User login
A change of heart
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
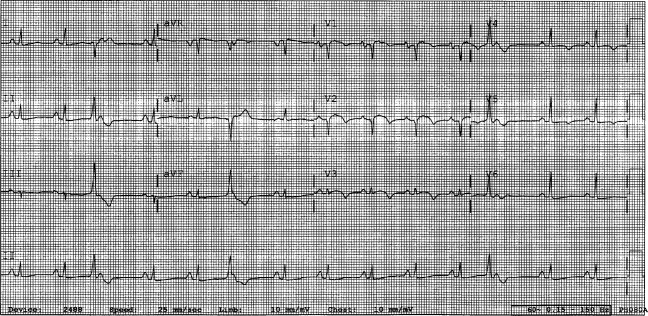
A 29‐year‐old man developed palpitations and dyspnea while loading boxes into a truck. In the emergency department, telemetry demonstrated a wide‐complex tachycardia at a rate of 204 beats per minute. The patient spontaneously cardioverted to sinus rhythm (Figure 1) before direct current cardioversion was performed.
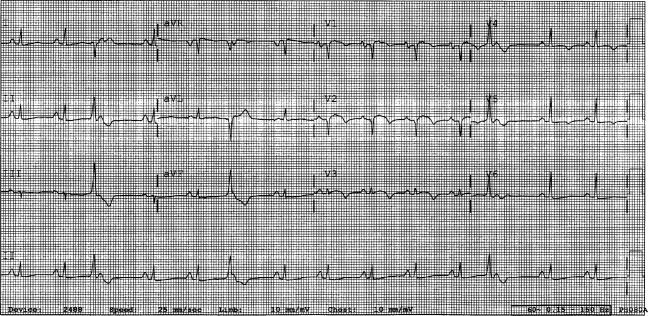
Wide‐complex tachycardia is usually explained by a supraventricular tachycardia with aberrant ventricular conduction or a ventricular tachycardia. Although algorithms exist to guide the clinician in parsing out those etiologies, often the knowledge of underlying structural cardiac disease is most informative. In patients with a history of myocardial infarction, greater than 95% of wide‐complex tachycardia is ventricular tachycardia. The ventricular ectopy, T‐wave inversion or flattening, and poor R‐wave progression are suggestive of a cardiomyopathy, either acute or chronic. A pressing concern, especially with the Q waves and concave ST morphology in V1 and V2, would be coronary ischemia. His age makes this less likely, but an aberrant coronary circulation or drug use could account for it.
Over the past 2 years, the patient had several episodes of sustained palpitations, which terminated after several minutes. Previously, the patient exercised frequently including playing rugby in college. However, over the past year he experienced difficulty climbing stairs due to shortness of breath, which he attributed to deconditioning and smoking. He had no significant medical history, was not taking any medications, nor did he use recreational stimulants. He drank alcohol occasionally. He had no risk factors for the human immunodeficiency virus (HIV). Both of the patient's parents were alive and well. There was no family history of sudden cardiac death.
The duration of symptoms suggests that this is a chronic cardiomyopathy rather than acute myocarditis or acute ischemia, acknowledging that either one could be superimposed. The absence of family history lowers the likelihood of heritable causes of arrhythmia that may accompany a structurally normal (eg, long QT syndrome) or abnormal (eg, hypertrophic cardiomyopathy) heart, although penetrance can be variable. What might account for a cardiomyopathy in a young person? Most cases are probably idiopathic, but etiologies that diverge from the usual suspects of coronary artery disease, hypertension, and valvular disease, which affect an older population, include antecedent viral myocarditis, substance abuse, HIV, or infiltrative disorders such as sarcoidosis.
The patient's pulse was 92 beats per minute and regular and the blood pressure was 96/52 mm Hg. The jugular venous pressure was elevated with prominent v‐waves, the point of maximal impulse was diffuse, there were no extra heart sounds or murmurs, and an enlarged liver was detected. An echocardiogram demonstrated left ventricular dysfunction with an ejection fraction of 30%, severe enlargement of the right atrium and right ventricle, and moderate tricuspid regurgitation. Cardiac catheterization revealed normal coronary arteries without evidence of pulmonary hypertension or intracardiac shunt.
The physical examination and echocardiographic findings of right‐sided failure are unusual given the absence of pulmonary hypertension or intracardiac shunt, and could prompt repeat of the hemodynamic measurements and/or investigations for pulmonary disease that may account for right‐sided pressure overload (in addition to that caused by left ventricular failure). An alternative explanation would be a cardiomyopathic process that preferentially involves the right side of the heart, such as arrhythmogenic right ventricular dysplasia (ARVD), but that would not satisfactorily explain the significant decline in left ventricular function. An acute right ventricular infarction could cause his acute symptoms and his examination and echocardiographic findings, but not the underlying chronic illness. It is common to see patients with long‐standing biventricular failure who present with prominent signs of right‐sided failure (elevated neck veins, hepatomegaly, and edema) but limited or no signs of left‐sided failure (rales) to match their degree of volume overload or dyspnea.
Cardiac magnetic resonance imaging (MRI) revealed a dilated right ventricle with extensive hyperenhancement, a right ventricular ejection fraction of 9%, and moderate left ventricular dysfunction (Figure 2). Electrophysiology testing induced both nonsustained polymorphic and monomorphic ventricular tachycardia. Late potentials were detected on a signal‐averaged electrocardiogram. A single‐chamber cardioverter defibrillator was implanted and the patient was discharged on carvedilol, lisinopril, and spironolactone. An HIV‐1 antibody was negative and a thyroid‐stimulating hormone concentration was within normal limits.
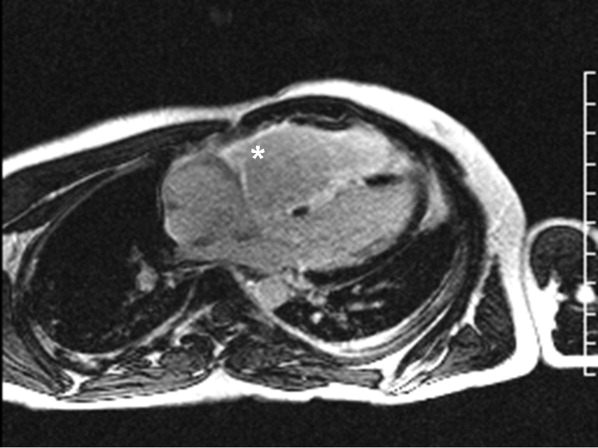
Assuming that accurate evaluation of the pulmonary circulation has been undertaken to exclude pulmonary hypertension, the enlarged and hyperenhanced right ventricle on MRI suggests a process that preferentially infiltrates the right ventricular myocardium, and may secondarily affect the left ventricle either by further infiltration or as a consequence of altered mechanics from the highly dysfunctional right ventricle. ARVD affects the right ventricle, but it is possible that another infiltrative cardiomyopathy, such as sarcoid or an antecedent viral infection, could be restricted in its distribution. Late‐potentials identified on signal average electrocardiograms indicate areas of abnormal conduction that may serve as substrate for reentrant ventricular arrhythmias. They are, however, nonspecific, as they are seen in a variety of myocardial diseases.
