User login
Vortioxetine for major depressive disorder
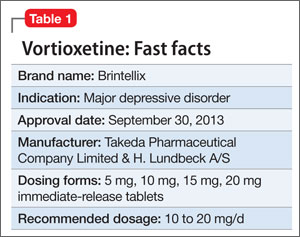
Vortioxetine is FDA-approved to treat major depressive disorder (MDD) (Table 1), having shown efficacy in relieving depressive symptoms in clinical trials.1 Vortioxetine’s mechanism of action enhances CNS serotonergic activity through inhibiting serotonin (5-HT) reuptake, agonizing the 5-HT1A receptor, partially agonizing the 5-HT1B receptor, and antagonizing the 5-HT3, 5-HT1D, and 5-HT7 receptors.
Clinical implications
It is hypothesized that depression is a heterogeneous disease caused by dysregulation of serotonin, norepinephrine, and dopamine, subsequently producing mood and neurovegetative symptoms of depression. Preclinical, in vivo studies indicate that vortioxetine enhances levels of serotonin, norepinephrine, dopamine, acetylcholine, and histamine in specific areas of the brain with the ability to improve depressive symptoms. Vortioxetine’s multimodal activity can be a useful alternative to other serotonergic antidepressants for some patients who are partial responders or non-responders to other treatment options. In addition, vortioxetine appears to have minimal effect on weight2 and sexual function—the latter being dose-dependent.3
How does it work?
Vortioxetine differs from other antidepressants in its multimodal activity (ie, affecting G-protein mode receptors, ion channel mode receptors, and neurotransmitter transporters). It inhibits the serotonin transporter (Ki = 1.6 nM), causing subsequent inhibition of serotonin reuptake into presynaptic neurons as well as selectively acting on the other subtypes of serotonergic receptors; however, activity on the norepinephrine transporter (Ki = 113 nM) and dopamine transporter (Ki > 1000 nM) is minimal. It is believed that mood-regulating effects of vortioxetine are caused by inhibition of serotonin reuptake, prolonged availability of serotonin to the postsynaptic neurons, its agonist activity on the 5-HT1A receptor (Ki = 15 nM), and partial agonist activity on the 5-HT1B receptor (Ki = 33 nM). Vortioxetine has strong affinity for the 5-HT3 receptor (Ki = 3.7 nM), which plays a role in modulation of centrally mediated nausea and vomiting. Positron emission tomography studies in humans determined that the occupancy of 5-HT transporter was 50% at 5 mg/d, 65% at 10 mg/d, and 80% at 20 mg/d.1,4 Human studies did not show that vortioxetine causes QTc prolongation.
Pharmacokinetics
Therapeutic activity of vortioxetine is thought to be due to the parent drug. It has a half-life of approximately 66 hours, and achieves steady state in 13.5 to 19 days. Bioavailability of vortioxetine is 75%; absorption does not depend on food; and 98% of drug is bound on plasma proteins.
Vortioxetine has linear pharmacokinetics, with maximum plasma concentration 7 to 11 hours after ingestion. The medication is metabolized primarily by oxidation through cytochrome P (CYP) 450: CYP2D6 (primary), CYP 3A4/5, CYP 2C19, CYP 2C9, CYP2A6, CYP2C8, and CYP2B6 with subsequent glucuronidation. This predisposes vortioxetine to potential pharmacokinetic drug-drug interaction warranting dose adjustment consideration when vortioxetine is coadministered with compounds inhibiting CYP2D6 or inducing CYP3A4 for ≥14 days, or for patients identified as poor 2D6 metabolizers.
In addition, coadministration of vortioxetine with serotonergic medications such as triptans, other antidepressants, and tramadol can cause potentially life-threatening serotonin syndrome, characterized by mental status changes, autonomic instability, neuromuscular aberrations, and GI symptoms. Concomitant use of vortioxetine and a nonsteroidal anti-inflammatory drug, aspirin, or warfarin can result in abnormal bleeding. Coadministration of vortioxetine with another highly protein-bound drug may increase or decrease the free concentration of either drug depending on the binding affinity of the drug for the protein.
Efficacy
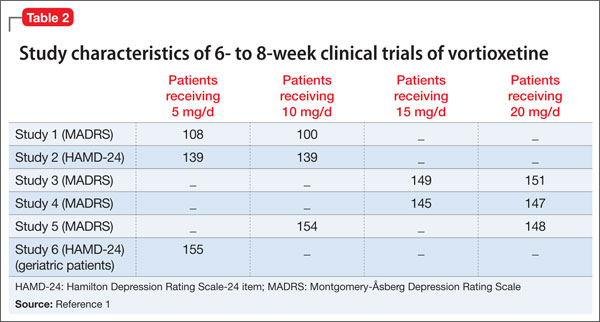
Vortioxetine reduced depressive symptoms in 6 positive, 6- to 8-week, double-blind, placebo controlled and randomized studies and 1 maintenance study.1 Subjects were adults (Studies 1 to 5) and geriatric patients from age 64 to 88 who had ≥1 depressive episode before age 60 (Study 6). All met DSM-IV-TR criteria for MDD. Subjects with cognitive impairment scoring <24 on the Mini-Mental Status Examination and children were excluded. Depending on the study, response to the treatment was primarily measured on the Montgomery-Åsberg Depression Rating Scale (MADRS) or Hamilton Depression Rating Scale (HAM-D).
See Table 2 for a description of the positive studies, including dosages. In all studies, vortioxetine was superior to placebo at least one dosage for treating depression. In the 6- to 8-week placebo-controlled studies, an effect of vortioxetine based on the primary efficacy measure was generally observed starting at Week 2; that effect increased in subsequent weeks with the full antidepressant effect of vortioxetine generally not seen until study Week 4 or later.1
The maintenance treatment study included 639 patients who met DSM-IV-TR criteria for MDD. This study lasted for as long as 64 weeks. The first 12-week period was open-label, during which patients were treated with vortioxetine, 5 mg/d or 10 mg/d, with a possibility to adjust the dosage in the first 8 weeks. By the end of Week 12, 396 subjects achieved remission (MADRS <10), 75% of whom were taking vortioxetine, 10 mg/d. These patients were then randomly assigned to placebo or the dosage of vortioxetine to which they had responded, and continued the study for as long as 64 weeks. Time to relapse (MADRS total score ≥22) or an insufficient therapeutic response (as judged by the investigator) was the primary efficacy outcome, and demonstrated that vortioxetine was superior to placebo.
Tolerability
The tolerability of vortioxetine is comparable with other serotonergic antidepressants. In pooled analysis of pre-marketing studies, 5% to 8% of patients receiving vortioxetine (5 to 20 mg/d) discontinued treatment because of adverse effects (AEs), compared with 4% in the placebo group. Nausea was the most commonly reported AE leading to discontinuation and appeared to be dose dependent.
AEs, such as nausea, constipation, and vomiting, most commonly occurred in the first week of treatment, with a median duration of 2 weeks.5 In the 6- to 8-week trials, the most common AEs were nausea, constipation, and vomiting. In longer trials (24 to 64 weeks), the most common AE was nausea.
In 6- to 8-week placebo-controlled studies, vortioxetine was not associated with any clinically significant effect on vital signs or laboratory values in hematology, urinalysis, or serum chemistry (except sodium). Hyponatremia, the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH), has occurred. The risk of developing SIADH and resultant hyponatremia is greater in geriatric patients and patients taking a diuretic.
Abruptly discontinuing vortioxetine can cause transient withdrawal symptoms, including headache and muscle tension, especially at a higher dosage (15 to 20 mg/d). Gradual tapering can reduce withdrawal symptoms.
Specific clinical issues
All antidepressants have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation and behaviors during the first months of treatment and with dosage changes.
Vortioxetine is categorized as pregnancy category C. Newborns exposed to a selective serotonin reuptake inhibitor (SSRI) in pregnancy may have an increased risk of persistent pulmonary hypertension during the neonatal period. When taken during the third trimester of pregnancy, SSRIs and serotonin-norepinephrine reuptake inhibitors can cause serious neonatal complications, including respiratory distress, cyanosis, apnea, and seizures, which may require longer hospitalization, respiratory support, or tube feeding for the infant. Consider risks and benefits of third-trimester use of an antidepressant. It is not known if vortioxetine is present in human breast milk.
Clinical studies on vortioxetine in pediatric patients have not been conducted.
No dosage adjustment is recommended on the basis of age for geriatric patients. No dose adjustment of vortioxetine is necessary on the basis of race, sex, ethnicity, renal function, or mild to moderate hepatic impairment. See Table 3 for practice points when prescribing vortioxetine. See Table 4 for contraindications to vortioxetine.


Dosing
The recommended starting dosage is 10 mg, administered orally once daily without regard to meals. Dosage should then be increased to 20 mg/d, as clinically warranted and tolerated. Consider a dosage decrease to 5 mg/d in patients who do not tolerate higher dosages or require drug adjustment because of drug-drug interaction or poor 2D6 metabolizer status.
Bottom Line
FDA-approved for major depressive disorder in adults, vortioxetine reduced depressive symptoms in 6 positive, double-blind, placebo-controlled, and randomized studies. The multimodal activity of vortioxeine can be a useful alternative to serotonergic antidepressants for some patients who are partial responders or nonresponders. Tolerability is comparable with other serotonergic antidepressants.
Related Resources
- Alam MY, Jacobsen PL, Chen Y, et al. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2014; 29(1):36-44.
- Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double-blind trial of 2.5 mg and 5 mg vortioxetine (Lu AA21004) versus placebo for 8 weeks in adults with major depressive disorder. Curr Med Res Opin. 2013;29(3):217-226.
Drug Brand Names
Linezolid • Zyvox Vortioxetine • Brintellix
Methylene blue • Urolene Blue Warfarin • Coumadin
Tramadol • Ultram
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Vortioxetine [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2013.
2. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a comprehensive review and meta-analysis. J Clin Psychopharmacol. 2009; 29(3):259-266.
4. Chen G, Lee R, Højer A, et al. Pharmacokinetic drug interactions involving vortioxetine (LU AA 21004), a multimodal antidepressant. Clin Drug Invetig. 2013; 33(10):727-736.
5. Citrome L. Vortioxetine for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Prac. 2014;68(1):60-82.
Vortioxetine is FDA-approved to treat major depressive disorder (MDD) (Table 1), having shown efficacy in relieving depressive symptoms in clinical trials.1 Vortioxetine’s mechanism of action enhances CNS serotonergic activity through inhibiting serotonin (5-HT) reuptake, agonizing the 5-HT1A receptor, partially agonizing the 5-HT1B receptor, and antagonizing the 5-HT3, 5-HT1D, and 5-HT7 receptors.
Clinical implications
It is hypothesized that depression is a heterogeneous disease caused by dysregulation of serotonin, norepinephrine, and dopamine, subsequently producing mood and neurovegetative symptoms of depression. Preclinical, in vivo studies indicate that vortioxetine enhances levels of serotonin, norepinephrine, dopamine, acetylcholine, and histamine in specific areas of the brain with the ability to improve depressive symptoms. Vortioxetine’s multimodal activity can be a useful alternative to other serotonergic antidepressants for some patients who are partial responders or non-responders to other treatment options. In addition, vortioxetine appears to have minimal effect on weight2 and sexual function—the latter being dose-dependent.3
How does it work?
Vortioxetine differs from other antidepressants in its multimodal activity (ie, affecting G-protein mode receptors, ion channel mode receptors, and neurotransmitter transporters). It inhibits the serotonin transporter (Ki = 1.6 nM), causing subsequent inhibition of serotonin reuptake into presynaptic neurons as well as selectively acting on the other subtypes of serotonergic receptors; however, activity on the norepinephrine transporter (Ki = 113 nM) and dopamine transporter (Ki > 1000 nM) is minimal. It is believed that mood-regulating effects of vortioxetine are caused by inhibition of serotonin reuptake, prolonged availability of serotonin to the postsynaptic neurons, its agonist activity on the 5-HT1A receptor (Ki = 15 nM), and partial agonist activity on the 5-HT1B receptor (Ki = 33 nM). Vortioxetine has strong affinity for the 5-HT3 receptor (Ki = 3.7 nM), which plays a role in modulation of centrally mediated nausea and vomiting. Positron emission tomography studies in humans determined that the occupancy of 5-HT transporter was 50% at 5 mg/d, 65% at 10 mg/d, and 80% at 20 mg/d.1,4 Human studies did not show that vortioxetine causes QTc prolongation.
Pharmacokinetics
Therapeutic activity of vortioxetine is thought to be due to the parent drug. It has a half-life of approximately 66 hours, and achieves steady state in 13.5 to 19 days. Bioavailability of vortioxetine is 75%; absorption does not depend on food; and 98% of drug is bound on plasma proteins.
Vortioxetine has linear pharmacokinetics, with maximum plasma concentration 7 to 11 hours after ingestion. The medication is metabolized primarily by oxidation through cytochrome P (CYP) 450: CYP2D6 (primary), CYP 3A4/5, CYP 2C19, CYP 2C9, CYP2A6, CYP2C8, and CYP2B6 with subsequent glucuronidation. This predisposes vortioxetine to potential pharmacokinetic drug-drug interaction warranting dose adjustment consideration when vortioxetine is coadministered with compounds inhibiting CYP2D6 or inducing CYP3A4 for ≥14 days, or for patients identified as poor 2D6 metabolizers.
In addition, coadministration of vortioxetine with serotonergic medications such as triptans, other antidepressants, and tramadol can cause potentially life-threatening serotonin syndrome, characterized by mental status changes, autonomic instability, neuromuscular aberrations, and GI symptoms. Concomitant use of vortioxetine and a nonsteroidal anti-inflammatory drug, aspirin, or warfarin can result in abnormal bleeding. Coadministration of vortioxetine with another highly protein-bound drug may increase or decrease the free concentration of either drug depending on the binding affinity of the drug for the protein.
Efficacy
Vortioxetine reduced depressive symptoms in 6 positive, 6- to 8-week, double-blind, placebo controlled and randomized studies and 1 maintenance study.1 Subjects were adults (Studies 1 to 5) and geriatric patients from age 64 to 88 who had ≥1 depressive episode before age 60 (Study 6). All met DSM-IV-TR criteria for MDD. Subjects with cognitive impairment scoring <24 on the Mini-Mental Status Examination and children were excluded. Depending on the study, response to the treatment was primarily measured on the Montgomery-Åsberg Depression Rating Scale (MADRS) or Hamilton Depression Rating Scale (HAM-D).
See Table 2 for a description of the positive studies, including dosages. In all studies, vortioxetine was superior to placebo at least one dosage for treating depression. In the 6- to 8-week placebo-controlled studies, an effect of vortioxetine based on the primary efficacy measure was generally observed starting at Week 2; that effect increased in subsequent weeks with the full antidepressant effect of vortioxetine generally not seen until study Week 4 or later.1
The maintenance treatment study included 639 patients who met DSM-IV-TR criteria for MDD. This study lasted for as long as 64 weeks. The first 12-week period was open-label, during which patients were treated with vortioxetine, 5 mg/d or 10 mg/d, with a possibility to adjust the dosage in the first 8 weeks. By the end of Week 12, 396 subjects achieved remission (MADRS <10), 75% of whom were taking vortioxetine, 10 mg/d. These patients were then randomly assigned to placebo or the dosage of vortioxetine to which they had responded, and continued the study for as long as 64 weeks. Time to relapse (MADRS total score ≥22) or an insufficient therapeutic response (as judged by the investigator) was the primary efficacy outcome, and demonstrated that vortioxetine was superior to placebo.
Tolerability
The tolerability of vortioxetine is comparable with other serotonergic antidepressants. In pooled analysis of pre-marketing studies, 5% to 8% of patients receiving vortioxetine (5 to 20 mg/d) discontinued treatment because of adverse effects (AEs), compared with 4% in the placebo group. Nausea was the most commonly reported AE leading to discontinuation and appeared to be dose dependent.
AEs, such as nausea, constipation, and vomiting, most commonly occurred in the first week of treatment, with a median duration of 2 weeks.5 In the 6- to 8-week trials, the most common AEs were nausea, constipation, and vomiting. In longer trials (24 to 64 weeks), the most common AE was nausea.
In 6- to 8-week placebo-controlled studies, vortioxetine was not associated with any clinically significant effect on vital signs or laboratory values in hematology, urinalysis, or serum chemistry (except sodium). Hyponatremia, the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH), has occurred. The risk of developing SIADH and resultant hyponatremia is greater in geriatric patients and patients taking a diuretic.
Abruptly discontinuing vortioxetine can cause transient withdrawal symptoms, including headache and muscle tension, especially at a higher dosage (15 to 20 mg/d). Gradual tapering can reduce withdrawal symptoms.
Specific clinical issues
All antidepressants have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation and behaviors during the first months of treatment and with dosage changes.
Vortioxetine is categorized as pregnancy category C. Newborns exposed to a selective serotonin reuptake inhibitor (SSRI) in pregnancy may have an increased risk of persistent pulmonary hypertension during the neonatal period. When taken during the third trimester of pregnancy, SSRIs and serotonin-norepinephrine reuptake inhibitors can cause serious neonatal complications, including respiratory distress, cyanosis, apnea, and seizures, which may require longer hospitalization, respiratory support, or tube feeding for the infant. Consider risks and benefits of third-trimester use of an antidepressant. It is not known if vortioxetine is present in human breast milk.
Clinical studies on vortioxetine in pediatric patients have not been conducted.
No dosage adjustment is recommended on the basis of age for geriatric patients. No dose adjustment of vortioxetine is necessary on the basis of race, sex, ethnicity, renal function, or mild to moderate hepatic impairment. See Table 3 for practice points when prescribing vortioxetine. See Table 4 for contraindications to vortioxetine.
Dosing
The recommended starting dosage is 10 mg, administered orally once daily without regard to meals. Dosage should then be increased to 20 mg/d, as clinically warranted and tolerated. Consider a dosage decrease to 5 mg/d in patients who do not tolerate higher dosages or require drug adjustment because of drug-drug interaction or poor 2D6 metabolizer status.
Bottom Line
FDA-approved for major depressive disorder in adults, vortioxetine reduced depressive symptoms in 6 positive, double-blind, placebo-controlled, and randomized studies. The multimodal activity of vortioxeine can be a useful alternative to serotonergic antidepressants for some patients who are partial responders or nonresponders. Tolerability is comparable with other serotonergic antidepressants.
Related Resources
- Alam MY, Jacobsen PL, Chen Y, et al. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2014; 29(1):36-44.
- Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double-blind trial of 2.5 mg and 5 mg vortioxetine (Lu AA21004) versus placebo for 8 weeks in adults with major depressive disorder. Curr Med Res Opin. 2013;29(3):217-226.
Drug Brand Names
Linezolid • Zyvox Vortioxetine • Brintellix
Methylene blue • Urolene Blue Warfarin • Coumadin
Tramadol • Ultram
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Vortioxetine is FDA-approved to treat major depressive disorder (MDD) (Table 1), having shown efficacy in relieving depressive symptoms in clinical trials.1 Vortioxetine’s mechanism of action enhances CNS serotonergic activity through inhibiting serotonin (5-HT) reuptake, agonizing the 5-HT1A receptor, partially agonizing the 5-HT1B receptor, and antagonizing the 5-HT3, 5-HT1D, and 5-HT7 receptors.
Clinical implications
It is hypothesized that depression is a heterogeneous disease caused by dysregulation of serotonin, norepinephrine, and dopamine, subsequently producing mood and neurovegetative symptoms of depression. Preclinical, in vivo studies indicate that vortioxetine enhances levels of serotonin, norepinephrine, dopamine, acetylcholine, and histamine in specific areas of the brain with the ability to improve depressive symptoms. Vortioxetine’s multimodal activity can be a useful alternative to other serotonergic antidepressants for some patients who are partial responders or non-responders to other treatment options. In addition, vortioxetine appears to have minimal effect on weight2 and sexual function—the latter being dose-dependent.3
How does it work?
Vortioxetine differs from other antidepressants in its multimodal activity (ie, affecting G-protein mode receptors, ion channel mode receptors, and neurotransmitter transporters). It inhibits the serotonin transporter (Ki = 1.6 nM), causing subsequent inhibition of serotonin reuptake into presynaptic neurons as well as selectively acting on the other subtypes of serotonergic receptors; however, activity on the norepinephrine transporter (Ki = 113 nM) and dopamine transporter (Ki > 1000 nM) is minimal. It is believed that mood-regulating effects of vortioxetine are caused by inhibition of serotonin reuptake, prolonged availability of serotonin to the postsynaptic neurons, its agonist activity on the 5-HT1A receptor (Ki = 15 nM), and partial agonist activity on the 5-HT1B receptor (Ki = 33 nM). Vortioxetine has strong affinity for the 5-HT3 receptor (Ki = 3.7 nM), which plays a role in modulation of centrally mediated nausea and vomiting. Positron emission tomography studies in humans determined that the occupancy of 5-HT transporter was 50% at 5 mg/d, 65% at 10 mg/d, and 80% at 20 mg/d.1,4 Human studies did not show that vortioxetine causes QTc prolongation.
Pharmacokinetics
Therapeutic activity of vortioxetine is thought to be due to the parent drug. It has a half-life of approximately 66 hours, and achieves steady state in 13.5 to 19 days. Bioavailability of vortioxetine is 75%; absorption does not depend on food; and 98% of drug is bound on plasma proteins.
Vortioxetine has linear pharmacokinetics, with maximum plasma concentration 7 to 11 hours after ingestion. The medication is metabolized primarily by oxidation through cytochrome P (CYP) 450: CYP2D6 (primary), CYP 3A4/5, CYP 2C19, CYP 2C9, CYP2A6, CYP2C8, and CYP2B6 with subsequent glucuronidation. This predisposes vortioxetine to potential pharmacokinetic drug-drug interaction warranting dose adjustment consideration when vortioxetine is coadministered with compounds inhibiting CYP2D6 or inducing CYP3A4 for ≥14 days, or for patients identified as poor 2D6 metabolizers.
