User login
Crusted Scabies Presenting as Erythroderma in a Patient With Iatrogenic Immunosuppression for Treatment of Granulomatosis With Polyangiitis
Scabies is caused by cutaneous ectoparasitic infection by the mite Sarcoptes scabiei var hominis. The infection is highly contagious via direct skin-to-skin contact or indirectly through infested bedding, clothing or fomites.1,2 Scabies occurs at all ages, in all ethnic groups, and at all socioeconomic levels.1 Analysis by the Global Burden of Disease estimates that 200 million individuals have been infected with scabies worldwide. The World Health Organization has declared scabies a neglected tropical disease.3
Crusted scabies is a severe and rare form of scabies, with hyperinfestation of thousands to millions of mites, and more commonly is associated with immunosuppressed states, including HIV and hematologic malignancies.1,2,4 Crusted scabies has a high mortality rate due to sepsis when left untreated.3,5
Occasionally, iatrogenic immunosuppression contributes to the development of crusted scabies.1,2 Iatrogenic immunosuppression leading to crusted scabies most commonly occurs secondary to immunosuppression after bone marrow or solid organ transplantation.6 Less often, crusted scabies is caused by iatrogenic immunosuppression from other clinical scenarios.1,2
We describe a patient with iatrogenic immunosuppression due to azathioprine-induced myelosuppression for the treatment of granulomatosis with polyangiitis (GPA) who developed crusted scabies that clinically presented as erythroderma. Crusted scabies should be included in the differential diagnosis of erythroderma, especially in the setting of iatrogenic immunosuppression, for timely and appropriate management.
Case Report
An 84-year-old man presented with worsening pruritus, erythema, and thick yellow scale that progressed to erythroderma over the last 2 weeks. He was diagnosed with GPA 6 months prior to presentation and was treated with azathioprine 150 mg/d, prednisone 10 mg/d, and sulfamethoxazole 800 mg plus trimethoprim 160 mg twice weekly for prophylaxis against Pneumocystis jirovecii pneumonia.
Three weeks prior to presentation, the patient was hospitalized for pancytopenia attributed to azathioprine-induced myelosuppression (hemoglobin, 6.1 g/dL [reference range, 13.5–18.0 g/dL]; hematocrit, 17.5% [reference range, 42%–52%]; white blood cell count, 1.66×103/μL [reference range, 4.0–10.5×103/μL]; platelet count, 146×103/μL [reference range, 150–450×103/μL]; absolute neutrophil count, 1.29×103/μL [reference range, 1.4–6.5×103/μL]). He was transferred to a skilled nursing facility after discharge and referred to dermatology for evaluation of the worsening pruritic rash.

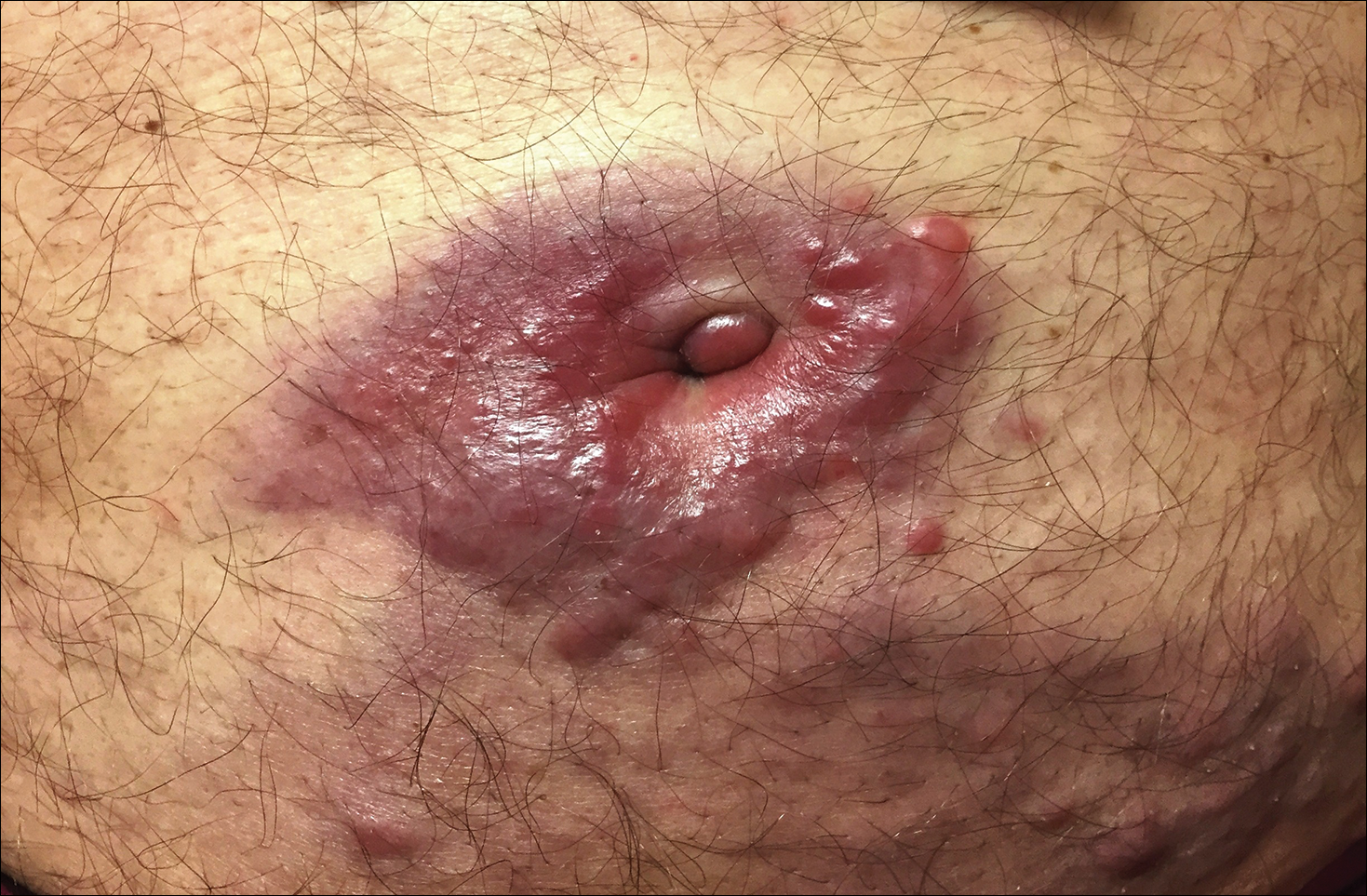
At the current presentation, the patient denied close contact with anyone who had a similar rash at home or at the skilled nursing facility. Physical examination revealed diffuse erythroderma with yellow scale on the scalp, trunk, arms, and legs (Figure 1). The palms showed scattered 2- to 3-mm pustules. The mucosal surfaces did not have lesions. A punch biopsy of a pustule from the right arm revealed focal spongiosis, parakeratosis, and acanthosis, as well as a perivascular and interstitial mixed inflammatory infiltrate with lymphocytes and eosinophils. Organisms morphologically compatible with scabies were found in the stratum corneum (Figure 2). Another punch biopsy of a pustule from the right arm was performed for direct immunofluorescence (DIF) and was negative for immunoglobulin deposition. Mineral oil preparation from pustules on the palm was positive for mites.
.")
The patient was treated with permethrin cream 5% and oral ivermectin 200 μg/kg on day 1 and day 10. The prednisone dosage was increased from 10 mg/d to 50 mg/d and tapered over 2 weeks to treat the symptomatic rash and GPA. He remains on maintenance rituximab for GPA, without recurrence of scabies.
Comment
Pathogenesis—As an obligate parasite, S scabiei spends its entire life cycle within the host. Impregnated female mites burrow into the epidermis after mating and lay eggs daily for 1 to 2 months. Eggs hatch 2 or 3 days later. Larvae then migrate to the skin surface; burrow into the stratum corneum, where they mature into adults; and then mate on the skin surface.1,4
Clinical Presentation and Sequelae—Typically, scabies presents 2 to 6 weeks after initial exposure with generalized and intense itching and inflammatory pruritic papules on the finger webs, wrists, elbows, axillae, buttocks, umbilicus, genitalia, and areolae.1 Burrows are specific for scabies but may not always be present. Often, there are nonspecific secondary lesions, including excoriations, dermatitis, and impetiginization.
Complications of scabies can be severe, with initial colonization and infection of the skin resulting in impetigo and cellulitis. Systematic sequelae from local skin infection include post-streptococcal glomerulonephritis, rheumatic fever, and sepsis. Mortality from sepsis in scabies can be high.3,5
Classic Crusted Scabies and Other Variants—Crusted scabies presents with psoriasiform hyperkeratotic plaques involving the hands and feet with potential nail involvement that can become more generalized.1 Alterations in CD4+ T-cell function have been implicated in the development of crusted scabies, in which an excessive helper T cell (TH2) response is elicited against the ectoparasite, which may help explain the intense pruritus of scabies.6 Occasionally, iatrogenic immunosuppression contributes to development of crusted scabies,1 as was the case with our patient. However, it is rare for crusted scabies to present with erythroderma.7
Other atypical presentations of scabies include a seborrheic dermatitis–like presentation in infants, nodular lesions in the groin and axillae in more chronic scabies, and vesicles or bullous lesions.1
Diagnosis—Identification of mites, eggs, or feces is necessary for definitive diagnosis of scabies.8 These materials can be obtained through skin scrapings with mineral oil and observed under light microscopy or direct dermoscopy. Multiple scrapings on many lesions should be performed because failure to identify mites can be common and does not rule out scabies. Dermoscopic examination of active lesions under low power also can be helpful, given that identification of dark brown triangular structures can correspond to visualization of the pigmented anterior section of the mite.9-11 A skin biopsy can help identify mites, but histopathology often shows a nonspecific hypersensitivity reaction.12 Therefore, empiric treatment often is necessary.
Differential Diagnosis—The differential diagnosis of erythroderma is broad and includes a drug eruption; Sézary syndrome; and pre-existing skin diseases, including psoriasis, atopic dermatitis, pityriasis rubra pilaris, pemphigus foliaceus, and bullous pemphigoid. Histopathology is critical to differentiate these diagnoses. Bullous pemphigoid and pemphigus foliaceus are immunobullous diseases that typically are positive for immunoglobulin deposition on DIF. In rare cases, scabies also can present with bullae and positive DIF test results.13
Treatment—First-line treatment of crusted scabies in the United States is permethrin cream 5%, followed by oral ivermectin 200 μg/kg.4,5,14,15 Other scabicides include topicals such as benzyl benzoate 10% to 25%; precipitated sulfur 2% to 10%; crotamiton 10%; malathion 0.5%; and lindane 1%.5 The association of neurotoxicity with lindane has considerably reduced the drug’s use.1
During treatment of scabies, it is important to isolate patients to mitigate the possibility of spread.4 Pruritus can persist for a few weeks after completion of therapy.5 Patients should be closely monitored to ensure that this symptom is secondary to skin inflammation and not incomplete treatment.
Treatment of crusted scabies may require repeated treatments to decrease the notable mite burden as well as the associated crusting and scale. Adding a keratolytic such as 5% to 10% salicylic acid in petrolatum to the treatment regimen may be useful for breaking up thick scale.5
Immunosuppression—With numerous immunomodulatory drugs for treating autoimmunity comes an increased risk for iatrogenic immunosuppression that may contribute to the development of crusted scabies.16 In a number of autoimmune diseases such as rheumatoid arthritis,17-19 psoriasis,20,21 pemphigus vulgaris,22 systemic lupus erythematosus,23 systemic sclerosis,22,24 bullous pemphigoid,25,26 and dermatomyositis,27 patients have developed crusted scabies secondary to treatment-related immunosuppression. These immunosuppressive therapies include systemic steroids,22-24,26-31 methotrexate,23 infliximab,18 adalimumab,21 toclizumab,19 and etanercept.20 In a case of drug-induced Stevens-Johnson syndrome, the patient developed crusted scabies during long-term use of oral steroids.22
Patients with a malignancy who are being treated with chemotherapy also can develop crusted scabies.28 Crusted scabies has even been associated with long-term topical steroid32-34 and topical calcineurin inhibitor use.16
Iatrogenic immunosuppression in our patient resulted from treatment of GPA with azathioprine, an immunosuppressive drug that acts as an antagonist of the breakdown of purines, leading to inhibition of DNA, RNA, and protein synthesis.35 On occasion, azathioprine can induce immunosuppression in the form of myelosuppression and resulting pancytopenia, as was the case with our patient.
Conclusion
Although scabies is designated as a neglected tropical disease by the World Health Organization, it still causes a notable burden worldwide, regardless of the economics. Our case highlights an unusual presentation of scabies as erythroderma in the setting of iatrogenic immunosuppression from azathioprine use. Dermatologists should consider crusted scabies in the differential diagnosis of erythroderma, especially in immunocompromised patients, to avoid delays in diagnosis and treatment. Immunosuppressive therapy is an important mainstay in the treatment of many conditions, but it is important to consider that these medications can place patients at an increased risk for rare opportunistic infections. Therefore, patients receiving such treatment should be closely monitored.
- Chosidow O. Clinical practices. Scabies. N Engl J Med. 2006;354:1718-1727. doi:10.1056/NEJMcp052784
- Salgado F, Elston DM. What’s eating you? scabies in the developing world. Cutis. 2017;100:287-289.
