User login
The Challenges of Precision Medicine and New Advances in Molecular Diagnostic Testing in Hematolymphoid Malignancies: Impact on the VHA (FULL)
In January 2015, President Obama introduced the Precision Medicine Initiative, a program set up to identify new biomedical discoveries for the development of a personalized knowledge base of disease entities and individualized treatments. Advances in precision medicine typically involve the use of targeted therapies tailored to individual genetic characteristics identified with molecular testing. The goals are to improve survival and reduce adverse effects. With an initial budget of $215 million, this initiative presented a unique opportunity to combine efforts in genomic discovery, bioinformatic analysis, and health information technology to move toward data-driven, evidence-based precision medicine.1
The VHA is the largest comprehensive health care system in the U. S. and has more than 1,700 care sites serving nearly 9 million veterans each year. The budget for this single-payer system is proposed by the President and approved by Congress. As the VHA must treat a diverse and aging veteran population in an environment of rising costs and budget constraints, limited resources must be monitored and appropriated for the most cost-effective health care delivery. Precision medicine offers a model in which physicians can select the most appropriate diagnostic tests in defined clinical settings to direct clinical care. It supports the testing needed to subdivide each disease category into distinct subcategories. Nevertheless, the need for fiscal responsibility in a capitated health care system recommends testing in cases in which it can change therapy or prognosis rather than for purely academic reasons.
Pathology and Laboratory Medicine Service
Given limited resources and an increasing number of requests for advanced molecular testing, the VA Pathology and Laboratory Medicine Service (P&LMS) formed the Molecular Genetics Pathology Workgroup (MGPW) in September 2013. The charter listed the tasks of the MGPW to “provide recommendations on how to effectively use molecular genetics tests, promote increased quality and availability of testing within the VHA, encourage internal referral testing, provide an organizational structure for Molecular Genetics Testing Consortia, and create a P&LMS policy for molecular genetic testing in general, specifically addressing the issues surrounding laboratory developed testing.” The MGPW has 4 subcommittees: molecular oncology, pharmacogenetics, hematopathology molecular genetics (HMG), and genetic medicine. Since its inception, the HMG subcommittee has had several objectives:
- Standardize the molecular testing nomenclature for and develop practice guidelines for acute myeloid leukemia (AML), myeloproliferative neoplasms (MPN), myelodysplastic syndrome (MDS), chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma, lymphoma, and plasma cell neoplasms;
- Develop standardized reporting guidelines for current VA molecular laboratories;
- Identify new tests as they are being reported in the literature and collaborate with hematology and oncology services to evaluate the clinical utility of these tests for VA patients;
- Network current VA molecular laboratories, perform fact-finding for these laboratories, and compile test menus; and
- Assess for the formation of VA-wide interfacility consultation services for hematopathology so that all VA facilities, regardless of their complexity, will be able to access the expertise of hematopathology-trained pathologists (Appendix).
The HMG subcommittee met monthly and discussed various diagnostic entities in hematopathology. For hematolymphoid malignancies, it was generally agreed that the traditional laboratory tools of morphology, flow cytometry, and immunohistochemistry (IHC) are standard in initial assessment and often in diagnosis. As the clinical molecular and cytogenetic assays of karyotype, fluorescence in situ hybridization (FISH), advanced DNA sequencing, microarray, and highly sensitive polymerase chain reaction (PCR) analysis affect diagnosis, subclassification, minimal residual disease (MRD) monitoring, prognosis, and therapy selection, their use is marked by a high degree of variability. As a result, standardization is needed. As each laboratory develops and reports ancillary testing, the variable reporting formats may generate postanalytic errors.
A detailed description of all molecular methodologies is beyond the scope of this article. For practicing pathologists, challenges remain in overall cost and reimbursement, extensive and time-consuming data analysis, and in some cases, interpretation differences.
Myeloid Neoplasms
Myeloid malignancies were divided into AML, MPN, and MDS. Next-generation sequencing (NGS) information for these malignancies was used to identify various contributory functional categories, including cell signaling (FLT3, KIT, JAK2, MPL, KRAS/NRAS, PTPN11, NF1, CSF3R); transcription (CEBPA, RUNX1, GATA1/GATA2, PHF6, ETV6); splicing (SF3B1, SRSF2, ZRSR2, U2AF1); epigenetics (DNMT3A, TET2, IDH1/IDH2, ASXL1, EZH2, SUZ12, KDM6A); cohesin complex (STAG2, SMC1A, SMC3, RAD21); and cell cycle (TP53, NPM1).2
Acute Myeloid Leukemia
The HMG subcommittee reviewed the literature on prognostically significant genes in myeloid leukemias. Karyotype abnormalities, such as t(8;21) and inv(16), collectively known as the core-binding factor (CBF) leukemias, t(15;17), t(11q23) (KMT2A/MLL), and so forth, are recurrent lesions in AML. Included in the minimum set of genes recommended by the National Comprehensive Cancer Network (NCCN) for AML prognosis evaluation are nucleolar protein nucleophosmin (NPM1), CCAAT/enhancer-binding protein
Some of the chromosomal translocations, such as inv(16)/t(16;16) in AML and t(15;17) in acute promyelocytic leukemia, can be monitored with FISH or reverse transcription–PCR (RT-PCR) analysis. As NPM1 mutations tend to be seen in recurrence, they can be used as molecular markers for MRD. Other mutations that provide important prognostic information in AML include:
- Activating insertions/duplications in the FLT3 receptor tyrosine kinase, which can be detected with PCR sizing assays;
- Mutations in the KIT receptor tyrosine kinase, which can be detected with DNA sequencing or more limited hotspot PCR;
- Mutations in the DNA methyltransferase, DNMT3A, a poor prognostic indicator seen in 22% of cases of AML, also detected with gene sequencing or more limited hotspot PCR; and
- Another set of genes, TET2, IDH1, IDH2, KRAS, NRAS, EZH2, and ASXL1, is mutated in MPN as well as AML and MDS, making a common molecular panel with next-generation sequencing useful in diagnosing and risk-stratifying all myeloid neoplasms.
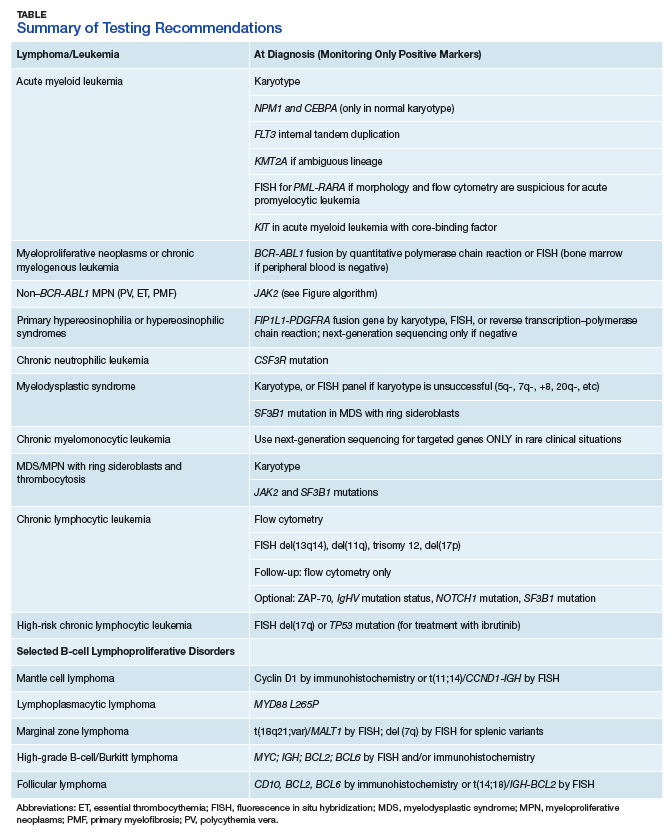
The HMG subcommittee agreed that, for de novo AML, chromosomal karyotype is the standard of care, necessary in detecting known cytogenetic abnormalities as well as a wide range of lesions that might indicate a diagnosis of AML with myelodysplasia-related changes at time of diagnosis. In addition, molecular analysis of FLT3 is useful in determining prognosis, and CEBPA (biallelic) and NPM1 mutations are good prognostic factors in normal-karyotype AML. KMT2A (MLL) rearrangements should be tested with FISH if the lineage is ambiguous. The PML-RARA fusion gene also should be tested with FISH if morphologic and flow cytometry results suggest acute promyelocytic leukemia (Table). At this time, testing for TP53, DNMT3A, RAS, and other such mutations is not recommended because it is not cost-effective for the VA.
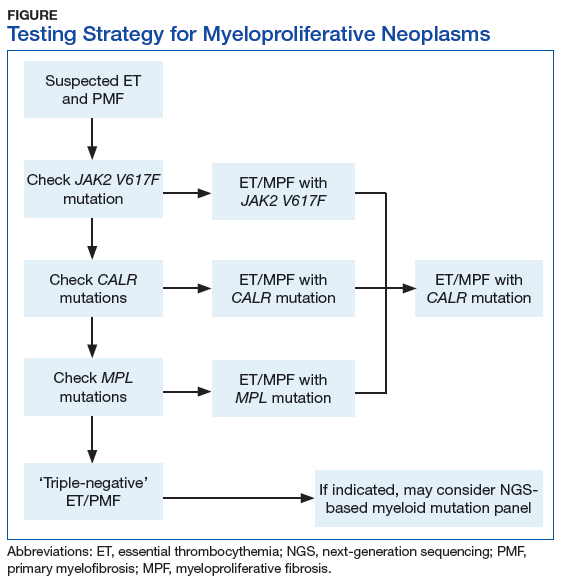
Myeloproliferative Neoplasms
Myeloproliferative neoplasms are clonal hematopoietic stem cell disorders characterized by proliferation of at least 1 myeloid lineage: granulocytic, erythroid, or megakaryocytic. Myeloproliferative neoplasms show a range of recurrent chromosomal translocations, such as BCR-ABL1 fusion in chronic myelogenous leukemia (CML) that can be detected with RT-PCR analysis as well as FISH. In CML, BCR-ABL1 fusion transcript levels detected by a quantitative PCR (qPCR) method are now used to monitor the course of CML therapy with tyrosine kinase inhibitors (TKIs) and to trigger a treatment change in drug-resistant cases. Given the importance of qPCR in clinical management, significant progress has been made in standardizing both the PCR protocol and the reference materials used to calibrate the BCR-ABL1 PCR assay. BCR-ABL1–negative MPN, including polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF), are most commonly associated with mutations in the tyrosine kinase JAK2. Mutations in CALR and MPL are seen in a subset of patients with ET and PMF as well, whereas PV is essentially exclusively a disease of JAK2 mutations.
Chronic myelogenous leukemia is the prototypical MPN. To establish the initial diagnosis, FISH and/or qPCR for BCR-ABL1 fusion should be used. If CML is confirmed, the sample can be reflexed to qPCR BCR-ABL1 on the initial peripheral blood and/or bone marrow sample(s) to establish the patient’s baseline. In addition, a bone marrow sample (aspirate) should be used for a complete karyotype and for morphologic confirmation of disease phase.
For follow-up assessment of CML patients’ response to TKI treatment, qPCR for BCR-ABL1 should be tested with a peripheral blood sample or a bone marrow sample every 3 months.4 A peripheral blood sample is more commonly used because it is conveniently obtained. Early molecular response as indicated by a BCR-ABL1 transcript ratio of < 10% on the International Scale at 3 months, has a strong prognostic value.5 Major molecular response as indicated by a BCR-ABL1 transcript ratio of < 0.1% on the International Scale at 12 to 18 months is also highly prognostic.5
After the peripheral blood sample becomes negative for BCR-ABL1 by qPCR, testing bone marrow samples may be considered. If important treatment response benchmarks are not achieved, or response is lost with rising BCR-ABL1 levels (TKI resistance), ABL1 kinase domain mutation analysis as well as repeat FISH (to assess for copy number multiplication) should be performed to guide further management. Patients with the ABL1 T315I mutation are resistant to all first-line TKIs but may respond to later third-generation TKIs.6
BCR-ABL1–negative MPNs include PV, ET, and PMF. Bone marrow morphology remains the cornerstone of ET and PMF diagnosis. The discovery of JAK2, CALR, and MPL mutations has contributed to how these disorders are diagnosed.7-12 Besides providing the clonality proof that is crucial for diagnosis, the molecular markers influence the prognosis. The JAK2 (p.V617F) or less common JAK2 exon 12 mutations, which are detected in more than 95% of PV cases, are used as molecular markers to confirm diagnosis.7 Further, the JAK2 (p.V617F), CALR (exon 9), and MPL (exon 10) mutations are detected in ET (~60%, 25%, and 3%-5%, respectively) and PMF (~55%, 30%, and 5%, respectively).12 If ET or PMF is suspected clinically, first JAK2 (p.V617F) mutation analysis should be performed, then CALR mutation analysis, and finally MPL mutation analysis. Although novel gain-of-function JAK2 and MPL mutations were recently discovered in triple-negative ET (negative for canonical mutations in JAK2, CALR, and MPL) and PMF by whole exome sequencing,13 clinical testing is not readily available. Besides its utility in the initial diagnosis of ET and PMF, the JAK2 or CALR mutation assay also may be considered for bone marrow transplantation follow-up (Table).14
Despite the continuing debate on the classification of eosinophilic myeloid disorders, the discovery of the FIP1L1-PDGFRA fusion represents a major milestone in the understanding of these disorders.15,16 Unlike PDGFRB (5q33) and FGFR1 (8p11) rearrangements, which can be detected with routine chromosomal analysis (cytogenetics), the cryptic FIP1L1-PDGFRA fusion must be detected with FISH (for CHIC2 deletion) or RT-PCR analysis. It should be pointed out that, as most eosinophilia is reactive or secondary, molecular testing for FIP1L1-PDGFRA fusion is indicated only when primary hypereosinophilia or hypereosinophilic syndrome (HES) is suspected. This is particularly the case in the following hypereosinophilia accompanying conditions: CML-like morphology, but BCR-ABL1–negative; chronic myelomonocytic leukemia (CMML)–like morphology with a normal karyotype; and new onset of cardiac damage or dysfunction.17
Primary eosinophilic myeloid disorders with PDGFRA or PDGFRB rearrangements can be treated with TKIs (eg, imatinib). Next-generation sequencing may be considered in cases of presumed HES when there is no identifiable karyotypic or FISH abnormality. Recent studies have found that cases of HES with somatic mutations indicating clonality had adverse clinical outcomes similar to those of cases of chronic eosinophilic leukemia.18
The discovery of CSF3R mutations offers a new molecular marker for the diagnosis of chronic neutrophilic leukemia (CNL), an MPN.19 The CSF3R (p.T618I) mutation or another activating CSF3R mutation is now used as a diagnostic criterion for CNL. Identification of specific CSF3R mutations may have therapeutic implications as well. The test should be ordered only for patients with clinical and morphologic findings suggestive of CNL; reactive neutrophilic leukocytosis (eg, infection, inflammation) should be ruled out before the test is ordered.
