User login
Sudden-Onset Blistering Rash
The Diagnosis: Generalized Bullous Fixed Drug Eruption
A punch biopsy from the left thigh revealed a vacuolar interface dermatitis with full-thickness necrosis of the epidermis and a patchy lichenoid inflammatory cell infiltrate in the superficial dermis consistent with a generalized bullous fixed drug eruption (GBFDE). The patient received supportive care and methylprednisolone with improvement of symptoms.
Generalized bullous fixed drug eruption is a rare, potentially life-threatening form of a fixed drug eruption (FDE), a cutaneous drug reaction that occurs in response to a causative medication. It typically presents with welldemarcated, dusky, erythematous patches or plaques that recur in the same sites with repeat exposure.1 The pathogenesis of FDE has been hypothesized to involve epidermal CD8+ T cells, which are activated by drug exposure and release cytotoxic molecules including Fas, Fas ligand, perforin, and granzyme B, resulting in lysis of the surrounding keratinocytes.1-3 Common eliciting drugs include nonsteroidal anti-inflammatory drugs, antibacterial agents (particularly trimethoprim-sulfamethoxazole), barbiturates, acetaminophen, and antimalarials.1 In addition to the findings seen in FDE, GBFDE is characterized by widespread bullous skin lesions.1-4 Typical histologic patterns seen in GBFDE are dispersed epidermal apoptotic keratinocytes, prominent dermal eosinophilic and lymphocytic infiltrates, and dermal melanophages.3 Discontinuing the causative agent and diligent prevention of re-exposure are the most important steps in management, as additional exposures can increase the number of lesions and overall severity. Symptoms typically resolve 7 to 14 days after drug discontinuation, often with postinflammatory hyperpigmentation.3
Generalized bullous fixed drug eruption presents a diagnostic challenge, as it sometimes involves the oral mucosa and can exhibit the Nikolsky sign. Thus, it often is confused with Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN).1,4 Stevens-Johnson syndrome and TEN are severe cutaneous drug eruptions that also can present with diffuse bullous skin lesions. Stevens-Johnson syndrome and TEN are thought to be a spectrum of the same disease that initially presents with dusky red macules that can coalesce, develop central blistering, and lead to skin detachment.5 Stevens-Johnson syndrome is defined as skin detachment of less than 10% body surface area (BSA); TEN is defined as skin detachment of more than 30% BSA. Stevens-Johnson syndrome/TEN overlap syndrome includes skin detachment of 10% to 30% BSA.5
Causative medications overlap substantially with GBFDE and include anticonvulsants, sulfa-containing drugs, antibiotics, nonsteroidal anti-inflammatory drugs, and uric acid–lowering agents. The histology of SJS/TEN also is quite similar to GBFDE, and these entities may be indistinguishable without clinical information.5 Lee et al1 found that absence of grouped necrotic keratinocytes (fire flag sign), deep inflammatory infiltrates, notable pigment incontinence, and higher eosinophil counts appear to be more common in GBFDE than SJS/TEN. Constitutional symptoms and mucosal involvement also were more frequent in SJS/TEN.
The timing of clinical presentation and medical history can be useful in differentiating between SJS/TEN and GBFDE. In SJS/TEN, drug exposure typically occurs 1 to 3 weeks before onset of symptoms vs 30 minutes to 24 hours in GBFDE.3 Additionally, a history of similar eruption in the same location is pathognomonic for GBFDE. Although GBFDE has been thought to have a better prognosis than SJS/TEN, more recent data suggest mortality rates may be similar.3 A case-control study found a mortality rate of 22% (13/58) in patients with GBFDE compared to 28% (n=170) in SJS/TEN patients.4
Erythema multiforme (EM) is an uncommon immunemediated disorder that typically presents as targetoid lesions with central epidermal necrosis in an acral distribution. Erythema multiforme can arise from a variety of factors, but up to 90% of cases are due to infection, most commonly herpes simplex virus; medications account for less than 10% of cases.6 Previously, EM has been thought to be on the same disease spectrum as SJS and TEN. It is now clear that EM is a separate entity with similar mucosal erosions but different cutaneous findings,6 mainly typical target lesions that differ from the atypical targets seen in SJS.
