User login
Benign Pneumatosis Intestinalis: A Case Report and Review of the Literature
Pneumatosis intestinalis (PI) is the finding of gas within the walls of the intestine on imaging. It is most commonly detected via radiograph or computed tomography (CT). The diseases leading to the accumulation of gas within the submucosal space of the gastrointestinal (GI) tract are heterogenous, and the finding of PI itself has a wide range of clinical implications from impending clinical deterioration to an incidental finding of minimal consequence.
We present the case of a veteran who had sustained a remote anoxic brain injury resulting in chronic dependence on a gastrostomy tube for enteral nutrition, found incidentally to have PI without signs of intra-abdominal catastrophe. An exclusion of other, more lifethreatening causes of PI led to a diagnosis of benign PI secondary to the presence of his gastrostomy tube. This case highlights the importance of interpreting the finding of PI in the clinical context of the specific patient and how conservative management may be appropriate in some cases.
Case Presentation
A 61-year-old male patient was admitted for fever. The patient had a remote history of cardiac arrest complicated by anoxic brain injury requiring tracheostomy, gastrostomy tube, and a suprapubic catheter with recurrent catheter-associated urinary tract infections (CAUTI), secondary seizure disorder, atrial fibrillation off anticoagulation due to recurrent GI bleeding, and treatment naive chronic hepatitis C virus. His ability to provide a clinical history was limited by his nonverbal status. He had no prior surgical history but had presented a month earlier for a high-grade small bowel obstruction (SBO) with pneumobilia that was managed conservatively as the surgical team deemed him a poor candidate for surgical intervention with his extensive comorbidities. A bioethics consultation at the time supported minimizing potential surgical risk in favor of conservative medical management; this was discussed with the patient’s surrogate decision maker, who also wished to avoid surgery. The SBO resolved with conservative management. He had been residing in a nursing home and doing well until 24 hours prior to admission when he developed fevers.
Vital signs on admission showed a temperature of 100.8 °F, heart rate 100 beats per minute, blood pressure 116/85, respiratory rate 22 per minute, and oxygen saturation of 100% on 6 L of oxygen via tracheostomy collar. His initial examination was notable for clear lung sounds, a nondistended nonrigid abdomen with an indwelling percutaneous gastrostomy tube, and absence of areas of skin breakdown or erythema. Notable laboratory studies showed a leukocytosis and urinalysis suggestive of CAUTI (Table). His urinary catheter was exchanged, he was fluid resuscitated and started on empiric vancomycin and piperacillin-tazobactam for management of sepsis due to CAUTI.
For the first 3 days of his hospitalization, he demonstrated clinical improvement on vancomycin and piperacillin-tazobactam while awaiting results from his urine bacterial culture. On hospital day 3, hedeveloped recurrent nonbloody, nonbilious emesis despite no change in the rate or formulation of his enteral nutrition. He also had 3 watery brown bowel movements. His vital signs remained within normal limits. His abdominal examination at this point showed mild distention and was hypertympanic to percussion, but there was no rigidity or involuntary guarding. On hospital day 4, he continued to have emesis with an unchanged abdominal examination. The differential diagnosis included recurrence of prior SBO, ileus, intestinal ischemia, enteral nutrition intolerance, Clostridioides difficile (C difficile) colitis, and GI dysmotility because of his anoxic brain injury.
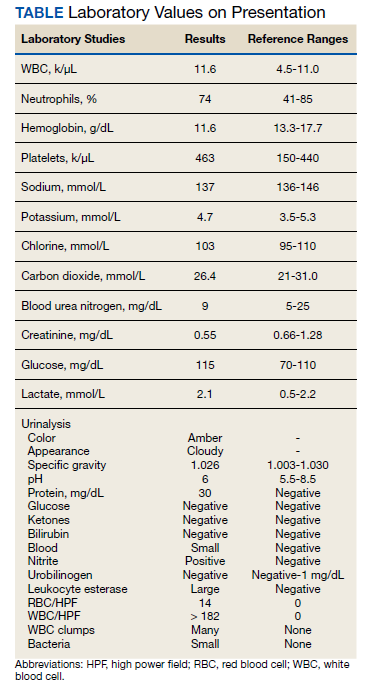
Testing for C difficile was negative. An abdominal radiograph was obtained and revealed no bowel obstruction but, alarmingly, showed extensive intramural bowel gas, suggestive of PI (Figure 1). His leukocyte count, serum bicarbonate, and serum lactate levels remained within normal limits. A CT with contrast of the abdomen and pelvis demonstrated no vascular obstruction but confirmed the presence of diffuse intramural gas in his stomach and proximal small bowel, as well as the presence of mesenteric and portal venous gas (Figures 2 and 3). Although his abdominal examination had not changed and did not suggest peritonitis, general surgery was consulted to discuss the need for surgical intervention. Given his overall clinical stability and high surgical risk due to his many comorbidities, surgery recommended a conservative approach.
Through the following hospital days, his enteral nutrition was held and serial abdominal examinations were performed without change. Serial laboratory studies, including serum lactate and leukocyte count, remained reassuringly within normal limits. His urine culture eventually revealed multidrugresistant Pseudomonas aeruginosa. Antimicrobial therapy was narrowed to piperacillintazobactam for a complete course. Enteral nutrition was gradually reintroduced at a low rate, ultimately reaching goal rate with return of bowel function by hospital day 9. Despite extensive workup, the etiology of his transient enteral nutrition intolerance remained uncertain, though an adverse effect of antibiotic therapy was thought possible. Follow-up abdominal radiographs demonstrated interval improvement of PI. He was discharged back to his skilled nursing facility on hospital day 11 without incident.
Discussion
PI is an incompletely understood condition seen in multiple diseases. Patients may present with highly variable symptoms, often more attributable to the underlying disease causing the PI than the presence of PI, as patients may be entirely asymptomatic. When symptoms are attributed to PI, those most reported are abdominal pain, bloody stools, and diarrhea.1 It is often detected on abdominal plain films. Alternative methods of diagnosis include ultrasonography, barium enema, and endoscopy although the last method has been known to occasionally lead to bowel perforation.2-6 The most sensitive method of detection is CT, which also provides additional information about abdominal pathology and may identify the underlying process responsible for the PI.7
While not fully understood, much information about PI and its pathogenesis is known. Understanding the mechanisms of PI is vital to direct the clinician’s evaluation of the patient for reversible conditions that may cause PI. Early descriptions of PI in the literature documented an association with pyloric stenosis, leading to the theory that gas from the intestinal lumen is driven into the submucosal space during episodes of forceful vomiting with increased intraluminal pressure.8 As PI was subsequently described in multiple other disease states not typically associated with increased intraluminal pressure such as inflammatory bowel disease, GI malignancy, cryptosporidiosis and CMV infection, additional theories about the pathogenesis of PI have arisen.9-24 There is now experimental data to support multiple mechanisms of intramural gas accumulation. It has become accepted that PI represents a common pathway shared across various pathologic states and results from multifactorial mechanisms of gas entry into the intestinal wall.25-29
Factors leading to the development of PI include bacterial production of gas, intraluminal GI gas compositions, increased intraluminal pressure, pulmonary gas tracking through vessels communicating with the thorax, and mucosal disruption. PI has been linked to bacterial infections of the GI tract in humans including C difficile, Klebsiella, and Whipple disease.14-18 In animal models, C difficile within the walls of rat intestine results in the appearance of pneumocysts, or discrete collections of submucosal gas, which are the hallmark feature of PI.30 It is thought that direct invasion of bacteria into intramural spaces can cause PI in humans, although bacteria have yet to be directly isolated from the pneumocysts. Translocation of luminal gas into pneumocysts found in PI is theorized to be driven by differences in partial pressures.31 The concentration of hydrogen within the intestinal lumen is high due to bacterial production. Hydrogen, diffusing along its partial pressure gradient between the lumen and blood, accumulates within the intestinal wall and causes the formation of pneumocysts. This phenomenon has been hypothesized to explain the tendency for pneumocysts to form around the mesenteric vasculature.
Gas from the lumen can also be forced into the intestinal wall during an abrupt increase in intra-abdominal pressure, such as that seen with forceful vomiting. The final possible origin of the gas is the lungs, as PI has been associated with lung disease. It was previously thought that gas from ruptured alveoli tracks along mediastinal vessels, below the diaphragm, and into the mesentery.30 Newer theories argue that increased intra-abdominal pressure, typical of patients with obstructive lung disease and frequent coughing, is the driver of PI by the mechanism previously described.32-34 Additionally, mucosal disruption leads to increased permeability and allows accumulation of gas within the intestinal walls. Mucosal abnormalities have been described in histopathologic studies of patients with PI and associated with conditions known to compromise mucosal integrity, such as immunodeficiencies, inflammatory bowel disease, and the receipt of cytotoxic chemotherapy.10,12,19-23
Our patient likely had mucosal disruption due to his gastrostomy tube as well as increased intraluminal pressure from recurrent vomiting, contributing to translocation of otherwise normal intraluminal gas. The presence of portal venous gas, as seen in this case, has historically portended a worse prognosis, with 37% mortality in one series.7,35,36 However, portal venous gas as well as pneumoperitoneum occur in benign etiologies of PI as well. It is thought that this occurs due to rupture of the submucosal pneumocysts through the wall opposite the intestinal lumen and thus does not result in a direct communication between the intestinal lumen and the peritoneal cavity.12
PI is not a diagnosis but a manifestation of an underlying disease. As such, the treatment of PI is targeted toward the underlying condition. Of note, the pattern and extent of PI seen on imaging has not been shown to correlate with the severity of the underlying pathologic process.35,37 Instead, assessment of the patient and their clinical trajectory should determine the appropriate treatment. The decision facing the clinician when PI is discovered is whether urgent surgery is indicated, as is the case in mesenteric ischemia, bowel necrosis, or intestinal perforation, conditions known to be associated with PI. Otherwise, there is no definitive treatment for PI. Bowel rest is almost universally pursued. There are reports of treating with supranormal levels of supplemental oxygen, maintaining arterial partial pressure of oxygen above 300 mm Hg, with a face mask and 8 L/min flow rate.38,39 The proposed mechanisms of benefit include establishing a favorable diffusion gradient for intramural gas to exit the pneumocysts as well as creating an inhospitable, aerobic environment for hydrogenproducing anaerobic enteric bacteria. A prudent approach for most cases of PI is conservative management with bowel rest and supplemental oxygen unless there is a definitive indication for urgent surgical intervention, such as peritonitis, abdominal sepsis, or perforation.40,41 Management recommendations suggest that up to 50% of cases can be successfully managed nonoperatively.42
Conclusions
PI is the radiographic finding of gas within the walls of the intestinal tract and has variable clinical significance. It can represent a benign incidental finding or a sequela of intraabdominal emergencies such as mesenteric ischemia or bowel necrosis. Because PI is seen in a variety of disorders, several proposed mechanisms are supported in the medical literature. These include bacterial production of gas, gas pressure gradients between the intestinal lumen and the blood, increased intraluminal pressure, pulmonary gas tracking from intrathoracic vessels, and mucosal disruption. The evaluation of a patient with PI must begin with an assessment for the need for urgent surgical intervention. Additional management measures include bowel rest, IV hydration, and supplemental oxygen administration. Because of its wide variety of etiologies of varying clinical urgency, placing the finding of PI in the context of the patient is paramount to selecting an appropriate management strategy.
1. Jamart J. Pneumatosis cystoides intestinalis. A statistical study of 919 cases. Acta Hepatogastroenterol (Stuttg). 1979;26(5):419-422.
2. Lafortune M, Trinh BC, Burns PN, et al. Air in the portal vein: sonographic and Doppler manifestations. Radiology. 1991;180(3):667-670. doi:10.1148/radiology.180.3.1871276
3. Kriegshauser JS, Reading CC, King BF, Welch TJ. Combined systemic and portal venous gas: sonographic and CT detection in two cases. AJR Am J Roentgenol. 1990;154(6):1219-1221. doi:10.2214/ajr.154.6.2110731
4. Goske MJ, Goldblum JR, Applegate KE, Mitchell CS, Bardo D. The “circle sign”: a new sonographic sign of pneumatosis intestinalis - clinical, pathologic and experimental findings. Pediatr Radiol. 1999;29(7):530-535. doi:10.1007/s002470050638
5. Marshak RH, Lindner AE, Maklansky D. Pneumatosis cystoides coli. Gastrointest Radiol. 1977;2(2):85-89. doi:10.1007/BF02256475
6. Jensen R, Gutnik SH. Pneumatosis cystoides intestinalis: a complication of colonoscopic polypectomy. S D J Med. 1991;44(7):177-179.
7. Knechtle SJ, Davidoff AM, Rice RP. Pneumatosis intestinalis. Surgical management and clinical outcome. Ann Surg. 1990;212(2):160-165. doi:10.1097/00000658-199008000-00008
8. Koss LG. Abdominal gas cysts (Pneumatosis cystoides intestinorum hominis); an analysis with a report of a case and a critical review of the literature. AMA Arch Pathol. 1952;53(6):523-549.
9. Jona JZ. Benign pneumatosis intestinalis coli after blunt trauma to the abdomen in a child. J Pediatr Surg. 2000;35(7):1109-1111. doi:10.1053/jpsu.2000.7837
10. Gagliardi G, Thompson IW, Hershman MJ, Forbes A, Hawley PR, Talbot IC. Pneumatosis coli: a proposed pathogenesis based on study of 25 cases and review of the literature. Int J Colorectal Dis. 1996;11(3):111-118. doi:10.1007/s003840050031
11. Seto T, Koide N, Taniuchi N, Yamada T, Hamaguchi M, Goto S. Pneumatosis cystoides intestinalis complicating carcinoma of the small intestine. Am J Surg. 2001;182(3):287-288. doi:10.1016/S0002-9610(01)00710-3
12. Galandiuk S, Fazio VW, Petras RE. Pneumatosis cystoides intestinalis in Crohn’s disease. Report of two cases. Dis Colon Rectum. 1985;28(12):951-956. doi:10.1007/BF02554315
13. Parra JA, Acinas O, Bueno J, Madrazo C, Fariñas C. An unusual form of pneumatosis intestinalis associated with appendicitis. Br J Radiol. 1998;71(843):326-328. doi:10.1259/bjr.71.843.9616245
14. Schenk P, Madl C, Kramer L, et al. Pneumatosis intestinalis with Clostridium difficile colitis as a cause of acute abdomen after lung transplantation. Dig Dis Sci. 1998;43(11):2455-2458. doi:10.1023/a:1026682131847
15. Kreiss C, Forohar F, Smithline AE, Brandt LJ. Pneumatosis intestinalis complicating C. difficile pseudomembranous colitis. Am J Gastroenterol. 1999;94(9):2560-2561. doi:10.1111/j.1572-0241.1999.01397.x
16. Day DL, Ramsay NK, Letourneau JG. Pneumatosis intestinalis after bone marrow transplantation. AJR Am J Roentgenol. 1988;151(1):85-87. doi:10.2214/ajr.151.1.85
17. Tahara S, Sakai Y, Katsuno H, Urano M, Kuroda M, Tsukamoto T. Pneumatosis intestinalis and hepatic portal venous gas associated with gas-forming bacterial translocation due to postoperative paralytic ileus: A case report. Medicine (Baltimore). 2019;98(2):e14079. doi:10.1097/MD.0000000000014079
18. Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of whipple’s disease in a patient mistakenly on anti-tnf therapy. ACG Case Rep J. 2013;1(1):25- 28. Published 2013 Oct 8. doi:10.14309/crj.2013.11
19. Burton EM, Mercado-Deane MG, Patel K. Pneumatosis intestinalis in a child with AIDS and pseudomembranous colitis. Pediatr Radiol. 1994;24(8):609-610. doi:10.1007/BF02012750
20. Berk RN, Wall SD, McArdle CB, et al. Cryptosporidiosis of the stomach and small intestine in patients with AIDS. AJR Am J Roentgenol. 1984;143(3):549-554. doi:10.2214/ajr.143.3.549
21. Samson VE, Brown WR. Pneumatosis cystoides intestinalis in AIDS-associated cryptosporidiosis. More than an incidental finding? J Clin Gastroenterol. 1996;22(4):311-312.doi:10.1097/00004836-199606000-00015
22. Tjon A Tham RT, Vlasveld LT, Willemze R. Gastrointestinal complications of cytosine-arabinoside chemotherapy: findings on plain abdominal radiographs. AJR Am J Roentgenol. 1990;154(1):95-98. doi:10.2214/ajr.154.1.2104733
23. Hashimoto S, Saitoh H, Wada K, et al. Pneumatosis cystoides intestinalis after chemotherapy for hematological malignancies: report of 4 cases. Intern Med. 1995;34(3):212-215. doi:10.2169/internalmedicine.34.212
24. Gelman SF, Brandt LJ. Pneumatosis intestinalis and AIDS: a case report and review of the literature. Am J Gastroenterol. 1998;93(4):646-650. doi:10.1111/j.1572-0241.1998.183_b.x
25. Gillon J, Tadesse K, Logan RF, Holt S, Sircus W. Breath hydrogen in pneumatosis cystoides intestinalis. Gut. 1979;20(11):1008-1011. doi:10.1136/gut.20.11.1008
26. Hughes DT, Gordon KC, Swann JC, Bolt GL. Pneumatosis cystoides intestinalis. Gut. 1966;7(5):553-557. doi:10.1136/gut.7.5.553
27. Read NW, Al-Janabi MN, Cann PA. Is raised breath hydrogen related to the pathogenesis of pneumatosis coli? Gut. 1984;25(8):839-845. doi:10.1136/gut.25.8.839
28. van der Linden W, Marsell R. Pneumatosis cystoides coli associated with high H2 excretion. Treatment with an elemental diet. Scand J Gastroenterol. 1979;14(2):173-174. doi:10.3109/00365527909179864
29. Christl SU, Gibson GR, Murgatroyd PR, Scheppach W, Cummings JH. Impaired hydrogen metabolism in pneumatosis cystoides intestinalis. Gastroenterology. 1993;104(2):392-397. doi:10.1016/0016-5085(93)90406-3
30. Keyting WS, Mccarver RR, Kovarik JL, Daywitt AL. Pneumatosis intestinalis: a new concept. Radiology. 1961;76:733-741. doi:10.1148/76.5.733
31. Florin TH, Hills BA. Does counterperfusion supersaturation cause gas cysts in pneumatosis cystoides coli, and can breathing heliox reduce them? Lancet. 1995;345(8959):1220-1222. doi:10.1016/S0140-6736(95)91996-1
32. Grieve DA, Unsworth IP. Pneumatosis cystoides intestinalis: an experience with hyperbaric oxygen treatment. Aust N Z J Surg. 1991;61(6):423-426.
33. Micklefield GH, Kuntz HD, May B. Pneumatosis cystoides intestinalis: case reports and review of the literature. Mater Med Pol. 1990;22(2):70-72.
34. Yale CE, Balish E, Wu JP. The bacterial etiology of pneumatosis cystoides intestinalis. Arch Surg. 1974;109(1):89- 94. doi:10.1001/archsurg.1974.01360010067017
35. Fenton LZ, Buonomo C. Benign pneumatosis in children. Pediatr Radiol. 2000;30(11):786-793. doi:10.1007/s002470000303
36. Tobias R, Coleman S, Helman CA. Pneumatosis coli simulating hepatomegaly. Am J Gastroenterol. 1985;80(2):146-149.
37. Feczko PJ, Mezwa DG, Farah MC, White BD. Clinical significance of pneumatosis of the bowel w a l l . Radiographics. 1992;12(6):1069-1078. doi:10.1148/radiographics.12.6.1439012
38. Masterson JS, Fratkin LB, Osler TR, Trapp WG. Treatment of pneumatosis cystoides intestinalis with hyperbaric oxygen. Ann Surg. 1978;187(3):245-247. doi:10.1097/00000658-197803000-00005
39. Höflin F, Linden W van der. Pneumatosis cystoides intestinalis treated by oxygen breathing. Scandinavian J Gastroenterol . 1974;9(5) :427-430. doi:10.1080/00365521.1974.12096852
40. St Peter SD, Abbas MA, Kelly KA. The spectrum of pneumatosis intestinalis. Arch Surg. 2003;138(1):68-75. doi:10.1001/archsurg.138.1.68
41. Ling F, Guo D, Zhu L. Pneumatosis cystoides intestinalis: a case report and literature review. BMC Gastroenterol. 2019;19(1):176. Published 2019 Nov 6. doi:10.1186/s12876-019-1087-9
42. Morris MS, Gee AC, Cho SD, et al. Management and outcome of pneumatosis intestinalis. Am J Surg. 2008;195(5):679-682. doi:10.1016/j.amjsurg.2008.01.011
Pneumatosis intestinalis (PI) is the finding of gas within the walls of the intestine on imaging. It is most commonly detected via radiograph or computed tomography (CT). The diseases leading to the accumulation of gas within the submucosal space of the gastrointestinal (GI) tract are heterogenous, and the finding of PI itself has a wide range of clinical implications from impending clinical deterioration to an incidental finding of minimal consequence.
We present the case of a veteran who had sustained a remote anoxic brain injury resulting in chronic dependence on a gastrostomy tube for enteral nutrition, found incidentally to have PI without signs of intra-abdominal catastrophe. An exclusion of other, more lifethreatening causes of PI led to a diagnosis of benign PI secondary to the presence of his gastrostomy tube. This case highlights the importance of interpreting the finding of PI in the clinical context of the specific patient and how conservative management may be appropriate in some cases.
