Quiz
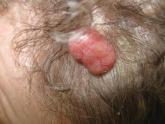
Lobular-Appearing Nodule on the Scalp
A 79-year-old woman presented with a lesion on the left side of the scalp of several years’ duration that had slowly increased in size. Despite...
Article
Hyperpigmented patches on the neck, shoulder, and back
A 17-year-old boy has had asymptomatic, unilateral hyperpigmented patches since birth. What is the diagnosis?