Myelodysplastic Syndrome
Myelodysplastic syndrome is a group of clonal bone marrow disorders characterized by ineffective hematopoiesis, manifested by morphologic dysplasia in ≥ 1 hematopoietic lineages and peripheral cytopenias (hemoglobin level, < 10 g/dL; platelet count, < 100×103/µL; absolute neutrophil count, < 1.8×103/µL). Diagnosis and classification of MDS depend mainly on the degree of morphologic dysplasia and blast percentages, as determined by examining well-prepared cellular bone marrow aspirate smears and/or biopsy touch preparations and peripheral blood smears.
Conventional karyotyping is an essential part of the diagnostic workup for all presumptive cases of MDS and is of both diagnostic and prognostic importance.20 About 60% of MDS cases have recurrent cytogenetic abnormalities, which can be detected with conventional karyotyping. If a high-quality cytogenetic analysis cannot be performed (eg, the bone marrow sample is inadequate), or if quick turnaround is required, an alternative FISH panel may be used to detect some of the common MDS-associated chromosomal abnormalities (eg, 5q deletion, 7q deletion/monosomy 7, +8, 20q deletion).21 Sequencing with FISH also can be useful for assessing MRD by detecting a previously identified chromosomal abnormality.
Targeted sequencing of a limited number of genes can detect mutations in the vast majority of patients with MDS. The most commonly mutated genes in MDS are SF3B1, TET2, SRSF2, ASXL1, DNMT3A, RUNX1, U2AF1, TP53, and EZH2. Mutations in SRSF2 cause RNA splicing abnormalities. In addition, mutations in TP53, EZH2, RUNX1, and ASXL1 are associated with poor prognosis,22,23 whereas mutations in SF3B1 confer better event-free survival.24 Despite these developments, the HMG subcommittee agreed that NGS-based mutation panels are not cost-effective for the VA population at this time and should not be included in a MDS workup. Only in rare situations and when clinically indicated (to change disease classification or patient management) should evaluation for specific gene mutations be considered—for instance, the SF3B1 mutation for patients with probable MDS with ring sideroblasts, if ring sideroblasts are < 15%.25
Myelodysplastic/Myeloproliferative Neoplasms
Myelodysplastic/myeloproliferative neoplasms are a group of myeloid neoplasms with clinical, laboratory, and morphologic features that overlap both MDS and MPN. In MDS/MPN, the karyotype is often normal or shows abnormalities in common with MDS.
In cases of unexplained monocytosis for which there is clinical concern for CMML, morphologic evaluation and conventional chromosomal karyotyping should be performed after other secondary causes and known myeloproliferative and myelodysplastic entities have been excluded. If concomitant hypereosinophilia is present and the karyotype is normal, FISH or PCR-based assay should be performed to rule out FIP1L1-PDGFRA rearrangements. BCR-ABL1, PDGFRB, FGFR1, and t(8;9)/PCM1-JAK2 rearrangements typically are detected with high-quality cytogenetic analysis and thus do not require targeted molecular assays. Although certain gene mutations (eg, SRSF2, TET2, ASXL1, CBL) are commonly detected in CMML, the HMG subcommittee does not recommend sequencing-based mutation panels, as there is insufficient information for testing for prognostic or treatment stratification.
If MDS/MPN with ring sideroblasts and thrombocytosis is suspected on the basis of the clinical and morphologic criteria, molecular tests for the JAK2 (p.V617F) and SF3B1 mutations may be considered in an effort to help confirm the diagnosis.
Atypical CML is a rare MDS/MPN subtype that is now better characterized molecularly with SETBP1 and/or ETNK1 mutations, which are detectable in up to a third of cases. If clinical suspicion is high, sequencing may be diagnostically helpful.
Lymphoid Neoplasms
Chronic Lymphocytic Leukemia
In CLL, recurrent chromosomal abnormalities (eg, deletions of 13q, trisomy 12, deletions of 11q, deletions of 17p) have clear prognostic value and can be detected with FISH. Other prognostic information, such as somatic mutation of immunoglobulin heavy chain variable (IgHV) genes, TP53 mutations, SF3B1, and NOTCH1 mutation, are mostly derived from PCR-based assays. The discovery of recurrently mutated genes in CLL has increased with the use of highly sensitive sequencing methods constructing a more detailed landscape of CLL at genetic, epigenetic, and cellular levels. A recent literature review summarizes the vast heterogeneity of CLL with recurrent pathogenetic findings in MYD88, SF3B1, TP53, ATM, and NOTCH1 signaling pathways.26 The treatment of CLL is rapidly evolving, and many clinical trials are proposing a change from the “watch and wait” paradigm to treatment upon initial presentation based on molecular findings. Additional testing based on new treatment options from current clinical trials will be recommended.
Flow cytometry and morphology are standard for CLL diagnosis. The HMG subcommittee recommends FISH for del(13q14), del(11q), trisomy 12, and del(17p) at time of diagnosis or immediately before therapy initiation. Zeta-chain (
Other B-Cell Lymphoproliferative Disorders
Unlike the common molecular changes in CLL, in other mature B-cell lymphomas, chromosomal translocations that juxtapose a variety of different oncogenes next to an Ig gene enhancer usually are—and those that switch regions less commonly are—important initiating events that can be detected with PCR, DNA sequencing, or FISH. In follicular lymphoma (FL), Burkitt lymphoma, marginal zone lymphoma (MZL), and mantle cell lymphoma (MCL), these oncogenes driven by an Ig gene enhancer typically include BCL2, MYC, MALT1, and CCND1 (cyclin D1), respectively. Molecular variants of these lymphomas that lack these classical translocations often activate homologous genes (eg, cyclin D3/CCND3 is activated in variants of MCL).
Morphology, flow cytometry, and IHC are routinely used for diagnosis. In inconclusive cases, Ig gene rearrangement by PCR may be used. The Table summarizes common molecular changes in B-cell lymphomas.
Mantle cell lymphoma. MCL is a non-Hodgkin lymphoma subtype characterized by t(11;14) (q13;q32) translocations that in the majority of cases lead to overexpression of cyclin D1 (BCL1). Recent molecular profiling has identified an MCL variant that is cyclin D1–negative but SOX11-positive and may have a more aggressive clinical course.30SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive MCL.
Lymphoplasmacytic lymphoma. Lymphoplasmacytic lymphoma (LPL), MZL, and CLL/small lymphocytic lymphoma are well-defined clinicopathologic entities. However, distinguishing LPL from MZL and atypical cases of CLL can sometimes be difficult because of overlapping clinical and morphologic features. Recent studies have identified a recurrent L265P mutation in the MYD88 gene in 90% to 95% of LPL cases with IgM paraprotein and in 40% to 50% of the rare non-IgM LPL cases. In contrast, the mutation is much less frequently present in MZL and other low-grade B-cell neoplasms (2%-7%).31 Therefore, testing for this abnormality can be a diagnostic aid in these difficult-to-classify cases. In addition, from a therapeutic perspective, presence or absence of MYD88 mutation may prove more significant than presence of a specific paraprotein or histopathologic features. Ibrutinib has shown efficacy in LPL and demonstrates improved response rates in patients with MYD88 mutation compared with that of their mutation-negative counterparts.32 Several MYD88 inhibitors are in clinical trials. This again indicates the need to more accurately identify and subclassify these non-IgM LPL cases to ensure appropriate molecular evaluation.
Hairy cell leukemia. Flow cytometry and morphology are usually sufficient for a hairy cell leukemia (HCL) diagnosis. However, rare cases are difficult to distinguish variant HCL from other mimics. The BRAF V600E mutation recently was described as a disease-defining molecular marker for HCL—present in nearly all HCL cases but virtually absent in HCL mimics. Therefore, detection of the BRAF mutation by IHC stain with specific antibody or PCR analysis is highly sensitive and specific for the diagnosis of HCL.33
Diffuse large B-cell lymphoma. Recent molecular analysis has created various risk stratification schemata for diffuse large B-cell lymphoma (DLBCL). The HGM subcommittee agrees that well-preserved morphology, IHC, flow cytometry, and FISH-specific markers (BCL2, BCL6, cMYC) are sufficient for diagnostic, prognostic, and therapeutic purposes. Although a wide range of genes have been implicated in the pathogenesis of DLBCL, sequencing and gene expression profiling are not cost-effective at this time and do not add benefit to patient treatment.
The MYD88 L265P mutation has been identified in DLBCL, particularly the activated B-cell-like type and primary central nervous system lymphoma (PCNSL), and may have implications for ibrutinib therapy. PCNSL commonly manifests aggressive clinical behavior and has a poor prognosis. It has been proposed that the MYD88 mutation can be used as a genetic hallmark for PCNSL to distinguish CNS involvement by systemic DLBCL from PCNSL.34
Plasma cell neoplasms. Flow cytometry is acceptable for the diagnosis of plasma cell neoplasms and for residual disease follow-up. Chromosomal karyotype or FISH for IGH/CCND1, IGH/MMSET, and IGH/CMAF dual fusion probes is recommended in conjunction with morphology, IHC, and flow cytometry. In plasma cell myeloma, several genetic mutations can be detected with NGS, including mutations in NRAS, KRAS, TP53, BCL7A, DIS3, and FAM46C.35 Less commonly, BRAF mutations, previously described in melanoma and several other solid tumors, can be detected with DNA sequencing in 4% of multiple myeloma cases, which may prove promising for targeted therapy with BRAF inhibitors. However, current therapeutic decisions are based on genetic and clinical factors, and sequence-based assays are not recommended at this time.
Follicular lymphoma. Cytology, histology, and IHC typically are sufficient for diagnosing FL. In difficult-to-diagnose cases and in cases with scant material, additional tests may help with diagnosis. Eighty to ninety percent of FL cases have t(14;18)(q32;q21), which places the BCL2 gene transcription under the control of the IGH promoter. In addition, about 10% of FL cases have 3q27 aberrancies at the BCL6 gene.36-38 More recently, cases of FL with bulky inguinal disease negative for IGH-BCL2 and BCL6 translocations were found to have 1p36 deletions. These 1p36-deleted FLs typically have a diffuse pattern and a good prognosis.39 For t(14;18), 3q27, or 1p36, FISH is a sensitive means for detecting these translocations, as is PCR for IGH-BCL2.40 There are reports that t(14;18) can be detected in a substantial fraction of otherwise healthy donors at levels and rates that depend on the type of detection test used.41-43 In addition, between one-fourth and one-third of de novo DLBCLs show t(14;18), and about one-third show BCL6 abnormalities at 3q27. Therefore, these genetic changes are not specific for FL and should not be used to subtype a lymphoma as follicular in origin.
Use of IGH-BCL2 as a marker for MRD is still controversial. Some studies have found that a postinduction and posttransplantation IGH-BCL2-positive finding by PCR predicted relapse.44,45 However, others studies have not found significance to postinduction IGH-BCL2 positivity.46 The NCCN guidelines recommend testing for IGH-BCL2 or BCL6 translocations or 1p36 deletion only if this testing is needed for diagnosis. The guidelines do not recommend using these genetic assays in follow-up biopsies, as the importance of treating early relapse has not been definitively demonstrated.
Therefore, if a lymphoma has morphologic, histologic, and IHC findings consistent with FL, then cytogenetic, FISH, or PCR testing is not needed for diagnosis but may be used as confirmation. Follow-up molecular and cytogenetic testing should be avoided if the original cytogenetic abnormality is unknown. That is, IGH-BCL2 FISH should be performed in follow-up samples only if the original lymphoma is known to contain the translocation. As follow-up genetic testing is of disputed clinical significance even in cases in which the original molecular change is known, the NCCN recommendations for therapy are no different. The HMG subcommittee does not recommend molecular or cytogenetic testing in FL beyond what is required for initial diagnosis.
T-Cell Lymphomas
Mature T-Cell Lymphoma and Leukemia
For mature T-cell lymphoma (TCL) and leukemia, the clinical and morphologic criteria have a very important role in the initial workup. However, IHC immunophenotyping is crucial for definitive diagnosis and subclassification. Flow cytometry is routinely used in diagnosing diseases such as T-cell prolymphocytic leukemia (TPLL), T-cell large granular lymphocytic (LGL) leukemia, and Sézary syndrome. T-cell clonality studies, preferably with BIOMED-II–validated primers against targets such as T-cell receptor
Significant advances in TCL classification have led to revisions and the inclusion of new provisional entities in the 2016 World Health Organization classification of lymphoid neoplasms.47 Many of these changes originated in studies of gene expression profiling and the genetic landscape of T-cell neoplasms. Even though subsets of peripheral TCL not otherwise specified (PTCL-NOS) have been recognized on the basis of phenotypic and molecular abnormalities with possible clinical implications, in most cases molecular testing is not part of routine practice. Typically, only a few cytogenetic abnormalities and genetic mutations are used in the evaluation of TCL and T-cell leukemia.
A group of T-cell lymphoproliferative disorders with expression of T follicular helper cell markers can be identified with IHC. These disorders include angioimmunoblastic TCL; follicular TCL, a new entity that is a PTCL-NOS subset; and primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder. The neoplastic cells should express at least 2 or 3 T follicular helper cell–related antigens, including CD279/PD1, CD10, BCL6, CXCL13, ICOS, SAP, and CCR5; the most commonly used are PD1, BCL6, and CD10. Recurrent fusion of ITK-SYK translocation t(5;9) or CTLA4-CD28 is also common in follicular TCL. Although recurrent mutation is found in these entities, conventional karyotyping or IHC should be sufficient for diagnosis.
Cutaneous γ -Δ T-Cell Lymphoma
Among cutaneous TCLs, primary cutaneous
Peripheral T-Cell Lymphoma
Gene expression profiling analysis of PTCLs has identified at least 3 subtypes characterized by overexpression of GATA3, TBX21, and cytotoxic genes and expression of the corresponding proteins with IHC.47 These subtypes are associated with different clinical behavior and therapy responses. The GATA3 subtype has an inferior prognosis and shows a high level of T helper type 2 cytokines, which can be identified with IHC. As IHC-stained GATA3 has been available as a marker of urothelial carcinoma at most IHC laboratories, GATA3 IHC staining also may be considered in the evaluation of PTCLs.
Many monoclonal antibody therapies are being used as primary or secondary regimens in the treatment of TCL. Clinical trials are working to establish their efficacy. If treatment with a monoclonal antibody is being considered, it is appropriate to conduct IHC to demonstrate the presence of the target antigen and at follow-up, to demonstrate the efficacy of treatment. These therapies include alemtuzumab, which targets CD52, and brentuximab, which targets CD30.
T-Cell Large Granular Lymphocytic Leukemia
T-cell LGL leukemia is a complex diagnosis that requires persistent clonal expansion of LGLs and clinically peripheral blood cytopenia. In many cases, the diagnosis is difficult to establish, as benign large granular lymphocytosis with clonal T cells may occur in conjunction with viral infections or autoimmune disorders. Somatic mutations in the STAT3 (signal transducer and activator of transcription 3) gene are found in 40% of patients with T-cell LGL leukemia.49 More recently, somatic mutations in the STAT5B gene were identified in 2% of T-cell LGL leukemia subsets. The clinical course of T-cell LGL leukemia in patients with the STAT5B mutation is aggressive and fatal, clearly different from the relatively favorable course of typical T-cell LGL leukemia.50 The HMG subcommittee recommends considering a STAT3 and STAT5B mutation study for selected cases in which it is difficult to distinguish true T-cell LGL leukemia from its reactive expansions.