Case Presentation
A 61-year-old male patient was admitted for fever. The patient had a remote history of cardiac arrest complicated by anoxic brain injury requiring tracheostomy, gastrostomy tube, and a suprapubic catheter with recurrent catheter-associated urinary tract infections (CAUTI), secondary seizure disorder, atrial fibrillation off anticoagulation due to recurrent GI bleeding, and treatment naive chronic hepatitis C virus. His ability to provide a clinical history was limited by his nonverbal status. He had no prior surgical history but had presented a month earlier for a high-grade small bowel obstruction (SBO) with pneumobilia that was managed conservatively as the surgical team deemed him a poor candidate for surgical intervention with his extensive comorbidities. A bioethics consultation at the time supported minimizing potential surgical risk in favor of conservative medical management; this was discussed with the patient’s surrogate decision maker, who also wished to avoid surgery. The SBO resolved with conservative management. He had been residing in a nursing home and doing well until 24 hours prior to admission when he developed fevers.
Vital signs on admission showed a temperature of 100.8 °F, heart rate 100 beats per minute, blood pressure 116/85, respiratory rate 22 per minute, and oxygen saturation of 100% on 6 L of oxygen via tracheostomy collar. His initial examination was notable for clear lung sounds, a nondistended nonrigid abdomen with an indwelling percutaneous gastrostomy tube, and absence of areas of skin breakdown or erythema. Notable laboratory studies showed a leukocytosis and urinalysis suggestive of CAUTI (Table). His urinary catheter was exchanged, he was fluid resuscitated and started on empiric vancomycin and piperacillin-tazobactam for management of sepsis due to CAUTI.
For the first 3 days of his hospitalization, he demonstrated clinical improvement on vancomycin and piperacillin-tazobactam while awaiting results from his urine bacterial culture. On hospital day 3, hedeveloped recurrent nonbloody, nonbilious emesis despite no change in the rate or formulation of his enteral nutrition. He also had 3 watery brown bowel movements. His vital signs remained within normal limits. His abdominal examination at this point showed mild distention and was hypertympanic to percussion, but there was no rigidity or involuntary guarding. On hospital day 4, he continued to have emesis with an unchanged abdominal examination. The differential diagnosis included recurrence of prior SBO, ileus, intestinal ischemia, enteral nutrition intolerance, Clostridioides difficile (C difficile) colitis, and GI dysmotility because of his anoxic brain injury.
Testing for C difficile was negative. An abdominal radiograph was obtained and revealed no bowel obstruction but, alarmingly, showed extensive intramural bowel gas, suggestive of PI (Figure 1). His leukocyte count, serum bicarbonate, and serum lactate levels remained within normal limits. A CT with contrast of the abdomen and pelvis demonstrated no vascular obstruction but confirmed the presence of diffuse intramural gas in his stomach and proximal small bowel, as well as the presence of mesenteric and portal venous gas (Figures 2 and 3). Although his abdominal examination had not changed and did not suggest peritonitis, general surgery was consulted to discuss the need for surgical intervention. Given his overall clinical stability and high surgical risk due to his many comorbidities, surgery recommended a conservative approach.
Through the following hospital days, his enteral nutrition was held and serial abdominal examinations were performed without change. Serial laboratory studies, including serum lactate and leukocyte count, remained reassuringly within normal limits. His urine culture eventually revealed multidrugresistant Pseudomonas aeruginosa. Antimicrobial therapy was narrowed to piperacillintazobactam for a complete course. Enteral nutrition was gradually reintroduced at a low rate, ultimately reaching goal rate with return of bowel function by hospital day 9. Despite extensive workup, the etiology of his transient enteral nutrition intolerance remained uncertain, though an adverse effect of antibiotic therapy was thought possible. Follow-up abdominal radiographs demonstrated interval improvement of PI. He was discharged back to his skilled nursing facility on hospital day 11 without incident.
Discussion
PI is an incompletely understood condition seen in multiple diseases. Patients may present with highly variable symptoms, often more attributable to the underlying disease causing the PI than the presence of PI, as patients may be entirely asymptomatic. When symptoms are attributed to PI, those most reported are abdominal pain, bloody stools, and diarrhea.1 It is often detected on abdominal plain films. Alternative methods of diagnosis include ultrasonography, barium enema, and endoscopy although the last method has been known to occasionally lead to bowel perforation.2-6 The most sensitive method of detection is CT, which also provides additional information about abdominal pathology and may identify the underlying process responsible for the PI.7
While not fully understood, much information about PI and its pathogenesis is known. Understanding the mechanisms of PI is vital to direct the clinician’s evaluation of the patient for reversible conditions that may cause PI. Early descriptions of PI in the literature documented an association with pyloric stenosis, leading to the theory that gas from the intestinal lumen is driven into the submucosal space during episodes of forceful vomiting with increased intraluminal pressure.8 As PI was subsequently described in multiple other disease states not typically associated with increased intraluminal pressure such as inflammatory bowel disease, GI malignancy, cryptosporidiosis and CMV infection, additional theories about the pathogenesis of PI have arisen.9-24 There is now experimental data to support multiple mechanisms of intramural gas accumulation. It has become accepted that PI represents a common pathway shared across various pathologic states and results from multifactorial mechanisms of gas entry into the intestinal wall.25-29
Factors leading to the development of PI include bacterial production of gas, intraluminal GI gas compositions, increased intraluminal pressure, pulmonary gas tracking through vessels communicating with the thorax, and mucosal disruption. PI has been linked to bacterial infections of the GI tract in humans including C difficile, Klebsiella, and Whipple disease.14-18 In animal models, C difficile within the walls of rat intestine results in the appearance of pneumocysts, or discrete collections of submucosal gas, which are the hallmark feature of PI.30 It is thought that direct invasion of bacteria into intramural spaces can cause PI in humans, although bacteria have yet to be directly isolated from the pneumocysts. Translocation of luminal gas into pneumocysts found in PI is theorized to be driven by differences in partial pressures.31 The concentration of hydrogen within the intestinal lumen is high due to bacterial production. Hydrogen, diffusing along its partial pressure gradient between the lumen and blood, accumulates within the intestinal wall and causes the formation of pneumocysts. This phenomenon has been hypothesized to explain the tendency for pneumocysts to form around the mesenteric vasculature.
Gas from the lumen can also be forced into the intestinal wall during an abrupt increase in intra-abdominal pressure, such as that seen with forceful vomiting. The final possible origin of the gas is the lungs, as PI has been associated with lung disease. It was previously thought that gas from ruptured alveoli tracks along mediastinal vessels, below the diaphragm, and into the mesentery.30 Newer theories argue that increased intra-abdominal pressure, typical of patients with obstructive lung disease and frequent coughing, is the driver of PI by the mechanism previously described.32-34 Additionally, mucosal disruption leads to increased permeability and allows accumulation of gas within the intestinal walls. Mucosal abnormalities have been described in histopathologic studies of patients with PI and associated with conditions known to compromise mucosal integrity, such as immunodeficiencies, inflammatory bowel disease, and the receipt of cytotoxic chemotherapy.10,12,19-23
Our patient likely had mucosal disruption due to his gastrostomy tube as well as increased intraluminal pressure from recurrent vomiting, contributing to translocation of otherwise normal intraluminal gas. The presence of portal venous gas, as seen in this case, has historically portended a worse prognosis, with 37% mortality in one series.7,35,36 However, portal venous gas as well as pneumoperitoneum occur in benign etiologies of PI as well. It is thought that this occurs due to rupture of the submucosal pneumocysts through the wall opposite the intestinal lumen and thus does not result in a direct communication between the intestinal lumen and the peritoneal cavity.12
PI is not a diagnosis but a manifestation of an underlying disease. As such, the treatment of PI is targeted toward the underlying condition. Of note, the pattern and extent of PI seen on imaging has not been shown to correlate with the severity of the underlying pathologic process.35,37 Instead, assessment of the patient and their clinical trajectory should determine the appropriate treatment. The decision facing the clinician when PI is discovered is whether urgent surgery is indicated, as is the case in mesenteric ischemia, bowel necrosis, or intestinal perforation, conditions known to be associated with PI. Otherwise, there is no definitive treatment for PI. Bowel rest is almost universally pursued. There are reports of treating with supranormal levels of supplemental oxygen, maintaining arterial partial pressure of oxygen above 300 mm Hg, with a face mask and 8 L/min flow rate.38,39 The proposed mechanisms of benefit include establishing a favorable diffusion gradient for intramural gas to exit the pneumocysts as well as creating an inhospitable, aerobic environment for hydrogenproducing anaerobic enteric bacteria. A prudent approach for most cases of PI is conservative management with bowel rest and supplemental oxygen unless there is a definitive indication for urgent surgical intervention, such as peritonitis, abdominal sepsis, or perforation.40,41 Management recommendations suggest that up to 50% of cases can be successfully managed nonoperatively.42
Conclusions
PI is the radiographic finding of gas within the walls of the intestinal tract and has variable clinical significance. It can represent a benign incidental finding or a sequela of intraabdominal emergencies such as mesenteric ischemia or bowel necrosis. Because PI is seen in a variety of disorders, several proposed mechanisms are supported in the medical literature. These include bacterial production of gas, gas pressure gradients between the intestinal lumen and the blood, increased intraluminal pressure, pulmonary gas tracking from intrathoracic vessels, and mucosal disruption. The evaluation of a patient with PI must begin with an assessment for the need for urgent surgical intervention. Additional management measures include bowel rest, IV hydration, and supplemental oxygen administration. Because of its wide variety of etiologies of varying clinical urgency, placing the finding of PI in the context of the patient is paramount to selecting an appropriate management strategy.
Pneumatosis intestinalis (PI) is the finding of gas within the walls of the intestine on imaging. It is most commonly detected via radiograph or computed tomography (CT). The diseases leading to the accumulation of gas within the submucosal space of the gastrointestinal (GI) tract are heterogenous, and the finding of PI itself has a wide range of clinical implications from impending clinical deterioration to an incidental finding of minimal consequence.
We present the case of a veteran who had sustained a remote anoxic brain injury resulting in chronic dependence on a gastrostomy tube for enteral nutrition, found incidentally to have PI without signs of intra-abdominal catastrophe. An exclusion of other, more lifethreatening causes of PI led to a diagnosis of benign PI secondary to the presence of his gastrostomy tube. This case highlights the importance of interpreting the finding of PI in the clinical context of the specific patient and how conservative management may be appropriate in some cases.
Case Presentation
A 61-year-old male patient was admitted for fever. The patient had a remote history of cardiac arrest complicated by anoxic brain injury requiring tracheostomy, gastrostomy tube, and a suprapubic catheter with recurrent catheter-associated urinary tract infections (CAUTI), secondary seizure disorder, atrial fibrillation off anticoagulation due to recurrent GI bleeding, and treatment naive chronic hepatitis C virus. His ability to provide a clinical history was limited by his nonverbal status. He had no prior surgical history but had presented a month earlier for a high-grade small bowel obstruction (SBO) with pneumobilia that was managed conservatively as the surgical team deemed him a poor candidate for surgical intervention with his extensive comorbidities. A bioethics consultation at the time supported minimizing potential surgical risk in favor of conservative medical management; this was discussed with the patient’s surrogate decision maker, who also wished to avoid surgery. The SBO resolved with conservative management. He had been residing in a nursing home and doing well until 24 hours prior to admission when he developed fevers.
Vital signs on admission showed a temperature of 100.8 °F, heart rate 100 beats per minute, blood pressure 116/85, respiratory rate 22 per minute, and oxygen saturation of 100% on 6 L of oxygen via tracheostomy collar. His initial examination was notable for clear lung sounds, a nondistended nonrigid abdomen with an indwelling percutaneous gastrostomy tube, and absence of areas of skin breakdown or erythema. Notable laboratory studies showed a leukocytosis and urinalysis suggestive of CAUTI (Table). His urinary catheter was exchanged, he was fluid resuscitated and started on empiric vancomycin and piperacillin-tazobactam for management of sepsis due to CAUTI.
For the first 3 days of his hospitalization, he demonstrated clinical improvement on vancomycin and piperacillin-tazobactam while awaiting results from his urine bacterial culture. On hospital day 3, hedeveloped recurrent nonbloody, nonbilious emesis despite no change in the rate or formulation of his enteral nutrition. He also had 3 watery brown bowel movements. His vital signs remained within normal limits. His abdominal examination at this point showed mild distention and was hypertympanic to percussion, but there was no rigidity or involuntary guarding. On hospital day 4, he continued to have emesis with an unchanged abdominal examination. The differential diagnosis included recurrence of prior SBO, ileus, intestinal ischemia, enteral nutrition intolerance, Clostridioides difficile (C difficile) colitis, and GI dysmotility because of his anoxic brain injury.
Testing for C difficile was negative. An abdominal radiograph was obtained and revealed no bowel obstruction but, alarmingly, showed extensive intramural bowel gas, suggestive of PI (Figure 1). His leukocyte count, serum bicarbonate, and serum lactate levels remained within normal limits. A CT with contrast of the abdomen and pelvis demonstrated no vascular obstruction but confirmed the presence of diffuse intramural gas in his stomach and proximal small bowel, as well as the presence of mesenteric and portal venous gas (Figures 2 and 3). Although his abdominal examination had not changed and did not suggest peritonitis, general surgery was consulted to discuss the need for surgical intervention. Given his overall clinical stability and high surgical risk due to his many comorbidities, surgery recommended a conservative approach.
Through the following hospital days, his enteral nutrition was held and serial abdominal examinations were performed without change. Serial laboratory studies, including serum lactate and leukocyte count, remained reassuringly within normal limits. His urine culture eventually revealed multidrugresistant Pseudomonas aeruginosa. Antimicrobial therapy was narrowed to piperacillintazobactam for a complete course. Enteral nutrition was gradually reintroduced at a low rate, ultimately reaching goal rate with return of bowel function by hospital day 9. Despite extensive workup, the etiology of his transient enteral nutrition intolerance remained uncertain, though an adverse effect of antibiotic therapy was thought possible. Follow-up abdominal radiographs demonstrated interval improvement of PI. He was discharged back to his skilled nursing facility on hospital day 11 without incident.
Discussion
PI is an incompletely understood condition seen in multiple diseases. Patients may present with highly variable symptoms, often more attributable to the underlying disease causing the PI than the presence of PI, as patients may be entirely asymptomatic. When symptoms are attributed to PI, those most reported are abdominal pain, bloody stools, and diarrhea.1 It is often detected on abdominal plain films. Alternative methods of diagnosis include ultrasonography, barium enema, and endoscopy although the last method has been known to occasionally lead to bowel perforation.2-6 The most sensitive method of detection is CT, which also provides additional information about abdominal pathology and may identify the underlying process responsible for the PI.7
While not fully understood, much information about PI and its pathogenesis is known. Understanding the mechanisms of PI is vital to direct the clinician’s evaluation of the patient for reversible conditions that may cause PI. Early descriptions of PI in the literature documented an association with pyloric stenosis, leading to the theory that gas from the intestinal lumen is driven into the submucosal space during episodes of forceful vomiting with increased intraluminal pressure.8 As PI was subsequently described in multiple other disease states not typically associated with increased intraluminal pressure such as inflammatory bowel disease, GI malignancy, cryptosporidiosis and CMV infection, additional theories about the pathogenesis of PI have arisen.9-24 There is now experimental data to support multiple mechanisms of intramural gas accumulation. It has become accepted that PI represents a common pathway shared across various pathologic states and results from multifactorial mechanisms of gas entry into the intestinal wall.25-29
Factors leading to the development of PI include bacterial production of gas, intraluminal GI gas compositions, increased intraluminal pressure, pulmonary gas tracking through vessels communicating with the thorax, and mucosal disruption. PI has been linked to bacterial infections of the GI tract in humans including C difficile, Klebsiella, and Whipple disease.14-18 In animal models, C difficile within the walls of rat intestine results in the appearance of pneumocysts, or discrete collections of submucosal gas, which are the hallmark feature of PI.30 It is thought that direct invasion of bacteria into intramural spaces can cause PI in humans, although bacteria have yet to be directly isolated from the pneumocysts. Translocation of luminal gas into pneumocysts found in PI is theorized to be driven by differences in partial pressures.31 The concentration of hydrogen within the intestinal lumen is high due to bacterial production. Hydrogen, diffusing along its partial pressure gradient between the lumen and blood, accumulates within the intestinal wall and causes the formation of pneumocysts. This phenomenon has been hypothesized to explain the tendency for pneumocysts to form around the mesenteric vasculature.
Gas from the lumen can also be forced into the intestinal wall during an abrupt increase in intra-abdominal pressure, such as that seen with forceful vomiting. The final possible origin of the gas is the lungs, as PI has been associated with lung disease. It was previously thought that gas from ruptured alveoli tracks along mediastinal vessels, below the diaphragm, and into the mesentery.30 Newer theories argue that increased intra-abdominal pressure, typical of patients with obstructive lung disease and frequent coughing, is the driver of PI by the mechanism previously described.32-34 Additionally, mucosal disruption leads to increased permeability and allows accumulation of gas within the intestinal walls. Mucosal abnormalities have been described in histopathologic studies of patients with PI and associated with conditions known to compromise mucosal integrity, such as immunodeficiencies, inflammatory bowel disease, and the receipt of cytotoxic chemotherapy.10,12,19-23
Our patient likely had mucosal disruption due to his gastrostomy tube as well as increased intraluminal pressure from recurrent vomiting, contributing to translocation of otherwise normal intraluminal gas. The presence of portal venous gas, as seen in this case, has historically portended a worse prognosis, with 37% mortality in one series.7,35,36 However, portal venous gas as well as pneumoperitoneum occur in benign etiologies of PI as well. It is thought that this occurs due to rupture of the submucosal pneumocysts through the wall opposite the intestinal lumen and thus does not result in a direct communication between the intestinal lumen and the peritoneal cavity.12
PI is not a diagnosis but a manifestation of an underlying disease. As such, the treatment of PI is targeted toward the underlying condition. Of note, the pattern and extent of PI seen on imaging has not been shown to correlate with the severity of the underlying pathologic process.35,37 Instead, assessment of the patient and their clinical trajectory should determine the appropriate treatment. The decision facing the clinician when PI is discovered is whether urgent surgery is indicated, as is the case in mesenteric ischemia, bowel necrosis, or intestinal perforation, conditions known to be associated with PI. Otherwise, there is no definitive treatment for PI. Bowel rest is almost universally pursued. There are reports of treating with supranormal levels of supplemental oxygen, maintaining arterial partial pressure of oxygen above 300 mm Hg, with a face mask and 8 L/min flow rate.38,39 The proposed mechanisms of benefit include establishing a favorable diffusion gradient for intramural gas to exit the pneumocysts as well as creating an inhospitable, aerobic environment for hydrogenproducing anaerobic enteric bacteria. A prudent approach for most cases of PI is conservative management with bowel rest and supplemental oxygen unless there is a definitive indication for urgent surgical intervention, such as peritonitis, abdominal sepsis, or perforation.40,41 Management recommendations suggest that up to 50% of cases can be successfully managed nonoperatively.42
Conclusions
PI is the radiographic finding of gas within the walls of the intestinal tract and has variable clinical significance. It can represent a benign incidental finding or a sequela of intraabdominal emergencies such as mesenteric ischemia or bowel necrosis. Because PI is seen in a variety of disorders, several proposed mechanisms are supported in the medical literature. These include bacterial production of gas, gas pressure gradients between the intestinal lumen and the blood, increased intraluminal pressure, pulmonary gas tracking from intrathoracic vessels, and mucosal disruption. The evaluation of a patient with PI must begin with an assessment for the need for urgent surgical intervention. Additional management measures include bowel rest, IV hydration, and supplemental oxygen administration. Because of its wide variety of etiologies of varying clinical urgency, placing the finding of PI in the context of the patient is paramount to selecting an appropriate management strategy.
1. Jamart J. Pneumatosis cystoides intestinalis. A statistical study of 919 cases. Acta Hepatogastroenterol (Stuttg). 1979;26(5):419-422.
2. Lafortune M, Trinh BC, Burns PN, et al. Air in the portal vein: sonographic and Doppler manifestations. Radiology. 1991;180(3):667-670. doi:10.1148/radiology.180.3.1871276
3. Kriegshauser JS, Reading CC, King BF, Welch TJ. Combined systemic and portal venous gas: sonographic and CT detection in two cases. AJR Am J Roentgenol. 1990;154(6):1219-1221. doi:10.2214/ajr.154.6.2110731
4. Goske MJ, Goldblum JR, Applegate KE, Mitchell CS, Bardo D. The “circle sign”: a new sonographic sign of pneumatosis intestinalis - clinical, pathologic and experimental findings. Pediatr Radiol. 1999;29(7):530-535. doi:10.1007/s002470050638
5. Marshak RH, Lindner AE, Maklansky D. Pneumatosis cystoides coli. Gastrointest Radiol. 1977;2(2):85-89. doi:10.1007/BF02256475
6. Jensen R, Gutnik SH. Pneumatosis cystoides intestinalis: a complication of colonoscopic polypectomy. S D J Med. 1991;44(7):177-179.
7. Knechtle SJ, Davidoff AM, Rice RP. Pneumatosis intestinalis. Surgical management and clinical outcome. Ann Surg. 1990;212(2):160-165. doi:10.1097/00000658-199008000-00008
8. Koss LG. Abdominal gas cysts (Pneumatosis cystoides intestinorum hominis); an analysis with a report of a case and a critical review of the literature. AMA Arch Pathol. 1952;53(6):523-549.