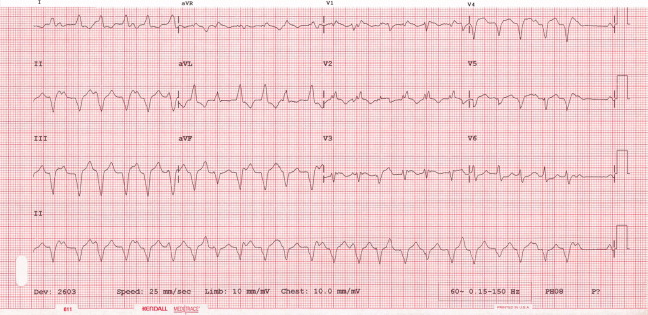
The patient continued to have progressive dyspnea and was readmitted after receiving an appropriate implantable cardioverter defibrillator shock for ventricular tachycardia. Recurrent slow ventricular tachycardia (Figure 3) was treated with supplemental beta‐blockade and amiodarone (10 g total). Repeat echocardiography demonstrated severe left ventricular dysfunction with an ejection fraction of less than 15%. There were no recurrences of ventricular arrhythmias and the patient was discharged and referred for cardiac transplant evaluation for ARVD.
This degree of left ventricular dysfunction is unlikely to be accounted for by altered mechanics and interactions from a failing right ventricle alone and frames this as a biventricular cardiomyopathy, which has an extensive differential diagnosis and requires information from the general medical evaluation.
On routine laboratory testing 6 months later, a serum aspartate aminotransferase of 79 units/L and a serum alanine aminotransferase of 118 units/L were found. Bilirubin, albumin, and alkaline phosphatase were normal. The transaminase levels had been normal on initial evaluation. The patient reported that 2 paternal uncles had end‐stage nonalcoholic cirrhosis. Transjugular liver biopsy was consistent with mild lobular hepatitis with mild portal fibrosis with a few lobular collections of mononuclear cells. There was no evidence of iron overload. The hepatic venogram and transhepatic pressure gradient (2 mm Hg) were normal.
The elevated transaminase levels could be due to amiodarone‐associated hepatotoxicity, hepatic congestion, or a primary liver disease. It is important to consider combined cardiohepatic syndromes such as hemochromatosis, sarcoidosis, or amyloidosis. The relatively normal liver histology and normal hepatic hemodynamics do not suggest a significant primary intrinsic liver disease. The 2 uncles with cirrhosis could suggest a heritable liver disease, although cirrhosis in multiple family members is frequently accounted for by shared habits such as alcohol consumption or excessive caloric intake. Liver disorders with a genetic component, such as hemochromatosis, Wilson's disease, and alpha‐1‐antitrypsin deficiency are mostly autosomal recessive, which would make this pattern of transmission unusual. Furthermore, aside from hemochromatosis, these genetic hepatic disorders have few cardiac manifestations. Right‐sided congestion and amiodarone appear to be the most likely explanations of his liver abnormalities.
Pulmonary function testing revealed normal lung volumes without obstruction, but the diffusing capacity for carbon monoxide was substantially reduced. Computed tomography of the chest identified scattered ground‐glass opacities as well as small nodules with an upper lobe distribution (Figure 4). Although not reported on the initial interpretation, review of a chest x‐ray taken 6 months previously also demonstrated small nodules in the upper lobe distribution. Bronchoscopic examination was normal. Bronchioalveolar lavage fluid stains and cultures for bacteria, mycobacteria, Pneumocystis, and fungus were negative. Transbronchial biopsies of the right middle lobe had no evidence of infection, malignancy, or granulomatous inflammation. The patient continued to have progressive New York Heart Association Class IV heart failure symptoms. Repeat right heart catheterization was notable for a cardiac index of 1.4 L/minute/m2. The mean pulmonary artery pressure was 20 mm Hg. An intraaortic balloon pump was placed for refractory cardiogenic shock.
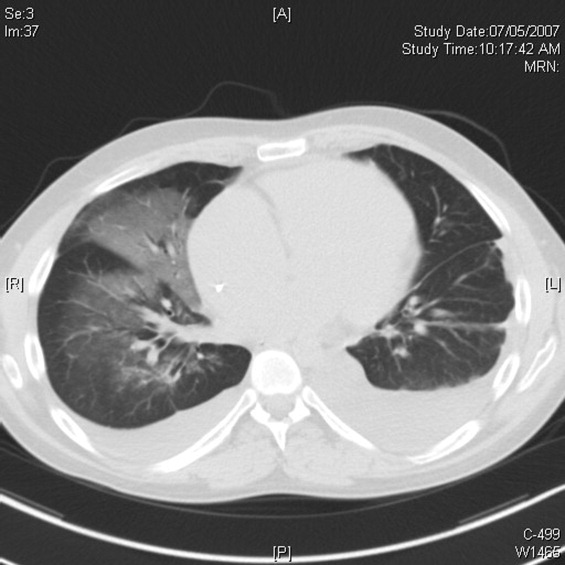
The reduced diffusion capacity and ground‐glass opacities suggest an interstitial process, which may have been missed on transbronchial biopsy because of sampling error. His pulmonary disease is likely another manifestation of his infiltrative cardiac disease. The constellation of cardiac, pulmonary, and hepatic involvement in the context of progressive dyspnea over 2 years is suggestive of sarcoidosis although the absence of hilar lymphadenopathy and 2 biopsy specimens without granulomas argue against the diagnosis, and the effects of amiodarone on the latter 2 organs cannot be ignored. On the limited menu of pharmacologic treatments that may treat this severe and progressive cardiomyopathy are steroids, which makes a diligent search for a steroid‐responsive syndrome important. Therefore, despite the negative studies, sarcoidosis must be investigated to the fullest extent with either an endomyocardial biopsy or surgical lung biopsy.
