User login
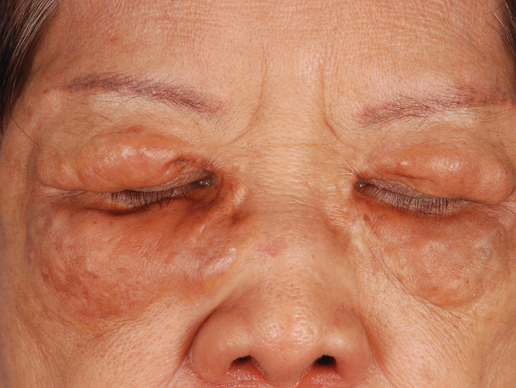
Yellowish Papulonodular Periorbital Eruption
The Diagnosis: Adult-Onset Xanthogranuloma
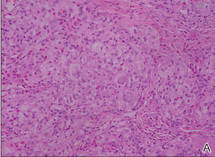
Biopsies of the lesions on the neck (Figure 1) and back were performed. Histologic analyses revealed a diffuse dermatitis consisting of foamy histiocytes admixed with a few Touton-type giant cells in the dermis (Figure 2), which was associated with an inflammatory infiltrate of eosinophils and lymphocytes. Laboratory investigations revealed mild thrombocytopenia with a platelet count of 134×109/L (reference range, 140–440×109/L). Other investigations including biochemistry, lipid, serum electrophoresis, and chest radiogram were normal. A bone marrow trephine biopsy and flow cytometry were performed and were normal. Magnetic resonance imaging revealed periorbital soft-tissue masses that did not extend into the orbits.
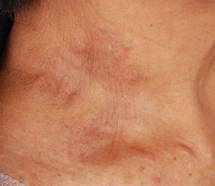 |
| Figure 1. Firm plaques with a yellowish tinge over the side of the neck |
 |
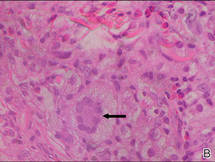 |
| Figure 2. A dense infiltrate of foamy histiocytes in the dermis (A) associated with a Touton-type giant cell (arrow) and an inflammatory infiltrate consisting of eosinophils and lymphocytes (B)(both H&E, original magnifications ×200 and ×400). |
Adult-onset xanthogranuloma (AXG) is a rare disease entity, usually presenting in the third to fourth decades of life. The condition typically presents as a red to yellow-brown nodular cutaneous lesion located on the scalp, face, neck, trunk, or limbs. The presentation typically consists of a solitary lesion, occurring in 70% to 89% of cases,1 but more rarely, as in this case, lesions can be multiple or even disseminated.
Histologically, AXG presents as a dense, well-circumscribed, histiocytic infiltrate consisting of lipophages possessing foamy cytoplasm and giant cells. The presence of histiocytic giant cells differentiates AXG from xanthelasma, a clinical differential diagnosis in this case, and xanthoma. In AXG, there are 4 main types of histiocytes: xanthomatized, spindle shaped, vacuolated, and oncocytic.2 They can be seen in variable proportions, together with different types of giant cells (eg, Touton, foreign body, ground glass, nonspecific). A mixed infiltrate of eosinophils, lymphocytes, plasma cells, and neutrophils also may be seen scattered throughout the lesion.2
Correlating with the clinical and histological features of xanthogranuloma, the firm plaques and nodules represent the dense dermal infiltration of histiocytes that may extend into the subcutis. The lesions demonstrate a time-dependent progression both clinically and histologically. Early lesions are comprised of a dense monomorphous nonlipid histiocytic inflammatory infiltrate, and they clinically appear more erythematous. In mature lesions, as in our patient, the infiltrate is predominantly composed of lipid-laden histiocytes with associated Touton giant cells. They appear more yellowish on clinical presentation.
