Article
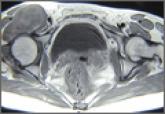
Giant Solitary Synovial Chondromatosis Mimicking Chondrosarcoma: Report of a Rare Histologic Presentation and Literature Review
Synovial chondromatosis is a benign lesion of the synovium, and giant solitary synovial chondromatosis (GSSCM) is a rare presentation of it. In...
Article
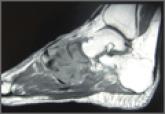
Unusual Form and Location of a Tumor: Multiosseous Ewing Sarcoma in the Foot
Ewing sarcoma, as the prototype of a small round blue cell tumor of bone, typically affects adolescents and young adults.