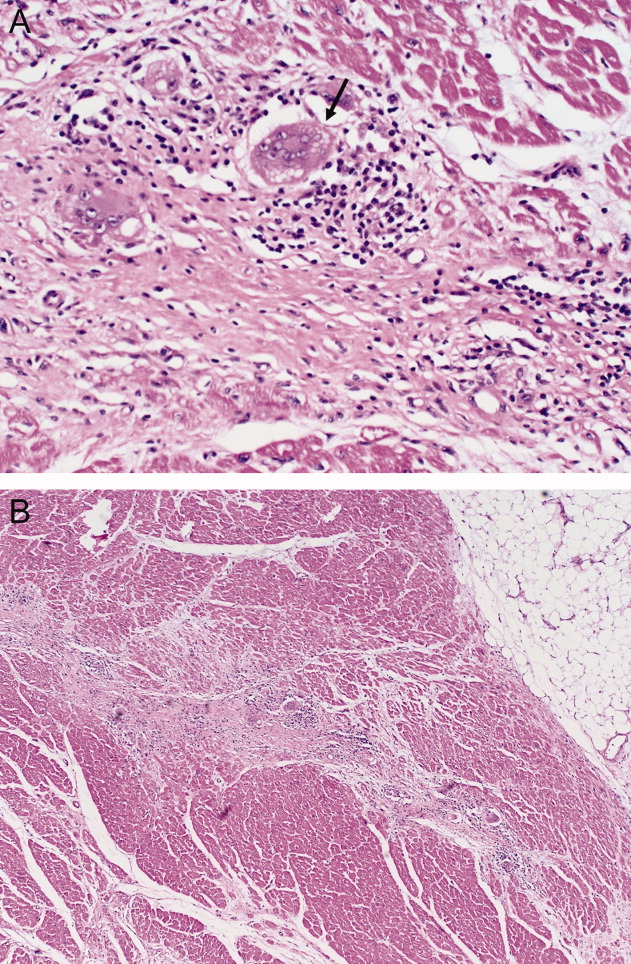
The patient underwent cardiac transplantation. The native heart was found to have right ventricular thinning, which was most notable at the right ventricular outflow tract. Microscopic examination revealed extensive fibrosis and granulomatous inflammation (Figure 5) with scarring typical of cardiac sarcoidosis. Six months after cardiac transplantation, the patient is doing well on prednisone, tacrolimus, and mycophenolate mofetil. Follow‐up chest x‐rays show resolution of the pulmonary nodules.
COMMENTARY
Cardiomyopathy in a young person is a relatively uncommon clinical event that prompts consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in older adults. This case highlights the challenges of arriving at a diagnosis in the absence of a gold standard, and the greater challenges of modifying initial diagnostic impressions as new clinical data become available.
After encountering ventricular tachycardia and right ventricular dysfunction in a young patient, the clinicians arrived at the diagnosis of ARVD. This rare and progressive disorder is associated with up to 20% of ventricular arrhythmias and sudden death in the young,1, 2 but can be challenging to diagnose. Despite common referrals for cardiac MRI to exclude ARVD, cardiac MRI is not the gold standard for diagnosis and is the most common method of misdiagnosis of ARVD.3 A diagnosis of ARVD requires the presence of 2 major, 1 major and 2 minor, or 4 minor International Task Force criteria (Table 1).4, 5 While the diagnostic criteria provide standardization across populations (eg, in clinical studies), additional considerations are needed in the management of individual patients. Scoring systems serve as a tool, but the final diagnosis requires balancing such criteria with competing hypotheses. This dilemma is familiar to clinicians considering other less common conditions such as amyotrophic lateral sclerosis (World Neurology Foundation), rheumatic fever (Jones criteria), or systemic lupus erythematosus (American College of Rheumatology). This patient's cardiac MRI findings, precordial T‐wave inversions, frequent ventricular ectopy, and late potentials on a signal‐averaged electrocardiogram fulfilled the International Task Force criteria for a diagnosis of ARVD. Discordant information included the right bundle branch pattern of the ventricular tachycardia, which suggested left ventricular origin, as opposed to the more common left bundle branch pattern observed in ARVD, and the absence of a family history. In addition, in U.S. populations only 25% of cases present with heart failure and fewer than 5% develop biventricular failure.6 Nonetheless, this patient's imaging evidence of right ventricular structural abnormalities and dysfunction and electrocardiographic abnormalities coupled with the absence of obvious systemic disease made ARVD the logical working diagnosis.
| Major | Minor | |
|---|---|---|
| ||
| I. Global and/or regional dysfunction and structural alterations | Severe dilation and reduction of right ventricular ejection fraction, localized right ventricular aneurysms | Mild right ventricular dilatation and/or reduced ejection fraction |
| II. Endomyocardial biopsy | Fibrofatty replacement of myocardium | |
| III. Repolarization abnormalities | T‐wave inversion in leads V1‐V3 or beyond | |
| IV. Depolarization/conduction abnormalities | Epsilon waves or localized QRS prolongation (>110 msec) in leads V1‐V3 | Late potentials on signal‐averaged electrocardiogram |
| V. Arrhythmias | Left bundle branch block‐type ventricular tachycardia (sustained and nonsustained) or frequent ventricular extra systoles (>1,000/24 hours) | |
| VI. Family history | Familial disease confirmed at necropsy or surgery | Familial history of premature sudden death (<35 years old) or clinical diagnosis based on present criteria |
When more widespread manifestations developed, namely hepatic and pulmonary abnormalities, each was investigated with imaging and biopsy. Once a multisystem illness became apparent, the discussant reframed the patient's illness to include other diagnostic possibilities. In practice it is difficult to reverse a working diagnosis despite contradictory evidence because of the common pitfall of anchoring bias. Tversky and Kahneman7 were the first to describe the cognitive processes behind probability assessment and decision making in time‐sensitive situations. Under these conditions, decision makers tend to focus on the first symptom, striking feature, or diagnosis and anchor subsequent probabilities to that initial presentation. Once a decision or diagnosis has been reached, clinicians tend to interpret subsequent findings in the context of the original diagnosis rather than reevaluating their initial impression. In the setting of a known diagnosis of ARVD, 3 separate diagnoses (ARVD, amiodarone‐associated lung injury, and amiodarone‐induced hepatic dysfunction) were considered by the treating physicians. The initial diagnosis of ARVD followed by the sequential rather than simultaneous manifestations of sarcoidosis made arriving at the revised diagnosis even more challenging.
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular dysplasia.8, 9 The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, myocarditis, idiopathic cardiomyopathy, and sarcoidosis. Cardiac sarcoidosis can present as ventricular ectopy, sustained ventricular arrhythmias, asymptomatic ventricular dysfunction, heart failure, or sudden death.10 Although 25% of patients with sarcoidosis have evidence of cardiac involvement at autopsy, only 5% have clinical manifestations.11 Those patients with clinical evidence of cardiac sarcoidosis have a wide range of clinical findings (Table 2). While the patient's cardiomyopathy was advanced, it is possible that earlier administration of corticosteroid therapy may have arrested his progressive biventricular failure. As clinicians, we should always remember to force ourselves to broaden our differential diagnosis when new findings become available, especially those that point to a systemicrather than an organ‐specificdisorder. In this case, while the original diagnostic findings were accurate and strongly suggested ARVD, a change of heart was needed to arrive at the ultimate diagnosis.
| Clinical Manifestation | Prevalence (%) |
|---|---|
| Atrioventricular block | 40 |
| Bundle branch block | 40 |
| Supraventricular tachycardia | 20 |
| Ventricular arrhythmias | 25 |
| Heart failure | 25 |
| Sudden cardiac death | 35 |
KEY POINTS FOR HOSPITALISTS
-
Cardiomyopathy in a young person requires consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in the elderly.
