User login
Pituitary Incidentaloma
Brian, 46, is referred to endocrinology for evaluation of a pituitary “mass.” The mass was an incidental finding of head and neck CT performed three months ago, when Brian went to an emergency department following a motor vehicle accident. He has fully recovered from the accident and feels well. He describes himself as a “completely healthy” person who has no chronic medical conditions and takes neither prescription nor OTC medications.
Brian denies significant headache, visual disturbance, change in appetite, unexplained weight change, skin rash (wide purple striae) or color changes (hyperpigmentation), polyuria or polydipsia, dizziness, syncopal episodes, low libido, erectile dysfunction, joint pain, and changes in ring or shoe size. He does not wear a hat or cap and is unaware of head size changes. He has not experienced changes in his facial features or trouble with chewing.
He is a happily married engineer with two healthy children and reports that he feels well except for this “brain tumor” finding that has been a shock to him and his family. There is no family history of pituitary adenoma or multiple endocrine neoplasia syndrome.
His vital signs, all within normal ranges, include a blood pressure of 103/65 mm Hg. His height is 6 ft and his weight, 180 lb. His BMI is 24.4.
HOW COMMON IS PITUITARY INCIDENTALOMA?
A pituitary incidentaloma is a lesion in the pituitary gland that was not previously suspected and was found through an imaging study ordered for other reasons. Pituitary incidentaloma is surprisingly common, with an average prevalence of 10.6% (as estimated from combined autopsy data), although it has been found in up to 20% of patients undergoing CT and 38% undergoing MRI.1,2 Most are microadenomas (< 1 cm in size).1
Continue for recommendations from the Endocrine Society >>
SHOULD AN ASYMPTOMATIC PATIENT BE EVALUATED FURTHER?
Endocrine Society guidelines2 recommend that all patients with pituitary incidentaloma, with or without symptoms, should undergo a complete history and physical examination and laboratory evaluation to exclude hypersecretion and hyposecretion of pituitary hormones.
The “classic” presentation of pituitary hormone hypersecretion—in the form of prolactinoma, adrenocorticotropic hormone (ACTH) excess (Cushing disease), growth hormone (GH) excess (gigantism/acromegaly), and TSH excess (secondary hyperthyroidism)—may be readily detectable on history and physical examination. Subtle cases, so-called subclinical disease, however, may exhibit little or no signs and symptoms initially but can be detrimental to the patient’s health if left untreated. For example, the estimated time from onset to diagnosis of acromegaly is approximately seven to 10 years—a delay that can significantly impact the patient’s morbidity and mortality.3
Prolactinoma can be more clinically apparent in premenopausal females due to irregular menstrual cycles (oligomenorrhea/amenorrhea). However, galactorrhea, or “milky” nipple discharge, occurs in only about 50% of women with prolactinoma and is extremely rare in men. Furthermore, the clinical presentation of prolactinoma in men is vague and related to hypogonadism, resulting from increased prolactin levels. Since men are essentially asymptomatic, these tumors can grow extensively (macroadenoma) and cause “mass effect,” such as headaches and visual impairment.
Therefore, without laboratory testing, abnormal pituitary function may go unrecognized.
WHAT LABS SHOULD I ORDER FOR THIS PATIENT?
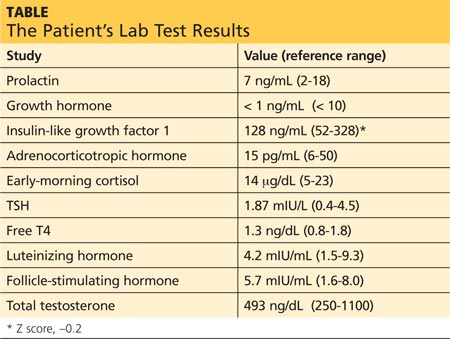
Guidelines suggest an initial screening panel of prolactin, GH, insulin-like growth factor 1 (IGF-1), ACTH, early-morning cortisol, TSH, free T4, luteinizing hormone (LH), follicle-stimulating hormone (FSH), and testosterone.2
Note the use of “suggest” rather than “recommend.” Even among guideline task force members, there were differences in opinion as to whether certain tests (eg, TSH, LH, and FSH) should be included in initial screening. Those tests can be ordered at the clinician’s discretion, according to the level of suspicion, or can be added later if necessary.
Brian’s sample for laboratory testing is drawn at 7:50 am. Results can be found in the table (previous page).
Next page: Caveats and concerns >>
ARE THERE ANY CAVEATS TO THE INTERPRETATION OF LAB VALUES?
It is important to note that in postmenopausal women who are not taking hormone replacement therapy, LH and FSH will be elevated and estradiol may provide an additional clue in the detection of abnormal function. Conversely, low LH and FSH in postmenopausal women should raise a flag for hypopituitarism.
Another caveat is that GH secretion is pulsatile and serum levels are undetectable between pulses. Therefore, low/undetectable GH does not necessarily suggest deficiency. The GH measurement would only be helpful if it is significantly elevated (suggestive of hypersecretion—gigantism/acromegaly). Otherwise, GH has little value as a screening test.
Instead, IGF-1, which is secreted from the liver in response to GH secretion, has a longer half-life and serves as a better screening tool. IGF-1 has age- and sex-adjusted reference ranges, which are often reported by the lab or given as a Z score.
WHAT IS THE PREFERRED IMAGING STUDY FOR THE PITUITARY GLAND?
The best choice is MRI of the pituitary gland (not the whole brain) with gadolinium. If the incidentaloma was initially diagnosed by a CT, additional testing with MRI should be performed, unless contraindicated.2
Brian is referred for MRI with gadolinium. The radiologist’s report describes a 5 x 4 x 4–mm pituitary microadenoma without sellar extension or involvement of the optic chiasm.
AT WHAT POINT SHOULD OPTIC CHIASM BE A CONCERN?
Since the pituitary gland is located directly beneath the optic chiasm, any compressive effect of growth against the optic nerve(s) can cause visual impairment. This includes bitemporal hemianopsia (loss of peripheral vision) or ophthalmoplegia (abnormal movement of the ocular muscle). Since clinical signs and symptoms can be subtle or absent, all patients with evidence of a pituitary lesion abutting or compressing the optic chiasm should have a formal visual field exam.2
Continue for surgical intervention >>
WHO REQUIRES SURGICAL INTERVENTION?
Patients with mass effect (headache, increased intracranial pressure, compromised optic chiasm) and those with hyperfunctioning nonprolactin adenomas, some (but not all) macroprolactinomas, or pituitary apoplexy should be referred for surgery.2 Almost all cases involve macroadenomas rather than microadenomas.
The preferred treatment for GH-secreting tumors and ACTH-secreting tumors is surgery. However, prolactinoma can be well controlled with pharmacologic agents (dopamine agonists) in most cases. For prolactinomas refractory to these medications, surgical resection is recommended. (Detailed treatment approaches are available elsewhere; those for hyperprolactinoma can be found on the Clinician Reviews website: http://bit.ly/1HOb9Jf.)
Pituitary apoplexy, a life-threatening emergency that requires prompt surgical decompression, is an infarction of the gland due to abrupt cessation of the blood supply, caused by either pituitary artery hemorrhage or sudden hypovolemia. Increased blood supply is needed due to the extra tissue and volume of the pituitary mass; this may stress the pituitary arteries, which are not equipped for this increased flow, causing them to rupture. Hemorrhage anywhere else in the body can lead to hypovolemia and decrease the blood supply to the pituitary gland. A classic example would be postpartum hemorrhage causing pituitary infarct, called Sheehan syndrome.
Due to increased estrogen levels, the pituitary gland doubles in size during pregnancy.4 A preexisting mass may further develop and compress the optic chiasm. Therefore, women of childbearing age should be engaged in discussion of the potential risks and benefits of decompression surgery before actively pursuing pregnancy—especially if the lesion is close to the optic chiasm.
Surgery can also be considered for patients with significant growth in adenoma size during monitoring, loss of endocrinologic function due to mass effect on other pituitary cells, or unremitting headache.2
Next: How should patients be monitored?
HOW SHOULD PATIENTS BE MONITORED?
Those who do not meet criteria for surgery can be closely monitored with periodic testing. Imaging can be repeated six months after the first scan for macroadenoma and in one year for microadenoma. If there is no change in the size of the mass, imaging can be done yearly for macroadenoma and for microadenoma, every one to two years for three years and then gradually less often thereafter.2
Unless the lesion is abutting the optic chiasm (seen via imaging) or the patient reports symptoms, visual field testing does not need to be repeated.
Lab testing should be repeated six months after initial testing for macroadenoma and yearly thereafter. No further testing is suggested for nonsecretory microadenoma, unless clinically indicated.2
If there are any changes in status—noted clinically or via imaging—more frequent testing is suggested.
Brian is reassured that pituitary adenoma is not an uncommon finding and that his adenoma is relatively small in size and nonsecretory. Repeat pituitary MRI in one year is recommended.
CONCLUSION
Most pituitary incidentalomas have no consequences to a patient’s health. However, patients often become highly anxious about the “brain tumor” they were told they have. Appropriate patient education and thorough evaluation can reassure patients and alleviate their concerns.
REFERENCES
1. Molitch ME. Nonfunctioning pituitary tumors and pituitary incidentalomas. Endocrinol Metab Clin North Am. 2008;37(1):151-171.
2. Freda PU, Beckers AM, Katznelson L, et al. Pituitary incidentaloma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96(4):894-904.
3. Katznelson L, Atkinson JL, Cook DM, et al. American Association of Clinical Endocrinologists Medical Guidelines For Clinical Practice For The Diagnosis And Treatment Of Acromegaly–2011 Update. Endocr Pract. 2011;17(suppl 4).
4. Jameson JL. Harrison’s Endocrinology. 2nd ed. China: McGraw-Hill; 2010:16-49.
Clinician Reviews in partnership with![]()
Clinician Reviews in partnership with![]()
Clinician Reviews in partnership with![]()
Brian, 46, is referred to endocrinology for evaluation of a pituitary “mass.” The mass was an incidental finding of head and neck CT performed three months ago, when Brian went to an emergency department following a motor vehicle accident. He has fully recovered from the accident and feels well. He describes himself as a “completely healthy” person who has no chronic medical conditions and takes neither prescription nor OTC medications.