- Karimkhani C, Colombara DV, Drucker AM, et al. The global burden of scabies: a cross-sectional analysis from the Global Burden of Disease Study 2015. Lancet Infect Dis. 2017;17:1247-1254. doi:10.1016/S1473-3099(17)30483-8
- Currie BJ, McCarthy JS. Permethrin and ivermectin for scabies. N Engl J Med. 2010;362:717-725. doi:10.1056/NEJMct0910329
- Thomas C, Coates SJ, Engelman D, et al. Ectoparasites: scabies. J Am Acad Dermatol. 2020;82:533-548. doi:10.1016/j.jaad.2019.05.109
- Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381. doi:10.1016/j.jinf.2004.08.033
- Wang X-D, Shen H, Liu Z-H. Contagious erythroderma. J Emerg Med. 2016;51:180-181. doi:10.1016/j.jemermed.2016.05.027
- Johnston G, Sladden M. Scabies: diagnosis and treatment. BMJ. 2005;331:619-622. doi:10.1136/bmj.331.7517.619
- Micali G, Lacarrubba F, Massimino D, et al. Dermatoscopy: alternative uses in daily clinical practice. J Am Acad Dermatol. 2011;64:1135-1146. doi:10.1016/j.jaad.2010.03.010
- Bollea Garlatti LA, Torre AC, Bollea Garlatti ML, et al.. Dermoscopy aids the diagnosis of crusted scabies in an erythrodermic patient. J Am Acad Dermatol. 2015;73:E93-E95. doi:10.1016/j.jaad.2015.04.061
- Tang J, You Z, Ran Y. Simple methods to enhance the diagnosis of scabies. J Am Acad Dermatol. 2019;80:E99-E100. doi:10.1016/j.jaad.2017.07.038
- Falk ES, Eide TJ. Histologic and clinical findings in human scabies. Int J Dermatol. 1981;20:600-605. doi:10.1111/j.1365-4362.1981.tb00844.x
- Shahab RKA, Loo DS. Bullous scabies. J Am Acad Dermatol. 2003;49:346-350. doi:10.1067/s0190-9622(03)00876-4
- Strong M, Johnstone P. Interventions for treating scabies. Cochrane Database Syst Rev. 2007:CD000320. doi:10.1002/14651858.CD000320.pub2
- Rosumeck S, Nast A, Dressler C. Evaluation of ivermectin vs permethrin for treating scabies—summary of a Cochrane Review. JAMA Dermatol. 2019;155:730-732. doi:10.1001/jamadermatol.2019.0279
- Ruiz-Maldonado R. Pimecrolimus related crusted scabies in an infant. Pediatr Dermatol. 2006;23:299-300. doi:10.1111/j.1525-1470.2006.00241.x
- Bu X, Fan J, Hu X, et al. Norwegian scabies in a patient treated with Tripterygium glycoside for rheumatoid arthritis. An Bras Dermatol. 2017;92:556-558. doi:10.1590/abd1806-4841.20174946
- Pipitone MA, Adams B, Sheth A, et al. Crusted scabies in a patient being treated with infliximab for juvenile rheumatoid arthritis. J Am Acad Dermatol. 2005;52:719-720. doi:10.1016/j.jaad.2004.12.039
- Baccouche K, Sellam J, Guegan S, et al. Crusted Norwegian scabies, an opportunistic infection, with tocilizumab in rheumatoid arthritis. Joint Bone Spine. 2011;78:402-404. doi:10.1016/j.jbspin.2011.02.008
- Saillard C, Darrieux L, Safa G. Crusted scabies complicates etanercept therapy in a patient with severe psoriasis. J Am Acad Dermatol. 2013;68:E138-E139. doi:10.1016/j.jaad.2012.09.049
- Belvisi V, Orsi GB, Del Borgo C, et al. Large nosocomial outbreakassociated with a Norwegian scabies index case undergoing TNF-α inhibitor treatment: management and control. Infect Control Hosp Epidemiol. 2015;36:1358-1360. doi:10.1017/ice.2015.188
- Nofal A. Variable response of crusted scabies to oral ivermectin: report on eight Egyptian patients. J Eur Acad Dermatol Venereol. 2009;23:793-797. doi:10.1111/j.1468-3083.2009.03177.x
- Yee BE, Carlos CA, Hata T. Crusted scabies of the scalp in a patient with systemic lupus erythematosus. Dermatol Online J. 2014;20:13030/qt9dm891gd.
- Bumb RA, Mehta RD. Crusted scabies in a patient of systemic sclerosis. Indian J Dermatol Venereol Leprol. 2000;66:143-144.
- Hylwa SA, Loss L, Grassi M. Crusted scabies and tinea corporis after treatment of presumed bullous pemphigoid. Cutis. 2013;92:193-198.
- Svecova D, Chmurova N, Pallova A, et al. Norwegian scabies in immunosuppressed patient misdiagnosed as an adverse drug reaction. Epidemiol Mikrobiol Imunol. 2009;58:121-123.
- Dourmishev AL, Serafimova DK, Dourmishev LA, et al. Crusted scabies of the scalp in dermatomyositis patients: three cases treated with oral ivermectin. Int J Dermatol. 1998;37:231-234. doi:10.1046/j.1365-4362.1998.00330.x
- Mortazavi H, Abedini R, Sadri F, et al. Crusted scabies in a patient with brain astrocytoma: report of a case. Int J Infect Dis. 2010;14:E526-E527. doi:10.1016/j.ijid.2009.06.011
- Lima FCDR, Cerqueira AMM, MBS, et al. Crusted scabies due to indiscriminate use of glucocorticoid therapy in infant. An Bras Dermatol. 2017;92:383-385. doi:10.1590/abd1806-4841.20174433
- Binic´ I, Jankovic´ A, Jovanovic´ D, et al. Crusted (Norwegian) scabies following systemic and topical corticosteroid therapy. J Korean Med Sci. 2010;25:188-191. doi:10.3346/jkms.2010.25.1.188
- Ohtaki N, Taniguchi H, Ohtomo H. Oral ivermectin treatment in two cases of scabies: effective in crusted scabies induced by corticosteroid but ineffective in nail scabies. J Dermatol. 2003;30:411-416. doi:10.1111/j.1346-8138.2003.tb00408.x
- Bilan P, Colin-Gorski AM, Chapelon E, et al. Crusted scabies induced by topical corticosteroids: a case report [in French]. Arch Pediatr. 2015;22:1292-1294. doi:10.1016/j.arcped.2015.09.004
- Marlière V, Roul S, C, et al. Crusted (Norwegian) scabies induced by use of topical corticosteroids and treated successfully with ivermectin. J Pediatr. 1999;135:122-124. doi:10.1016/s0022-3476(99)70342-2
- Jaramillo-Ayerbe F, J. Ivermectin for crusted Norwegian scabies induced by use of topical steroids. Arch Dermatol. 1998;134:143-145. doi:10.1001/archderm.134.2.143
- Elion GB. The purine path to chemotherapy. Science. 1989;244:41-47. doi:10.1126/science.2649979
Scabies is caused by cutaneous ectoparasitic infection by the mite Sarcoptes scabiei var hominis. The infection is highly contagious via direct skin-to-skin contact or indirectly through infested bedding, clothing or fomites.1,2 Scabies occurs at all ages, in all ethnic groups, and at all socioeconomic levels.1 Analysis by the Global Burden of Disease estimates that 200 million individuals have been infected with scabies worldwide. The World Health Organization has declared scabies a neglected tropical disease.3
Crusted scabies is a severe and rare form of scabies, with hyperinfestation of thousands to millions of mites, and more commonly is associated with immunosuppressed states, including HIV and hematologic malignancies.1,2,4 Crusted scabies has a high mortality rate due to sepsis when left untreated.3,5
Occasionally, iatrogenic immunosuppression contributes to the development of crusted scabies.1,2 Iatrogenic immunosuppression leading to crusted scabies most commonly occurs secondary to immunosuppression after bone marrow or solid organ transplantation.6 Less often, crusted scabies is caused by iatrogenic immunosuppression from other clinical scenarios.1,2
We describe a patient with iatrogenic immunosuppression due to azathioprine-induced myelosuppression for the treatment of granulomatosis with polyangiitis (GPA) who developed crusted scabies that clinically presented as erythroderma. Crusted scabies should be included in the differential diagnosis of erythroderma, especially in the setting of iatrogenic immunosuppression, for timely and appropriate management.
Case Report
An 84-year-old man presented with worsening pruritus, erythema, and thick yellow scale that progressed to erythroderma over the last 2 weeks. He was diagnosed with GPA 6 months prior to presentation and was treated with azathioprine 150 mg/d, prednisone 10 mg/d, and sulfamethoxazole 800 mg plus trimethoprim 160 mg twice weekly for prophylaxis against Pneumocystis jirovecii pneumonia.
Three weeks prior to presentation, the patient was hospitalized for pancytopenia attributed to azathioprine-induced myelosuppression (hemoglobin, 6.1 g/dL [reference range, 13.5–18.0 g/dL]; hematocrit, 17.5% [reference range, 42%–52%]; white blood cell count, 1.66×103/μL [reference range, 4.0–10.5×103/μL]; platelet count, 146×103/μL [reference range, 150–450×103/μL]; absolute neutrophil count, 1.29×103/μL [reference range, 1.4–6.5×103/μL]). He was transferred to a skilled nursing facility after discharge and referred to dermatology for evaluation of the worsening pruritic rash.
At the current presentation, the patient denied close contact with anyone who had a similar rash at home or at the skilled nursing facility. Physical examination revealed diffuse erythroderma with yellow scale on the scalp, trunk, arms, and legs (Figure 1). The palms showed scattered 2- to 3-mm pustules. The mucosal surfaces did not have lesions. A punch biopsy of a pustule from the right arm revealed focal spongiosis, parakeratosis, and acanthosis, as well as a perivascular and interstitial mixed inflammatory infiltrate with lymphocytes and eosinophils. Organisms morphologically compatible with scabies were found in the stratum corneum (Figure 2). Another punch biopsy of a pustule from the right arm was performed for direct immunofluorescence (DIF) and was negative for immunoglobulin deposition. Mineral oil preparation from pustules on the palm was positive for mites.
The patient was treated with permethrin cream 5% and oral ivermectin 200 μg/kg on day 1 and day 10. The prednisone dosage was increased from 10 mg/d to 50 mg/d and tapered over 2 weeks to treat the symptomatic rash and GPA. He remains on maintenance rituximab for GPA, without recurrence of scabies.
Comment
Pathogenesis—As an obligate parasite, S scabiei spends its entire life cycle within the host. Impregnated female mites burrow into the epidermis after mating and lay eggs daily for 1 to 2 months. Eggs hatch 2 or 3 days later. Larvae then migrate to the skin surface; burrow into the stratum corneum, where they mature into adults; and then mate on the skin surface.1,4
Clinical Presentation and Sequelae—Typically, scabies presents 2 to 6 weeks after initial exposure with generalized and intense itching and inflammatory pruritic papules on the finger webs, wrists, elbows, axillae, buttocks, umbilicus, genitalia, and areolae.1 Burrows are specific for scabies but may not always be present. Often, there are nonspecific secondary lesions, including excoriations, dermatitis, and impetiginization.
Complications of scabies can be severe, with initial colonization and infection of the skin resulting in impetigo and cellulitis. Systematic sequelae from local skin infection include post-streptococcal glomerulonephritis, rheumatic fever, and sepsis. Mortality from sepsis in scabies can be high.3,5
Classic Crusted Scabies and Other Variants—Crusted scabies presents with psoriasiform hyperkeratotic plaques involving the hands and feet with potential nail involvement that can become more generalized.1 Alterations in CD4+ T-cell function have been implicated in the development of crusted scabies, in which an excessive helper T cell (TH2) response is elicited against the ectoparasite, which may help explain the intense pruritus of scabies.6 Occasionally, iatrogenic immunosuppression contributes to development of crusted scabies,1 as was the case with our patient. However, it is rare for crusted scabies to present with erythroderma.7
Other atypical presentations of scabies include a seborrheic dermatitis–like presentation in infants, nodular lesions in the groin and axillae in more chronic scabies, and vesicles or bullous lesions.1
Diagnosis—Identification of mites, eggs, or feces is necessary for definitive diagnosis of scabies.8 These materials can be obtained through skin scrapings with mineral oil and observed under light microscopy or direct dermoscopy. Multiple scrapings on many lesions should be performed because failure to identify mites can be common and does not rule out scabies. Dermoscopic examination of active lesions under low power also can be helpful, given that identification of dark brown triangular structures can correspond to visualization of the pigmented anterior section of the mite.9-11 A skin biopsy can help identify mites, but histopathology often shows a nonspecific hypersensitivity reaction.12 Therefore, empiric treatment often is necessary.
Differential Diagnosis—The differential diagnosis of erythroderma is broad and includes a drug eruption; Sézary syndrome; and pre-existing skin diseases, including psoriasis, atopic dermatitis, pityriasis rubra pilaris, pemphigus foliaceus, and bullous pemphigoid. Histopathology is critical to differentiate these diagnoses. Bullous pemphigoid and pemphigus foliaceus are immunobullous diseases that typically are positive for immunoglobulin deposition on DIF. In rare cases, scabies also can present with bullae and positive DIF test results.13
Treatment—First-line treatment of crusted scabies in the United States is permethrin cream 5%, followed by oral ivermectin 200 μg/kg.4,5,14,15 Other scabicides include topicals such as benzyl benzoate 10% to 25%; precipitated sulfur 2% to 10%; crotamiton 10%; malathion 0.5%; and lindane 1%.5 The association of neurotoxicity with lindane has considerably reduced the drug’s use.1
During treatment of scabies, it is important to isolate patients to mitigate the possibility of spread.4 Pruritus can persist for a few weeks after completion of therapy.5 Patients should be closely monitored to ensure that this symptom is secondary to skin inflammation and not incomplete treatment.
Treatment of crusted scabies may require repeated treatments to decrease the notable mite burden as well as the associated crusting and scale. Adding a keratolytic such as 5% to 10% salicylic acid in petrolatum to the treatment regimen may be useful for breaking up thick scale.5
Immunosuppression—With numerous immunomodulatory drugs for treating autoimmunity comes an increased risk for iatrogenic immunosuppression that may contribute to the development of crusted scabies.16 In a number of autoimmune diseases such as rheumatoid arthritis,17-19 psoriasis,20,21 pemphigus vulgaris,22 systemic lupus erythematosus,23 systemic sclerosis,22,24 bullous pemphigoid,25,26 and dermatomyositis,27 patients have developed crusted scabies secondary to treatment-related immunosuppression. These immunosuppressive therapies include systemic steroids,22-24,26-31 methotrexate,23 infliximab,18 adalimumab,21 toclizumab,19 and etanercept.20 In a case of drug-induced Stevens-Johnson syndrome, the patient developed crusted scabies during long-term use of oral steroids.22
Patients with a malignancy who are being treated with chemotherapy also can develop crusted scabies.28 Crusted scabies has even been associated with long-term topical steroid32-34 and topical calcineurin inhibitor use.16
Iatrogenic immunosuppression in our patient resulted from treatment of GPA with azathioprine, an immunosuppressive drug that acts as an antagonist of the breakdown of purines, leading to inhibition of DNA, RNA, and protein synthesis.35 On occasion, azathioprine can induce immunosuppression in the form of myelosuppression and resulting pancytopenia, as was the case with our patient.
Conclusion
Although scabies is designated as a neglected tropical disease by the World Health Organization, it still causes a notable burden worldwide, regardless of the economics. Our case highlights an unusual presentation of scabies as erythroderma in the setting of iatrogenic immunosuppression from azathioprine use. Dermatologists should consider crusted scabies in the differential diagnosis of erythroderma, especially in immunocompromised patients, to avoid delays in diagnosis and treatment. Immunosuppressive therapy is an important mainstay in the treatment of many conditions, but it is important to consider that these medications can place patients at an increased risk for rare opportunistic infections. Therefore, patients receiving such treatment should be closely monitored.
Scabies is caused by cutaneous ectoparasitic infection by the mite Sarcoptes scabiei var hominis. The infection is highly contagious via direct skin-to-skin contact or indirectly through infested bedding, clothing or fomites.1,2 Scabies occurs at all ages, in all ethnic groups, and at all socioeconomic levels.1 Analysis by the Global Burden of Disease estimates that 200 million individuals have been infected with scabies worldwide. The World Health Organization has declared scabies a neglected tropical disease.3
Crusted scabies is a severe and rare form of scabies, with hyperinfestation of thousands to millions of mites, and more commonly is associated with immunosuppressed states, including HIV and hematologic malignancies.1,2,4 Crusted scabies has a high mortality rate due to sepsis when left untreated.3,5
Occasionally, iatrogenic immunosuppression contributes to the development of crusted scabies.1,2 Iatrogenic immunosuppression leading to crusted scabies most commonly occurs secondary to immunosuppression after bone marrow or solid organ transplantation.6 Less often, crusted scabies is caused by iatrogenic immunosuppression from other clinical scenarios.1,2
We describe a patient with iatrogenic immunosuppression due to azathioprine-induced myelosuppression for the treatment of granulomatosis with polyangiitis (GPA) who developed crusted scabies that clinically presented as erythroderma. Crusted scabies should be included in the differential diagnosis of erythroderma, especially in the setting of iatrogenic immunosuppression, for timely and appropriate management.