In addition, coadministration of vortioxetine with serotonergic medications such as triptans, other antidepressants, and tramadol can cause potentially life-threatening serotonin syndrome, characterized by mental status changes, autonomic instability, neuromuscular aberrations, and GI symptoms. Concomitant use of vortioxetine and a nonsteroidal anti-inflammatory drug, aspirin, or warfarin can result in abnormal bleeding. Coadministration of vortioxetine with another highly protein-bound drug may increase or decrease the free concentration of either drug depending on the binding affinity of the drug for the protein.
Efficacy
Vortioxetine reduced depressive symptoms in 6 positive, 6- to 8-week, double-blind, placebo controlled and randomized studies and 1 maintenance study.1 Subjects were adults (Studies 1 to 5) and geriatric patients from age 64 to 88 who had ≥1 depressive episode before age 60 (Study 6). All met DSM-IV-TR criteria for MDD. Subjects with cognitive impairment scoring <24 on the Mini-Mental Status Examination and children were excluded. Depending on the study, response to the treatment was primarily measured on the Montgomery-Åsberg Depression Rating Scale (MADRS) or Hamilton Depression Rating Scale (HAM-D).
See Table 2 for a description of the positive studies, including dosages. In all studies, vortioxetine was superior to placebo at least one dosage for treating depression. In the 6- to 8-week placebo-controlled studies, an effect of vortioxetine based on the primary efficacy measure was generally observed starting at Week 2; that effect increased in subsequent weeks with the full antidepressant effect of vortioxetine generally not seen until study Week 4 or later.1
The maintenance treatment study included 639 patients who met DSM-IV-TR criteria for MDD. This study lasted for as long as 64 weeks. The first 12-week period was open-label, during which patients were treated with vortioxetine, 5 mg/d or 10 mg/d, with a possibility to adjust the dosage in the first 8 weeks. By the end of Week 12, 396 subjects achieved remission (MADRS <10), 75% of whom were taking vortioxetine, 10 mg/d. These patients were then randomly assigned to placebo or the dosage of vortioxetine to which they had responded, and continued the study for as long as 64 weeks. Time to relapse (MADRS total score ≥22) or an insufficient therapeutic response (as judged by the investigator) was the primary efficacy outcome, and demonstrated that vortioxetine was superior to placebo.
Tolerability
The tolerability of vortioxetine is comparable with other serotonergic antidepressants. In pooled analysis of pre-marketing studies, 5% to 8% of patients receiving vortioxetine (5 to 20 mg/d) discontinued treatment because of adverse effects (AEs), compared with 4% in the placebo group. Nausea was the most commonly reported AE leading to discontinuation and appeared to be dose dependent.
AEs, such as nausea, constipation, and vomiting, most commonly occurred in the first week of treatment, with a median duration of 2 weeks.5 In the 6- to 8-week trials, the most common AEs were nausea, constipation, and vomiting. In longer trials (24 to 64 weeks), the most common AE was nausea.
In 6- to 8-week placebo-controlled studies, vortioxetine was not associated with any clinically significant effect on vital signs or laboratory values in hematology, urinalysis, or serum chemistry (except sodium). Hyponatremia, the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH), has occurred. The risk of developing SIADH and resultant hyponatremia is greater in geriatric patients and patients taking a diuretic.
Abruptly discontinuing vortioxetine can cause transient withdrawal symptoms, including headache and muscle tension, especially at a higher dosage (15 to 20 mg/d). Gradual tapering can reduce withdrawal symptoms.
Specific clinical issues
All antidepressants have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation and behaviors during the first months of treatment and with dosage changes.
Vortioxetine is categorized as pregnancy category C. Newborns exposed to a selective serotonin reuptake inhibitor (SSRI) in pregnancy may have an increased risk of persistent pulmonary hypertension during the neonatal period. When taken during the third trimester of pregnancy, SSRIs and serotonin-norepinephrine reuptake inhibitors can cause serious neonatal complications, including respiratory distress, cyanosis, apnea, and seizures, which may require longer hospitalization, respiratory support, or tube feeding for the infant. Consider risks and benefits of third-trimester use of an antidepressant. It is not known if vortioxetine is present in human breast milk.
Clinical studies on vortioxetine in pediatric patients have not been conducted.
No dosage adjustment is recommended on the basis of age for geriatric patients. No dose adjustment of vortioxetine is necessary on the basis of race, sex, ethnicity, renal function, or mild to moderate hepatic impairment. See Table 3 for practice points when prescribing vortioxetine. See Table 4 for contraindications to vortioxetine.
Dosing
The recommended starting dosage is 10 mg, administered orally once daily without regard to meals. Dosage should then be increased to 20 mg/d, as clinically warranted and tolerated. Consider a dosage decrease to 5 mg/d in patients who do not tolerate higher dosages or require drug adjustment because of drug-drug interaction or poor 2D6 metabolizer status.
Bottom Line
FDA-approved for major depressive disorder in adults, vortioxetine reduced depressive symptoms in 6 positive, double-blind, placebo-controlled, and randomized studies. The multimodal activity of vortioxeine can be a useful alternative to serotonergic antidepressants for some patients who are partial responders or nonresponders. Tolerability is comparable with other serotonergic antidepressants.
Related Resources
- Alam MY, Jacobsen PL, Chen Y, et al. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2014; 29(1):36-44.
- Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double-blind trial of 2.5 mg and 5 mg vortioxetine (Lu AA21004) versus placebo for 8 weeks in adults with major depressive disorder. Curr Med Res Opin. 2013;29(3):217-226.
Drug Brand Names
Linezolid • Zyvox Vortioxetine • Brintellix
Methylene blue • Urolene Blue Warfarin • Coumadin
Tramadol • Ultram
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Vortioxetine [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2013.
2. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a comprehensive review and meta-analysis. J Clin Psychopharmacol. 2009; 29(3):259-266.
4. Chen G, Lee R, Højer A, et al. Pharmacokinetic drug interactions involving vortioxetine (LU AA 21004), a multimodal antidepressant. Clin Drug Invetig. 2013; 33(10):727-736.
5. Citrome L. Vortioxetine for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Prac. 2014;68(1):60-82.
1. Vortioxetine [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2013.
2. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a comprehensive review and meta-analysis. J Clin Psychopharmacol. 2009; 29(3):259-266.
4. Chen G, Lee R, Højer A, et al. Pharmacokinetic drug interactions involving vortioxetine (LU AA 21004), a multimodal antidepressant. Clin Drug Invetig. 2013; 33(10):727-736.
5. Citrome L. Vortioxetine for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Prac. 2014;68(1):60-82.
Long-acting injectable aripiprazole for adult schizophrenia
In February 2013, the FDA approved a long-acting IM aripiprazole formulation for treating adult schizophrenia (Table 1).1 It is the fourth second-generation antipsychotic (SGA) depot formulation approved for treating schizophrenia, and the sixth depot antipsychotic if haloperidol and fluphenazine decanoate are considered.
Table 1
Depot aripiprazole: Fast facts
| Brand name: Abilify Maintena |
| Class: Atypical antipsychotic |
| Indication: Adult schizophrenia |
| Approval date: February 28, 2013 |
| Availability date: March 18, 2013 |
| Manufacturer: Otsuka Pharmaceutical and Lundbeck |
| Dosing form: IM long-acting injection |
| Recommended dosage: 400 mg IM once a month; 200 to 300 mg IM if drug-drug interactions, poor cytochrome P450 2D6 metabolism, or adverse effects |
| Source: Reference 1 |
Clinical implications
Depot medications can improve treatment adherence2; however, long-term antipsychotic use can lead to irreversible adverse effects (dyskinesias), which in some cases were reduced by using newer antipsychotics.3
How it works
Similar to other SGAs, aripiprazole’s mechanism of action is unknown. Aripiprazole was developed based on the dopamine theory, in which dopamine hyperactivity in mesolimbic pathways of the brain leads to hallucinations, delusions, disorganization, and catatonia, and dopamine hypoactivity in mesocortical pathways and the prefrontal cortex causes alogia, anhedonia, autism, avolition, and problems with attention and abstract thinking.
Aripiprazole’s proposed mechanism of action on dopamine receptors is that of partial agonism,1 rather than antagonism, as is the case for other SGAs. In theory, aripiprazole antagonizes postsynaptic D2 receptors and activates presynaptic D2 autoreceptors, with subsequently decreased dopamine production and further stabilization of the dopamine system.4 Its antagonism of 5-HT2A is similar to other SGAs.5
Pharmacokinetics
After depot aripiprazole is injected into the gluteal muscle, the active moiety slowly is released into circulation. The effectiveness of depot aripiprazole is attributable to its active parent drug, aripiprazole monohydrate, and its active metabolite, dehydro-aripiprazole, which is the same as oral aripiprazole. Depot aripiprazole reaches maximum concentration in 5 to 7 days. The elimination half-life of depot aripiprazole is 29.9 days for a 300-mg dose and 46.5 days for a 400-mg dose if administered monthly.1
Aripiprazole does not undergo direct glucuronidation. It is metabolized predominantly through cytochrome P450 (CYP) 2D6 and 3A4 enzymes, which predisposes it to significant drug-drug interactions and may require dose adjustment (Table 2).1
Table 2
Dose adjustments of depot aripiprazole
| Drug-drug interaction | Adjusted dose |
|---|---|
| CYP2D6 poor metabolizers | 300 mg |
| CYP2D6 poor metabolizers taking CYP3A4 inhibitors (ketoconazole, itraconazole, erythromycin, grapefruit juice) | 200 mg |
| Lithium, valproate, desvenlafaxine, venlafaxine, escitalopram, dextromethorphan, omeprazole, warfarin | No significant interaction No dose adjustment |
| Sex, race, liver impairment, renal impairment, tobacco smokers | No dose adjustment |
| Patients taking 400 mg of depot aripiprazole with: | |
| 300 mg |
| 200 mg |
| Avoid use |
| Patients taking 300 mg of depot aripiprazole with: | |
| 200 mg |
| 160 mg |
| Avoid use |
| CYP: cytochrome P450 Source: Adapted from reference 1 | |
Efficacy
The ability of depot aripiprazole to sustain long-term symptom control in adult patients with schizophrenia was demonstrated in a randomized-withdrawal, double-blind, placebo-controlled trial.1 Adults included had a DSM-IV-TR diagnosis of schizophrenia, had ≥3-year history of the illness, had undergone treatment with ≥1 antipsychotic, and had a history of relapse or symptom exacerbation when not receiving antipsychotics. Psychopathology was measured by the Positive and Negative Syndrome Scale (PANSS), the Clinical Global Impression-Severity scale, the Clinical Global Impression-Improvement (CGI-I) scale, and the Clinical Global Impression-Severity of Suicide (CGI-SS) scale.1
The trial lasted 52 weeks, was divided into 4 phases, and concluded early because of demonstrated efficacy.
Phase I: Conversion phase switched patients from a different antipsychotic to oral aripiprazole. This phase lasted 4 to 6 weeks and included 633 patients. An additional 210 patients already receiving aripiprazole were entered directly into Phase II.
Phase II: Open-label, oral stabilization phase included 710 patients (60% males) age 18 to 60 who had a mean PANSS score 66. Patients received 10 to 20 mg/d of oral aripiprazole until they achieved stabilization, defined as PANSS score
Phase III: IM depot stabilization (uncontrolled single blind) included 576 patients. Patients were started on depot aripiprazole, 400 mg monthly, and continued to take 10 to 20 mg/d of oral aripiprazole for 14 consecutive days. Depot aripiprazole was decreased to 300 mg monthly if a patient developed adverse effects. Patients continued to the double-blind phase when stabilization was achieved, as evidenced by PANSS score
Phase IV: Maintenance (double-blind, randomized, placebo-controlled) included 403 patients. Two-thirds of patients continued to take the same dose of depot aripiprazole they took in Phase III. One-third of patients were switched to placebo. The primary efficacy endpoint was time to impending relapse, defined as the first occurrence of ≥1 criteria: hospitalization due to psychosis; violence toward self, others, or property; CGI-SS score ≥4 on part I or ≥7 on part II; or CGI-I score ≥5 and any individual PANSS score >4 for disorganization, hallucinations, suspiciousness, or abnormal thought content.1
Patients randomized to continue depot aripiprazole took longer to relapse or worsening of symptoms compared with the placebo group. Of 403 patients, 10% taking an active drug and 39.6% taking placebo relapsed within 360 days of randomization. This difference was statistically significant (P 1
Tolerability
One possible problem with any long-acting medication is increased duration of adverse effects (AEs), if they develop. Therefore, assessment of safety and tolerability is more important in depot formulations than in oral drugs. During the clinical trial, depot aripiprazole was well tolerated.6
During clinical trials, the most common AEs—insomnia (>5%), anxiety, and tremors—were mild to moderate and occurred within the first 4 weeks. Discontinuation of the medication because of AEs was low, and pain at the injection site was minimal.6 There were 2 deaths during the trial, which were unrelated to depot aripiprazole.6
Aripiprazole’s activity on the D2 receptor can cause extrapyramidal AEs. In head-to-head trials, patients taking aripiprazole had fewer extrapyramidal AEs than those taking risperidone or ziprasidone, but more than patients receiving olanzapine.7 Its moderate antagonism on α-adrenergic and histamine 1 (H1) receptors translates to low orthostatic hypotension, H1-mediated weight gain, and sedation. In clinical trials, weight gain and metabolic changes were comparable with placebo. In head-to-head trials, aripiprazole caused less weight gain and a higher incidence of increased cholesterol than olanzapine and risperidone, and less increase in blood glucose than olanzapine, but more than risperidone.8 Muscarinic 1-mediated cognitive impairment, dry mouth, constipation, urinary retention, and increased intraocular pressure were low.8 See Table 3 for aripiprazole's receptor binding profile.
Table 3
Aripiprazole’s receptor binding profile
| Affinity | Ki (nM)a | Effects associated with activity on the receptor | |
|---|---|---|---|
| D2 | High | 0.34 | Partial agonist |
| D3 | High | 0.8 | Partial agonist |
| 5-HT1A | High | 1.7 | Partial agonist |
| 5-HT2A | High | 3.4 | Antagonist |
| 5-HT2C | Moderate | 15 | Partial agonist |
| 5-HT7 | Moderate | 39 | Antagonist |
| D4 | Moderate | 44 | Partial agonist |
| α1-adrenergic | Moderate | 57 | Antagonist |
| H1 | Moderate | 61 | Antagonist |
| M1 | No appreciable activity | >1,000 | No appreciable activity |
| aKi dissociation constant: lower numbers indicate higher affinity of the compound for the receptor H1: histamine 1; M1: muscarinic 1 Source: References 1,6 | |||
Unique clinical issues
Clinical features for depot aripiprazole can be partially deduced based on data on oral aripiprazole. Advantages over other depot SGAs might include aripiprazole’s more favorable weight and metabolic profile.
Contraindications
Depot aripiprazole is contraindicated in patients with known sensitivity to aripiprazole or other components of the formulation. Because of pharmacokinetic drug-drug interactions, using depot aripiprazole should be avoided in patients taking strong CYP3A4 inducers (eg, rifampin and carbamazepine). Dose adjustment is recommended in patients who are taking moderate CYP2D6 and 3A4 inhibitors, such as paroxetine, fluoxetine, ketoconazole, or erythromycin.1 A “black-box” warning of increased mortality in older patients with dementia-related psychosis applies for depot aripiprazole as well as for other atypical antipsychotics.1
Depot aripiprazole is pregnancy category C and should be used in pregnant or breastfeeding mothers only when benefits outweigh the risks. Use of depot aripiprazole in geriatric and pediatric populations has not been studied; however, patients age ≥65 who received oral aripiprazole, 15 mg/d, showed decreased clearance by 20%.1
Dosing
Depot aripiprazole is available as a lyophilized powder that needs to be reconstituted in sterile water. The drug can be stored at room temperature. The kit includes two 21-gauge needles, a 1.5-inch needle for non-obese patients and a 2-inch needle for obese patients. Depot aripiprazole should be given to patients who demonstrate tolerability to oral aripiprazole. The starting and maintenance dose of depot aripiprazole is 400 mg injected into the gluteal muscle, once a month. If a patient develops an AE, decrease the monthly dose to 300 mg. Rotate the injection site between gluteal muscles to reduce AEs from injection.
Because of the potential for significant pharmacokinetic drug-drug interactions, dose adjustment is recommended for patients who are CYP2D6 poor metabolizers and those taking certain other medications (Table 4).1 See Table 4 for the recommended dosage adjustment in the case of missed doses.
Table 4
Adjusting depot aripiprazole after missed doses
| Doses missed since last injection | ||||
|---|---|---|---|---|
| Second or third dose | Fourth or subsequent dose | |||
| >4 weeks and | >5 weeks | >4 weeks and | >6 weeks | |
| Oral aripiprazole | Administer for 14 days | Administer for 14 days | ||
| Depot aripiprazole | Administer as soon as possible | Administer next injection | Administer as soon as possible | Administer next injection |
| Source: Reference 1 | ||||
After depot aripiprazole is injected into the gluteal muscle, the patient receives 10 to 20 mg/d of oral aripiprazole for 14 consecutive days to avoid a drop in plasma concentrations into subtherapeutic levels.
Related Resource
- Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
Drug Brand Names
- Aripiprazole • Abilify
- Aripiprazole depot • Maintena
- Carbamazepine • Tegretol
- Desvenlafaxine • Pristiq
- Dextromethorphan • Delsym
- Erythromycin • E-Mycin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Itraconazole • Sporanox
- Ketoconazole • Nizoral
- Lithium • Eskalith, Lithobid
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Paliperidone • Invega
- Paroxetine • Paxil
- Quinidine • Quinidex
- Rifampin • Rifadin
- Risperidone • Risperdal
- Valproate • Depakote
- Venlafaxine • Effexor
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
Dr. Lincoln receives grant or research support from the Wichita Center for Graduate Medical Education.
1. Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
2. Leucht C, Heres S, Kane JM, et al. Oral versus depot antipsychotic drugs for schizophrenia—a critical systematic review and meta-analysis of randomized long-term trials. Schizophr Res. 2011;127(1-3):83-92.
3. de Araújo AN, de Sena EP, de Oliveira IR, et al. Antipsychotic agents: efficacy and safety in schizophrenia. Drug Healthc Patient Saf. 2012;4:173-180.
4. Mailman RB, Murty V. Third generation antipsychotic drugs: partial agonism or receptor functional selectivity? Curr Pharm Des. 2010;16(5):488-501.
5. Roth BL, Meltzer HY. The role of serotonin in schizophrenia. http://www.acnp.org/g4/GN401000117/CH115.html. Published 2000. Accessed March 27 2013.
6. Kane JM, Sanchez R, Perry PP, et al. Aripiprazole intramuscular depot as maintenance treatment in patients with schizophrenia: a 52-week, multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2012;73(5):617-624.
7. Rummel-Kluge C, Komossa K, Schwarz S, et al. Second-generation antipsychotic drugs and extrapyramidal side effects: a systematic review and meta-analysis of head-to-head comparisons. Schizophr Bull. 2012;38(1):167-177.
8. Rummel-Kluge C, Komossa K, Schwarz S, et al. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2010;123(2-3):225-233.
In February 2013, the FDA approved a long-acting IM aripiprazole formulation for treating adult schizophrenia (Table 1).1 It is the fourth second-generation antipsychotic (SGA) depot formulation approved for treating schizophrenia, and the sixth depot antipsychotic if haloperidol and fluphenazine decanoate are considered.
Table 1
Depot aripiprazole: Fast facts
| Brand name: Abilify Maintena |
| Class: Atypical antipsychotic |
| Indication: Adult schizophrenia |
| Approval date: February 28, 2013 |
| Availability date: March 18, 2013 |
| Manufacturer: Otsuka Pharmaceutical and Lundbeck |
| Dosing form: IM long-acting injection |
| Recommended dosage: 400 mg IM once a month; 200 to 300 mg IM if drug-drug interactions, poor cytochrome P450 2D6 metabolism, or adverse effects |
| Source: Reference 1 |
Clinical implications
Depot medications can improve treatment adherence2; however, long-term antipsychotic use can lead to irreversible adverse effects (dyskinesias), which in some cases were reduced by using newer antipsychotics.3
How it works
Similar to other SGAs, aripiprazole’s mechanism of action is unknown. Aripiprazole was developed based on the dopamine theory, in which dopamine hyperactivity in mesolimbic pathways of the brain leads to hallucinations, delusions, disorganization, and catatonia, and dopamine hypoactivity in mesocortical pathways and the prefrontal cortex causes alogia, anhedonia, autism, avolition, and problems with attention and abstract thinking.