Brian denies significant headache, visual disturbance, change in appetite, unexplained weight change, skin rash (wide purple striae) or color changes (hyperpigmentation), polyuria or polydipsia, dizziness, syncopal episodes, low libido, erectile dysfunction, joint pain, and changes in ring or shoe size. He does not wear a hat or cap and is unaware of head size changes. He has not experienced changes in his facial features or trouble with chewing.
He is a happily married engineer with two healthy children and reports that he feels well except for this “brain tumor” finding that has been a shock to him and his family. There is no family history of pituitary adenoma or multiple endocrine neoplasia syndrome.
His vital signs, all within normal ranges, include a blood pressure of 103/65 mm Hg. His height is 6 ft and his weight, 180 lb. His BMI is 24.4.
HOW COMMON IS PITUITARY INCIDENTALOMA?
A pituitary incidentaloma is a lesion in the pituitary gland that was not previously suspected and was found through an imaging study ordered for other reasons. Pituitary incidentaloma is surprisingly common, with an average prevalence of 10.6% (as estimated from combined autopsy data), although it has been found in up to 20% of patients undergoing CT and 38% undergoing MRI.1,2 Most are microadenomas (< 1 cm in size).1
Continue for recommendations from the Endocrine Society >>
SHOULD AN ASYMPTOMATIC PATIENT BE EVALUATED FURTHER?
Endocrine Society guidelines2 recommend that all patients with pituitary incidentaloma, with or without symptoms, should undergo a complete history and physical examination and laboratory evaluation to exclude hypersecretion and hyposecretion of pituitary hormones.
The “classic” presentation of pituitary hormone hypersecretion—in the form of prolactinoma, adrenocorticotropic hormone (ACTH) excess (Cushing disease), growth hormone (GH) excess (gigantism/acromegaly), and TSH excess (secondary hyperthyroidism)—may be readily detectable on history and physical examination. Subtle cases, so-called subclinical disease, however, may exhibit little or no signs and symptoms initially but can be detrimental to the patient’s health if left untreated. For example, the estimated time from onset to diagnosis of acromegaly is approximately seven to 10 years—a delay that can significantly impact the patient’s morbidity and mortality.3
Prolactinoma can be more clinically apparent in premenopausal females due to irregular menstrual cycles (oligomenorrhea/amenorrhea). However, galactorrhea, or “milky” nipple discharge, occurs in only about 50% of women with prolactinoma and is extremely rare in men. Furthermore, the clinical presentation of prolactinoma in men is vague and related to hypogonadism, resulting from increased prolactin levels. Since men are essentially asymptomatic, these tumors can grow extensively (macroadenoma) and cause “mass effect,” such as headaches and visual impairment.
Therefore, without laboratory testing, abnormal pituitary function may go unrecognized.
WHAT LABS SHOULD I ORDER FOR THIS PATIENT?
Guidelines suggest an initial screening panel of prolactin, GH, insulin-like growth factor 1 (IGF-1), ACTH, early-morning cortisol, TSH, free T4, luteinizing hormone (LH), follicle-stimulating hormone (FSH), and testosterone.2
Note the use of “suggest” rather than “recommend.” Even among guideline task force members, there were differences in opinion as to whether certain tests (eg, TSH, LH, and FSH) should be included in initial screening. Those tests can be ordered at the clinician’s discretion, according to the level of suspicion, or can be added later if necessary.
Brian’s sample for laboratory testing is drawn at 7:50 am. Results can be found in the table (previous page).
Next page: Caveats and concerns >>
ARE THERE ANY CAVEATS TO THE INTERPRETATION OF LAB VALUES?
It is important to note that in postmenopausal women who are not taking hormone replacement therapy, LH and FSH will be elevated and estradiol may provide an additional clue in the detection of abnormal function. Conversely, low LH and FSH in postmenopausal women should raise a flag for hypopituitarism.
Another caveat is that GH secretion is pulsatile and serum levels are undetectable between pulses. Therefore, low/undetectable GH does not necessarily suggest deficiency. The GH measurement would only be helpful if it is significantly elevated (suggestive of hypersecretion—gigantism/acromegaly). Otherwise, GH has little value as a screening test.
Instead, IGF-1, which is secreted from the liver in response to GH secretion, has a longer half-life and serves as a better screening tool. IGF-1 has age- and sex-adjusted reference ranges, which are often reported by the lab or given as a Z score.
WHAT IS THE PREFERRED IMAGING STUDY FOR THE PITUITARY GLAND?
The best choice is MRI of the pituitary gland (not the whole brain) with gadolinium. If the incidentaloma was initially diagnosed by a CT, additional testing with MRI should be performed, unless contraindicated.2
Brian is referred for MRI with gadolinium. The radiologist’s report describes a 5 x 4 x 4–mm pituitary microadenoma without sellar extension or involvement of the optic chiasm.
AT WHAT POINT SHOULD OPTIC CHIASM BE A CONCERN?
Since the pituitary gland is located directly beneath the optic chiasm, any compressive effect of growth against the optic nerve(s) can cause visual impairment. This includes bitemporal hemianopsia (loss of peripheral vision) or ophthalmoplegia (abnormal movement of the ocular muscle). Since clinical signs and symptoms can be subtle or absent, all patients with evidence of a pituitary lesion abutting or compressing the optic chiasm should have a formal visual field exam.2
Continue for surgical intervention >>
WHO REQUIRES SURGICAL INTERVENTION?
Patients with mass effect (headache, increased intracranial pressure, compromised optic chiasm) and those with hyperfunctioning nonprolactin adenomas, some (but not all) macroprolactinomas, or pituitary apoplexy should be referred for surgery.2 Almost all cases involve macroadenomas rather than microadenomas.
The preferred treatment for GH-secreting tumors and ACTH-secreting tumors is surgery. However, prolactinoma can be well controlled with pharmacologic agents (dopamine agonists) in most cases. For prolactinomas refractory to these medications, surgical resection is recommended. (Detailed treatment approaches are available elsewhere; those for hyperprolactinoma can be found on the Clinician Reviews website: http://bit.ly/1HOb9Jf.)
Pituitary apoplexy, a life-threatening emergency that requires prompt surgical decompression, is an infarction of the gland due to abrupt cessation of the blood supply, caused by either pituitary artery hemorrhage or sudden hypovolemia. Increased blood supply is needed due to the extra tissue and volume of the pituitary mass; this may stress the pituitary arteries, which are not equipped for this increased flow, causing them to rupture. Hemorrhage anywhere else in the body can lead to hypovolemia and decrease the blood supply to the pituitary gland. A classic example would be postpartum hemorrhage causing pituitary infarct, called Sheehan syndrome.
Due to increased estrogen levels, the pituitary gland doubles in size during pregnancy.4 A preexisting mass may further develop and compress the optic chiasm. Therefore, women of childbearing age should be engaged in discussion of the potential risks and benefits of decompression surgery before actively pursuing pregnancy—especially if the lesion is close to the optic chiasm.
Surgery can also be considered for patients with significant growth in adenoma size during monitoring, loss of endocrinologic function due to mass effect on other pituitary cells, or unremitting headache.2
Next: How should patients be monitored?
HOW SHOULD PATIENTS BE MONITORED?
Those who do not meet criteria for surgery can be closely monitored with periodic testing. Imaging can be repeated six months after the first scan for macroadenoma and in one year for microadenoma. If there is no change in the size of the mass, imaging can be done yearly for macroadenoma and for microadenoma, every one to two years for three years and then gradually less often thereafter.2
Unless the lesion is abutting the optic chiasm (seen via imaging) or the patient reports symptoms, visual field testing does not need to be repeated.
Lab testing should be repeated six months after initial testing for macroadenoma and yearly thereafter. No further testing is suggested for nonsecretory microadenoma, unless clinically indicated.2
If there are any changes in status—noted clinically or via imaging—more frequent testing is suggested.
Brian is reassured that pituitary adenoma is not an uncommon finding and that his adenoma is relatively small in size and nonsecretory. Repeat pituitary MRI in one year is recommended.
CONCLUSION
Most pituitary incidentalomas have no consequences to a patient’s health. However, patients often become highly anxious about the “brain tumor” they were told they have. Appropriate patient education and thorough evaluation can reassure patients and alleviate their concerns.
REFERENCES
1. Molitch ME. Nonfunctioning pituitary tumors and pituitary incidentalomas. Endocrinol Metab Clin North Am. 2008;37(1):151-171.
2. Freda PU, Beckers AM, Katznelson L, et al. Pituitary incidentaloma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96(4):894-904.
3. Katznelson L, Atkinson JL, Cook DM, et al. American Association of Clinical Endocrinologists Medical Guidelines For Clinical Practice For The Diagnosis And Treatment Of Acromegaly–2011 Update. Endocr Pract. 2011;17(suppl 4).
4. Jameson JL. Harrison’s Endocrinology. 2nd ed. China: McGraw-Hill; 2010:16-49.
Brian, 46, is referred to endocrinology for evaluation of a pituitary “mass.” The mass was an incidental finding of head and neck CT performed three months ago, when Brian went to an emergency department following a motor vehicle accident. He has fully recovered from the accident and feels well. He describes himself as a “completely healthy” person who has no chronic medical conditions and takes neither prescription nor OTC medications.
Brian denies significant headache, visual disturbance, change in appetite, unexplained weight change, skin rash (wide purple striae) or color changes (hyperpigmentation), polyuria or polydipsia, dizziness, syncopal episodes, low libido, erectile dysfunction, joint pain, and changes in ring or shoe size. He does not wear a hat or cap and is unaware of head size changes. He has not experienced changes in his facial features or trouble with chewing.
He is a happily married engineer with two healthy children and reports that he feels well except for this “brain tumor” finding that has been a shock to him and his family. There is no family history of pituitary adenoma or multiple endocrine neoplasia syndrome.
His vital signs, all within normal ranges, include a blood pressure of 103/65 mm Hg. His height is 6 ft and his weight, 180 lb. His BMI is 24.4.
HOW COMMON IS PITUITARY INCIDENTALOMA?
