User login
Bacterial signals set the stage for PMP

Preclinical research suggests bacterial signals are crucial to the development of pre-leukemic myeloproliferation (PMP).
Researchers found that bacterial translocation leads to increased production of interleukin-6 (IL-6), which prompts PMP development in mice with Tet2 deficiency.
However, antibiotics and blockade of IL-6 were able to reverse PMP in the mice.
Bana Jabri, MD, PhD, of the University of Chicago in Illinois, and her colleagues reported these findings in Nature.
The researchers noted that, in humans and mice, TET2 deficiency leads to increased self-renewal of hematopoietic stem cells favoring the myeloid lineage, and this can lead to PMP.
However, not all humans or mice with TET2 deficiency actually develop PMP, which suggests other factors are at play.
With this in mind, the researchers studied Tet2-deficient mice. The team found that loss of Tet2 expression leads to defects in the intestinal barrier, although it isn’t clear how this occurs.
The intestinal defects allow bacteria living in the gut to spread into the blood and peripheral organs. The spread of bacteria prompts an increase in IL-6. This promotes proliferation of granulocyte–macrophage progenitors that express high levels of IL-6Rα in the absence of Tet2, and this leads to PMP.
The researchers found they could induce PMP in symptom-free Tet2−/− mice by disrupting intestinal barrier integrity. PMP also developed in response to systemic bacterial stimuli.
However, antibiotics and blockade of IL-6 signals could reverse PMP in mice that developed symptoms. And germ-free Tet2−/− mice did not develop symptoms, which supports the idea that bacteria must be present to drive the development of PMP.
Dr Jabri said the next step is to conduct studies in humans to see if patients with PMP also have signs of bacterial translocation. Then, clinical trials could test whether treatments that target aberrant IL-6 signals in response to bacteria can reverse the course of PMP.
Preclinical research suggests bacterial signals are crucial to the development of pre-leukemic myeloproliferation (PMP).
Researchers found that bacterial translocation leads to increased production of interleukin-6 (IL-6), which prompts PMP development in mice with Tet2 deficiency.
However, antibiotics and blockade of IL-6 were able to reverse PMP in the mice.
Bana Jabri, MD, PhD, of the University of Chicago in Illinois, and her colleagues reported these findings in Nature.
The researchers noted that, in humans and mice, TET2 deficiency leads to increased self-renewal of hematopoietic stem cells favoring the myeloid lineage, and this can lead to PMP.
However, not all humans or mice with TET2 deficiency actually develop PMP, which suggests other factors are at play.
With this in mind, the researchers studied Tet2-deficient mice. The team found that loss of Tet2 expression leads to defects in the intestinal barrier, although it isn’t clear how this occurs.
The intestinal defects allow bacteria living in the gut to spread into the blood and peripheral organs. The spread of bacteria prompts an increase in IL-6. This promotes proliferation of granulocyte–macrophage progenitors that express high levels of IL-6Rα in the absence of Tet2, and this leads to PMP.
The researchers found they could induce PMP in symptom-free Tet2−/− mice by disrupting intestinal barrier integrity. PMP also developed in response to systemic bacterial stimuli.
However, antibiotics and blockade of IL-6 signals could reverse PMP in mice that developed symptoms. And germ-free Tet2−/− mice did not develop symptoms, which supports the idea that bacteria must be present to drive the development of PMP.
Dr Jabri said the next step is to conduct studies in humans to see if patients with PMP also have signs of bacterial translocation. Then, clinical trials could test whether treatments that target aberrant IL-6 signals in response to bacteria can reverse the course of PMP.
Preclinical research suggests bacterial signals are crucial to the development of pre-leukemic myeloproliferation (PMP).
Researchers found that bacterial translocation leads to increased production of interleukin-6 (IL-6), which prompts PMP development in mice with Tet2 deficiency.
However, antibiotics and blockade of IL-6 were able to reverse PMP in the mice.
Bana Jabri, MD, PhD, of the University of Chicago in Illinois, and her colleagues reported these findings in Nature.
The researchers noted that, in humans and mice, TET2 deficiency leads to increased self-renewal of hematopoietic stem cells favoring the myeloid lineage, and this can lead to PMP.
However, not all humans or mice with TET2 deficiency actually develop PMP, which suggests other factors are at play.
With this in mind, the researchers studied Tet2-deficient mice. The team found that loss of Tet2 expression leads to defects in the intestinal barrier, although it isn’t clear how this occurs.
The intestinal defects allow bacteria living in the gut to spread into the blood and peripheral organs. The spread of bacteria prompts an increase in IL-6. This promotes proliferation of granulocyte–macrophage progenitors that express high levels of IL-6Rα in the absence of Tet2, and this leads to PMP.
The researchers found they could induce PMP in symptom-free Tet2−/− mice by disrupting intestinal barrier integrity. PMP also developed in response to systemic bacterial stimuli.
However, antibiotics and blockade of IL-6 signals could reverse PMP in mice that developed symptoms. And germ-free Tet2−/− mice did not develop symptoms, which supports the idea that bacteria must be present to drive the development of PMP.
Dr Jabri said the next step is to conduct studies in humans to see if patients with PMP also have signs of bacterial translocation. Then, clinical trials could test whether treatments that target aberrant IL-6 signals in response to bacteria can reverse the course of PMP.
EHRs enhance clinical trial follow-up

Electronic health records (EHRs) can enhance results from randomized controlled trials (RCTs), according to research published in Scientific Reports.
Researchers found EHRs could be used to track trial participants, enabling long-term monitoring of medical interventions and health outcomes and providing new insights into population health.
“In this study, we reported on the feasibility and efficiency of electronic follow-up and compared it with traditional trial follow-up,” said study author Sue Jordan, PhD, MB BCh, of Swansea University in Swansea, UK.
“We gained new insights from outcomes electronically recorded 3 years after the end of the trial and could then identify the differences between trial data and electronic data.”
Dr Jordan and her colleagues followed up on RCT participants using EHRs in the Secure Anonymised Information Linkage (SAIL) databank at Swansea University Medical School.
In this RCT, investigators had assessed the impact of probiotics on asthma and eczema in children born from 2005 to 2007. The trial had 2 years of traditional fieldwork follow-up.
Dr Jordan and her colleagues compared field results to EHR results at 2 years in 93% of trial participants.
The researchers said EHRs improved retention of children from lower socio-economic groups, which helped reduce volunteer bias.
The team also performed electronic follow-up at 5 years, which provided the “first robust analysis of asthma endpoints.”
The researchers said the asthma endpoints are “generally more reliable” in the 5-year EHR data for 2 reasons. The first is that the children are older, and asthma typically appears after 2 years of age.
The second reason is that, with the fieldwork follow-up, parents or guardians may have mistakenly identified symptoms as asthma without a child actually having an asthma diagnosis.
“Trial data are vulnerable to misunderstandings of questionnaires or definitions of illness,” Dr Jordan noted.
She and her colleagues also pointed out that retention was still high (82%) and free of bias in socio-economic status with the 5-year EHR data.
“These results lead us to conclude that using electronic health records have benefits relating to the cost-effective, long-term monitoring of complex interventions, which could have a positive impact for future clinical trial design,” said study author Michael Gravenor, DPhil, of Swansea University.
Electronic health records (EHRs) can enhance results from randomized controlled trials (RCTs), according to research published in Scientific Reports.
Researchers found EHRs could be used to track trial participants, enabling long-term monitoring of medical interventions and health outcomes and providing new insights into population health.
“In this study, we reported on the feasibility and efficiency of electronic follow-up and compared it with traditional trial follow-up,” said study author Sue Jordan, PhD, MB BCh, of Swansea University in Swansea, UK.
“We gained new insights from outcomes electronically recorded 3 years after the end of the trial and could then identify the differences between trial data and electronic data.”
Dr Jordan and her colleagues followed up on RCT participants using EHRs in the Secure Anonymised Information Linkage (SAIL) databank at Swansea University Medical School.
In this RCT, investigators had assessed the impact of probiotics on asthma and eczema in children born from 2005 to 2007. The trial had 2 years of traditional fieldwork follow-up.
Dr Jordan and her colleagues compared field results to EHR results at 2 years in 93% of trial participants.
The researchers said EHRs improved retention of children from lower socio-economic groups, which helped reduce volunteer bias.
The team also performed electronic follow-up at 5 years, which provided the “first robust analysis of asthma endpoints.”
The researchers said the asthma endpoints are “generally more reliable” in the 5-year EHR data for 2 reasons. The first is that the children are older, and asthma typically appears after 2 years of age.
The second reason is that, with the fieldwork follow-up, parents or guardians may have mistakenly identified symptoms as asthma without a child actually having an asthma diagnosis.
“Trial data are vulnerable to misunderstandings of questionnaires or definitions of illness,” Dr Jordan noted.
She and her colleagues also pointed out that retention was still high (82%) and free of bias in socio-economic status with the 5-year EHR data.
“These results lead us to conclude that using electronic health records have benefits relating to the cost-effective, long-term monitoring of complex interventions, which could have a positive impact for future clinical trial design,” said study author Michael Gravenor, DPhil, of Swansea University.