Staphylococcal scalded skin syndrome is a blistering skin disorder associated with local Staphylococcus aureus infection. It most commonly is seen in children and rarely occurs in adults who are not on dialysis. Some Staphylococcus strains produce exfoliative toxins A and B, which are serine proteases that target and cleave desmoglein 1, a mediator of keratinocyte adhesion. Staphylococcal scalded skin syndrome initially presents with erythema accentuated in the skin folds that becomes generalized. The disruption of keratinocyte adhesion leads to bullae formation in areas of erythema and diffuse sheetlike desquamation. Pathology reveals subcorneal rather than subepidermal blistering, which is seen in GBFDE and SJS/TEN. Treatment involves antistaphylococcal antibiotics and supportive care. With proper treatment, most cases resolve within 2 to 3 weeks.7
Mycoplasma pneumoniae–induced rash and mucositis presents with prominent mucositis and can have cutaneous findings of sparse vesiculobullous or targetoid eruption.8Mycoplasma pneumoniae typically infects the lungs and is a leading cause of community-acquired pneumonia. However, a subset of patients can have extrapulmonary disease presenting as mucocutaneous eruptions, which is preceded by an approximately weeklong prodrome of fever, cough, and malaise.7Mycoplasma pneumoniae–induced rash and mucositis also affect children and young patients and is more common in males.8
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15. doi:10.1016/j.dsi.2012.02.002
- Cho Y-T, Lin J-W, Chen Y-C, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548. doi:10.1016/j.jaad.2013.11.015
- Mitre V, Applebaum DS, Albahrani Y, et al. Generalized bullous fixed drug eruption imitating toxic epidermal necrolysis: a case report and literature review. Dermatol Online J. 2017;23: 13030/qt25v009gs.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with StevensJohnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732. doi:10.1111/bjd.12133
- Cho Y-T, Chu C-Y. Treatments for severe cutaneous adverse reactions [published online December 27, 2017]. J Immunol Res. doi:10.1155/2017/1503709
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902. doi:10.1111/j.1365-4632.2011.05348.x
- Leung AKC, Barankin B, Leong KF. Staphylococcal-scalded skin syndrome: evaluation, diagnosis, and management. World J Pediatr. 2018;14:116-120.
- Canavan TN, Mathes EF, Frieden I, et al. Mycoplasma pneumoniae–induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme: a systematic review. J Am Acad Dermatol. 2015;72:239-245. doi:10.1016/j .jaad.2014.06.026
The Diagnosis: Generalized Bullous Fixed Drug Eruption
A punch biopsy from the left thigh revealed a vacuolar interface dermatitis with full-thickness necrosis of the epidermis and a patchy lichenoid inflammatory cell infiltrate in the superficial dermis consistent with a generalized bullous fixed drug eruption (GBFDE). The patient received supportive care and methylprednisolone with improvement of symptoms.
