User login
Papules and Telangiectases on the Distal Fingers of a Child
The Diagnosis: Juvenile Dermatomyositis
Juvenile dermatomyositis (JDM) is a rare idiopathic inflammatory myopathy of childhood that is autoimmune in nature with an annual incidence ranging from 2.5 to 4.1 cases per million children. Its peak incidence is between 5 and 10 years of age, and it affects girls more than boys at a 2-fold to 5-fold greater rate.1 Juvenile dermatomyositis is characterized by skeletal muscle weakness in the presence of distinctive rashes, including Gottron papules and heliotrope erythema. Muscle weakness typically is proximal and symmetrical, and eventually patients may have trouble rising from a seated position or lifting objects overhead. Other skin manifestations include nail fold capillary changes, calcinosis cutis, and less commonly ulcerations signifying vasculopathy of the skin.2 A subset of patients will present with juvenile amyopathic dermatomyositis. These children have the characteristic skin changes without the muscle weakness or elevated muscle enzymes for more than 6 months; however, one-quarter may go on to develop mysositis.3
Diagnosis of JDM traditionally was based on the following 5 diagnostic criteria: characteristic skin rash, proximal muscle weakness, elevated muscle enzymes, myopathic changes on electromyogram, and typical muscle biopsy.1 Current practice shows a broadening of diagnostic criteria using new techniques in the diagnosis of JDM. To make the diagnosis, the patient must have the characteristic skin manifestations with a minimum of 3 other criteria.4 A 2006 international consensus survey expanded the list of criteria to include typical findings on magnetic resonance imaging (MRI), nail fold capillaroscopy abnormalities, calcinosis, and
dysphonia.5
To assess muscle disease, MRI is utilized because it is a reliable noninvasive tool to assess muscle inflammation. Muscle biopsy is only recommended if the diagnosis is unclear.5 The results of the MRI in our patient displayed symmetric mild fatty atrophy of the gluteus maximus muscle, as well as edema in the right rectus femoris and left vastus lateralis muscles, suggesting early findings of myositis. Muscle enzymes may not be diagnostic because they are not always elevated at diagnosis. Our patient had a normal creatinine kinase level (92 U/L [reference range, <190 U/L]), and both aldolase and lactate dehydrogenase also were within reference range. Conversely, antinuclear antibodies frequently are positive in patients with JDM, such as in our patient at a 1:320 dilution, but are nonspecific and nondiagnostic. It is recommended to include nail fold capillaroscopy to evaluate periungual capillary changes because nailfold capillary density is a sensitive measure of both skin and muscle disease.5 Using dermoscopy, nail fold capillary dilation was observed in our patient.
Other differential diagnoses can have somewhat similar clinical features to JDM. Infantile papular acrodermatitis, commonly referred to as Gianotti-Crosti syndrome, is a viral exanthem that affects children (median age, 2 years).6 The rash appears as monomorphous, flat-topped, pink to brown papules affecting the face, buttocks, and arms; it typically spontaneously resolves in 10 days.6
Juvenile-onset lupus is a chronic autoimmune disorder that can involve any organ system and typically affects children aged 11 to 12 years with a female preponderance. Skin manifestations are similar to adult-onset lupus and include malar rash, discoid rash, oral ulcerations, petechiae, palpable purpura, and digital telangiectasia and ulcers. 7
Juvenile scleroderma is rare connective-tissue disorder that also has multiple organ involvement. Cutaneous involvement can range from isolated morphealike plaques to diffuse sclerotic lesions with growth disturbances, contractures, and facial atrophy.8
Verrucae planae, commonly referred to as flat warts, are papules caused primarily by human papillomavirus types 3, 10, 28, and 41. Children and young adults commonly are affected, and warts can appear on the hands, as in our patient.6
Treatment of JDM depends on disease severity at initial presentation and requires a multidisciplinary approach. The mainstay of treatment is high-dose oral prednisone in combination with disease-modifying drugs such as methotrexate and cyclosporin A. Patients with more severe presentations (eg, ulcerative skin disease) or life-threatening organ involvement are treated with cyclophosphamide, usually in combination with high-dose glucocorticoids.9
Early detection with aggressive treatment is vital to reduce morbidity and mortality from organ damage and disease complications. Mortality rates have dropped to 3%10 in recent decades with the use of systemic glucocorticoids. Delayed treatment is associated with a prolonged disease course and poorer outcomes. Disease complications in children with JDM include osteoporosis, calcinosis, and intestinal perforation; however, with early treatment, children with JDM can expect full recovery and to live a normal life as compared to adults with dermatomyositis.10
Prior to our patient's diagnosis, the family was assigned to move to an overseas location through the US Military with no direct access to advanced medical care. Early detection and diagnosis of JDM through an astute clinical examination allowed the patient and her family to remain in the continental United States to continue receiving specialty care.