Adult-onset xanthogranuloma is part of a rare heterogenous group of disorders termed adult orbital xanthogranulomatous disease, which includes 3 other syndromes: necrobiotic xanthogranuloma, adult-onset asthma and periocular xanthogranuloma, and Erdheim-Chester disease.
Necrobiotic xanthogranuloma is clinically characterized by the presence of subcutaneous lesions that ulcerate in approximately 40% to 50% of cases and is histologically characterized by necrobiosis with palisading epithelioid histiocytes. It also is systemically associated with paraproteinemia and multiple myeloma.3
Adult-onset asthma and periocular xanthogranuloma is characterized by yellowish papules and nodules predominantly over the lower eyelids that are histologically comprised of lymphoid follicles with reactive germinal centers. It is associated with asthma, which normally is severe and often occurs almost simultaneously with the periorbital lesions.4
There are no definite diagnostic criteria for Erdheim-Chester disease and the diagnosis is usually based on radiologic findings of osteosclerosis and histopathologic evidence of foamy histiocytic infiltration. Systemic manifestations are common with lymphohistiocytic infiltration of the heart, lungs, pericardium, bones, and intestines. Prognosis is uniformly dismal.
Based on the clinical presentation of a nonulcerative papulonodular eruption and the absence of systemic involvement including asthma, we made the diagnosis of AXG. In view of the heterogeneity among these clinical entities as well as the time-based evolution of the lesions involved, we continued to monitor the patient for 2 years and there was no development of other systemic manifestations and hematologic abnormalities.
In contrast to the more common form of juvenile-onset xanthogranuloma, the adult type is not associated with widespread visceral lesions. Hence extensive screening investigations for systemic disease generally are not necessary. Another difference is that AXG has been associated with hematologic abnormalities, including essential thrombocytosis, chronic lymphocytic leukemia, large B-cell lymphoma, and monoclonal gammopathy.5,6 In our patient, the presence of thrombocytopenia and older age caused us to be concerned about an associated hematologic malignancy; therefore, a bone marrow biopsy and flow cytometry were performed.
Adult-onset xanthogranuloma typically is an asymptomatic and self-healing disease and therefore treatment usually is not required. Spontaneous regression of xanthogranuloma was observed to occur in 54% of cases with a median duration of 22 months,7 though lesions were noted to last as long as 15 years.8 When treatment is necessary, a combination of local and systemic corticosteroids, cytotoxic agents, and radiotherapy have been routinely used. In particular, intralesional corticosteroid therapy has been found to be efficacious in controlling symptoms and signs of AXG affecting the eyelids and orbits while avoiding the systemic side effects of other agents.9
Because our patient’s lesions were longstanding and disfiguring, we opted for active intervention with intralesional triamcinolone, which resulted in only a slight reduction in size of the lesions. The lesions remain largely unchanged in 2 years of follow-up.
1. Chang SE, Cho S, Choi JC, et al. Clinicohistopathologic comparison of adult type and juvenile type xanthogranulomas in Korea. J Dermatol. 2001;28:413-418.
2. Gelmetti C. Non-Langerhans cell histiocytosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
3. Mehregan DA, Winkelmann RK. Necrobiotic xanthogranuloma. Arch Dermatol. 1992;128:94-100.
4. Jakobiec FA, Mills MD, Hidayat AA, et al. Periocular xanthogranulomas associated with severe adult-onset asthma. Trans Am Ophthalmol Soc. 1993;91:99-125.
5. Shoo BA, Shinkai K, McCalmont TH, et al. Xanthogranulomas associated with hematologic malignancy in adulthood. J Am Acad Dermatol. 2008;59:488-493.
6. Chiou CC, Wang PN, Yang LC, et al. Disseminated xanthogranulomas associated with adult T-cell leukaemia/lymphoma: a case report and review the association of haematologic malignancies. J Eur Acad Dermatol Venereol. 2007;21:532-535.
7. Robinson HM, Harmon CE, Firminger HI. Multiple lipoidal histiocytomas with regression. Arch Dermatol. 1963;88:660-667.
