1136/bmj.b4909
- Saremi A, Bahn GD, Reaven PD; Veterans Affairs Diabetes Trial (VADT). A link between hypoglycemia and progression of atherosclerosis in the Veterans Affairs Diabetes Trial (VADT). Diabetes Care 2016; 39(3):448–454. doi:10.2337/dc15-2107
- American Diabetes Association. 8. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes—2018. Diabetes Care 2018; 41(suppl 1):S73–S85. doi:10.2337/dc18-S008
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359(15):1577–1589. doi:10.1056/NEJMoa0806470
- US Food and Drug Administration. FDA drug safety communication: FDA revises warnings regarding use of the diabetes medicine metformin in certain patients with reduced kidney function. www.fda.gov/Drugs/DrugSafety/ucm493244.htm. Accessed August 5, 2019.
- Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP; Sitagliptin Study 024 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9(2):194–205. doi:10.1111/j.1463-1326.2006.00704.x
- Gangji AS, Cukierman T, Gerstein HC, Goldsmith CH, Clase CM. A systematic review and meta-analysis of hypoglycemia and cardiovascular events: a comparison of glyburide with other secretagogues and with insulin. Diabetes Care 2007; 30(2):389–394. doi:10.2337/dc06-1789
- Nauck M, Frid A, Hermansen K, et al; LEAD-2 Study Group. Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care 2009; 32(1):84–90. doi:10.2337/dc08-1355
- Centers for Disease Control and Prevention. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed August 5, 2019.
- Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr 2010; 8:29. doi:10.1186/1478-7954-8-29
- Sinclair AJ, Paolisso G, Castro M, Bourdel-Marchasson I, Gadsby R, Rodriguez Mañas L; European Diabetes Working Party for Older People. European Diabetes Working Party for Older People 2011 clinical guidelines for type 2 diabetes mellitus. Executive summary. Diabetes Metab 2011; 37(suppl 3):S27–S38. doi:10.1016/S1262-3636(11)70962-4
- American Geriatrics Society Expert Panel on Care of Older Adults with Diabetes Mellitus; Moreno G, Mangione CM, Kimbro L, Vaisberg E. Guidelines abstracted from the American Geriatrics Society Guidelines for Improving the Care of Older Adults with Diabetes Mellitus: 2013 update. J Am Geriatr Soc 2013; 61(11):2020–2026. doi:10.1111/jgs.12514
When scientists discovered the band of hemoglobin A1c during electrophoresis in the 1950s and 1960s and discerned it was elevated in patients with diabetes, little did they know the important role it would play in the diagnosis and treatment of diabetes in the decades to come.1–3 Despite some caveats, a hemoglobin A1c level of 6.5% or higher is diagnostic of diabetes across most populations, and hemoglobin A1c goals ranging from 6.5% to 7.5% have been set for different subsets of patients depending on comorbidities, complications, risk of hypoglycemia, life expectancy, disease duration, patient preferences, and available resources.4
With a growing number of medications for diabetes—insulin in its various formulations and 11 other classes—hemoglobin A1c targets can now be tailored to fit individual patient profiles. Although helping patients attain their glycemic goals is paramount, other factors should be considered when prescribing or changing a drug treatment regimen, such as cardiovascular risk reduction, weight control, avoidance of hypoglycemia, and minimizing out-of-pocket drug costs (Table 1).
CARDIOVASCULAR BENEFIT
Patients with type 2 diabetes have a 2 to 3 times higher risk of clinical atherosclerotic disease, according to 20 years of surveillance data from the Framingham cohort.5
Mixed results with intensive treatment
Reducing cardiovascular risk remains an important goal in diabetes management, but unfortunately, data from the long-term clinical trials aimed at reducing macrovascular risk with intensive glycemic management have been conflicting.
The United Kingdom Prospective Diabetes Study (UKPDS),6 which enrolled more than 4,000 patients with newly diagnosed type 2 diabetes, did not initially show a statistically significant difference in the incidence of myocardial infarction with intensive control vs conventional control, although intensive treatment did reduce the incidence of microvascular disease. However, 10 years after the trial ended, the incidence was 15% lower in the intensive-treatment group than in the conventional-treatment group, and the difference was statistically significant.7
A 10-year follow-up analysis of the Veterans Affairs Diabetes Trial (VADT)8 showed that patients who had been randomly assigned to intensive glucose control for 5.6 years had 8.6 fewer major cardiovascular events per 1,000 person-years than those assigned to standard therapy, but no improvement in median overall survival. The hemoglobin A1c levels achieved during the trial were 6.9% and 8.4%, respectively.
In 2008, the US Food and Drug Administration (FDA)9 mandated that all new applications for diabetes drugs must include cardiovascular outcome studies. Therefore, we now have data on the cardiovascular benefits of two antihyperglycemic drug classes—incretins and sodium-glucose cotransporter 2 (SGLT2) inhibitors, making them attractive medications to target both cardiac and glucose concerns.
Incretins
The incretin drugs comprise 2 classes, glucagon-like peptide 1 (GLP-1) receptor agonists and dipeptidyl peptidase 4 (DPP-4) inhibitors.
Liraglutide. The Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results (LEADER) trial10 compared liraglutide (a GLP-1 receptor agonist) and placebo in 9,000 patients with diabetes who either had or were at high risk of cardiovascular disease. Patients in the liraglutide group had a lower risk of the primary composite end point of death from cardiovascular causes or the first episode of nonfatal (including silent) myocardial infarction or nonfatal stroke, and a lower risk of cardiovascular death, all-cause mortality, and microvascular events than those in the placebo group. The number of patients who would need to be treated to prevent 1 event in 3 years was 66 in the analysis of the primary outcome and 98 in the analysis of death from any cause.9
Lixisenatide. The Evaluation of Lixisenatide in Acute Coronary Syndrome (ELIXA) trial11 studied the effect of the once-daily GLP-1 receptor agonist lixisenatide on cardiovascular outcomes in 6,000 patients with type 2 diabetes with a recent coronary event. In contrast to LEADER, ELIXA did not show a cardiovascular benefit over placebo.
Exenatide. The Exenatide Study of Cardiovascular Event Lowering (EXSCEL)12 assessed another GLP-1 extended-release drug, exenatide, in 14,000 patients, 73% of whom had established cardiovascular disease. In those patients, the drug had a modest benefit in terms of first occurrence of any component of the composite outcome of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke (3-component major adverse cardiac event [MACE] outcome) in a time-to-event analysis, but the results were not statistically significant. However, the drug did significantly reduce all-cause mortality.
Semaglutide, another GLP-1 receptor agonist recently approved by the FDA, also showed benefit in patients who had cardiovascular disease or were at high risk, with significant reduction in the primary composite end point of death from cardiovascular causes or the first occurrence of nonfatal myocardial infarction (including silent) or nonfatal stroke.13
Dulaglutide, a newer GLP-1 drug, was associated with significantly reduced major adverse cardiovascular events (a composite end point of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke) in about 9,900 patients with diabetes, with a median follow-up of more than 5 years. Only 31% of the patients in the trial had established cardiovascular disease.14
Comment. GLP-1 drugs as a class are a good option for patients with diabetes who require weight loss, and liraglutide is now FDA-approved for reduction of cardiovascular events in patients with type 2 diabetes with established cardiovascular disease. However, other factors should be considered when prescribing these drugs: they have adverse gastrointestinal effects, the cardiovascular benefit was not a class effect, they are relatively expensive, and they must be injected. Also, they should not be prescribed concurrently with a DPP-4 inhibitor because they target the same pathway.
SGLT2 inhibitors
The other class of diabetes drugs that have shown cardiovascular benefit are the SGLT2 inhibitors.
Empagliflozin. The Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients (EMPA-REG)15 compared the efficacy of empagliflozin vs placebo in 7,000 patients with diabetes and cardiovascular disease and showed relative risk reductions of 38% in death from cardiovascular death, 31% in sudden death, and 35% in heart failure hospitalizations. Empagliflozin also showed benefit in terms of progression of kidney disease and occurrence of clinically relevant renal events in this population.16
Canagliflozin also has cardiovascular outcome data and showed significant benefit when compared with placebo in the primary outcome of the composite of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke, but no significant effects on cardiovascular death or all-cause mortality.17 Data from this trial also suggested a nonsignificant benefit of canagliflozin in decreasing progression of albuminuria and in the composite outcome of a sustained 40% reduction in the estimated glomerular filtration rate (eGFR), the need for renal replacement therapy, or death from renal causes.
The above data led to an additional indication from the FDA for empagliflozin—and recently, canagliflozin—to prevent cardiovascular death in patients with diabetes with established disease, but other factors should be considered when prescribing them. Patients taking canagliflozin showed a significantly increased risk of amputation. SGLT2 inhibitors as a class also increase the risk of genital infections in men and women; this is an important consideration since patients with diabetes complain of vaginal fungal and urinary tract infections even without the use of these drugs. A higher incidence of fractures with canagliflozin should also be considered when using these medications in elderly and osteoporosis-prone patients at high risk of falling.
Dapagliflozin, the third drug in this class, was associated with a lower rate of hospitalization for heart failure in about 17,160 patients—including 10,186 without atherosclerotic cardiovascular disease—who were followed for a median of 4.2 years.18 It did not show benefit for the primary safety outcome, a composite of major adverse cardiovascular events defined as cardiovascular death, myocardial infarction, or ischemic stroke.
WEIGHT MANAGEMENT
Weight loss can help overweight patients reach their hemoglobin A1c target.
Metformin should be continued as other drugs are added because it does not induce weight gain and may help with weight loss of up to 2 kg as shown in the Diabetes Prevention Program Outcomes Study.19
GLP-1 receptor agonists and SGLT2 inhibitors help with weight loss and are good additions to a basal insulin regimen to minimize weight gain.
Liraglutide was associated with a mean weight loss of 2.3 kg over 36 months of treatment compared with placebo in the LEADER trial.10
In the Trial to Evaluate Cardiovascular and Other Long-term Outcomes With Semaglutide in Subjects With Type 2 Diabetes (SUSTAIN-6),20 the mean body weight in the semaglutide group, compared with the placebo group, was 2.9 kg lower in the group receiving a lower dose and 4.3 kg lower in the group receiving a higher dose of the drug.
In a 24-week trial in 182 patients with type 2 diabetes inadequately controlled on metformin, dapagliflozin produced a statistically significant weight reduction of 2.08 kg (95% confidence interval 2.84–1.31; P < .0001) compared with placebo.21
Lifestyle changes aimed at weight management should be emphasized and discussed at every visit.
HYPOGLYCEMIA RISK
Hypoglycemia is a major consideration when tailoring hemoglobin A1c targets. In the Action to Control Cardiovascular Risk (ACCORD) trial,22 severe, symptomatic hypoglycemia increased the risk of death in both the intensive and conventional treatment groups. In VADT, the occurrence of a recent severe hypoglycemic event was the strongest independent predictor of death within 90 days. Further analysis showed that even though serious hypoglycemia occurred more often in the intensive therapy group, it was associated with progression of coronary artery calcification in the standard therapy group.23 Hence, it is imperative that tight glycemic control not be achieved at the cost of severe or recurrent hypoglycemia.
In terms of hypoglycemia, metformin is an excellent medication. The American Diabetes Association24 recommends metformin as the first-line therapy for newly diagnosed diabetes. Long-term follow-up data from UKPDS showed that metformin decreased mortality and the incidence of myocardial infarction and lowered treatment costs as well as the overall risk of hypoglycemia.25 When prescribed, it should be titrated to the highest dose.
The FDA26 has changed the prescribing information for metformin in patients with renal impairment. Metformin should not be started if the eGFR is less than 45 mL/min/1.73 m2, but it can be continued if the patient is already receiving it and the eGFR is between 30 and 45. Previously, creatinine levels were used to define renal impairment and suitability for metformin. This change has increased the number of patients who can benefit from this medication.
In patients who have a contraindication to metformin, DPP-4 inhibitors can be considered, as they carry a low risk of hypoglycemia as well. Sulfonylureas should be used with caution in these patients, especially if their oral intake is variable. When sulfonylureas were compared to the DPP-4 inhibitor sitagliptin as an add-on to metformin, the rate of hypoglycemia was 32% in the sulfonylurea group vs 5% in the sitagliptin group.27
Of the sulfonylureas, glipizide and glimepiride are better than glyburide because of a comparatively lower risk of hypoglycemia and a higher selectivity for binding the KATP channel on the pancreatic beta cell.28
Meglitinides can be a good option for patients who skip meals, but they are more expensive than other generic oral hypoglycemic agents and require multiple daily dosing.
GLP-1 analogues also have a low risk of hypoglycemia but are only available in injectable formulations. Patients must be willing and able to perform the injections themselves.29
LOOSER TARGETS FOR OLDER PATIENTS
In 2010, among US residents age 65 and older, 10.9 million (about 27%) had diabetes,30 and this number is projected to increase to 26.7 million by 2050.31 This population is prone to hypoglycemia when treated with insulin and sulfonylureas. An injury sustained by a fall induced by hypoglycemia can be life-altering. In addition, no randomized clinical trials show the effect of tight glycemic control on complications in older patients with diabetes because patients older than 80 are often excluded.
A reasonable goal suggested by the European Diabetes Working Party for Older People 201132 and reiterated by the American Geriatrics Society in 201333 is a hemoglobin A1c between 7% and 7.5% for relatively healthy older patients and 7.5% to 8% or 8.5% in frail elderly patients with diabetes.
Consider prescribing medications that carry a low risk of hypoglycemia, can be dose-adjusted for kidney function, and do not rely on manual dexterity for administration (ie, do not require patients to give themselves injections). These include metformin and DPP-4 inhibitors.
DRUG COMBINATIONS
Polypharmacy is a concern for all patients with diabetes, especially since it increases the risk of drug interactions and adverse effects, increases out-of-pocket costs, and decreases the likelihood that patients will remain adherent to their treatment regimen. The use of combination medications can reduce the number of pills or injections required, as well as copayments.
Due to concern for multiple drug-drug interactions (and also due to the progressive nature of diabetes), many people with type 2 diabetes are given insulin in lieu of pills to lower their blood glucose. In addition to premixed insulin combinations (such as combinations of neutral protamine Hagedorn and regular insulin or combinations of insulin analogues), long-acting basal insulins can now be prescribed with a GLP-1 drug in fixed-dose combinations such as insulin glargine plus lixisenatide and insulin degludec plus liraglutide.
COST CONSIDERATIONS
It is important to discuss medication cost with patients, because many newer diabetic drugs are expensive and add to the financial burden of patients already paying for multiple medications, such as antihypertensives and statins.
Metformin and sulfonylureas are less expensive alternatives for patients who cannot afford GLP-1 analogues or SGLT2 inhibitors. Even within the same drug class, the formulary-preferred drug may be cheaper than the nonformulary alternative. Thus, it is helpful to research formulary alternatives before discussing treatment regimens with patients.
When scientists discovered the band of hemoglobin A1c during electrophoresis in the 1950s and 1960s and discerned it was elevated in patients with diabetes, little did they know the important role it would play in the diagnosis and treatment of diabetes in the decades to come.1–3 Despite some caveats, a hemoglobin A1c level of 6.5% or higher is diagnostic of diabetes across most populations, and hemoglobin A1c goals ranging from 6.5% to 7.5% have been set for different subsets of patients depending on comorbidities, complications, risk of hypoglycemia, life expectancy, disease duration, patient preferences, and available resources.4
With a growing number of medications for diabetes—insulin in its various formulations and 11 other classes—hemoglobin A1c targets can now be tailored to fit individual patient profiles. Although helping patients attain their glycemic goals is paramount, other factors should be considered when prescribing or changing a drug treatment regimen, such as cardiovascular risk reduction, weight control, avoidance of hypoglycemia, and minimizing out-of-pocket drug costs (Table 1).
CARDIOVASCULAR BENEFIT
Patients with type 2 diabetes have a 2 to 3 times higher risk of clinical atherosclerotic disease, according to 20 years of surveillance data from the Framingham cohort.5
Mixed results with intensive treatment
Reducing cardiovascular risk remains an important goal in diabetes management, but unfortunately, data from the long-term clinical trials aimed at reducing macrovascular risk with intensive glycemic management have been conflicting.
The United Kingdom Prospective Diabetes Study (UKPDS),6 which enrolled more than 4,000 patients with newly diagnosed type 2 diabetes, did not initially show a statistically significant difference in the incidence of myocardial infarction with intensive control vs conventional control, although intensive treatment did reduce the incidence of microvascular disease. However, 10 years after the trial ended, the incidence was 15% lower in the intensive-treatment group than in the conventional-treatment group, and the difference was statistically significant.7
A 10-year follow-up analysis of the Veterans Affairs Diabetes Trial (VADT)8 showed that patients who had been randomly assigned to intensive glucose control for 5.6 years had 8.6 fewer major cardiovascular events per 1,000 person-years than those assigned to standard therapy, but no improvement in median overall survival. The hemoglobin A1c levels achieved during the trial were 6.9% and 8.4%, respectively.
In 2008, the US Food and Drug Administration (FDA)9 mandated that all new applications for diabetes drugs must include cardiovascular outcome studies. Therefore, we now have data on the cardiovascular benefits of two antihyperglycemic drug classes—incretins and sodium-glucose cotransporter 2 (SGLT2) inhibitors, making them attractive medications to target both cardiac and glucose concerns.
Incretins
The incretin drugs comprise 2 classes, glucagon-like peptide 1 (GLP-1) receptor agonists and dipeptidyl peptidase 4 (DPP-4) inhibitors.
Liraglutide. The Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results (LEADER) trial10 compared liraglutide (a GLP-1 receptor agonist) and placebo in 9,000 patients with diabetes who either had or were at high risk of cardiovascular disease. Patients in the liraglutide group had a lower risk of the primary composite end point of death from cardiovascular causes or the first episode of nonfatal (including silent) myocardial infarction or nonfatal stroke, and a lower risk of cardiovascular death, all-cause mortality, and microvascular events than those in the placebo group. The number of patients who would need to be treated to prevent 1 event in 3 years was 66 in the analysis of the primary outcome and 98 in the analysis of death from any cause.9
Lixisenatide. The Evaluation of Lixisenatide in Acute Coronary Syndrome (ELIXA) trial11 studied the effect of the once-daily GLP-1 receptor agonist lixisenatide on cardiovascular outcomes in 6,000 patients with type 2 diabetes with a recent coronary event. In contrast to LEADER, ELIXA did not show a cardiovascular benefit over placebo.
Exenatide. The Exenatide Study of Cardiovascular Event Lowering (EXSCEL)12 assessed another GLP-1 extended-release drug, exenatide, in 14,000 patients, 73% of whom had established cardiovascular disease. In those patients, the drug had a modest benefit in terms of first occurrence of any component of the composite outcome of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke (3-component major adverse cardiac event [MACE] outcome) in a time-to-event analysis, but the results were not statistically significant. However, the drug did significantly reduce all-cause mortality.
Semaglutide, another GLP-1 receptor agonist recently approved by the FDA, also showed benefit in patients who had cardiovascular disease or were at high risk, with significant reduction in the primary composite end point of death from cardiovascular causes or the first occurrence of nonfatal myocardial infarction (including silent) or nonfatal stroke.13
Dulaglutide, a newer GLP-1 drug, was associated with significantly reduced major adverse cardiovascular events (a composite end point of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke) in about 9,900 patients with diabetes, with a median follow-up of more than 5 years. Only 31% of the patients in the trial had established cardiovascular disease.14
Comment. GLP-1 drugs as a class are a good option for patients with diabetes who require weight loss, and liraglutide is now FDA-approved for reduction of cardiovascular events in patients with type 2 diabetes with established cardiovascular disease. However, other factors should be considered when prescribing these drugs: they have adverse gastrointestinal effects, the cardiovascular benefit was not a class effect, they are relatively expensive, and they must be injected. Also, they should not be prescribed concurrently with a DPP-4 inhibitor because they target the same pathway.
SGLT2 inhibitors
The other class of diabetes drugs that have shown cardiovascular benefit are the SGLT2 inhibitors.
Empagliflozin. The Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients (EMPA-REG)15 compared the efficacy of empagliflozin vs placebo in 7,000 patients with diabetes and cardiovascular disease and showed relative risk reductions of 38% in death from cardiovascular death, 31% in sudden death, and 35% in heart failure hospitalizations. Empagliflozin also showed benefit in terms of progression of kidney disease and occurrence of clinically relevant renal events in this population.16
Canagliflozin also has cardiovascular outcome data and showed significant benefit when compared with placebo in the primary outcome of the composite of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke, but no significant effects on cardiovascular death or all-cause mortality.17 Data from this trial also suggested a nonsignificant benefit of canagliflozin in decreasing progression of albuminuria and in the composite outcome of a sustained 40% reduction in the estimated glomerular filtration rate (eGFR), the need for renal replacement therapy, or death from renal causes.
The above data led to an additional indication from the FDA for empagliflozin—and recently, canagliflozin—to prevent cardiovascular death in patients with diabetes with established disease, but other factors should be considered when prescribing them. Patients taking canagliflozin showed a significantly increased risk of amputation. SGLT2 inhibitors as a class also increase the risk of genital infections in men and women; this is an important consideration since patients with diabetes complain of vaginal fungal and urinary tract infections even without the use of these drugs. A higher incidence of fractures with canagliflozin should also be considered when using these medications in elderly and osteoporosis-prone patients at high risk of falling.
Dapagliflozin, the third drug in this class, was associated with a lower rate of hospitalization for heart failure in about 17,160 patients—including 10,186 without atherosclerotic cardiovascular disease—who were followed for a median of 4.2 years.18 It did not show benefit for the primary safety outcome, a composite of major adverse cardiovascular events defined as cardiovascular death, myocardial infarction, or ischemic stroke.
WEIGHT MANAGEMENT
Weight loss can help overweight patients reach their hemoglobin A1c target.
Metformin should be continued as other drugs are added because it does not induce weight gain and may help with weight loss of up to 2 kg as shown in the Diabetes Prevention Program Outcomes Study.19
GLP-1 receptor agonists and SGLT2 inhibitors help with weight loss and are good additions to a basal insulin regimen to minimize weight gain.
Liraglutide was associated with a mean weight loss of 2.3 kg over 36 months of treatment compared with placebo in the LEADER trial.10
In the Trial to Evaluate Cardiovascular and Other Long-term Outcomes With Semaglutide in Subjects With Type 2 Diabetes (SUSTAIN-6),20 the mean body weight in the semaglutide group, compared with the placebo group, was 2.9 kg lower in the group receiving a lower dose and 4.3 kg lower in the group receiving a higher dose of the drug.
In a 24-week trial in 182 patients with type 2 diabetes inadequately controlled on metformin, dapagliflozin produced a statistically significant weight reduction of 2.08 kg (95% confidence interval 2.84–1.31; P < .0001) compared with placebo.21
Lifestyle changes aimed at weight management should be emphasized and discussed at every visit.
HYPOGLYCEMIA RISK
Hypoglycemia is a major consideration when tailoring hemoglobin A1c targets. In the Action to Control Cardiovascular Risk (ACCORD) trial,22 severe, symptomatic hypoglycemia increased the risk of death in both the intensive and conventional treatment groups. In VADT, the occurrence of a recent severe hypoglycemic event was the strongest independent predictor of death within 90 days. Further analysis showed that even though serious hypoglycemia occurred more often in the intensive therapy group, it was associated with progression of coronary artery calcification in the standard therapy group.23 Hence, it is imperative that tight glycemic control not be achieved at the cost of severe or recurrent hypoglycemia.
In terms of hypoglycemia, metformin is an excellent medication. The American Diabetes Association24 recommends metformin as the first-line therapy for newly diagnosed diabetes. Long-term follow-up data from UKPDS showed that metformin decreased mortality and the incidence of myocardial infarction and lowered treatment costs as well as the overall risk of hypoglycemia.25 When prescribed, it should be titrated to the highest dose.
The FDA26 has changed the prescribing information for metformin in patients with renal impairment. Metformin should not be started if the eGFR is less than 45 mL/min/1.73 m2, but it can be continued if the patient is already receiving it and the eGFR is between 30 and 45. Previously, creatinine levels were used to define renal impairment and suitability for metformin. This change has increased the number of patients who can benefit from this medication.
In patients who have a contraindication to metformin, DPP-4 inhibitors can be considered, as they carry a low risk of hypoglycemia as well. Sulfonylureas should be used with caution in these patients, especially if their oral intake is variable. When sulfonylureas were compared to the DPP-4 inhibitor sitagliptin as an add-on to metformin, the rate of hypoglycemia was 32% in the sulfonylurea group vs 5% in the sitagliptin group.27
Of the sulfonylureas, glipizide and glimepiride are better than glyburide because of a comparatively lower risk of hypoglycemia and a higher selectivity for binding the KATP channel on the pancreatic beta cell.28
Meglitinides can be a good option for patients who skip meals, but they are more expensive than other generic oral hypoglycemic agents and require multiple daily dosing.
GLP-1 analogues also have a low risk of hypoglycemia but are only available in injectable formulations. Patients must be willing and able to perform the injections themselves.29
LOOSER TARGETS FOR OLDER PATIENTS
In 2010, among US residents age 65 and older, 10.9 million (about 27%) had diabetes,30 and this number is projected to increase to 26.7 million by 2050.31 This population is prone to hypoglycemia when treated with insulin and sulfonylureas. An injury sustained by a fall induced by hypoglycemia can be life-altering. In addition, no randomized clinical trials show the effect of tight glycemic control on complications in older patients with diabetes because patients older than 80 are often excluded.
A reasonable goal suggested by the European Diabetes Working Party for Older People 201132 and reiterated by the American Geriatrics Society in 201333 is a hemoglobin A1c between 7% and 7.5% for relatively healthy older patients and 7.5% to 8% or 8.5% in frail elderly patients with diabetes.
Consider prescribing medications that carry a low risk of hypoglycemia, can be dose-adjusted for kidney function, and do not rely on manual dexterity for administration (ie, do not require patients to give themselves injections). These include metformin and DPP-4 inhibitors.
DRUG COMBINATIONS
Polypharmacy is a concern for all patients with diabetes, especially since it increases the risk of drug interactions and adverse effects, increases out-of-pocket costs, and decreases the likelihood that patients will remain adherent to their treatment regimen. The use of combination medications can reduce the number of pills or injections required, as well as copayments.
Due to concern for multiple drug-drug interactions (and also due to the progressive nature of diabetes), many people with type 2 diabetes are given insulin in lieu of pills to lower their blood glucose. In addition to premixed insulin combinations (such as combinations of neutral protamine Hagedorn and regular insulin or combinations of insulin analogues), long-acting basal insulins can now be prescribed with a GLP-1 drug in fixed-dose combinations such as insulin glargine plus lixisenatide and insulin degludec plus liraglutide.
COST CONSIDERATIONS
It is important to discuss medication cost with patients, because many newer diabetic drugs are expensive and add to the financial burden of patients already paying for multiple medications, such as antihypertensives and statins.
Metformin and sulfonylureas are less expensive alternatives for patients who cannot afford GLP-1 analogues or SGLT2 inhibitors. Even within the same drug class, the formulary-preferred drug may be cheaper than the nonformulary alternative. Thus, it is helpful to research formulary alternatives before discussing treatment regimens with patients.
- Allen DW, Schroeder WA, Balog J. Observations on the chromatographic heterogeneity of normal adult and fetal human hemoglobin: a study of the effects of crystallization and chromatography on the heterogeneity and isoleucine content. J Amer Chem Soc 1958; 80(7):1628–1634. doi:10.1021/ja01540a030
- Huisman TH, Dozy AM. Studies on the heterogeneity of hemoglobin. V. Binding of hemoglobin with oxidized glutathione. J Lab Clin Med 1962; 60:302–319. pmid:14449875
- Rahbar S, Blumenfeld O, Ranney HM. Studies of an unusual hemoglobin in patients with diabetes mellitus. Biochem Biophys Res Commun 1969; 36(5):838–843. pmid:5808299
- American Diabetes Association. 6. Glycemic targets: standards of medical care in diabetes—2018. Diabetes Care 2018; 41(suppl 1):S55–S64. doi:10.2337/dc18-S006
- Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA 1979; 241(19):2035–2038. pmid:430798
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352(9131):837–853. [Erratum in Lancet 1999; 354:602.] pmid:9742976
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359(15):1577–1589. doi:10.1056/NEJMoa0806470
- Hayward RA, Reaven PD, Wiitala WL, et al; VADT Investigators. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015; 372(23):2197–2206. doi:10.1056/NEJMoa1414266
- US Food and Drug Administration. Guidance for industry: diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. https://www.govinfo.gov/content/pkg/FR-2008-12-19/pdf/E8-30086.pdf. Accessed August 6, 2019.
- Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Steering Committee; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016; 375(4):311–322. doi:10.1056/NEJMoa1603827
- Pfeffer MA, Claggett B, Diaz R, et al; ELIXA Investigators. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med 2015; 373(23):2247–2257. doi:10.1056/NEJMoa1509225
- Holman RR, Bethel MA, Mentz RJ, et al; EXSCEL Study Group. Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2017; 377(13):1228–1239. doi:10.1056/NEJMoa1612917
- Cosmi F, Laini R, Nicolucci A. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2017; 376(9):890. doi:10.1056/NEJMc1615712
- Gerstein HC, Colhoun HM, Dagenais GR, et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet 2019; 394(10193):121–130. doi:10.1016/S0140-6736(19)31149-3
- Zinman B, Wanner C, Lachin JM, et al; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015; 373(22):2117–2128. doi:10.1056/NEJMoa1504720
- Wanner C, Inzucchi SE, Lachin JM, et al; EMPA-REG OUTCOME Investigators. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 2016; 375(4):323–334. doi:10.1056/NEJMoa1515920
- Neal B, Perkovic V, Mahaffey KW, et al; CANVAS Program Collaborative Group. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 2017; 377(7):644–657. doi:10.1056/NEJMoa1611925
- Wiviott SD, Raz I, Bonaca MP, et al; DECLARE–TIMI 58 Investigators. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2018. [Epub ahead of print] doi:10.1056/NEJMoa1812389
- Diabetes Prevention Program Research Group; Knowler WC, Fowler SE, Hamman RF, et al. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009; 374(9702):1677–1686. doi:10.1016/S0140-6736(09)61457-4
- Marso SP, Bain SC, Consoli A, et al, for the SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2016; 375:1834–1844. doi:10.1056/NEJMoa1607141
- Bolinder J, Ljunggren Ö, Kullberg J, et al. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab 2012; 97(3):1020–1031. doi:10.1210/jc.2011-2260
- Bonds DE, Miller ME, Bergenstal RM, et al. The association between symptomatic, severe hypoglycaemia and mortality in type 2 diabetes: retrospective epidemiological analysis of the ACCORD study. BMJ 2010; 340:b4909. doi:10.1136/bmj.b4909
- Saremi A, Bahn GD, Reaven PD; Veterans Affairs Diabetes Trial (VADT). A link between hypoglycemia and progression of atherosclerosis in the Veterans Affairs Diabetes Trial (VADT). Diabetes Care 2016; 39(3):448–454. doi:10.2337/dc15-2107
- American Diabetes Association. 8. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes—2018. Diabetes Care 2018; 41(suppl 1):S73–S85. doi:10.2337/dc18-S008
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359(15):1577–1589. doi:10.1056/NEJMoa0806470
- US Food and Drug Administration. FDA drug safety communication: FDA revises warnings regarding use of the diabetes medicine metformin in certain patients with reduced kidney function. www.fda.gov/Drugs/DrugSafety/ucm493244.htm. Accessed August 5, 2019.
- Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP; Sitagliptin Study 024 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9(2):194–205. doi:10.1111/j.1463-1326.2006.00704.x
- Gangji AS, Cukierman T, Gerstein HC, Goldsmith CH, Clase CM. A systematic review and meta-analysis of hypoglycemia and cardiovascular events: a comparison of glyburide with other secretagogues and with insulin. Diabetes Care 2007; 30(2):389–394. doi:10.2337/dc06-1789
- Nauck M, Frid A, Hermansen K, et al; LEAD-2 Study Group. Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care 2009; 32(1):84–90. doi:10.2337/dc08-1355
- Centers for Disease Control and Prevention. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed August 5, 2019.
- Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr 2010; 8:29. doi:10.1186/1478-7954-8-29
- Sinclair AJ, Paolisso G, Castro M, Bourdel-Marchasson I, Gadsby R, Rodriguez Mañas L; European Diabetes Working Party for Older People. European Diabetes Working Party for Older People 2011 clinical guidelines for type 2 diabetes mellitus. Executive summary. Diabetes Metab 2011; 37(suppl 3):S27–S38. doi:10.1016/S1262-3636(11)70962-4
- American Geriatrics Society Expert Panel on Care of Older Adults with Diabetes Mellitus; Moreno G, Mangione CM, Kimbro L, Vaisberg E. Guidelines abstracted from the American Geriatrics Society Guidelines for Improving the Care of Older Adults with Diabetes Mellitus: 2013 update. J Am Geriatr Soc 2013; 61(11):2020–2026. doi:10.1111/jgs.12514
- Allen DW, Schroeder WA, Balog J. Observations on the chromatographic heterogeneity of normal adult and fetal human hemoglobin: a study of the effects of crystallization and chromatography on the heterogeneity and isoleucine content. J Amer Chem Soc 1958; 80(7):1628–1634. doi:10.1021/ja01540a030
- Huisman TH, Dozy AM. Studies on the heterogeneity of hemoglobin. V. Binding of hemoglobin with oxidized glutathione. J Lab Clin Med 1962; 60:302–319. pmid:14449875
- Rahbar S, Blumenfeld O, Ranney HM. Studies of an unusual hemoglobin in patients with diabetes mellitus. Biochem Biophys Res Commun 1969; 36(5):838–843. pmid:5808299
- American Diabetes Association. 6. Glycemic targets: standards of medical care in diabetes—2018. Diabetes Care 2018; 41(suppl 1):S55–S64. doi:10.2337/dc18-S006
- Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA 1979; 241(19):2035–2038. pmid:430798
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352(9131):837–853. [Erratum in Lancet 1999; 354:602.] pmid:9742976
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359(15):1577–1589. doi:10.1056/NEJMoa0806470
- Hayward RA, Reaven PD, Wiitala WL, et al; VADT Investigators. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015; 372(23):2197–2206. doi:10.1056/NEJMoa1414266
- US Food and Drug Administration. Guidance for industry: diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. https://www.govinfo.gov/content/pkg/FR-2008-12-19/pdf/E8-30086.pdf. Accessed August 6, 2019.
- Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Steering Committee; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016; 375(4):311–322. doi:10.1056/NEJMoa1603827
- Pfeffer MA, Claggett B, Diaz R, et al; ELIXA Investigators. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med 2015; 373(23):2247–2257. doi:10.1056/NEJMoa1509225
- Holman RR, Bethel MA, Mentz RJ, et al; EXSCEL Study Group. Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2017; 377(13):1228–1239. doi:10.1056/NEJMoa1612917
- Cosmi F, Laini R, Nicolucci A. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2017; 376(9):890. doi:10.1056/NEJMc1615712
- Gerstein HC, Colhoun HM, Dagenais GR, et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet 2019; 394(10193):121–130. doi:10.1016/S0140-6736(19)31149-3
- Zinman B, Wanner C, Lachin JM, et al; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015; 373(22):2117–2128. doi:10.1056/NEJMoa1504720
- Wanner C, Inzucchi SE, Lachin JM, et al; EMPA-REG OUTCOME Investigators. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 2016; 375(4):323–334. doi:10.1056/NEJMoa1515920
- Neal B, Perkovic V, Mahaffey KW, et al; CANVAS Program Collaborative Group. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 2017; 377(7):644–657. doi:10.1056/NEJMoa1611925
- Wiviott SD, Raz I, Bonaca MP, et al; DECLARE–TIMI 58 Investigators. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2018. [Epub ahead of print] doi:10.1056/NEJMoa1812389
- Diabetes Prevention Program Research Group; Knowler WC, Fowler SE, Hamman RF, et al. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009; 374(9702):1677–1686. doi:10.1016/S0140-6736(09)61457-4
- Marso SP, Bain SC, Consoli A, et al, for the SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2016; 375:1834–1844. doi:10.1056/NEJMoa1607141
- Bolinder J, Ljunggren Ö, Kullberg J, et al. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab 2012; 97(3):1020–1031. doi:10.1210/jc.2011-2260
- Bonds DE, Miller ME, Bergenstal RM, et al. The association between symptomatic, severe hypoglycaemia and mortality in type 2 diabetes: retrospective epidemiological analysis of the ACCORD study. BMJ 2010; 340:b4909. doi:10.1136/bmj.b4909
- Saremi A, Bahn GD, Reaven PD; Veterans Affairs Diabetes Trial (VADT). A link between hypoglycemia and progression of atherosclerosis in the Veterans Affairs Diabetes Trial (VADT). Diabetes Care 2016; 39(3):448–454. doi:10.2337/dc15-2107
- American Diabetes Association. 8. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes—2018. Diabetes Care 2018; 41(suppl 1):S73–S85. doi:10.2337/dc18-S008
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359(15):1577–1589. doi:10.1056/NEJMoa0806470
- US Food and Drug Administration. FDA drug safety communication: FDA revises warnings regarding use of the diabetes medicine metformin in certain patients with reduced kidney function. www.fda.gov/Drugs/DrugSafety/ucm493244.htm. Accessed August 5, 2019.
- Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP; Sitagliptin Study 024 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9(2):194–205. doi:10.1111/j.1463-1326.2006.00704.x
- Gangji AS, Cukierman T, Gerstein HC, Goldsmith CH, Clase CM. A systematic review and meta-analysis of hypoglycemia and cardiovascular events: a comparison of glyburide with other secretagogues and with insulin. Diabetes Care 2007; 30(2):389–394. doi:10.2337/dc06-1789
- Nauck M, Frid A, Hermansen K, et al; LEAD-2 Study Group. Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care 2009; 32(1):84–90. doi:10.2337/dc08-1355
- Centers for Disease Control and Prevention. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed August 5, 2019.
- Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr 2010; 8:29. doi:10.1186/1478-7954-8-29
- Sinclair AJ, Paolisso G, Castro M, Bourdel-Marchasson I, Gadsby R, Rodriguez Mañas L; European Diabetes Working Party for Older People. European Diabetes Working Party for Older People 2011 clinical guidelines for type 2 diabetes mellitus. Executive summary. Diabetes Metab 2011; 37(suppl 3):S27–S38. doi:10.1016/S1262-3636(11)70962-4
- American Geriatrics Society Expert Panel on Care of Older Adults with Diabetes Mellitus; Moreno G, Mangione CM, Kimbro L, Vaisberg E. Guidelines abstracted from the American Geriatrics Society Guidelines for Improving the Care of Older Adults with Diabetes Mellitus: 2013 update. J Am Geriatr Soc 2013; 61(11):2020–2026. doi:10.1111/jgs.12514
KEY POINTS
- Some glucagon-like peptide 1 (GLP-1) receptor agonists have been shown to reduce cardiovascular risk, and liraglutide carries an indication for this use.
- The sodium-glucose cotransporter 2 inhibitors empaglifozin and canaglifozin carry indications to prevent cardiovascular death in patients with diabetes with established cardiovascular disease.
- Metformin, GLP-1 receptor agonists, and dipeptidyl peptidase 4 inhibitors are beneficial in terms of promoting weight loss—or at least not causing weight gain.
- Disadvantages and adverse effects of various drugs must also be considered.
ACE inhibitors and ARBs: Managing potassium and renal function
A highly active, water- and alcohol-soluble, basic pressor substance is formed when renin and renin-activator interact, for which we suggest the name “angiotonin.”
—Irvine H. Page and O.M. Helmer, 1940.1
The renin-angiotensin-aldosterone system regulates salt and, in part, water homeostasis, and therefore blood pressure and fluid balance through its actions on the heart, kidneys, and blood vessels.2 Drugs that target this system—angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs)—are used primarily to treat hypertension and also to treat chronic kidney disease and heart failure with reduced ejection fraction.
Controlling blood pressure is important, as hypertension increases the risk of myocardial infarction, cerebrovascular events, and progression of chronic kidney disease, which itself is a risk factor for cardiovascular disease. However, the benefit of these drugs is only partly due to their effect on blood pressure. They also reduce proteinuria, which is a graded risk factor for progression of kidney disease as well as morbidity and death from vascular events.3
Despite the benefits of ACE inhibitors and ARBs, concern about their adverse effects—especially hyperkalemia and a decline in renal function—has led to their underuse in patients likely to derive the greatest benefit.3
ACE INHIBITORS AND ARBs

ACE inhibitors, as their name indicates, inhibit conversion of angiotensin I to angiotensin II by ACE, resulting in vasodilation of the efferent arteriole and a drop in blood pressure. Inhibition of ACE, a kininase, also results in a rise in kinins. One of these, bradykinin, is associated with some of the side effects of this class of drugs such as cough, which affects 5% to 20% of patients.4 Elevation of bradykinin is also believed to account for ACE inhibitor-induced angioedema, an uncommon but potentially serious side effect. Kinins are also associated with desirable effects such as lowering blood pressure, increasing insulin sensitivity, and dilating blood vessels.
ARBs were developed as an alternative for patients unable to tolerate the adverse effects of ACE inhibitors. While ACE inhibitors reduce the activity of angiotensin II at both the AT1 and AT2 receptors, ARBs block only the AT1 receptors, thereby inhibiting their vasoconstricting activity on smooth muscle. ARBs also raise the levels of renin, angiotensin I, and angiotensin II as a result of feedback inhibition. Angiotensin II is associated with release of inflammatory mediators such as tumor necrosis factor alpha, cytokines, and chemokines, the consequences of which are also inhibited by ARBs, further preventing renal fibrosis and scarring from chronic inflammation.3
What is the evidence supporting the use of ACE inhibitors and ARBs?
ACE inhibitors and ARBs, used singly, reduce blood pressure and proteinuria, slow progression of kidney disease, and improve outcomes in patients who have heart failure, diabetes mellitus, or a history of myocardial infarction.5–11
While dual blockade with the combination of an ACE inhibitor and an ARB lowers blood pressure and proteinuria to a greater degree than monotherapy, dual blockade has been associated with higher rates of complications, including hyperkalemia.12–17
RISK FACTORS FOR HYPERKALEMIA
ACE inhibitors and ARBs raise potassium, especially when used in combination. Other risk factors for hyperkalemia include the following—and note that some of them are also indications for ACE inhibitors and ARBs:
Renal insufficiency. The kidneys are responsible for over 90% of potassium removal in healthy individuals,18,19 and the lower the GFR, the higher the risk of hyperkalemia.3,20,21
Heart failure
Diabetes mellitus6,21–23
Endogenous potassium load due to hemolysis, rhabdomyolysis, insulin deficiency, lactic acidosis, or gastrointestinal bleeding
Exogenous potassium load due to dietary consumption or blood products
Other medications, eg, sacubitril-valsartan, aldosterone antagonists, mineralocorticoid receptor antagonists, potassium-sparing diuretics, beta-adrenergic antagonists, nonsteroidal anti-inflammatory drugs, heparin, cyclosporine, trimethoprim, digoxin
Hypertension
Hypoaldosteronism (including type 4 renal tubular acidosis)
Addison disease
Advanced age
Lower body mass index.
Both hypokalemia and hyperkalemia are associated with a higher risk of death,20,21,24 but in patients with heart failure, the survival benefit from ACE inhibitors, ARBs, and mineralocorticoid receptor antagonists outweighs the risk of hyperkalemia.25–27 Weir and Rolfe28 concluded that patients with heart failure and chronic kidney disease are at greatest risk of hyperkalemia from renin-angiotensin-aldosterone system inhibition, but the increases in potassium levels are small (about 0.1 to 0.3 mmol/L) and unlikely to be clinically significant.
Hyperkalemia tends to recur. Einhorn et al20 found that nearly half of patients with chronic kidney disease who had an episode of hyperkalemia had 1 or more recurrent episodes within a year.
ACE INHIBITORS, ARBs, ABD RENAL FUNCTION
Another concern about using ACE inhibitors and ARBs, especially in patients with chronic kidney disease, is that the serum creatinine level tends to rise when starting these drugs,29 although several studies have shown that an acute rise in creatinine may demonstrate that the drug is actually protecting the kidney.30,31 Hirsch32 described this phenomenon as “prerenal success,” proposing that the decline in GFR is hemodynamic, secondary to a fall in intraglomerular pressure as a result of efferent vasodilation, and therefore should not be reversed.
Schmidt et al,33,34 in a study in 122,363 patients who began ACE inhibitor or ARB therapy, found that cardiorenal outcomes were worse, with higher rates of end-stage renal disease, myocardial infarction, heart failure, and death, in those in whom creatinine rose by 30% or more since starting treatment. This trend was also seen, to a lesser degree, in those with a smaller increase in creatinine, suggesting that even this group of patients should receive close monitoring.
Whether renin-angiotensin-aldosterone system inhibitors provide a benefit in advanced progressive chronic kidney disease remains unclear.35–37 The Angiotensin Converting Enzyme Inhibitor (ACEi)/Angiotensin Receptor Blocker (ARB) Withdrawal in Advanced Renal Disease trial (STOP-ACEi),38 currently under way, will provide valuable data to help close this gap in our knowledge. This open-label randomized controlled trial is testing the hypothesis that stopping ACE inhibitor or ARB treatment, or a combination of both, compared with continuing these treatments, will improve or stabilize renal function in patients with progressive stage 4 or 5 chronic kidney disease.
NEED FOR MONITORING
Taken together, the above data suggest close and regular monitoring is required in patients receiving these drugs. However, monitoring tends to be lax.34,37,39 A 2017 study of adherence to the guidelines for monitoring serum creatinine and potassium after starting an ACE inhibitor or ARB and subsequent discontinuation found that fewer than 10% of patients had follow-up within the recommended 2 weeks after starting these drugs.34 Most patients with a creatinine rise of 30% or more or a potassium level higher than 6.0 mmol/L continued treatment. There was also no evidence of increased monitoring in those deemed at higher risk of these complications.
WHAT DO THE GUIDELINES SUGGEST?
ACE inhibitors and ARBs in chronic kidney disease and hypertension
Target blood pressures vary in guidelines from different organizations.4,40–45 The 2017 joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA)40 recommend a target blood pressure of 130/80 mm Hg or less in all patients irrespective of the level of proteinuria and whether they have diabetes mellitus, based on several studies.46–48 In the elderly, other factors such as the risk of hypotension and falls must be taken into consideration in establishing the most appropriate blood pressure target.
In general, a renin-angiotensin-aldosterone system inhibitor is recommended if the patient has diabetes, stage 1, 2, or 3 chronic kidney disease, or proteinuria. For example, the guidelines recommend a renin-angiotensin-aldosterone system inhibitor in diabetic patients with albuminuria.
None of the guidelines recommend routine use of combination therapy.
ACE inhibitors and ARBs in heart failure
The 2017 ACC/AHA and Heart Failure Society of America (HFSA) guidelines for heart failure49 recommend an ACE inhibitor or ARB for patients with stage C (symptomatic) heart failure with reduced ejection fraction, in view of the known cardiovascular morbidity and mortality benefits.
The European Society of Cardiology50 recommends ACE inhibitors for patients with symptomatic heart failure with reduced ejection fraction, as well as those with asymptomatic left ventricular systolic dysfunction. In patients with stable coronary artery disease, an ACE inhibitor should be considered even with normal left ventricular function.
ARBs should be used as alternatives in those unable to tolerate ACE inhibitors.