Generalized bullous fixed drug eruption is a rare, potentially life-threatening form of a fixed drug eruption (FDE), a cutaneous drug reaction that occurs in response to a causative medication. It typically presents with welldemarcated, dusky, erythematous patches or plaques that recur in the same sites with repeat exposure.1 The pathogenesis of FDE has been hypothesized to involve epidermal CD8+ T cells, which are activated by drug exposure and release cytotoxic molecules including Fas, Fas ligand, perforin, and granzyme B, resulting in lysis of the surrounding keratinocytes.1-3 Common eliciting drugs include nonsteroidal anti-inflammatory drugs, antibacterial agents (particularly trimethoprim-sulfamethoxazole), barbiturates, acetaminophen, and antimalarials.1 In addition to the findings seen in FDE, GBFDE is characterized by widespread bullous skin lesions.1-4 Typical histologic patterns seen in GBFDE are dispersed epidermal apoptotic keratinocytes, prominent dermal eosinophilic and lymphocytic infiltrates, and dermal melanophages.3 Discontinuing the causative agent and diligent prevention of re-exposure are the most important steps in management, as additional exposures can increase the number of lesions and overall severity. Symptoms typically resolve 7 to 14 days after drug discontinuation, often with postinflammatory hyperpigmentation.3
Generalized bullous fixed drug eruption presents a diagnostic challenge, as it sometimes involves the oral mucosa and can exhibit the Nikolsky sign. Thus, it often is confused with Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN).1,4 Stevens-Johnson syndrome and TEN are severe cutaneous drug eruptions that also can present with diffuse bullous skin lesions. Stevens-Johnson syndrome and TEN are thought to be a spectrum of the same disease that initially presents with dusky red macules that can coalesce, develop central blistering, and lead to skin detachment.5 Stevens-Johnson syndrome is defined as skin detachment of less than 10% body surface area (BSA); TEN is defined as skin detachment of more than 30% BSA. Stevens-Johnson syndrome/TEN overlap syndrome includes skin detachment of 10% to 30% BSA.5
Causative medications overlap substantially with GBFDE and include anticonvulsants, sulfa-containing drugs, antibiotics, nonsteroidal anti-inflammatory drugs, and uric acid–lowering agents. The histology of SJS/TEN also is quite similar to GBFDE, and these entities may be indistinguishable without clinical information.5 Lee et al1 found that absence of grouped necrotic keratinocytes (fire flag sign), deep inflammatory infiltrates, notable pigment incontinence, and higher eosinophil counts appear to be more common in GBFDE than SJS/TEN. Constitutional symptoms and mucosal involvement also were more frequent in SJS/TEN.
The timing of clinical presentation and medical history can be useful in differentiating between SJS/TEN and GBFDE. In SJS/TEN, drug exposure typically occurs 1 to 3 weeks before onset of symptoms vs 30 minutes to 24 hours in GBFDE.3 Additionally, a history of similar eruption in the same location is pathognomonic for GBFDE. Although GBFDE has been thought to have a better prognosis than SJS/TEN, more recent data suggest mortality rates may be similar.3 A case-control study found a mortality rate of 22% (13/58) in patients with GBFDE compared to 28% (n=170) in SJS/TEN patients.4
Erythema multiforme (EM) is an uncommon immunemediated disorder that typically presents as targetoid lesions with central epidermal necrosis in an acral distribution. Erythema multiforme can arise from a variety of factors, but up to 90% of cases are due to infection, most commonly herpes simplex virus; medications account for less than 10% of cases.6 Previously, EM has been thought to be on the same disease spectrum as SJS and TEN. It is now clear that EM is a separate entity with similar mucosal erosions but different cutaneous findings,6 mainly typical target lesions that differ from the atypical targets seen in SJS.
Staphylococcal scalded skin syndrome is a blistering skin disorder associated with local Staphylococcus aureus infection. It most commonly is seen in children and rarely occurs in adults who are not on dialysis. Some Staphylococcus strains produce exfoliative toxins A and B, which are serine proteases that target and cleave desmoglein 1, a mediator of keratinocyte adhesion. Staphylococcal scalded skin syndrome initially presents with erythema accentuated in the skin folds that becomes generalized. The disruption of keratinocyte adhesion leads to bullae formation in areas of erythema and diffuse sheetlike desquamation. Pathology reveals subcorneal rather than subepidermal blistering, which is seen in GBFDE and SJS/TEN. Treatment involves antistaphylococcal antibiotics and supportive care. With proper treatment, most cases resolve within 2 to 3 weeks.7
Mycoplasma pneumoniae–induced rash and mucositis presents with prominent mucositis and can have cutaneous findings of sparse vesiculobullous or targetoid eruption.8Mycoplasma pneumoniae typically infects the lungs and is a leading cause of community-acquired pneumonia. However, a subset of patients can have extrapulmonary disease presenting as mucocutaneous eruptions, which is preceded by an approximately weeklong prodrome of fever, cough, and malaise.7Mycoplasma pneumoniae–induced rash and mucositis also affect children and young patients and is more common in males.8
The Diagnosis: Generalized Bullous Fixed Drug Eruption
A punch biopsy from the left thigh revealed a vacuolar interface dermatitis with full-thickness necrosis of the epidermis and a patchy lichenoid inflammatory cell infiltrate in the superficial dermis consistent with a generalized bullous fixed drug eruption (GBFDE). The patient received supportive care and methylprednisolone with improvement of symptoms.
