User login
Blueberry Muffin Rash Secondary to Hereditary Spherocytosis
The term blueberry muffin rash historically was used to describe the cutaneous manifestations observed in congenital rubella. The term traditionally describes the result of a postnatal dermal extramedullary hematopoiesis. Today, TORCH (toxoplasmosis, other agents, rubella, cytomegalovirus, herpes) infections and plasma cell dyscrasias are all potential causes of extramedullary hematopoiesis. Herein, we present a unique case of a neonate born with a blueberry muffin rash secondary to extramedullary hematopoiesis induced by hereditary spherocytosis.
Case Report
The dermatology department was consulted to evaluate a 2-day-old male neonate born with a “rash.” The patient was born to a 34-year-old gravida 3, para 2, woman at 39 weeks’ gestation. The mother’s prenatal laboratory values were within reference range and ultrasounds were normal, and she was compliant with her prenatal care. She underwent a normal spontaneous vaginal delivery 3 hours after rupture of membranes without complication. The amniotic fluid and umbilical cord both were clear. There was no use of forceps or any other external aiding devices during the delivery. At the time of delivery, the consulting physician noted that the patient had “skin lesions from head to toe.”
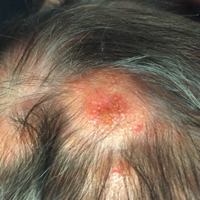
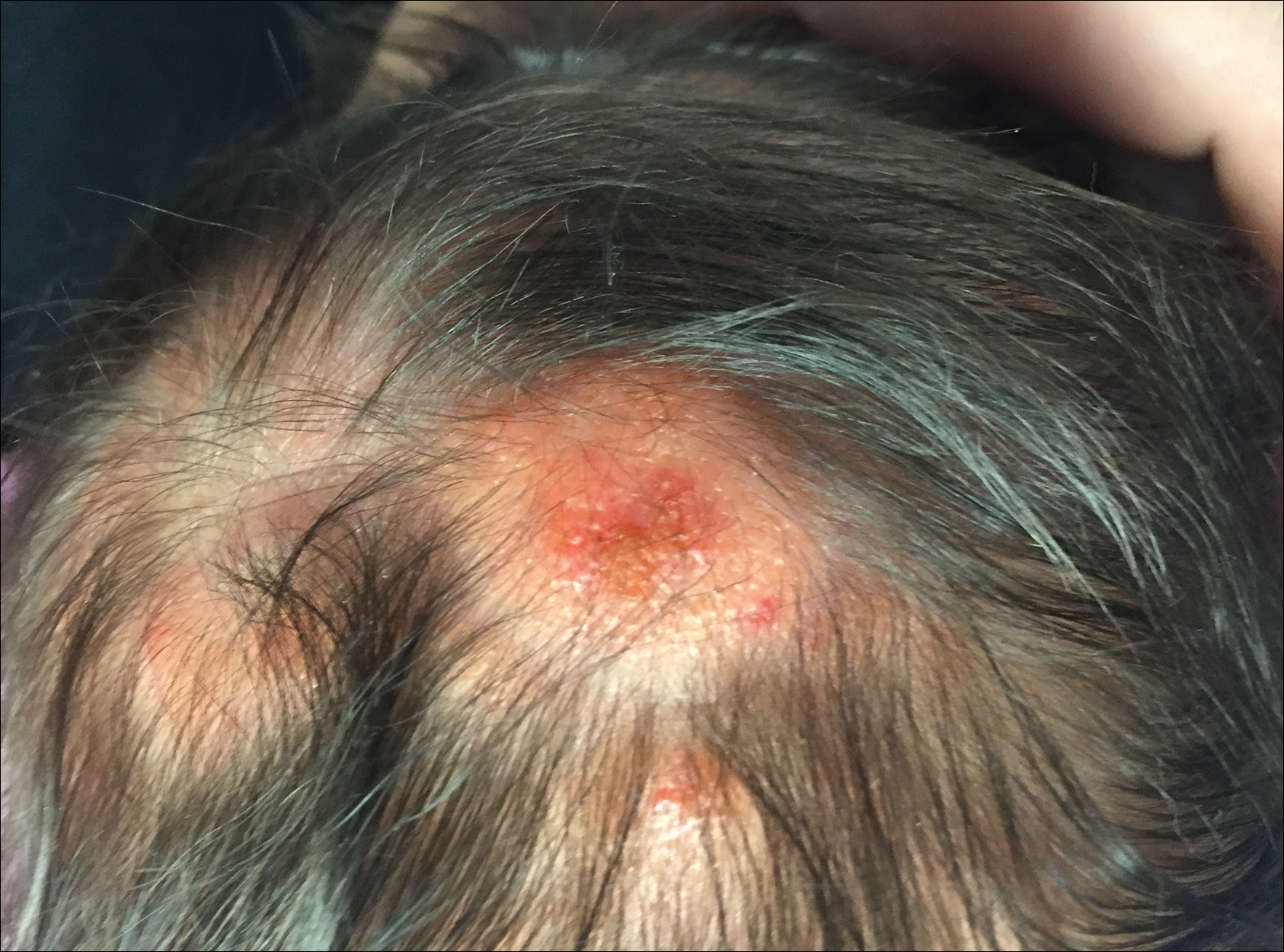
The patient’s parents reported that the rash did not seem to cause any discomfort for the patient. In the 24 hours after birth, the parents reported that the erythema seemed to slightly fade. Physical examination revealed many scattered erythematous to violaceous, nonblanching papulonodules affecting the scalp (Figure 1), face, arms, hands (Figure 2A), back (Figure 2B), buttocks, legs, and feet. Some of the papulonodules were soft while others were firm and indurated. Several lesions had a yellowish hue with some overlying crust. There was no mucosal, genital, or ocular involvement. No erosions, ulcerations, petechiae, ecchymoses, or hepatosplenomegaly were noted on examination.