Aripiprazole’s proposed mechanism of action on dopamine receptors is that of partial agonism,1 rather than antagonism, as is the case for other SGAs. In theory, aripiprazole antagonizes postsynaptic D2 receptors and activates presynaptic D2 autoreceptors, with subsequently decreased dopamine production and further stabilization of the dopamine system.4 Its antagonism of 5-HT2A is similar to other SGAs.5
Pharmacokinetics
After depot aripiprazole is injected into the gluteal muscle, the active moiety slowly is released into circulation. The effectiveness of depot aripiprazole is attributable to its active parent drug, aripiprazole monohydrate, and its active metabolite, dehydro-aripiprazole, which is the same as oral aripiprazole. Depot aripiprazole reaches maximum concentration in 5 to 7 days. The elimination half-life of depot aripiprazole is 29.9 days for a 300-mg dose and 46.5 days for a 400-mg dose if administered monthly.1
Aripiprazole does not undergo direct glucuronidation. It is metabolized predominantly through cytochrome P450 (CYP) 2D6 and 3A4 enzymes, which predisposes it to significant drug-drug interactions and may require dose adjustment (Table 2).1
Table 2
Dose adjustments of depot aripiprazole
| Drug-drug interaction | Adjusted dose |
|---|---|
| CYP2D6 poor metabolizers | 300 mg |
| CYP2D6 poor metabolizers taking CYP3A4 inhibitors (ketoconazole, itraconazole, erythromycin, grapefruit juice) | 200 mg |
| Lithium, valproate, desvenlafaxine, venlafaxine, escitalopram, dextromethorphan, omeprazole, warfarin | No significant interaction No dose adjustment |
| Sex, race, liver impairment, renal impairment, tobacco smokers | No dose adjustment |
| Patients taking 400 mg of depot aripiprazole with: | |
| 300 mg |
| 200 mg |
| Avoid use |
| Patients taking 300 mg of depot aripiprazole with: | |
| 200 mg |
| 160 mg |
| Avoid use |
| CYP: cytochrome P450 Source: Adapted from reference 1 | |
Efficacy
The ability of depot aripiprazole to sustain long-term symptom control in adult patients with schizophrenia was demonstrated in a randomized-withdrawal, double-blind, placebo-controlled trial.1 Adults included had a DSM-IV-TR diagnosis of schizophrenia, had ≥3-year history of the illness, had undergone treatment with ≥1 antipsychotic, and had a history of relapse or symptom exacerbation when not receiving antipsychotics. Psychopathology was measured by the Positive and Negative Syndrome Scale (PANSS), the Clinical Global Impression-Severity scale, the Clinical Global Impression-Improvement (CGI-I) scale, and the Clinical Global Impression-Severity of Suicide (CGI-SS) scale.1
The trial lasted 52 weeks, was divided into 4 phases, and concluded early because of demonstrated efficacy.
Phase I: Conversion phase switched patients from a different antipsychotic to oral aripiprazole. This phase lasted 4 to 6 weeks and included 633 patients. An additional 210 patients already receiving aripiprazole were entered directly into Phase II.
Phase II: Open-label, oral stabilization phase included 710 patients (60% males) age 18 to 60 who had a mean PANSS score 66. Patients received 10 to 20 mg/d of oral aripiprazole until they achieved stabilization, defined as PANSS score
Phase III: IM depot stabilization (uncontrolled single blind) included 576 patients. Patients were started on depot aripiprazole, 400 mg monthly, and continued to take 10 to 20 mg/d of oral aripiprazole for 14 consecutive days. Depot aripiprazole was decreased to 300 mg monthly if a patient developed adverse effects. Patients continued to the double-blind phase when stabilization was achieved, as evidenced by PANSS score
Phase IV: Maintenance (double-blind, randomized, placebo-controlled) included 403 patients. Two-thirds of patients continued to take the same dose of depot aripiprazole they took in Phase III. One-third of patients were switched to placebo. The primary efficacy endpoint was time to impending relapse, defined as the first occurrence of ≥1 criteria: hospitalization due to psychosis; violence toward self, others, or property; CGI-SS score ≥4 on part I or ≥7 on part II; or CGI-I score ≥5 and any individual PANSS score >4 for disorganization, hallucinations, suspiciousness, or abnormal thought content.1
Patients randomized to continue depot aripiprazole took longer to relapse or worsening of symptoms compared with the placebo group. Of 403 patients, 10% taking an active drug and 39.6% taking placebo relapsed within 360 days of randomization. This difference was statistically significant (P 1
Tolerability
One possible problem with any long-acting medication is increased duration of adverse effects (AEs), if they develop. Therefore, assessment of safety and tolerability is more important in depot formulations than in oral drugs. During the clinical trial, depot aripiprazole was well tolerated.6
During clinical trials, the most common AEs—insomnia (>5%), anxiety, and tremors—were mild to moderate and occurred within the first 4 weeks. Discontinuation of the medication because of AEs was low, and pain at the injection site was minimal.6 There were 2 deaths during the trial, which were unrelated to depot aripiprazole.6
Aripiprazole’s activity on the D2 receptor can cause extrapyramidal AEs. In head-to-head trials, patients taking aripiprazole had fewer extrapyramidal AEs than those taking risperidone or ziprasidone, but more than patients receiving olanzapine.7 Its moderate antagonism on α-adrenergic and histamine 1 (H1) receptors translates to low orthostatic hypotension, H1-mediated weight gain, and sedation. In clinical trials, weight gain and metabolic changes were comparable with placebo. In head-to-head trials, aripiprazole caused less weight gain and a higher incidence of increased cholesterol than olanzapine and risperidone, and less increase in blood glucose than olanzapine, but more than risperidone.8 Muscarinic 1-mediated cognitive impairment, dry mouth, constipation, urinary retention, and increased intraocular pressure were low.8 See Table 3 for aripiprazole's receptor binding profile.
Table 3
Aripiprazole’s receptor binding profile
| Affinity | Ki (nM)a | Effects associated with activity on the receptor | |
|---|---|---|---|
| D2 | High | 0.34 | Partial agonist |
| D3 | High | 0.8 | Partial agonist |
| 5-HT1A | High | 1.7 | Partial agonist |
| 5-HT2A | High | 3.4 | Antagonist |
| 5-HT2C | Moderate | 15 | Partial agonist |
| 5-HT7 | Moderate | 39 | Antagonist |
| D4 | Moderate | 44 | Partial agonist |
| α1-adrenergic | Moderate | 57 | Antagonist |
| H1 | Moderate | 61 | Antagonist |
| M1 | No appreciable activity | >1,000 | No appreciable activity |
| aKi dissociation constant: lower numbers indicate higher affinity of the compound for the receptor H1: histamine 1; M1: muscarinic 1 Source: References 1,6 | |||
Unique clinical issues
Clinical features for depot aripiprazole can be partially deduced based on data on oral aripiprazole. Advantages over other depot SGAs might include aripiprazole’s more favorable weight and metabolic profile.
Contraindications
Depot aripiprazole is contraindicated in patients with known sensitivity to aripiprazole or other components of the formulation. Because of pharmacokinetic drug-drug interactions, using depot aripiprazole should be avoided in patients taking strong CYP3A4 inducers (eg, rifampin and carbamazepine). Dose adjustment is recommended in patients who are taking moderate CYP2D6 and 3A4 inhibitors, such as paroxetine, fluoxetine, ketoconazole, or erythromycin.1 A “black-box” warning of increased mortality in older patients with dementia-related psychosis applies for depot aripiprazole as well as for other atypical antipsychotics.1
Depot aripiprazole is pregnancy category C and should be used in pregnant or breastfeeding mothers only when benefits outweigh the risks. Use of depot aripiprazole in geriatric and pediatric populations has not been studied; however, patients age ≥65 who received oral aripiprazole, 15 mg/d, showed decreased clearance by 20%.1
Dosing
Depot aripiprazole is available as a lyophilized powder that needs to be reconstituted in sterile water. The drug can be stored at room temperature. The kit includes two 21-gauge needles, a 1.5-inch needle for non-obese patients and a 2-inch needle for obese patients. Depot aripiprazole should be given to patients who demonstrate tolerability to oral aripiprazole. The starting and maintenance dose of depot aripiprazole is 400 mg injected into the gluteal muscle, once a month. If a patient develops an AE, decrease the monthly dose to 300 mg. Rotate the injection site between gluteal muscles to reduce AEs from injection.
Because of the potential for significant pharmacokinetic drug-drug interactions, dose adjustment is recommended for patients who are CYP2D6 poor metabolizers and those taking certain other medications (Table 4).1 See Table 4 for the recommended dosage adjustment in the case of missed doses.
Table 4
Adjusting depot aripiprazole after missed doses
| Doses missed since last injection | ||||
|---|---|---|---|---|
| Second or third dose | Fourth or subsequent dose | |||
| >4 weeks and | >5 weeks | >4 weeks and | >6 weeks | |
| Oral aripiprazole | Administer for 14 days | Administer for 14 days | ||
| Depot aripiprazole | Administer as soon as possible | Administer next injection | Administer as soon as possible | Administer next injection |
| Source: Reference 1 | ||||
After depot aripiprazole is injected into the gluteal muscle, the patient receives 10 to 20 mg/d of oral aripiprazole for 14 consecutive days to avoid a drop in plasma concentrations into subtherapeutic levels.
Related Resource
- Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
Drug Brand Names
- Aripiprazole • Abilify
- Aripiprazole depot • Maintena
- Carbamazepine • Tegretol
- Desvenlafaxine • Pristiq
- Dextromethorphan • Delsym
- Erythromycin • E-Mycin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Itraconazole • Sporanox
- Ketoconazole • Nizoral
- Lithium • Eskalith, Lithobid
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Paliperidone • Invega
- Paroxetine • Paxil
- Quinidine • Quinidex
- Rifampin • Rifadin
- Risperidone • Risperdal
- Valproate • Depakote
- Venlafaxine • Effexor
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
Dr. Lincoln receives grant or research support from the Wichita Center for Graduate Medical Education.
In February 2013, the FDA approved a long-acting IM aripiprazole formulation for treating adult schizophrenia (Table 1).1 It is the fourth second-generation antipsychotic (SGA) depot formulation approved for treating schizophrenia, and the sixth depot antipsychotic if haloperidol and fluphenazine decanoate are considered.
Table 1
Depot aripiprazole: Fast facts
| Brand name: Abilify Maintena |
| Class: Atypical antipsychotic |
| Indication: Adult schizophrenia |
| Approval date: February 28, 2013 |
| Availability date: March 18, 2013 |
| Manufacturer: Otsuka Pharmaceutical and Lundbeck |
| Dosing form: IM long-acting injection |
| Recommended dosage: 400 mg IM once a month; 200 to 300 mg IM if drug-drug interactions, poor cytochrome P450 2D6 metabolism, or adverse effects |
| Source: Reference 1 |
Clinical implications
Depot medications can improve treatment adherence2; however, long-term antipsychotic use can lead to irreversible adverse effects (dyskinesias), which in some cases were reduced by using newer antipsychotics.3
How it works
Similar to other SGAs, aripiprazole’s mechanism of action is unknown. Aripiprazole was developed based on the dopamine theory, in which dopamine hyperactivity in mesolimbic pathways of the brain leads to hallucinations, delusions, disorganization, and catatonia, and dopamine hypoactivity in mesocortical pathways and the prefrontal cortex causes alogia, anhedonia, autism, avolition, and problems with attention and abstract thinking.
Aripiprazole’s proposed mechanism of action on dopamine receptors is that of partial agonism,1 rather than antagonism, as is the case for other SGAs. In theory, aripiprazole antagonizes postsynaptic D2 receptors and activates presynaptic D2 autoreceptors, with subsequently decreased dopamine production and further stabilization of the dopamine system.4 Its antagonism of 5-HT2A is similar to other SGAs.5
Pharmacokinetics
After depot aripiprazole is injected into the gluteal muscle, the active moiety slowly is released into circulation. The effectiveness of depot aripiprazole is attributable to its active parent drug, aripiprazole monohydrate, and its active metabolite, dehydro-aripiprazole, which is the same as oral aripiprazole. Depot aripiprazole reaches maximum concentration in 5 to 7 days. The elimination half-life of depot aripiprazole is 29.9 days for a 300-mg dose and 46.5 days for a 400-mg dose if administered monthly.1
Aripiprazole does not undergo direct glucuronidation. It is metabolized predominantly through cytochrome P450 (CYP) 2D6 and 3A4 enzymes, which predisposes it to significant drug-drug interactions and may require dose adjustment (Table 2).1
Table 2
Dose adjustments of depot aripiprazole
| Drug-drug interaction | Adjusted dose |
|---|---|
| CYP2D6 poor metabolizers | 300 mg |
| CYP2D6 poor metabolizers taking CYP3A4 inhibitors (ketoconazole, itraconazole, erythromycin, grapefruit juice) | 200 mg |
| Lithium, valproate, desvenlafaxine, venlafaxine, escitalopram, dextromethorphan, omeprazole, warfarin | No significant interaction No dose adjustment |
| Sex, race, liver impairment, renal impairment, tobacco smokers | No dose adjustment |
| Patients taking 400 mg of depot aripiprazole with: | |
| 300 mg |
| 200 mg |
| Avoid use |
| Patients taking 300 mg of depot aripiprazole with: | |
| 200 mg |
| 160 mg |
| Avoid use |
| CYP: cytochrome P450 Source: Adapted from reference 1 | |
Efficacy
The ability of depot aripiprazole to sustain long-term symptom control in adult patients with schizophrenia was demonstrated in a randomized-withdrawal, double-blind, placebo-controlled trial.1 Adults included had a DSM-IV-TR diagnosis of schizophrenia, had ≥3-year history of the illness, had undergone treatment with ≥1 antipsychotic, and had a history of relapse or symptom exacerbation when not receiving antipsychotics. Psychopathology was measured by the Positive and Negative Syndrome Scale (PANSS), the Clinical Global Impression-Severity scale, the Clinical Global Impression-Improvement (CGI-I) scale, and the Clinical Global Impression-Severity of Suicide (CGI-SS) scale.1
The trial lasted 52 weeks, was divided into 4 phases, and concluded early because of demonstrated efficacy.
Phase I: Conversion phase switched patients from a different antipsychotic to oral aripiprazole. This phase lasted 4 to 6 weeks and included 633 patients. An additional 210 patients already receiving aripiprazole were entered directly into Phase II.
Phase II: Open-label, oral stabilization phase included 710 patients (60% males) age 18 to 60 who had a mean PANSS score 66. Patients received 10 to 20 mg/d of oral aripiprazole until they achieved stabilization, defined as PANSS score
Phase III: IM depot stabilization (uncontrolled single blind) included 576 patients. Patients were started on depot aripiprazole, 400 mg monthly, and continued to take 10 to 20 mg/d of oral aripiprazole for 14 consecutive days. Depot aripiprazole was decreased to 300 mg monthly if a patient developed adverse effects. Patients continued to the double-blind phase when stabilization was achieved, as evidenced by PANSS score
Phase IV: Maintenance (double-blind, randomized, placebo-controlled) included 403 patients. Two-thirds of patients continued to take the same dose of depot aripiprazole they took in Phase III. One-third of patients were switched to placebo. The primary efficacy endpoint was time to impending relapse, defined as the first occurrence of ≥1 criteria: hospitalization due to psychosis; violence toward self, others, or property; CGI-SS score ≥4 on part I or ≥7 on part II; or CGI-I score ≥5 and any individual PANSS score >4 for disorganization, hallucinations, suspiciousness, or abnormal thought content.1
Patients randomized to continue depot aripiprazole took longer to relapse or worsening of symptoms compared with the placebo group. Of 403 patients, 10% taking an active drug and 39.6% taking placebo relapsed within 360 days of randomization. This difference was statistically significant (P 1
Tolerability
One possible problem with any long-acting medication is increased duration of adverse effects (AEs), if they develop. Therefore, assessment of safety and tolerability is more important in depot formulations than in oral drugs. During the clinical trial, depot aripiprazole was well tolerated.6
During clinical trials, the most common AEs—insomnia (>5%), anxiety, and tremors—were mild to moderate and occurred within the first 4 weeks. Discontinuation of the medication because of AEs was low, and pain at the injection site was minimal.6 There were 2 deaths during the trial, which were unrelated to depot aripiprazole.6
Aripiprazole’s activity on the D2 receptor can cause extrapyramidal AEs. In head-to-head trials, patients taking aripiprazole had fewer extrapyramidal AEs than those taking risperidone or ziprasidone, but more than patients receiving olanzapine.7 Its moderate antagonism on α-adrenergic and histamine 1 (H1) receptors translates to low orthostatic hypotension, H1-mediated weight gain, and sedation. In clinical trials, weight gain and metabolic changes were comparable with placebo. In head-to-head trials, aripiprazole caused less weight gain and a higher incidence of increased cholesterol than olanzapine and risperidone, and less increase in blood glucose than olanzapine, but more than risperidone.8 Muscarinic 1-mediated cognitive impairment, dry mouth, constipation, urinary retention, and increased intraocular pressure were low.8 See Table 3 for aripiprazole's receptor binding profile.
Table 3
Aripiprazole’s receptor binding profile
| Affinity | Ki (nM)a | Effects associated with activity on the receptor | |
|---|---|---|---|
| D2 | High | 0.34 | Partial agonist |
| D3 | High | 0.8 | Partial agonist |
| 5-HT1A | High | 1.7 | Partial agonist |
| 5-HT2A | High | 3.4 | Antagonist |
| 5-HT2C | Moderate | 15 | Partial agonist |
| 5-HT7 | Moderate | 39 | Antagonist |
| D4 | Moderate | 44 | Partial agonist |
| α1-adrenergic | Moderate | 57 | Antagonist |
| H1 | Moderate | 61 | Antagonist |
| M1 | No appreciable activity | >1,000 | No appreciable activity |
| aKi dissociation constant: lower numbers indicate higher affinity of the compound for the receptor H1: histamine 1; M1: muscarinic 1 Source: References 1,6 | |||
Unique clinical issues
Clinical features for depot aripiprazole can be partially deduced based on data on oral aripiprazole. Advantages over other depot SGAs might include aripiprazole’s more favorable weight and metabolic profile.
Contraindications
Depot aripiprazole is contraindicated in patients with known sensitivity to aripiprazole or other components of the formulation. Because of pharmacokinetic drug-drug interactions, using depot aripiprazole should be avoided in patients taking strong CYP3A4 inducers (eg, rifampin and carbamazepine). Dose adjustment is recommended in patients who are taking moderate CYP2D6 and 3A4 inhibitors, such as paroxetine, fluoxetine, ketoconazole, or erythromycin.1 A “black-box” warning of increased mortality in older patients with dementia-related psychosis applies for depot aripiprazole as well as for other atypical antipsychotics.1
Depot aripiprazole is pregnancy category C and should be used in pregnant or breastfeeding mothers only when benefits outweigh the risks. Use of depot aripiprazole in geriatric and pediatric populations has not been studied; however, patients age ≥65 who received oral aripiprazole, 15 mg/d, showed decreased clearance by 20%.1
Dosing
Depot aripiprazole is available as a lyophilized powder that needs to be reconstituted in sterile water. The drug can be stored at room temperature. The kit includes two 21-gauge needles, a 1.5-inch needle for non-obese patients and a 2-inch needle for obese patients. Depot aripiprazole should be given to patients who demonstrate tolerability to oral aripiprazole. The starting and maintenance dose of depot aripiprazole is 400 mg injected into the gluteal muscle, once a month. If a patient develops an AE, decrease the monthly dose to 300 mg. Rotate the injection site between gluteal muscles to reduce AEs from injection.
Because of the potential for significant pharmacokinetic drug-drug interactions, dose adjustment is recommended for patients who are CYP2D6 poor metabolizers and those taking certain other medications (Table 4).1 See Table 4 for the recommended dosage adjustment in the case of missed doses.
Table 4
Adjusting depot aripiprazole after missed doses
| Doses missed since last injection | ||||
|---|---|---|---|---|
| Second or third dose | Fourth or subsequent dose | |||
| >4 weeks and | >5 weeks | >4 weeks and | >6 weeks | |
| Oral aripiprazole | Administer for 14 days | Administer for 14 days | ||
| Depot aripiprazole | Administer as soon as possible | Administer next injection | Administer as soon as possible | Administer next injection |
| Source: Reference 1 | ||||
After depot aripiprazole is injected into the gluteal muscle, the patient receives 10 to 20 mg/d of oral aripiprazole for 14 consecutive days to avoid a drop in plasma concentrations into subtherapeutic levels.
Related Resource
- Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
Drug Brand Names
- Aripiprazole • Abilify
- Aripiprazole depot • Maintena
- Carbamazepine • Tegretol
- Desvenlafaxine • Pristiq
- Dextromethorphan • Delsym
- Erythromycin • E-Mycin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Itraconazole • Sporanox
- Ketoconazole • Nizoral
- Lithium • Eskalith, Lithobid
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Paliperidone • Invega
- Paroxetine • Paxil
- Quinidine • Quinidex
- Rifampin • Rifadin
- Risperidone • Risperdal
- Valproate • Depakote
- Venlafaxine • Effexor
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
Dr. Lincoln receives grant or research support from the Wichita Center for Graduate Medical Education.
1. Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
2. Leucht C, Heres S, Kane JM, et al. Oral versus depot antipsychotic drugs for schizophrenia—a critical systematic review and meta-analysis of randomized long-term trials. Schizophr Res. 2011;127(1-3):83-92.
3. de Araújo AN, de Sena EP, de Oliveira IR, et al. Antipsychotic agents: efficacy and safety in schizophrenia. Drug Healthc Patient Saf. 2012;4:173-180.
4. Mailman RB, Murty V. Third generation antipsychotic drugs: partial agonism or receptor functional selectivity? Curr Pharm Des. 2010;16(5):488-501.
5. Roth BL, Meltzer HY. The role of serotonin in schizophrenia. http://www.acnp.org/g4/GN401000117/CH115.html. Published 2000. Accessed March 27 2013.
6. Kane JM, Sanchez R, Perry PP, et al. Aripiprazole intramuscular depot as maintenance treatment in patients with schizophrenia: a 52-week, multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2012;73(5):617-624.
7. Rummel-Kluge C, Komossa K, Schwarz S, et al. Second-generation antipsychotic drugs and extrapyramidal side effects: a systematic review and meta-analysis of head-to-head comparisons. Schizophr Bull. 2012;38(1):167-177.
8. Rummel-Kluge C, Komossa K, Schwarz S, et al. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2010;123(2-3):225-233.
1. Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
2. Leucht C, Heres S, Kane JM, et al. Oral versus depot antipsychotic drugs for schizophrenia—a critical systematic review and meta-analysis of randomized long-term trials. Schizophr Res. 2011;127(1-3):83-92.
3. de Araújo AN, de Sena EP, de Oliveira IR, et al. Antipsychotic agents: efficacy and safety in schizophrenia. Drug Healthc Patient Saf. 2012;4:173-180.
4. Mailman RB, Murty V. Third generation antipsychotic drugs: partial agonism or receptor functional selectivity? Curr Pharm Des. 2010;16(5):488-501.