A pituitary incidentaloma is a lesion in the pituitary gland that was not previously suspected and was found through an imaging study ordered for other reasons. Pituitary incidentaloma is surprisingly common, with an average prevalence of 10.6% (as estimated from combined autopsy data), although it has been found in up to 20% of patients undergoing CT and 38% undergoing MRI.1,2 Most are microadenomas (< 1 cm in size).1
Continue for recommendations from the Endocrine Society >>
SHOULD AN ASYMPTOMATIC PATIENT BE EVALUATED FURTHER?
Endocrine Society guidelines2 recommend that all patients with pituitary incidentaloma, with or without symptoms, should undergo a complete history and physical examination and laboratory evaluation to exclude hypersecretion and hyposecretion of pituitary hormones.
The “classic” presentation of pituitary hormone hypersecretion—in the form of prolactinoma, adrenocorticotropic hormone (ACTH) excess (Cushing disease), growth hormone (GH) excess (gigantism/acromegaly), and TSH excess (secondary hyperthyroidism)—may be readily detectable on history and physical examination. Subtle cases, so-called subclinical disease, however, may exhibit little or no signs and symptoms initially but can be detrimental to the patient’s health if left untreated. For example, the estimated time from onset to diagnosis of acromegaly is approximately seven to 10 years—a delay that can significantly impact the patient’s morbidity and mortality.3
Prolactinoma can be more clinically apparent in premenopausal females due to irregular menstrual cycles (oligomenorrhea/amenorrhea). However, galactorrhea, or “milky” nipple discharge, occurs in only about 50% of women with prolactinoma and is extremely rare in men. Furthermore, the clinical presentation of prolactinoma in men is vague and related to hypogonadism, resulting from increased prolactin levels. Since men are essentially asymptomatic, these tumors can grow extensively (macroadenoma) and cause “mass effect,” such as headaches and visual impairment.
Therefore, without laboratory testing, abnormal pituitary function may go unrecognized.
WHAT LABS SHOULD I ORDER FOR THIS PATIENT?
Guidelines suggest an initial screening panel of prolactin, GH, insulin-like growth factor 1 (IGF-1), ACTH, early-morning cortisol, TSH, free T4, luteinizing hormone (LH), follicle-stimulating hormone (FSH), and testosterone.2
Note the use of “suggest” rather than “recommend.” Even among guideline task force members, there were differences in opinion as to whether certain tests (eg, TSH, LH, and FSH) should be included in initial screening. Those tests can be ordered at the clinician’s discretion, according to the level of suspicion, or can be added later if necessary.
Brian’s sample for laboratory testing is drawn at 7:50 am. Results can be found in the table (previous page).
Next page: Caveats and concerns >>
ARE THERE ANY CAVEATS TO THE INTERPRETATION OF LAB VALUES?
It is important to note that in postmenopausal women who are not taking hormone replacement therapy, LH and FSH will be elevated and estradiol may provide an additional clue in the detection of abnormal function. Conversely, low LH and FSH in postmenopausal women should raise a flag for hypopituitarism.
Another caveat is that GH secretion is pulsatile and serum levels are undetectable between pulses. Therefore, low/undetectable GH does not necessarily suggest deficiency. The GH measurement would only be helpful if it is significantly elevated (suggestive of hypersecretion—gigantism/acromegaly). Otherwise, GH has little value as a screening test.
Instead, IGF-1, which is secreted from the liver in response to GH secretion, has a longer half-life and serves as a better screening tool. IGF-1 has age- and sex-adjusted reference ranges, which are often reported by the lab or given as a Z score.
WHAT IS THE PREFERRED IMAGING STUDY FOR THE PITUITARY GLAND?
The best choice is MRI of the pituitary gland (not the whole brain) with gadolinium. If the incidentaloma was initially diagnosed by a CT, additional testing with MRI should be performed, unless contraindicated.2
Brian is referred for MRI with gadolinium. The radiologist’s report describes a 5 x 4 x 4–mm pituitary microadenoma without sellar extension or involvement of the optic chiasm.
AT WHAT POINT SHOULD OPTIC CHIASM BE A CONCERN?
Since the pituitary gland is located directly beneath the optic chiasm, any compressive effect of growth against the optic nerve(s) can cause visual impairment. This includes bitemporal hemianopsia (loss of peripheral vision) or ophthalmoplegia (abnormal movement of the ocular muscle). Since clinical signs and symptoms can be subtle or absent, all patients with evidence of a pituitary lesion abutting or compressing the optic chiasm should have a formal visual field exam.2
Continue for surgical intervention >>
WHO REQUIRES SURGICAL INTERVENTION?
Patients with mass effect (headache, increased intracranial pressure, compromised optic chiasm) and those with hyperfunctioning nonprolactin adenomas, some (but not all) macroprolactinomas, or pituitary apoplexy should be referred for surgery.2 Almost all cases involve macroadenomas rather than microadenomas.
The preferred treatment for GH-secreting tumors and ACTH-secreting tumors is surgery. However, prolactinoma can be well controlled with pharmacologic agents (dopamine agonists) in most cases. For prolactinomas refractory to these medications, surgical resection is recommended. (Detailed treatment approaches are available elsewhere; those for hyperprolactinoma can be found on the Clinician Reviews website: http://bit.ly/1HOb9Jf.)
Pituitary apoplexy, a life-threatening emergency that requires prompt surgical decompression, is an infarction of the gland due to abrupt cessation of the blood supply, caused by either pituitary artery hemorrhage or sudden hypovolemia. Increased blood supply is needed due to the extra tissue and volume of the pituitary mass; this may stress the pituitary arteries, which are not equipped for this increased flow, causing them to rupture. Hemorrhage anywhere else in the body can lead to hypovolemia and decrease the blood supply to the pituitary gland. A classic example would be postpartum hemorrhage causing pituitary infarct, called Sheehan syndrome.
Due to increased estrogen levels, the pituitary gland doubles in size during pregnancy.4 A preexisting mass may further develop and compress the optic chiasm. Therefore, women of childbearing age should be engaged in discussion of the potential risks and benefits of decompression surgery before actively pursuing pregnancy—especially if the lesion is close to the optic chiasm.
Surgery can also be considered for patients with significant growth in adenoma size during monitoring, loss of endocrinologic function due to mass effect on other pituitary cells, or unremitting headache.2
Next: How should patients be monitored?
HOW SHOULD PATIENTS BE MONITORED?
Those who do not meet criteria for surgery can be closely monitored with periodic testing. Imaging can be repeated six months after the first scan for macroadenoma and in one year for microadenoma. If there is no change in the size of the mass, imaging can be done yearly for macroadenoma and for microadenoma, every one to two years for three years and then gradually less often thereafter.2
Unless the lesion is abutting the optic chiasm (seen via imaging) or the patient reports symptoms, visual field testing does not need to be repeated.
Lab testing should be repeated six months after initial testing for macroadenoma and yearly thereafter. No further testing is suggested for nonsecretory microadenoma, unless clinically indicated.2
If there are any changes in status—noted clinically or via imaging—more frequent testing is suggested.
Brian is reassured that pituitary adenoma is not an uncommon finding and that his adenoma is relatively small in size and nonsecretory. Repeat pituitary MRI in one year is recommended.
CONCLUSION
Most pituitary incidentalomas have no consequences to a patient’s health. However, patients often become highly anxious about the “brain tumor” they were told they have. Appropriate patient education and thorough evaluation can reassure patients and alleviate their concerns.
REFERENCES
1. Molitch ME. Nonfunctioning pituitary tumors and pituitary incidentalomas. Endocrinol Metab Clin North Am. 2008;37(1):151-171.
2. Freda PU, Beckers AM, Katznelson L, et al. Pituitary incidentaloma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96(4):894-904.
3. Katznelson L, Atkinson JL, Cook DM, et al. American Association of Clinical Endocrinologists Medical Guidelines For Clinical Practice For The Diagnosis And Treatment Of Acromegaly–2011 Update. Endocr Pract. 2011;17(suppl 4).
4. Jameson JL. Harrison’s Endocrinology. 2nd ed. China: McGraw-Hill; 2010:16-49.
Targeting the Kidneys to Improve Glycemic Control
A 37-year-old woman with a history of papillary carcinoma (status post total thyroidectomy 12 years ago, with negative recurrence) presents for a check-up. She also has polycystic ovarian syndrome (PCOS) with obesity and is taking metformin XR (one 500-mg tablet bid). Her visit is uneventful, and she leaves the office with an order for labwork.
Results indicate normal thyroid function and negative thyroglobulin. However, her serum glucose level is 350 mg/dL, so the patient is called and informed of the result. She denies polyphagia, polydipsia, and polyuria. Repeat blood work confirms overt hyperglycemia (320 mg/dL) with an A1C of 13%, undetectable C-peptide, and negative glutamic acid decarboxylase 65 (GAD65) and islet cell antibodies.
She is advised to increase her metformin dose (to two 500-mg tablets bid) and is started on insulin detemir (20 U every evening), with instructions to increase the latter by three units every two to three days until a target fasting glucose level of 100 to 140 mg/dL is achieved. She is also advised to follow a low-carbohydrate diet and increase her exercise.
The patient returns in two weeks for follow-up. She remains asymptomatic and has now increased her insulin detemir to 34 U bid (she started splitting the dosage after it reached 50 U/d). However, her glucose is still in the low 200s in the morning and the high 200s during the day (after lunch and dinner).
Her overt hyperglycemia is most likely a result of her longstanding insulin resistance, essential lack of b-cell function, and PCOS-associated obesity. Once diabetes from autoimmunity is ruled out by laboratory findings (negative antibodies) and clinical assessment (classic metabolic syndrome features), we focus on her glycemic control.
Even with nearly 70 U/d of insulin, the patient’s glycemic improvement is disappointing, suggesting significant insulin resistance and glucose toxicity. Living in an era with numerous classes of antidiabetic medications, we have lengthy discussions on treatment options. Canagliflozin, recently (at the time) approved, is included. The patient is interested in this new medication, and it is a reasonable choice to get her out of the glucotoxic phase.