- Mendez EP, Lipton R, Ramsey-Goldman R, et al. US incidence of juvenile dermatomyositis,1995-1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum. 2003;49:300-305.
- Shah M, Mamyrova G, Targoff IN, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine. 2013;92:25-41.
- Gerami P, Walling HW, Lewis J, et al. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. Br J Dermatol. 2007;57:637-644.
- Enders FB, Bader-Meunier B, Baildam E, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017;76:329-340.
- Brown VE, Pilkington CA, Feldman BM, et al. An international consensus survey of the diagnostic criteria for juvenile dermatomyositis (JDM). Rheumatology (Oxford). 2006;45:990-993.
- William JD, Berger TG, Elston DM. Viral diseases. In: William JD, Berger TG, Elston DM. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:360-413.
- Levy DM, Kamphuis S. Systemic lupus erythematosus in children and adolescents. Pediatr Clin North Am. 2012;59:345-364.
- Li SC, Torok KS, Pope E, et al; Childhood Arthritis and Rheumatology Research Alliance (CARRA) Localized Scleroderma Workgroup. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res (Hoboken). 2012;64:1175-1185.
- Stringer E, Ota S, Bohnsack J, et al. Treatment approaches to juvenile dermatomyositis (JDM) across North America: the Childhood Arthritis and Rheumatology Research Alliance (CARRA) JDM treatment study. J Rhematol. 2010;37:S1953-S1961.
- Huber AM, Feldman BM. Long-term outcomes in juvenile dermatomyositis: how did we get here and where are we going? Curr Rheumatol Rep. 2005;7:441-446.
The Diagnosis: Juvenile Dermatomyositis
Juvenile dermatomyositis (JDM) is a rare idiopathic inflammatory myopathy of childhood that is autoimmune in nature with an annual incidence ranging from 2.5 to 4.1 cases per million children. Its peak incidence is between 5 and 10 years of age, and it affects girls more than boys at a 2-fold to 5-fold greater rate.1 Juvenile dermatomyositis is characterized by skeletal muscle weakness in the presence of distinctive rashes, including Gottron papules and heliotrope erythema. Muscle weakness typically is proximal and symmetrical, and eventually patients may have trouble rising from a seated position or lifting objects overhead. Other skin manifestations include nail fold capillary changes, calcinosis cutis, and less commonly ulcerations signifying vasculopathy of the skin.2 A subset of patients will present with juvenile amyopathic dermatomyositis. These children have the characteristic skin changes without the muscle weakness or elevated muscle enzymes for more than 6 months; however, one-quarter may go on to develop mysositis.3
Diagnosis of JDM traditionally was based on the following 5 diagnostic criteria: characteristic skin rash, proximal muscle weakness, elevated muscle enzymes, myopathic changes on electromyogram, and typical muscle biopsy.1 Current practice shows a broadening of diagnostic criteria using new techniques in the diagnosis of JDM. To make the diagnosis, the patient must have the characteristic skin manifestations with a minimum of 3 other criteria.4 A 2006 international consensus survey expanded the list of criteria to include typical findings on magnetic resonance imaging (MRI), nail fold capillaroscopy abnormalities, calcinosis, and
dysphonia.5
To assess muscle disease, MRI is utilized because it is a reliable noninvasive tool to assess muscle inflammation. Muscle biopsy is only recommended if the diagnosis is unclear.5 The results of the MRI in our patient displayed symmetric mild fatty atrophy of the gluteus maximus muscle, as well as edema in the right rectus femoris and left vastus lateralis muscles, suggesting early findings of myositis. Muscle enzymes may not be diagnostic because they are not always elevated at diagnosis. Our patient had a normal creatinine kinase level (92 U/L [reference range, <190 U/L]), and both aldolase and lactate dehydrogenase also were within reference range. Conversely, antinuclear antibodies frequently are positive in patients with JDM, such as in our patient at a 1:320 dilution, but are nonspecific and nondiagnostic. It is recommended to include nail fold capillaroscopy to evaluate periungual capillary changes because nailfold capillary density is a sensitive measure of both skin and muscle disease.5 Using dermoscopy, nail fold capillary dilation was observed in our patient.