Case Report
An 84-year-old man presented with worsening pruritus, erythema, and thick yellow scale that progressed to erythroderma over the last 2 weeks. He was diagnosed with GPA 6 months prior to presentation and was treated with azathioprine 150 mg/d, prednisone 10 mg/d, and sulfamethoxazole 800 mg plus trimethoprim 160 mg twice weekly for prophylaxis against Pneumocystis jirovecii pneumonia.
Three weeks prior to presentation, the patient was hospitalized for pancytopenia attributed to azathioprine-induced myelosuppression (hemoglobin, 6.1 g/dL [reference range, 13.5–18.0 g/dL]; hematocrit, 17.5% [reference range, 42%–52%]; white blood cell count, 1.66×103/μL [reference range, 4.0–10.5×103/μL]; platelet count, 146×103/μL [reference range, 150–450×103/μL]; absolute neutrophil count, 1.29×103/μL [reference range, 1.4–6.5×103/μL]). He was transferred to a skilled nursing facility after discharge and referred to dermatology for evaluation of the worsening pruritic rash.
At the current presentation, the patient denied close contact with anyone who had a similar rash at home or at the skilled nursing facility. Physical examination revealed diffuse erythroderma with yellow scale on the scalp, trunk, arms, and legs (Figure 1). The palms showed scattered 2- to 3-mm pustules. The mucosal surfaces did not have lesions. A punch biopsy of a pustule from the right arm revealed focal spongiosis, parakeratosis, and acanthosis, as well as a perivascular and interstitial mixed inflammatory infiltrate with lymphocytes and eosinophils. Organisms morphologically compatible with scabies were found in the stratum corneum (Figure 2). Another punch biopsy of a pustule from the right arm was performed for direct immunofluorescence (DIF) and was negative for immunoglobulin deposition. Mineral oil preparation from pustules on the palm was positive for mites.
The patient was treated with permethrin cream 5% and oral ivermectin 200 μg/kg on day 1 and day 10. The prednisone dosage was increased from 10 mg/d to 50 mg/d and tapered over 2 weeks to treat the symptomatic rash and GPA. He remains on maintenance rituximab for GPA, without recurrence of scabies.
Comment
Pathogenesis—As an obligate parasite, S scabiei spends its entire life cycle within the host. Impregnated female mites burrow into the epidermis after mating and lay eggs daily for 1 to 2 months. Eggs hatch 2 or 3 days later. Larvae then migrate to the skin surface; burrow into the stratum corneum, where they mature into adults; and then mate on the skin surface.1,4
Clinical Presentation and Sequelae—Typically, scabies presents 2 to 6 weeks after initial exposure with generalized and intense itching and inflammatory pruritic papules on the finger webs, wrists, elbows, axillae, buttocks, umbilicus, genitalia, and areolae.1 Burrows are specific for scabies but may not always be present. Often, there are nonspecific secondary lesions, including excoriations, dermatitis, and impetiginization.
Complications of scabies can be severe, with initial colonization and infection of the skin resulting in impetigo and cellulitis. Systematic sequelae from local skin infection include post-streptococcal glomerulonephritis, rheumatic fever, and sepsis. Mortality from sepsis in scabies can be high.3,5
Classic Crusted Scabies and Other Variants—Crusted scabies presents with psoriasiform hyperkeratotic plaques involving the hands and feet with potential nail involvement that can become more generalized.1 Alterations in CD4+ T-cell function have been implicated in the development of crusted scabies, in which an excessive helper T cell (TH2) response is elicited against the ectoparasite, which may help explain the intense pruritus of scabies.6 Occasionally, iatrogenic immunosuppression contributes to development of crusted scabies,1 as was the case with our patient. However, it is rare for crusted scabies to present with erythroderma.7
Other atypical presentations of scabies include a seborrheic dermatitis–like presentation in infants, nodular lesions in the groin and axillae in more chronic scabies, and vesicles or bullous lesions.1
Diagnosis—Identification of mites, eggs, or feces is necessary for definitive diagnosis of scabies.8 These materials can be obtained through skin scrapings with mineral oil and observed under light microscopy or direct dermoscopy. Multiple scrapings on many lesions should be performed because failure to identify mites can be common and does not rule out scabies. Dermoscopic examination of active lesions under low power also can be helpful, given that identification of dark brown triangular structures can correspond to visualization of the pigmented anterior section of the mite.9-11 A skin biopsy can help identify mites, but histopathology often shows a nonspecific hypersensitivity reaction.12 Therefore, empiric treatment often is necessary.
Differential Diagnosis—The differential diagnosis of erythroderma is broad and includes a drug eruption; Sézary syndrome; and pre-existing skin diseases, including psoriasis, atopic dermatitis, pityriasis rubra pilaris, pemphigus foliaceus, and bullous pemphigoid. Histopathology is critical to differentiate these diagnoses. Bullous pemphigoid and pemphigus foliaceus are immunobullous diseases that typically are positive for immunoglobulin deposition on DIF. In rare cases, scabies also can present with bullae and positive DIF test results.13
Treatment—First-line treatment of crusted scabies in the United States is permethrin cream 5%, followed by oral ivermectin 200 μg/kg.4,5,14,15 Other scabicides include topicals such as benzyl benzoate 10% to 25%; precipitated sulfur 2% to 10%; crotamiton 10%; malathion 0.5%; and lindane 1%.5 The association of neurotoxicity with lindane has considerably reduced the drug’s use.1
During treatment of scabies, it is important to isolate patients to mitigate the possibility of spread.4 Pruritus can persist for a few weeks after completion of therapy.5 Patients should be closely monitored to ensure that this symptom is secondary to skin inflammation and not incomplete treatment.
Treatment of crusted scabies may require repeated treatments to decrease the notable mite burden as well as the associated crusting and scale. Adding a keratolytic such as 5% to 10% salicylic acid in petrolatum to the treatment regimen may be useful for breaking up thick scale.5
Immunosuppression—With numerous immunomodulatory drugs for treating autoimmunity comes an increased risk for iatrogenic immunosuppression that may contribute to the development of crusted scabies.16 In a number of autoimmune diseases such as rheumatoid arthritis,17-19 psoriasis,20,21 pemphigus vulgaris,22 systemic lupus erythematosus,23 systemic sclerosis,22,24 bullous pemphigoid,25,26 and dermatomyositis,27 patients have developed crusted scabies secondary to treatment-related immunosuppression. These immunosuppressive therapies include systemic steroids,22-24,26-31 methotrexate,23 infliximab,18 adalimumab,21 toclizumab,19 and etanercept.20 In a case of drug-induced Stevens-Johnson syndrome, the patient developed crusted scabies during long-term use of oral steroids.22
Patients with a malignancy who are being treated with chemotherapy also can develop crusted scabies.28 Crusted scabies has even been associated with long-term topical steroid32-34 and topical calcineurin inhibitor use.16
Iatrogenic immunosuppression in our patient resulted from treatment of GPA with azathioprine, an immunosuppressive drug that acts as an antagonist of the breakdown of purines, leading to inhibition of DNA, RNA, and protein synthesis.35 On occasion, azathioprine can induce immunosuppression in the form of myelosuppression and resulting pancytopenia, as was the case with our patient.
Conclusion
Although scabies is designated as a neglected tropical disease by the World Health Organization, it still causes a notable burden worldwide, regardless of the economics. Our case highlights an unusual presentation of scabies as erythroderma in the setting of iatrogenic immunosuppression from azathioprine use. Dermatologists should consider crusted scabies in the differential diagnosis of erythroderma, especially in immunocompromised patients, to avoid delays in diagnosis and treatment. Immunosuppressive therapy is an important mainstay in the treatment of many conditions, but it is important to consider that these medications can place patients at an increased risk for rare opportunistic infections. Therefore, patients receiving such treatment should be closely monitored.
- Chosidow O. Clinical practices. Scabies. N Engl J Med. 2006;354:1718-1727. doi:10.1056/NEJMcp052784
- Salgado F, Elston DM. What’s eating you? scabies in the developing world. Cutis. 2017;100:287-289.
- Karimkhani C, Colombara DV, Drucker AM, et al. The global burden of scabies: a cross-sectional analysis from the Global Burden of Disease Study 2015. Lancet Infect Dis. 2017;17:1247-1254. doi:10.1016/S1473-3099(17)30483-8
- Currie BJ, McCarthy JS. Permethrin and ivermectin for scabies. N Engl J Med. 2010;362:717-725. doi:10.1056/NEJMct0910329
- Thomas C, Coates SJ, Engelman D, et al. Ectoparasites: scabies. J Am Acad Dermatol. 2020;82:533-548. doi:10.1016/j.jaad.2019.05.109
- Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381. doi:10.1016/j.jinf.2004.08.033
- Wang X-D, Shen H, Liu Z-H. Contagious erythroderma. J Emerg Med. 2016;51:180-181. doi:10.1016/j.jemermed.2016.05.027
- Johnston G, Sladden M. Scabies: diagnosis and treatment. BMJ. 2005;331:619-622. doi:10.1136/bmj.331.7517.619
- Micali G, Lacarrubba F, Massimino D, et al. Dermatoscopy: alternative uses in daily clinical practice. J Am Acad Dermatol. 2011;64:1135-1146. doi:10.1016/j.jaad.2010.03.010
- Bollea Garlatti LA, Torre AC, Bollea Garlatti ML, et al.. Dermoscopy aids the diagnosis of crusted scabies in an erythrodermic patient. J Am Acad Dermatol. 2015;73:E93-E95. doi:10.1016/j.jaad.2015.04.061
- Tang J, You Z, Ran Y. Simple methods to enhance the diagnosis of scabies. J Am Acad Dermatol. 2019;80:E99-E100. doi:10.1016/j.jaad.2017.07.038
- Falk ES, Eide TJ. Histologic and clinical findings in human scabies. Int J Dermatol. 1981;20:600-605. doi:10.1111/j.1365-4362.1981.tb00844.x
- Shahab RKA, Loo DS. Bullous scabies. J Am Acad Dermatol. 2003;49:346-350. doi:10.1067/s0190-9622(03)00876-4
- Strong M, Johnstone P. Interventions for treating scabies. Cochrane Database Syst Rev. 2007:CD000320. doi:10.1002/14651858.CD000320.pub2
- Rosumeck S, Nast A, Dressler C. Evaluation of ivermectin vs permethrin for treating scabies—summary of a Cochrane Review. JAMA Dermatol. 2019;155:730-732. doi:10.1001/jamadermatol.2019.0279
- Ruiz-Maldonado R. Pimecrolimus related crusted scabies in an infant. Pediatr Dermatol. 2006;23:299-300. doi:10.1111/j.1525-1470.2006.00241.x
- Bu X, Fan J, Hu X, et al. Norwegian scabies in a patient treated with Tripterygium glycoside for rheumatoid arthritis. An Bras Dermatol. 2017;92:556-558. doi:10.1590/abd1806-4841.20174946
- Pipitone MA, Adams B, Sheth A, et al. Crusted scabies in a patient being treated with infliximab for juvenile rheumatoid arthritis. J Am Acad Dermatol. 2005;52:719-720. doi:10.1016/j.jaad.2004.12.039
- Baccouche K, Sellam J, Guegan S, et al. Crusted Norwegian scabies, an opportunistic infection, with tocilizumab in rheumatoid arthritis. Joint Bone Spine. 2011;78:402-404. doi:10.1016/j.jbspin.2011.02.008
- Saillard C, Darrieux L, Safa G. Crusted scabies complicates etanercept therapy in a patient with severe psoriasis. J Am Acad Dermatol. 2013;68:E138-E139. doi:10.1016/j.jaad.2012.09.049
- Belvisi V, Orsi GB, Del Borgo C, et al. Large nosocomial outbreakassociated with a Norwegian scabies index case undergoing TNF-α inhibitor treatment: management and control. Infect Control Hosp Epidemiol. 2015;36:1358-1360. doi:10.1017/ice.2015.188
- Nofal A. Variable response of crusted scabies to oral ivermectin: report on eight Egyptian patients. J Eur Acad Dermatol Venereol. 2009;23:793-797. doi:10.1111/j.1468-3083.2009.03177.x
- Yee BE, Carlos CA, Hata T. Crusted scabies of the scalp in a patient with systemic lupus erythematosus. Dermatol Online J. 2014;20:13030/qt9dm891gd.
- Bumb RA, Mehta RD. Crusted scabies in a patient of systemic sclerosis. Indian J Dermatol Venereol Leprol. 2000;66:143-144.
- Hylwa SA, Loss L, Grassi M. Crusted scabies and tinea corporis after treatment of presumed bullous pemphigoid. Cutis. 2013;92:193-198.
- Svecova D, Chmurova N, Pallova A, et al. Norwegian scabies in immunosuppressed patient misdiagnosed as an adverse drug reaction. Epidemiol Mikrobiol Imunol. 2009;58:121-123.
- Dourmishev AL, Serafimova DK, Dourmishev LA, et al. Crusted scabies of the scalp in dermatomyositis patients: three cases treated with oral ivermectin. Int J Dermatol. 1998;37:231-234. doi:10.1046/j.1365-4362.1998.00330.x
- Mortazavi H, Abedini R, Sadri F, et al. Crusted scabies in a patient with brain astrocytoma: report of a case. Int J Infect Dis. 2010;14:E526-E527. doi:10.1016/j.ijid.2009.06.011
- Lima FCDR, Cerqueira AMM, MBS, et al. Crusted scabies due to indiscriminate use of glucocorticoid therapy in infant. An Bras Dermatol. 2017;92:383-385. doi:10.1590/abd1806-4841.20174433
- Binic´ I, Jankovic´ A, Jovanovic´ D, et al. Crusted (Norwegian) scabies following systemic and topical corticosteroid therapy. J Korean Med Sci. 2010;25:188-191. doi:10.3346/jkms.2010.25.1.188
- Ohtaki N, Taniguchi H, Ohtomo H. Oral ivermectin treatment in two cases of scabies: effective in crusted scabies induced by corticosteroid but ineffective in nail scabies. J Dermatol. 2003;30:411-416. doi:10.1111/j.1346-8138.2003.tb00408.x
- Bilan P, Colin-Gorski AM, Chapelon E, et al. Crusted scabies induced by topical corticosteroids: a case report [in French]. Arch Pediatr. 2015;22:1292-1294. doi:10.1016/j.arcped.2015.09.004
- Marlière V, Roul S, C, et al. Crusted (Norwegian) scabies induced by use of topical corticosteroids and treated successfully with ivermectin. J Pediatr. 1999;135:122-124. doi:10.1016/s0022-3476(99)70342-2
- Jaramillo-Ayerbe F, J. Ivermectin for crusted Norwegian scabies induced by use of topical steroids. Arch Dermatol. 1998;134:143-145. doi:10.1001/archderm.134.2.143
- Elion GB. The purine path to chemotherapy. Science. 1989;244:41-47. doi:10.1126/science.2649979
- Chosidow O. Clinical practices. Scabies. N Engl J Med. 2006;354:1718-1727. doi:10.1056/NEJMcp052784
- Salgado F, Elston DM. What’s eating you? scabies in the developing world. Cutis. 2017;100:287-289.