5. Roth BL, Meltzer HY. The role of serotonin in schizophrenia. http://www.acnp.org/g4/GN401000117/CH115.html. Published 2000. Accessed March 27 2013.
6. Kane JM, Sanchez R, Perry PP, et al. Aripiprazole intramuscular depot as maintenance treatment in patients with schizophrenia: a 52-week, multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2012;73(5):617-624.
7. Rummel-Kluge C, Komossa K, Schwarz S, et al. Second-generation antipsychotic drugs and extrapyramidal side effects: a systematic review and meta-analysis of head-to-head comparisons. Schizophr Bull. 2012;38(1):167-177.
8. Rummel-Kluge C, Komossa K, Schwarz S, et al. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2010;123(2-3):225-233.
Lurasidone for schizophrenia
In October 2010, the FDA approved lurasidone for the acute treatment of schizophrenia at a dose of 40 or 80 mg/d administered once daily with food (Table 1).
Table 1
Lurasidone: Fast facts
| Brand name: Latuda |
| Indication: Schizophrenia in adults |
| Approval date: October 28, 2010 |
| Availability date: February 2011 |
| Manufacturer: Sunovion Pharmaceuticals, Inc. |
| Dosing forms: 40 mg and 80 mg tablets |
| Recommended dosage: Starting dose: 40 mg/d. Maximum dose: 80 mg/d |
How it works
Although the drug’s exact mechanism of action is not known, it is thought that lurasidone’s antipsychotic properties are related to its antagonism at serotonin 2A (5-HT2A) and dopamine D2 receptors.1
Similar to most other atypical antipsychotics, lurasidone has high binding affinity for 5-HT2A and D2. Lurasidone has also high binding affinity for 5-HT7, 5-HT1A, and α2C-adrenergic receptors, low affinity for α-1 receptors, and virtually no affinity for H1 and M1 receptors (Table 2). Activity on 5-HT7, 5-HT1A, and α2C-adrenergic receptors is believed to enhance cognition, and 5-HT7 is being studied for a potential role in mood regulation and sensory processing.2,3 Lurasidone’s low activity on α-1, H1, and M1 receptors suggests a low risk of orthostatic hypotension, H1-mediated sedation and weight gain, and H1- and M1-mediated cognitive blunting.
Pharmacokinetics
Lurasidone is absorbed in the gastrointestinal tract. It reaches maximum concentration (Cmax) in 1 to 3 hours. Cmax doubles when lurasidone is administered with food (≥350 calories), but absorption is independent of the meal’s fat content.4 After absorption, the drug is highly bound (99%) to serum proteins (albumin and α-1-glycoprotein). The elimination half-life is 18 hours and steady-state concentration is reached within 7 days.1 Lurasidone is eliminated predominantly through cytochrome P450 (CYP) 3A4 metabolism in the liver.
Efficacy
Lurasidone’s efficacy for treatment of acute schizophrenia was established in four 6-week, randomized placebo-controlled clinical trials.1 The patients were adults (mean age: 38.8; range: 18 to 72) who met DSM-IVTR criteria for schizophrenia, didn’t abuse drugs or alcohol, and had not taken any investigational drug for ≥1 month. Symptoms were measured on the Positive and Negative Syndrome Scale (PANSS); Brief Psychiatric Rating Scale as derived from the PANSS (BPRSd); and the Clinical Global Impressions-Severity scale (CGI-S).
In the first clinical trial, 145 patients were randomized to lurasidone, 40 mg/d or 120 mg/d, or placebo. Treatment with either dose of lurasidone was superior to treatment with placebo on the BPRSd (Least Squares Mean [LSM] difference from placebo in change from baseline: -5.6 on lurasidone 40 mg/d, -6.7 on lurasidone 120 mg/d) and CGI-S.1,5
The second trial randomized 180 patients to lurasidone, 80 mg/d, or placebo. Lurasidone, 80 mg/d, was superior to placebo as measured on the BPRSd (LSM difference from placebo in change from baseline: -4.7 on lurasidone 80 mg/d) and CGI-S.1,6
The third trial randomized 489 patients to lurasidone, 40 mg/d, 80 mg/d, 120 mg/d, or placebo. All lurasidone arms were superior to placebo on PANSS (LSM difference from placebo in change from baseline: -2.1 on 40 mg/d, -6.4 on 80 mg/d, and -3.5 on 120 mg/d) and CGI-S scores. This study also showed that lurasidone appears to have a rapid onset of action (day 3 to 4) and provides sustained improvement of symptoms.1
In the fourth trial, 473 individuals were randomized to lurasidone, 40 mg/d or 120 mg/d, olanzapine, 15 mg/d, or placebo. Olanzapine and both dosages of lurasidone were superior to placebo in improving PANSS scores (LSM difference from placebo in change from baseline: -9.7 on lurasidone 40 mg/d, -7.5 on lurasidone 120 mg/d, and -12.6 on olanzapine 15 mg/d) and CGI-S.1,7 Both doses of lurasidone were not superior to olanzapine but had less negative impact on lipid profile, weight gain, and glycemia.
Tolerability
Tolerability information is extracted from a clinical study database consisting of 2,096 patients with schizophrenia who participated in premarketing clinical trials and were exposed to single or multiple doses of lurasidone, 20 mg, 40 mg, 80 mg, or 120 mg.1 Overall, lurasidone was well tolerated. The rate of discontinuation from clinical trials because of adverse effects was 9.4% for lurasidone vs 5.9% for placebo. Somnolence, akathisia, nausea, parkinsonism, and agitation were the most commonly reported adverse reactions; somnolence and akathisia appear dose-related. Other adverse effects associated with lurasidone were nausea, vomiting, dyspepsia, dystonia, dizziness, insomnia, agitation, and anxiety (Table 2).
Metabolic changes (hyperlipidemia, hyperglycemia, and increased body weight) associated with cardiovascular risk in patients treated with atypical antipsychotics were studied in short-term placebo-controlled trials. Lurasidone is considered to be weight-neutral and does not have significant effects on serum lipids or glucose.2 As is the case with other D2 antagonists, lurasidone is associated with increased prolactin, which appears to be greater in females and is dose-dependent. Lurasidone is not associated with significant QTc prolongation, seizures, transaminases increase, or changes in serum chemistry, hematology, or urinalysis.
Table 2
Lurasidone receptor binding profile and receptor-related effects
| Ki (nM)* | Effects associated with activity on the receptor | |
|---|---|---|
| D2 | 0.994 | Antipsychotic effects. Akathisia (15%), parkinsonism (11%), dystonia (5%), hyperprolactinemia (8.3% for women, 1.9% for men) |
| 5-HT2A | 0.47 | Antipsychotic effects. Improves extrapyramidal symptoms |
| 5-HT7 | 0.495 | Antipsychotic effects. Improves cognition, mood |
| 5-HT1A | 6.38 | Improves cognition, mood. Nausea (12%), vomiting (8%) |
| α-1 | 48 | Orthostatic hypotension (5%), sedation (22%) |
| α-2C | 10.8 | Improves cognition |
| H1 | >1000 | No significant adverse effects mediated through H1 receptor because of low binding affinity |
| M1 | >1000 | No significant adverse effects mediated through M1 receptor because of low binding affinity |
| *Ki dissociation constant; the lower the number, the higher affinity of the compound for the receptor Source: Adapted from reference 1, expert opinion, and lurasidone data on file, 2008 | ||
Contraindications
Lurasidone is contraindicated in patients with known sensitivity to lurasidone hydrochloride. Because of the risk for pharmacokinetic drug-drug interactions, lurasidone is contraindicated for patients who are taking strong CYP3A4 inhibitors (eg, ketoconazole) or inducers (eg, rifampin).
Similar to other medications in its class, lurasidone carries a “black-box” warning of increased mortality in elderly patients with dementia-related psychosis and it is not FDA-approved for treating this condition. Animal teratogenicity studies using lurasidone, 25 mg/kg/d and 50 mg/kg/d, did not show adverse effects during organogenesis, and lurasidone is classified as pregnancy category B (animal studies failed to demonstrate risk to the fetus and there are no adequate and well-controlled studies in pregnant women, or animal studies have shown an adverse effect, but adequate and well-controlled studies in pregnant women have failed to demonstrate a risk to the fetus in any trimester). The use of lurasidone in geriatric and pediatric populations was not studied.1
Dosing
Lurasidone is manufactured as 40 mg and 80 mg tablets. The recommended starting dose is 40 mg/d and the maximum recommended dose is 80 mg/d.1 In clinical trials, lurasidone, 120 mg/d, was associated with increased incidence of adverse effects without added benefit.
Lurasidone doesn’t require initial dose titration and should be given with food that provides ≥350 calories to improve medication absorption. Dose adjustment is recommended for use in patients with moderate or severe renal or hepatic impairment and when coadministered with CYP3A4 moderate inhibitors; the dose in these patients should not exceed 40 mg/d.
Related Resource
- Citrome L. Lurasidone for schizophrenia: a review of the efficacy and safety profile for this newly approved second-generation antipsychotic. Int J Clin Pract. 2010 Epub ahead of print.
Drug Brand Names
- Ketoconazole • Nizoral
- Lurasidone • Latuda
- Olanzapine • Zyprexa
- Rifampin • Rifadin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Latuda [package insert]. Marlborough, MA: Sunovion Pharmaceuticals, Inc.; 2010.
2. Meyer JM, Loebel AD, Schweizer E. Lurasidone: a new drug in development for schizophrenia. Expert Opin Investig Drugs. 2009;18(11):1715-1726.
3. Terry AV, Jr, Buccafusco JJ, Wilson C. Cognitive dysfunction in neuropsychiatric, disorders: selected serotonin receptor subtypes as therapeutic targets. Behav Brain Res. 2008;195(1):30-38.
4. Preskorn SH, Yu-Yuan CH, Sarubbi D, et al. Lurasidone pharmacokinetics: Assessment of potential for drug-drug interaction. Abstract presented at: The American College of Neuropsychopharmacology 49th Annual Meeting; December 5-9, 2010; Miami Beach, FL.
5. Loebel A, Cucchiaro J, Ogasa M, et al. Lurasidone for schizophrenia: symptomatic remission during short-term treatment. Abstract presented at: 162nd Annual Meeting of American Psychiatric Association; May 16-21, 2009; San Francisco, CA. Abstract NR1-054.
6. Nakamura M, Ogasa M, Guarino J, et al. Lurasidone in the treatment of acute schizophrenia: a double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(6):829-836.
7. Meltzer H, Cucchiaro J, Silva R, et al. Lurasidone in the treatment of acute schizophrenia: results of the double-blind, placebo-controlled, PEARL 2 trial. Abstract presented at: 48th Annual Meeting of American College of Neuropsychopharmacology; December 6-10, 2009; Hollywood, FL. Abstract 76.
In October 2010, the FDA approved lurasidone for the acute treatment of schizophrenia at a dose of 40 or 80 mg/d administered once daily with food (Table 1).
Table 1
Lurasidone: Fast facts
| Brand name: Latuda |
| Indication: Schizophrenia in adults |
| Approval date: October 28, 2010 |
| Availability date: February 2011 |
| Manufacturer: Sunovion Pharmaceuticals, Inc. |
| Dosing forms: 40 mg and 80 mg tablets |
| Recommended dosage: Starting dose: 40 mg/d. Maximum dose: 80 mg/d |
How it works
Although the drug’s exact mechanism of action is not known, it is thought that lurasidone’s antipsychotic properties are related to its antagonism at serotonin 2A (5-HT2A) and dopamine D2 receptors.1
Similar to most other atypical antipsychotics, lurasidone has high binding affinity for 5-HT2A and D2. Lurasidone has also high binding affinity for 5-HT7, 5-HT1A, and α2C-adrenergic receptors, low affinity for α-1 receptors, and virtually no affinity for H1 and M1 receptors (Table 2). Activity on 5-HT7, 5-HT1A, and α2C-adrenergic receptors is believed to enhance cognition, and 5-HT7 is being studied for a potential role in mood regulation and sensory processing.2,3 Lurasidone’s low activity on α-1, H1, and M1 receptors suggests a low risk of orthostatic hypotension, H1-mediated sedation and weight gain, and H1- and M1-mediated cognitive blunting.
Pharmacokinetics
Lurasidone is absorbed in the gastrointestinal tract. It reaches maximum concentration (Cmax) in 1 to 3 hours. Cmax doubles when lurasidone is administered with food (≥350 calories), but absorption is independent of the meal’s fat content.4 After absorption, the drug is highly bound (99%) to serum proteins (albumin and α-1-glycoprotein). The elimination half-life is 18 hours and steady-state concentration is reached within 7 days.1 Lurasidone is eliminated predominantly through cytochrome P450 (CYP) 3A4 metabolism in the liver.
Efficacy
Lurasidone’s efficacy for treatment of acute schizophrenia was established in four 6-week, randomized placebo-controlled clinical trials.1 The patients were adults (mean age: 38.8; range: 18 to 72) who met DSM-IVTR criteria for schizophrenia, didn’t abuse drugs or alcohol, and had not taken any investigational drug for ≥1 month. Symptoms were measured on the Positive and Negative Syndrome Scale (PANSS); Brief Psychiatric Rating Scale as derived from the PANSS (BPRSd); and the Clinical Global Impressions-Severity scale (CGI-S).
In the first clinical trial, 145 patients were randomized to lurasidone, 40 mg/d or 120 mg/d, or placebo. Treatment with either dose of lurasidone was superior to treatment with placebo on the BPRSd (Least Squares Mean [LSM] difference from placebo in change from baseline: -5.6 on lurasidone 40 mg/d, -6.7 on lurasidone 120 mg/d) and CGI-S.1,5
The second trial randomized 180 patients to lurasidone, 80 mg/d, or placebo. Lurasidone, 80 mg/d, was superior to placebo as measured on the BPRSd (LSM difference from placebo in change from baseline: -4.7 on lurasidone 80 mg/d) and CGI-S.1,6
The third trial randomized 489 patients to lurasidone, 40 mg/d, 80 mg/d, 120 mg/d, or placebo. All lurasidone arms were superior to placebo on PANSS (LSM difference from placebo in change from baseline: -2.1 on 40 mg/d, -6.4 on 80 mg/d, and -3.5 on 120 mg/d) and CGI-S scores. This study also showed that lurasidone appears to have a rapid onset of action (day 3 to 4) and provides sustained improvement of symptoms.1
In the fourth trial, 473 individuals were randomized to lurasidone, 40 mg/d or 120 mg/d, olanzapine, 15 mg/d, or placebo. Olanzapine and both dosages of lurasidone were superior to placebo in improving PANSS scores (LSM difference from placebo in change from baseline: -9.7 on lurasidone 40 mg/d, -7.5 on lurasidone 120 mg/d, and -12.6 on olanzapine 15 mg/d) and CGI-S.1,7 Both doses of lurasidone were not superior to olanzapine but had less negative impact on lipid profile, weight gain, and glycemia.
Tolerability
Tolerability information is extracted from a clinical study database consisting of 2,096 patients with schizophrenia who participated in premarketing clinical trials and were exposed to single or multiple doses of lurasidone, 20 mg, 40 mg, 80 mg, or 120 mg.1 Overall, lurasidone was well tolerated. The rate of discontinuation from clinical trials because of adverse effects was 9.4% for lurasidone vs 5.9% for placebo. Somnolence, akathisia, nausea, parkinsonism, and agitation were the most commonly reported adverse reactions; somnolence and akathisia appear dose-related. Other adverse effects associated with lurasidone were nausea, vomiting, dyspepsia, dystonia, dizziness, insomnia, agitation, and anxiety (Table 2).
Metabolic changes (hyperlipidemia, hyperglycemia, and increased body weight) associated with cardiovascular risk in patients treated with atypical antipsychotics were studied in short-term placebo-controlled trials. Lurasidone is considered to be weight-neutral and does not have significant effects on serum lipids or glucose.2 As is the case with other D2 antagonists, lurasidone is associated with increased prolactin, which appears to be greater in females and is dose-dependent. Lurasidone is not associated with significant QTc prolongation, seizures, transaminases increase, or changes in serum chemistry, hematology, or urinalysis.
Table 2
Lurasidone receptor binding profile and receptor-related effects
| Ki (nM)* | Effects associated with activity on the receptor | |
|---|---|---|
| D2 | 0.994 | Antipsychotic effects. Akathisia (15%), parkinsonism (11%), dystonia (5%), hyperprolactinemia (8.3% for women, 1.9% for men) |
| 5-HT2A | 0.47 | Antipsychotic effects. Improves extrapyramidal symptoms |
| 5-HT7 | 0.495 | Antipsychotic effects. Improves cognition, mood |
| 5-HT1A | 6.38 | Improves cognition, mood. Nausea (12%), vomiting (8%) |
| α-1 | 48 | Orthostatic hypotension (5%), sedation (22%) |
| α-2C | 10.8 | Improves cognition |
| H1 | >1000 | No significant adverse effects mediated through H1 receptor because of low binding affinity |
| M1 | >1000 | No significant adverse effects mediated through M1 receptor because of low binding affinity |
| *Ki dissociation constant; the lower the number, the higher affinity of the compound for the receptor Source: Adapted from reference 1, expert opinion, and lurasidone data on file, 2008 | ||
Contraindications
Lurasidone is contraindicated in patients with known sensitivity to lurasidone hydrochloride. Because of the risk for pharmacokinetic drug-drug interactions, lurasidone is contraindicated for patients who are taking strong CYP3A4 inhibitors (eg, ketoconazole) or inducers (eg, rifampin).
Similar to other medications in its class, lurasidone carries a “black-box” warning of increased mortality in elderly patients with dementia-related psychosis and it is not FDA-approved for treating this condition. Animal teratogenicity studies using lurasidone, 25 mg/kg/d and 50 mg/kg/d, did not show adverse effects during organogenesis, and lurasidone is classified as pregnancy category B (animal studies failed to demonstrate risk to the fetus and there are no adequate and well-controlled studies in pregnant women, or animal studies have shown an adverse effect, but adequate and well-controlled studies in pregnant women have failed to demonstrate a risk to the fetus in any trimester). The use of lurasidone in geriatric and pediatric populations was not studied.1
Dosing
Lurasidone is manufactured as 40 mg and 80 mg tablets. The recommended starting dose is 40 mg/d and the maximum recommended dose is 80 mg/d.1 In clinical trials, lurasidone, 120 mg/d, was associated with increased incidence of adverse effects without added benefit.
Lurasidone doesn’t require initial dose titration and should be given with food that provides ≥350 calories to improve medication absorption. Dose adjustment is recommended for use in patients with moderate or severe renal or hepatic impairment and when coadministered with CYP3A4 moderate inhibitors; the dose in these patients should not exceed 40 mg/d.
Related Resource
- Citrome L. Lurasidone for schizophrenia: a review of the efficacy and safety profile for this newly approved second-generation antipsychotic. Int J Clin Pract. 2010 Epub ahead of print.
Drug Brand Names
- Ketoconazole • Nizoral
- Lurasidone • Latuda
- Olanzapine • Zyprexa
- Rifampin • Rifadin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
In October 2010, the FDA approved lurasidone for the acute treatment of schizophrenia at a dose of 40 or 80 mg/d administered once daily with food (Table 1).
Table 1
Lurasidone: Fast facts
| Brand name: Latuda |
| Indication: Schizophrenia in adults |
| Approval date: October 28, 2010 |
| Availability date: February 2011 |
| Manufacturer: Sunovion Pharmaceuticals, Inc. |
| Dosing forms: 40 mg and 80 mg tablets |
| Recommended dosage: Starting dose: 40 mg/d. Maximum dose: 80 mg/d |
How it works
Although the drug’s exact mechanism of action is not known, it is thought that lurasidone’s antipsychotic properties are related to its antagonism at serotonin 2A (5-HT2A) and dopamine D2 receptors.1
Similar to most other atypical antipsychotics, lurasidone has high binding affinity for 5-HT2A and D2. Lurasidone has also high binding affinity for 5-HT7, 5-HT1A, and α2C-adrenergic receptors, low affinity for α-1 receptors, and virtually no affinity for H1 and M1 receptors (Table 2). Activity on 5-HT7, 5-HT1A, and α2C-adrenergic receptors is believed to enhance cognition, and 5-HT7 is being studied for a potential role in mood regulation and sensory processing.2,3 Lurasidone’s low activity on α-1, H1, and M1 receptors suggests a low risk of orthostatic hypotension, H1-mediated sedation and weight gain, and H1- and M1-mediated cognitive blunting.
Pharmacokinetics
Lurasidone is absorbed in the gastrointestinal tract. It reaches maximum concentration (Cmax) in 1 to 3 hours. Cmax doubles when lurasidone is administered with food (≥350 calories), but absorption is independent of the meal’s fat content.4 After absorption, the drug is highly bound (99%) to serum proteins (albumin and α-1-glycoprotein). The elimination half-life is 18 hours and steady-state concentration is reached within 7 days.1 Lurasidone is eliminated predominantly through cytochrome P450 (CYP) 3A4 metabolism in the liver.
Efficacy
Lurasidone’s efficacy for treatment of acute schizophrenia was established in four 6-week, randomized placebo-controlled clinical trials.1 The patients were adults (mean age: 38.8; range: 18 to 72) who met DSM-IVTR criteria for schizophrenia, didn’t abuse drugs or alcohol, and had not taken any investigational drug for ≥1 month. Symptoms were measured on the Positive and Negative Syndrome Scale (PANSS); Brief Psychiatric Rating Scale as derived from the PANSS (BPRSd); and the Clinical Global Impressions-Severity scale (CGI-S).