-
Anchoring bias is a common pitfall in clinical decision making. When new or contradictory findings are uncovered, clinicians should reevaluate their initial impression to ensure it remains the most likely diagnosis.
-
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular cardiomyopathy. The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, right ventricular outflow tract tachycardia, myocarditis, idiopathic dilated cardiomyopathy, and sarcoidosis.
- ,,, et al.Right ventricular dysplasia: a report of 24 adult cases.Circulation.1982;65:384–398.
- ,,,,.Right ventricular cardiomyopathy and sudden death in young people.N Engl J Med.1988;318:129–133.
- ,,, et al.Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy.J Cardiovasc Electrophysiol.2004;15:300–306.
- ,,, et al.Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology.Br Heart J.1994;71:215–218.
- ,,, et al.Predictors of appropriate implantable defibrillator therapies in patients with arrhythmogenic right ventricular dysplasia.Heart Rhythm.2005;2:1188–1194.
- ,,, et al.Arrhythmogenic right ventricular dysplasia: a United States experience.Circulation.2005;112:3823–3832.
- ,.Judgment under uncertainty: heuristics and biases.Science.1974;185:1124–1131.
- ,,.Unusual presentation of cardiac sarcoidosis.Congest Heart Fail.2007;13:116–118.
- ,,, et al.Cardiac sarcoidosis mimicking right ventricular dysplasia.Circ J.2003;67:169–171.
- ,,,,.Refractory ventricular tachycardia secondary to cardiac sarcoid: electrophysiologic characteristics, mapping, and ablation.Heart Rhythm.2006;3:924–929.
- ,.Sarcoid heart disease: clinical course and treatment.Int J Cardiol.2004;97:173–182.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 29‐year‐old man developed palpitations and dyspnea while loading boxes into a truck. In the emergency department, telemetry demonstrated a wide‐complex tachycardia at a rate of 204 beats per minute. The patient spontaneously cardioverted to sinus rhythm (Figure 1) before direct current cardioversion was performed.
Wide‐complex tachycardia is usually explained by a supraventricular tachycardia with aberrant ventricular conduction or a ventricular tachycardia. Although algorithms exist to guide the clinician in parsing out those etiologies, often the knowledge of underlying structural cardiac disease is most informative. In patients with a history of myocardial infarction, greater than 95% of wide‐complex tachycardia is ventricular tachycardia. The ventricular ectopy, T‐wave inversion or flattening, and poor R‐wave progression are suggestive of a cardiomyopathy, either acute or chronic. A pressing concern, especially with the Q waves and concave ST morphology in V1 and V2, would be coronary ischemia. His age makes this less likely, but an aberrant coronary circulation or drug use could account for it.
Over the past 2 years, the patient had several episodes of sustained palpitations, which terminated after several minutes. Previously, the patient exercised frequently including playing rugby in college. However, over the past year he experienced difficulty climbing stairs due to shortness of breath, which he attributed to deconditioning and smoking. He had no significant medical history, was not taking any medications, nor did he use recreational stimulants. He drank alcohol occasionally. He had no risk factors for the human immunodeficiency virus (HIV). Both of the patient's parents were alive and well. There was no family history of sudden cardiac death.
The duration of symptoms suggests that this is a chronic cardiomyopathy rather than acute myocarditis or acute ischemia, acknowledging that either one could be superimposed. The absence of family history lowers the likelihood of heritable causes of arrhythmia that may accompany a structurally normal (eg, long QT syndrome) or abnormal (eg, hypertrophic cardiomyopathy) heart, although penetrance can be variable. What might account for a cardiomyopathy in a young person? Most cases are probably idiopathic, but etiologies that diverge from the usual suspects of coronary artery disease, hypertension, and valvular disease, which affect an older population, include antecedent viral myocarditis, substance abuse, HIV, or infiltrative disorders such as sarcoidosis.
The patient's pulse was 92 beats per minute and regular and the blood pressure was 96/52 mm Hg. The jugular venous pressure was elevated with prominent v‐waves, the point of maximal impulse was diffuse, there were no extra heart sounds or murmurs, and an enlarged liver was detected. An echocardiogram demonstrated left ventricular dysfunction with an ejection fraction of 30%, severe enlargement of the right atrium and right ventricle, and moderate tricuspid regurgitation. Cardiac catheterization revealed normal coronary arteries without evidence of pulmonary hypertension or intracardiac shunt.
The physical examination and echocardiographic findings of right‐sided failure are unusual given the absence of pulmonary hypertension or intracardiac shunt, and could prompt repeat of the hemodynamic measurements and/or investigations for pulmonary disease that may account for right‐sided pressure overload (in addition to that caused by left ventricular failure). An alternative explanation would be a cardiomyopathic process that preferentially involves the right side of the heart, such as arrhythmogenic right ventricular dysplasia (ARVD), but that would not satisfactorily explain the significant decline in left ventricular function. An acute right ventricular infarction could cause his acute symptoms and his examination and echocardiographic findings, but not the underlying chronic illness. It is common to see patients with long‐standing biventricular failure who present with prominent signs of right‐sided failure (elevated neck veins, hepatomegaly, and edema) but limited or no signs of left‐sided failure (rales) to match their degree of volume overload or dyspnea.
Cardiac magnetic resonance imaging (MRI) revealed a dilated right ventricle with extensive hyperenhancement, a right ventricular ejection fraction of 9%, and moderate left ventricular dysfunction (Figure 2). Electrophysiology testing induced both nonsustained polymorphic and monomorphic ventricular tachycardia. Late potentials were detected on a signal‐averaged electrocardiogram. A single‐chamber cardioverter defibrillator was implanted and the patient was discharged on carvedilol, lisinopril, and spironolactone. An HIV‐1 antibody was negative and a thyroid‐stimulating hormone concentration was within normal limits.
Assuming that accurate evaluation of the pulmonary circulation has been undertaken to exclude pulmonary hypertension, the enlarged and hyperenhanced right ventricle on MRI suggests a process that preferentially infiltrates the right ventricular myocardium, and may secondarily affect the left ventricle either by further infiltration or as a consequence of altered mechanics from the highly dysfunctional right ventricle. ARVD affects the right ventricle, but it is possible that another infiltrative cardiomyopathy, such as sarcoid or an antecedent viral infection, could be restricted in its distribution. Late‐potentials identified on signal average electrocardiograms indicate areas of abnormal conduction that may serve as substrate for reentrant ventricular arrhythmias. They are, however, nonspecific, as they are seen in a variety of myocardial diseases.