After a discussion of benefits and potential adverse effects, she is placed on canagliflozin 100 mg/d. Her glucose log in one week shows fasting glucose values in the range of 140 to 160 mg/dL and postprandial glucose values in the 180s. As a result, she lowers her insulin to 25 U bid. Her renal panel shows a potassium level of 4.3 mEq/L (reference range, 3.5 to 5.3) and a glomerular filtration rate (GFR) of 103 mL/min/1.73 m2. She is advised to further increase her canagliflozin to 300 mg and slowly titrate her insulin down as needed, with a target fasting glucose level of 80 to 110 mg/dL and a postprandial target of 100 to 140 mg/dL.
What are SGLT2 inhibitors, and how do they work?
What are SGLT2 inhibitors, and how do they work?
Sodium-GLucose co-Transporter 2 (SGLT2) inhibitors are a new class of antihyperglycemic agent. The first, canagliflozin, was approved by the FDA in March 2013, followed by dapagliflozin (January 2014) and empagliflozin (August 2014).
As glucose is filtered through the nephrons of the kidney, about 90% is reabsorbed via SGLT2 in the proximal tubule (SGLT1 is responsible for the remaining 10%) so that glucose calories are not eliminated through urine.1 In a healthy person, the renal glucose threshold is about 180 mg/dL.1 When blood glucose exceeds this level, glucose is excreted into the urine. However, in diabetic patients, this threshold is higher due to the up-regulation of SGLT2s (and other glucose transporters), which worsens hyperglycemia.1 SGLT2 inhibitors will reset the threshold, which in turn will increase glucosuria and thereby lower serum glucose.1
SGLT2 inhibitors lower A1C by about 0.7% to 0.8%.2 Independent of other mechanisms such as the degree of b-cell function or insulin resistance, these agents can be used regardless of the duration of diabetes3 if the GFR is intact (≥ 45 mL/min/1.73 m2 for canagliflozin and empagliflozin, ≥ 60 mL/min/ 1.73 m2 for dapagliflozin).4,5
What are the risks and benefits associated with these agents?
What are the risks and benefits associated with these agents?
Modest weight loss is seen with the use of SGLT2 inhibitors. Initial weight loss is believed to be related to volume loss, but more sustained weight loss is thought to be from loss of fat mass.6 This is not surprising, as excreting glucose means excreting calories through urine.
Risk for hypoglycemia is extremely low, which makes this therapeutic class an attractive option. However, caution should be exercised when SGLT2 inhibitors are combined with other agents known to cause hypoglycemia (sulfonylureas and insulin).6
The most common adverse effect is genital mycotic infection. Women with a history of recurrent genital mycotic infection and uncircumcised men are at the greatest risk.6
Due to increased glycosuria, which results in an osmotic diuresis, modest blood pressure improvement has been seen (3 to 4 mm Hg systolic and 1 to 2 mm Hg diastolic7,8) in patients taking SGLT2 inhibitors, which is an additional benefit for hypertensive diabetic patients.6 On the other hand, use of SGLT2 inhibitors can also cause dehydration and volume depletion and can raise serum creatinine in patients who are already taking diuretics (particularly loop diuretics).6 Drug tolerance and adherence can be improved by advising patients to expect transient increased urination (approximately 135 to 350 mL/d increase from baseline5,9) and emphasizing the importance of good hydration and maintaining good genital hygiene.
A slight increase in LDL cholesterol was seen in clinical trials of the SGLT2 inhibitors, although this phenomenon is poorly understood. However, HDL cholesterol increased as well, maintaining the LDL:HDL ratio.6 No long-term cardiovascular outcome data are available at this time; as with any new antidiabetic medication, postmarketing studies, as required by the FDA, are currently ongoing.6
What are the options in this therapeutic category, and how are they distinct?
What are the options in this therapeutic category, and how are they distinct?
As mentioned previously, there are currently three SGLT2 inhibitors on the market: canagliflozin, dapagliflozin, and empagliflozin. There are subtle clinical differences among these three agents, which might direct the clinician’s choice.
First, canagliflozin is available in dosages of 100 and 300 mg. The starting dosage is 100 mg, which can be titrated to 300 mg in patients with a GFR ≥ 60 mL/min/1.73 m2 who require a greater glucose-lowering effect. Those with a GFR < 60 mL/min/1.73 m2 but ≥ 45 mL/min/1.73 m2 are limited to the 100-mg dosage. Dapagliflozin is available in 5-mg and 10-mg dosages, the former being the starting dosage. But dapagliflozin is not recommended in patients whose GFR is < 60 mL/min/1.73 m2.4
Empagliflozin is available in dosages of 10 and 25 mg. The starting dosage of 10 mg can be increased to 25 mg if the patient has not achieved his/her target glucose level. Either can be used in patients with a GFR ≥ 45 mL/min/1.73 m2.5
Second, hyperkalemia was seen in patients taking canagliflozin but not in those taking dapagliflozin or empagliflozin. Therefore, serum potassium should be monitored and caution used, especially when patients are being treated with potassium-sparing diuretics and/or ACE inhibitors or angiotensin II receptor blockers.6
Third, dapagliflozin carries a warning for bladder cancer, as higher rates of newly diagnosed bladder cancer were seen with this drug compared with placebo or comparator drugs (0.17% vs 0.03%, respectively).4 However, this finding may have resulted from a randomization imbalance of patients in the study, and further research is needed to clarify this risk.6 It is not recommended that dapagliflozin be used in patients with active or a history of bladder cancer at this time.
With these agents, there is a paradoxical rise in glucagon that increases endogenous glucose production from the liver.10 The mechanism is poorly understood, but it might be due to the body’s compensatory (survival) mechanism to “make up” the loss of glucose through urine by increasing hepatic gluconeogenesis.
Using an incretin agent, such as dipeptidyl peptidase 4 (DPP-4) inhibitors or glucagon-like peptide 1 (GLP-1) receptor agonists, in conjunction with an SGLT2 inhibitor, has been suggested as a way to potentiate the glucose-lowering effect, as it may attenuate the paradoxical rise in glucagon.10 Since the incretin class is weight neutral (DPP-4 inhibitors) or associated with weight loss (GLP-1 agonists), using incretins with SGLT2 inhibitors might produce more significant weight loss, which has numerous additional benefits for diabetic patients.
SGLT2 inhibitors are currently approved as an adjunct to diet and exercise for patients with type 2 diabetes. They are not approved for those with type 1 diabetes, although the mechanism of action of these drugs (which is independent of the b-cell function) might make them effective in this population. Active pilot studies of this patient population are in progress.11
Conclusion
In summary, SGLT2 inhibitors are an exciting new class of antidiabetic medication that offers a unique mechanism to lower serum glucose. It is the only medication that will actually remove glucose from the body; by contrast, all other existing antidiabetic medications move glucose within the body (to liver, fat, muscle, etc).
There is no curative medication for diabetes. But with an increasing diabetic population and an emphasis on individualizing antihyperglycemic regimens, we always welcome medications with novel mechanisms of action. Due to SLGT2 inhibitors’ recent approval, however, short-term and long-term adverse effects are unknown, and ongoing postmarketing surveillance should be closely followed.
References
1. Abdul-Ghani MA, DeFronzo RA. Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus. Endocr Pract. 2008;14:782-790.
2. Berhan A, Barker A. Sodium glucose co-transport 2 inhibitors in the treatment of type 2 diabetes mellitus: a meta-analysis of randomized double-blind controlled trials. BMC Endocr Disord. 2013;13(1):58.
3. Wilding JP, Norwood P, T’joen C, et al. A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers. Diabetes Care. 2009;32:1656-1662.
4. Taylor JR. Dapagliflozin offers differences from other SGLT2 inhibitors. Endocrine Today. May 2014.
5. Jardiance [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; 2014.
6. Bakris G, Fonseca VA, Peters AL, Wysham CH. Clinical perspectives on the role of the kidney in the pathophysiology of T2DM: emerging options for treatment [video series]. 2013. www.thedoctorschannel.com/view/the-kid ney-in-t2dm-cme-part-1/. Accessed September 12, 2014.
7. Vercruysse F. Efficacy and safety of canagliflozin in subjects with type 2 diabetes mellitus inadequately controlled with metformin plus sulphonylurea over 52 weeks [abstract 934]. Presented at the 49th European Association for the Study of Diabetes Annual Meeting: Barcelona; September 24, 2013.
8. Hach T. Empagliflozin improves glycaemic parameters and cardiovascular risk factors in patients with type 2 diabetes: pooled data from four pivotal phase III trials [abstract 943]. Presented at the 49th European Association for the Study of Diabetes Annual Meeting: Barcelona; September 24, 2013.
9. List JF, Woo V, Morales E, et al. Sodium-glucose co-transport inhibition with dapagliflozin in type 2 diabetes mellitus. Diabetes Care. 2009;32(4):650-657.
10. Merovci A, Solis-Herrera C, Daniele G, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. 2014;124(5):2287.
11. Perkins BA, Cherney DZ, Partridge H, et al. Sodium-glucose cotransporter 2 inhibition and glycemic control in type 1 diabetes: results of an 8-week open-label proof-of-concept trial. Diabetes Care. 2014;37(5):1480-1483.
Ji Hyun Chun practices at Endocrinology Associates in Scottsdale, Arizona, and is a faculty member in the Arizona School of Health Sciences at AT Still University.
Clinician Reviews in partnership with![]()
Ji Hyun Chun practices at Endocrinology Associates in Scottsdale, Arizona, and is a faculty member in the Arizona School of Health Sciences at AT Still University.
Clinician Reviews in partnership with![]()
Ji Hyun Chun practices at Endocrinology Associates in Scottsdale, Arizona, and is a faculty member in the Arizona School of Health Sciences at AT Still University.
Clinician Reviews in partnership with![]()
A 37-year-old woman with a history of papillary carcinoma (status post total thyroidectomy 12 years ago, with negative recurrence) presents for a check-up. She also has polycystic ovarian syndrome (PCOS) with obesity and is taking metformin XR (one 500-mg tablet bid). Her visit is uneventful, and she leaves the office with an order for labwork.
Results indicate normal thyroid function and negative thyroglobulin. However, her serum glucose level is 350 mg/dL, so the patient is called and informed of the result. She denies polyphagia, polydipsia, and polyuria. Repeat blood work confirms overt hyperglycemia (320 mg/dL) with an A1C of 13%, undetectable C-peptide, and negative glutamic acid decarboxylase 65 (GAD65) and islet cell antibodies.