8. Winkelmann RK. Cutaneous syndromes of non-X histiocytosis: a review of the macrophage-histiocyte diseases of the skin. Arch Dermatol. 1981;117:667-672.
9. Elner VM, Mintz R, Demirci H, et al. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Ophthal Plast Reconstr Surg. 2006;22:36-40.
The Diagnosis: Adult-Onset Xanthogranuloma
Biopsies of the lesions on the neck (Figure 1) and back were performed. Histologic analyses revealed a diffuse dermatitis consisting of foamy histiocytes admixed with a few Touton-type giant cells in the dermis (Figure 2), which was associated with an inflammatory infiltrate of eosinophils and lymphocytes. Laboratory investigations revealed mild thrombocytopenia with a platelet count of 134×109/L (reference range, 140–440×109/L). Other investigations including biochemistry, lipid, serum electrophoresis, and chest radiogram were normal. A bone marrow trephine biopsy and flow cytometry were performed and were normal. Magnetic resonance imaging revealed periorbital soft-tissue masses that did not extend into the orbits.
|
| Figure 1. Firm plaques with a yellowish tinge over the side of the neck |
|
|
| Figure 2. A dense infiltrate of foamy histiocytes in the dermis (A) associated with a Touton-type giant cell (arrow) and an inflammatory infiltrate consisting of eosinophils and lymphocytes (B)(both H&E, original magnifications ×200 and ×400). |
Adult-onset xanthogranuloma (AXG) is a rare disease entity, usually presenting in the third to fourth decades of life. The condition typically presents as a red to yellow-brown nodular cutaneous lesion located on the scalp, face, neck, trunk, or limbs. The presentation typically consists of a solitary lesion, occurring in 70% to 89% of cases,1 but more rarely, as in this case, lesions can be multiple or even disseminated.
Histologically, AXG presents as a dense, well-circumscribed, histiocytic infiltrate consisting of lipophages possessing foamy cytoplasm and giant cells. The presence of histiocytic giant cells differentiates AXG from xanthelasma, a clinical differential diagnosis in this case, and xanthoma. In AXG, there are 4 main types of histiocytes: xanthomatized, spindle shaped, vacuolated, and oncocytic.2 They can be seen in variable proportions, together with different types of giant cells (eg, Touton, foreign body, ground glass, nonspecific). A mixed infiltrate of eosinophils, lymphocytes, plasma cells, and neutrophils also may be seen scattered throughout the lesion.2
Correlating with the clinical and histological features of xanthogranuloma, the firm plaques and nodules represent the dense dermal infiltration of histiocytes that may extend into the subcutis. The lesions demonstrate a time-dependent progression both clinically and histologically. Early lesions are comprised of a dense monomorphous nonlipid histiocytic inflammatory infiltrate, and they clinically appear more erythematous. In mature lesions, as in our patient, the infiltrate is predominantly composed of lipid-laden histiocytes with associated Touton giant cells. They appear more yellowish on clinical presentation.
Adult-onset xanthogranuloma is part of a rare heterogenous group of disorders termed adult orbital xanthogranulomatous disease, which includes 3 other syndromes: necrobiotic xanthogranuloma, adult-onset asthma and periocular xanthogranuloma, and Erdheim-Chester disease.
Necrobiotic xanthogranuloma is clinically characterized by the presence of subcutaneous lesions that ulcerate in approximately 40% to 50% of cases and is histologically characterized by necrobiosis with palisading epithelioid histiocytes. It also is systemically associated with paraproteinemia and multiple myeloma.3
Adult-onset asthma and periocular xanthogranuloma is characterized by yellowish papules and nodules predominantly over the lower eyelids that are histologically comprised of lymphoid follicles with reactive germinal centers. It is associated with asthma, which normally is severe and often occurs almost simultaneously with the periorbital lesions.4
There are no definite diagnostic criteria for Erdheim-Chester disease and the diagnosis is usually based on radiologic findings of osteosclerosis and histopathologic evidence of foamy histiocytic infiltration. Systemic manifestations are common with lymphohistiocytic infiltration of the heart, lungs, pericardium, bones, and intestines. Prognosis is uniformly dismal.