The patient was otherwise healthy with an Apgar score of 8/9 at 1 and 5 minutes. His birth weight, length, and head circumference were within normal limits. There was no evidence of ABO blood group or Rhesus factor incompatibility. His temperature, vital signs, laboratory values (including calcium level and TORCH titers, which included cytomegalovirus, rubella, toxoplasmosis, and herpes simplex virus), and review of systems all were within reference range. A bone survey of the skull, spine, ribs, arms, pelvis, legs, and feet was within normal limits.
The mother’s placenta was sent for pathology and revealed a lymphoplasmacytic chronic deciduitis and acute subchorionitis consistent with a nonspecific inflammatory response, unlikely to be from an infectious etiology.
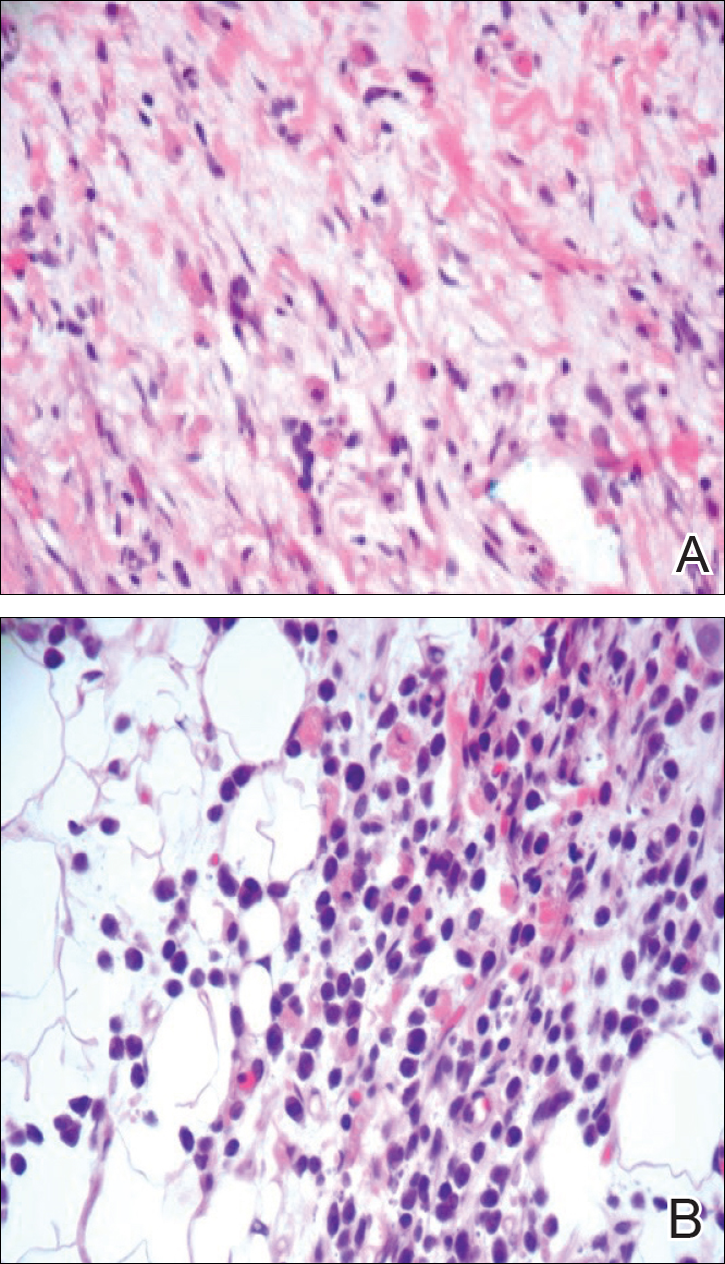
A 4-mm punch biopsy was taken from the left thigh and revealed a predominately lymphocytic infiltrate with rare eosinophils and erythrocyte precursors (Figure 3). Immunohistochemical staining was performed showing that the majority of the lymphocytes represented T lymphocytes, which stained positive for CD45 and CD3 and negative for S-100, CD1a, CD30, and CD117. There were scattered CD34+ cells, and scattered cells stained positive for myeloperoxidase. No significant CD20 immunoreactivity was noted. There were scattered eosinophils and rare normoblasts but no megakaryocytes. A complete blood cell count (CBC) with differential and reticulocyte count was within reference range.