9. Jona JZ. Benign pneumatosis intestinalis coli after blunt trauma to the abdomen in a child. J Pediatr Surg. 2000;35(7):1109-1111. doi:10.1053/jpsu.2000.7837
10. Gagliardi G, Thompson IW, Hershman MJ, Forbes A, Hawley PR, Talbot IC. Pneumatosis coli: a proposed pathogenesis based on study of 25 cases and review of the literature. Int J Colorectal Dis. 1996;11(3):111-118. doi:10.1007/s003840050031
11. Seto T, Koide N, Taniuchi N, Yamada T, Hamaguchi M, Goto S. Pneumatosis cystoides intestinalis complicating carcinoma of the small intestine. Am J Surg. 2001;182(3):287-288. doi:10.1016/S0002-9610(01)00710-3
12. Galandiuk S, Fazio VW, Petras RE. Pneumatosis cystoides intestinalis in Crohn’s disease. Report of two cases. Dis Colon Rectum. 1985;28(12):951-956. doi:10.1007/BF02554315
13. Parra JA, Acinas O, Bueno J, Madrazo C, Fariñas C. An unusual form of pneumatosis intestinalis associated with appendicitis. Br J Radiol. 1998;71(843):326-328. doi:10.1259/bjr.71.843.9616245
14. Schenk P, Madl C, Kramer L, et al. Pneumatosis intestinalis with Clostridium difficile colitis as a cause of acute abdomen after lung transplantation. Dig Dis Sci. 1998;43(11):2455-2458. doi:10.1023/a:1026682131847
15. Kreiss C, Forohar F, Smithline AE, Brandt LJ. Pneumatosis intestinalis complicating C. difficile pseudomembranous colitis. Am J Gastroenterol. 1999;94(9):2560-2561. doi:10.1111/j.1572-0241.1999.01397.x
16. Day DL, Ramsay NK, Letourneau JG. Pneumatosis intestinalis after bone marrow transplantation. AJR Am J Roentgenol. 1988;151(1):85-87. doi:10.2214/ajr.151.1.85
17. Tahara S, Sakai Y, Katsuno H, Urano M, Kuroda M, Tsukamoto T. Pneumatosis intestinalis and hepatic portal venous gas associated with gas-forming bacterial translocation due to postoperative paralytic ileus: A case report. Medicine (Baltimore). 2019;98(2):e14079. doi:10.1097/MD.0000000000014079
18. Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of whipple’s disease in a patient mistakenly on anti-tnf therapy. ACG Case Rep J. 2013;1(1):25- 28. Published 2013 Oct 8. doi:10.14309/crj.2013.11
19. Burton EM, Mercado-Deane MG, Patel K. Pneumatosis intestinalis in a child with AIDS and pseudomembranous colitis. Pediatr Radiol. 1994;24(8):609-610. doi:10.1007/BF02012750
20. Berk RN, Wall SD, McArdle CB, et al. Cryptosporidiosis of the stomach and small intestine in patients with AIDS. AJR Am J Roentgenol. 1984;143(3):549-554. doi:10.2214/ajr.143.3.549
21. Samson VE, Brown WR. Pneumatosis cystoides intestinalis in AIDS-associated cryptosporidiosis. More than an incidental finding? J Clin Gastroenterol. 1996;22(4):311-312.doi:10.1097/00004836-199606000-00015
22. Tjon A Tham RT, Vlasveld LT, Willemze R. Gastrointestinal complications of cytosine-arabinoside chemotherapy: findings on plain abdominal radiographs. AJR Am J Roentgenol. 1990;154(1):95-98. doi:10.2214/ajr.154.1.2104733
23. Hashimoto S, Saitoh H, Wada K, et al. Pneumatosis cystoides intestinalis after chemotherapy for hematological malignancies: report of 4 cases. Intern Med. 1995;34(3):212-215. doi:10.2169/internalmedicine.34.212
24. Gelman SF, Brandt LJ. Pneumatosis intestinalis and AIDS: a case report and review of the literature. Am J Gastroenterol. 1998;93(4):646-650. doi:10.1111/j.1572-0241.1998.183_b.x
25. Gillon J, Tadesse K, Logan RF, Holt S, Sircus W. Breath hydrogen in pneumatosis cystoides intestinalis. Gut. 1979;20(11):1008-1011. doi:10.1136/gut.20.11.1008
26. Hughes DT, Gordon KC, Swann JC, Bolt GL. Pneumatosis cystoides intestinalis. Gut. 1966;7(5):553-557. doi:10.1136/gut.7.5.553
27. Read NW, Al-Janabi MN, Cann PA. Is raised breath hydrogen related to the pathogenesis of pneumatosis coli? Gut. 1984;25(8):839-845. doi:10.1136/gut.25.8.839
28. van der Linden W, Marsell R. Pneumatosis cystoides coli associated with high H2 excretion. Treatment with an elemental diet. Scand J Gastroenterol. 1979;14(2):173-174. doi:10.3109/00365527909179864
29. Christl SU, Gibson GR, Murgatroyd PR, Scheppach W, Cummings JH. Impaired hydrogen metabolism in pneumatosis cystoides intestinalis. Gastroenterology. 1993;104(2):392-397. doi:10.1016/0016-5085(93)90406-3
30. Keyting WS, Mccarver RR, Kovarik JL, Daywitt AL. Pneumatosis intestinalis: a new concept. Radiology. 1961;76:733-741. doi:10.1148/76.5.733
31. Florin TH, Hills BA. Does counterperfusion supersaturation cause gas cysts in pneumatosis cystoides coli, and can breathing heliox reduce them? Lancet. 1995;345(8959):1220-1222. doi:10.1016/S0140-6736(95)91996-1
32. Grieve DA, Unsworth IP. Pneumatosis cystoides intestinalis: an experience with hyperbaric oxygen treatment. Aust N Z J Surg. 1991;61(6):423-426.
33. Micklefield GH, Kuntz HD, May B. Pneumatosis cystoides intestinalis: case reports and review of the literature. Mater Med Pol. 1990;22(2):70-72.
34. Yale CE, Balish E, Wu JP. The bacterial etiology of pneumatosis cystoides intestinalis. Arch Surg. 1974;109(1):89- 94. doi:10.1001/archsurg.1974.01360010067017
35. Fenton LZ, Buonomo C. Benign pneumatosis in children. Pediatr Radiol. 2000;30(11):786-793. doi:10.1007/s002470000303
36. Tobias R, Coleman S, Helman CA. Pneumatosis coli simulating hepatomegaly. Am J Gastroenterol. 1985;80(2):146-149.
37. Feczko PJ, Mezwa DG, Farah MC, White BD. Clinical significance of pneumatosis of the bowel w a l l . Radiographics. 1992;12(6):1069-1078. doi:10.1148/radiographics.12.6.1439012
38. Masterson JS, Fratkin LB, Osler TR, Trapp WG. Treatment of pneumatosis cystoides intestinalis with hyperbaric oxygen. Ann Surg. 1978;187(3):245-247. doi:10.1097/00000658-197803000-00005
39. Höflin F, Linden W van der. Pneumatosis cystoides intestinalis treated by oxygen breathing. Scandinavian J Gastroenterol . 1974;9(5) :427-430. doi:10.1080/00365521.1974.12096852
40. St Peter SD, Abbas MA, Kelly KA. The spectrum of pneumatosis intestinalis. Arch Surg. 2003;138(1):68-75. doi:10.1001/archsurg.138.1.68
41. Ling F, Guo D, Zhu L. Pneumatosis cystoides intestinalis: a case report and literature review. BMC Gastroenterol. 2019;19(1):176. Published 2019 Nov 6. doi:10.1186/s12876-019-1087-9
42. Morris MS, Gee AC, Cho SD, et al. Management and outcome of pneumatosis intestinalis. Am J Surg. 2008;195(5):679-682. doi:10.1016/j.amjsurg.2008.01.011
1. Jamart J. Pneumatosis cystoides intestinalis. A statistical study of 919 cases. Acta Hepatogastroenterol (Stuttg). 1979;26(5):419-422.
2. Lafortune M, Trinh BC, Burns PN, et al. Air in the portal vein: sonographic and Doppler manifestations. Radiology. 1991;180(3):667-670. doi:10.1148/radiology.180.3.1871276
3. Kriegshauser JS, Reading CC, King BF, Welch TJ. Combined systemic and portal venous gas: sonographic and CT detection in two cases. AJR Am J Roentgenol. 1990;154(6):1219-1221. doi:10.2214/ajr.154.6.2110731
4. Goske MJ, Goldblum JR, Applegate KE, Mitchell CS, Bardo D. The “circle sign”: a new sonographic sign of pneumatosis intestinalis - clinical, pathologic and experimental findings. Pediatr Radiol. 1999;29(7):530-535. doi:10.1007/s002470050638
5. Marshak RH, Lindner AE, Maklansky D. Pneumatosis cystoides coli. Gastrointest Radiol. 1977;2(2):85-89. doi:10.1007/BF02256475
6. Jensen R, Gutnik SH. Pneumatosis cystoides intestinalis: a complication of colonoscopic polypectomy. S D J Med. 1991;44(7):177-179.
7. Knechtle SJ, Davidoff AM, Rice RP. Pneumatosis intestinalis. Surgical management and clinical outcome. Ann Surg. 1990;212(2):160-165. doi:10.1097/00000658-199008000-00008
8. Koss LG. Abdominal gas cysts (Pneumatosis cystoides intestinorum hominis); an analysis with a report of a case and a critical review of the literature. AMA Arch Pathol. 1952;53(6):523-549.
9. Jona JZ. Benign pneumatosis intestinalis coli after blunt trauma to the abdomen in a child. J Pediatr Surg. 2000;35(7):1109-1111. doi:10.1053/jpsu.2000.7837
10. Gagliardi G, Thompson IW, Hershman MJ, Forbes A, Hawley PR, Talbot IC. Pneumatosis coli: a proposed pathogenesis based on study of 25 cases and review of the literature. Int J Colorectal Dis. 1996;11(3):111-118. doi:10.1007/s003840050031
11. Seto T, Koide N, Taniuchi N, Yamada T, Hamaguchi M, Goto S. Pneumatosis cystoides intestinalis complicating carcinoma of the small intestine. Am J Surg. 2001;182(3):287-288. doi:10.1016/S0002-9610(01)00710-3
12. Galandiuk S, Fazio VW, Petras RE. Pneumatosis cystoides intestinalis in Crohn’s disease. Report of two cases. Dis Colon Rectum. 1985;28(12):951-956. doi:10.1007/BF02554315
13. Parra JA, Acinas O, Bueno J, Madrazo C, Fariñas C. An unusual form of pneumatosis intestinalis associated with appendicitis. Br J Radiol. 1998;71(843):326-328. doi:10.1259/bjr.71.843.9616245
14. Schenk P, Madl C, Kramer L, et al. Pneumatosis intestinalis with Clostridium difficile colitis as a cause of acute abdomen after lung transplantation. Dig Dis Sci. 1998;43(11):2455-2458. doi:10.1023/a:1026682131847
15. Kreiss C, Forohar F, Smithline AE, Brandt LJ. Pneumatosis intestinalis complicating C. difficile pseudomembranous colitis. Am J Gastroenterol. 1999;94(9):2560-2561. doi:10.1111/j.1572-0241.1999.01397.x
16. Day DL, Ramsay NK, Letourneau JG. Pneumatosis intestinalis after bone marrow transplantation. AJR Am J Roentgenol. 1988;151(1):85-87. doi:10.2214/ajr.151.1.85
17. Tahara S, Sakai Y, Katsuno H, Urano M, Kuroda M, Tsukamoto T. Pneumatosis intestinalis and hepatic portal venous gas associated with gas-forming bacterial translocation due to postoperative paralytic ileus: A case report. Medicine (Baltimore). 2019;98(2):e14079. doi:10.1097/MD.0000000000014079
18. Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of whipple’s disease in a patient mistakenly on anti-tnf therapy. ACG Case Rep J. 2013;1(1):25- 28. Published 2013 Oct 8. doi:10.14309/crj.2013.11
19. Burton EM, Mercado-Deane MG, Patel K. Pneumatosis intestinalis in a child with AIDS and pseudomembranous colitis. Pediatr Radiol. 1994;24(8):609-610. doi:10.1007/BF02012750
20. Berk RN, Wall SD, McArdle CB, et al. Cryptosporidiosis of the stomach and small intestine in patients with AIDS. AJR Am J Roentgenol. 1984;143(3):549-554. doi:10.2214/ajr.143.3.549
21. Samson VE, Brown WR. Pneumatosis cystoides intestinalis in AIDS-associated cryptosporidiosis. More than an incidental finding? J Clin Gastroenterol. 1996;22(4):311-312.doi:10.1097/00004836-199606000-00015
22. Tjon A Tham RT, Vlasveld LT, Willemze R. Gastrointestinal complications of cytosine-arabinoside chemotherapy: findings on plain abdominal radiographs. AJR Am J Roentgenol. 1990;154(1):95-98. doi:10.2214/ajr.154.1.2104733
23. Hashimoto S, Saitoh H, Wada K, et al. Pneumatosis cystoides intestinalis after chemotherapy for hematological malignancies: report of 4 cases. Intern Med. 1995;34(3):212-215. doi:10.2169/internalmedicine.34.212
24. Gelman SF, Brandt LJ. Pneumatosis intestinalis and AIDS: a case report and review of the literature. Am J Gastroenterol. 1998;93(4):646-650. doi:10.1111/j.1572-0241.1998.183_b.x
25. Gillon J, Tadesse K, Logan RF, Holt S, Sircus W. Breath hydrogen in pneumatosis cystoides intestinalis. Gut. 1979;20(11):1008-1011. doi:10.1136/gut.20.11.1008
26. Hughes DT, Gordon KC, Swann JC, Bolt GL. Pneumatosis cystoides intestinalis. Gut. 1966;7(5):553-557. doi:10.1136/gut.7.5.553
27. Read NW, Al-Janabi MN, Cann PA. Is raised breath hydrogen related to the pathogenesis of pneumatosis coli? Gut. 1984;25(8):839-845. doi:10.1136/gut.25.8.839
28. van der Linden W, Marsell R. Pneumatosis cystoides coli associated with high H2 excretion. Treatment with an elemental diet. Scand J Gastroenterol. 1979;14(2):173-174. doi:10.3109/00365527909179864
29. Christl SU, Gibson GR, Murgatroyd PR, Scheppach W, Cummings JH. Impaired hydrogen metabolism in pneumatosis cystoides intestinalis. Gastroenterology. 1993;104(2):392-397. doi:10.1016/0016-5085(93)90406-3
30. Keyting WS, Mccarver RR, Kovarik JL, Daywitt AL. Pneumatosis intestinalis: a new concept. Radiology. 1961;76:733-741. doi:10.1148/76.5.733
31. Florin TH, Hills BA. Does counterperfusion supersaturation cause gas cysts in pneumatosis cystoides coli, and can breathing heliox reduce them? Lancet. 1995;345(8959):1220-1222. doi:10.1016/S0140-6736(95)91996-1
32. Grieve DA, Unsworth IP. Pneumatosis cystoides intestinalis: an experience with hyperbaric oxygen treatment. Aust N Z J Surg. 1991;61(6):423-426.
33. Micklefield GH, Kuntz HD, May B. Pneumatosis cystoides intestinalis: case reports and review of the literature. Mater Med Pol. 1990;22(2):70-72.
34. Yale CE, Balish E, Wu JP. The bacterial etiology of pneumatosis cystoides intestinalis. Arch Surg. 1974;109(1):89- 94. doi:10.1001/archsurg.1974.01360010067017
35. Fenton LZ, Buonomo C. Benign pneumatosis in children. Pediatr Radiol. 2000;30(11):786-793. doi:10.1007/s002470000303
36. Tobias R, Coleman S, Helman CA. Pneumatosis coli simulating hepatomegaly. Am J Gastroenterol. 1985;80(2):146-149.
37. Feczko PJ, Mezwa DG, Farah MC, White BD. Clinical significance of pneumatosis of the bowel w a l l . Radiographics. 1992;12(6):1069-1078. doi:10.1148/radiographics.12.6.1439012
38. Masterson JS, Fratkin LB, Osler TR, Trapp WG. Treatment of pneumatosis cystoides intestinalis with hyperbaric oxygen. Ann Surg. 1978;187(3):245-247. doi:10.1097/00000658-197803000-00005
39. Höflin F, Linden W van der. Pneumatosis cystoides intestinalis treated by oxygen breathing. Scandinavian J Gastroenterol . 1974;9(5) :427-430. doi:10.1080/00365521.1974.12096852
40. St Peter SD, Abbas MA, Kelly KA. The spectrum of pneumatosis intestinalis. Arch Surg. 2003;138(1):68-75. doi:10.1001/archsurg.138.1.68
41. Ling F, Guo D, Zhu L. Pneumatosis cystoides intestinalis: a case report and literature review. BMC Gastroenterol. 2019;19(1):176. Published 2019 Nov 6. doi:10.1186/s12876-019-1087-9
42. Morris MS, Gee AC, Cho SD, et al. Management and outcome of pneumatosis intestinalis. Am J Surg. 2008;195(5):679-682. doi:10.1016/j.amjsurg.2008.01.011
The Natural History of a Patient With COVID-19 Pneumonia and Silent Hypoxemia
In less than a year, COVID-19 has infected nearly 100 million people worldwide and caused more than 2 million deaths and counting. Although the infection fatality rate is estimated to be 1% and the case fatality rate between 2% and 3%, COVID-19 has had a disproportionate effect on the older population and those with comorbidities. Some of these findings are mirrored in the US Department of Veterans Affairs (VA) population, which has seen a higher case fatality rate.1-4
As a respiratory tract infection, the most dreaded presentation is severe pneumonia with acute hypoxemia, which may rapidly deteriorate to acute respiratory distress syndrome (ARDS) and respiratory failure.5-7 This possibility has led to early intubation strategies aimed at preempting this rapid deterioration and minimizing viral exposure to health care workers. Intubation rates have varied widely with extremes of 6 to 88%.8,9
However, this early intubation strategy has waned as some of the rationale behind its endorsement has been called into question. Early intubation bypasses alternatives to intubation; high-flow nasal cannula oxygen, noninvasive ventilation, and awake proning are all effective maneuvers in the appropriate patient.10,11 The use of first-line high-flow nasal cannula oxygen and noninvasive ventilation has been widely reported. Reports of first-line use of high-flow nasal cannula oxygen has not demonstrated inferior outcomes, nor has the timing of intubation, suggesting a significant portion of patients could benefit from a trial of therapy and eventually avoid intubation.11-14 Other therapies, such as systemic corticosteroids, confer a mortality benefit in those patients with COVID-19 who require oxygen or mechanical ventilation, but their impact on the progression of respiratory failure and need for intubation are undetermined.
There also are reports of patients who report no signs of respiratory distress or dyspnea with their COVID-19 pneumonia despite profound hypoxemia or high oxygen requirements. Various terms, including silent hypoxemia or happy hypoxia, are descriptive of the demeanor of these patients, and treatment has invariably included oxygen.15,16 Nevertheless, low oxygen measurements have generally prompted higher levels of supplemental oxygen or more invasive therapies.
Treatment rendered may obscure the trajectory of response, which is important to understand to better position options for invasive therapies and other therapeutics. We recently encountered a patient with a course of illness that represented the natural history of COVID-19 pneumonia with low oxygen levels (referred to as hypoxemia for consistency) that highlighted several issues of management.
Case Presentation
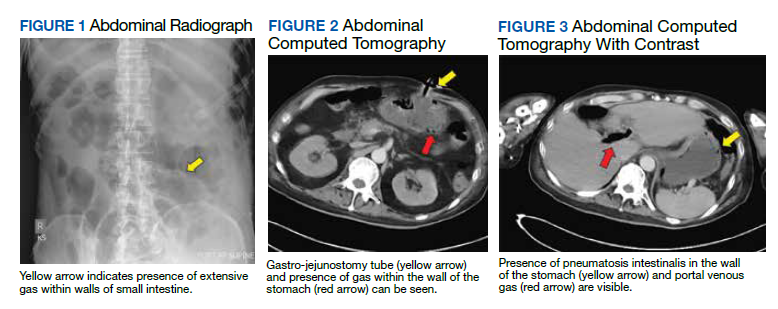
A 62-year-old undomiciled woman with morbid obesity, prediabetes mellitus, long-standing schizophrenia, and bipolar disorder presented to our facility for evaluation of dry cough and need for tuberculosis clearance for admittance to a shelter. She appeared comfortable and was afebrile with blood pressure 111/74 mm Hg, heart rate 82 beats per minute. Her respiratory rate was 18 breaths per minute, but the pulse oximetry showed oxygen saturation of 70 to 75% on room air at rest. A chest X-ray showed bibasilar infiltrates (Figure 1), and a rapid COVID-19 nasopharyngeal polymerase chain reaction (PCR) test returned positive, confirmed by a second PCR test. Baseline inflammatory markers were elevated (Figure 2). In addition, the serum interleukin-6 also was elevated to 66.1 pg/mL (normal < 5.0), erythrocyte sedimentation rate elevated to 69 mm/h, but serum procalcitonin was essentially normal (0.22 ng/mL; normal < 20 ng/mL) as was the serum lactate (1.4 mmol/L).