Generalized bullous fixed drug eruption is a rare, potentially life-threatening form of a fixed drug eruption (FDE), a cutaneous drug reaction that occurs in response to a causative medication. It typically presents with welldemarcated, dusky, erythematous patches or plaques that recur in the same sites with repeat exposure.1 The pathogenesis of FDE has been hypothesized to involve epidermal CD8+ T cells, which are activated by drug exposure and release cytotoxic molecules including Fas, Fas ligand, perforin, and granzyme B, resulting in lysis of the surrounding keratinocytes.1-3 Common eliciting drugs include nonsteroidal anti-inflammatory drugs, antibacterial agents (particularly trimethoprim-sulfamethoxazole), barbiturates, acetaminophen, and antimalarials.1 In addition to the findings seen in FDE, GBFDE is characterized by widespread bullous skin lesions.1-4 Typical histologic patterns seen in GBFDE are dispersed epidermal apoptotic keratinocytes, prominent dermal eosinophilic and lymphocytic infiltrates, and dermal melanophages.3 Discontinuing the causative agent and diligent prevention of re-exposure are the most important steps in management, as additional exposures can increase the number of lesions and overall severity. Symptoms typically resolve 7 to 14 days after drug discontinuation, often with postinflammatory hyperpigmentation.3
Generalized bullous fixed drug eruption presents a diagnostic challenge, as it sometimes involves the oral mucosa and can exhibit the Nikolsky sign. Thus, it often is confused with Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN).1,4 Stevens-Johnson syndrome and TEN are severe cutaneous drug eruptions that also can present with diffuse bullous skin lesions. Stevens-Johnson syndrome and TEN are thought to be a spectrum of the same disease that initially presents with dusky red macules that can coalesce, develop central blistering, and lead to skin detachment.5 Stevens-Johnson syndrome is defined as skin detachment of less than 10% body surface area (BSA); TEN is defined as skin detachment of more than 30% BSA. Stevens-Johnson syndrome/TEN overlap syndrome includes skin detachment of 10% to 30% BSA.5
Causative medications overlap substantially with GBFDE and include anticonvulsants, sulfa-containing drugs, antibiotics, nonsteroidal anti-inflammatory drugs, and uric acid–lowering agents. The histology of SJS/TEN also is quite similar to GBFDE, and these entities may be indistinguishable without clinical information.5 Lee et al1 found that absence of grouped necrotic keratinocytes (fire flag sign), deep inflammatory infiltrates, notable pigment incontinence, and higher eosinophil counts appear to be more common in GBFDE than SJS/TEN. Constitutional symptoms and mucosal involvement also were more frequent in SJS/TEN.
The timing of clinical presentation and medical history can be useful in differentiating between SJS/TEN and GBFDE. In SJS/TEN, drug exposure typically occurs 1 to 3 weeks before onset of symptoms vs 30 minutes to 24 hours in GBFDE.3 Additionally, a history of similar eruption in the same location is pathognomonic for GBFDE. Although GBFDE has been thought to have a better prognosis than SJS/TEN, more recent data suggest mortality rates may be similar.3 A case-control study found a mortality rate of 22% (13/58) in patients with GBFDE compared to 28% (n=170) in SJS/TEN patients.4
Erythema multiforme (EM) is an uncommon immunemediated disorder that typically presents as targetoid lesions with central epidermal necrosis in an acral distribution. Erythema multiforme can arise from a variety of factors, but up to 90% of cases are due to infection, most commonly herpes simplex virus; medications account for less than 10% of cases.6 Previously, EM has been thought to be on the same disease spectrum as SJS and TEN. It is now clear that EM is a separate entity with similar mucosal erosions but different cutaneous findings,6 mainly typical target lesions that differ from the atypical targets seen in SJS.