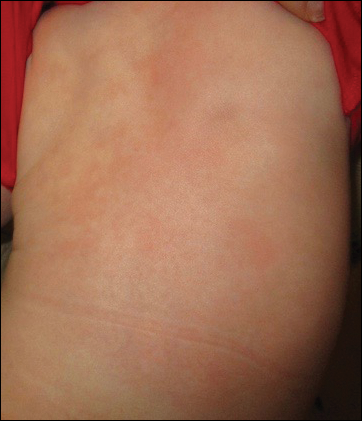
At 1-, 3-, 8-, 12-, and 28-week follow-up visits, the patient continued to grow and feed appropriately. No new lesions developed during this time, and the preexisting lesions continued to fade into slightly hyperpigmented patches without induration (Figure 4). At 6 months of age, a CBC performed at the time of an upper respiratory infection and otitis media revealed normocytic anemia with a hemoglobin level of 9.9 g/dL (reference range, 14.0–17.5 g/dL), a reticulocyte count of 0.8% (reference range, 0.5%–1.5%), and a lactate dehydrogenase level of 424 U/L (reference range, 100–200 U/L). All red blood cell (RBC) indices were within reference range. Flow cytometry, eosin-5-maleimide, and ektacytometry were performed with results consistent with mild hereditary spherocytosis.
Comment
Dermal extramedullary hematopoiesis is a normal component of embryologic development up until the fifth month of gestation.1 The term blueberry muffin rash typically is used to describe the cutaneous manifestations of extramedullary hematopoiesis, which commonly is caused by a TORCH infection or hematologic dyscrasia.2 It has been suggested that the term be expanded to include neoplastic processes (eg, neuroblastomas) and vascular processes (eg, multiple hemangiomas, blue rubber bleb nevus syndrome, glomangiomas, multifocal lymphangionendotheliomatosis), which although not associated with an extramedullary hematopoiesis, can clinically resemble a blueberry muffin rash.
Because of the potential for serious systemic complications, a cause must be sought for all newborns presenting with a blueberry muffin rash. Our patient’s lack of cardiovascular, otic, and ocular involvement combined with a negative TORCH screen and normal CBC strongly suggested against a TORCH infection. In addition, a normal bone survey and CBC, as well as a lack of petechiae, ecchymoses, and hepatosplenomegaly, were evidence against congenital leukemia.3 With the spontaneously resolving lesions and apparent clinical resolution, a bone marrow biopsy was not performed. The skin biopsy revealed negative staining for S-100 and CD1a, making the diagnosis of congenital self-healing reticulohistiocytosis unlikely. No panniculitis was noted and calcium levels were normal, ruling out subcutaneous fat necrosis of the newborn. The predominantly T-cell lymphocytic infiltrate demonstrated on skin biopsy led us to a differential diagnosis of aleukemic leukemia cutis versus idiopathic dermal extramedullary hematopoiesis; however, normocytic anemia was later identified when the patient’s hemoglobin level dropped to 9.9 g/dL. The abnormal eosin-5-maleimide and ektacytometry results unmasked a hereditary spherocytosis.
Hereditary spherocytosis typically is inherited in an autosomal-dominant manner and may be caused by mutations in ankyrin-1, band 3, spectrin, or protein 4.2 on the erythrocyte membrane. It is the third leading cause of hemolytic anemia in newborns and the leading cause of direct Coombs-negative hemolytic anemia requiring blood transfusion in neonates. It is most common in neonates of Northern European ancestry, affecting 1 in every 1000 to 2000 births.4 Presentation may range from asymptomatic to severe anemia with hydrops fetalis. Most neonates have an elevated mean corpuscular hemoglobin and low mean corpuscular volume. Acute illness may cause hemolytic or aplastic crises, possibly explaining our patient’s normocytic anemia discovered on a CBC during an episode of an upper respiratory infection and otitis media.
Treatment options for hereditary spherocytosis include phototherapy for jaundiced neonates, folate supplementation, packed erythrocyte transfusions for symptomatic anemia, and recombinant erythropoietin in neonates.4 Splenectomy is curative for the majority of patients and requires immunization against Streptococcus pneumoniae, Haemophilus influenzae type b, and Neisseria meningitidis several weeks preoperatively. Patients with symptomatic gallstones may be treated with cholecystectomy at the time of splenectomy or by laparoscopic cholecystectomy, endoscopic sphincterotomy, cholecystostomy, or extracorporeal cholecystolithotripsy.5
Although a PubMed search of articles indexed for MEDLINE using the terms dermal hematopoiesis, extramedullary hematopoiesis, hereditary spherocytosis, and blueberry muffin rash yielded only 1 other known case of blueberry muffin rash caused by hereditary spherocytosis,6 other case reports demonstrate extramedullary hematopoiesis in hereditary spherocytosis patients in locations other than the skin. Calhoun et al7 described a case of a 9-year-old boy with hereditary spherocytosis who presented with jaundice. Pathologic examination revealed a 5-cm suprarenal mass demonstrating extramedullary hematopoiesis.7 A case reported by Xiros et al8 described a 64-year-old man with a history of hereditary spherocytosis who presented with hemothorax from paravertebral extramedullary hematopoiesis. De Backer et al9 reported a case of a 60-year-old man diagnosed with hereditary spherocytosis after an abnormal CBC who was subsequently found to have paravertebral masses containing extramedullary hematopoiesis.