The patient was admitted to the intensive care unit (ICU) for close monitoring in anticipation of the possibility of decompensation based on her age, hypoxia, and elevated inflammatory markers.17 Besides a subsequent low-grade fever (100.4 oF) and lymphopenia (manual count 550/uL), she remained clinically unchanged. Throughout her hospitalization, she maintained a persistent psychotic delusion that she did not have COVID-19, refusing all medical interventions, including a peripheral IV line and supplemental oxygen for the entire duration. Extensive efforts to identify family or a surrogate decision maker were unsuccessful. After consultation with Psychiatry, Bio-Ethics, and hospital leadership, the patient was deemed to lack decision-making capacity regarding treatment or disposition and was placed on a psychiatric hold. However, since any interventions against her will would require sedation, IV access, and potentially increase the risk of nosocomial COVID-19 transmission, she was allowed to remain untreated and was closely monitored for symptoms of worsening respiratory failure.
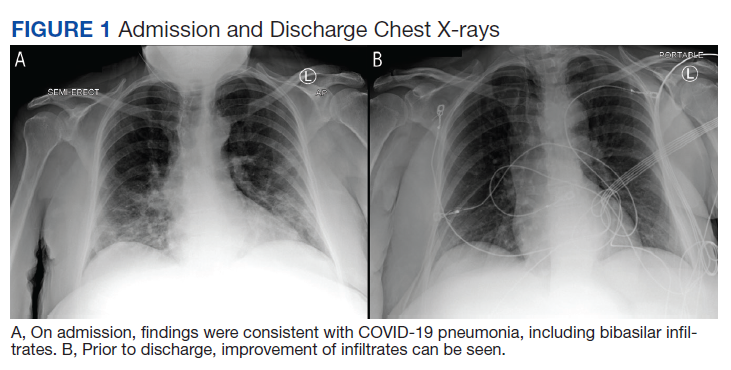
Over the next 2 weeks, her hypoxemia, inflammatory markers, and the infiltrates on imaging resolved (Figure 2). The lowest daily awake room air pulse oximetry readings are reported, initially with consistent readings in the low 80% range, but on day 12, readings were > 90% and remained > 90% for the remainder of her hospitalization. Therefore, shortly after hospital day 12, she was clinically stable for discharge from acute care to a subacute facility, but this required documentation of the clearance of her viral infection. She refused to undergo a subsequent nasopharyngeal swab but allowed an oropharyngeal COVID-19 PCR swab, which was negative. She remained stable and unchanged for the remainder of her hospitalization, awaiting identification of a receiving facility and was able to be discharged to transitional housing on day 38.
Discussion
The initial reports of COVID-19 pneumonia focused on ARDS and respiratory failure requiring mechanical ventilation with less emphasis on those with lower severity of illness. This was heightened by health care systems that were overwhelmed with large number of patients while faced with limited supplies and equipment. Given the risk to patients and providers of crash intubations, some recommended early intubation strategies.3 However, the natural history of COVID-19 pneumonia and the threshold for intubation of these patients remain poorly defined despite the creation of prognostic tools.17 This patient’s persistent hypoxemia and elevated inflammatory markers certainly met markers of disease associated with a high risk of progression.
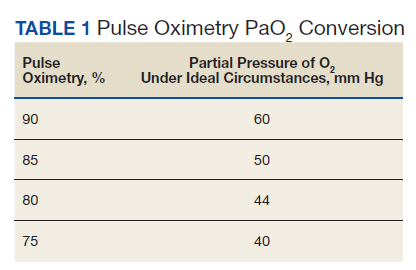
The greatest concern would have been her level of hypoxemia. Acceptable thresholds of hypoxemia vary, but general consensus would classify pulse oximetry < 90% as hypoxemia and a threshold for administering supplemental oxygen. It is important to recognize how pulse oximetry readings translate to partial pressure of oxygen (PaO2) measurements (Table 1). Pulse oximetry readings of 90% corresponds to a PaO2 readings of 60 mm Hg in ideal conditions without the influence of acidosis, PaCO2, or temperature. While lower readings are of concern, these do not represent absolute indications for assisted ventilatory support as lower levels are well tolerated in a variety of conditions. A common example are patients with chronic obstructive pulmonary disease. Long-term mortality benefits of continuous supplemental oxygen are well established in specific populations, but the threshold for correction in the acute setting remains a case-by-case decision. This decision is complex and is based on more than an absolute number or the amount of oxygen required to achieve a threshold level of oxygenation.
The PaO2/FIO2 (fraction of inspired oxygen) is a common measure used to address severity of disease and oxygen requirements. It also has been used to define the severity of ARDS, but the ratio is based on intubated and mechanically ventilated patients and may not translate well to those not on assisted ventilation. Treatment with supplemental oxygen also involves entrained air with associated imprecision in oxygen delivery.18 For this discussion, the patient’s admission PaO2/FIO2 on room air would have been between 190 and 260. Coupled with the bilateral infiltrates on imaging, there was justified concern for progression to severe ARDS. Her presentation would have met most of the epidemiologic criteria used in initial case finding for severe COVID-19 cases, including a blood oxygen saturation ≤ 93%, PaO2/FIO2 < 300 with infiltrates involving close to if not exceeding 50% of the lung.
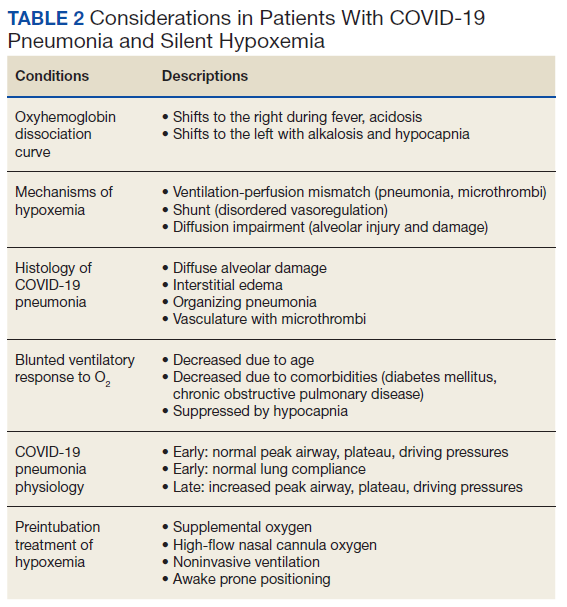
With COVID-19 pneumonia, the pathologic injury to the alveoli resembles that of any viral pneumonia with recruitment of predominantly lymphocytic inflammatory cells that fill the alveoli, derangements in ventilation/perfusion mismatch as the core mechanism of hypoxemia with interstitial edema and shuntlike physiology developing at the extremes of involvement. In later stages, the histologic appearance is similar to ARDS, including hyaline membrane formation and thickened alveolar septa with perivascular lymphocytic-plasmocytic infiltration. In addition, there also are findings of organizing pneumonia with fibroblastic proliferation, thrombosis, and diffuse alveolar damage, a constellation of findings similar to that seen in the latter stages of ARDS.2
Although these histologic findings resemble ARDS, many patients with respiratory failure due to COVID-19 have a different physiologic profile compared with those with typical ARDS, with the most striking finding of lungs with low elastance or high compliance. From the critical care standpoint, this meant that the lungs were relatively easy to ventilate with lower peak airway and plateau pressures and low driving pressures. This condition suggested that there was relatively less lung that could be recruited with positive end expiratory pressure; therefore, a somewhat different entity from that associated with ARDS.19 These findings were often noted early in the course of respiratory failure, and although there is debate about whether this represents a different phenotype or timepoint in the spectrum of disease, it clearly represents a subset that is distinct from that which had been previously encountered.
On the other hand, the clinical features seen in those patients with COVID-19 pneumonia who progressed to advanced respiratory failure were essentially indistinguishable from those patients with traditional ARDS. Other explanations for this respiratory failure have included a disrupted vasoregulatory response to hypoxemia with failed hypoxic vasoconstriction, intravascular microthrombi, and impaired diffusion, all contributing to impaired gas exchange and hypoxemia.19-21 This can lead to shuntlike conditions that neither respond well to supplemental oxygen nor manifest the type of physiologic response seen with other causes of hypoxemia.
The severity of hypoxemia manifested by this patient may have elicited additional findings of respiratory distress, such as dyspnea and tachypnea. However, in patients with severe COVID-19 pneumonia, dyspnea was not a universal finding, reported in the 20 to 60% range of cohorts, higher in those with ARDS and mechanical ventilation, although some report near universal dyspnea in their series.1,4,8,22,23 Tachypnea is another symptom of interest. Using a threshold of > 24 breaths/min, tachypnea was noted in 16 to 29% of patients with a much greater proportion (63%) in nonsurvivors.6,24 Several explanations have been proposed for the discordance between the presence and severity of hypoxemia and lack of symptoms of dyspnea and tachypnea. It is important to recognize that misclassification of the severity of hypoxemia can occur due to technical issues and potential errors involving pulse oximetry measurement and shifts in the oxyhemoglobin dissociation curve. However, this is more pertinent for those with mild disease as the severity of hypoxemia in severe pneumonia is beyond what can be attributed to technical issues.
More important, the ventilatory response curve to hypoxemia may not be normal for some patients, blunted by as much as 50% in older patients, especially in those with diabetes mellitus.7,25,26 In addition, the ventilatory response varies widely even among normal individuals. This would translate to lower levels of minute ventilation (less tachypnea or respiratory effort) with hypoxemia. Hypocapnic hypoxemia also blunts the ventilatory response to hypoxemia. Subjects do not increase their minute ventilation if the PaCO2 remains low despite oxygen desaturation to < 70%, especially if PaCO2 < 30 mm Hg or alternatively, increases in minute ventilation are not seen until the PaCO2 exceeds 39 mm Hg.27 Both scenarios occur in those with COVID-19 pneumonia and provide another explanation for the absence of respiratory symptoms or signs of respiratory distress in some patients.
The observation of more compliant lungs may help in the understanding of the variable presentation of these patients. Compliant lungs do not require the increased pressure needed to achieve a specific tidal volume that, in turn, may increase the work of breathing. This may add to the explanation of seemingly paradoxical silent hypoxemia in those patients where the combination of a blunted ventilatory response, hypocapnia, shunt physiology, and normal respiratory system compliance is represented by the absence of increased breathing effort despite severe hypoxemia.
If not for the patient’s refusal of medical services, this patient quite possibly would have been intubated due to hypoxemia and health care providers’ concern for her risk of deterioration. Reported intubation and mechanical ventilation rates have varied widely from extremes of from < 5 to 88% in severely ill patients.9,22 About 75% will need oxygen, but many can be treated and recover without the need for intubation and mechanical ventilation.
As previously mentioned, options for treatment include standard and high-flow oxygen delivery, noninvasive ventilation, and awake prone ventilation. Their role in patient management has been recently outlined, and instead of an early intubation strategy, represents gradual escalation of support that may be sufficient to treat hypoxemia and avoid the need for intubation and mechanical ventilation (Table 2).
In addition, the patient’s hospital course was notable for the decline in known markers of active inflammation that mirrored the resolution of her hypoxemia and pneumonia. This included elevated lactate dehydrogenase, D-dimer, ferritin, and C-reactive protein with all but the latter rising and decreasing over 2 weeks. These findings provide additional information of the time for recovery and supports the use of these markers to monitor the course of pneumonia.
The patient declined all intervention, including oxygen, and recovered to her presumed prehospitalization condition. This experiment of nature due to unique circumstances may shed light on the natural time course of untreated hypoxemic COVID-19 pneumonia that has not previously been well appreciated. It is important to recognize that recovery occurred over 2 weeks. This is close to the observed and expected time for recovery that has been reported for those with severe COVID-19 pneumonia.
Conclusions
Since the emergence of the COVID-19, evidence has accumulated for the benefit of several adjunctive therapies in the treatment of this type of pneumonia, with corticosteroids providing a mortality benefit. Although unknown whether this patient’s experience can be generalized to others or whether it represents her unique response, this case provides another perspective for comparison of treatments and reinforces the need for prospective, randomized clinical trials to establish treatment efficacy. The exact nature of silent hypoxemia of COVID-19 remains incompletely understood; however, this case highlights the importance of treating the individual instead of clinical markers and provides a time course for recovery from pneumonia and severe hypoxemia that occurs without oxygen or any other treatment over about 2 weeks.
1. Ioannou GN, Locke E, Green P, et al. Risk factors for hospitalization, mechanical ventilation, or death among 10131 US veterans with SARS-CoV-2 infection. JAMA Netw Open. 2020;3(9):e2022310. doi:10.1001/jamanetworkopen.2020.22310
2. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA. 2020;324(8):782-793. doi:10.1001/jama.2020.12839
3. Alhazzani W, Moller MH, Arabi YM, et al. Surviving sepsis campaign: guidelines on the management of critically ill adults with coronavirus disease 2019 (COVID-19). Crit Care Med. 2020;48(6):e440-e469. doi:10.1097/CCM.0000000000004363
4. Ziehr DR, Alladina J, Petri CR, et al. Respiratory pathophysiology of mechanically ventilated patients with COVID-19: a cohort study. Am J Respir Crit Care Med. 2020;201(12):1560-1564. doi:10.1164/rccm.202004-1163LE
5. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239-1242. doi:10.1001/jama.2020.2648
6. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054-1062. doi:10.1016/S01406736(20)30566-3
7. Tobin MJ, Laghi F, Jubran A. Why COVID-19 silent hypoxemia is baffling to physicians. Am J Respir Crit Care Med. 2020;202(3):356-360. doi:10.1164/rccm.202006-2157CP
8. Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708-1720. doi:10.1056/NEJMoa2002032
9. Grasselli G, Zangrillo A, Zanella A, et al. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy Region, Italy. JAMA. 2020;323(16):1574-1581. doi:10.1001/jama.2020.5394
10. Raoof S, Nava S, Carpati C, Hill NS. High-flow, noninvasive ventilation and awake (nonintubation) proning in patients with coronavirus disease 2019 with respiratory failure. Chest. 2020;158(5):1992-2002. doi:10.1016/j.chest.2020.07.013
11. Ackermann M, Mentzer SJ, Jonigk D. Pulmonary vascular pathology in COVID-19. Reply. N Engl J Med. 2020;383(9):888-889. doi:10.1056/NEJMc2022068
12. McDonough G, Khaing P, Treacy T, McGrath C, Yoo EJ. The use of high-flow nasal oxygen in the ICU as a first-line therapy for acute hypoxemic respiratory failure secondary to coronavirus disease 2019. Crit Care Explor. 2020;2(10):e0257. doi:10.1097/CCE.0000000000000257
13. Hernandez-Romieu AC, Adelman MW, et al. Timing of intubation and mortality among critically ill coronavirus disease 2019 patients: a single-center cohort study. Crit Care Med. 2020;48(11):e1045-e1053. doi:10.1097/CCM.0000000000004600
14. Cummings MJ, Baldwin MR, Abrams D, et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study. Lancet. 2020;395(10239):1763-1770. doi:10.1016/S0140-6736(20)31189-2
15. Dhont S, Derom E, Van Braeckel E, Depuydt P, Lambrecht BN. The pathophysiology of ‘happy’ hypoxemia in COVID-19. Respir Res. 2020;21(1):198. doi:10.1186/s12931-020-01462-5
16. Wilkerson RG, Adler JD, Shah NG, Brown R. Silent hypoxia: a harbinger of clinical deterioration in patients with COVID-19. Am J Emerg Med. 2020;38(10):2243.e5-2243.e6. doi:10.1016/j.ajem.2020.05.044
17. Gong J, Ou J, Qiu X, et al. A tool for early prediction of severe coronavirus disease 2019 (COVID-19): a multicenter study using the risk nomogram in Wuhan and Guangdong, China. Clin Infect Dis. 2020;71(15):833-840. doi:10.1093/cid/ciaa443
18. Force ADT, Ranieri VM, Rubenfeld GD, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526-2533. doi:10.1001/jama.2012.5669
19. Marini JJ, Gattinoni L. Management of COVID-19 respiratory distress. JAMA. 2020;323(22):2329-2330. doi:10.1001/jama.2020.6825
20. Schaller T, Hirschbuhl K, Burkhardt K, et al. Postmortem examination of patients with COVID-19. JAMA. 2020;323(24):2518-2520. doi:10.1001/jama.2020.8907
21. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383(2):120-128. doi:10.1056/NEJMoa2015432
22. Wu C, Chen X, Cai Y, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934-943. doi:10.1001/jamainternmed.2020.0994. Published correction appeared May 11, 2020. Errors in data and units of measure. doi:10.1001/jamainternmed.2020.1429
23. Yang J, Zheng Y, Gou X, et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int J Infect Dis. 2020;94:91-95. doi:10.1016/j.ijid.2020.03.017
24. Richardson S, Hirsch JS, Narasimhan M, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA. 2020;323(20):2052-2059. doi:10.1001/jama.2020.6775
25. Tobin MJ, Jubran A, Laghi F. Misconceptions of pathophysiology of happy hypoxemia and implications for management of COVID-19. Respir Res. 2020;21(1):249. doi:10.1186/s12931-020-01520-y
26. Bickler PE, Feiner JR, Lipnick MS, McKleroy W. “Silent” presentation of hypoxemia and cardiorespiratory compensation in COVID-19. Anesthesiology. 2020;134(2):262-269. doi:10.1097/ALN.0000000000003578
27. Jounieaux V, Parreira VF, Aubert G, Dury M, Delguste P, Rodenstein DO. Effects of hypocapnic hyperventilation on the response to hypoxia in normal subjects receiving intermittent positive-pressure ventilation. Chest. 2002;121(4):1141-1148. doi:10.1378/chest.121.4.1141
In less than a year, COVID-19 has infected nearly 100 million people worldwide and caused more than 2 million deaths and counting. Although the infection fatality rate is estimated to be 1% and the case fatality rate between 2% and 3%, COVID-19 has had a disproportionate effect on the older population and those with comorbidities. Some of these findings are mirrored in the US Department of Veterans Affairs (VA) population, which has seen a higher case fatality rate.1-4
As a respiratory tract infection, the most dreaded presentation is severe pneumonia with acute hypoxemia, which may rapidly deteriorate to acute respiratory distress syndrome (ARDS) and respiratory failure.5-7 This possibility has led to early intubation strategies aimed at preempting this rapid deterioration and minimizing viral exposure to health care workers. Intubation rates have varied widely with extremes of 6 to 88%.8,9
However, this early intubation strategy has waned as some of the rationale behind its endorsement has been called into question. Early intubation bypasses alternatives to intubation; high-flow nasal cannula oxygen, noninvasive ventilation, and awake proning are all effective maneuvers in the appropriate patient.10,11 The use of first-line high-flow nasal cannula oxygen and noninvasive ventilation has been widely reported. Reports of first-line use of high-flow nasal cannula oxygen has not demonstrated inferior outcomes, nor has the timing of intubation, suggesting a significant portion of patients could benefit from a trial of therapy and eventually avoid intubation.11-14 Other therapies, such as systemic corticosteroids, confer a mortality benefit in those patients with COVID-19 who require oxygen or mechanical ventilation, but their impact on the progression of respiratory failure and need for intubation are undetermined.
There also are reports of patients who report no signs of respiratory distress or dyspnea with their COVID-19 pneumonia despite profound hypoxemia or high oxygen requirements. Various terms, including silent hypoxemia or happy hypoxia, are descriptive of the demeanor of these patients, and treatment has invariably included oxygen.15,16 Nevertheless, low oxygen measurements have generally prompted higher levels of supplemental oxygen or more invasive therapies.
Treatment rendered may obscure the trajectory of response, which is important to understand to better position options for invasive therapies and other therapeutics. We recently encountered a patient with a course of illness that represented the natural history of COVID-19 pneumonia with low oxygen levels (referred to as hypoxemia for consistency) that highlighted several issues of management.
Case Presentation
A 62-year-old undomiciled woman with morbid obesity, prediabetes mellitus, long-standing schizophrenia, and bipolar disorder presented to our facility for evaluation of dry cough and need for tuberculosis clearance for admittance to a shelter. She appeared comfortable and was afebrile with blood pressure 111/74 mm Hg, heart rate 82 beats per minute. Her respiratory rate was 18 breaths per minute, but the pulse oximetry showed oxygen saturation of 70 to 75% on room air at rest. A chest X-ray showed bibasilar infiltrates (Figure 1), and a rapid COVID-19 nasopharyngeal polymerase chain reaction (PCR) test returned positive, confirmed by a second PCR test. Baseline inflammatory markers were elevated (Figure 2). In addition, the serum interleukin-6 also was elevated to 66.1 pg/mL (normal < 5.0), erythrocyte sedimentation rate elevated to 69 mm/h, but serum procalcitonin was essentially normal (0.22 ng/mL; normal < 20 ng/mL) as was the serum lactate (1.4 mmol/L).
The patient was admitted to the intensive care unit (ICU) for close monitoring in anticipation of the possibility of decompensation based on her age, hypoxia, and elevated inflammatory markers.17 Besides a subsequent low-grade fever (100.4 oF) and lymphopenia (manual count 550/uL), she remained clinically unchanged. Throughout her hospitalization, she maintained a persistent psychotic delusion that she did not have COVID-19, refusing all medical interventions, including a peripheral IV line and supplemental oxygen for the entire duration. Extensive efforts to identify family or a surrogate decision maker were unsuccessful. After consultation with Psychiatry, Bio-Ethics, and hospital leadership, the patient was deemed to lack decision-making capacity regarding treatment or disposition and was placed on a psychiatric hold. However, since any interventions against her will would require sedation, IV access, and potentially increase the risk of nosocomial COVID-19 transmission, she was allowed to remain untreated and was closely monitored for symptoms of worsening respiratory failure.