T-Cell Prolymphocytic Leukemia
T-cell prolymphocytic leukemia (T-PLL) is a rare, aggressive disease and is most commonly associated with a prolymphocytic morphology and expression of CD4. However, since a specific immunophenotypic profile of T-PLL has not been identified, flow cytometry is not adequate in isolation for definitive classification as T-PLL.51 A diagnosis of T-PLL often requires cytogenetics or a FISH study to confirm a suspected case. Most TPLL cases harbor characteristic chromosomal abnormalities involving 14q11.2 (TCR
Anaplastic Large Cell Lymphoma
The World Health Organization recognizes 3 distinct types of anaplastic large cell lymphoma (ALCL): systemic anaplastic lymphoma kinase (ALK)–positive ALCL, systemic ALK-negative ALCL, and primary cutaneous ALCL. Systemic ALK-positive ALCLs consistently have ALK gene rearrangements and favorable outcomes. The most common translocation is the t(2;5) rearrangement of NPM1 and ALK, though other ALK partners are also possible. In contrast, systemic ALK-negative ALCLs lack ALK gene rearrangements and as a whole have outcomes inferior to those of systemic ALK-positive ALCLs. However, studies have found systemic ALK-negative ALCL to be a genetically and clinically heterogeneous entity.54 About 30% of cases have rearrangements of the DUSP22-IRF4 locus on 6p25.3 (DUSP22 rearrangement), and these cases have favorable outcomes similar to those of systemic ALK-positive ALCL.55 Only 8% of patients have TP63 rearrangements and very poor outcomes. The remaining cases lack ALK, DUSP22, and TP63 rearrangements and have intermediate outcomes. The HMG subcommittee recommends considering DUSP22 rearrangement by FISH in the evaluation of systemic ALK-negative ALCL.
Conclusion
The pathologic diagnosis, classification, and risk stratification of lymphoma and leukemia require an approach that integrates morphology, flow cytometry, cytogenetics, and molecular pathology. Rapidly evolving molecular techniques currently allow for detailed description of the molecular defects in lymphoma and leukemia, including driver mutations, amplification/deletion events, and clonal evolution. Unfortunately, the technical ability to catalogue the molecular defects in lymphoma and leukemia, often at great expense, is outpacing the ability to use this detailed information in treating patients with hematologic malignancies. The challenge, then, is to identify best practices for the diagnosis and classification of lymphoma and leukemia in VHA hospitals that incorporate the most useful molecular tests without wasting financial resources.
In this report, the HMG subcommittee of the MGPW has presented its recommendations for molecular testing in AML, MPN, MDS, and lymphomas in the context of standard morphologic and immunophenotypic approaches to hematopathology diagnosis and classification. Adoption of these recommendations by VHA hospitals and clinics should help ensure that all VA patients with hematologic malignancies benefit from the latest advances in precision medicine.
Within the vast and comprehensive national VHA health care system are multiple centers of expertise in hematopathology. In addition, multiple VA clinical molecular diagnostic laboratories are performing state-of-the-art testing. The HMG subcommittee proposes that, to make best use of these expert resources, the VHA should establish an interfacility hematopathology consultation service. This service would allow any VA pathologist to consult a board-certified hematopathologist regarding use of ancillary molecular genetic testing in the diagnosis of hematologic malignancy.
In addition, the HMG subcommittee recommends consolidating VA molecular diagnostic reference laboratories and having them perform molecular testing for other VA hospitals rather than using commercial reference laboratories, where testing standards are not uniform and results may be difficult to interpret. Several well-established VA clinical laboratories with technical expertise and informatics support are already performing selected molecular diagnostic testing. These laboratories’ resources should be expanded, where practical, to cost-effectively provide VA expertise to all veterans and to improve access to appropriate molecular diagnostic testing.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of
Click here to read the digital edition.
1. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793-795.
2. Matynia AP, Szankasi P, Shen W, Kelley TW. Molecular genetic biomarkers in myeloid malignancies. Arch Pathol Lab Med. 2015;139(5):594-601.
3. Wang ML, Bailey NG. Acute myeloid leukemia genetics: risk stratification and implications for therapy. Arch Pathol Lab Med. 2015;139(10):1215-1223.
4. Marum JE, Branford S. Current developments in molecular monitoring in chronic myeloid leukemia. Ther Adv Hematol. 2016;7(5):237-251.
5. Hughes TP, Saglio G, Kantarjian HM, et al. Early molecular response predicts outcomes in patients with chronic myeloid leukemia in chronic phase treated with frontline nilotinib or imatinib. Blood. 2014;123(9):1353-1360.
6. Pallera A, Altman JK, Berman E, et al. Guidelines insights: chronic myeloid leukemia, version 1.2017. J Natl Compr Canc Netw. 2016;14:1505-1512.
7. James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144-1148.
8. Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779-1790.
9. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008;112(1):141-149.
10. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood. 2008;112(3):844-847.
11. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391-2405.
12. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379-2390.
13. Harrison CN, Vannucchi AM. Closing the gap: genetic landscape of MPN. Blood. 2016;127(3):276-278.
14. Tefferi A, Barbui T. Essential thrombocythemia and polycythemia vera: focus on clinical practice. Mayo Clin Proc. 2015;90(9):1283-1293.
15. Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348(13):1201-1214.
16. Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130(3):607-612.e609.
17. Bain B, Billiland D, Horny H, Verdiman J. Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. Vol 2. Lyon, France: IRAC Press; 2008:68-73.
18. Wang SA, Tam W, Tsai AG, et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod Pathol. 2016;29(8):854-864.
19. Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781-1790.
20. Schanz J, Tüchler H, Solé F, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30(8):820-829.
21. Marshall D, Roboz GJ. Standardizing the initial evaluation for myelodysplastic syndromes. Curr Hematol Malig Rep. 2013;8(4):361-369.
22. Damm F, Chesnais V, Nagata Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122(18):3169-3177.
23. Malcovati L, Papaemmanuil E, Ambaglio I, et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood. 2014;124(9):1513-1521.
24. Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384-1395.
25. Patnaik MM, Hanson CA, Sulai NH, et al. Prognostic irrelevance of ring sideroblast percentage in World Health Organization-defined myelodysplastic syndromes without excess blasts. Blood. 2012;119(24):5674-5677.
26. Guièze R, Wu CJ. Genomic and epigenomic heterogeneity in chronic lymphocytic leukemia. Blood. 2015;126(4):445-453.
27. Jeromin S, Weissmann S, Haferlach C, et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia. 2014;28(1):108-117.
28. Del Giudice I, Rossi D, Chiaretti S, et al. NOTCH1 mutations in +12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of +12 CLL. Haematologica. 2012;97(3):437-441.
29. Weissmann S, Roller A, Jeromin S, et al. Prognostic impact and landscape of NOTCH1 mutations in chronic lymphocytic leukemia (CLL): a study on 852 patients. Leukemia. 2013;27(12):2393-2396.
30. Vegliante MC, Palomero J, Pérez-Galán P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood. 2013;121(12):2175-2185.
31. King RL, Gonsalves WI, Ansell SM, et al. Lymphoplasmacytic lymphoma with a non-IgM paraprotein shows clinical and pathologic heterogeneity and may harbor MYD88 L265P mutations. Am J Clin Pathol. 2016;145(6):843-851.
32. Insuasti-Beltran G, Gale JM, Wilson CS, Foucar K, Czuchlewski DR. Significance of MYD88 L265P mutation status in the subclassification of low-grade B-cell lymphoma/leukemia. Arch Pathol Lab Med. 2015;139(8):1035-1041.
33. Wang XJ, Kim A, Li S. Immunohistochemical analysis using a BRAF V600E mutation specific antibody is highly sensitive and specific for the diagnosis of hairy cell leukemia. Int J Clin Exp Pathol. 2014;7(7):4323-4328.
34. Nakamura T, Tateishi K, Niwa T, et al. Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathol Appl Neurobiol. 2016;42(3):279-290.
35. Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467-472.
36. Bosga-Bouwer AG, van Imhoff GW, Boonstra R, et al. Follicular lymphoma grade 3B includes 3 cytogenetically defined subgroups with primary t(14;18), 3q27, or other translocations: t(14;18) and 3q27 are mutually exclusive. Blood. 2003;101(3):1149-1154.
37. Gu K, Fu K, Jain S, et al. t(14;18)-negative follicular lymphomas are associated with a high frequency of BCL6 rearrangement at the alternative breakpoint region. Mod Pathol. 2009;22(9):1251-1257.
38. Katzenberger T, Ott G, Klein T, Kalla J, Müller-Hermelink HK, Ott MM. Cytogenetic alterations affecting BCL6 are predominantly found in follicular lymphomas grade 3B with a diffuse large B-cell component. Am J Pathol. 2004;165(2):481-490.
39. Katzenberger T, Kalla J, Leich E, et al. A distinctive subtype of t(14;18)-negative nodal follicular non-Hodgkin lymphoma characterized by a predominantly diffuse growth pattern and deletions in the chromosomal region 1p36. Blood. 2009;113(5):1053-1061.
40. Belaud-Rotureau MA, Parrens M, Carrere N, et al. Interphase fluorescence in situ hybridization is more sensitive than BIOMED-2 polymerase chain reaction protocol in detecting IGH-BCL2 rearrangement in both fixed and frozen lymph node with follicular lymphoma. Hum Pathol. 2007;38(2):365-372.
41. Limpens J, Stad R, Vos C, et al. Lymphoma-associated translocation t(14;18) in blood B cells of normal individuals. Blood. 1995;85(9):2528-2536.
42. Schmitt C, Balogh B, Grundt A, et al. The bcl-2/IgH rearrangement in a population of 204 healthy individuals: occurrence, age and gender distribution, breakpoints, and detection method validity. Leuk Res. 2006;30(6):745-750.
43. Summers KE, Goff LK, Wilson AG, Gupta RK, Lister TA, Fitzgibbon J. Frequency of the Bcl-2/IgH rearrangement in normal individuals: implications for the monitoring of disease in patients with follicular lymphoma. J Clin Oncol. 2001;19(2):420-424.
44. Galimberti S, Luminari S, Ciabatti E, et al. Minimal residual disease after conventional treatment significantly impacts on progression-free survival of patients with follicular lymphoma: the FIL FOLL05 trial. Clin Cancer Res. 2014;20(24):6398-6405.
45. Ladetto M, Lobetti-Bodoni C, Mantoan B, et al; Fondazione Italiana Linfomi. Persistence of minimal residual disease in bone marrow predicts outcome in follicular lymphomas treated with a rituximab-intensive program. Blood. 2013;122(23):3759-3766.
46. van Oers MHJ, Tönnissen E, Van Glabbeke M, et al. BCL-2/IgH polymerase chain reaction status at the end of induction treatment is not predictive for progression-free survival in relapsed/resistant follicular lymphoma: results of a prospective randomized EORTC 20981 phase III intergroup study. J Clin Oncol. 2010;28(13):2246-2252.
47. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390.
48. Dewar R, Andea AA, Guitart J, Arber DA, Weiss LM. Best practices in diagnostic immunohistochemistry: workup of cutaneous lymphoid lesions in the diagnosis of primary cutaneous lymphoma. Arch Pathol Lab Med. 2015;139(3):338-350.
49. Koskela HLM, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366(20):1905-1913.
50. Rajala HL, Eldfors S, Kuusanmäki H, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121(22):4541-4550.
51. Chen X, Cherian S. Immunophenotypic characterization of T-cell prolymphocytic leukemia. Am J Clin Pathol. 2013;140(5):727-735.
52. Delgado P, Starshak P, Rao N, Tirado C. A comprehensive update on molecular and cytogenetic abnormalities in T-cell prolymphocytic leukemia (T-PLL). J Assoc Genet Technol. 2012;38(4):193-198.
53. Stengel A, Kern W, Zenger M, et al. Genetic characterization of T-PLL reveals two major biologic subgroups and JAK3 mutations as prognostic marker. Genes Chromosomes Cancer. 2016;55(1):82-94.
54. Thompson MA, Stumph J, Henrickson SE, et al. Differential gene expression in anaplastic lymphoma kinase-positive and anaplastic lymphoma kinase-negative anaplastic large cell lymphomas. Hum Pathol. 2005;36(5):494-504.
55. King R, Dao L, McPhail E, et al. Morphologic features of ALK-negative anaplastic large cell lymphomas with DUSP22 rearrangements. Am J Surg Pathol. 2016;40(1):36-43.
In January 2015, President Obama introduced the Precision Medicine Initiative, a program set up to identify new biomedical discoveries for the development of a personalized knowledge base of disease entities and individualized treatments. Advances in precision medicine typically involve the use of targeted therapies tailored to individual genetic characteristics identified with molecular testing. The goals are to improve survival and reduce adverse effects. With an initial budget of $215 million, this initiative presented a unique opportunity to combine efforts in genomic discovery, bioinformatic analysis, and health information technology to move toward data-driven, evidence-based precision medicine.1
The VHA is the largest comprehensive health care system in the U. S. and has more than 1,700 care sites serving nearly 9 million veterans each year. The budget for this single-payer system is proposed by the President and approved by Congress. As the VHA must treat a diverse and aging veteran population in an environment of rising costs and budget constraints, limited resources must be monitored and appropriated for the most cost-effective health care delivery. Precision medicine offers a model in which physicians can select the most appropriate diagnostic tests in defined clinical settings to direct clinical care. It supports the testing needed to subdivide each disease category into distinct subcategories. Nevertheless, the need for fiscal responsibility in a capitated health care system recommends testing in cases in which it can change therapy or prognosis rather than for purely academic reasons.
Pathology and Laboratory Medicine Service
Given limited resources and an increasing number of requests for advanced molecular testing, the VA Pathology and Laboratory Medicine Service (P&LMS) formed the Molecular Genetics Pathology Workgroup (MGPW) in September 2013. The charter listed the tasks of the MGPW to “provide recommendations on how to effectively use molecular genetics tests, promote increased quality and availability of testing within the VHA, encourage internal referral testing, provide an organizational structure for Molecular Genetics Testing Consortia, and create a P&LMS policy for molecular genetic testing in general, specifically addressing the issues surrounding laboratory developed testing.” The MGPW has 4 subcommittees: molecular oncology, pharmacogenetics, hematopathology molecular genetics (HMG), and genetic medicine. Since its inception, the HMG subcommittee has had several objectives:
- Standardize the molecular testing nomenclature for and develop practice guidelines for acute myeloid leukemia (AML), myeloproliferative neoplasms (MPN), myelodysplastic syndrome (MDS), chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma, lymphoma, and plasma cell neoplasms;
- Develop standardized reporting guidelines for current VA molecular laboratories;
- Identify new tests as they are being reported in the literature and collaborate with hematology and oncology services to evaluate the clinical utility of these tests for VA patients;
- Network current VA molecular laboratories, perform fact-finding for these laboratories, and compile test menus; and
- Assess for the formation of VA-wide interfacility consultation services for hematopathology so that all VA facilities, regardless of their complexity, will be able to access the expertise of hematopathology-trained pathologists (Appendix).
The HMG subcommittee met monthly and discussed various diagnostic entities in hematopathology. For hematolymphoid malignancies, it was generally agreed that the traditional laboratory tools of morphology, flow cytometry, and immunohistochemistry (IHC) are standard in initial assessment and often in diagnosis. As the clinical molecular and cytogenetic assays of karyotype, fluorescence in situ hybridization (FISH), advanced DNA sequencing, microarray, and highly sensitive polymerase chain reaction (PCR) analysis affect diagnosis, subclassification, minimal residual disease (MRD) monitoring, prognosis, and therapy selection, their use is marked by a high degree of variability. As a result, standardization is needed. As each laboratory develops and reports ancillary testing, the variable reporting formats may generate postanalytic errors.