Other differential diagnoses can have somewhat similar clinical features to JDM. Infantile papular acrodermatitis, commonly referred to as Gianotti-Crosti syndrome, is a viral exanthem that affects children (median age, 2 years).6 The rash appears as monomorphous, flat-topped, pink to brown papules affecting the face, buttocks, and arms; it typically spontaneously resolves in 10 days.6
Juvenile-onset lupus is a chronic autoimmune disorder that can involve any organ system and typically affects children aged 11 to 12 years with a female preponderance. Skin manifestations are similar to adult-onset lupus and include malar rash, discoid rash, oral ulcerations, petechiae, palpable purpura, and digital telangiectasia and ulcers. 7
Juvenile scleroderma is rare connective-tissue disorder that also has multiple organ involvement. Cutaneous involvement can range from isolated morphealike plaques to diffuse sclerotic lesions with growth disturbances, contractures, and facial atrophy.8
Verrucae planae, commonly referred to as flat warts, are papules caused primarily by human papillomavirus types 3, 10, 28, and 41. Children and young adults commonly are affected, and warts can appear on the hands, as in our patient.6
Treatment of JDM depends on disease severity at initial presentation and requires a multidisciplinary approach. The mainstay of treatment is high-dose oral prednisone in combination with disease-modifying drugs such as methotrexate and cyclosporin A. Patients with more severe presentations (eg, ulcerative skin disease) or life-threatening organ involvement are treated with cyclophosphamide, usually in combination with high-dose glucocorticoids.9
Early detection with aggressive treatment is vital to reduce morbidity and mortality from organ damage and disease complications. Mortality rates have dropped to 3%10 in recent decades with the use of systemic glucocorticoids. Delayed treatment is associated with a prolonged disease course and poorer outcomes. Disease complications in children with JDM include osteoporosis, calcinosis, and intestinal perforation; however, with early treatment, children with JDM can expect full recovery and to live a normal life as compared to adults with dermatomyositis.10
Prior to our patient's diagnosis, the family was assigned to move to an overseas location through the US Military with no direct access to advanced medical care. Early detection and diagnosis of JDM through an astute clinical examination allowed the patient and her family to remain in the continental United States to continue receiving specialty care.