Electronic health records (EHRs) can enhance results from randomized controlled trials (RCTs), according to research published in Scientific Reports.
Researchers found EHRs could be used to track trial participants, enabling long-term monitoring of medical interventions and health outcomes and providing new insights into population health.
“In this study, we reported on the feasibility and efficiency of electronic follow-up and compared it with traditional trial follow-up,” said study author Sue Jordan, PhD, MB BCh, of Swansea University in Swansea, UK.
“We gained new insights from outcomes electronically recorded 3 years after the end of the trial and could then identify the differences between trial data and electronic data.”
Dr Jordan and her colleagues followed up on RCT participants using EHRs in the Secure Anonymised Information Linkage (SAIL) databank at Swansea University Medical School.
In this RCT, investigators had assessed the impact of probiotics on asthma and eczema in children born from 2005 to 2007. The trial had 2 years of traditional fieldwork follow-up.
Dr Jordan and her colleagues compared field results to EHR results at 2 years in 93% of trial participants.
The researchers said EHRs improved retention of children from lower socio-economic groups, which helped reduce volunteer bias.
The team also performed electronic follow-up at 5 years, which provided the “first robust analysis of asthma endpoints.”
The researchers said the asthma endpoints are “generally more reliable” in the 5-year EHR data for 2 reasons. The first is that the children are older, and asthma typically appears after 2 years of age.
The second reason is that, with the fieldwork follow-up, parents or guardians may have mistakenly identified symptoms as asthma without a child actually having an asthma diagnosis.
“Trial data are vulnerable to misunderstandings of questionnaires or definitions of illness,” Dr Jordan noted.
She and her colleagues also pointed out that retention was still high (82%) and free of bias in socio-economic status with the 5-year EHR data.
“These results lead us to conclude that using electronic health records have benefits relating to the cost-effective, long-term monitoring of complex interventions, which could have a positive impact for future clinical trial design,” said study author Michael Gravenor, DPhil, of Swansea University.
Regimen can improve DFS in newly diagnosed T-ALL

The addition of nelarabine can improve treatment outcomes for certain patients with T-cell acute lymphoblastic leukemia (T-ALL), according to a phase 3 trial.
Patients with newly diagnosed, intermediate- or high-risk T-ALL had a significant improvement in 4-year disease-free survival (DFS) if they received nelarabine in addition to chemotherapy and cranial irradiation.
The DFS benefit with nelarabine was significant for patients who received high-dose methotrexate but not for those who received escalating-dose methotrexate.
This study also included patients with T-cell lymphoblastic lymphoma (T-LL), and they did not experience an improvement in DFS with the addition of nelarabine.
Kimberly Dunsmore, MD, of Virginia Tech Carilion School of Medicine in Roanoke, presented these results in a press briefing in advance of the 2018 ASCO Annual Meeting. Additional results are scheduled to be presented at the meeting as abstract 10500.
This research was supported by the National Cancer Institute/National Institutes of Health and St. Baldrick’s Foundation. The researchers’ disclosures are listed with the abstract.
Patients and treatment
The trial enrolled 1895 patients, ages 1 to 30, who were newly diagnosed with T-ALL (94%) or T-LL (6%).
Patients received standard 4-drug induction chemotherapy, and 1307 of these patients were then randomized to 1 of 4 treatment arms.
Regardless of which arm they were randomized to, patients received an 11-drug chemotherapy regimen—the augmented Berlin-Frankfurt-Munster regimen. Intermediate- and high-risk patients in all 4 arms also received cranial irradiation.
In the first treatment arm, T-LL (n=58) and T-ALL (n=372) patients received escalating-dose methotrexate without leucovorin rescue and pegaspargase (C-MTX).
In the second treatment arm, patients with intermediate- and high-risk T-ALL (n=147) and T-LL (n=60) received C-MTX plus nelarabine (six 5-day courses at 650 mg/m2/day).
In the third arm, T-ALL patients (n=451) received high-dose methotrexate with leucovorin rescue (HD-MTX). T-LL patients were not eligible for this arm or the fourth treatment arm.
In the fourth arm, intermediate- and high-risk T-ALL patients (n=219) received HD-MTX and nelarabine (same schedule as above). This included 43 T-ALL patients who had induction failure and were assigned to this arm non-randomly.
Results
For T-ALL patients, the 4-year disease-free survival (DFS) rate was 84%, and the 4-year overall survival rate was 90%.
There was a significant improvement in DFS for T-ALL patients who received nelarabine compared to those who did not—89% and 83%, respectively (P=0.0332).
“Historically, about 80% of people [with T-ALL] live at least 4 years after being treated for their disease, but we felt we could and must do better,” Dr Dunsmore said. “Our trial shows that we could further increase survival rates by about 10%, which is very encouraging.”
Dr Dunsmore also noted that patients who received nelarabine had fewer central nervous system relapses.
Among T-ALL patients who received C-MTX, there was no significant difference in DFS for those who received nelarabine and those who did not—92% and 90%, respectively (P=0.3825).
However, for patients who received HD-MTX, the difference in DFS was significant. The DFS rate was 86% in patients who received nelarabine and 78% in those who did not (P=0.024).
For the T-ALL patients who failed induction and were assigned to HD-MTX and nelarabine, the 4-year DFS rate was 55%.
Patients with T-LL did not benefit from the addition of nelarabine. The 4-year DFS rate was 85% in the nelarabine recipients and 89% in non-recipients (P=0.2788).
There were no significant differences in overall toxicity or peripheral neurotoxicity between the treatment arms.
Dr Dunsmore said the next step with this research will be to examine the implications and potential benefits of using nelarabine in treatment protocols that do not include cranial radiation.
The addition of nelarabine can improve treatment outcomes for certain patients with T-cell acute lymphoblastic leukemia (T-ALL), according to a phase 3 trial.
Patients with newly diagnosed, intermediate- or high-risk T-ALL had a significant improvement in 4-year disease-free survival (DFS) if they received nelarabine in addition to chemotherapy and cranial irradiation.
The DFS benefit with nelarabine was significant for patients who received high-dose methotrexate but not for those who received escalating-dose methotrexate.
This study also included patients with T-cell lymphoblastic lymphoma (T-LL), and they did not experience an improvement in DFS with the addition of nelarabine.
Kimberly Dunsmore, MD, of Virginia Tech Carilion School of Medicine in Roanoke, presented these results in a press briefing in advance of the 2018 ASCO Annual Meeting. Additional results are scheduled to be presented at the meeting as abstract 10500.
This research was supported by the National Cancer Institute/National Institutes of Health and St. Baldrick’s Foundation. The researchers’ disclosures are listed with the abstract.
Patients and treatment
The trial enrolled 1895 patients, ages 1 to 30, who were newly diagnosed with T-ALL (94%) or T-LL (6%).
Patients received standard 4-drug induction chemotherapy, and 1307 of these patients were then randomized to 1 of 4 treatment arms.
Regardless of which arm they were randomized to, patients received an 11-drug chemotherapy regimen—the augmented Berlin-Frankfurt-Munster regimen. Intermediate- and high-risk patients in all 4 arms also received cranial irradiation.
In the first treatment arm, T-LL (n=58) and T-ALL (n=372) patients received escalating-dose methotrexate without leucovorin rescue and pegaspargase (C-MTX).
In the second treatment arm, patients with intermediate- and high-risk T-ALL (n=147) and T-LL (n=60) received C-MTX plus nelarabine (six 5-day courses at 650 mg/m2/day).
In the third arm, T-ALL patients (n=451) received high-dose methotrexate with leucovorin rescue (HD-MTX). T-LL patients were not eligible for this arm or the fourth treatment arm.
In the fourth arm, intermediate- and high-risk T-ALL patients (n=219) received HD-MTX and nelarabine (same schedule as above). This included 43 T-ALL patients who had induction failure and were assigned to this arm non-randomly.
Results
For T-ALL patients, the 4-year disease-free survival (DFS) rate was 84%, and the 4-year overall survival rate was 90%.
There was a significant improvement in DFS for T-ALL patients who received nelarabine compared to those who did not—89% and 83%, respectively (P=0.0332).
“Historically, about 80% of people [with T-ALL] live at least 4 years after being treated for their disease, but we felt we could and must do better,” Dr Dunsmore said. “Our trial shows that we could further increase survival rates by about 10%, which is very encouraging.”
Dr Dunsmore also noted that patients who received nelarabine had fewer central nervous system relapses.
Among T-ALL patients who received C-MTX, there was no significant difference in DFS for those who received nelarabine and those who did not—92% and 90%, respectively (P=0.3825).
However, for patients who received HD-MTX, the difference in DFS was significant. The DFS rate was 86% in patients who received nelarabine and 78% in those who did not (P=0.024).
For the T-ALL patients who failed induction and were assigned to HD-MTX and nelarabine, the 4-year DFS rate was 55%.
Patients with T-LL did not benefit from the addition of nelarabine. The 4-year DFS rate was 85% in the nelarabine recipients and 89% in non-recipients (P=0.2788).