The patient continued to have progressive dyspnea and was readmitted after receiving an appropriate implantable cardioverter defibrillator shock for ventricular tachycardia. Recurrent slow ventricular tachycardia (Figure 3) was treated with supplemental beta‐blockade and amiodarone (10 g total). Repeat echocardiography demonstrated severe left ventricular dysfunction with an ejection fraction of less than 15%. There were no recurrences of ventricular arrhythmias and the patient was discharged and referred for cardiac transplant evaluation for ARVD.
This degree of left ventricular dysfunction is unlikely to be accounted for by altered mechanics and interactions from a failing right ventricle alone and frames this as a biventricular cardiomyopathy, which has an extensive differential diagnosis and requires information from the general medical evaluation.
On routine laboratory testing 6 months later, a serum aspartate aminotransferase of 79 units/L and a serum alanine aminotransferase of 118 units/L were found. Bilirubin, albumin, and alkaline phosphatase were normal. The transaminase levels had been normal on initial evaluation. The patient reported that 2 paternal uncles had end‐stage nonalcoholic cirrhosis. Transjugular liver biopsy was consistent with mild lobular hepatitis with mild portal fibrosis with a few lobular collections of mononuclear cells. There was no evidence of iron overload. The hepatic venogram and transhepatic pressure gradient (2 mm Hg) were normal.
The elevated transaminase levels could be due to amiodarone‐associated hepatotoxicity, hepatic congestion, or a primary liver disease. It is important to consider combined cardiohepatic syndromes such as hemochromatosis, sarcoidosis, or amyloidosis. The relatively normal liver histology and normal hepatic hemodynamics do not suggest a significant primary intrinsic liver disease. The 2 uncles with cirrhosis could suggest a heritable liver disease, although cirrhosis in multiple family members is frequently accounted for by shared habits such as alcohol consumption or excessive caloric intake. Liver disorders with a genetic component, such as hemochromatosis, Wilson's disease, and alpha‐1‐antitrypsin deficiency are mostly autosomal recessive, which would make this pattern of transmission unusual. Furthermore, aside from hemochromatosis, these genetic hepatic disorders have few cardiac manifestations. Right‐sided congestion and amiodarone appear to be the most likely explanations of his liver abnormalities.
Pulmonary function testing revealed normal lung volumes without obstruction, but the diffusing capacity for carbon monoxide was substantially reduced. Computed tomography of the chest identified scattered ground‐glass opacities as well as small nodules with an upper lobe distribution (Figure 4). Although not reported on the initial interpretation, review of a chest x‐ray taken 6 months previously also demonstrated small nodules in the upper lobe distribution. Bronchoscopic examination was normal. Bronchioalveolar lavage fluid stains and cultures for bacteria, mycobacteria, Pneumocystis, and fungus were negative. Transbronchial biopsies of the right middle lobe had no evidence of infection, malignancy, or granulomatous inflammation. The patient continued to have progressive New York Heart Association Class IV heart failure symptoms. Repeat right heart catheterization was notable for a cardiac index of 1.4 L/minute/m2. The mean pulmonary artery pressure was 20 mm Hg. An intraaortic balloon pump was placed for refractory cardiogenic shock.
The reduced diffusion capacity and ground‐glass opacities suggest an interstitial process, which may have been missed on transbronchial biopsy because of sampling error. His pulmonary disease is likely another manifestation of his infiltrative cardiac disease. The constellation of cardiac, pulmonary, and hepatic involvement in the context of progressive dyspnea over 2 years is suggestive of sarcoidosis although the absence of hilar lymphadenopathy and 2 biopsy specimens without granulomas argue against the diagnosis, and the effects of amiodarone on the latter 2 organs cannot be ignored. On the limited menu of pharmacologic treatments that may treat this severe and progressive cardiomyopathy are steroids, which makes a diligent search for a steroid‐responsive syndrome important. Therefore, despite the negative studies, sarcoidosis must be investigated to the fullest extent with either an endomyocardial biopsy or surgical lung biopsy.
The patient underwent cardiac transplantation. The native heart was found to have right ventricular thinning, which was most notable at the right ventricular outflow tract. Microscopic examination revealed extensive fibrosis and granulomatous inflammation (Figure 5) with scarring typical of cardiac sarcoidosis. Six months after cardiac transplantation, the patient is doing well on prednisone, tacrolimus, and mycophenolate mofetil. Follow‐up chest x‐rays show resolution of the pulmonary nodules.
COMMENTARY
Cardiomyopathy in a young person is a relatively uncommon clinical event that prompts consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in older adults. This case highlights the challenges of arriving at a diagnosis in the absence of a gold standard, and the greater challenges of modifying initial diagnostic impressions as new clinical data become available.
After encountering ventricular tachycardia and right ventricular dysfunction in a young patient, the clinicians arrived at the diagnosis of ARVD. This rare and progressive disorder is associated with up to 20% of ventricular arrhythmias and sudden death in the young,1, 2 but can be challenging to diagnose. Despite common referrals for cardiac MRI to exclude ARVD, cardiac MRI is not the gold standard for diagnosis and is the most common method of misdiagnosis of ARVD.3 A diagnosis of ARVD requires the presence of 2 major, 1 major and 2 minor, or 4 minor International Task Force criteria (Table 1).4, 5 While the diagnostic criteria provide standardization across populations (eg, in clinical studies), additional considerations are needed in the management of individual patients. Scoring systems serve as a tool, but the final diagnosis requires balancing such criteria with competing hypotheses. This dilemma is familiar to clinicians considering other less common conditions such as amyotrophic lateral sclerosis (World Neurology Foundation), rheumatic fever (Jones criteria), or systemic lupus erythematosus (American College of Rheumatology). This patient's cardiac MRI findings, precordial T‐wave inversions, frequent ventricular ectopy, and late potentials on a signal‐averaged electrocardiogram fulfilled the International Task Force criteria for a diagnosis of ARVD. Discordant information included the right bundle branch pattern of the ventricular tachycardia, which suggested left ventricular origin, as opposed to the more common left bundle branch pattern observed in ARVD, and the absence of a family history. In addition, in U.S. populations only 25% of cases present with heart failure and fewer than 5% develop biventricular failure.6 Nonetheless, this patient's imaging evidence of right ventricular structural abnormalities and dysfunction and electrocardiographic abnormalities coupled with the absence of obvious systemic disease made ARVD the logical working diagnosis.