There is one known case of a blueberry muffin rash caused by hereditary spherocytosis.6 A female neonate was born at 38 weeks’ gestation with multiple petechiae and faint purpuric papules. Initial complications included intracranial ventricular hemorrhage, hyperbilirubinemia, and anemia requiring blood transfusions on the first day of life. TORCH titers were negative and a skin biopsy demonstrated a diffuse infiltrate of mature RBCs, normoblasts, and pronormoblasts in the reticular dermis. She was healthy until 3 months of age when she had several days of vomiting and diarrhea. Laboratory workup revealed a hematocrit level of 20.5% (reference range, 41%–50%); a reticulocyte count of 22.6% (reference range, 0.5%–1.5%); and a peripheral blood smear demonstrating polychromatophilia, anisocytosis, and spherocytosis. She was then diagnosed with hereditary spherocytosis.6
Hereditary spherocytosis is a known, albeit rare, cause of extramedullary hematopoiesis presenting as blueberry muffin rash. Patients with mild hereditary spherocytosis may have a compensated hemolysis without anemia or spherocytes on peripheral smear, which may explain the lack of severe hemolytic anemia or RBC-predominant pathology in our patient.5 Argyle and Zone6 proposed that severe hemolysis and hypoxia were the cause of extramedullary hematopoiesis in their patient. Because our patient did not experience a notable hemolytic episode until he had an upper respiratory infection and otitis media at 6 months of age, the pathophysiology is less clear; a compensated hemolytic process may underlie the extramedullary hematopoiesis and normal RBC indices.
Regardless of the precise cause of extramedullary hematopoiesis in our patient, this case of a T lymphocyte–dominant cutaneous infiltrate in a patient with mild hereditary spherocytosis is exceptionally rare and leads us to consider that perhaps there are causes of this pathology that are unknown to us.
- Zhang IH, Zane LT, Braun BS, et al. Congenital leukemia cutis with subsequent development of leukemia. J Am Acad Dermatol. 2006;54(2 suppl):S22–S27.
- Karmegaraj B, Vijayakumar S, Ramanathan R, et al. Extramedullary haematopoiesis resembling a blueberry muffin, in a neonate. BMJ Case Rep. pii: bcr2014208473. doi: 10.1136/bcr-2014-208473.
- Handler MZ, Schwartz RA. Neonatal leukaemia cutis. J Eur Acad Dermatol Venereol. 2015;29:1884-1889.
- Christensen RD, Yaish HM, Gallagher PG. A pediatrician’s practical guide to diagnosing and treating hereditary spherocytosis in neonates. Pediatrics. 2015;135:1107-1114.
- Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411-1426.
- Argyle JC, Zone JJ. Dermal erythropoiesis in a neonate. Arch Dermatol. 1981;117:492-494.
- Calhoun SK, Murphy RC, Shariati N, et al. Extramedullary hematopoiesis in a child with hereditary spherocytosis: an uncommon cause of an adrenal mass. Pediatr Radiol. 2001;31:879-881.
- Xiros N, Economopoulos T, Papageorgiou E, et al. Massive hemothorax due to intrathoracic extramedullary hematopoiesis in a patient with hereditary spherocytosis. Ann Hematol. 2001;80:38-40.
- De Backer AI, Zachée P, Vanschoubroeck IJ, et al. Extramedullary paraspinal hematopoiesis in hereditary spherocytosis. JBR-BTR. 2002;85:206-208.
The term blueberry muffin rash historically was used to describe the cutaneous manifestations observed in congenital rubella. The term traditionally describes the result of a postnatal dermal extramedullary hematopoiesis. Today, TORCH (toxoplasmosis, other agents, rubella, cytomegalovirus, herpes) infections and plasma cell dyscrasias are all potential causes of extramedullary hematopoiesis. Herein, we present a unique case of a neonate born with a blueberry muffin rash secondary to extramedullary hematopoiesis induced by hereditary spherocytosis.
Case Report
The dermatology department was consulted to evaluate a 2-day-old male neonate born with a “rash.” The patient was born to a 34-year-old gravida 3, para 2, woman at 39 weeks’ gestation. The mother’s prenatal laboratory values were within reference range and ultrasounds were normal, and she was compliant with her prenatal care. She underwent a normal spontaneous vaginal delivery 3 hours after rupture of membranes without complication. The amniotic fluid and umbilical cord both were clear. There was no use of forceps or any other external aiding devices during the delivery. At the time of delivery, the consulting physician noted that the patient had “skin lesions from head to toe.”
The patient’s parents reported that the rash did not seem to cause any discomfort for the patient. In the 24 hours after birth, the parents reported that the erythema seemed to slightly fade. Physical examination revealed many scattered erythematous to violaceous, nonblanching papulonodules affecting the scalp (Figure 1), face, arms, hands (Figure 2A), back (Figure 2B), buttocks, legs, and feet. Some of the papulonodules were soft while others were firm and indurated. Several lesions had a yellowish hue with some overlying crust. There was no mucosal, genital, or ocular involvement. No erosions, ulcerations, petechiae, ecchymoses, or hepatosplenomegaly were noted on examination.
The patient was otherwise healthy with an Apgar score of 8/9 at 1 and 5 minutes. His birth weight, length, and head circumference were within normal limits. There was no evidence of ABO blood group or Rhesus factor incompatibility. His temperature, vital signs, laboratory values (including calcium level and TORCH titers, which included cytomegalovirus, rubella, toxoplasmosis, and herpes simplex virus), and review of systems all were within reference range. A bone survey of the skull, spine, ribs, arms, pelvis, legs, and feet was within normal limits.