Based on the clinical presentation of a nonulcerative papulonodular eruption and the absence of systemic involvement including asthma, we made the diagnosis of AXG. In view of the heterogeneity among these clinical entities as well as the time-based evolution of the lesions involved, we continued to monitor the patient for 2 years and there was no development of other systemic manifestations and hematologic abnormalities.
In contrast to the more common form of juvenile-onset xanthogranuloma, the adult type is not associated with widespread visceral lesions. Hence extensive screening investigations for systemic disease generally are not necessary. Another difference is that AXG has been associated with hematologic abnormalities, including essential thrombocytosis, chronic lymphocytic leukemia, large B-cell lymphoma, and monoclonal gammopathy.5,6 In our patient, the presence of thrombocytopenia and older age caused us to be concerned about an associated hematologic malignancy; therefore, a bone marrow biopsy and flow cytometry were performed.
Adult-onset xanthogranuloma typically is an asymptomatic and self-healing disease and therefore treatment usually is not required. Spontaneous regression of xanthogranuloma was observed to occur in 54% of cases with a median duration of 22 months,7 though lesions were noted to last as long as 15 years.8 When treatment is necessary, a combination of local and systemic corticosteroids, cytotoxic agents, and radiotherapy have been routinely used. In particular, intralesional corticosteroid therapy has been found to be efficacious in controlling symptoms and signs of AXG affecting the eyelids and orbits while avoiding the systemic side effects of other agents.9
Because our patient’s lesions were longstanding and disfiguring, we opted for active intervention with intralesional triamcinolone, which resulted in only a slight reduction in size of the lesions. The lesions remain largely unchanged in 2 years of follow-up.
The Diagnosis: Adult-Onset Xanthogranuloma
Biopsies of the lesions on the neck (Figure 1) and back were performed. Histologic analyses revealed a diffuse dermatitis consisting of foamy histiocytes admixed with a few Touton-type giant cells in the dermis (Figure 2), which was associated with an inflammatory infiltrate of eosinophils and lymphocytes. Laboratory investigations revealed mild thrombocytopenia with a platelet count of 134×109/L (reference range, 140–440×109/L). Other investigations including biochemistry, lipid, serum electrophoresis, and chest radiogram were normal. A bone marrow trephine biopsy and flow cytometry were performed and were normal. Magnetic resonance imaging revealed periorbital soft-tissue masses that did not extend into the orbits.
|
| Figure 1. Firm plaques with a yellowish tinge over the side of the neck |
|
|
| Figure 2. A dense infiltrate of foamy histiocytes in the dermis (A) associated with a Touton-type giant cell (arrow) and an inflammatory infiltrate consisting of eosinophils and lymphocytes (B)(both H&E, original magnifications ×200 and ×400). |
Adult-onset xanthogranuloma (AXG) is a rare disease entity, usually presenting in the third to fourth decades of life. The condition typically presents as a red to yellow-brown nodular cutaneous lesion located on the scalp, face, neck, trunk, or limbs. The presentation typically consists of a solitary lesion, occurring in 70% to 89% of cases,1 but more rarely, as in this case, lesions can be multiple or even disseminated.