| Major | Minor | |
|---|---|---|
| ||
| I. Global and/or regional dysfunction and structural alterations | Severe dilation and reduction of right ventricular ejection fraction, localized right ventricular aneurysms | Mild right ventricular dilatation and/or reduced ejection fraction |
| II. Endomyocardial biopsy | Fibrofatty replacement of myocardium | |
| III. Repolarization abnormalities | T‐wave inversion in leads V1‐V3 or beyond | |
| IV. Depolarization/conduction abnormalities | Epsilon waves or localized QRS prolongation (>110 msec) in leads V1‐V3 | Late potentials on signal‐averaged electrocardiogram |
| V. Arrhythmias | Left bundle branch block‐type ventricular tachycardia (sustained and nonsustained) or frequent ventricular extra systoles (>1,000/24 hours) | |
| VI. Family history | Familial disease confirmed at necropsy or surgery | Familial history of premature sudden death (<35 years old) or clinical diagnosis based on present criteria |
When more widespread manifestations developed, namely hepatic and pulmonary abnormalities, each was investigated with imaging and biopsy. Once a multisystem illness became apparent, the discussant reframed the patient's illness to include other diagnostic possibilities. In practice it is difficult to reverse a working diagnosis despite contradictory evidence because of the common pitfall of anchoring bias. Tversky and Kahneman7 were the first to describe the cognitive processes behind probability assessment and decision making in time‐sensitive situations. Under these conditions, decision makers tend to focus on the first symptom, striking feature, or diagnosis and anchor subsequent probabilities to that initial presentation. Once a decision or diagnosis has been reached, clinicians tend to interpret subsequent findings in the context of the original diagnosis rather than reevaluating their initial impression. In the setting of a known diagnosis of ARVD, 3 separate diagnoses (ARVD, amiodarone‐associated lung injury, and amiodarone‐induced hepatic dysfunction) were considered by the treating physicians. The initial diagnosis of ARVD followed by the sequential rather than simultaneous manifestations of sarcoidosis made arriving at the revised diagnosis even more challenging.
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular dysplasia.8, 9 The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, myocarditis, idiopathic cardiomyopathy, and sarcoidosis. Cardiac sarcoidosis can present as ventricular ectopy, sustained ventricular arrhythmias, asymptomatic ventricular dysfunction, heart failure, or sudden death.10 Although 25% of patients with sarcoidosis have evidence of cardiac involvement at autopsy, only 5% have clinical manifestations.11 Those patients with clinical evidence of cardiac sarcoidosis have a wide range of clinical findings (Table 2). While the patient's cardiomyopathy was advanced, it is possible that earlier administration of corticosteroid therapy may have arrested his progressive biventricular failure. As clinicians, we should always remember to force ourselves to broaden our differential diagnosis when new findings become available, especially those that point to a systemicrather than an organ‐specificdisorder. In this case, while the original diagnostic findings were accurate and strongly suggested ARVD, a change of heart was needed to arrive at the ultimate diagnosis.
| Clinical Manifestation | Prevalence (%) |
|---|---|
| Atrioventricular block | 40 |
| Bundle branch block | 40 |
| Supraventricular tachycardia | 20 |
| Ventricular arrhythmias | 25 |
| Heart failure | 25 |
| Sudden cardiac death | 35 |
KEY POINTS FOR HOSPITALISTS
-
Cardiomyopathy in a young person requires consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in the elderly.
-
Anchoring bias is a common pitfall in clinical decision making. When new or contradictory findings are uncovered, clinicians should reevaluate their initial impression to ensure it remains the most likely diagnosis.
-
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular cardiomyopathy. The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, right ventricular outflow tract tachycardia, myocarditis, idiopathic dilated cardiomyopathy, and sarcoidosis.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 29‐year‐old man developed palpitations and dyspnea while loading boxes into a truck. In the emergency department, telemetry demonstrated a wide‐complex tachycardia at a rate of 204 beats per minute. The patient spontaneously cardioverted to sinus rhythm (Figure 1) before direct current cardioversion was performed.
Wide‐complex tachycardia is usually explained by a supraventricular tachycardia with aberrant ventricular conduction or a ventricular tachycardia. Although algorithms exist to guide the clinician in parsing out those etiologies, often the knowledge of underlying structural cardiac disease is most informative. In patients with a history of myocardial infarction, greater than 95% of wide‐complex tachycardia is ventricular tachycardia. The ventricular ectopy, T‐wave inversion or flattening, and poor R‐wave progression are suggestive of a cardiomyopathy, either acute or chronic. A pressing concern, especially with the Q waves and concave ST morphology in V1 and V2, would be coronary ischemia. His age makes this less likely, but an aberrant coronary circulation or drug use could account for it.
Over the past 2 years, the patient had several episodes of sustained palpitations, which terminated after several minutes. Previously, the patient exercised frequently including playing rugby in college. However, over the past year he experienced difficulty climbing stairs due to shortness of breath, which he attributed to deconditioning and smoking. He had no significant medical history, was not taking any medications, nor did he use recreational stimulants. He drank alcohol occasionally. He had no risk factors for the human immunodeficiency virus (HIV). Both of the patient's parents were alive and well. There was no family history of sudden cardiac death.
The duration of symptoms suggests that this is a chronic cardiomyopathy rather than acute myocarditis or acute ischemia, acknowledging that either one could be superimposed. The absence of family history lowers the likelihood of heritable causes of arrhythmia that may accompany a structurally normal (eg, long QT syndrome) or abnormal (eg, hypertrophic cardiomyopathy) heart, although penetrance can be variable. What might account for a cardiomyopathy in a young person? Most cases are probably idiopathic, but etiologies that diverge from the usual suspects of coronary artery disease, hypertension, and valvular disease, which affect an older population, include antecedent viral myocarditis, substance abuse, HIV, or infiltrative disorders such as sarcoidosis.
The patient's pulse was 92 beats per minute and regular and the blood pressure was 96/52 mm Hg. The jugular venous pressure was elevated with prominent v‐waves, the point of maximal impulse was diffuse, there were no extra heart sounds or murmurs, and an enlarged liver was detected. An echocardiogram demonstrated left ventricular dysfunction with an ejection fraction of 30%, severe enlargement of the right atrium and right ventricle, and moderate tricuspid regurgitation. Cardiac catheterization revealed normal coronary arteries without evidence of pulmonary hypertension or intracardiac shunt.
The physical examination and echocardiographic findings of right‐sided failure are unusual given the absence of pulmonary hypertension or intracardiac shunt, and could prompt repeat of the hemodynamic measurements and/or investigations for pulmonary disease that may account for right‐sided pressure overload (in addition to that caused by left ventricular failure). An alternative explanation would be a cardiomyopathic process that preferentially involves the right side of the heart, such as arrhythmogenic right ventricular dysplasia (ARVD), but that would not satisfactorily explain the significant decline in left ventricular function. An acute right ventricular infarction could cause his acute symptoms and his examination and echocardiographic findings, but not the underlying chronic illness. It is common to see patients with long‐standing biventricular failure who present with prominent signs of right‐sided failure (elevated neck veins, hepatomegaly, and edema) but limited or no signs of left‐sided failure (rales) to match their degree of volume overload or dyspnea.
Cardiac magnetic resonance imaging (MRI) revealed a dilated right ventricle with extensive hyperenhancement, a right ventricular ejection fraction of 9%, and moderate left ventricular dysfunction (Figure 2). Electrophysiology testing induced both nonsustained polymorphic and monomorphic ventricular tachycardia. Late potentials were detected on a signal‐averaged electrocardiogram. A single‐chamber cardioverter defibrillator was implanted and the patient was discharged on carvedilol, lisinopril, and spironolactone. An HIV‐1 antibody was negative and a thyroid‐stimulating hormone concentration was within normal limits.