She is advised to increase her metformin dose (to two 500-mg tablets bid) and is started on insulin detemir (20 U every evening), with instructions to increase the latter by three units every two to three days until a target fasting glucose level of 100 to 140 mg/dL is achieved. She is also advised to follow a low-carbohydrate diet and increase her exercise.
The patient returns in two weeks for follow-up. She remains asymptomatic and has now increased her insulin detemir to 34 U bid (she started splitting the dosage after it reached 50 U/d). However, her glucose is still in the low 200s in the morning and the high 200s during the day (after lunch and dinner).
Her overt hyperglycemia is most likely a result of her longstanding insulin resistance, essential lack of b-cell function, and PCOS-associated obesity. Once diabetes from autoimmunity is ruled out by laboratory findings (negative antibodies) and clinical assessment (classic metabolic syndrome features), we focus on her glycemic control.
Even with nearly 70 U/d of insulin, the patient’s glycemic improvement is disappointing, suggesting significant insulin resistance and glucose toxicity. Living in an era with numerous classes of antidiabetic medications, we have lengthy discussions on treatment options. Canagliflozin, recently (at the time) approved, is included. The patient is interested in this new medication, and it is a reasonable choice to get her out of the glucotoxic phase.
After a discussion of benefits and potential adverse effects, she is placed on canagliflozin 100 mg/d. Her glucose log in one week shows fasting glucose values in the range of 140 to 160 mg/dL and postprandial glucose values in the 180s. As a result, she lowers her insulin to 25 U bid. Her renal panel shows a potassium level of 4.3 mEq/L (reference range, 3.5 to 5.3) and a glomerular filtration rate (GFR) of 103 mL/min/1.73 m2. She is advised to further increase her canagliflozin to 300 mg and slowly titrate her insulin down as needed, with a target fasting glucose level of 80 to 110 mg/dL and a postprandial target of 100 to 140 mg/dL.
What are SGLT2 inhibitors, and how do they work?
What are SGLT2 inhibitors, and how do they work?
Sodium-GLucose co-Transporter 2 (SGLT2) inhibitors are a new class of antihyperglycemic agent. The first, canagliflozin, was approved by the FDA in March 2013, followed by dapagliflozin (January 2014) and empagliflozin (August 2014).
As glucose is filtered through the nephrons of the kidney, about 90% is reabsorbed via SGLT2 in the proximal tubule (SGLT1 is responsible for the remaining 10%) so that glucose calories are not eliminated through urine.1 In a healthy person, the renal glucose threshold is about 180 mg/dL.1 When blood glucose exceeds this level, glucose is excreted into the urine. However, in diabetic patients, this threshold is higher due to the up-regulation of SGLT2s (and other glucose transporters), which worsens hyperglycemia.1 SGLT2 inhibitors will reset the threshold, which in turn will increase glucosuria and thereby lower serum glucose.1
SGLT2 inhibitors lower A1C by about 0.7% to 0.8%.2 Independent of other mechanisms such as the degree of b-cell function or insulin resistance, these agents can be used regardless of the duration of diabetes3 if the GFR is intact (≥ 45 mL/min/1.73 m2 for canagliflozin and empagliflozin, ≥ 60 mL/min/ 1.73 m2 for dapagliflozin).4,5
What are the risks and benefits associated with these agents?
What are the risks and benefits associated with these agents?
Modest weight loss is seen with the use of SGLT2 inhibitors. Initial weight loss is believed to be related to volume loss, but more sustained weight loss is thought to be from loss of fat mass.6 This is not surprising, as excreting glucose means excreting calories through urine.
Risk for hypoglycemia is extremely low, which makes this therapeutic class an attractive option. However, caution should be exercised when SGLT2 inhibitors are combined with other agents known to cause hypoglycemia (sulfonylureas and insulin).6
The most common adverse effect is genital mycotic infection. Women with a history of recurrent genital mycotic infection and uncircumcised men are at the greatest risk.6
Due to increased glycosuria, which results in an osmotic diuresis, modest blood pressure improvement has been seen (3 to 4 mm Hg systolic and 1 to 2 mm Hg diastolic7,8) in patients taking SGLT2 inhibitors, which is an additional benefit for hypertensive diabetic patients.6 On the other hand, use of SGLT2 inhibitors can also cause dehydration and volume depletion and can raise serum creatinine in patients who are already taking diuretics (particularly loop diuretics).6 Drug tolerance and adherence can be improved by advising patients to expect transient increased urination (approximately 135 to 350 mL/d increase from baseline5,9) and emphasizing the importance of good hydration and maintaining good genital hygiene.
A slight increase in LDL cholesterol was seen in clinical trials of the SGLT2 inhibitors, although this phenomenon is poorly understood. However, HDL cholesterol increased as well, maintaining the LDL:HDL ratio.6 No long-term cardiovascular outcome data are available at this time; as with any new antidiabetic medication, postmarketing studies, as required by the FDA, are currently ongoing.6
What are the options in this therapeutic category, and how are they distinct?
What are the options in this therapeutic category, and how are they distinct?
As mentioned previously, there are currently three SGLT2 inhibitors on the market: canagliflozin, dapagliflozin, and empagliflozin. There are subtle clinical differences among these three agents, which might direct the clinician’s choice.
First, canagliflozin is available in dosages of 100 and 300 mg. The starting dosage is 100 mg, which can be titrated to 300 mg in patients with a GFR ≥ 60 mL/min/1.73 m2 who require a greater glucose-lowering effect. Those with a GFR < 60 mL/min/1.73 m2 but ≥ 45 mL/min/1.73 m2 are limited to the 100-mg dosage. Dapagliflozin is available in 5-mg and 10-mg dosages, the former being the starting dosage. But dapagliflozin is not recommended in patients whose GFR is < 60 mL/min/1.73 m2.4
Empagliflozin is available in dosages of 10 and 25 mg. The starting dosage of 10 mg can be increased to 25 mg if the patient has not achieved his/her target glucose level. Either can be used in patients with a GFR ≥ 45 mL/min/1.73 m2.5
Second, hyperkalemia was seen in patients taking canagliflozin but not in those taking dapagliflozin or empagliflozin. Therefore, serum potassium should be monitored and caution used, especially when patients are being treated with potassium-sparing diuretics and/or ACE inhibitors or angiotensin II receptor blockers.6
Third, dapagliflozin carries a warning for bladder cancer, as higher rates of newly diagnosed bladder cancer were seen with this drug compared with placebo or comparator drugs (0.17% vs 0.03%, respectively).4 However, this finding may have resulted from a randomization imbalance of patients in the study, and further research is needed to clarify this risk.6 It is not recommended that dapagliflozin be used in patients with active or a history of bladder cancer at this time.
With these agents, there is a paradoxical rise in glucagon that increases endogenous glucose production from the liver.10 The mechanism is poorly understood, but it might be due to the body’s compensatory (survival) mechanism to “make up” the loss of glucose through urine by increasing hepatic gluconeogenesis.
Using an incretin agent, such as dipeptidyl peptidase 4 (DPP-4) inhibitors or glucagon-like peptide 1 (GLP-1) receptor agonists, in conjunction with an SGLT2 inhibitor, has been suggested as a way to potentiate the glucose-lowering effect, as it may attenuate the paradoxical rise in glucagon.10 Since the incretin class is weight neutral (DPP-4 inhibitors) or associated with weight loss (GLP-1 agonists), using incretins with SGLT2 inhibitors might produce more significant weight loss, which has numerous additional benefits for diabetic patients.
SGLT2 inhibitors are currently approved as an adjunct to diet and exercise for patients with type 2 diabetes. They are not approved for those with type 1 diabetes, although the mechanism of action of these drugs (which is independent of the b-cell function) might make them effective in this population. Active pilot studies of this patient population are in progress.11
Conclusion
In summary, SGLT2 inhibitors are an exciting new class of antidiabetic medication that offers a unique mechanism to lower serum glucose. It is the only medication that will actually remove glucose from the body; by contrast, all other existing antidiabetic medications move glucose within the body (to liver, fat, muscle, etc).
There is no curative medication for diabetes. But with an increasing diabetic population and an emphasis on individualizing antihyperglycemic regimens, we always welcome medications with novel mechanisms of action. Due to SLGT2 inhibitors’ recent approval, however, short-term and long-term adverse effects are unknown, and ongoing postmarketing surveillance should be closely followed.
References
1. Abdul-Ghani MA, DeFronzo RA. Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus. Endocr Pract. 2008;14:782-790.
2. Berhan A, Barker A. Sodium glucose co-transport 2 inhibitors in the treatment of type 2 diabetes mellitus: a meta-analysis of randomized double-blind controlled trials. BMC Endocr Disord. 2013;13(1):58.
3. Wilding JP, Norwood P, T’joen C, et al. A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers. Diabetes Care. 2009;32:1656-1662.
4. Taylor JR. Dapagliflozin offers differences from other SGLT2 inhibitors. Endocrine Today. May 2014.
5. Jardiance [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; 2014.
6. Bakris G, Fonseca VA, Peters AL, Wysham CH. Clinical perspectives on the role of the kidney in the pathophysiology of T2DM: emerging options for treatment [video series]. 2013. www.thedoctorschannel.com/view/the-kid ney-in-t2dm-cme-part-1/. Accessed September 12, 2014.
7. Vercruysse F. Efficacy and safety of canagliflozin in subjects with type 2 diabetes mellitus inadequately controlled with metformin plus sulphonylurea over 52 weeks [abstract 934]. Presented at the 49th European Association for the Study of Diabetes Annual Meeting: Barcelona; September 24, 2013.
8. Hach T. Empagliflozin improves glycaemic parameters and cardiovascular risk factors in patients with type 2 diabetes: pooled data from four pivotal phase III trials [abstract 943]. Presented at the 49th European Association for the Study of Diabetes Annual Meeting: Barcelona; September 24, 2013.
9. List JF, Woo V, Morales E, et al. Sodium-glucose co-transport inhibition with dapagliflozin in type 2 diabetes mellitus. Diabetes Care. 2009;32(4):650-657.