Over the next 2 weeks, her hypoxemia, inflammatory markers, and the infiltrates on imaging resolved (Figure 2). The lowest daily awake room air pulse oximetry readings are reported, initially with consistent readings in the low 80% range, but on day 12, readings were > 90% and remained > 90% for the remainder of her hospitalization. Therefore, shortly after hospital day 12, she was clinically stable for discharge from acute care to a subacute facility, but this required documentation of the clearance of her viral infection. She refused to undergo a subsequent nasopharyngeal swab but allowed an oropharyngeal COVID-19 PCR swab, which was negative. She remained stable and unchanged for the remainder of her hospitalization, awaiting identification of a receiving facility and was able to be discharged to transitional housing on day 38.
Discussion
The initial reports of COVID-19 pneumonia focused on ARDS and respiratory failure requiring mechanical ventilation with less emphasis on those with lower severity of illness. This was heightened by health care systems that were overwhelmed with large number of patients while faced with limited supplies and equipment. Given the risk to patients and providers of crash intubations, some recommended early intubation strategies.3 However, the natural history of COVID-19 pneumonia and the threshold for intubation of these patients remain poorly defined despite the creation of prognostic tools.17 This patient’s persistent hypoxemia and elevated inflammatory markers certainly met markers of disease associated with a high risk of progression.
The greatest concern would have been her level of hypoxemia. Acceptable thresholds of hypoxemia vary, but general consensus would classify pulse oximetry < 90% as hypoxemia and a threshold for administering supplemental oxygen. It is important to recognize how pulse oximetry readings translate to partial pressure of oxygen (PaO2) measurements (Table 1). Pulse oximetry readings of 90% corresponds to a PaO2 readings of 60 mm Hg in ideal conditions without the influence of acidosis, PaCO2, or temperature. While lower readings are of concern, these do not represent absolute indications for assisted ventilatory support as lower levels are well tolerated in a variety of conditions. A common example are patients with chronic obstructive pulmonary disease. Long-term mortality benefits of continuous supplemental oxygen are well established in specific populations, but the threshold for correction in the acute setting remains a case-by-case decision. This decision is complex and is based on more than an absolute number or the amount of oxygen required to achieve a threshold level of oxygenation.
The PaO2/FIO2 (fraction of inspired oxygen) is a common measure used to address severity of disease and oxygen requirements. It also has been used to define the severity of ARDS, but the ratio is based on intubated and mechanically ventilated patients and may not translate well to those not on assisted ventilation. Treatment with supplemental oxygen also involves entrained air with associated imprecision in oxygen delivery.18 For this discussion, the patient’s admission PaO2/FIO2 on room air would have been between 190 and 260. Coupled with the bilateral infiltrates on imaging, there was justified concern for progression to severe ARDS. Her presentation would have met most of the epidemiologic criteria used in initial case finding for severe COVID-19 cases, including a blood oxygen saturation ≤ 93%, PaO2/FIO2 < 300 with infiltrates involving close to if not exceeding 50% of the lung.
With COVID-19 pneumonia, the pathologic injury to the alveoli resembles that of any viral pneumonia with recruitment of predominantly lymphocytic inflammatory cells that fill the alveoli, derangements in ventilation/perfusion mismatch as the core mechanism of hypoxemia with interstitial edema and shuntlike physiology developing at the extremes of involvement. In later stages, the histologic appearance is similar to ARDS, including hyaline membrane formation and thickened alveolar septa with perivascular lymphocytic-plasmocytic infiltration. In addition, there also are findings of organizing pneumonia with fibroblastic proliferation, thrombosis, and diffuse alveolar damage, a constellation of findings similar to that seen in the latter stages of ARDS.2
Although these histologic findings resemble ARDS, many patients with respiratory failure due to COVID-19 have a different physiologic profile compared with those with typical ARDS, with the most striking finding of lungs with low elastance or high compliance. From the critical care standpoint, this meant that the lungs were relatively easy to ventilate with lower peak airway and plateau pressures and low driving pressures. This condition suggested that there was relatively less lung that could be recruited with positive end expiratory pressure; therefore, a somewhat different entity from that associated with ARDS.19 These findings were often noted early in the course of respiratory failure, and although there is debate about whether this represents a different phenotype or timepoint in the spectrum of disease, it clearly represents a subset that is distinct from that which had been previously encountered.
On the other hand, the clinical features seen in those patients with COVID-19 pneumonia who progressed to advanced respiratory failure were essentially indistinguishable from those patients with traditional ARDS. Other explanations for this respiratory failure have included a disrupted vasoregulatory response to hypoxemia with failed hypoxic vasoconstriction, intravascular microthrombi, and impaired diffusion, all contributing to impaired gas exchange and hypoxemia.19-21 This can lead to shuntlike conditions that neither respond well to supplemental oxygen nor manifest the type of physiologic response seen with other causes of hypoxemia.
The severity of hypoxemia manifested by this patient may have elicited additional findings of respiratory distress, such as dyspnea and tachypnea. However, in patients with severe COVID-19 pneumonia, dyspnea was not a universal finding, reported in the 20 to 60% range of cohorts, higher in those with ARDS and mechanical ventilation, although some report near universal dyspnea in their series.1,4,8,22,23 Tachypnea is another symptom of interest. Using a threshold of > 24 breaths/min, tachypnea was noted in 16 to 29% of patients with a much greater proportion (63%) in nonsurvivors.6,24 Several explanations have been proposed for the discordance between the presence and severity of hypoxemia and lack of symptoms of dyspnea and tachypnea. It is important to recognize that misclassification of the severity of hypoxemia can occur due to technical issues and potential errors involving pulse oximetry measurement and shifts in the oxyhemoglobin dissociation curve. However, this is more pertinent for those with mild disease as the severity of hypoxemia in severe pneumonia is beyond what can be attributed to technical issues.
More important, the ventilatory response curve to hypoxemia may not be normal for some patients, blunted by as much as 50% in older patients, especially in those with diabetes mellitus.7,25,26 In addition, the ventilatory response varies widely even among normal individuals. This would translate to lower levels of minute ventilation (less tachypnea or respiratory effort) with hypoxemia. Hypocapnic hypoxemia also blunts the ventilatory response to hypoxemia. Subjects do not increase their minute ventilation if the PaCO2 remains low despite oxygen desaturation to < 70%, especially if PaCO2 < 30 mm Hg or alternatively, increases in minute ventilation are not seen until the PaCO2 exceeds 39 mm Hg.27 Both scenarios occur in those with COVID-19 pneumonia and provide another explanation for the absence of respiratory symptoms or signs of respiratory distress in some patients.
The observation of more compliant lungs may help in the understanding of the variable presentation of these patients. Compliant lungs do not require the increased pressure needed to achieve a specific tidal volume that, in turn, may increase the work of breathing. This may add to the explanation of seemingly paradoxical silent hypoxemia in those patients where the combination of a blunted ventilatory response, hypocapnia, shunt physiology, and normal respiratory system compliance is represented by the absence of increased breathing effort despite severe hypoxemia.
If not for the patient’s refusal of medical services, this patient quite possibly would have been intubated due to hypoxemia and health care providers’ concern for her risk of deterioration. Reported intubation and mechanical ventilation rates have varied widely from extremes of from < 5 to 88% in severely ill patients.9,22 About 75% will need oxygen, but many can be treated and recover without the need for intubation and mechanical ventilation.
As previously mentioned, options for treatment include standard and high-flow oxygen delivery, noninvasive ventilation, and awake prone ventilation. Their role in patient management has been recently outlined, and instead of an early intubation strategy, represents gradual escalation of support that may be sufficient to treat hypoxemia and avoid the need for intubation and mechanical ventilation (Table 2).
In addition, the patient’s hospital course was notable for the decline in known markers of active inflammation that mirrored the resolution of her hypoxemia and pneumonia. This included elevated lactate dehydrogenase, D-dimer, ferritin, and C-reactive protein with all but the latter rising and decreasing over 2 weeks. These findings provide additional information of the time for recovery and supports the use of these markers to monitor the course of pneumonia.
The patient declined all intervention, including oxygen, and recovered to her presumed prehospitalization condition. This experiment of nature due to unique circumstances may shed light on the natural time course of untreated hypoxemic COVID-19 pneumonia that has not previously been well appreciated. It is important to recognize that recovery occurred over 2 weeks. This is close to the observed and expected time for recovery that has been reported for those with severe COVID-19 pneumonia.
Conclusions
Since the emergence of the COVID-19, evidence has accumulated for the benefit of several adjunctive therapies in the treatment of this type of pneumonia, with corticosteroids providing a mortality benefit. Although unknown whether this patient’s experience can be generalized to others or whether it represents her unique response, this case provides another perspective for comparison of treatments and reinforces the need for prospective, randomized clinical trials to establish treatment efficacy. The exact nature of silent hypoxemia of COVID-19 remains incompletely understood; however, this case highlights the importance of treating the individual instead of clinical markers and provides a time course for recovery from pneumonia and severe hypoxemia that occurs without oxygen or any other treatment over about 2 weeks.
In less than a year, COVID-19 has infected nearly 100 million people worldwide and caused more than 2 million deaths and counting. Although the infection fatality rate is estimated to be 1% and the case fatality rate between 2% and 3%, COVID-19 has had a disproportionate effect on the older population and those with comorbidities. Some of these findings are mirrored in the US Department of Veterans Affairs (VA) population, which has seen a higher case fatality rate.1-4
As a respiratory tract infection, the most dreaded presentation is severe pneumonia with acute hypoxemia, which may rapidly deteriorate to acute respiratory distress syndrome (ARDS) and respiratory failure.5-7 This possibility has led to early intubation strategies aimed at preempting this rapid deterioration and minimizing viral exposure to health care workers. Intubation rates have varied widely with extremes of 6 to 88%.8,9
However, this early intubation strategy has waned as some of the rationale behind its endorsement has been called into question. Early intubation bypasses alternatives to intubation; high-flow nasal cannula oxygen, noninvasive ventilation, and awake proning are all effective maneuvers in the appropriate patient.10,11 The use of first-line high-flow nasal cannula oxygen and noninvasive ventilation has been widely reported. Reports of first-line use of high-flow nasal cannula oxygen has not demonstrated inferior outcomes, nor has the timing of intubation, suggesting a significant portion of patients could benefit from a trial of therapy and eventually avoid intubation.11-14 Other therapies, such as systemic corticosteroids, confer a mortality benefit in those patients with COVID-19 who require oxygen or mechanical ventilation, but their impact on the progression of respiratory failure and need for intubation are undetermined.
There also are reports of patients who report no signs of respiratory distress or dyspnea with their COVID-19 pneumonia despite profound hypoxemia or high oxygen requirements. Various terms, including silent hypoxemia or happy hypoxia, are descriptive of the demeanor of these patients, and treatment has invariably included oxygen.15,16 Nevertheless, low oxygen measurements have generally prompted higher levels of supplemental oxygen or more invasive therapies.
Treatment rendered may obscure the trajectory of response, which is important to understand to better position options for invasive therapies and other therapeutics. We recently encountered a patient with a course of illness that represented the natural history of COVID-19 pneumonia with low oxygen levels (referred to as hypoxemia for consistency) that highlighted several issues of management.
Case Presentation
A 62-year-old undomiciled woman with morbid obesity, prediabetes mellitus, long-standing schizophrenia, and bipolar disorder presented to our facility for evaluation of dry cough and need for tuberculosis clearance for admittance to a shelter. She appeared comfortable and was afebrile with blood pressure 111/74 mm Hg, heart rate 82 beats per minute. Her respiratory rate was 18 breaths per minute, but the pulse oximetry showed oxygen saturation of 70 to 75% on room air at rest. A chest X-ray showed bibasilar infiltrates (Figure 1), and a rapid COVID-19 nasopharyngeal polymerase chain reaction (PCR) test returned positive, confirmed by a second PCR test. Baseline inflammatory markers were elevated (Figure 2). In addition, the serum interleukin-6 also was elevated to 66.1 pg/mL (normal < 5.0), erythrocyte sedimentation rate elevated to 69 mm/h, but serum procalcitonin was essentially normal (0.22 ng/mL; normal < 20 ng/mL) as was the serum lactate (1.4 mmol/L).
The patient was admitted to the intensive care unit (ICU) for close monitoring in anticipation of the possibility of decompensation based on her age, hypoxia, and elevated inflammatory markers.17 Besides a subsequent low-grade fever (100.4 oF) and lymphopenia (manual count 550/uL), she remained clinically unchanged. Throughout her hospitalization, she maintained a persistent psychotic delusion that she did not have COVID-19, refusing all medical interventions, including a peripheral IV line and supplemental oxygen for the entire duration. Extensive efforts to identify family or a surrogate decision maker were unsuccessful. After consultation with Psychiatry, Bio-Ethics, and hospital leadership, the patient was deemed to lack decision-making capacity regarding treatment or disposition and was placed on a psychiatric hold. However, since any interventions against her will would require sedation, IV access, and potentially increase the risk of nosocomial COVID-19 transmission, she was allowed to remain untreated and was closely monitored for symptoms of worsening respiratory failure.
Over the next 2 weeks, her hypoxemia, inflammatory markers, and the infiltrates on imaging resolved (Figure 2). The lowest daily awake room air pulse oximetry readings are reported, initially with consistent readings in the low 80% range, but on day 12, readings were > 90% and remained > 90% for the remainder of her hospitalization. Therefore, shortly after hospital day 12, she was clinically stable for discharge from acute care to a subacute facility, but this required documentation of the clearance of her viral infection. She refused to undergo a subsequent nasopharyngeal swab but allowed an oropharyngeal COVID-19 PCR swab, which was negative. She remained stable and unchanged for the remainder of her hospitalization, awaiting identification of a receiving facility and was able to be discharged to transitional housing on day 38.
Discussion
The initial reports of COVID-19 pneumonia focused on ARDS and respiratory failure requiring mechanical ventilation with less emphasis on those with lower severity of illness. This was heightened by health care systems that were overwhelmed with large number of patients while faced with limited supplies and equipment. Given the risk to patients and providers of crash intubations, some recommended early intubation strategies.3 However, the natural history of COVID-19 pneumonia and the threshold for intubation of these patients remain poorly defined despite the creation of prognostic tools.17 This patient’s persistent hypoxemia and elevated inflammatory markers certainly met markers of disease associated with a high risk of progression.
The greatest concern would have been her level of hypoxemia. Acceptable thresholds of hypoxemia vary, but general consensus would classify pulse oximetry < 90% as hypoxemia and a threshold for administering supplemental oxygen. It is important to recognize how pulse oximetry readings translate to partial pressure of oxygen (PaO2) measurements (Table 1). Pulse oximetry readings of 90% corresponds to a PaO2 readings of 60 mm Hg in ideal conditions without the influence of acidosis, PaCO2, or temperature. While lower readings are of concern, these do not represent absolute indications for assisted ventilatory support as lower levels are well tolerated in a variety of conditions. A common example are patients with chronic obstructive pulmonary disease. Long-term mortality benefits of continuous supplemental oxygen are well established in specific populations, but the threshold for correction in the acute setting remains a case-by-case decision. This decision is complex and is based on more than an absolute number or the amount of oxygen required to achieve a threshold level of oxygenation.
The PaO2/FIO2 (fraction of inspired oxygen) is a common measure used to address severity of disease and oxygen requirements. It also has been used to define the severity of ARDS, but the ratio is based on intubated and mechanically ventilated patients and may not translate well to those not on assisted ventilation. Treatment with supplemental oxygen also involves entrained air with associated imprecision in oxygen delivery.18 For this discussion, the patient’s admission PaO2/FIO2 on room air would have been between 190 and 260. Coupled with the bilateral infiltrates on imaging, there was justified concern for progression to severe ARDS. Her presentation would have met most of the epidemiologic criteria used in initial case finding for severe COVID-19 cases, including a blood oxygen saturation ≤ 93%, PaO2/FIO2 < 300 with infiltrates involving close to if not exceeding 50% of the lung.
With COVID-19 pneumonia, the pathologic injury to the alveoli resembles that of any viral pneumonia with recruitment of predominantly lymphocytic inflammatory cells that fill the alveoli, derangements in ventilation/perfusion mismatch as the core mechanism of hypoxemia with interstitial edema and shuntlike physiology developing at the extremes of involvement. In later stages, the histologic appearance is similar to ARDS, including hyaline membrane formation and thickened alveolar septa with perivascular lymphocytic-plasmocytic infiltration. In addition, there also are findings of organizing pneumonia with fibroblastic proliferation, thrombosis, and diffuse alveolar damage, a constellation of findings similar to that seen in the latter stages of ARDS.2
Although these histologic findings resemble ARDS, many patients with respiratory failure due to COVID-19 have a different physiologic profile compared with those with typical ARDS, with the most striking finding of lungs with low elastance or high compliance. From the critical care standpoint, this meant that the lungs were relatively easy to ventilate with lower peak airway and plateau pressures and low driving pressures. This condition suggested that there was relatively less lung that could be recruited with positive end expiratory pressure; therefore, a somewhat different entity from that associated with ARDS.19 These findings were often noted early in the course of respiratory failure, and although there is debate about whether this represents a different phenotype or timepoint in the spectrum of disease, it clearly represents a subset that is distinct from that which had been previously encountered.
On the other hand, the clinical features seen in those patients with COVID-19 pneumonia who progressed to advanced respiratory failure were essentially indistinguishable from those patients with traditional ARDS. Other explanations for this respiratory failure have included a disrupted vasoregulatory response to hypoxemia with failed hypoxic vasoconstriction, intravascular microthrombi, and impaired diffusion, all contributing to impaired gas exchange and hypoxemia.19-21 This can lead to shuntlike conditions that neither respond well to supplemental oxygen nor manifest the type of physiologic response seen with other causes of hypoxemia.
The severity of hypoxemia manifested by this patient may have elicited additional findings of respiratory distress, such as dyspnea and tachypnea. However, in patients with severe COVID-19 pneumonia, dyspnea was not a universal finding, reported in the 20 to 60% range of cohorts, higher in those with ARDS and mechanical ventilation, although some report near universal dyspnea in their series.1,4,8,22,23 Tachypnea is another symptom of interest. Using a threshold of > 24 breaths/min, tachypnea was noted in 16 to 29% of patients with a much greater proportion (63%) in nonsurvivors.6,24 Several explanations have been proposed for the discordance between the presence and severity of hypoxemia and lack of symptoms of dyspnea and tachypnea. It is important to recognize that misclassification of the severity of hypoxemia can occur due to technical issues and potential errors involving pulse oximetry measurement and shifts in the oxyhemoglobin dissociation curve. However, this is more pertinent for those with mild disease as the severity of hypoxemia in severe pneumonia is beyond what can be attributed to technical issues.
More important, the ventilatory response curve to hypoxemia may not be normal for some patients, blunted by as much as 50% in older patients, especially in those with diabetes mellitus.7,25,26 In addition, the ventilatory response varies widely even among normal individuals. This would translate to lower levels of minute ventilation (less tachypnea or respiratory effort) with hypoxemia. Hypocapnic hypoxemia also blunts the ventilatory response to hypoxemia. Subjects do not increase their minute ventilation if the PaCO2 remains low despite oxygen desaturation to < 70%, especially if PaCO2 < 30 mm Hg or alternatively, increases in minute ventilation are not seen until the PaCO2 exceeds 39 mm Hg.27 Both scenarios occur in those with COVID-19 pneumonia and provide another explanation for the absence of respiratory symptoms or signs of respiratory distress in some patients.
The observation of more compliant lungs may help in the understanding of the variable presentation of these patients. Compliant lungs do not require the increased pressure needed to achieve a specific tidal volume that, in turn, may increase the work of breathing. This may add to the explanation of seemingly paradoxical silent hypoxemia in those patients where the combination of a blunted ventilatory response, hypocapnia, shunt physiology, and normal respiratory system compliance is represented by the absence of increased breathing effort despite severe hypoxemia.
If not for the patient’s refusal of medical services, this patient quite possibly would have been intubated due to hypoxemia and health care providers’ concern for her risk of deterioration. Reported intubation and mechanical ventilation rates have varied widely from extremes of from < 5 to 88% in severely ill patients.9,22 About 75% will need oxygen, but many can be treated and recover without the need for intubation and mechanical ventilation.
As previously mentioned, options for treatment include standard and high-flow oxygen delivery, noninvasive ventilation, and awake prone ventilation. Their role in patient management has been recently outlined, and instead of an early intubation strategy, represents gradual escalation of support that may be sufficient to treat hypoxemia and avoid the need for intubation and mechanical ventilation (Table 2).
In addition, the patient’s hospital course was notable for the decline in known markers of active inflammation that mirrored the resolution of her hypoxemia and pneumonia. This included elevated lactate dehydrogenase, D-dimer, ferritin, and C-reactive protein with all but the latter rising and decreasing over 2 weeks. These findings provide additional information of the time for recovery and supports the use of these markers to monitor the course of pneumonia.
The patient declined all intervention, including oxygen, and recovered to her presumed prehospitalization condition. This experiment of nature due to unique circumstances may shed light on the natural time course of untreated hypoxemic COVID-19 pneumonia that has not previously been well appreciated. It is important to recognize that recovery occurred over 2 weeks. This is close to the observed and expected time for recovery that has been reported for those with severe COVID-19 pneumonia.