Combination therapy should be avoided due to the increased risk of renal impairment and hyperkalemia but may be considered in patients with heart failure and reduced ejection fraction in whom other treatments are unsuitable. These include patients on beta-blockers who cannot tolerate mineralocorticoid receptor antagonists such as spironolactone. Combination therapy should be done only under strict supervision.50
Starting ACE or ARB therapy
Close monitoring of serum potassium is recommended during ACE inhibitor or ARB use. Those at greatest risk of hyperkalemia include elderly patients, those taking other medications associated with hyperkalemia, and diabetic patients, because of their higher risk of renovascular disease.
Caution is advised when starting ACE inhibitor or ARB therapy in these high-risk groups as well as in patients with potassium levels higher than 5.0 mmol/L at baseline, at high risk of prerenal acute kidney injury, with known renal insufficiency, and with previous deterioration in renal function on these medications.3,41,51
Before starting therapy, ensure that patients are volume-replete and measure baseline serum electrolytes and creatinine.41,51
The ACC/AHA and HFSA recommend starting at a low dose and titrating upward slowly. If maximal doses are not tolerated, then a lower dose should be maintained.49 The European Society of Cardiology guidelines52 suggest increasing the dose at no less than every 2 weeks unless in an inpatient setting. Blood testing should be done 7 to 14 days after starting therapy, after any titration in dosage, and every 4 months thereafter.53
The guidelines generally agree that a rise in creatinine of up to 30% and a fall in eGFR of up to 25% is acceptable, with the need for regular monitoring, particularly in high-risk groups.40–42,51,52
What if serum potassium or creatinine rises during treatment?
If hyperkalemia arises or renal function declines by a significant amount, one should first address contributing factors. If no improvement is seen, then the dose of the ACE inhibitor or ARB should be reduced by 50% and blood work repeated in 1 to 2 weeks. If the laboratory values do not return to an acceptable level, reducing the dose further or stopping the drug is advised.
Give dietary advice to all patients with chronic kidney disease being considered for a renin-angiotensin-aldosterone system inhibitor or for an increase in dose with a potassium level higher than 4.5 mmol/L. A low-potassium diet should aim for potassium intake of less than 50 or 75 mmol/day and sodium intake of less than 60 mmol/day for hypertensive patients with chronic kidney disease.
Review the patient’s medications if the baseline potassium level is higher than 5.0 mmol/L. Consider stopping potassium-sparing agents, digoxin, trimethoprim, and nonsteroidal anti-inflammatory drugs. Also think about starting a non–potassium-sparing diuretic as well as sodium bicarbonate to reduce potassium levels. Blood work should be repeated within 2 weeks after these changes.
Do not start a renin-angiotensin-aldosterone system inhibitor, or do not increase the dose, if the potassium level is elevated until measures have been taken to reduce the degree of hyperkalemia.51
In renal transplant recipients, renin-angiotensin-aldosterone system inhibitors are often preferred to manage hypertension in those who have proteinuria or cardiovascular disease. However, the risk of hyperkalemia is also greater with concomitant use of immunosuppressive drugs such as tacrolimus and cyclosporine. Management of complications should be approached according to guidelines discussed above.51
Monitor renal function, potassium. The National Institute for Health and Care Excellence guideline54 advocates that baseline renal function testing should be followed by repeat blood testing 1 to 2 weeks after starting renin-angiotensin-aldosterone system inhibitors in patients with ischemic heart disease. The advice is similar when starting therapy in patients with chronic heart failure, emphasizing the need to monitor after each dose increment and to use clinical judgment when deciding to start treatment. The AHA advises caution in patients with renal insufficiency or a potassium level above 5.0 mmol/L.49
Sick day rules. The National Institute for Health and Care Excellence encourages discussing “sick day rules” with patients starting renin-angiotensin-aldosterone system inhibitors. This means patients should be advised to temporarily stop taking nephrotoxic medications, including over-the-counter nonsteroidal anti-inflammatory drugs, in any potential state of illness or dehydration, such as diarrhea and vomiting. There is, however, little evidence that this advice can actually reduce the incidence of acute kidney injury.55,56

OUR RECOMMENDATIONS
Our advice for managing patients receiving ACE inhibitors or ARBs is summarized in Table 1.
- Page IH, Helmer OM. A crystalline pressor substance (angiotonin) resulting from the reaction between renin and renin-activator. Exp Med 1940; 71(1):29–42. doi:10.1084/jem.71.1.29
- Steddon S, Ashman N, Chesser A, Cunningham J. Oxford Handbook of Nephrology and Hypertension. 2nd ed. Oxford: Oxford University Press; 2016:203–206, 508–509.
- Barratt J, Topham P, Harris K. Oxford Desk Reference. 1st ed. Oxford: Oxford University Press; 2008.
- International Kidney Foundation. KDIGO clinical practice guideline for the management of blood pressure in chronic kidney disease. http://www.kdigo.org/clinical_practice_guidelines/pdf/KDIGO_BP_GL.pdf. Accessed April 3, 2019.
- Heart Outcomes Prevention Evaluation Study Investigators; Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med 2000; 342(3):145–153. doi:10.1056/NEJM200001203420301
- Swedberg K, Kjekshus J. Effects of enalapril on mortality in severe congestive heart failure: results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). Am J Cardiol 1988; 62(2):60A–66A. pmid:2839019
- Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345(12):861–869. doi:10.1056/NEJMoa011161
- Pfeffer MA, McMurray JJ, Velazquez EJ, et al. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003; 349(20):1893–1906. doi:10.1056/NEJMoa032292
- Epstein M. Reduction of cardiovascular risk in chronic kidney disease by mineralocorticoid receptor antagonism. Lancet Diabetes Endocrinol 2015; 3(12):993–1003. doi:10.1016/S2213-8587(15)00289-2
- SOLVD Investigators; Yusuf S, Pitt B, Davis CE, Hood WB, Cohn JN. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med 1991; 325(5):293–302. doi:10.1056/NEJM199108013250501
- Jafar TH, Stark PC, Schmid CH, et al; AIPRD Study Group; Angiotensin-Converting Enzymne Inhibition and Progression of Renal Disease. Proteinuria as a modifiable risk factor for the progression of non-diabetic renal disease. Kidney Int 2001; 60(3):1131–1140. doi:10.1046/j.1523-1755.2001.0600031131.x
- Palmer SC, Mavridis D, Navarese E, et al. Comparative efficacy and safety of blood pressure-lowering agents in adults with diabetes and kidney disease: a network meta-analysis. Lancet 2015; 385(9982):2047–2056. doi:10.1016/S0140-6736(14)62459-4
- Ruggenenti P, Perticucci E, Cravedi P, et al. Role of remission clinics in the longitudinal treatment of CKD. J Am Soc Nephrol 2008; 19(6):1213–1224. doi:10.1681/ASN.2007090970
- Makani H, Bangalore S, Desouza KA, Shah A, Messerli FH. Efficacy and safety of dual blockade of the renin-angiotensin system: meta-analysis of randomised trials. BMJ 2013; 346:f360. doi:10.1136/bmj.f360
- ONTARGET Investigators; Yusuf S, Teo KK, Pogue J, et al. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008; 358(15):1547–1559. doi:10.1056/NEJMoa0801317
- Fried LF, Emanuele N, Zhang JH, et al; VA NEPHRON-D Investigators. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med 2013; 369(20):1892–1903.
doi:10.1056/NEJMoa1303154 - Catalá-López F, Macías Saint-Gerons D, González-Bermejo D, et al. Cardiovascular and renal outcomes of renin-angiotensin system blockade in adult patients with diabetes mellitus: a systematic review with network meta-analyses. PLoS Med 2016; 13(3):e1001971. doi:10.1371/journal.pmed.1001971
- Agarwal R, Afzalpurkar R, Fordtran JS. Pathophysiology of potassium absorption and secretion by the human intestine. Gastroenterology 1994; 107(2):548–571. pmid:8039632
- Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10(6):1050–1060. doi:10.2215/CJN.08580813
- Einhorn LM, Zhan M, Hsu VD, et al. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Intern Med 2009; 169(12):1156–1162. doi:10.1001/archinternmed.2009.132
- Nakhoul GN, Huang H, Arrigain S, et al. Serum potassium, end-stage renal disease and mortality in chronic kidney disease. Am J Nephrol 2015; 41(6):456–463. doi:10.1159/000437151
- Acker CG, Johnson JP, Palevsky PM, Greenberg A. Hyperkalemia in hospitalized patients: causes, adequacy of treatment, and results of an attempt to improve physician compliance with published therapy guidelines. Arch Intern Med 1998; 158(8):917–924. pmid:9570179
- Desai AS, Swedberg K, McMurray JJ, et al; CHARM Program Investigators. Incidence and predictors of hyperkalemia in patients with heart failure: an analysis of the CHARM Program. J Am Coll Cardiol 2007; 50(20):1959–1966. doi:10.1016/j.jacc.2007.07.067
- Cheungpasitporn W, Thongprayoon C, Kittanamongkolchai W, Sakhuja A, Mao MA, Erickson SB. Impact of admission serum potassium on mortality in patients with chronic kidney disease and cardiovascular disease. QJM 2017; 110(11):713–719. doi:10.1093/qjmed/hcx118
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364(1):11–21. doi:10.1056/NEJMoa1009492
- Rossignol P, Dobre D, McMurray JJ, et al. Incidence, determinants, and prognostic significance of hyperkalemia and worsening renal function in patients with heart failure receiving the mineralocorticoid receptor antagonist eplerenone or placebo in addition to optimal medical therapy: results from the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF). Circ Heart Fail 2014; 7(1):51–58. doi:10.1161/CIRCHEARTFAILURE.113.000792
- Testani JM, Kimmel SE, Dries DL, Coca SG. Prognostic importance of early worsening renal function after initiation of angiotensin-converting enzyme inhibitor therapy in patients with cardiac dysfunction. Circ Heart Fail 2011; 4(6):685–691. doi:10.1161/CIRCHEARTFAILURE.111.963256
- Weir M, Rolfe M. Potassium homeostasis and renin-angiotensin-aldosterone system inhibitors. Clin J Am Soc Nephrol 2010; 5(3):531–548. doi:10.2215/CJN.07821109
- Valente M, Bhandari S. Renal function after new treatment with renin-angiotensin system blockers. BMJ 2017; 356:j1122. doi:10.1136/bmj.j1122
- Bakris G, Weir M. Angiotensin-converting enzyme inhibitor–associated elevations in serum creatinine. Arch Intern Med 2000; 160(5):685–693. pmid:10724055
- Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345(12):861–869. doi:10.1056/NEJMoa011161
- Hirsch S. Pre-renal success. Kidney Int 2012; 81(6):596. doi:10.1038/ki.2011.418
- Schmidt M, Mansfield KE, Bhaskaran K, et al. Serum creatinine elevation after renin-angiotensin system blockade and long term cardiorenal risks: cohort study. BMJ 2017; 356:j791. doi:10.1136/bmj.j791
- Schmidt M, Mansfield KE, Bhaskaran K, et al. Adherence to guidelines for creatinine and potassium monitoring and discontinuation following renin–angiotensin system blockade: a UK general practice-based cohort study. BMJ Open 2017; 7(1):e012818. doi:10.1136/bmjopen-2016-012818
- Lund LH, Carrero JJ, Farahmand B, et al. Association between enrollment in a heart failure quality registry and subsequent mortality—a nationwide cohort study. Eur J Heart Fail 2017; 19(9):1107–1116. doi:10.1002/ejhf.762
- Edner M, Benson L, Dahlstrom U, Lund LH. Association between renin-angiotensin system antagonist use and mortality in heart failure with severe renal insuffuciency: a prospective propensity score-matched cohort study. Eur Heart J 2015; 36(34):2318–2326. doi:10.1093/eurheartj/ehv268
- Epstein M, Reaven NL, Funk SE, McGaughey KJ, Oestreicher N, Knispel J. Evaluation of the treatment gap between clinical guidelines and the utilization of renin-angiotensin-aldosterone system inhibitors. Am J Manag Care 2015; 21(suppl 11):S212–S220. pmid:26619183
- Bhandari S, Ives N, Brettell EA, et al. Multicentre randomized controlled trial of angiotensin-converting enzyme inhibitor/angiotensin receptor blocker withdrawal in advanced renal disease: the STOP-ACEi trial. Nephrol Dial Transplant 2016; 31(2):255–261. doi:10.1093/ndt/gfv346
- Raebel MA, Ross C, Xu S, et al. Diabetes and drug-associated hyperkalemia: effect of potassium monitoring. J Gen Intern Med 2010; 25(4):326–333. doi:10.1007/s11606-009-1228-x
- Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018; 71(6):e13–e115. doi:10.1161/HYP.0000000000000065
- The Renal Association. The UK eCKD Guide. https://renal.org/information-resources/the-uk-eckd-guide. Accessed August 12, 2019.
- National Institute for Health and Care Excellence (NICE). Chronic kidney disease in adults: assessment and management. https://www.nice.org.uk/guidance/cg182. Accessed August 12, 2019.
- National Institute for Health and Care Excellence (NICE). Hypertension in adults: diagnosis and management. https://www.nice.org.uk/Guidance/CG127. Accessed August 12, 2019.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34(28):2159–2219. doi:10.1093/eurheartj/eht151
- International Kidney Foundation. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. https://www.sciencedirect.com/journal/kidney-international-supplements/vol/3/issue/1. Accessed August 12, 2019.
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373(22):2103–2116. doi:10.1056/NEJMoa1511939
- Wright J, Bakris G, Greene T. Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease. Results from the AASK trial. ACC Current Journal Review 2003; 12(2):37–38. doi:10.1016/s1062-1458(03)00035-7
- Ku E, Bakris G, Johansen K, et al. Acute declines in renal function during intensive BP lowering: implications for future ESRD risk. J Am Soc Nephrol 2017; 28(9):2794–2801. doi:10.1681/ASN.2017010040
- Yancy CW, Jessup M, Bozkurt B, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2017; 136(6):e137–e161. doi:10.1161/CIR.0000000000000509
- Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016; 37(27):2129–2200. doi:10.1093/eurheartj/ehw128
- Kidney Disease Outcomes Quality Initiative (K/DOQI). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 51):S1–S290. pmid:15114537
- Asenjo RM, Bueno H, Mcintosh M. Angiotensin converting enzyme inhibitors (ACE inhibitors) and angiotensin II receptor blockers (ARBs). ACE inhibitors and ARBs, a cornerstone in the prevention and treatment of cardiovascular disease. www.escardio.org/Education/ESC-Prevention-of-CVD-Programme/Treatment-goals/Cardio-Protective-drugs/angiotensin-converting-enzyme-inhibitors-ace-inhibitors-and-angiotensin-ii-rec. Accessed August 12, 2019.
- López-Sendón J, Swedberg K, McMurray J, et al; Task Force on ACE-inhibitors of the European Society of Cardiology. Expert consensus document on angiotensin converting enzyme inhibitors in cardiovascular disease. The Task Force on ACE-inhibitors of the European Society of Cardiology. Eur Heart J 2004; 25(16):1454–1470. doi:10.1016/j.ehj.2004.06.003
- National Institute for Health and Care Excellence (NICE). Myocardial infarction: cardiac rehabilitation and prevention of further cardiovascular disease. https://www.nice.org.uk/Guidance/CG172. Accessed April 3, 2019.
- National Institute for Health and Care Excellence (NICE). Acute kidney injury: prevention, detection and management. https://www.nice.org.uk/Guidance/CG169. Accessed August 12, 2019.
- Think Kidneys. “Sick day” guidance in patients at risk of acute kidney injury: a position statement from the Think Kidneys Board. https://www.thinkkidneys.nhs.uk/aki/wp-content/uploads/sites/2/2018/01/Think-Kidneys-Sick-Day-Guidance-2018.pdf. Accessed August 12, 2019.
- Meaney CJ, Beccari MV, Yang Y, Zhao J. Systematic review and meta-analysis of patiromer and sodium zirconium cyclosilicate: a new armamentarium for the treatment of hyperkalemia. Pharmacotherapy 2017; 37(4):401–411. doi:10.1002/phar.1906
A highly active, water- and alcohol-soluble, basic pressor substance is formed when renin and renin-activator interact, for which we suggest the name “angiotonin.”
—Irvine H. Page and O.M. Helmer, 1940.1
The renin-angiotensin-aldosterone system regulates salt and, in part, water homeostasis, and therefore blood pressure and fluid balance through its actions on the heart, kidneys, and blood vessels.2 Drugs that target this system—angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs)—are used primarily to treat hypertension and also to treat chronic kidney disease and heart failure with reduced ejection fraction.
Controlling blood pressure is important, as hypertension increases the risk of myocardial infarction, cerebrovascular events, and progression of chronic kidney disease, which itself is a risk factor for cardiovascular disease. However, the benefit of these drugs is only partly due to their effect on blood pressure. They also reduce proteinuria, which is a graded risk factor for progression of kidney disease as well as morbidity and death from vascular events.3
Despite the benefits of ACE inhibitors and ARBs, concern about their adverse effects—especially hyperkalemia and a decline in renal function—has led to their underuse in patients likely to derive the greatest benefit.3
ACE INHIBITORS AND ARBs
ACE inhibitors, as their name indicates, inhibit conversion of angiotensin I to angiotensin II by ACE, resulting in vasodilation of the efferent arteriole and a drop in blood pressure. Inhibition of ACE, a kininase, also results in a rise in kinins. One of these, bradykinin, is associated with some of the side effects of this class of drugs such as cough, which affects 5% to 20% of patients.4 Elevation of bradykinin is also believed to account for ACE inhibitor-induced angioedema, an uncommon but potentially serious side effect. Kinins are also associated with desirable effects such as lowering blood pressure, increasing insulin sensitivity, and dilating blood vessels.
ARBs were developed as an alternative for patients unable to tolerate the adverse effects of ACE inhibitors. While ACE inhibitors reduce the activity of angiotensin II at both the AT1 and AT2 receptors, ARBs block only the AT1 receptors, thereby inhibiting their vasoconstricting activity on smooth muscle. ARBs also raise the levels of renin, angiotensin I, and angiotensin II as a result of feedback inhibition. Angiotensin II is associated with release of inflammatory mediators such as tumor necrosis factor alpha, cytokines, and chemokines, the consequences of which are also inhibited by ARBs, further preventing renal fibrosis and scarring from chronic inflammation.3
What is the evidence supporting the use of ACE inhibitors and ARBs?
ACE inhibitors and ARBs, used singly, reduce blood pressure and proteinuria, slow progression of kidney disease, and improve outcomes in patients who have heart failure, diabetes mellitus, or a history of myocardial infarction.5–11
While dual blockade with the combination of an ACE inhibitor and an ARB lowers blood pressure and proteinuria to a greater degree than monotherapy, dual blockade has been associated with higher rates of complications, including hyperkalemia.12–17
RISK FACTORS FOR HYPERKALEMIA
ACE inhibitors and ARBs raise potassium, especially when used in combination. Other risk factors for hyperkalemia include the following—and note that some of them are also indications for ACE inhibitors and ARBs:
Renal insufficiency. The kidneys are responsible for over 90% of potassium removal in healthy individuals,18,19 and the lower the GFR, the higher the risk of hyperkalemia.3,20,21
Heart failure
Diabetes mellitus6,21–23
Endogenous potassium load due to hemolysis, rhabdomyolysis, insulin deficiency, lactic acidosis, or gastrointestinal bleeding
Exogenous potassium load due to dietary consumption or blood products
Other medications, eg, sacubitril-valsartan, aldosterone antagonists, mineralocorticoid receptor antagonists, potassium-sparing diuretics, beta-adrenergic antagonists, nonsteroidal anti-inflammatory drugs, heparin, cyclosporine, trimethoprim, digoxin
Hypertension
Hypoaldosteronism (including type 4 renal tubular acidosis)
Addison disease
Advanced age
Lower body mass index.
Both hypokalemia and hyperkalemia are associated with a higher risk of death,20,21,24 but in patients with heart failure, the survival benefit from ACE inhibitors, ARBs, and mineralocorticoid receptor antagonists outweighs the risk of hyperkalemia.25–27 Weir and Rolfe28 concluded that patients with heart failure and chronic kidney disease are at greatest risk of hyperkalemia from renin-angiotensin-aldosterone system inhibition, but the increases in potassium levels are small (about 0.1 to 0.3 mmol/L) and unlikely to be clinically significant.
Hyperkalemia tends to recur. Einhorn et al20 found that nearly half of patients with chronic kidney disease who had an episode of hyperkalemia had 1 or more recurrent episodes within a year.
ACE INHIBITORS, ARBs, ABD RENAL FUNCTION
Another concern about using ACE inhibitors and ARBs, especially in patients with chronic kidney disease, is that the serum creatinine level tends to rise when starting these drugs,29 although several studies have shown that an acute rise in creatinine may demonstrate that the drug is actually protecting the kidney.30,31 Hirsch32 described this phenomenon as “prerenal success,” proposing that the decline in GFR is hemodynamic, secondary to a fall in intraglomerular pressure as a result of efferent vasodilation, and therefore should not be reversed.
Schmidt et al,33,34 in a study in 122,363 patients who began ACE inhibitor or ARB therapy, found that cardiorenal outcomes were worse, with higher rates of end-stage renal disease, myocardial infarction, heart failure, and death, in those in whom creatinine rose by 30% or more since starting treatment. This trend was also seen, to a lesser degree, in those with a smaller increase in creatinine, suggesting that even this group of patients should receive close monitoring.
Whether renin-angiotensin-aldosterone system inhibitors provide a benefit in advanced progressive chronic kidney disease remains unclear.35–37 The Angiotensin Converting Enzyme Inhibitor (ACEi)/Angiotensin Receptor Blocker (ARB) Withdrawal in Advanced Renal Disease trial (STOP-ACEi),38 currently under way, will provide valuable data to help close this gap in our knowledge. This open-label randomized controlled trial is testing the hypothesis that stopping ACE inhibitor or ARB treatment, or a combination of both, compared with continuing these treatments, will improve or stabilize renal function in patients with progressive stage 4 or 5 chronic kidney disease.
NEED FOR MONITORING
Taken together, the above data suggest close and regular monitoring is required in patients receiving these drugs. However, monitoring tends to be lax.34,37,39 A 2017 study of adherence to the guidelines for monitoring serum creatinine and potassium after starting an ACE inhibitor or ARB and subsequent discontinuation found that fewer than 10% of patients had follow-up within the recommended 2 weeks after starting these drugs.34 Most patients with a creatinine rise of 30% or more or a potassium level higher than 6.0 mmol/L continued treatment. There was also no evidence of increased monitoring in those deemed at higher risk of these complications.
WHAT DO THE GUIDELINES SUGGEST?
ACE inhibitors and ARBs in chronic kidney disease and hypertension
Target blood pressures vary in guidelines from different organizations.4,40–45 The 2017 joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA)40 recommend a target blood pressure of 130/80 mm Hg or less in all patients irrespective of the level of proteinuria and whether they have diabetes mellitus, based on several studies.46–48 In the elderly, other factors such as the risk of hypotension and falls must be taken into consideration in establishing the most appropriate blood pressure target.
In general, a renin-angiotensin-aldosterone system inhibitor is recommended if the patient has diabetes, stage 1, 2, or 3 chronic kidney disease, or proteinuria. For example, the guidelines recommend a renin-angiotensin-aldosterone system inhibitor in diabetic patients with albuminuria.
None of the guidelines recommend routine use of combination therapy.
ACE inhibitors and ARBs in heart failure
The 2017 ACC/AHA and Heart Failure Society of America (HFSA) guidelines for heart failure49 recommend an ACE inhibitor or ARB for patients with stage C (symptomatic) heart failure with reduced ejection fraction, in view of the known cardiovascular morbidity and mortality benefits.
The European Society of Cardiology50 recommends ACE inhibitors for patients with symptomatic heart failure with reduced ejection fraction, as well as those with asymptomatic left ventricular systolic dysfunction. In patients with stable coronary artery disease, an ACE inhibitor should be considered even with normal left ventricular function.
ARBs should be used as alternatives in those unable to tolerate ACE inhibitors.
Combination therapy should be avoided due to the increased risk of renal impairment and hyperkalemia but may be considered in patients with heart failure and reduced ejection fraction in whom other treatments are unsuitable. These include patients on beta-blockers who cannot tolerate mineralocorticoid receptor antagonists such as spironolactone. Combination therapy should be done only under strict supervision.50
Starting ACE or ARB therapy
Close monitoring of serum potassium is recommended during ACE inhibitor or ARB use. Those at greatest risk of hyperkalemia include elderly patients, those taking other medications associated with hyperkalemia, and diabetic patients, because of their higher risk of renovascular disease.
Caution is advised when starting ACE inhibitor or ARB therapy in these high-risk groups as well as in patients with potassium levels higher than 5.0 mmol/L at baseline, at high risk of prerenal acute kidney injury, with known renal insufficiency, and with previous deterioration in renal function on these medications.3,41,51
Before starting therapy, ensure that patients are volume-replete and measure baseline serum electrolytes and creatinine.41,51
The ACC/AHA and HFSA recommend starting at a low dose and titrating upward slowly. If maximal doses are not tolerated, then a lower dose should be maintained.49 The European Society of Cardiology guidelines52 suggest increasing the dose at no less than every 2 weeks unless in an inpatient setting. Blood testing should be done 7 to 14 days after starting therapy, after any titration in dosage, and every 4 months thereafter.53
The guidelines generally agree that a rise in creatinine of up to 30% and a fall in eGFR of up to 25% is acceptable, with the need for regular monitoring, particularly in high-risk groups.40–42,51,52
What if serum potassium or creatinine rises during treatment?
If hyperkalemia arises or renal function declines by a significant amount, one should first address contributing factors. If no improvement is seen, then the dose of the ACE inhibitor or ARB should be reduced by 50% and blood work repeated in 1 to 2 weeks. If the laboratory values do not return to an acceptable level, reducing the dose further or stopping the drug is advised.
Give dietary advice to all patients with chronic kidney disease being considered for a renin-angiotensin-aldosterone system inhibitor or for an increase in dose with a potassium level higher than 4.5 mmol/L. A low-potassium diet should aim for potassium intake of less than 50 or 75 mmol/day and sodium intake of less than 60 mmol/day for hypertensive patients with chronic kidney disease.
Review the patient’s medications if the baseline potassium level is higher than 5.0 mmol/L. Consider stopping potassium-sparing agents, digoxin, trimethoprim, and nonsteroidal anti-inflammatory drugs. Also think about starting a non–potassium-sparing diuretic as well as sodium bicarbonate to reduce potassium levels. Blood work should be repeated within 2 weeks after these changes.
Do not start a renin-angiotensin-aldosterone system inhibitor, or do not increase the dose, if the potassium level is elevated until measures have been taken to reduce the degree of hyperkalemia.51
In renal transplant recipients, renin-angiotensin-aldosterone system inhibitors are often preferred to manage hypertension in those who have proteinuria or cardiovascular disease. However, the risk of hyperkalemia is also greater with concomitant use of immunosuppressive drugs such as tacrolimus and cyclosporine. Management of complications should be approached according to guidelines discussed above.51
Monitor renal function, potassium. The National Institute for Health and Care Excellence guideline54 advocates that baseline renal function testing should be followed by repeat blood testing 1 to 2 weeks after starting renin-angiotensin-aldosterone system inhibitors in patients with ischemic heart disease. The advice is similar when starting therapy in patients with chronic heart failure, emphasizing the need to monitor after each dose increment and to use clinical judgment when deciding to start treatment. The AHA advises caution in patients with renal insufficiency or a potassium level above 5.0 mmol/L.49
Sick day rules. The National Institute for Health and Care Excellence encourages discussing “sick day rules” with patients starting renin-angiotensin-aldosterone system inhibitors. This means patients should be advised to temporarily stop taking nephrotoxic medications, including over-the-counter nonsteroidal anti-inflammatory drugs, in any potential state of illness or dehydration, such as diarrhea and vomiting. There is, however, little evidence that this advice can actually reduce the incidence of acute kidney injury.55,56
OUR RECOMMENDATIONS
Our advice for managing patients receiving ACE inhibitors or ARBs is summarized in Table 1.
A highly active, water- and alcohol-soluble, basic pressor substance is formed when renin and renin-activator interact, for which we suggest the name “angiotonin.”
—Irvine H. Page and O.M. Helmer, 1940.1
The renin-angiotensin-aldosterone system regulates salt and, in part, water homeostasis, and therefore blood pressure and fluid balance through its actions on the heart, kidneys, and blood vessels.2 Drugs that target this system—angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs)—are used primarily to treat hypertension and also to treat chronic kidney disease and heart failure with reduced ejection fraction.
Controlling blood pressure is important, as hypertension increases the risk of myocardial infarction, cerebrovascular events, and progression of chronic kidney disease, which itself is a risk factor for cardiovascular disease. However, the benefit of these drugs is only partly due to their effect on blood pressure. They also reduce proteinuria, which is a graded risk factor for progression of kidney disease as well as morbidity and death from vascular events.3
Despite the benefits of ACE inhibitors and ARBs, concern about their adverse effects—especially hyperkalemia and a decline in renal function—has led to their underuse in patients likely to derive the greatest benefit.3
ACE INHIBITORS AND ARBs
ACE inhibitors, as their name indicates, inhibit conversion of angiotensin I to angiotensin II by ACE, resulting in vasodilation of the efferent arteriole and a drop in blood pressure. Inhibition of ACE, a kininase, also results in a rise in kinins. One of these, bradykinin, is associated with some of the side effects of this class of drugs such as cough, which affects 5% to 20% of patients.4 Elevation of bradykinin is also believed to account for ACE inhibitor-induced angioedema, an uncommon but potentially serious side effect. Kinins are also associated with desirable effects such as lowering blood pressure, increasing insulin sensitivity, and dilating blood vessels.
ARBs were developed as an alternative for patients unable to tolerate the adverse effects of ACE inhibitors. While ACE inhibitors reduce the activity of angiotensin II at both the AT1 and AT2 receptors, ARBs block only the AT1 receptors, thereby inhibiting their vasoconstricting activity on smooth muscle. ARBs also raise the levels of renin, angiotensin I, and angiotensin II as a result of feedback inhibition. Angiotensin II is associated with release of inflammatory mediators such as tumor necrosis factor alpha, cytokines, and chemokines, the consequences of which are also inhibited by ARBs, further preventing renal fibrosis and scarring from chronic inflammation.3
What is the evidence supporting the use of ACE inhibitors and ARBs?
ACE inhibitors and ARBs, used singly, reduce blood pressure and proteinuria, slow progression of kidney disease, and improve outcomes in patients who have heart failure, diabetes mellitus, or a history of myocardial infarction.5–11
While dual blockade with the combination of an ACE inhibitor and an ARB lowers blood pressure and proteinuria to a greater degree than monotherapy, dual blockade has been associated with higher rates of complications, including hyperkalemia.12–17
RISK FACTORS FOR HYPERKALEMIA
ACE inhibitors and ARBs raise potassium, especially when used in combination. Other risk factors for hyperkalemia include the following—and note that some of them are also indications for ACE inhibitors and ARBs:
Renal insufficiency. The kidneys are responsible for over 90% of potassium removal in healthy individuals,18,19 and the lower the GFR, the higher the risk of hyperkalemia.3,20,21
Heart failure
Diabetes mellitus6,21–23
Endogenous potassium load due to hemolysis, rhabdomyolysis, insulin deficiency, lactic acidosis, or gastrointestinal bleeding
Exogenous potassium load due to dietary consumption or blood products
Other medications, eg, sacubitril-valsartan, aldosterone antagonists, mineralocorticoid receptor antagonists, potassium-sparing diuretics, beta-adrenergic antagonists, nonsteroidal anti-inflammatory drugs, heparin, cyclosporine, trimethoprim, digoxin
Hypertension
Hypoaldosteronism (including type 4 renal tubular acidosis)
Addison disease
Advanced age
Lower body mass index.
Both hypokalemia and hyperkalemia are associated with a higher risk of death,20,21,24 but in patients with heart failure, the survival benefit from ACE inhibitors, ARBs, and mineralocorticoid receptor antagonists outweighs the risk of hyperkalemia.25–27 Weir and Rolfe28 concluded that patients with heart failure and chronic kidney disease are at greatest risk of hyperkalemia from renin-angiotensin-aldosterone system inhibition, but the increases in potassium levels are small (about 0.1 to 0.3 mmol/L) and unlikely to be clinically significant.
Hyperkalemia tends to recur. Einhorn et al20 found that nearly half of patients with chronic kidney disease who had an episode of hyperkalemia had 1 or more recurrent episodes within a year.
ACE INHIBITORS, ARBs, ABD RENAL FUNCTION
Another concern about using ACE inhibitors and ARBs, especially in patients with chronic kidney disease, is that the serum creatinine level tends to rise when starting these drugs,29 although several studies have shown that an acute rise in creatinine may demonstrate that the drug is actually protecting the kidney.30,31 Hirsch32 described this phenomenon as “prerenal success,” proposing that the decline in GFR is hemodynamic, secondary to a fall in intraglomerular pressure as a result of efferent vasodilation, and therefore should not be reversed.
Schmidt et al,33,34 in a study in 122,363 patients who began ACE inhibitor or ARB therapy, found that cardiorenal outcomes were worse, with higher rates of end-stage renal disease, myocardial infarction, heart failure, and death, in those in whom creatinine rose by 30% or more since starting treatment. This trend was also seen, to a lesser degree, in those with a smaller increase in creatinine, suggesting that even this group of patients should receive close monitoring.
Whether renin-angiotensin-aldosterone system inhibitors provide a benefit in advanced progressive chronic kidney disease remains unclear.35–37 The Angiotensin Converting Enzyme Inhibitor (ACEi)/Angiotensin Receptor Blocker (ARB) Withdrawal in Advanced Renal Disease trial (STOP-ACEi),38 currently under way, will provide valuable data to help close this gap in our knowledge. This open-label randomized controlled trial is testing the hypothesis that stopping ACE inhibitor or ARB treatment, or a combination of both, compared with continuing these treatments, will improve or stabilize renal function in patients with progressive stage 4 or 5 chronic kidney disease.
NEED FOR MONITORING
Taken together, the above data suggest close and regular monitoring is required in patients receiving these drugs. However, monitoring tends to be lax.34,37,39 A 2017 study of adherence to the guidelines for monitoring serum creatinine and potassium after starting an ACE inhibitor or ARB and subsequent discontinuation found that fewer than 10% of patients had follow-up within the recommended 2 weeks after starting these drugs.34 Most patients with a creatinine rise of 30% or more or a potassium level higher than 6.0 mmol/L continued treatment. There was also no evidence of increased monitoring in those deemed at higher risk of these complications.
WHAT DO THE GUIDELINES SUGGEST?
ACE inhibitors and ARBs in chronic kidney disease and hypertension
Target blood pressures vary in guidelines from different organizations.4,40–45 The 2017 joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA)40 recommend a target blood pressure of 130/80 mm Hg or less in all patients irrespective of the level of proteinuria and whether they have diabetes mellitus, based on several studies.46–48 In the elderly, other factors such as the risk of hypotension and falls must be taken into consideration in establishing the most appropriate blood pressure target.
In general, a renin-angiotensin-aldosterone system inhibitor is recommended if the patient has diabetes, stage 1, 2, or 3 chronic kidney disease, or proteinuria. For example, the guidelines recommend a renin-angiotensin-aldosterone system inhibitor in diabetic patients with albuminuria.
None of the guidelines recommend routine use of combination therapy.
ACE inhibitors and ARBs in heart failure
The 2017 ACC/AHA and Heart Failure Society of America (HFSA) guidelines for heart failure49 recommend an ACE inhibitor or ARB for patients with stage C (symptomatic) heart failure with reduced ejection fraction, in view of the known cardiovascular morbidity and mortality benefits.
The European Society of Cardiology50 recommends ACE inhibitors for patients with symptomatic heart failure with reduced ejection fraction, as well as those with asymptomatic left ventricular systolic dysfunction. In patients with stable coronary artery disease, an ACE inhibitor should be considered even with normal left ventricular function.
ARBs should be used as alternatives in those unable to tolerate ACE inhibitors.
Combination therapy should be avoided due to the increased risk of renal impairment and hyperkalemia but may be considered in patients with heart failure and reduced ejection fraction in whom other treatments are unsuitable. These include patients on beta-blockers who cannot tolerate mineralocorticoid receptor antagonists such as spironolactone. Combination therapy should be done only under strict supervision.50
Starting ACE or ARB therapy
Close monitoring of serum potassium is recommended during ACE inhibitor or ARB use. Those at greatest risk of hyperkalemia include elderly patients, those taking other medications associated with hyperkalemia, and diabetic patients, because of their higher risk of renovascular disease.
Caution is advised when starting ACE inhibitor or ARB therapy in these high-risk groups as well as in patients with potassium levels higher than 5.0 mmol/L at baseline, at high risk of prerenal acute kidney injury, with known renal insufficiency, and with previous deterioration in renal function on these medications.3,41,51
Before starting therapy, ensure that patients are volume-replete and measure baseline serum electrolytes and creatinine.41,51
The ACC/AHA and HFSA recommend starting at a low dose and titrating upward slowly. If maximal doses are not tolerated, then a lower dose should be maintained.49 The European Society of Cardiology guidelines52 suggest increasing the dose at no less than every 2 weeks unless in an inpatient setting. Blood testing should be done 7 to 14 days after starting therapy, after any titration in dosage, and every 4 months thereafter.53
The guidelines generally agree that a rise in creatinine of up to 30% and a fall in eGFR of up to 25% is acceptable, with the need for regular monitoring, particularly in high-risk groups.40–42,51,52
What if serum potassium or creatinine rises during treatment?
If hyperkalemia arises or renal function declines by a significant amount, one should first address contributing factors. If no improvement is seen, then the dose of the ACE inhibitor or ARB should be reduced by 50% and blood work repeated in 1 to 2 weeks. If the laboratory values do not return to an acceptable level, reducing the dose further or stopping the drug is advised.
Give dietary advice to all patients with chronic kidney disease being considered for a renin-angiotensin-aldosterone system inhibitor or for an increase in dose with a potassium level higher than 4.5 mmol/L. A low-potassium diet should aim for potassium intake of less than 50 or 75 mmol/day and sodium intake of less than 60 mmol/day for hypertensive patients with chronic kidney disease.
Review the patient’s medications if the baseline potassium level is higher than 5.0 mmol/L. Consider stopping potassium-sparing agents, digoxin, trimethoprim, and nonsteroidal anti-inflammatory drugs. Also think about starting a non–potassium-sparing diuretic as well as sodium bicarbonate to reduce potassium levels. Blood work should be repeated within 2 weeks after these changes.
Do not start a renin-angiotensin-aldosterone system inhibitor, or do not increase the dose, if the potassium level is elevated until measures have been taken to reduce the degree of hyperkalemia.51
In renal transplant recipients, renin-angiotensin-aldosterone system inhibitors are often preferred to manage hypertension in those who have proteinuria or cardiovascular disease. However, the risk of hyperkalemia is also greater with concomitant use of immunosuppressive drugs such as tacrolimus and cyclosporine. Management of complications should be approached according to guidelines discussed above.51
Monitor renal function, potassium. The National Institute for Health and Care Excellence guideline54 advocates that baseline renal function testing should be followed by repeat blood testing 1 to 2 weeks after starting renin-angiotensin-aldosterone system inhibitors in patients with ischemic heart disease. The advice is similar when starting therapy in patients with chronic heart failure, emphasizing the need to monitor after each dose increment and to use clinical judgment when deciding to start treatment. The AHA advises caution in patients with renal insufficiency or a potassium level above 5.0 mmol/L.49
Sick day rules. The National Institute for Health and Care Excellence encourages discussing “sick day rules” with patients starting renin-angiotensin-aldosterone system inhibitors. This means patients should be advised to temporarily stop taking nephrotoxic medications, including over-the-counter nonsteroidal anti-inflammatory drugs, in any potential state of illness or dehydration, such as diarrhea and vomiting. There is, however, little evidence that this advice can actually reduce the incidence of acute kidney injury.55,56
OUR RECOMMENDATIONS
Our advice for managing patients receiving ACE inhibitors or ARBs is summarized in Table 1.
- Page IH, Helmer OM. A crystalline pressor substance (angiotonin) resulting from the reaction between renin and renin-activator. Exp Med 1940; 71(1):29–42. doi:10.1084/jem.71.1.29
- Steddon S, Ashman N, Chesser A, Cunningham J. Oxford Handbook of Nephrology and Hypertension. 2nd ed. Oxford: Oxford University Press; 2016:203–206, 508–509.
- Barratt J, Topham P, Harris K. Oxford Desk Reference. 1st ed. Oxford: Oxford University Press; 2008.
- International Kidney Foundation. KDIGO clinical practice guideline for the management of blood pressure in chronic kidney disease. http://www.kdigo.org/clinical_practice_guidelines/pdf/KDIGO_BP_GL.pdf. Accessed April 3, 2019.
- Heart Outcomes Prevention Evaluation Study Investigators; Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med 2000; 342(3):145–153. doi:10.1056/NEJM200001203420301
- Swedberg K, Kjekshus J. Effects of enalapril on mortality in severe congestive heart failure: results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). Am J Cardiol 1988; 62(2):60A–66A. pmid:2839019
- Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345(12):861–869. doi:10.1056/NEJMoa011161
- Pfeffer MA, McMurray JJ, Velazquez EJ, et al. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003; 349(20):1893–1906. doi:10.1056/NEJMoa032292
- Epstein M. Reduction of cardiovascular risk in chronic kidney disease by mineralocorticoid receptor antagonism. Lancet Diabetes Endocrinol 2015; 3(12):993–1003. doi:10.1016/S2213-8587(15)00289-2
- SOLVD Investigators; Yusuf S, Pitt B, Davis CE, Hood WB, Cohn JN. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med 1991; 325(5):293–302. doi:10.1056/NEJM199108013250501
- Jafar TH, Stark PC, Schmid CH, et al; AIPRD Study Group; Angiotensin-Converting Enzymne Inhibition and Progression of Renal Disease. Proteinuria as a modifiable risk factor for the progression of non-diabetic renal disease. Kidney Int 2001; 60(3):1131–1140. doi:10.1046/j.1523-1755.2001.0600031131.x
- Palmer SC, Mavridis D, Navarese E, et al. Comparative efficacy and safety of blood pressure-lowering agents in adults with diabetes and kidney disease: a network meta-analysis. Lancet 2015; 385(9982):2047–2056. doi:10.1016/S0140-6736(14)62459-4
- Ruggenenti P, Perticucci E, Cravedi P, et al. Role of remission clinics in the longitudinal treatment of CKD. J Am Soc Nephrol 2008; 19(6):1213–1224. doi:10.1681/ASN.2007090970
- Makani H, Bangalore S, Desouza KA, Shah A, Messerli FH. Efficacy and safety of dual blockade of the renin-angiotensin system: meta-analysis of randomised trials. BMJ 2013; 346:f360. doi:10.1136/bmj.f360
- ONTARGET Investigators; Yusuf S, Teo KK, Pogue J, et al. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008; 358(15):1547–1559. doi:10.1056/NEJMoa0801317
- Fried LF, Emanuele N, Zhang JH, et al; VA NEPHRON-D Investigators. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med 2013; 369(20):1892–1903.
doi:10.1056/NEJMoa1303154 - Catalá-López F, Macías Saint-Gerons D, González-Bermejo D, et al. Cardiovascular and renal outcomes of renin-angiotensin system blockade in adult patients with diabetes mellitus: a systematic review with network meta-analyses. PLoS Med 2016; 13(3):e1001971. doi:10.1371/journal.pmed.1001971
- Agarwal R, Afzalpurkar R, Fordtran JS. Pathophysiology of potassium absorption and secretion by the human intestine. Gastroenterology 1994; 107(2):548–571. pmid:8039632
- Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10(6):1050–1060. doi:10.2215/CJN.08580813
- Einhorn LM, Zhan M, Hsu VD, et al. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Intern Med 2009; 169(12):1156–1162. doi:10.1001/archinternmed.2009.132
- Nakhoul GN, Huang H, Arrigain S, et al. Serum potassium, end-stage renal disease and mortality in chronic kidney disease. Am J Nephrol 2015; 41(6):456–463. doi:10.1159/000437151
- Acker CG, Johnson JP, Palevsky PM, Greenberg A. Hyperkalemia in hospitalized patients: causes, adequacy of treatment, and results of an attempt to improve physician compliance with published therapy guidelines. Arch Intern Med 1998; 158(8):917–924. pmid:9570179
- Desai AS, Swedberg K, McMurray JJ, et al; CHARM Program Investigators. Incidence and predictors of hyperkalemia in patients with heart failure: an analysis of the CHARM Program. J Am Coll Cardiol 2007; 50(20):1959–1966. doi:10.1016/j.jacc.2007.07.067
- Cheungpasitporn W, Thongprayoon C, Kittanamongkolchai W, Sakhuja A, Mao MA, Erickson SB. Impact of admission serum potassium on mortality in patients with chronic kidney disease and cardiovascular disease. QJM 2017; 110(11):713–719. doi:10.1093/qjmed/hcx118
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364(1):11–21. doi:10.1056/NEJMoa1009492
- Rossignol P, Dobre D, McMurray JJ, et al. Incidence, determinants, and prognostic significance of hyperkalemia and worsening renal function in patients with heart failure receiving the mineralocorticoid receptor antagonist eplerenone or placebo in addition to optimal medical therapy: results from the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF). Circ Heart Fail 2014; 7(1):51–58. doi:10.1161/CIRCHEARTFAILURE.113.000792
- Testani JM, Kimmel SE, Dries DL, Coca SG. Prognostic importance of early worsening renal function after initiation of angiotensin-converting enzyme inhibitor therapy in patients with cardiac dysfunction. Circ Heart Fail 2011; 4(6):685–691. doi:10.1161/CIRCHEARTFAILURE.111.963256
- Weir M, Rolfe M. Potassium homeostasis and renin-angiotensin-aldosterone system inhibitors. Clin J Am Soc Nephrol 2010; 5(3):531–548. doi:10.2215/CJN.07821109
- Valente M, Bhandari S. Renal function after new treatment with renin-angiotensin system blockers. BMJ 2017; 356:j1122. doi:10.1136/bmj.j1122
- Bakris G, Weir M. Angiotensin-converting enzyme inhibitor–associated elevations in serum creatinine. Arch Intern Med 2000; 160(5):685–693. pmid:10724055
- Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345(12):861–869. doi:10.1056/NEJMoa011161
- Hirsch S. Pre-renal success. Kidney Int 2012; 81(6):596. doi:10.1038/ki.2011.418
- Schmidt M, Mansfield KE, Bhaskaran K, et al. Serum creatinine elevation after renin-angiotensin system blockade and long term cardiorenal risks: cohort study. BMJ 2017; 356:j791. doi:10.1136/bmj.j791
- Schmidt M, Mansfield KE, Bhaskaran K, et al. Adherence to guidelines for creatinine and potassium monitoring and discontinuation following renin–angiotensin system blockade: a UK general practice-based cohort study. BMJ Open 2017; 7(1):e012818. doi:10.1136/bmjopen-2016-012818
- Lund LH, Carrero JJ, Farahmand B, et al. Association between enrollment in a heart failure quality registry and subsequent mortality—a nationwide cohort study. Eur J Heart Fail 2017; 19(9):1107–1116. doi:10.1002/ejhf.762
- Edner M, Benson L, Dahlstrom U, Lund LH. Association between renin-angiotensin system antagonist use and mortality in heart failure with severe renal insuffuciency: a prospective propensity score-matched cohort study. Eur Heart J 2015; 36(34):2318–2326. doi:10.1093/eurheartj/ehv268
- Epstein M, Reaven NL, Funk SE, McGaughey KJ, Oestreicher N, Knispel J. Evaluation of the treatment gap between clinical guidelines and the utilization of renin-angiotensin-aldosterone system inhibitors. Am J Manag Care 2015; 21(suppl 11):S212–S220. pmid:26619183
- Bhandari S, Ives N, Brettell EA, et al. Multicentre randomized controlled trial of angiotensin-converting enzyme inhibitor/angiotensin receptor blocker withdrawal in advanced renal disease: the STOP-ACEi trial. Nephrol Dial Transplant 2016; 31(2):255–261. doi:10.1093/ndt/gfv346
- Raebel MA, Ross C, Xu S, et al. Diabetes and drug-associated hyperkalemia: effect of potassium monitoring. J Gen Intern Med 2010; 25(4):326–333. doi:10.1007/s11606-009-1228-x
- Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018; 71(6):e13–e115. doi:10.1161/HYP.0000000000000065
- The Renal Association. The UK eCKD Guide. https://renal.org/information-resources/the-uk-eckd-guide. Accessed August 12, 2019.