A detailed description of all molecular methodologies is beyond the scope of this article. For practicing pathologists, challenges remain in overall cost and reimbursement, extensive and time-consuming data analysis, and in some cases, interpretation differences.
Myeloid Neoplasms
Myeloid malignancies were divided into AML, MPN, and MDS. Next-generation sequencing (NGS) information for these malignancies was used to identify various contributory functional categories, including cell signaling (FLT3, KIT, JAK2, MPL, KRAS/NRAS, PTPN11, NF1, CSF3R); transcription (CEBPA, RUNX1, GATA1/GATA2, PHF6, ETV6); splicing (SF3B1, SRSF2, ZRSR2, U2AF1); epigenetics (DNMT3A, TET2, IDH1/IDH2, ASXL1, EZH2, SUZ12, KDM6A); cohesin complex (STAG2, SMC1A, SMC3, RAD21); and cell cycle (TP53, NPM1).2
Acute Myeloid Leukemia
The HMG subcommittee reviewed the literature on prognostically significant genes in myeloid leukemias. Karyotype abnormalities, such as t(8;21) and inv(16), collectively known as the core-binding factor (CBF) leukemias, t(15;17), t(11q23) (KMT2A/MLL), and so forth, are recurrent lesions in AML. Included in the minimum set of genes recommended by the National Comprehensive Cancer Network (NCCN) for AML prognosis evaluation are nucleolar protein nucleophosmin (NPM1), CCAAT/enhancer-binding protein
Some of the chromosomal translocations, such as inv(16)/t(16;16) in AML and t(15;17) in acute promyelocytic leukemia, can be monitored with FISH or reverse transcription–PCR (RT-PCR) analysis. As NPM1 mutations tend to be seen in recurrence, they can be used as molecular markers for MRD. Other mutations that provide important prognostic information in AML include:
- Activating insertions/duplications in the FLT3 receptor tyrosine kinase, which can be detected with PCR sizing assays;
- Mutations in the KIT receptor tyrosine kinase, which can be detected with DNA sequencing or more limited hotspot PCR;
- Mutations in the DNA methyltransferase, DNMT3A, a poor prognostic indicator seen in 22% of cases of AML, also detected with gene sequencing or more limited hotspot PCR; and
- Another set of genes, TET2, IDH1, IDH2, KRAS, NRAS, EZH2, and ASXL1, is mutated in MPN as well as AML and MDS, making a common molecular panel with next-generation sequencing useful in diagnosing and risk-stratifying all myeloid neoplasms.
The HMG subcommittee agreed that, for de novo AML, chromosomal karyotype is the standard of care, necessary in detecting known cytogenetic abnormalities as well as a wide range of lesions that might indicate a diagnosis of AML with myelodysplasia-related changes at time of diagnosis. In addition, molecular analysis of FLT3 is useful in determining prognosis, and CEBPA (biallelic) and NPM1 mutations are good prognostic factors in normal-karyotype AML. KMT2A (MLL) rearrangements should be tested with FISH if the lineage is ambiguous. The PML-RARA fusion gene also should be tested with FISH if morphologic and flow cytometry results suggest acute promyelocytic leukemia (Table). At this time, testing for TP53, DNMT3A, RAS, and other such mutations is not recommended because it is not cost-effective for the VA.
Myeloproliferative Neoplasms
Myeloproliferative neoplasms are clonal hematopoietic stem cell disorders characterized by proliferation of at least 1 myeloid lineage: granulocytic, erythroid, or megakaryocytic. Myeloproliferative neoplasms show a range of recurrent chromosomal translocations, such as BCR-ABL1 fusion in chronic myelogenous leukemia (CML) that can be detected with RT-PCR analysis as well as FISH. In CML, BCR-ABL1 fusion transcript levels detected by a quantitative PCR (qPCR) method are now used to monitor the course of CML therapy with tyrosine kinase inhibitors (TKIs) and to trigger a treatment change in drug-resistant cases. Given the importance of qPCR in clinical management, significant progress has been made in standardizing both the PCR protocol and the reference materials used to calibrate the BCR-ABL1 PCR assay. BCR-ABL1–negative MPN, including polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF), are most commonly associated with mutations in the tyrosine kinase JAK2. Mutations in CALR and MPL are seen in a subset of patients with ET and PMF as well, whereas PV is essentially exclusively a disease of JAK2 mutations.
Chronic myelogenous leukemia is the prototypical MPN. To establish the initial diagnosis, FISH and/or qPCR for BCR-ABL1 fusion should be used. If CML is confirmed, the sample can be reflexed to qPCR BCR-ABL1 on the initial peripheral blood and/or bone marrow sample(s) to establish the patient’s baseline. In addition, a bone marrow sample (aspirate) should be used for a complete karyotype and for morphologic confirmation of disease phase.
For follow-up assessment of CML patients’ response to TKI treatment, qPCR for BCR-ABL1 should be tested with a peripheral blood sample or a bone marrow sample every 3 months.4 A peripheral blood sample is more commonly used because it is conveniently obtained. Early molecular response as indicated by a BCR-ABL1 transcript ratio of < 10% on the International Scale at 3 months, has a strong prognostic value.5 Major molecular response as indicated by a BCR-ABL1 transcript ratio of < 0.1% on the International Scale at 12 to 18 months is also highly prognostic.5
After the peripheral blood sample becomes negative for BCR-ABL1 by qPCR, testing bone marrow samples may be considered. If important treatment response benchmarks are not achieved, or response is lost with rising BCR-ABL1 levels (TKI resistance), ABL1 kinase domain mutation analysis as well as repeat FISH (to assess for copy number multiplication) should be performed to guide further management. Patients with the ABL1 T315I mutation are resistant to all first-line TKIs but may respond to later third-generation TKIs.6
BCR-ABL1–negative MPNs include PV, ET, and PMF. Bone marrow morphology remains the cornerstone of ET and PMF diagnosis. The discovery of JAK2, CALR, and MPL mutations has contributed to how these disorders are diagnosed.7-12 Besides providing the clonality proof that is crucial for diagnosis, the molecular markers influence the prognosis. The JAK2 (p.V617F) or less common JAK2 exon 12 mutations, which are detected in more than 95% of PV cases, are used as molecular markers to confirm diagnosis.7 Further, the JAK2 (p.V617F), CALR (exon 9), and MPL (exon 10) mutations are detected in ET (~60%, 25%, and 3%-5%, respectively) and PMF (~55%, 30%, and 5%, respectively).12 If ET or PMF is suspected clinically, first JAK2 (p.V617F) mutation analysis should be performed, then CALR mutation analysis, and finally MPL mutation analysis. Although novel gain-of-function JAK2 and MPL mutations were recently discovered in triple-negative ET (negative for canonical mutations in JAK2, CALR, and MPL) and PMF by whole exome sequencing,13 clinical testing is not readily available. Besides its utility in the initial diagnosis of ET and PMF, the JAK2 or CALR mutation assay also may be considered for bone marrow transplantation follow-up (Table).14
Despite the continuing debate on the classification of eosinophilic myeloid disorders, the discovery of the FIP1L1-PDGFRA fusion represents a major milestone in the understanding of these disorders.15,16 Unlike PDGFRB (5q33) and FGFR1 (8p11) rearrangements, which can be detected with routine chromosomal analysis (cytogenetics), the cryptic FIP1L1-PDGFRA fusion must be detected with FISH (for CHIC2 deletion) or RT-PCR analysis. It should be pointed out that, as most eosinophilia is reactive or secondary, molecular testing for FIP1L1-PDGFRA fusion is indicated only when primary hypereosinophilia or hypereosinophilic syndrome (HES) is suspected. This is particularly the case in the following hypereosinophilia accompanying conditions: CML-like morphology, but BCR-ABL1–negative; chronic myelomonocytic leukemia (CMML)–like morphology with a normal karyotype; and new onset of cardiac damage or dysfunction.17
Primary eosinophilic myeloid disorders with PDGFRA or PDGFRB rearrangements can be treated with TKIs (eg, imatinib). Next-generation sequencing may be considered in cases of presumed HES when there is no identifiable karyotypic or FISH abnormality. Recent studies have found that cases of HES with somatic mutations indicating clonality had adverse clinical outcomes similar to those of cases of chronic eosinophilic leukemia.18
The discovery of CSF3R mutations offers a new molecular marker for the diagnosis of chronic neutrophilic leukemia (CNL), an MPN.19 The CSF3R (p.T618I) mutation or another activating CSF3R mutation is now used as a diagnostic criterion for CNL. Identification of specific CSF3R mutations may have therapeutic implications as well. The test should be ordered only for patients with clinical and morphologic findings suggestive of CNL; reactive neutrophilic leukocytosis (eg, infection, inflammation) should be ruled out before the test is ordered.
Myelodysplastic Syndrome
Myelodysplastic syndrome is a group of clonal bone marrow disorders characterized by ineffective hematopoiesis, manifested by morphologic dysplasia in ≥ 1 hematopoietic lineages and peripheral cytopenias (hemoglobin level, < 10 g/dL; platelet count, < 100×103/µL; absolute neutrophil count, < 1.8×103/µL). Diagnosis and classification of MDS depend mainly on the degree of morphologic dysplasia and blast percentages, as determined by examining well-prepared cellular bone marrow aspirate smears and/or biopsy touch preparations and peripheral blood smears.
Conventional karyotyping is an essential part of the diagnostic workup for all presumptive cases of MDS and is of both diagnostic and prognostic importance.20 About 60% of MDS cases have recurrent cytogenetic abnormalities, which can be detected with conventional karyotyping. If a high-quality cytogenetic analysis cannot be performed (eg, the bone marrow sample is inadequate), or if quick turnaround is required, an alternative FISH panel may be used to detect some of the common MDS-associated chromosomal abnormalities (eg, 5q deletion, 7q deletion/monosomy 7, +8, 20q deletion).21 Sequencing with FISH also can be useful for assessing MRD by detecting a previously identified chromosomal abnormality.
Targeted sequencing of a limited number of genes can detect mutations in the vast majority of patients with MDS. The most commonly mutated genes in MDS are SF3B1, TET2, SRSF2, ASXL1, DNMT3A, RUNX1, U2AF1, TP53, and EZH2. Mutations in SRSF2 cause RNA splicing abnormalities. In addition, mutations in TP53, EZH2, RUNX1, and ASXL1 are associated with poor prognosis,22,23 whereas mutations in SF3B1 confer better event-free survival.24 Despite these developments, the HMG subcommittee agreed that NGS-based mutation panels are not cost-effective for the VA population at this time and should not be included in a MDS workup. Only in rare situations and when clinically indicated (to change disease classification or patient management) should evaluation for specific gene mutations be considered—for instance, the SF3B1 mutation for patients with probable MDS with ring sideroblasts, if ring sideroblasts are < 15%.25
Myelodysplastic/Myeloproliferative Neoplasms
Myelodysplastic/myeloproliferative neoplasms are a group of myeloid neoplasms with clinical, laboratory, and morphologic features that overlap both MDS and MPN. In MDS/MPN, the karyotype is often normal or shows abnormalities in common with MDS.
In cases of unexplained monocytosis for which there is clinical concern for CMML, morphologic evaluation and conventional chromosomal karyotyping should be performed after other secondary causes and known myeloproliferative and myelodysplastic entities have been excluded. If concomitant hypereosinophilia is present and the karyotype is normal, FISH or PCR-based assay should be performed to rule out FIP1L1-PDGFRA rearrangements. BCR-ABL1, PDGFRB, FGFR1, and t(8;9)/PCM1-JAK2 rearrangements typically are detected with high-quality cytogenetic analysis and thus do not require targeted molecular assays. Although certain gene mutations (eg, SRSF2, TET2, ASXL1, CBL) are commonly detected in CMML, the HMG subcommittee does not recommend sequencing-based mutation panels, as there is insufficient information for testing for prognostic or treatment stratification.
If MDS/MPN with ring sideroblasts and thrombocytosis is suspected on the basis of the clinical and morphologic criteria, molecular tests for the JAK2 (p.V617F) and SF3B1 mutations may be considered in an effort to help confirm the diagnosis.
Atypical CML is a rare MDS/MPN subtype that is now better characterized molecularly with SETBP1 and/or ETNK1 mutations, which are detectable in up to a third of cases. If clinical suspicion is high, sequencing may be diagnostically helpful.
Lymphoid Neoplasms
Chronic Lymphocytic Leukemia
In CLL, recurrent chromosomal abnormalities (eg, deletions of 13q, trisomy 12, deletions of 11q, deletions of 17p) have clear prognostic value and can be detected with FISH. Other prognostic information, such as somatic mutation of immunoglobulin heavy chain variable (IgHV) genes, TP53 mutations, SF3B1, and NOTCH1 mutation, are mostly derived from PCR-based assays. The discovery of recurrently mutated genes in CLL has increased with the use of highly sensitive sequencing methods constructing a more detailed landscape of CLL at genetic, epigenetic, and cellular levels. A recent literature review summarizes the vast heterogeneity of CLL with recurrent pathogenetic findings in MYD88, SF3B1, TP53, ATM, and NOTCH1 signaling pathways.26 The treatment of CLL is rapidly evolving, and many clinical trials are proposing a change from the “watch and wait” paradigm to treatment upon initial presentation based on molecular findings. Additional testing based on new treatment options from current clinical trials will be recommended.
Flow cytometry and morphology are standard for CLL diagnosis. The HMG subcommittee recommends FISH for del(13q14), del(11q), trisomy 12, and del(17p) at time of diagnosis or immediately before therapy initiation. Zeta-chain (
Other B-Cell Lymphoproliferative Disorders
Unlike the common molecular changes in CLL, in other mature B-cell lymphomas, chromosomal translocations that juxtapose a variety of different oncogenes next to an Ig gene enhancer usually are—and those that switch regions less commonly are—important initiating events that can be detected with PCR, DNA sequencing, or FISH. In follicular lymphoma (FL), Burkitt lymphoma, marginal zone lymphoma (MZL), and mantle cell lymphoma (MCL), these oncogenes driven by an Ig gene enhancer typically include BCL2, MYC, MALT1, and CCND1 (cyclin D1), respectively. Molecular variants of these lymphomas that lack these classical translocations often activate homologous genes (eg, cyclin D3/CCND3 is activated in variants of MCL).
Morphology, flow cytometry, and IHC are routinely used for diagnosis. In inconclusive cases, Ig gene rearrangement by PCR may be used. The Table summarizes common molecular changes in B-cell lymphomas.
Mantle cell lymphoma. MCL is a non-Hodgkin lymphoma subtype characterized by t(11;14) (q13;q32) translocations that in the majority of cases lead to overexpression of cyclin D1 (BCL1). Recent molecular profiling has identified an MCL variant that is cyclin D1–negative but SOX11-positive and may have a more aggressive clinical course.30SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive MCL.