Histologically, AXG presents as a dense, well-circumscribed, histiocytic infiltrate consisting of lipophages possessing foamy cytoplasm and giant cells. The presence of histiocytic giant cells differentiates AXG from xanthelasma, a clinical differential diagnosis in this case, and xanthoma. In AXG, there are 4 main types of histiocytes: xanthomatized, spindle shaped, vacuolated, and oncocytic.2 They can be seen in variable proportions, together with different types of giant cells (eg, Touton, foreign body, ground glass, nonspecific). A mixed infiltrate of eosinophils, lymphocytes, plasma cells, and neutrophils also may be seen scattered throughout the lesion.2
Correlating with the clinical and histological features of xanthogranuloma, the firm plaques and nodules represent the dense dermal infiltration of histiocytes that may extend into the subcutis. The lesions demonstrate a time-dependent progression both clinically and histologically. Early lesions are comprised of a dense monomorphous nonlipid histiocytic inflammatory infiltrate, and they clinically appear more erythematous. In mature lesions, as in our patient, the infiltrate is predominantly composed of lipid-laden histiocytes with associated Touton giant cells. They appear more yellowish on clinical presentation.
Adult-onset xanthogranuloma is part of a rare heterogenous group of disorders termed adult orbital xanthogranulomatous disease, which includes 3 other syndromes: necrobiotic xanthogranuloma, adult-onset asthma and periocular xanthogranuloma, and Erdheim-Chester disease.
Necrobiotic xanthogranuloma is clinically characterized by the presence of subcutaneous lesions that ulcerate in approximately 40% to 50% of cases and is histologically characterized by necrobiosis with palisading epithelioid histiocytes. It also is systemically associated with paraproteinemia and multiple myeloma.3
Adult-onset asthma and periocular xanthogranuloma is characterized by yellowish papules and nodules predominantly over the lower eyelids that are histologically comprised of lymphoid follicles with reactive germinal centers. It is associated with asthma, which normally is severe and often occurs almost simultaneously with the periorbital lesions.4
There are no definite diagnostic criteria for Erdheim-Chester disease and the diagnosis is usually based on radiologic findings of osteosclerosis and histopathologic evidence of foamy histiocytic infiltration. Systemic manifestations are common with lymphohistiocytic infiltration of the heart, lungs, pericardium, bones, and intestines. Prognosis is uniformly dismal.
Based on the clinical presentation of a nonulcerative papulonodular eruption and the absence of systemic involvement including asthma, we made the diagnosis of AXG. In view of the heterogeneity among these clinical entities as well as the time-based evolution of the lesions involved, we continued to monitor the patient for 2 years and there was no development of other systemic manifestations and hematologic abnormalities.
In contrast to the more common form of juvenile-onset xanthogranuloma, the adult type is not associated with widespread visceral lesions. Hence extensive screening investigations for systemic disease generally are not necessary. Another difference is that AXG has been associated with hematologic abnormalities, including essential thrombocytosis, chronic lymphocytic leukemia, large B-cell lymphoma, and monoclonal gammopathy.5,6 In our patient, the presence of thrombocytopenia and older age caused us to be concerned about an associated hematologic malignancy; therefore, a bone marrow biopsy and flow cytometry were performed.
Adult-onset xanthogranuloma typically is an asymptomatic and self-healing disease and therefore treatment usually is not required. Spontaneous regression of xanthogranuloma was observed to occur in 54% of cases with a median duration of 22 months,7 though lesions were noted to last as long as 15 years.8 When treatment is necessary, a combination of local and systemic corticosteroids, cytotoxic agents, and radiotherapy have been routinely used. In particular, intralesional corticosteroid therapy has been found to be efficacious in controlling symptoms and signs of AXG affecting the eyelids and orbits while avoiding the systemic side effects of other agents.9
Because our patient’s lesions were longstanding and disfiguring, we opted for active intervention with intralesional triamcinolone, which resulted in only a slight reduction in size of the lesions. The lesions remain largely unchanged in 2 years of follow-up.
1. Chang SE, Cho S, Choi JC, et al. Clinicohistopathologic comparison of adult type and juvenile type xanthogranulomas in Korea. J Dermatol. 2001;28:413-418.