- National Institute for Health and Care Excellence (NICE). Chronic kidney disease in adults: assessment and management. https://www.nice.org.uk/guidance/cg182. Accessed August 12, 2019.
- National Institute for Health and Care Excellence (NICE). Hypertension in adults: diagnosis and management. https://www.nice.org.uk/Guidance/CG127. Accessed August 12, 2019.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34(28):2159–2219. doi:10.1093/eurheartj/eht151
- International Kidney Foundation. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. https://www.sciencedirect.com/journal/kidney-international-supplements/vol/3/issue/1. Accessed August 12, 2019.
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373(22):2103–2116. doi:10.1056/NEJMoa1511939
- Wright J, Bakris G, Greene T. Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease. Results from the AASK trial. ACC Current Journal Review 2003; 12(2):37–38. doi:10.1016/s1062-1458(03)00035-7
- Ku E, Bakris G, Johansen K, et al. Acute declines in renal function during intensive BP lowering: implications for future ESRD risk. J Am Soc Nephrol 2017; 28(9):2794–2801. doi:10.1681/ASN.2017010040
- Yancy CW, Jessup M, Bozkurt B, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2017; 136(6):e137–e161. doi:10.1161/CIR.0000000000000509
- Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016; 37(27):2129–2200. doi:10.1093/eurheartj/ehw128
- Kidney Disease Outcomes Quality Initiative (K/DOQI). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 51):S1–S290. pmid:15114537
- Asenjo RM, Bueno H, Mcintosh M. Angiotensin converting enzyme inhibitors (ACE inhibitors) and angiotensin II receptor blockers (ARBs). ACE inhibitors and ARBs, a cornerstone in the prevention and treatment of cardiovascular disease. www.escardio.org/Education/ESC-Prevention-of-CVD-Programme/Treatment-goals/Cardio-Protective-drugs/angiotensin-converting-enzyme-inhibitors-ace-inhibitors-and-angiotensin-ii-rec. Accessed August 12, 2019.
- López-Sendón J, Swedberg K, McMurray J, et al; Task Force on ACE-inhibitors of the European Society of Cardiology. Expert consensus document on angiotensin converting enzyme inhibitors in cardiovascular disease. The Task Force on ACE-inhibitors of the European Society of Cardiology. Eur Heart J 2004; 25(16):1454–1470. doi:10.1016/j.ehj.2004.06.003
- National Institute for Health and Care Excellence (NICE). Myocardial infarction: cardiac rehabilitation and prevention of further cardiovascular disease. https://www.nice.org.uk/Guidance/CG172. Accessed April 3, 2019.
- National Institute for Health and Care Excellence (NICE). Acute kidney injury: prevention, detection and management. https://www.nice.org.uk/Guidance/CG169. Accessed August 12, 2019.
- Think Kidneys. “Sick day” guidance in patients at risk of acute kidney injury: a position statement from the Think Kidneys Board. https://www.thinkkidneys.nhs.uk/aki/wp-content/uploads/sites/2/2018/01/Think-Kidneys-Sick-Day-Guidance-2018.pdf. Accessed August 12, 2019.
- Meaney CJ, Beccari MV, Yang Y, Zhao J. Systematic review and meta-analysis of patiromer and sodium zirconium cyclosilicate: a new armamentarium for the treatment of hyperkalemia. Pharmacotherapy 2017; 37(4):401–411. doi:10.1002/phar.1906
- Page IH, Helmer OM. A crystalline pressor substance (angiotonin) resulting from the reaction between renin and renin-activator. Exp Med 1940; 71(1):29–42. doi:10.1084/jem.71.1.29
- Steddon S, Ashman N, Chesser A, Cunningham J. Oxford Handbook of Nephrology and Hypertension. 2nd ed. Oxford: Oxford University Press; 2016:203–206, 508–509.
- Barratt J, Topham P, Harris K. Oxford Desk Reference. 1st ed. Oxford: Oxford University Press; 2008.
- International Kidney Foundation. KDIGO clinical practice guideline for the management of blood pressure in chronic kidney disease. http://www.kdigo.org/clinical_practice_guidelines/pdf/KDIGO_BP_GL.pdf. Accessed April 3, 2019.
- Heart Outcomes Prevention Evaluation Study Investigators; Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med 2000; 342(3):145–153. doi:10.1056/NEJM200001203420301
- Swedberg K, Kjekshus J. Effects of enalapril on mortality in severe congestive heart failure: results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). Am J Cardiol 1988; 62(2):60A–66A. pmid:2839019
- Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345(12):861–869. doi:10.1056/NEJMoa011161
- Pfeffer MA, McMurray JJ, Velazquez EJ, et al. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003; 349(20):1893–1906. doi:10.1056/NEJMoa032292
- Epstein M. Reduction of cardiovascular risk in chronic kidney disease by mineralocorticoid receptor antagonism. Lancet Diabetes Endocrinol 2015; 3(12):993–1003. doi:10.1016/S2213-8587(15)00289-2
- SOLVD Investigators; Yusuf S, Pitt B, Davis CE, Hood WB, Cohn JN. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med 1991; 325(5):293–302. doi:10.1056/NEJM199108013250501
- Jafar TH, Stark PC, Schmid CH, et al; AIPRD Study Group; Angiotensin-Converting Enzymne Inhibition and Progression of Renal Disease. Proteinuria as a modifiable risk factor for the progression of non-diabetic renal disease. Kidney Int 2001; 60(3):1131–1140. doi:10.1046/j.1523-1755.2001.0600031131.x
- Palmer SC, Mavridis D, Navarese E, et al. Comparative efficacy and safety of blood pressure-lowering agents in adults with diabetes and kidney disease: a network meta-analysis. Lancet 2015; 385(9982):2047–2056. doi:10.1016/S0140-6736(14)62459-4
- Ruggenenti P, Perticucci E, Cravedi P, et al. Role of remission clinics in the longitudinal treatment of CKD. J Am Soc Nephrol 2008; 19(6):1213–1224. doi:10.1681/ASN.2007090970
- Makani H, Bangalore S, Desouza KA, Shah A, Messerli FH. Efficacy and safety of dual blockade of the renin-angiotensin system: meta-analysis of randomised trials. BMJ 2013; 346:f360. doi:10.1136/bmj.f360
- ONTARGET Investigators; Yusuf S, Teo KK, Pogue J, et al. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008; 358(15):1547–1559. doi:10.1056/NEJMoa0801317
- Fried LF, Emanuele N, Zhang JH, et al; VA NEPHRON-D Investigators. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med 2013; 369(20):1892–1903.
doi:10.1056/NEJMoa1303154 - Catalá-López F, Macías Saint-Gerons D, González-Bermejo D, et al. Cardiovascular and renal outcomes of renin-angiotensin system blockade in adult patients with diabetes mellitus: a systematic review with network meta-analyses. PLoS Med 2016; 13(3):e1001971. doi:10.1371/journal.pmed.1001971
- Agarwal R, Afzalpurkar R, Fordtran JS. Pathophysiology of potassium absorption and secretion by the human intestine. Gastroenterology 1994; 107(2):548–571. pmid:8039632
- Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10(6):1050–1060. doi:10.2215/CJN.08580813
- Einhorn LM, Zhan M, Hsu VD, et al. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Intern Med 2009; 169(12):1156–1162. doi:10.1001/archinternmed.2009.132
- Nakhoul GN, Huang H, Arrigain S, et al. Serum potassium, end-stage renal disease and mortality in chronic kidney disease. Am J Nephrol 2015; 41(6):456–463. doi:10.1159/000437151
- Acker CG, Johnson JP, Palevsky PM, Greenberg A. Hyperkalemia in hospitalized patients: causes, adequacy of treatment, and results of an attempt to improve physician compliance with published therapy guidelines. Arch Intern Med 1998; 158(8):917–924. pmid:9570179
- Desai AS, Swedberg K, McMurray JJ, et al; CHARM Program Investigators. Incidence and predictors of hyperkalemia in patients with heart failure: an analysis of the CHARM Program. J Am Coll Cardiol 2007; 50(20):1959–1966. doi:10.1016/j.jacc.2007.07.067
- Cheungpasitporn W, Thongprayoon C, Kittanamongkolchai W, Sakhuja A, Mao MA, Erickson SB. Impact of admission serum potassium on mortality in patients with chronic kidney disease and cardiovascular disease. QJM 2017; 110(11):713–719. doi:10.1093/qjmed/hcx118
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364(1):11–21. doi:10.1056/NEJMoa1009492
- Rossignol P, Dobre D, McMurray JJ, et al. Incidence, determinants, and prognostic significance of hyperkalemia and worsening renal function in patients with heart failure receiving the mineralocorticoid receptor antagonist eplerenone or placebo in addition to optimal medical therapy: results from the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF). Circ Heart Fail 2014; 7(1):51–58. doi:10.1161/CIRCHEARTFAILURE.113.000792
- Testani JM, Kimmel SE, Dries DL, Coca SG. Prognostic importance of early worsening renal function after initiation of angiotensin-converting enzyme inhibitor therapy in patients with cardiac dysfunction. Circ Heart Fail 2011; 4(6):685–691. doi:10.1161/CIRCHEARTFAILURE.111.963256
- Weir M, Rolfe M. Potassium homeostasis and renin-angiotensin-aldosterone system inhibitors. Clin J Am Soc Nephrol 2010; 5(3):531–548. doi:10.2215/CJN.07821109
- Valente M, Bhandari S. Renal function after new treatment with renin-angiotensin system blockers. BMJ 2017; 356:j1122. doi:10.1136/bmj.j1122
- Bakris G, Weir M. Angiotensin-converting enzyme inhibitor–associated elevations in serum creatinine. Arch Intern Med 2000; 160(5):685–693. pmid:10724055
- Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345(12):861–869. doi:10.1056/NEJMoa011161
- Hirsch S. Pre-renal success. Kidney Int 2012; 81(6):596. doi:10.1038/ki.2011.418
- Schmidt M, Mansfield KE, Bhaskaran K, et al. Serum creatinine elevation after renin-angiotensin system blockade and long term cardiorenal risks: cohort study. BMJ 2017; 356:j791. doi:10.1136/bmj.j791
- Schmidt M, Mansfield KE, Bhaskaran K, et al. Adherence to guidelines for creatinine and potassium monitoring and discontinuation following renin–angiotensin system blockade: a UK general practice-based cohort study. BMJ Open 2017; 7(1):e012818. doi:10.1136/bmjopen-2016-012818
- Lund LH, Carrero JJ, Farahmand B, et al. Association between enrollment in a heart failure quality registry and subsequent mortality—a nationwide cohort study. Eur J Heart Fail 2017; 19(9):1107–1116. doi:10.1002/ejhf.762
- Edner M, Benson L, Dahlstrom U, Lund LH. Association between renin-angiotensin system antagonist use and mortality in heart failure with severe renal insuffuciency: a prospective propensity score-matched cohort study. Eur Heart J 2015; 36(34):2318–2326. doi:10.1093/eurheartj/ehv268
- Epstein M, Reaven NL, Funk SE, McGaughey KJ, Oestreicher N, Knispel J. Evaluation of the treatment gap between clinical guidelines and the utilization of renin-angiotensin-aldosterone system inhibitors. Am J Manag Care 2015; 21(suppl 11):S212–S220. pmid:26619183
- Bhandari S, Ives N, Brettell EA, et al. Multicentre randomized controlled trial of angiotensin-converting enzyme inhibitor/angiotensin receptor blocker withdrawal in advanced renal disease: the STOP-ACEi trial. Nephrol Dial Transplant 2016; 31(2):255–261. doi:10.1093/ndt/gfv346
- Raebel MA, Ross C, Xu S, et al. Diabetes and drug-associated hyperkalemia: effect of potassium monitoring. J Gen Intern Med 2010; 25(4):326–333. doi:10.1007/s11606-009-1228-x
- Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018; 71(6):e13–e115. doi:10.1161/HYP.0000000000000065
- The Renal Association. The UK eCKD Guide. https://renal.org/information-resources/the-uk-eckd-guide. Accessed August 12, 2019.
- National Institute for Health and Care Excellence (NICE). Chronic kidney disease in adults: assessment and management. https://www.nice.org.uk/guidance/cg182. Accessed August 12, 2019.
- National Institute for Health and Care Excellence (NICE). Hypertension in adults: diagnosis and management. https://www.nice.org.uk/Guidance/CG127. Accessed August 12, 2019.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34(28):2159–2219. doi:10.1093/eurheartj/eht151
- International Kidney Foundation. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. https://www.sciencedirect.com/journal/kidney-international-supplements/vol/3/issue/1. Accessed August 12, 2019.
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373(22):2103–2116. doi:10.1056/NEJMoa1511939
- Wright J, Bakris G, Greene T. Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease. Results from the AASK trial. ACC Current Journal Review 2003; 12(2):37–38. doi:10.1016/s1062-1458(03)00035-7
- Ku E, Bakris G, Johansen K, et al. Acute declines in renal function during intensive BP lowering: implications for future ESRD risk. J Am Soc Nephrol 2017; 28(9):2794–2801. doi:10.1681/ASN.2017010040
- Yancy CW, Jessup M, Bozkurt B, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2017; 136(6):e137–e161. doi:10.1161/CIR.0000000000000509
- Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016; 37(27):2129–2200. doi:10.1093/eurheartj/ehw128
- Kidney Disease Outcomes Quality Initiative (K/DOQI). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 51):S1–S290. pmid:15114537
- Asenjo RM, Bueno H, Mcintosh M. Angiotensin converting enzyme inhibitors (ACE inhibitors) and angiotensin II receptor blockers (ARBs). ACE inhibitors and ARBs, a cornerstone in the prevention and treatment of cardiovascular disease. www.escardio.org/Education/ESC-Prevention-of-CVD-Programme/Treatment-goals/Cardio-Protective-drugs/angiotensin-converting-enzyme-inhibitors-ace-inhibitors-and-angiotensin-ii-rec. Accessed August 12, 2019.
- López-Sendón J, Swedberg K, McMurray J, et al; Task Force on ACE-inhibitors of the European Society of Cardiology. Expert consensus document on angiotensin converting enzyme inhibitors in cardiovascular disease. The Task Force on ACE-inhibitors of the European Society of Cardiology. Eur Heart J 2004; 25(16):1454–1470. doi:10.1016/j.ehj.2004.06.003
- National Institute for Health and Care Excellence (NICE). Myocardial infarction: cardiac rehabilitation and prevention of further cardiovascular disease. https://www.nice.org.uk/Guidance/CG172. Accessed April 3, 2019.
- National Institute for Health and Care Excellence (NICE). Acute kidney injury: prevention, detection and management. https://www.nice.org.uk/Guidance/CG169. Accessed August 12, 2019.
- Think Kidneys. “Sick day” guidance in patients at risk of acute kidney injury: a position statement from the Think Kidneys Board. https://www.thinkkidneys.nhs.uk/aki/wp-content/uploads/sites/2/2018/01/Think-Kidneys-Sick-Day-Guidance-2018.pdf. Accessed August 12, 2019.
- Meaney CJ, Beccari MV, Yang Y, Zhao J. Systematic review and meta-analysis of patiromer and sodium zirconium cyclosilicate: a new armamentarium for the treatment of hyperkalemia. Pharmacotherapy 2017; 37(4):401–411. doi:10.1002/phar.1906
KEY POINTS
- ACE inhibitors and ARBs reduce proteinuria by lowering the intraglomerular pressure, reducing hyperfiltration.
- These drugs tend to raise the serum potassium level and reduce the glomerular filtration rate (GFR). Monitoring the serum potassium and creatinine levels and the GFR is therefore imperative.
- Despite the benefits, concern for adverse effects including hyperkalemia and a rise in serum creatinine has led to reluctance to prescribe these drugs, and they are underused in the patients who may derive the greatest benefit.
Hydroxychloroquine: An old drug with new relevance
A 29-year-old African American woman presents with a photosensitive malar rash, fatigue, morning stiffness, and swelling in her hands. She is found to have elevated antinuclear antibody at a titer of 1:320. A complete blood cell count demonstrates leukopenia and thrombocytopenia. Results of renal function testing and urinalysis are within normal limits. She has no other medical problems and no history of blood clots or pregnancy loss.
Her arthritis and rash suggest systemic lupus erythematosus. She is counseled to avoid sun exposure, and treatment with hydroxychloroquine is considered.
WHAT IS HYDROXYCHLOROQUINE?
Hydroxychloroquine was developed to treat malaria but was later found to have immunomodulatory properties. It is now approved by the US Food and Drug Administration for treatment of discoid lupus, systemic lupus erythematosus, and rheumatoid arthritis. It is also approved to treat malaria; however, of the several malarial parasites, only Plasmodium falciparum can still be cured by hydroxychloroquine, and growing resistance limits the geographic locations where this drug can be used effectively.1,2
HISTORICAL BACKGROUND
Antimalarial drugs were discovered shortly before World War II. Their production was industrialized during the war because malaria was a leading cause of disease among soldiers, especially those deployed to the South Pacific.3
Atabrine (quinacrine), the first antimalarial widely used, had numerous side effects including yellowing of the skin. Aggressive research efforts to develop an alternative led to field testing of one of its derivative compounds, chloroquine, by the US Army in 1943. Continued chemical modification would create hydroxychloroquine, introduced in 1955.
A serendipitous consequence of the mass use of antimalarials during World War II was the discovery that they could be used to treat inflammatory arthritis and lupus. Eight years after the war ended, Shee4 reported that chloroquine had a beneficial effect on lupus and rheumatoid arthritis in US soldiers. Hydroxychloroquine is now the most commonly prescribed antimalarial for treatment of autoimmune disease.
HOW HYDROXYCHLOROQUINE WORKS
The primary mechanism by which hydroxychloroquine modulates systemic lupus erythematosus is by suppressing activation of Toll-like receptors, which exist on the surface of endosomes and play a significant role in the innate immune response and in autoimmune disease. Their activation is necessary for the expression of interferon-regulated genes and production of tumor necrosis factor alpha, which are key in the cell-mediated inflammatory response.
Antimalarial drugs such as hydroxychloroquine prevent Toll-like receptor activation by binding directly to nucleic acids in the activation pathway.5 In vitro studies show that blocking this pathway blunts the body’s primary cell-mediated inflammatory response; in vivo studies show that use of hydroxychloroquine is strongly correlated with a reduction in interferon alpha levels.6 The powerful effect of hydroxychloroquine on the cell-mediated pattern of inflammation found in lupus is consistent with this theory.
It was previously hypothesized that the immune-modulating effects of hydroxychloroquine were associated with a more general dysregulation of cellular lysosomes through inhibition of proteolysis or changes in cellular pH.7 This theory has since been displaced by the more specific and elegant mechanism described above.5
HOW WELL DOES IT WORK?
Benefit in systemic lupus erythematosus
Hydroxychloroquine has consistently demonstrated significant and multifaceted benefit in patients with systemic lupus erythematosus.
A systematic review of 95 articles8 concluded that this drug decreases lupus flares and decreases mortality rates in lupus patients by at least 50%, with a high level of evidence. Beneficial effects that had a moderate level of evidence were an increase in bone mineral density, fewer thrombotic events, and fewer cases of irreversible organ damage.
The preventive effect of hydroxychloroquine on thrombosis in lupus patients has been consistently demonstrated and is one of the key reasons the drug is considered a cornerstone of therapy in this disease.9 A nested case-control study of patients with lupus and thromboembolism demonstrated an odds ratio of 0.31 and relative risk reduction of 68% for those using antimalarials.10
Benefit in antiphospholipid antibody syndrome
Hydroxychloroquine prevents thrombosis in other diseases as well. For example, it has been shown to reduce the incidence of thrombotic events in patients with primary antiphospholipid syndrome.
In a retrospective cohort study in 114 patients with this disease, hydroxychloroquine significantly reduced the incidence of arterial thrombotic events over 10 years of follow-up (recurrence incidence 0 in those treated with hydroxychloroquine vs 1.14% in those not treated).11 The study also tracked levels of antiphospholipid antibodies and reported that hydroxychloroquine significantly reduced the levels of antibodies to cardiolipin and beta-2 glycoprotein 1, both implicated in the pathology of thrombosis.11
In vitro studies have also demonstrated that hydroxychloroquine can modulate a dysregulated inflammatory system to reduce thrombosis. For example, it has been shown that hydroxychloroquine can reverse platelet activation by antiphospholipid antibodies, prevent linking of antibody complexes to cell membranes, and promote proper membrane protein expression, thereby reducing the thrombotic qualities of antiphospholipid antibodies and even improving clearance times of antiphospholipid-related thrombi.12
Benefit in rheumatoid arthritis
Though there is less evidence, hydroxychloroquine has also shown benefit in rheumatoid arthritis, where it can be used by itself in mild disease or as part of combination therapy with active arthritis. Compared with biologic therapy in patients with early aggressive rheumatoid arthritis, triple therapy with methotrexate, sulfasalazine, and hydroxychloroquine was nearly as effective in terms of quality of life, and it cost only one-third as much, saving $20,000 per year of therapy per patient.13
Hydroxychloroquine has also been compared directly with chloroquine, its closest relation, in a large study incorporating patients with rheumatoid arthritis and patients with systemic lupus erythematosus. Patients using chloroquine experienced significantly more side effects, though it did prove marginally more effective.14
No benefit shown in Sjögren syndrome
Unfortunately, despite widespread use, hydroxychloroquine has not demonstrated positive clinical effects when used to treat primary Sjögren syndrome. Most notably, a 2014 randomized controlled trial of hydroxychloroquine vs placebo in 120 Sjögren patients found no significant improvement in primary symptoms of dryness, pain, or fatigue after 6 months of therapy.15
Metabolic benefits
Unexpectedly, hydroxychloroquine is associated with multiple metabolic benefits including improved lipid profiles and lower blood glucose levels. These findings, in addition to a reduced incidence of thrombosis, were initially reported in the Baltimore Lupus Cohort in 1996.16 Specifically, longitudinal evaluation of a cohort of lupus patients showed that hydroxychloroquine use was associated with a 7.6% reduction in total cholesterol and a 13.7% reduction in low-density lipoprotein cholesterol (LDL-C) over 3 months of therapy.17
Similar findings, including a reduction in LDL-C and an increase in high-density lipoprotein cholesterol, were strongly associated with the addition of hydroxychloroquine to methotrexate or to methotrexate and etanercept in a large cohort of rheumatoid arthritis patients followed over 2 years of therapy.18
In nondiabetic women with systemic lupus erythematosus or rheumatoid arthritis, average blood glucose was significantly lower in those taking hydroxychloroquine than in nonusers. The incidence of insulin resistance was also lower, but the difference was not statistically significant.19
Some have suggested that hydroxychloroquine may prevent diabetes mellitus. In a retrospective case series, compared with rheumatoid arthritis patients not taking the drug, patients treated with hydroxychloroquine for more than 4 years had a 25% lower risk of developing diabetes mellitus.20
In view of these metabolic benefits, especially regarding lipid regulation, and the above described antithrombotic properties of hydroxychloroquine, some researchers have recently hypothesized that hydroxychloroquine may be of benefit in patients with coronary artery disease.21 They suggested that the inflammatory contribution to the mechanism of coronary artery disease could be lessened by hydroxychloroquine even in patients without lupus erythematosus or rheumatoid arthritis.
PHARMACOLOGIC PROPERTIES
Understanding the pharmacologic properties of hydroxychloroquine is key to using it appropriately in clinical practice.
The half-life of elimination of hydroxychloroquine is 40 to 50 days, with half of the drug excreted renally in a concentration-dependent fashion.22,23 The drug reaches 95% of its steady-state concentration by about 6 months of therapy. Shorter durations of therapy do not provide adequate time for the drug to achieve steady-state concentration and may not allow patients and providers time to see its full clinical results. Therefore, its manufacturers recommend a 6-month trial of therapy to adequately determine if the drug improves symptoms.1
The oral bioavailability of hydroxychloroquine is about 75%, but pharmacokinetics vary among individuals.22,23 It has been suggested that this variability affects the efficacy of hydroxychloroquine. In a study of 300 patients with cutaneous lupus erythematosus, those whose treatment failed had significantly lower blood concentrations of hydroxychloroquine, while those who achieved complete remission had significantly higher concentrations.24
Another study found that titrating doses to target therapeutic blood concentrations can reduce disease activity in cutaneous lupus erythematosus.25 Measuring the blood concentration of hydroxychloroquine is not common in clinical practice but may have a role in select patients in whom initial therapy using a standard dosing regimen does not produce the desired results.
HOW SAFE IS HYDROXYCHLOROQUINE?
Hydroxychloroquine has numerous adverse effects, necessitating vigilance on the part of the prescriber. Most commonly reported are retinopathy, hyperpigmentation, myopathy, and skin reactions.1
Retinopathy
Retinopathy’s irreversibility—the threat of permanent vision loss—and its substantial prevalence in patients with a large drug exposure history, have marked retinopathy as the most concerning potential toxicity. The risk of ocular toxicity increases with the cumulative hydroxychloroquine dose. The prevalence of retinopathy in those using the drug less than 10 years is less than 2%; in contrast, the prevalence in patients with more than 20 years of exposure is reported to be as high as 20%.26
The American Academy of Ophthalmology has long stated that retinopathy is a significant risk of hydroxychloroquine therapy and that patients taking hydroxychloroquine should therefore undergo routine retinal and visual field screening by an ophthalmologist.

Currently, initial screening followed by yearly screening beginning 5 years thereafter is recommended for patients at low risk of toxicity (Table 1).27 Patients determined by an ophthalmologist to be at higher risk of retinopathy should be screened yearly. As identified by the American Academy of Ophthalmology, major risk factors for retinopathy include duration of use, concomitant tamoxifen exposure, significant renal disease, and preexisting retinal and macular disease.26,28
Recommendations for hydroxychloroquine dosing and screening were recently revised, for 2 reasons. Initially, its manufacturers recommended that hydroxychloroquine dosage be no higher than 6.5 mg/kg of ideal body weight to prevent retinopathy.1,29,30 However, it has recently been demonstrated that real body weight is a better predictor of risk of retinopathy than ideal body weight when dosing hydroxychloroquine, perhaps because of the increasing variance of real body weight in our patient population.26

Further, an atypical pattern of retinopathy called pericentral retinopathy is more common in Asians. A study of about 200 patients with a history of hydroxychloroquine retinopathy, including 36 Asian patients, found that the pericentral pattern occurred in half the Asian patients but only 2% of the white patients.31 The mechanism for this finding is unclear, but because pericentral retinopathy spares the macula, it can be missed using standard screening methods. Therefore, the American Academy of Ophthalmology now recommends that the dose limit be reduced from 6.5 mg/kg of ideal body weight to no more than 5.0 mg/kg of real body weight (Table 2).28
It is also recommended that screening methods such as automated visual fields and optical coherence tomography extend their fields beyond the macula in Asian patients to ensure that pericentral retinopathy is not missed.28
Optical coherence tomography is a particularly useful tool in the ocular evaluation of patients taking hydroxychloroquine. It can detect subtle changes such as thinning of the foveal photoreceptor outer segment, thickening of the retinal pigment epithelium, and loss of the macular ganglion cell–inner plexiform layer before there are visible signs of retinopathy and before symptoms arise.32
Currently, these guidelines are underutilized in clinical practice. Physician adherence to ophthalmologic guidelines is reported at about 50%.33 This statistic is jarring, given the potential for permanent loss of vision in those with hydroxychloroquine-mediated retinopathy, and demonstrates the importance of reinforcing proper understanding of the use of hydroxychloroquine in clinical practice.
Other adverse effects
Cutaneous hyperpigmentation can occur with hydroxychloroquine use (Figure 1). The hyperpigmentation appears to be due to local bruising following deposition of iron in the soft tissue.

While the pigmentation may persist permanently and cause an undesirable cosmetic effect, it has not been associated with other adverse outcomes.
Myopathy is a rare adverse effect. In one case series, 3 of 214 patients treated with hydroxychloroquine developed hydroxychloroquine-induced myopathy.35 Over the duration of their therapy, this was equivalent to an incidence of 1 case of myopathy in 100 patient-years of therapy. Myopathy improves with discontinuation of therapy, though it can persist for weeks, likely because of hydroxychloroquine’s prolonged elimination half-life.
Cardiomyopathy, specifically neurocardiomyopathy, is also an extremely rare adverse effect of hydroxychloroquine use. The mechanism is believed to be associated with the effect of hydroxychloroquine on lysosomal action, leading to an acquired lysosomal storage disorder with the typical cardiac hypertrophy and conduction abnormalities associated with this family of diseases.36
Acute generalized exanthematous pustulosis is another rare complication of hydroxychloroquine therapy. The appearance of the reaction is similar to that of pustular psoriasis, with pustules overlying flaking and scaling skin. It usually resolves within 2 weeks after cessation of hydroxychloroquine therapy. In a select few cases, the reaction persists or waxes and wanes over a period of weeks to months, and longer durations of recovery are thought to be due to hydroxychloroquine’s long half-life, as in hydroxychloroquine-induced myopathy.37
In view of this rare reaction, manufacturers of hydroxychloroquine recommend caution when using the drug in patients with psoriasis.1
Hematologic abnormalities. In very rare cases, hydroxychloroquine is associated with hematologic abnormalities including agranulocytosis, anemia, aplastic anemia, leukopenia, and thrombocytopenia.1
While no specific guidelines exist, caution is warranted when using hydroxychloroquine in patients with porphyria. Additionally, hydroxychloroquine and other antimalarials including primaquine have been associated with hemolysis in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. The risk of hemolysis is generally considered low except at high hydroxychloroquine doses in patients with severe G6PD deficiency.38

For the above reasons, manufacturers recommended baseline and routine blood counts, and some providers screen patients for G6PD deficiency when prescribing hydroxychloroquine (Table 3).
PREGNANCY
Hydroxychloroquine is in pregnancy category C. Information is limited, and in view of the risks, the manufacturer says that it should be avoided in pregnancy.1 Nevertheless, it is generally considered safe during pregnancy, and its benefits may make it acceptable to continue in a patient who becomes pregnant, in spite of the possible risks.
We favor continuing hydroxychloroquine. This drug has been associated with improved maternal and fetal outcomes in lupus patients. Its use during pregnancy has not been associated with congenital malformations. The adverse visual effects of long-term hydroxychloroquine use, namely retinopathy, have never been reported in children as a consequence of exposure in utero.
In addition, hydroxychloroquine is transmitted only in minute quantities in breast milk.39 In pregnant women with systemic lupus erythematosus, hydroxychloroquine was associated with a lower risk of adverse outcomes, including preterm delivery and intrauterine growth restriction.40 However, hydroxychloroquine is far more toxic when ingested directly by infants than in adults.1
Maternal outcomes are also improved with the use of hydroxychloroquine. Stopping hydroxychloroquine during pregnancy in women with systemic lupus erythematosus is associated with significantly higher disease activity—fully twice as high as in those who continue hydroxychloroquine.41 These study results were corroborated in a small randomized trial in which pregnant women with lupus on placebo had significantly higher lupus disease activity scores than those pregnant women who were given hydroxychloroquine.42 The women taking hydroxychloroquine experienced no severe lupus flares for the duration of their pregnancies.
These findings suggest not only that hydroxychloroquine is safe in pregnancy, but also that it should be continued in lupus patients during pregnancy to prevent disease flares and adverse fetal outcomes.
AREAS OF UNCERTAINTY
Benefit in preclinical lupus?
Hydroxychloroquine has a consistently profound effect on outcomes in systemic lupus erythematosus. These findings, in addition to the more widespread use of antibody screening, have led to suggestions that hydroxychloroquine could be of benefit even before systemic lupus erythematosus is diagnosed.
A study in US military personnel found that patients taking hydroxychloroquine experienced a significantly longer lag time between first reported clinical symptoms of lupus and official diagnosis compared with matched controls who also went on to develop the disease, averaging 1.08 vs 0.29 years to disease classification.43 Those who used hydroxychloroquine also had lower rates of autoantibody accumulation. Therefore, hydroxychloroquine could be of benefit in carefully selected candidates at high risk of developing systemic lupus erythematosus.
The beneficial effects of hydroxychloroquine on patients with lupus and rheumatoid arthritis, in terms of primary measures of disease activity and secondary outcomes, were discovered fortuitously and were not the original intended targets of the drug. Because of its versatility, there are numerous other disease states in which hydroxychloroquine has shown a degree of benefit or has shown a potential for benefit.
Antiviral activity?
It has been suggested that antimalarial drugs could serve as adjunctive therapies against filoviruses such as Marburg and Ebola. There is a small body of in vitro and in vivo evidence that hydroxychloroquine could temper severe systemic inflammatory responses to filoviruses both through dysregulation of lysosomes and lysosomal pH (filoviruses have a pH-dependent mechanism of action) and through decreased production of tumor necrosis factor alpha and interferons. Heavy burdens of interferons and tumor necrosis factor alpha are associated with increased mortality rates in those infected with filoviruses.44
Antineoplastic activity?
Hydroxychloroquine has undergone in vitro testing as an adjunct to cancer therapies. There are several mechanisms by which it is theorized that hydroxychloroquine could target malignant cells, including inhibition of multidrug resistance pumps or autophagy, improvement of chemotherapy cell penetration, potentiation of presentation of major histocompatibility complexes, or even intercalation directly into DNA.45,46 However, it can also impair natural anticancer immunity and may allow cancer cells better nutrient supply through vascular effects.
In vitro studies have shown tumoricidal effects in lymphoma and melanoma, and inhibition of growth in lung, colon, breast, cervix, larynx, liver, and prostate cancers. In vivo studies have shown that hydroxychloroquine in high doses can prolong survival in glioblastoma.45
Unfortunately, all of these theorized or observed effects are dose-dependent and likely require doses that exceed currently recommended maximums.
Negative chronotropic effect?
Hydroxychloroquine has been found to decrease the resting heart rate in a cumulative dose-dependent fashion.47 Further, hydroxychloroquine has been known to increase digoxin levels, and the medications should not be used in combination.1
Whether the decrease in resting heart rate is associated with harm or benefit and whether the effect is significant enough to be considered when implementing therapy remain unanswered and deserve further investigation, as does the primary use of hydroxychloroquine for beneficial lipid and glucose reduction in patients who are otherwise healthy.
CASE CONCLUSION
The patient described at the beginning of this article was provided with information on the risks and benefits of hydroxychloroquine for treatment of her arthritis and rash suggestive of mild systemic lupus, and she opted to begin therapy. Her baseline eye screening was within normal limits. Based on her weight of 62 kg, she was started on 300 mg of hydroxychloroquine daily.
She had no significant adverse effects from the medication and reported slow improvement in her rash and joint complaints over the next 2 months. She remained on hydroxychloroquine over the next year without adverse effects or new evidence of autoimmune disease.
- Sanofi-Aventis. Product monograph: Plaquenil. http://products.sanofi.ca/en/plaquenil.pdf. Accessed May 2, 2018.
- Centers for Disease Control and Prevention (CDC). Malaria information and prophylaxis, by country. www.cdc.gov/malaria/travelers/country_table/a.html. Accessed May 2, 2018.
- Wallace DJ. The history of antimalarials. Lupus 1996; 5(suppl 1):S2–S3. pmid:8803902
- Shee JC. Lupus erythematosus treated with chloroquine. Lancet 1953; 265(6778):201–202. pmid:13070595
- Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol 2011; 186:4794–4804. doi:10.4049/jimmunol.1000702
- Willis R, Seif AM, McGwin G Jr, et al. Effect of hydroxychloroquine treatment on pro-inflammatory cytokines and disease activity in SLE patients: data from LUMINA, a multiethnic US cohort. Lupus 2012; 21(8):830–835. doi:10.1177/0961203312437270
- Fox R. Anti-malarial drugs: possible mechanisms of action in autoimmune disease and prospects for drug development. Lupus 1996; 5(suppl 1):S4–S10. pmid:8803903
- Ruiz-Irastorza G, Ramos-Casals M, Brito-Zeron P, Khamashta MA. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis 2010; 69(1):20–28. doi:10.1136/ard.2008.101766
- Lam NC, Ghetu MV, Bieniek ML. Systemic lupus erythematosus: primary care approach to diagnosis and management. Am Fam Physician 2016; 94(4):284–294. pmid:27548593
- Jung H, Bobba R, Su J, et al. The protective effect of antimalarial drugs on thrombovascular events in systemic lupus erythematosus. Arthritis Rheum 2010; 62(3):863–868. doi:10.1002/art.27289
- Nuri E, Taraborelli M, Andreoli L, et al. Long-term use of hydroxychloroquine reduces antiphospholipid antibodies levels in patients with primary antiphospholipid syndrome. Immunol Res 2017; 65(1):17–24. doi:10.1007/s12026-016-8812-z
- Erkan D, Aguiar CL, Andrade D, et al. 14th International Congress on Antiphospholipid Antibodies: task force report on antiphospholipid syndrome treatment trends. Autoimmun Rev 2014; 13(6):685–696. doi:10.1016/j.autrev.2014.01.053
- Jalal H, O’Dell JR, Bridges SL Jr, et al. Cost-effectiveness of triple therapy versus etanercept plus methotrexate in early aggressive rheumatoid arthritis. Arthritis Care Res (Hoboken) 2016; 68(12):1751–1757. doi:10.1002/acr.22895
- Avina-Zubieta JA, Galindo-Rodriguez G, Newman S, Suarez-Almazor ME, Russell AS. Long-term effectiveness of antimalarial drugs in rheumatic diseases. Ann Rheum Dis 1998; 57(10):582–587. pmid:9893568
- Gottenberg JE, Ravaud P, Puechal X, et al. Effects of hydroxychloroquine on symptomatic improvement in primary Sjogren syndrome. JAMA 2014; 312(3):249–258. doi:10.1001/jama.2014.7682
- Petri M. Hydroxychloroquine use in the Baltimore Lupus Cohort: effects on lipids, glucose and thrombosis. Lupus 1996; 5(suppl 1):S16–S22. pmid:8803905
- Cairoli E, Rebella M, Danese N, Garra V, Borba EF. Hydroxychloroquine reduces low-density lipoprotein cholesterol levels in systemic lupus erythematosus: a longitudinal evaluation of the lipid-lowering effect. Lupus 2012; 21(11):1178–1182. doi:10.1177/0961203312450084
- Charles-Schoeman C, Wang X, Lee YY, et al. Association of triple therapy with improvement in cholesterol profiles over two-year followup in the treatment of early aggressive rheumatoid arthritis trial. Arthritis Rheumatol 2016; 68(3):577–586. doi:10.1002/art.39502
- Penn SK, Kao AH, Schott LL, et al. Hydroxychloroquine and glycemia in women with rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol 2010; 37(6):1136–1142. doi:10.3899/jrheum.090994
- Wasko MC, Hubert HB, Lingala VB, et al. Hydroxychloroquine and risk of diabetes in patients with rheumatoid arthritis. JAMA 2007; 298(2):187–193. doi:10.1001/jama.298.2.187
- Sun L, Liu M, Li R, et al. Hydroxychloroquine, a promising choice for coronary artery disease? Med Hypotheses 2016; 93:5–7. doi:10.1016/j.mehy.2016.04.045
- Tett SE, Cutler DJ, Day RO, Brown KF. Bioavailability of hydroxychloroquine tablets in healthy volunteers. Br J Clin Pharmacol 1989; 27(6):771–779. pmid:2757893
- Furst DE. Pharmacokinetics of hydroxychloroquine and chloroquine during treatment of rheumatic diseases. Lupus 1996; 5(suppl 1):S11–S15. pmid:8803904
- Frances C, Cosnes A, Duhaut P, et al. Low blood concentration of hydroxychloroquine in patients with refractory cutaneous lupus erythematosus. Arch Dermatol 2012; 148(4):479–484. doi:10.1001/archdermatol.2011.2558
- Chasset F, Arnaud L, Costedoat-Chalumeau N, Zahr N, Bessis D, Francès C. The effect of increasing the dose of hydroxychloroquine in patients with refractory cutaneous lupus erythematosus: an open-label prospective pilot study. J Am Acad Dermatol 2016; 74(4):693–699.e3. doi:10.1016/j.jaad.2015.09.064
- Melles RB, Marmor MF. The risk of toxic retinopathy in patients on long-term hydroxychloroquine therapy. JAMA Ophthalmol 2014; 132(12):1453–1460. doi:10.1001/jamaophthalmol.2014.3459
- Committee on Rheumatologic Care. American College of Rheumatology position statement. Screening for hydroxychloroquine retinopathy. www.rheumatology.org/Portals/0/Files/Screening-for-Hydroxychloroquine-Retinopathy-Position-Statement.pdf. Accessed April 2, 2018.
- Marmor MF, Kellner U, Lai TY, Melles RB, Mieler WF; American Academy of Ophthalmology. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology 2016; 123(6):1386–1394. doi:10.1016/j.ophtha.2016.01.058
- Mackenzie AH. Antimalarial drugs for rheumatoid arthritis. Am J Med 1983; 75(6A):48–58. pmid:6362406
- Mackenzie AH. Dose refinements in long-term therapy of rheumatoid arthritis with antimalarials. Am J Med 1983; 75(1A):40–45. pmid:6869410
- Melles RB, Marmor MF. Pericentral retinopathy and racial differences in hydroxychloroquine toxicity. Ophthalmology 2015; 122(1):110–116. doi:10.1016/j.ophtha.2014.07.018
- Uslu H, Gurler B, Yildirim A, et al. Effect of hydroxychloroquine on the retinal layers: a quantitative evaluation with spectral-domain optical coherence tomography. J Ophthalmol 2016; 2016:8643174. doi:10.1155/2016/8643174
- Au A, Parikh V, Modi YS, Ehlers JP, Schachat AP, Singh RP. Hydroxychloroquine screening practice patterns within a large multispecialty ophthalmic practice. Am J Ophthalmol 2015; 160(3):561–568.e2. doi:10.1016/j.ajo.2015.06.009
- Jallouli M, Frances C, Plette JC, et al; Plaquenil Lupus Systemic Study Group. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus. JAMA Dermatol 2013; 149(8):935–940. doi:10.1001/jamadermatol.2013.709
- Avina-Zubieta JA, Johnson ES, Suarez-Almazor ME, Russell AS. Incidence of myopathy in patients treated with antimalarials: a report of three cases and review of the literature. Br J Rheumatol 1995; 34(2):166–170. pmid:7704464
- Yogasundaram H, Putko BN, Tien J, et al. Hydroxychloroquine-induced cardiomyopathy: case report, pathophysiology, diagnosis, and treatment. Can J Cardiol 2014; 30:1706–1715. doi:10.1016/j.cjca.2014.08.016
- Pearson KC, Morrell DS, Runge SR, Jolly P. Prolonged pustular eruption from hydroxychloroquine: an unusual case of acute generalized exanthematous pustulosis. Cutis 2016; 97(3):212–216. pmid:27023083
- Youngster I, Arcavi L, Schechmaster R, et al. Medications and glucose-6-phosphate dehydrogenase deficiency: an evidence-based review. Drug Saf 2010; 33(9):713–726. doi:10.2165/11536520-000000000-00000
- Ostensen M, Khamashta M, Lockshin M, et al. Anti-inflammatory and immunosuppressive drugs and reproduction. Arthritis Res Ther 2006; 8(3):209. doi:10.1186/ar1957
- Leroux M, Desveaux C, Parcevaux M, et al. Impact of hydroxychloroquine on preterm delivery and intrauterine growth restriction in pregnant women with systemic lupus erythematosus: a descriptive cohort study. Lupus 2015; 24(13):1384–1391. doi:10.1177/0961203315591027
- Clowse MEB, Magder L, Witter F, Petri M. Hydroxychloroquine in lupus pregnancy. Arthritis Rheum 2006; 54(11):3640–3647. doi:10.1002/art.22159
- Levy RA, Vilela VS, Cataldo MJ, et al. Hydroxychloroquine in lupus pregnancy: double-blind and placebo-controlled study. Lupus 2001; 10(6):401–404. doi:10.1191/096120301678646137
- James JA, Kim-Howard XR, Bruner BF, et al. Hydroxychloroquine sulfate treatment is associated with later onset of systemic lupus erythematosus. Lupus 2007; 16(6):401–409. doi:10.1177/0961203307078579
- Akpovwa H. Chloroquine could be used for the treatment of filoviral infections and other viral infections that emerge or emerged from viruses requiring an acidic pH for infectivity. Cell Biochem Funct 2016; 34(4):191–196. doi:10.1002/cbf.3182
- Pascolo S. Time to use a dose of chloroquine as an adjuvant to anti-cancer chemotherapies. Eur J Pharmacol 2016; 771:139–144. doi:10.1016/j.ejphar.2015.12.017
- Furlong HC, Wessels JM, Guerra MT, Stämpfli MR, Foster WG. Hydroxychloroquine attenuates cigarette smoke induced autophagic signaling in the mouse ovary. Reprod Toxicol 2016; 61:105–113. doi:10.1016/j.reprotox.2016.03.044
- Cairoli E, Danese N, Teliz M, et al. Cumulative dose of hydroxychloroquine is associated with a decrease of resting heart rate in patients with systemic lupus erythematosus: a pilot study. Lupus 2015; 24(11):1204–1209. doi:10.1177/0961203315580870
A 29-year-old African American woman presents with a photosensitive malar rash, fatigue, morning stiffness, and swelling in her hands. She is found to have elevated antinuclear antibody at a titer of 1:320. A complete blood cell count demonstrates leukopenia and thrombocytopenia. Results of renal function testing and urinalysis are within normal limits. She has no other medical problems and no history of blood clots or pregnancy loss.
Her arthritis and rash suggest systemic lupus erythematosus. She is counseled to avoid sun exposure, and treatment with hydroxychloroquine is considered.
WHAT IS HYDROXYCHLOROQUINE?
Hydroxychloroquine was developed to treat malaria but was later found to have immunomodulatory properties. It is now approved by the US Food and Drug Administration for treatment of discoid lupus, systemic lupus erythematosus, and rheumatoid arthritis. It is also approved to treat malaria; however, of the several malarial parasites, only Plasmodium falciparum can still be cured by hydroxychloroquine, and growing resistance limits the geographic locations where this drug can be used effectively.1,2
HISTORICAL BACKGROUND
Antimalarial drugs were discovered shortly before World War II. Their production was industrialized during the war because malaria was a leading cause of disease among soldiers, especially those deployed to the South Pacific.3
Atabrine (quinacrine), the first antimalarial widely used, had numerous side effects including yellowing of the skin. Aggressive research efforts to develop an alternative led to field testing of one of its derivative compounds, chloroquine, by the US Army in 1943. Continued chemical modification would create hydroxychloroquine, introduced in 1955.