Assuming that accurate evaluation of the pulmonary circulation has been undertaken to exclude pulmonary hypertension, the enlarged and hyperenhanced right ventricle on MRI suggests a process that preferentially infiltrates the right ventricular myocardium, and may secondarily affect the left ventricle either by further infiltration or as a consequence of altered mechanics from the highly dysfunctional right ventricle. ARVD affects the right ventricle, but it is possible that another infiltrative cardiomyopathy, such as sarcoid or an antecedent viral infection, could be restricted in its distribution. Late‐potentials identified on signal average electrocardiograms indicate areas of abnormal conduction that may serve as substrate for reentrant ventricular arrhythmias. They are, however, nonspecific, as they are seen in a variety of myocardial diseases.
The patient continued to have progressive dyspnea and was readmitted after receiving an appropriate implantable cardioverter defibrillator shock for ventricular tachycardia. Recurrent slow ventricular tachycardia (Figure 3) was treated with supplemental beta‐blockade and amiodarone (10 g total). Repeat echocardiography demonstrated severe left ventricular dysfunction with an ejection fraction of less than 15%. There were no recurrences of ventricular arrhythmias and the patient was discharged and referred for cardiac transplant evaluation for ARVD.
This degree of left ventricular dysfunction is unlikely to be accounted for by altered mechanics and interactions from a failing right ventricle alone and frames this as a biventricular cardiomyopathy, which has an extensive differential diagnosis and requires information from the general medical evaluation.
On routine laboratory testing 6 months later, a serum aspartate aminotransferase of 79 units/L and a serum alanine aminotransferase of 118 units/L were found. Bilirubin, albumin, and alkaline phosphatase were normal. The transaminase levels had been normal on initial evaluation. The patient reported that 2 paternal uncles had end‐stage nonalcoholic cirrhosis. Transjugular liver biopsy was consistent with mild lobular hepatitis with mild portal fibrosis with a few lobular collections of mononuclear cells. There was no evidence of iron overload. The hepatic venogram and transhepatic pressure gradient (2 mm Hg) were normal.
The elevated transaminase levels could be due to amiodarone‐associated hepatotoxicity, hepatic congestion, or a primary liver disease. It is important to consider combined cardiohepatic syndromes such as hemochromatosis, sarcoidosis, or amyloidosis. The relatively normal liver histology and normal hepatic hemodynamics do not suggest a significant primary intrinsic liver disease. The 2 uncles with cirrhosis could suggest a heritable liver disease, although cirrhosis in multiple family members is frequently accounted for by shared habits such as alcohol consumption or excessive caloric intake. Liver disorders with a genetic component, such as hemochromatosis, Wilson's disease, and alpha‐1‐antitrypsin deficiency are mostly autosomal recessive, which would make this pattern of transmission unusual. Furthermore, aside from hemochromatosis, these genetic hepatic disorders have few cardiac manifestations. Right‐sided congestion and amiodarone appear to be the most likely explanations of his liver abnormalities.
Pulmonary function testing revealed normal lung volumes without obstruction, but the diffusing capacity for carbon monoxide was substantially reduced. Computed tomography of the chest identified scattered ground‐glass opacities as well as small nodules with an upper lobe distribution (Figure 4). Although not reported on the initial interpretation, review of a chest x‐ray taken 6 months previously also demonstrated small nodules in the upper lobe distribution. Bronchoscopic examination was normal. Bronchioalveolar lavage fluid stains and cultures for bacteria, mycobacteria, Pneumocystis, and fungus were negative. Transbronchial biopsies of the right middle lobe had no evidence of infection, malignancy, or granulomatous inflammation. The patient continued to have progressive New York Heart Association Class IV heart failure symptoms. Repeat right heart catheterization was notable for a cardiac index of 1.4 L/minute/m2. The mean pulmonary artery pressure was 20 mm Hg. An intraaortic balloon pump was placed for refractory cardiogenic shock.
The reduced diffusion capacity and ground‐glass opacities suggest an interstitial process, which may have been missed on transbronchial biopsy because of sampling error. His pulmonary disease is likely another manifestation of his infiltrative cardiac disease. The constellation of cardiac, pulmonary, and hepatic involvement in the context of progressive dyspnea over 2 years is suggestive of sarcoidosis although the absence of hilar lymphadenopathy and 2 biopsy specimens without granulomas argue against the diagnosis, and the effects of amiodarone on the latter 2 organs cannot be ignored. On the limited menu of pharmacologic treatments that may treat this severe and progressive cardiomyopathy are steroids, which makes a diligent search for a steroid‐responsive syndrome important. Therefore, despite the negative studies, sarcoidosis must be investigated to the fullest extent with either an endomyocardial biopsy or surgical lung biopsy.
The patient underwent cardiac transplantation. The native heart was found to have right ventricular thinning, which was most notable at the right ventricular outflow tract. Microscopic examination revealed extensive fibrosis and granulomatous inflammation (Figure 5) with scarring typical of cardiac sarcoidosis. Six months after cardiac transplantation, the patient is doing well on prednisone, tacrolimus, and mycophenolate mofetil. Follow‐up chest x‐rays show resolution of the pulmonary nodules.
COMMENTARY
Cardiomyopathy in a young person is a relatively uncommon clinical event that prompts consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in older adults. This case highlights the challenges of arriving at a diagnosis in the absence of a gold standard, and the greater challenges of modifying initial diagnostic impressions as new clinical data become available.
After encountering ventricular tachycardia and right ventricular dysfunction in a young patient, the clinicians arrived at the diagnosis of ARVD. This rare and progressive disorder is associated with up to 20% of ventricular arrhythmias and sudden death in the young,1, 2 but can be challenging to diagnose. Despite common referrals for cardiac MRI to exclude ARVD, cardiac MRI is not the gold standard for diagnosis and is the most common method of misdiagnosis of ARVD.3 A diagnosis of ARVD requires the presence of 2 major, 1 major and 2 minor, or 4 minor International Task Force criteria (Table 1).4, 5 While the diagnostic criteria provide standardization across populations (eg, in clinical studies), additional considerations are needed in the management of individual patients. Scoring systems serve as a tool, but the final diagnosis requires balancing such criteria with competing hypotheses. This dilemma is familiar to clinicians considering other less common conditions such as amyotrophic lateral sclerosis (World Neurology Foundation), rheumatic fever (Jones criteria), or systemic lupus erythematosus (American College of Rheumatology). This patient's cardiac MRI findings, precordial T‐wave inversions, frequent ventricular ectopy, and late potentials on a signal‐averaged electrocardiogram fulfilled the International Task Force criteria for a diagnosis of ARVD. Discordant information included the right bundle branch pattern of the ventricular tachycardia, which suggested left ventricular origin, as opposed to the more common left bundle branch pattern observed in ARVD, and the absence of a family history. In addition, in U.S. populations only 25% of cases present with heart failure and fewer than 5% develop biventricular failure.6 Nonetheless, this patient's imaging evidence of right ventricular structural abnormalities and dysfunction and electrocardiographic abnormalities coupled with the absence of obvious systemic disease made ARVD the logical working diagnosis.