Conclusions
Since the emergence of the COVID-19, evidence has accumulated for the benefit of several adjunctive therapies in the treatment of this type of pneumonia, with corticosteroids providing a mortality benefit. Although unknown whether this patient’s experience can be generalized to others or whether it represents her unique response, this case provides another perspective for comparison of treatments and reinforces the need for prospective, randomized clinical trials to establish treatment efficacy. The exact nature of silent hypoxemia of COVID-19 remains incompletely understood; however, this case highlights the importance of treating the individual instead of clinical markers and provides a time course for recovery from pneumonia and severe hypoxemia that occurs without oxygen or any other treatment over about 2 weeks.
1. Ioannou GN, Locke E, Green P, et al. Risk factors for hospitalization, mechanical ventilation, or death among 10131 US veterans with SARS-CoV-2 infection. JAMA Netw Open. 2020;3(9):e2022310. doi:10.1001/jamanetworkopen.2020.22310
2. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA. 2020;324(8):782-793. doi:10.1001/jama.2020.12839
3. Alhazzani W, Moller MH, Arabi YM, et al. Surviving sepsis campaign: guidelines on the management of critically ill adults with coronavirus disease 2019 (COVID-19). Crit Care Med. 2020;48(6):e440-e469. doi:10.1097/CCM.0000000000004363
4. Ziehr DR, Alladina J, Petri CR, et al. Respiratory pathophysiology of mechanically ventilated patients with COVID-19: a cohort study. Am J Respir Crit Care Med. 2020;201(12):1560-1564. doi:10.1164/rccm.202004-1163LE
5. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239-1242. doi:10.1001/jama.2020.2648
6. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054-1062. doi:10.1016/S01406736(20)30566-3
7. Tobin MJ, Laghi F, Jubran A. Why COVID-19 silent hypoxemia is baffling to physicians. Am J Respir Crit Care Med. 2020;202(3):356-360. doi:10.1164/rccm.202006-2157CP
8. Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708-1720. doi:10.1056/NEJMoa2002032
9. Grasselli G, Zangrillo A, Zanella A, et al. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy Region, Italy. JAMA. 2020;323(16):1574-1581. doi:10.1001/jama.2020.5394
10. Raoof S, Nava S, Carpati C, Hill NS. High-flow, noninvasive ventilation and awake (nonintubation) proning in patients with coronavirus disease 2019 with respiratory failure. Chest. 2020;158(5):1992-2002. doi:10.1016/j.chest.2020.07.013
11. Ackermann M, Mentzer SJ, Jonigk D. Pulmonary vascular pathology in COVID-19. Reply. N Engl J Med. 2020;383(9):888-889. doi:10.1056/NEJMc2022068
12. McDonough G, Khaing P, Treacy T, McGrath C, Yoo EJ. The use of high-flow nasal oxygen in the ICU as a first-line therapy for acute hypoxemic respiratory failure secondary to coronavirus disease 2019. Crit Care Explor. 2020;2(10):e0257. doi:10.1097/CCE.0000000000000257
13. Hernandez-Romieu AC, Adelman MW, et al. Timing of intubation and mortality among critically ill coronavirus disease 2019 patients: a single-center cohort study. Crit Care Med. 2020;48(11):e1045-e1053. doi:10.1097/CCM.0000000000004600
14. Cummings MJ, Baldwin MR, Abrams D, et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study. Lancet. 2020;395(10239):1763-1770. doi:10.1016/S0140-6736(20)31189-2
15. Dhont S, Derom E, Van Braeckel E, Depuydt P, Lambrecht BN. The pathophysiology of ‘happy’ hypoxemia in COVID-19. Respir Res. 2020;21(1):198. doi:10.1186/s12931-020-01462-5
16. Wilkerson RG, Adler JD, Shah NG, Brown R. Silent hypoxia: a harbinger of clinical deterioration in patients with COVID-19. Am J Emerg Med. 2020;38(10):2243.e5-2243.e6. doi:10.1016/j.ajem.2020.05.044
17. Gong J, Ou J, Qiu X, et al. A tool for early prediction of severe coronavirus disease 2019 (COVID-19): a multicenter study using the risk nomogram in Wuhan and Guangdong, China. Clin Infect Dis. 2020;71(15):833-840. doi:10.1093/cid/ciaa443
18. Force ADT, Ranieri VM, Rubenfeld GD, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526-2533. doi:10.1001/jama.2012.5669
19. Marini JJ, Gattinoni L. Management of COVID-19 respiratory distress. JAMA. 2020;323(22):2329-2330. doi:10.1001/jama.2020.6825
20. Schaller T, Hirschbuhl K, Burkhardt K, et al. Postmortem examination of patients with COVID-19. JAMA. 2020;323(24):2518-2520. doi:10.1001/jama.2020.8907
21. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383(2):120-128. doi:10.1056/NEJMoa2015432
22. Wu C, Chen X, Cai Y, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934-943. doi:10.1001/jamainternmed.2020.0994. Published correction appeared May 11, 2020. Errors in data and units of measure. doi:10.1001/jamainternmed.2020.1429
23. Yang J, Zheng Y, Gou X, et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int J Infect Dis. 2020;94:91-95. doi:10.1016/j.ijid.2020.03.017
24. Richardson S, Hirsch JS, Narasimhan M, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA. 2020;323(20):2052-2059. doi:10.1001/jama.2020.6775
25. Tobin MJ, Jubran A, Laghi F. Misconceptions of pathophysiology of happy hypoxemia and implications for management of COVID-19. Respir Res. 2020;21(1):249. doi:10.1186/s12931-020-01520-y
26. Bickler PE, Feiner JR, Lipnick MS, McKleroy W. “Silent” presentation of hypoxemia and cardiorespiratory compensation in COVID-19. Anesthesiology. 2020;134(2):262-269. doi:10.1097/ALN.0000000000003578
27. Jounieaux V, Parreira VF, Aubert G, Dury M, Delguste P, Rodenstein DO. Effects of hypocapnic hyperventilation on the response to hypoxia in normal subjects receiving intermittent positive-pressure ventilation. Chest. 2002;121(4):1141-1148. doi:10.1378/chest.121.4.1141
1. Ioannou GN, Locke E, Green P, et al. Risk factors for hospitalization, mechanical ventilation, or death among 10131 US veterans with SARS-CoV-2 infection. JAMA Netw Open. 2020;3(9):e2022310. doi:10.1001/jamanetworkopen.2020.22310
2. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA. 2020;324(8):782-793. doi:10.1001/jama.2020.12839
3. Alhazzani W, Moller MH, Arabi YM, et al. Surviving sepsis campaign: guidelines on the management of critically ill adults with coronavirus disease 2019 (COVID-19). Crit Care Med. 2020;48(6):e440-e469. doi:10.1097/CCM.0000000000004363
4. Ziehr DR, Alladina J, Petri CR, et al. Respiratory pathophysiology of mechanically ventilated patients with COVID-19: a cohort study. Am J Respir Crit Care Med. 2020;201(12):1560-1564. doi:10.1164/rccm.202004-1163LE
5. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239-1242. doi:10.1001/jama.2020.2648
6. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054-1062. doi:10.1016/S01406736(20)30566-3
7. Tobin MJ, Laghi F, Jubran A. Why COVID-19 silent hypoxemia is baffling to physicians. Am J Respir Crit Care Med. 2020;202(3):356-360. doi:10.1164/rccm.202006-2157CP
8. Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708-1720. doi:10.1056/NEJMoa2002032
9. Grasselli G, Zangrillo A, Zanella A, et al. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy Region, Italy. JAMA. 2020;323(16):1574-1581. doi:10.1001/jama.2020.5394
10. Raoof S, Nava S, Carpati C, Hill NS. High-flow, noninvasive ventilation and awake (nonintubation) proning in patients with coronavirus disease 2019 with respiratory failure. Chest. 2020;158(5):1992-2002. doi:10.1016/j.chest.2020.07.013
11. Ackermann M, Mentzer SJ, Jonigk D. Pulmonary vascular pathology in COVID-19. Reply. N Engl J Med. 2020;383(9):888-889. doi:10.1056/NEJMc2022068
12. McDonough G, Khaing P, Treacy T, McGrath C, Yoo EJ. The use of high-flow nasal oxygen in the ICU as a first-line therapy for acute hypoxemic respiratory failure secondary to coronavirus disease 2019. Crit Care Explor. 2020;2(10):e0257. doi:10.1097/CCE.0000000000000257
13. Hernandez-Romieu AC, Adelman MW, et al. Timing of intubation and mortality among critically ill coronavirus disease 2019 patients: a single-center cohort study. Crit Care Med. 2020;48(11):e1045-e1053. doi:10.1097/CCM.0000000000004600
14. Cummings MJ, Baldwin MR, Abrams D, et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study. Lancet. 2020;395(10239):1763-1770. doi:10.1016/S0140-6736(20)31189-2
15. Dhont S, Derom E, Van Braeckel E, Depuydt P, Lambrecht BN. The pathophysiology of ‘happy’ hypoxemia in COVID-19. Respir Res. 2020;21(1):198. doi:10.1186/s12931-020-01462-5
16. Wilkerson RG, Adler JD, Shah NG, Brown R. Silent hypoxia: a harbinger of clinical deterioration in patients with COVID-19. Am J Emerg Med. 2020;38(10):2243.e5-2243.e6. doi:10.1016/j.ajem.2020.05.044
17. Gong J, Ou J, Qiu X, et al. A tool for early prediction of severe coronavirus disease 2019 (COVID-19): a multicenter study using the risk nomogram in Wuhan and Guangdong, China. Clin Infect Dis. 2020;71(15):833-840. doi:10.1093/cid/ciaa443
18. Force ADT, Ranieri VM, Rubenfeld GD, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526-2533. doi:10.1001/jama.2012.5669
19. Marini JJ, Gattinoni L. Management of COVID-19 respiratory distress. JAMA. 2020;323(22):2329-2330. doi:10.1001/jama.2020.6825
20. Schaller T, Hirschbuhl K, Burkhardt K, et al. Postmortem examination of patients with COVID-19. JAMA. 2020;323(24):2518-2520. doi:10.1001/jama.2020.8907
21. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383(2):120-128. doi:10.1056/NEJMoa2015432
22. Wu C, Chen X, Cai Y, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934-943. doi:10.1001/jamainternmed.2020.0994. Published correction appeared May 11, 2020. Errors in data and units of measure. doi:10.1001/jamainternmed.2020.1429
23. Yang J, Zheng Y, Gou X, et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int J Infect Dis. 2020;94:91-95. doi:10.1016/j.ijid.2020.03.017
24. Richardson S, Hirsch JS, Narasimhan M, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA. 2020;323(20):2052-2059. doi:10.1001/jama.2020.6775
25. Tobin MJ, Jubran A, Laghi F. Misconceptions of pathophysiology of happy hypoxemia and implications for management of COVID-19. Respir Res. 2020;21(1):249. doi:10.1186/s12931-020-01520-y
26. Bickler PE, Feiner JR, Lipnick MS, McKleroy W. “Silent” presentation of hypoxemia and cardiorespiratory compensation in COVID-19. Anesthesiology. 2020;134(2):262-269. doi:10.1097/ALN.0000000000003578
27. Jounieaux V, Parreira VF, Aubert G, Dury M, Delguste P, Rodenstein DO. Effects of hypocapnic hyperventilation on the response to hypoxia in normal subjects receiving intermittent positive-pressure ventilation. Chest. 2002;121(4):1141-1148. doi:10.1378/chest.121.4.1141
Valproate-Induced Lower Extremity Swelling
Bilateral lower extremity edema is a common condition with a broad differential diagnosis. New, severe peripheral edema implies a more nefarious underlying etiology than chronic venous insufficiency and should prompt a thorough evaluation for underlying conditions, such as congestive heart failure (CHF), cirrhosis, nephrotic syndrome, hypoalbuminemia, or lymphatic or venous obstruction. We present a case of a patient with sudden onset new bilateral lower extremity edema due to a rare adverse drug reaction (ADR) from valproate.
Case Presentation
A 63-year-old male with a history of seizures, bipolar disorder type I, and memory impairment due to traumatic brain injury (TBI) from a gunshot wound 24 years prior presented to the emergency department for witnessed seizure activity in the community. The patient had been incarcerated for the past 20 years, throughout which he had been taking the antiepileptic drugs (AEDs) phenytoin and divalproex and did not have any seizure activity. No records prior to his incarceration were available for review.
The patient recently had been released from prison and was nonadherent with his AEDs, leading to a witnessed seizure. This episode was described as preceded by an electric sensation, followed by rhythmic shaking of the right upper extremity without loss of consciousness. His regimen prior to admission included divalproex 1,000 mg daily and phenytoin 200 mg daily. His only other medication was folic acid.
Neurology was consulted on admission. An awake and asleep 4-hour electroencephalogram showed intermittent focal slowing of the right frontocentral region and frequent epileptiform discharges in the right prefrontal region during sleep, corresponding to areas of chronic right anterior frontal and temporal encephalomalacia seen on brain imaging. His seizures were thought likely to be secondary to prior head trauma. While the described seizure activity involving the right upper extremity was not consistent with the location of his prior TBI, neurology considered that he might have simple partial seizures with multiple foci or that his seizure event prior to admission was not accurately described. The neurology consult recommended switching from phenytoin 200 mg daily to lacosamide 100 mg twice daily on admission. His prior dose of divalproex 1,000 mg daily also was resumed for its antiepileptic effect and the added benefit of mood stabilization, as the patient reported elevated mood and decreased need for sleep on admission.
Eight days after changing his AED regimen, the patient was found to have new onset bilateral grade 1+ pitting edema to the level of his shins. He had no history of dyspnea, orthopnea, paroxysmal nocturnal dyspnea, dysuria, or changes in his urination. Although medical records from his incarceration were not available for review, the patient reported that he had never had peripheral edema.
On physical examination, the patient had no periorbital edema, jugular venous pressure of 8 cm H2O, negative hepatojugular reflex, unremarkable cardiac and lung examination, and grade 2+ posterior tibial and dorsalis pedis pulses bilaterally. He underwent extensive laboratory evaluation for potential underlying causes, including nephrotic syndrome, cirrhosis, hypothyroidism, and CHF (Table). Valproate levels were initially subtherapeutic on admission (< 10 µg/mL, reference range 50-125 µg/mL) then rose to within therapeutic range (54 µg/mL-80 µg/mL throughout admission) after neurology recommended increasing the dose from 1,000 mg daily to 1,500 mg daily. His measured valproate levels were never supratherapeutic.
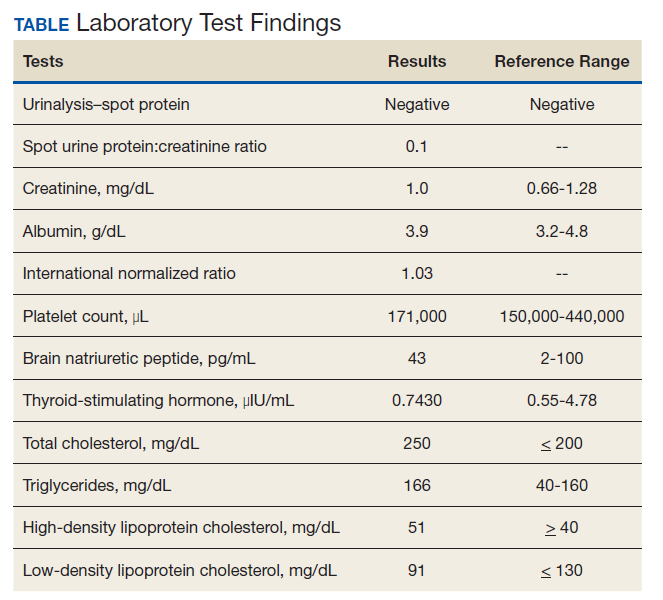
An electrocardiogram showed normal sinus rhythm unchanged from admission. Transthoracic echocardiogram showed normal left ventricular (LV) size and estimated LV ejection fraction of 55 to 60%. Abdominal ultrasound showed no evidence of cirrhosis and normal portal vein flow. Ultrasound of the lower extremities showed no deep venous thrombosis or valvular insufficiency. The patient was prescribed compression stockings. However, due to memory impairment, he was relatively nonadherent, and his lower extremity edema worsened to grade 3+ over several days. Due to the progressive swelling with no identified cause, a computed tomographic venogram of the abdomen and pelvis was performed to determine whether an inferior vena cava (IVC) thrombus was present. This study was unremarkable and did not show any external IVC compression.
After extensive evaluation did not reveal any other cause, the temporal course of events suggested an association between the patient’s peripheral edema and resumption of divalproex. His swelling remained stable. Discontinuation of divalproex was considered, but the patient’s mood remained euthymic, and he had no further seizure activity while on this medication, so the benefit of continuation was felt to outweigh any risks of switching to another agent.
Discussion
Valproate and its related forms, such as divalproex, often are used in the treatment of generalized or partial seizures, psychiatric disorders, and the prophylaxis of migraine headaches. Common ADRs include gastrointestinal symptoms, sedation, and dose-related thrombocytopenia, among many others. Rare ADRs include fulminant hepatitis, pancreatitis, hyperammonemia, and peripheral edema.1 There have been case reports of valproate-induced peripheral edema, which seems to be an idiosyncratic ADR that occurs after long-term administration of the medication.2,3 Early studies reported valproate-related edema in the context of valproate-induced hepatic injury.4 However, in more recent case reports, valproate-related edema has been found in patients without hepatotoxicity or supratherapeutic drug levels.1,2
The exact mechanism by which valproate causes peripheral edema is unknown. It has been reported that medications affecting the γ-aminobutyric acid (GABA) system such as benzodiazepines, for example, can cause this rare ADR.5 Unlike benzodiazepines, valproate has an indirect effect on the GABA system, through increasing availability of GABA.6 GABA receptors have been identified on peripheral tissues, suggesting that GABAergic medications also may have an effect on regional vascular resistance.7 This mechanism was proposed by prior case reports but has yet to be proven in studies.2
In this case, initiation of lacosamide temporally coinciding with development of the patient’s edema leads one to question whether lacosamide may have caused this ADR. Other medications commonly used in seizure management (such as benzodiazepines and gabapentin) have been reported to cause new onset peripheral edema.5,8 To date, however, there are no reported cases of peripheral edema due to lacosamide. While there are known interactions between various AEDs that may impact drug levels of valproate, there are no reported drug-drug interactions between lacosamide and valproate.9
Conclusions
Our case adds to the small but growing body of literature that suggests peripheral edema is a rare but clinically significant ADR of valproate. With its broad differential diagnosis, new onset peripheral edema is a concern that often warrants an extensive evaluation for underlying causes. Clinicians should be aware of this ADR as use of valproate becomes increasingly common so that an extensive workup is not always performed on patients with peripheral edema.
1. Prajapati H, Kansal D, Negi R. Magnesium valproate-induced pedal edema on chronic therapy: a rare adverse drug reaction. Indian J Pharmacol. 2017;49(5):399. doi:10.4103/ijp.IJP_239_17
2. Lin ST, Chen CS, Yen CF, Tsei JH, Wang SY. Valproate-related peripheral oedema: a manageable but probably neglected condition. Int J Neuropsychopharmacol. 2009;12(7):991-993. doi:10.1017/S1461145709000509
3. Ettinger A, Moshe S, Shinnar S. Edema associated with long‐term valproate therapy. Epilepsia. 1990;31(2):211-213. doi:10.1111/j.1528-1167.1990.tb06308.x
4. Zimmerman HJ, Ishak KG. Valproate‐induced hepatic injury: analyses of 23 fatal cases. Hepatology. 1982;2(5):591S-597S. doi:10.1002/hep.1840020513
5. Mathew T, D’Souza D, Nadimpally US, Nadig R. Clobazam‐induced pedal edema: “an unrecognized side effect of a common antiepileptic drug.” Epilepsia. 2016;57(3): 524-525. doi:10.1111/epi.13316
6. Bourin M, Chenu F, Hascoët M. The role of sodium channels in the mechanism of action of antidepressants and mood stabilizers. Curr Drug Targets. 2009;10(11):1052-1060. doi:10.2174/138945009789735138
7. Takemoto Y. Effects of gamma‐aminobutyric acid on regional vascular resistances of conscious spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 1995;22(suppl):S102-Sl04. doi:10.1111/j.1440-1681.1995.tb02839.x
8. Bidaki R, Sadeghi Z, Shafizadegan S, et al. Gabapentin induces edema, hyperesthesia and scaling in a depressed patient; a diagnostic challenge. Adv Biomed Res. 2016;5:1. doi:10.4103/2277-9175.174955
9. Cawello W, Nickel B, Eggert‐Formella A. No pharmacokinetic interaction between lacosamide and carbamazepine in healthy volunteers. J Clin Pharmacol. 2010;50(4):459-471. doi:10.1177/0091270009347675
Bilateral lower extremity edema is a common condition with a broad differential diagnosis. New, severe peripheral edema implies a more nefarious underlying etiology than chronic venous insufficiency and should prompt a thorough evaluation for underlying conditions, such as congestive heart failure (CHF), cirrhosis, nephrotic syndrome, hypoalbuminemia, or lymphatic or venous obstruction. We present a case of a patient with sudden onset new bilateral lower extremity edema due to a rare adverse drug reaction (ADR) from valproate.