In the first clinical trial, 145 patients were randomized to lurasidone, 40 mg/d or 120 mg/d, or placebo. Treatment with either dose of lurasidone was superior to treatment with placebo on the BPRSd (Least Squares Mean [LSM] difference from placebo in change from baseline: -5.6 on lurasidone 40 mg/d, -6.7 on lurasidone 120 mg/d) and CGI-S.1,5
The second trial randomized 180 patients to lurasidone, 80 mg/d, or placebo. Lurasidone, 80 mg/d, was superior to placebo as measured on the BPRSd (LSM difference from placebo in change from baseline: -4.7 on lurasidone 80 mg/d) and CGI-S.1,6
The third trial randomized 489 patients to lurasidone, 40 mg/d, 80 mg/d, 120 mg/d, or placebo. All lurasidone arms were superior to placebo on PANSS (LSM difference from placebo in change from baseline: -2.1 on 40 mg/d, -6.4 on 80 mg/d, and -3.5 on 120 mg/d) and CGI-S scores. This study also showed that lurasidone appears to have a rapid onset of action (day 3 to 4) and provides sustained improvement of symptoms.1
In the fourth trial, 473 individuals were randomized to lurasidone, 40 mg/d or 120 mg/d, olanzapine, 15 mg/d, or placebo. Olanzapine and both dosages of lurasidone were superior to placebo in improving PANSS scores (LSM difference from placebo in change from baseline: -9.7 on lurasidone 40 mg/d, -7.5 on lurasidone 120 mg/d, and -12.6 on olanzapine 15 mg/d) and CGI-S.1,7 Both doses of lurasidone were not superior to olanzapine but had less negative impact on lipid profile, weight gain, and glycemia.
Tolerability
Tolerability information is extracted from a clinical study database consisting of 2,096 patients with schizophrenia who participated in premarketing clinical trials and were exposed to single or multiple doses of lurasidone, 20 mg, 40 mg, 80 mg, or 120 mg.1 Overall, lurasidone was well tolerated. The rate of discontinuation from clinical trials because of adverse effects was 9.4% for lurasidone vs 5.9% for placebo. Somnolence, akathisia, nausea, parkinsonism, and agitation were the most commonly reported adverse reactions; somnolence and akathisia appear dose-related. Other adverse effects associated with lurasidone were nausea, vomiting, dyspepsia, dystonia, dizziness, insomnia, agitation, and anxiety (Table 2).
Metabolic changes (hyperlipidemia, hyperglycemia, and increased body weight) associated with cardiovascular risk in patients treated with atypical antipsychotics were studied in short-term placebo-controlled trials. Lurasidone is considered to be weight-neutral and does not have significant effects on serum lipids or glucose.2 As is the case with other D2 antagonists, lurasidone is associated with increased prolactin, which appears to be greater in females and is dose-dependent. Lurasidone is not associated with significant QTc prolongation, seizures, transaminases increase, or changes in serum chemistry, hematology, or urinalysis.
Table 2
Lurasidone receptor binding profile and receptor-related effects
| Ki (nM)* | Effects associated with activity on the receptor | |
|---|---|---|
| D2 | 0.994 | Antipsychotic effects. Akathisia (15%), parkinsonism (11%), dystonia (5%), hyperprolactinemia (8.3% for women, 1.9% for men) |
| 5-HT2A | 0.47 | Antipsychotic effects. Improves extrapyramidal symptoms |
| 5-HT7 | 0.495 | Antipsychotic effects. Improves cognition, mood |
| 5-HT1A | 6.38 | Improves cognition, mood. Nausea (12%), vomiting (8%) |
| α-1 | 48 | Orthostatic hypotension (5%), sedation (22%) |
| α-2C | 10.8 | Improves cognition |
| H1 | >1000 | No significant adverse effects mediated through H1 receptor because of low binding affinity |
| M1 | >1000 | No significant adverse effects mediated through M1 receptor because of low binding affinity |
| *Ki dissociation constant; the lower the number, the higher affinity of the compound for the receptor Source: Adapted from reference 1, expert opinion, and lurasidone data on file, 2008 | ||
Contraindications
Lurasidone is contraindicated in patients with known sensitivity to lurasidone hydrochloride. Because of the risk for pharmacokinetic drug-drug interactions, lurasidone is contraindicated for patients who are taking strong CYP3A4 inhibitors (eg, ketoconazole) or inducers (eg, rifampin).
Similar to other medications in its class, lurasidone carries a “black-box” warning of increased mortality in elderly patients with dementia-related psychosis and it is not FDA-approved for treating this condition. Animal teratogenicity studies using lurasidone, 25 mg/kg/d and 50 mg/kg/d, did not show adverse effects during organogenesis, and lurasidone is classified as pregnancy category B (animal studies failed to demonstrate risk to the fetus and there are no adequate and well-controlled studies in pregnant women, or animal studies have shown an adverse effect, but adequate and well-controlled studies in pregnant women have failed to demonstrate a risk to the fetus in any trimester). The use of lurasidone in geriatric and pediatric populations was not studied.1
Dosing
Lurasidone is manufactured as 40 mg and 80 mg tablets. The recommended starting dose is 40 mg/d and the maximum recommended dose is 80 mg/d.1 In clinical trials, lurasidone, 120 mg/d, was associated with increased incidence of adverse effects without added benefit.
Lurasidone doesn’t require initial dose titration and should be given with food that provides ≥350 calories to improve medication absorption. Dose adjustment is recommended for use in patients with moderate or severe renal or hepatic impairment and when coadministered with CYP3A4 moderate inhibitors; the dose in these patients should not exceed 40 mg/d.
Related Resource
- Citrome L. Lurasidone for schizophrenia: a review of the efficacy and safety profile for this newly approved second-generation antipsychotic. Int J Clin Pract. 2010 Epub ahead of print.
Drug Brand Names
- Ketoconazole • Nizoral
- Lurasidone • Latuda
- Olanzapine • Zyprexa
- Rifampin • Rifadin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Latuda [package insert]. Marlborough, MA: Sunovion Pharmaceuticals, Inc.; 2010.
2. Meyer JM, Loebel AD, Schweizer E. Lurasidone: a new drug in development for schizophrenia. Expert Opin Investig Drugs. 2009;18(11):1715-1726.
3. Terry AV, Jr, Buccafusco JJ, Wilson C. Cognitive dysfunction in neuropsychiatric, disorders: selected serotonin receptor subtypes as therapeutic targets. Behav Brain Res. 2008;195(1):30-38.
4. Preskorn SH, Yu-Yuan CH, Sarubbi D, et al. Lurasidone pharmacokinetics: Assessment of potential for drug-drug interaction. Abstract presented at: The American College of Neuropsychopharmacology 49th Annual Meeting; December 5-9, 2010; Miami Beach, FL.
5. Loebel A, Cucchiaro J, Ogasa M, et al. Lurasidone for schizophrenia: symptomatic remission during short-term treatment. Abstract presented at: 162nd Annual Meeting of American Psychiatric Association; May 16-21, 2009; San Francisco, CA. Abstract NR1-054.
6. Nakamura M, Ogasa M, Guarino J, et al. Lurasidone in the treatment of acute schizophrenia: a double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(6):829-836.
7. Meltzer H, Cucchiaro J, Silva R, et al. Lurasidone in the treatment of acute schizophrenia: results of the double-blind, placebo-controlled, PEARL 2 trial. Abstract presented at: 48th Annual Meeting of American College of Neuropsychopharmacology; December 6-10, 2009; Hollywood, FL. Abstract 76.
1. Latuda [package insert]. Marlborough, MA: Sunovion Pharmaceuticals, Inc.; 2010.
2. Meyer JM, Loebel AD, Schweizer E. Lurasidone: a new drug in development for schizophrenia. Expert Opin Investig Drugs. 2009;18(11):1715-1726.
3. Terry AV, Jr, Buccafusco JJ, Wilson C. Cognitive dysfunction in neuropsychiatric, disorders: selected serotonin receptor subtypes as therapeutic targets. Behav Brain Res. 2008;195(1):30-38.
4. Preskorn SH, Yu-Yuan CH, Sarubbi D, et al. Lurasidone pharmacokinetics: Assessment of potential for drug-drug interaction. Abstract presented at: The American College of Neuropsychopharmacology 49th Annual Meeting; December 5-9, 2010; Miami Beach, FL.
5. Loebel A, Cucchiaro J, Ogasa M, et al. Lurasidone for schizophrenia: symptomatic remission during short-term treatment. Abstract presented at: 162nd Annual Meeting of American Psychiatric Association; May 16-21, 2009; San Francisco, CA. Abstract NR1-054.
6. Nakamura M, Ogasa M, Guarino J, et al. Lurasidone in the treatment of acute schizophrenia: a double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(6):829-836.
7. Meltzer H, Cucchiaro J, Silva R, et al. Lurasidone in the treatment of acute schizophrenia: results of the double-blind, placebo-controlled, PEARL 2 trial. Abstract presented at: 48th Annual Meeting of American College of Neuropsychopharmacology; December 6-10, 2009; Hollywood, FL. Abstract 76.
Asenapine for schizophrenia and bipolar I disorder
In August 2009, the FDA approved asenapine for treating acute exacerbation of schizophrenia and acute manic or mixed episodes of bipolar disorder with or without psychosis in adults (Table 1). Asenapine is the first psychotropic to obtain simultaneous FDA approval for schizophrenia and bipolar disorder. The drug’s unique receptor binding profile shows promise in treatment of positive and negative symptoms of schizophrenia with a low risk of extrapyramidal and anticholinergic side effects.
Table 1
Asenapine: Fast facts
| Brand name: Saphris |
| Indications: Acute schizophrenia in adults; acute mixed or manic episodes with or without psychosis associated with bipolar I disorder in adults |
| Approval date: August 2009 |
| Availability date: Late 2009 |
| Manufacturer: Schering-Plough |
| Dosing forms: 5-mg and 10-mg sublingual dissolvable tablets |
| Recommended dose: Schizophrenia: 5 mg twice daily; bipolar disorder: 10 mg twice daily |
How it works
Asenapine is an atypical antipsychotic. Although the exact mechanism of these medications’ efficacy is unknown, their antipsychotic and antimanic activity is thought to be the result of antagonism of central dopamine receptors. According to dopamine theory proposed in the 1960s:
- dopaminergic hyperactivity in mesolimbic dopaminergic pathways contributes to positive symptoms of schizophrenia—hallucinations, delusions, disorganized thoughts and behaviors, and catatonia
- dopaminergic hypoactivity in mesocortical dopaminergic pathways (prefrontal cortex) contributes to negative symptoms of schizophrenia—alogia, avolition, anhedonia, autism, social withdrawal, attention problems, blunted affect, and abstract thinking difficulty.
Asenapine has high affinity for multiple dopamine, serotonin, noradrenergic α1 and α2, and histamine H1 receptors, where it works as an antagonist. Asenapine’s affinity for several serotonin, noradrenergic, and dopaminergic D3 and D4 receptors is higher than its affinity for D2 receptors (Table 2),1 which distinguishes asenapine from other atypical antipsychotics except clozapine.
Blockade of 5-HT2A and 5-HT2C receptors in prefrontal cortex increases dopamine release in this area; theoretically, this effect should improve negative symptoms. Another mechanism that possibly improves cognition and negative symptoms is asenapine’s antagonism at central α2 noradrenergic receptors. Central α1 noradrenergic receptor antagonism also might be helpful in improving positive symptoms of schizophrenia.1
Asenapine’s affinity for the muscarinic-1 cholinergic receptors is quite low, and adverse effects associated with antagonism at these receptors—dry mouth, blurred vision, constipation, and urinary retention—are minimal.2
Table 2
Asenapine’s binding affinity for receptor subtypes*
| Receptor substype | Affinity [Ki (nM)] |
|---|---|
| 5-HT2A | 0.06 |
| 5-HT2C | 0.03 |
| D1 | 1.4 |
| D2 | 1.3 |
| D3 | 0.42 |
| D4 | 1.1 |
| α1 | 1.2 |
| α2 | 1.2 |
| H1 | 1.0 |
| M1 | 8128 |
| *Lower numbers indicate higher affinity | |
| 5-HT: serotonin receptors; D1-4: dopamine receptors; α1, α2: noradrenergic receptors; H1: histamine receptor; M1: muscarinic (cholinergic) receptor | |
| Source: Reference 1 | |
Pharmacokinetics
Absorption of asenapine after oral (swallowed) administration is 2%. To increase total bioavailability to 35%, the drug is manufactured as sublingual dissolvable tablets. After sublingual administration, asenapine is readily absorbed and achieves peak plasma concentration in approximately 1 hour. After absorption, 95% of asenapine binds to transport proteins albumin and α1 acid glycoprotein. The half-life of the medication is approximately 24 hours, and steady state usually is achieved in 3 days.
Metabolism creates about 40 metabolites via multiple metabolic pathways; the main ones are glucuronidation by UGT1A4 and oxidative metabolism by cytochrome P450 (CYP)1A2. Asenapine is a weak inhibitor of CYP2D6, so coadministration of asenapine with other drugs that are substrates or inhibitors of CYP1A2 (eg, fluvoxamine) or CYP2D6 (eg, paroxetine, fluoxetine) should be done cautiously. Because asenapine elimination is biphasic, twice-daily dosing is recommended.3
Efficacy in clinical trials
Schizophrenia. Asenapine’s efficacy for treating schizophrenia was evaluated in 3 fixed-dose, 6-week, randomized, double-blind, placebo- and active- (haloperidol, olanzapine, and risperidone) controlled clinical trials in adults.3-5 Subjects in these studies met DSM-IV criteria for schizophrenia and had acute exacerbation of their illness, with Positive and Negative Syndrome Scale (PANSS) total scores ≥60. Symptom improvement was measured after 6 weeks by PANSS total score, PANSS positive subscale, and Clinical Global Impression scale (CGI).
The first trial (n=174) compared asenapine, 5 mg twice daily, to placebo and risperidone, 3 mg twice daily.3-5 Asenapine was superior to placebo as demonstrated by symptom improvement on all 3 scales. Risperidone showed statistically significant symptom improvement on PANSS positive subscale and CGI but not on PANSS total score.
In the second trial (n=448), 2 fixed doses of asenapine (5 mg twice daily and 10 mg twice daily) and olanzapine, 15 mg/d, were compared with placebo.3,5 The only statistically significant symptom improvement in the asenapine group compared with placebo was on the PANSS positive subscale among subjects receiving 5 mg twice daily. Improvements measured by CGI and PANSS total score were not statistically significant.
Olanzapine showed statistically significant symptom improvement on all 3 scales compared with placebo. This study is a negative trial for asenapine; asenapine failed to separate from placebo, whereas olanzapine—the active comparator—did.
The third trial (n=448) compared asenapine, 5 mg twice daily and 10 mg twice daily, with placebo and haloperidol, 4 mg twice daily.3,5 Compared with placebo, asenapine at both doses and haloperidol improved symptoms on all 3 scales. The 10-mg twice-daily dosage did not provide any additional benefits compared with the 5 mg twice-daily dosage.
Bipolar disorder. Asenapine’s efficacy for bipolar disorder was established in two 3-week, randomized, double-blind, placebo- and olanzapine-controlled studies in adults with acute manic or mixed episodes with or without psychosis.3,6-9 Symptoms were assessed using the Young Mania Rating Scale (YMRS) and Clinical Global Impression-Bipolar (CGI-BP) scale.
In both studies, subjects were randomly assigned to receive asenapine, 10 mg twice daily; olanzapine, 5 to 20 mg/d; or placebo. Depending on efficacy and tolerability, the asenapine dose could be adjusted within the dosing range of 5 mg to 10 mg twice daily starting on day 2. Ninety percent of subjects stayed on the 10 mg twice-daily dose. In both studies, asenapine and olanzapine were statistically superior to placebo on YMRS and CGI-BP severity of illness scores.
Currently no evidence supports asenapine’s efficacy for maintenance treatment of schizophrenia or bipolar disorder. American Psychiatric Association practice guidelines recommend continuing treatment for a minimum of 6 months after stabilization of acute episodes of schizophrenia or bipolar disorder to prevent recurrence.10
Tolerability in clinical trials
Tolerability information provided in this article was obtained from a Clinical Trial Database consisting of 3,350 subjects:11
- 1,953 patients participated in multiple dose effectiveness trials (1,480 with schizophrenia and 473 with bipolar disorder manic/mixed episodes)
- 486 subjects were treated for at least 24 weeks
- 293 subjects were treated for at least 52 weeks.
Overall, asenapine was well tolerated (Table 3).11 The most common adverse effects in schizophrenia trials were akathisia, oral hypoesthesia, and somnolence. The discontinuation rate due to adverse effects in schizophrenia trials was 9% in the asenapine group vs 10% in the placebo group.
Among patients with bipolar disorder, the most common side effects were somnolence, dizziness, extrapyramidal symptoms other than akathisia, and increased weight. The discontinuation rate for subjects treated with asenapine was 10% vs 6% with placebo. The most common adverse reactions associated with discontinuation were anxiety and oral hypoesthesia. Oral hypoesthesia did not occur in the placebo group, and akathisia was the only dose-dependent adverse reaction.
Dizziness and weight gain. Clinically important adverse effects of asenapine include dizziness and weight gain. Dizziness is possibly related to orthostatic hypotension caused by the drug’s activity at the α1 receptor (antagonist). To prevent ischemic events or falls with subsequent injuries, use asenapine with caution in hypotensive patients and those with cardiovascular or cerebrovascular problems.
In clinical trials investigating asenapine’s efficacy, mean weight gain was greater in patients receiving asenapine than those receiving placebo. In short-term studies, mean weight gain in patients treated with asenapine was 1.1 kg for subjects with schizophrenia and 1.3 kg for subjects with bipolar mania.3 Mean weight gain in patients treated with placebo was 0.1 kg for subjects with schizophrenia and 0.2 kg for those with bipolar mania.
In a 52-week comparator study of patients with schizophrenia and schizoaffective disorder, mean weight gain was 0.9 kg in the asenapine group vs 4.2 kg in the olanzapine group.3 In both groups, the greatest weight increase occurred in subjects with body mass index <23.
There were no clinically relevant mean changes in serum fasting glucose, serum fasting triglycerides, fasting cholesterol, transaminases, and prolactin. Thrombocytopenia, anemia, tachycardia, temporary bundle branch block, visual accommodation disorder, oral paresthesia, glossodynia, swollen tongue, hyponatremia, and dysarthria occurred in 1 in 100 to 1 in 1,000 patients.
Table 3
Percentages of clinical trial patients who experienced adverse effects with asenapine vs placebo
| Schizophrenia | Bipolar disorder (mania/mixed) | |||||
|---|---|---|---|---|---|---|
| Adverse effect | Placebo (n=378) | Asenapine, 5 mg bid (n=274) | Asenapine, 10 mg bid (n=208) | Asenapine, 5 or 10 mg bid (n=572) | Placebo (n=203) | Asenapine, 5 or 10 mg bid (n=379) |
| Oral hypoesthesia | 1 | 6 | 7 | 5 | <1 | 4 |
| Weight gain | <1 | 2 | 2 | 3 | <1 | 5 |
| Increased appetite | <1 | 3 | 0 | 2 | 1 | 4 |
| Anxiety | 2 | 4 | ||||
| Akathisia | 3 | 4 | 11 | 6 | 2 | 4 |
| Other EPS (excluding akathisia) | 7 | 9 | 12 | 10 | 2 | 7 |
| Insomnia | 13 | 16 | 15 | 15 | 5 | 6 |
| Somnolence | 7 | 15 | 13 | 13 | 6 | 24 |
| Dizziness | 4 | 7 | 3 | 5 | 3 | 11 |
| EPS: extrapyramidal symptoms | ||||||
| Source: Reference 11 | ||||||
Contraindications
There are no absolute contraindications to asenapine use; however, the medication is not recommended for treating:
- women who are pregnant if the risks of treatment outweigh the benefits (pregnancy risk C)
- breast-feeding mothers
- patients with severe hepatic impairment (Child-Pugh C).
Asenapine carries the same class warnings and precautions as other antipsychotic medications, including a “black box” warning of increased mortality risk in elderly patients with dementia-related psychosis. Other class warnings include an increased risk of transient ischemic attack and cerebrovascular accidents in elderly patients with dementia-related psychosis; neuroleptic malignant syndrome; tardive dyskinesia; glycemia/diabetes mellitus; hyperprolactinemia; leukopenia; neutropenia; and agranulocytosis.
Because asenapine is associated with QT prolongation, do not administer it with other QT-prolonging agents, such as procainamide, sotalol, quinidine, erythromycin, clarithromycin, methadone, or other antipsychotics.
Dosing
Asenapine is manufactured as 5-mg and 10-mg sublingual tablets. Advise patients to avoid eating or drinking for 10 minutes after taking asenapine.
The recommended starting and target dosage for patients with schizophrenia is 5 mg twice daily. The recommended starting dosage for patients with an acute mixed or manic episode of bipolar I disorder is 10 mg twice daily; however, this can be reduced to 5 mg twice daily if the patient experiences intolerable side effects.
Related resource
- Asenapine (Saphris) prescribing information. www.spfiles.com/pisaphrisv1.pdf.
Drug brand names
- Asenapine • Saphris
- Clarithromycin • Biaxin
- Clozapine • Clozaril
- Erythromycin • ERY-C, Ery-Tab
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Methadone • Dolophine, Methadose
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Procainamide • Procanbid
- Quinidine • Quinidine
- Risperidone • Risperdal
- Sotalol • Betapace, Sorine
Disclosures
Dr. Lincoln reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Preskorn receives grant/research support from AstraZeneca, Biovail, Boehringer-Ingleheim, Cyberonics, Eli Lilly and Company, EnVivo, GlaxoSmithKline, UNC Chapel Hill, and Wyeth. He is a consultant to Allergan, Covidien, Eli Lilly and Company, Evotec, Lundbeck/Takeda, Transcept, and Wyeth.
1. Bishara D, Taylor D. Upcoming agents for the treatment of schizophrenia: mechanism of action, efficacy and tolerability. Drugs. 2008;68(16):2269-2292.