There were no significant differences in overall toxicity or peripheral neurotoxicity between the treatment arms.
Dr Dunsmore said the next step with this research will be to examine the implications and potential benefits of using nelarabine in treatment protocols that do not include cranial radiation.
The addition of nelarabine can improve treatment outcomes for certain patients with T-cell acute lymphoblastic leukemia (T-ALL), according to a phase 3 trial.
Patients with newly diagnosed, intermediate- or high-risk T-ALL had a significant improvement in 4-year disease-free survival (DFS) if they received nelarabine in addition to chemotherapy and cranial irradiation.
The DFS benefit with nelarabine was significant for patients who received high-dose methotrexate but not for those who received escalating-dose methotrexate.
This study also included patients with T-cell lymphoblastic lymphoma (T-LL), and they did not experience an improvement in DFS with the addition of nelarabine.
Kimberly Dunsmore, MD, of Virginia Tech Carilion School of Medicine in Roanoke, presented these results in a press briefing in advance of the 2018 ASCO Annual Meeting. Additional results are scheduled to be presented at the meeting as abstract 10500.
This research was supported by the National Cancer Institute/National Institutes of Health and St. Baldrick’s Foundation. The researchers’ disclosures are listed with the abstract.
Patients and treatment
The trial enrolled 1895 patients, ages 1 to 30, who were newly diagnosed with T-ALL (94%) or T-LL (6%).
Patients received standard 4-drug induction chemotherapy, and 1307 of these patients were then randomized to 1 of 4 treatment arms.
Regardless of which arm they were randomized to, patients received an 11-drug chemotherapy regimen—the augmented Berlin-Frankfurt-Munster regimen. Intermediate- and high-risk patients in all 4 arms also received cranial irradiation.
In the first treatment arm, T-LL (n=58) and T-ALL (n=372) patients received escalating-dose methotrexate without leucovorin rescue and pegaspargase (C-MTX).
In the second treatment arm, patients with intermediate- and high-risk T-ALL (n=147) and T-LL (n=60) received C-MTX plus nelarabine (six 5-day courses at 650 mg/m2/day).
In the third arm, T-ALL patients (n=451) received high-dose methotrexate with leucovorin rescue (HD-MTX). T-LL patients were not eligible for this arm or the fourth treatment arm.
In the fourth arm, intermediate- and high-risk T-ALL patients (n=219) received HD-MTX and nelarabine (same schedule as above). This included 43 T-ALL patients who had induction failure and were assigned to this arm non-randomly.
Results
For T-ALL patients, the 4-year disease-free survival (DFS) rate was 84%, and the 4-year overall survival rate was 90%.
There was a significant improvement in DFS for T-ALL patients who received nelarabine compared to those who did not—89% and 83%, respectively (P=0.0332).
“Historically, about 80% of people [with T-ALL] live at least 4 years after being treated for their disease, but we felt we could and must do better,” Dr Dunsmore said. “Our trial shows that we could further increase survival rates by about 10%, which is very encouraging.”
Dr Dunsmore also noted that patients who received nelarabine had fewer central nervous system relapses.
Among T-ALL patients who received C-MTX, there was no significant difference in DFS for those who received nelarabine and those who did not—92% and 90%, respectively (P=0.3825).
However, for patients who received HD-MTX, the difference in DFS was significant. The DFS rate was 86% in patients who received nelarabine and 78% in those who did not (P=0.024).
For the T-ALL patients who failed induction and were assigned to HD-MTX and nelarabine, the 4-year DFS rate was 55%.
Patients with T-LL did not benefit from the addition of nelarabine. The 4-year DFS rate was 85% in the nelarabine recipients and 89% in non-recipients (P=0.2788).
There were no significant differences in overall toxicity or peripheral neurotoxicity between the treatment arms.
Dr Dunsmore said the next step with this research will be to examine the implications and potential benefits of using nelarabine in treatment protocols that do not include cranial radiation.
Treating insomnia in cancer survivors

Treatment with acupuncture or cognitive behavioral therapy (CBT) can decrease the severity of insomnia among cancer survivors, according to new research.
Overall, improvements in insomnia were greatest in patients treated with CBT, and patients with mild insomnia at baseline had a significantly greater improvement with CBT than with acupuncture.
However, among patients who had moderate to severe insomnia at baseline, there was no significant difference in results with CBT and acupuncture.
Jun J. Mao, MD, of Memorial Sloan Kettering Cancer Center in New York, New York, presented these results in a press briefing in advance of the 2018 ASCO Annual Meeting.
Additional results are scheduled to be presented at the meeting as abstract 10001.
“Up to 60% of cancer survivors have some form of insomnia, but it is often underdiagnosed and undertreated,” Dr Mao said. “Our trial showed that both CBT-I [CBT for insomnia] and acupuncture were effective in treating moderate to severe insomnia, although CBT-I was more effective for those with mild symptoms of insomnia. Now, patients have more choices to manage their insomnia.”
Dr Mao and his colleagues studied 160 individuals who had completed cancer treatment. Their mean time since cancer diagnosis was about 6 years.
The subjects had received treatment for hematologic malignancies (8%) as well as breast (31%), prostate (23%), head and neck (7%), colorectal (6%), gynecological (4%), and other cancers (14%). Six percent of subjects had been treated for more than 1 type of cancer.
All subjects had been clinically diagnosed with insomnia. Seventy-nine percent had moderate (n=94) to severe (n=33) insomnia, and 21% had mild insomnia (n=33).
Interventions
Subjects were randomized to receive CBT or acupuncture for 8 weeks. Those who received CBT worked with a therapist to re-establish a restorative sleep schedule by:
- Reducing the amount of time spent in bed
- Limiting activities performed in bed to sleep and sexual activity
- Modifying unhelpful beliefs about sleep
- Promoting good sleep hygiene (setting a regular sleep schedule and avoiding activities that include light from tablets and cellphones, eating too late, and performing vigorous activities).
The study’s primary outcome was reduction in insomnia severity, as measured by the Insomnia Severity Index (ISI), from study entry to the end of treatment at week 8. Subjects were also reassessed at 20 weeks.
The ISI is a questionnaire that asks people to rate the severity of insomnia problems, such as difficulty falling asleep and staying asleep, and the impact of insomnia on their daily functioning and quality of life.
ISI scoring ranges from 0 to 28. Scores of 0 to 7 denote no clinically significant insomnia, 8 to 14 denote mild insomnia, 15 to 21 denote moderate insomnia, and 22 to 28 denote severe insomnia.
Results
At 8 weeks, the mean ISI score fell 10.9 points (from 18.5 to 7.5) for subjects who received CBT and 8.3 points (from 17.55 to 9.23) for those who received acupuncture (P=0.0007).
Among subjects with mild insomnia at baseline, far more responded to CBT than to acupuncture—85% and 18%, respectively (P<0.0001).
However, response rates were similar in subjects who had moderate to severe insomnia at baseline. Seventy-five percent of those who received CBT achieved a response, as did 66% of those who received acupuncture (P=0.26).
Both treatment groups had few mild adverse events, and all subjects maintained their improvements in insomnia at the 20-week assessment.
This study was funded by the Patient-Centered Outcomes Research Institute. The researchers’ disclosures are listed with the abstract.
Treatment with acupuncture or cognitive behavioral therapy (CBT) can decrease the severity of insomnia among cancer survivors, according to new research.
Overall, improvements in insomnia were greatest in patients treated with CBT, and patients with mild insomnia at baseline had a significantly greater improvement with CBT than with acupuncture.
However, among patients who had moderate to severe insomnia at baseline, there was no significant difference in results with CBT and acupuncture.
Jun J. Mao, MD, of Memorial Sloan Kettering Cancer Center in New York, New York, presented these results in a press briefing in advance of the 2018 ASCO Annual Meeting.
Additional results are scheduled to be presented at the meeting as abstract 10001.
“Up to 60% of cancer survivors have some form of insomnia, but it is often underdiagnosed and undertreated,” Dr Mao said. “Our trial showed that both CBT-I [CBT for insomnia] and acupuncture were effective in treating moderate to severe insomnia, although CBT-I was more effective for those with mild symptoms of insomnia. Now, patients have more choices to manage their insomnia.”
Dr Mao and his colleagues studied 160 individuals who had completed cancer treatment. Their mean time since cancer diagnosis was about 6 years.
The subjects had received treatment for hematologic malignancies (8%) as well as breast (31%), prostate (23%), head and neck (7%), colorectal (6%), gynecological (4%), and other cancers (14%). Six percent of subjects had been treated for more than 1 type of cancer.
All subjects had been clinically diagnosed with insomnia. Seventy-nine percent had moderate (n=94) to severe (n=33) insomnia, and 21% had mild insomnia (n=33).
Interventions
Subjects were randomized to receive CBT or acupuncture for 8 weeks. Those who received CBT worked with a therapist to re-establish a restorative sleep schedule by:
- Reducing the amount of time spent in bed
- Limiting activities performed in bed to sleep and sexual activity
- Modifying unhelpful beliefs about sleep
- Promoting good sleep hygiene (setting a regular sleep schedule and avoiding activities that include light from tablets and cellphones, eating too late, and performing vigorous activities).