Case Presentation
A 63-year-old male with a history of seizures, bipolar disorder type I, and memory impairment due to traumatic brain injury (TBI) from a gunshot wound 24 years prior presented to the emergency department for witnessed seizure activity in the community. The patient had been incarcerated for the past 20 years, throughout which he had been taking the antiepileptic drugs (AEDs) phenytoin and divalproex and did not have any seizure activity. No records prior to his incarceration were available for review.
The patient recently had been released from prison and was nonadherent with his AEDs, leading to a witnessed seizure. This episode was described as preceded by an electric sensation, followed by rhythmic shaking of the right upper extremity without loss of consciousness. His regimen prior to admission included divalproex 1,000 mg daily and phenytoin 200 mg daily. His only other medication was folic acid.
Neurology was consulted on admission. An awake and asleep 4-hour electroencephalogram showed intermittent focal slowing of the right frontocentral region and frequent epileptiform discharges in the right prefrontal region during sleep, corresponding to areas of chronic right anterior frontal and temporal encephalomalacia seen on brain imaging. His seizures were thought likely to be secondary to prior head trauma. While the described seizure activity involving the right upper extremity was not consistent with the location of his prior TBI, neurology considered that he might have simple partial seizures with multiple foci or that his seizure event prior to admission was not accurately described. The neurology consult recommended switching from phenytoin 200 mg daily to lacosamide 100 mg twice daily on admission. His prior dose of divalproex 1,000 mg daily also was resumed for its antiepileptic effect and the added benefit of mood stabilization, as the patient reported elevated mood and decreased need for sleep on admission.
Eight days after changing his AED regimen, the patient was found to have new onset bilateral grade 1+ pitting edema to the level of his shins. He had no history of dyspnea, orthopnea, paroxysmal nocturnal dyspnea, dysuria, or changes in his urination. Although medical records from his incarceration were not available for review, the patient reported that he had never had peripheral edema.
On physical examination, the patient had no periorbital edema, jugular venous pressure of 8 cm H2O, negative hepatojugular reflex, unremarkable cardiac and lung examination, and grade 2+ posterior tibial and dorsalis pedis pulses bilaterally. He underwent extensive laboratory evaluation for potential underlying causes, including nephrotic syndrome, cirrhosis, hypothyroidism, and CHF (Table). Valproate levels were initially subtherapeutic on admission (< 10 µg/mL, reference range 50-125 µg/mL) then rose to within therapeutic range (54 µg/mL-80 µg/mL throughout admission) after neurology recommended increasing the dose from 1,000 mg daily to 1,500 mg daily. His measured valproate levels were never supratherapeutic.
An electrocardiogram showed normal sinus rhythm unchanged from admission. Transthoracic echocardiogram showed normal left ventricular (LV) size and estimated LV ejection fraction of 55 to 60%. Abdominal ultrasound showed no evidence of cirrhosis and normal portal vein flow. Ultrasound of the lower extremities showed no deep venous thrombosis or valvular insufficiency. The patient was prescribed compression stockings. However, due to memory impairment, he was relatively nonadherent, and his lower extremity edema worsened to grade 3+ over several days. Due to the progressive swelling with no identified cause, a computed tomographic venogram of the abdomen and pelvis was performed to determine whether an inferior vena cava (IVC) thrombus was present. This study was unremarkable and did not show any external IVC compression.
After extensive evaluation did not reveal any other cause, the temporal course of events suggested an association between the patient’s peripheral edema and resumption of divalproex. His swelling remained stable. Discontinuation of divalproex was considered, but the patient’s mood remained euthymic, and he had no further seizure activity while on this medication, so the benefit of continuation was felt to outweigh any risks of switching to another agent.
Discussion
Valproate and its related forms, such as divalproex, often are used in the treatment of generalized or partial seizures, psychiatric disorders, and the prophylaxis of migraine headaches. Common ADRs include gastrointestinal symptoms, sedation, and dose-related thrombocytopenia, among many others. Rare ADRs include fulminant hepatitis, pancreatitis, hyperammonemia, and peripheral edema.1 There have been case reports of valproate-induced peripheral edema, which seems to be an idiosyncratic ADR that occurs after long-term administration of the medication.2,3 Early studies reported valproate-related edema in the context of valproate-induced hepatic injury.4 However, in more recent case reports, valproate-related edema has been found in patients without hepatotoxicity or supratherapeutic drug levels.1,2
The exact mechanism by which valproate causes peripheral edema is unknown. It has been reported that medications affecting the γ-aminobutyric acid (GABA) system such as benzodiazepines, for example, can cause this rare ADR.5 Unlike benzodiazepines, valproate has an indirect effect on the GABA system, through increasing availability of GABA.6 GABA receptors have been identified on peripheral tissues, suggesting that GABAergic medications also may have an effect on regional vascular resistance.7 This mechanism was proposed by prior case reports but has yet to be proven in studies.2
In this case, initiation of lacosamide temporally coinciding with development of the patient’s edema leads one to question whether lacosamide may have caused this ADR. Other medications commonly used in seizure management (such as benzodiazepines and gabapentin) have been reported to cause new onset peripheral edema.5,8 To date, however, there are no reported cases of peripheral edema due to lacosamide. While there are known interactions between various AEDs that may impact drug levels of valproate, there are no reported drug-drug interactions between lacosamide and valproate.9
Conclusions
Our case adds to the small but growing body of literature that suggests peripheral edema is a rare but clinically significant ADR of valproate. With its broad differential diagnosis, new onset peripheral edema is a concern that often warrants an extensive evaluation for underlying causes. Clinicians should be aware of this ADR as use of valproate becomes increasingly common so that an extensive workup is not always performed on patients with peripheral edema.
Bilateral lower extremity edema is a common condition with a broad differential diagnosis. New, severe peripheral edema implies a more nefarious underlying etiology than chronic venous insufficiency and should prompt a thorough evaluation for underlying conditions, such as congestive heart failure (CHF), cirrhosis, nephrotic syndrome, hypoalbuminemia, or lymphatic or venous obstruction. We present a case of a patient with sudden onset new bilateral lower extremity edema due to a rare adverse drug reaction (ADR) from valproate.
Case Presentation
A 63-year-old male with a history of seizures, bipolar disorder type I, and memory impairment due to traumatic brain injury (TBI) from a gunshot wound 24 years prior presented to the emergency department for witnessed seizure activity in the community. The patient had been incarcerated for the past 20 years, throughout which he had been taking the antiepileptic drugs (AEDs) phenytoin and divalproex and did not have any seizure activity. No records prior to his incarceration were available for review.
The patient recently had been released from prison and was nonadherent with his AEDs, leading to a witnessed seizure. This episode was described as preceded by an electric sensation, followed by rhythmic shaking of the right upper extremity without loss of consciousness. His regimen prior to admission included divalproex 1,000 mg daily and phenytoin 200 mg daily. His only other medication was folic acid.
Neurology was consulted on admission. An awake and asleep 4-hour electroencephalogram showed intermittent focal slowing of the right frontocentral region and frequent epileptiform discharges in the right prefrontal region during sleep, corresponding to areas of chronic right anterior frontal and temporal encephalomalacia seen on brain imaging. His seizures were thought likely to be secondary to prior head trauma. While the described seizure activity involving the right upper extremity was not consistent with the location of his prior TBI, neurology considered that he might have simple partial seizures with multiple foci or that his seizure event prior to admission was not accurately described. The neurology consult recommended switching from phenytoin 200 mg daily to lacosamide 100 mg twice daily on admission. His prior dose of divalproex 1,000 mg daily also was resumed for its antiepileptic effect and the added benefit of mood stabilization, as the patient reported elevated mood and decreased need for sleep on admission.
Eight days after changing his AED regimen, the patient was found to have new onset bilateral grade 1+ pitting edema to the level of his shins. He had no history of dyspnea, orthopnea, paroxysmal nocturnal dyspnea, dysuria, or changes in his urination. Although medical records from his incarceration were not available for review, the patient reported that he had never had peripheral edema.
On physical examination, the patient had no periorbital edema, jugular venous pressure of 8 cm H2O, negative hepatojugular reflex, unremarkable cardiac and lung examination, and grade 2+ posterior tibial and dorsalis pedis pulses bilaterally. He underwent extensive laboratory evaluation for potential underlying causes, including nephrotic syndrome, cirrhosis, hypothyroidism, and CHF (Table). Valproate levels were initially subtherapeutic on admission (< 10 µg/mL, reference range 50-125 µg/mL) then rose to within therapeutic range (54 µg/mL-80 µg/mL throughout admission) after neurology recommended increasing the dose from 1,000 mg daily to 1,500 mg daily. His measured valproate levels were never supratherapeutic.
An electrocardiogram showed normal sinus rhythm unchanged from admission. Transthoracic echocardiogram showed normal left ventricular (LV) size and estimated LV ejection fraction of 55 to 60%. Abdominal ultrasound showed no evidence of cirrhosis and normal portal vein flow. Ultrasound of the lower extremities showed no deep venous thrombosis or valvular insufficiency. The patient was prescribed compression stockings. However, due to memory impairment, he was relatively nonadherent, and his lower extremity edema worsened to grade 3+ over several days. Due to the progressive swelling with no identified cause, a computed tomographic venogram of the abdomen and pelvis was performed to determine whether an inferior vena cava (IVC) thrombus was present. This study was unremarkable and did not show any external IVC compression.
After extensive evaluation did not reveal any other cause, the temporal course of events suggested an association between the patient’s peripheral edema and resumption of divalproex. His swelling remained stable. Discontinuation of divalproex was considered, but the patient’s mood remained euthymic, and he had no further seizure activity while on this medication, so the benefit of continuation was felt to outweigh any risks of switching to another agent.
Discussion
Valproate and its related forms, such as divalproex, often are used in the treatment of generalized or partial seizures, psychiatric disorders, and the prophylaxis of migraine headaches. Common ADRs include gastrointestinal symptoms, sedation, and dose-related thrombocytopenia, among many others. Rare ADRs include fulminant hepatitis, pancreatitis, hyperammonemia, and peripheral edema.1 There have been case reports of valproate-induced peripheral edema, which seems to be an idiosyncratic ADR that occurs after long-term administration of the medication.2,3 Early studies reported valproate-related edema in the context of valproate-induced hepatic injury.4 However, in more recent case reports, valproate-related edema has been found in patients without hepatotoxicity or supratherapeutic drug levels.1,2
The exact mechanism by which valproate causes peripheral edema is unknown. It has been reported that medications affecting the γ-aminobutyric acid (GABA) system such as benzodiazepines, for example, can cause this rare ADR.5 Unlike benzodiazepines, valproate has an indirect effect on the GABA system, through increasing availability of GABA.6 GABA receptors have been identified on peripheral tissues, suggesting that GABAergic medications also may have an effect on regional vascular resistance.7 This mechanism was proposed by prior case reports but has yet to be proven in studies.2
In this case, initiation of lacosamide temporally coinciding with development of the patient’s edema leads one to question whether lacosamide may have caused this ADR. Other medications commonly used in seizure management (such as benzodiazepines and gabapentin) have been reported to cause new onset peripheral edema.5,8 To date, however, there are no reported cases of peripheral edema due to lacosamide. While there are known interactions between various AEDs that may impact drug levels of valproate, there are no reported drug-drug interactions between lacosamide and valproate.9
Conclusions
Our case adds to the small but growing body of literature that suggests peripheral edema is a rare but clinically significant ADR of valproate. With its broad differential diagnosis, new onset peripheral edema is a concern that often warrants an extensive evaluation for underlying causes. Clinicians should be aware of this ADR as use of valproate becomes increasingly common so that an extensive workup is not always performed on patients with peripheral edema.
1. Prajapati H, Kansal D, Negi R. Magnesium valproate-induced pedal edema on chronic therapy: a rare adverse drug reaction. Indian J Pharmacol. 2017;49(5):399. doi:10.4103/ijp.IJP_239_17
2. Lin ST, Chen CS, Yen CF, Tsei JH, Wang SY. Valproate-related peripheral oedema: a manageable but probably neglected condition. Int J Neuropsychopharmacol. 2009;12(7):991-993. doi:10.1017/S1461145709000509
3. Ettinger A, Moshe S, Shinnar S. Edema associated with long‐term valproate therapy. Epilepsia. 1990;31(2):211-213. doi:10.1111/j.1528-1167.1990.tb06308.x
4. Zimmerman HJ, Ishak KG. Valproate‐induced hepatic injury: analyses of 23 fatal cases. Hepatology. 1982;2(5):591S-597S. doi:10.1002/hep.1840020513
5. Mathew T, D’Souza D, Nadimpally US, Nadig R. Clobazam‐induced pedal edema: “an unrecognized side effect of a common antiepileptic drug.” Epilepsia. 2016;57(3): 524-525. doi:10.1111/epi.13316
6. Bourin M, Chenu F, Hascoët M. The role of sodium channels in the mechanism of action of antidepressants and mood stabilizers. Curr Drug Targets. 2009;10(11):1052-1060. doi:10.2174/138945009789735138
7. Takemoto Y. Effects of gamma‐aminobutyric acid on regional vascular resistances of conscious spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 1995;22(suppl):S102-Sl04. doi:10.1111/j.1440-1681.1995.tb02839.x
8. Bidaki R, Sadeghi Z, Shafizadegan S, et al. Gabapentin induces edema, hyperesthesia and scaling in a depressed patient; a diagnostic challenge. Adv Biomed Res. 2016;5:1. doi:10.4103/2277-9175.174955
9. Cawello W, Nickel B, Eggert‐Formella A. No pharmacokinetic interaction between lacosamide and carbamazepine in healthy volunteers. J Clin Pharmacol. 2010;50(4):459-471. doi:10.1177/0091270009347675
1. Prajapati H, Kansal D, Negi R. Magnesium valproate-induced pedal edema on chronic therapy: a rare adverse drug reaction. Indian J Pharmacol. 2017;49(5):399. doi:10.4103/ijp.IJP_239_17
2. Lin ST, Chen CS, Yen CF, Tsei JH, Wang SY. Valproate-related peripheral oedema: a manageable but probably neglected condition. Int J Neuropsychopharmacol. 2009;12(7):991-993. doi:10.1017/S1461145709000509
3. Ettinger A, Moshe S, Shinnar S. Edema associated with long‐term valproate therapy. Epilepsia. 1990;31(2):211-213. doi:10.1111/j.1528-1167.1990.tb06308.x
4. Zimmerman HJ, Ishak KG. Valproate‐induced hepatic injury: analyses of 23 fatal cases. Hepatology. 1982;2(5):591S-597S. doi:10.1002/hep.1840020513
5. Mathew T, D’Souza D, Nadimpally US, Nadig R. Clobazam‐induced pedal edema: “an unrecognized side effect of a common antiepileptic drug.” Epilepsia. 2016;57(3): 524-525. doi:10.1111/epi.13316
6. Bourin M, Chenu F, Hascoët M. The role of sodium channels in the mechanism of action of antidepressants and mood stabilizers. Curr Drug Targets. 2009;10(11):1052-1060. doi:10.2174/138945009789735138
7. Takemoto Y. Effects of gamma‐aminobutyric acid on regional vascular resistances of conscious spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 1995;22(suppl):S102-Sl04. doi:10.1111/j.1440-1681.1995.tb02839.x
8. Bidaki R, Sadeghi Z, Shafizadegan S, et al. Gabapentin induces edema, hyperesthesia and scaling in a depressed patient; a diagnostic challenge. Adv Biomed Res. 2016;5:1. doi:10.4103/2277-9175.174955
9. Cawello W, Nickel B, Eggert‐Formella A. No pharmacokinetic interaction between lacosamide and carbamazepine in healthy volunteers. J Clin Pharmacol. 2010;50(4):459-471. doi:10.1177/0091270009347675
Nephrogenic Systemic Fibrosis in a Patient With Multiple Inflammatory Disorders
First described in 2000 in a case series of 15 patients, nephrogenic systemic fibrosis (NSF) is a rare scleroderma-like fibrosing skin condition associated with gadolinium exposure in end stage renal disease (ESRD).1 Patients with advanced chronic kidney disease (CKD) or ESRD are at the highest risk for this condition when exposed to gadolinium-based contrast dyes.
Nephrogenic systemic fibrosis is a devastating and rapidly progressive condition, making its prevention in at-risk populations of utmost importance. In this article, the authors describe a case of a patient who developed NSF in the setting of gadolinium exposure and multiple inflammatory dermatologic conditions. This case illustrates the possible role of a pro-inflammatory state in predisposing to NSF, which may help further elucidate its mechanism of action.
Case Presentation
A 61-year-old Hispanic male with a history of IV heroin use with ESRD secondary to membranous glomerulonephritis on hemodialysis and chronic hepatitis C infection presented to the West Los Angeles VAMC with fevers and night sweats that had persisted for 2 weeks. His physical examination was notable for diffuse tender palpable purpura and petechiae (including his palms and soles), altered mental status, and diffuse myoclonic jerks, which necessitated endotracheal intubation and mechanical ventilation for airway protection. Blood cultures were positive for methicillin-sensitive Staphylococcus aureus (MSSA). Laboratory results were notable for an elevated sedimentation rate of 53 mm/h (0-10 mm/h), C-reactive protein of 19.8 mg/L (< 0.744 mg/dL), and albumin of 1.2 g/dL (3.2-4.8 g/dL). An extensive rheumatologic workup was unrevealing, and a lumbar puncture was unremarkable. A biopsy of his skin lesions was consistent with leukocytoclastic vasculitis.
The patient’s prior hemodialysis access, a tunneled dialysis catheter in the right subclavian vein, was removed given concern for line infection and replaced with an internal jugular temporary hemodialysis line. Given his altered mental status and myoclonic jerks, the decision was made to pursue a magnetic resonance imaging (MRI) scan of the brain and spine with gadolinium contrast to evaluate for cerebral vasculitis and/or septic emboli to the brain.
The patient received 15 mL of gadoversetamide contrast in accordance with hospital imaging protocol. The MRI revealed only chronic ischemic changes. The patient underwent hemodialysis about 18 hours later. The patient was treated with a 6-week course of IV penicillin G. His altered mental status and myoclonic jerks resolved without intervention, and he was then discharged to an acute rehabilitation unit.
Eight weeks after his initial presentation the patient developed a purulent wound on his right forearm (Figure 1) 
The patient was discharged to continue physical and occupational therapy to preserve his functional mobility, as no other treatment options were available. 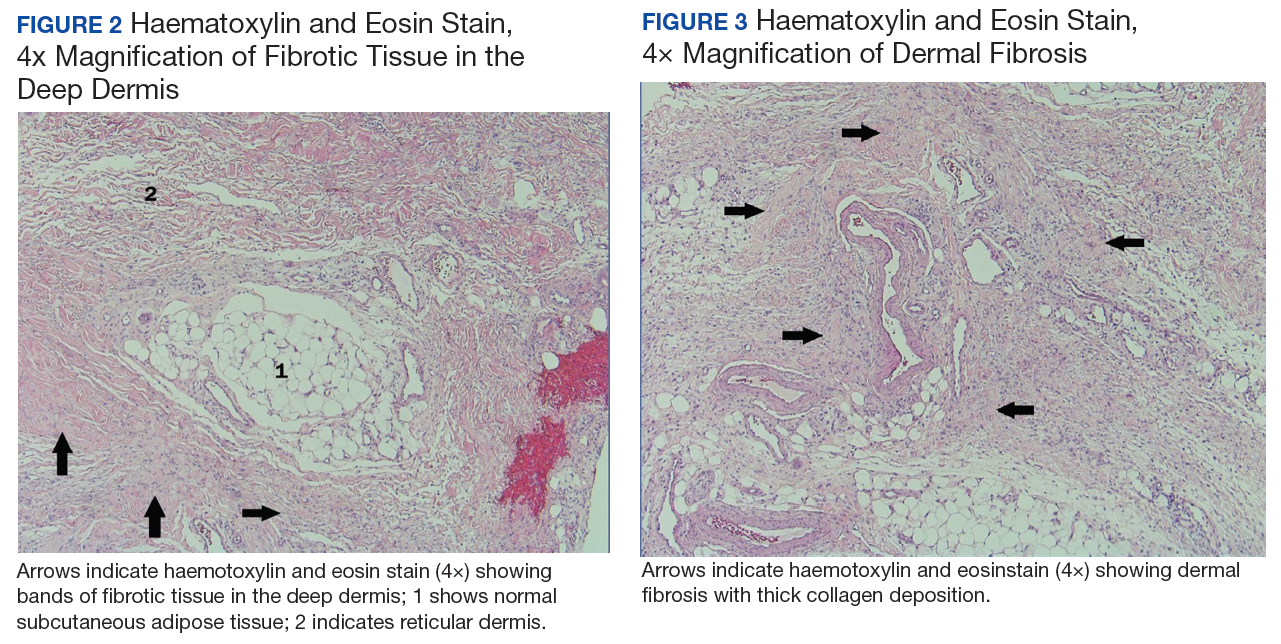
Discussion
Nephrogenic systemic fibrosis is a poorly understood inflammatory condition that produces diffuse fibrosis of the skin. Typically, the disease begins with progressive skin induration of the extremities. Systemic involvement may occur, leading to fibrosis of skeletal muscle, fascia, and multiple organs. Flexion contractures may develop that limit physical function. Fibrosis can become apparent within days to months after exposure to gadolinium contrast.