2. Shahid M, Walker GB, Zorn SH, et al. Asenapine: a novel psychopharmacologic agent with a unique human receptor signature. J Psychopharmacol. 2009;23(1):65-73.
3. Kowalski R, Potkin S, Szeged A, et al. Psychopharmacologic Drugs Advisory Committee: Saphris (asenapine) sublingual tablets. NDA 22-117. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PsychopharmacologicDrugsAdvisoryCommittee/UCM179975.pdf. Accessed November 3, 2009.
4. Potkin SG, Cohen M, Panagides J. Efficacy and tolerability of asenapine in acute schizophrenia: a placebo- and risperidone-controlled trial. J Clin Psychiatry. 2007;68(10):1492-1500.
5. Potkin SG, Kane JM, Emsley RA, et al. Asenapine in schizophrenia: an overview of clinical trials in the Olympia program. Abstract 80. Presented at: Annual Meeting of the American Psychiatric Association; May 8, 2008; Washington, DC.
6. McIntyre RS, Hirschfeld R, Calabrese J, et al. Asenapine in bipolar disorder: an overview of clinical trials in the Olympia program. Abstract 44. Presented at: Annual Meeting of the American Psychiatric Association; May 6, 2008; Washington, DC.
7. McIntyre RS, Cohen M, Zhao J, et al. A 3-week, randomized, placebo-controlled trial of asenapine in the treatment of acute mania in bipolar mania and mixed states. Bipolar Disord. 2009;11(7):673-686.
8. McIntyre R, Hirschfeld R, Alphs L, et al. Asenapine in the treatment of acute mania in bipolar I disorder: outcomes from two randomized and placebo-controlled trials. J Affect Disord. 2008;107(suppl 1):S56.-
9. McIntyre R, Panagides J, Alphs L, et al. Treatment of mania in bipolar I disorder: a placebo and olanzapine-controlled trial of asenapine (ARES 7501005). Eur Neuropsychopharmacol. 2007;17(suppl 4):S383.-
10. American Psychiatric Association Work Group on Bipolar Disorder. Practice guideline for the treatment of patients with bipolar disorder. 2nd ed. Arlington, VA: American Psychiatric Association; 2002. Available at: http://www.psychiatryonline.com/pracGuide/loadGuidelinePdf.aspx?file=Bipolar2e_Inactivated_04-16-09. Accessed November 3, 2009.
11. Saphris [package insert]. Kenilworth, NJ: Schering-Plough; 2009.
In August 2009, the FDA approved asenapine for treating acute exacerbation of schizophrenia and acute manic or mixed episodes of bipolar disorder with or without psychosis in adults (Table 1). Asenapine is the first psychotropic to obtain simultaneous FDA approval for schizophrenia and bipolar disorder. The drug’s unique receptor binding profile shows promise in treatment of positive and negative symptoms of schizophrenia with a low risk of extrapyramidal and anticholinergic side effects.
Table 1
Asenapine: Fast facts
| Brand name: Saphris |
| Indications: Acute schizophrenia in adults; acute mixed or manic episodes with or without psychosis associated with bipolar I disorder in adults |
| Approval date: August 2009 |
| Availability date: Late 2009 |
| Manufacturer: Schering-Plough |
| Dosing forms: 5-mg and 10-mg sublingual dissolvable tablets |
| Recommended dose: Schizophrenia: 5 mg twice daily; bipolar disorder: 10 mg twice daily |
How it works
Asenapine is an atypical antipsychotic. Although the exact mechanism of these medications’ efficacy is unknown, their antipsychotic and antimanic activity is thought to be the result of antagonism of central dopamine receptors. According to dopamine theory proposed in the 1960s:
- dopaminergic hyperactivity in mesolimbic dopaminergic pathways contributes to positive symptoms of schizophrenia—hallucinations, delusions, disorganized thoughts and behaviors, and catatonia
- dopaminergic hypoactivity in mesocortical dopaminergic pathways (prefrontal cortex) contributes to negative symptoms of schizophrenia—alogia, avolition, anhedonia, autism, social withdrawal, attention problems, blunted affect, and abstract thinking difficulty.
Asenapine has high affinity for multiple dopamine, serotonin, noradrenergic α1 and α2, and histamine H1 receptors, where it works as an antagonist. Asenapine’s affinity for several serotonin, noradrenergic, and dopaminergic D3 and D4 receptors is higher than its affinity for D2 receptors (Table 2),1 which distinguishes asenapine from other atypical antipsychotics except clozapine.
Blockade of 5-HT2A and 5-HT2C receptors in prefrontal cortex increases dopamine release in this area; theoretically, this effect should improve negative symptoms. Another mechanism that possibly improves cognition and negative symptoms is asenapine’s antagonism at central α2 noradrenergic receptors. Central α1 noradrenergic receptor antagonism also might be helpful in improving positive symptoms of schizophrenia.1
Asenapine’s affinity for the muscarinic-1 cholinergic receptors is quite low, and adverse effects associated with antagonism at these receptors—dry mouth, blurred vision, constipation, and urinary retention—are minimal.2
Table 2
Asenapine’s binding affinity for receptor subtypes*
| Receptor substype | Affinity [Ki (nM)] |
|---|---|
| 5-HT2A | 0.06 |
| 5-HT2C | 0.03 |
| D1 | 1.4 |
| D2 | 1.3 |
| D3 | 0.42 |
| D4 | 1.1 |
| α1 | 1.2 |
| α2 | 1.2 |
| H1 | 1.0 |
| M1 | 8128 |
| *Lower numbers indicate higher affinity | |
| 5-HT: serotonin receptors; D1-4: dopamine receptors; α1, α2: noradrenergic receptors; H1: histamine receptor; M1: muscarinic (cholinergic) receptor | |
| Source: Reference 1 | |
Pharmacokinetics
Absorption of asenapine after oral (swallowed) administration is 2%. To increase total bioavailability to 35%, the drug is manufactured as sublingual dissolvable tablets. After sublingual administration, asenapine is readily absorbed and achieves peak plasma concentration in approximately 1 hour. After absorption, 95% of asenapine binds to transport proteins albumin and α1 acid glycoprotein. The half-life of the medication is approximately 24 hours, and steady state usually is achieved in 3 days.
Metabolism creates about 40 metabolites via multiple metabolic pathways; the main ones are glucuronidation by UGT1A4 and oxidative metabolism by cytochrome P450 (CYP)1A2. Asenapine is a weak inhibitor of CYP2D6, so coadministration of asenapine with other drugs that are substrates or inhibitors of CYP1A2 (eg, fluvoxamine) or CYP2D6 (eg, paroxetine, fluoxetine) should be done cautiously. Because asenapine elimination is biphasic, twice-daily dosing is recommended.3
Efficacy in clinical trials
Schizophrenia. Asenapine’s efficacy for treating schizophrenia was evaluated in 3 fixed-dose, 6-week, randomized, double-blind, placebo- and active- (haloperidol, olanzapine, and risperidone) controlled clinical trials in adults.3-5 Subjects in these studies met DSM-IV criteria for schizophrenia and had acute exacerbation of their illness, with Positive and Negative Syndrome Scale (PANSS) total scores ≥60. Symptom improvement was measured after 6 weeks by PANSS total score, PANSS positive subscale, and Clinical Global Impression scale (CGI).
The first trial (n=174) compared asenapine, 5 mg twice daily, to placebo and risperidone, 3 mg twice daily.3-5 Asenapine was superior to placebo as demonstrated by symptom improvement on all 3 scales. Risperidone showed statistically significant symptom improvement on PANSS positive subscale and CGI but not on PANSS total score.
In the second trial (n=448), 2 fixed doses of asenapine (5 mg twice daily and 10 mg twice daily) and olanzapine, 15 mg/d, were compared with placebo.3,5 The only statistically significant symptom improvement in the asenapine group compared with placebo was on the PANSS positive subscale among subjects receiving 5 mg twice daily. Improvements measured by CGI and PANSS total score were not statistically significant.
Olanzapine showed statistically significant symptom improvement on all 3 scales compared with placebo. This study is a negative trial for asenapine; asenapine failed to separate from placebo, whereas olanzapine—the active comparator—did.
The third trial (n=448) compared asenapine, 5 mg twice daily and 10 mg twice daily, with placebo and haloperidol, 4 mg twice daily.3,5 Compared with placebo, asenapine at both doses and haloperidol improved symptoms on all 3 scales. The 10-mg twice-daily dosage did not provide any additional benefits compared with the 5 mg twice-daily dosage.
Bipolar disorder. Asenapine’s efficacy for bipolar disorder was established in two 3-week, randomized, double-blind, placebo- and olanzapine-controlled studies in adults with acute manic or mixed episodes with or without psychosis.3,6-9 Symptoms were assessed using the Young Mania Rating Scale (YMRS) and Clinical Global Impression-Bipolar (CGI-BP) scale.
In both studies, subjects were randomly assigned to receive asenapine, 10 mg twice daily; olanzapine, 5 to 20 mg/d; or placebo. Depending on efficacy and tolerability, the asenapine dose could be adjusted within the dosing range of 5 mg to 10 mg twice daily starting on day 2. Ninety percent of subjects stayed on the 10 mg twice-daily dose. In both studies, asenapine and olanzapine were statistically superior to placebo on YMRS and CGI-BP severity of illness scores.
Currently no evidence supports asenapine’s efficacy for maintenance treatment of schizophrenia or bipolar disorder. American Psychiatric Association practice guidelines recommend continuing treatment for a minimum of 6 months after stabilization of acute episodes of schizophrenia or bipolar disorder to prevent recurrence.10
Tolerability in clinical trials
Tolerability information provided in this article was obtained from a Clinical Trial Database consisting of 3,350 subjects:11
- 1,953 patients participated in multiple dose effectiveness trials (1,480 with schizophrenia and 473 with bipolar disorder manic/mixed episodes)
- 486 subjects were treated for at least 24 weeks
- 293 subjects were treated for at least 52 weeks.
Overall, asenapine was well tolerated (Table 3).11 The most common adverse effects in schizophrenia trials were akathisia, oral hypoesthesia, and somnolence. The discontinuation rate due to adverse effects in schizophrenia trials was 9% in the asenapine group vs 10% in the placebo group.
Among patients with bipolar disorder, the most common side effects were somnolence, dizziness, extrapyramidal symptoms other than akathisia, and increased weight. The discontinuation rate for subjects treated with asenapine was 10% vs 6% with placebo. The most common adverse reactions associated with discontinuation were anxiety and oral hypoesthesia. Oral hypoesthesia did not occur in the placebo group, and akathisia was the only dose-dependent adverse reaction.
Dizziness and weight gain. Clinically important adverse effects of asenapine include dizziness and weight gain. Dizziness is possibly related to orthostatic hypotension caused by the drug’s activity at the α1 receptor (antagonist). To prevent ischemic events or falls with subsequent injuries, use asenapine with caution in hypotensive patients and those with cardiovascular or cerebrovascular problems.
In clinical trials investigating asenapine’s efficacy, mean weight gain was greater in patients receiving asenapine than those receiving placebo. In short-term studies, mean weight gain in patients treated with asenapine was 1.1 kg for subjects with schizophrenia and 1.3 kg for subjects with bipolar mania.3 Mean weight gain in patients treated with placebo was 0.1 kg for subjects with schizophrenia and 0.2 kg for those with bipolar mania.
In a 52-week comparator study of patients with schizophrenia and schizoaffective disorder, mean weight gain was 0.9 kg in the asenapine group vs 4.2 kg in the olanzapine group.3 In both groups, the greatest weight increase occurred in subjects with body mass index <23.
There were no clinically relevant mean changes in serum fasting glucose, serum fasting triglycerides, fasting cholesterol, transaminases, and prolactin. Thrombocytopenia, anemia, tachycardia, temporary bundle branch block, visual accommodation disorder, oral paresthesia, glossodynia, swollen tongue, hyponatremia, and dysarthria occurred in 1 in 100 to 1 in 1,000 patients.
Table 3
Percentages of clinical trial patients who experienced adverse effects with asenapine vs placebo
| Schizophrenia | Bipolar disorder (mania/mixed) | |||||
|---|---|---|---|---|---|---|
| Adverse effect | Placebo (n=378) | Asenapine, 5 mg bid (n=274) | Asenapine, 10 mg bid (n=208) | Asenapine, 5 or 10 mg bid (n=572) | Placebo (n=203) | Asenapine, 5 or 10 mg bid (n=379) |
| Oral hypoesthesia | 1 | 6 | 7 | 5 | <1 | 4 |
| Weight gain | <1 | 2 | 2 | 3 | <1 | 5 |
| Increased appetite | <1 | 3 | 0 | 2 | 1 | 4 |
| Anxiety | 2 | 4 | ||||
| Akathisia | 3 | 4 | 11 | 6 | 2 | 4 |
| Other EPS (excluding akathisia) | 7 | 9 | 12 | 10 | 2 | 7 |
| Insomnia | 13 | 16 | 15 | 15 | 5 | 6 |
| Somnolence | 7 | 15 | 13 | 13 | 6 | 24 |
| Dizziness | 4 | 7 | 3 | 5 | 3 | 11 |
| EPS: extrapyramidal symptoms | ||||||
| Source: Reference 11 | ||||||
Contraindications
There are no absolute contraindications to asenapine use; however, the medication is not recommended for treating:
- women who are pregnant if the risks of treatment outweigh the benefits (pregnancy risk C)
- breast-feeding mothers
- patients with severe hepatic impairment (Child-Pugh C).
Asenapine carries the same class warnings and precautions as other antipsychotic medications, including a “black box” warning of increased mortality risk in elderly patients with dementia-related psychosis. Other class warnings include an increased risk of transient ischemic attack and cerebrovascular accidents in elderly patients with dementia-related psychosis; neuroleptic malignant syndrome; tardive dyskinesia; glycemia/diabetes mellitus; hyperprolactinemia; leukopenia; neutropenia; and agranulocytosis.
Because asenapine is associated with QT prolongation, do not administer it with other QT-prolonging agents, such as procainamide, sotalol, quinidine, erythromycin, clarithromycin, methadone, or other antipsychotics.
Dosing
Asenapine is manufactured as 5-mg and 10-mg sublingual tablets. Advise patients to avoid eating or drinking for 10 minutes after taking asenapine.
The recommended starting and target dosage for patients with schizophrenia is 5 mg twice daily. The recommended starting dosage for patients with an acute mixed or manic episode of bipolar I disorder is 10 mg twice daily; however, this can be reduced to 5 mg twice daily if the patient experiences intolerable side effects.
Related resource
- Asenapine (Saphris) prescribing information. www.spfiles.com/pisaphrisv1.pdf.
Drug brand names
- Asenapine • Saphris
- Clarithromycin • Biaxin
- Clozapine • Clozaril
- Erythromycin • ERY-C, Ery-Tab
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Methadone • Dolophine, Methadose
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Procainamide • Procanbid
- Quinidine • Quinidine
- Risperidone • Risperdal
- Sotalol • Betapace, Sorine
Disclosures
Dr. Lincoln reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Preskorn receives grant/research support from AstraZeneca, Biovail, Boehringer-Ingleheim, Cyberonics, Eli Lilly and Company, EnVivo, GlaxoSmithKline, UNC Chapel Hill, and Wyeth. He is a consultant to Allergan, Covidien, Eli Lilly and Company, Evotec, Lundbeck/Takeda, Transcept, and Wyeth.
In August 2009, the FDA approved asenapine for treating acute exacerbation of schizophrenia and acute manic or mixed episodes of bipolar disorder with or without psychosis in adults (Table 1). Asenapine is the first psychotropic to obtain simultaneous FDA approval for schizophrenia and bipolar disorder. The drug’s unique receptor binding profile shows promise in treatment of positive and negative symptoms of schizophrenia with a low risk of extrapyramidal and anticholinergic side effects.
Table 1
Asenapine: Fast facts
| Brand name: Saphris |
| Indications: Acute schizophrenia in adults; acute mixed or manic episodes with or without psychosis associated with bipolar I disorder in adults |
| Approval date: August 2009 |
| Availability date: Late 2009 |
| Manufacturer: Schering-Plough |
| Dosing forms: 5-mg and 10-mg sublingual dissolvable tablets |
| Recommended dose: Schizophrenia: 5 mg twice daily; bipolar disorder: 10 mg twice daily |
How it works
Asenapine is an atypical antipsychotic. Although the exact mechanism of these medications’ efficacy is unknown, their antipsychotic and antimanic activity is thought to be the result of antagonism of central dopamine receptors. According to dopamine theory proposed in the 1960s:
- dopaminergic hyperactivity in mesolimbic dopaminergic pathways contributes to positive symptoms of schizophrenia—hallucinations, delusions, disorganized thoughts and behaviors, and catatonia
- dopaminergic hypoactivity in mesocortical dopaminergic pathways (prefrontal cortex) contributes to negative symptoms of schizophrenia—alogia, avolition, anhedonia, autism, social withdrawal, attention problems, blunted affect, and abstract thinking difficulty.
Asenapine has high affinity for multiple dopamine, serotonin, noradrenergic α1 and α2, and histamine H1 receptors, where it works as an antagonist. Asenapine’s affinity for several serotonin, noradrenergic, and dopaminergic D3 and D4 receptors is higher than its affinity for D2 receptors (Table 2),1 which distinguishes asenapine from other atypical antipsychotics except clozapine.
Blockade of 5-HT2A and 5-HT2C receptors in prefrontal cortex increases dopamine release in this area; theoretically, this effect should improve negative symptoms. Another mechanism that possibly improves cognition and negative symptoms is asenapine’s antagonism at central α2 noradrenergic receptors. Central α1 noradrenergic receptor antagonism also might be helpful in improving positive symptoms of schizophrenia.1
Asenapine’s affinity for the muscarinic-1 cholinergic receptors is quite low, and adverse effects associated with antagonism at these receptors—dry mouth, blurred vision, constipation, and urinary retention—are minimal.2
Table 2
Asenapine’s binding affinity for receptor subtypes*
| Receptor substype | Affinity [Ki (nM)] |
|---|---|
| 5-HT2A | 0.06 |
| 5-HT2C | 0.03 |
| D1 | 1.4 |
| D2 | 1.3 |
| D3 | 0.42 |
| D4 | 1.1 |
| α1 | 1.2 |
| α2 | 1.2 |
| H1 | 1.0 |
| M1 | 8128 |
| *Lower numbers indicate higher affinity | |
| 5-HT: serotonin receptors; D1-4: dopamine receptors; α1, α2: noradrenergic receptors; H1: histamine receptor; M1: muscarinic (cholinergic) receptor | |
| Source: Reference 1 | |
Pharmacokinetics
Absorption of asenapine after oral (swallowed) administration is 2%. To increase total bioavailability to 35%, the drug is manufactured as sublingual dissolvable tablets. After sublingual administration, asenapine is readily absorbed and achieves peak plasma concentration in approximately 1 hour. After absorption, 95% of asenapine binds to transport proteins albumin and α1 acid glycoprotein. The half-life of the medication is approximately 24 hours, and steady state usually is achieved in 3 days.
Metabolism creates about 40 metabolites via multiple metabolic pathways; the main ones are glucuronidation by UGT1A4 and oxidative metabolism by cytochrome P450 (CYP)1A2. Asenapine is a weak inhibitor of CYP2D6, so coadministration of asenapine with other drugs that are substrates or inhibitors of CYP1A2 (eg, fluvoxamine) or CYP2D6 (eg, paroxetine, fluoxetine) should be done cautiously. Because asenapine elimination is biphasic, twice-daily dosing is recommended.3
Efficacy in clinical trials
Schizophrenia. Asenapine’s efficacy for treating schizophrenia was evaluated in 3 fixed-dose, 6-week, randomized, double-blind, placebo- and active- (haloperidol, olanzapine, and risperidone) controlled clinical trials in adults.3-5 Subjects in these studies met DSM-IV criteria for schizophrenia and had acute exacerbation of their illness, with Positive and Negative Syndrome Scale (PANSS) total scores ≥60. Symptom improvement was measured after 6 weeks by PANSS total score, PANSS positive subscale, and Clinical Global Impression scale (CGI).
The first trial (n=174) compared asenapine, 5 mg twice daily, to placebo and risperidone, 3 mg twice daily.3-5 Asenapine was superior to placebo as demonstrated by symptom improvement on all 3 scales. Risperidone showed statistically significant symptom improvement on PANSS positive subscale and CGI but not on PANSS total score.
In the second trial (n=448), 2 fixed doses of asenapine (5 mg twice daily and 10 mg twice daily) and olanzapine, 15 mg/d, were compared with placebo.3,5 The only statistically significant symptom improvement in the asenapine group compared with placebo was on the PANSS positive subscale among subjects receiving 5 mg twice daily. Improvements measured by CGI and PANSS total score were not statistically significant.
Olanzapine showed statistically significant symptom improvement on all 3 scales compared with placebo. This study is a negative trial for asenapine; asenapine failed to separate from placebo, whereas olanzapine—the active comparator—did.
The third trial (n=448) compared asenapine, 5 mg twice daily and 10 mg twice daily, with placebo and haloperidol, 4 mg twice daily.3,5 Compared with placebo, asenapine at both doses and haloperidol improved symptoms on all 3 scales. The 10-mg twice-daily dosage did not provide any additional benefits compared with the 5 mg twice-daily dosage.
Bipolar disorder. Asenapine’s efficacy for bipolar disorder was established in two 3-week, randomized, double-blind, placebo- and olanzapine-controlled studies in adults with acute manic or mixed episodes with or without psychosis.3,6-9 Symptoms were assessed using the Young Mania Rating Scale (YMRS) and Clinical Global Impression-Bipolar (CGI-BP) scale.
In both studies, subjects were randomly assigned to receive asenapine, 10 mg twice daily; olanzapine, 5 to 20 mg/d; or placebo. Depending on efficacy and tolerability, the asenapine dose could be adjusted within the dosing range of 5 mg to 10 mg twice daily starting on day 2. Ninety percent of subjects stayed on the 10 mg twice-daily dose. In both studies, asenapine and olanzapine were statistically superior to placebo on YMRS and CGI-BP severity of illness scores.