Lymphoplasmacytic lymphoma. Lymphoplasmacytic lymphoma (LPL), MZL, and CLL/small lymphocytic lymphoma are well-defined clinicopathologic entities. However, distinguishing LPL from MZL and atypical cases of CLL can sometimes be difficult because of overlapping clinical and morphologic features. Recent studies have identified a recurrent L265P mutation in the MYD88 gene in 90% to 95% of LPL cases with IgM paraprotein and in 40% to 50% of the rare non-IgM LPL cases. In contrast, the mutation is much less frequently present in MZL and other low-grade B-cell neoplasms (2%-7%).31 Therefore, testing for this abnormality can be a diagnostic aid in these difficult-to-classify cases. In addition, from a therapeutic perspective, presence or absence of MYD88 mutation may prove more significant than presence of a specific paraprotein or histopathologic features. Ibrutinib has shown efficacy in LPL and demonstrates improved response rates in patients with MYD88 mutation compared with that of their mutation-negative counterparts.32 Several MYD88 inhibitors are in clinical trials. This again indicates the need to more accurately identify and subclassify these non-IgM LPL cases to ensure appropriate molecular evaluation.
Hairy cell leukemia. Flow cytometry and morphology are usually sufficient for a hairy cell leukemia (HCL) diagnosis. However, rare cases are difficult to distinguish variant HCL from other mimics. The BRAF V600E mutation recently was described as a disease-defining molecular marker for HCL—present in nearly all HCL cases but virtually absent in HCL mimics. Therefore, detection of the BRAF mutation by IHC stain with specific antibody or PCR analysis is highly sensitive and specific for the diagnosis of HCL.33
Diffuse large B-cell lymphoma. Recent molecular analysis has created various risk stratification schemata for diffuse large B-cell lymphoma (DLBCL). The HGM subcommittee agrees that well-preserved morphology, IHC, flow cytometry, and FISH-specific markers (BCL2, BCL6, cMYC) are sufficient for diagnostic, prognostic, and therapeutic purposes. Although a wide range of genes have been implicated in the pathogenesis of DLBCL, sequencing and gene expression profiling are not cost-effective at this time and do not add benefit to patient treatment.
The MYD88 L265P mutation has been identified in DLBCL, particularly the activated B-cell-like type and primary central nervous system lymphoma (PCNSL), and may have implications for ibrutinib therapy. PCNSL commonly manifests aggressive clinical behavior and has a poor prognosis. It has been proposed that the MYD88 mutation can be used as a genetic hallmark for PCNSL to distinguish CNS involvement by systemic DLBCL from PCNSL.34
Plasma cell neoplasms. Flow cytometry is acceptable for the diagnosis of plasma cell neoplasms and for residual disease follow-up. Chromosomal karyotype or FISH for IGH/CCND1, IGH/MMSET, and IGH/CMAF dual fusion probes is recommended in conjunction with morphology, IHC, and flow cytometry. In plasma cell myeloma, several genetic mutations can be detected with NGS, including mutations in NRAS, KRAS, TP53, BCL7A, DIS3, and FAM46C.35 Less commonly, BRAF mutations, previously described in melanoma and several other solid tumors, can be detected with DNA sequencing in 4% of multiple myeloma cases, which may prove promising for targeted therapy with BRAF inhibitors. However, current therapeutic decisions are based on genetic and clinical factors, and sequence-based assays are not recommended at this time.
Follicular lymphoma. Cytology, histology, and IHC typically are sufficient for diagnosing FL. In difficult-to-diagnose cases and in cases with scant material, additional tests may help with diagnosis. Eighty to ninety percent of FL cases have t(14;18)(q32;q21), which places the BCL2 gene transcription under the control of the IGH promoter. In addition, about 10% of FL cases have 3q27 aberrancies at the BCL6 gene.36-38 More recently, cases of FL with bulky inguinal disease negative for IGH-BCL2 and BCL6 translocations were found to have 1p36 deletions. These 1p36-deleted FLs typically have a diffuse pattern and a good prognosis.39 For t(14;18), 3q27, or 1p36, FISH is a sensitive means for detecting these translocations, as is PCR for IGH-BCL2.40 There are reports that t(14;18) can be detected in a substantial fraction of otherwise healthy donors at levels and rates that depend on the type of detection test used.41-43 In addition, between one-fourth and one-third of de novo DLBCLs show t(14;18), and about one-third show BCL6 abnormalities at 3q27. Therefore, these genetic changes are not specific for FL and should not be used to subtype a lymphoma as follicular in origin.
Use of IGH-BCL2 as a marker for MRD is still controversial. Some studies have found that a postinduction and posttransplantation IGH-BCL2-positive finding by PCR predicted relapse.44,45 However, others studies have not found significance to postinduction IGH-BCL2 positivity.46 The NCCN guidelines recommend testing for IGH-BCL2 or BCL6 translocations or 1p36 deletion only if this testing is needed for diagnosis. The guidelines do not recommend using these genetic assays in follow-up biopsies, as the importance of treating early relapse has not been definitively demonstrated.
Therefore, if a lymphoma has morphologic, histologic, and IHC findings consistent with FL, then cytogenetic, FISH, or PCR testing is not needed for diagnosis but may be used as confirmation. Follow-up molecular and cytogenetic testing should be avoided if the original cytogenetic abnormality is unknown. That is, IGH-BCL2 FISH should be performed in follow-up samples only if the original lymphoma is known to contain the translocation. As follow-up genetic testing is of disputed clinical significance even in cases in which the original molecular change is known, the NCCN recommendations for therapy are no different. The HMG subcommittee does not recommend molecular or cytogenetic testing in FL beyond what is required for initial diagnosis.
T-Cell Lymphomas
Mature T-Cell Lymphoma and Leukemia
For mature T-cell lymphoma (TCL) and leukemia, the clinical and morphologic criteria have a very important role in the initial workup. However, IHC immunophenotyping is crucial for definitive diagnosis and subclassification. Flow cytometry is routinely used in diagnosing diseases such as T-cell prolymphocytic leukemia (TPLL), T-cell large granular lymphocytic (LGL) leukemia, and Sézary syndrome. T-cell clonality studies, preferably with BIOMED-II–validated primers against targets such as T-cell receptor
Significant advances in TCL classification have led to revisions and the inclusion of new provisional entities in the 2016 World Health Organization classification of lymphoid neoplasms.47 Many of these changes originated in studies of gene expression profiling and the genetic landscape of T-cell neoplasms. Even though subsets of peripheral TCL not otherwise specified (PTCL-NOS) have been recognized on the basis of phenotypic and molecular abnormalities with possible clinical implications, in most cases molecular testing is not part of routine practice. Typically, only a few cytogenetic abnormalities and genetic mutations are used in the evaluation of TCL and T-cell leukemia.
A group of T-cell lymphoproliferative disorders with expression of T follicular helper cell markers can be identified with IHC. These disorders include angioimmunoblastic TCL; follicular TCL, a new entity that is a PTCL-NOS subset; and primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder. The neoplastic cells should express at least 2 or 3 T follicular helper cell–related antigens, including CD279/PD1, CD10, BCL6, CXCL13, ICOS, SAP, and CCR5; the most commonly used are PD1, BCL6, and CD10. Recurrent fusion of ITK-SYK translocation t(5;9) or CTLA4-CD28 is also common in follicular TCL. Although recurrent mutation is found in these entities, conventional karyotyping or IHC should be sufficient for diagnosis.
Cutaneous γ -Δ T-Cell Lymphoma
Among cutaneous TCLs, primary cutaneous
Peripheral T-Cell Lymphoma
Gene expression profiling analysis of PTCLs has identified at least 3 subtypes characterized by overexpression of GATA3, TBX21, and cytotoxic genes and expression of the corresponding proteins with IHC.47 These subtypes are associated with different clinical behavior and therapy responses. The GATA3 subtype has an inferior prognosis and shows a high level of T helper type 2 cytokines, which can be identified with IHC. As IHC-stained GATA3 has been available as a marker of urothelial carcinoma at most IHC laboratories, GATA3 IHC staining also may be considered in the evaluation of PTCLs.
Many monoclonal antibody therapies are being used as primary or secondary regimens in the treatment of TCL. Clinical trials are working to establish their efficacy. If treatment with a monoclonal antibody is being considered, it is appropriate to conduct IHC to demonstrate the presence of the target antigen and at follow-up, to demonstrate the efficacy of treatment. These therapies include alemtuzumab, which targets CD52, and brentuximab, which targets CD30.
T-Cell Large Granular Lymphocytic Leukemia
T-cell LGL leukemia is a complex diagnosis that requires persistent clonal expansion of LGLs and clinically peripheral blood cytopenia. In many cases, the diagnosis is difficult to establish, as benign large granular lymphocytosis with clonal T cells may occur in conjunction with viral infections or autoimmune disorders. Somatic mutations in the STAT3 (signal transducer and activator of transcription 3) gene are found in 40% of patients with T-cell LGL leukemia.49 More recently, somatic mutations in the STAT5B gene were identified in 2% of T-cell LGL leukemia subsets. The clinical course of T-cell LGL leukemia in patients with the STAT5B mutation is aggressive and fatal, clearly different from the relatively favorable course of typical T-cell LGL leukemia.50 The HMG subcommittee recommends considering a STAT3 and STAT5B mutation study for selected cases in which it is difficult to distinguish true T-cell LGL leukemia from its reactive expansions.
T-Cell Prolymphocytic Leukemia
T-cell prolymphocytic leukemia (T-PLL) is a rare, aggressive disease and is most commonly associated with a prolymphocytic morphology and expression of CD4. However, since a specific immunophenotypic profile of T-PLL has not been identified, flow cytometry is not adequate in isolation for definitive classification as T-PLL.51 A diagnosis of T-PLL often requires cytogenetics or a FISH study to confirm a suspected case. Most TPLL cases harbor characteristic chromosomal abnormalities involving 14q11.2 (TCR
Anaplastic Large Cell Lymphoma
The World Health Organization recognizes 3 distinct types of anaplastic large cell lymphoma (ALCL): systemic anaplastic lymphoma kinase (ALK)–positive ALCL, systemic ALK-negative ALCL, and primary cutaneous ALCL. Systemic ALK-positive ALCLs consistently have ALK gene rearrangements and favorable outcomes. The most common translocation is the t(2;5) rearrangement of NPM1 and ALK, though other ALK partners are also possible. In contrast, systemic ALK-negative ALCLs lack ALK gene rearrangements and as a whole have outcomes inferior to those of systemic ALK-positive ALCLs. However, studies have found systemic ALK-negative ALCL to be a genetically and clinically heterogeneous entity.54 About 30% of cases have rearrangements of the DUSP22-IRF4 locus on 6p25.3 (DUSP22 rearrangement), and these cases have favorable outcomes similar to those of systemic ALK-positive ALCL.55 Only 8% of patients have TP63 rearrangements and very poor outcomes. The remaining cases lack ALK, DUSP22, and TP63 rearrangements and have intermediate outcomes. The HMG subcommittee recommends considering DUSP22 rearrangement by FISH in the evaluation of systemic ALK-negative ALCL.
Conclusion
The pathologic diagnosis, classification, and risk stratification of lymphoma and leukemia require an approach that integrates morphology, flow cytometry, cytogenetics, and molecular pathology. Rapidly evolving molecular techniques currently allow for detailed description of the molecular defects in lymphoma and leukemia, including driver mutations, amplification/deletion events, and clonal evolution. Unfortunately, the technical ability to catalogue the molecular defects in lymphoma and leukemia, often at great expense, is outpacing the ability to use this detailed information in treating patients with hematologic malignancies. The challenge, then, is to identify best practices for the diagnosis and classification of lymphoma and leukemia in VHA hospitals that incorporate the most useful molecular tests without wasting financial resources.
In this report, the HMG subcommittee of the MGPW has presented its recommendations for molecular testing in AML, MPN, MDS, and lymphomas in the context of standard morphologic and immunophenotypic approaches to hematopathology diagnosis and classification. Adoption of these recommendations by VHA hospitals and clinics should help ensure that all VA patients with hematologic malignancies benefit from the latest advances in precision medicine.
Within the vast and comprehensive national VHA health care system are multiple centers of expertise in hematopathology. In addition, multiple VA clinical molecular diagnostic laboratories are performing state-of-the-art testing. The HMG subcommittee proposes that, to make best use of these expert resources, the VHA should establish an interfacility hematopathology consultation service. This service would allow any VA pathologist to consult a board-certified hematopathologist regarding use of ancillary molecular genetic testing in the diagnosis of hematologic malignancy.
In addition, the HMG subcommittee recommends consolidating VA molecular diagnostic reference laboratories and having them perform molecular testing for other VA hospitals rather than using commercial reference laboratories, where testing standards are not uniform and results may be difficult to interpret. Several well-established VA clinical laboratories with technical expertise and informatics support are already performing selected molecular diagnostic testing. These laboratories’ resources should be expanded, where practical, to cost-effectively provide VA expertise to all veterans and to improve access to appropriate molecular diagnostic testing.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of
Click here to read the digital edition.
In January 2015, President Obama introduced the Precision Medicine Initiative, a program set up to identify new biomedical discoveries for the development of a personalized knowledge base of disease entities and individualized treatments. Advances in precision medicine typically involve the use of targeted therapies tailored to individual genetic characteristics identified with molecular testing. The goals are to improve survival and reduce adverse effects. With an initial budget of $215 million, this initiative presented a unique opportunity to combine efforts in genomic discovery, bioinformatic analysis, and health information technology to move toward data-driven, evidence-based precision medicine.1
The VHA is the largest comprehensive health care system in the U. S. and has more than 1,700 care sites serving nearly 9 million veterans each year. The budget for this single-payer system is proposed by the President and approved by Congress. As the VHA must treat a diverse and aging veteran population in an environment of rising costs and budget constraints, limited resources must be monitored and appropriated for the most cost-effective health care delivery. Precision medicine offers a model in which physicians can select the most appropriate diagnostic tests in defined clinical settings to direct clinical care. It supports the testing needed to subdivide each disease category into distinct subcategories. Nevertheless, the need for fiscal responsibility in a capitated health care system recommends testing in cases in which it can change therapy or prognosis rather than for purely academic reasons.
Pathology and Laboratory Medicine Service
Given limited resources and an increasing number of requests for advanced molecular testing, the VA Pathology and Laboratory Medicine Service (P&LMS) formed the Molecular Genetics Pathology Workgroup (MGPW) in September 2013. The charter listed the tasks of the MGPW to “provide recommendations on how to effectively use molecular genetics tests, promote increased quality and availability of testing within the VHA, encourage internal referral testing, provide an organizational structure for Molecular Genetics Testing Consortia, and create a P&LMS policy for molecular genetic testing in general, specifically addressing the issues surrounding laboratory developed testing.” The MGPW has 4 subcommittees: molecular oncology, pharmacogenetics, hematopathology molecular genetics (HMG), and genetic medicine. Since its inception, the HMG subcommittee has had several objectives:
- Standardize the molecular testing nomenclature for and develop practice guidelines for acute myeloid leukemia (AML), myeloproliferative neoplasms (MPN), myelodysplastic syndrome (MDS), chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma, lymphoma, and plasma cell neoplasms;
- Develop standardized reporting guidelines for current VA molecular laboratories;
- Identify new tests as they are being reported in the literature and collaborate with hematology and oncology services to evaluate the clinical utility of these tests for VA patients;
- Network current VA molecular laboratories, perform fact-finding for these laboratories, and compile test menus; and
- Assess for the formation of VA-wide interfacility consultation services for hematopathology so that all VA facilities, regardless of their complexity, will be able to access the expertise of hematopathology-trained pathologists (Appendix).