The mother’s placenta was sent for pathology and revealed a lymphoplasmacytic chronic deciduitis and acute subchorionitis consistent with a nonspecific inflammatory response, unlikely to be from an infectious etiology.
A 4-mm punch biopsy was taken from the left thigh and revealed a predominately lymphocytic infiltrate with rare eosinophils and erythrocyte precursors (Figure 3). Immunohistochemical staining was performed showing that the majority of the lymphocytes represented T lymphocytes, which stained positive for CD45 and CD3 and negative for S-100, CD1a, CD30, and CD117. There were scattered CD34+ cells, and scattered cells stained positive for myeloperoxidase. No significant CD20 immunoreactivity was noted. There were scattered eosinophils and rare normoblasts but no megakaryocytes. A complete blood cell count (CBC) with differential and reticulocyte count was within reference range.
At 1-, 3-, 8-, 12-, and 28-week follow-up visits, the patient continued to grow and feed appropriately. No new lesions developed during this time, and the preexisting lesions continued to fade into slightly hyperpigmented patches without induration (Figure 4). At 6 months of age, a CBC performed at the time of an upper respiratory infection and otitis media revealed normocytic anemia with a hemoglobin level of 9.9 g/dL (reference range, 14.0–17.5 g/dL), a reticulocyte count of 0.8% (reference range, 0.5%–1.5%), and a lactate dehydrogenase level of 424 U/L (reference range, 100–200 U/L). All red blood cell (RBC) indices were within reference range. Flow cytometry, eosin-5-maleimide, and ektacytometry were performed with results consistent with mild hereditary spherocytosis.
Comment
Dermal extramedullary hematopoiesis is a normal component of embryologic development up until the fifth month of gestation.1 The term blueberry muffin rash typically is used to describe the cutaneous manifestations of extramedullary hematopoiesis, which commonly is caused by a TORCH infection or hematologic dyscrasia.2 It has been suggested that the term be expanded to include neoplastic processes (eg, neuroblastomas) and vascular processes (eg, multiple hemangiomas, blue rubber bleb nevus syndrome, glomangiomas, multifocal lymphangionendotheliomatosis), which although not associated with an extramedullary hematopoiesis, can clinically resemble a blueberry muffin rash.
Because of the potential for serious systemic complications, a cause must be sought for all newborns presenting with a blueberry muffin rash. Our patient’s lack of cardiovascular, otic, and ocular involvement combined with a negative TORCH screen and normal CBC strongly suggested against a TORCH infection. In addition, a normal bone survey and CBC, as well as a lack of petechiae, ecchymoses, and hepatosplenomegaly, were evidence against congenital leukemia.3 With the spontaneously resolving lesions and apparent clinical resolution, a bone marrow biopsy was not performed. The skin biopsy revealed negative staining for S-100 and CD1a, making the diagnosis of congenital self-healing reticulohistiocytosis unlikely. No panniculitis was noted and calcium levels were normal, ruling out subcutaneous fat necrosis of the newborn. The predominantly T-cell lymphocytic infiltrate demonstrated on skin biopsy led us to a differential diagnosis of aleukemic leukemia cutis versus idiopathic dermal extramedullary hematopoiesis; however, normocytic anemia was later identified when the patient’s hemoglobin level dropped to 9.9 g/dL. The abnormal eosin-5-maleimide and ektacytometry results unmasked a hereditary spherocytosis.
Hereditary spherocytosis typically is inherited in an autosomal-dominant manner and may be caused by mutations in ankyrin-1, band 3, spectrin, or protein 4.2 on the erythrocyte membrane. It is the third leading cause of hemolytic anemia in newborns and the leading cause of direct Coombs-negative hemolytic anemia requiring blood transfusion in neonates. It is most common in neonates of Northern European ancestry, affecting 1 in every 1000 to 2000 births.4 Presentation may range from asymptomatic to severe anemia with hydrops fetalis. Most neonates have an elevated mean corpuscular hemoglobin and low mean corpuscular volume. Acute illness may cause hemolytic or aplastic crises, possibly explaining our patient’s normocytic anemia discovered on a CBC during an episode of an upper respiratory infection and otitis media.
Treatment options for hereditary spherocytosis include phototherapy for jaundiced neonates, folate supplementation, packed erythrocyte transfusions for symptomatic anemia, and recombinant erythropoietin in neonates.4 Splenectomy is curative for the majority of patients and requires immunization against Streptococcus pneumoniae, Haemophilus influenzae type b, and Neisseria meningitidis several weeks preoperatively. Patients with symptomatic gallstones may be treated with cholecystectomy at the time of splenectomy or by laparoscopic cholecystectomy, endoscopic sphincterotomy, cholecystostomy, or extracorporeal cholecystolithotripsy.5
Although a PubMed search of articles indexed for MEDLINE using the terms dermal hematopoiesis, extramedullary hematopoiesis, hereditary spherocytosis, and blueberry muffin rash yielded only 1 other known case of blueberry muffin rash caused by hereditary spherocytosis,6 other case reports demonstrate extramedullary hematopoiesis in hereditary spherocytosis patients in locations other than the skin. Calhoun et al7 described a case of a 9-year-old boy with hereditary spherocytosis who presented with jaundice. Pathologic examination revealed a 5-cm suprarenal mass demonstrating extramedullary hematopoiesis.7 A case reported by Xiros et al8 described a 64-year-old man with a history of hereditary spherocytosis who presented with hemothorax from paravertebral extramedullary hematopoiesis. De Backer et al9 reported a case of a 60-year-old man diagnosed with hereditary spherocytosis after an abnormal CBC who was subsequently found to have paravertebral masses containing extramedullary hematopoiesis.