A serendipitous consequence of the mass use of antimalarials during World War II was the discovery that they could be used to treat inflammatory arthritis and lupus. Eight years after the war ended, Shee4 reported that chloroquine had a beneficial effect on lupus and rheumatoid arthritis in US soldiers. Hydroxychloroquine is now the most commonly prescribed antimalarial for treatment of autoimmune disease.
HOW HYDROXYCHLOROQUINE WORKS
The primary mechanism by which hydroxychloroquine modulates systemic lupus erythematosus is by suppressing activation of Toll-like receptors, which exist on the surface of endosomes and play a significant role in the innate immune response and in autoimmune disease. Their activation is necessary for the expression of interferon-regulated genes and production of tumor necrosis factor alpha, which are key in the cell-mediated inflammatory response.
Antimalarial drugs such as hydroxychloroquine prevent Toll-like receptor activation by binding directly to nucleic acids in the activation pathway.5 In vitro studies show that blocking this pathway blunts the body’s primary cell-mediated inflammatory response; in vivo studies show that use of hydroxychloroquine is strongly correlated with a reduction in interferon alpha levels.6 The powerful effect of hydroxychloroquine on the cell-mediated pattern of inflammation found in lupus is consistent with this theory.
It was previously hypothesized that the immune-modulating effects of hydroxychloroquine were associated with a more general dysregulation of cellular lysosomes through inhibition of proteolysis or changes in cellular pH.7 This theory has since been displaced by the more specific and elegant mechanism described above.5
HOW WELL DOES IT WORK?
Benefit in systemic lupus erythematosus
Hydroxychloroquine has consistently demonstrated significant and multifaceted benefit in patients with systemic lupus erythematosus.
A systematic review of 95 articles8 concluded that this drug decreases lupus flares and decreases mortality rates in lupus patients by at least 50%, with a high level of evidence. Beneficial effects that had a moderate level of evidence were an increase in bone mineral density, fewer thrombotic events, and fewer cases of irreversible organ damage.
The preventive effect of hydroxychloroquine on thrombosis in lupus patients has been consistently demonstrated and is one of the key reasons the drug is considered a cornerstone of therapy in this disease.9 A nested case-control study of patients with lupus and thromboembolism demonstrated an odds ratio of 0.31 and relative risk reduction of 68% for those using antimalarials.10
Benefit in antiphospholipid antibody syndrome
Hydroxychloroquine prevents thrombosis in other diseases as well. For example, it has been shown to reduce the incidence of thrombotic events in patients with primary antiphospholipid syndrome.
In a retrospective cohort study in 114 patients with this disease, hydroxychloroquine significantly reduced the incidence of arterial thrombotic events over 10 years of follow-up (recurrence incidence 0 in those treated with hydroxychloroquine vs 1.14% in those not treated).11 The study also tracked levels of antiphospholipid antibodies and reported that hydroxychloroquine significantly reduced the levels of antibodies to cardiolipin and beta-2 glycoprotein 1, both implicated in the pathology of thrombosis.11
In vitro studies have also demonstrated that hydroxychloroquine can modulate a dysregulated inflammatory system to reduce thrombosis. For example, it has been shown that hydroxychloroquine can reverse platelet activation by antiphospholipid antibodies, prevent linking of antibody complexes to cell membranes, and promote proper membrane protein expression, thereby reducing the thrombotic qualities of antiphospholipid antibodies and even improving clearance times of antiphospholipid-related thrombi.12
Benefit in rheumatoid arthritis
Though there is less evidence, hydroxychloroquine has also shown benefit in rheumatoid arthritis, where it can be used by itself in mild disease or as part of combination therapy with active arthritis. Compared with biologic therapy in patients with early aggressive rheumatoid arthritis, triple therapy with methotrexate, sulfasalazine, and hydroxychloroquine was nearly as effective in terms of quality of life, and it cost only one-third as much, saving $20,000 per year of therapy per patient.13
Hydroxychloroquine has also been compared directly with chloroquine, its closest relation, in a large study incorporating patients with rheumatoid arthritis and patients with systemic lupus erythematosus. Patients using chloroquine experienced significantly more side effects, though it did prove marginally more effective.14
No benefit shown in Sjögren syndrome
Unfortunately, despite widespread use, hydroxychloroquine has not demonstrated positive clinical effects when used to treat primary Sjögren syndrome. Most notably, a 2014 randomized controlled trial of hydroxychloroquine vs placebo in 120 Sjögren patients found no significant improvement in primary symptoms of dryness, pain, or fatigue after 6 months of therapy.15
Metabolic benefits
Unexpectedly, hydroxychloroquine is associated with multiple metabolic benefits including improved lipid profiles and lower blood glucose levels. These findings, in addition to a reduced incidence of thrombosis, were initially reported in the Baltimore Lupus Cohort in 1996.16 Specifically, longitudinal evaluation of a cohort of lupus patients showed that hydroxychloroquine use was associated with a 7.6% reduction in total cholesterol and a 13.7% reduction in low-density lipoprotein cholesterol (LDL-C) over 3 months of therapy.17
Similar findings, including a reduction in LDL-C and an increase in high-density lipoprotein cholesterol, were strongly associated with the addition of hydroxychloroquine to methotrexate or to methotrexate and etanercept in a large cohort of rheumatoid arthritis patients followed over 2 years of therapy.18
In nondiabetic women with systemic lupus erythematosus or rheumatoid arthritis, average blood glucose was significantly lower in those taking hydroxychloroquine than in nonusers. The incidence of insulin resistance was also lower, but the difference was not statistically significant.19
Some have suggested that hydroxychloroquine may prevent diabetes mellitus. In a retrospective case series, compared with rheumatoid arthritis patients not taking the drug, patients treated with hydroxychloroquine for more than 4 years had a 25% lower risk of developing diabetes mellitus.20
In view of these metabolic benefits, especially regarding lipid regulation, and the above described antithrombotic properties of hydroxychloroquine, some researchers have recently hypothesized that hydroxychloroquine may be of benefit in patients with coronary artery disease.21 They suggested that the inflammatory contribution to the mechanism of coronary artery disease could be lessened by hydroxychloroquine even in patients without lupus erythematosus or rheumatoid arthritis.
PHARMACOLOGIC PROPERTIES
Understanding the pharmacologic properties of hydroxychloroquine is key to using it appropriately in clinical practice.
The half-life of elimination of hydroxychloroquine is 40 to 50 days, with half of the drug excreted renally in a concentration-dependent fashion.22,23 The drug reaches 95% of its steady-state concentration by about 6 months of therapy. Shorter durations of therapy do not provide adequate time for the drug to achieve steady-state concentration and may not allow patients and providers time to see its full clinical results. Therefore, its manufacturers recommend a 6-month trial of therapy to adequately determine if the drug improves symptoms.1
The oral bioavailability of hydroxychloroquine is about 75%, but pharmacokinetics vary among individuals.22,23 It has been suggested that this variability affects the efficacy of hydroxychloroquine. In a study of 300 patients with cutaneous lupus erythematosus, those whose treatment failed had significantly lower blood concentrations of hydroxychloroquine, while those who achieved complete remission had significantly higher concentrations.24
Another study found that titrating doses to target therapeutic blood concentrations can reduce disease activity in cutaneous lupus erythematosus.25 Measuring the blood concentration of hydroxychloroquine is not common in clinical practice but may have a role in select patients in whom initial therapy using a standard dosing regimen does not produce the desired results.
HOW SAFE IS HYDROXYCHLOROQUINE?
Hydroxychloroquine has numerous adverse effects, necessitating vigilance on the part of the prescriber. Most commonly reported are retinopathy, hyperpigmentation, myopathy, and skin reactions.1
Retinopathy
Retinopathy’s irreversibility—the threat of permanent vision loss—and its substantial prevalence in patients with a large drug exposure history, have marked retinopathy as the most concerning potential toxicity. The risk of ocular toxicity increases with the cumulative hydroxychloroquine dose. The prevalence of retinopathy in those using the drug less than 10 years is less than 2%; in contrast, the prevalence in patients with more than 20 years of exposure is reported to be as high as 20%.26
The American Academy of Ophthalmology has long stated that retinopathy is a significant risk of hydroxychloroquine therapy and that patients taking hydroxychloroquine should therefore undergo routine retinal and visual field screening by an ophthalmologist.
Currently, initial screening followed by yearly screening beginning 5 years thereafter is recommended for patients at low risk of toxicity (Table 1).27 Patients determined by an ophthalmologist to be at higher risk of retinopathy should be screened yearly. As identified by the American Academy of Ophthalmology, major risk factors for retinopathy include duration of use, concomitant tamoxifen exposure, significant renal disease, and preexisting retinal and macular disease.26,28
Recommendations for hydroxychloroquine dosing and screening were recently revised, for 2 reasons. Initially, its manufacturers recommended that hydroxychloroquine dosage be no higher than 6.5 mg/kg of ideal body weight to prevent retinopathy.1,29,30 However, it has recently been demonstrated that real body weight is a better predictor of risk of retinopathy than ideal body weight when dosing hydroxychloroquine, perhaps because of the increasing variance of real body weight in our patient population.26
Further, an atypical pattern of retinopathy called pericentral retinopathy is more common in Asians. A study of about 200 patients with a history of hydroxychloroquine retinopathy, including 36 Asian patients, found that the pericentral pattern occurred in half the Asian patients but only 2% of the white patients.31 The mechanism for this finding is unclear, but because pericentral retinopathy spares the macula, it can be missed using standard screening methods. Therefore, the American Academy of Ophthalmology now recommends that the dose limit be reduced from 6.5 mg/kg of ideal body weight to no more than 5.0 mg/kg of real body weight (Table 2).28
It is also recommended that screening methods such as automated visual fields and optical coherence tomography extend their fields beyond the macula in Asian patients to ensure that pericentral retinopathy is not missed.28
Optical coherence tomography is a particularly useful tool in the ocular evaluation of patients taking hydroxychloroquine. It can detect subtle changes such as thinning of the foveal photoreceptor outer segment, thickening of the retinal pigment epithelium, and loss of the macular ganglion cell–inner plexiform layer before there are visible signs of retinopathy and before symptoms arise.32
Currently, these guidelines are underutilized in clinical practice. Physician adherence to ophthalmologic guidelines is reported at about 50%.33 This statistic is jarring, given the potential for permanent loss of vision in those with hydroxychloroquine-mediated retinopathy, and demonstrates the importance of reinforcing proper understanding of the use of hydroxychloroquine in clinical practice.
Other adverse effects
Cutaneous hyperpigmentation can occur with hydroxychloroquine use (Figure 1). The hyperpigmentation appears to be due to local bruising following deposition of iron in the soft tissue.
While the pigmentation may persist permanently and cause an undesirable cosmetic effect, it has not been associated with other adverse outcomes.
Myopathy is a rare adverse effect. In one case series, 3 of 214 patients treated with hydroxychloroquine developed hydroxychloroquine-induced myopathy.35 Over the duration of their therapy, this was equivalent to an incidence of 1 case of myopathy in 100 patient-years of therapy. Myopathy improves with discontinuation of therapy, though it can persist for weeks, likely because of hydroxychloroquine’s prolonged elimination half-life.
Cardiomyopathy, specifically neurocardiomyopathy, is also an extremely rare adverse effect of hydroxychloroquine use. The mechanism is believed to be associated with the effect of hydroxychloroquine on lysosomal action, leading to an acquired lysosomal storage disorder with the typical cardiac hypertrophy and conduction abnormalities associated with this family of diseases.36
Acute generalized exanthematous pustulosis is another rare complication of hydroxychloroquine therapy. The appearance of the reaction is similar to that of pustular psoriasis, with pustules overlying flaking and scaling skin. It usually resolves within 2 weeks after cessation of hydroxychloroquine therapy. In a select few cases, the reaction persists or waxes and wanes over a period of weeks to months, and longer durations of recovery are thought to be due to hydroxychloroquine’s long half-life, as in hydroxychloroquine-induced myopathy.37
In view of this rare reaction, manufacturers of hydroxychloroquine recommend caution when using the drug in patients with psoriasis.1
Hematologic abnormalities. In very rare cases, hydroxychloroquine is associated with hematologic abnormalities including agranulocytosis, anemia, aplastic anemia, leukopenia, and thrombocytopenia.1
While no specific guidelines exist, caution is warranted when using hydroxychloroquine in patients with porphyria. Additionally, hydroxychloroquine and other antimalarials including primaquine have been associated with hemolysis in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. The risk of hemolysis is generally considered low except at high hydroxychloroquine doses in patients with severe G6PD deficiency.38
For the above reasons, manufacturers recommended baseline and routine blood counts, and some providers screen patients for G6PD deficiency when prescribing hydroxychloroquine (Table 3).
PREGNANCY
Hydroxychloroquine is in pregnancy category C. Information is limited, and in view of the risks, the manufacturer says that it should be avoided in pregnancy.1 Nevertheless, it is generally considered safe during pregnancy, and its benefits may make it acceptable to continue in a patient who becomes pregnant, in spite of the possible risks.
We favor continuing hydroxychloroquine. This drug has been associated with improved maternal and fetal outcomes in lupus patients. Its use during pregnancy has not been associated with congenital malformations. The adverse visual effects of long-term hydroxychloroquine use, namely retinopathy, have never been reported in children as a consequence of exposure in utero.
In addition, hydroxychloroquine is transmitted only in minute quantities in breast milk.39 In pregnant women with systemic lupus erythematosus, hydroxychloroquine was associated with a lower risk of adverse outcomes, including preterm delivery and intrauterine growth restriction.40 However, hydroxychloroquine is far more toxic when ingested directly by infants than in adults.1
Maternal outcomes are also improved with the use of hydroxychloroquine. Stopping hydroxychloroquine during pregnancy in women with systemic lupus erythematosus is associated with significantly higher disease activity—fully twice as high as in those who continue hydroxychloroquine.41 These study results were corroborated in a small randomized trial in which pregnant women with lupus on placebo had significantly higher lupus disease activity scores than those pregnant women who were given hydroxychloroquine.42 The women taking hydroxychloroquine experienced no severe lupus flares for the duration of their pregnancies.
These findings suggest not only that hydroxychloroquine is safe in pregnancy, but also that it should be continued in lupus patients during pregnancy to prevent disease flares and adverse fetal outcomes.
AREAS OF UNCERTAINTY
Benefit in preclinical lupus?
Hydroxychloroquine has a consistently profound effect on outcomes in systemic lupus erythematosus. These findings, in addition to the more widespread use of antibody screening, have led to suggestions that hydroxychloroquine could be of benefit even before systemic lupus erythematosus is diagnosed.
A study in US military personnel found that patients taking hydroxychloroquine experienced a significantly longer lag time between first reported clinical symptoms of lupus and official diagnosis compared with matched controls who also went on to develop the disease, averaging 1.08 vs 0.29 years to disease classification.43 Those who used hydroxychloroquine also had lower rates of autoantibody accumulation. Therefore, hydroxychloroquine could be of benefit in carefully selected candidates at high risk of developing systemic lupus erythematosus.
The beneficial effects of hydroxychloroquine on patients with lupus and rheumatoid arthritis, in terms of primary measures of disease activity and secondary outcomes, were discovered fortuitously and were not the original intended targets of the drug. Because of its versatility, there are numerous other disease states in which hydroxychloroquine has shown a degree of benefit or has shown a potential for benefit.
Antiviral activity?
It has been suggested that antimalarial drugs could serve as adjunctive therapies against filoviruses such as Marburg and Ebola. There is a small body of in vitro and in vivo evidence that hydroxychloroquine could temper severe systemic inflammatory responses to filoviruses both through dysregulation of lysosomes and lysosomal pH (filoviruses have a pH-dependent mechanism of action) and through decreased production of tumor necrosis factor alpha and interferons. Heavy burdens of interferons and tumor necrosis factor alpha are associated with increased mortality rates in those infected with filoviruses.44
Antineoplastic activity?
Hydroxychloroquine has undergone in vitro testing as an adjunct to cancer therapies. There are several mechanisms by which it is theorized that hydroxychloroquine could target malignant cells, including inhibition of multidrug resistance pumps or autophagy, improvement of chemotherapy cell penetration, potentiation of presentation of major histocompatibility complexes, or even intercalation directly into DNA.45,46 However, it can also impair natural anticancer immunity and may allow cancer cells better nutrient supply through vascular effects.
In vitro studies have shown tumoricidal effects in lymphoma and melanoma, and inhibition of growth in lung, colon, breast, cervix, larynx, liver, and prostate cancers. In vivo studies have shown that hydroxychloroquine in high doses can prolong survival in glioblastoma.45
Unfortunately, all of these theorized or observed effects are dose-dependent and likely require doses that exceed currently recommended maximums.
Negative chronotropic effect?
Hydroxychloroquine has been found to decrease the resting heart rate in a cumulative dose-dependent fashion.47 Further, hydroxychloroquine has been known to increase digoxin levels, and the medications should not be used in combination.1
Whether the decrease in resting heart rate is associated with harm or benefit and whether the effect is significant enough to be considered when implementing therapy remain unanswered and deserve further investigation, as does the primary use of hydroxychloroquine for beneficial lipid and glucose reduction in patients who are otherwise healthy.
CASE CONCLUSION
The patient described at the beginning of this article was provided with information on the risks and benefits of hydroxychloroquine for treatment of her arthritis and rash suggestive of mild systemic lupus, and she opted to begin therapy. Her baseline eye screening was within normal limits. Based on her weight of 62 kg, she was started on 300 mg of hydroxychloroquine daily.
She had no significant adverse effects from the medication and reported slow improvement in her rash and joint complaints over the next 2 months. She remained on hydroxychloroquine over the next year without adverse effects or new evidence of autoimmune disease.
A 29-year-old African American woman presents with a photosensitive malar rash, fatigue, morning stiffness, and swelling in her hands. She is found to have elevated antinuclear antibody at a titer of 1:320. A complete blood cell count demonstrates leukopenia and thrombocytopenia. Results of renal function testing and urinalysis are within normal limits. She has no other medical problems and no history of blood clots or pregnancy loss.
Her arthritis and rash suggest systemic lupus erythematosus. She is counseled to avoid sun exposure, and treatment with hydroxychloroquine is considered.
WHAT IS HYDROXYCHLOROQUINE?
Hydroxychloroquine was developed to treat malaria but was later found to have immunomodulatory properties. It is now approved by the US Food and Drug Administration for treatment of discoid lupus, systemic lupus erythematosus, and rheumatoid arthritis. It is also approved to treat malaria; however, of the several malarial parasites, only Plasmodium falciparum can still be cured by hydroxychloroquine, and growing resistance limits the geographic locations where this drug can be used effectively.1,2
HISTORICAL BACKGROUND
Antimalarial drugs were discovered shortly before World War II. Their production was industrialized during the war because malaria was a leading cause of disease among soldiers, especially those deployed to the South Pacific.3
Atabrine (quinacrine), the first antimalarial widely used, had numerous side effects including yellowing of the skin. Aggressive research efforts to develop an alternative led to field testing of one of its derivative compounds, chloroquine, by the US Army in 1943. Continued chemical modification would create hydroxychloroquine, introduced in 1955.
A serendipitous consequence of the mass use of antimalarials during World War II was the discovery that they could be used to treat inflammatory arthritis and lupus. Eight years after the war ended, Shee4 reported that chloroquine had a beneficial effect on lupus and rheumatoid arthritis in US soldiers. Hydroxychloroquine is now the most commonly prescribed antimalarial for treatment of autoimmune disease.
HOW HYDROXYCHLOROQUINE WORKS
The primary mechanism by which hydroxychloroquine modulates systemic lupus erythematosus is by suppressing activation of Toll-like receptors, which exist on the surface of endosomes and play a significant role in the innate immune response and in autoimmune disease. Their activation is necessary for the expression of interferon-regulated genes and production of tumor necrosis factor alpha, which are key in the cell-mediated inflammatory response.
Antimalarial drugs such as hydroxychloroquine prevent Toll-like receptor activation by binding directly to nucleic acids in the activation pathway.5 In vitro studies show that blocking this pathway blunts the body’s primary cell-mediated inflammatory response; in vivo studies show that use of hydroxychloroquine is strongly correlated with a reduction in interferon alpha levels.6 The powerful effect of hydroxychloroquine on the cell-mediated pattern of inflammation found in lupus is consistent with this theory.
It was previously hypothesized that the immune-modulating effects of hydroxychloroquine were associated with a more general dysregulation of cellular lysosomes through inhibition of proteolysis or changes in cellular pH.7 This theory has since been displaced by the more specific and elegant mechanism described above.5
HOW WELL DOES IT WORK?
Benefit in systemic lupus erythematosus
Hydroxychloroquine has consistently demonstrated significant and multifaceted benefit in patients with systemic lupus erythematosus.
A systematic review of 95 articles8 concluded that this drug decreases lupus flares and decreases mortality rates in lupus patients by at least 50%, with a high level of evidence. Beneficial effects that had a moderate level of evidence were an increase in bone mineral density, fewer thrombotic events, and fewer cases of irreversible organ damage.
The preventive effect of hydroxychloroquine on thrombosis in lupus patients has been consistently demonstrated and is one of the key reasons the drug is considered a cornerstone of therapy in this disease.9 A nested case-control study of patients with lupus and thromboembolism demonstrated an odds ratio of 0.31 and relative risk reduction of 68% for those using antimalarials.10
Benefit in antiphospholipid antibody syndrome
Hydroxychloroquine prevents thrombosis in other diseases as well. For example, it has been shown to reduce the incidence of thrombotic events in patients with primary antiphospholipid syndrome.
In a retrospective cohort study in 114 patients with this disease, hydroxychloroquine significantly reduced the incidence of arterial thrombotic events over 10 years of follow-up (recurrence incidence 0 in those treated with hydroxychloroquine vs 1.14% in those not treated).11 The study also tracked levels of antiphospholipid antibodies and reported that hydroxychloroquine significantly reduced the levels of antibodies to cardiolipin and beta-2 glycoprotein 1, both implicated in the pathology of thrombosis.11
In vitro studies have also demonstrated that hydroxychloroquine can modulate a dysregulated inflammatory system to reduce thrombosis. For example, it has been shown that hydroxychloroquine can reverse platelet activation by antiphospholipid antibodies, prevent linking of antibody complexes to cell membranes, and promote proper membrane protein expression, thereby reducing the thrombotic qualities of antiphospholipid antibodies and even improving clearance times of antiphospholipid-related thrombi.12
Benefit in rheumatoid arthritis
Though there is less evidence, hydroxychloroquine has also shown benefit in rheumatoid arthritis, where it can be used by itself in mild disease or as part of combination therapy with active arthritis. Compared with biologic therapy in patients with early aggressive rheumatoid arthritis, triple therapy with methotrexate, sulfasalazine, and hydroxychloroquine was nearly as effective in terms of quality of life, and it cost only one-third as much, saving $20,000 per year of therapy per patient.13
Hydroxychloroquine has also been compared directly with chloroquine, its closest relation, in a large study incorporating patients with rheumatoid arthritis and patients with systemic lupus erythematosus. Patients using chloroquine experienced significantly more side effects, though it did prove marginally more effective.14
No benefit shown in Sjögren syndrome
Unfortunately, despite widespread use, hydroxychloroquine has not demonstrated positive clinical effects when used to treat primary Sjögren syndrome. Most notably, a 2014 randomized controlled trial of hydroxychloroquine vs placebo in 120 Sjögren patients found no significant improvement in primary symptoms of dryness, pain, or fatigue after 6 months of therapy.15
Metabolic benefits
Unexpectedly, hydroxychloroquine is associated with multiple metabolic benefits including improved lipid profiles and lower blood glucose levels. These findings, in addition to a reduced incidence of thrombosis, were initially reported in the Baltimore Lupus Cohort in 1996.16 Specifically, longitudinal evaluation of a cohort of lupus patients showed that hydroxychloroquine use was associated with a 7.6% reduction in total cholesterol and a 13.7% reduction in low-density lipoprotein cholesterol (LDL-C) over 3 months of therapy.17
Similar findings, including a reduction in LDL-C and an increase in high-density lipoprotein cholesterol, were strongly associated with the addition of hydroxychloroquine to methotrexate or to methotrexate and etanercept in a large cohort of rheumatoid arthritis patients followed over 2 years of therapy.18
In nondiabetic women with systemic lupus erythematosus or rheumatoid arthritis, average blood glucose was significantly lower in those taking hydroxychloroquine than in nonusers. The incidence of insulin resistance was also lower, but the difference was not statistically significant.19
Some have suggested that hydroxychloroquine may prevent diabetes mellitus. In a retrospective case series, compared with rheumatoid arthritis patients not taking the drug, patients treated with hydroxychloroquine for more than 4 years had a 25% lower risk of developing diabetes mellitus.20
In view of these metabolic benefits, especially regarding lipid regulation, and the above described antithrombotic properties of hydroxychloroquine, some researchers have recently hypothesized that hydroxychloroquine may be of benefit in patients with coronary artery disease.21 They suggested that the inflammatory contribution to the mechanism of coronary artery disease could be lessened by hydroxychloroquine even in patients without lupus erythematosus or rheumatoid arthritis.
PHARMACOLOGIC PROPERTIES
Understanding the pharmacologic properties of hydroxychloroquine is key to using it appropriately in clinical practice.
The half-life of elimination of hydroxychloroquine is 40 to 50 days, with half of the drug excreted renally in a concentration-dependent fashion.22,23 The drug reaches 95% of its steady-state concentration by about 6 months of therapy. Shorter durations of therapy do not provide adequate time for the drug to achieve steady-state concentration and may not allow patients and providers time to see its full clinical results. Therefore, its manufacturers recommend a 6-month trial of therapy to adequately determine if the drug improves symptoms.1
The oral bioavailability of hydroxychloroquine is about 75%, but pharmacokinetics vary among individuals.22,23 It has been suggested that this variability affects the efficacy of hydroxychloroquine. In a study of 300 patients with cutaneous lupus erythematosus, those whose treatment failed had significantly lower blood concentrations of hydroxychloroquine, while those who achieved complete remission had significantly higher concentrations.24
Another study found that titrating doses to target therapeutic blood concentrations can reduce disease activity in cutaneous lupus erythematosus.25 Measuring the blood concentration of hydroxychloroquine is not common in clinical practice but may have a role in select patients in whom initial therapy using a standard dosing regimen does not produce the desired results.
HOW SAFE IS HYDROXYCHLOROQUINE?
Hydroxychloroquine has numerous adverse effects, necessitating vigilance on the part of the prescriber. Most commonly reported are retinopathy, hyperpigmentation, myopathy, and skin reactions.1
Retinopathy
Retinopathy’s irreversibility—the threat of permanent vision loss—and its substantial prevalence in patients with a large drug exposure history, have marked retinopathy as the most concerning potential toxicity. The risk of ocular toxicity increases with the cumulative hydroxychloroquine dose. The prevalence of retinopathy in those using the drug less than 10 years is less than 2%; in contrast, the prevalence in patients with more than 20 years of exposure is reported to be as high as 20%.26
The American Academy of Ophthalmology has long stated that retinopathy is a significant risk of hydroxychloroquine therapy and that patients taking hydroxychloroquine should therefore undergo routine retinal and visual field screening by an ophthalmologist.
Currently, initial screening followed by yearly screening beginning 5 years thereafter is recommended for patients at low risk of toxicity (Table 1).27 Patients determined by an ophthalmologist to be at higher risk of retinopathy should be screened yearly. As identified by the American Academy of Ophthalmology, major risk factors for retinopathy include duration of use, concomitant tamoxifen exposure, significant renal disease, and preexisting retinal and macular disease.26,28
Recommendations for hydroxychloroquine dosing and screening were recently revised, for 2 reasons. Initially, its manufacturers recommended that hydroxychloroquine dosage be no higher than 6.5 mg/kg of ideal body weight to prevent retinopathy.1,29,30 However, it has recently been demonstrated that real body weight is a better predictor of risk of retinopathy than ideal body weight when dosing hydroxychloroquine, perhaps because of the increasing variance of real body weight in our patient population.26
Further, an atypical pattern of retinopathy called pericentral retinopathy is more common in Asians. A study of about 200 patients with a history of hydroxychloroquine retinopathy, including 36 Asian patients, found that the pericentral pattern occurred in half the Asian patients but only 2% of the white patients.31 The mechanism for this finding is unclear, but because pericentral retinopathy spares the macula, it can be missed using standard screening methods. Therefore, the American Academy of Ophthalmology now recommends that the dose limit be reduced from 6.5 mg/kg of ideal body weight to no more than 5.0 mg/kg of real body weight (Table 2).28
It is also recommended that screening methods such as automated visual fields and optical coherence tomography extend their fields beyond the macula in Asian patients to ensure that pericentral retinopathy is not missed.28
Optical coherence tomography is a particularly useful tool in the ocular evaluation of patients taking hydroxychloroquine. It can detect subtle changes such as thinning of the foveal photoreceptor outer segment, thickening of the retinal pigment epithelium, and loss of the macular ganglion cell–inner plexiform layer before there are visible signs of retinopathy and before symptoms arise.32
Currently, these guidelines are underutilized in clinical practice. Physician adherence to ophthalmologic guidelines is reported at about 50%.33 This statistic is jarring, given the potential for permanent loss of vision in those with hydroxychloroquine-mediated retinopathy, and demonstrates the importance of reinforcing proper understanding of the use of hydroxychloroquine in clinical practice.
Other adverse effects
Cutaneous hyperpigmentation can occur with hydroxychloroquine use (Figure 1). The hyperpigmentation appears to be due to local bruising following deposition of iron in the soft tissue.
While the pigmentation may persist permanently and cause an undesirable cosmetic effect, it has not been associated with other adverse outcomes.
Myopathy is a rare adverse effect. In one case series, 3 of 214 patients treated with hydroxychloroquine developed hydroxychloroquine-induced myopathy.35 Over the duration of their therapy, this was equivalent to an incidence of 1 case of myopathy in 100 patient-years of therapy. Myopathy improves with discontinuation of therapy, though it can persist for weeks, likely because of hydroxychloroquine’s prolonged elimination half-life.
Cardiomyopathy, specifically neurocardiomyopathy, is also an extremely rare adverse effect of hydroxychloroquine use. The mechanism is believed to be associated with the effect of hydroxychloroquine on lysosomal action, leading to an acquired lysosomal storage disorder with the typical cardiac hypertrophy and conduction abnormalities associated with this family of diseases.36
Acute generalized exanthematous pustulosis is another rare complication of hydroxychloroquine therapy. The appearance of the reaction is similar to that of pustular psoriasis, with pustules overlying flaking and scaling skin. It usually resolves within 2 weeks after cessation of hydroxychloroquine therapy. In a select few cases, the reaction persists or waxes and wanes over a period of weeks to months, and longer durations of recovery are thought to be due to hydroxychloroquine’s long half-life, as in hydroxychloroquine-induced myopathy.37
In view of this rare reaction, manufacturers of hydroxychloroquine recommend caution when using the drug in patients with psoriasis.1
Hematologic abnormalities. In very rare cases, hydroxychloroquine is associated with hematologic abnormalities including agranulocytosis, anemia, aplastic anemia, leukopenia, and thrombocytopenia.1
While no specific guidelines exist, caution is warranted when using hydroxychloroquine in patients with porphyria. Additionally, hydroxychloroquine and other antimalarials including primaquine have been associated with hemolysis in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. The risk of hemolysis is generally considered low except at high hydroxychloroquine doses in patients with severe G6PD deficiency.38
For the above reasons, manufacturers recommended baseline and routine blood counts, and some providers screen patients for G6PD deficiency when prescribing hydroxychloroquine (Table 3).
PREGNANCY
Hydroxychloroquine is in pregnancy category C. Information is limited, and in view of the risks, the manufacturer says that it should be avoided in pregnancy.1 Nevertheless, it is generally considered safe during pregnancy, and its benefits may make it acceptable to continue in a patient who becomes pregnant, in spite of the possible risks.
We favor continuing hydroxychloroquine. This drug has been associated with improved maternal and fetal outcomes in lupus patients. Its use during pregnancy has not been associated with congenital malformations. The adverse visual effects of long-term hydroxychloroquine use, namely retinopathy, have never been reported in children as a consequence of exposure in utero.
In addition, hydroxychloroquine is transmitted only in minute quantities in breast milk.39 In pregnant women with systemic lupus erythematosus, hydroxychloroquine was associated with a lower risk of adverse outcomes, including preterm delivery and intrauterine growth restriction.40 However, hydroxychloroquine is far more toxic when ingested directly by infants than in adults.1
Maternal outcomes are also improved with the use of hydroxychloroquine. Stopping hydroxychloroquine during pregnancy in women with systemic lupus erythematosus is associated with significantly higher disease activity—fully twice as high as in those who continue hydroxychloroquine.41 These study results were corroborated in a small randomized trial in which pregnant women with lupus on placebo had significantly higher lupus disease activity scores than those pregnant women who were given hydroxychloroquine.42 The women taking hydroxychloroquine experienced no severe lupus flares for the duration of their pregnancies.
These findings suggest not only that hydroxychloroquine is safe in pregnancy, but also that it should be continued in lupus patients during pregnancy to prevent disease flares and adverse fetal outcomes.
AREAS OF UNCERTAINTY
Benefit in preclinical lupus?
Hydroxychloroquine has a consistently profound effect on outcomes in systemic lupus erythematosus. These findings, in addition to the more widespread use of antibody screening, have led to suggestions that hydroxychloroquine could be of benefit even before systemic lupus erythematosus is diagnosed.
A study in US military personnel found that patients taking hydroxychloroquine experienced a significantly longer lag time between first reported clinical symptoms of lupus and official diagnosis compared with matched controls who also went on to develop the disease, averaging 1.08 vs 0.29 years to disease classification.43 Those who used hydroxychloroquine also had lower rates of autoantibody accumulation. Therefore, hydroxychloroquine could be of benefit in carefully selected candidates at high risk of developing systemic lupus erythematosus.
The beneficial effects of hydroxychloroquine on patients with lupus and rheumatoid arthritis, in terms of primary measures of disease activity and secondary outcomes, were discovered fortuitously and were not the original intended targets of the drug. Because of its versatility, there are numerous other disease states in which hydroxychloroquine has shown a degree of benefit or has shown a potential for benefit.
Antiviral activity?
It has been suggested that antimalarial drugs could serve as adjunctive therapies against filoviruses such as Marburg and Ebola. There is a small body of in vitro and in vivo evidence that hydroxychloroquine could temper severe systemic inflammatory responses to filoviruses both through dysregulation of lysosomes and lysosomal pH (filoviruses have a pH-dependent mechanism of action) and through decreased production of tumor necrosis factor alpha and interferons. Heavy burdens of interferons and tumor necrosis factor alpha are associated with increased mortality rates in those infected with filoviruses.44
Antineoplastic activity?
Hydroxychloroquine has undergone in vitro testing as an adjunct to cancer therapies. There are several mechanisms by which it is theorized that hydroxychloroquine could target malignant cells, including inhibition of multidrug resistance pumps or autophagy, improvement of chemotherapy cell penetration, potentiation of presentation of major histocompatibility complexes, or even intercalation directly into DNA.45,46 However, it can also impair natural anticancer immunity and may allow cancer cells better nutrient supply through vascular effects.
In vitro studies have shown tumoricidal effects in lymphoma and melanoma, and inhibition of growth in lung, colon, breast, cervix, larynx, liver, and prostate cancers. In vivo studies have shown that hydroxychloroquine in high doses can prolong survival in glioblastoma.45
Unfortunately, all of these theorized or observed effects are dose-dependent and likely require doses that exceed currently recommended maximums.
Negative chronotropic effect?
Hydroxychloroquine has been found to decrease the resting heart rate in a cumulative dose-dependent fashion.47 Further, hydroxychloroquine has been known to increase digoxin levels, and the medications should not be used in combination.1
Whether the decrease in resting heart rate is associated with harm or benefit and whether the effect is significant enough to be considered when implementing therapy remain unanswered and deserve further investigation, as does the primary use of hydroxychloroquine for beneficial lipid and glucose reduction in patients who are otherwise healthy.
CASE CONCLUSION
The patient described at the beginning of this article was provided with information on the risks and benefits of hydroxychloroquine for treatment of her arthritis and rash suggestive of mild systemic lupus, and she opted to begin therapy. Her baseline eye screening was within normal limits. Based on her weight of 62 kg, she was started on 300 mg of hydroxychloroquine daily.
She had no significant adverse effects from the medication and reported slow improvement in her rash and joint complaints over the next 2 months. She remained on hydroxychloroquine over the next year without adverse effects or new evidence of autoimmune disease.
- Sanofi-Aventis. Product monograph: Plaquenil. http://products.sanofi.ca/en/plaquenil.pdf. Accessed May 2, 2018.
- Centers for Disease Control and Prevention (CDC). Malaria information and prophylaxis, by country. www.cdc.gov/malaria/travelers/country_table/a.html. Accessed May 2, 2018.
- Wallace DJ. The history of antimalarials. Lupus 1996; 5(suppl 1):S2–S3. pmid:8803902
- Shee JC. Lupus erythematosus treated with chloroquine. Lancet 1953; 265(6778):201–202. pmid:13070595
- Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol 2011; 186:4794–4804. doi:10.4049/jimmunol.1000702
- Willis R, Seif AM, McGwin G Jr, et al. Effect of hydroxychloroquine treatment on pro-inflammatory cytokines and disease activity in SLE patients: data from LUMINA, a multiethnic US cohort. Lupus 2012; 21(8):830–835. doi:10.1177/0961203312437270
- Fox R. Anti-malarial drugs: possible mechanisms of action in autoimmune disease and prospects for drug development. Lupus 1996; 5(suppl 1):S4–S10. pmid:8803903
- Ruiz-Irastorza G, Ramos-Casals M, Brito-Zeron P, Khamashta MA. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis 2010; 69(1):20–28. doi:10.1136/ard.2008.101766
- Lam NC, Ghetu MV, Bieniek ML. Systemic lupus erythematosus: primary care approach to diagnosis and management. Am Fam Physician 2016; 94(4):284–294. pmid:27548593
- Jung H, Bobba R, Su J, et al. The protective effect of antimalarial drugs on thrombovascular events in systemic lupus erythematosus. Arthritis Rheum 2010; 62(3):863–868. doi:10.1002/art.27289
- Nuri E, Taraborelli M, Andreoli L, et al. Long-term use of hydroxychloroquine reduces antiphospholipid antibodies levels in patients with primary antiphospholipid syndrome. Immunol Res 2017; 65(1):17–24. doi:10.1007/s12026-016-8812-z
- Erkan D, Aguiar CL, Andrade D, et al. 14th International Congress on Antiphospholipid Antibodies: task force report on antiphospholipid syndrome treatment trends. Autoimmun Rev 2014; 13(6):685–696. doi:10.1016/j.autrev.2014.01.053
- Jalal H, O’Dell JR, Bridges SL Jr, et al. Cost-effectiveness of triple therapy versus etanercept plus methotrexate in early aggressive rheumatoid arthritis. Arthritis Care Res (Hoboken) 2016; 68(12):1751–1757. doi:10.1002/acr.22895
- Avina-Zubieta JA, Galindo-Rodriguez G, Newman S, Suarez-Almazor ME, Russell AS. Long-term effectiveness of antimalarial drugs in rheumatic diseases. Ann Rheum Dis 1998; 57(10):582–587. pmid:9893568
- Gottenberg JE, Ravaud P, Puechal X, et al. Effects of hydroxychloroquine on symptomatic improvement in primary Sjogren syndrome. JAMA 2014; 312(3):249–258. doi:10.1001/jama.2014.7682
- Petri M. Hydroxychloroquine use in the Baltimore Lupus Cohort: effects on lipids, glucose and thrombosis. Lupus 1996; 5(suppl 1):S16–S22. pmid:8803905
- Cairoli E, Rebella M, Danese N, Garra V, Borba EF. Hydroxychloroquine reduces low-density lipoprotein cholesterol levels in systemic lupus erythematosus: a longitudinal evaluation of the lipid-lowering effect. Lupus 2012; 21(11):1178–1182. doi:10.1177/0961203312450084
- Charles-Schoeman C, Wang X, Lee YY, et al. Association of triple therapy with improvement in cholesterol profiles over two-year followup in the treatment of early aggressive rheumatoid arthritis trial. Arthritis Rheumatol 2016; 68(3):577–586. doi:10.1002/art.39502
- Penn SK, Kao AH, Schott LL, et al. Hydroxychloroquine and glycemia in women with rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol 2010; 37(6):1136–1142. doi:10.3899/jrheum.090994
- Wasko MC, Hubert HB, Lingala VB, et al. Hydroxychloroquine and risk of diabetes in patients with rheumatoid arthritis. JAMA 2007; 298(2):187–193. doi:10.1001/jama.298.2.187
- Sun L, Liu M, Li R, et al. Hydroxychloroquine, a promising choice for coronary artery disease? Med Hypotheses 2016; 93:5–7. doi:10.1016/j.mehy.2016.04.045
- Tett SE, Cutler DJ, Day RO, Brown KF. Bioavailability of hydroxychloroquine tablets in healthy volunteers. Br J Clin Pharmacol 1989; 27(6):771–779. pmid:2757893
- Furst DE. Pharmacokinetics of hydroxychloroquine and chloroquine during treatment of rheumatic diseases. Lupus 1996; 5(suppl 1):S11–S15. pmid:8803904
- Frances C, Cosnes A, Duhaut P, et al. Low blood concentration of hydroxychloroquine in patients with refractory cutaneous lupus erythematosus. Arch Dermatol 2012; 148(4):479–484. doi:10.1001/archdermatol.2011.2558
- Chasset F, Arnaud L, Costedoat-Chalumeau N, Zahr N, Bessis D, Francès C. The effect of increasing the dose of hydroxychloroquine in patients with refractory cutaneous lupus erythematosus: an open-label prospective pilot study. J Am Acad Dermatol 2016; 74(4):693–699.e3. doi:10.1016/j.jaad.2015.09.064
- Melles RB, Marmor MF. The risk of toxic retinopathy in patients on long-term hydroxychloroquine therapy. JAMA Ophthalmol 2014; 132(12):1453–1460. doi:10.1001/jamaophthalmol.2014.3459
- Committee on Rheumatologic Care. American College of Rheumatology position statement. Screening for hydroxychloroquine retinopathy. www.rheumatology.org/Portals/0/Files/Screening-for-Hydroxychloroquine-Retinopathy-Position-Statement.pdf. Accessed April 2, 2018.
- Marmor MF, Kellner U, Lai TY, Melles RB, Mieler WF; American Academy of Ophthalmology. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology 2016; 123(6):1386–1394. doi:10.1016/j.ophtha.2016.01.058
- Mackenzie AH. Antimalarial drugs for rheumatoid arthritis. Am J Med 1983; 75(6A):48–58. pmid:6362406
- Mackenzie AH. Dose refinements in long-term therapy of rheumatoid arthritis with antimalarials. Am J Med 1983; 75(1A):40–45. pmid:6869410
- Melles RB, Marmor MF. Pericentral retinopathy and racial differences in hydroxychloroquine toxicity. Ophthalmology 2015; 122(1):110–116. doi:10.1016/j.ophtha.2014.07.018
- Uslu H, Gurler B, Yildirim A, et al. Effect of hydroxychloroquine on the retinal layers: a quantitative evaluation with spectral-domain optical coherence tomography. J Ophthalmol 2016; 2016:8643174. doi:10.1155/2016/8643174
- Au A, Parikh V, Modi YS, Ehlers JP, Schachat AP, Singh RP. Hydroxychloroquine screening practice patterns within a large multispecialty ophthalmic practice. Am J Ophthalmol 2015; 160(3):561–568.e2. doi:10.1016/j.ajo.2015.06.009
- Jallouli M, Frances C, Plette JC, et al; Plaquenil Lupus Systemic Study Group. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus. JAMA Dermatol 2013; 149(8):935–940. doi:10.1001/jamadermatol.2013.709
- Avina-Zubieta JA, Johnson ES, Suarez-Almazor ME, Russell AS. Incidence of myopathy in patients treated with antimalarials: a report of three cases and review of the literature. Br J Rheumatol 1995; 34(2):166–170. pmid:7704464
- Yogasundaram H, Putko BN, Tien J, et al. Hydroxychloroquine-induced cardiomyopathy: case report, pathophysiology, diagnosis, and treatment. Can J Cardiol 2014; 30:1706–1715. doi:10.1016/j.cjca.2014.08.016
- Pearson KC, Morrell DS, Runge SR, Jolly P. Prolonged pustular eruption from hydroxychloroquine: an unusual case of acute generalized exanthematous pustulosis. Cutis 2016; 97(3):212–216. pmid:27023083
- Youngster I, Arcavi L, Schechmaster R, et al. Medications and glucose-6-phosphate dehydrogenase deficiency: an evidence-based review. Drug Saf 2010; 33(9):713–726. doi:10.2165/11536520-000000000-00000
- Ostensen M, Khamashta M, Lockshin M, et al. Anti-inflammatory and immunosuppressive drugs and reproduction. Arthritis Res Ther 2006; 8(3):209. doi:10.1186/ar1957
- Leroux M, Desveaux C, Parcevaux M, et al. Impact of hydroxychloroquine on preterm delivery and intrauterine growth restriction in pregnant women with systemic lupus erythematosus: a descriptive cohort study. Lupus 2015; 24(13):1384–1391. doi:10.1177/0961203315591027
- Clowse MEB, Magder L, Witter F, Petri M. Hydroxychloroquine in lupus pregnancy. Arthritis Rheum 2006; 54(11):3640–3647. doi:10.1002/art.22159
- Levy RA, Vilela VS, Cataldo MJ, et al. Hydroxychloroquine in lupus pregnancy: double-blind and placebo-controlled study. Lupus 2001; 10(6):401–404. doi:10.1191/096120301678646137
- James JA, Kim-Howard XR, Bruner BF, et al. Hydroxychloroquine sulfate treatment is associated with later onset of systemic lupus erythematosus. Lupus 2007; 16(6):401–409. doi:10.1177/0961203307078579
- Akpovwa H. Chloroquine could be used for the treatment of filoviral infections and other viral infections that emerge or emerged from viruses requiring an acidic pH for infectivity. Cell Biochem Funct 2016; 34(4):191–196. doi:10.1002/cbf.3182
- Pascolo S. Time to use a dose of chloroquine as an adjuvant to anti-cancer chemotherapies. Eur J Pharmacol 2016; 771:139–144. doi:10.1016/j.ejphar.2015.12.017
- Furlong HC, Wessels JM, Guerra MT, Stämpfli MR, Foster WG. Hydroxychloroquine attenuates cigarette smoke induced autophagic signaling in the mouse ovary. Reprod Toxicol 2016; 61:105–113. doi:10.1016/j.reprotox.2016.03.044
- Cairoli E, Danese N, Teliz M, et al. Cumulative dose of hydroxychloroquine is associated with a decrease of resting heart rate in patients with systemic lupus erythematosus: a pilot study. Lupus 2015; 24(11):1204–1209. doi:10.1177/0961203315580870
- Sanofi-Aventis. Product monograph: Plaquenil. http://products.sanofi.ca/en/plaquenil.pdf. Accessed May 2, 2018.
- Centers for Disease Control and Prevention (CDC). Malaria information and prophylaxis, by country. www.cdc.gov/malaria/travelers/country_table/a.html. Accessed May 2, 2018.