The HMG subcommittee met monthly and discussed various diagnostic entities in hematopathology. For hematolymphoid malignancies, it was generally agreed that the traditional laboratory tools of morphology, flow cytometry, and immunohistochemistry (IHC) are standard in initial assessment and often in diagnosis. As the clinical molecular and cytogenetic assays of karyotype, fluorescence in situ hybridization (FISH), advanced DNA sequencing, microarray, and highly sensitive polymerase chain reaction (PCR) analysis affect diagnosis, subclassification, minimal residual disease (MRD) monitoring, prognosis, and therapy selection, their use is marked by a high degree of variability. As a result, standardization is needed. As each laboratory develops and reports ancillary testing, the variable reporting formats may generate postanalytic errors.
A detailed description of all molecular methodologies is beyond the scope of this article. For practicing pathologists, challenges remain in overall cost and reimbursement, extensive and time-consuming data analysis, and in some cases, interpretation differences.
Myeloid Neoplasms
Myeloid malignancies were divided into AML, MPN, and MDS. Next-generation sequencing (NGS) information for these malignancies was used to identify various contributory functional categories, including cell signaling (FLT3, KIT, JAK2, MPL, KRAS/NRAS, PTPN11, NF1, CSF3R); transcription (CEBPA, RUNX1, GATA1/GATA2, PHF6, ETV6); splicing (SF3B1, SRSF2, ZRSR2, U2AF1); epigenetics (DNMT3A, TET2, IDH1/IDH2, ASXL1, EZH2, SUZ12, KDM6A); cohesin complex (STAG2, SMC1A, SMC3, RAD21); and cell cycle (TP53, NPM1).2
Acute Myeloid Leukemia
The HMG subcommittee reviewed the literature on prognostically significant genes in myeloid leukemias. Karyotype abnormalities, such as t(8;21) and inv(16), collectively known as the core-binding factor (CBF) leukemias, t(15;17), t(11q23) (KMT2A/MLL), and so forth, are recurrent lesions in AML. Included in the minimum set of genes recommended by the National Comprehensive Cancer Network (NCCN) for AML prognosis evaluation are nucleolar protein nucleophosmin (NPM1), CCAAT/enhancer-binding protein
Some of the chromosomal translocations, such as inv(16)/t(16;16) in AML and t(15;17) in acute promyelocytic leukemia, can be monitored with FISH or reverse transcription–PCR (RT-PCR) analysis. As NPM1 mutations tend to be seen in recurrence, they can be used as molecular markers for MRD. Other mutations that provide important prognostic information in AML include:
- Activating insertions/duplications in the FLT3 receptor tyrosine kinase, which can be detected with PCR sizing assays;
- Mutations in the KIT receptor tyrosine kinase, which can be detected with DNA sequencing or more limited hotspot PCR;
- Mutations in the DNA methyltransferase, DNMT3A, a poor prognostic indicator seen in 22% of cases of AML, also detected with gene sequencing or more limited hotspot PCR; and
- Another set of genes, TET2, IDH1, IDH2, KRAS, NRAS, EZH2, and ASXL1, is mutated in MPN as well as AML and MDS, making a common molecular panel with next-generation sequencing useful in diagnosing and risk-stratifying all myeloid neoplasms.
The HMG subcommittee agreed that, for de novo AML, chromosomal karyotype is the standard of care, necessary in detecting known cytogenetic abnormalities as well as a wide range of lesions that might indicate a diagnosis of AML with myelodysplasia-related changes at time of diagnosis. In addition, molecular analysis of FLT3 is useful in determining prognosis, and CEBPA (biallelic) and NPM1 mutations are good prognostic factors in normal-karyotype AML. KMT2A (MLL) rearrangements should be tested with FISH if the lineage is ambiguous. The PML-RARA fusion gene also should be tested with FISH if morphologic and flow cytometry results suggest acute promyelocytic leukemia (Table). At this time, testing for TP53, DNMT3A, RAS, and other such mutations is not recommended because it is not cost-effective for the VA.
Myeloproliferative Neoplasms
Myeloproliferative neoplasms are clonal hematopoietic stem cell disorders characterized by proliferation of at least 1 myeloid lineage: granulocytic, erythroid, or megakaryocytic. Myeloproliferative neoplasms show a range of recurrent chromosomal translocations, such as BCR-ABL1 fusion in chronic myelogenous leukemia (CML) that can be detected with RT-PCR analysis as well as FISH. In CML, BCR-ABL1 fusion transcript levels detected by a quantitative PCR (qPCR) method are now used to monitor the course of CML therapy with tyrosine kinase inhibitors (TKIs) and to trigger a treatment change in drug-resistant cases. Given the importance of qPCR in clinical management, significant progress has been made in standardizing both the PCR protocol and the reference materials used to calibrate the BCR-ABL1 PCR assay. BCR-ABL1–negative MPN, including polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF), are most commonly associated with mutations in the tyrosine kinase JAK2. Mutations in CALR and MPL are seen in a subset of patients with ET and PMF as well, whereas PV is essentially exclusively a disease of JAK2 mutations.
Chronic myelogenous leukemia is the prototypical MPN. To establish the initial diagnosis, FISH and/or qPCR for BCR-ABL1 fusion should be used. If CML is confirmed, the sample can be reflexed to qPCR BCR-ABL1 on the initial peripheral blood and/or bone marrow sample(s) to establish the patient’s baseline. In addition, a bone marrow sample (aspirate) should be used for a complete karyotype and for morphologic confirmation of disease phase.
For follow-up assessment of CML patients’ response to TKI treatment, qPCR for BCR-ABL1 should be tested with a peripheral blood sample or a bone marrow sample every 3 months.4 A peripheral blood sample is more commonly used because it is conveniently obtained. Early molecular response as indicated by a BCR-ABL1 transcript ratio of < 10% on the International Scale at 3 months, has a strong prognostic value.5 Major molecular response as indicated by a BCR-ABL1 transcript ratio of < 0.1% on the International Scale at 12 to 18 months is also highly prognostic.5
After the peripheral blood sample becomes negative for BCR-ABL1 by qPCR, testing bone marrow samples may be considered. If important treatment response benchmarks are not achieved, or response is lost with rising BCR-ABL1 levels (TKI resistance), ABL1 kinase domain mutation analysis as well as repeat FISH (to assess for copy number multiplication) should be performed to guide further management. Patients with the ABL1 T315I mutation are resistant to all first-line TKIs but may respond to later third-generation TKIs.6
BCR-ABL1–negative MPNs include PV, ET, and PMF. Bone marrow morphology remains the cornerstone of ET and PMF diagnosis. The discovery of JAK2, CALR, and MPL mutations has contributed to how these disorders are diagnosed.7-12 Besides providing the clonality proof that is crucial for diagnosis, the molecular markers influence the prognosis. The JAK2 (p.V617F) or less common JAK2 exon 12 mutations, which are detected in more than 95% of PV cases, are used as molecular markers to confirm diagnosis.7 Further, the JAK2 (p.V617F), CALR (exon 9), and MPL (exon 10) mutations are detected in ET (~60%, 25%, and 3%-5%, respectively) and PMF (~55%, 30%, and 5%, respectively).12 If ET or PMF is suspected clinically, first JAK2 (p.V617F) mutation analysis should be performed, then CALR mutation analysis, and finally MPL mutation analysis. Although novel gain-of-function JAK2 and MPL mutations were recently discovered in triple-negative ET (negative for canonical mutations in JAK2, CALR, and MPL) and PMF by whole exome sequencing,13 clinical testing is not readily available. Besides its utility in the initial diagnosis of ET and PMF, the JAK2 or CALR mutation assay also may be considered for bone marrow transplantation follow-up (Table).14
Despite the continuing debate on the classification of eosinophilic myeloid disorders, the discovery of the FIP1L1-PDGFRA fusion represents a major milestone in the understanding of these disorders.15,16 Unlike PDGFRB (5q33) and FGFR1 (8p11) rearrangements, which can be detected with routine chromosomal analysis (cytogenetics), the cryptic FIP1L1-PDGFRA fusion must be detected with FISH (for CHIC2 deletion) or RT-PCR analysis. It should be pointed out that, as most eosinophilia is reactive or secondary, molecular testing for FIP1L1-PDGFRA fusion is indicated only when primary hypereosinophilia or hypereosinophilic syndrome (HES) is suspected. This is particularly the case in the following hypereosinophilia accompanying conditions: CML-like morphology, but BCR-ABL1–negative; chronic myelomonocytic leukemia (CMML)–like morphology with a normal karyotype; and new onset of cardiac damage or dysfunction.17
Primary eosinophilic myeloid disorders with PDGFRA or PDGFRB rearrangements can be treated with TKIs (eg, imatinib). Next-generation sequencing may be considered in cases of presumed HES when there is no identifiable karyotypic or FISH abnormality. Recent studies have found that cases of HES with somatic mutations indicating clonality had adverse clinical outcomes similar to those of cases of chronic eosinophilic leukemia.18
The discovery of CSF3R mutations offers a new molecular marker for the diagnosis of chronic neutrophilic leukemia (CNL), an MPN.19 The CSF3R (p.T618I) mutation or another activating CSF3R mutation is now used as a diagnostic criterion for CNL. Identification of specific CSF3R mutations may have therapeutic implications as well. The test should be ordered only for patients with clinical and morphologic findings suggestive of CNL; reactive neutrophilic leukocytosis (eg, infection, inflammation) should be ruled out before the test is ordered.
Myelodysplastic Syndrome
Myelodysplastic syndrome is a group of clonal bone marrow disorders characterized by ineffective hematopoiesis, manifested by morphologic dysplasia in ≥ 1 hematopoietic lineages and peripheral cytopenias (hemoglobin level, < 10 g/dL; platelet count, < 100×103/µL; absolute neutrophil count, < 1.8×103/µL). Diagnosis and classification of MDS depend mainly on the degree of morphologic dysplasia and blast percentages, as determined by examining well-prepared cellular bone marrow aspirate smears and/or biopsy touch preparations and peripheral blood smears.
Conventional karyotyping is an essential part of the diagnostic workup for all presumptive cases of MDS and is of both diagnostic and prognostic importance.20 About 60% of MDS cases have recurrent cytogenetic abnormalities, which can be detected with conventional karyotyping. If a high-quality cytogenetic analysis cannot be performed (eg, the bone marrow sample is inadequate), or if quick turnaround is required, an alternative FISH panel may be used to detect some of the common MDS-associated chromosomal abnormalities (eg, 5q deletion, 7q deletion/monosomy 7, +8, 20q deletion).21 Sequencing with FISH also can be useful for assessing MRD by detecting a previously identified chromosomal abnormality.
Targeted sequencing of a limited number of genes can detect mutations in the vast majority of patients with MDS. The most commonly mutated genes in MDS are SF3B1, TET2, SRSF2, ASXL1, DNMT3A, RUNX1, U2AF1, TP53, and EZH2. Mutations in SRSF2 cause RNA splicing abnormalities. In addition, mutations in TP53, EZH2, RUNX1, and ASXL1 are associated with poor prognosis,22,23 whereas mutations in SF3B1 confer better event-free survival.24 Despite these developments, the HMG subcommittee agreed that NGS-based mutation panels are not cost-effective for the VA population at this time and should not be included in a MDS workup. Only in rare situations and when clinically indicated (to change disease classification or patient management) should evaluation for specific gene mutations be considered—for instance, the SF3B1 mutation for patients with probable MDS with ring sideroblasts, if ring sideroblasts are < 15%.25
Myelodysplastic/Myeloproliferative Neoplasms
Myelodysplastic/myeloproliferative neoplasms are a group of myeloid neoplasms with clinical, laboratory, and morphologic features that overlap both MDS and MPN. In MDS/MPN, the karyotype is often normal or shows abnormalities in common with MDS.
In cases of unexplained monocytosis for which there is clinical concern for CMML, morphologic evaluation and conventional chromosomal karyotyping should be performed after other secondary causes and known myeloproliferative and myelodysplastic entities have been excluded. If concomitant hypereosinophilia is present and the karyotype is normal, FISH or PCR-based assay should be performed to rule out FIP1L1-PDGFRA rearrangements. BCR-ABL1, PDGFRB, FGFR1, and t(8;9)/PCM1-JAK2 rearrangements typically are detected with high-quality cytogenetic analysis and thus do not require targeted molecular assays. Although certain gene mutations (eg, SRSF2, TET2, ASXL1, CBL) are commonly detected in CMML, the HMG subcommittee does not recommend sequencing-based mutation panels, as there is insufficient information for testing for prognostic or treatment stratification.
If MDS/MPN with ring sideroblasts and thrombocytosis is suspected on the basis of the clinical and morphologic criteria, molecular tests for the JAK2 (p.V617F) and SF3B1 mutations may be considered in an effort to help confirm the diagnosis.
Atypical CML is a rare MDS/MPN subtype that is now better characterized molecularly with SETBP1 and/or ETNK1 mutations, which are detectable in up to a third of cases. If clinical suspicion is high, sequencing may be diagnostically helpful.
Lymphoid Neoplasms
Chronic Lymphocytic Leukemia
In CLL, recurrent chromosomal abnormalities (eg, deletions of 13q, trisomy 12, deletions of 11q, deletions of 17p) have clear prognostic value and can be detected with FISH. Other prognostic information, such as somatic mutation of immunoglobulin heavy chain variable (IgHV) genes, TP53 mutations, SF3B1, and NOTCH1 mutation, are mostly derived from PCR-based assays. The discovery of recurrently mutated genes in CLL has increased with the use of highly sensitive sequencing methods constructing a more detailed landscape of CLL at genetic, epigenetic, and cellular levels. A recent literature review summarizes the vast heterogeneity of CLL with recurrent pathogenetic findings in MYD88, SF3B1, TP53, ATM, and NOTCH1 signaling pathways.26 The treatment of CLL is rapidly evolving, and many clinical trials are proposing a change from the “watch and wait” paradigm to treatment upon initial presentation based on molecular findings. Additional testing based on new treatment options from current clinical trials will be recommended.
Flow cytometry and morphology are standard for CLL diagnosis. The HMG subcommittee recommends FISH for del(13q14), del(11q), trisomy 12, and del(17p) at time of diagnosis or immediately before therapy initiation. Zeta-chain (
Other B-Cell Lymphoproliferative Disorders
Unlike the common molecular changes in CLL, in other mature B-cell lymphomas, chromosomal translocations that juxtapose a variety of different oncogenes next to an Ig gene enhancer usually are—and those that switch regions less commonly are—important initiating events that can be detected with PCR, DNA sequencing, or FISH. In follicular lymphoma (FL), Burkitt lymphoma, marginal zone lymphoma (MZL), and mantle cell lymphoma (MCL), these oncogenes driven by an Ig gene enhancer typically include BCL2, MYC, MALT1, and CCND1 (cyclin D1), respectively. Molecular variants of these lymphomas that lack these classical translocations often activate homologous genes (eg, cyclin D3/CCND3 is activated in variants of MCL).
Morphology, flow cytometry, and IHC are routinely used for diagnosis. In inconclusive cases, Ig gene rearrangement by PCR may be used. The Table summarizes common molecular changes in B-cell lymphomas.