There is one known case of a blueberry muffin rash caused by hereditary spherocytosis.6 A female neonate was born at 38 weeks’ gestation with multiple petechiae and faint purpuric papules. Initial complications included intracranial ventricular hemorrhage, hyperbilirubinemia, and anemia requiring blood transfusions on the first day of life. TORCH titers were negative and a skin biopsy demonstrated a diffuse infiltrate of mature RBCs, normoblasts, and pronormoblasts in the reticular dermis. She was healthy until 3 months of age when she had several days of vomiting and diarrhea. Laboratory workup revealed a hematocrit level of 20.5% (reference range, 41%–50%); a reticulocyte count of 22.6% (reference range, 0.5%–1.5%); and a peripheral blood smear demonstrating polychromatophilia, anisocytosis, and spherocytosis. She was then diagnosed with hereditary spherocytosis.6
Hereditary spherocytosis is a known, albeit rare, cause of extramedullary hematopoiesis presenting as blueberry muffin rash. Patients with mild hereditary spherocytosis may have a compensated hemolysis without anemia or spherocytes on peripheral smear, which may explain the lack of severe hemolytic anemia or RBC-predominant pathology in our patient.5 Argyle and Zone6 proposed that severe hemolysis and hypoxia were the cause of extramedullary hematopoiesis in their patient. Because our patient did not experience a notable hemolytic episode until he had an upper respiratory infection and otitis media at 6 months of age, the pathophysiology is less clear; a compensated hemolytic process may underlie the extramedullary hematopoiesis and normal RBC indices.
Regardless of the precise cause of extramedullary hematopoiesis in our patient, this case of a T lymphocyte–dominant cutaneous infiltrate in a patient with mild hereditary spherocytosis is exceptionally rare and leads us to consider that perhaps there are causes of this pathology that are unknown to us.
The term blueberry muffin rash historically was used to describe the cutaneous manifestations observed in congenital rubella. The term traditionally describes the result of a postnatal dermal extramedullary hematopoiesis. Today, TORCH (toxoplasmosis, other agents, rubella, cytomegalovirus, herpes) infections and plasma cell dyscrasias are all potential causes of extramedullary hematopoiesis. Herein, we present a unique case of a neonate born with a blueberry muffin rash secondary to extramedullary hematopoiesis induced by hereditary spherocytosis.
Case Report
The dermatology department was consulted to evaluate a 2-day-old male neonate born with a “rash.” The patient was born to a 34-year-old gravida 3, para 2, woman at 39 weeks’ gestation. The mother’s prenatal laboratory values were within reference range and ultrasounds were normal, and she was compliant with her prenatal care. She underwent a normal spontaneous vaginal delivery 3 hours after rupture of membranes without complication. The amniotic fluid and umbilical cord both were clear. There was no use of forceps or any other external aiding devices during the delivery. At the time of delivery, the consulting physician noted that the patient had “skin lesions from head to toe.”
The patient’s parents reported that the rash did not seem to cause any discomfort for the patient. In the 24 hours after birth, the parents reported that the erythema seemed to slightly fade. Physical examination revealed many scattered erythematous to violaceous, nonblanching papulonodules affecting the scalp (Figure 1), face, arms, hands (Figure 2A), back (Figure 2B), buttocks, legs, and feet. Some of the papulonodules were soft while others were firm and indurated. Several lesions had a yellowish hue with some overlying crust. There was no mucosal, genital, or ocular involvement. No erosions, ulcerations, petechiae, ecchymoses, or hepatosplenomegaly were noted on examination.
The patient was otherwise healthy with an Apgar score of 8/9 at 1 and 5 minutes. His birth weight, length, and head circumference were within normal limits. There was no evidence of ABO blood group or Rhesus factor incompatibility. His temperature, vital signs, laboratory values (including calcium level and TORCH titers, which included cytomegalovirus, rubella, toxoplasmosis, and herpes simplex virus), and review of systems all were within reference range. A bone survey of the skull, spine, ribs, arms, pelvis, legs, and feet was within normal limits.
The mother’s placenta was sent for pathology and revealed a lymphoplasmacytic chronic deciduitis and acute subchorionitis consistent with a nonspecific inflammatory response, unlikely to be from an infectious etiology.
A 4-mm punch biopsy was taken from the left thigh and revealed a predominately lymphocytic infiltrate with rare eosinophils and erythrocyte precursors (Figure 3). Immunohistochemical staining was performed showing that the majority of the lymphocytes represented T lymphocytes, which stained positive for CD45 and CD3 and negative for S-100, CD1a, CD30, and CD117. There were scattered CD34+ cells, and scattered cells stained positive for myeloperoxidase. No significant CD20 immunoreactivity was noted. There were scattered eosinophils and rare normoblasts but no megakaryocytes. A complete blood cell count (CBC) with differential and reticulocyte count was within reference range.