10. Merovci A, Solis-Herrera C, Daniele G, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. 2014;124(5):2287.
11. Perkins BA, Cherney DZ, Partridge H, et al. Sodium-glucose cotransporter 2 inhibition and glycemic control in type 1 diabetes: results of an 8-week open-label proof-of-concept trial. Diabetes Care. 2014;37(5):1480-1483.
A 37-year-old woman with a history of papillary carcinoma (status post total thyroidectomy 12 years ago, with negative recurrence) presents for a check-up. She also has polycystic ovarian syndrome (PCOS) with obesity and is taking metformin XR (one 500-mg tablet bid). Her visit is uneventful, and she leaves the office with an order for labwork.
Results indicate normal thyroid function and negative thyroglobulin. However, her serum glucose level is 350 mg/dL, so the patient is called and informed of the result. She denies polyphagia, polydipsia, and polyuria. Repeat blood work confirms overt hyperglycemia (320 mg/dL) with an A1C of 13%, undetectable C-peptide, and negative glutamic acid decarboxylase 65 (GAD65) and islet cell antibodies.
She is advised to increase her metformin dose (to two 500-mg tablets bid) and is started on insulin detemir (20 U every evening), with instructions to increase the latter by three units every two to three days until a target fasting glucose level of 100 to 140 mg/dL is achieved. She is also advised to follow a low-carbohydrate diet and increase her exercise.
The patient returns in two weeks for follow-up. She remains asymptomatic and has now increased her insulin detemir to 34 U bid (she started splitting the dosage after it reached 50 U/d). However, her glucose is still in the low 200s in the morning and the high 200s during the day (after lunch and dinner).
Her overt hyperglycemia is most likely a result of her longstanding insulin resistance, essential lack of b-cell function, and PCOS-associated obesity. Once diabetes from autoimmunity is ruled out by laboratory findings (negative antibodies) and clinical assessment (classic metabolic syndrome features), we focus on her glycemic control.
Even with nearly 70 U/d of insulin, the patient’s glycemic improvement is disappointing, suggesting significant insulin resistance and glucose toxicity. Living in an era with numerous classes of antidiabetic medications, we have lengthy discussions on treatment options. Canagliflozin, recently (at the time) approved, is included. The patient is interested in this new medication, and it is a reasonable choice to get her out of the glucotoxic phase.
After a discussion of benefits and potential adverse effects, she is placed on canagliflozin 100 mg/d. Her glucose log in one week shows fasting glucose values in the range of 140 to 160 mg/dL and postprandial glucose values in the 180s. As a result, she lowers her insulin to 25 U bid. Her renal panel shows a potassium level of 4.3 mEq/L (reference range, 3.5 to 5.3) and a glomerular filtration rate (GFR) of 103 mL/min/1.73 m2. She is advised to further increase her canagliflozin to 300 mg and slowly titrate her insulin down as needed, with a target fasting glucose level of 80 to 110 mg/dL and a postprandial target of 100 to 140 mg/dL.
What are SGLT2 inhibitors, and how do they work?
What are SGLT2 inhibitors, and how do they work?
Sodium-GLucose co-Transporter 2 (SGLT2) inhibitors are a new class of antihyperglycemic agent. The first, canagliflozin, was approved by the FDA in March 2013, followed by dapagliflozin (January 2014) and empagliflozin (August 2014).
As glucose is filtered through the nephrons of the kidney, about 90% is reabsorbed via SGLT2 in the proximal tubule (SGLT1 is responsible for the remaining 10%) so that glucose calories are not eliminated through urine.1 In a healthy person, the renal glucose threshold is about 180 mg/dL.1 When blood glucose exceeds this level, glucose is excreted into the urine. However, in diabetic patients, this threshold is higher due to the up-regulation of SGLT2s (and other glucose transporters), which worsens hyperglycemia.1 SGLT2 inhibitors will reset the threshold, which in turn will increase glucosuria and thereby lower serum glucose.1
SGLT2 inhibitors lower A1C by about 0.7% to 0.8%.2 Independent of other mechanisms such as the degree of b-cell function or insulin resistance, these agents can be used regardless of the duration of diabetes3 if the GFR is intact (≥ 45 mL/min/1.73 m2 for canagliflozin and empagliflozin, ≥ 60 mL/min/ 1.73 m2 for dapagliflozin).4,5
What are the risks and benefits associated with these agents?
What are the risks and benefits associated with these agents?
Modest weight loss is seen with the use of SGLT2 inhibitors. Initial weight loss is believed to be related to volume loss, but more sustained weight loss is thought to be from loss of fat mass.6 This is not surprising, as excreting glucose means excreting calories through urine.
Risk for hypoglycemia is extremely low, which makes this therapeutic class an attractive option. However, caution should be exercised when SGLT2 inhibitors are combined with other agents known to cause hypoglycemia (sulfonylureas and insulin).6
The most common adverse effect is genital mycotic infection. Women with a history of recurrent genital mycotic infection and uncircumcised men are at the greatest risk.6
Due to increased glycosuria, which results in an osmotic diuresis, modest blood pressure improvement has been seen (3 to 4 mm Hg systolic and 1 to 2 mm Hg diastolic7,8) in patients taking SGLT2 inhibitors, which is an additional benefit for hypertensive diabetic patients.6 On the other hand, use of SGLT2 inhibitors can also cause dehydration and volume depletion and can raise serum creatinine in patients who are already taking diuretics (particularly loop diuretics).6 Drug tolerance and adherence can be improved by advising patients to expect transient increased urination (approximately 135 to 350 mL/d increase from baseline5,9) and emphasizing the importance of good hydration and maintaining good genital hygiene.
A slight increase in LDL cholesterol was seen in clinical trials of the SGLT2 inhibitors, although this phenomenon is poorly understood. However, HDL cholesterol increased as well, maintaining the LDL:HDL ratio.6 No long-term cardiovascular outcome data are available at this time; as with any new antidiabetic medication, postmarketing studies, as required by the FDA, are currently ongoing.6
What are the options in this therapeutic category, and how are they distinct?
What are the options in this therapeutic category, and how are they distinct?
As mentioned previously, there are currently three SGLT2 inhibitors on the market: canagliflozin, dapagliflozin, and empagliflozin. There are subtle clinical differences among these three agents, which might direct the clinician’s choice.
First, canagliflozin is available in dosages of 100 and 300 mg. The starting dosage is 100 mg, which can be titrated to 300 mg in patients with a GFR ≥ 60 mL/min/1.73 m2 who require a greater glucose-lowering effect. Those with a GFR < 60 mL/min/1.73 m2 but ≥ 45 mL/min/1.73 m2 are limited to the 100-mg dosage. Dapagliflozin is available in 5-mg and 10-mg dosages, the former being the starting dosage. But dapagliflozin is not recommended in patients whose GFR is < 60 mL/min/1.73 m2.4
Empagliflozin is available in dosages of 10 and 25 mg. The starting dosage of 10 mg can be increased to 25 mg if the patient has not achieved his/her target glucose level. Either can be used in patients with a GFR ≥ 45 mL/min/1.73 m2.5
Second, hyperkalemia was seen in patients taking canagliflozin but not in those taking dapagliflozin or empagliflozin. Therefore, serum potassium should be monitored and caution used, especially when patients are being treated with potassium-sparing diuretics and/or ACE inhibitors or angiotensin II receptor blockers.6
Third, dapagliflozin carries a warning for bladder cancer, as higher rates of newly diagnosed bladder cancer were seen with this drug compared with placebo or comparator drugs (0.17% vs 0.03%, respectively).4 However, this finding may have resulted from a randomization imbalance of patients in the study, and further research is needed to clarify this risk.6 It is not recommended that dapagliflozin be used in patients with active or a history of bladder cancer at this time.
With these agents, there is a paradoxical rise in glucagon that increases endogenous glucose production from the liver.10 The mechanism is poorly understood, but it might be due to the body’s compensatory (survival) mechanism to “make up” the loss of glucose through urine by increasing hepatic gluconeogenesis.
Using an incretin agent, such as dipeptidyl peptidase 4 (DPP-4) inhibitors or glucagon-like peptide 1 (GLP-1) receptor agonists, in conjunction with an SGLT2 inhibitor, has been suggested as a way to potentiate the glucose-lowering effect, as it may attenuate the paradoxical rise in glucagon.10 Since the incretin class is weight neutral (DPP-4 inhibitors) or associated with weight loss (GLP-1 agonists), using incretins with SGLT2 inhibitors might produce more significant weight loss, which has numerous additional benefits for diabetic patients.
SGLT2 inhibitors are currently approved as an adjunct to diet and exercise for patients with type 2 diabetes. They are not approved for those with type 1 diabetes, although the mechanism of action of these drugs (which is independent of the b-cell function) might make them effective in this population. Active pilot studies of this patient population are in progress.11
Conclusion
In summary, SGLT2 inhibitors are an exciting new class of antidiabetic medication that offers a unique mechanism to lower serum glucose. It is the only medication that will actually remove glucose from the body; by contrast, all other existing antidiabetic medications move glucose within the body (to liver, fat, muscle, etc).
There is no curative medication for diabetes. But with an increasing diabetic population and an emphasis on individualizing antihyperglycemic regimens, we always welcome medications with novel mechanisms of action. Due to SLGT2 inhibitors’ recent approval, however, short-term and long-term adverse effects are unknown, and ongoing postmarketing surveillance should be closely followed.
References
1. Abdul-Ghani MA, DeFronzo RA. Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus. Endocr Pract. 2008;14:782-790.
2. Berhan A, Barker A. Sodium glucose co-transport 2 inhibitors in the treatment of type 2 diabetes mellitus: a meta-analysis of randomized double-blind controlled trials. BMC Endocr Disord. 2013;13(1):58.
3. Wilding JP, Norwood P, T’joen C, et al. A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers. Diabetes Care. 2009;32:1656-1662.
4. Taylor JR. Dapagliflozin offers differences from other SGLT2 inhibitors. Endocrine Today. May 2014.
5. Jardiance [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; 2014.
6. Bakris G, Fonseca VA, Peters AL, Wysham CH. Clinical perspectives on the role of the kidney in the pathophysiology of T2DM: emerging options for treatment [video series]. 2013. www.thedoctorschannel.com/view/the-kid ney-in-t2dm-cme-part-1/. Accessed September 12, 2014.