| Major | Minor | |
|---|---|---|
| ||
| I. Global and/or regional dysfunction and structural alterations | Severe dilation and reduction of right ventricular ejection fraction, localized right ventricular aneurysms | Mild right ventricular dilatation and/or reduced ejection fraction |
| II. Endomyocardial biopsy | Fibrofatty replacement of myocardium | |
| III. Repolarization abnormalities | T‐wave inversion in leads V1‐V3 or beyond | |
| IV. Depolarization/conduction abnormalities | Epsilon waves or localized QRS prolongation (>110 msec) in leads V1‐V3 | Late potentials on signal‐averaged electrocardiogram |
| V. Arrhythmias | Left bundle branch block‐type ventricular tachycardia (sustained and nonsustained) or frequent ventricular extra systoles (>1,000/24 hours) | |
| VI. Family history | Familial disease confirmed at necropsy or surgery | Familial history of premature sudden death (<35 years old) or clinical diagnosis based on present criteria |
When more widespread manifestations developed, namely hepatic and pulmonary abnormalities, each was investigated with imaging and biopsy. Once a multisystem illness became apparent, the discussant reframed the patient's illness to include other diagnostic possibilities. In practice it is difficult to reverse a working diagnosis despite contradictory evidence because of the common pitfall of anchoring bias. Tversky and Kahneman7 were the first to describe the cognitive processes behind probability assessment and decision making in time‐sensitive situations. Under these conditions, decision makers tend to focus on the first symptom, striking feature, or diagnosis and anchor subsequent probabilities to that initial presentation. Once a decision or diagnosis has been reached, clinicians tend to interpret subsequent findings in the context of the original diagnosis rather than reevaluating their initial impression. In the setting of a known diagnosis of ARVD, 3 separate diagnoses (ARVD, amiodarone‐associated lung injury, and amiodarone‐induced hepatic dysfunction) were considered by the treating physicians. The initial diagnosis of ARVD followed by the sequential rather than simultaneous manifestations of sarcoidosis made arriving at the revised diagnosis even more challenging.
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular dysplasia.8, 9 The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, myocarditis, idiopathic cardiomyopathy, and sarcoidosis. Cardiac sarcoidosis can present as ventricular ectopy, sustained ventricular arrhythmias, asymptomatic ventricular dysfunction, heart failure, or sudden death.10 Although 25% of patients with sarcoidosis have evidence of cardiac involvement at autopsy, only 5% have clinical manifestations.11 Those patients with clinical evidence of cardiac sarcoidosis have a wide range of clinical findings (Table 2). While the patient's cardiomyopathy was advanced, it is possible that earlier administration of corticosteroid therapy may have arrested his progressive biventricular failure. As clinicians, we should always remember to force ourselves to broaden our differential diagnosis when new findings become available, especially those that point to a systemicrather than an organ‐specificdisorder. In this case, while the original diagnostic findings were accurate and strongly suggested ARVD, a change of heart was needed to arrive at the ultimate diagnosis.
| Clinical Manifestation | Prevalence (%) |
|---|---|
| Atrioventricular block | 40 |
| Bundle branch block | 40 |
| Supraventricular tachycardia | 20 |
| Ventricular arrhythmias | 25 |
| Heart failure | 25 |
| Sudden cardiac death | 35 |
KEY POINTS FOR HOSPITALISTS
-
Cardiomyopathy in a young person requires consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in the elderly.
-
Anchoring bias is a common pitfall in clinical decision making. When new or contradictory findings are uncovered, clinicians should reevaluate their initial impression to ensure it remains the most likely diagnosis.
-
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular cardiomyopathy. The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, right ventricular outflow tract tachycardia, myocarditis, idiopathic dilated cardiomyopathy, and sarcoidosis.
- ,,, et al.Right ventricular dysplasia: a report of 24 adult cases.Circulation.1982;65:384–398.
- ,,,,.Right ventricular cardiomyopathy and sudden death in young people.N Engl J Med.1988;318:129–133.
- ,,, et al.Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy.J Cardiovasc Electrophysiol.2004;15:300–306.
- ,,, et al.Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology.Br Heart J.1994;71:215–218.
- ,,, et al.Predictors of appropriate implantable defibrillator therapies in patients with arrhythmogenic right ventricular dysplasia.Heart Rhythm.2005;2:1188–1194.
- ,,, et al.Arrhythmogenic right ventricular dysplasia: a United States experience.Circulation.2005;112:3823–3832.
- ,.Judgment under uncertainty: heuristics and biases.Science.1974;185:1124–1131.
- ,,.Unusual presentation of cardiac sarcoidosis.Congest Heart Fail.2007;13:116–118.
- ,,, et al.Cardiac sarcoidosis mimicking right ventricular dysplasia.Circ J.2003;67:169–171.
- ,,,,.Refractory ventricular tachycardia secondary to cardiac sarcoid: electrophysiologic characteristics, mapping, and ablation.Heart Rhythm.2006;3:924–929.
- ,.Sarcoid heart disease: clinical course and treatment.Int J Cardiol.2004;97:173–182.
- ,,, et al.Right ventricular dysplasia: a report of 24 adult cases.Circulation.1982;65:384–398.
- ,,,,.Right ventricular cardiomyopathy and sudden death in young people.N Engl J Med.1988;318:129–133.
- ,,, et al.Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy.J Cardiovasc Electrophysiol.2004;15:300–306.
- ,,, et al.Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology.Br Heart J.1994;71:215–218.
- ,,, et al.Predictors of appropriate implantable defibrillator therapies in patients with arrhythmogenic right ventricular dysplasia.Heart Rhythm.2005;2:1188–1194.
- ,,, et al.Arrhythmogenic right ventricular dysplasia: a United States experience.Circulation.2005;112:3823–3832.
- ,.Judgment under uncertainty: heuristics and biases.Science.1974;185:1124–1131.
- ,,.Unusual presentation of cardiac sarcoidosis.Congest Heart Fail.2007;13:116–118.
- ,,, et al.Cardiac sarcoidosis mimicking right ventricular dysplasia.Circ J.2003;67:169–171.
- ,,,,.Refractory ventricular tachycardia secondary to cardiac sarcoid: electrophysiologic characteristics, mapping, and ablation.Heart Rhythm.2006;3:924–929.
- ,.Sarcoid heart disease: clinical course and treatment.Int J Cardiol.2004;97:173–182.