At 1-, 3-, 8-, 12-, and 28-week follow-up visits, the patient continued to grow and feed appropriately. No new lesions developed during this time, and the preexisting lesions continued to fade into slightly hyperpigmented patches without induration (Figure 4). At 6 months of age, a CBC performed at the time of an upper respiratory infection and otitis media revealed normocytic anemia with a hemoglobin level of 9.9 g/dL (reference range, 14.0–17.5 g/dL), a reticulocyte count of 0.8% (reference range, 0.5%–1.5%), and a lactate dehydrogenase level of 424 U/L (reference range, 100–200 U/L). All red blood cell (RBC) indices were within reference range. Flow cytometry, eosin-5-maleimide, and ektacytometry were performed with results consistent with mild hereditary spherocytosis.
Comment
Dermal extramedullary hematopoiesis is a normal component of embryologic development up until the fifth month of gestation.1 The term blueberry muffin rash typically is used to describe the cutaneous manifestations of extramedullary hematopoiesis, which commonly is caused by a TORCH infection or hematologic dyscrasia.2 It has been suggested that the term be expanded to include neoplastic processes (eg, neuroblastomas) and vascular processes (eg, multiple hemangiomas, blue rubber bleb nevus syndrome, glomangiomas, multifocal lymphangionendotheliomatosis), which although not associated with an extramedullary hematopoiesis, can clinically resemble a blueberry muffin rash.
Because of the potential for serious systemic complications, a cause must be sought for all newborns presenting with a blueberry muffin rash. Our patient’s lack of cardiovascular, otic, and ocular involvement combined with a negative TORCH screen and normal CBC strongly suggested against a TORCH infection. In addition, a normal bone survey and CBC, as well as a lack of petechiae, ecchymoses, and hepatosplenomegaly, were evidence against congenital leukemia.3 With the spontaneously resolving lesions and apparent clinical resolution, a bone marrow biopsy was not performed. The skin biopsy revealed negative staining for S-100 and CD1a, making the diagnosis of congenital self-healing reticulohistiocytosis unlikely. No panniculitis was noted and calcium levels were normal, ruling out subcutaneous fat necrosis of the newborn. The predominantly T-cell lymphocytic infiltrate demonstrated on skin biopsy led us to a differential diagnosis of aleukemic leukemia cutis versus idiopathic dermal extramedullary hematopoiesis; however, normocytic anemia was later identified when the patient’s hemoglobin level dropped to 9.9 g/dL. The abnormal eosin-5-maleimide and ektacytometry results unmasked a hereditary spherocytosis.
Hereditary spherocytosis typically is inherited in an autosomal-dominant manner and may be caused by mutations in ankyrin-1, band 3, spectrin, or protein 4.2 on the erythrocyte membrane. It is the third leading cause of hemolytic anemia in newborns and the leading cause of direct Coombs-negative hemolytic anemia requiring blood transfusion in neonates. It is most common in neonates of Northern European ancestry, affecting 1 in every 1000 to 2000 births.4 Presentation may range from asymptomatic to severe anemia with hydrops fetalis. Most neonates have an elevated mean corpuscular hemoglobin and low mean corpuscular volume. Acute illness may cause hemolytic or aplastic crises, possibly explaining our patient’s normocytic anemia discovered on a CBC during an episode of an upper respiratory infection and otitis media.
Treatment options for hereditary spherocytosis include phototherapy for jaundiced neonates, folate supplementation, packed erythrocyte transfusions for symptomatic anemia, and recombinant erythropoietin in neonates.4 Splenectomy is curative for the majority of patients and requires immunization against Streptococcus pneumoniae, Haemophilus influenzae type b, and Neisseria meningitidis several weeks preoperatively. Patients with symptomatic gallstones may be treated with cholecystectomy at the time of splenectomy or by laparoscopic cholecystectomy, endoscopic sphincterotomy, cholecystostomy, or extracorporeal cholecystolithotripsy.5
Although a PubMed search of articles indexed for MEDLINE using the terms dermal hematopoiesis, extramedullary hematopoiesis, hereditary spherocytosis, and blueberry muffin rash yielded only 1 other known case of blueberry muffin rash caused by hereditary spherocytosis,6 other case reports demonstrate extramedullary hematopoiesis in hereditary spherocytosis patients in locations other than the skin. Calhoun et al7 described a case of a 9-year-old boy with hereditary spherocytosis who presented with jaundice. Pathologic examination revealed a 5-cm suprarenal mass demonstrating extramedullary hematopoiesis.7 A case reported by Xiros et al8 described a 64-year-old man with a history of hereditary spherocytosis who presented with hemothorax from paravertebral extramedullary hematopoiesis. De Backer et al9 reported a case of a 60-year-old man diagnosed with hereditary spherocytosis after an abnormal CBC who was subsequently found to have paravertebral masses containing extramedullary hematopoiesis.