Mantle cell lymphoma. MCL is a non-Hodgkin lymphoma subtype characterized by t(11;14) (q13;q32) translocations that in the majority of cases lead to overexpression of cyclin D1 (BCL1). Recent molecular profiling has identified an MCL variant that is cyclin D1–negative but SOX11-positive and may have a more aggressive clinical course.30SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive MCL.
Lymphoplasmacytic lymphoma. Lymphoplasmacytic lymphoma (LPL), MZL, and CLL/small lymphocytic lymphoma are well-defined clinicopathologic entities. However, distinguishing LPL from MZL and atypical cases of CLL can sometimes be difficult because of overlapping clinical and morphologic features. Recent studies have identified a recurrent L265P mutation in the MYD88 gene in 90% to 95% of LPL cases with IgM paraprotein and in 40% to 50% of the rare non-IgM LPL cases. In contrast, the mutation is much less frequently present in MZL and other low-grade B-cell neoplasms (2%-7%).31 Therefore, testing for this abnormality can be a diagnostic aid in these difficult-to-classify cases. In addition, from a therapeutic perspective, presence or absence of MYD88 mutation may prove more significant than presence of a specific paraprotein or histopathologic features. Ibrutinib has shown efficacy in LPL and demonstrates improved response rates in patients with MYD88 mutation compared with that of their mutation-negative counterparts.32 Several MYD88 inhibitors are in clinical trials. This again indicates the need to more accurately identify and subclassify these non-IgM LPL cases to ensure appropriate molecular evaluation.
Hairy cell leukemia. Flow cytometry and morphology are usually sufficient for a hairy cell leukemia (HCL) diagnosis. However, rare cases are difficult to distinguish variant HCL from other mimics. The BRAF V600E mutation recently was described as a disease-defining molecular marker for HCL—present in nearly all HCL cases but virtually absent in HCL mimics. Therefore, detection of the BRAF mutation by IHC stain with specific antibody or PCR analysis is highly sensitive and specific for the diagnosis of HCL.33
Diffuse large B-cell lymphoma. Recent molecular analysis has created various risk stratification schemata for diffuse large B-cell lymphoma (DLBCL). The HGM subcommittee agrees that well-preserved morphology, IHC, flow cytometry, and FISH-specific markers (BCL2, BCL6, cMYC) are sufficient for diagnostic, prognostic, and therapeutic purposes. Although a wide range of genes have been implicated in the pathogenesis of DLBCL, sequencing and gene expression profiling are not cost-effective at this time and do not add benefit to patient treatment.
The MYD88 L265P mutation has been identified in DLBCL, particularly the activated B-cell-like type and primary central nervous system lymphoma (PCNSL), and may have implications for ibrutinib therapy. PCNSL commonly manifests aggressive clinical behavior and has a poor prognosis. It has been proposed that the MYD88 mutation can be used as a genetic hallmark for PCNSL to distinguish CNS involvement by systemic DLBCL from PCNSL.34
Plasma cell neoplasms. Flow cytometry is acceptable for the diagnosis of plasma cell neoplasms and for residual disease follow-up. Chromosomal karyotype or FISH for IGH/CCND1, IGH/MMSET, and IGH/CMAF dual fusion probes is recommended in conjunction with morphology, IHC, and flow cytometry. In plasma cell myeloma, several genetic mutations can be detected with NGS, including mutations in NRAS, KRAS, TP53, BCL7A, DIS3, and FAM46C.35 Less commonly, BRAF mutations, previously described in melanoma and several other solid tumors, can be detected with DNA sequencing in 4% of multiple myeloma cases, which may prove promising for targeted therapy with BRAF inhibitors. However, current therapeutic decisions are based on genetic and clinical factors, and sequence-based assays are not recommended at this time.
Follicular lymphoma. Cytology, histology, and IHC typically are sufficient for diagnosing FL. In difficult-to-diagnose cases and in cases with scant material, additional tests may help with diagnosis. Eighty to ninety percent of FL cases have t(14;18)(q32;q21), which places the BCL2 gene transcription under the control of the IGH promoter. In addition, about 10% of FL cases have 3q27 aberrancies at the BCL6 gene.36-38 More recently, cases of FL with bulky inguinal disease negative for IGH-BCL2 and BCL6 translocations were found to have 1p36 deletions. These 1p36-deleted FLs typically have a diffuse pattern and a good prognosis.39 For t(14;18), 3q27, or 1p36, FISH is a sensitive means for detecting these translocations, as is PCR for IGH-BCL2.40 There are reports that t(14;18) can be detected in a substantial fraction of otherwise healthy donors at levels and rates that depend on the type of detection test used.41-43 In addition, between one-fourth and one-third of de novo DLBCLs show t(14;18), and about one-third show BCL6 abnormalities at 3q27. Therefore, these genetic changes are not specific for FL and should not be used to subtype a lymphoma as follicular in origin.
Use of IGH-BCL2 as a marker for MRD is still controversial. Some studies have found that a postinduction and posttransplantation IGH-BCL2-positive finding by PCR predicted relapse.44,45 However, others studies have not found significance to postinduction IGH-BCL2 positivity.46 The NCCN guidelines recommend testing for IGH-BCL2 or BCL6 translocations or 1p36 deletion only if this testing is needed for diagnosis. The guidelines do not recommend using these genetic assays in follow-up biopsies, as the importance of treating early relapse has not been definitively demonstrated.
Therefore, if a lymphoma has morphologic, histologic, and IHC findings consistent with FL, then cytogenetic, FISH, or PCR testing is not needed for diagnosis but may be used as confirmation. Follow-up molecular and cytogenetic testing should be avoided if the original cytogenetic abnormality is unknown. That is, IGH-BCL2 FISH should be performed in follow-up samples only if the original lymphoma is known to contain the translocation. As follow-up genetic testing is of disputed clinical significance even in cases in which the original molecular change is known, the NCCN recommendations for therapy are no different. The HMG subcommittee does not recommend molecular or cytogenetic testing in FL beyond what is required for initial diagnosis.
T-Cell Lymphomas
Mature T-Cell Lymphoma and Leukemia
For mature T-cell lymphoma (TCL) and leukemia, the clinical and morphologic criteria have a very important role in the initial workup. However, IHC immunophenotyping is crucial for definitive diagnosis and subclassification. Flow cytometry is routinely used in diagnosing diseases such as T-cell prolymphocytic leukemia (TPLL), T-cell large granular lymphocytic (LGL) leukemia, and Sézary syndrome. T-cell clonality studies, preferably with BIOMED-II–validated primers against targets such as T-cell receptor
Significant advances in TCL classification have led to revisions and the inclusion of new provisional entities in the 2016 World Health Organization classification of lymphoid neoplasms.47 Many of these changes originated in studies of gene expression profiling and the genetic landscape of T-cell neoplasms. Even though subsets of peripheral TCL not otherwise specified (PTCL-NOS) have been recognized on the basis of phenotypic and molecular abnormalities with possible clinical implications, in most cases molecular testing is not part of routine practice. Typically, only a few cytogenetic abnormalities and genetic mutations are used in the evaluation of TCL and T-cell leukemia.
A group of T-cell lymphoproliferative disorders with expression of T follicular helper cell markers can be identified with IHC. These disorders include angioimmunoblastic TCL; follicular TCL, a new entity that is a PTCL-NOS subset; and primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder. The neoplastic cells should express at least 2 or 3 T follicular helper cell–related antigens, including CD279/PD1, CD10, BCL6, CXCL13, ICOS, SAP, and CCR5; the most commonly used are PD1, BCL6, and CD10. Recurrent fusion of ITK-SYK translocation t(5;9) or CTLA4-CD28 is also common in follicular TCL. Although recurrent mutation is found in these entities, conventional karyotyping or IHC should be sufficient for diagnosis.
Cutaneous γ -Δ T-Cell Lymphoma
Among cutaneous TCLs, primary cutaneous
Peripheral T-Cell Lymphoma
Gene expression profiling analysis of PTCLs has identified at least 3 subtypes characterized by overexpression of GATA3, TBX21, and cytotoxic genes and expression of the corresponding proteins with IHC.47 These subtypes are associated with different clinical behavior and therapy responses. The GATA3 subtype has an inferior prognosis and shows a high level of T helper type 2 cytokines, which can be identified with IHC. As IHC-stained GATA3 has been available as a marker of urothelial carcinoma at most IHC laboratories, GATA3 IHC staining also may be considered in the evaluation of PTCLs.
Many monoclonal antibody therapies are being used as primary or secondary regimens in the treatment of TCL. Clinical trials are working to establish their efficacy. If treatment with a monoclonal antibody is being considered, it is appropriate to conduct IHC to demonstrate the presence of the target antigen and at follow-up, to demonstrate the efficacy of treatment. These therapies include alemtuzumab, which targets CD52, and brentuximab, which targets CD30.
T-Cell Large Granular Lymphocytic Leukemia
T-cell LGL leukemia is a complex diagnosis that requires persistent clonal expansion of LGLs and clinically peripheral blood cytopenia. In many cases, the diagnosis is difficult to establish, as benign large granular lymphocytosis with clonal T cells may occur in conjunction with viral infections or autoimmune disorders. Somatic mutations in the STAT3 (signal transducer and activator of transcription 3) gene are found in 40% of patients with T-cell LGL leukemia.49 More recently, somatic mutations in the STAT5B gene were identified in 2% of T-cell LGL leukemia subsets. The clinical course of T-cell LGL leukemia in patients with the STAT5B mutation is aggressive and fatal, clearly different from the relatively favorable course of typical T-cell LGL leukemia.50 The HMG subcommittee recommends considering a STAT3 and STAT5B mutation study for selected cases in which it is difficult to distinguish true T-cell LGL leukemia from its reactive expansions.
T-Cell Prolymphocytic Leukemia
T-cell prolymphocytic leukemia (T-PLL) is a rare, aggressive disease and is most commonly associated with a prolymphocytic morphology and expression of CD4. However, since a specific immunophenotypic profile of T-PLL has not been identified, flow cytometry is not adequate in isolation for definitive classification as T-PLL.51 A diagnosis of T-PLL often requires cytogenetics or a FISH study to confirm a suspected case. Most TPLL cases harbor characteristic chromosomal abnormalities involving 14q11.2 (TCR
Anaplastic Large Cell Lymphoma
The World Health Organization recognizes 3 distinct types of anaplastic large cell lymphoma (ALCL): systemic anaplastic lymphoma kinase (ALK)–positive ALCL, systemic ALK-negative ALCL, and primary cutaneous ALCL. Systemic ALK-positive ALCLs consistently have ALK gene rearrangements and favorable outcomes. The most common translocation is the t(2;5) rearrangement of NPM1 and ALK, though other ALK partners are also possible. In contrast, systemic ALK-negative ALCLs lack ALK gene rearrangements and as a whole have outcomes inferior to those of systemic ALK-positive ALCLs. However, studies have found systemic ALK-negative ALCL to be a genetically and clinically heterogeneous entity.54 About 30% of cases have rearrangements of the DUSP22-IRF4 locus on 6p25.3 (DUSP22 rearrangement), and these cases have favorable outcomes similar to those of systemic ALK-positive ALCL.55 Only 8% of patients have TP63 rearrangements and very poor outcomes. The remaining cases lack ALK, DUSP22, and TP63 rearrangements and have intermediate outcomes. The HMG subcommittee recommends considering DUSP22 rearrangement by FISH in the evaluation of systemic ALK-negative ALCL.
Conclusion
The pathologic diagnosis, classification, and risk stratification of lymphoma and leukemia require an approach that integrates morphology, flow cytometry, cytogenetics, and molecular pathology. Rapidly evolving molecular techniques currently allow for detailed description of the molecular defects in lymphoma and leukemia, including driver mutations, amplification/deletion events, and clonal evolution. Unfortunately, the technical ability to catalogue the molecular defects in lymphoma and leukemia, often at great expense, is outpacing the ability to use this detailed information in treating patients with hematologic malignancies. The challenge, then, is to identify best practices for the diagnosis and classification of lymphoma and leukemia in VHA hospitals that incorporate the most useful molecular tests without wasting financial resources.
In this report, the HMG subcommittee of the MGPW has presented its recommendations for molecular testing in AML, MPN, MDS, and lymphomas in the context of standard morphologic and immunophenotypic approaches to hematopathology diagnosis and classification. Adoption of these recommendations by VHA hospitals and clinics should help ensure that all VA patients with hematologic malignancies benefit from the latest advances in precision medicine.
Within the vast and comprehensive national VHA health care system are multiple centers of expertise in hematopathology. In addition, multiple VA clinical molecular diagnostic laboratories are performing state-of-the-art testing. The HMG subcommittee proposes that, to make best use of these expert resources, the VHA should establish an interfacility hematopathology consultation service. This service would allow any VA pathologist to consult a board-certified hematopathologist regarding use of ancillary molecular genetic testing in the diagnosis of hematologic malignancy.
In addition, the HMG subcommittee recommends consolidating VA molecular diagnostic reference laboratories and having them perform molecular testing for other VA hospitals rather than using commercial reference laboratories, where testing standards are not uniform and results may be difficult to interpret. Several well-established VA clinical laboratories with technical expertise and informatics support are already performing selected molecular diagnostic testing. These laboratories’ resources should be expanded, where practical, to cost-effectively provide VA expertise to all veterans and to improve access to appropriate molecular diagnostic testing.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of
Click here to read the digital edition.
1. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793-795.
2. Matynia AP, Szankasi P, Shen W, Kelley TW. Molecular genetic biomarkers in myeloid malignancies. Arch Pathol Lab Med. 2015;139(5):594-601.
3. Wang ML, Bailey NG. Acute myeloid leukemia genetics: risk stratification and implications for therapy. Arch Pathol Lab Med. 2015;139(10):1215-1223.
4. Marum JE, Branford S. Current developments in molecular monitoring in chronic myeloid leukemia. Ther Adv Hematol. 2016;7(5):237-251.
5. Hughes TP, Saglio G, Kantarjian HM, et al. Early molecular response predicts outcomes in patients with chronic myeloid leukemia in chronic phase treated with frontline nilotinib or imatinib. Blood. 2014;123(9):1353-1360.
6. Pallera A, Altman JK, Berman E, et al. Guidelines insights: chronic myeloid leukemia, version 1.2017. J Natl Compr Canc Netw. 2016;14:1505-1512.
7. James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144-1148.
8. Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779-1790.
9. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008;112(1):141-149.
10. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood. 2008;112(3):844-847.
11. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391-2405.
12. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379-2390.
13. Harrison CN, Vannucchi AM. Closing the gap: genetic landscape of MPN. Blood. 2016;127(3):276-278.
14. Tefferi A, Barbui T. Essential thrombocythemia and polycythemia vera: focus on clinical practice. Mayo Clin Proc. 2015;90(9):1283-1293.
15. Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348(13):1201-1214.
16. Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130(3):607-612.e609.
17. Bain B, Billiland D, Horny H, Verdiman J. Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. Vol 2. Lyon, France: IRAC Press; 2008:68-73.
18. Wang SA, Tam W, Tsai AG, et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod Pathol. 2016;29(8):854-864.
19. Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781-1790.
20. Schanz J, Tüchler H, Solé F, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30(8):820-829.
21. Marshall D, Roboz GJ. Standardizing the initial evaluation for myelodysplastic syndromes. Curr Hematol Malig Rep. 2013;8(4):361-369.
