Article
Basal Cell Carcinoma: Analysis of Factors Associated With Incomplete Excision at a Referral Hospital in Southern Spain
Basal cell carcinoma (BCC) is the most prevalent malignancy, with excision as the best therapeutic approach. Incomplete excision of nonmelanoma...
Article
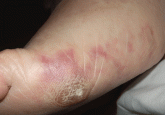
A skin lesion after cardiac catheterization
Two days after coronary angiography, the patient developed pain, cyanosis, and lesions of the sole of his foot. What is the most likely diagnosis...
Article

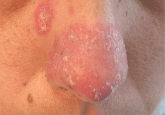
A nodule on a woman’s face
It is firm, pink, 10 mm in diameter, with surrounding telangiectasias. What is it?
Article
An erythematous plaque on the nose
A 38-year-old woman presents with a lesion that appears during periods of cold weather. What is the most likely diagnosis?
Article
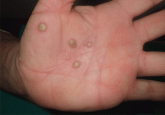
Palmoplantar eruption
A 38-year-old woman with a history of episodes of arthritis presents with pustules on the palms and on the soles of her feet. What is the most...
Article

An ulcerated plaque on the hand
A 73-year-old farmer has a lesion on the dorsum of his hand that bleeds intermittently. What is it?
Article

Painful red nodule on the right hand
A healthy man presents with a tender subcutaneous nodule on his hand that appeared after cleaning his aquarium. What is the most likely diagnosis...
Article
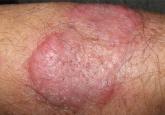
An erythematous plaque on the arm
A 68-year-old farmer presents with an asymptomatic lesion that appeared spontaneously 5 months ago and has grown progressively. What is the...
Article
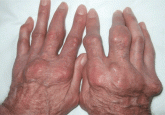
Giant nodules on the hands
A 78-year-old man with hyperuricemia treated with allopurinol presents with asymptomatic nodules on both hands. What is the most likely diagnosis...
Article
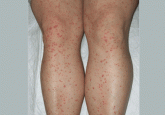
Palpable purpura
A healthy 47-year-old woman presents with a 3-day history of widespread asymptomatic lesions in the extremities, fever, arthralgias, and mild...