- Karimkhani C, Colombara DV, Drucker AM, et al. The global burden of scabies: a cross-sectional analysis from the Global Burden of Disease Study 2015. Lancet Infect Dis. 2017;17:1247-1254. doi:10.1016/S1473-3099(17)30483-8
- Currie BJ, McCarthy JS. Permethrin and ivermectin for scabies. N Engl J Med. 2010;362:717-725. doi:10.1056/NEJMct0910329
- Thomas C, Coates SJ, Engelman D, et al. Ectoparasites: scabies. J Am Acad Dermatol. 2020;82:533-548. doi:10.1016/j.jaad.2019.05.109
- Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381. doi:10.1016/j.jinf.2004.08.033
- Wang X-D, Shen H, Liu Z-H. Contagious erythroderma. J Emerg Med. 2016;51:180-181. doi:10.1016/j.jemermed.2016.05.027
- Johnston G, Sladden M. Scabies: diagnosis and treatment. BMJ. 2005;331:619-622. doi:10.1136/bmj.331.7517.619
- Micali G, Lacarrubba F, Massimino D, et al. Dermatoscopy: alternative uses in daily clinical practice. J Am Acad Dermatol. 2011;64:1135-1146. doi:10.1016/j.jaad.2010.03.010
- Bollea Garlatti LA, Torre AC, Bollea Garlatti ML, et al.. Dermoscopy aids the diagnosis of crusted scabies in an erythrodermic patient. J Am Acad Dermatol. 2015;73:E93-E95. doi:10.1016/j.jaad.2015.04.061
- Tang J, You Z, Ran Y. Simple methods to enhance the diagnosis of scabies. J Am Acad Dermatol. 2019;80:E99-E100. doi:10.1016/j.jaad.2017.07.038
- Falk ES, Eide TJ. Histologic and clinical findings in human scabies. Int J Dermatol. 1981;20:600-605. doi:10.1111/j.1365-4362.1981.tb00844.x
- Shahab RKA, Loo DS. Bullous scabies. J Am Acad Dermatol. 2003;49:346-350. doi:10.1067/s0190-9622(03)00876-4
- Strong M, Johnstone P. Interventions for treating scabies. Cochrane Database Syst Rev. 2007:CD000320. doi:10.1002/14651858.CD000320.pub2
- Rosumeck S, Nast A, Dressler C. Evaluation of ivermectin vs permethrin for treating scabies—summary of a Cochrane Review. JAMA Dermatol. 2019;155:730-732. doi:10.1001/jamadermatol.2019.0279
- Ruiz-Maldonado R. Pimecrolimus related crusted scabies in an infant. Pediatr Dermatol. 2006;23:299-300. doi:10.1111/j.1525-1470.2006.00241.x
- Bu X, Fan J, Hu X, et al. Norwegian scabies in a patient treated with Tripterygium glycoside for rheumatoid arthritis. An Bras Dermatol. 2017;92:556-558. doi:10.1590/abd1806-4841.20174946
- Pipitone MA, Adams B, Sheth A, et al. Crusted scabies in a patient being treated with infliximab for juvenile rheumatoid arthritis. J Am Acad Dermatol. 2005;52:719-720. doi:10.1016/j.jaad.2004.12.039
- Baccouche K, Sellam J, Guegan S, et al. Crusted Norwegian scabies, an opportunistic infection, with tocilizumab in rheumatoid arthritis. Joint Bone Spine. 2011;78:402-404. doi:10.1016/j.jbspin.2011.02.008
- Saillard C, Darrieux L, Safa G. Crusted scabies complicates etanercept therapy in a patient with severe psoriasis. J Am Acad Dermatol. 2013;68:E138-E139. doi:10.1016/j.jaad.2012.09.049
- Belvisi V, Orsi GB, Del Borgo C, et al. Large nosocomial outbreakassociated with a Norwegian scabies index case undergoing TNF-α inhibitor treatment: management and control. Infect Control Hosp Epidemiol. 2015;36:1358-1360. doi:10.1017/ice.2015.188
- Nofal A. Variable response of crusted scabies to oral ivermectin: report on eight Egyptian patients. J Eur Acad Dermatol Venereol. 2009;23:793-797. doi:10.1111/j.1468-3083.2009.03177.x
- Yee BE, Carlos CA, Hata T. Crusted scabies of the scalp in a patient with systemic lupus erythematosus. Dermatol Online J. 2014;20:13030/qt9dm891gd.
- Bumb RA, Mehta RD. Crusted scabies in a patient of systemic sclerosis. Indian J Dermatol Venereol Leprol. 2000;66:143-144.
- Hylwa SA, Loss L, Grassi M. Crusted scabies and tinea corporis after treatment of presumed bullous pemphigoid. Cutis. 2013;92:193-198.
- Svecova D, Chmurova N, Pallova A, et al. Norwegian scabies in immunosuppressed patient misdiagnosed as an adverse drug reaction. Epidemiol Mikrobiol Imunol. 2009;58:121-123.
- Dourmishev AL, Serafimova DK, Dourmishev LA, et al. Crusted scabies of the scalp in dermatomyositis patients: three cases treated with oral ivermectin. Int J Dermatol. 1998;37:231-234. doi:10.1046/j.1365-4362.1998.00330.x
- Mortazavi H, Abedini R, Sadri F, et al. Crusted scabies in a patient with brain astrocytoma: report of a case. Int J Infect Dis. 2010;14:E526-E527. doi:10.1016/j.ijid.2009.06.011
- Lima FCDR, Cerqueira AMM, MBS, et al. Crusted scabies due to indiscriminate use of glucocorticoid therapy in infant. An Bras Dermatol. 2017;92:383-385. doi:10.1590/abd1806-4841.20174433
- Binic´ I, Jankovic´ A, Jovanovic´ D, et al. Crusted (Norwegian) scabies following systemic and topical corticosteroid therapy. J Korean Med Sci. 2010;25:188-191. doi:10.3346/jkms.2010.25.1.188
- Ohtaki N, Taniguchi H, Ohtomo H. Oral ivermectin treatment in two cases of scabies: effective in crusted scabies induced by corticosteroid but ineffective in nail scabies. J Dermatol. 2003;30:411-416. doi:10.1111/j.1346-8138.2003.tb00408.x
- Bilan P, Colin-Gorski AM, Chapelon E, et al. Crusted scabies induced by topical corticosteroids: a case report [in French]. Arch Pediatr. 2015;22:1292-1294. doi:10.1016/j.arcped.2015.09.004
- Marlière V, Roul S, C, et al. Crusted (Norwegian) scabies induced by use of topical corticosteroids and treated successfully with ivermectin. J Pediatr. 1999;135:122-124. doi:10.1016/s0022-3476(99)70342-2
- Jaramillo-Ayerbe F, J. Ivermectin for crusted Norwegian scabies induced by use of topical steroids. Arch Dermatol. 1998;134:143-145. doi:10.1001/archderm.134.2.143
- Elion GB. The purine path to chemotherapy. Science. 1989;244:41-47. doi:10.1126/science.2649979
Practice Points
- Crusted scabies is a highly contagious, severe cutaneous ectoparasitic infection that can present atypically in the form of erythroderma.
- Immunomodulatory drugs for the treatment of autoimmune disease can predispose patients to infection, including ectoparasitic infection.
- Dermatologists should be familiar with the full scope of the clinical presentations of scabies and should especially consider this condition in the differential diagnosis of patients who present in an immunosuppressed state.

Violaceous Plaques and Papulonodules on the Umbilicus
The Diagnosis: Cutaneous Deposits Of Myeloma
Cutaneous deposits of myeloma are a rare skin manifestation of multiple myeloma that typically occur in less than 5% of patients.1,2 The lesions represent monoclonal proliferations of plasma cells and arise from direct extension of a neoplastic mass or less commonly from hematogenous or lymphatic spread. This secondary cutaneous involvement by plasma cell myeloma has been referred to in the literature as metastatic or extramedullary cutaneous plasmacytoma.1,2 This condition must be distinguished from cutaneous plasma cell infiltrates without underlying bone marrow involvement, classified by the World Health Organization as primary cutaneous marginal zone B-cell lymphoma and previously referred to as primary cutaneous plasmacytoma.3
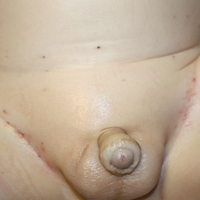
Clinically, cutaneous deposits of myeloma manifest as erythematous to violaceous papules, plaques, or nodules with a smooth surface and firm consistency.1,2 The lesions typically occur on the trunk and less commonly on the head, neck, arms, and legs. In a review of 83 cases of metastatic cutaneous plasmacytoma and primary cutaneous plasmacytoma in multiple myeloma, Kato et al4 found that 52% (43/83) of cases occurred in IgG myelomas and 23% (19/83) in IgA myelomas.
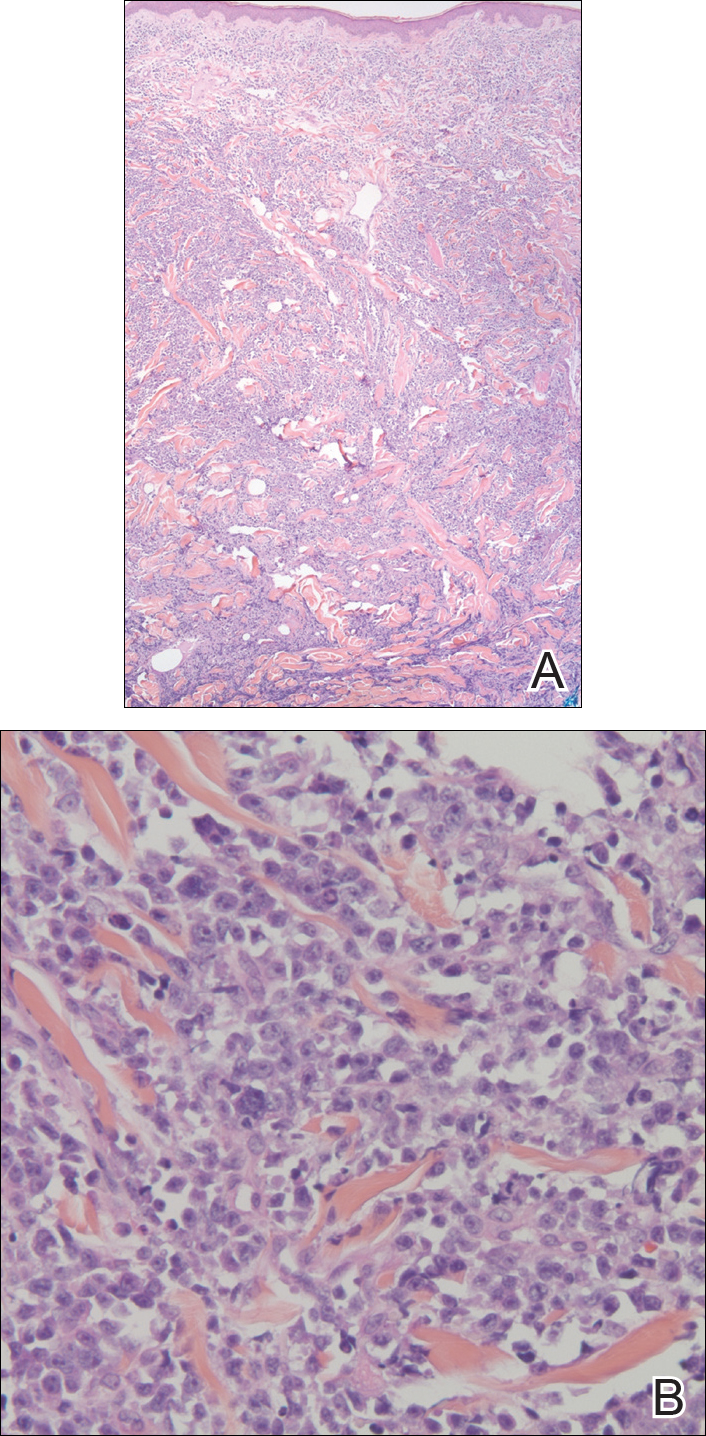
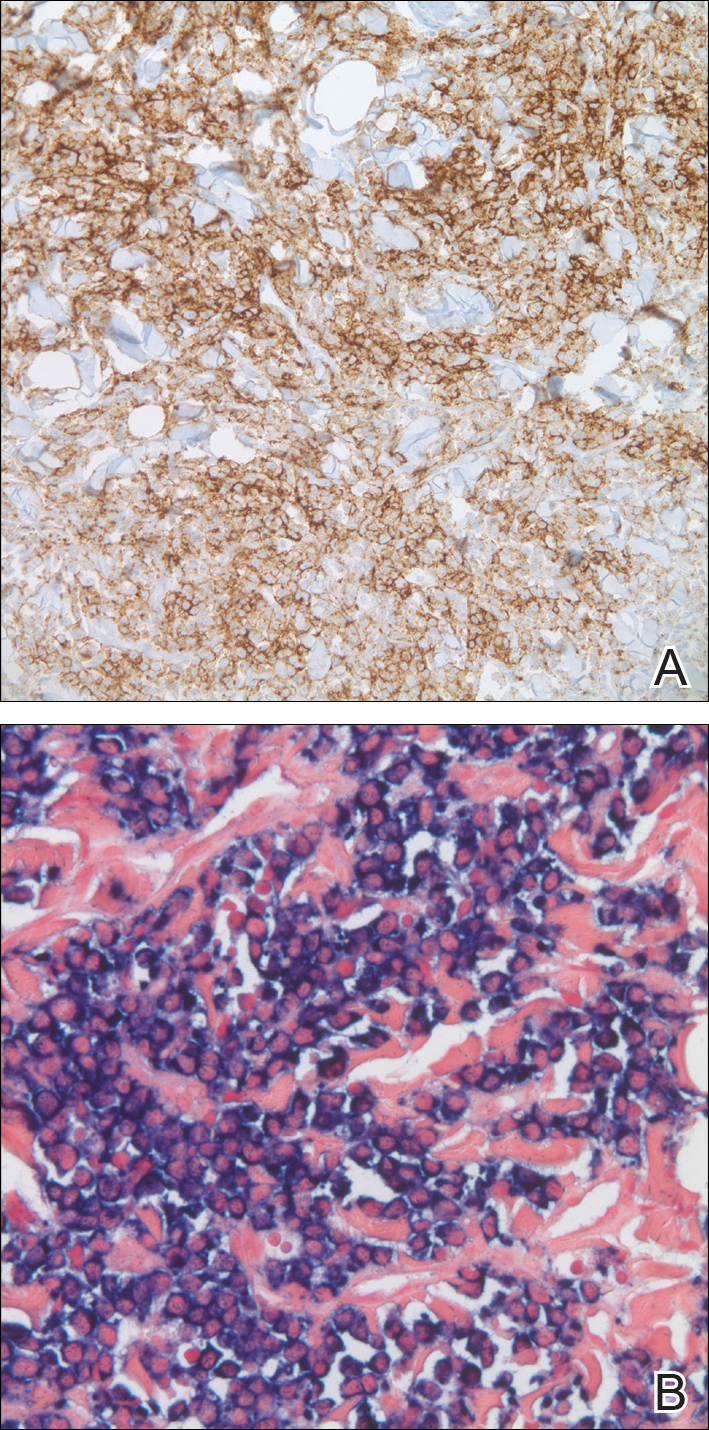
In our patient, a 4-mm punch biopsy of an umbilical plaque demonstrated a dense infiltrate of atypical plasmacytoid cells through the full thickness of the dermis with nuclear pleomorphism, prominent nucleoli, and frequent mitoses (Figure 1). Immunohistochemical staining was positive for IgA λ light chain (Figure 2A) and CD138 (Figure 2B) and was negative for CD20, which was consistent with the patient's known plasma cell myeloma. Positron emission tomography revealed progression of underlying disease compared to prior studies with hypermetabolic mediastinal, retroperitoneal, and pelvic side wall lymphadenopathy, as well as extensive hypermetabolic soft tissue masses with involvement of the periumbilical region.