Beyond renal insufficiency, it is unclear what other risk factors predispose patients to developing this condition. Only a minority of patients with CKD stages 1 through 4 will develop NSF on exposure to gadolinium contrast. However, the incidence of NSF among patients with CKD stage 5 who are exposed to gadolinium has been estimated to be about 13.4% in a prospective study involving 18 patients.2
In a 2015 meta-analysis by Zhang and colleagues, the only clear risk factor identified for the development of NSF, aside from gadolinium exposure, was severe renal insufficiency with a glomerular filtration rate of < 30 mL/min/1.75m2.3 Due to the limited number of patients identified with this disease, it is difficult to identify other risk factors associated with the development of NSF. Based on in vitro studies, it has been postulated that a pro-inflammatory state predisposes patients to develop NSF.4,5 The proposed mechanism for NSF involves extravasation of gadolinium in the setting of vascular endothelial permeability.5,6 Gadolinium then interacts with tissue macrophages, which induce the release of inflammatory cytokines and the secretion of smooth muscle actin by dermal fibroblasts.6,7
Treatment of NSF has been largely unsuccessful. Multiple modalities of treatment that included topical and oral steroids, immunosuppression, plasmapheresis, and ultraviolent therapy have been attempted, none of which have been proven to consistently limit progression of the disease.8 The most effective intervention is early physical therapy to preserve functionality and prevent contracture formation. For patients who are eligible, early renal transplantation may offer the best chance of improved mobility. In a case series review by Cuffy and colleagues, 5 of 6 patients who underwent renal transplantation after the development of NSF experienced softening of the involved skin, and 2 patients had improved mobility of joints.9
Conclusion
The case presented here illustrates a possible association between a pro-inflammatory state and the development of NSF. This patient had multiple inflammatory conditions, including MSSA bacteremia, leukocytoclastic vasculitis, and pyoderma gangrenosum (the latter 2 conditions were thought to be associated with his underlying chronic hepatitis C infection), which the authors believe predisposed him to endothelial permeability and risk for developing NSF. The risk of developing NSF in at-risk patients with each episode of gadolinium exposure is estimated around 2.4%, or an incidence of 4.3 cases per 1,000 patient-years, leading the American College of Radiologists to recommend against the administration of gadolinium-based contrast except in cases in which benefits clearly outweigh risks.10 However, an MRI with gadolinium contrast can offer high diagnostic yield in cases such as the one presented here in which a diagnosis remains elusive. Moreover, the use of linear gadolinium-based contrast agents such as gadoversetamide, as in this case, has been reported to be associated with higher incidence of NSF.5 Since this case, the West Los Angeles VAMC has switched to gadobutrol contrast for its MRI protocol, which has been purported to be a lower risk agent compared with that of linear gadolinium-based contrast agents (although several cases of NSF have been reported with gadobutrol in the literature).11
Providers weighing the decision to administer gadolinium contrast to patients with ESRD should discuss the risks and benefits thoroughly, especially in patients with preexisting inflammatory conditions. In addition, although it has not been shown to effectively reduce the risk of NSF after administration of gadolinium, hemodialysis is recommended 2 hours after contrast administration for individuals at risk (the study patient received hemodialysis approximately 18 hours after).12 Given the lack of effective treatment options for NSF, prevention is key. A deeper understanding of the pathophysiology of NSF and identification of its risk factors is paramount to the prevention of this devastating disease.
1. Cowper SE, Robin HS, Steinberg SM, Su LD, Gupta S, LeBoit PE. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients. Lancet. 2000;356(9234):1000-1001.
2. Todd DJ, Kagan A, Chibnik LB, Kay J. Cutaneous changes of nephrogenic systemic fibrosis. Arthritis Rheum. 2007;56(10):3433-3441.
3. Zhang B, Liang L, Chen W, Liang C, Zhang S. An updated study to determine association between gadolinium-based contrast agents and nephrogenic systemic fibrosis. PLoS One. 2015;10(6):e0129720.
4. Wermuth PJ, Del Galdo F, Jiménez SA. Induction of the expression of profibrotic cytokines and growth factors in normal human peripheral blood monocytes by gadolinium contrast agents. Arthritis Rheum. 2009;60(5):1508-1518.
5. Daftari Besheli L, Aran S, Shaqdan K, Kay J, Abujudeh H. Current status of nephrogenic systemic fibrosis. Clin Radiol. 2014;69(7):661-668.
6. Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;31(1):F1-F11.
7. Idée JM, Fretellier N, Robic C, Corot C. The role of gadolinium chelates in the mechanism of nephrogenic systemic fibrosis: a critical update. Crit Rev Toxicol. 2014;44(10):895-913.
8. Mendoza FA, Artlett CM, Sandorfi N, Latinis K, Piera-Velazquez S, Jimenez SA. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35(4):238-249.
9. Cuffy MC, Singh M, Formica R, et al. Renal transplantation for nephrogenic systemic fibrosis: a case report and review of the literature. Nephrol Dial Transplant. 2011;26(3):1099-1109.
10. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development of gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267
11. Elmholdt TR, Jørgensen B, Ramsing M, Pedersen M, Olesen AB. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287.
12. Abu-Alfa AK. Nephrogenic systemic fibrosis and gadolinium-based contrast agents. Adv Chronic Kidney Dis. 2011;18(3);188-198.
First described in 2000 in a case series of 15 patients, nephrogenic systemic fibrosis (NSF) is a rare scleroderma-like fibrosing skin condition associated with gadolinium exposure in end stage renal disease (ESRD).1 Patients with advanced chronic kidney disease (CKD) or ESRD are at the highest risk for this condition when exposed to gadolinium-based contrast dyes.
Nephrogenic systemic fibrosis is a devastating and rapidly progressive condition, making its prevention in at-risk populations of utmost importance. In this article, the authors describe a case of a patient who developed NSF in the setting of gadolinium exposure and multiple inflammatory dermatologic conditions. This case illustrates the possible role of a pro-inflammatory state in predisposing to NSF, which may help further elucidate its mechanism of action.
Case Presentation
A 61-year-old Hispanic male with a history of IV heroin use with ESRD secondary to membranous glomerulonephritis on hemodialysis and chronic hepatitis C infection presented to the West Los Angeles VAMC with fevers and night sweats that had persisted for 2 weeks. His physical examination was notable for diffuse tender palpable purpura and petechiae (including his palms and soles), altered mental status, and diffuse myoclonic jerks, which necessitated endotracheal intubation and mechanical ventilation for airway protection. Blood cultures were positive for methicillin-sensitive Staphylococcus aureus (MSSA). Laboratory results were notable for an elevated sedimentation rate of 53 mm/h (0-10 mm/h), C-reactive protein of 19.8 mg/L (< 0.744 mg/dL), and albumin of 1.2 g/dL (3.2-4.8 g/dL). An extensive rheumatologic workup was unrevealing, and a lumbar puncture was unremarkable. A biopsy of his skin lesions was consistent with leukocytoclastic vasculitis.
The patient’s prior hemodialysis access, a tunneled dialysis catheter in the right subclavian vein, was removed given concern for line infection and replaced with an internal jugular temporary hemodialysis line. Given his altered mental status and myoclonic jerks, the decision was made to pursue a magnetic resonance imaging (MRI) scan of the brain and spine with gadolinium contrast to evaluate for cerebral vasculitis and/or septic emboli to the brain.
The patient received 15 mL of gadoversetamide contrast in accordance with hospital imaging protocol. The MRI revealed only chronic ischemic changes. The patient underwent hemodialysis about 18 hours later. The patient was treated with a 6-week course of IV penicillin G. His altered mental status and myoclonic jerks resolved without intervention, and he was then discharged to an acute rehabilitation unit.
Eight weeks after his initial presentation the patient developed a purulent wound on his right forearm (Figure 1)
The patient was discharged to continue physical and occupational therapy to preserve his functional mobility, as no other treatment options were available.
Discussion
Nephrogenic systemic fibrosis is a poorly understood inflammatory condition that produces diffuse fibrosis of the skin. Typically, the disease begins with progressive skin induration of the extremities. Systemic involvement may occur, leading to fibrosis of skeletal muscle, fascia, and multiple organs. Flexion contractures may develop that limit physical function. Fibrosis can become apparent within days to months after exposure to gadolinium contrast.
Beyond renal insufficiency, it is unclear what other risk factors predispose patients to developing this condition. Only a minority of patients with CKD stages 1 through 4 will develop NSF on exposure to gadolinium contrast. However, the incidence of NSF among patients with CKD stage 5 who are exposed to gadolinium has been estimated to be about 13.4% in a prospective study involving 18 patients.2
In a 2015 meta-analysis by Zhang and colleagues, the only clear risk factor identified for the development of NSF, aside from gadolinium exposure, was severe renal insufficiency with a glomerular filtration rate of < 30 mL/min/1.75m2.3 Due to the limited number of patients identified with this disease, it is difficult to identify other risk factors associated with the development of NSF. Based on in vitro studies, it has been postulated that a pro-inflammatory state predisposes patients to develop NSF.4,5 The proposed mechanism for NSF involves extravasation of gadolinium in the setting of vascular endothelial permeability.5,6 Gadolinium then interacts with tissue macrophages, which induce the release of inflammatory cytokines and the secretion of smooth muscle actin by dermal fibroblasts.6,7
Treatment of NSF has been largely unsuccessful. Multiple modalities of treatment that included topical and oral steroids, immunosuppression, plasmapheresis, and ultraviolent therapy have been attempted, none of which have been proven to consistently limit progression of the disease.8 The most effective intervention is early physical therapy to preserve functionality and prevent contracture formation. For patients who are eligible, early renal transplantation may offer the best chance of improved mobility. In a case series review by Cuffy and colleagues, 5 of 6 patients who underwent renal transplantation after the development of NSF experienced softening of the involved skin, and 2 patients had improved mobility of joints.9
Conclusion
The case presented here illustrates a possible association between a pro-inflammatory state and the development of NSF. This patient had multiple inflammatory conditions, including MSSA bacteremia, leukocytoclastic vasculitis, and pyoderma gangrenosum (the latter 2 conditions were thought to be associated with his underlying chronic hepatitis C infection), which the authors believe predisposed him to endothelial permeability and risk for developing NSF. The risk of developing NSF in at-risk patients with each episode of gadolinium exposure is estimated around 2.4%, or an incidence of 4.3 cases per 1,000 patient-years, leading the American College of Radiologists to recommend against the administration of gadolinium-based contrast except in cases in which benefits clearly outweigh risks.10 However, an MRI with gadolinium contrast can offer high diagnostic yield in cases such as the one presented here in which a diagnosis remains elusive. Moreover, the use of linear gadolinium-based contrast agents such as gadoversetamide, as in this case, has been reported to be associated with higher incidence of NSF.5 Since this case, the West Los Angeles VAMC has switched to gadobutrol contrast for its MRI protocol, which has been purported to be a lower risk agent compared with that of linear gadolinium-based contrast agents (although several cases of NSF have been reported with gadobutrol in the literature).11
Providers weighing the decision to administer gadolinium contrast to patients with ESRD should discuss the risks and benefits thoroughly, especially in patients with preexisting inflammatory conditions. In addition, although it has not been shown to effectively reduce the risk of NSF after administration of gadolinium, hemodialysis is recommended 2 hours after contrast administration for individuals at risk (the study patient received hemodialysis approximately 18 hours after).12 Given the lack of effective treatment options for NSF, prevention is key. A deeper understanding of the pathophysiology of NSF and identification of its risk factors is paramount to the prevention of this devastating disease.
First described in 2000 in a case series of 15 patients, nephrogenic systemic fibrosis (NSF) is a rare scleroderma-like fibrosing skin condition associated with gadolinium exposure in end stage renal disease (ESRD).1 Patients with advanced chronic kidney disease (CKD) or ESRD are at the highest risk for this condition when exposed to gadolinium-based contrast dyes.
Nephrogenic systemic fibrosis is a devastating and rapidly progressive condition, making its prevention in at-risk populations of utmost importance. In this article, the authors describe a case of a patient who developed NSF in the setting of gadolinium exposure and multiple inflammatory dermatologic conditions. This case illustrates the possible role of a pro-inflammatory state in predisposing to NSF, which may help further elucidate its mechanism of action.
Case Presentation
A 61-year-old Hispanic male with a history of IV heroin use with ESRD secondary to membranous glomerulonephritis on hemodialysis and chronic hepatitis C infection presented to the West Los Angeles VAMC with fevers and night sweats that had persisted for 2 weeks. His physical examination was notable for diffuse tender palpable purpura and petechiae (including his palms and soles), altered mental status, and diffuse myoclonic jerks, which necessitated endotracheal intubation and mechanical ventilation for airway protection. Blood cultures were positive for methicillin-sensitive Staphylococcus aureus (MSSA). Laboratory results were notable for an elevated sedimentation rate of 53 mm/h (0-10 mm/h), C-reactive protein of 19.8 mg/L (< 0.744 mg/dL), and albumin of 1.2 g/dL (3.2-4.8 g/dL). An extensive rheumatologic workup was unrevealing, and a lumbar puncture was unremarkable. A biopsy of his skin lesions was consistent with leukocytoclastic vasculitis.
The patient’s prior hemodialysis access, a tunneled dialysis catheter in the right subclavian vein, was removed given concern for line infection and replaced with an internal jugular temporary hemodialysis line. Given his altered mental status and myoclonic jerks, the decision was made to pursue a magnetic resonance imaging (MRI) scan of the brain and spine with gadolinium contrast to evaluate for cerebral vasculitis and/or septic emboli to the brain.
The patient received 15 mL of gadoversetamide contrast in accordance with hospital imaging protocol. The MRI revealed only chronic ischemic changes. The patient underwent hemodialysis about 18 hours later. The patient was treated with a 6-week course of IV penicillin G. His altered mental status and myoclonic jerks resolved without intervention, and he was then discharged to an acute rehabilitation unit.
Eight weeks after his initial presentation the patient developed a purulent wound on his right forearm (Figure 1)
The patient was discharged to continue physical and occupational therapy to preserve his functional mobility, as no other treatment options were available.
Discussion
Nephrogenic systemic fibrosis is a poorly understood inflammatory condition that produces diffuse fibrosis of the skin. Typically, the disease begins with progressive skin induration of the extremities. Systemic involvement may occur, leading to fibrosis of skeletal muscle, fascia, and multiple organs. Flexion contractures may develop that limit physical function. Fibrosis can become apparent within days to months after exposure to gadolinium contrast.
Beyond renal insufficiency, it is unclear what other risk factors predispose patients to developing this condition. Only a minority of patients with CKD stages 1 through 4 will develop NSF on exposure to gadolinium contrast. However, the incidence of NSF among patients with CKD stage 5 who are exposed to gadolinium has been estimated to be about 13.4% in a prospective study involving 18 patients.2
In a 2015 meta-analysis by Zhang and colleagues, the only clear risk factor identified for the development of NSF, aside from gadolinium exposure, was severe renal insufficiency with a glomerular filtration rate of < 30 mL/min/1.75m2.3 Due to the limited number of patients identified with this disease, it is difficult to identify other risk factors associated with the development of NSF. Based on in vitro studies, it has been postulated that a pro-inflammatory state predisposes patients to develop NSF.4,5 The proposed mechanism for NSF involves extravasation of gadolinium in the setting of vascular endothelial permeability.5,6 Gadolinium then interacts with tissue macrophages, which induce the release of inflammatory cytokines and the secretion of smooth muscle actin by dermal fibroblasts.6,7
Treatment of NSF has been largely unsuccessful. Multiple modalities of treatment that included topical and oral steroids, immunosuppression, plasmapheresis, and ultraviolent therapy have been attempted, none of which have been proven to consistently limit progression of the disease.8 The most effective intervention is early physical therapy to preserve functionality and prevent contracture formation. For patients who are eligible, early renal transplantation may offer the best chance of improved mobility. In a case series review by Cuffy and colleagues, 5 of 6 patients who underwent renal transplantation after the development of NSF experienced softening of the involved skin, and 2 patients had improved mobility of joints.9
Conclusion
The case presented here illustrates a possible association between a pro-inflammatory state and the development of NSF. This patient had multiple inflammatory conditions, including MSSA bacteremia, leukocytoclastic vasculitis, and pyoderma gangrenosum (the latter 2 conditions were thought to be associated with his underlying chronic hepatitis C infection), which the authors believe predisposed him to endothelial permeability and risk for developing NSF. The risk of developing NSF in at-risk patients with each episode of gadolinium exposure is estimated around 2.4%, or an incidence of 4.3 cases per 1,000 patient-years, leading the American College of Radiologists to recommend against the administration of gadolinium-based contrast except in cases in which benefits clearly outweigh risks.10 However, an MRI with gadolinium contrast can offer high diagnostic yield in cases such as the one presented here in which a diagnosis remains elusive. Moreover, the use of linear gadolinium-based contrast agents such as gadoversetamide, as in this case, has been reported to be associated with higher incidence of NSF.5 Since this case, the West Los Angeles VAMC has switched to gadobutrol contrast for its MRI protocol, which has been purported to be a lower risk agent compared with that of linear gadolinium-based contrast agents (although several cases of NSF have been reported with gadobutrol in the literature).11
Providers weighing the decision to administer gadolinium contrast to patients with ESRD should discuss the risks and benefits thoroughly, especially in patients with preexisting inflammatory conditions. In addition, although it has not been shown to effectively reduce the risk of NSF after administration of gadolinium, hemodialysis is recommended 2 hours after contrast administration for individuals at risk (the study patient received hemodialysis approximately 18 hours after).12 Given the lack of effective treatment options for NSF, prevention is key. A deeper understanding of the pathophysiology of NSF and identification of its risk factors is paramount to the prevention of this devastating disease.
1. Cowper SE, Robin HS, Steinberg SM, Su LD, Gupta S, LeBoit PE. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients. Lancet. 2000;356(9234):1000-1001.
2. Todd DJ, Kagan A, Chibnik LB, Kay J. Cutaneous changes of nephrogenic systemic fibrosis. Arthritis Rheum. 2007;56(10):3433-3441.
3. Zhang B, Liang L, Chen W, Liang C, Zhang S. An updated study to determine association between gadolinium-based contrast agents and nephrogenic systemic fibrosis. PLoS One. 2015;10(6):e0129720.
4. Wermuth PJ, Del Galdo F, Jiménez SA. Induction of the expression of profibrotic cytokines and growth factors in normal human peripheral blood monocytes by gadolinium contrast agents. Arthritis Rheum. 2009;60(5):1508-1518.
5. Daftari Besheli L, Aran S, Shaqdan K, Kay J, Abujudeh H. Current status of nephrogenic systemic fibrosis. Clin Radiol. 2014;69(7):661-668.
6. Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;31(1):F1-F11.
7. Idée JM, Fretellier N, Robic C, Corot C. The role of gadolinium chelates in the mechanism of nephrogenic systemic fibrosis: a critical update. Crit Rev Toxicol. 2014;44(10):895-913.
8. Mendoza FA, Artlett CM, Sandorfi N, Latinis K, Piera-Velazquez S, Jimenez SA. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35(4):238-249.
9. Cuffy MC, Singh M, Formica R, et al. Renal transplantation for nephrogenic systemic fibrosis: a case report and review of the literature. Nephrol Dial Transplant. 2011;26(3):1099-1109.
10. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development of gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267
11. Elmholdt TR, Jørgensen B, Ramsing M, Pedersen M, Olesen AB. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287.
12. Abu-Alfa AK. Nephrogenic systemic fibrosis and gadolinium-based contrast agents. Adv Chronic Kidney Dis. 2011;18(3);188-198.
1. Cowper SE, Robin HS, Steinberg SM, Su LD, Gupta S, LeBoit PE. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients. Lancet. 2000;356(9234):1000-1001.
2. Todd DJ, Kagan A, Chibnik LB, Kay J. Cutaneous changes of nephrogenic systemic fibrosis. Arthritis Rheum. 2007;56(10):3433-3441.
3. Zhang B, Liang L, Chen W, Liang C, Zhang S. An updated study to determine association between gadolinium-based contrast agents and nephrogenic systemic fibrosis. PLoS One. 2015;10(6):e0129720.
4. Wermuth PJ, Del Galdo F, Jiménez SA. Induction of the expression of profibrotic cytokines and growth factors in normal human peripheral blood monocytes by gadolinium contrast agents. Arthritis Rheum. 2009;60(5):1508-1518.
5. Daftari Besheli L, Aran S, Shaqdan K, Kay J, Abujudeh H. Current status of nephrogenic systemic fibrosis. Clin Radiol. 2014;69(7):661-668.
6. Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;31(1):F1-F11.
7. Idée JM, Fretellier N, Robic C, Corot C. The role of gadolinium chelates in the mechanism of nephrogenic systemic fibrosis: a critical update. Crit Rev Toxicol. 2014;44(10):895-913.
8. Mendoza FA, Artlett CM, Sandorfi N, Latinis K, Piera-Velazquez S, Jimenez SA. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35(4):238-249.
9. Cuffy MC, Singh M, Formica R, et al. Renal transplantation for nephrogenic systemic fibrosis: a case report and review of the literature. Nephrol Dial Transplant. 2011;26(3):1099-1109.
10. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development of gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267
11. Elmholdt TR, Jørgensen B, Ramsing M, Pedersen M, Olesen AB. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287.
12. Abu-Alfa AK. Nephrogenic systemic fibrosis and gadolinium-based contrast agents. Adv Chronic Kidney Dis. 2011;18(3);188-198.