- Wallace DJ. The history of antimalarials. Lupus 1996; 5(suppl 1):S2–S3. pmid:8803902
- Shee JC. Lupus erythematosus treated with chloroquine. Lancet 1953; 265(6778):201–202. pmid:13070595
- Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol 2011; 186:4794–4804. doi:10.4049/jimmunol.1000702
- Willis R, Seif AM, McGwin G Jr, et al. Effect of hydroxychloroquine treatment on pro-inflammatory cytokines and disease activity in SLE patients: data from LUMINA, a multiethnic US cohort. Lupus 2012; 21(8):830–835. doi:10.1177/0961203312437270
- Fox R. Anti-malarial drugs: possible mechanisms of action in autoimmune disease and prospects for drug development. Lupus 1996; 5(suppl 1):S4–S10. pmid:8803903
- Ruiz-Irastorza G, Ramos-Casals M, Brito-Zeron P, Khamashta MA. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis 2010; 69(1):20–28. doi:10.1136/ard.2008.101766
- Lam NC, Ghetu MV, Bieniek ML. Systemic lupus erythematosus: primary care approach to diagnosis and management. Am Fam Physician 2016; 94(4):284–294. pmid:27548593
- Jung H, Bobba R, Su J, et al. The protective effect of antimalarial drugs on thrombovascular events in systemic lupus erythematosus. Arthritis Rheum 2010; 62(3):863–868. doi:10.1002/art.27289
- Nuri E, Taraborelli M, Andreoli L, et al. Long-term use of hydroxychloroquine reduces antiphospholipid antibodies levels in patients with primary antiphospholipid syndrome. Immunol Res 2017; 65(1):17–24. doi:10.1007/s12026-016-8812-z
- Erkan D, Aguiar CL, Andrade D, et al. 14th International Congress on Antiphospholipid Antibodies: task force report on antiphospholipid syndrome treatment trends. Autoimmun Rev 2014; 13(6):685–696. doi:10.1016/j.autrev.2014.01.053
- Jalal H, O’Dell JR, Bridges SL Jr, et al. Cost-effectiveness of triple therapy versus etanercept plus methotrexate in early aggressive rheumatoid arthritis. Arthritis Care Res (Hoboken) 2016; 68(12):1751–1757. doi:10.1002/acr.22895
- Avina-Zubieta JA, Galindo-Rodriguez G, Newman S, Suarez-Almazor ME, Russell AS. Long-term effectiveness of antimalarial drugs in rheumatic diseases. Ann Rheum Dis 1998; 57(10):582–587. pmid:9893568
- Gottenberg JE, Ravaud P, Puechal X, et al. Effects of hydroxychloroquine on symptomatic improvement in primary Sjogren syndrome. JAMA 2014; 312(3):249–258. doi:10.1001/jama.2014.7682
- Petri M. Hydroxychloroquine use in the Baltimore Lupus Cohort: effects on lipids, glucose and thrombosis. Lupus 1996; 5(suppl 1):S16–S22. pmid:8803905
- Cairoli E, Rebella M, Danese N, Garra V, Borba EF. Hydroxychloroquine reduces low-density lipoprotein cholesterol levels in systemic lupus erythematosus: a longitudinal evaluation of the lipid-lowering effect. Lupus 2012; 21(11):1178–1182. doi:10.1177/0961203312450084
- Charles-Schoeman C, Wang X, Lee YY, et al. Association of triple therapy with improvement in cholesterol profiles over two-year followup in the treatment of early aggressive rheumatoid arthritis trial. Arthritis Rheumatol 2016; 68(3):577–586. doi:10.1002/art.39502
- Penn SK, Kao AH, Schott LL, et al. Hydroxychloroquine and glycemia in women with rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol 2010; 37(6):1136–1142. doi:10.3899/jrheum.090994
- Wasko MC, Hubert HB, Lingala VB, et al. Hydroxychloroquine and risk of diabetes in patients with rheumatoid arthritis. JAMA 2007; 298(2):187–193. doi:10.1001/jama.298.2.187
- Sun L, Liu M, Li R, et al. Hydroxychloroquine, a promising choice for coronary artery disease? Med Hypotheses 2016; 93:5–7. doi:10.1016/j.mehy.2016.04.045
- Tett SE, Cutler DJ, Day RO, Brown KF. Bioavailability of hydroxychloroquine tablets in healthy volunteers. Br J Clin Pharmacol 1989; 27(6):771–779. pmid:2757893
- Furst DE. Pharmacokinetics of hydroxychloroquine and chloroquine during treatment of rheumatic diseases. Lupus 1996; 5(suppl 1):S11–S15. pmid:8803904
- Frances C, Cosnes A, Duhaut P, et al. Low blood concentration of hydroxychloroquine in patients with refractory cutaneous lupus erythematosus. Arch Dermatol 2012; 148(4):479–484. doi:10.1001/archdermatol.2011.2558
- Chasset F, Arnaud L, Costedoat-Chalumeau N, Zahr N, Bessis D, Francès C. The effect of increasing the dose of hydroxychloroquine in patients with refractory cutaneous lupus erythematosus: an open-label prospective pilot study. J Am Acad Dermatol 2016; 74(4):693–699.e3. doi:10.1016/j.jaad.2015.09.064
- Melles RB, Marmor MF. The risk of toxic retinopathy in patients on long-term hydroxychloroquine therapy. JAMA Ophthalmol 2014; 132(12):1453–1460. doi:10.1001/jamaophthalmol.2014.3459
- Committee on Rheumatologic Care. American College of Rheumatology position statement. Screening for hydroxychloroquine retinopathy. www.rheumatology.org/Portals/0/Files/Screening-for-Hydroxychloroquine-Retinopathy-Position-Statement.pdf. Accessed April 2, 2018.
- Marmor MF, Kellner U, Lai TY, Melles RB, Mieler WF; American Academy of Ophthalmology. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology 2016; 123(6):1386–1394. doi:10.1016/j.ophtha.2016.01.058
- Mackenzie AH. Antimalarial drugs for rheumatoid arthritis. Am J Med 1983; 75(6A):48–58. pmid:6362406
- Mackenzie AH. Dose refinements in long-term therapy of rheumatoid arthritis with antimalarials. Am J Med 1983; 75(1A):40–45. pmid:6869410
- Melles RB, Marmor MF. Pericentral retinopathy and racial differences in hydroxychloroquine toxicity. Ophthalmology 2015; 122(1):110–116. doi:10.1016/j.ophtha.2014.07.018
- Uslu H, Gurler B, Yildirim A, et al. Effect of hydroxychloroquine on the retinal layers: a quantitative evaluation with spectral-domain optical coherence tomography. J Ophthalmol 2016; 2016:8643174. doi:10.1155/2016/8643174
- Au A, Parikh V, Modi YS, Ehlers JP, Schachat AP, Singh RP. Hydroxychloroquine screening practice patterns within a large multispecialty ophthalmic practice. Am J Ophthalmol 2015; 160(3):561–568.e2. doi:10.1016/j.ajo.2015.06.009
- Jallouli M, Frances C, Plette JC, et al; Plaquenil Lupus Systemic Study Group. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus. JAMA Dermatol 2013; 149(8):935–940. doi:10.1001/jamadermatol.2013.709
- Avina-Zubieta JA, Johnson ES, Suarez-Almazor ME, Russell AS. Incidence of myopathy in patients treated with antimalarials: a report of three cases and review of the literature. Br J Rheumatol 1995; 34(2):166–170. pmid:7704464
- Yogasundaram H, Putko BN, Tien J, et al. Hydroxychloroquine-induced cardiomyopathy: case report, pathophysiology, diagnosis, and treatment. Can J Cardiol 2014; 30:1706–1715. doi:10.1016/j.cjca.2014.08.016
- Pearson KC, Morrell DS, Runge SR, Jolly P. Prolonged pustular eruption from hydroxychloroquine: an unusual case of acute generalized exanthematous pustulosis. Cutis 2016; 97(3):212–216. pmid:27023083
- Youngster I, Arcavi L, Schechmaster R, et al. Medications and glucose-6-phosphate dehydrogenase deficiency: an evidence-based review. Drug Saf 2010; 33(9):713–726. doi:10.2165/11536520-000000000-00000
- Ostensen M, Khamashta M, Lockshin M, et al. Anti-inflammatory and immunosuppressive drugs and reproduction. Arthritis Res Ther 2006; 8(3):209. doi:10.1186/ar1957
- Leroux M, Desveaux C, Parcevaux M, et al. Impact of hydroxychloroquine on preterm delivery and intrauterine growth restriction in pregnant women with systemic lupus erythematosus: a descriptive cohort study. Lupus 2015; 24(13):1384–1391. doi:10.1177/0961203315591027
- Clowse MEB, Magder L, Witter F, Petri M. Hydroxychloroquine in lupus pregnancy. Arthritis Rheum 2006; 54(11):3640–3647. doi:10.1002/art.22159
- Levy RA, Vilela VS, Cataldo MJ, et al. Hydroxychloroquine in lupus pregnancy: double-blind and placebo-controlled study. Lupus 2001; 10(6):401–404. doi:10.1191/096120301678646137
- James JA, Kim-Howard XR, Bruner BF, et al. Hydroxychloroquine sulfate treatment is associated with later onset of systemic lupus erythematosus. Lupus 2007; 16(6):401–409. doi:10.1177/0961203307078579
- Akpovwa H. Chloroquine could be used for the treatment of filoviral infections and other viral infections that emerge or emerged from viruses requiring an acidic pH for infectivity. Cell Biochem Funct 2016; 34(4):191–196. doi:10.1002/cbf.3182
- Pascolo S. Time to use a dose of chloroquine as an adjuvant to anti-cancer chemotherapies. Eur J Pharmacol 2016; 771:139–144. doi:10.1016/j.ejphar.2015.12.017
- Furlong HC, Wessels JM, Guerra MT, Stämpfli MR, Foster WG. Hydroxychloroquine attenuates cigarette smoke induced autophagic signaling in the mouse ovary. Reprod Toxicol 2016; 61:105–113. doi:10.1016/j.reprotox.2016.03.044
- Cairoli E, Danese N, Teliz M, et al. Cumulative dose of hydroxychloroquine is associated with a decrease of resting heart rate in patients with systemic lupus erythematosus: a pilot study. Lupus 2015; 24(11):1204–1209. doi:10.1177/0961203315580870
KEY POINTS
- Hydroxychloroquine acts by suppressing Toll-like receptors to trigger important immunomodulatory effects.
- Hydroxychloroquine is a well-established and effective therapy for systemic and cutaneous lupus and other autoimmune diseases.
- Patients with systemic lupus erythematosus treated with hydroxychloroquine have lower mortality rates and a lower risk of lupus nephritis.
- Retinal toxicity is the most serious potential complication of hydroxychloroquine therapy. Adherence to current ophthalmologic screening recommendations and proper dosing protocols lowers this risk.

Pharmacotherapy for obesity: What you need to know
Weight-loss drugs are not magic pills, but they can help patients lose about 10 to 25 more pounds than they otherwise could, when used in a program that includes diet, exercise, and other lifestyle changes.

HALF OF ADULTS MAY BE OBESE BY 2030
Obesity is a major public health challenge in the United States, with nearly 37% of adults classified as obese.1 The prevalence has increased more than 75% since 1980,2 and it is estimated that 51% of US adults will be obese by 2030.3 Obesity is the second-leading cause of preventable deaths, after smoking.4
Obesity increases the risk of many chronic medical conditions, including type 2 diabetes mellitus, heart disease, hypertension, stroke, nonalcoholic fatty liver disease, osteoarthritis, and cancers of the breast, colon, endometrium, and kidney.5
WHEN IS DRUG THERAPY INDICATED?
Guidelines from the major obesity societies recommend that all weight-loss programs have a lifestyle component that includes a low-calorie diet, increased physical activity, and behavioral therapy, to which pharmacotherapy may be added as an adjunct.6–8
Weight-loss medications are indicated for patients who have a body mass index (BMI) of at least 30 kg/m2 or who have obesity-associated comorbidities and a BMI of at least 27 kg/m2. However, the best results are achieved when pharmacotherapy is combined with lifestyle modification.9

HISTORY OF WEIGHT-LOSS DRUGS: NOT A PRETTY PICTURE
The earliest drugs to induce weight loss, which worked mainly by increasing metabolism, included thyroid hormone, amphetamines (which also suppress appetite), and dinitrophenol (a pesticide). Adverse reactions limited their usefulness: cardiovascular effects with thyroid hormones, abuse potential with amphetamines, and neuropathy and cataracts with dinitrophenol.
Researchers then looked to drugs that could suppress appetite like amphetamines do, but without the potential for abuse. Medications that increased levels of norepinephrine and serotonin, both by increasing release and decreasing reuptake of these neuromodulators, had some success. But again, serious adverse effects occurred, and several drugs had to be withdrawn from the market.
The most publicized of these withdrawals was for the combination fenfluramine and phentermine (“fen-phen”) and its cousin dexfenfluramine (Redux). Up to 30% of patients taking fenfluramine-phentermine developed echocardiographic evidence of valvular heart disease.11 Fenfluramine also increased the risk of pulmonary hypertension. These findings led to the 1997 withdrawal of these drugs from the US market.
Sibutramine (Meridia), a norepinephrine and serotonin reuptake inhibitor, was approved for weight loss in 1997. Increases in blood pressure and heart rate were noted in the initial trial,12 and then a postmarketing study found increased rates of nonfatal myocardial infarction and stroke in patients with preexisting cardiovascular disease or diabetes mellitus.13 Based on these results, sibutramine was withdrawn from both US and European markets.
Rimonabant (Acomplia, Zimulti), a cannabinoid-receptor inhibitor, was approved in Europe in 2006, but its approval was withdrawn just 2 years later because of increased suicidality in a postmarketing study.14 It was never approved for use in the United States.
NORADRENERGIC SYMPATHOMIMETICS: FOR SHORT-TERM USE
Several noradrenergic sympathomimetic drugs are FDA-approved for short-term weight loss, but phentermine is by far the most commonly prescribed drug in this class. In fact, it is the most commonly prescribed drug for obesity in the United States.15
Phentermine
Phentermine is an atypical amphetamine analogue that suppresses appetite by norepinephrine agonism in the central nervous system. The FDA approved it for short-term weight management in 1959, and its use became widespread in the 1960s, followed by decades of popularity.
Dosage. Phentermine is prescribed at an oral dose of 15, 30, or 37.5 mg daily, either before breakfast or 1 to 2 hours after. It is a schedule IV controlled substance, based on its similarity to amphetamine. (The 5 US controlled substance schedules range from schedule I, which includes heroin, amphetamine, and cannabis, to schedule V, which includes cough syrups containing no more than 200 mg of codeine per 100 mL.) However, concerns about addiction and dependence with phentermine are largely unfounded, and abrupt cessation of the drug has not been shown to cause amphetamine-like withdrawal.16
Adverse effects. Common adverse reactions include nervousness, insomnia, and dry mouth, but these effects tend to wane with continued use.
Contraindications. Cardiovascular disease is a contraindication to phentermine because of concerns about increased blood pressure and pulse rate, although these concerns seem to be more theoretic than observed.16 Other contraindications include hyperthyroidism, glaucoma, agitation, a history of drug abuse, pregnancy, breastfeeding, and current or recent use of a monoamine oxidase inhibitor. No serious adverse events have been reported in trials of phentermine.
Efficacy. In a pooled analysis of 6 trials lasting 2 to 24 weeks completed between 1975 and 1999, phentermine-treated patients lost an average of 3.6 kg more weight than placebo recipients.17 More than 80% of study participants were women.
In a 36-week study in 108 women,18 participants lost a mean of 12.2 kg with continuous phentermine use, 13.0 kg with intermittent use (4 weeks on, 4 weeks off; the difference was not significant), and 4.8 kg with placebo.
Minimal data exist on long-term efficacy of phentermine monotherapy.
DRUGS FOR LONG-TERM THERAPY
Orlistat
Orlistat was approved as a prescription drug (Xenical, 120 mg) in 1999 and as an over-the-counter medication (Alli, 60 mg) in 2007.
Orlistat works by inhibiting pancreatic and gastric lipase, causing incomplete hydrolysis of ingested fat, thereby increasing fecal fat excretion in a dose-dependent manner. It is a good choice for weight-loss drug therapy because of its safe cardiovascular risk profile and beneficial effects on lipid levels. However, its long-term effect on weight is only modest.19,20
Dosage. The dosage for prescription orlistat is 120 mg 3 times per day, in addition to a low-fat diet (< 30% of daily calories from fat). To prevent potential deficiencies of fat-soluble vitamins, a daily multivitamin supplement is recommended, but it should not be taken with meals.
Efficacy. In a 2014 systematic review, 35% to 73% of patients treated with orlistat 120 mg had lost at least 5% of their body weight at 1 year, and 14% to 41% had lost at least 10%.21 At the end of the second year, orlistat-treated patients had lost about 3.3 kg more than placebo recipients.
In a randomized trial,22 4 years of treatment with orlistat vs placebo led to a significant (37.3%) risk reduction in the incidence of type 2 diabetes mellitus in obese participants, as well as significant improvements in cardiovascular risk factors. Mean weight loss at 1 year was significantly greater with orlistat than with placebo (10.6 vs 6.2 kg), and it remained greater at 4 years (5.8 vs 3.0 kg; P < .001).
Adverse effects. Long-term orlistat use is hampered by adverse reactions. A population-based, retrospective cohort analysis showed that fewer than 10% of patients were still using it at 1 year, and only 2% were using it at 2 years, although reasons for discontinuation were not reported.23
Adverse reactions are predominantly gastrointestinal, attributed to the high content of undigested fat in stools. Patients who do not limit their dietary fat intake are affected the most. Other reported adverse reactions include hepatotoxicity and oxalate-induced nephropathy.
Orlistat has been reported to interfere with some drugs, particularly those that are lipophilic. Drugs that should be closely monitored with orlistat are warfarin, amiodarone, cyclosporine, certain antiepileptic drugs, and levothyroxine.
Phentermine-topiramate
The combination of phentermine and topiramate was approved by the FDA in 2012 and is available under the brand name Qsymia.
Topiramate had been approved for treating seizure disorder in 1996 and as migraine prophylaxis in 2004. It is not approved as monotherapy for obesity; however, patients taking it for seizures or for psychiatric disorders (eg, binge eating, borderline personality disorder) have reported weight loss during treatment.
How topiramate promotes weight loss is not known. Proposed mechanisms include taste inhibition by carbonic anhydrase, influences on gamma-aminobutyric acid transmission causing appetite suppression, sensitization of insulin activity, and adiponectin secretion in the peripheral tissues.24,25
Phentermine-topiramate therapy has an advantage over monotherapy because lower doses of each medication can be used to achieve the same benefit, thus avoiding dose-related adverse reactions.
Dosage. Phentermine-topiramate is available in capsules containing 3.75/23, 7.5/46, 11.25/69, and 15/92 mg. The recommended starting dosage is 3.75/23 mg/day for 14 days, increasing to 7.5/46 mg/day. If patients do not lose at least 3% of their body weight after 12 weeks, the dose can be increased to 11.25/69 mg daily for 14 days, followed by 15/92 mg daily.26 Phentermine-topiramate is a schedule IV controlled substance with a low potential for abuse and dependence.
Efficacy. Approval of phentermine-topiramate for treating obesity was primarily based on 3 clinical trials.27–29 In 1 of these trials,28 at 1 year, patients had lost 9.9 kg with the medium dose and 12.9 kg with the high dose.
Adverse effects. Phentermine-topiramate was well tolerated in the trials. The most commonly reported adverse reactions were dry mouth, dizziness, constipation, insomnia, dysgeusia, paresthesia, and increased resting heart rate.28,29 Acute myopia and angle-closure glaucoma also have been reported with topiramate.30 Topiramate monotherapy has been associated with dose-dependent neuropsychiatric adverse effects, including memory symptoms and depression. However, across all 3 trials of phentermine-topiramate therapy, symptoms of depression improved over time, and no significant increase in suicide risk was identified.27–29
Recommended monitoring for patients on phentermine-topiramate includes a blood chemistry panel, resting heart rate, blood pressure, and depression screening.
Because topiramate has teratogenic potential (craniofacial abnormalities), it is labeled as pregnancy category X (contraindicated). A negative pregnancy test is needed before women of childbearing age take the drug and monthly thereafter. Women should be counseled to use effective birth control. A home pregnancy test is an alternative to laboratory testing, but this option should be left to the prescribing clinician’s judgment and be based on reliability of the test and patient compliance.
Lorcaserin
Lorcaserin (Belviq) was approved by the FDA in 2012 for chronic weight management. It suppresses appetite by activating the serotonin 2C receptor in the brain. Because it is selective for the 2C receptor, it does not appear to have the same detrimental effects on heart valves as occurred with less-selective serotonergic agents such as fenfluramine and dexfenfluramine.31
Dosage. The recommended dosage for lorcaserin is 10 mg twice daily. Lorcaserin is a schedule IV controlled substance because of studies that showed increases in positive subjective measures such as euphoria in patients taking the drug. The incidence of euphoria was similar to that seen with zolpidem.32
Efficacy. Lorcaserin was approved on the basis of 2 trials in nondiabetic obese and overweight adults who did not have diabetes but who had a weight-related condition,33,34 and in a third trial in obese and overweight adults with type 2 diabetes mellitus who were taking oral hypoglycemic agents.35 In these trials, lorcaserin use resulted in a modest 4.7- to 5.8-kg weight loss compared with 1.6 to 2.2 kg in the placebo group.33–35 There was a high dropout rate in all 3 of these studies (33% to 45% of participants).
A pilot study that added phentermine to lorcaserin yielded double the weight loss from lorcaserin alone.36 This drug combination warrants further investigation.
Contraindications. Lorcaserin should not be given to patients who have severe renal insufficiency (creatinine clearance < 30 mL/min) or severe hepatic impairment, or who are pregnant.
Adverse effects. Common adverse reactions include dry mouth, dizziness, somnolence, headache, and gastrointestinal disturbances (nausea, constipation, or diarrhea).37
Patients with type 2 diabetes mellitus should be monitored for hypoglycemia.
Lorcaserin should be used with extreme caution in patients taking other serotonergic agents because of the risk of the serotonin syndrome.
A theoretic potential for increased risk of breast cancer also exists with lorcaserin. When rats were given supraphysiologic doses of lorcaserin (more than 50 times higher than recommended in humans), fibroadenomas and adenocarcinomas occurred at higher rates.38 Breast cancer data were not reported in the 3 randomized trials discussed above.33–35
Naltrexone-bupropion
The combination of naltrexone and bupropion was approved by the FDA in 2014 under the brand name Contrave. Both drugs are approved for monotherapy in conditions other than obesity.
Naltrexone is a mu opioid receptor antagonist approved to treat alcohol and opioid dependency. Bupropion is a dopamine-norepinephrine reuptake inhibitor approved to treat depression and to help with smoking cessation. Combining the drugs produces weight loss and metabolic benefits through effects on 2 areas of the brain that regulate food intake: the hypothalamus (appetite) and the mesolimbic dopamine circuit (reward system).
Dosage. Naltrexone-bupropion comes as an extended-release tablet of 8/90 mg. The maintenance dose of 2 tablets twice daily is reached at week 4 through a specific dose-titration regimen (Table 1). The dose should be adjusted if patients have renal or hepatic impairment or if they are also taking a CYP2B6 inhibitor.
Efficacy. FDA approval was based on the results of 4 clinical trials.39–42 Using a modified intention-to-treat analysis, Yanovski and Yanovski43 calculated that at 1 year, placebo-subtracted mean weight loss was 4.6% (4.9 kg), and mean total weight loss was 6.8% (7.3 kg) across the studies. Attrition rates, however, were high, ranging from 42% to 50%.
Cardiometabolic effects in 2 of the trials40,41 included decreased waist circumference, triglyceride levels, and C-reactive protein levels, and increased high-density lipoprotein levels at the initial dose. At the maintenance dose, additional lowering of fasting plasma insulin and glucose levels occurred along with lower levels of the homeostatic model assessment of insulin resistance. In the COR-Diabetes Study Group trial, patients with type 2 diabetes mellitus had decreased hemoglobin A1c levels without an increase in hypoglycemia and an increased likelihood of reaching the target hemoglobin A1c level below 7%.39
Contraindications. Naltrexone-bupropion is contraindicated for patients who have uncontrolled hypertension, seizure disorder, eating disorder, or end-stage renal failure; who are pregnant; or who have been treated with a monoamine oxidase inhibitor within 14 days. It should not be used with other bupropion-containing products or in patients who have taken opioids chronically or have acute opiate withdrawal.
Because of its bupropion component, this product carries an FDA black-box warning about possible suicidal thoughts and behaviors and neuropsychiatric reactions.
Adverse effects. The adverse reactions most commonly associated with naltrexone-bupropion were nausea (32.5%), constipation (19.2%), headache (17.6%), vomiting (10.7%), dizziness (9.9%), insomnia (9.2%), dry mouth (8.1%), and diarrhea (7.1%).44
Liraglutide
Liraglutide, previously FDA-approved to treat type 2 diabetes mellitus under the brand name Victoza, received approval in 2014 in a higher-dose formulation (Saxenda) to treat obesity.
Liraglutide is a glucagon-like peptide-1 receptor agonist that stimulates glucose-dependent insulin release from the pancreatic islet cells, slows gastric emptying, regulates postprandial glucagon, and reduces food intake.
Dosage. Liraglutide is given as a once-daily injection in the abdomen, thigh, or arm. The initial dosage is 0.6 mg daily for the first week and can be titrated up by 0.6 mg weekly to a target dose of 3 mg daily. If a patient does not lose 4% of baseline body weight after 16 weeks on the target dose, the drug should be discontinued because it is unlikely to lead to clinically significant weight loss.
Efficacy. Liraglutide for weight management (3 mg once daily) was evaluated in a large (N = 3,731), randomized, double-blind, placebo-controlled international trial.45 Participants did not have diabetes mellitus, but 60% had prediabetes. Liraglutide or placebo was given for 56 weeks, along with lifestyle counseling. At the end of the study, the liraglutide group had lost a mean of 8.4 kg vs 2.8 kg in the placebo group. Additionally, 63% of the liraglutide group lost at least 5% of body weight vs 27% in the placebo group, and 33% lost at least 10% of body weight vs 10% in the placebo group.
A 2-year extension found systolic blood pressure decreased with no change in pulse, and the prevalence of prediabetes and metabolic syndrome decreased by 52% and 59%, respectively.46 At 2 years, mean scores for physical function, self-esteem, and work had improved more in the liraglutide group than the placebo group.47
Adverse effects. The most common adverse reactions with liraglutide were nausea, vomiting, diarrhea, constipation, hypoglycemia, and loss of appetite. In most cases, nausea and vomiting were tolerable, transient, and associated with greater weight loss but not with decreased quality-of-life scores. Serious adverse reactions included pancreatitis, gallbladder disease, renal impairment, and suicidal thoughts.
CHOOSING A DRUG
For obese patients, when lifestyle modifications do not result in the desired weight loss, pharmacotherapy is an option. Practitioners have several FDA-approved options for weight management. Because of evidence that these drugs can postpone the onset of other complications and improve metabolic and cardiovascular parameters, they should be considered.
In phase 3 trials, these drugs caused modest weight loss of 5% to 10% of body weight. More weight was lost with the combination of phentermine-topiramate than with the other drugs.
In a 2016 meta-analysis, these drugs were associated with at least 5% weight reduction compared with placebo.48 Phentermine-topiramate and liraglutide were most likely to produce at least a 5% weight loss, while liraglutide and naltrexone-bupropion were most likely to be discontinued because of adverse events. Combination drugs may have the advantages of synergistic effects on weight loss and fewer adverse reactions because lower doses of the individual drug components are used.
Response to therapy with most of these drugs should be evaluated at 12 weeks on the maintenance dose. If less than 5% weight loss has been achieved, the medication should be discontinued.
Adverse-effect profiles, drug interactions, abuse, misuse, and overdose potential should be considered when prescribing these drugs. Weight-loss drugs are contraindicated in pregnancy because they offer no potential benefit to a pregnant woman and may harm the fetus.
The development of new drugs and better drug combinations is expected to provide more effective therapeutic strategies, which are essential for combating the obesity epidemic.
- Ogden CL, Carroll MD, Fryar CD, Flegal KM. Prevalence of obesity among adults and youth: United States, 2011-2014. NCHS Data Brief 2015; 219:1–8.
- Yanovski SZ, Yanovski JA. Obesity. N Engl J Med 2002; 346:591–602.
- Finkelstein EA, Khavjou OA, Thompson H, et al. Obesity and severe obesity forecasts through 2030. Am J Prev Med 2012; 42:563–570.
- Hill JO, Wyatt H. Outpatient management of obesity: a primary care perspective. Obes Res 2002; 10(suppl 2):124S–130S.
- US Department of Health and Human Services. National Institute of Diabetes and Digestive and Kidney Diseases. Overweight and obesity statistics. www.niddk.nih.gov/health-information/health-statistics/Pages/overweight-obesity-statistics.aspx#overweight. Accessed October 10, 2017.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society. J Am Coll Cardiol 2014; 63:2985–3023.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society. Circulation 2014; 129(suppl 2):S102–S138.
- American Association of Clinical Endocrinologists. AACE/ACE algorithm for the medical care of patients with obesity. www.aace.com/files/guidelines/ObesityAlgorithm.pdf. Accessed July 25, 2017.
- Wadden TA, Berkowitz RI, Womble LG, et al. Randomized trial of lifestyle modification and pharmacotherapy for obesity. N Engl J Med 2005; 353:2111–2120.
- Mechanick JI, Youdim A, Jones DB, et al. Clinical practice guidelines for the perioperative nutritional, metabolic, and nonsurgical support of the bariatric surgery patient, 2013 update: cosponsored by American Association of Clinical Endocrinologists, the Obesity Society, and American Society for Metabolic and Bariatric Surgery. Surg Obes Relat Dis 2013; 9:159–191.
- Connolly HM, Crary JL, McGoon MD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med 1997; 337:581–588.
- Kim SH, Lee YM, Jee SH, et al. Effect of sibutramine on weight loss and blood pressure: a meta-analysis of controlled trials. Obes Res 2003; 11:1116–1123.
- James WP, Caterson ID, Coutinho W, et al; SCOUT Investigators. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N Engl J Med 2010; 363:905–917.
- Nissen SE, Nicholls SJ, Wolski K, et al; STRADIVARIUS Investigators. Effect of rimonabant on progression of atherosclerosis in patients with abdominal obesity and coronary artery disease: the STRADIVARIUS randomized controlled trial. JAMA 2008; 299:1547–1560.
- Ryan DH, Bray GA. Pharmacologic treatment options for obesity: what is old is new again. Curr Hypertens Rep 2013; 15:182–189.
- Hendricks EJ, Greenway FL, Westman EC, Gupta AK. Blood pressure and heart rate effects, weight loss and maintenance during long-term phentermine pharmacotherapy for obesity. Obesity (Silver Spring) 2011; 19:2351–2360.
- Li Z, Maglione M, Tu W, et al. Meta-analysis: pharmacologic treatment of obesity. Ann Intern Med 2005; 142:532–546.
- Munro JF, MacCuish AC, Wilson EM, Duncan LJ. Comparison of continuous and intermittent anorectic therapy in obesity. Br Med J 1968; 1:352–354.
- Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med 2000; 9:160–167.
- Rossner S, Sjostrom L, Noack R, Meinders AE, Noseda G. Weight loss, weight maintenance, and improved cardiovascular risk factors after 2 years treatment with orlistat for obesity. European Orlistat Obesity Study Group. Obes Res 2000; 8:49–61.
- Yanovski SZ, Yanovski JA. Long-term drug treatment for obesity: a systematic and clinical review. JAMA 2014; 311:74–86.
- Torgerson JS, Hauptman J, Boldrin MN, Sjostrom L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2004; 27:155–161.
- Padwal R, Kezouh A, Levine M, Etminan M. Long-term persistence with orlistat and sibutramine in a population-based cohort. Int J Obes (Lond) 2007; 31:1567–1570.
- Xiong GL, Gadde KM. Combination phentermine-topiramate for obesity treatment in primary care: a review. Postgrad Med 2014; 126:110–116.
- Pucci A, Finer N. New medications for treatment of obesity: metabolic and cardiovascular effects. Can J Cardiol 2015; 31:142–152.
- Smith SM, Meyer M, Trinkley KE. Phentermine-topiramate for the treatment of obesity. Ann Pharmacother 2013; 47:340–349.
- Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine-topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring) 2012; 20:330–342.
- Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet 2011; 377:1341–1352.
- Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine-topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr 2012; 95:297–308.
- Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs 2010; 24:501–526.
- Weissman NJ, Sanchez M, Koch GG, Smith SR, Shanahan WR, Anderson CM. Echocardiographic assessment of cardiac valvular regurgitation with lorcaserin from analysis of 3 phase 3 clinical trials. Circ Cardiovasc Imaging 2013; 6:560–567.
- US Department of Justice Drug Enforcement Administration. Schedules of controlled substances: placement of lorcaserin into Schedule IV. Federal Register 2013; 78:26701–26705.
- Smith SR, Weissman NJ, Anderson CM, et al; Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med 2010; 363:245–256.
- Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab 2011; 96:3067–3077.
- O’Neil PM, Smith SR, Weissman NJ, et al. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM study. Obesity (Silver Spring) 2012; 20:1426–1436.
- Kumar RB, Aronne LJ. Efficacy comparison of medications approved for chronic weight management. Obesity (Silver Spring) 2015; 23(suppl 1):S4–S7.
- Chan EW, He Y, Chui CS, Wong AY, Lau WC, Wong IC. Efficacy and safety of lorcaserin in obese adults: a meta-analysis of 1-year randomized controlled trials (RCTs) and narrative review on short-term RCTs. Obes Rev 2013; 14:383–392.
- Miller LE. Lorcaserin for weight loss: insights into US Food and Drug Administration approval. J Acad Nutr Diet 2013; 113:25–30.
- Hollander P, Gupta AK, Plodkowski R, et al; COR-Diabetes Study Group. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care 2013; 36:4022–4029.
- Apovian CM, Aronne L, Rubino D, et al; COR-II Study Group. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Spring) 2013; 21:935–943.
- Greenway FL, Fujioka K, Plodkowski RA, et al; COR-I Study Group. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2010; 376:595–605.
- Wadden TA, Foreyt JP, Foster GD, et al. Weight loss with naltrexone SR/bupropion SR combination therapy as an adjunct to behavior modification: the COR-BMOD trial. Obesity (Silver Spring) 2011; 19:110–120.
- Yanovski SZ, Yanovski JA. Naltrexone extended-release plus bupropion extended-release for treatment of obesity. JAMA 2015; 313:1213–1214.
- Contrave (naltrexone HC1 and bupropion HC1) extended release tablets [package insert]. Orexigen Therapeutics, 2017. https://contrave.com/wp-content/uploads/2017/05/Contrave_PI.pdf. Accessed November 7, 2017.
- Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med 2015; 373:11–22.
- Astrup A, Carraro R, Finer N, et al; NN8022-1807 Investigators. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes (Lond) 2012; 36:843–854.
- Lean ME, Carraro R, Finer N, et al; NN8022-1807 Investigators. Tolerability of nausea and vomiting and associations with weight loss in a randomized trial of liraglutide in obese, non-diabetic adults. Int J Obes (Lond) 2014; 38:689–697.
- Khera R, Murad MH, Chandar AK, et al. Association of pharmacological treatments for obesity with weight loss and adverse events: a systematic review and meta-analysis. JAMA 2016; 315:2424–2434.
Weight-loss drugs are not magic pills, but they can help patients lose about 10 to 25 more pounds than they otherwise could, when used in a program that includes diet, exercise, and other lifestyle changes.
HALF OF ADULTS MAY BE OBESE BY 2030
Obesity is a major public health challenge in the United States, with nearly 37% of adults classified as obese.1 The prevalence has increased more than 75% since 1980,2 and it is estimated that 51% of US adults will be obese by 2030.3 Obesity is the second-leading cause of preventable deaths, after smoking.4
Obesity increases the risk of many chronic medical conditions, including type 2 diabetes mellitus, heart disease, hypertension, stroke, nonalcoholic fatty liver disease, osteoarthritis, and cancers of the breast, colon, endometrium, and kidney.5
WHEN IS DRUG THERAPY INDICATED?
Guidelines from the major obesity societies recommend that all weight-loss programs have a lifestyle component that includes a low-calorie diet, increased physical activity, and behavioral therapy, to which pharmacotherapy may be added as an adjunct.6–8
Weight-loss medications are indicated for patients who have a body mass index (BMI) of at least 30 kg/m2 or who have obesity-associated comorbidities and a BMI of at least 27 kg/m2. However, the best results are achieved when pharmacotherapy is combined with lifestyle modification.9
HISTORY OF WEIGHT-LOSS DRUGS: NOT A PRETTY PICTURE
The earliest drugs to induce weight loss, which worked mainly by increasing metabolism, included thyroid hormone, amphetamines (which also suppress appetite), and dinitrophenol (a pesticide). Adverse reactions limited their usefulness: cardiovascular effects with thyroid hormones, abuse potential with amphetamines, and neuropathy and cataracts with dinitrophenol.
Researchers then looked to drugs that could suppress appetite like amphetamines do, but without the potential for abuse. Medications that increased levels of norepinephrine and serotonin, both by increasing release and decreasing reuptake of these neuromodulators, had some success. But again, serious adverse effects occurred, and several drugs had to be withdrawn from the market.
The most publicized of these withdrawals was for the combination fenfluramine and phentermine (“fen-phen”) and its cousin dexfenfluramine (Redux). Up to 30% of patients taking fenfluramine-phentermine developed echocardiographic evidence of valvular heart disease.11 Fenfluramine also increased the risk of pulmonary hypertension. These findings led to the 1997 withdrawal of these drugs from the US market.
Sibutramine (Meridia), a norepinephrine and serotonin reuptake inhibitor, was approved for weight loss in 1997. Increases in blood pressure and heart rate were noted in the initial trial,12 and then a postmarketing study found increased rates of nonfatal myocardial infarction and stroke in patients with preexisting cardiovascular disease or diabetes mellitus.13 Based on these results, sibutramine was withdrawn from both US and European markets.
Rimonabant (Acomplia, Zimulti), a cannabinoid-receptor inhibitor, was approved in Europe in 2006, but its approval was withdrawn just 2 years later because of increased suicidality in a postmarketing study.14 It was never approved for use in the United States.
NORADRENERGIC SYMPATHOMIMETICS: FOR SHORT-TERM USE
Several noradrenergic sympathomimetic drugs are FDA-approved for short-term weight loss, but phentermine is by far the most commonly prescribed drug in this class. In fact, it is the most commonly prescribed drug for obesity in the United States.15
Phentermine
Phentermine is an atypical amphetamine analogue that suppresses appetite by norepinephrine agonism in the central nervous system. The FDA approved it for short-term weight management in 1959, and its use became widespread in the 1960s, followed by decades of popularity.
Dosage. Phentermine is prescribed at an oral dose of 15, 30, or 37.5 mg daily, either before breakfast or 1 to 2 hours after. It is a schedule IV controlled substance, based on its similarity to amphetamine. (The 5 US controlled substance schedules range from schedule I, which includes heroin, amphetamine, and cannabis, to schedule V, which includes cough syrups containing no more than 200 mg of codeine per 100 mL.) However, concerns about addiction and dependence with phentermine are largely unfounded, and abrupt cessation of the drug has not been shown to cause amphetamine-like withdrawal.16
Adverse effects. Common adverse reactions include nervousness, insomnia, and dry mouth, but these effects tend to wane with continued use.
Contraindications. Cardiovascular disease is a contraindication to phentermine because of concerns about increased blood pressure and pulse rate, although these concerns seem to be more theoretic than observed.16 Other contraindications include hyperthyroidism, glaucoma, agitation, a history of drug abuse, pregnancy, breastfeeding, and current or recent use of a monoamine oxidase inhibitor. No serious adverse events have been reported in trials of phentermine.
Efficacy. In a pooled analysis of 6 trials lasting 2 to 24 weeks completed between 1975 and 1999, phentermine-treated patients lost an average of 3.6 kg more weight than placebo recipients.17 More than 80% of study participants were women.
In a 36-week study in 108 women,18 participants lost a mean of 12.2 kg with continuous phentermine use, 13.0 kg with intermittent use (4 weeks on, 4 weeks off; the difference was not significant), and 4.8 kg with placebo.
Minimal data exist on long-term efficacy of phentermine monotherapy.
DRUGS FOR LONG-TERM THERAPY
Orlistat
Orlistat was approved as a prescription drug (Xenical, 120 mg) in 1999 and as an over-the-counter medication (Alli, 60 mg) in 2007.
Orlistat works by inhibiting pancreatic and gastric lipase, causing incomplete hydrolysis of ingested fat, thereby increasing fecal fat excretion in a dose-dependent manner. It is a good choice for weight-loss drug therapy because of its safe cardiovascular risk profile and beneficial effects on lipid levels. However, its long-term effect on weight is only modest.19,20
Dosage. The dosage for prescription orlistat is 120 mg 3 times per day, in addition to a low-fat diet (< 30% of daily calories from fat). To prevent potential deficiencies of fat-soluble vitamins, a daily multivitamin supplement is recommended, but it should not be taken with meals.
Efficacy. In a 2014 systematic review, 35% to 73% of patients treated with orlistat 120 mg had lost at least 5% of their body weight at 1 year, and 14% to 41% had lost at least 10%.21 At the end of the second year, orlistat-treated patients had lost about 3.3 kg more than placebo recipients.
In a randomized trial,22 4 years of treatment with orlistat vs placebo led to a significant (37.3%) risk reduction in the incidence of type 2 diabetes mellitus in obese participants, as well as significant improvements in cardiovascular risk factors. Mean weight loss at 1 year was significantly greater with orlistat than with placebo (10.6 vs 6.2 kg), and it remained greater at 4 years (5.8 vs 3.0 kg; P < .001).
Adverse effects. Long-term orlistat use is hampered by adverse reactions. A population-based, retrospective cohort analysis showed that fewer than 10% of patients were still using it at 1 year, and only 2% were using it at 2 years, although reasons for discontinuation were not reported.23
Adverse reactions are predominantly gastrointestinal, attributed to the high content of undigested fat in stools. Patients who do not limit their dietary fat intake are affected the most. Other reported adverse reactions include hepatotoxicity and oxalate-induced nephropathy.
Orlistat has been reported to interfere with some drugs, particularly those that are lipophilic. Drugs that should be closely monitored with orlistat are warfarin, amiodarone, cyclosporine, certain antiepileptic drugs, and levothyroxine.
Phentermine-topiramate
The combination of phentermine and topiramate was approved by the FDA in 2012 and is available under the brand name Qsymia.
Topiramate had been approved for treating seizure disorder in 1996 and as migraine prophylaxis in 2004. It is not approved as monotherapy for obesity; however, patients taking it for seizures or for psychiatric disorders (eg, binge eating, borderline personality disorder) have reported weight loss during treatment.
How topiramate promotes weight loss is not known. Proposed mechanisms include taste inhibition by carbonic anhydrase, influences on gamma-aminobutyric acid transmission causing appetite suppression, sensitization of insulin activity, and adiponectin secretion in the peripheral tissues.24,25
Phentermine-topiramate therapy has an advantage over monotherapy because lower doses of each medication can be used to achieve the same benefit, thus avoiding dose-related adverse reactions.
Dosage. Phentermine-topiramate is available in capsules containing 3.75/23, 7.5/46, 11.25/69, and 15/92 mg. The recommended starting dosage is 3.75/23 mg/day for 14 days, increasing to 7.5/46 mg/day. If patients do not lose at least 3% of their body weight after 12 weeks, the dose can be increased to 11.25/69 mg daily for 14 days, followed by 15/92 mg daily.26 Phentermine-topiramate is a schedule IV controlled substance with a low potential for abuse and dependence.
Efficacy. Approval of phentermine-topiramate for treating obesity was primarily based on 3 clinical trials.27–29 In 1 of these trials,28 at 1 year, patients had lost 9.9 kg with the medium dose and 12.9 kg with the high dose.
Adverse effects. Phentermine-topiramate was well tolerated in the trials. The most commonly reported adverse reactions were dry mouth, dizziness, constipation, insomnia, dysgeusia, paresthesia, and increased resting heart rate.28,29 Acute myopia and angle-closure glaucoma also have been reported with topiramate.30 Topiramate monotherapy has been associated with dose-dependent neuropsychiatric adverse effects, including memory symptoms and depression. However, across all 3 trials of phentermine-topiramate therapy, symptoms of depression improved over time, and no significant increase in suicide risk was identified.27–29
Recommended monitoring for patients on phentermine-topiramate includes a blood chemistry panel, resting heart rate, blood pressure, and depression screening.
Because topiramate has teratogenic potential (craniofacial abnormalities), it is labeled as pregnancy category X (contraindicated). A negative pregnancy test is needed before women of childbearing age take the drug and monthly thereafter. Women should be counseled to use effective birth control. A home pregnancy test is an alternative to laboratory testing, but this option should be left to the prescribing clinician’s judgment and be based on reliability of the test and patient compliance.
Lorcaserin
Lorcaserin (Belviq) was approved by the FDA in 2012 for chronic weight management. It suppresses appetite by activating the serotonin 2C receptor in the brain. Because it is selective for the 2C receptor, it does not appear to have the same detrimental effects on heart valves as occurred with less-selective serotonergic agents such as fenfluramine and dexfenfluramine.31
Dosage. The recommended dosage for lorcaserin is 10 mg twice daily. Lorcaserin is a schedule IV controlled substance because of studies that showed increases in positive subjective measures such as euphoria in patients taking the drug. The incidence of euphoria was similar to that seen with zolpidem.32
Efficacy. Lorcaserin was approved on the basis of 2 trials in nondiabetic obese and overweight adults who did not have diabetes but who had a weight-related condition,33,34 and in a third trial in obese and overweight adults with type 2 diabetes mellitus who were taking oral hypoglycemic agents.35 In these trials, lorcaserin use resulted in a modest 4.7- to 5.8-kg weight loss compared with 1.6 to 2.2 kg in the placebo group.33–35 There was a high dropout rate in all 3 of these studies (33% to 45% of participants).
A pilot study that added phentermine to lorcaserin yielded double the weight loss from lorcaserin alone.36 This drug combination warrants further investigation.
Contraindications. Lorcaserin should not be given to patients who have severe renal insufficiency (creatinine clearance < 30 mL/min) or severe hepatic impairment, or who are pregnant.
Adverse effects. Common adverse reactions include dry mouth, dizziness, somnolence, headache, and gastrointestinal disturbances (nausea, constipation, or diarrhea).37
Patients with type 2 diabetes mellitus should be monitored for hypoglycemia.
Lorcaserin should be used with extreme caution in patients taking other serotonergic agents because of the risk of the serotonin syndrome.
A theoretic potential for increased risk of breast cancer also exists with lorcaserin. When rats were given supraphysiologic doses of lorcaserin (more than 50 times higher than recommended in humans), fibroadenomas and adenocarcinomas occurred at higher rates.38 Breast cancer data were not reported in the 3 randomized trials discussed above.33–35
Naltrexone-bupropion
The combination of naltrexone and bupropion was approved by the FDA in 2014 under the brand name Contrave. Both drugs are approved for monotherapy in conditions other than obesity.
Naltrexone is a mu opioid receptor antagonist approved to treat alcohol and opioid dependency. Bupropion is a dopamine-norepinephrine reuptake inhibitor approved to treat depression and to help with smoking cessation. Combining the drugs produces weight loss and metabolic benefits through effects on 2 areas of the brain that regulate food intake: the hypothalamus (appetite) and the mesolimbic dopamine circuit (reward system).
Dosage. Naltrexone-bupropion comes as an extended-release tablet of 8/90 mg. The maintenance dose of 2 tablets twice daily is reached at week 4 through a specific dose-titration regimen (Table 1). The dose should be adjusted if patients have renal or hepatic impairment or if they are also taking a CYP2B6 inhibitor.