The study’s primary outcome was reduction in insomnia severity, as measured by the Insomnia Severity Index (ISI), from study entry to the end of treatment at week 8. Subjects were also reassessed at 20 weeks.
The ISI is a questionnaire that asks people to rate the severity of insomnia problems, such as difficulty falling asleep and staying asleep, and the impact of insomnia on their daily functioning and quality of life.
ISI scoring ranges from 0 to 28. Scores of 0 to 7 denote no clinically significant insomnia, 8 to 14 denote mild insomnia, 15 to 21 denote moderate insomnia, and 22 to 28 denote severe insomnia.
Results
At 8 weeks, the mean ISI score fell 10.9 points (from 18.5 to 7.5) for subjects who received CBT and 8.3 points (from 17.55 to 9.23) for those who received acupuncture (P=0.0007).
Among subjects with mild insomnia at baseline, far more responded to CBT than to acupuncture—85% and 18%, respectively (P<0.0001).
However, response rates were similar in subjects who had moderate to severe insomnia at baseline. Seventy-five percent of those who received CBT achieved a response, as did 66% of those who received acupuncture (P=0.26).
Both treatment groups had few mild adverse events, and all subjects maintained their improvements in insomnia at the 20-week assessment.
This study was funded by the Patient-Centered Outcomes Research Institute. The researchers’ disclosures are listed with the abstract.
Treatment with acupuncture or cognitive behavioral therapy (CBT) can decrease the severity of insomnia among cancer survivors, according to new research.
Overall, improvements in insomnia were greatest in patients treated with CBT, and patients with mild insomnia at baseline had a significantly greater improvement with CBT than with acupuncture.
However, among patients who had moderate to severe insomnia at baseline, there was no significant difference in results with CBT and acupuncture.
Jun J. Mao, MD, of Memorial Sloan Kettering Cancer Center in New York, New York, presented these results in a press briefing in advance of the 2018 ASCO Annual Meeting.
Additional results are scheduled to be presented at the meeting as abstract 10001.
“Up to 60% of cancer survivors have some form of insomnia, but it is often underdiagnosed and undertreated,” Dr Mao said. “Our trial showed that both CBT-I [CBT for insomnia] and acupuncture were effective in treating moderate to severe insomnia, although CBT-I was more effective for those with mild symptoms of insomnia. Now, patients have more choices to manage their insomnia.”
Dr Mao and his colleagues studied 160 individuals who had completed cancer treatment. Their mean time since cancer diagnosis was about 6 years.
The subjects had received treatment for hematologic malignancies (8%) as well as breast (31%), prostate (23%), head and neck (7%), colorectal (6%), gynecological (4%), and other cancers (14%). Six percent of subjects had been treated for more than 1 type of cancer.
All subjects had been clinically diagnosed with insomnia. Seventy-nine percent had moderate (n=94) to severe (n=33) insomnia, and 21% had mild insomnia (n=33).
Interventions
Subjects were randomized to receive CBT or acupuncture for 8 weeks. Those who received CBT worked with a therapist to re-establish a restorative sleep schedule by:
- Reducing the amount of time spent in bed
- Limiting activities performed in bed to sleep and sexual activity
- Modifying unhelpful beliefs about sleep
- Promoting good sleep hygiene (setting a regular sleep schedule and avoiding activities that include light from tablets and cellphones, eating too late, and performing vigorous activities).
The study’s primary outcome was reduction in insomnia severity, as measured by the Insomnia Severity Index (ISI), from study entry to the end of treatment at week 8. Subjects were also reassessed at 20 weeks.
The ISI is a questionnaire that asks people to rate the severity of insomnia problems, such as difficulty falling asleep and staying asleep, and the impact of insomnia on their daily functioning and quality of life.
ISI scoring ranges from 0 to 28. Scores of 0 to 7 denote no clinically significant insomnia, 8 to 14 denote mild insomnia, 15 to 21 denote moderate insomnia, and 22 to 28 denote severe insomnia.
Results
At 8 weeks, the mean ISI score fell 10.9 points (from 18.5 to 7.5) for subjects who received CBT and 8.3 points (from 17.55 to 9.23) for those who received acupuncture (P=0.0007).
Among subjects with mild insomnia at baseline, far more responded to CBT than to acupuncture—85% and 18%, respectively (P<0.0001).
However, response rates were similar in subjects who had moderate to severe insomnia at baseline. Seventy-five percent of those who received CBT achieved a response, as did 66% of those who received acupuncture (P=0.26).
Both treatment groups had few mild adverse events, and all subjects maintained their improvements in insomnia at the 20-week assessment.
This study was funded by the Patient-Centered Outcomes Research Institute. The researchers’ disclosures are listed with the abstract.
FDA approves first epoetin alfa biosimilar

The US Food and Drug Administration (FDA) has approved epoetin alfa-epbx (Retacrit), a biosimilar to epoetin alfa (Epogen/Procrit).
Epoetin alfa-epbx is approved for the treatment of anemia caused by chronic kidney disease, the use of zidovudine in patients with HIV infection, and myelosuppressive chemotherapy in patients who have a minimum of 2 additional months of planned chemotherapy.
Epoetin alfa-epbx is also approved for use before and after surgery to reduce the chance that red blood cell transfusions will be needed because of blood loss during elective, noncardiac, or nonvascular surgery.
As with epoetin alfa, the prescribing information for epoetin alfa-epbx contains a Boxed Warning noting that erythropoiesis-stimulating agents increase the risk of death, myocardial infarction, stroke, venous thromboembolism, thrombosis of vascular access, and tumor progression or recurrence.
The FDA granted approval of epoetin alfa-epbx to Hospira Inc., a Pfizer company.
The agency’s approval is based on a review of evidence that included structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data.
This evidence is available in an FDA briefing document on the biologics license application for epoetin alfa-epbx.
The US Food and Drug Administration (FDA) has approved epoetin alfa-epbx (Retacrit), a biosimilar to epoetin alfa (Epogen/Procrit).
Epoetin alfa-epbx is approved for the treatment of anemia caused by chronic kidney disease, the use of zidovudine in patients with HIV infection, and myelosuppressive chemotherapy in patients who have a minimum of 2 additional months of planned chemotherapy.
Epoetin alfa-epbx is also approved for use before and after surgery to reduce the chance that red blood cell transfusions will be needed because of blood loss during elective, noncardiac, or nonvascular surgery.
As with epoetin alfa, the prescribing information for epoetin alfa-epbx contains a Boxed Warning noting that erythropoiesis-stimulating agents increase the risk of death, myocardial infarction, stroke, venous thromboembolism, thrombosis of vascular access, and tumor progression or recurrence.
The FDA granted approval of epoetin alfa-epbx to Hospira Inc., a Pfizer company.
The agency’s approval is based on a review of evidence that included structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data.
This evidence is available in an FDA briefing document on the biologics license application for epoetin alfa-epbx.
The US Food and Drug Administration (FDA) has approved epoetin alfa-epbx (Retacrit), a biosimilar to epoetin alfa (Epogen/Procrit).
Epoetin alfa-epbx is approved for the treatment of anemia caused by chronic kidney disease, the use of zidovudine in patients with HIV infection, and myelosuppressive chemotherapy in patients who have a minimum of 2 additional months of planned chemotherapy.
Epoetin alfa-epbx is also approved for use before and after surgery to reduce the chance that red blood cell transfusions will be needed because of blood loss during elective, noncardiac, or nonvascular surgery.
As with epoetin alfa, the prescribing information for epoetin alfa-epbx contains a Boxed Warning noting that erythropoiesis-stimulating agents increase the risk of death, myocardial infarction, stroke, venous thromboembolism, thrombosis of vascular access, and tumor progression or recurrence.
The FDA granted approval of epoetin alfa-epbx to Hospira Inc., a Pfizer company.
The agency’s approval is based on a review of evidence that included structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data.
This evidence is available in an FDA briefing document on the biologics license application for epoetin alfa-epbx.
Team analyzes skin odor to detect malaria
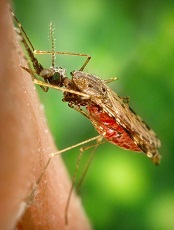
Changes in skin odor can reveal malaria infection in patients with no external symptoms, according to research published in PNAS.
Researchers examined chemical compounds released from the skin of Kenyan children and discovered characteristic patterns in these compounds that identified patients with acute and asymptomatic malaria infections.
“Our previous work in a mouse model found that malaria infection altered the odors of infected mice in ways that made them more attractive to mosquitoes, particularly at a stage of infection where the transmissible stage of the parasite was present at high levels,” said study author Consuelo De Moraes, PhD, of ETH Zurich in Switzerland.
“We also found long-term changes in the odor profiles of infected mice. [So] we had reason to hope that similar changes in human odors might provide biomarkers that could be used for diagnosis.”