Currently no evidence supports asenapine’s efficacy for maintenance treatment of schizophrenia or bipolar disorder. American Psychiatric Association practice guidelines recommend continuing treatment for a minimum of 6 months after stabilization of acute episodes of schizophrenia or bipolar disorder to prevent recurrence.10
Tolerability in clinical trials
Tolerability information provided in this article was obtained from a Clinical Trial Database consisting of 3,350 subjects:11
- 1,953 patients participated in multiple dose effectiveness trials (1,480 with schizophrenia and 473 with bipolar disorder manic/mixed episodes)
- 486 subjects were treated for at least 24 weeks
- 293 subjects were treated for at least 52 weeks.
Overall, asenapine was well tolerated (Table 3).11 The most common adverse effects in schizophrenia trials were akathisia, oral hypoesthesia, and somnolence. The discontinuation rate due to adverse effects in schizophrenia trials was 9% in the asenapine group vs 10% in the placebo group.
Among patients with bipolar disorder, the most common side effects were somnolence, dizziness, extrapyramidal symptoms other than akathisia, and increased weight. The discontinuation rate for subjects treated with asenapine was 10% vs 6% with placebo. The most common adverse reactions associated with discontinuation were anxiety and oral hypoesthesia. Oral hypoesthesia did not occur in the placebo group, and akathisia was the only dose-dependent adverse reaction.
Dizziness and weight gain. Clinically important adverse effects of asenapine include dizziness and weight gain. Dizziness is possibly related to orthostatic hypotension caused by the drug’s activity at the α1 receptor (antagonist). To prevent ischemic events or falls with subsequent injuries, use asenapine with caution in hypotensive patients and those with cardiovascular or cerebrovascular problems.
In clinical trials investigating asenapine’s efficacy, mean weight gain was greater in patients receiving asenapine than those receiving placebo. In short-term studies, mean weight gain in patients treated with asenapine was 1.1 kg for subjects with schizophrenia and 1.3 kg for subjects with bipolar mania.3 Mean weight gain in patients treated with placebo was 0.1 kg for subjects with schizophrenia and 0.2 kg for those with bipolar mania.
In a 52-week comparator study of patients with schizophrenia and schizoaffective disorder, mean weight gain was 0.9 kg in the asenapine group vs 4.2 kg in the olanzapine group.3 In both groups, the greatest weight increase occurred in subjects with body mass index <23.
There were no clinically relevant mean changes in serum fasting glucose, serum fasting triglycerides, fasting cholesterol, transaminases, and prolactin. Thrombocytopenia, anemia, tachycardia, temporary bundle branch block, visual accommodation disorder, oral paresthesia, glossodynia, swollen tongue, hyponatremia, and dysarthria occurred in 1 in 100 to 1 in 1,000 patients.
Table 3
Percentages of clinical trial patients who experienced adverse effects with asenapine vs placebo
| Schizophrenia | Bipolar disorder (mania/mixed) | |||||
|---|---|---|---|---|---|---|
| Adverse effect | Placebo (n=378) | Asenapine, 5 mg bid (n=274) | Asenapine, 10 mg bid (n=208) | Asenapine, 5 or 10 mg bid (n=572) | Placebo (n=203) | Asenapine, 5 or 10 mg bid (n=379) |
| Oral hypoesthesia | 1 | 6 | 7 | 5 | <1 | 4 |
| Weight gain | <1 | 2 | 2 | 3 | <1 | 5 |
| Increased appetite | <1 | 3 | 0 | 2 | 1 | 4 |
| Anxiety | 2 | 4 | ||||
| Akathisia | 3 | 4 | 11 | 6 | 2 | 4 |
| Other EPS (excluding akathisia) | 7 | 9 | 12 | 10 | 2 | 7 |
| Insomnia | 13 | 16 | 15 | 15 | 5 | 6 |
| Somnolence | 7 | 15 | 13 | 13 | 6 | 24 |
| Dizziness | 4 | 7 | 3 | 5 | 3 | 11 |
| EPS: extrapyramidal symptoms | ||||||
| Source: Reference 11 | ||||||
Contraindications
There are no absolute contraindications to asenapine use; however, the medication is not recommended for treating:
- women who are pregnant if the risks of treatment outweigh the benefits (pregnancy risk C)
- breast-feeding mothers
- patients with severe hepatic impairment (Child-Pugh C).
Asenapine carries the same class warnings and precautions as other antipsychotic medications, including a “black box” warning of increased mortality risk in elderly patients with dementia-related psychosis. Other class warnings include an increased risk of transient ischemic attack and cerebrovascular accidents in elderly patients with dementia-related psychosis; neuroleptic malignant syndrome; tardive dyskinesia; glycemia/diabetes mellitus; hyperprolactinemia; leukopenia; neutropenia; and agranulocytosis.
Because asenapine is associated with QT prolongation, do not administer it with other QT-prolonging agents, such as procainamide, sotalol, quinidine, erythromycin, clarithromycin, methadone, or other antipsychotics.
Dosing
Asenapine is manufactured as 5-mg and 10-mg sublingual tablets. Advise patients to avoid eating or drinking for 10 minutes after taking asenapine.
The recommended starting and target dosage for patients with schizophrenia is 5 mg twice daily. The recommended starting dosage for patients with an acute mixed or manic episode of bipolar I disorder is 10 mg twice daily; however, this can be reduced to 5 mg twice daily if the patient experiences intolerable side effects.
Related resource
- Asenapine (Saphris) prescribing information. www.spfiles.com/pisaphrisv1.pdf.
Drug brand names
- Asenapine • Saphris
- Clarithromycin • Biaxin
- Clozapine • Clozaril
- Erythromycin • ERY-C, Ery-Tab
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Methadone • Dolophine, Methadose
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Procainamide • Procanbid
- Quinidine • Quinidine
- Risperidone • Risperdal
- Sotalol • Betapace, Sorine
Disclosures
Dr. Lincoln reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Preskorn receives grant/research support from AstraZeneca, Biovail, Boehringer-Ingleheim, Cyberonics, Eli Lilly and Company, EnVivo, GlaxoSmithKline, UNC Chapel Hill, and Wyeth. He is a consultant to Allergan, Covidien, Eli Lilly and Company, Evotec, Lundbeck/Takeda, Transcept, and Wyeth.
1. Bishara D, Taylor D. Upcoming agents for the treatment of schizophrenia: mechanism of action, efficacy and tolerability. Drugs. 2008;68(16):2269-2292.
2. Shahid M, Walker GB, Zorn SH, et al. Asenapine: a novel psychopharmacologic agent with a unique human receptor signature. J Psychopharmacol. 2009;23(1):65-73.
3. Kowalski R, Potkin S, Szeged A, et al. Psychopharmacologic Drugs Advisory Committee: Saphris (asenapine) sublingual tablets. NDA 22-117. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PsychopharmacologicDrugsAdvisoryCommittee/UCM179975.pdf. Accessed November 3, 2009.
4. Potkin SG, Cohen M, Panagides J. Efficacy and tolerability of asenapine in acute schizophrenia: a placebo- and risperidone-controlled trial. J Clin Psychiatry. 2007;68(10):1492-1500.
5. Potkin SG, Kane JM, Emsley RA, et al. Asenapine in schizophrenia: an overview of clinical trials in the Olympia program. Abstract 80. Presented at: Annual Meeting of the American Psychiatric Association; May 8, 2008; Washington, DC.
6. McIntyre RS, Hirschfeld R, Calabrese J, et al. Asenapine in bipolar disorder: an overview of clinical trials in the Olympia program. Abstract 44. Presented at: Annual Meeting of the American Psychiatric Association; May 6, 2008; Washington, DC.
7. McIntyre RS, Cohen M, Zhao J, et al. A 3-week, randomized, placebo-controlled trial of asenapine in the treatment of acute mania in bipolar mania and mixed states. Bipolar Disord. 2009;11(7):673-686.
8. McIntyre R, Hirschfeld R, Alphs L, et al. Asenapine in the treatment of acute mania in bipolar I disorder: outcomes from two randomized and placebo-controlled trials. J Affect Disord. 2008;107(suppl 1):S56.-
9. McIntyre R, Panagides J, Alphs L, et al. Treatment of mania in bipolar I disorder: a placebo and olanzapine-controlled trial of asenapine (ARES 7501005). Eur Neuropsychopharmacol. 2007;17(suppl 4):S383.-
10. American Psychiatric Association Work Group on Bipolar Disorder. Practice guideline for the treatment of patients with bipolar disorder. 2nd ed. Arlington, VA: American Psychiatric Association; 2002. Available at: http://www.psychiatryonline.com/pracGuide/loadGuidelinePdf.aspx?file=Bipolar2e_Inactivated_04-16-09. Accessed November 3, 2009.
11. Saphris [package insert]. Kenilworth, NJ: Schering-Plough; 2009.
1. Bishara D, Taylor D. Upcoming agents for the treatment of schizophrenia: mechanism of action, efficacy and tolerability. Drugs. 2008;68(16):2269-2292.
2. Shahid M, Walker GB, Zorn SH, et al. Asenapine: a novel psychopharmacologic agent with a unique human receptor signature. J Psychopharmacol. 2009;23(1):65-73.
3. Kowalski R, Potkin S, Szeged A, et al. Psychopharmacologic Drugs Advisory Committee: Saphris (asenapine) sublingual tablets. NDA 22-117. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PsychopharmacologicDrugsAdvisoryCommittee/UCM179975.pdf. Accessed November 3, 2009.
4. Potkin SG, Cohen M, Panagides J. Efficacy and tolerability of asenapine in acute schizophrenia: a placebo- and risperidone-controlled trial. J Clin Psychiatry. 2007;68(10):1492-1500.
5. Potkin SG, Kane JM, Emsley RA, et al. Asenapine in schizophrenia: an overview of clinical trials in the Olympia program. Abstract 80. Presented at: Annual Meeting of the American Psychiatric Association; May 8, 2008; Washington, DC.
6. McIntyre RS, Hirschfeld R, Calabrese J, et al. Asenapine in bipolar disorder: an overview of clinical trials in the Olympia program. Abstract 44. Presented at: Annual Meeting of the American Psychiatric Association; May 6, 2008; Washington, DC.
7. McIntyre RS, Cohen M, Zhao J, et al. A 3-week, randomized, placebo-controlled trial of asenapine in the treatment of acute mania in bipolar mania and mixed states. Bipolar Disord. 2009;11(7):673-686.
8. McIntyre R, Hirschfeld R, Alphs L, et al. Asenapine in the treatment of acute mania in bipolar I disorder: outcomes from two randomized and placebo-controlled trials. J Affect Disord. 2008;107(suppl 1):S56.-
9. McIntyre R, Panagides J, Alphs L, et al. Treatment of mania in bipolar I disorder: a placebo and olanzapine-controlled trial of asenapine (ARES 7501005). Eur Neuropsychopharmacol. 2007;17(suppl 4):S383.-
10. American Psychiatric Association Work Group on Bipolar Disorder. Practice guideline for the treatment of patients with bipolar disorder. 2nd ed. Arlington, VA: American Psychiatric Association; 2002. Available at: http://www.psychiatryonline.com/pracGuide/loadGuidelinePdf.aspx?file=Bipolar2e_Inactivated_04-16-09. Accessed November 3, 2009.
11. Saphris [package insert]. Kenilworth, NJ: Schering-Plough; 2009.
Desvenlafaxine for depression
Compared with other antidepressants, desvenlafaxine might have more predictable effects and a lower risk of drug-drug interactions because of the way it is metabolized. The FDA approved this selective serotonin-norepinephrine reuptake inhibitor (SNRI)—a major active metabolite of venlafaxine—for treating major depressive disorder (MDD, Table 1). In clinical trials, desvenlafaxine was more effective than placebo in improving patients’ scores on scales of depressive symptoms and overall improvement.1
Table 1
Desvenlafaxine: Fast facts
| Brand name: Pristiq |
| Class: Serotonin-norepinephrine reuptake inhibitor |
| Indication: Major depressive disorder |
| Approval date: February 2008 |
| Availability date: May 2008 |
| Manufacturer: Wyeth Pharmaceuticals |
| Dosing forms: 50- and 100-mg tablets |
| Recommended dose: 50 to 100 mg/d |
Clinical implications
Unlike other SNRIs (venlafaxine and duloxetine), desvenlafaxine does not depend on cytochrome P450 (CYP) 2D6 for bio-transformation. As a result plasma concentrations vary less among individual patients, which should result in more predictable efficacy and tolerability. In addition, unlike bupropion, duloxetine, fluoxetine, and paroxetine, desvenlafaxine does not affect the functional activity of CYP 2D6. This translates into a lower risk of drug-drug interactions and more predictable effects on coadministered drugs that are cleared by CYP 2D6.
How it works
Serotonin, norepinephrine, and dopamine in the CNS are involved in mood and neurovegetative functions that are disturbed in patients with MDD. Desvenlafaxine selectively inhibits serotonin and norepinephrine reuptake pumps, therefore increasing serotonin and norepinephrine concentration in the synaptic cleft.2 The drug has weak binding affinity for the dopamine transporter and does not cause substantial changes in extracellular dopamine concentration. Decreased presynaptic serotonin and norepinephrine uptake increases the synaptic concentration of these neurotransmitters. These effects are thought to be responsible for desvenlafaxine’s antidepressant efficacy.
Pharmacokinetics
Desvenlafaxine’s single-dose pharmacokinetics are linear and dose-proportional over the recommended 50 to 100 mg/d dosing range. The half-life is approximately 11 hours. Steady-state plasma concentration is achieved in 4 to 5 days with once-daily dosing.
Food does not affect intestinal absorption. Bioavailability after oral administration is 80%, and time to reach maximum concentration (Tmax) is 7.5 hours. Plasma protein binding is 30% and is independent of desvenlafaxine concentration.1
Desvenlafaxine is excreted renally:
- unchanged (45% at 72 hours after administration)
- as desvenlafaxine-glucuronide
- as N-desvenlafaxine-glucuronide.
Desvenlafaxine-glucuronide is the final metabolite of conjugation reaction with glucuronic acid. N-desvenlafaxine-glucuronide is an end product of a 2-step metabolic reaction that starts with oxidation by CYP 3A4 to produce N-desvenlafaxine, which is conjugated with glucuronic acid to create N-desvenlafaxine-glucuronide. As a result of these metabolic and elimination pathways, dosing adjustment is recommended for patients with severe renal impairment or who are taking a CYP 3A4 inhibitor.
Dosing
Desvenlafaxine is available as 50-mg and 100-mg tablets. The recommended dosage is 50 mg/d, and the maximum recommended dosage in patients with hepatic impairment is 100 mg/d.
No dosing adjustment is necessary for patients with moderate renal impairment. The recommend regimen for those with severe renal impairment or end-stage renal disease is 50 mg every other day.
Instruct patients to take desvenlafaxine at approximately the same time each day, with or without food. Tell them not to discontinue the drug abruptly and to immediately report any adverse effects (AEs).
Efficacy
Desvenlafaxine’s antidepressant efficacy was established in four 8-week, randomized, double-blind, placebo-controlled, fixed-dose (50 mg to 400 mg once daily) studies in adult outpatients who met DSM-IV-TR criteria for MDD.1,2,4
In the first study,3 461 patients received desvenlafaxine, 100 mg, 200 mg, or 400 mg, or placebo. In the second study,4 369 patients received 200 mg or 400 mg or placebo. In 2 additional studies, a total of 930 patients received 50 mg or 100 mg or placebo.1
All studies used the 17-item Hamilton Rating Scale for Depression (HAM-D17) to measure depressive symptom improvement and the Clinical Global Impressions-Improvement (CGI-I) scale to measure overall improvement. Desvenlafaxine was more effective than placebo in HAM-D17 score improvement in all 4 studies and in CGI-I score improvement in 3 studies.
In studies comparing 50 mg/d with 100 mg/d, doses >50 mg/d provided no additional benefit. Higher starting fixed doses were associated with more frequent AEs and discontinuation.
Gender or age had no effect on treatment outcome. There was no difference in safety in elderly vs younger patients. Data are insufficient to establish a relationship between race and desvenlafaxine responsiveness. Desvenlafaxine’s safety and effectiveness in children and adolescents was not evaluated, and the drug is not approved for these patients.
Tolerability and safety
Desvenlafaxine’s tolerability is comparable to that of other SNRIs. In premarketing studies, 12% of patients receiving desvenlafaxine (50 mg/d to 400 mg/d) discontinued treatment because of AEs, compared with 3% in the placebo group. The discontinuation rate in patients receiving 100 mg/d was 8.7% compared with 4.1% in patients taking 50 mg/d.
AEs generally occur during the first week of treatment. In the 8-week trials, the most common AEs were nausea and dizziness (Table 2). In a long-term study (up to 9 months), the most common AE was vomiting. Although the recommended starting dose is 50 mg/d, to avoid AEs consider beginning with every-other-day dosing.
Table 2
Desvenlafaxine trials: Rates of adverse effects
| Desvenlafaxine dose | |||
|---|---|---|---|
| Adverse effect | 50 mg/d | 100 mg/d | Placebo |
| Nausea | 22% | 26% | 10% |
| Dizziness | 13% | 10% | 5% |
| Insomnia | 9% | 12% | 6% |
| Hyperhidrosis | 10% | 11% | 4% |
| Constipation | 9% | 9% | 4% |
| Somnolence | 4% | 9% | 4% |
| Decreased appetite | 5% | 8% | 2% |
| Erectile dysfunction | 3% | 6% | 1% |
| Decreased libido | 4% | 5% | 1% |
| Anxiety | 3% | 5% | 2% |
| Source: Reference 1 | |||
Abruptly discontinuing desvenlafaxine can cause withdrawal symptoms, including dizziness, nausea, headache, irritability, insomnia, diarrhea, anxiety, abnormal dreams, fatigue, and hyperhidrosis. The frequency of withdrawal symptoms is higher with longer treatment duration. Gradually reducing the dose by administering 50 mg of desvenlafaxine less often can reduce withdrawal symptoms.
Clinical issues
All SNRIs and selective serotonin reuptake inhibitors (SSRIs) have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation/behaviors during the first months of treatment and with dose changes.
When taken in the third trimester of pregnancy, SNRIs and SSRIs can cause serious neonatal complications—including respiratory distress, cyanosis, apnea, and seizures—that may require longer hospitalization, respiratory support, or tube feeding for the infant. Carefully consider risks and benefits of third-trimester antidepressant use.5 Desvenlafaxine is excreted in breast milk and may cause AEs in infants who are breast-fed.
In clinical trials, patients taking desvenlafaxine experienced increased cholesterol, triglycerides, and blood pressure. Monitor these parameters closely in patients taking desvenlafaxine, and use the drug with caution in patients with cerebrovascular and cardiovascular disease.
Other concerns in patients taking desvenlafaxine include:
- Antidepressant medications can trigger hypomania or mania in patients with bipolar disorder.
- Patients—particularly those who are elderly or taking diuretics—may develop hyponatremia as a result of syndrome of in-appropriate antidiuretic hormone.
- Patients with an increased risk of glaucoma need to be monitored because of the drug’s effect on blood pressure.
Drug interactions. Coadministering desvenlafaxine with serotonergic medications— such as triptans, other antidepressants, and tramadol—can cause serotonin syndrome, a potentially life-threatening condition characterized by mental status changes, autonomic instability, neuromuscular aberrations, and gastrointestinal symptoms. Concomitant use of desvenlafaxine and blood-thinning medications such as warfarin, aspirin, and nonsteroidal anti-inflammatory drugs may result in abnormal bleeding. Patients taking a potent CYP 3A4 inhibitor such as ketoconazole may have increased desvenlafaxine concentration.
Contraindications
Do not prescribe desvenlafaxine to patients who are:
- hypersensitive to venlafaxine chloride, desvenlafaxine succinate, or any parts of the desvenlafaxine formulation
- taking a monoamine oxidase inhibitor (MAOI), or have discontinued an MAOI within 14 days.
Patients who stop taking desvenlafaxine should wait 7 days before starting an MAOI.
Drug brand names
- Bupropion • Wellbutrin
- Desvenlafaxine • Pristiq
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Ketoconazole • Nizoral
- Paroxetine • Prozac
- Tramadol • Ultram
- Venlafaxine • Effexor
- Warfarin • Coumadin
Disclosures
Dr. Lincoln reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Preskorn has in the past year received research/ grant support from and served as a speaker for Wyeth Pharmaceuticals. Previously, he has received research/grant support from or served as a speaker for or consultant to Abbott Laboratories, AstraZeneca, Aventis, Biovail, Boehringer Ingleheim, Bristol-Myers Squibb, Eisai, Eli Lilly and Company, GlaxoSmithKline, Hoffman LaRoche, Janssen, L.P., Johnson & Johnson, Lundbeck, Merck, Novartis, Organon, Otsuka, Pfizer Inc., Solvay, Somerset, Sumitomo, and Yamanouchi.
1. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals; 2008.
2. Deecher DC, Beyer CE, Johnston G, et al. Desvenlafaxine succinate: a new serotonin and norepinephrine reuptake inhibitor. J Pharmacol Exp Ther 2006;318(2):657-65.
3. DeMartinis NA, Yeung PP, Entsuah R, Manley AL. A double-blind, placebo-controlled study of the efficacy and safety of desvenlafaxine succinate in the treatment of major depressive disorder. J Clin Psychiatry 2007;68(5):677-88.
4. Septien-Velez L, Pitrosky B, Padmanabhan SK, et al. A randomized, double-blind, placebo-controlled trial of desvenlafaxine succinate in the treatment of major depressive disorder. Int Clin Psychopharmacol 2007;22(6):338-47.
5. Lennestål R, Källén B. Delivery outcome in relation to maternal use of some recently introduced antidepressants. J Clin Psychopharmacol 2007;27(6):607-13.