Staphylococcal scalded skin syndrome is a blistering skin disorder associated with local Staphylococcus aureus infection. It most commonly is seen in children and rarely occurs in adults who are not on dialysis. Some Staphylococcus strains produce exfoliative toxins A and B, which are serine proteases that target and cleave desmoglein 1, a mediator of keratinocyte adhesion. Staphylococcal scalded skin syndrome initially presents with erythema accentuated in the skin folds that becomes generalized. The disruption of keratinocyte adhesion leads to bullae formation in areas of erythema and diffuse sheetlike desquamation. Pathology reveals subcorneal rather than subepidermal blistering, which is seen in GBFDE and SJS/TEN. Treatment involves antistaphylococcal antibiotics and supportive care. With proper treatment, most cases resolve within 2 to 3 weeks.7
Mycoplasma pneumoniae–induced rash and mucositis presents with prominent mucositis and can have cutaneous findings of sparse vesiculobullous or targetoid eruption.8Mycoplasma pneumoniae typically infects the lungs and is a leading cause of community-acquired pneumonia. However, a subset of patients can have extrapulmonary disease presenting as mucocutaneous eruptions, which is preceded by an approximately weeklong prodrome of fever, cough, and malaise.7Mycoplasma pneumoniae–induced rash and mucositis also affect children and young patients and is more common in males.8
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15. doi:10.1016/j.dsi.2012.02.002
- Cho Y-T, Lin J-W, Chen Y-C, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548. doi:10.1016/j.jaad.2013.11.015
- Mitre V, Applebaum DS, Albahrani Y, et al. Generalized bullous fixed drug eruption imitating toxic epidermal necrolysis: a case report and literature review. Dermatol Online J. 2017;23: 13030/qt25v009gs.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with StevensJohnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732. doi:10.1111/bjd.12133
- Cho Y-T, Chu C-Y. Treatments for severe cutaneous adverse reactions [published online December 27, 2017]. J Immunol Res. doi:10.1155/2017/1503709
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902. doi:10.1111/j.1365-4632.2011.05348.x
- Leung AKC, Barankin B, Leong KF. Staphylococcal-scalded skin syndrome: evaluation, diagnosis, and management. World J Pediatr. 2018;14:116-120.
- Canavan TN, Mathes EF, Frieden I, et al. Mycoplasma pneumoniae–induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme: a systematic review. J Am Acad Dermatol. 2015;72:239-245. doi:10.1016/j .jaad.2014.06.026
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15. doi:10.1016/j.dsi.2012.02.002
- Cho Y-T, Lin J-W, Chen Y-C, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548. doi:10.1016/j.jaad.2013.11.015
- Mitre V, Applebaum DS, Albahrani Y, et al. Generalized bullous fixed drug eruption imitating toxic epidermal necrolysis: a case report and literature review. Dermatol Online J. 2017;23: 13030/qt25v009gs.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with StevensJohnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732. doi:10.1111/bjd.12133
- Cho Y-T, Chu C-Y. Treatments for severe cutaneous adverse reactions [published online December 27, 2017]. J Immunol Res. doi:10.1155/2017/1503709
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902. doi:10.1111/j.1365-4632.2011.05348.x
- Leung AKC, Barankin B, Leong KF. Staphylococcal-scalded skin syndrome: evaluation, diagnosis, and management. World J Pediatr. 2018;14:116-120.