To test this theory, Dr De Moraes and her colleagues studied more than 400 Kenyan school children. The researchers collected blood samples as well as samples of volatile substances released from the subjects’ skin.
The team used the blood samples to test for malaria, first via light microscopy and an SD Bioline Rapid Diagnostic Test, then using polymerase chain reaction (PCR) methods to confirm the initial results.
There were 330 subjects who were clearly positive or negative for malaria, and there were 66 subjects who were positive by PCR but negative by microscopy. The researchers compared these findings to results from the skin tests.
To assess the subjects’ skin, the researchers placed each child’s foot and arm into sealed Teflon bags and passed an air current over the skin for about 1 hour. The air was then channeled through special filters that collected the volatile compounds.
Using gas chromatography and mass spectrometry, the researchers then determined the identity and quantity of each compound to generate odor profiles for infected and uninfected children.
Further analysis of these profiles revealed volatile biomarkers that enabled the researchers to accurately identify whether a child was infected with the malaria parasite. Even for asymptomatic infections, the detection rate was close to 100%.
“This high detection rate was encouraging,” Dr De Moraes said. “Initially, we weren’t sure which chemical compounds we should be looking for.”
The researchers noted that malaria infection does not create new chemical compounds, but it alters the amounts of compounds that are already present in the odors of healthy people.
Odor profiles were different for malaria-infected and uninfected subjects, but profiles were also different for patients with acute and asymptomatic infections.
The researchers hope the biomarkers they identified could be used to develop a new tool for the early detection of malaria.
“These new volatile biomarkers are an important first step,” said Mark Mescher, of ETH Zurich. “Now, someone needs to develop an application that can be used cheaply and reliably in the field.”
“In the near-term, our goal is to refine the current findings to find the most reliable and effective biomarkers we can. There is still a lot more work to be done to develop a practical diagnostic assay.”
Changes in skin odor can reveal malaria infection in patients with no external symptoms, according to research published in PNAS.
Researchers examined chemical compounds released from the skin of Kenyan children and discovered characteristic patterns in these compounds that identified patients with acute and asymptomatic malaria infections.
“Our previous work in a mouse model found that malaria infection altered the odors of infected mice in ways that made them more attractive to mosquitoes, particularly at a stage of infection where the transmissible stage of the parasite was present at high levels,” said study author Consuelo De Moraes, PhD, of ETH Zurich in Switzerland.
“We also found long-term changes in the odor profiles of infected mice. [So] we had reason to hope that similar changes in human odors might provide biomarkers that could be used for diagnosis.”
To test this theory, Dr De Moraes and her colleagues studied more than 400 Kenyan school children. The researchers collected blood samples as well as samples of volatile substances released from the subjects’ skin.
The team used the blood samples to test for malaria, first via light microscopy and an SD Bioline Rapid Diagnostic Test, then using polymerase chain reaction (PCR) methods to confirm the initial results.
There were 330 subjects who were clearly positive or negative for malaria, and there were 66 subjects who were positive by PCR but negative by microscopy. The researchers compared these findings to results from the skin tests.
To assess the subjects’ skin, the researchers placed each child’s foot and arm into sealed Teflon bags and passed an air current over the skin for about 1 hour. The air was then channeled through special filters that collected the volatile compounds.
Using gas chromatography and mass spectrometry, the researchers then determined the identity and quantity of each compound to generate odor profiles for infected and uninfected children.
Further analysis of these profiles revealed volatile biomarkers that enabled the researchers to accurately identify whether a child was infected with the malaria parasite. Even for asymptomatic infections, the detection rate was close to 100%.
“This high detection rate was encouraging,” Dr De Moraes said. “Initially, we weren’t sure which chemical compounds we should be looking for.”
The researchers noted that malaria infection does not create new chemical compounds, but it alters the amounts of compounds that are already present in the odors of healthy people.
Odor profiles were different for malaria-infected and uninfected subjects, but profiles were also different for patients with acute and asymptomatic infections.
The researchers hope the biomarkers they identified could be used to develop a new tool for the early detection of malaria.
“These new volatile biomarkers are an important first step,” said Mark Mescher, of ETH Zurich. “Now, someone needs to develop an application that can be used cheaply and reliably in the field.”
“In the near-term, our goal is to refine the current findings to find the most reliable and effective biomarkers we can. There is still a lot more work to be done to develop a practical diagnostic assay.”
Changes in skin odor can reveal malaria infection in patients with no external symptoms, according to research published in PNAS.
Researchers examined chemical compounds released from the skin of Kenyan children and discovered characteristic patterns in these compounds that identified patients with acute and asymptomatic malaria infections.
“Our previous work in a mouse model found that malaria infection altered the odors of infected mice in ways that made them more attractive to mosquitoes, particularly at a stage of infection where the transmissible stage of the parasite was present at high levels,” said study author Consuelo De Moraes, PhD, of ETH Zurich in Switzerland.
“We also found long-term changes in the odor profiles of infected mice. [So] we had reason to hope that similar changes in human odors might provide biomarkers that could be used for diagnosis.”
To test this theory, Dr De Moraes and her colleagues studied more than 400 Kenyan school children. The researchers collected blood samples as well as samples of volatile substances released from the subjects’ skin.
The team used the blood samples to test for malaria, first via light microscopy and an SD Bioline Rapid Diagnostic Test, then using polymerase chain reaction (PCR) methods to confirm the initial results.
There were 330 subjects who were clearly positive or negative for malaria, and there were 66 subjects who were positive by PCR but negative by microscopy. The researchers compared these findings to results from the skin tests.
To assess the subjects’ skin, the researchers placed each child’s foot and arm into sealed Teflon bags and passed an air current over the skin for about 1 hour. The air was then channeled through special filters that collected the volatile compounds.
Using gas chromatography and mass spectrometry, the researchers then determined the identity and quantity of each compound to generate odor profiles for infected and uninfected children.
Further analysis of these profiles revealed volatile biomarkers that enabled the researchers to accurately identify whether a child was infected with the malaria parasite. Even for asymptomatic infections, the detection rate was close to 100%.
“This high detection rate was encouraging,” Dr De Moraes said. “Initially, we weren’t sure which chemical compounds we should be looking for.”
The researchers noted that malaria infection does not create new chemical compounds, but it alters the amounts of compounds that are already present in the odors of healthy people.
Odor profiles were different for malaria-infected and uninfected subjects, but profiles were also different for patients with acute and asymptomatic infections.
The researchers hope the biomarkers they identified could be used to develop a new tool for the early detection of malaria.
“These new volatile biomarkers are an important first step,” said Mark Mescher, of ETH Zurich. “Now, someone needs to develop an application that can be used cheaply and reliably in the field.”
“In the near-term, our goal is to refine the current findings to find the most reliable and effective biomarkers we can. There is still a lot more work to be done to develop a practical diagnostic assay.”
Umbralisib has ‘distinct’ safety profile

Phase 1 trial results suggest umbralisib, a PI3Kδ/CK1ε inhibitor, can be safe and active in patients with relapsed or refractory B-cell malignancies.
Researchers said the safety profile of umbralisib “was distinct from that of other PI3Kδ inhibitors,” as it produced few immune-mediated adverse events (AEs).
Umbralisib also produced an objective response rate of 37% in the entire study cohort, 80% in patients with chronic lymphocytic leukemia (CLL), 53% in patients with follicular lymphoma (FL), and 31% in patients with diffuse large B-cell lymphoma (DLBCL).
These results were published in The Lancet Oncology. The study was sponsored by TG Therapeutics, Inc.
The trial enrolled 90 patients between January 17, 2013, and January 14, 2016.
There were 24 patients with CLL, 22 with FL, 16 with DLBCL, 11 with Hodgkin lymphoma, 6 with mantle cell lymphoma, 5 with marginal zone lymphoma, 3 with Waldenstrom’s macroglobulinemia, 2 with T-cell lymphoma, and 1 with hairy cell leukemia.
The median number of prior therapies was 3 (range, 2-5), and 49% of patients were refractory to previous therapy.
Treatment
Patients took umbralisib once daily in 28-day cycles until disease progression, unacceptable toxicity, or withdrawal of consent.
Initially, patients took the drug in a fasting state at doses of 50 mg, 100 mg, 200 mg, 400 mg, 800 mg, 1200 mg, or 1800 mg.
In April 2014, the researchers did a second dose-escalation with a micronized formulation of umbralisib, taken with food, at doses of 200 mg, 400 mg, 800 mg, 1200 mg, or 1800 mg.
In August, 2014, all patients who were still on the study transitioned to the 800 mg dose of the micronized formulation. This was the recommended phase 2 dose.
At the data cutoff in November 2016, 44 patients (49%) had received umbralisib for more than 6 cycles, and 23 (26%) had received the drug for more than 12 cycles. Thirteen patients (14%) were still taking umbralisib at the end of the study.
Most patients who stopped treatment did so because of disease progression (n=50, 56%) or AEs (n=9, 10%).
“We are pleased to have treated the first patient ever with umbralisib over 5 years ago and believe it has an important place in the treatment landscape for patients with hematologic malignancies,” said study author Howard A. Burris, MD, of the Sarah Cannon Research Institute in Nashville, Tennessee.