Dr. Lincoln is a clinical instructor and Dr. Preskorn is a professor of psychiatry, University of Kansas Medical Center, Wichita. Dr. Preskorn also is the psychopharmacology Section Editor of CURRENT PSYCHIATRY and president and CEO of Clinical Research Institute, Wichita.
Compared with other antidepressants, desvenlafaxine might have more predictable effects and a lower risk of drug-drug interactions because of the way it is metabolized. The FDA approved this selective serotonin-norepinephrine reuptake inhibitor (SNRI)—a major active metabolite of venlafaxine—for treating major depressive disorder (MDD, Table 1). In clinical trials, desvenlafaxine was more effective than placebo in improving patients’ scores on scales of depressive symptoms and overall improvement.1
Table 1
Desvenlafaxine: Fast facts
| Brand name: Pristiq |
| Class: Serotonin-norepinephrine reuptake inhibitor |
| Indication: Major depressive disorder |
| Approval date: February 2008 |
| Availability date: May 2008 |
| Manufacturer: Wyeth Pharmaceuticals |
| Dosing forms: 50- and 100-mg tablets |
| Recommended dose: 50 to 100 mg/d |
Clinical implications
Unlike other SNRIs (venlafaxine and duloxetine), desvenlafaxine does not depend on cytochrome P450 (CYP) 2D6 for bio-transformation. As a result plasma concentrations vary less among individual patients, which should result in more predictable efficacy and tolerability. In addition, unlike bupropion, duloxetine, fluoxetine, and paroxetine, desvenlafaxine does not affect the functional activity of CYP 2D6. This translates into a lower risk of drug-drug interactions and more predictable effects on coadministered drugs that are cleared by CYP 2D6.
How it works
Serotonin, norepinephrine, and dopamine in the CNS are involved in mood and neurovegetative functions that are disturbed in patients with MDD. Desvenlafaxine selectively inhibits serotonin and norepinephrine reuptake pumps, therefore increasing serotonin and norepinephrine concentration in the synaptic cleft.2 The drug has weak binding affinity for the dopamine transporter and does not cause substantial changes in extracellular dopamine concentration. Decreased presynaptic serotonin and norepinephrine uptake increases the synaptic concentration of these neurotransmitters. These effects are thought to be responsible for desvenlafaxine’s antidepressant efficacy.
Pharmacokinetics
Desvenlafaxine’s single-dose pharmacokinetics are linear and dose-proportional over the recommended 50 to 100 mg/d dosing range. The half-life is approximately 11 hours. Steady-state plasma concentration is achieved in 4 to 5 days with once-daily dosing.
Food does not affect intestinal absorption. Bioavailability after oral administration is 80%, and time to reach maximum concentration (Tmax) is 7.5 hours. Plasma protein binding is 30% and is independent of desvenlafaxine concentration.1
Desvenlafaxine is excreted renally:
- unchanged (45% at 72 hours after administration)
- as desvenlafaxine-glucuronide
- as N-desvenlafaxine-glucuronide.
Desvenlafaxine-glucuronide is the final metabolite of conjugation reaction with glucuronic acid. N-desvenlafaxine-glucuronide is an end product of a 2-step metabolic reaction that starts with oxidation by CYP 3A4 to produce N-desvenlafaxine, which is conjugated with glucuronic acid to create N-desvenlafaxine-glucuronide. As a result of these metabolic and elimination pathways, dosing adjustment is recommended for patients with severe renal impairment or who are taking a CYP 3A4 inhibitor.
Dosing
Desvenlafaxine is available as 50-mg and 100-mg tablets. The recommended dosage is 50 mg/d, and the maximum recommended dosage in patients with hepatic impairment is 100 mg/d.
No dosing adjustment is necessary for patients with moderate renal impairment. The recommend regimen for those with severe renal impairment or end-stage renal disease is 50 mg every other day.
Instruct patients to take desvenlafaxine at approximately the same time each day, with or without food. Tell them not to discontinue the drug abruptly and to immediately report any adverse effects (AEs).
Efficacy
Desvenlafaxine’s antidepressant efficacy was established in four 8-week, randomized, double-blind, placebo-controlled, fixed-dose (50 mg to 400 mg once daily) studies in adult outpatients who met DSM-IV-TR criteria for MDD.1,2,4
In the first study,3 461 patients received desvenlafaxine, 100 mg, 200 mg, or 400 mg, or placebo. In the second study,4 369 patients received 200 mg or 400 mg or placebo. In 2 additional studies, a total of 930 patients received 50 mg or 100 mg or placebo.1
All studies used the 17-item Hamilton Rating Scale for Depression (HAM-D17) to measure depressive symptom improvement and the Clinical Global Impressions-Improvement (CGI-I) scale to measure overall improvement. Desvenlafaxine was more effective than placebo in HAM-D17 score improvement in all 4 studies and in CGI-I score improvement in 3 studies.
In studies comparing 50 mg/d with 100 mg/d, doses >50 mg/d provided no additional benefit. Higher starting fixed doses were associated with more frequent AEs and discontinuation.
Gender or age had no effect on treatment outcome. There was no difference in safety in elderly vs younger patients. Data are insufficient to establish a relationship between race and desvenlafaxine responsiveness. Desvenlafaxine’s safety and effectiveness in children and adolescents was not evaluated, and the drug is not approved for these patients.
Tolerability and safety
Desvenlafaxine’s tolerability is comparable to that of other SNRIs. In premarketing studies, 12% of patients receiving desvenlafaxine (50 mg/d to 400 mg/d) discontinued treatment because of AEs, compared with 3% in the placebo group. The discontinuation rate in patients receiving 100 mg/d was 8.7% compared with 4.1% in patients taking 50 mg/d.
AEs generally occur during the first week of treatment. In the 8-week trials, the most common AEs were nausea and dizziness (Table 2). In a long-term study (up to 9 months), the most common AE was vomiting. Although the recommended starting dose is 50 mg/d, to avoid AEs consider beginning with every-other-day dosing.
Table 2
Desvenlafaxine trials: Rates of adverse effects
| Desvenlafaxine dose | |||
|---|---|---|---|
| Adverse effect | 50 mg/d | 100 mg/d | Placebo |
| Nausea | 22% | 26% | 10% |
| Dizziness | 13% | 10% | 5% |
| Insomnia | 9% | 12% | 6% |
| Hyperhidrosis | 10% | 11% | 4% |
| Constipation | 9% | 9% | 4% |
| Somnolence | 4% | 9% | 4% |
| Decreased appetite | 5% | 8% | 2% |
| Erectile dysfunction | 3% | 6% | 1% |
| Decreased libido | 4% | 5% | 1% |
| Anxiety | 3% | 5% | 2% |
| Source: Reference 1 | |||
Abruptly discontinuing desvenlafaxine can cause withdrawal symptoms, including dizziness, nausea, headache, irritability, insomnia, diarrhea, anxiety, abnormal dreams, fatigue, and hyperhidrosis. The frequency of withdrawal symptoms is higher with longer treatment duration. Gradually reducing the dose by administering 50 mg of desvenlafaxine less often can reduce withdrawal symptoms.
Clinical issues
All SNRIs and selective serotonin reuptake inhibitors (SSRIs) have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation/behaviors during the first months of treatment and with dose changes.
When taken in the third trimester of pregnancy, SNRIs and SSRIs can cause serious neonatal complications—including respiratory distress, cyanosis, apnea, and seizures—that may require longer hospitalization, respiratory support, or tube feeding for the infant. Carefully consider risks and benefits of third-trimester antidepressant use.5 Desvenlafaxine is excreted in breast milk and may cause AEs in infants who are breast-fed.
In clinical trials, patients taking desvenlafaxine experienced increased cholesterol, triglycerides, and blood pressure. Monitor these parameters closely in patients taking desvenlafaxine, and use the drug with caution in patients with cerebrovascular and cardiovascular disease.
Other concerns in patients taking desvenlafaxine include:
- Antidepressant medications can trigger hypomania or mania in patients with bipolar disorder.
- Patients—particularly those who are elderly or taking diuretics—may develop hyponatremia as a result of syndrome of in-appropriate antidiuretic hormone.
- Patients with an increased risk of glaucoma need to be monitored because of the drug’s effect on blood pressure.
Drug interactions. Coadministering desvenlafaxine with serotonergic medications— such as triptans, other antidepressants, and tramadol—can cause serotonin syndrome, a potentially life-threatening condition characterized by mental status changes, autonomic instability, neuromuscular aberrations, and gastrointestinal symptoms. Concomitant use of desvenlafaxine and blood-thinning medications such as warfarin, aspirin, and nonsteroidal anti-inflammatory drugs may result in abnormal bleeding. Patients taking a potent CYP 3A4 inhibitor such as ketoconazole may have increased desvenlafaxine concentration.
Contraindications
Do not prescribe desvenlafaxine to patients who are:
- hypersensitive to venlafaxine chloride, desvenlafaxine succinate, or any parts of the desvenlafaxine formulation
- taking a monoamine oxidase inhibitor (MAOI), or have discontinued an MAOI within 14 days.
Patients who stop taking desvenlafaxine should wait 7 days before starting an MAOI.
Drug brand names
- Bupropion • Wellbutrin
- Desvenlafaxine • Pristiq
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Ketoconazole • Nizoral
- Paroxetine • Prozac
- Tramadol • Ultram
- Venlafaxine • Effexor
- Warfarin • Coumadin
Disclosures
Dr. Lincoln reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Preskorn has in the past year received research/ grant support from and served as a speaker for Wyeth Pharmaceuticals. Previously, he has received research/grant support from or served as a speaker for or consultant to Abbott Laboratories, AstraZeneca, Aventis, Biovail, Boehringer Ingleheim, Bristol-Myers Squibb, Eisai, Eli Lilly and Company, GlaxoSmithKline, Hoffman LaRoche, Janssen, L.P., Johnson & Johnson, Lundbeck, Merck, Novartis, Organon, Otsuka, Pfizer Inc., Solvay, Somerset, Sumitomo, and Yamanouchi.
Compared with other antidepressants, desvenlafaxine might have more predictable effects and a lower risk of drug-drug interactions because of the way it is metabolized. The FDA approved this selective serotonin-norepinephrine reuptake inhibitor (SNRI)—a major active metabolite of venlafaxine—for treating major depressive disorder (MDD, Table 1). In clinical trials, desvenlafaxine was more effective than placebo in improving patients’ scores on scales of depressive symptoms and overall improvement.1
Table 1
Desvenlafaxine: Fast facts
| Brand name: Pristiq |
| Class: Serotonin-norepinephrine reuptake inhibitor |
| Indication: Major depressive disorder |
| Approval date: February 2008 |
| Availability date: May 2008 |
| Manufacturer: Wyeth Pharmaceuticals |
| Dosing forms: 50- and 100-mg tablets |
| Recommended dose: 50 to 100 mg/d |
Clinical implications
Unlike other SNRIs (venlafaxine and duloxetine), desvenlafaxine does not depend on cytochrome P450 (CYP) 2D6 for bio-transformation. As a result plasma concentrations vary less among individual patients, which should result in more predictable efficacy and tolerability. In addition, unlike bupropion, duloxetine, fluoxetine, and paroxetine, desvenlafaxine does not affect the functional activity of CYP 2D6. This translates into a lower risk of drug-drug interactions and more predictable effects on coadministered drugs that are cleared by CYP 2D6.
How it works
Serotonin, norepinephrine, and dopamine in the CNS are involved in mood and neurovegetative functions that are disturbed in patients with MDD. Desvenlafaxine selectively inhibits serotonin and norepinephrine reuptake pumps, therefore increasing serotonin and norepinephrine concentration in the synaptic cleft.2 The drug has weak binding affinity for the dopamine transporter and does not cause substantial changes in extracellular dopamine concentration. Decreased presynaptic serotonin and norepinephrine uptake increases the synaptic concentration of these neurotransmitters. These effects are thought to be responsible for desvenlafaxine’s antidepressant efficacy.
Pharmacokinetics
Desvenlafaxine’s single-dose pharmacokinetics are linear and dose-proportional over the recommended 50 to 100 mg/d dosing range. The half-life is approximately 11 hours. Steady-state plasma concentration is achieved in 4 to 5 days with once-daily dosing.
Food does not affect intestinal absorption. Bioavailability after oral administration is 80%, and time to reach maximum concentration (Tmax) is 7.5 hours. Plasma protein binding is 30% and is independent of desvenlafaxine concentration.1
Desvenlafaxine is excreted renally:
- unchanged (45% at 72 hours after administration)
- as desvenlafaxine-glucuronide
- as N-desvenlafaxine-glucuronide.
Desvenlafaxine-glucuronide is the final metabolite of conjugation reaction with glucuronic acid. N-desvenlafaxine-glucuronide is an end product of a 2-step metabolic reaction that starts with oxidation by CYP 3A4 to produce N-desvenlafaxine, which is conjugated with glucuronic acid to create N-desvenlafaxine-glucuronide. As a result of these metabolic and elimination pathways, dosing adjustment is recommended for patients with severe renal impairment or who are taking a CYP 3A4 inhibitor.
Dosing
Desvenlafaxine is available as 50-mg and 100-mg tablets. The recommended dosage is 50 mg/d, and the maximum recommended dosage in patients with hepatic impairment is 100 mg/d.
No dosing adjustment is necessary for patients with moderate renal impairment. The recommend regimen for those with severe renal impairment or end-stage renal disease is 50 mg every other day.
Instruct patients to take desvenlafaxine at approximately the same time each day, with or without food. Tell them not to discontinue the drug abruptly and to immediately report any adverse effects (AEs).
Efficacy
Desvenlafaxine’s antidepressant efficacy was established in four 8-week, randomized, double-blind, placebo-controlled, fixed-dose (50 mg to 400 mg once daily) studies in adult outpatients who met DSM-IV-TR criteria for MDD.1,2,4
In the first study,3 461 patients received desvenlafaxine, 100 mg, 200 mg, or 400 mg, or placebo. In the second study,4 369 patients received 200 mg or 400 mg or placebo. In 2 additional studies, a total of 930 patients received 50 mg or 100 mg or placebo.1
All studies used the 17-item Hamilton Rating Scale for Depression (HAM-D17) to measure depressive symptom improvement and the Clinical Global Impressions-Improvement (CGI-I) scale to measure overall improvement. Desvenlafaxine was more effective than placebo in HAM-D17 score improvement in all 4 studies and in CGI-I score improvement in 3 studies.
In studies comparing 50 mg/d with 100 mg/d, doses >50 mg/d provided no additional benefit. Higher starting fixed doses were associated with more frequent AEs and discontinuation.
Gender or age had no effect on treatment outcome. There was no difference in safety in elderly vs younger patients. Data are insufficient to establish a relationship between race and desvenlafaxine responsiveness. Desvenlafaxine’s safety and effectiveness in children and adolescents was not evaluated, and the drug is not approved for these patients.
Tolerability and safety
Desvenlafaxine’s tolerability is comparable to that of other SNRIs. In premarketing studies, 12% of patients receiving desvenlafaxine (50 mg/d to 400 mg/d) discontinued treatment because of AEs, compared with 3% in the placebo group. The discontinuation rate in patients receiving 100 mg/d was 8.7% compared with 4.1% in patients taking 50 mg/d.
AEs generally occur during the first week of treatment. In the 8-week trials, the most common AEs were nausea and dizziness (Table 2). In a long-term study (up to 9 months), the most common AE was vomiting. Although the recommended starting dose is 50 mg/d, to avoid AEs consider beginning with every-other-day dosing.
Table 2
Desvenlafaxine trials: Rates of adverse effects
| Desvenlafaxine dose | |||
|---|---|---|---|
| Adverse effect | 50 mg/d | 100 mg/d | Placebo |
| Nausea | 22% | 26% | 10% |
| Dizziness | 13% | 10% | 5% |
| Insomnia | 9% | 12% | 6% |
| Hyperhidrosis | 10% | 11% | 4% |
| Constipation | 9% | 9% | 4% |
| Somnolence | 4% | 9% | 4% |
| Decreased appetite | 5% | 8% | 2% |
| Erectile dysfunction | 3% | 6% | 1% |
| Decreased libido | 4% | 5% | 1% |
| Anxiety | 3% | 5% | 2% |
| Source: Reference 1 | |||
Abruptly discontinuing desvenlafaxine can cause withdrawal symptoms, including dizziness, nausea, headache, irritability, insomnia, diarrhea, anxiety, abnormal dreams, fatigue, and hyperhidrosis. The frequency of withdrawal symptoms is higher with longer treatment duration. Gradually reducing the dose by administering 50 mg of desvenlafaxine less often can reduce withdrawal symptoms.
Clinical issues
All SNRIs and selective serotonin reuptake inhibitors (SSRIs) have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation/behaviors during the first months of treatment and with dose changes.
When taken in the third trimester of pregnancy, SNRIs and SSRIs can cause serious neonatal complications—including respiratory distress, cyanosis, apnea, and seizures—that may require longer hospitalization, respiratory support, or tube feeding for the infant. Carefully consider risks and benefits of third-trimester antidepressant use.5 Desvenlafaxine is excreted in breast milk and may cause AEs in infants who are breast-fed.
In clinical trials, patients taking desvenlafaxine experienced increased cholesterol, triglycerides, and blood pressure. Monitor these parameters closely in patients taking desvenlafaxine, and use the drug with caution in patients with cerebrovascular and cardiovascular disease.
Other concerns in patients taking desvenlafaxine include:
- Antidepressant medications can trigger hypomania or mania in patients with bipolar disorder.
- Patients—particularly those who are elderly or taking diuretics—may develop hyponatremia as a result of syndrome of in-appropriate antidiuretic hormone.
- Patients with an increased risk of glaucoma need to be monitored because of the drug’s effect on blood pressure.
Drug interactions. Coadministering desvenlafaxine with serotonergic medications— such as triptans, other antidepressants, and tramadol—can cause serotonin syndrome, a potentially life-threatening condition characterized by mental status changes, autonomic instability, neuromuscular aberrations, and gastrointestinal symptoms. Concomitant use of desvenlafaxine and blood-thinning medications such as warfarin, aspirin, and nonsteroidal anti-inflammatory drugs may result in abnormal bleeding. Patients taking a potent CYP 3A4 inhibitor such as ketoconazole may have increased desvenlafaxine concentration.
Contraindications
Do not prescribe desvenlafaxine to patients who are:
- hypersensitive to venlafaxine chloride, desvenlafaxine succinate, or any parts of the desvenlafaxine formulation
- taking a monoamine oxidase inhibitor (MAOI), or have discontinued an MAOI within 14 days.
Patients who stop taking desvenlafaxine should wait 7 days before starting an MAOI.
Drug brand names
- Bupropion • Wellbutrin
- Desvenlafaxine • Pristiq
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Ketoconazole • Nizoral
- Paroxetine • Prozac
- Tramadol • Ultram
- Venlafaxine • Effexor
- Warfarin • Coumadin
Disclosures
Dr. Lincoln reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Preskorn has in the past year received research/ grant support from and served as a speaker for Wyeth Pharmaceuticals. Previously, he has received research/grant support from or served as a speaker for or consultant to Abbott Laboratories, AstraZeneca, Aventis, Biovail, Boehringer Ingleheim, Bristol-Myers Squibb, Eisai, Eli Lilly and Company, GlaxoSmithKline, Hoffman LaRoche, Janssen, L.P., Johnson & Johnson, Lundbeck, Merck, Novartis, Organon, Otsuka, Pfizer Inc., Solvay, Somerset, Sumitomo, and Yamanouchi.
1. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals; 2008.
2. Deecher DC, Beyer CE, Johnston G, et al. Desvenlafaxine succinate: a new serotonin and norepinephrine reuptake inhibitor. J Pharmacol Exp Ther 2006;318(2):657-65.
3. DeMartinis NA, Yeung PP, Entsuah R, Manley AL. A double-blind, placebo-controlled study of the efficacy and safety of desvenlafaxine succinate in the treatment of major depressive disorder. J Clin Psychiatry 2007;68(5):677-88.
4. Septien-Velez L, Pitrosky B, Padmanabhan SK, et al. A randomized, double-blind, placebo-controlled trial of desvenlafaxine succinate in the treatment of major depressive disorder. Int Clin Psychopharmacol 2007;22(6):338-47.
5. Lennestål R, Källén B. Delivery outcome in relation to maternal use of some recently introduced antidepressants. J Clin Psychopharmacol 2007;27(6):607-13.
Dr. Lincoln is a clinical instructor and Dr. Preskorn is a professor of psychiatry, University of Kansas Medical Center, Wichita. Dr. Preskorn also is the psychopharmacology Section Editor of CURRENT PSYCHIATRY and president and CEO of Clinical Research Institute, Wichita.
1. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals; 2008.
2. Deecher DC, Beyer CE, Johnston G, et al. Desvenlafaxine succinate: a new serotonin and norepinephrine reuptake inhibitor. J Pharmacol Exp Ther 2006;318(2):657-65.
3. DeMartinis NA, Yeung PP, Entsuah R, Manley AL. A double-blind, placebo-controlled study of the efficacy and safety of desvenlafaxine succinate in the treatment of major depressive disorder. J Clin Psychiatry 2007;68(5):677-88.
4. Septien-Velez L, Pitrosky B, Padmanabhan SK, et al. A randomized, double-blind, placebo-controlled trial of desvenlafaxine succinate in the treatment of major depressive disorder. Int Clin Psychopharmacol 2007;22(6):338-47.
5. Lennestål R, Källén B. Delivery outcome in relation to maternal use of some recently introduced antidepressants. J Clin Psychopharmacol 2007;27(6):607-13.
Dr. Lincoln is a clinical instructor and Dr. Preskorn is a professor of psychiatry, University of Kansas Medical Center, Wichita. Dr. Preskorn also is the psychopharmacology Section Editor of CURRENT PSYCHIATRY and president and CEO of Clinical Research Institute, Wichita.