The differential diagnosis for violaceous periumbilical plaques includes cutaneous marginal zone B-cell lymphoma (primary or secondary) or T-cell lymphoma (primary or secondary), cutaneous metastases from solid organ or hematologic malignancies (eg, Sister Mary Joseph nodule), AIDS-associated Kaposi sarcoma (plum-colored plaques that may be extensive), and cutaneous endometriosis (umbilical nodules that may develop in women after surgical excision of endometrial tissue).
The mainstay of therapy for secondary cutaneous involvement of plasma cell myeloma includes treatment with chemotherapy and local radiotherapy.1,2,5 After the diagnosis of cutaneous deposits of myeloma was made in our patient, he was treated with bortezomib, cyclophosphamide with dexamethasone, and local radiotherapy to symptomatic bony lesions; however, he was unresponsive to therapy and the disease progressed with numerous extramedullary lesions of the mediastinum, gastrointestinal tract, and retroperitoneum 2 months later. The patient developed hydronephrosis from external renal compression necessitating nephrostomy tube and malignant pleural effusions requiring intubation. He experienced rapid clinical decline and died 3 months after the initial presentation due to multiorgan failure.
Cutaneous deposits of myeloma are a sign of underlying disease progression in plasma cell myeloma and often herald a fulminant course (eg, death within 12 months of presentation), as seen in our patient.5 Clinicians should be aware of this rare manifestation of plasma cell myeloma and pursue aggressive therapy given the poor prognostic nature of these cutaneous findings.
- Jorizzo JL, Gammon WR, Briggaman RA. Cutaneous plasmacytomas: a review and presentation of an unusual case. J Am Acad Dermatol. 1979;1:59-66.
- Bayer-Garner IB, Smoller BR. The spectrum of cutaneous disease in multiple myeloma. J Am Acad Dermatol. 2003;48:497-507.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Kato N, Kimura K, Yasukawa K, et al. Metastatic cutaneous plasmacytoma: a case report associated with IgA lambda multiple myeloma and a review of the literature of metastatic cutaneous plasmacytomas associated with multiple myeloma and primary cutaneous plasmacytomas. J Dermatol. 1999;26:587-594.
- Sanal SM, Yaylaci M, Mangold KA, et al. Extensive extramedullary disease in myeloma. an uncommon variant with features of poor prognosis and dedifferentiation. Cancer. 1996;77:1298-1302.
The Diagnosis: Cutaneous Deposits Of Myeloma
Cutaneous deposits of myeloma are a rare skin manifestation of multiple myeloma that typically occur in less than 5% of patients.1,2 The lesions represent monoclonal proliferations of plasma cells and arise from direct extension of a neoplastic mass or less commonly from hematogenous or lymphatic spread. This secondary cutaneous involvement by plasma cell myeloma has been referred to in the literature as metastatic or extramedullary cutaneous plasmacytoma.1,2 This condition must be distinguished from cutaneous plasma cell infiltrates without underlying bone marrow involvement, classified by the World Health Organization as primary cutaneous marginal zone B-cell lymphoma and previously referred to as primary cutaneous plasmacytoma.3
Clinically, cutaneous deposits of myeloma manifest as erythematous to violaceous papules, plaques, or nodules with a smooth surface and firm consistency.1,2 The lesions typically occur on the trunk and less commonly on the head, neck, arms, and legs. In a review of 83 cases of metastatic cutaneous plasmacytoma and primary cutaneous plasmacytoma in multiple myeloma, Kato et al4 found that 52% (43/83) of cases occurred in IgG myelomas and 23% (19/83) in IgA myelomas.
In our patient, a 4-mm punch biopsy of an umbilical plaque demonstrated a dense infiltrate of atypical plasmacytoid cells through the full thickness of the dermis with nuclear pleomorphism, prominent nucleoli, and frequent mitoses (Figure 1). Immunohistochemical staining was positive for IgA λ light chain (Figure 2A) and CD138 (Figure 2B) and was negative for CD20, which was consistent with the patient's known plasma cell myeloma. Positron emission tomography revealed progression of underlying disease compared to prior studies with hypermetabolic mediastinal, retroperitoneal, and pelvic side wall lymphadenopathy, as well as extensive hypermetabolic soft tissue masses with involvement of the periumbilical region.
The differential diagnosis for violaceous periumbilical plaques includes cutaneous marginal zone B-cell lymphoma (primary or secondary) or T-cell lymphoma (primary or secondary), cutaneous metastases from solid organ or hematologic malignancies (eg, Sister Mary Joseph nodule), AIDS-associated Kaposi sarcoma (plum-colored plaques that may be extensive), and cutaneous endometriosis (umbilical nodules that may develop in women after surgical excision of endometrial tissue).
The mainstay of therapy for secondary cutaneous involvement of plasma cell myeloma includes treatment with chemotherapy and local radiotherapy.1,2,5 After the diagnosis of cutaneous deposits of myeloma was made in our patient, he was treated with bortezomib, cyclophosphamide with dexamethasone, and local radiotherapy to symptomatic bony lesions; however, he was unresponsive to therapy and the disease progressed with numerous extramedullary lesions of the mediastinum, gastrointestinal tract, and retroperitoneum 2 months later. The patient developed hydronephrosis from external renal compression necessitating nephrostomy tube and malignant pleural effusions requiring intubation. He experienced rapid clinical decline and died 3 months after the initial presentation due to multiorgan failure.
Cutaneous deposits of myeloma are a sign of underlying disease progression in plasma cell myeloma and often herald a fulminant course (eg, death within 12 months of presentation), as seen in our patient.5 Clinicians should be aware of this rare manifestation of plasma cell myeloma and pursue aggressive therapy given the poor prognostic nature of these cutaneous findings.
The Diagnosis: Cutaneous Deposits Of Myeloma
Cutaneous deposits of myeloma are a rare skin manifestation of multiple myeloma that typically occur in less than 5% of patients.1,2 The lesions represent monoclonal proliferations of plasma cells and arise from direct extension of a neoplastic mass or less commonly from hematogenous or lymphatic spread. This secondary cutaneous involvement by plasma cell myeloma has been referred to in the literature as metastatic or extramedullary cutaneous plasmacytoma.1,2 This condition must be distinguished from cutaneous plasma cell infiltrates without underlying bone marrow involvement, classified by the World Health Organization as primary cutaneous marginal zone B-cell lymphoma and previously referred to as primary cutaneous plasmacytoma.3
Clinically, cutaneous deposits of myeloma manifest as erythematous to violaceous papules, plaques, or nodules with a smooth surface and firm consistency.1,2 The lesions typically occur on the trunk and less commonly on the head, neck, arms, and legs. In a review of 83 cases of metastatic cutaneous plasmacytoma and primary cutaneous plasmacytoma in multiple myeloma, Kato et al4 found that 52% (43/83) of cases occurred in IgG myelomas and 23% (19/83) in IgA myelomas.
In our patient, a 4-mm punch biopsy of an umbilical plaque demonstrated a dense infiltrate of atypical plasmacytoid cells through the full thickness of the dermis with nuclear pleomorphism, prominent nucleoli, and frequent mitoses (Figure 1). Immunohistochemical staining was positive for IgA λ light chain (Figure 2A) and CD138 (Figure 2B) and was negative for CD20, which was consistent with the patient's known plasma cell myeloma. Positron emission tomography revealed progression of underlying disease compared to prior studies with hypermetabolic mediastinal, retroperitoneal, and pelvic side wall lymphadenopathy, as well as extensive hypermetabolic soft tissue masses with involvement of the periumbilical region.
The differential diagnosis for violaceous periumbilical plaques includes cutaneous marginal zone B-cell lymphoma (primary or secondary) or T-cell lymphoma (primary or secondary), cutaneous metastases from solid organ or hematologic malignancies (eg, Sister Mary Joseph nodule), AIDS-associated Kaposi sarcoma (plum-colored plaques that may be extensive), and cutaneous endometriosis (umbilical nodules that may develop in women after surgical excision of endometrial tissue).
The mainstay of therapy for secondary cutaneous involvement of plasma cell myeloma includes treatment with chemotherapy and local radiotherapy.1,2,5 After the diagnosis of cutaneous deposits of myeloma was made in our patient, he was treated with bortezomib, cyclophosphamide with dexamethasone, and local radiotherapy to symptomatic bony lesions; however, he was unresponsive to therapy and the disease progressed with numerous extramedullary lesions of the mediastinum, gastrointestinal tract, and retroperitoneum 2 months later. The patient developed hydronephrosis from external renal compression necessitating nephrostomy tube and malignant pleural effusions requiring intubation. He experienced rapid clinical decline and died 3 months after the initial presentation due to multiorgan failure.
Cutaneous deposits of myeloma are a sign of underlying disease progression in plasma cell myeloma and often herald a fulminant course (eg, death within 12 months of presentation), as seen in our patient.5 Clinicians should be aware of this rare manifestation of plasma cell myeloma and pursue aggressive therapy given the poor prognostic nature of these cutaneous findings.
- Jorizzo JL, Gammon WR, Briggaman RA. Cutaneous plasmacytomas: a review and presentation of an unusual case. J Am Acad Dermatol. 1979;1:59-66.
- Bayer-Garner IB, Smoller BR. The spectrum of cutaneous disease in multiple myeloma. J Am Acad Dermatol. 2003;48:497-507.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Kato N, Kimura K, Yasukawa K, et al. Metastatic cutaneous plasmacytoma: a case report associated with IgA lambda multiple myeloma and a review of the literature of metastatic cutaneous plasmacytomas associated with multiple myeloma and primary cutaneous plasmacytomas. J Dermatol. 1999;26:587-594.
- Sanal SM, Yaylaci M, Mangold KA, et al. Extensive extramedullary disease in myeloma. an uncommon variant with features of poor prognosis and dedifferentiation. Cancer. 1996;77:1298-1302.
- Jorizzo JL, Gammon WR, Briggaman RA. Cutaneous plasmacytomas: a review and presentation of an unusual case. J Am Acad Dermatol. 1979;1:59-66.
- Bayer-Garner IB, Smoller BR. The spectrum of cutaneous disease in multiple myeloma. J Am Acad Dermatol. 2003;48:497-507.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Kato N, Kimura K, Yasukawa K, et al. Metastatic cutaneous plasmacytoma: a case report associated with IgA lambda multiple myeloma and a review of the literature of metastatic cutaneous plasmacytomas associated with multiple myeloma and primary cutaneous plasmacytomas. J Dermatol. 1999;26:587-594.
- Sanal SM, Yaylaci M, Mangold KA, et al. Extensive extramedullary disease in myeloma. an uncommon variant with features of poor prognosis and dedifferentiation. Cancer. 1996;77:1298-1302.
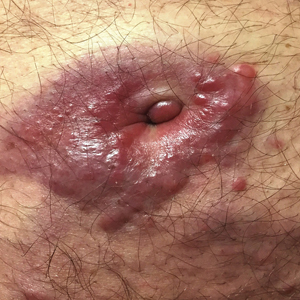
A 75-year-old man presented for evaluation of lesions on the umbilicus and lower abdomen that had developed over the past 4 weeks and were asymptomatic. His medical history was notable for plasma cell myeloma (stage III, IgA λ light chain restricted), deep vein thrombosis, and a 30-year history of smoking (20 packs per year). On physical examination, violaceous plaques and papulonodules were noted on the umbilicus. The lesions had a firm consistency and smooth surface without epidermal change. Violaceous papulonodules and subcutaneous plaques were noted on the lower abdomen. The lesions were nontender to palpation. Bilateral edema of the legs also was noted. The remainder of the skin was normal and there was no cervical, axillary, or inguinal lymphadenopathy.
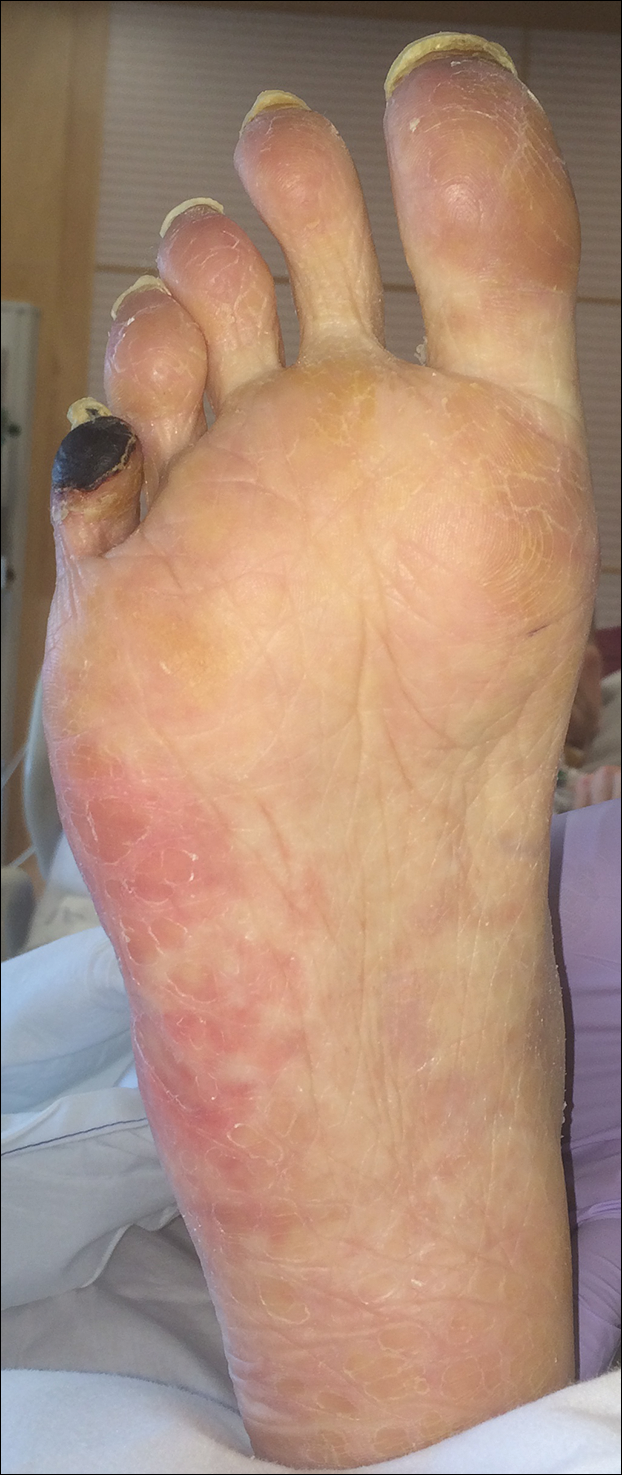
Cyanosis of the Foot
The Diagnosis: Antiphospholipid Antibody Syndrome
A biopsy demonstrated scattered intravascular thrombi in the dermis and subcutis, intact vascular walls, and scant lymphocytic inflammation in a background of stasis (Figure 1). A periodic acid-Schiff stain was negative for fungal elements and highlighted the intravascular thrombi. Histologic findings were consistent with thrombotic vasculopathy. On further laboratory workup, lupus anticoagulant studies, including a mixing study, diluted Russell viper venom test, and hexagonal phase phospholipid neutralization test, were abnormal. Titers of anticardiolipin and β2-glycoprotein I antibodies were elevated (anticardiolipin IgG, 137.7 calculated units [normal, <15 calculated units]; β2-glycoprotein I IgG, 256.4 calculated units [normal, <20 calculated units]). Tissue cultures showed no growth of microorganisms and studies for cryoglobulinemia were negative.

