Article
Inspection of Deep Tumor Margins for Accurate Cutaneous Squamous Cell Carcinoma Staging
To the Editor:
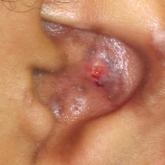
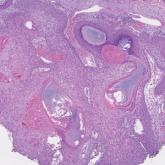
Histopathologic analysis of debulk specimens in Mohs micrographic surgery (MMS) may augment identification of high-risk...
Article
Complex Regional Pain Syndrome Type II After a Brachial Plexus and C6 Nerve Root Injury
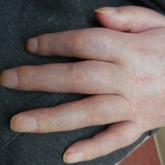
Complex regional pain syndrome (CRPS) is a neuropathic disorder of the extremities characterized by pain, a variety of autonomic and motor...