Article

Clustered Vesicles on the Neck
A 6-year-old girl presented to the dermatology clinic with a rash on the right side of the neck that was noted at birth as a small raised lesion...
Article
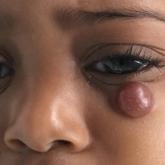
Rubbery Nodule on the Face of an Infant
A 10-month-old girl presented with a facial nodule of 7 months’ duration that started as a small lesion. On physical examination, a single 10×10-...