“Several patients from this phase 1 study are still on study today, approaching 5 years of continuous daily therapy, speaking to both the safety and efficacy profile of this unique agent.”
Safety
Dose-limiting toxicities (DLTs) occurred in 4 patients. One DLT was grade 3 maculopapular rash in a patient receiving the 800 mg dose of the initial formulation.
Another DLT was grade 3 hypokalemia in a patient receiving 1800 mg of the initial formulation. A third DLT was grade 3 fatigue, which occurred in 2 patients receiving 1800 mg of the micronized formulation.
Because of these toxicities, the maximum tolerated dose was 1200 mg of the micronized formulation.
The most common treatment-emergent AEs were diarrhea (43%), nausea (42%), and fatigue (31%). The most common grade 3/4 AEs were neutropenia (13%), anemia (9%), and thrombocytopenia (7%).
Serious AEs considered at least possibly related to umbralisib were pneumonia (3%), lung infection (1%), febrile neutropenia (1%), and colitis (2%).
Treatment discontinuation due to AEs considered at least possibly related to umbralisib occurred in 6 patients (7%). Two patients had grade 3 colitis, 2 had increased ALT/AST (grade 1 and grade 4), 1 had grade 2 diarrhea, and 1 had grade 3 fatigue.
There were no treatment-related deaths.
The researchers said the safety profile of umbralisib was distinct from that of other PI3Kδ inhibitors, as patients in this trial had fewer occurrences of autoimmune-like toxicities, such as colitis.
“Preclinically, umbralisib has a very unique profile, selectively inhibiting both PI3Kδ and CK1ε,” said study author Owen O’Connor, MD, PhD, of Columbia Presbyterian Medical Center in New York, New York.
“The clinical results in this paper support our thesis that the differentiated preclinical profile explains the differences seen in the clinic between umbralisib and the other PI3Kδ inhibitors.”
Response
The objective response rate was 37%, with 33 patients achieving a response and 3 patients having a complete response (CR).
Sixteen CLL patients responded (80%), all with partial responses (PRs). Four DLBCL patients responded (31%), all with PRs. And 9 FL patients responded (53%), 2 with CRs.
The remaining CR occurred in a Hodgkin lymphoma patient, and this was the only response in this patient group.
One patient with marginal zone lymphoma had a PR, as did 1 patient with mantle cell lymphoma. All other patients had stable disease or progressed.
The mean duration of response was 13.4 months in the CLL patients, 6.4 months in the DLBCL patients, and 9.3 months in the FL patients.
Phase 1 trial results suggest umbralisib, a PI3Kδ/CK1ε inhibitor, can be safe and active in patients with relapsed or refractory B-cell malignancies.
Researchers said the safety profile of umbralisib “was distinct from that of other PI3Kδ inhibitors,” as it produced few immune-mediated adverse events (AEs).
Umbralisib also produced an objective response rate of 37% in the entire study cohort, 80% in patients with chronic lymphocytic leukemia (CLL), 53% in patients with follicular lymphoma (FL), and 31% in patients with diffuse large B-cell lymphoma (DLBCL).
These results were published in The Lancet Oncology. The study was sponsored by TG Therapeutics, Inc.
The trial enrolled 90 patients between January 17, 2013, and January 14, 2016.
There were 24 patients with CLL, 22 with FL, 16 with DLBCL, 11 with Hodgkin lymphoma, 6 with mantle cell lymphoma, 5 with marginal zone lymphoma, 3 with Waldenstrom’s macroglobulinemia, 2 with T-cell lymphoma, and 1 with hairy cell leukemia.
The median number of prior therapies was 3 (range, 2-5), and 49% of patients were refractory to previous therapy.
Treatment
Patients took umbralisib once daily in 28-day cycles until disease progression, unacceptable toxicity, or withdrawal of consent.
Initially, patients took the drug in a fasting state at doses of 50 mg, 100 mg, 200 mg, 400 mg, 800 mg, 1200 mg, or 1800 mg.
In April 2014, the researchers did a second dose-escalation with a micronized formulation of umbralisib, taken with food, at doses of 200 mg, 400 mg, 800 mg, 1200 mg, or 1800 mg.
In August, 2014, all patients who were still on the study transitioned to the 800 mg dose of the micronized formulation. This was the recommended phase 2 dose.
At the data cutoff in November 2016, 44 patients (49%) had received umbralisib for more than 6 cycles, and 23 (26%) had received the drug for more than 12 cycles. Thirteen patients (14%) were still taking umbralisib at the end of the study.
Most patients who stopped treatment did so because of disease progression (n=50, 56%) or AEs (n=9, 10%).
“We are pleased to have treated the first patient ever with umbralisib over 5 years ago and believe it has an important place in the treatment landscape for patients with hematologic malignancies,” said study author Howard A. Burris, MD, of the Sarah Cannon Research Institute in Nashville, Tennessee.
“Several patients from this phase 1 study are still on study today, approaching 5 years of continuous daily therapy, speaking to both the safety and efficacy profile of this unique agent.”
Safety
Dose-limiting toxicities (DLTs) occurred in 4 patients. One DLT was grade 3 maculopapular rash in a patient receiving the 800 mg dose of the initial formulation.
Another DLT was grade 3 hypokalemia in a patient receiving 1800 mg of the initial formulation. A third DLT was grade 3 fatigue, which occurred in 2 patients receiving 1800 mg of the micronized formulation.
Because of these toxicities, the maximum tolerated dose was 1200 mg of the micronized formulation.
The most common treatment-emergent AEs were diarrhea (43%), nausea (42%), and fatigue (31%). The most common grade 3/4 AEs were neutropenia (13%), anemia (9%), and thrombocytopenia (7%).
Serious AEs considered at least possibly related to umbralisib were pneumonia (3%), lung infection (1%), febrile neutropenia (1%), and colitis (2%).
Treatment discontinuation due to AEs considered at least possibly related to umbralisib occurred in 6 patients (7%). Two patients had grade 3 colitis, 2 had increased ALT/AST (grade 1 and grade 4), 1 had grade 2 diarrhea, and 1 had grade 3 fatigue.
There were no treatment-related deaths.
The researchers said the safety profile of umbralisib was distinct from that of other PI3Kδ inhibitors, as patients in this trial had fewer occurrences of autoimmune-like toxicities, such as colitis.
“Preclinically, umbralisib has a very unique profile, selectively inhibiting both PI3Kδ and CK1ε,” said study author Owen O’Connor, MD, PhD, of Columbia Presbyterian Medical Center in New York, New York.
“The clinical results in this paper support our thesis that the differentiated preclinical profile explains the differences seen in the clinic between umbralisib and the other PI3Kδ inhibitors.”
Response
The objective response rate was 37%, with 33 patients achieving a response and 3 patients having a complete response (CR).
Sixteen CLL patients responded (80%), all with partial responses (PRs). Four DLBCL patients responded (31%), all with PRs. And 9 FL patients responded (53%), 2 with CRs.
The remaining CR occurred in a Hodgkin lymphoma patient, and this was the only response in this patient group.
One patient with marginal zone lymphoma had a PR, as did 1 patient with mantle cell lymphoma. All other patients had stable disease or progressed.
The mean duration of response was 13.4 months in the CLL patients, 6.4 months in the DLBCL patients, and 9.3 months in the FL patients.
Phase 1 trial results suggest umbralisib, a PI3Kδ/CK1ε inhibitor, can be safe and active in patients with relapsed or refractory B-cell malignancies.
Researchers said the safety profile of umbralisib “was distinct from that of other PI3Kδ inhibitors,” as it produced few immune-mediated adverse events (AEs).
Umbralisib also produced an objective response rate of 37% in the entire study cohort, 80% in patients with chronic lymphocytic leukemia (CLL), 53% in patients with follicular lymphoma (FL), and 31% in patients with diffuse large B-cell lymphoma (DLBCL).
These results were published in The Lancet Oncology. The study was sponsored by TG Therapeutics, Inc.
The trial enrolled 90 patients between January 17, 2013, and January 14, 2016.
There were 24 patients with CLL, 22 with FL, 16 with DLBCL, 11 with Hodgkin lymphoma, 6 with mantle cell lymphoma, 5 with marginal zone lymphoma, 3 with Waldenstrom’s macroglobulinemia, 2 with T-cell lymphoma, and 1 with hairy cell leukemia.
The median number of prior therapies was 3 (range, 2-5), and 49% of patients were refractory to previous therapy.
Treatment
Patients took umbralisib once daily in 28-day cycles until disease progression, unacceptable toxicity, or withdrawal of consent.
Initially, patients took the drug in a fasting state at doses of 50 mg, 100 mg, 200 mg, 400 mg, 800 mg, 1200 mg, or 1800 mg.
In April 2014, the researchers did a second dose-escalation with a micronized formulation of umbralisib, taken with food, at doses of 200 mg, 400 mg, 800 mg, 1200 mg, or 1800 mg.