2. Gelmetti C. Non-Langerhans cell histiocytosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
3. Mehregan DA, Winkelmann RK. Necrobiotic xanthogranuloma. Arch Dermatol. 1992;128:94-100.
4. Jakobiec FA, Mills MD, Hidayat AA, et al. Periocular xanthogranulomas associated with severe adult-onset asthma. Trans Am Ophthalmol Soc. 1993;91:99-125.
5. Shoo BA, Shinkai K, McCalmont TH, et al. Xanthogranulomas associated with hematologic malignancy in adulthood. J Am Acad Dermatol. 2008;59:488-493.
6. Chiou CC, Wang PN, Yang LC, et al. Disseminated xanthogranulomas associated with adult T-cell leukaemia/lymphoma: a case report and review the association of haematologic malignancies. J Eur Acad Dermatol Venereol. 2007;21:532-535.
7. Robinson HM, Harmon CE, Firminger HI. Multiple lipoidal histiocytomas with regression. Arch Dermatol. 1963;88:660-667.
8. Winkelmann RK. Cutaneous syndromes of non-X histiocytosis: a review of the macrophage-histiocyte diseases of the skin. Arch Dermatol. 1981;117:667-672.
9. Elner VM, Mintz R, Demirci H, et al. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Ophthal Plast Reconstr Surg. 2006;22:36-40.
1. Chang SE, Cho S, Choi JC, et al. Clinicohistopathologic comparison of adult type and juvenile type xanthogranulomas in Korea. J Dermatol. 2001;28:413-418.
2. Gelmetti C. Non-Langerhans cell histiocytosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
3. Mehregan DA, Winkelmann RK. Necrobiotic xanthogranuloma. Arch Dermatol. 1992;128:94-100.
4. Jakobiec FA, Mills MD, Hidayat AA, et al. Periocular xanthogranulomas associated with severe adult-onset asthma. Trans Am Ophthalmol Soc. 1993;91:99-125.
5. Shoo BA, Shinkai K, McCalmont TH, et al. Xanthogranulomas associated with hematologic malignancy in adulthood. J Am Acad Dermatol. 2008;59:488-493.
6. Chiou CC, Wang PN, Yang LC, et al. Disseminated xanthogranulomas associated with adult T-cell leukaemia/lymphoma: a case report and review the association of haematologic malignancies. J Eur Acad Dermatol Venereol. 2007;21:532-535.
7. Robinson HM, Harmon CE, Firminger HI. Multiple lipoidal histiocytomas with regression. Arch Dermatol. 1963;88:660-667.
8. Winkelmann RK. Cutaneous syndromes of non-X histiocytosis: a review of the macrophage-histiocyte diseases of the skin. Arch Dermatol. 1981;117:667-672.
9. Elner VM, Mintz R, Demirci H, et al. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Ophthal Plast Reconstr Surg. 2006;22:36-40.
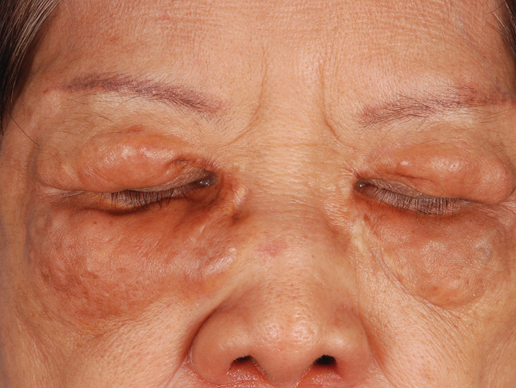
A 66-year-old woman with a history of type 2 diabetes mellitus and mild dyslipidemia presented with persistent lesions over the eyelids and cheeks of 10 years’ duration. Systemic review was unremarkable. There was no family or personal history of atopy, asthma, or other dermatologic disorders. Physical examination revealed confluent yellowish plaques and nodules over the periorbital regions as well as yellowish plaques over the neck and back. The lesions were firm to palpation and the epidermis appeared unaffected. The ophthalmic examination was normal and other mucosal surfaces were unaffected.