Efficacy. FDA approval was based on the results of 4 clinical trials.39–42 Using a modified intention-to-treat analysis, Yanovski and Yanovski43 calculated that at 1 year, placebo-subtracted mean weight loss was 4.6% (4.9 kg), and mean total weight loss was 6.8% (7.3 kg) across the studies. Attrition rates, however, were high, ranging from 42% to 50%.
Cardiometabolic effects in 2 of the trials40,41 included decreased waist circumference, triglyceride levels, and C-reactive protein levels, and increased high-density lipoprotein levels at the initial dose. At the maintenance dose, additional lowering of fasting plasma insulin and glucose levels occurred along with lower levels of the homeostatic model assessment of insulin resistance. In the COR-Diabetes Study Group trial, patients with type 2 diabetes mellitus had decreased hemoglobin A1c levels without an increase in hypoglycemia and an increased likelihood of reaching the target hemoglobin A1c level below 7%.39
Contraindications. Naltrexone-bupropion is contraindicated for patients who have uncontrolled hypertension, seizure disorder, eating disorder, or end-stage renal failure; who are pregnant; or who have been treated with a monoamine oxidase inhibitor within 14 days. It should not be used with other bupropion-containing products or in patients who have taken opioids chronically or have acute opiate withdrawal.
Because of its bupropion component, this product carries an FDA black-box warning about possible suicidal thoughts and behaviors and neuropsychiatric reactions.
Adverse effects. The adverse reactions most commonly associated with naltrexone-bupropion were nausea (32.5%), constipation (19.2%), headache (17.6%), vomiting (10.7%), dizziness (9.9%), insomnia (9.2%), dry mouth (8.1%), and diarrhea (7.1%).44
Liraglutide
Liraglutide, previously FDA-approved to treat type 2 diabetes mellitus under the brand name Victoza, received approval in 2014 in a higher-dose formulation (Saxenda) to treat obesity.
Liraglutide is a glucagon-like peptide-1 receptor agonist that stimulates glucose-dependent insulin release from the pancreatic islet cells, slows gastric emptying, regulates postprandial glucagon, and reduces food intake.
Dosage. Liraglutide is given as a once-daily injection in the abdomen, thigh, or arm. The initial dosage is 0.6 mg daily for the first week and can be titrated up by 0.6 mg weekly to a target dose of 3 mg daily. If a patient does not lose 4% of baseline body weight after 16 weeks on the target dose, the drug should be discontinued because it is unlikely to lead to clinically significant weight loss.
Efficacy. Liraglutide for weight management (3 mg once daily) was evaluated in a large (N = 3,731), randomized, double-blind, placebo-controlled international trial.45 Participants did not have diabetes mellitus, but 60% had prediabetes. Liraglutide or placebo was given for 56 weeks, along with lifestyle counseling. At the end of the study, the liraglutide group had lost a mean of 8.4 kg vs 2.8 kg in the placebo group. Additionally, 63% of the liraglutide group lost at least 5% of body weight vs 27% in the placebo group, and 33% lost at least 10% of body weight vs 10% in the placebo group.
A 2-year extension found systolic blood pressure decreased with no change in pulse, and the prevalence of prediabetes and metabolic syndrome decreased by 52% and 59%, respectively.46 At 2 years, mean scores for physical function, self-esteem, and work had improved more in the liraglutide group than the placebo group.47
Adverse effects. The most common adverse reactions with liraglutide were nausea, vomiting, diarrhea, constipation, hypoglycemia, and loss of appetite. In most cases, nausea and vomiting were tolerable, transient, and associated with greater weight loss but not with decreased quality-of-life scores. Serious adverse reactions included pancreatitis, gallbladder disease, renal impairment, and suicidal thoughts.
CHOOSING A DRUG
For obese patients, when lifestyle modifications do not result in the desired weight loss, pharmacotherapy is an option. Practitioners have several FDA-approved options for weight management. Because of evidence that these drugs can postpone the onset of other complications and improve metabolic and cardiovascular parameters, they should be considered.
In phase 3 trials, these drugs caused modest weight loss of 5% to 10% of body weight. More weight was lost with the combination of phentermine-topiramate than with the other drugs.
In a 2016 meta-analysis, these drugs were associated with at least 5% weight reduction compared with placebo.48 Phentermine-topiramate and liraglutide were most likely to produce at least a 5% weight loss, while liraglutide and naltrexone-bupropion were most likely to be discontinued because of adverse events. Combination drugs may have the advantages of synergistic effects on weight loss and fewer adverse reactions because lower doses of the individual drug components are used.
Response to therapy with most of these drugs should be evaluated at 12 weeks on the maintenance dose. If less than 5% weight loss has been achieved, the medication should be discontinued.
Adverse-effect profiles, drug interactions, abuse, misuse, and overdose potential should be considered when prescribing these drugs. Weight-loss drugs are contraindicated in pregnancy because they offer no potential benefit to a pregnant woman and may harm the fetus.
The development of new drugs and better drug combinations is expected to provide more effective therapeutic strategies, which are essential for combating the obesity epidemic.
Weight-loss drugs are not magic pills, but they can help patients lose about 10 to 25 more pounds than they otherwise could, when used in a program that includes diet, exercise, and other lifestyle changes.
HALF OF ADULTS MAY BE OBESE BY 2030
Obesity is a major public health challenge in the United States, with nearly 37% of adults classified as obese.1 The prevalence has increased more than 75% since 1980,2 and it is estimated that 51% of US adults will be obese by 2030.3 Obesity is the second-leading cause of preventable deaths, after smoking.4
Obesity increases the risk of many chronic medical conditions, including type 2 diabetes mellitus, heart disease, hypertension, stroke, nonalcoholic fatty liver disease, osteoarthritis, and cancers of the breast, colon, endometrium, and kidney.5
WHEN IS DRUG THERAPY INDICATED?
Guidelines from the major obesity societies recommend that all weight-loss programs have a lifestyle component that includes a low-calorie diet, increased physical activity, and behavioral therapy, to which pharmacotherapy may be added as an adjunct.6–8
Weight-loss medications are indicated for patients who have a body mass index (BMI) of at least 30 kg/m2 or who have obesity-associated comorbidities and a BMI of at least 27 kg/m2. However, the best results are achieved when pharmacotherapy is combined with lifestyle modification.9
HISTORY OF WEIGHT-LOSS DRUGS: NOT A PRETTY PICTURE
The earliest drugs to induce weight loss, which worked mainly by increasing metabolism, included thyroid hormone, amphetamines (which also suppress appetite), and dinitrophenol (a pesticide). Adverse reactions limited their usefulness: cardiovascular effects with thyroid hormones, abuse potential with amphetamines, and neuropathy and cataracts with dinitrophenol.
Researchers then looked to drugs that could suppress appetite like amphetamines do, but without the potential for abuse. Medications that increased levels of norepinephrine and serotonin, both by increasing release and decreasing reuptake of these neuromodulators, had some success. But again, serious adverse effects occurred, and several drugs had to be withdrawn from the market.
The most publicized of these withdrawals was for the combination fenfluramine and phentermine (“fen-phen”) and its cousin dexfenfluramine (Redux). Up to 30% of patients taking fenfluramine-phentermine developed echocardiographic evidence of valvular heart disease.11 Fenfluramine also increased the risk of pulmonary hypertension. These findings led to the 1997 withdrawal of these drugs from the US market.
Sibutramine (Meridia), a norepinephrine and serotonin reuptake inhibitor, was approved for weight loss in 1997. Increases in blood pressure and heart rate were noted in the initial trial,12 and then a postmarketing study found increased rates of nonfatal myocardial infarction and stroke in patients with preexisting cardiovascular disease or diabetes mellitus.13 Based on these results, sibutramine was withdrawn from both US and European markets.
Rimonabant (Acomplia, Zimulti), a cannabinoid-receptor inhibitor, was approved in Europe in 2006, but its approval was withdrawn just 2 years later because of increased suicidality in a postmarketing study.14 It was never approved for use in the United States.
NORADRENERGIC SYMPATHOMIMETICS: FOR SHORT-TERM USE
Several noradrenergic sympathomimetic drugs are FDA-approved for short-term weight loss, but phentermine is by far the most commonly prescribed drug in this class. In fact, it is the most commonly prescribed drug for obesity in the United States.15
Phentermine
Phentermine is an atypical amphetamine analogue that suppresses appetite by norepinephrine agonism in the central nervous system. The FDA approved it for short-term weight management in 1959, and its use became widespread in the 1960s, followed by decades of popularity.
Dosage. Phentermine is prescribed at an oral dose of 15, 30, or 37.5 mg daily, either before breakfast or 1 to 2 hours after. It is a schedule IV controlled substance, based on its similarity to amphetamine. (The 5 US controlled substance schedules range from schedule I, which includes heroin, amphetamine, and cannabis, to schedule V, which includes cough syrups containing no more than 200 mg of codeine per 100 mL.) However, concerns about addiction and dependence with phentermine are largely unfounded, and abrupt cessation of the drug has not been shown to cause amphetamine-like withdrawal.16
Adverse effects. Common adverse reactions include nervousness, insomnia, and dry mouth, but these effects tend to wane with continued use.
Contraindications. Cardiovascular disease is a contraindication to phentermine because of concerns about increased blood pressure and pulse rate, although these concerns seem to be more theoretic than observed.16 Other contraindications include hyperthyroidism, glaucoma, agitation, a history of drug abuse, pregnancy, breastfeeding, and current or recent use of a monoamine oxidase inhibitor. No serious adverse events have been reported in trials of phentermine.
Efficacy. In a pooled analysis of 6 trials lasting 2 to 24 weeks completed between 1975 and 1999, phentermine-treated patients lost an average of 3.6 kg more weight than placebo recipients.17 More than 80% of study participants were women.
In a 36-week study in 108 women,18 participants lost a mean of 12.2 kg with continuous phentermine use, 13.0 kg with intermittent use (4 weeks on, 4 weeks off; the difference was not significant), and 4.8 kg with placebo.
Minimal data exist on long-term efficacy of phentermine monotherapy.
DRUGS FOR LONG-TERM THERAPY
Orlistat
Orlistat was approved as a prescription drug (Xenical, 120 mg) in 1999 and as an over-the-counter medication (Alli, 60 mg) in 2007.
Orlistat works by inhibiting pancreatic and gastric lipase, causing incomplete hydrolysis of ingested fat, thereby increasing fecal fat excretion in a dose-dependent manner. It is a good choice for weight-loss drug therapy because of its safe cardiovascular risk profile and beneficial effects on lipid levels. However, its long-term effect on weight is only modest.19,20
Dosage. The dosage for prescription orlistat is 120 mg 3 times per day, in addition to a low-fat diet (< 30% of daily calories from fat). To prevent potential deficiencies of fat-soluble vitamins, a daily multivitamin supplement is recommended, but it should not be taken with meals.
Efficacy. In a 2014 systematic review, 35% to 73% of patients treated with orlistat 120 mg had lost at least 5% of their body weight at 1 year, and 14% to 41% had lost at least 10%.21 At the end of the second year, orlistat-treated patients had lost about 3.3 kg more than placebo recipients.
In a randomized trial,22 4 years of treatment with orlistat vs placebo led to a significant (37.3%) risk reduction in the incidence of type 2 diabetes mellitus in obese participants, as well as significant improvements in cardiovascular risk factors. Mean weight loss at 1 year was significantly greater with orlistat than with placebo (10.6 vs 6.2 kg), and it remained greater at 4 years (5.8 vs 3.0 kg; P < .001).
Adverse effects. Long-term orlistat use is hampered by adverse reactions. A population-based, retrospective cohort analysis showed that fewer than 10% of patients were still using it at 1 year, and only 2% were using it at 2 years, although reasons for discontinuation were not reported.23
Adverse reactions are predominantly gastrointestinal, attributed to the high content of undigested fat in stools. Patients who do not limit their dietary fat intake are affected the most. Other reported adverse reactions include hepatotoxicity and oxalate-induced nephropathy.
Orlistat has been reported to interfere with some drugs, particularly those that are lipophilic. Drugs that should be closely monitored with orlistat are warfarin, amiodarone, cyclosporine, certain antiepileptic drugs, and levothyroxine.
Phentermine-topiramate
The combination of phentermine and topiramate was approved by the FDA in 2012 and is available under the brand name Qsymia.
Topiramate had been approved for treating seizure disorder in 1996 and as migraine prophylaxis in 2004. It is not approved as monotherapy for obesity; however, patients taking it for seizures or for psychiatric disorders (eg, binge eating, borderline personality disorder) have reported weight loss during treatment.
How topiramate promotes weight loss is not known. Proposed mechanisms include taste inhibition by carbonic anhydrase, influences on gamma-aminobutyric acid transmission causing appetite suppression, sensitization of insulin activity, and adiponectin secretion in the peripheral tissues.24,25
Phentermine-topiramate therapy has an advantage over monotherapy because lower doses of each medication can be used to achieve the same benefit, thus avoiding dose-related adverse reactions.
Dosage. Phentermine-topiramate is available in capsules containing 3.75/23, 7.5/46, 11.25/69, and 15/92 mg. The recommended starting dosage is 3.75/23 mg/day for 14 days, increasing to 7.5/46 mg/day. If patients do not lose at least 3% of their body weight after 12 weeks, the dose can be increased to 11.25/69 mg daily for 14 days, followed by 15/92 mg daily.26 Phentermine-topiramate is a schedule IV controlled substance with a low potential for abuse and dependence.
Efficacy. Approval of phentermine-topiramate for treating obesity was primarily based on 3 clinical trials.27–29 In 1 of these trials,28 at 1 year, patients had lost 9.9 kg with the medium dose and 12.9 kg with the high dose.
Adverse effects. Phentermine-topiramate was well tolerated in the trials. The most commonly reported adverse reactions were dry mouth, dizziness, constipation, insomnia, dysgeusia, paresthesia, and increased resting heart rate.28,29 Acute myopia and angle-closure glaucoma also have been reported with topiramate.30 Topiramate monotherapy has been associated with dose-dependent neuropsychiatric adverse effects, including memory symptoms and depression. However, across all 3 trials of phentermine-topiramate therapy, symptoms of depression improved over time, and no significant increase in suicide risk was identified.27–29
Recommended monitoring for patients on phentermine-topiramate includes a blood chemistry panel, resting heart rate, blood pressure, and depression screening.
Because topiramate has teratogenic potential (craniofacial abnormalities), it is labeled as pregnancy category X (contraindicated). A negative pregnancy test is needed before women of childbearing age take the drug and monthly thereafter. Women should be counseled to use effective birth control. A home pregnancy test is an alternative to laboratory testing, but this option should be left to the prescribing clinician’s judgment and be based on reliability of the test and patient compliance.
Lorcaserin
Lorcaserin (Belviq) was approved by the FDA in 2012 for chronic weight management. It suppresses appetite by activating the serotonin 2C receptor in the brain. Because it is selective for the 2C receptor, it does not appear to have the same detrimental effects on heart valves as occurred with less-selective serotonergic agents such as fenfluramine and dexfenfluramine.31
Dosage. The recommended dosage for lorcaserin is 10 mg twice daily. Lorcaserin is a schedule IV controlled substance because of studies that showed increases in positive subjective measures such as euphoria in patients taking the drug. The incidence of euphoria was similar to that seen with zolpidem.32
Efficacy. Lorcaserin was approved on the basis of 2 trials in nondiabetic obese and overweight adults who did not have diabetes but who had a weight-related condition,33,34 and in a third trial in obese and overweight adults with type 2 diabetes mellitus who were taking oral hypoglycemic agents.35 In these trials, lorcaserin use resulted in a modest 4.7- to 5.8-kg weight loss compared with 1.6 to 2.2 kg in the placebo group.33–35 There was a high dropout rate in all 3 of these studies (33% to 45% of participants).
A pilot study that added phentermine to lorcaserin yielded double the weight loss from lorcaserin alone.36 This drug combination warrants further investigation.
Contraindications. Lorcaserin should not be given to patients who have severe renal insufficiency (creatinine clearance < 30 mL/min) or severe hepatic impairment, or who are pregnant.
Adverse effects. Common adverse reactions include dry mouth, dizziness, somnolence, headache, and gastrointestinal disturbances (nausea, constipation, or diarrhea).37
Patients with type 2 diabetes mellitus should be monitored for hypoglycemia.
Lorcaserin should be used with extreme caution in patients taking other serotonergic agents because of the risk of the serotonin syndrome.
A theoretic potential for increased risk of breast cancer also exists with lorcaserin. When rats were given supraphysiologic doses of lorcaserin (more than 50 times higher than recommended in humans), fibroadenomas and adenocarcinomas occurred at higher rates.38 Breast cancer data were not reported in the 3 randomized trials discussed above.33–35
Naltrexone-bupropion
The combination of naltrexone and bupropion was approved by the FDA in 2014 under the brand name Contrave. Both drugs are approved for monotherapy in conditions other than obesity.
Naltrexone is a mu opioid receptor antagonist approved to treat alcohol and opioid dependency. Bupropion is a dopamine-norepinephrine reuptake inhibitor approved to treat depression and to help with smoking cessation. Combining the drugs produces weight loss and metabolic benefits through effects on 2 areas of the brain that regulate food intake: the hypothalamus (appetite) and the mesolimbic dopamine circuit (reward system).
Dosage. Naltrexone-bupropion comes as an extended-release tablet of 8/90 mg. The maintenance dose of 2 tablets twice daily is reached at week 4 through a specific dose-titration regimen (Table 1). The dose should be adjusted if patients have renal or hepatic impairment or if they are also taking a CYP2B6 inhibitor.
Efficacy. FDA approval was based on the results of 4 clinical trials.39–42 Using a modified intention-to-treat analysis, Yanovski and Yanovski43 calculated that at 1 year, placebo-subtracted mean weight loss was 4.6% (4.9 kg), and mean total weight loss was 6.8% (7.3 kg) across the studies. Attrition rates, however, were high, ranging from 42% to 50%.
Cardiometabolic effects in 2 of the trials40,41 included decreased waist circumference, triglyceride levels, and C-reactive protein levels, and increased high-density lipoprotein levels at the initial dose. At the maintenance dose, additional lowering of fasting plasma insulin and glucose levels occurred along with lower levels of the homeostatic model assessment of insulin resistance. In the COR-Diabetes Study Group trial, patients with type 2 diabetes mellitus had decreased hemoglobin A1c levels without an increase in hypoglycemia and an increased likelihood of reaching the target hemoglobin A1c level below 7%.39
Contraindications. Naltrexone-bupropion is contraindicated for patients who have uncontrolled hypertension, seizure disorder, eating disorder, or end-stage renal failure; who are pregnant; or who have been treated with a monoamine oxidase inhibitor within 14 days. It should not be used with other bupropion-containing products or in patients who have taken opioids chronically or have acute opiate withdrawal.
Because of its bupropion component, this product carries an FDA black-box warning about possible suicidal thoughts and behaviors and neuropsychiatric reactions.
Adverse effects. The adverse reactions most commonly associated with naltrexone-bupropion were nausea (32.5%), constipation (19.2%), headache (17.6%), vomiting (10.7%), dizziness (9.9%), insomnia (9.2%), dry mouth (8.1%), and diarrhea (7.1%).44
Liraglutide
Liraglutide, previously FDA-approved to treat type 2 diabetes mellitus under the brand name Victoza, received approval in 2014 in a higher-dose formulation (Saxenda) to treat obesity.
Liraglutide is a glucagon-like peptide-1 receptor agonist that stimulates glucose-dependent insulin release from the pancreatic islet cells, slows gastric emptying, regulates postprandial glucagon, and reduces food intake.
Dosage. Liraglutide is given as a once-daily injection in the abdomen, thigh, or arm. The initial dosage is 0.6 mg daily for the first week and can be titrated up by 0.6 mg weekly to a target dose of 3 mg daily. If a patient does not lose 4% of baseline body weight after 16 weeks on the target dose, the drug should be discontinued because it is unlikely to lead to clinically significant weight loss.
Efficacy. Liraglutide for weight management (3 mg once daily) was evaluated in a large (N = 3,731), randomized, double-blind, placebo-controlled international trial.45 Participants did not have diabetes mellitus, but 60% had prediabetes. Liraglutide or placebo was given for 56 weeks, along with lifestyle counseling. At the end of the study, the liraglutide group had lost a mean of 8.4 kg vs 2.8 kg in the placebo group. Additionally, 63% of the liraglutide group lost at least 5% of body weight vs 27% in the placebo group, and 33% lost at least 10% of body weight vs 10% in the placebo group.
A 2-year extension found systolic blood pressure decreased with no change in pulse, and the prevalence of prediabetes and metabolic syndrome decreased by 52% and 59%, respectively.46 At 2 years, mean scores for physical function, self-esteem, and work had improved more in the liraglutide group than the placebo group.47
Adverse effects. The most common adverse reactions with liraglutide were nausea, vomiting, diarrhea, constipation, hypoglycemia, and loss of appetite. In most cases, nausea and vomiting were tolerable, transient, and associated with greater weight loss but not with decreased quality-of-life scores. Serious adverse reactions included pancreatitis, gallbladder disease, renal impairment, and suicidal thoughts.
CHOOSING A DRUG
For obese patients, when lifestyle modifications do not result in the desired weight loss, pharmacotherapy is an option. Practitioners have several FDA-approved options for weight management. Because of evidence that these drugs can postpone the onset of other complications and improve metabolic and cardiovascular parameters, they should be considered.
In phase 3 trials, these drugs caused modest weight loss of 5% to 10% of body weight. More weight was lost with the combination of phentermine-topiramate than with the other drugs.
In a 2016 meta-analysis, these drugs were associated with at least 5% weight reduction compared with placebo.48 Phentermine-topiramate and liraglutide were most likely to produce at least a 5% weight loss, while liraglutide and naltrexone-bupropion were most likely to be discontinued because of adverse events. Combination drugs may have the advantages of synergistic effects on weight loss and fewer adverse reactions because lower doses of the individual drug components are used.
Response to therapy with most of these drugs should be evaluated at 12 weeks on the maintenance dose. If less than 5% weight loss has been achieved, the medication should be discontinued.
Adverse-effect profiles, drug interactions, abuse, misuse, and overdose potential should be considered when prescribing these drugs. Weight-loss drugs are contraindicated in pregnancy because they offer no potential benefit to a pregnant woman and may harm the fetus.
The development of new drugs and better drug combinations is expected to provide more effective therapeutic strategies, which are essential for combating the obesity epidemic.
- Ogden CL, Carroll MD, Fryar CD, Flegal KM. Prevalence of obesity among adults and youth: United States, 2011-2014. NCHS Data Brief 2015; 219:1–8.
- Yanovski SZ, Yanovski JA. Obesity. N Engl J Med 2002; 346:591–602.
- Finkelstein EA, Khavjou OA, Thompson H, et al. Obesity and severe obesity forecasts through 2030. Am J Prev Med 2012; 42:563–570.
- Hill JO, Wyatt H. Outpatient management of obesity: a primary care perspective. Obes Res 2002; 10(suppl 2):124S–130S.
- US Department of Health and Human Services. National Institute of Diabetes and Digestive and Kidney Diseases. Overweight and obesity statistics. www.niddk.nih.gov/health-information/health-statistics/Pages/overweight-obesity-statistics.aspx#overweight. Accessed October 10, 2017.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society. J Am Coll Cardiol 2014; 63:2985–3023.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society. Circulation 2014; 129(suppl 2):S102–S138.
- American Association of Clinical Endocrinologists. AACE/ACE algorithm for the medical care of patients with obesity. www.aace.com/files/guidelines/ObesityAlgorithm.pdf. Accessed July 25, 2017.
- Wadden TA, Berkowitz RI, Womble LG, et al. Randomized trial of lifestyle modification and pharmacotherapy for obesity. N Engl J Med 2005; 353:2111–2120.
- Mechanick JI, Youdim A, Jones DB, et al. Clinical practice guidelines for the perioperative nutritional, metabolic, and nonsurgical support of the bariatric surgery patient, 2013 update: cosponsored by American Association of Clinical Endocrinologists, the Obesity Society, and American Society for Metabolic and Bariatric Surgery. Surg Obes Relat Dis 2013; 9:159–191.
- Connolly HM, Crary JL, McGoon MD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med 1997; 337:581–588.
- Kim SH, Lee YM, Jee SH, et al. Effect of sibutramine on weight loss and blood pressure: a meta-analysis of controlled trials. Obes Res 2003; 11:1116–1123.
- James WP, Caterson ID, Coutinho W, et al; SCOUT Investigators. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N Engl J Med 2010; 363:905–917.
- Nissen SE, Nicholls SJ, Wolski K, et al; STRADIVARIUS Investigators. Effect of rimonabant on progression of atherosclerosis in patients with abdominal obesity and coronary artery disease: the STRADIVARIUS randomized controlled trial. JAMA 2008; 299:1547–1560.
- Ryan DH, Bray GA. Pharmacologic treatment options for obesity: what is old is new again. Curr Hypertens Rep 2013; 15:182–189.
- Hendricks EJ, Greenway FL, Westman EC, Gupta AK. Blood pressure and heart rate effects, weight loss and maintenance during long-term phentermine pharmacotherapy for obesity. Obesity (Silver Spring) 2011; 19:2351–2360.
- Li Z, Maglione M, Tu W, et al. Meta-analysis: pharmacologic treatment of obesity. Ann Intern Med 2005; 142:532–546.
- Munro JF, MacCuish AC, Wilson EM, Duncan LJ. Comparison of continuous and intermittent anorectic therapy in obesity. Br Med J 1968; 1:352–354.
- Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med 2000; 9:160–167.
- Rossner S, Sjostrom L, Noack R, Meinders AE, Noseda G. Weight loss, weight maintenance, and improved cardiovascular risk factors after 2 years treatment with orlistat for obesity. European Orlistat Obesity Study Group. Obes Res 2000; 8:49–61.
- Yanovski SZ, Yanovski JA. Long-term drug treatment for obesity: a systematic and clinical review. JAMA 2014; 311:74–86.
- Torgerson JS, Hauptman J, Boldrin MN, Sjostrom L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2004; 27:155–161.
- Padwal R, Kezouh A, Levine M, Etminan M. Long-term persistence with orlistat and sibutramine in a population-based cohort. Int J Obes (Lond) 2007; 31:1567–1570.
- Xiong GL, Gadde KM. Combination phentermine-topiramate for obesity treatment in primary care: a review. Postgrad Med 2014; 126:110–116.
- Pucci A, Finer N. New medications for treatment of obesity: metabolic and cardiovascular effects. Can J Cardiol 2015; 31:142–152.
- Smith SM, Meyer M, Trinkley KE. Phentermine-topiramate for the treatment of obesity. Ann Pharmacother 2013; 47:340–349.
- Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine-topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring) 2012; 20:330–342.
- Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet 2011; 377:1341–1352.
- Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine-topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr 2012; 95:297–308.
- Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs 2010; 24:501–526.
- Weissman NJ, Sanchez M, Koch GG, Smith SR, Shanahan WR, Anderson CM. Echocardiographic assessment of cardiac valvular regurgitation with lorcaserin from analysis of 3 phase 3 clinical trials. Circ Cardiovasc Imaging 2013; 6:560–567.
- US Department of Justice Drug Enforcement Administration. Schedules of controlled substances: placement of lorcaserin into Schedule IV. Federal Register 2013; 78:26701–26705.
- Smith SR, Weissman NJ, Anderson CM, et al; Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med 2010; 363:245–256.
- Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab 2011; 96:3067–3077.
- O’Neil PM, Smith SR, Weissman NJ, et al. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM study. Obesity (Silver Spring) 2012; 20:1426–1436.
- Kumar RB, Aronne LJ. Efficacy comparison of medications approved for chronic weight management. Obesity (Silver Spring) 2015; 23(suppl 1):S4–S7.
- Chan EW, He Y, Chui CS, Wong AY, Lau WC, Wong IC. Efficacy and safety of lorcaserin in obese adults: a meta-analysis of 1-year randomized controlled trials (RCTs) and narrative review on short-term RCTs. Obes Rev 2013; 14:383–392.
- Miller LE. Lorcaserin for weight loss: insights into US Food and Drug Administration approval. J Acad Nutr Diet 2013; 113:25–30.
- Hollander P, Gupta AK, Plodkowski R, et al; COR-Diabetes Study Group. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care 2013; 36:4022–4029.
- Apovian CM, Aronne L, Rubino D, et al; COR-II Study Group. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Spring) 2013; 21:935–943.
- Greenway FL, Fujioka K, Plodkowski RA, et al; COR-I Study Group. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2010; 376:595–605.
- Wadden TA, Foreyt JP, Foster GD, et al. Weight loss with naltrexone SR/bupropion SR combination therapy as an adjunct to behavior modification: the COR-BMOD trial. Obesity (Silver Spring) 2011; 19:110–120.
- Yanovski SZ, Yanovski JA. Naltrexone extended-release plus bupropion extended-release for treatment of obesity. JAMA 2015; 313:1213–1214.
- Contrave (naltrexone HC1 and bupropion HC1) extended release tablets [package insert]. Orexigen Therapeutics, 2017. https://contrave.com/wp-content/uploads/2017/05/Contrave_PI.pdf. Accessed November 7, 2017.
- Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med 2015; 373:11–22.
- Astrup A, Carraro R, Finer N, et al; NN8022-1807 Investigators. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes (Lond) 2012; 36:843–854.
- Lean ME, Carraro R, Finer N, et al; NN8022-1807 Investigators. Tolerability of nausea and vomiting and associations with weight loss in a randomized trial of liraglutide in obese, non-diabetic adults. Int J Obes (Lond) 2014; 38:689–697.
- Khera R, Murad MH, Chandar AK, et al. Association of pharmacological treatments for obesity with weight loss and adverse events: a systematic review and meta-analysis. JAMA 2016; 315:2424–2434.
- Ogden CL, Carroll MD, Fryar CD, Flegal KM. Prevalence of obesity among adults and youth: United States, 2011-2014. NCHS Data Brief 2015; 219:1–8.
- Yanovski SZ, Yanovski JA. Obesity. N Engl J Med 2002; 346:591–602.
- Finkelstein EA, Khavjou OA, Thompson H, et al. Obesity and severe obesity forecasts through 2030. Am J Prev Med 2012; 42:563–570.
- Hill JO, Wyatt H. Outpatient management of obesity: a primary care perspective. Obes Res 2002; 10(suppl 2):124S–130S.
- US Department of Health and Human Services. National Institute of Diabetes and Digestive and Kidney Diseases. Overweight and obesity statistics. www.niddk.nih.gov/health-information/health-statistics/Pages/overweight-obesity-statistics.aspx#overweight. Accessed October 10, 2017.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society. J Am Coll Cardiol 2014; 63:2985–3023.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society. Circulation 2014; 129(suppl 2):S102–S138.
- American Association of Clinical Endocrinologists. AACE/ACE algorithm for the medical care of patients with obesity. www.aace.com/files/guidelines/ObesityAlgorithm.pdf. Accessed July 25, 2017.
- Wadden TA, Berkowitz RI, Womble LG, et al. Randomized trial of lifestyle modification and pharmacotherapy for obesity. N Engl J Med 2005; 353:2111–2120.
- Mechanick JI, Youdim A, Jones DB, et al. Clinical practice guidelines for the perioperative nutritional, metabolic, and nonsurgical support of the bariatric surgery patient, 2013 update: cosponsored by American Association of Clinical Endocrinologists, the Obesity Society, and American Society for Metabolic and Bariatric Surgery. Surg Obes Relat Dis 2013; 9:159–191.
- Connolly HM, Crary JL, McGoon MD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med 1997; 337:581–588.
- Kim SH, Lee YM, Jee SH, et al. Effect of sibutramine on weight loss and blood pressure: a meta-analysis of controlled trials. Obes Res 2003; 11:1116–1123.
- James WP, Caterson ID, Coutinho W, et al; SCOUT Investigators. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N Engl J Med 2010; 363:905–917.
- Nissen SE, Nicholls SJ, Wolski K, et al; STRADIVARIUS Investigators. Effect of rimonabant on progression of atherosclerosis in patients with abdominal obesity and coronary artery disease: the STRADIVARIUS randomized controlled trial. JAMA 2008; 299:1547–1560.
- Ryan DH, Bray GA. Pharmacologic treatment options for obesity: what is old is new again. Curr Hypertens Rep 2013; 15:182–189.
- Hendricks EJ, Greenway FL, Westman EC, Gupta AK. Blood pressure and heart rate effects, weight loss and maintenance during long-term phentermine pharmacotherapy for obesity. Obesity (Silver Spring) 2011; 19:2351–2360.
- Li Z, Maglione M, Tu W, et al. Meta-analysis: pharmacologic treatment of obesity. Ann Intern Med 2005; 142:532–546.
- Munro JF, MacCuish AC, Wilson EM, Duncan LJ. Comparison of continuous and intermittent anorectic therapy in obesity. Br Med J 1968; 1:352–354.
- Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med 2000; 9:160–167.
- Rossner S, Sjostrom L, Noack R, Meinders AE, Noseda G. Weight loss, weight maintenance, and improved cardiovascular risk factors after 2 years treatment with orlistat for obesity. European Orlistat Obesity Study Group. Obes Res 2000; 8:49–61.
- Yanovski SZ, Yanovski JA. Long-term drug treatment for obesity: a systematic and clinical review. JAMA 2014; 311:74–86.
- Torgerson JS, Hauptman J, Boldrin MN, Sjostrom L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2004; 27:155–161.
- Padwal R, Kezouh A, Levine M, Etminan M. Long-term persistence with orlistat and sibutramine in a population-based cohort. Int J Obes (Lond) 2007; 31:1567–1570.
- Xiong GL, Gadde KM. Combination phentermine-topiramate for obesity treatment in primary care: a review. Postgrad Med 2014; 126:110–116.
- Pucci A, Finer N. New medications for treatment of obesity: metabolic and cardiovascular effects. Can J Cardiol 2015; 31:142–152.
- Smith SM, Meyer M, Trinkley KE. Phentermine-topiramate for the treatment of obesity. Ann Pharmacother 2013; 47:340–349.
- Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine-topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring) 2012; 20:330–342.
- Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet 2011; 377:1341–1352.
- Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine-topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr 2012; 95:297–308.
- Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs 2010; 24:501–526.
- Weissman NJ, Sanchez M, Koch GG, Smith SR, Shanahan WR, Anderson CM. Echocardiographic assessment of cardiac valvular regurgitation with lorcaserin from analysis of 3 phase 3 clinical trials. Circ Cardiovasc Imaging 2013; 6:560–567.
- US Department of Justice Drug Enforcement Administration. Schedules of controlled substances: placement of lorcaserin into Schedule IV. Federal Register 2013; 78:26701–26705.
- Smith SR, Weissman NJ, Anderson CM, et al; Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med 2010; 363:245–256.
- Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab 2011; 96:3067–3077.
- O’Neil PM, Smith SR, Weissman NJ, et al. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM study. Obesity (Silver Spring) 2012; 20:1426–1436.
- Kumar RB, Aronne LJ. Efficacy comparison of medications approved for chronic weight management. Obesity (Silver Spring) 2015; 23(suppl 1):S4–S7.
- Chan EW, He Y, Chui CS, Wong AY, Lau WC, Wong IC. Efficacy and safety of lorcaserin in obese adults: a meta-analysis of 1-year randomized controlled trials (RCTs) and narrative review on short-term RCTs. Obes Rev 2013; 14:383–392.
- Miller LE. Lorcaserin for weight loss: insights into US Food and Drug Administration approval. J Acad Nutr Diet 2013; 113:25–30.
- Hollander P, Gupta AK, Plodkowski R, et al; COR-Diabetes Study Group. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care 2013; 36:4022–4029.
- Apovian CM, Aronne L, Rubino D, et al; COR-II Study Group. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Spring) 2013; 21:935–943.
- Greenway FL, Fujioka K, Plodkowski RA, et al; COR-I Study Group. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2010; 376:595–605.
- Wadden TA, Foreyt JP, Foster GD, et al. Weight loss with naltrexone SR/bupropion SR combination therapy as an adjunct to behavior modification: the COR-BMOD trial. Obesity (Silver Spring) 2011; 19:110–120.
- Yanovski SZ, Yanovski JA. Naltrexone extended-release plus bupropion extended-release for treatment of obesity. JAMA 2015; 313:1213–1214.
- Contrave (naltrexone HC1 and bupropion HC1) extended release tablets [package insert]. Orexigen Therapeutics, 2017. https://contrave.com/wp-content/uploads/2017/05/Contrave_PI.pdf. Accessed November 7, 2017.
- Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med 2015; 373:11–22.
- Astrup A, Carraro R, Finer N, et al; NN8022-1807 Investigators. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes (Lond) 2012; 36:843–854.
- Lean ME, Carraro R, Finer N, et al; NN8022-1807 Investigators. Tolerability of nausea and vomiting and associations with weight loss in a randomized trial of liraglutide in obese, non-diabetic adults. Int J Obes (Lond) 2014; 38:689–697.
- Khera R, Murad MH, Chandar AK, et al. Association of pharmacological treatments for obesity with weight loss and adverse events: a systematic review and meta-analysis. JAMA 2016; 315:2424–2434.
KEY POINTS
- Weight-loss drugs should only be used in combination with lifestyle modification.
- Preparations that combine 2 drugs have greater weight-loss benefits and better side-effect profiles.
- Weight-loss drugs should be discontinued if substantial (5%) weight loss has not occurred by 12 weeks.
- All weight-loss drugs are contraindicated in pregnancy.

Dual antiplatelet therapy for acute coronary syndromes: How long to continue?
Percutaneous coronary intervention for acute coronary syndromes has evolved, and so, hand in hand, has antiplatelet therapy. With the advent of clopidogrel and newer agents, several studies demonstrated the benefits of dual antiplatelet therapy in preventing major vascular ischemic complications. The findings culminated in a guideline recommendation for at least 12 months of dual antiplatelet therapy after placement of a drug-eluting stent, when feasible—a class I recommendation (treatment should be given), level of evidence B (limited populations evaluated).1,2 But extending dual antiplatelet therapy beyond 12 months had no strong favorable evidence until the recent Dual Antiplatelet Therapy (DAPT) study3 shed light on this topic.
Here, we review the evidence thus far on the optimal duration of dual antiplatelet therapy in the secondary prevention of coronary artery disease.
PLATELETS IN ACUTE CORONARY SYNDROMES AND STENT THROMBOSIS
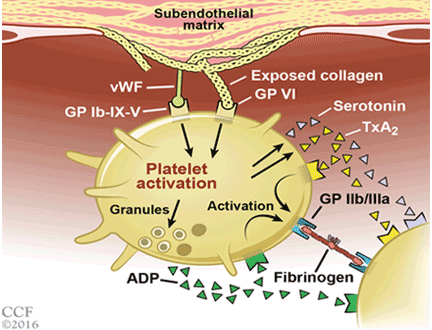
Acute coronary syndromes begin with fissuring or ulceration of a vulnerable atherosclerotic plaque, followed by thrombosis and occlusion, mediated by platelet adhesion, activation, and aggregation (Figure 1). Transient occlusion results in unstable angina or non-ST-elevation myocardial infarction, while total occlusion usually results in ST-elevation myocardial infarction.
Platelet aggregation is prominent among the mechanisms leading to stent thrombosis and vaso-occlusive ischemic complications after percutaneous coronary intervention. Thus, antiplatelet agents play a vital role in both primary and secondary prevention of cardiovascular events.4–6
Adhesion, activation, and aggregation
Adhesion. Disruption of the vascular endothelium as a result of vulnerable plaque fissuring or ulceration exposes subendothelial thrombogenic collagen and von Willebrand factor to blood. Collagen engages platelets through their glycoprotein (GP) Ia, IIa, and VI receptors, and von Willebrand factor binds platelets through the GP Ib-IX-V receptor.
Activation. Once platelets adhere to the subendothelium, they undergo a conformational change and become activated. Simultaneous release of various autocrine and paracrine mediators including adenosine diphosphate, serotonin, epinephrine, thromboxane, and various ligand-receptor interactions all contribute to the activation cascade. Adenosine diphosphate binds to the platelet receptor P2Y1, leading to an increase in intracellular calcium, and it binds to P2Y12, leading to a decrease in cyclic adenosine monophosphate, both of which cause GP IIb/IIIa receptor activation. Thromboxane A2 released by platelets by cyclo-oxygenase 1 binds to alpha or beta variant receptors and contributes to GP IIb/IIIa activation through elevation of intracellular calcium levels.
Aggregation and thrombosis. Exposure of tissue factor to plasma following plaque rupture activates the coagulation cascade via the extrinsic pathway, which generates thrombin, a powerful platelet activator that causes thrombus formation via fibrin. Thrombin binds to protease-activated receptors PAR-1 and PAR-4 on platelets, causing an increase in intracellular calcium and a decrease in cyclic adenosine monophosphate with subsequent GP IIb/IIIa activation. GP IIb/IIIa facilitates platelet aggregation by binding to fibrinogen and forming a stable platelet thrombus.
In the early stages of thrombus formation, platelets predominate (“white” thrombi); further organization with fibrin results in older “red” thrombi. The stages of thrombi vary in non-ST-elevation and ST-elevation myocardial infarction and are prognostic markers of death.4–8
PERCUTANEOUS INTERVENTION, RESTENOSIS, AND STENT THROMBOSIS
Percutaneous coronary intervention, the preferred means of revascularization for many patients, is performed emergently in patients with ST-elevation myocardial infarction, urgently in those with acute coronary syndromes without ST elevation, and electively in those with stable ischemic symptoms.
Percutaneous revascularization techniques have evolved from balloon angioplasty to bare-metal stents to drug-eluting stents, but each of these procedures has been associated with a periprocedural and postprocedural risk of thrombosis.

Balloon angioplasty was associated with vascular intimal injury, inciting elastic vascular recoil and smooth muscle cell proliferation leading to restenosis.
Bare-metal stents reduced the restenosis rate by eliminating vascular recoil, although restenosis still occurred within the stent because of neointimal proliferation of vascular smooth muscle cells. This was an important limitation, as both acute and subacute stent thrombosis were refractory to aggressive anticoagulation regimens that were associated with major bleeding complications and longer hospital length of stay. Stenting became mainstream practice only after the ISAR9 and STARS10 trials showed that dual antiplatelet therapy controlled stent thrombosis.
Drug-eluting stents coated with anti-proliferative and anti-inflammatory polymers markedly reduced in-stent restenosis rates by suppressing the initial vascular smooth-muscle proliferative response. However, they were still associated with late and very late stent thrombosis with incomplete endothelialization, even up to 40 months after implantation. Proposed mechanisms include incomplete stent apposition and inflammatory hypersensitivity reactions to the polymer coating. Incomplete stent apposition associated with low-velocity blood flow at the junction of the stent strut and vessel wall, together with delayed endothelialization, promotes platelet adhesion and aggregation, followed by thrombus formation.11
Second-generation drug-eluting stents have thinner struts and more biocompatible polymers and are thought to favor more complete re-endothelialization, reducing the rates of stent thrombosis.8,12,13
Predictors of early stent thrombosis
The Dutch Stent Thrombosis Registry and other studies looked at risk factors for stent thrombosis.14,15
Procedure-related factors included:
- Stent undersizing
- Residual uncovered dissections after angioplasty
- Longer stents
- Low flow after angioplasty (< 3 on the 0–3 Thrombolysis in Myocardial Infarction [TIMI] scale).
Lesion-related factors included:
- Intermediate coronary artery disease both proximal and distal to the culprit lesions
- Bifurcation lesions.
Patient-related factors included:
- Low left ventricular ejection fraction
- Diabetes mellitus
- Peripheral arterial disease Premature discontinuation of clopidogrel.
ANTIPLATELET AGENTS: MECHANISM OF ACTION
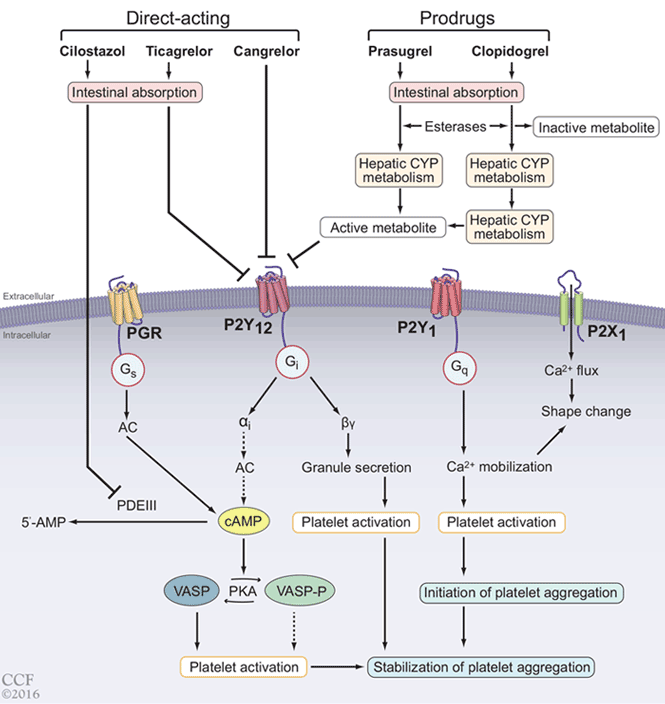
Various pathways play synergistic roles in platelet activation and aggregation and thrombus formation, and different antiplatelet agents inhibit these specific pathways, thus complementing each other and having additive effects (Figure 2, Table 1).5,16–21
Aspirin inhibits cyclo-oxygenase 1
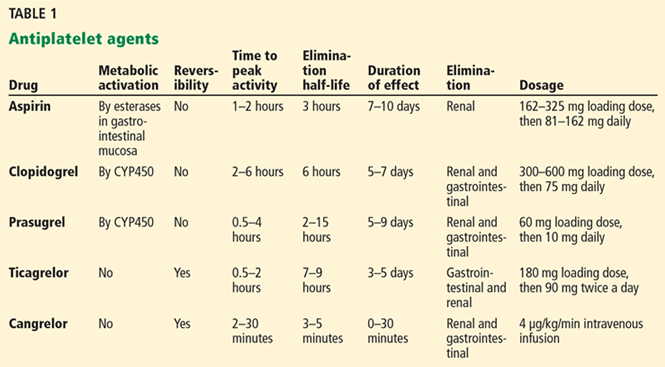
Cyclo-oxygenase 1, found in platelets, endothelial cells, and other cells, catalyzes the conversion of arachidonic acid to thromboxane A2. Aspirin irreversibly inhibits cyclo-oxygenase 1 by acetylating its serine residue, preventing formation of thromboxane A2 and preventing platelet activation and aggregation.
P2Y12 ADP receptor antagonists
Clopidogrel and prasugrel are thienopyridine agents that irreversibly inhibit the P2Y12 receptor, thereby preventing binding of adenosine diphosphate and the subsequent platelet activation-aggregation cascade. They are both prodrugs and require conversion by cytochrome P450 enzymes to active metabolites. Prasugrel is 10 times more potent than clopidogrel due to more efficient formation of its active metabolite, and it achieves a comparable effect on platelet inhibition 30 minutes faster than the peak effect of clopidogrel at 6 hours. The overall peak inhibitory effect of prasugrel is twice that of clopidogrel.22
Ticagrelor, a cyclopentyl-triazolo-pyrimidine, directly and reversibly inhibits the P2Y12 ADP receptor. Unlike clopidogrel and prasugrel, it does not need to be converted to an active metabolite, and it noncompetitively inhibits P2Y12 at a site different from the adenosine diphosphate binding site.23 Like prasugrel, ticagrelor inhibits platelet function more rapidly and more completely than clopidogrel.