In August, 2014, all patients who were still on the study transitioned to the 800 mg dose of the micronized formulation. This was the recommended phase 2 dose.
At the data cutoff in November 2016, 44 patients (49%) had received umbralisib for more than 6 cycles, and 23 (26%) had received the drug for more than 12 cycles. Thirteen patients (14%) were still taking umbralisib at the end of the study.
Most patients who stopped treatment did so because of disease progression (n=50, 56%) or AEs (n=9, 10%).
“We are pleased to have treated the first patient ever with umbralisib over 5 years ago and believe it has an important place in the treatment landscape for patients with hematologic malignancies,” said study author Howard A. Burris, MD, of the Sarah Cannon Research Institute in Nashville, Tennessee.
“Several patients from this phase 1 study are still on study today, approaching 5 years of continuous daily therapy, speaking to both the safety and efficacy profile of this unique agent.”
Safety
Dose-limiting toxicities (DLTs) occurred in 4 patients. One DLT was grade 3 maculopapular rash in a patient receiving the 800 mg dose of the initial formulation.
Another DLT was grade 3 hypokalemia in a patient receiving 1800 mg of the initial formulation. A third DLT was grade 3 fatigue, which occurred in 2 patients receiving 1800 mg of the micronized formulation.
Because of these toxicities, the maximum tolerated dose was 1200 mg of the micronized formulation.
The most common treatment-emergent AEs were diarrhea (43%), nausea (42%), and fatigue (31%). The most common grade 3/4 AEs were neutropenia (13%), anemia (9%), and thrombocytopenia (7%).
Serious AEs considered at least possibly related to umbralisib were pneumonia (3%), lung infection (1%), febrile neutropenia (1%), and colitis (2%).
Treatment discontinuation due to AEs considered at least possibly related to umbralisib occurred in 6 patients (7%). Two patients had grade 3 colitis, 2 had increased ALT/AST (grade 1 and grade 4), 1 had grade 2 diarrhea, and 1 had grade 3 fatigue.
There were no treatment-related deaths.
The researchers said the safety profile of umbralisib was distinct from that of other PI3Kδ inhibitors, as patients in this trial had fewer occurrences of autoimmune-like toxicities, such as colitis.
“Preclinically, umbralisib has a very unique profile, selectively inhibiting both PI3Kδ and CK1ε,” said study author Owen O’Connor, MD, PhD, of Columbia Presbyterian Medical Center in New York, New York.
“The clinical results in this paper support our thesis that the differentiated preclinical profile explains the differences seen in the clinic between umbralisib and the other PI3Kδ inhibitors.”
Response
The objective response rate was 37%, with 33 patients achieving a response and 3 patients having a complete response (CR).
Sixteen CLL patients responded (80%), all with partial responses (PRs). Four DLBCL patients responded (31%), all with PRs. And 9 FL patients responded (53%), 2 with CRs.
The remaining CR occurred in a Hodgkin lymphoma patient, and this was the only response in this patient group.
One patient with marginal zone lymphoma had a PR, as did 1 patient with mantle cell lymphoma. All other patients had stable disease or progressed.
The mean duration of response was 13.4 months in the CLL patients, 6.4 months in the DLBCL patients, and 9.3 months in the FL patients.
FDA approves new use for Zika test
The US Food and Drug Administration (FDA) has approved an additional use for the cobas Zika test.
The approval allows for the streamlined screening of multiple individual blood or plasma donations that have been pooled together.
The cobas Zika test is a qualitative in vitro nucleic acid screening test for the direct detection of Zika virus RNA in plasma specimens from blood donors.
The test is used with the cobas 6800/8800 systems, which are fully automated, high-volume systems that perform sample pooling, sample preparation (nucleic acid extraction and purification), and polymerase chain reaction amplification and detection.
Automated data management is performed by the cobas 6800/8800 software, which assigns a test result of non-reactive, reactive, or invalid.
Roche deployed the cobas Zika test in April 2016 under the FDA’s investigational new drug application protocol to screen blood donations collected in Puerto Rico.
This initial testing protocol enabled the reinstatement of blood services in Puerto Rico after concerns over high rates of Zika infection posed a threat to the blood supply.
The cobas Zika test received commercial approval from the FDA in October 2017, enabling routine use of the test to support individual donor screening efforts throughout Puerto Rico and the continental US.
“More than 6 million blood donations from the United States and Puerto Rico have been screened with the cobas Zika test since its initial release under the investigational new drug application protocol in 2016 and subsequent commercial approval in 2017,” said Uwe Oberlaender, head of Roche Molecular Diagnostics.
The US Food and Drug Administration (FDA) has approved an additional use for the cobas Zika test.
The approval allows for the streamlined screening of multiple individual blood or plasma donations that have been pooled together.
The cobas Zika test is a qualitative in vitro nucleic acid screening test for the direct detection of Zika virus RNA in plasma specimens from blood donors.
The test is used with the cobas 6800/8800 systems, which are fully automated, high-volume systems that perform sample pooling, sample preparation (nucleic acid extraction and purification), and polymerase chain reaction amplification and detection.
Automated data management is performed by the cobas 6800/8800 software, which assigns a test result of non-reactive, reactive, or invalid.
Roche deployed the cobas Zika test in April 2016 under the FDA’s investigational new drug application protocol to screen blood donations collected in Puerto Rico.
This initial testing protocol enabled the reinstatement of blood services in Puerto Rico after concerns over high rates of Zika infection posed a threat to the blood supply.
The cobas Zika test received commercial approval from the FDA in October 2017, enabling routine use of the test to support individual donor screening efforts throughout Puerto Rico and the continental US.
“More than 6 million blood donations from the United States and Puerto Rico have been screened with the cobas Zika test since its initial release under the investigational new drug application protocol in 2016 and subsequent commercial approval in 2017,” said Uwe Oberlaender, head of Roche Molecular Diagnostics.
The US Food and Drug Administration (FDA) has approved an additional use for the cobas Zika test.
The approval allows for the streamlined screening of multiple individual blood or plasma donations that have been pooled together.
The cobas Zika test is a qualitative in vitro nucleic acid screening test for the direct detection of Zika virus RNA in plasma specimens from blood donors.
The test is used with the cobas 6800/8800 systems, which are fully automated, high-volume systems that perform sample pooling, sample preparation (nucleic acid extraction and purification), and polymerase chain reaction amplification and detection.
Automated data management is performed by the cobas 6800/8800 software, which assigns a test result of non-reactive, reactive, or invalid.
Roche deployed the cobas Zika test in April 2016 under the FDA’s investigational new drug application protocol to screen blood donations collected in Puerto Rico.
This initial testing protocol enabled the reinstatement of blood services in Puerto Rico after concerns over high rates of Zika infection posed a threat to the blood supply.
The cobas Zika test received commercial approval from the FDA in October 2017, enabling routine use of the test to support individual donor screening efforts throughout Puerto Rico and the continental US.
“More than 6 million blood donations from the United States and Puerto Rico have been screened with the cobas Zika test since its initial release under the investigational new drug application protocol in 2016 and subsequent commercial approval in 2017,” said Uwe Oberlaender, head of Roche Molecular Diagnostics.
Study reveals gender imbalance in cancer research funding

A new study suggests male investigators in the UK receive more funding for cancer research than their female counterparts.
Researchers analyzed funding for more than 4000 studies and found that male primary investigators (PIs) were consistently awarded more funding.
The total investment value was 3.6 times greater for male PIs than for female PIs.
The mean award value was 1.6 times greater, and the median award value was 1.3 times greater for males.
Rifat Atun, MBBS, of Harvard T. H. Chan School of Public Health in Boston, Massachusetts, and his colleagues reported these findings in BMJ Open.
The researchers analyzed data on public and philanthropic cancer research funding awarded to UK institutions between 2000 and 2013.
From this data, the team identified 4186 eligible studies with a total investment value of £2.33 billion.
The researchers compared the total investment, number of awards, and mean and median award value between male and female PIs.
Male PIs were awarded 2890 (69%) grants with a total value of £1.8 billion (78%), while female PIs were awarded 1296 (31%) grants with a total value of £0.5 billion (22%).
The median award value was £252,647 (interquartile range, £127,343–£553,560) for men and £198,485 (interquartile range, £99,317–£382,650) for women.
The mean award value was £630,324 (standard deviation, £1,662,559) for men and £394,730 (standard deviation, £666,574) for women.
Dr Atun and his colleagues acknowledged that this study was dependent on the accuracy of original investment data from the funding bodies, and the team could not openly access data of private sector research funding nor obtain disaggregated data from Cancer Research UK, one of the largest funders of cancer research.
The researchers also noted that the gender discrepancies they observed “are likely multifactorial,” and the team was unable to “postulate the underlying mechanisms responsible.” Still, the data do suggest a “substantial” imbalance, which should be investigated.
A new study suggests male investigators in the UK receive more funding for cancer research than their female counterparts.
Researchers analyzed funding for more than 4000 studies and found that male primary investigators (PIs) were consistently awarded more funding.
The total investment value was 3.6 times greater for male PIs than for female PIs.