7. Vercruysse F. Efficacy and safety of canagliflozin in subjects with type 2 diabetes mellitus inadequately controlled with metformin plus sulphonylurea over 52 weeks [abstract 934]. Presented at the 49th European Association for the Study of Diabetes Annual Meeting: Barcelona; September 24, 2013.
8. Hach T. Empagliflozin improves glycaemic parameters and cardiovascular risk factors in patients with type 2 diabetes: pooled data from four pivotal phase III trials [abstract 943]. Presented at the 49th European Association for the Study of Diabetes Annual Meeting: Barcelona; September 24, 2013.
9. List JF, Woo V, Morales E, et al. Sodium-glucose co-transport inhibition with dapagliflozin in type 2 diabetes mellitus. Diabetes Care. 2009;32(4):650-657.
10. Merovci A, Solis-Herrera C, Daniele G, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. 2014;124(5):2287.
11. Perkins BA, Cherney DZ, Partridge H, et al. Sodium-glucose cotransporter 2 inhibition and glycemic control in type 1 diabetes: results of an 8-week open-label proof-of-concept trial. Diabetes Care. 2014;37(5):1480-1483.
Hyperprolactinemia: Causes and Treatments
A 31-year-old woman is referred by her Ob-Gyn for elevated prolactin. She initially presented with a three-month history of amenorrhea, a negative home pregnancy test, and 100% compliance with condom use. She denies hirsutism and acne but admits to thin milky nipple discharge upon squeezing (but not spontaneous).
Two weeks ago, her Ob-Gyn ordered labs; results were negative for serum beta human chorionic gonadotropin and within normal ranges for thyroid-stimulating hormone (TSH), luteinizing hormone, follicle-stimulating hormone, estradiol, free and total testosterone, dehydroepiandrosterone sulfate (DHEAs), complete chemistry panel, and complete blood count. Her serum prolactin level was 110 ng/mL (normal, 3 to 27 ng/mL).
Q: How is prolactin physiologically regulated?
The primary role of prolactin, which is produced by lactotroph cells in the anterior pituitary gland, is to stimulate lactation and breast development. Prolactin is regulated by dopamine (also known as prolactin inhibitory hormone), which is secreted from the hypothalamus via an inhibitory pathway unique to the hypothalamus-pituitary hormone system. Dopamine essentially suppresses prolactin.
Other hormones can have a stimulatory effect on the anterior pituitary gland and thus increase prolactin levels. Estrogen can induce lactotroph hyperplasia and elevated prolactin; however, this is only clinically relevant in the context of estrogen surge during pregnancy. (Estrogen therapy, such as oral contraception or hormone replacement therapy, on the other hand, is targeted to “normal” estrogen levels.) Thyrotropin-releasing hormone (TRH) from the hypothalamus also stimulates the anterior pituitary gland, so patients with inadequately treated or untreated primary hypothyroidism will have mildly elevated prolactin.
Neurogenic stimuli of the chest wall, through nipple suckling or varicella zoster infection (shingles), can also increase prolactin secretion. And since prolactin is eliminated by the liver (75%) and the kidney (25%), significant liver disease and/or renal insufficiency can raise prolactin levels, due to decreased clearance.
What are the possible etiologies for elevated prolactin? See answer on the next page...
Q: What are the possible etiologies for elevated prolactin?
The causes of hyperprolactinemia fall into three categories: physiologic, pharmacologic, and pathologic.2 The table provides examples from each category.
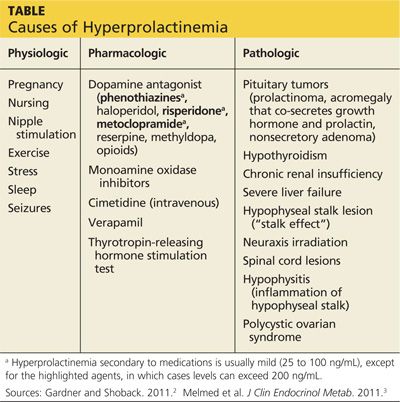
A nonsecretory pituitary adenoma or any lesion in the brain that would disrupt the hypophyseal stalk may interfere with dopamine’s inhibitory control and thereby increase prolactin. This is called the stalk effect. It is important to note that not all MRI-proven pituitary adenomas are prolactin secreting, even in the presence of hyperprolactinemia. According to an autopsy series, about 12% of the general population had pituitary microadenoma.3
There is rough correlation between prolactinoma size and level of prolactin. Large nonsecretory pituitary adenomas have prolactin levels less than 150 ng/mL. Microprolactinomas (< 1 cm) are usually in the range of 100 to 250 ng/mL, while macroprolactinomas (> 1 cm) are generally
≥ 250 ng/mL. If the tumor is very large and invades the cavernous sinus, prolactin can measure in the 1,000s.3
Polycystic ovarian syndrome (PCOS) is a common disorder affecting women of reproductive age and the most common cause of underlying ovulatory problems. Patients with PCOS can have mildly elevated prolactin; the exact mechanism of hyperprolactinemia in PCOS is unknown. One theory is that constant high levels of estrogen experienced in PCOS would stimulate prolactin production. It is important to rule out other causes of hyperprolactinemia before making the diagnosis of PCOS.
What is the clinical significance of elevated prolactin? Why do we have to work up and treat it? See answer on the next page...
Q: What is the clinical significance of elevated prolactin? Why do we have to work up and treat it?
By physiologic mechanisms not completely understood, hyperprolactinemia can interrupt the gonadal axis, leading to hypogonadism. In women, it can cause irregular menstrual cycles, oligomenorrhea, amenorrhea, and infertility. In men, it can lower testosterone levels. Long-term effects include declining bone mineral density due to insufficient estrogen in women or testosterone in men.
With macroadenoma, the size of the tumor can have a mass effect such as headache and visual defect by compressing the optic chiasm (bitemporal hemianopsia), which may lead to permanent vision loss if left untreated. Referral to an ophthalmologist may be necessary for formal visual field examination.
How is hyperprolactinemia treated? See answer on the next page...
Q: How is hyperprolactinemia treated?
There are three options for treatment: medication, surgery, and radiation.
Dopamine agonists (bromocriptine, cabergoline) are effective in normalizing prolactin and reducing the size of the tumor in the majority of cases. However, some patients may require long-term treatment. Bromocriptine has been used since the late 1970s, but, due to better tolerance and less frequent dosing, cabergoline is the preferred agent.3
Transsphenoidal surgery is indicated for patients who are intolerant to medication, who have a medication-resistant tumor or significant mass effect, or who prefer definitive treatment. Women of childbearing age with a macroadenoma might consider surgery due to the risk for tumor expansion during pregnancy (estrogen effect) and risk for pituitary apoplexy (hemorrhage or infarct of the pituitary gland). Surgical risk is usually low with a neurosurgeon who has extensive experience.
Radiation can be considered for large tumors that are resistant to medication. It can be used as adjunctive therapy to surgery, since reducing the size of the tumor can make the surgical field smaller. In some medication-resistant tumors, radiation can raise sensitivity to medication.
What does follow-up entail? See next page for answer...
Q: What does follow-up entail?
Once medication is initiated or dosage is adjusted, have the patient follow up in one month and recheck the prolactin level to assess responsiveness to medication (as well as medication adherence). When a therapeutic prolactin level is achieved, recheck the prolactin and have the patient follow up at three and six months and then every six months thereafter.3
MRI of the pituitary gland should be performed at baseline, then in six months to assess tumor response to medication, and then at 12 and 24 months.3 If tumor regression has stabilized or if the tumor has shrunk to a nondetectable size, consider discontinuing the dopamine agonist. If medication is discontinued, recheck prolactin every three months for the first year; if it remains in normal reference range, simply check serum prolactin annually.3
See next page for summary.

See next page for references.
REFERENCES
1. Jameson JL. Harrison’s Endocrinology. 18th ed. China: McGraw-Hill; 2010.
2. Gardner D, Shoback D. Greenspan’s Basic & Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.
A 31-year-old woman is referred by her Ob-Gyn for elevated prolactin. She initially presented with a three-month history of amenorrhea, a negative home pregnancy test, and 100% compliance with condom use. She denies hirsutism and acne but admits to thin milky nipple discharge upon squeezing (but not spontaneous).
Two weeks ago, her Ob-Gyn ordered labs; results were negative for serum beta human chorionic gonadotropin and within normal ranges for thyroid-stimulating hormone (TSH), luteinizing hormone, follicle-stimulating hormone, estradiol, free and total testosterone, dehydroepiandrosterone sulfate (DHEAs), complete chemistry panel, and complete blood count. Her serum prolactin level was 110 ng/mL (normal, 3 to 27 ng/mL).
Q: How is prolactin physiologically regulated?
The primary role of prolactin, which is produced by lactotroph cells in the anterior pituitary gland, is to stimulate lactation and breast development. Prolactin is regulated by dopamine (also known as prolactin inhibitory hormone), which is secreted from the hypothalamus via an inhibitory pathway unique to the hypothalamus-pituitary hormone system. Dopamine essentially suppresses prolactin.
Other hormones can have a stimulatory effect on the anterior pituitary gland and thus increase prolactin levels. Estrogen can induce lactotroph hyperplasia and elevated prolactin; however, this is only clinically relevant in the context of estrogen surge during pregnancy. (Estrogen therapy, such as oral contraception or hormone replacement therapy, on the other hand, is targeted to “normal” estrogen levels.) Thyrotropin-releasing hormone (TRH) from the hypothalamus also stimulates the anterior pituitary gland, so patients with inadequately treated or untreated primary hypothyroidism will have mildly elevated prolactin.
Neurogenic stimuli of the chest wall, through nipple suckling or varicella zoster infection (shingles), can also increase prolactin secretion. And since prolactin is eliminated by the liver (75%) and the kidney (25%), significant liver disease and/or renal insufficiency can raise prolactin levels, due to decreased clearance.
What are the possible etiologies for elevated prolactin? See answer on the next page...
Q: What are the possible etiologies for elevated prolactin?
The causes of hyperprolactinemia fall into three categories: physiologic, pharmacologic, and pathologic.2 The table provides examples from each category.