Cangrelor, an intravenously administered analogue of adenosine triphosphate, reversibly inhibits the P2Y12 receptor. It has undergone phase 3 trials but is not yet approved for clinical use.24
WHY DUAL ANTIPLATELET THERAPY?
Aspirin is good, clopidogrel is better
Aspirin has a well-validated role in both primary and secondary prevention of coronary and noncoronary atherosclerotic vascular disease.
The CAPRIE trial found clopidogrel monotherapy to be superior to aspirin monotherapy in patients with established atherosclerotic vascular disease.25
After stenting, short-term dual therapy is better than short-term warfarin
Thrombotic complications in the early postprocedural period were a major limitation of stenting, and existing anticoagulation regimens were ineffective in preventing them.26,27
The ISAR trial studied the benefit of combined antiplatelet vs anticoagulant therapy after stent placement. Patients randomized to receive combined aspirin plus ticlopidine (an early P2Y12 inhibitor) had significantly lower rates of primary cardiac, hemorrhagic, and vascular events at 30 days.9 Two other trials confirmed this finding.28,29
STARS10 also confirmed the benefit of aspirin and ticlopidine after stenting. Patients were randomly assigned to aspirin alone, aspirin plus warfarin, or aspirin plus ticlopidine after stent placement. The rate of stent thrombosis at 30 days was significantly lower in the dual antiplatelet group than in the other two groups. The dual antiplatelet group had a higher rate of bleeding than the aspirin-alone group, but the rate was similar to that of the aspirin-plus-warfarin group.
Long-term dual antiplatelet therapy is beneficial in several situations
ISAR and STARS were landmark trials that showed stent thrombosis could be reduced by dual antiplatelet therapy for a 30-day period. However, the long-term role of dual antiplatelet therapy was still unknown.
The CURE trial30–32 randomized patients presenting with acute coronary syndromes without ST elevation to receive clopidogrel plus aspirin or placebo plus aspirin for 3 to 12 months. The rate of the primary end point (cardiac death, nonfatal myocardial infarction, or stroke) was significantly lower in the clopidogrel-plus-aspirin group. A similar benefit of dual antiplatelet therapy was seen in the subgroup of patients who underwent percutaneous coronary intervention. Both pretreatment with clopidogrel plus aspirin for a median of 10 days prior to percutaneous intervention and continuing it for a mean of 9 months reduced major adverse cardiovascular events.
The CREDO trial20 found that the combination of clopidogrel and aspirin significantly reduced the incidence of death, myocardial infarction, or stroke at 1 year after percutaneous coronary intervention. A subgroup of patients in this trial who had a longer pretreatment interval with a loading clopidogrel dose showed a benefit at 28 days, which was not as evident with a shorter loading dose interval.
The CLARITY-TIMI 28 trial33,34 showed the advantage of adding clopidogrel to aspirin in patients receiving fibrinolytic therapy for ST-elevation myocardial infarction. Adding clopidogrel both improved the patency of the infarct-related artery and reduced ischemic complications. In patients who subsequently underwent percutaneous coronary intervention and stenting, clopidogrel pretreatment was associated with a significant decrease in ischemic complications before and after the procedure. There was no significant increase in bleeding complications in either group.
COMMIT/CCS 235 also showed the benefit of dual antiplatelet therapy in patients with ST-elevation myocardial infarction. Clopidogrel added to aspirin during the short-term in-hospital or postdischarge treatment period significantly reduced a composite end point of reinfarction, death, or stroke as well as death from any cause.
The CHARISMA trial36–38 aimed to determine if patients who were more stable (ie, no recent acute coronary syndrome event or percutaneous coronary intervention) would benefit. Overall, CHARISMA showed no benefit of adding clopidogrel to aspirin compared with aspirin alone in a broad population of patients with established vascular disease (secondary prevention) or risk factors for vascular disease (primary prevention).
But importantly, though no benefit was seen in the primary prevention group, the large subgroup of patients with established atherosclerotic vascular disease (12,153 of the 15,603 patients in the trial) did benefit from dual antiplatelet therapy.36,37 This subgroup showed an overall reduction in absolute risk of 1.5% (relative risk 0.88, P = .046) over a median follow-up of 27.6 months. This benefit was even more apparent in the 9,478 patients with prior myocardial infarction, stroke, or peripheral artery disease, for whom the relative risk reduction was 17.1% (P = .01) and the reduction in absolute risk 1.5%.38
These results are comparable to the 2% absolute risk reduction in the CURE trial for similar end points over 9 months. In both studies, there was no significant increase in the risk of major bleeding or intracranial bleeding in the clopidogrel-plus-aspirin groups, although minor bleeding was increased by dual antiplatelet therapy.
The rate of severe bleeding, which was the primary safety end point in CHARISMA, was not significantly different in the clopidogrel-plus-aspirin group compared with the placebo-plus-aspirin group (relative risk 1.25, 95% CI 0.97–1.61, P = .09).
Thus, although the CHARISMA findings were negative overall, the positive finding observed in the predominant subgroup of patients with established vascular disease can therefore be considered supportive of the results of the subsequent trials discussed below.
The PEGASUS-TIMI 54 trial39 studied the benefit of adding ticagrelor (60 or 90 mg) to low-dose aspirin in patients with stable coronary artery disease who had had a myocardial infarction 1 to 3 years earlier.
Confirming the results of the CHARISMA subgroup analysis, the incidence of the ischemic primary efficacy end point (a composite of cardiovascular death, myocardial infarction, and stroke) was significantly lower in both groups receiving ticagrelor plus aspirin compared with those receiving placebo plus aspirin. The Kaplan-Meier rate at 3 years for the ticagrelor 90 mg-plus-aspirin group was 7.85% vs 9.04% for the placebo-plus-aspirin group (hazard ratio 0.85, 95% confidence interval [CI] 0.75–0.96, P = .008). The rate for the ticagrelor 60 mg-plus-aspirin group was 7.77% vs 9.04% for the placebo-plus-aspirin group (hazard ratio 0.84, 95% CI 0.74–0.95, P = .004).
The rates of all TIMI major and minor bleeding, as well as bleeding requiring transfusion or discontinuation of the study drug, were significantly higher in both ticagrelor dosing groups than in the placebo group (P < .01 for both groups vs placebo). The rates of fatal bleeding and nonfatal intracranial hemorrhage were not significantly higher. Although there was an overall reduction in ischemic end points with the addition of ticagrelor, there was also a significantly higher incidence of bleeding in this group.
Comment. Thus, with or without percutaneous coronary intervention in acute coronary syndrome as well as in stable coronary artery disease, dual antiplatelet therapy was shown to improve outcomes and decrease ischemic complications compared with aspirin alone. It provided benefit in the setting of acute coronary syndrome (in the CURE trial) and percutaneous coronary intervention (in the CREDO trial) for up to 1 year.
Major questions remained to be addressed:
- Do the results of CREDO, which was performed before the current interventional era and the use of drug-eluting stents, reflect outcomes after current interventional practice?
- Could shorter periods of dual antiplatelet therapy be sufficient, especially with newer stents with less risk of late thrombosis?
- Does the benefit of dual antiplatelet therapy extend beyond the 1-year time period tested in those trials to date?
RECOMMENDATIONS FOR DOSING
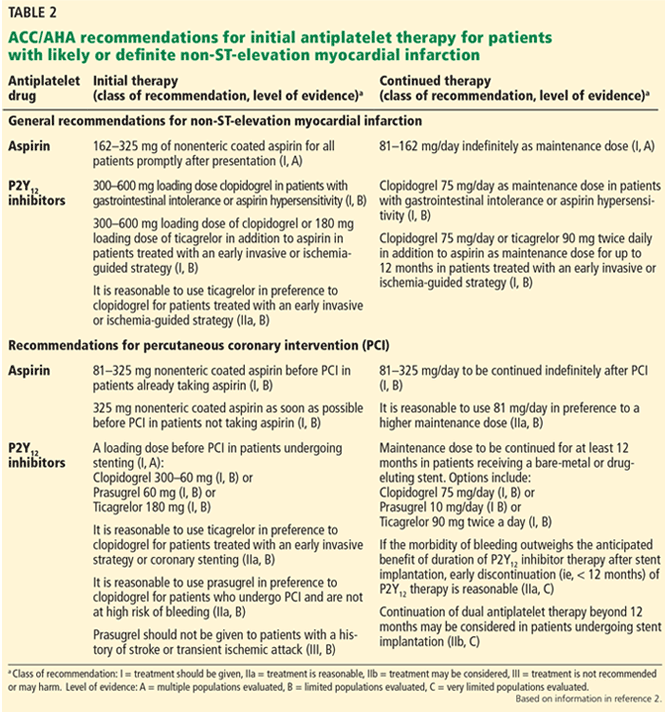
The American College of Cardiology Foundation/American Heart Association guidelines for dosing of antiplatelet agents for non-ST-elevation myocardial infarction are summarized in Table 2, and those for ST-elevation myocardial infarction are summarized in Table 3.1,2
WOULD SHORTER THERAPY AFTER STENTING WORK AS WELL?
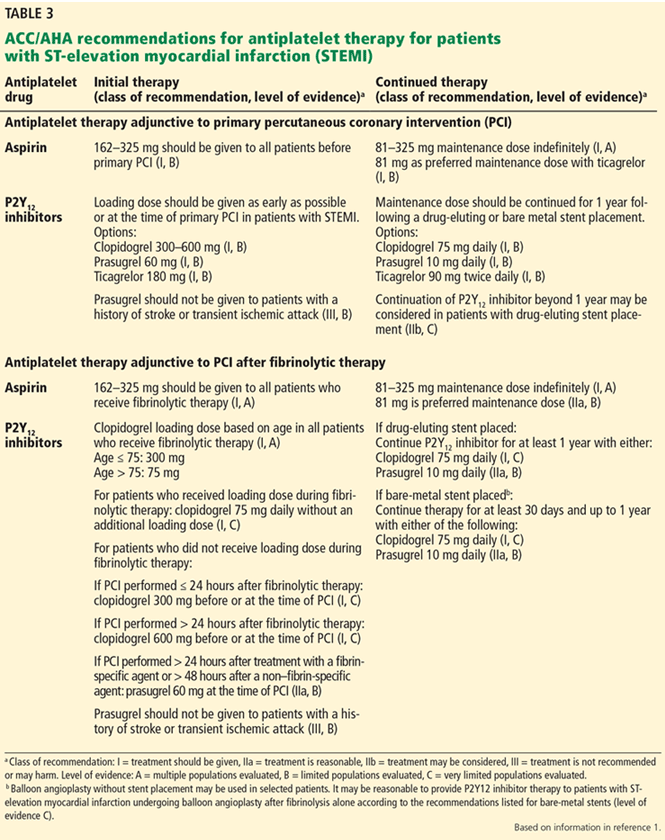
The American College of Cardiology Foundation/American Heart Association currently recommend dual antiplatelet therapy for at least 12 months after drug-eluting stent placement, with shorter courses appropriate for patients who develop excessive bleeding complications or who are at high risk of bleeding.
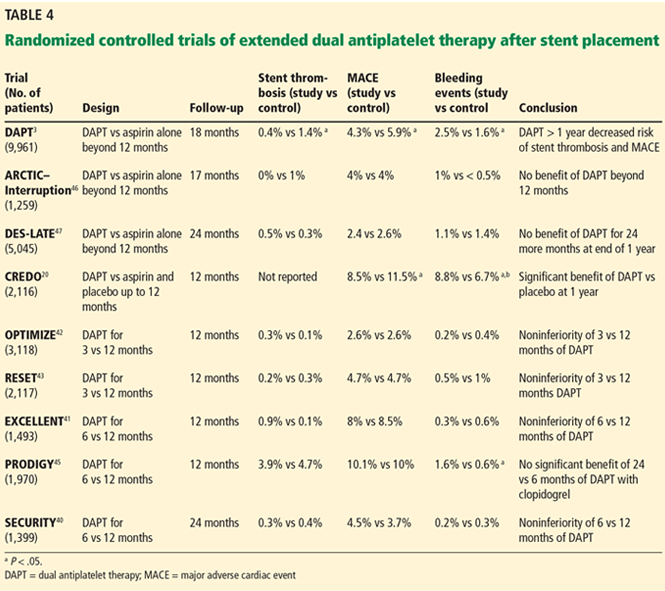
Four trials (Table 4) evaluated whether shorter durations of dual antiplatelet therapy would suffice: SECURITY,40 EXCELLENT,41 OPTIMIZE,42 and RESET.43 All of them showed that short-duration therapy was not inferior to standard-duration therapy.44 These studies were comparable in that:
- Patients were randomized at the time of percutaneous coronary intervention or within 24 hours of it.
- Most patients received a second-generation drug-eluting stent, with the following exceptions: in EXCELLENT,41 one-fourth of patients received a Cypher first-generation drug-eluting stent, and in RESET,43 approximately one-fourth of the patients received a sirolimus-eluting stent in the standard-duration group for short lesions. Those patients with longer lesions in the RESET standard-duration group received an everolimus drug-eluting stent.
- The second antiplatelet added to aspirin in all studies was clopidogrel, with the exception of the SECURITY trial, in which fewer than 2% of patients received ticagrelor or prasugrel.40
- All the trials except RESET excluded patients who had had a myocardial infarction within 72 hours, and thus most patients studied had a lower risk profile.
- All of the trials sought to study noninferiority of short- vs standard-duration dual antiplatelet therapy, defined as the occurrence of a primary end point at 1 year (a composite of cardiovascular death, myocardial infarction, stroke, stent thrombosis, target vessel failure or revascularization, or bleeding).
Their low-risk patient populations and infrequent end points rendered these studies underpowered to make definitive conclusions about the relative efficacy of 6-months vs 12-months of dual antiplatelet therapy.
WOULD LONGER THERAPY BE BETTER?
The PRODIGY trial45 assessed durations of dual antiplatelet therapy both shorter and longer than the conventional 1 year, randomizing patients undergoing placement of a bare-metal stent, first-generation drug-eluting stent, or second-generation drug-eluting stent to receive aspirin and clopidogrel for either 6 months or 24 months. The study showed no significant difference in primary outcomes in the short- or long-duration groups.
Other trials that compared the standard 12 months of dual antiplatelet therapy with extended duration beyond 12 months were DAPT,3 ARCTIC-Interruption,46 and DES-LATE.47 The trials were comparable in that:
- All patients were randomized after completing 12 months of dual antiplatelet therapy following drug-eluting stent placement.
- All patients who were included had been free of major cardiac ischemic events or bleeding during the 12 months following stent placement.
- The primary aim of all three studies was to compare primary end points in groups receiving aspirin alone vs extended dual antiplatelet therapy. The primary end point was a composite of death due to a cardiovascular cause, nonfatal myocardial infarction, stroke, or stent thrombosis.
- The principal safety end point was bleeding.
Although the two earlier studies (ARCTIC-Interruption and DES-LATE) did not show any benefit of extended dual antiplatelet therapy compared with the standard 12-month duration, the recent DAPT study did.
The DAPT study
The DAPT study3 was an international, multicenter, placebo-controlled, double-blind randomized trial designed to examine the benefit of dual antiplatelet therapy beyond 1 year in a patient population large enough to provide definitive assessment of benefit and risk.
A total of 9,961 patients who received drug-eluting stents were randomized after 12 months of dual antiplatelet therapy to receive either a thienopyridine (clopidogrel or prasugrel) plus aspirin or placebo plus aspirin. They were followed for an additional 18 months. The coprimary efficacy end points were stent thrombosis and a composite of death, myocardial infarction, or stroke, while the primary safety end point was moderate or severe bleeding. The patients were also observed from months 30 to 33 on aspirin alone after stopping the thienopyridine.
Results. Longer therapy substantially reduced the risks of stent thrombosis (hazard ratio [HR] 0.29, 95% confidence interval [CI] 0.17–0.48) and the composite ischemic end point (HR 0.71, 95% CI 0.59–0.85). Follow-up during the 3-month thienopyridine discontinuation phase starting at 30 months revealed convergence of the ischemic event-rate curves in the two groups, which suggested that continuing dual antiplatelet therapy beyond 30 months might have been beneficial. Myocardial infarction unrelated to stent thrombosis accounted for 55% of the treatment benefit of dual antiplatelet therapy.
The risk of bleeding was higher in the thienopyridine group during the treatment period (2.5% vs 1.6%, P = .001). There was also a higher rate of noncardiovascular mortality in the thienopyridine group, although this difference may have been due to chance.3,48
Why were the results different?
All three trials included first- and second-generation drug-eluting stents, with different proportions in different trials. In ARCTIC-Interruption,46 43% of the patients in the continuation group had a first-generation stent, as did 64% of the patients in the dual antiplatelet group of DES-LATE.47 In the DAPT trial,3 38% of the patients in the longer-duration arm had a first-generation stent, and in 26% of cases it was a paclitaxel-eluting stent.
Only clopidogrel was used as the second antiplatelet agent in DES-LATE, whereas prasugrel was used in 10% of patients in ARCTIC-Interruption and 35% in DAPT.
Yet none of these differences seem to explain the differences in outcome among the studies. ARCTIC-Interruption and DES-LATE did not show any benefit of continued dual antiplatelet therapy beyond 12 months. DAPT showed benefit of extended therapy with prasugrel or with clopidogrel, and with first-generation or second-generation drug-eluting stents. The most likely explanation for the different results was that DAPT was the only trial sufficiently powered to definitively assess the end points, including stent thrombosis.
A balance between ischemic efficacy and bleeding risk is the major consideration with any antithrombotic and antiplatelet therapy. In the three largest trials we discussed (the vascular disease subgroups of CHARISMA,38 PEGASUS,39 and DAPT3), comparison of the prespecified efficacy and safety end points of each trial suggests that dual antiplatelet therapy has a net benefit, particularly given the irreversible nature of ischemic end points.
In CHARISMA,38 60 cardiovascular deaths, myocardial infarctions, or strokes were prevented per year per 10,000 patients treated, at the cost of 28 excess moderate bleeding events.
In PEGASUS,39 42 cardiovascular deaths, myocardial infarctions, or strokes were prevented, at the cost of 79 excess bleeding events requiring transfusion.
In DAPT (a selected population who had tolerated dual antiplatelet therapy for 1 year), 106 deaths, myocardial infarctions, or stroke events were prevented, at the cost of 47 excess moderate bleeding events.3
Indirect comparisons between trials are problematic, given different end point definitions, populations, and background therapies. But their results suggest that less-intensive inhibition with clopidogrel as the second antiplatelet long-term (as in CHARISMA) may provide the best balance of benefit vs risk.
BALANCING RISK AND BENEFIT
The evidence is unequivocal that dual antiplatelet therapy suppresses coronary ischemic complications resulting from thrombosis at sites of spontaneous plaque rupture following acute coronary syndromes or mechanical plaque disruption and foreign body implantation associated with percutaneous coronary intervention.
Three large-scale trials (DAPT,3 PEGASUS,39 and the secondary prevention subgroup of CHARISMA38) showed that the protective effect of dual antiplatelet therapy continues with prolonged therapy in patients who have experienced an acute coronary syndrome event or have received a drug-eluting stent. That benefit seems to be due to the action of these therapies on the culprit vessel (the one that caused the acute coronary syndrome or the site of stenting), as well as nonculprit arteries, emphasizing that dual antiplatelet therapy protects against atherosclerosis progression and future plaque rupture events.
For the durations studied in the longest trials thus far, 30 months (DAPT3) and 36 months (PEGASUS39), event curves continue to diverge, indicating that the advantage of dual antiplatelet therapy may persist for an indefinite period of time. Thus, indefinite therapy with dual antiplatelet agents can be supported, particularly in patients with advanced coronary artery disease or those who have had multiple coronary events.
We believe that the balance of evidence suggests that smaller studies that failed to show a benefit of longer-term therapy were underpowered to do so.
The ischemic protection is associated with the adverse effect of increased bleeding risk. Unfortunately, there has been little success in guiding dual antiplatelet therapy based on ischemic vs bleeding risk, in part because the same factors that predict risk of ischemic complications seem to predict increased susceptibility to bleeding. Nevertheless, indirect comparisons between studies suggest that for longer-term therapy clopidogrel may be superior to ticagrelor or prasugrel: the absolute excess bleeding risk with dual antiplatelet therapy vs aspirin in the CHARISMA secondary prevention subgroup was less than that in PEGASUS, with similar absolute reductions in ischemic events. So while the TRITON-TIMI 3822 and PLATO23 trials support the superiority of prasugrel or ticagrelor over clopidogrel for the first year after acute coronary syndrome, subsequent years of therapy may best be provided with clopidogrel.
Some patients may have identifiable factors that place them at very high risk of bleeding—need for surgical procedures, need for anticoagulation, or occurrence of bleeding complications or excessive “nuisance bleeding.” In those patients, the data suggest that dual antiplatelet therapy could be discontinued after 6 months, or perhaps even 3 months in the highest bleeding risk circumstances after second-generation drug-eluting stent placement.
WOEST49 was an open-label randomized controlled trial that studied the safety of antiplatelet regimens in patients on anticoagulation requiring percutaneous coronary interventions. Patients were randomized to double therapy with anticoagulant and clopidogrel vs triple therapy with additional aspirin following percutaneous coronary intervention. The primary end point was bleeding events within 1 year. Clopidogrel without aspirin was associated with significantly fewer bleeding events compared with triple therapy, with no increase in adverse ischemic events. The strategy tested in the WOEST trial seems reasonable in the specific group of patients who require ongoing anticoagulant therapy after drug-eluting stent placement, recognizing that the trial was somewhat underpowered to make definitive conclusions, particularly in patients at high risk for stent thrombosis.
Based on the results of PEGASUS and the CHARISMA subgroup with established ischemic burden, in which dual antiplatelet therapy was started after an interruption following the index coronary event, it is also reasonable to restart long-term dual antiplatelet therapy in patients who require interruption for short-term indications such as a surgical procedure.
- American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:e78–e140.
- Amsterdam EA, Wenger NK, Brindis RG, et al; ACC/AHA Task Force Members. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 130:e344–e426.
- Mauri L, Kereiakes DJ, Yeh RW, et al; DAPT Study Investigators. Twelve or 30 months of dual antiplatelet therapy after drug-eluting stents. N Engl J Med 2014; 371:2155–2166.
- Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J 2010; 74:597–607.
- Papp J, Kenyeres P, Toth K. Clinical importance of antiplatelet drugs in cardiovascular diseases. Clin Hemorheol Microcirc 2013; 53:81–96.
- Showkathali R, Natarajan A. Antiplatelet and antithrombin strategies in acute coronary syndrome: state-of-the-art review. Curr Cardiol Rev 2012; 8:239–249.
- Angiolillo DJ. The evolution of antiplatelet therapy in the treatment of acute coronary syndromes: from aspirin to the present day. Drugs 2012; 72:2087–2116.
- Claessen BE, Henriques JP, Jaffer FA, Mehran R, Piek JJ, Dangas GD. Stent thrombosis: a clinical perspective. JACC Cardiovasc Interv 2014; 7:1081–1092.
- Schomig A, Neumann FJ, Kastrati A, et al. A randomized comparison of antiplatelet and anticoagulant therapy after the placement of coronary-artery stents. N Engl J Med 1996; 334:1084–1089.
- Leon MB, Baim DS, Popma JJ, et al. A clinical trial comparing three antithrombotic-drug regimens after coronary-artery stenting. Stent Anticoagulation Restenosis Study Investigators. N Engl J Med 1998; 339:1665–1671.
- Joner M, Finn AV, Farb A, et al. Pathology of drug-eluting stents in humans: delayed healing and late thrombotic risk. J Am Coll Cardiol 2006; 48:193–202.
- Nikam N, Steinberg TB, Steinberg DH. Advances in stent technologies and their effect on clinical efficacy and safety. Med Devices (Auckl) 2014; 7:165–178.
- Simard T, Hibbert B, Ramirez FD, Froeschl M, Chen YX, O’Brien ER. The evolution of coronary stents: a brief review. Can J Cardiol 2014; 30:35–45.
- Byrne RA, Joner M, Kastrati A. Stent thrombosis and restenosis: what have we learned and where are we going? The Andreas Gruntzig Lecture ESC 2014. Eur Heart J 2015; 36:3320–3331.
- van Werkum JW, Heestermans AA, Zomer AC, et al. Predictors of coronary stent thrombosis: the Dutch Stent Thrombosis Registry. J Am Coll Cardiol 2009; 53:1399–1409.
- Berger JS. Aspirin, clopidogrel, and ticagrelor in acute coronary syndromes. Am J Cardiol 2013; 112:737–745.
- Franchi F, Angiolillo DJ. Novel antiplatelet agents in acute coronary syndrome. Nat Rev Cardiol 2015; 12:30–47.
- Patrono C, Rocca B. The future of antiplatelet therapy in cardiovascular disease. Annu Rev Med 2010; 61:49–61.
- Park SJ, Kang SM, Park DW. Dual antiplatelet therapy after drug-eluting stents: defining the proper duration. Coron Artery Dis 2014; 25:83–89.
- Steinhubl SR, Berger PB, Mann JT 3rd, et al; CREDO Investigators. Clopidogrel for the reduction of events during observation. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Nusca A, Patti G. Platelet function and inhibition in ischemic heart disease. Curr Cardiol Rep 2012; 14:457–467.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Genereux P, Stone GW, Harrington RA, et al; CHAMPION PHOENIX Investigators. Impact of intraprocedural stent thrombosis during percutaneous coronary intervention: Insights from the CHAMPION PHOENIX Trial (Clinical Trial Comparing Cangrelor to Clopidogrel Standard of Care Therapy in Subjects Who Require Percutaneous Coronary Intervention). J Am Coll Cardiol 2014; 63:619–629.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Brilakis ES, Patel VG, Banerjee S. Medical management after coronary stent implantation: a review. JAMA 2013; 310:189–198.
- Warren J, Baber U, Mehran R. Antiplatelet therapy after drug-eluting stent implantation. J Cardiol 2015; 65:98–104.
- Urban P, Macaya C, Rupprecht HJ, et al. Randomized evaluation of anticoagulation versus antiplatelet therapy after coronary stent implantation in high-risk patients: the Multicenter Aspirin and Ticlopidine Trial After Intracoronary Stenting (MATTIS). Circulation 1998; 98:2126–2132.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: The PCI-CURE study. Lancet 2001; 358:527–533.
- Morais J. Insights from CURE: using clopidogrel on top of standard therapy. Cerebrovasc Dis 2002; 13(suppl 1):17–21.
- Ferguson JJ. Clopidogrel plus aspirin in patients with acute myocardial infarction treated with fibrinolytic therapy—CLARITY-TIMI 28. Future Cardiol 2005; 1:605–610.
- Sabatine MS, Cannon CP, Gibson CM, et al; Clopidogrel as Adjunctive Reperfusion Therapy (CLARITY)-Thrombolysis in Myocardial Infarction (TIMI) 28 Investigators. Effect of clopidogrel pretreatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005; 294:1224–1232.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bonaca MP, Bhatt DL, Cohen M, et al; PEGASUS-TIMI 54 Steering Committee and Investigators. Long-term use of ticagrelor in patients with prior myocardial infarction. N Engl J Med 2015; 372:1791–1800.
- Colombo A, Chieffo A, Frasheri A, et al. Second-generation drug-eluting stent implantation followed by 6- versus 12-month dual antiplatelet therapy: the SECURITY randomized clinical trial. J Am Coll Cardiol 2014; 64:2086–2097.
- Gwon HC, Hahn JY, Park KW, et al. Six-month versus 12-month dual antiplatelet therapy after implantation of drug-eluting stents: the Efficacy of Xience/Promus versus Cypher to Reduce Late Loss After Stenting (EXCELLENT) randomized, multicenter study. Circulation 2012; 125:505–513.
- Feres F, Costa RA, Abizaid A, et al; OPTIMIZE Trial Investigators. Three vs twelve months of dual antiplatelet therapy after zotarolimus-eluting stents: the OPTIMIZE randomized trial. JAMA 2013; 310:2510–2522.
- Kim BK, Hong MK, Shin DH, et al; RESET Investigators. A new strategy for discontinuation of dual antiplatelet therapy: the RESET Trial (REal Safety and Efficacy of 3-month dual antiplatelet Therapy following endeavor zotarolimus-eluting stent implantation). J Am Coll Cardiol 2012; 60:1340–1348.
- El-Hayek G, Messerli F, Bangalore S, et al. Meta-analysis of randomized clinical trials comparing short-term versus long-term dual antiplatelet therapy following drug-eluting stents. Am J Cardiol 2014; 114:236–242.
- Valgimigli M, Campo G, Monti M, et al; Prolonging Dual Antiplatelet Treatment After Grading Stent-Induced Intimal Hyperplasia Study (PRODIGY) Investigators. Short- versus long-term duration of dual-antiplatelet therapy after coronary stenting: a randomized multicenter trial. Circulation 2012; 125:2015–2026.
- Collet JP, Silvain J, Barthelemy O, et al; ARCTIC investigators. Dual-antiplatelet treatment beyond 1 year after drug-eluting stent implantation (ARCTIC-Interruption): a randomised trial. Lancet 2014; 384:1577–1585.
- Lee CW, Ahn JM, Park DW, et al. Optimal duration of dual antiplatelet therapy after drug-eluting stent implantation: a randomized, controlled trial. Circulation 2014; 129:304–312.
- Kwok CS, Bulluck H, Ryding AD, Loke YK. Benefits and harms of extending the duration of dual antiplatelet therapy after percutaneous coronary intervention with drug-eluting stents: a meta-analysis. ScientificWorldJournal 2014; 2014:794078.
- Dewilde WJ, Oirbans T, Verheugt FW, et al; WOEST study investigators. Use of clopidogrel with or without aspirin in patients taking oral anticoagulant therapy and undergoing percutaneous coronary intervention: an open-label, randomised, controlled trial. Lancet 2013; 381:1107–1115.
Percutaneous coronary intervention for acute coronary syndromes has evolved, and so, hand in hand, has antiplatelet therapy. With the advent of clopidogrel and newer agents, several studies demonstrated the benefits of dual antiplatelet therapy in preventing major vascular ischemic complications. The findings culminated in a guideline recommendation for at least 12 months of dual antiplatelet therapy after placement of a drug-eluting stent, when feasible—a class I recommendation (treatment should be given), level of evidence B (limited populations evaluated).1,2 But extending dual antiplatelet therapy beyond 12 months had no strong favorable evidence until the recent Dual Antiplatelet Therapy (DAPT) study3 shed light on this topic.
Here, we review the evidence thus far on the optimal duration of dual antiplatelet therapy in the secondary prevention of coronary artery disease.
PLATELETS IN ACUTE CORONARY SYNDROMES AND STENT THROMBOSIS
Acute coronary syndromes begin with fissuring or ulceration of a vulnerable atherosclerotic plaque, followed by thrombosis and occlusion, mediated by platelet adhesion, activation, and aggregation (Figure 1). Transient occlusion results in unstable angina or non-ST-elevation myocardial infarction, while total occlusion usually results in ST-elevation myocardial infarction.
Platelet aggregation is prominent among the mechanisms leading to stent thrombosis and vaso-occlusive ischemic complications after percutaneous coronary intervention. Thus, antiplatelet agents play a vital role in both primary and secondary prevention of cardiovascular events.4–6
Adhesion, activation, and aggregation
Adhesion. Disruption of the vascular endothelium as a result of vulnerable plaque fissuring or ulceration exposes subendothelial thrombogenic collagen and von Willebrand factor to blood. Collagen engages platelets through their glycoprotein (GP) Ia, IIa, and VI receptors, and von Willebrand factor binds platelets through the GP Ib-IX-V receptor.
Activation. Once platelets adhere to the subendothelium, they undergo a conformational change and become activated. Simultaneous release of various autocrine and paracrine mediators including adenosine diphosphate, serotonin, epinephrine, thromboxane, and various ligand-receptor interactions all contribute to the activation cascade. Adenosine diphosphate binds to the platelet receptor P2Y1, leading to an increase in intracellular calcium, and it binds to P2Y12, leading to a decrease in cyclic adenosine monophosphate, both of which cause GP IIb/IIIa receptor activation. Thromboxane A2 released by platelets by cyclo-oxygenase 1 binds to alpha or beta variant receptors and contributes to GP IIb/IIIa activation through elevation of intracellular calcium levels.
Aggregation and thrombosis. Exposure of tissue factor to plasma following plaque rupture activates the coagulation cascade via the extrinsic pathway, which generates thrombin, a powerful platelet activator that causes thrombus formation via fibrin. Thrombin binds to protease-activated receptors PAR-1 and PAR-4 on platelets, causing an increase in intracellular calcium and a decrease in cyclic adenosine monophosphate with subsequent GP IIb/IIIa activation. GP IIb/IIIa facilitates platelet aggregation by binding to fibrinogen and forming a stable platelet thrombus.
In the early stages of thrombus formation, platelets predominate (“white” thrombi); further organization with fibrin results in older “red” thrombi. The stages of thrombi vary in non-ST-elevation and ST-elevation myocardial infarction and are prognostic markers of death.4–8
PERCUTANEOUS INTERVENTION, RESTENOSIS, AND STENT THROMBOSIS
Percutaneous coronary intervention, the preferred means of revascularization for many patients, is performed emergently in patients with ST-elevation myocardial infarction, urgently in those with acute coronary syndromes without ST elevation, and electively in those with stable ischemic symptoms.
Percutaneous revascularization techniques have evolved from balloon angioplasty to bare-metal stents to drug-eluting stents, but each of these procedures has been associated with a periprocedural and postprocedural risk of thrombosis.
Balloon angioplasty was associated with vascular intimal injury, inciting elastic vascular recoil and smooth muscle cell proliferation leading to restenosis.
Bare-metal stents reduced the restenosis rate by eliminating vascular recoil, although restenosis still occurred within the stent because of neointimal proliferation of vascular smooth muscle cells. This was an important limitation, as both acute and subacute stent thrombosis were refractory to aggressive anticoagulation regimens that were associated with major bleeding complications and longer hospital length of stay. Stenting became mainstream practice only after the ISAR9 and STARS10 trials showed that dual antiplatelet therapy controlled stent thrombosis.
Drug-eluting stents coated with anti-proliferative and anti-inflammatory polymers markedly reduced in-stent restenosis rates by suppressing the initial vascular smooth-muscle proliferative response. However, they were still associated with late and very late stent thrombosis with incomplete endothelialization, even up to 40 months after implantation. Proposed mechanisms include incomplete stent apposition and inflammatory hypersensitivity reactions to the polymer coating. Incomplete stent apposition associated with low-velocity blood flow at the junction of the stent strut and vessel wall, together with delayed endothelialization, promotes platelet adhesion and aggregation, followed by thrombus formation.11
Second-generation drug-eluting stents have thinner struts and more biocompatible polymers and are thought to favor more complete re-endothelialization, reducing the rates of stent thrombosis.8,12,13
Predictors of early stent thrombosis
The Dutch Stent Thrombosis Registry and other studies looked at risk factors for stent thrombosis.14,15
Procedure-related factors included:
- Stent undersizing
- Residual uncovered dissections after angioplasty
- Longer stents
- Low flow after angioplasty (< 3 on the 0–3 Thrombolysis in Myocardial Infarction [TIMI] scale).
Lesion-related factors included:
- Intermediate coronary artery disease both proximal and distal to the culprit lesions
- Bifurcation lesions.
Patient-related factors included:
- Low left ventricular ejection fraction
- Diabetes mellitus
- Peripheral arterial disease Premature discontinuation of clopidogrel.
ANTIPLATELET AGENTS: MECHANISM OF ACTION
Various pathways play synergistic roles in platelet activation and aggregation and thrombus formation, and different antiplatelet agents inhibit these specific pathways, thus complementing each other and having additive effects (Figure 2, Table 1).5,16–21
Aspirin inhibits cyclo-oxygenase 1
Cyclo-oxygenase 1, found in platelets, endothelial cells, and other cells, catalyzes the conversion of arachidonic acid to thromboxane A2. Aspirin irreversibly inhibits cyclo-oxygenase 1 by acetylating its serine residue, preventing formation of thromboxane A2 and preventing platelet activation and aggregation.
P2Y12 ADP receptor antagonists
Clopidogrel and prasugrel are thienopyridine agents that irreversibly inhibit the P2Y12 receptor, thereby preventing binding of adenosine diphosphate and the subsequent platelet activation-aggregation cascade. They are both prodrugs and require conversion by cytochrome P450 enzymes to active metabolites. Prasugrel is 10 times more potent than clopidogrel due to more efficient formation of its active metabolite, and it achieves a comparable effect on platelet inhibition 30 minutes faster than the peak effect of clopidogrel at 6 hours. The overall peak inhibitory effect of prasugrel is twice that of clopidogrel.22
Ticagrelor, a cyclopentyl-triazolo-pyrimidine, directly and reversibly inhibits the P2Y12 ADP receptor. Unlike clopidogrel and prasugrel, it does not need to be converted to an active metabolite, and it noncompetitively inhibits P2Y12 at a site different from the adenosine diphosphate binding site.23 Like prasugrel, ticagrelor inhibits platelet function more rapidly and more completely than clopidogrel.
Cangrelor, an intravenously administered analogue of adenosine triphosphate, reversibly inhibits the P2Y12 receptor. It has undergone phase 3 trials but is not yet approved for clinical use.24
WHY DUAL ANTIPLATELET THERAPY?
Aspirin is good, clopidogrel is better
Aspirin has a well-validated role in both primary and secondary prevention of coronary and noncoronary atherosclerotic vascular disease.
The CAPRIE trial found clopidogrel monotherapy to be superior to aspirin monotherapy in patients with established atherosclerotic vascular disease.25
After stenting, short-term dual therapy is better than short-term warfarin
Thrombotic complications in the early postprocedural period were a major limitation of stenting, and existing anticoagulation regimens were ineffective in preventing them.26,27
The ISAR trial studied the benefit of combined antiplatelet vs anticoagulant therapy after stent placement. Patients randomized to receive combined aspirin plus ticlopidine (an early P2Y12 inhibitor) had significantly lower rates of primary cardiac, hemorrhagic, and vascular events at 30 days.9 Two other trials confirmed this finding.28,29
STARS10 also confirmed the benefit of aspirin and ticlopidine after stenting. Patients were randomly assigned to aspirin alone, aspirin plus warfarin, or aspirin plus ticlopidine after stent placement. The rate of stent thrombosis at 30 days was significantly lower in the dual antiplatelet group than in the other two groups. The dual antiplatelet group had a higher rate of bleeding than the aspirin-alone group, but the rate was similar to that of the aspirin-plus-warfarin group.
Long-term dual antiplatelet therapy is beneficial in several situations
ISAR and STARS were landmark trials that showed stent thrombosis could be reduced by dual antiplatelet therapy for a 30-day period. However, the long-term role of dual antiplatelet therapy was still unknown.
The CURE trial30–32 randomized patients presenting with acute coronary syndromes without ST elevation to receive clopidogrel plus aspirin or placebo plus aspirin for 3 to 12 months. The rate of the primary end point (cardiac death, nonfatal myocardial infarction, or stroke) was significantly lower in the clopidogrel-plus-aspirin group. A similar benefit of dual antiplatelet therapy was seen in the subgroup of patients who underwent percutaneous coronary intervention. Both pretreatment with clopidogrel plus aspirin for a median of 10 days prior to percutaneous intervention and continuing it for a mean of 9 months reduced major adverse cardiovascular events.
The CREDO trial20 found that the combination of clopidogrel and aspirin significantly reduced the incidence of death, myocardial infarction, or stroke at 1 year after percutaneous coronary intervention. A subgroup of patients in this trial who had a longer pretreatment interval with a loading clopidogrel dose showed a benefit at 28 days, which was not as evident with a shorter loading dose interval.
The CLARITY-TIMI 28 trial33,34 showed the advantage of adding clopidogrel to aspirin in patients receiving fibrinolytic therapy for ST-elevation myocardial infarction. Adding clopidogrel both improved the patency of the infarct-related artery and reduced ischemic complications. In patients who subsequently underwent percutaneous coronary intervention and stenting, clopidogrel pretreatment was associated with a significant decrease in ischemic complications before and after the procedure. There was no significant increase in bleeding complications in either group.
COMMIT/CCS 235 also showed the benefit of dual antiplatelet therapy in patients with ST-elevation myocardial infarction. Clopidogrel added to aspirin during the short-term in-hospital or postdischarge treatment period significantly reduced a composite end point of reinfarction, death, or stroke as well as death from any cause.
The CHARISMA trial36–38 aimed to determine if patients who were more stable (ie, no recent acute coronary syndrome event or percutaneous coronary intervention) would benefit. Overall, CHARISMA showed no benefit of adding clopidogrel to aspirin compared with aspirin alone in a broad population of patients with established vascular disease (secondary prevention) or risk factors for vascular disease (primary prevention).
But importantly, though no benefit was seen in the primary prevention group, the large subgroup of patients with established atherosclerotic vascular disease (12,153 of the 15,603 patients in the trial) did benefit from dual antiplatelet therapy.36,37 This subgroup showed an overall reduction in absolute risk of 1.5% (relative risk 0.88, P = .046) over a median follow-up of 27.6 months. This benefit was even more apparent in the 9,478 patients with prior myocardial infarction, stroke, or peripheral artery disease, for whom the relative risk reduction was 17.1% (P = .01) and the reduction in absolute risk 1.5%.38
These results are comparable to the 2% absolute risk reduction in the CURE trial for similar end points over 9 months. In both studies, there was no significant increase in the risk of major bleeding or intracranial bleeding in the clopidogrel-plus-aspirin groups, although minor bleeding was increased by dual antiplatelet therapy.
The rate of severe bleeding, which was the primary safety end point in CHARISMA, was not significantly different in the clopidogrel-plus-aspirin group compared with the placebo-plus-aspirin group (relative risk 1.25, 95% CI 0.97–1.61, P = .09).
Thus, although the CHARISMA findings were negative overall, the positive finding observed in the predominant subgroup of patients with established vascular disease can therefore be considered supportive of the results of the subsequent trials discussed below.
The PEGASUS-TIMI 54 trial39 studied the benefit of adding ticagrelor (60 or 90 mg) to low-dose aspirin in patients with stable coronary artery disease who had had a myocardial infarction 1 to 3 years earlier.
Confirming the results of the CHARISMA subgroup analysis, the incidence of the ischemic primary efficacy end point (a composite of cardiovascular death, myocardial infarction, and stroke) was significantly lower in both groups receiving ticagrelor plus aspirin compared with those receiving placebo plus aspirin. The Kaplan-Meier rate at 3 years for the ticagrelor 90 mg-plus-aspirin group was 7.85% vs 9.04% for the placebo-plus-aspirin group (hazard ratio 0.85, 95% confidence interval [CI] 0.75–0.96, P = .008). The rate for the ticagrelor 60 mg-plus-aspirin group was 7.77% vs 9.04% for the placebo-plus-aspirin group (hazard ratio 0.84, 95% CI 0.74–0.95, P = .004).
The rates of all TIMI major and minor bleeding, as well as bleeding requiring transfusion or discontinuation of the study drug, were significantly higher in both ticagrelor dosing groups than in the placebo group (P < .01 for both groups vs placebo). The rates of fatal bleeding and nonfatal intracranial hemorrhage were not significantly higher. Although there was an overall reduction in ischemic end points with the addition of ticagrelor, there was also a significantly higher incidence of bleeding in this group.
Comment. Thus, with or without percutaneous coronary intervention in acute coronary syndrome as well as in stable coronary artery disease, dual antiplatelet therapy was shown to improve outcomes and decrease ischemic complications compared with aspirin alone. It provided benefit in the setting of acute coronary syndrome (in the CURE trial) and percutaneous coronary intervention (in the CREDO trial) for up to 1 year.
Major questions remained to be addressed:
- Do the results of CREDO, which was performed before the current interventional era and the use of drug-eluting stents, reflect outcomes after current interventional practice?
- Could shorter periods of dual antiplatelet therapy be sufficient, especially with newer stents with less risk of late thrombosis?
- Does the benefit of dual antiplatelet therapy extend beyond the 1-year time period tested in those trials to date?
RECOMMENDATIONS FOR DOSING
The American College of Cardiology Foundation/American Heart Association guidelines for dosing of antiplatelet agents for non-ST-elevation myocardial infarction are summarized in Table 2, and those for ST-elevation myocardial infarction are summarized in Table 3.1,2
WOULD SHORTER THERAPY AFTER STENTING WORK AS WELL?
The American College of Cardiology Foundation/American Heart Association currently recommend dual antiplatelet therapy for at least 12 months after drug-eluting stent placement, with shorter courses appropriate for patients who develop excessive bleeding complications or who are at high risk of bleeding.
Four trials (Table 4) evaluated whether shorter durations of dual antiplatelet therapy would suffice: SECURITY,40 EXCELLENT,41 OPTIMIZE,42 and RESET.43 All of them showed that short-duration therapy was not inferior to standard-duration therapy.44 These studies were comparable in that:
- Patients were randomized at the time of percutaneous coronary intervention or within 24 hours of it.
- Most patients received a second-generation drug-eluting stent, with the following exceptions: in EXCELLENT,41 one-fourth of patients received a Cypher first-generation drug-eluting stent, and in RESET,43 approximately one-fourth of the patients received a sirolimus-eluting stent in the standard-duration group for short lesions. Those patients with longer lesions in the RESET standard-duration group received an everolimus drug-eluting stent.
- The second antiplatelet added to aspirin in all studies was clopidogrel, with the exception of the SECURITY trial, in which fewer than 2% of patients received ticagrelor or prasugrel.40
- All the trials except RESET excluded patients who had had a myocardial infarction within 72 hours, and thus most patients studied had a lower risk profile.
- All of the trials sought to study noninferiority of short- vs standard-duration dual antiplatelet therapy, defined as the occurrence of a primary end point at 1 year (a composite of cardiovascular death, myocardial infarction, stroke, stent thrombosis, target vessel failure or revascularization, or bleeding).
Their low-risk patient populations and infrequent end points rendered these studies underpowered to make definitive conclusions about the relative efficacy of 6-months vs 12-months of dual antiplatelet therapy.
WOULD LONGER THERAPY BE BETTER?
The PRODIGY trial45 assessed durations of dual antiplatelet therapy both shorter and longer than the conventional 1 year, randomizing patients undergoing placement of a bare-metal stent, first-generation drug-eluting stent, or second-generation drug-eluting stent to receive aspirin and clopidogrel for either 6 months or 24 months. The study showed no significant difference in primary outcomes in the short- or long-duration groups.
Other trials that compared the standard 12 months of dual antiplatelet therapy with extended duration beyond 12 months were DAPT,3 ARCTIC-Interruption,46 and DES-LATE.47 The trials were comparable in that:
- All patients were randomized after completing 12 months of dual antiplatelet therapy following drug-eluting stent placement.
- All patients who were included had been free of major cardiac ischemic events or bleeding during the 12 months following stent placement.
- The primary aim of all three studies was to compare primary end points in groups receiving aspirin alone vs extended dual antiplatelet therapy. The primary end point was a composite of death due to a cardiovascular cause, nonfatal myocardial infarction, stroke, or stent thrombosis.
- The principal safety end point was bleeding.
Although the two earlier studies (ARCTIC-Interruption and DES-LATE) did not show any benefit of extended dual antiplatelet therapy compared with the standard 12-month duration, the recent DAPT study did.
The DAPT study
The DAPT study3 was an international, multicenter, placebo-controlled, double-blind randomized trial designed to examine the benefit of dual antiplatelet therapy beyond 1 year in a patient population large enough to provide definitive assessment of benefit and risk.