The patient was diagnosed with primary antiphospholipid syndrome (APS). He remained on anticoagulation therapy with fondaparinux as an inpatient and was treated with pulse-dose intravenous (IV) corticosteroids followed by a slow oral taper, daily plasmapheresis for 1 week, IV immunoglobulin (0.5 g/kg) for 3 doses, and 4 weekly doses of rituximab (375 mg/m2). His cutaneous findings slowly improved over the next several weeks (Figure 2).
Antiphospholipid syndrome is an autoimmune disorder characterized by thrombotic events and the presence of autoantibodies. The syndrome is defined by 2 major criteria: (1) the occurrence of at least 1 clinical feature of either an episode of vascular thrombosis or pregnancy morbidity such as unexplained fetal death beyond 10 weeks of gestation or recurrent unexplained pregnancy losses; and (2) the presence of at least 1 type of autoantibody, including lupus anticoagulant, anticardiolipin, or β2-glycoprotein antibodies, on 2 separate occasions at least 12 weeks apart.1 Antiphospholipid syndrome can either be primary with no identifiable associated rheumatologic disease or secondary to another autoimmune disease such as systemic lupus erythematosus. Cutaneous manifestations are common and frequently are the first sign of disease in 30% to 40% of patients.2 The most common skin finding is persistent livedo reticularis, which can be seen in 20% to 25% of patients. Patients also may develop skin necrosis, ulcerations, digital gangrene, splinter hemorrhages, and livedoid vasculopathy.2 Systemic manifestations of APS include thrombocytopenia, nephropathy, cognitive dysfunction, and cardiac valve abnormalities.
The exact pathogenesis of APS remains unknown. It is thought to be due to the combination of an inflammatory stimulus that has yet to be characterized in conjunction with autoantibodies that affect multiple target cells including monocytes, platelets, and endothelial cells, which results in activation of the complement system and clotting cascade.3 In rare cases, the disorder can progress to catastrophic antiphospholipid syndrome (CAPS), which requires fulfillment of 4 criteria: (1) evidence of involvement of 3 organs, tissues, or systems; (2) development of manifestations simultaneously or in less than 1 week; (3) laboratory confirmation of the presence of antiphospholipid antibodies; and (4) confirmation by histopathology of small vessel occlusion.4 Probable CAPS is diagnosed when 3 of 4 criteria are present. Our patient met criteria for probable CAPS, as his antibody titers remained elevated 15 weeks after initial presentation. Precipitating factors that can lead to CAPS are thought to include infection, surgical procedures, medications, or discontinuation of anticoagulation drugs.2 Although the mainstay of management of APS is anticoagulation therapy with warfarin and antiplatelet agents such as aspirin, first-line treatment of CAPS involves high-dose systemic glucocorticoids and plasma exchange. Intravenous immunoglobulin also may be employed in treatment. Data from the CAPS registry demonstrate a role for rituximab, an anti-CD20 antibody, at 375 mg/m2 weekly for 4 weeks (the regimen described in our case) or 1 g every 14 days for 2 sessions.5 A majority of the registry patients treated with rituximab recovered (75% [15/20]) and had no recurrent thrombosis (87% [13/15]) at follow-up.5 Data also are emerging on the role of eculizumab, an anti-C5 antibody that inhibits the terminal complement cascade, as a therapy in difficult-to-treat or refractory CAPS.6-8 The prognosis for CAPS patients without treatment is poor, and mortality has been reported in up to 44% of patients. However, with intervention mortality is reduced by more than 2-fold.9,10
It is important to recognize that acral cyanosis with persistent livedo reticularis and digital gangrene can be a presenting manifestation of APS. These cutaneous manifestations should prompt histologic evaluation for thrombotic vasculopathy in addition to serologic tests for APS autoantibodies. Although APS may be treated with anticoagulants and antiplatelet agents, CAPS may require more aggressive therapy with systemic steroids, plasma exchange, IV immunoglobulin, rituximab, and/or eculizumab.
- Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309-1311.
- Pinto-Almeida T, Caetano M, Sanches M, et al. Cutaneous manifestations of antiphospholipid syndrome: a review of the clinical features, diagnosis and management. Acta Reumatol Port. 2013;38:10-18.
- Meroni PL, Chighizola CB, Rovelli F, et al. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther. 2014;16:209.
- Asherson RA, Cervera R, de Grott PG, et al; Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12:530-534.
- Berman H, Rodríguez-Pintó I, Cervera R, et al. Rituximab use in the catastrophic antiphospholipid syndrome: descriptive analysis of the CAPS registry patients receiving rituximab [published online June 15, 2013]. Autoimmun Rev. 2013;12:1085-1090.
- Shapira I, Andrade D, Allen SL, et al. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64:2719-2723.
- Strakhan M, Hurtado-Sbordoni M, Galeas N, et al. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. 2014;2014:704371.
- Lonze BE, Zachary AA, Magro CM, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14:459-465.
- Bucciarelli S, Espinosa G, Cervera R, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54:2568-2576.
- Asherson RA, Cervera R, Piette JC, et al. Catastrophic antiphospholipid syndrome. clinical and laboratory features of 50 patients. Medicine (Baltimore). 1998;77:195-207.
The Diagnosis: Antiphospholipid Antibody Syndrome
A biopsy demonstrated scattered intravascular thrombi in the dermis and subcutis, intact vascular walls, and scant lymphocytic inflammation in a background of stasis (Figure 1). A periodic acid-Schiff stain was negative for fungal elements and highlighted the intravascular thrombi. Histologic findings were consistent with thrombotic vasculopathy. On further laboratory workup, lupus anticoagulant studies, including a mixing study, diluted Russell viper venom test, and hexagonal phase phospholipid neutralization test, were abnormal. Titers of anticardiolipin and β2-glycoprotein I antibodies were elevated (anticardiolipin IgG, 137.7 calculated units [normal, <15 calculated units]; β2-glycoprotein I IgG, 256.4 calculated units [normal, <20 calculated units]). Tissue cultures showed no growth of microorganisms and studies for cryoglobulinemia were negative.
The patient was diagnosed with primary antiphospholipid syndrome (APS). He remained on anticoagulation therapy with fondaparinux as an inpatient and was treated with pulse-dose intravenous (IV) corticosteroids followed by a slow oral taper, daily plasmapheresis for 1 week, IV immunoglobulin (0.5 g/kg) for 3 doses, and 4 weekly doses of rituximab (375 mg/m2). His cutaneous findings slowly improved over the next several weeks (Figure 2).
Antiphospholipid syndrome is an autoimmune disorder characterized by thrombotic events and the presence of autoantibodies. The syndrome is defined by 2 major criteria: (1) the occurrence of at least 1 clinical feature of either an episode of vascular thrombosis or pregnancy morbidity such as unexplained fetal death beyond 10 weeks of gestation or recurrent unexplained pregnancy losses; and (2) the presence of at least 1 type of autoantibody, including lupus anticoagulant, anticardiolipin, or β2-glycoprotein antibodies, on 2 separate occasions at least 12 weeks apart.1 Antiphospholipid syndrome can either be primary with no identifiable associated rheumatologic disease or secondary to another autoimmune disease such as systemic lupus erythematosus. Cutaneous manifestations are common and frequently are the first sign of disease in 30% to 40% of patients.2 The most common skin finding is persistent livedo reticularis, which can be seen in 20% to 25% of patients. Patients also may develop skin necrosis, ulcerations, digital gangrene, splinter hemorrhages, and livedoid vasculopathy.2 Systemic manifestations of APS include thrombocytopenia, nephropathy, cognitive dysfunction, and cardiac valve abnormalities.
The exact pathogenesis of APS remains unknown. It is thought to be due to the combination of an inflammatory stimulus that has yet to be characterized in conjunction with autoantibodies that affect multiple target cells including monocytes, platelets, and endothelial cells, which results in activation of the complement system and clotting cascade.3 In rare cases, the disorder can progress to catastrophic antiphospholipid syndrome (CAPS), which requires fulfillment of 4 criteria: (1) evidence of involvement of 3 organs, tissues, or systems; (2) development of manifestations simultaneously or in less than 1 week; (3) laboratory confirmation of the presence of antiphospholipid antibodies; and (4) confirmation by histopathology of small vessel occlusion.4 Probable CAPS is diagnosed when 3 of 4 criteria are present. Our patient met criteria for probable CAPS, as his antibody titers remained elevated 15 weeks after initial presentation. Precipitating factors that can lead to CAPS are thought to include infection, surgical procedures, medications, or discontinuation of anticoagulation drugs.2 Although the mainstay of management of APS is anticoagulation therapy with warfarin and antiplatelet agents such as aspirin, first-line treatment of CAPS involves high-dose systemic glucocorticoids and plasma exchange. Intravenous immunoglobulin also may be employed in treatment. Data from the CAPS registry demonstrate a role for rituximab, an anti-CD20 antibody, at 375 mg/m2 weekly for 4 weeks (the regimen described in our case) or 1 g every 14 days for 2 sessions.5 A majority of the registry patients treated with rituximab recovered (75% [15/20]) and had no recurrent thrombosis (87% [13/15]) at follow-up.5 Data also are emerging on the role of eculizumab, an anti-C5 antibody that inhibits the terminal complement cascade, as a therapy in difficult-to-treat or refractory CAPS.6-8 The prognosis for CAPS patients without treatment is poor, and mortality has been reported in up to 44% of patients. However, with intervention mortality is reduced by more than 2-fold.9,10
It is important to recognize that acral cyanosis with persistent livedo reticularis and digital gangrene can be a presenting manifestation of APS. These cutaneous manifestations should prompt histologic evaluation for thrombotic vasculopathy in addition to serologic tests for APS autoantibodies. Although APS may be treated with anticoagulants and antiplatelet agents, CAPS may require more aggressive therapy with systemic steroids, plasma exchange, IV immunoglobulin, rituximab, and/or eculizumab.
The Diagnosis: Antiphospholipid Antibody Syndrome
A biopsy demonstrated scattered intravascular thrombi in the dermis and subcutis, intact vascular walls, and scant lymphocytic inflammation in a background of stasis (Figure 1). A periodic acid-Schiff stain was negative for fungal elements and highlighted the intravascular thrombi. Histologic findings were consistent with thrombotic vasculopathy. On further laboratory workup, lupus anticoagulant studies, including a mixing study, diluted Russell viper venom test, and hexagonal phase phospholipid neutralization test, were abnormal. Titers of anticardiolipin and β2-glycoprotein I antibodies were elevated (anticardiolipin IgG, 137.7 calculated units [normal, <15 calculated units]; β2-glycoprotein I IgG, 256.4 calculated units [normal, <20 calculated units]). Tissue cultures showed no growth of microorganisms and studies for cryoglobulinemia were negative.
The patient was diagnosed with primary antiphospholipid syndrome (APS). He remained on anticoagulation therapy with fondaparinux as an inpatient and was treated with pulse-dose intravenous (IV) corticosteroids followed by a slow oral taper, daily plasmapheresis for 1 week, IV immunoglobulin (0.5 g/kg) for 3 doses, and 4 weekly doses of rituximab (375 mg/m2). His cutaneous findings slowly improved over the next several weeks (Figure 2).
Antiphospholipid syndrome is an autoimmune disorder characterized by thrombotic events and the presence of autoantibodies. The syndrome is defined by 2 major criteria: (1) the occurrence of at least 1 clinical feature of either an episode of vascular thrombosis or pregnancy morbidity such as unexplained fetal death beyond 10 weeks of gestation or recurrent unexplained pregnancy losses; and (2) the presence of at least 1 type of autoantibody, including lupus anticoagulant, anticardiolipin, or β2-glycoprotein antibodies, on 2 separate occasions at least 12 weeks apart.1 Antiphospholipid syndrome can either be primary with no identifiable associated rheumatologic disease or secondary to another autoimmune disease such as systemic lupus erythematosus. Cutaneous manifestations are common and frequently are the first sign of disease in 30% to 40% of patients.2 The most common skin finding is persistent livedo reticularis, which can be seen in 20% to 25% of patients. Patients also may develop skin necrosis, ulcerations, digital gangrene, splinter hemorrhages, and livedoid vasculopathy.2 Systemic manifestations of APS include thrombocytopenia, nephropathy, cognitive dysfunction, and cardiac valve abnormalities.
The exact pathogenesis of APS remains unknown. It is thought to be due to the combination of an inflammatory stimulus that has yet to be characterized in conjunction with autoantibodies that affect multiple target cells including monocytes, platelets, and endothelial cells, which results in activation of the complement system and clotting cascade.3 In rare cases, the disorder can progress to catastrophic antiphospholipid syndrome (CAPS), which requires fulfillment of 4 criteria: (1) evidence of involvement of 3 organs, tissues, or systems; (2) development of manifestations simultaneously or in less than 1 week; (3) laboratory confirmation of the presence of antiphospholipid antibodies; and (4) confirmation by histopathology of small vessel occlusion.4 Probable CAPS is diagnosed when 3 of 4 criteria are present. Our patient met criteria for probable CAPS, as his antibody titers remained elevated 15 weeks after initial presentation. Precipitating factors that can lead to CAPS are thought to include infection, surgical procedures, medications, or discontinuation of anticoagulation drugs.2 Although the mainstay of management of APS is anticoagulation therapy with warfarin and antiplatelet agents such as aspirin, first-line treatment of CAPS involves high-dose systemic glucocorticoids and plasma exchange. Intravenous immunoglobulin also may be employed in treatment. Data from the CAPS registry demonstrate a role for rituximab, an anti-CD20 antibody, at 375 mg/m2 weekly for 4 weeks (the regimen described in our case) or 1 g every 14 days for 2 sessions.5 A majority of the registry patients treated with rituximab recovered (75% [15/20]) and had no recurrent thrombosis (87% [13/15]) at follow-up.5 Data also are emerging on the role of eculizumab, an anti-C5 antibody that inhibits the terminal complement cascade, as a therapy in difficult-to-treat or refractory CAPS.6-8 The prognosis for CAPS patients without treatment is poor, and mortality has been reported in up to 44% of patients. However, with intervention mortality is reduced by more than 2-fold.9,10
It is important to recognize that acral cyanosis with persistent livedo reticularis and digital gangrene can be a presenting manifestation of APS. These cutaneous manifestations should prompt histologic evaluation for thrombotic vasculopathy in addition to serologic tests for APS autoantibodies. Although APS may be treated with anticoagulants and antiplatelet agents, CAPS may require more aggressive therapy with systemic steroids, plasma exchange, IV immunoglobulin, rituximab, and/or eculizumab.
- Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309-1311.
- Pinto-Almeida T, Caetano M, Sanches M, et al. Cutaneous manifestations of antiphospholipid syndrome: a review of the clinical features, diagnosis and management. Acta Reumatol Port. 2013;38:10-18.
- Meroni PL, Chighizola CB, Rovelli F, et al. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther. 2014;16:209.
- Asherson RA, Cervera R, de Grott PG, et al; Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12:530-534.
- Berman H, Rodríguez-Pintó I, Cervera R, et al. Rituximab use in the catastrophic antiphospholipid syndrome: descriptive analysis of the CAPS registry patients receiving rituximab [published online June 15, 2013]. Autoimmun Rev. 2013;12:1085-1090.
- Shapira I, Andrade D, Allen SL, et al. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64:2719-2723.
- Strakhan M, Hurtado-Sbordoni M, Galeas N, et al. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. 2014;2014:704371.
- Lonze BE, Zachary AA, Magro CM, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14:459-465.
- Bucciarelli S, Espinosa G, Cervera R, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54:2568-2576.
- Asherson RA, Cervera R, Piette JC, et al. Catastrophic antiphospholipid syndrome. clinical and laboratory features of 50 patients. Medicine (Baltimore). 1998;77:195-207.
- Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309-1311.
- Pinto-Almeida T, Caetano M, Sanches M, et al. Cutaneous manifestations of antiphospholipid syndrome: a review of the clinical features, diagnosis and management. Acta Reumatol Port. 2013;38:10-18.
- Meroni PL, Chighizola CB, Rovelli F, et al. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther. 2014;16:209.
- Asherson RA, Cervera R, de Grott PG, et al; Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12:530-534.
- Berman H, Rodríguez-Pintó I, Cervera R, et al. Rituximab use in the catastrophic antiphospholipid syndrome: descriptive analysis of the CAPS registry patients receiving rituximab [published online June 15, 2013]. Autoimmun Rev. 2013;12:1085-1090.
- Shapira I, Andrade D, Allen SL, et al. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64:2719-2723.
- Strakhan M, Hurtado-Sbordoni M, Galeas N, et al. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. 2014;2014:704371.
- Lonze BE, Zachary AA, Magro CM, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14:459-465.
- Bucciarelli S, Espinosa G, Cervera R, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54:2568-2576.
- Asherson RA, Cervera R, Piette JC, et al. Catastrophic antiphospholipid syndrome. clinical and laboratory features of 50 patients. Medicine (Baltimore). 1998;77:195-207.
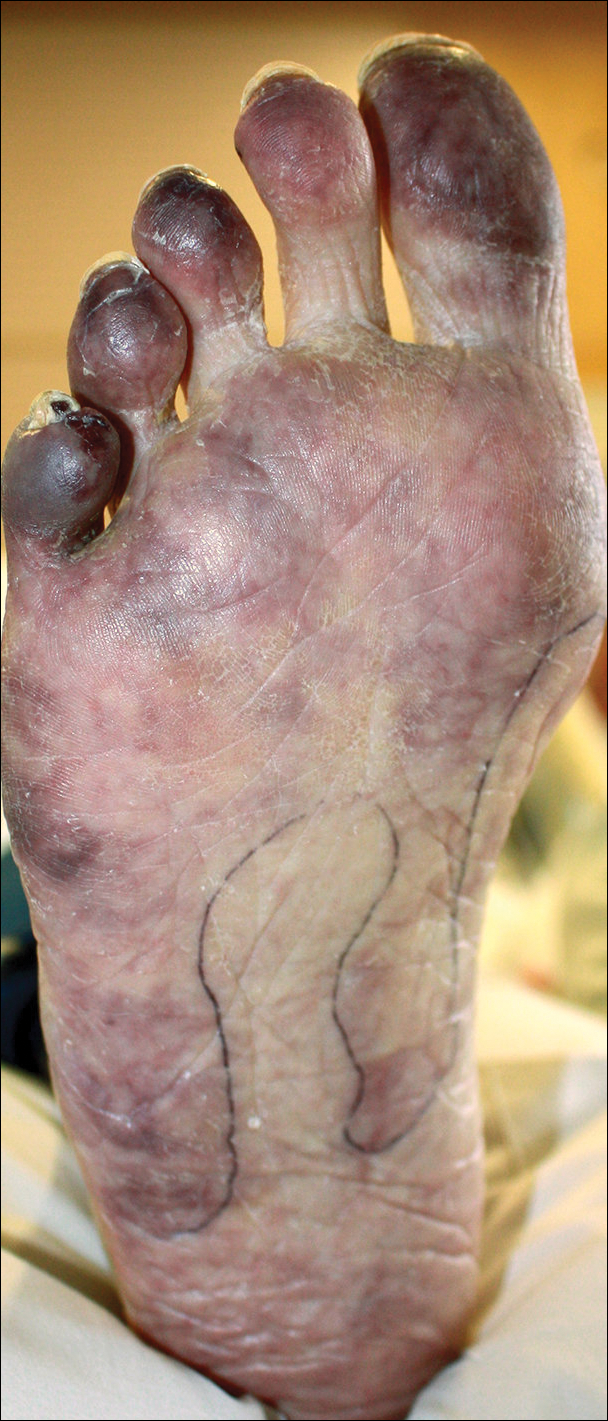
A man in his 50s with a medical history of arterial thrombosis of the right arm, multiple deep vein thromboses (DVTs) of the legs on long-term warfarin, ischemic stroke, atrial fibrillation, and peripheral arterial disease presented with discoloration of the right foot and increasing tenderness of 1 month's duration. There was no history of trauma or recent change in outpatient medications. A family history was notable for an aunt and 2 cousins with DVTs and protein S deficiency. Physical examination revealed livedo reticularis on the sole and lateral aspect of the right foot. There was violaceous discoloration of the volar aspects of all 5 toes and a focal area of ulceration on the fifth toe. Pulses were palpable bilaterally. Initial laboratory evaluation was notable for thrombocytopenia, and preliminary blood cultures revealed no growth of bacterial or fungal organisms. Imaging studies revealed increased arterial stenosis of the right leg as well as DVT of the right great saphenous vein. A punch biopsy of the right medial foot was performed for hematoxylin and eosin stain as well as tissue culture.
Purpuric Lesions of the Scalp, Axillae, and Groin of an Infant
The Diagnosis: Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH) is a clonal proliferative disorder of Langerhans cells that can affect any organ, most commonly the skin and bones. It typically develops in children aged 1 to 3 years, with a male to female ratio of 2 to 1.1 Skin manifestations include purpuric papules, pustules, vesicles, erosions, and fissuring distributed predominantly on the scalp and flexural sites. Mucosal sites, particularly the oral mucosa, may be involved and usually present as erosions associated with underlying bone lesions.1 Langerhans cell histiocytosis should be considered in the differential diagnosis of recalcitrant diaper dermatitis in an infant, especially when there is purpura and erosions, as seen in our patient. Common conditions in infants such as cutaneous candidiasis (intense erythema with superficial erosions, peripheral scale and satellite pustules on flexural areas, potassium hydroxide microscopy revealing yeast forms and pseudohyphae) and seborrheic dermatitis (well-defined pink to red, moist, and often scaly patches favoring the folds) may be distinguished clinically from Hailey-Hailey disease (malodorous plaques with fissures and erosions favoring the folds), which is rare in infancy, and acrodermatitis enteropathica (erythema and erosions with scale-crust and desquamation on periorificial, acral, and intertriginous skin).
Histopathologic evaluation is instrumental in diagnosing the skin lesions of LCH. Further evaluation for systemic involvement is necessary once the diagnosis is made. Skin biopsy of the scalp and right inguinal fold revealed a wedge-shaped infiltrate of histiocytes with slightly folded nuclear contours in our patient (Figure 1). CD1a (Figure 2) and S-100 stains were markedly positive, which is characteristic of LCH. Complete blood cell count, renal function, liver function, urinalysis, and flow cytometry results were within reference range. A skeletal survey and echocardiogram were unremarkable; however, mild hepatosplenomegaly was noted on abdominal ultrasonography.
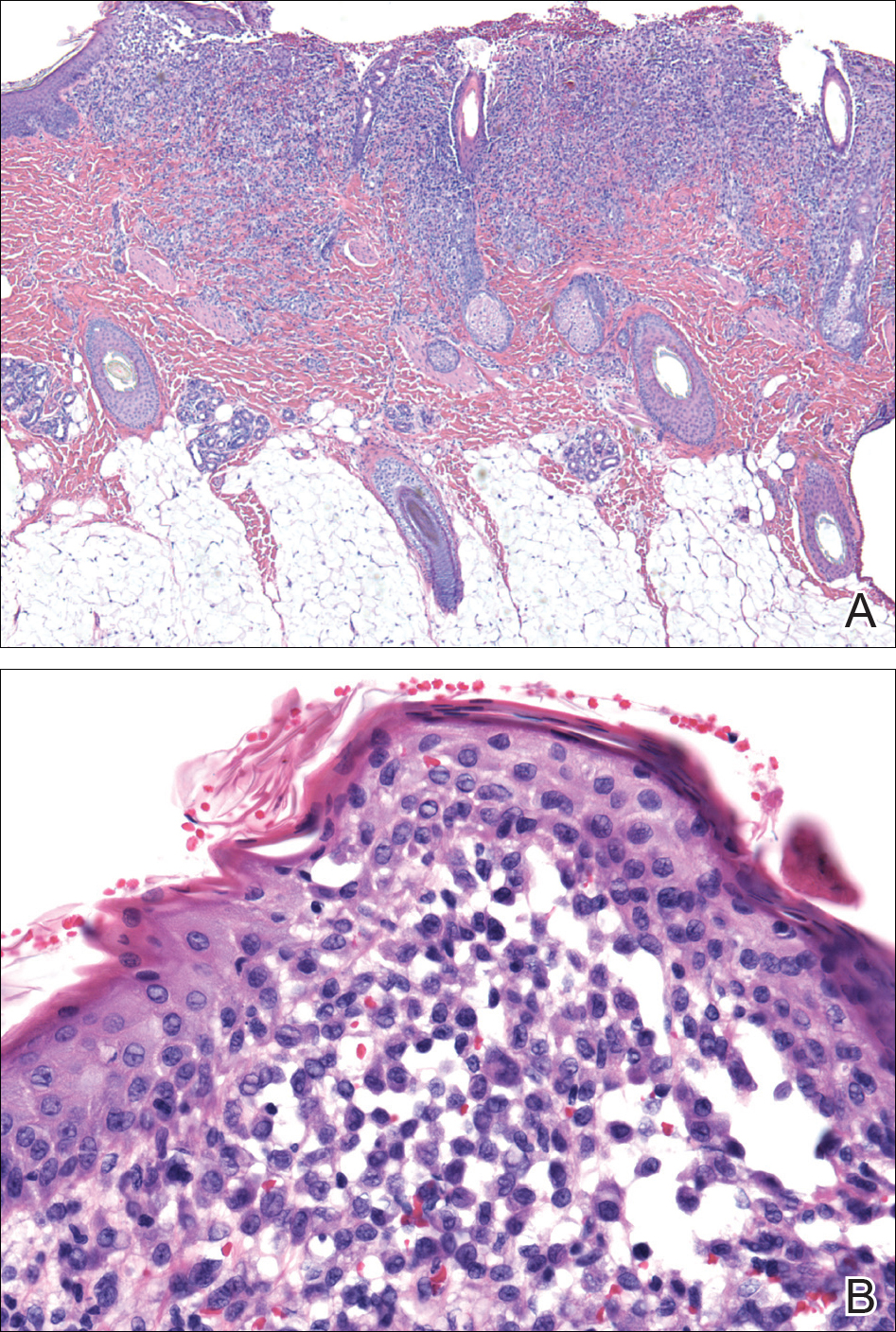
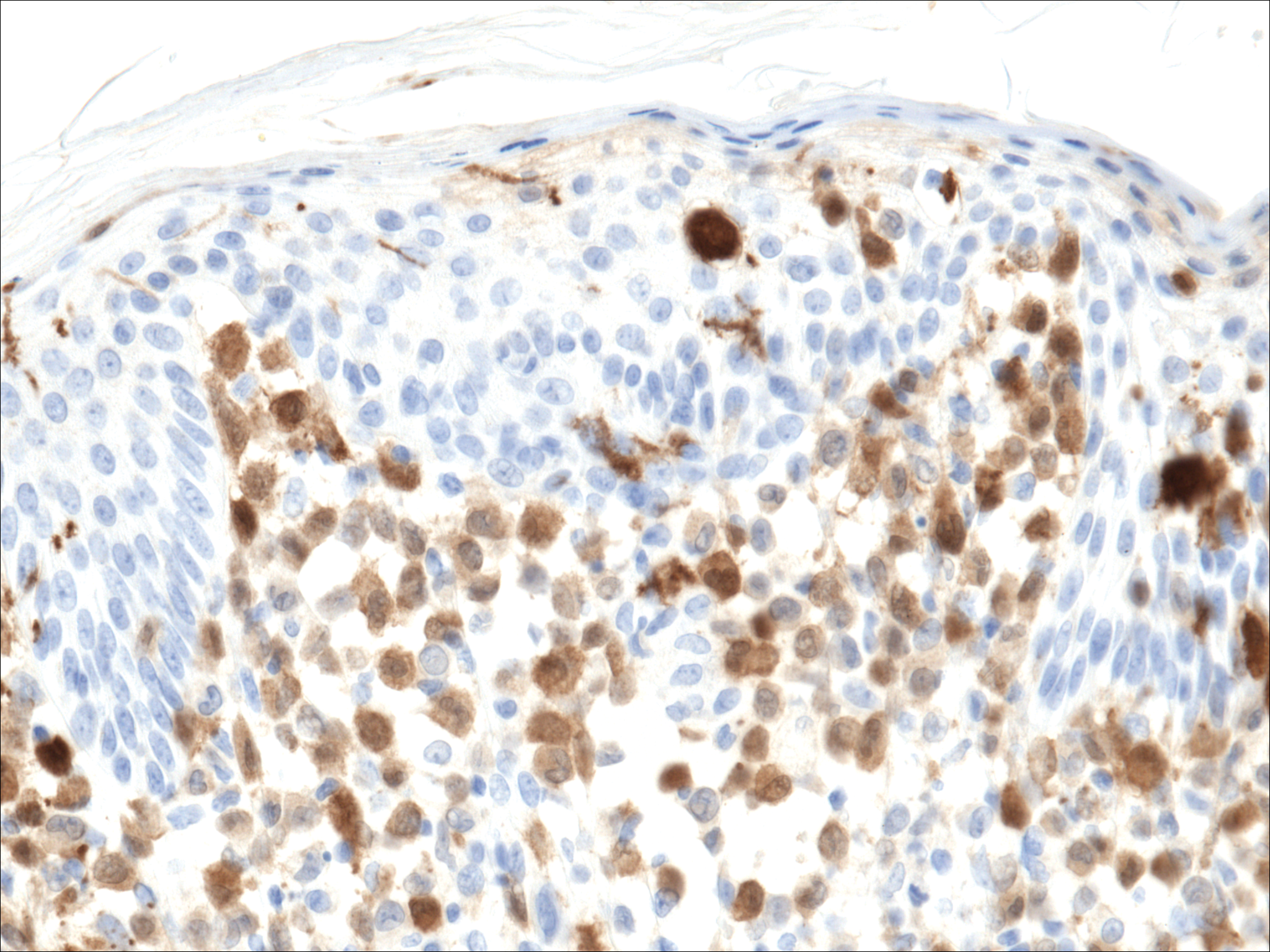
Treatment of LCH varies based on the extent of organ involvement. For isolated cutaneous disease, topical steroids, topical nitrogen mustard, phototherapy, and thalidomide may be employed.2 Multisystem disease requires chemotherapeutic agents including vinblastine and prednisone.2,3 Because more than half of patients with LCH have oncogenic BRAF V600E mutations,4 vemurafenib may have a therapeutic role in treatment. Rare case reports have documented disease response in patients with LCH and Erdheim-Chester disease.5,6
Prognosis varies based on age and extent of systemic involvement. Children younger than 2 years with multiorgan involvement have a poor prognosis (35%-55% mortality rate) compared to older children without hematopoietic, hepatosplenic, or lung involvement (100% survival rate). Additionally, response to treatment affects prognosis, as there is a 66% mortality rate in those who do not respond to treatment after 6 weeks.3 Long-term sequelae of LCH include endocrine dysfunction (ie, diabetes insipidus, growth hormone deficiencies), hearing impairment, orthopedic impairment, and neuropsychological disease; thus, multidisciplinary care often is neccessary.7
Given the multisystem involvement in our patient, he was treated with vinblastine, 6-mercaptopurine, and prednisolone with only partial and transient disease response. He was then treated with clofarabine with dramatic resolution of the mediastinal mass on follow-up positron emission tomography. The cutaneous lesions persisted and were managed with topical corticosteroids.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years [published online October 25, 2012]. Pediatr Blood Cancer. 2013;60:175-184.