The Diagnosis: Juvenile Dermatomyositis
Juvenile dermatomyositis (JDM) is a rare idiopathic inflammatory myopathy of childhood that is autoimmune in nature with an annual incidence ranging from 2.5 to 4.1 cases per million children. Its peak incidence is between 5 and 10 years of age, and it affects girls more than boys at a 2-fold to 5-fold greater rate.1 Juvenile dermatomyositis is characterized by skeletal muscle weakness in the presence of distinctive rashes, including Gottron papules and heliotrope erythema. Muscle weakness typically is proximal and symmetrical, and eventually patients may have trouble rising from a seated position or lifting objects overhead. Other skin manifestations include nail fold capillary changes, calcinosis cutis, and less commonly ulcerations signifying vasculopathy of the skin.2 A subset of patients will present with juvenile amyopathic dermatomyositis. These children have the characteristic skin changes without the muscle weakness or elevated muscle enzymes for more than 6 months; however, one-quarter may go on to develop mysositis.3
Diagnosis of JDM traditionally was based on the following 5 diagnostic criteria: characteristic skin rash, proximal muscle weakness, elevated muscle enzymes, myopathic changes on electromyogram, and typical muscle biopsy.1 Current practice shows a broadening of diagnostic criteria using new techniques in the diagnosis of JDM. To make the diagnosis, the patient must have the characteristic skin manifestations with a minimum of 3 other criteria.4 A 2006 international consensus survey expanded the list of criteria to include typical findings on magnetic resonance imaging (MRI), nail fold capillaroscopy abnormalities, calcinosis, and
dysphonia.5
To assess muscle disease, MRI is utilized because it is a reliable noninvasive tool to assess muscle inflammation. Muscle biopsy is only recommended if the diagnosis is unclear.5 The results of the MRI in our patient displayed symmetric mild fatty atrophy of the gluteus maximus muscle, as well as edema in the right rectus femoris and left vastus lateralis muscles, suggesting early findings of myositis. Muscle enzymes may not be diagnostic because they are not always elevated at diagnosis. Our patient had a normal creatinine kinase level (92 U/L [reference range, <190 U/L]), and both aldolase and lactate dehydrogenase also were within reference range. Conversely, antinuclear antibodies frequently are positive in patients with JDM, such as in our patient at a 1:320 dilution, but are nonspecific and nondiagnostic. It is recommended to include nail fold capillaroscopy to evaluate periungual capillary changes because nailfold capillary density is a sensitive measure of both skin and muscle disease.5 Using dermoscopy, nail fold capillary dilation was observed in our patient.
Other differential diagnoses can have somewhat similar clinical features to JDM. Infantile papular acrodermatitis, commonly referred to as Gianotti-Crosti syndrome, is a viral exanthem that affects children (median age, 2 years).6 The rash appears as monomorphous, flat-topped, pink to brown papules affecting the face, buttocks, and arms; it typically spontaneously resolves in 10 days.6
Juvenile-onset lupus is a chronic autoimmune disorder that can involve any organ system and typically affects children aged 11 to 12 years with a female preponderance. Skin manifestations are similar to adult-onset lupus and include malar rash, discoid rash, oral ulcerations, petechiae, palpable purpura, and digital telangiectasia and ulcers. 7
Juvenile scleroderma is rare connective-tissue disorder that also has multiple organ involvement. Cutaneous involvement can range from isolated morphealike plaques to diffuse sclerotic lesions with growth disturbances, contractures, and facial atrophy.8
Verrucae planae, commonly referred to as flat warts, are papules caused primarily by human papillomavirus types 3, 10, 28, and 41. Children and young adults commonly are affected, and warts can appear on the hands, as in our patient.6
Treatment of JDM depends on disease severity at initial presentation and requires a multidisciplinary approach. The mainstay of treatment is high-dose oral prednisone in combination with disease-modifying drugs such as methotrexate and cyclosporin A. Patients with more severe presentations (eg, ulcerative skin disease) or life-threatening organ involvement are treated with cyclophosphamide, usually in combination with high-dose glucocorticoids.9
Early detection with aggressive treatment is vital to reduce morbidity and mortality from organ damage and disease complications. Mortality rates have dropped to 3%10 in recent decades with the use of systemic glucocorticoids. Delayed treatment is associated with a prolonged disease course and poorer outcomes. Disease complications in children with JDM include osteoporosis, calcinosis, and intestinal perforation; however, with early treatment, children with JDM can expect full recovery and to live a normal life as compared to adults with dermatomyositis.10
Prior to our patient's diagnosis, the family was assigned to move to an overseas location through the US Military with no direct access to advanced medical care. Early detection and diagnosis of JDM through an astute clinical examination allowed the patient and her family to remain in the continental United States to continue receiving specialty care.
- Mendez EP, Lipton R, Ramsey-Goldman R, et al. US incidence of juvenile dermatomyositis,1995-1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum. 2003;49:300-305.