- Canavan TN, Mathes EF, Frieden I, et al. Mycoplasma pneumoniae–induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme: a systematic review. J Am Acad Dermatol. 2015;72:239-245. doi:10.1016/j .jaad.2014.06.026
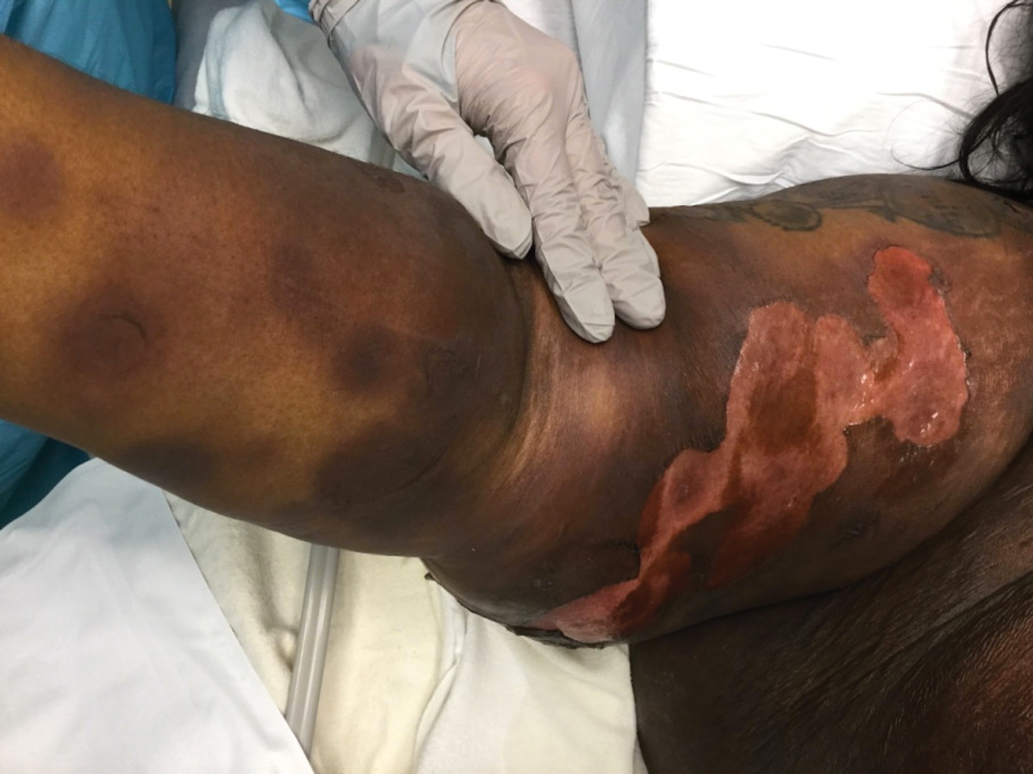
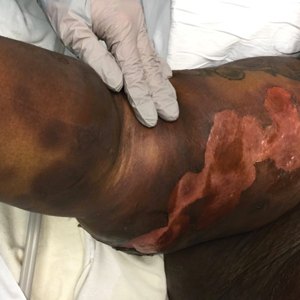
A 45-year-old woman presented with a diffuse rash 2 days after receiving ondansetron. She developed blisters on the arms, legs, trunk, and face 2 hours after exposure. There was no oral or vaginal involvement. She reported a history of leg blisters after prior exposure to ondansetron that were not as severe or numerous as the current episode. Physical examination revealed innumerable coalescing, ovoid and circular, dusky patches, some with central flaccid bullae, along with large areas of denuded skin on the trunk, arms, legs, and face. There were erosions on the lower eyelids without conjunctival or other mucosal involvement.
VEXAS syndrome: Implications for dermatologists
When I was a medical student, I always found it gratifying when there was a unifying mechanism that explained the symptoms of a disease. Part of the reason I chose dermatology as a specialty was how frequently we are able to “see” these mechanisms in the skin, both clinically and histologically. What’s even more interesting is that this condition is caused by a postzygotic somatic mutation, an apparently underrecognized cause of disease that we are just now beginning to understand. An example of a postzygotic somatic mutation causing a disease that we all learned about in medical school is paroxysmal nocturnal hemoglobinuria.