22. Damm F, Chesnais V, Nagata Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122(18):3169-3177.
23. Malcovati L, Papaemmanuil E, Ambaglio I, et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood. 2014;124(9):1513-1521.
24. Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384-1395.
25. Patnaik MM, Hanson CA, Sulai NH, et al. Prognostic irrelevance of ring sideroblast percentage in World Health Organization-defined myelodysplastic syndromes without excess blasts. Blood. 2012;119(24):5674-5677.
26. Guièze R, Wu CJ. Genomic and epigenomic heterogeneity in chronic lymphocytic leukemia. Blood. 2015;126(4):445-453.
27. Jeromin S, Weissmann S, Haferlach C, et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia. 2014;28(1):108-117.
28. Del Giudice I, Rossi D, Chiaretti S, et al. NOTCH1 mutations in +12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of +12 CLL. Haematologica. 2012;97(3):437-441.
29. Weissmann S, Roller A, Jeromin S, et al. Prognostic impact and landscape of NOTCH1 mutations in chronic lymphocytic leukemia (CLL): a study on 852 patients. Leukemia. 2013;27(12):2393-2396.
30. Vegliante MC, Palomero J, Pérez-Galán P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood. 2013;121(12):2175-2185.
31. King RL, Gonsalves WI, Ansell SM, et al. Lymphoplasmacytic lymphoma with a non-IgM paraprotein shows clinical and pathologic heterogeneity and may harbor MYD88 L265P mutations. Am J Clin Pathol. 2016;145(6):843-851.
32. Insuasti-Beltran G, Gale JM, Wilson CS, Foucar K, Czuchlewski DR. Significance of MYD88 L265P mutation status in the subclassification of low-grade B-cell lymphoma/leukemia. Arch Pathol Lab Med. 2015;139(8):1035-1041.
33. Wang XJ, Kim A, Li S. Immunohistochemical analysis using a BRAF V600E mutation specific antibody is highly sensitive and specific for the diagnosis of hairy cell leukemia. Int J Clin Exp Pathol. 2014;7(7):4323-4328.
34. Nakamura T, Tateishi K, Niwa T, et al. Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathol Appl Neurobiol. 2016;42(3):279-290.
35. Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467-472.
36. Bosga-Bouwer AG, van Imhoff GW, Boonstra R, et al. Follicular lymphoma grade 3B includes 3 cytogenetically defined subgroups with primary t(14;18), 3q27, or other translocations: t(14;18) and 3q27 are mutually exclusive. Blood. 2003;101(3):1149-1154.
37. Gu K, Fu K, Jain S, et al. t(14;18)-negative follicular lymphomas are associated with a high frequency of BCL6 rearrangement at the alternative breakpoint region. Mod Pathol. 2009;22(9):1251-1257.
38. Katzenberger T, Ott G, Klein T, Kalla J, Müller-Hermelink HK, Ott MM. Cytogenetic alterations affecting BCL6 are predominantly found in follicular lymphomas grade 3B with a diffuse large B-cell component. Am J Pathol. 2004;165(2):481-490.
39. Katzenberger T, Kalla J, Leich E, et al. A distinctive subtype of t(14;18)-negative nodal follicular non-Hodgkin lymphoma characterized by a predominantly diffuse growth pattern and deletions in the chromosomal region 1p36. Blood. 2009;113(5):1053-1061.
40. Belaud-Rotureau MA, Parrens M, Carrere N, et al. Interphase fluorescence in situ hybridization is more sensitive than BIOMED-2 polymerase chain reaction protocol in detecting IGH-BCL2 rearrangement in both fixed and frozen lymph node with follicular lymphoma. Hum Pathol. 2007;38(2):365-372.
41. Limpens J, Stad R, Vos C, et al. Lymphoma-associated translocation t(14;18) in blood B cells of normal individuals. Blood. 1995;85(9):2528-2536.
42. Schmitt C, Balogh B, Grundt A, et al. The bcl-2/IgH rearrangement in a population of 204 healthy individuals: occurrence, age and gender distribution, breakpoints, and detection method validity. Leuk Res. 2006;30(6):745-750.
43. Summers KE, Goff LK, Wilson AG, Gupta RK, Lister TA, Fitzgibbon J. Frequency of the Bcl-2/IgH rearrangement in normal individuals: implications for the monitoring of disease in patients with follicular lymphoma. J Clin Oncol. 2001;19(2):420-424.
44. Galimberti S, Luminari S, Ciabatti E, et al. Minimal residual disease after conventional treatment significantly impacts on progression-free survival of patients with follicular lymphoma: the FIL FOLL05 trial. Clin Cancer Res. 2014;20(24):6398-6405.
45. Ladetto M, Lobetti-Bodoni C, Mantoan B, et al; Fondazione Italiana Linfomi. Persistence of minimal residual disease in bone marrow predicts outcome in follicular lymphomas treated with a rituximab-intensive program. Blood. 2013;122(23):3759-3766.
46. van Oers MHJ, Tönnissen E, Van Glabbeke M, et al. BCL-2/IgH polymerase chain reaction status at the end of induction treatment is not predictive for progression-free survival in relapsed/resistant follicular lymphoma: results of a prospective randomized EORTC 20981 phase III intergroup study. J Clin Oncol. 2010;28(13):2246-2252.
47. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390.
48. Dewar R, Andea AA, Guitart J, Arber DA, Weiss LM. Best practices in diagnostic immunohistochemistry: workup of cutaneous lymphoid lesions in the diagnosis of primary cutaneous lymphoma. Arch Pathol Lab Med. 2015;139(3):338-350.
49. Koskela HLM, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366(20):1905-1913.
50. Rajala HL, Eldfors S, Kuusanmäki H, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121(22):4541-4550.
51. Chen X, Cherian S. Immunophenotypic characterization of T-cell prolymphocytic leukemia. Am J Clin Pathol. 2013;140(5):727-735.
52. Delgado P, Starshak P, Rao N, Tirado C. A comprehensive update on molecular and cytogenetic abnormalities in T-cell prolymphocytic leukemia (T-PLL). J Assoc Genet Technol. 2012;38(4):193-198.
53. Stengel A, Kern W, Zenger M, et al. Genetic characterization of T-PLL reveals two major biologic subgroups and JAK3 mutations as prognostic marker. Genes Chromosomes Cancer. 2016;55(1):82-94.
54. Thompson MA, Stumph J, Henrickson SE, et al. Differential gene expression in anaplastic lymphoma kinase-positive and anaplastic lymphoma kinase-negative anaplastic large cell lymphomas. Hum Pathol. 2005;36(5):494-504.
55. King R, Dao L, McPhail E, et al. Morphologic features of ALK-negative anaplastic large cell lymphomas with DUSP22 rearrangements. Am J Surg Pathol. 2016;40(1):36-43.
1. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793-795.
2. Matynia AP, Szankasi P, Shen W, Kelley TW. Molecular genetic biomarkers in myeloid malignancies. Arch Pathol Lab Med. 2015;139(5):594-601.
3. Wang ML, Bailey NG. Acute myeloid leukemia genetics: risk stratification and implications for therapy. Arch Pathol Lab Med. 2015;139(10):1215-1223.
4. Marum JE, Branford S. Current developments in molecular monitoring in chronic myeloid leukemia. Ther Adv Hematol. 2016;7(5):237-251.
5. Hughes TP, Saglio G, Kantarjian HM, et al. Early molecular response predicts outcomes in patients with chronic myeloid leukemia in chronic phase treated with frontline nilotinib or imatinib. Blood. 2014;123(9):1353-1360.
6. Pallera A, Altman JK, Berman E, et al. Guidelines insights: chronic myeloid leukemia, version 1.2017. J Natl Compr Canc Netw. 2016;14:1505-1512.
7. James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144-1148.
8. Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779-1790.
9. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008;112(1):141-149.
10. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood. 2008;112(3):844-847.
11. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391-2405.
12. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379-2390.
13. Harrison CN, Vannucchi AM. Closing the gap: genetic landscape of MPN. Blood. 2016;127(3):276-278.
14. Tefferi A, Barbui T. Essential thrombocythemia and polycythemia vera: focus on clinical practice. Mayo Clin Proc. 2015;90(9):1283-1293.
15. Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348(13):1201-1214.
16. Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130(3):607-612.e609.
17. Bain B, Billiland D, Horny H, Verdiman J. Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. Vol 2. Lyon, France: IRAC Press; 2008:68-73.
18. Wang SA, Tam W, Tsai AG, et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod Pathol. 2016;29(8):854-864.
19. Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781-1790.
20. Schanz J, Tüchler H, Solé F, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30(8):820-829.
21. Marshall D, Roboz GJ. Standardizing the initial evaluation for myelodysplastic syndromes. Curr Hematol Malig Rep. 2013;8(4):361-369.
22. Damm F, Chesnais V, Nagata Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122(18):3169-3177.
23. Malcovati L, Papaemmanuil E, Ambaglio I, et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood. 2014;124(9):1513-1521.
24. Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384-1395.
25. Patnaik MM, Hanson CA, Sulai NH, et al. Prognostic irrelevance of ring sideroblast percentage in World Health Organization-defined myelodysplastic syndromes without excess blasts. Blood. 2012;119(24):5674-5677.
26. Guièze R, Wu CJ. Genomic and epigenomic heterogeneity in chronic lymphocytic leukemia. Blood. 2015;126(4):445-453.
27. Jeromin S, Weissmann S, Haferlach C, et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia. 2014;28(1):108-117.
28. Del Giudice I, Rossi D, Chiaretti S, et al. NOTCH1 mutations in +12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of +12 CLL. Haematologica. 2012;97(3):437-441.
29. Weissmann S, Roller A, Jeromin S, et al. Prognostic impact and landscape of NOTCH1 mutations in chronic lymphocytic leukemia (CLL): a study on 852 patients. Leukemia. 2013;27(12):2393-2396.
30. Vegliante MC, Palomero J, Pérez-Galán P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood. 2013;121(12):2175-2185.
31. King RL, Gonsalves WI, Ansell SM, et al. Lymphoplasmacytic lymphoma with a non-IgM paraprotein shows clinical and pathologic heterogeneity and may harbor MYD88 L265P mutations. Am J Clin Pathol. 2016;145(6):843-851.
32. Insuasti-Beltran G, Gale JM, Wilson CS, Foucar K, Czuchlewski DR. Significance of MYD88 L265P mutation status in the subclassification of low-grade B-cell lymphoma/leukemia. Arch Pathol Lab Med. 2015;139(8):1035-1041.
33. Wang XJ, Kim A, Li S. Immunohistochemical analysis using a BRAF V600E mutation specific antibody is highly sensitive and specific for the diagnosis of hairy cell leukemia. Int J Clin Exp Pathol. 2014;7(7):4323-4328.
34. Nakamura T, Tateishi K, Niwa T, et al. Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathol Appl Neurobiol. 2016;42(3):279-290.
35. Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467-472.
36. Bosga-Bouwer AG, van Imhoff GW, Boonstra R, et al. Follicular lymphoma grade 3B includes 3 cytogenetically defined subgroups with primary t(14;18), 3q27, or other translocations: t(14;18) and 3q27 are mutually exclusive. Blood. 2003;101(3):1149-1154.
37. Gu K, Fu K, Jain S, et al. t(14;18)-negative follicular lymphomas are associated with a high frequency of BCL6 rearrangement at the alternative breakpoint region. Mod Pathol. 2009;22(9):1251-1257.
38. Katzenberger T, Ott G, Klein T, Kalla J, Müller-Hermelink HK, Ott MM. Cytogenetic alterations affecting BCL6 are predominantly found in follicular lymphomas grade 3B with a diffuse large B-cell component. Am J Pathol. 2004;165(2):481-490.
39. Katzenberger T, Kalla J, Leich E, et al. A distinctive subtype of t(14;18)-negative nodal follicular non-Hodgkin lymphoma characterized by a predominantly diffuse growth pattern and deletions in the chromosomal region 1p36. Blood. 2009;113(5):1053-1061.
40. Belaud-Rotureau MA, Parrens M, Carrere N, et al. Interphase fluorescence in situ hybridization is more sensitive than BIOMED-2 polymerase chain reaction protocol in detecting IGH-BCL2 rearrangement in both fixed and frozen lymph node with follicular lymphoma. Hum Pathol. 2007;38(2):365-372.
41. Limpens J, Stad R, Vos C, et al. Lymphoma-associated translocation t(14;18) in blood B cells of normal individuals. Blood. 1995;85(9):2528-2536.
42. Schmitt C, Balogh B, Grundt A, et al. The bcl-2/IgH rearrangement in a population of 204 healthy individuals: occurrence, age and gender distribution, breakpoints, and detection method validity. Leuk Res. 2006;30(6):745-750.
43. Summers KE, Goff LK, Wilson AG, Gupta RK, Lister TA, Fitzgibbon J. Frequency of the Bcl-2/IgH rearrangement in normal individuals: implications for the monitoring of disease in patients with follicular lymphoma. J Clin Oncol. 2001;19(2):420-424.
44. Galimberti S, Luminari S, Ciabatti E, et al. Minimal residual disease after conventional treatment significantly impacts on progression-free survival of patients with follicular lymphoma: the FIL FOLL05 trial. Clin Cancer Res. 2014;20(24):6398-6405.
45. Ladetto M, Lobetti-Bodoni C, Mantoan B, et al; Fondazione Italiana Linfomi. Persistence of minimal residual disease in bone marrow predicts outcome in follicular lymphomas treated with a rituximab-intensive program. Blood. 2013;122(23):3759-3766.
46. van Oers MHJ, Tönnissen E, Van Glabbeke M, et al. BCL-2/IgH polymerase chain reaction status at the end of induction treatment is not predictive for progression-free survival in relapsed/resistant follicular lymphoma: results of a prospective randomized EORTC 20981 phase III intergroup study. J Clin Oncol. 2010;28(13):2246-2252.
47. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390.
48. Dewar R, Andea AA, Guitart J, Arber DA, Weiss LM. Best practices in diagnostic immunohistochemistry: workup of cutaneous lymphoid lesions in the diagnosis of primary cutaneous lymphoma. Arch Pathol Lab Med. 2015;139(3):338-350.
49. Koskela HLM, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366(20):1905-1913.
50. Rajala HL, Eldfors S, Kuusanmäki H, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121(22):4541-4550.
51. Chen X, Cherian S. Immunophenotypic characterization of T-cell prolymphocytic leukemia. Am J Clin Pathol. 2013;140(5):727-735.
52. Delgado P, Starshak P, Rao N, Tirado C. A comprehensive update on molecular and cytogenetic abnormalities in T-cell prolymphocytic leukemia (T-PLL). J Assoc Genet Technol. 2012;38(4):193-198.
53. Stengel A, Kern W, Zenger M, et al. Genetic characterization of T-PLL reveals two major biologic subgroups and JAK3 mutations as prognostic marker. Genes Chromosomes Cancer. 2016;55(1):82-94.
54. Thompson MA, Stumph J, Henrickson SE, et al. Differential gene expression in anaplastic lymphoma kinase-positive and anaplastic lymphoma kinase-negative anaplastic large cell lymphomas. Hum Pathol. 2005;36(5):494-504.
55. King R, Dao L, McPhail E, et al. Morphologic features of ALK-negative anaplastic large cell lymphomas with DUSP22 rearrangements. Am J Surg Pathol. 2016;40(1):36-43.