A nonsecretory pituitary adenoma or any lesion in the brain that would disrupt the hypophyseal stalk may interfere with dopamine’s inhibitory control and thereby increase prolactin. This is called the stalk effect. It is important to note that not all MRI-proven pituitary adenomas are prolactin secreting, even in the presence of hyperprolactinemia. According to an autopsy series, about 12% of the general population had pituitary microadenoma.3
There is rough correlation between prolactinoma size and level of prolactin. Large nonsecretory pituitary adenomas have prolactin levels less than 150 ng/mL. Microprolactinomas (< 1 cm) are usually in the range of 100 to 250 ng/mL, while macroprolactinomas (> 1 cm) are generally
≥ 250 ng/mL. If the tumor is very large and invades the cavernous sinus, prolactin can measure in the 1,000s.3
Polycystic ovarian syndrome (PCOS) is a common disorder affecting women of reproductive age and the most common cause of underlying ovulatory problems. Patients with PCOS can have mildly elevated prolactin; the exact mechanism of hyperprolactinemia in PCOS is unknown. One theory is that constant high levels of estrogen experienced in PCOS would stimulate prolactin production. It is important to rule out other causes of hyperprolactinemia before making the diagnosis of PCOS.
What is the clinical significance of elevated prolactin? Why do we have to work up and treat it? See answer on the next page...
Q: What is the clinical significance of elevated prolactin? Why do we have to work up and treat it?
By physiologic mechanisms not completely understood, hyperprolactinemia can interrupt the gonadal axis, leading to hypogonadism. In women, it can cause irregular menstrual cycles, oligomenorrhea, amenorrhea, and infertility. In men, it can lower testosterone levels. Long-term effects include declining bone mineral density due to insufficient estrogen in women or testosterone in men.
With macroadenoma, the size of the tumor can have a mass effect such as headache and visual defect by compressing the optic chiasm (bitemporal hemianopsia), which may lead to permanent vision loss if left untreated. Referral to an ophthalmologist may be necessary for formal visual field examination.
How is hyperprolactinemia treated? See answer on the next page...
Q: How is hyperprolactinemia treated?
There are three options for treatment: medication, surgery, and radiation.
Dopamine agonists (bromocriptine, cabergoline) are effective in normalizing prolactin and reducing the size of the tumor in the majority of cases. However, some patients may require long-term treatment. Bromocriptine has been used since the late 1970s, but, due to better tolerance and less frequent dosing, cabergoline is the preferred agent.3
Transsphenoidal surgery is indicated for patients who are intolerant to medication, who have a medication-resistant tumor or significant mass effect, or who prefer definitive treatment. Women of childbearing age with a macroadenoma might consider surgery due to the risk for tumor expansion during pregnancy (estrogen effect) and risk for pituitary apoplexy (hemorrhage or infarct of the pituitary gland). Surgical risk is usually low with a neurosurgeon who has extensive experience.
Radiation can be considered for large tumors that are resistant to medication. It can be used as adjunctive therapy to surgery, since reducing the size of the tumor can make the surgical field smaller. In some medication-resistant tumors, radiation can raise sensitivity to medication.
What does follow-up entail? See next page for answer...
Q: What does follow-up entail?
Once medication is initiated or dosage is adjusted, have the patient follow up in one month and recheck the prolactin level to assess responsiveness to medication (as well as medication adherence). When a therapeutic prolactin level is achieved, recheck the prolactin and have the patient follow up at three and six months and then every six months thereafter.3
MRI of the pituitary gland should be performed at baseline, then in six months to assess tumor response to medication, and then at 12 and 24 months.3 If tumor regression has stabilized or if the tumor has shrunk to a nondetectable size, consider discontinuing the dopamine agonist. If medication is discontinued, recheck prolactin every three months for the first year; if it remains in normal reference range, simply check serum prolactin annually.3
See next page for summary.
See next page for references.
REFERENCES
1. Jameson JL. Harrison’s Endocrinology. 18th ed. China: McGraw-Hill; 2010.
2. Gardner D, Shoback D. Greenspan’s Basic & Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.
A 31-year-old woman is referred by her Ob-Gyn for elevated prolactin. She initially presented with a three-month history of amenorrhea, a negative home pregnancy test, and 100% compliance with condom use. She denies hirsutism and acne but admits to thin milky nipple discharge upon squeezing (but not spontaneous).
Two weeks ago, her Ob-Gyn ordered labs; results were negative for serum beta human chorionic gonadotropin and within normal ranges for thyroid-stimulating hormone (TSH), luteinizing hormone, follicle-stimulating hormone, estradiol, free and total testosterone, dehydroepiandrosterone sulfate (DHEAs), complete chemistry panel, and complete blood count. Her serum prolactin level was 110 ng/mL (normal, 3 to 27 ng/mL).
Q: How is prolactin physiologically regulated?
The primary role of prolactin, which is produced by lactotroph cells in the anterior pituitary gland, is to stimulate lactation and breast development. Prolactin is regulated by dopamine (also known as prolactin inhibitory hormone), which is secreted from the hypothalamus via an inhibitory pathway unique to the hypothalamus-pituitary hormone system. Dopamine essentially suppresses prolactin.
Other hormones can have a stimulatory effect on the anterior pituitary gland and thus increase prolactin levels. Estrogen can induce lactotroph hyperplasia and elevated prolactin; however, this is only clinically relevant in the context of estrogen surge during pregnancy. (Estrogen therapy, such as oral contraception or hormone replacement therapy, on the other hand, is targeted to “normal” estrogen levels.) Thyrotropin-releasing hormone (TRH) from the hypothalamus also stimulates the anterior pituitary gland, so patients with inadequately treated or untreated primary hypothyroidism will have mildly elevated prolactin.
Neurogenic stimuli of the chest wall, through nipple suckling or varicella zoster infection (shingles), can also increase prolactin secretion. And since prolactin is eliminated by the liver (75%) and the kidney (25%), significant liver disease and/or renal insufficiency can raise prolactin levels, due to decreased clearance.
What are the possible etiologies for elevated prolactin? See answer on the next page...
Q: What are the possible etiologies for elevated prolactin?
The causes of hyperprolactinemia fall into three categories: physiologic, pharmacologic, and pathologic.2 The table provides examples from each category.
A nonsecretory pituitary adenoma or any lesion in the brain that would disrupt the hypophyseal stalk may interfere with dopamine’s inhibitory control and thereby increase prolactin. This is called the stalk effect. It is important to note that not all MRI-proven pituitary adenomas are prolactin secreting, even in the presence of hyperprolactinemia. According to an autopsy series, about 12% of the general population had pituitary microadenoma.3
There is rough correlation between prolactinoma size and level of prolactin. Large nonsecretory pituitary adenomas have prolactin levels less than 150 ng/mL. Microprolactinomas (< 1 cm) are usually in the range of 100 to 250 ng/mL, while macroprolactinomas (> 1 cm) are generally
≥ 250 ng/mL. If the tumor is very large and invades the cavernous sinus, prolactin can measure in the 1,000s.3
Polycystic ovarian syndrome (PCOS) is a common disorder affecting women of reproductive age and the most common cause of underlying ovulatory problems. Patients with PCOS can have mildly elevated prolactin; the exact mechanism of hyperprolactinemia in PCOS is unknown. One theory is that constant high levels of estrogen experienced in PCOS would stimulate prolactin production. It is important to rule out other causes of hyperprolactinemia before making the diagnosis of PCOS.
What is the clinical significance of elevated prolactin? Why do we have to work up and treat it? See answer on the next page...
Q: What is the clinical significance of elevated prolactin? Why do we have to work up and treat it?
By physiologic mechanisms not completely understood, hyperprolactinemia can interrupt the gonadal axis, leading to hypogonadism. In women, it can cause irregular menstrual cycles, oligomenorrhea, amenorrhea, and infertility. In men, it can lower testosterone levels. Long-term effects include declining bone mineral density due to insufficient estrogen in women or testosterone in men.
With macroadenoma, the size of the tumor can have a mass effect such as headache and visual defect by compressing the optic chiasm (bitemporal hemianopsia), which may lead to permanent vision loss if left untreated. Referral to an ophthalmologist may be necessary for formal visual field examination.
How is hyperprolactinemia treated? See answer on the next page...
Q: How is hyperprolactinemia treated?
There are three options for treatment: medication, surgery, and radiation.
Dopamine agonists (bromocriptine, cabergoline) are effective in normalizing prolactin and reducing the size of the tumor in the majority of cases. However, some patients may require long-term treatment. Bromocriptine has been used since the late 1970s, but, due to better tolerance and less frequent dosing, cabergoline is the preferred agent.3
Transsphenoidal surgery is indicated for patients who are intolerant to medication, who have a medication-resistant tumor or significant mass effect, or who prefer definitive treatment. Women of childbearing age with a macroadenoma might consider surgery due to the risk for tumor expansion during pregnancy (estrogen effect) and risk for pituitary apoplexy (hemorrhage or infarct of the pituitary gland). Surgical risk is usually low with a neurosurgeon who has extensive experience.
Radiation can be considered for large tumors that are resistant to medication. It can be used as adjunctive therapy to surgery, since reducing the size of the tumor can make the surgical field smaller. In some medication-resistant tumors, radiation can raise sensitivity to medication.
What does follow-up entail? See next page for answer...
Q: What does follow-up entail?
Once medication is initiated or dosage is adjusted, have the patient follow up in one month and recheck the prolactin level to assess responsiveness to medication (as well as medication adherence). When a therapeutic prolactin level is achieved, recheck the prolactin and have the patient follow up at three and six months and then every six months thereafter.3
MRI of the pituitary gland should be performed at baseline, then in six months to assess tumor response to medication, and then at 12 and 24 months.3 If tumor regression has stabilized or if the tumor has shrunk to a nondetectable size, consider discontinuing the dopamine agonist. If medication is discontinued, recheck prolactin every three months for the first year; if it remains in normal reference range, simply check serum prolactin annually.3
See next page for summary.
See next page for references.
REFERENCES
1. Jameson JL. Harrison’s Endocrinology. 18th ed. China: McGraw-Hill; 2010.
2. Gardner D, Shoback D. Greenspan’s Basic & Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.