A total of 9,961 patients who received drug-eluting stents were randomized after 12 months of dual antiplatelet therapy to receive either a thienopyridine (clopidogrel or prasugrel) plus aspirin or placebo plus aspirin. They were followed for an additional 18 months. The coprimary efficacy end points were stent thrombosis and a composite of death, myocardial infarction, or stroke, while the primary safety end point was moderate or severe bleeding. The patients were also observed from months 30 to 33 on aspirin alone after stopping the thienopyridine.
Results. Longer therapy substantially reduced the risks of stent thrombosis (hazard ratio [HR] 0.29, 95% confidence interval [CI] 0.17–0.48) and the composite ischemic end point (HR 0.71, 95% CI 0.59–0.85). Follow-up during the 3-month thienopyridine discontinuation phase starting at 30 months revealed convergence of the ischemic event-rate curves in the two groups, which suggested that continuing dual antiplatelet therapy beyond 30 months might have been beneficial. Myocardial infarction unrelated to stent thrombosis accounted for 55% of the treatment benefit of dual antiplatelet therapy.
The risk of bleeding was higher in the thienopyridine group during the treatment period (2.5% vs 1.6%, P = .001). There was also a higher rate of noncardiovascular mortality in the thienopyridine group, although this difference may have been due to chance.3,48
Why were the results different?
All three trials included first- and second-generation drug-eluting stents, with different proportions in different trials. In ARCTIC-Interruption,46 43% of the patients in the continuation group had a first-generation stent, as did 64% of the patients in the dual antiplatelet group of DES-LATE.47 In the DAPT trial,3 38% of the patients in the longer-duration arm had a first-generation stent, and in 26% of cases it was a paclitaxel-eluting stent.
Only clopidogrel was used as the second antiplatelet agent in DES-LATE, whereas prasugrel was used in 10% of patients in ARCTIC-Interruption and 35% in DAPT.
Yet none of these differences seem to explain the differences in outcome among the studies. ARCTIC-Interruption and DES-LATE did not show any benefit of continued dual antiplatelet therapy beyond 12 months. DAPT showed benefit of extended therapy with prasugrel or with clopidogrel, and with first-generation or second-generation drug-eluting stents. The most likely explanation for the different results was that DAPT was the only trial sufficiently powered to definitively assess the end points, including stent thrombosis.
A balance between ischemic efficacy and bleeding risk is the major consideration with any antithrombotic and antiplatelet therapy. In the three largest trials we discussed (the vascular disease subgroups of CHARISMA,38 PEGASUS,39 and DAPT3), comparison of the prespecified efficacy and safety end points of each trial suggests that dual antiplatelet therapy has a net benefit, particularly given the irreversible nature of ischemic end points.
In CHARISMA,38 60 cardiovascular deaths, myocardial infarctions, or strokes were prevented per year per 10,000 patients treated, at the cost of 28 excess moderate bleeding events.
In PEGASUS,39 42 cardiovascular deaths, myocardial infarctions, or strokes were prevented, at the cost of 79 excess bleeding events requiring transfusion.
In DAPT (a selected population who had tolerated dual antiplatelet therapy for 1 year), 106 deaths, myocardial infarctions, or stroke events were prevented, at the cost of 47 excess moderate bleeding events.3
Indirect comparisons between trials are problematic, given different end point definitions, populations, and background therapies. But their results suggest that less-intensive inhibition with clopidogrel as the second antiplatelet long-term (as in CHARISMA) may provide the best balance of benefit vs risk.
BALANCING RISK AND BENEFIT
The evidence is unequivocal that dual antiplatelet therapy suppresses coronary ischemic complications resulting from thrombosis at sites of spontaneous plaque rupture following acute coronary syndromes or mechanical plaque disruption and foreign body implantation associated with percutaneous coronary intervention.
Three large-scale trials (DAPT,3 PEGASUS,39 and the secondary prevention subgroup of CHARISMA38) showed that the protective effect of dual antiplatelet therapy continues with prolonged therapy in patients who have experienced an acute coronary syndrome event or have received a drug-eluting stent. That benefit seems to be due to the action of these therapies on the culprit vessel (the one that caused the acute coronary syndrome or the site of stenting), as well as nonculprit arteries, emphasizing that dual antiplatelet therapy protects against atherosclerosis progression and future plaque rupture events.
For the durations studied in the longest trials thus far, 30 months (DAPT3) and 36 months (PEGASUS39), event curves continue to diverge, indicating that the advantage of dual antiplatelet therapy may persist for an indefinite period of time. Thus, indefinite therapy with dual antiplatelet agents can be supported, particularly in patients with advanced coronary artery disease or those who have had multiple coronary events.
We believe that the balance of evidence suggests that smaller studies that failed to show a benefit of longer-term therapy were underpowered to do so.
The ischemic protection is associated with the adverse effect of increased bleeding risk. Unfortunately, there has been little success in guiding dual antiplatelet therapy based on ischemic vs bleeding risk, in part because the same factors that predict risk of ischemic complications seem to predict increased susceptibility to bleeding. Nevertheless, indirect comparisons between studies suggest that for longer-term therapy clopidogrel may be superior to ticagrelor or prasugrel: the absolute excess bleeding risk with dual antiplatelet therapy vs aspirin in the CHARISMA secondary prevention subgroup was less than that in PEGASUS, with similar absolute reductions in ischemic events. So while the TRITON-TIMI 3822 and PLATO23 trials support the superiority of prasugrel or ticagrelor over clopidogrel for the first year after acute coronary syndrome, subsequent years of therapy may best be provided with clopidogrel.
Some patients may have identifiable factors that place them at very high risk of bleeding—need for surgical procedures, need for anticoagulation, or occurrence of bleeding complications or excessive “nuisance bleeding.” In those patients, the data suggest that dual antiplatelet therapy could be discontinued after 6 months, or perhaps even 3 months in the highest bleeding risk circumstances after second-generation drug-eluting stent placement.
WOEST49 was an open-label randomized controlled trial that studied the safety of antiplatelet regimens in patients on anticoagulation requiring percutaneous coronary interventions. Patients were randomized to double therapy with anticoagulant and clopidogrel vs triple therapy with additional aspirin following percutaneous coronary intervention. The primary end point was bleeding events within 1 year. Clopidogrel without aspirin was associated with significantly fewer bleeding events compared with triple therapy, with no increase in adverse ischemic events. The strategy tested in the WOEST trial seems reasonable in the specific group of patients who require ongoing anticoagulant therapy after drug-eluting stent placement, recognizing that the trial was somewhat underpowered to make definitive conclusions, particularly in patients at high risk for stent thrombosis.
Based on the results of PEGASUS and the CHARISMA subgroup with established ischemic burden, in which dual antiplatelet therapy was started after an interruption following the index coronary event, it is also reasonable to restart long-term dual antiplatelet therapy in patients who require interruption for short-term indications such as a surgical procedure.
Percutaneous coronary intervention for acute coronary syndromes has evolved, and so, hand in hand, has antiplatelet therapy. With the advent of clopidogrel and newer agents, several studies demonstrated the benefits of dual antiplatelet therapy in preventing major vascular ischemic complications. The findings culminated in a guideline recommendation for at least 12 months of dual antiplatelet therapy after placement of a drug-eluting stent, when feasible—a class I recommendation (treatment should be given), level of evidence B (limited populations evaluated).1,2 But extending dual antiplatelet therapy beyond 12 months had no strong favorable evidence until the recent Dual Antiplatelet Therapy (DAPT) study3 shed light on this topic.
Here, we review the evidence thus far on the optimal duration of dual antiplatelet therapy in the secondary prevention of coronary artery disease.
PLATELETS IN ACUTE CORONARY SYNDROMES AND STENT THROMBOSIS
Acute coronary syndromes begin with fissuring or ulceration of a vulnerable atherosclerotic plaque, followed by thrombosis and occlusion, mediated by platelet adhesion, activation, and aggregation (Figure 1). Transient occlusion results in unstable angina or non-ST-elevation myocardial infarction, while total occlusion usually results in ST-elevation myocardial infarction.
Platelet aggregation is prominent among the mechanisms leading to stent thrombosis and vaso-occlusive ischemic complications after percutaneous coronary intervention. Thus, antiplatelet agents play a vital role in both primary and secondary prevention of cardiovascular events.4–6
Adhesion, activation, and aggregation
Adhesion. Disruption of the vascular endothelium as a result of vulnerable plaque fissuring or ulceration exposes subendothelial thrombogenic collagen and von Willebrand factor to blood. Collagen engages platelets through their glycoprotein (GP) Ia, IIa, and VI receptors, and von Willebrand factor binds platelets through the GP Ib-IX-V receptor.
Activation. Once platelets adhere to the subendothelium, they undergo a conformational change and become activated. Simultaneous release of various autocrine and paracrine mediators including adenosine diphosphate, serotonin, epinephrine, thromboxane, and various ligand-receptor interactions all contribute to the activation cascade. Adenosine diphosphate binds to the platelet receptor P2Y1, leading to an increase in intracellular calcium, and it binds to P2Y12, leading to a decrease in cyclic adenosine monophosphate, both of which cause GP IIb/IIIa receptor activation. Thromboxane A2 released by platelets by cyclo-oxygenase 1 binds to alpha or beta variant receptors and contributes to GP IIb/IIIa activation through elevation of intracellular calcium levels.
Aggregation and thrombosis. Exposure of tissue factor to plasma following plaque rupture activates the coagulation cascade via the extrinsic pathway, which generates thrombin, a powerful platelet activator that causes thrombus formation via fibrin. Thrombin binds to protease-activated receptors PAR-1 and PAR-4 on platelets, causing an increase in intracellular calcium and a decrease in cyclic adenosine monophosphate with subsequent GP IIb/IIIa activation. GP IIb/IIIa facilitates platelet aggregation by binding to fibrinogen and forming a stable platelet thrombus.
In the early stages of thrombus formation, platelets predominate (“white” thrombi); further organization with fibrin results in older “red” thrombi. The stages of thrombi vary in non-ST-elevation and ST-elevation myocardial infarction and are prognostic markers of death.4–8
PERCUTANEOUS INTERVENTION, RESTENOSIS, AND STENT THROMBOSIS
Percutaneous coronary intervention, the preferred means of revascularization for many patients, is performed emergently in patients with ST-elevation myocardial infarction, urgently in those with acute coronary syndromes without ST elevation, and electively in those with stable ischemic symptoms.
Percutaneous revascularization techniques have evolved from balloon angioplasty to bare-metal stents to drug-eluting stents, but each of these procedures has been associated with a periprocedural and postprocedural risk of thrombosis.
Balloon angioplasty was associated with vascular intimal injury, inciting elastic vascular recoil and smooth muscle cell proliferation leading to restenosis.
Bare-metal stents reduced the restenosis rate by eliminating vascular recoil, although restenosis still occurred within the stent because of neointimal proliferation of vascular smooth muscle cells. This was an important limitation, as both acute and subacute stent thrombosis were refractory to aggressive anticoagulation regimens that were associated with major bleeding complications and longer hospital length of stay. Stenting became mainstream practice only after the ISAR9 and STARS10 trials showed that dual antiplatelet therapy controlled stent thrombosis.
Drug-eluting stents coated with anti-proliferative and anti-inflammatory polymers markedly reduced in-stent restenosis rates by suppressing the initial vascular smooth-muscle proliferative response. However, they were still associated with late and very late stent thrombosis with incomplete endothelialization, even up to 40 months after implantation. Proposed mechanisms include incomplete stent apposition and inflammatory hypersensitivity reactions to the polymer coating. Incomplete stent apposition associated with low-velocity blood flow at the junction of the stent strut and vessel wall, together with delayed endothelialization, promotes platelet adhesion and aggregation, followed by thrombus formation.11
Second-generation drug-eluting stents have thinner struts and more biocompatible polymers and are thought to favor more complete re-endothelialization, reducing the rates of stent thrombosis.8,12,13
Predictors of early stent thrombosis
The Dutch Stent Thrombosis Registry and other studies looked at risk factors for stent thrombosis.14,15
Procedure-related factors included:
- Stent undersizing
- Residual uncovered dissections after angioplasty
- Longer stents
- Low flow after angioplasty (< 3 on the 0–3 Thrombolysis in Myocardial Infarction [TIMI] scale).
Lesion-related factors included:
- Intermediate coronary artery disease both proximal and distal to the culprit lesions
- Bifurcation lesions.
Patient-related factors included:
- Low left ventricular ejection fraction
- Diabetes mellitus
- Peripheral arterial disease Premature discontinuation of clopidogrel.
ANTIPLATELET AGENTS: MECHANISM OF ACTION
Various pathways play synergistic roles in platelet activation and aggregation and thrombus formation, and different antiplatelet agents inhibit these specific pathways, thus complementing each other and having additive effects (Figure 2, Table 1).5,16–21
Aspirin inhibits cyclo-oxygenase 1
Cyclo-oxygenase 1, found in platelets, endothelial cells, and other cells, catalyzes the conversion of arachidonic acid to thromboxane A2. Aspirin irreversibly inhibits cyclo-oxygenase 1 by acetylating its serine residue, preventing formation of thromboxane A2 and preventing platelet activation and aggregation.
P2Y12 ADP receptor antagonists
Clopidogrel and prasugrel are thienopyridine agents that irreversibly inhibit the P2Y12 receptor, thereby preventing binding of adenosine diphosphate and the subsequent platelet activation-aggregation cascade. They are both prodrugs and require conversion by cytochrome P450 enzymes to active metabolites. Prasugrel is 10 times more potent than clopidogrel due to more efficient formation of its active metabolite, and it achieves a comparable effect on platelet inhibition 30 minutes faster than the peak effect of clopidogrel at 6 hours. The overall peak inhibitory effect of prasugrel is twice that of clopidogrel.22
Ticagrelor, a cyclopentyl-triazolo-pyrimidine, directly and reversibly inhibits the P2Y12 ADP receptor. Unlike clopidogrel and prasugrel, it does not need to be converted to an active metabolite, and it noncompetitively inhibits P2Y12 at a site different from the adenosine diphosphate binding site.23 Like prasugrel, ticagrelor inhibits platelet function more rapidly and more completely than clopidogrel.
Cangrelor, an intravenously administered analogue of adenosine triphosphate, reversibly inhibits the P2Y12 receptor. It has undergone phase 3 trials but is not yet approved for clinical use.24
WHY DUAL ANTIPLATELET THERAPY?
Aspirin is good, clopidogrel is better
Aspirin has a well-validated role in both primary and secondary prevention of coronary and noncoronary atherosclerotic vascular disease.
The CAPRIE trial found clopidogrel monotherapy to be superior to aspirin monotherapy in patients with established atherosclerotic vascular disease.25
After stenting, short-term dual therapy is better than short-term warfarin
Thrombotic complications in the early postprocedural period were a major limitation of stenting, and existing anticoagulation regimens were ineffective in preventing them.26,27
The ISAR trial studied the benefit of combined antiplatelet vs anticoagulant therapy after stent placement. Patients randomized to receive combined aspirin plus ticlopidine (an early P2Y12 inhibitor) had significantly lower rates of primary cardiac, hemorrhagic, and vascular events at 30 days.9 Two other trials confirmed this finding.28,29
STARS10 also confirmed the benefit of aspirin and ticlopidine after stenting. Patients were randomly assigned to aspirin alone, aspirin plus warfarin, or aspirin plus ticlopidine after stent placement. The rate of stent thrombosis at 30 days was significantly lower in the dual antiplatelet group than in the other two groups. The dual antiplatelet group had a higher rate of bleeding than the aspirin-alone group, but the rate was similar to that of the aspirin-plus-warfarin group.
Long-term dual antiplatelet therapy is beneficial in several situations
ISAR and STARS were landmark trials that showed stent thrombosis could be reduced by dual antiplatelet therapy for a 30-day period. However, the long-term role of dual antiplatelet therapy was still unknown.
The CURE trial30–32 randomized patients presenting with acute coronary syndromes without ST elevation to receive clopidogrel plus aspirin or placebo plus aspirin for 3 to 12 months. The rate of the primary end point (cardiac death, nonfatal myocardial infarction, or stroke) was significantly lower in the clopidogrel-plus-aspirin group. A similar benefit of dual antiplatelet therapy was seen in the subgroup of patients who underwent percutaneous coronary intervention. Both pretreatment with clopidogrel plus aspirin for a median of 10 days prior to percutaneous intervention and continuing it for a mean of 9 months reduced major adverse cardiovascular events.
The CREDO trial20 found that the combination of clopidogrel and aspirin significantly reduced the incidence of death, myocardial infarction, or stroke at 1 year after percutaneous coronary intervention. A subgroup of patients in this trial who had a longer pretreatment interval with a loading clopidogrel dose showed a benefit at 28 days, which was not as evident with a shorter loading dose interval.
The CLARITY-TIMI 28 trial33,34 showed the advantage of adding clopidogrel to aspirin in patients receiving fibrinolytic therapy for ST-elevation myocardial infarction. Adding clopidogrel both improved the patency of the infarct-related artery and reduced ischemic complications. In patients who subsequently underwent percutaneous coronary intervention and stenting, clopidogrel pretreatment was associated with a significant decrease in ischemic complications before and after the procedure. There was no significant increase in bleeding complications in either group.
COMMIT/CCS 235 also showed the benefit of dual antiplatelet therapy in patients with ST-elevation myocardial infarction. Clopidogrel added to aspirin during the short-term in-hospital or postdischarge treatment period significantly reduced a composite end point of reinfarction, death, or stroke as well as death from any cause.
The CHARISMA trial36–38 aimed to determine if patients who were more stable (ie, no recent acute coronary syndrome event or percutaneous coronary intervention) would benefit. Overall, CHARISMA showed no benefit of adding clopidogrel to aspirin compared with aspirin alone in a broad population of patients with established vascular disease (secondary prevention) or risk factors for vascular disease (primary prevention).
But importantly, though no benefit was seen in the primary prevention group, the large subgroup of patients with established atherosclerotic vascular disease (12,153 of the 15,603 patients in the trial) did benefit from dual antiplatelet therapy.36,37 This subgroup showed an overall reduction in absolute risk of 1.5% (relative risk 0.88, P = .046) over a median follow-up of 27.6 months. This benefit was even more apparent in the 9,478 patients with prior myocardial infarction, stroke, or peripheral artery disease, for whom the relative risk reduction was 17.1% (P = .01) and the reduction in absolute risk 1.5%.38
These results are comparable to the 2% absolute risk reduction in the CURE trial for similar end points over 9 months. In both studies, there was no significant increase in the risk of major bleeding or intracranial bleeding in the clopidogrel-plus-aspirin groups, although minor bleeding was increased by dual antiplatelet therapy.
The rate of severe bleeding, which was the primary safety end point in CHARISMA, was not significantly different in the clopidogrel-plus-aspirin group compared with the placebo-plus-aspirin group (relative risk 1.25, 95% CI 0.97–1.61, P = .09).
Thus, although the CHARISMA findings were negative overall, the positive finding observed in the predominant subgroup of patients with established vascular disease can therefore be considered supportive of the results of the subsequent trials discussed below.
The PEGASUS-TIMI 54 trial39 studied the benefit of adding ticagrelor (60 or 90 mg) to low-dose aspirin in patients with stable coronary artery disease who had had a myocardial infarction 1 to 3 years earlier.
Confirming the results of the CHARISMA subgroup analysis, the incidence of the ischemic primary efficacy end point (a composite of cardiovascular death, myocardial infarction, and stroke) was significantly lower in both groups receiving ticagrelor plus aspirin compared with those receiving placebo plus aspirin. The Kaplan-Meier rate at 3 years for the ticagrelor 90 mg-plus-aspirin group was 7.85% vs 9.04% for the placebo-plus-aspirin group (hazard ratio 0.85, 95% confidence interval [CI] 0.75–0.96, P = .008). The rate for the ticagrelor 60 mg-plus-aspirin group was 7.77% vs 9.04% for the placebo-plus-aspirin group (hazard ratio 0.84, 95% CI 0.74–0.95, P = .004).
The rates of all TIMI major and minor bleeding, as well as bleeding requiring transfusion or discontinuation of the study drug, were significantly higher in both ticagrelor dosing groups than in the placebo group (P < .01 for both groups vs placebo). The rates of fatal bleeding and nonfatal intracranial hemorrhage were not significantly higher. Although there was an overall reduction in ischemic end points with the addition of ticagrelor, there was also a significantly higher incidence of bleeding in this group.
Comment. Thus, with or without percutaneous coronary intervention in acute coronary syndrome as well as in stable coronary artery disease, dual antiplatelet therapy was shown to improve outcomes and decrease ischemic complications compared with aspirin alone. It provided benefit in the setting of acute coronary syndrome (in the CURE trial) and percutaneous coronary intervention (in the CREDO trial) for up to 1 year.
Major questions remained to be addressed:
- Do the results of CREDO, which was performed before the current interventional era and the use of drug-eluting stents, reflect outcomes after current interventional practice?
- Could shorter periods of dual antiplatelet therapy be sufficient, especially with newer stents with less risk of late thrombosis?
- Does the benefit of dual antiplatelet therapy extend beyond the 1-year time period tested in those trials to date?
RECOMMENDATIONS FOR DOSING
The American College of Cardiology Foundation/American Heart Association guidelines for dosing of antiplatelet agents for non-ST-elevation myocardial infarction are summarized in Table 2, and those for ST-elevation myocardial infarction are summarized in Table 3.1,2
WOULD SHORTER THERAPY AFTER STENTING WORK AS WELL?
The American College of Cardiology Foundation/American Heart Association currently recommend dual antiplatelet therapy for at least 12 months after drug-eluting stent placement, with shorter courses appropriate for patients who develop excessive bleeding complications or who are at high risk of bleeding.
Four trials (Table 4) evaluated whether shorter durations of dual antiplatelet therapy would suffice: SECURITY,40 EXCELLENT,41 OPTIMIZE,42 and RESET.43 All of them showed that short-duration therapy was not inferior to standard-duration therapy.44 These studies were comparable in that:
- Patients were randomized at the time of percutaneous coronary intervention or within 24 hours of it.
- Most patients received a second-generation drug-eluting stent, with the following exceptions: in EXCELLENT,41 one-fourth of patients received a Cypher first-generation drug-eluting stent, and in RESET,43 approximately one-fourth of the patients received a sirolimus-eluting stent in the standard-duration group for short lesions. Those patients with longer lesions in the RESET standard-duration group received an everolimus drug-eluting stent.
- The second antiplatelet added to aspirin in all studies was clopidogrel, with the exception of the SECURITY trial, in which fewer than 2% of patients received ticagrelor or prasugrel.40
- All the trials except RESET excluded patients who had had a myocardial infarction within 72 hours, and thus most patients studied had a lower risk profile.
- All of the trials sought to study noninferiority of short- vs standard-duration dual antiplatelet therapy, defined as the occurrence of a primary end point at 1 year (a composite of cardiovascular death, myocardial infarction, stroke, stent thrombosis, target vessel failure or revascularization, or bleeding).
Their low-risk patient populations and infrequent end points rendered these studies underpowered to make definitive conclusions about the relative efficacy of 6-months vs 12-months of dual antiplatelet therapy.
WOULD LONGER THERAPY BE BETTER?
The PRODIGY trial45 assessed durations of dual antiplatelet therapy both shorter and longer than the conventional 1 year, randomizing patients undergoing placement of a bare-metal stent, first-generation drug-eluting stent, or second-generation drug-eluting stent to receive aspirin and clopidogrel for either 6 months or 24 months. The study showed no significant difference in primary outcomes in the short- or long-duration groups.
Other trials that compared the standard 12 months of dual antiplatelet therapy with extended duration beyond 12 months were DAPT,3 ARCTIC-Interruption,46 and DES-LATE.47 The trials were comparable in that:
- All patients were randomized after completing 12 months of dual antiplatelet therapy following drug-eluting stent placement.
- All patients who were included had been free of major cardiac ischemic events or bleeding during the 12 months following stent placement.
- The primary aim of all three studies was to compare primary end points in groups receiving aspirin alone vs extended dual antiplatelet therapy. The primary end point was a composite of death due to a cardiovascular cause, nonfatal myocardial infarction, stroke, or stent thrombosis.
- The principal safety end point was bleeding.
Although the two earlier studies (ARCTIC-Interruption and DES-LATE) did not show any benefit of extended dual antiplatelet therapy compared with the standard 12-month duration, the recent DAPT study did.
The DAPT study
The DAPT study3 was an international, multicenter, placebo-controlled, double-blind randomized trial designed to examine the benefit of dual antiplatelet therapy beyond 1 year in a patient population large enough to provide definitive assessment of benefit and risk.
A total of 9,961 patients who received drug-eluting stents were randomized after 12 months of dual antiplatelet therapy to receive either a thienopyridine (clopidogrel or prasugrel) plus aspirin or placebo plus aspirin. They were followed for an additional 18 months. The coprimary efficacy end points were stent thrombosis and a composite of death, myocardial infarction, or stroke, while the primary safety end point was moderate or severe bleeding. The patients were also observed from months 30 to 33 on aspirin alone after stopping the thienopyridine.
Results. Longer therapy substantially reduced the risks of stent thrombosis (hazard ratio [HR] 0.29, 95% confidence interval [CI] 0.17–0.48) and the composite ischemic end point (HR 0.71, 95% CI 0.59–0.85). Follow-up during the 3-month thienopyridine discontinuation phase starting at 30 months revealed convergence of the ischemic event-rate curves in the two groups, which suggested that continuing dual antiplatelet therapy beyond 30 months might have been beneficial. Myocardial infarction unrelated to stent thrombosis accounted for 55% of the treatment benefit of dual antiplatelet therapy.
The risk of bleeding was higher in the thienopyridine group during the treatment period (2.5% vs 1.6%, P = .001). There was also a higher rate of noncardiovascular mortality in the thienopyridine group, although this difference may have been due to chance.3,48
Why were the results different?
All three trials included first- and second-generation drug-eluting stents, with different proportions in different trials. In ARCTIC-Interruption,46 43% of the patients in the continuation group had a first-generation stent, as did 64% of the patients in the dual antiplatelet group of DES-LATE.47 In the DAPT trial,3 38% of the patients in the longer-duration arm had a first-generation stent, and in 26% of cases it was a paclitaxel-eluting stent.
Only clopidogrel was used as the second antiplatelet agent in DES-LATE, whereas prasugrel was used in 10% of patients in ARCTIC-Interruption and 35% in DAPT.
Yet none of these differences seem to explain the differences in outcome among the studies. ARCTIC-Interruption and DES-LATE did not show any benefit of continued dual antiplatelet therapy beyond 12 months. DAPT showed benefit of extended therapy with prasugrel or with clopidogrel, and with first-generation or second-generation drug-eluting stents. The most likely explanation for the different results was that DAPT was the only trial sufficiently powered to definitively assess the end points, including stent thrombosis.
A balance between ischemic efficacy and bleeding risk is the major consideration with any antithrombotic and antiplatelet therapy. In the three largest trials we discussed (the vascular disease subgroups of CHARISMA,38 PEGASUS,39 and DAPT3), comparison of the prespecified efficacy and safety end points of each trial suggests that dual antiplatelet therapy has a net benefit, particularly given the irreversible nature of ischemic end points.
In CHARISMA,38 60 cardiovascular deaths, myocardial infarctions, or strokes were prevented per year per 10,000 patients treated, at the cost of 28 excess moderate bleeding events.
In PEGASUS,39 42 cardiovascular deaths, myocardial infarctions, or strokes were prevented, at the cost of 79 excess bleeding events requiring transfusion.
In DAPT (a selected population who had tolerated dual antiplatelet therapy for 1 year), 106 deaths, myocardial infarctions, or stroke events were prevented, at the cost of 47 excess moderate bleeding events.3
Indirect comparisons between trials are problematic, given different end point definitions, populations, and background therapies. But their results suggest that less-intensive inhibition with clopidogrel as the second antiplatelet long-term (as in CHARISMA) may provide the best balance of benefit vs risk.
BALANCING RISK AND BENEFIT
The evidence is unequivocal that dual antiplatelet therapy suppresses coronary ischemic complications resulting from thrombosis at sites of spontaneous plaque rupture following acute coronary syndromes or mechanical plaque disruption and foreign body implantation associated with percutaneous coronary intervention.
Three large-scale trials (DAPT,3 PEGASUS,39 and the secondary prevention subgroup of CHARISMA38) showed that the protective effect of dual antiplatelet therapy continues with prolonged therapy in patients who have experienced an acute coronary syndrome event or have received a drug-eluting stent. That benefit seems to be due to the action of these therapies on the culprit vessel (the one that caused the acute coronary syndrome or the site of stenting), as well as nonculprit arteries, emphasizing that dual antiplatelet therapy protects against atherosclerosis progression and future plaque rupture events.
For the durations studied in the longest trials thus far, 30 months (DAPT3) and 36 months (PEGASUS39), event curves continue to diverge, indicating that the advantage of dual antiplatelet therapy may persist for an indefinite period of time. Thus, indefinite therapy with dual antiplatelet agents can be supported, particularly in patients with advanced coronary artery disease or those who have had multiple coronary events.
We believe that the balance of evidence suggests that smaller studies that failed to show a benefit of longer-term therapy were underpowered to do so.
The ischemic protection is associated with the adverse effect of increased bleeding risk. Unfortunately, there has been little success in guiding dual antiplatelet therapy based on ischemic vs bleeding risk, in part because the same factors that predict risk of ischemic complications seem to predict increased susceptibility to bleeding. Nevertheless, indirect comparisons between studies suggest that for longer-term therapy clopidogrel may be superior to ticagrelor or prasugrel: the absolute excess bleeding risk with dual antiplatelet therapy vs aspirin in the CHARISMA secondary prevention subgroup was less than that in PEGASUS, with similar absolute reductions in ischemic events. So while the TRITON-TIMI 3822 and PLATO23 trials support the superiority of prasugrel or ticagrelor over clopidogrel for the first year after acute coronary syndrome, subsequent years of therapy may best be provided with clopidogrel.
Some patients may have identifiable factors that place them at very high risk of bleeding—need for surgical procedures, need for anticoagulation, or occurrence of bleeding complications or excessive “nuisance bleeding.” In those patients, the data suggest that dual antiplatelet therapy could be discontinued after 6 months, or perhaps even 3 months in the highest bleeding risk circumstances after second-generation drug-eluting stent placement.
WOEST49 was an open-label randomized controlled trial that studied the safety of antiplatelet regimens in patients on anticoagulation requiring percutaneous coronary interventions. Patients were randomized to double therapy with anticoagulant and clopidogrel vs triple therapy with additional aspirin following percutaneous coronary intervention. The primary end point was bleeding events within 1 year. Clopidogrel without aspirin was associated with significantly fewer bleeding events compared with triple therapy, with no increase in adverse ischemic events. The strategy tested in the WOEST trial seems reasonable in the specific group of patients who require ongoing anticoagulant therapy after drug-eluting stent placement, recognizing that the trial was somewhat underpowered to make definitive conclusions, particularly in patients at high risk for stent thrombosis.
Based on the results of PEGASUS and the CHARISMA subgroup with established ischemic burden, in which dual antiplatelet therapy was started after an interruption following the index coronary event, it is also reasonable to restart long-term dual antiplatelet therapy in patients who require interruption for short-term indications such as a surgical procedure.
- American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:e78–e140.
- Amsterdam EA, Wenger NK, Brindis RG, et al; ACC/AHA Task Force Members. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 130:e344–e426.
- Mauri L, Kereiakes DJ, Yeh RW, et al; DAPT Study Investigators. Twelve or 30 months of dual antiplatelet therapy after drug-eluting stents. N Engl J Med 2014; 371:2155–2166.
- Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J 2010; 74:597–607.
- Papp J, Kenyeres P, Toth K. Clinical importance of antiplatelet drugs in cardiovascular diseases. Clin Hemorheol Microcirc 2013; 53:81–96.
- Showkathali R, Natarajan A. Antiplatelet and antithrombin strategies in acute coronary syndrome: state-of-the-art review. Curr Cardiol Rev 2012; 8:239–249.
- Angiolillo DJ. The evolution of antiplatelet therapy in the treatment of acute coronary syndromes: from aspirin to the present day. Drugs 2012; 72:2087–2116.
- Claessen BE, Henriques JP, Jaffer FA, Mehran R, Piek JJ, Dangas GD. Stent thrombosis: a clinical perspective. JACC Cardiovasc Interv 2014; 7:1081–1092.
- Schomig A, Neumann FJ, Kastrati A, et al. A randomized comparison of antiplatelet and anticoagulant therapy after the placement of coronary-artery stents. N Engl J Med 1996; 334:1084–1089.
- Leon MB, Baim DS, Popma JJ, et al. A clinical trial comparing three antithrombotic-drug regimens after coronary-artery stenting. Stent Anticoagulation Restenosis Study Investigators. N Engl J Med 1998; 339:1665–1671.
- Joner M, Finn AV, Farb A, et al. Pathology of drug-eluting stents in humans: delayed healing and late thrombotic risk. J Am Coll Cardiol 2006; 48:193–202.
- Nikam N, Steinberg TB, Steinberg DH. Advances in stent technologies and their effect on clinical efficacy and safety. Med Devices (Auckl) 2014; 7:165–178.
- Simard T, Hibbert B, Ramirez FD, Froeschl M, Chen YX, O’Brien ER. The evolution of coronary stents: a brief review. Can J Cardiol 2014; 30:35–45.
- Byrne RA, Joner M, Kastrati A. Stent thrombosis and restenosis: what have we learned and where are we going? The Andreas Gruntzig Lecture ESC 2014. Eur Heart J 2015; 36:3320–3331.
- van Werkum JW, Heestermans AA, Zomer AC, et al. Predictors of coronary stent thrombosis: the Dutch Stent Thrombosis Registry. J Am Coll Cardiol 2009; 53:1399–1409.
- Berger JS. Aspirin, clopidogrel, and ticagrelor in acute coronary syndromes. Am J Cardiol 2013; 112:737–745.
- Franchi F, Angiolillo DJ. Novel antiplatelet agents in acute coronary syndrome. Nat Rev Cardiol 2015; 12:30–47.
- Patrono C, Rocca B. The future of antiplatelet therapy in cardiovascular disease. Annu Rev Med 2010; 61:49–61.
- Park SJ, Kang SM, Park DW. Dual antiplatelet therapy after drug-eluting stents: defining the proper duration. Coron Artery Dis 2014; 25:83–89.
- Steinhubl SR, Berger PB, Mann JT 3rd, et al; CREDO Investigators. Clopidogrel for the reduction of events during observation. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Nusca A, Patti G. Platelet function and inhibition in ischemic heart disease. Curr Cardiol Rep 2012; 14:457–467.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Genereux P, Stone GW, Harrington RA, et al; CHAMPION PHOENIX Investigators. Impact of intraprocedural stent thrombosis during percutaneous coronary intervention: Insights from the CHAMPION PHOENIX Trial (Clinical Trial Comparing Cangrelor to Clopidogrel Standard of Care Therapy in Subjects Who Require Percutaneous Coronary Intervention). J Am Coll Cardiol 2014; 63:619–629.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Brilakis ES, Patel VG, Banerjee S. Medical management after coronary stent implantation: a review. JAMA 2013; 310:189–198.
- Warren J, Baber U, Mehran R. Antiplatelet therapy after drug-eluting stent implantation. J Cardiol 2015; 65:98–104.
- Urban P, Macaya C, Rupprecht HJ, et al. Randomized evaluation of anticoagulation versus antiplatelet therapy after coronary stent implantation in high-risk patients: the Multicenter Aspirin and Ticlopidine Trial After Intracoronary Stenting (MATTIS). Circulation 1998; 98:2126–2132.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: The PCI-CURE study. Lancet 2001; 358:527–533.
- Morais J. Insights from CURE: using clopidogrel on top of standard therapy. Cerebrovasc Dis 2002; 13(suppl 1):17–21.
- Ferguson JJ. Clopidogrel plus aspirin in patients with acute myocardial infarction treated with fibrinolytic therapy—CLARITY-TIMI 28. Future Cardiol 2005; 1:605–610.
- Sabatine MS, Cannon CP, Gibson CM, et al; Clopidogrel as Adjunctive Reperfusion Therapy (CLARITY)-Thrombolysis in Myocardial Infarction (TIMI) 28 Investigators. Effect of clopidogrel pretreatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005; 294:1224–1232.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bonaca MP, Bhatt DL, Cohen M, et al; PEGASUS-TIMI 54 Steering Committee and Investigators. Long-term use of ticagrelor in patients with prior myocardial infarction. N Engl J Med 2015; 372:1791–1800.
- Colombo A, Chieffo A, Frasheri A, et al. Second-generation drug-eluting stent implantation followed by 6- versus 12-month dual antiplatelet therapy: the SECURITY randomized clinical trial. J Am Coll Cardiol 2014; 64:2086–2097.
- Gwon HC, Hahn JY, Park KW, et al. Six-month versus 12-month dual antiplatelet therapy after implantation of drug-eluting stents: the Efficacy of Xience/Promus versus Cypher to Reduce Late Loss After Stenting (EXCELLENT) randomized, multicenter study. Circulation 2012; 125:505–513.
- Feres F, Costa RA, Abizaid A, et al; OPTIMIZE Trial Investigators. Three vs twelve months of dual antiplatelet therapy after zotarolimus-eluting stents: the OPTIMIZE randomized trial. JAMA 2013; 310:2510–2522.
- Kim BK, Hong MK, Shin DH, et al; RESET Investigators. A new strategy for discontinuation of dual antiplatelet therapy: the RESET Trial (REal Safety and Efficacy of 3-month dual antiplatelet Therapy following endeavor zotarolimus-eluting stent implantation). J Am Coll Cardiol 2012; 60:1340–1348.
- El-Hayek G, Messerli F, Bangalore S, et al. Meta-analysis of randomized clinical trials comparing short-term versus long-term dual antiplatelet therapy following drug-eluting stents. Am J Cardiol 2014; 114:236–242.
- Valgimigli M, Campo G, Monti M, et al; Prolonging Dual Antiplatelet Treatment After Grading Stent-Induced Intimal Hyperplasia Study (PRODIGY) Investigators. Short- versus long-term duration of dual-antiplatelet therapy after coronary stenting: a randomized multicenter trial. Circulation 2012; 125:2015–2026.
- Collet JP, Silvain J, Barthelemy O, et al; ARCTIC investigators. Dual-antiplatelet treatment beyond 1 year after drug-eluting stent implantation (ARCTIC-Interruption): a randomised trial. Lancet 2014; 384:1577–1585.
- Lee CW, Ahn JM, Park DW, et al. Optimal duration of dual antiplatelet therapy after drug-eluting stent implantation: a randomized, controlled trial. Circulation 2014; 129:304–312.
- Kwok CS, Bulluck H, Ryding AD, Loke YK. Benefits and harms of extending the duration of dual antiplatelet therapy after percutaneous coronary intervention with drug-eluting stents: a meta-analysis. ScientificWorldJournal 2014; 2014:794078.
- Dewilde WJ, Oirbans T, Verheugt FW, et al; WOEST study investigators. Use of clopidogrel with or without aspirin in patients taking oral anticoagulant therapy and undergoing percutaneous coronary intervention: an open-label, randomised, controlled trial. Lancet 2013; 381:1107–1115.
- American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:e78–e140.
- Amsterdam EA, Wenger NK, Brindis RG, et al; ACC/AHA Task Force Members. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 130:e344–e426.
- Mauri L, Kereiakes DJ, Yeh RW, et al; DAPT Study Investigators. Twelve or 30 months of dual antiplatelet therapy after drug-eluting stents. N Engl J Med 2014; 371:2155–2166.
- Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J 2010; 74:597–607.
- Papp J, Kenyeres P, Toth K. Clinical importance of antiplatelet drugs in cardiovascular diseases. Clin Hemorheol Microcirc 2013; 53:81–96.
- Showkathali R, Natarajan A. Antiplatelet and antithrombin strategies in acute coronary syndrome: state-of-the-art review. Curr Cardiol Rev 2012; 8:239–249.
- Angiolillo DJ. The evolution of antiplatelet therapy in the treatment of acute coronary syndromes: from aspirin to the present day. Drugs 2012; 72:2087–2116.
- Claessen BE, Henriques JP, Jaffer FA, Mehran R, Piek JJ, Dangas GD. Stent thrombosis: a clinical perspective. JACC Cardiovasc Interv 2014; 7:1081–1092.
- Schomig A, Neumann FJ, Kastrati A, et al. A randomized comparison of antiplatelet and anticoagulant therapy after the placement of coronary-artery stents. N Engl J Med 1996; 334:1084–1089.
- Leon MB, Baim DS, Popma JJ, et al. A clinical trial comparing three antithrombotic-drug regimens after coronary-artery stenting. Stent Anticoagulation Restenosis Study Investigators. N Engl J Med 1998; 339:1665–1671.
- Joner M, Finn AV, Farb A, et al. Pathology of drug-eluting stents in humans: delayed healing and late thrombotic risk. J Am Coll Cardiol 2006; 48:193–202.
- Nikam N, Steinberg TB, Steinberg DH. Advances in stent technologies and their effect on clinical efficacy and safety. Med Devices (Auckl) 2014; 7:165–178.
- Simard T, Hibbert B, Ramirez FD, Froeschl M, Chen YX, O’Brien ER. The evolution of coronary stents: a brief review. Can J Cardiol 2014; 30:35–45.
- Byrne RA, Joner M, Kastrati A. Stent thrombosis and restenosis: what have we learned and where are we going? The Andreas Gruntzig Lecture ESC 2014. Eur Heart J 2015; 36:3320–3331.
- van Werkum JW, Heestermans AA, Zomer AC, et al. Predictors of coronary stent thrombosis: the Dutch Stent Thrombosis Registry. J Am Coll Cardiol 2009; 53:1399–1409.
- Berger JS. Aspirin, clopidogrel, and ticagrelor in acute coronary syndromes. Am J Cardiol 2013; 112:737–745.
- Franchi F, Angiolillo DJ. Novel antiplatelet agents in acute coronary syndrome. Nat Rev Cardiol 2015; 12:30–47.
- Patrono C, Rocca B. The future of antiplatelet therapy in cardiovascular disease. Annu Rev Med 2010; 61:49–61.
- Park SJ, Kang SM, Park DW. Dual antiplatelet therapy after drug-eluting stents: defining the proper duration. Coron Artery Dis 2014; 25:83–89.
- Steinhubl SR, Berger PB, Mann JT 3rd, et al; CREDO Investigators. Clopidogrel for the reduction of events during observation. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Nusca A, Patti G. Platelet function and inhibition in ischemic heart disease. Curr Cardiol Rep 2012; 14:457–467.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Genereux P, Stone GW, Harrington RA, et al; CHAMPION PHOENIX Investigators. Impact of intraprocedural stent thrombosis during percutaneous coronary intervention: Insights from the CHAMPION PHOENIX Trial (Clinical Trial Comparing Cangrelor to Clopidogrel Standard of Care Therapy in Subjects Who Require Percutaneous Coronary Intervention). J Am Coll Cardiol 2014; 63:619–629.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Brilakis ES, Patel VG, Banerjee S. Medical management after coronary stent implantation: a review. JAMA 2013; 310:189–198.
- Warren J, Baber U, Mehran R. Antiplatelet therapy after drug-eluting stent implantation. J Cardiol 2015; 65:98–104.
- Urban P, Macaya C, Rupprecht HJ, et al. Randomized evaluation of anticoagulation versus antiplatelet therapy after coronary stent implantation in high-risk patients: the Multicenter Aspirin and Ticlopidine Trial After Intracoronary Stenting (MATTIS). Circulation 1998; 98:2126–2132.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: The PCI-CURE study. Lancet 2001; 358:527–533.
- Morais J. Insights from CURE: using clopidogrel on top of standard therapy. Cerebrovasc Dis 2002; 13(suppl 1):17–21.
- Ferguson JJ. Clopidogrel plus aspirin in patients with acute myocardial infarction treated with fibrinolytic therapy—CLARITY-TIMI 28. Future Cardiol 2005; 1:605–610.
- Sabatine MS, Cannon CP, Gibson CM, et al; Clopidogrel as Adjunctive Reperfusion Therapy (CLARITY)-Thrombolysis in Myocardial Infarction (TIMI) 28 Investigators. Effect of clopidogrel pretreatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005; 294:1224–1232.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bonaca MP, Bhatt DL, Cohen M, et al; PEGASUS-TIMI 54 Steering Committee and Investigators. Long-term use of ticagrelor in patients with prior myocardial infarction. N Engl J Med 2015; 372:1791–1800.
- Colombo A, Chieffo A, Frasheri A, et al. Second-generation drug-eluting stent implantation followed by 6- versus 12-month dual antiplatelet therapy: the SECURITY randomized clinical trial. J Am Coll Cardiol 2014; 64:2086–2097.
- Gwon HC, Hahn JY, Park KW, et al. Six-month versus 12-month dual antiplatelet therapy after implantation of drug-eluting stents: the Efficacy of Xience/Promus versus Cypher to Reduce Late Loss After Stenting (EXCELLENT) randomized, multicenter study. Circulation 2012; 125:505–513.
- Feres F, Costa RA, Abizaid A, et al; OPTIMIZE Trial Investigators. Three vs twelve months of dual antiplatelet therapy after zotarolimus-eluting stents: the OPTIMIZE randomized trial. JAMA 2013; 310:2510–2522.
- Kim BK, Hong MK, Shin DH, et al; RESET Investigators. A new strategy for discontinuation of dual antiplatelet therapy: the RESET Trial (REal Safety and Efficacy of 3-month dual antiplatelet Therapy following endeavor zotarolimus-eluting stent implantation). J Am Coll Cardiol 2012; 60:1340–1348.
- El-Hayek G, Messerli F, Bangalore S, et al. Meta-analysis of randomized clinical trials comparing short-term versus long-term dual antiplatelet therapy following drug-eluting stents. Am J Cardiol 2014; 114:236–242.
- Valgimigli M, Campo G, Monti M, et al; Prolonging Dual Antiplatelet Treatment After Grading Stent-Induced Intimal Hyperplasia Study (PRODIGY) Investigators. Short- versus long-term duration of dual-antiplatelet therapy after coronary stenting: a randomized multicenter trial. Circulation 2012; 125:2015–2026.
- Collet JP, Silvain J, Barthelemy O, et al; ARCTIC investigators. Dual-antiplatelet treatment beyond 1 year after drug-eluting stent implantation (ARCTIC-Interruption): a randomised trial. Lancet 2014; 384:1577–1585.
- Lee CW, Ahn JM, Park DW, et al. Optimal duration of dual antiplatelet therapy after drug-eluting stent implantation: a randomized, controlled trial. Circulation 2014; 129:304–312.
- Kwok CS, Bulluck H, Ryding AD, Loke YK. Benefits and harms of extending the duration of dual antiplatelet therapy after percutaneous coronary intervention with drug-eluting stents: a meta-analysis. ScientificWorldJournal 2014; 2014:794078.
- Dewilde WJ, Oirbans T, Verheugt FW, et al; WOEST study investigators. Use of clopidogrel with or without aspirin in patients taking oral anticoagulant therapy and undergoing percutaneous coronary intervention: an open-label, randomised, controlled trial. Lancet 2013; 381:1107–1115.
KEY POINTS
- The outcomes of patients with acute coronary syndrome events have been improving as percutaneous coronary intervention and its accompanying medical therapy have evolved.
- Newer, more potent antiplatelet agents are preferred over clopidogrel when possible.
- Two earlier studies showed no advantage of extended dual antiplatelet therapy over the standard 12-month duration, but the recent Dual Antiplatelet Therapy trial did.
- The protection against ischemia afforded by dual antiplatelet therapy comes at the price of increased risk of bleeding.