- Gadner H, Grois N, Arico M, et al; Histiocyte Society. A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr. 2001;138:728-734.
- Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121:1495-1500.
- Charles J, Beani JC, Fiandrino G, et al. Major response to vemurafenib in patient with severe cutaneous Langerhans cell histiocytosis harboring BRAF V600E mutation. J Am Acad Dermatol. 2014;71:E97-E99.
- Martin A, Macmillan S, Murphy D, et al. Langerhans cell histiocytosis: 23 years' paediatric experience highlights severe long-term sequelae. Scott Med J. 2014;59:149-157.
The Diagnosis: Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH) is a clonal proliferative disorder of Langerhans cells that can affect any organ, most commonly the skin and bones. It typically develops in children aged 1 to 3 years, with a male to female ratio of 2 to 1.1 Skin manifestations include purpuric papules, pustules, vesicles, erosions, and fissuring distributed predominantly on the scalp and flexural sites. Mucosal sites, particularly the oral mucosa, may be involved and usually present as erosions associated with underlying bone lesions.1 Langerhans cell histiocytosis should be considered in the differential diagnosis of recalcitrant diaper dermatitis in an infant, especially when there is purpura and erosions, as seen in our patient. Common conditions in infants such as cutaneous candidiasis (intense erythema with superficial erosions, peripheral scale and satellite pustules on flexural areas, potassium hydroxide microscopy revealing yeast forms and pseudohyphae) and seborrheic dermatitis (well-defined pink to red, moist, and often scaly patches favoring the folds) may be distinguished clinically from Hailey-Hailey disease (malodorous plaques with fissures and erosions favoring the folds), which is rare in infancy, and acrodermatitis enteropathica (erythema and erosions with scale-crust and desquamation on periorificial, acral, and intertriginous skin).
Histopathologic evaluation is instrumental in diagnosing the skin lesions of LCH. Further evaluation for systemic involvement is necessary once the diagnosis is made. Skin biopsy of the scalp and right inguinal fold revealed a wedge-shaped infiltrate of histiocytes with slightly folded nuclear contours in our patient (Figure 1). CD1a (Figure 2) and S-100 stains were markedly positive, which is characteristic of LCH. Complete blood cell count, renal function, liver function, urinalysis, and flow cytometry results were within reference range. A skeletal survey and echocardiogram were unremarkable; however, mild hepatosplenomegaly was noted on abdominal ultrasonography.
Treatment of LCH varies based on the extent of organ involvement. For isolated cutaneous disease, topical steroids, topical nitrogen mustard, phototherapy, and thalidomide may be employed.2 Multisystem disease requires chemotherapeutic agents including vinblastine and prednisone.2,3 Because more than half of patients with LCH have oncogenic BRAF V600E mutations,4 vemurafenib may have a therapeutic role in treatment. Rare case reports have documented disease response in patients with LCH and Erdheim-Chester disease.5,6
Prognosis varies based on age and extent of systemic involvement. Children younger than 2 years with multiorgan involvement have a poor prognosis (35%-55% mortality rate) compared to older children without hematopoietic, hepatosplenic, or lung involvement (100% survival rate). Additionally, response to treatment affects prognosis, as there is a 66% mortality rate in those who do not respond to treatment after 6 weeks.3 Long-term sequelae of LCH include endocrine dysfunction (ie, diabetes insipidus, growth hormone deficiencies), hearing impairment, orthopedic impairment, and neuropsychological disease; thus, multidisciplinary care often is neccessary.7
Given the multisystem involvement in our patient, he was treated with vinblastine, 6-mercaptopurine, and prednisolone with only partial and transient disease response. He was then treated with clofarabine with dramatic resolution of the mediastinal mass on follow-up positron emission tomography. The cutaneous lesions persisted and were managed with topical corticosteroids.
The Diagnosis: Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH) is a clonal proliferative disorder of Langerhans cells that can affect any organ, most commonly the skin and bones. It typically develops in children aged 1 to 3 years, with a male to female ratio of 2 to 1.1 Skin manifestations include purpuric papules, pustules, vesicles, erosions, and fissuring distributed predominantly on the scalp and flexural sites. Mucosal sites, particularly the oral mucosa, may be involved and usually present as erosions associated with underlying bone lesions.1 Langerhans cell histiocytosis should be considered in the differential diagnosis of recalcitrant diaper dermatitis in an infant, especially when there is purpura and erosions, as seen in our patient. Common conditions in infants such as cutaneous candidiasis (intense erythema with superficial erosions, peripheral scale and satellite pustules on flexural areas, potassium hydroxide microscopy revealing yeast forms and pseudohyphae) and seborrheic dermatitis (well-defined pink to red, moist, and often scaly patches favoring the folds) may be distinguished clinically from Hailey-Hailey disease (malodorous plaques with fissures and erosions favoring the folds), which is rare in infancy, and acrodermatitis enteropathica (erythema and erosions with scale-crust and desquamation on periorificial, acral, and intertriginous skin).
Histopathologic evaluation is instrumental in diagnosing the skin lesions of LCH. Further evaluation for systemic involvement is necessary once the diagnosis is made. Skin biopsy of the scalp and right inguinal fold revealed a wedge-shaped infiltrate of histiocytes with slightly folded nuclear contours in our patient (Figure 1). CD1a (Figure 2) and S-100 stains were markedly positive, which is characteristic of LCH. Complete blood cell count, renal function, liver function, urinalysis, and flow cytometry results were within reference range. A skeletal survey and echocardiogram were unremarkable; however, mild hepatosplenomegaly was noted on abdominal ultrasonography.
Treatment of LCH varies based on the extent of organ involvement. For isolated cutaneous disease, topical steroids, topical nitrogen mustard, phototherapy, and thalidomide may be employed.2 Multisystem disease requires chemotherapeutic agents including vinblastine and prednisone.2,3 Because more than half of patients with LCH have oncogenic BRAF V600E mutations,4 vemurafenib may have a therapeutic role in treatment. Rare case reports have documented disease response in patients with LCH and Erdheim-Chester disease.5,6
Prognosis varies based on age and extent of systemic involvement. Children younger than 2 years with multiorgan involvement have a poor prognosis (35%-55% mortality rate) compared to older children without hematopoietic, hepatosplenic, or lung involvement (100% survival rate). Additionally, response to treatment affects prognosis, as there is a 66% mortality rate in those who do not respond to treatment after 6 weeks.3 Long-term sequelae of LCH include endocrine dysfunction (ie, diabetes insipidus, growth hormone deficiencies), hearing impairment, orthopedic impairment, and neuropsychological disease; thus, multidisciplinary care often is neccessary.7
Given the multisystem involvement in our patient, he was treated with vinblastine, 6-mercaptopurine, and prednisolone with only partial and transient disease response. He was then treated with clofarabine with dramatic resolution of the mediastinal mass on follow-up positron emission tomography. The cutaneous lesions persisted and were managed with topical corticosteroids.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years [published online October 25, 2012]. Pediatr Blood Cancer. 2013;60:175-184.
- Gadner H, Grois N, Arico M, et al; Histiocyte Society. A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr. 2001;138:728-734.
- Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121:1495-1500.
- Charles J, Beani JC, Fiandrino G, et al. Major response to vemurafenib in patient with severe cutaneous Langerhans cell histiocytosis harboring BRAF V600E mutation. J Am Acad Dermatol. 2014;71:E97-E99.
- Martin A, Macmillan S, Murphy D, et al. Langerhans cell histiocytosis: 23 years' paediatric experience highlights severe long-term sequelae. Scott Med J. 2014;59:149-157.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years [published online October 25, 2012]. Pediatr Blood Cancer. 2013;60:175-184.
- Gadner H, Grois N, Arico M, et al; Histiocyte Society. A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr. 2001;138:728-734.
- Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121:1495-1500.
- Charles J, Beani JC, Fiandrino G, et al. Major response to vemurafenib in patient with severe cutaneous Langerhans cell histiocytosis harboring BRAF V600E mutation. J Am Acad Dermatol. 2014;71:E97-E99.
- Martin A, Macmillan S, Murphy D, et al. Langerhans cell histiocytosis: 23 years' paediatric experience highlights severe long-term sequelae. Scott Med J. 2014;59:149-157.
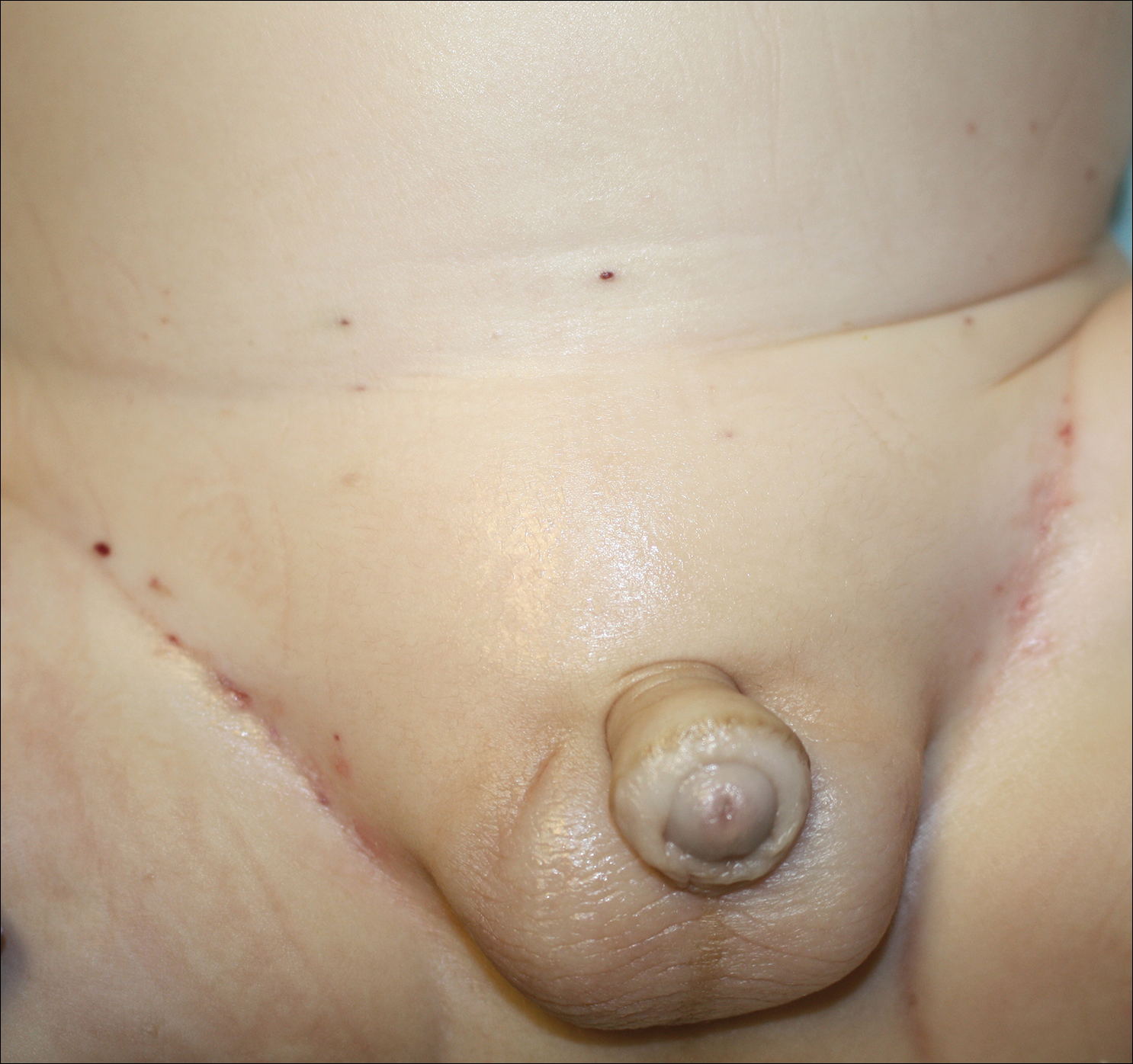
A 7-month-old boy admitted to the hospital with new-onset respiratory stridor was found to have a rash of the scalp, axillae, and groin of 1 month's duration that was unresponsive to treatment with mineral oil. Bronchoscopy revealed tracheal compression, and urgent magnetic resonance imaging of the chest demonstrated an anterior mediastinal mass. Prior to presentation, the patient was otherwise healthy with normal growth and development. On physical examination, scattered red-brown and purpuric papules with hemorrhagic crust were noted on the scalp. There were well-defined pink erosive patches and purpuric papules in the inguinal folds bilaterally and similar erosive patches in the axillae. Numerous punched out ulcerations were noted on the lower gingiva. There was no palpable lymphadenopathy. The hands, feet, penis, scrotum, and perianal area were spared. Biopsies of the skin and mediastinal mass were performed.