Article
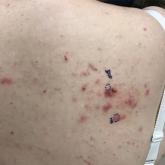
Anti–PD1 Immune Checkpoint Inhibitor–Induced Bullous Pemphigoid in Metastatic Melanoma and Non–Small Cell Lung Cancer
Anti–programmed cell death 1 targeted therapies improve survival in solid and hematologic malignancies but are associated with autoimmune side...
Article
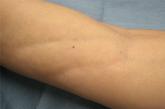
Linear Depressions and Progressive Tightening of the Extremities
A 50-year-old woman presented with progressive tightening of her extremities of 2 months’ duration with eventual involvement of her trunk.