The mean award value was 1.6 times greater, and the median award value was 1.3 times greater for males.
Rifat Atun, MBBS, of Harvard T. H. Chan School of Public Health in Boston, Massachusetts, and his colleagues reported these findings in BMJ Open.
The researchers analyzed data on public and philanthropic cancer research funding awarded to UK institutions between 2000 and 2013.
From this data, the team identified 4186 eligible studies with a total investment value of £2.33 billion.
The researchers compared the total investment, number of awards, and mean and median award value between male and female PIs.
Male PIs were awarded 2890 (69%) grants with a total value of £1.8 billion (78%), while female PIs were awarded 1296 (31%) grants with a total value of £0.5 billion (22%).
The median award value was £252,647 (interquartile range, £127,343–£553,560) for men and £198,485 (interquartile range, £99,317–£382,650) for women.
The mean award value was £630,324 (standard deviation, £1,662,559) for men and £394,730 (standard deviation, £666,574) for women.
Dr Atun and his colleagues acknowledged that this study was dependent on the accuracy of original investment data from the funding bodies, and the team could not openly access data of private sector research funding nor obtain disaggregated data from Cancer Research UK, one of the largest funders of cancer research.
The researchers also noted that the gender discrepancies they observed “are likely multifactorial,” and the team was unable to “postulate the underlying mechanisms responsible.” Still, the data do suggest a “substantial” imbalance, which should be investigated.
A new study suggests male investigators in the UK receive more funding for cancer research than their female counterparts.
Researchers analyzed funding for more than 4000 studies and found that male primary investigators (PIs) were consistently awarded more funding.
The total investment value was 3.6 times greater for male PIs than for female PIs.
The mean award value was 1.6 times greater, and the median award value was 1.3 times greater for males.
Rifat Atun, MBBS, of Harvard T. H. Chan School of Public Health in Boston, Massachusetts, and his colleagues reported these findings in BMJ Open.
The researchers analyzed data on public and philanthropic cancer research funding awarded to UK institutions between 2000 and 2013.
From this data, the team identified 4186 eligible studies with a total investment value of £2.33 billion.
The researchers compared the total investment, number of awards, and mean and median award value between male and female PIs.
Male PIs were awarded 2890 (69%) grants with a total value of £1.8 billion (78%), while female PIs were awarded 1296 (31%) grants with a total value of £0.5 billion (22%).
The median award value was £252,647 (interquartile range, £127,343–£553,560) for men and £198,485 (interquartile range, £99,317–£382,650) for women.
The mean award value was £630,324 (standard deviation, £1,662,559) for men and £394,730 (standard deviation, £666,574) for women.
Dr Atun and his colleagues acknowledged that this study was dependent on the accuracy of original investment data from the funding bodies, and the team could not openly access data of private sector research funding nor obtain disaggregated data from Cancer Research UK, one of the largest funders of cancer research.
The researchers also noted that the gender discrepancies they observed “are likely multifactorial,” and the team was unable to “postulate the underlying mechanisms responsible.” Still, the data do suggest a “substantial” imbalance, which should be investigated.
NCCN releases guidelines for AML patients

The National Comprehensive Cancer Network (NCCN) has released guidelines for patients with acute myeloid leukemia (AML).
The guidelines are intended to help patients make better informed decision about their care.
The guidelines explain what AML is, describe testing procedures and treatment options, and provide tips to help patients choose the best treatment.
The guidelines also include a list of suggested questions for patients to bring up with their doctors, illustrations, and a glossary of key terms and acronyms.
“People with AML often aren’t aware that this isn’t a single disease but many different diseases that share fundamental characteristics,” said Martin Tallman, MD, a hematologic oncologist at Memorial Sloan Kettering Cancer Center in New York, New York, and vice-chair of the NCCN Guidelines Panel for AML.
“The good news is that the future for AML is getting a lot brighter. After 40 years without much progress, 4 new medications were just approved last year*, and more new treatment courses are in development as we speak.”
The guidelines are available for free on the NCCN website or via the NCCN Patient Guides for Cancer app. Printed versions of the guidelines can be purchased at Amazon.com.
NCCN also has guidelines for patients with acute lymphoblastic leukemia, chronic lymphocytic leukemia, chronic myelogenous leukemia, Hodgkin lymphoma, multiple myeloma, myelodysplastic syndromes, myeloproliferative neoplasms, non-Hodgkin lymphomas, and Waldenström’s macroglobulinemia, as well as solid tumor malignancies.
There are patient guidelines for adolescents and young adults with cancer and guidelines on distress/supportive care, and nausea and vomiting/supportive care as well.
*(1) Midostaurin (Rydapt) was approved for adults with newly diagnosed, FLT3-positive AML
(2) Gemtuzumab ozogamicin (Mylotarg) was approved for adults with newly diagnosed AML and patients age 2 and older with relapsed/refractory AML
(3) Liposomal daunorubicin/cytarabine (Vyxeos) was approved for AML with myelodysplasia-related changes and newly diagnosed, therapy-related AML
(4) Enasidenib (Idhifa) was approved for adults with relapsed/refractory AML and an IDH2 mutation.
The National Comprehensive Cancer Network (NCCN) has released guidelines for patients with acute myeloid leukemia (AML).
The guidelines are intended to help patients make better informed decision about their care.
The guidelines explain what AML is, describe testing procedures and treatment options, and provide tips to help patients choose the best treatment.
The guidelines also include a list of suggested questions for patients to bring up with their doctors, illustrations, and a glossary of key terms and acronyms.
“People with AML often aren’t aware that this isn’t a single disease but many different diseases that share fundamental characteristics,” said Martin Tallman, MD, a hematologic oncologist at Memorial Sloan Kettering Cancer Center in New York, New York, and vice-chair of the NCCN Guidelines Panel for AML.
“The good news is that the future for AML is getting a lot brighter. After 40 years without much progress, 4 new medications were just approved last year*, and more new treatment courses are in development as we speak.”
The guidelines are available for free on the NCCN website or via the NCCN Patient Guides for Cancer app. Printed versions of the guidelines can be purchased at Amazon.com.
NCCN also has guidelines for patients with acute lymphoblastic leukemia, chronic lymphocytic leukemia, chronic myelogenous leukemia, Hodgkin lymphoma, multiple myeloma, myelodysplastic syndromes, myeloproliferative neoplasms, non-Hodgkin lymphomas, and Waldenström’s macroglobulinemia, as well as solid tumor malignancies.
There are patient guidelines for adolescents and young adults with cancer and guidelines on distress/supportive care, and nausea and vomiting/supportive care as well.
*(1) Midostaurin (Rydapt) was approved for adults with newly diagnosed, FLT3-positive AML
(2) Gemtuzumab ozogamicin (Mylotarg) was approved for adults with newly diagnosed AML and patients age 2 and older with relapsed/refractory AML
(3) Liposomal daunorubicin/cytarabine (Vyxeos) was approved for AML with myelodysplasia-related changes and newly diagnosed, therapy-related AML
(4) Enasidenib (Idhifa) was approved for adults with relapsed/refractory AML and an IDH2 mutation.
The National Comprehensive Cancer Network (NCCN) has released guidelines for patients with acute myeloid leukemia (AML).
The guidelines are intended to help patients make better informed decision about their care.
The guidelines explain what AML is, describe testing procedures and treatment options, and provide tips to help patients choose the best treatment.
The guidelines also include a list of suggested questions for patients to bring up with their doctors, illustrations, and a glossary of key terms and acronyms.
“People with AML often aren’t aware that this isn’t a single disease but many different diseases that share fundamental characteristics,” said Martin Tallman, MD, a hematologic oncologist at Memorial Sloan Kettering Cancer Center in New York, New York, and vice-chair of the NCCN Guidelines Panel for AML.
“The good news is that the future for AML is getting a lot brighter. After 40 years without much progress, 4 new medications were just approved last year*, and more new treatment courses are in development as we speak.”
The guidelines are available for free on the NCCN website or via the NCCN Patient Guides for Cancer app. Printed versions of the guidelines can be purchased at Amazon.com.
NCCN also has guidelines for patients with acute lymphoblastic leukemia, chronic lymphocytic leukemia, chronic myelogenous leukemia, Hodgkin lymphoma, multiple myeloma, myelodysplastic syndromes, myeloproliferative neoplasms, non-Hodgkin lymphomas, and Waldenström’s macroglobulinemia, as well as solid tumor malignancies.
There are patient guidelines for adolescents and young adults with cancer and guidelines on distress/supportive care, and nausea and vomiting/supportive care as well.
*(1) Midostaurin (Rydapt) was approved for adults with newly diagnosed, FLT3-positive AML
(2) Gemtuzumab ozogamicin (Mylotarg) was approved for adults with newly diagnosed AML and patients age 2 and older with relapsed/refractory AML
(3) Liposomal daunorubicin/cytarabine (Vyxeos) was approved for AML with myelodysplasia-related changes and newly diagnosed, therapy-related AML
(4) Enasidenib (Idhifa) was approved for adults with relapsed/refractory AML and an IDH2 mutation.