- Shah M, Mamyrova G, Targoff IN, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine. 2013;92:25-41.
- Gerami P, Walling HW, Lewis J, et al. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. Br J Dermatol. 2007;57:637-644.
- Enders FB, Bader-Meunier B, Baildam E, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017;76:329-340.
- Brown VE, Pilkington CA, Feldman BM, et al. An international consensus survey of the diagnostic criteria for juvenile dermatomyositis (JDM). Rheumatology (Oxford). 2006;45:990-993.
- William JD, Berger TG, Elston DM. Viral diseases. In: William JD, Berger TG, Elston DM. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:360-413.
- Levy DM, Kamphuis S. Systemic lupus erythematosus in children and adolescents. Pediatr Clin North Am. 2012;59:345-364.
- Li SC, Torok KS, Pope E, et al; Childhood Arthritis and Rheumatology Research Alliance (CARRA) Localized Scleroderma Workgroup. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res (Hoboken). 2012;64:1175-1185.
- Stringer E, Ota S, Bohnsack J, et al. Treatment approaches to juvenile dermatomyositis (JDM) across North America: the Childhood Arthritis and Rheumatology Research Alliance (CARRA) JDM treatment study. J Rhematol. 2010;37:S1953-S1961.
- Huber AM, Feldman BM. Long-term outcomes in juvenile dermatomyositis: how did we get here and where are we going? Curr Rheumatol Rep. 2005;7:441-446.
- Mendez EP, Lipton R, Ramsey-Goldman R, et al. US incidence of juvenile dermatomyositis,1995-1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum. 2003;49:300-305.
- Shah M, Mamyrova G, Targoff IN, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine. 2013;92:25-41.
- Gerami P, Walling HW, Lewis J, et al. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. Br J Dermatol. 2007;57:637-644.
- Enders FB, Bader-Meunier B, Baildam E, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017;76:329-340.
- Brown VE, Pilkington CA, Feldman BM, et al. An international consensus survey of the diagnostic criteria for juvenile dermatomyositis (JDM). Rheumatology (Oxford). 2006;45:990-993.
- William JD, Berger TG, Elston DM. Viral diseases. In: William JD, Berger TG, Elston DM. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:360-413.
- Levy DM, Kamphuis S. Systemic lupus erythematosus in children and adolescents. Pediatr Clin North Am. 2012;59:345-364.
- Li SC, Torok KS, Pope E, et al; Childhood Arthritis and Rheumatology Research Alliance (CARRA) Localized Scleroderma Workgroup. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res (Hoboken). 2012;64:1175-1185.
- Stringer E, Ota S, Bohnsack J, et al. Treatment approaches to juvenile dermatomyositis (JDM) across North America: the Childhood Arthritis and Rheumatology Research Alliance (CARRA) JDM treatment study. J Rhematol. 2010;37:S1953-S1961.
- Huber AM, Feldman BM. Long-term outcomes in juvenile dermatomyositis: how did we get here and where are we going? Curr Rheumatol Rep. 2005;7:441-446.