Using a “bottom-up” approach, researchers at the National Institutes of Health and in the United Kingdom identified 25 patients with somatic UBA1 mutations and noticed that they had strikingly similar autoinflammatory syndromes. UBA1 encodes ubiquitin E1, which is part of the pathway the breaks down proteins as part of the normal cellular machine. It is localized to the X chromosome, so all 25 affected patients were males, and most were aged between 40 and 70 years. These patients had an autoinflammatory syndrome characterized by fever, chondritis (similar to relapsing polychondritis), vasculitis, and neutrophilic dermatoses. Many patients also had features of myelodysplastic syndrome and plasma cell dyscrasia. The inflammatory pattern in this condition seems to show elevations in tumor necrosis factor, interleukin-6, and interferon-gamma.
So why is this syndrome relevant to dermatology? We are often asked to evaluate patients for neutrophilic dermatosis and vasculitis, and many affected patients had clinical and histologic findings compatible with polyarteritis nodosa and Sweet syndrome. When confronted with a neutrophilic dermatosis, we’ve all been taught to evaluate for myelodysplastic syndrome, which many of these patients appeared to have, at least on the surface. When bone marrow biopsies were done, the myeloid cell precursors that give rise to neutrophils were noted to have prominent cytoplasmic vacuoles, hence the “V” in VEXAS.
In reading the article describing 25 patients with this syndrome, which was published in the New England Journal of Medicine, I was struck by how refractory they were to treatment. Most patients had been treated with systemic steroids, multiple biologics, and several nonbiologic medications that are mainstays of treatment for neutrophilic dermatosis like dapsone and colchicine. I was fortunate enough to speak to Amanda Ombrello, MD, of the National Human Genome Research Institute, one of the lead authors of the paper, who drew my attention to the supplementary appendix, which showed the marked injection-site reactions some patients had to anakinra – yet another reason why a patient might end up in a dermatology clinic. In my mind, all of these features could be a clue to a diagnosis of VEXAS syndrome.
Many patients seemed to fare poorly, with 40% of patients dying before the completion of the study. When it comes to extremely rare diseases, it seems that the more physicians who are aware of the existence of a particular syndrome, the more likely it is a patient will come under our care and be correctly diagnosed.
Dr. Saardi is a dermatologist and internist, and is director of the inpatient dermatology service at the George Washington University Hospital, Washington. He has no disclosures.
When I was a medical student, I always found it gratifying when there was a unifying mechanism that explained the symptoms of a disease. Part of the reason I chose dermatology as a specialty was how frequently we are able to “see” these mechanisms in the skin, both clinically and histologically. What’s even more interesting is that this condition is caused by a postzygotic somatic mutation, an apparently underrecognized cause of disease that we are just now beginning to understand. An example of a postzygotic somatic mutation causing a disease that we all learned about in medical school is paroxysmal nocturnal hemoglobinuria.
Using a “bottom-up” approach, researchers at the National Institutes of Health and in the United Kingdom identified 25 patients with somatic UBA1 mutations and noticed that they had strikingly similar autoinflammatory syndromes. UBA1 encodes ubiquitin E1, which is part of the pathway the breaks down proteins as part of the normal cellular machine. It is localized to the X chromosome, so all 25 affected patients were males, and most were aged between 40 and 70 years. These patients had an autoinflammatory syndrome characterized by fever, chondritis (similar to relapsing polychondritis), vasculitis, and neutrophilic dermatoses. Many patients also had features of myelodysplastic syndrome and plasma cell dyscrasia. The inflammatory pattern in this condition seems to show elevations in tumor necrosis factor, interleukin-6, and interferon-gamma.
So why is this syndrome relevant to dermatology? We are often asked to evaluate patients for neutrophilic dermatosis and vasculitis, and many affected patients had clinical and histologic findings compatible with polyarteritis nodosa and Sweet syndrome. When confronted with a neutrophilic dermatosis, we’ve all been taught to evaluate for myelodysplastic syndrome, which many of these patients appeared to have, at least on the surface. When bone marrow biopsies were done, the myeloid cell precursors that give rise to neutrophils were noted to have prominent cytoplasmic vacuoles, hence the “V” in VEXAS.