There is one known case of a blueberry muffin rash caused by hereditary spherocytosis.6 A female neonate was born at 38 weeks’ gestation with multiple petechiae and faint purpuric papules. Initial complications included intracranial ventricular hemorrhage, hyperbilirubinemia, and anemia requiring blood transfusions on the first day of life. TORCH titers were negative and a skin biopsy demonstrated a diffuse infiltrate of mature RBCs, normoblasts, and pronormoblasts in the reticular dermis. She was healthy until 3 months of age when she had several days of vomiting and diarrhea. Laboratory workup revealed a hematocrit level of 20.5% (reference range, 41%–50%); a reticulocyte count of 22.6% (reference range, 0.5%–1.5%); and a peripheral blood smear demonstrating polychromatophilia, anisocytosis, and spherocytosis. She was then diagnosed with hereditary spherocytosis.6
Hereditary spherocytosis is a known, albeit rare, cause of extramedullary hematopoiesis presenting as blueberry muffin rash. Patients with mild hereditary spherocytosis may have a compensated hemolysis without anemia or spherocytes on peripheral smear, which may explain the lack of severe hemolytic anemia or RBC-predominant pathology in our patient.5 Argyle and Zone6 proposed that severe hemolysis and hypoxia were the cause of extramedullary hematopoiesis in their patient. Because our patient did not experience a notable hemolytic episode until he had an upper respiratory infection and otitis media at 6 months of age, the pathophysiology is less clear; a compensated hemolytic process may underlie the extramedullary hematopoiesis and normal RBC indices.
Regardless of the precise cause of extramedullary hematopoiesis in our patient, this case of a T lymphocyte–dominant cutaneous infiltrate in a patient with mild hereditary spherocytosis is exceptionally rare and leads us to consider that perhaps there are causes of this pathology that are unknown to us.
- Zhang IH, Zane LT, Braun BS, et al. Congenital leukemia cutis with subsequent development of leukemia. J Am Acad Dermatol. 2006;54(2 suppl):S22–S27.
- Karmegaraj B, Vijayakumar S, Ramanathan R, et al. Extramedullary haematopoiesis resembling a blueberry muffin, in a neonate. BMJ Case Rep. pii: bcr2014208473. doi: 10.1136/bcr-2014-208473.
- Handler MZ, Schwartz RA. Neonatal leukaemia cutis. J Eur Acad Dermatol Venereol. 2015;29:1884-1889.
- Christensen RD, Yaish HM, Gallagher PG. A pediatrician’s practical guide to diagnosing and treating hereditary spherocytosis in neonates. Pediatrics. 2015;135:1107-1114.
- Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411-1426.
- Argyle JC, Zone JJ. Dermal erythropoiesis in a neonate. Arch Dermatol. 1981;117:492-494.
- Calhoun SK, Murphy RC, Shariati N, et al. Extramedullary hematopoiesis in a child with hereditary spherocytosis: an uncommon cause of an adrenal mass. Pediatr Radiol. 2001;31:879-881.
- Xiros N, Economopoulos T, Papageorgiou E, et al. Massive hemothorax due to intrathoracic extramedullary hematopoiesis in a patient with hereditary spherocytosis. Ann Hematol. 2001;80:38-40.
- De Backer AI, Zachée P, Vanschoubroeck IJ, et al. Extramedullary paraspinal hematopoiesis in hereditary spherocytosis. JBR-BTR. 2002;85:206-208.
- Zhang IH, Zane LT, Braun BS, et al. Congenital leukemia cutis with subsequent development of leukemia. J Am Acad Dermatol. 2006;54(2 suppl):S22–S27.
- Karmegaraj B, Vijayakumar S, Ramanathan R, et al. Extramedullary haematopoiesis resembling a blueberry muffin, in a neonate. BMJ Case Rep. pii: bcr2014208473. doi: 10.1136/bcr-2014-208473.
- Handler MZ, Schwartz RA. Neonatal leukaemia cutis. J Eur Acad Dermatol Venereol. 2015;29:1884-1889.
- Christensen RD, Yaish HM, Gallagher PG. A pediatrician’s practical guide to diagnosing and treating hereditary spherocytosis in neonates. Pediatrics. 2015;135:1107-1114.
- Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411-1426.
- Argyle JC, Zone JJ. Dermal erythropoiesis in a neonate. Arch Dermatol. 1981;117:492-494.
- Calhoun SK, Murphy RC, Shariati N, et al. Extramedullary hematopoiesis in a child with hereditary spherocytosis: an uncommon cause of an adrenal mass. Pediatr Radiol. 2001;31:879-881.
- Xiros N, Economopoulos T, Papageorgiou E, et al. Massive hemothorax due to intrathoracic extramedullary hematopoiesis in a patient with hereditary spherocytosis. Ann Hematol. 2001;80:38-40.
- De Backer AI, Zachée P, Vanschoubroeck IJ, et al. Extramedullary paraspinal hematopoiesis in hereditary spherocytosis. JBR-BTR. 2002;85:206-208.
Practice Points
- The term blueberry muffin rash is used to describe the clinical presentation of dermal extramedullary hematopoiesis. The common culprits of this rash include a TORCH (toxoplasmosis, other agents, rubella, cytomegalovirus, herpes) infection or hematologic dyscrasia.
- Because of the potential for serious systemic complications, a cause must be sought for all newborns presenting with a blueberry muffin rash.
- Hereditary spherocytosis typically is inherited in an autosomal-dominant manner and may be caused by mutations in ankyrin-1, band 3, spectrin, or protein 4.2 on the erythrocyte membrane. It is the third leading cause of hemolytic anemia in newborns and the leading cause of direct Coombs-negative hemolytic anemia requiring blood transfusion in neonates.
- Treatment options for hereditary spherocytosis include phototherapy for jaundiced neonates, folate supplementation, packed erythrocyte transfusions for symptomatic anemia, and recombinant erythropoietin in neonates.