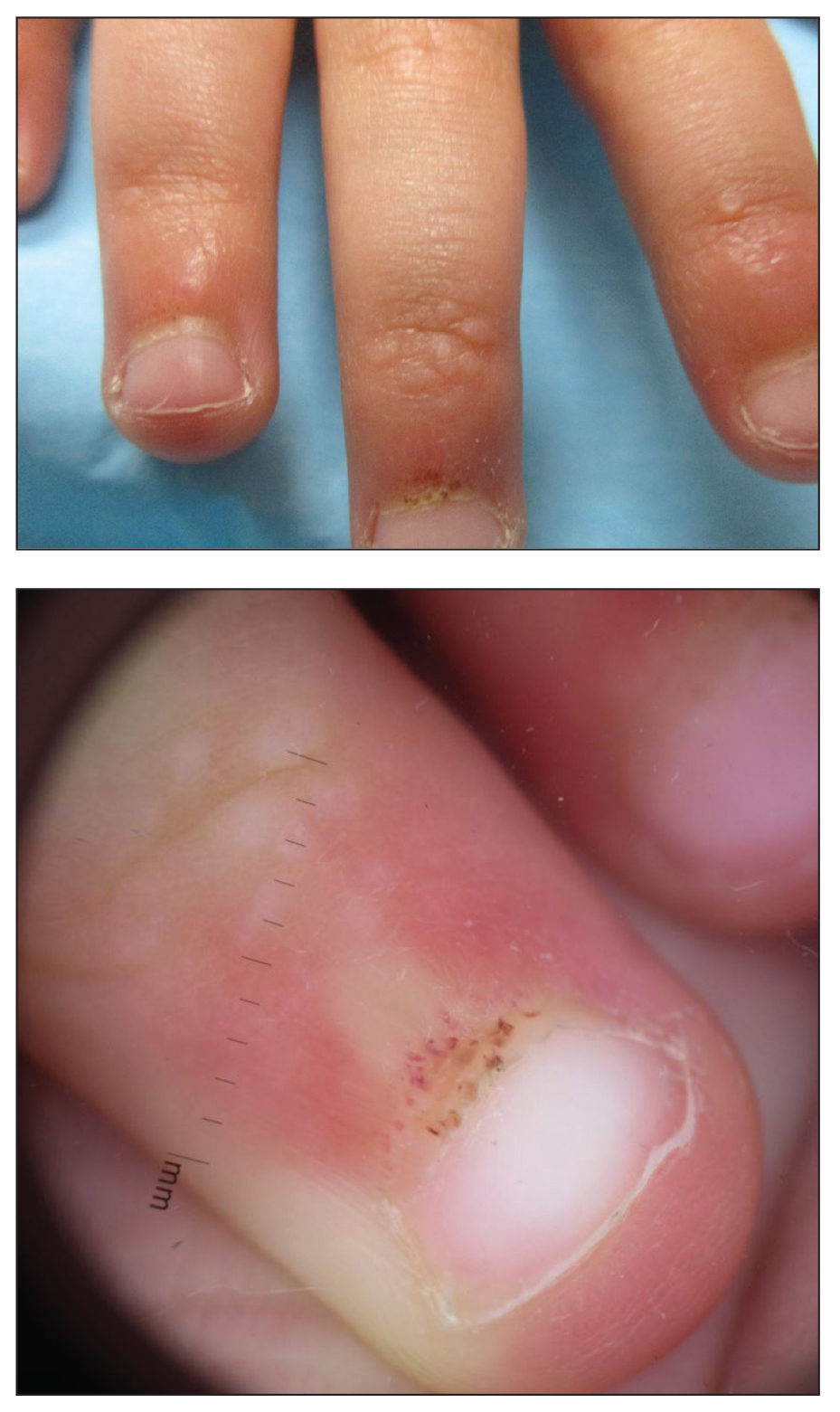
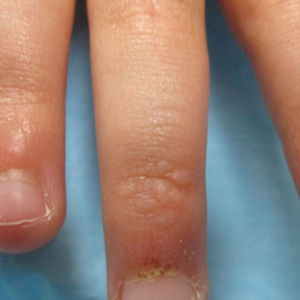
A 4-year-old girl presented to our dermatology clinic with asymptomatic flesh-colored bumps on the fingers of 2 to 3 months’ duration. Prior to presentation the patient was otherwise healthy with normal growth and development. She was referred to dermatology for recommended treatment options for suspected flat warts. On physical examination, grouped 1- to 3-mm, smooth, flat-topped papules were found on the dorsal aspects of the distal interphalangeal joints of all fingers (top). The papules were nonpruritic. Additionally, there were nail findings of ragged cuticles and dilated capillary loops in the proximal nail folds (bottom). The patient did not bite her nails, per the mother’s report, and no other rashes were noted. There were no systemic symptoms or reports of muscle fatigue. She was positive for antinuclear antibodies at 1:320 dilution. Magnetic resonance imaging of the thighs and pelvis was ordered.