In reading the article describing 25 patients with this syndrome, which was published in the New England Journal of Medicine, I was struck by how refractory they were to treatment. Most patients had been treated with systemic steroids, multiple biologics, and several nonbiologic medications that are mainstays of treatment for neutrophilic dermatosis like dapsone and colchicine. I was fortunate enough to speak to Amanda Ombrello, MD, of the National Human Genome Research Institute, one of the lead authors of the paper, who drew my attention to the supplementary appendix, which showed the marked injection-site reactions some patients had to anakinra – yet another reason why a patient might end up in a dermatology clinic. In my mind, all of these features could be a clue to a diagnosis of VEXAS syndrome.
Many patients seemed to fare poorly, with 40% of patients dying before the completion of the study. When it comes to extremely rare diseases, it seems that the more physicians who are aware of the existence of a particular syndrome, the more likely it is a patient will come under our care and be correctly diagnosed.
Dr. Saardi is a dermatologist and internist, and is director of the inpatient dermatology service at the George Washington University Hospital, Washington. He has no disclosures.
When I was a medical student, I always found it gratifying when there was a unifying mechanism that explained the symptoms of a disease. Part of the reason I chose dermatology as a specialty was how frequently we are able to “see” these mechanisms in the skin, both clinically and histologically. What’s even more interesting is that this condition is caused by a postzygotic somatic mutation, an apparently underrecognized cause of disease that we are just now beginning to understand. An example of a postzygotic somatic mutation causing a disease that we all learned about in medical school is paroxysmal nocturnal hemoglobinuria.
Using a “bottom-up” approach, researchers at the National Institutes of Health and in the United Kingdom identified 25 patients with somatic UBA1 mutations and noticed that they had strikingly similar autoinflammatory syndromes. UBA1 encodes ubiquitin E1, which is part of the pathway the breaks down proteins as part of the normal cellular machine. It is localized to the X chromosome, so all 25 affected patients were males, and most were aged between 40 and 70 years. These patients had an autoinflammatory syndrome characterized by fever, chondritis (similar to relapsing polychondritis), vasculitis, and neutrophilic dermatoses. Many patients also had features of myelodysplastic syndrome and plasma cell dyscrasia. The inflammatory pattern in this condition seems to show elevations in tumor necrosis factor, interleukin-6, and interferon-gamma.
So why is this syndrome relevant to dermatology? We are often asked to evaluate patients for neutrophilic dermatosis and vasculitis, and many affected patients had clinical and histologic findings compatible with polyarteritis nodosa and Sweet syndrome. When confronted with a neutrophilic dermatosis, we’ve all been taught to evaluate for myelodysplastic syndrome, which many of these patients appeared to have, at least on the surface. When bone marrow biopsies were done, the myeloid cell precursors that give rise to neutrophils were noted to have prominent cytoplasmic vacuoles, hence the “V” in VEXAS.
In reading the article describing 25 patients with this syndrome, which was published in the New England Journal of Medicine, I was struck by how refractory they were to treatment. Most patients had been treated with systemic steroids, multiple biologics, and several nonbiologic medications that are mainstays of treatment for neutrophilic dermatosis like dapsone and colchicine. I was fortunate enough to speak to Amanda Ombrello, MD, of the National Human Genome Research Institute, one of the lead authors of the paper, who drew my attention to the supplementary appendix, which showed the marked injection-site reactions some patients had to anakinra – yet another reason why a patient might end up in a dermatology clinic. In my mind, all of these features could be a clue to a diagnosis of VEXAS syndrome.
Many patients seemed to fare poorly, with 40% of patients dying before the completion of the study. When it comes to extremely rare diseases, it seems that the more physicians who are aware of the existence of a particular syndrome, the more likely it is a patient will come under our care and be correctly diagnosed.
Dr. Saardi is a dermatologist and internist, and is director of the inpatient dermatology service at the George Washington University Hospital, Washington. He has no disclosures.